
Home » Posts tagged 'Phase3 drugs' (Page 4)
Tag Archives: Phase3 drugs
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GSK/Isis rare disease drug moves into Phase II/III

feb20,2013
GlaxoSmithKline is paying out $7.5 million to partner Isis Pharmaceuticals as an antisense drug being developed for transthyretin amyloidosis, “a severe and rare genetic disease,” goes into a Phase II/III study.
TTR amyloidosis is characterised by progressive dysfunction of peripheral nerve and/or heart tissues and affects 50,000 patients worldwide and current treatments are limited. The 15-month study of the drug, known as ISIS-TTRRx, will involve some 200 patients with familial amyloid polyneuropathy who experience TTR build-up in their peripheral nerves and experience the loss of motor functions, such as walking.
Lynne Parshall, chief operating officer at Isis, said that “the rapid development of ISIS-TTRRx from a research-stage programme to a drug in late-stage clinical development in just over two years represents the strong commitment of both teams”. She added that the encouraging data from a Phase I study, “in which our drug was well tolerated and produced significant reductions in TTR protein, supported the advancement of ISIS-TTRRx directly into this registration-directed Phase II/III study”.
The drug is part of an Isis-GSK strategic alliance to develop RNA therapeutics for rare and infectious diseases. That pact was recently amended and apart from the $7.5 million fee, Isis is eligible to earn an additional $50 million to support the study, plus regulatory and sales milestone payments, plus double-digit royalties.
AbbVie Announces First Long-term, Patient-Reported Health Outcomes Data for Use of HUMIRA® (Adalimumab) in Patients with Pediatric Crohn’s Disease
Adalimumab
monoclonal antibody
http://www.nature.com/nrd/journal/v2/n9/fig_tab/nrd1182_F1.html
VIENNA, Feb. 15, 2013 AbbVie announced the first long-term, patient-reported health outcomes data from analyses of the Phase 3 IMAgINE-1 trial. The analyses assessed improvements in health-related quality of life (HRQOL) measures for pediatric patients aged 6 to 17 years with severe active Crohn’s disease, taking HUMIRA, who had an inadequate response, were intolerant or had contraindications to conventional therapy, as well as the work productivity of their caregivers throughout the 52-week study. The results of these analyses are being presented this week at the European Crohn’s and Colitis Organisation (ECCO) 8th Annual Congress.
Adalimumab (HUMIRA, Abbott) is the third TNF inhibitor, after infliximab and etanercept, to be approved in the United States. Like infliximab and etanercept, adalimumab binds to Tumor necrosis factor-alpha (TNFα), preventing it from activating TNF receptors. Adalimumab was constructed from a fully human monoclonal antibody, while infliximab is a mouse-human chimeric antibody and etanercept is a TNF receptor-IgG fusion protein. TNFα inactivation has proven to be important in downregulating the inflammatory reactions associated with autoimmune diseases. As of 2008 adalimumab has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, moderate to severe chronic psoriasis and juvenile idiopathic arthritis. Although only approved for ulcerative colitis from late 2012 by the FDA in the disease’s management, it has been used for several years in cases that have not responded to conventional treatment at standard dosing for Crohn’s Disease.

