
Home » Posts tagged 'PHASE 3' (Page 16)
Tag Archives: PHASE 3
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PIRODAVIR



A mixture of 10.4 parts of 3-chloro-6-methylpyridazine, 22.4 parts of ethyl 4-[2-(4-piperidinyl)ethoxy]benzoate butanedioate (1:1), 8.6 parts of sodium carbonate and 0.9 parts of N,N-dimethylformamide was stirred for 3 hours in an oil bath at .+-.150.degree. C. After cooling, water and dichloromethane were added and the layers were separated. The organic layer was dried, filtered and evaporated. The residue was purified by column chromatography over silica gel using a mixture of trichloromethane and ethanol (99:1 by volume) as eluent. The pure fractions were collected and the eluent was evaporated. The residue was crystallized from a mixture of 2,2′-oxybispropane and 2-propanone (75:25 by volume). The precipitated product was filtered off and dried, yielding 17 parts (56.8%) of ethyl 4-[2-[1-(6-methyl-3-pyridazinyl)-4-piperidinyl]-ethoxy]benzoate; mp. 130.1.degree. C. (comp. 1).

Scheme 1. Synthesis of Pirodavir (3) and Related Compounds
| US2985657 * | Oct 12, 1959 | May 23, 1961 | Paul A J Janssen | 1-(aroylalkyl)-4-heterocyclylpiperazines |
| US4068383 * | Sep 30, 1976 | Jan 17, 1978 | Hoechstmass Balzer Gmbh & Co. | Tape measure reel |
| US4451476 * | Oct 17, 1983 | May 29, 1984 | Sterling Drug Inc. | Isoxazoles as antiviral agents |
| US4604127 * | May 15, 1985 | Aug 5, 1986 | Eli Lilly And Company | Herbicidal pyridazinylimidazolidinone compounds |
| EP0137242A2 * | Aug 20, 1984 | Apr 17, 1985 | Sterling Winthrop Inc. | (Substituted) Phenyl-aliphatic-isoxazoles useful as antiviral agents and preparation thereof |
| EP0156433A2 * | Mar 15, 1985 | Oct 2, 1985 | Janssen Pharmaceutica N.V. | Anti-virally active pyridazinamines |
| EP0211457A2 * | Jul 9, 1986 | Feb 25, 1987 | Janssen Pharmaceutica N.V. | Novel (4-substituted-piperazinyl)pyridazines |
| JPS5877866A * | Title not available |
BARDOXOLONE METHYL

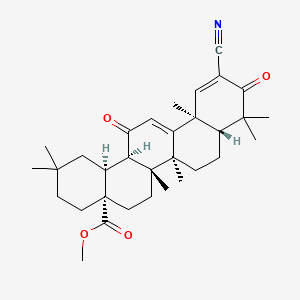
BARDOXOLONE METHYL
- Molecular FormulaC32H43NO4
- Average mass505.688 Da
Methyl 2-cyano-3,12-dioxooleana-1,9(11)dien-28-oate
methyl 2-cyano-3, 12-dioxooleana-1,9(11)-dien-28-oate
2-Cyano-3,12-dioxoolean-1,9(11)-dien-28-oic acid methyl ester
(6aR,6bS,8aR,12aS,14aR,14bS)-11-Cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,3,4,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-hexadecahydropicene-4a(2H)-carboxylic acid methyl ester
BARD
CDDO-Me
Methyl-CDDO
NSC-713200
RTA-402
TP-155C
218600-53-4 CAS
218600-44-3 (free acid)
Treatment of pulmonary arterial hypertension (PAH), diabetic nephropathies and hereditary nephritis, Phase 3

Compounds were synthesized as below:

Scheme 1

Scheme 2
a: HCO2Et/MeONa/THF,b: PhSeCl/AcOEt; 30%H202/THF,c: NH2OH-HCI EtOH/H2O, d: MeONa/MeOH/Et2O,e: KOH/MeOH,f: Jones,g:HCO2Et/MeONa/PhH,h: Lil/DMF Compound 10 was prepared by formylation of OA (Compound 9) (Simonsen and Ross, 1957) with ethyl formate in the presence of sodium methoxide in THF (Clinton et al., 1961). Compound 7 was obtained by introduction of a double bond at C-l of Compound 10 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30%) hydrogen peroxide (Sharpless et al, 1973). Compound 11 was synthesized from Compound 10 by addition of hydroxylamine in aqueous ethanol; cleavage of Compound 11 with sodium methoxide gave Compound 12 (Johnson and Shelberg, 1945). Compound 14 was prepared from Compound 13 (Picard et al, 1939) by alkali hydrolysis followed by Jones oxidation. Compound 15 was prepared by formylation of Compound 14 with ethyl formate in the presence of sodium methoxide in benzene. Compound 16 was synthesized from Compound 15 by addition of hydroxylamine. Cleavage of 16 with sodium methoxide gave Compound 17. Compound 6 (CDDO) was prepared by introduction of a double bond at C-l of Compound 17 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30% hydrogen peroxide, followed by halogenolysis with lithium iodide in DMF (Dean, P.D.G., 1965).
Bardoxolone methyl (also known as “RTA 402” and “CDDO-methyl ester”) is an orally-available first-in-class synthetic triterpenoid. It is an inducer of the Nrf2 pathway, which can suppress oxidative stress and inflammation, and is undergoing clinical development for the treatment of advanced chronic kidney disease (CKD) in type 2 diabetes mellitus patients.
Bardoxolone methyl was previously being investigated by Reata Pharmaceuticals, Inc. in partnership with Abbott Laboratories and Kyowa Hakko Kirin, as an experimental therapy for advanced chronic kidney disease (CKD) in type 2 diabetes mellitus patients. Reata, in consultation with the BEACON Steering Committee, has decided to terminate the Phase 3 BEACON trial of bardoxolone methyl in patients with stage 4 chronic kidney disease and type 2 diabetes. This decision was made based upon a recommendation of the Independent Data Monitoring Committee (IDMC) to stop the trial “for safety concerns due to excess serious adverse events and mortality in the bardoxolone methyl arm.” [1][2][3][4]
RTA-402 is a triterpenoid anti-inflammatory agent in phase II trials at Reata Pharmaceuticals for the treatment of pulmonary arterial hypertension.
This company and M.D. Anderson Cancer Center had been evaluating clinically the product for the treatment of lymphoma. Reata had been evaluating the compound in combination with gemcitabine in patients with unresectable pancreatic cancer and melanoma. Preclinical studies were also being conducted by Reata for the treatment of inflammatory bowel disease (IBD) and autoimmune disease. Reata Pharmaceuticals and Kyowa Hakko Kirin had been conducting phase II clinical studies for the treatment of diabetic nephropathy. Reata and Abbott also had been conducting phase III clinical trials for delaying progression to end-stage renal disease in patients with chronic kidney disease and type 2 diabetes; however, in 2012 these trials were discontinued due to serious adverse events and mortality. Phase II clinical trials for this indication were discontinued by Kyowa Hakko Kirin in Japan. The compound had been in early clinical studies for the treatment of multiple myeloma; however, no recent development has been reported for this indication. Phase I clinical trials for the treatment of solid tumors have been completed.
RTA-402 has demonstrated a wide variety of potentially therapeutic mechanisms, including inhibition of inducible nitric oxide synthase and cyclooxygenase expression, stimulation of expression of cytoprotective enzymes such as NAD(P)H quinine oxidoreductase and hemeoxygenase-1, and reduction in pSTAT3 levels. In cancer patients, the drug candidate exploits fundamental physiological differences between cancerous and non-cancerous cells by modulating oxidative stress response pathways. Due to this mechanism, RTA-402 is toxic to cancer cells, but induces protective antioxidant and anti-inflammatory responses in normal cells. In previous studies, the compound was shown to inhibit growth and cause regression of cancerous tumors as a single agent and, in combination with radiation and chemotherapy, to suppress radiation and chemotherapy-induced toxicities in normal tissues and cause minimal toxicity in non-human primates when dosed orally at very high doses for 28 consecutive days.
An analog of RTA-401, RTA-402 is a compound found in medicinal plants with a greater potency than the natural product.
RTA-401 was originally developed at Dartmouth College and M.D. Anderson Cancer Center. In November 2004, Reata completed a license agreement with these organizations, and was granted exclusive worldwide rights to this new class of anticancer compounds. In 2008, orphan drug designation was assigned by the FDA for the treatment of pancreatic cancer. In 2010, the compound was licensed to Kyowa Hakko Kirin by Reata Pharmaceuticals in China, Japan, Korea, Thailand and Southeast Asian countries for the treatment of chronic kidney disease. Abbott acquired rights to develop and commercialize the drug outside US, excluding certain Asian markets.
Phase 1
Bardoxolone methyl was first advanced into the clinic to assess its anticancer properties. In two Phase 1 trials that included 81 oncology patients, bardoxolone methyl reduced serum creatinine levels, with a corresponding improvement in estimated glomerular filtration rate (eGFR). Improvements were more pronounced in a subset of patients with established CKD and were maintained over time in patients who continued on bardoxolone methyl therapy for 5 months. Based on these observed effects and the well-described role of oxidative stress and inflammation in CKD, especially in type 2 diabetes, it was hypothesized that bardoxolone methyl could improve renal function in CKD patients with type 2 diabetes.[5]
Phase 2
A multi-center, double-blind, placebo-controlled Phase 2b clinical trial (BEAM) conducted in the US studied 227 patients with moderate to severe CKD (eGFR 20 – 45 ml/min/1.73m²) and type 2 diabetes. The primary endpoint was change in estimated GFR following 24 weeks of treatment. Following 24 weeks, patients treated with bardoxolone methyl experienced a mean increase in estimated GFR of over 10 ml/min/1.73m², compared with no change in the placebo group. Approximately three-quarters of bardoxolone methyl treated patients experienced an improvement in eGFR of 10 percent or more, including one-quarter who saw a significant improvement of 50% or more compared to less than 2% of patients on placebo. Adverse events were generally manageable and mild to moderate in severity. The most frequently reported adverse event in the bardoxolone methyl group was muscle spasm. Final data was published in The New England Journal of Medicine.
Concerns have been raised whether there is a true improvement in kidney function because of the significant weight loss of the patients in the active-treatment-group that ranged from 7.7-10.1 kg (7-10% of the initial body weight) and whether this weight loss in patients receiving bardoxolone included muscle wasting with a commensurate decrease in the serum creatinine level. In that case the decrease in creatinine would not necessarily be a true improvement in kidney function.[6][7][8][9][10]
Phase 3
A multinational, double-blind, placebo-controlled Phase 3 outcomes study (BEACON) was started in June 2011, testing bardoxolone methyl’s impact on progression to ESRD or cardiovascular death in 1600 patients with Stage 4 CKD (eGFR 15 – 30 ml/min/1.73m²) and type 2 diabetes. This phase 3 trail was halted in October 2012 because of adverse effects (namely a higher cardiovascular mortality in the treatment arm).[11]
Mechanism of action
Bardoxolone methyl is an inducer of the KEAP1–Nrf2 pathway.
PAPER
http://modernsteroid.blogspot.com/2012/04/synthetic-oleane-triterpenoids-as.html