However, because TNFα is part of the immune system that protects the body from infection, prolonged treatment with adalimumab may slightly increase the risk of developing infections.
HUMIRA (“Human Monoclonal Antibody in Rheumatoid Arthritis”) is marketed in both preloaded 0.8 mL syringes and also in preloaded pen devices (called Humira Pen), both injected subcutaneously, typically by the patient at home.
Adalimumab was discovered as a result of the collaboration between BASF Bioresearch Corporation (Worcester, Massachusetts, a unit of BASF) and Cambridge Antibody Technology which began in 1993.[4]
The drug candidate was discovered initially using CAT’s phage display technology and named D2E7.[2] The key components of the drug were found by guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen TNF alpha.[5] The ultimate clinical candidate, D2E7, was created and manufactured at BASF Bioresearch Corporation and taken through most of the drug development process by BASF Knoll, then further development, manufacturing and marketing by Abbott Laboratories, after Abbott acquired the pharmaceutical arm of BASF Knoll.[6]
Adalimumab was the first fully human monoclonal antibody drug approved by the FDA. It was derived from phage display,[1] and was discovered through a collaboration between BASF Bioresearch Corporation (Worcester, Massachusetts, a unit of BASF) and Cambridge Antibody Technology as D2E7,[2] then further manufactured at BASF Bioresearch Corporation and developed by BASF Knoll (BASF Pharma) and, ultimately, manufactured and marketed by Abbott Laboratories after the acquisition of BASF Pharma by Abbott.
In 2009, HUMIRA had over $5 billion in annual sales.[3]

Components of a Humira autoinjector pen
- Brekke OH , Sandlie I (January 2003). “Therapeutic antibodies for human diseases at the dawn of the twenty-first century”. Nat Rev Drug Discov 2 (1): 52–62. doi:10.1038/nrd984. PMID 12509759.
- Kempeni J (January 1999). “Preliminary results of early clinical trials with the fully human anti-TNFα monoclonal antibody D2E7”. Ann Rheum Dis 58 (suppl 1): I70–2. doi:10.1136/ard.58.2008.i70. PMC 1766582. PMID 10577977.
- http://www.abbott.com/static/content/microsite/annual_report/2006/humira.html
- [1] Cambridge Antibody Technology website
- Jespers LS, Roberts A, Mahler SM, Winter G, Hoogenboom HR (September 1994). “Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen”. Biotechnology (N.Y.) 12 (9): 899–903. doi:10.1038/nbt0994-899. PMID 7521646.
- http://www2.basf.us/corporate/news2000/newsknoll_pharma_121500.html
- http://www.prnewswire.com/cgi-bin/stories.pl?ACCT=104&STORY=/www/story/11-12-2001/0001613559&EDATE=
- Rau R (January 2002). “Adalimumab (a fully human anti-tumour necrosis factor α monoclonal antibody) in the treatment of active rheumatoid arthritis: the initial results of five trials”. Ann Rheum Dis 61 (Suppl 2): ii70–3. doi:10.1136/ard.61.suppl_2.ii70. PMC 1766697. PMID 12379628.

Genzyme Announces Positive New Data from Two Phase 3 Studies for Oral Eliglustat Tartrate for Gaucher Disease
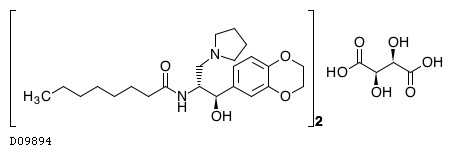
Eliglustat tartrate (USAN)
ENGAGE Study Results:
In ENGAGE, a Phase 3 trial to evaluate the safety and efficacy of eliglustat tartrate in 40 treatment-naïve patients with Gaucher disease type 1, improvements were observed across all primary and secondary efficacy endpoints over the 9-month study period. Results were reported today at the WORLD Symposium by Pramod Mistry, MD, PhD, FRCP, Professor of Pediatrics & Internal Medicine at Yale University School of Medicine, and an investigator in the trial.
The randomized, double-blind, placebo-controlled study had a primary efficacy endpoint of improvement in spleen size in patients treated with eliglustat tartrate. Patients were stratified at baseline by spleen volume. In the study, a statistically significant improvement in spleen size was observed at nine months in patients treated with eliglustat tartrate compared with placebo. Spleen volume in patients treated with eliglustat tartrate decreased from baseline by a mean of 28 percent compared with a mean increase of two percent in placebo patients, for an absolute difference of 30 percent (p<0.0001).
Eliglustat tartate (Genz-112638)
What is Eliglustat?
- Eliglustat is a new investigational phase 3 compound from Genzyme Corporation that is being studied for type 1 Gaucher Disease.
- Eliglustat works as a substrate reduction therapy by reducing glucocerebroside. formation.
- This product is an oral agent (i.e. a pill) that is taken once or twice a day in contrast to an IV infusion for enzyme replacement therapy. Enzyme replacement therapy focuses on replenishing the enzyme that is deficient in Gaucher Disease and breaks down glucocerebroside that accumulates.
- The clinical trials for eliglustat tartate are sponsored by Genzyme Corporation.


Dermasciences-Diabetic foot ulcer Phase 3 trials of DSC-127, Nle3 A(l-7)
DSC-127
feb 2013
Dermasciences-Diabetic foot ulcer Phase 3 trials of DSC-127, Nle3 A(l-7)
University of Southern California
A method for treating a subject that has suffered combined (i) exposure to total body ionizing irradiation and (ii) burns, comprising administering to the subject an amount effective to treat the radiation effects and/or the burn of a peptide comprising at least 5 amino acids of a peptide of SEQ ID NO: l (Asp- Arg-Nle-Tyr-Ile-His-Pro), or a pharmaceutical salt thereof.
WO-2012106427,
A method for treating diabetic foot ulcers, comprising administering to a human patient suffering from a diabetic foot ulcer an amount of a peptide of at least 5 contiguous amino acids of Nle3 A(l-7) effective to treat the diabetic foot ulcer
DSC127 is an analog of a naturally occurring peptide, Angiotensin. It has been shown to increase keratinocyte proliferation, increase extracellular matrix production, and increase vascularization. Additionally, histological examination has shown thatDSC127 accelerated collagen deposition six-fold. All these help to accelerate dermal tissue repair. One potential method of action is the up-regulation of mesenchymal stem cells (MSCs) at the site of injury. MSCs originate in the human embryo and are considered to be multipotent — a type of stem cell that has not yet adopted a specific cellular phenotype. Such cells have the ability to differentiate into various types of cells found within the human body, including fibroblasts, adipose cells, muscle cells, bone cells, and skin cells.
The patented amino acid peptide DSC127 optimizes the well published wound healing capabilities of Angiotensin while removing all blood pressure effects of the compound.
Derma Sciences, under license from the University of Southern California (USC), is developing DSC-127 (USB-001), a topical formulation of an angiotensin analog NorLeu3-A(1-7) that recruits mesenchymal stem cells to the sites of tissue injury, for the potential treatment of diabetic foot ulcer
Wound Repair and Regeneration
Volume 20, Issue 4, pages 482–490, July-August 2012
http://onlinelibrary.wiley.com/doi/10.1111/j.1524-475X.2012.00804.x/full
NorLeu3-A(1–7) stimulation of diabetic foot ulcer healing: …………
Gilead Initiates Phase 3 Clinical Program for Tenofovir Alafenamide,TAF a Novel Low-Dose Prodrug for the Treatment of HIV