Click to access ol400399x_si_001.pdf

1. To a stirred solution of oleanolic acid (22.8 grams, 0.05 mol, 1.0 equiv) in dimethyl formamide (200 mL) was
added powdered K2CO3 (20.7 grams, 0.15 mol, 3.0 equiv) slowly upon stirring, and the reaction mixture was allowed to
cool to 0 o
C. To the stirred suspension was added iodomethane (3.4 mL, 0.055 mol, 1.1 equiv) slowly, and after the
completion of addition, the reaction was allowed to warm to room temperature overnight. After the completion of the
reaction, dimethyl formamide was removed by distillation. The resulting solid mixture was dissolved in methylene
chloride (1 L) and washed with water (4 x 100 mL) and brine (1 x 100 mL). The organics was dried over Na2SO4 and the
solvent was removed to give the crude product 8 as a white solid, which was used directly for the next step without
further purifications.
2. To a stirred suspension of ester 8 (11.8 grams, 0.025 mol, 1.0 equiv) obtained above in anhydrous dimethyl
sulfoxide (250 mL) was added iodoxybenzoic acid (21.0 grams, 0.075 mol, 3.0 equiv) and fluorobenzene (5 mL). The
resulting suspension was heated to 85 o
C under nitrogen for 24 hours. After the completion of the reaction, it was
quenched with 20% aqueous sodium thiosulfate (200 mL). The resulting mixture was extracted with methylene chloride
(4 x 150 mL), the combined organic extracts were washed with saturated NaHCO3 (100 mL) and brine (100 mL), and
dried over Na2SO4. The solvent was removed to give the crude product 14 as yellowish solid, which was used directly for
the next step without further purifications.
3. To a stirred solution of 14 (9.32 grams, 0.02 mol, 1.0 equiv) in methylene chloride (100 mL) was slowly added mchloroperbenzoic
acid (6.4 grams, ~70% purity, 0.026 mol, 1.3 equiv) at 0 o
C. After the completion of addition, the
reaction was allowed to warm to room temperature and kept stirring for 24 hours. After the completion of the reaction,
the reaction mixture was diluted with methylene chloride (300 mL), and the resulting mixture was washed with 20%
aqueous sodium thiosulfate (3 x 100 mL), 10% potassium carbonate (2 x 100 mL), and brine (100 mL). The organics were
dried over Na2SO4 and the solvent was removed to give crude mixture of 15 and 16 as yellowish solid, which was used
directly for the next step without purifications.
4. To the resulting solution of 15 and 16 obtained above in acetic acid (50 mL) was added dropwise hydrobromic
acid (1.0 mL, 0.009 mol, 0.44 equiv) at room temperature. The reaction mixture was then heated to 35 o
C, and bromine
(5.8 mL, 0.05 mol, 2.4 equiv) was thus added dropwise. The resulting reaction mixture was kept stirring for another 24 h.
After completion of the reaction, the acid was removed under vacuum. And the residue was then quenched with 20%
aqueous sodium thiosulfate (100 mL), and extracted with methylene chloride (4 x 100 mL). The combined organic
extracts were washed with saturated sodium bicarbonate (2 x 50 mL), brine (1 x 50 mL), and dried over Na2SO4. The
solvent was removed to give crude bromo enone 17 as yellowish to yellow solid, which can be used directly for the next
step without further purification or subjected to flash column chromatography to give pure bromo enone 17 as a
yellowish solid.
5. To a stirred solution of bromo enone 17 (5.8 grams, 10.0 mmol, 1.0 equiv) in anhydrous dimethyl formamide (80
mL) was added copper (I) cyanide (1.0 grams, 11.0 mmol, 1.1 equiv) and potassium iodide (328 mg, 2.0 mmol, 0.20
equiv), and the resulting reaction mixture was heated to 120 o
C for 24 h. After the completion of reaction, it was cooled
to room temperature, quenched with water (200 mL), and diluted with ethyl acetate (500 mL). The organic phase was
washed with saturated NaHCO3 (2 x 80 mL), brine (80 mL), and dried over Na2SO4. Removal of solvent and flash column
chromatography over silica gel using hexanes:EtOAc (2:1) to give bardoxolone methyl (1) as a yellowish solid.
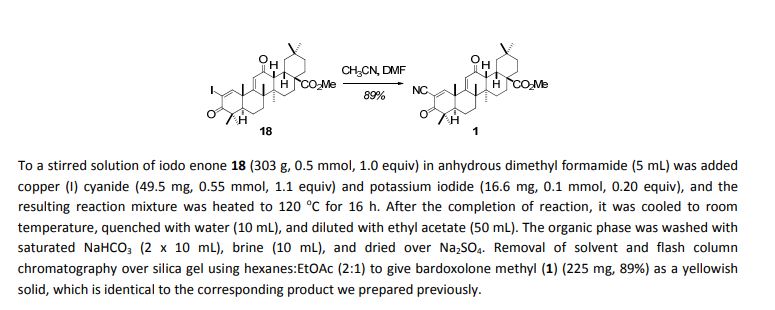
After the completion of the reaction, it was cooled to room temperature and
quenched with 20% aqueous sodium thiosulfate (20 mL). It was extracted with methylene chloride (3 x 20 mL), the
combined organic extracts were washed with saturated aqueous NaHCO3 (10 mL), brine (10 mL), and dried over Na2SO4.
Removal of solvent and flash column chromatography over silica gel using hexanes:EtOAc (4:1 & 2:1) to give iodo enone
18 (509 mg, 84%) as a yellowish solid. 1H NMR (500 MHz, CDCl3) δ 8.12 (s, 1H), 6.00 (s, 1H), 3.70 (s, 3H), 3.04 (dd, 1H, J1 =
10.0 Hz, J2 = 3.7 Hz), 2.92 (d, 1H, J = 4.6 Hz), 1.63-1.94 (m, 9H), 1.46-1.62 (m, 3H), 1.43 (s, 3H), 1.18-1.36 (m, 3H), 1.30 (s,
3H), 1.23 (s, 3H), 1.17 (s, 3H), 1.02 (s, 3H), 1.00 (s, 3H), 0.90 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 199.6, 196.9, 178.4,
170.3, 163.5, 124.1, 102.3, 52.1, 49.9, 48.4, 47.4, 46.4, 45.9, 45.4, 42.3, 36.0, 34.7, 33.5, 33.0, 32.1, 31.7, 30.9, 28.3, 28.2,
27.3, 24.8, 23.3, 22.9, 22.4, 21.9, 18.8; FT-IR (solution, CDCl3, cm-1): 2952, 2869, 2253, 1717, 1659, 1469, 1386, 907, 732,
651, 623, 443; HRMS-ESI (calcd. for C31H44IO4 [M+H]+
) 607.2284, found 607.2280.
CLIP

PATENT
In a preferred embodiment, such compounds include derivatives of ursolic acid and oleanoic acid. In a particularly preferred embodiment, derivatives of OA, e.g., 2-cyano-3,12-dioxoolean-l,9-dien-28oic acid (CDDO):

have been found to be effective in suppression of human breast cancer cell growth, and highly potent in many vitro assay systems such as: suppression of nitric oxide and prostaglandin production in macrophages, inhibition of growth of human breast cancer cells, suppression of nitric oxide formation in rat prostate cells, and suppression of prostaglandin formation in human colon fibroblasts, as detailed in the Figures.
Compounds were synthesized as below:
Scheme 1
Scheme 2
a: HCO2Et/MeONa/THF,b: PhSeCl/AcOEt; 30%H202/THF,c: NH2OH-HCI EtOH/H2O, d: MeONa/MeOH/Et2O,e: KOH/MeOH,f: Jones,g:HCO2Et/MeONa/PhH,h: Lil/DMF Compound 10 was prepared by formylation of OA (Compound 9) (Simonsen and Ross, 1957) with ethyl formate in the presence of sodium methoxide in THF (Clinton et al., 1961). Compound 7 was obtained by introduction of a double bond at C-l of Compound 10 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30%) hydrogen peroxide (Sharpless et al, 1973). Compound 11 was synthesized from Compound 10 by addition of hydroxylamine in aqueous ethanol; cleavage of Compound 11 with sodium methoxide gave Compound 12 (Johnson and Shelberg, 1945). Compound 14 was prepared from Compound 13 (Picard et al, 1939) by alkali hydrolysis followed by Jones oxidation. Compound 15 was prepared by formylation of Compound 14 with ethyl formate in the presence of sodium methoxide in benzene. Compound 16 was synthesized from Compound 15 by addition of hydroxylamine. Cleavage of 16 with sodium methoxide gave Compound 17. Compound 6 (CDDO) was prepared by introduction of a double bond at C-l of Compound 17 with phenylselenenyl chloride in ethyl acetate and sequential addition of 30% hydrogen peroxide, followed by halogenolysis with lithium iodide in DMF (Dean, P.D.G., 1965).
PATENT
WO2009/146216 A2,

Compounds 401, 402, 404, 402-04, 402-35 and 402-56 can be prepared according to the methods taught by Honda et al. (1998), Honda et al. (2000b), Honda et al. (2002), Yates et al. (2007), and U.S. Patent 6,974,801, which are all incorporated herein by reference. The synthesis of the other compounds are disclosed in the following applications, each of which is incorporated herein by reference: U.S. Application Nos. 61/046,332, 61/046,342, 61/046,363, 61/046,366, 61/111,333, 61/111,269, and 61/111,294. The synthesis of the other compounds are also disclosed in the following separate applications filed concurrently herewith, each of which is incorporated herein by reference in their entireties: U.S. Patent Application by Eric Anderson, Xin Jiang, Xiaofeng Liu; Melean Visnick, entitled “Antioxidant Inflammation Modulators: Oleanolic Acid Derivatives With Saturation in the C- Ring,” filed April 20, 2009; U.S. Patent Application by Eric Anderson, Xin Jiang and Melean Visnick, entitled “Antioxidant Inflammation Modulators: Oleanolic Acid Derivatives with Amino and Other Modifications At C-17,” filed April 20, 2009; U.S. Patent Application by Xin Jiang, Xioafeng Liu, Jack Greiner, Stephen S. Szucs, Melean Visnick entitled, “Antioxidant Inflammation Modulators: C-17 Homologated Oleanolic Acid Derivatives,” filed April 20, 2009.
PAPER
Chemical Communications, 2011 , vol. 47, 33 p. 9495 – 9497
http://pubs.rsc.org/en/Content/ArticleLanding/2011/CC/c1cc11633a#!divAbstract
http://www.rsc.org/suppdata/cc/c1/c1cc11633a/c1cc11633a.pdf NMR GIVEN
2-Cyano-3,12-dioxooleana-1,9(11)-dien-28-oate (CDDO)
A mixture of 1 (0.25 g, 0.51 mmol) and DDQ (0.12 g, 0.51 mmol) in anhydrous benzene (20 mL) was
refluxed for 15 min. After filtration, the filtrate was evaporated in vacuo to give a residue, which was
subjected to flash column chromatography (petroleum ether/EtOAc) to give CDDO as an amorphous
solid (0.23 g, 91%). The title compound was known as CAS 218600-44-3
m.p. 180-182 °C;
ESI-MS: 490 [M-H]-, 492 [M+H]+;
1H NMR (300M Hz, CDCl3, 25 °C, TMS): δ 8.05 (1H, s), 5.99 (1H, s), 3.03-2.98 (2H, m), 1.55,1.38,
1.34, 1.22, 1.00, 0.91, 0.85 (each 3H,s ,CH3) ppm.
PAPER
SYNTHESIS
Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
http://pubs.acs.org/doi/full/10.1021/jm0002230

Bioorganic and Medicinal Chemistry Letters, 1998 , vol. 8, 19 p. 2711 – 2714
http://www.sciencedirect.com/science/article/pii/S0960894X9800479X

PAPER
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, # 9 p. 2215 – 2219
http://www.sciencedirect.com/science/article/pii/S0960894X05003306

PATENT
Method of synthesis of CDDO. CDDO may be synthesized by the scheme outlined below.

Methyl-CDDO. Methyl-CDDO (CDDO-Me), the C-28 methyl ester of CDDO, also exerts strong antiproliferative and apoptotic effects on leukemic cell lines and in primary AML samples in vitro as well as induces monocytic differentiation of leukemic cell lines and some primary AMLs. Thus, CDDO-Me provides chemotherapy for the treatment of leukemias. The present invention demonstrates that this effect is profoundly increased by combination of CDDO-Me with other chemotherapeutic agents. These include retinoids such as ATRA, 9-cis retinoic acid, , LG100268, LGD1069 (Targretin, bexarotene), fenretinide [N-(4- hydroxyphenyl)retinamide, 4-HPR], CD437 and other RXR and RAR-specific ligands. This combination also increases ara-C cytotoxicity, further reduces AML colony formation, inhibits ERK phosphorylation and promotes Bcl-2 dephosphorylation, and inhibits in vitro angiogenesis. The ability of CDDO-Me in combination with retinoids to induce differentiation in leukemic cells in vitro show that these compounds may have similar in vivo effects. The anti-angiogenic properties of CDDO-Me further increase its potent anti-leukemia activity in combination with retinoids. Furthermore, CDDO-Me was found to be more potent at lower concentrations than CDDO.
Method of synthesis of CDDO-Me.
CDDO-Me may be synthesized by the scheme outlined below.