Tenofovir Alafenamide
(S)-Isopropyl 2-(((S)-((((R)-1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)(phenoxy)phosphoryl)amino)propanoate
| cas no | 379270-37-8 |
|---|---|
http://www.ama-assn.org/resources/doc/usan/tenofovir-alafenamide.pdf
Gilead Sciences, Inc.on January 24, 2013 announced the initiation of the first of two Phase 3 clinical trials (Study 104) evaluating a single tablet regimen containing tenofovir alafenamide (TAF) for the treatment of HIV-1 infection in treatment-naïve adults. TAF is a novel prodrug of tenofovir, the active agent in Viread® (tenofovir disoproxil fumarate). The Phase 3 studies will examine a once-daily single tablet regimen of TAF 10 mg/elvitegravir 150 mg/cobicistat 150 mg/emtricitabine 200 mg compared to Gilead’s Stribild® (elvitegravir 150 mg/cobicistat 150 mg/emtricitabine 200 mg/tenofovir disoproxil fumarate 300 mg) among patients new to HIV therapy. The second Phase 3 study (Study 111) will be initiated later this quarter.
We are pleased to move TAF into Phase 3 clinical research,” said Norbert Bischofberger, PhD, Executive Vice President, Research and Development and Chief Scientific Officer, Gilead Sciences. “We believe that TAF’s smaller milligram size has the potential to offer safety and tolerability advantages over existing therapies, and may enable the creation of new single tablet regimens for HIV.”
In October 2012, Gilead announced topline results from a Phase 2 study comparing the TAF/elvitegravir/cobicistat/emtricitabine single tablet regimen to Stribild. The study found that the TAF-based regimen met its primary objective based on the proportion of patients with HIV RNA (viral load) levels < 50 copies/mL at 24 weeks of therapy. In addition, statistically significant differences in bone and renal safety were observed between the two arms in favor of the TAF-containing regimen. Both the type and frequency of laboratory abnormalities and adverse events were otherwise comparable between study arms. Full results from the Phase 2 study will be presented at an upcoming medical conference.
Stribild was approved by the U.S. Food and Drug Administration (FDA) in August 2012 and is Gilead’s third single tablet regimen for HIV. A marketing application for Stribild is currently pending in Europe.
Tenofovir alafenamide fumarate (TAF), or GS 7340, is a nucleotide reverse transcriptase inhibitor and a novel prodrug of tenofovir. It is under development by Gilead Sciences for use in the treatment of HIV infection. Closely related to the commonly used reverse-transcriptase inhibitor tenofovir disoproxil fumarate (Viread), TAF has greater antiviral activity and better distribution into lymphoid tissues than that agent.[1][2] Gilead has announced a phase 3 clinical trial evaluating a single-tablet regimen combining GS-7340 with cobicistat, emtricitabine and elvitegravir[3] and plans to coformulate the drug with cobicistat, emtricitabine and the protease inhibitor darunavir.[4][5][6] In a 48 week study comparing Elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate toelvitegravir/cobicistat/emtricitabine/GS 7340, the results showed the prodrug to be non inferior to the established agent, but at much lower dosages and with lower incidence of adverse side effects such as impaired kidney function.[7]
References
- Eisenberg, E. J.; He, G. X.; Lee, W. A. (2001). “Metabolism of Gs-7340, A Novel Phenyl Monophosphoramidate Intracellular Prodrug of Pmpa, in Blood”. Nucleosides, Nucleotides and Nucleic Acids 20 (4–7): 1091–1098. doi:10.1081/NCN-100002496.PMID 11562963. edit
- M Markowitz, A Zolopa, et al. GS-7340 Demonstrates Greater Declines in HIV-1 RNA than Tenofovir Disoproxil Fumarate During 14 Days of Monotherapy in HIV-1 Infected Subjects. 18th Conference on Retroviruses and Opportunistic Infections 2 Mar 2011. Paper # 152LB
- “Gilead Initiates Phase 3 Clinical Program for Tenofovir Alafenamide” (Press release). Gilead. January 2013.
- McQueen, Courtney (16 Nov 2011). “Gilead And Tibotec To Develop Single-Pill Protease Inhibitor-Based Combination Regimen”. The AIDS Beacon.
- GS 7340 Packs Greater HIV Punch, Potentially Better Safety, Versus Viread Horn, Tim. 15 Mar 2012. AIDSmeds.com
- Pharmacokinetics of a Novel EVG/COBI/FTC/GS-7340 Single Tablet Regimen. 13th International Workshop on Clinical Pharmacology of HIV Therapy. Barcelona, Spain. April 16-18, 2012.
- Once-Daily Tenofovir Prodrug Combo Pill as Effective as Stribild. AIDSmeds.com 1 Nov 2012.
New oral anticancer drug, TS-1 (S-1)
New oral anticancer drug, TS-1 (S-1)
http://www.ncbi.nlm.nih.gov/pubmed/11432358
novel oral anticancer agent, S-1, as a combination drug with a molar ratio of 1:0.4:1 for FT, CDHP, and Oxo, respectively
components, an oral fluoropyrimidine agent, tegafur (FT);
a DPD inhibitor (CDHP: 5-chloro-2, 4-dihydroxypyridine) which is about 200-fold more potent than uracil used in UFT;
and
an ORTC inhibitor (Oxo: potassium oxonate) which is localized in the gastrointestinal tract
TS-1 and (TS-), the gastric cancer (oral anti-cancer agent that is used, for example, an anti-cancer agent is a kind of), is classified as an antimetabolite.
Product names, TS- , the common name tegafur, gimeracil, oteracil potassium in, manufacturing, vendors are Taiho Pharmaceutical Co., Ltd
Tegafur (tegafur, international generic name ) is a cancer to thechemotherapy used in the fluorouracil of prodrug is. Tegafur-uracil ( UFTcomponents of). Tegafur is metabolized fluorouracil shows anti-cancer activity