The present invention provides combinations of CDDO-compounds and chemotherapeutic agents that are useful as treatments for cancers and hematological malignancies. In one embodiment, the chemotherapeutics are retinoids. As CDDO- compounds are PPARγ ligands and PPARγ is known to be altered in many types of cancers, the inventors contemplate, that ligation of PPARγ in combination with retinoids such as, RXR-specific ligands, provides a mechanistic basis for maximal increase in transcriptional activity of the target genes that control apoptosis and differentiation. The CDDO-compounds and retinoids in combination demonstrate an increased ability to induce differentiation, induce cytotoxicity, induce apoptosis, induce cell killing, reduce colony formation and inhibit the growth of several types of leukemic cells.
PAPER
Efficient and scalable synthesis of bardoxolone methyl (cddo-methyl ester).
Bardoxolone methyl (2-cyano-3,12-dioxooleane-1,9(11)-dien-28-oic acid methyl ester; CDDO-Me) (1), a synthetic oleanane triterpenoid with highly potent anti-inflammatory activity (levels below 1 nM), has completed a successful phase I clinical trial for the treatment of cancer and a successful phase II trial for the treatment of chronic kidney disease in type 2 diabetes patients. Our synthesis of bardoxolone methyl (1) proceeds in ∼50% overall yield in five steps from oleanolic acid (2), requires only one to two chromatographic purifications, and can provide gram quantities of 1.

References
- “Bardoxolone methyl – Oral, Once Daily AIM for Renal/Cardiovascular/Metabolic Diseases”. Reata Pharmaceuticals. Archived from the original on 15 July 2011. Retrieved June 2, 2011.
- “Abbott and Reata Pharmaceuticals Announce Agreement to Develop and Commercialize Bardoxolone Methyl for Chronic Kidney Disease Outside the U.S.” (Press release). Reata Pharmaceuticals. September 23, 2010. Retrieved June 2, 2011.
- “Reata Pharmaceuticals Licenses Chronic Kidney Disease Drug Bardoxolone Methyl to Kyowa Hakko Kirin”(Press release). Reata Pharmaceuticals. January 7, 2010. Retrieved June 2, 2011.
- “Company Statement: Termination of Beacon Trial”.Reata Pharmaceuticals. Retrieved October 18, 2012.
- Pergola, P. E.; Krauth, M.; Huff, J. W.; Ferguson, D. A.; Ruiz, S.; Meyer, C. J.; Warnock, D. G. (2011). “Effect of Bardoxolone Methyl on Kidney Function in Patients with T2D and Stage 3b–4 CKD”. American Journal of Nephrology 33 (5): 469–476. doi:10.1159/000327599. PMID 21508635.
- Pergola, P. E.; Raskin, P.; Toto, R. D.; Meyer, C. J.; Huff, J. W.; Grossman, E. B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; Wittes, J.; Warnock, D. G.; Beam Study, I. (2011). “Bardoxolone Methyl and Kidney Function in CKD with Type 2 Diabetes” (pdf). New England Journal of Medicine 365 (4): 327–336.doi:10.1056/NEJMoa1105351. PMID 21699484. edit
- van Laecke, S.; Vanholder, R. (2011). “Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1745, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047578.
- Rogacev, K. S.; Bittenbring, J. T.; Fliser, D. (2011).“Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1745–1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047579.
- Upadhyay, A.; Sarnak, M. J.; Levey, A. S. (2011).“Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047580.
- McMahon, G. M.; Forman, J. P. (2011). “Communication: Bardoxolone methyl, chronic kidney disease, and type 2 diabetes”. New England Journal of Medicine 365 (18): 1746, author reply 1746–1747.doi:10.1056/NEJMc1110239. PMID 22047581.
- ClinicalTrials.gov NCT01351675 Bardoxolone Methyl Evaluation in Patients With Chronic Kidney Disease and Type 2 Diabetes (BEACON)
- Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages
Bioorg Med Chem Lett 1998, 8(19): 2711 - Novel synthetic oleanate triterpenoids: A series of highly active inhibitors of nitric production in mouse macrophages
Bioorg Med Chem Lett 1999, 9(24): 3429 - WO 1999065478
- WO 2013169553
- CN 102875634
- US 2012330050
- US 2012071684
- WO 2011130302
- WO 2010093944
- WO 2009089545
- WO 2009023232
- WO 2008111497
- Anderson, Amy C.; Browning, R. Greg; Couch, Robin D.; Gribble, Gordon W.; Honda, Tadashi; Wright, Dennis L.; Sporn, Michael B.
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, 9 p. 2215 – 2219 - Journal of Medicinal Chemistry, 2004 , vol. 47, 20 p. 4923 – 4932
- Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
- Bioorganic and Medicinal Chemistry Letters, 2002 , vol. 12, 7 p. 1027 – 1030
- Journal of Medicinal Chemistry, 2000 , vol. 43, 22 p. 4233 – 4246
- Chemical Communications, 2011 , vol. 47, 33 p. 9495 – 9497
- Reata Pharmaceuticals, Inc. – bardoxolone methyl
- Bardoxolone Methyl and Kidney Function in CKD with Type 2 Diabetes
- Effect of Bardoxolone Methyl on Kidney Function in Patients with T2D and Stage 3b – 4 CKD
- American Diabetes Association presentation
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8440854 * | Jan 23, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino acid and other modifications at C-17 |
| US8513436 | Dec 19, 2011 | Aug 20, 2013 | Reata Pharmaceuticals, Inc. | Pyrazolyl and pyrimidinyl tricyclic enones as antioxidant inflammation modulators |
| WO2002047611A2 * | Nov 28, 2001 | Jun 20, 2002 | Univ Texas | Cddo-compounds and combination therapies thereof |
| WO2008064132A2 * | Nov 16, 2007 | May 29, 2008 | Dartmouth College | Synthetic triterpenoids and tricyclic-bis-enones for use in stimulating bone and cartilage growth |
| WO2009118441A1 * | Feb 12, 2009 | Oct 1, 2009 | Consejo Superior De Investigaciones Cientifícas | Use of pentacyclic triterpene for the preparation of a pharmaceutical compound intended for the treatment of multiple sclerosis |
| WO2013083659A1 | Dec 5, 2012 | Jun 13, 2013 | Cambridge Enterprise Limited | Combination treatment comprising ho – 1 inhibitor and immunotherapeutic agent |
| US7176237 | Jan 15, 2003 | Feb 13, 2007 | The Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US7435755 | Nov 28, 2001 | Oct 14, 2008 | The Trustees Of Dartmouth College | CDDO-compounds and combination therapies thereof |
| US7678830 | Feb 7, 2007 | Mar 16, 2010 | Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US7714012 | Nov 16, 2007 | May 11, 2010 | Trustees Of Dartmouth University | Synthesis and biological activities of new tricyclic-bis-enones (TBEs) |
| US7795305 | Oct 10, 2008 | Sep 14, 2010 | Board Of Regents, The University Of Texas System | CDDO-compounds and combination therapies thereof |
| US7863327 | May 3, 2005 | Jan 4, 2011 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
| US7915402 | Apr 20, 2009 | Mar 29, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US7943778 | Apr 20, 2009 | May 17, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: C-17 homologated oleanolic acid derivatives |
| US8034955 | Oct 29, 2007 | Oct 11, 2011 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
| US8067394 | May 10, 2010 | Nov 29, 2011 | Trustees Of Dartmouth College | Synthesis and biological activities of new tricyclic-bis-enones (TBEs) |
| US8067465 | Mar 11, 2010 | Nov 29, 2011 | The Trustees Of Dartmouth College | Tricyclic-bis-enone derivatives and methods of use thereof |
| US8071632 | Apr 20, 2009 | Dec 6, 2011 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: novel derivatives of oleanolic acid |
| US8124656 | Feb 23, 2011 | Feb 28, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US8124799 | Apr 20, 2009 | Feb 28, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino and other modifications at C-17 |
| US8129429 | Jan 12, 2009 | Mar 6, 2012 | Reata Pharmaceuticals, Inc. | Synthetic triterpenoids and methods of use in the treatment of disease |
| US8258329 | Apr 20, 2009 | Sep 4, 2012 | Reata Pharmaceuticals, Inc. | Dehydroandrosterone analogs including an anti-inflammatory pharmacore and methods of use |
| US8299046 | Nov 16, 2007 | Oct 30, 2012 | Trustees Of Dartmouth College | Synthetic triterpenoids and tricyclic-bis-enones for use in stimulating bone and cartilage growth |
| US8314137 | Jul 22, 2009 | Nov 20, 2012 | Trustess Of Dartmouth College | Monocyclic cyanoenones and methods of use thereof |
| US8338618 | Nov 11, 2011 | Dec 25, 2012 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: novel derivatives of oleanolic acid |
| US8394967 | Feb 23, 2011 | Mar 12, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: C-17 homologated oleanolic acid derivatives |
| US8440820 | Jan 11, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with saturation in the C-ring |
| US8440854 | Jan 23, 2012 | May 14, 2013 | Reata Pharmaceuticals, Inc. | Antioxidant inflammation modulators: oleanolic acid derivatives with amino acid and other modifications at C-17 |
| US8455544 | Jan 26, 2012 | Jun 4, 2013 | Reata Pharmaecuticals, Inc. | Synthetic triterpenoids and methods of use in the treatment of disease |
| US8513436 | Dec 19, 2011 | Aug 20, 2013 | Reata Pharmaceuticals, Inc. | Pyrazolyl and pyrimidinyl tricyclic enones as antioxidant inflammation modulators |
| US8586775 | Aug 24, 2011 | Nov 19, 2013 | Trustees Of Dartmouth College | Therapeutic compounds and methods of use |
 |
| Professor Honda received his B.S. degree in Chemistry in 1974, his M.S. degree in Organic Chemistry in 1976, and his Ph.D. in Organic Chemistry in 1979 from the University of Tokyo. In 1979, he joined the Department of Drug Discovery Chemistry at Suntory Institute for Biomedical Research in Japan and worked there as a drug synthetic chemist (finally senior researcher) for 13 years. In 1991, he joined the Central Pharmaceutical Research Institute at Japan Tobacco Inc. and worked as a chief senior researcher for 3 years. In 1995, he joined Dr. Gribble’s laboratory at Dartmouth College as a research associate. In 1998, he joined the research faculty of Dartmouth College. In 2005, he was promoted to Research Associate Professor. |
Dr. Honda and his collaborators have further explored new structures based on CDDO and different five-ringed triterpenoids.
During the course of these investigations, Dr. Honda has designed three-ringed compounds with similar enone functionalities in rings A and C to those of CDDO, but having a much simpler structure than five-ringed triterpenoids. He and his collaborators have found that they are also a novel class of potent anti-inflammatory, cytoprotective, growth suppressive, and pro-apoptotic compounds. Amongst such three-ringed compounds, TBE-31 with the C-8a ethynyl group is much more potent than CDDO in various bioassays in vitro and in vivo. Thus, further investigation (design, synthesis, biological evaluation, etc.) of new TBE-31 analogues is currently being performed in order to discover analogues having different and/or better features than TBE-31, for example, higher potency and lower toxicity, better bioavailability and different distributions in organs, high water-solubility and so on.