5-fluoro-1-(tetrahydrofuran-2-yl) pyrimidine-2, four (1 H , three H )-dione
Wednesday, 30 January 2013
ASCO GU – Another Breakthrough in Pancreatic Cancer – TS-1 superior to Gemcitabine in improving overall survival in patients with resected Pancreatic Cancer
Roche’s Phase III leukemia drug Obinutuzumab (GA101) yields positive results
- GA101 is the first glycoengineered, type II anti-CD20 mAb.

Roche’s Phase III leukemia drug Obinutuzumab (GA101) yields positive results
Obinutuzumab (GA101)
| Formula | C6512H10060N1712O2020S44 |
|---|
GA101 is the first glycoengineered, type II anti-CD20 monoclonal antibody (mAb) that has been designed for increased antibody-dependent cellular cytotoxicity (ADCC) and Direct CellDeath.1 This agent is being investigated in collaboration with Biogen Idec.
Swiss pharmaceutical company Roche has announced that its early Phase III trial of Leukemia drug obinutuzumab (GA101) demonstrated significantly improved progression-free survival in people with chronic lymphocytic leukemia (CLL).
The positive results yield from stage 1 of a three-arm study called CLL11, designed to investigate the efficacy and safety profile of obinutuzumab (GA101) plus chlorambucil, a chemotherapy, compared with chlorambucil alone in people with previously untreated chronic lymphocytic leukemia (CLL).
This phase of the study met its primary endpoint and an improvement in progression-free survival was achieved; obinutuzumab plus chlorambucil significantly reduced the risk of disease worsening or death compared to chlorambucil alone.
Roche chief medical officer and global product development head Hal Barron said; “the improvement in progression-free survival seen with GA101 is encouraging for people with CLL, a chronic illness of older people for which new treatment options are needed.”
“GA101 demonstrates our ongoing commitment to the research and development of new medicines for this disease.”
Obinutuzumab is Roche’s most advanced drug in development for the treatment of hematological malignancies.
It has been specifically designed as the first glycoengineered, type 2 anti-CD20 monoclonal antibody in development for B cell malignancies.
Afutuzumab is a monoclonal antibody being developed by Hoffmann-La Roche Inc. for the treatment of lymphoma.[1] It acts as an immunomodulator.[2][3] It was renamed obinutuzumab in 2009.[4]
References
- Robak, T (2009). “GA-101, a third-generation, humanized and glyco-engineered anti-CD20 mAb for the treatment of B-cell lymphoid malignancies”. Current opinion in investigational drugs (London, England : 2000) 10 (6): 588–96. PMID 19513948.
- Statement On A Nonproprietary Name Adopted By The Usan Council – Afutuzumab,American Medical Association.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
- International Nonproprietary Names for Pharmaceutical Substances (INN), World Health Organization.
-
obinutuzumab isMonoclonal antibody Type Whole antibody Source Humanized (from mouse) Target CD20

{kind=link}