Mechanism studies suggest that CDDO regulates various molecules regarding inflammation, differentiation, apoptosis, and proliferation by reversible Michael addition between the cyano enone functionality of CDDO and the sulfhydryl groups of cysteine moieties on these molecules. Based on this fact and the structure of TBE-31, Dr. Honda has designed single-ringed compounds, which represent the ideal simple structure. The synthesis of these new compounds is currently in progress.

 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| ChEMBL | |
| ECHA InfoCard | 100.132.153 |
| Chemical and physical data | |
| Formula | C32H43NO4 |
| Molar mass | 505.69 g/mol |
| 3D model (JSmol) | |
///////////////Bardoxolone Methyl, CDDO-Me; CDDO methyl ester; 218600-53-4; Bardoxolone (methyl); RTA 402 CDDO-Me, CDDO methyl ester, 218600-53-4, Bardoxolone (methyl), RTA 402 , PHASE 3,NSC 713200
CC1(CCC2(CCC3(C(C2C1)C(=O)C=C4C3(CCC5C4(C=C(C(=O)C5(C)C)C#N)C)C)C)C(=O)OC)C
Actelion’s novel antibiotic Cadazolid receives US FDA Qualified Infectious Disease Product designation for the treatment of Clostridium difficile-associated diarrhea .

CADAZOLID, ACT-179811
1-Cyclopropyl-6-fluoro-7-[4-({2-fluoro-4-[(5R)-5-(hydroxymethyl)-2-oxo-1,3-oxazolidin-3-yl]phenoxy}methyl)-4-hydroxypiperidin-1-yl]-4-oxo-1,4-dihydroquinolin-3-carboxylic acid
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-(R)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid
| Formula | C29H29F2N3O8 |
|---|---|
| Mol. mass | 585.55 g/mol |
Actelion Pharmaceuticals Ltd / Actelion’s novel antibiotic cadazolid receives US FDA Qualified Infectious Disease Product designation for the treatment of Clostridium difficile-associated diarrhea .
ALLSCHWIL/BASEL, SWITZERLAND – 27 February 2014 – Actelion Ltd (six:ATLN) today announced that the US Food and Drug Administration (FDA) has designated cadazolid as both a Qualified Infectious Disease Product (QIDP) and a Fast Track development program for the treatment of Clostridium difficile-associated diarrhea (CDAD).
The QIDP designation for cadazolid means that – among other incentives – cadazolid would receive a nine-month priority review upon successful completion of the ongoing global Phase III IMPACT program. The Fast Track designation is intended to promote communication and collaboration between the FDA and the Company on the development of the drug.
The designations are based on the 2012 US Generating Antibiotic Incentives Now (GAIN) Act. The GAIN act is a legislative effort to incentivize the development of new antibiotic agents that target serious life-threatening infections.
Guy Braunstein, M.D. and Head of Clinical Development commented: “Clostridium difficile-associated diarrhea is a very serious and potentially life-threatening infection. There is a great need for an antibiotic that allows effective treatment of CDAD with low recurrence rates, particularly in infections caused by hypervirulent strains. The GAIN act highlights the importance of research in this area and we are very happy to receive the advantages that this designation for cadazolid will afford us.”
ABOUT THE IMPACT PROGRAM
IMPACT is an International Multi-center Program Assessing Cadazolid Treatment in patients suffering from Clostridium difficile-associated diarrhea (CDAD). The program comprises two Phase III studies comparing the efficacy and safety of cadazolid (250 mg administered orally twice daily for 10 days) versus vancomycin (125 mg administered orally four times daily for 10 days).
The IMPACT studies are designed to determine whether the clinical response after administration of cadazolid is non-inferior to vancomycin in subjects with CDAD, and whether administration of cadazolid is superior to vancomycin in the sustained clinical response. The program is expected to enroll approximately 1’280 subjects worldwide, and commenced enrollment in the fourth quarter of 2013.
ABOUT CADAZOLID
The novel antibiotic cadazolid is a strong inhibitor of Clostridium difficile protein synthesis leading to strong suppression of toxin and spore formation. In preclinical studies cadazolid showed potent in vitro activity against Clostridium difficile clinical isolates and a low propensity for resistance development. In a human gut model of CDAD, cadazolid had a very limited impact on the normal gut microflora.
Cadazolid absorption is negligible resulting in high gut lumen concentrations and low systemic exposure, even in severe cases of CDAD where the gut wall can be severely damaged and permeability to drugs potentially increased.
Cadazolid is an experimental antibiotic of the oxazolidinone class made by Actelion Pharmaceuticals Ltd. which is effective against Clostridium difficile, a major cause of drug resistant diarrhea in the elderly.[1] Current drug treatments for this infection involve orally delivered antibiotics, principally fidaxomicin, metronidazole and vancomycin; the last two drugs are the principal therapeutic agents in use, but fail in approximately 20 to 45% of the cases. The drug is presently in Phase III trials.[1] The drug works by inhibiting synthesis of proteins in the bacteria, thus inhibiting the production of toxins and the formation of spores.[2]
Structure
The chemical structure of cadazolid combines the pharmacophores of oxazolidinone and fluoroquinolone.[2]
In a study published in the journal Anaerobe, cadazolid has been shown to be effective in vitro against 133 strains of Clostridium difficile all collected from Sweden.[3]
In phase I tests, sixty four male patients reacted favourably to cadazolid which primarily acted and remained in the colon while displaying little toxicity even in regimes involving large doses.[1]
ABOUT CADAZOLID IN THE PHASE II STUDY
Cadazolid was studied in a Phase II multi-center, double-blind, randomized, active reference, parallel group, therapeutic exploratory study. The study evaluated the efficacy, safety and tolerability of a 10-day, twice daily oral administration of 3 doses (250 mg, 500 mg or 1,000 mg b.i.d.) of cadazolid in subjects with Clostridium difficile-associated diarrhea (CDAD). As the current standard of care for CDAD, oral vancomycin (125 mg qid for 10 days) was used as the active reference. The study was completed in December of 2012, after having enrolled 84 subjects with CDAD.
The results of the Phase II study indicate that the effect of all doses of cadazolid were numerically similar to, or better than vancomycin on key endpoints including CDAD clinical cure rates as well as sustained cure rates. Clinical cure rate was defined as the resolution of diarrhea and no further need for CDAD therapy at test-of-cure 24 to 72 hours after the last dose of treatment, while sustained cure rate was defined as clinical cure with no recurrence of CDAD up to 4 weeks post-treatment. Recurrence rates were numerically lower for all doses of cadazolid as compared to vancomycin. Cadazolid was safe and well tolerated.
ABOUT THE GAIN ACT (INCLUDING FAST TRACK DESIGNATION)
The Food and Drug Administration Safety and Innovation Act (FDASIA) was signed into law in July 2012. The GAIN Act is Title VIII to FDASIA. The purpose of the GAIN Act is to encourage pharmaceutical research of certain antibiotics by designation of products as QIDPs. These products are intended to treat serious or life-threatening infections and include those to treat certain specifically identified pathogens, which are listed in the GAIN Act. C. difficile is one such specifically identified pathogen and drugs to treat CDAD would be eligible for designation as a QIDP.
The GAIN Act also provides that qualifying drugs (QIDPs) are eligible for inclusion in the FDA’s Fast Track program. This program is intended to facilitate development and expedite review of new drugs and includes close early communication between the FDA and a drug’s sponsor.
ABOUT FAST TRACK DRUG DEVELOPMENT PROGRAMS
For further information regarding Fast Track Drug Development Programs, please refer to the FDA document “Guidance for Industry on Fast Track Drug Development Programs: Designation, Development, and Application Review”. This document is available on the Internet at:
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079736.pdf
ABOUT CLOSTRIDIUM DIFFICILE-ASSOCIATED DIARRHEA
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacterium that is the leading cause of nosocomial diarrhea. Clostridium difficile-associated diarrhea (CDAD or CDI for Clostridium difficile infection) can be a severe and life-threatening disease and results from the overgrowth in the colon of toxigenic strains of Clostridium difficile, generally during or after therapy with broad-spectrum antibiotics. CDAD is a major healthcare problem and a leading cause of morbidity in elderly hospitalized patients. The frequency and severity of CDAD in the western world has increased in recent years, and new hypervirulent and epidemic strains of Clostridium difficile have been discovered that are characterized by overproduction of toxins and other virulence factors, and by acquired resistance to fluoroquinolones such as moxifloxacin.
Current antibiotic therapy for CDAD includes vancomycin and metronidazole. While clinical cure rates are generally 85-90%, recurrences rates of 15-30 % with either drug are problematic as Clostridium difficile produces spores that are resistant to antibiotic treatment and routine disinfection. Spores surviving in the gut of patients and/or in the hospital environment may play a major role in re-infection and recurrence of CDAD after antibiotic treatment. Vancomycin and metronidazole are reported to promote spore formation in vitro at sub-inhibitory concentrations.
Actelion Ltd.
Actelion Ltd. is a leading biopharmaceutical company focused on the discovery, development and commercialization of innovative drugs for diseases with significant unmet medical needs.
Actelion is a leader in the field of pulmonary arterial hypertension (PAH). Our portfolio of PAH treatments covers the spectrum of disease, from WHO Functional Class (FC) II through to FC IV, with oral, inhaled and intravenous medications. Although not available in all countries, Actelion has treatments approved by health authorities for a number of specialist diseases including Type 1 Gaucher disease, Niemann-Pick type C disease, Digital Ulcers in patients suffering from systemic sclerosis, and mycosis fungoides in patients with cutaneous T-cell lymphoma.
Founded in late 1997, with now over 2,400 dedicated professionals covering all key markets around the world including the US, Japan, China, Russia and Mexico, Actelion has its corporate headquarters in Allschwil / Basel, Switzerland
…………………..
Preparation of the compound of formula II
The compound of formula II can be obtained by hydrogenation of the compound of formula VIII

VIII
over a noble metal catalyst such as palladium or platinum on charcoal in a solvent such as THF, MeOH or EA between 00C and 400C or by hydrolysis of in presence of a solution of HBr in water or AcOH between 00C and 800C in a solvent such as AcOH.
The compounds of formula III can be prepared as summarized in Scheme 1 hereafter.

IX VI IIIA: R1= H IIIS: ^ = SO2R5
Scheme 1
The compounds of formula V can be prepared as summarized in Scheme 2 hereafter.

II X XI

Scheme 2
The compounds of formula X can be prepared from the methylidene derivatives of formula XII as summarized in Scheme 3 hereafter.

Xc XII Xa: R1 = H

Scheme 3
Example 1:
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((/f)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid:
1 i. (R)-3-(3-fluoro-4-hydroxy-phenyl)-5-hydroxymethyl-oxazolidin-2-one:
A solution of (7?y)-3-(4-benzyloxy-3-fluoro-phenyl)-5-hydroxymethyl-oxazolidin-2-one (6.34 g, prepared according to WO 2004/096221) in THF/MeOH (1 :1; 200 ml) was hydrogenated over Pd/C 10% (1 g) overnight. The catalyst was filtered off, the filtrate evaporated under reduced pressure and the residue stirred in EA. The crystals were collected by filtration, affording 3.16 g (70% yield) of a colourless solid. 1H NMR (DMSOd6; δ ppm): 3.5 (m, IH), 3.64 (m, IH), 3.74 (dd, J = 8.8, 6.4, IH), 3.99 (t, J = 8.8, IH), 4.64 (m, IH), 5.16 (t, J = 5.6, IH), 6.93 (dd, J = 9.7, 8.8, IH), 7.08 (ddd, J = 8.8, 2.6, 1.2, IH), 7.45 (dd, J = 13.5, 2.6, IH), 9.66 (s, IH). MS (ESI): 228.1.
1. ii. 4-[2-fluoro-4- ((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]- 4-hydroxy-piperidine-l-carboxylic acid benzyl ester:
A solution of intermediate l.i (1.27 g) and l-oxa-6-aza-spiro[2.5]octane-6-carboxylic acid benzyl ester (1.60 g; prepared according to US 4244961) were dissolved in DMF (15 ml) and treated with Na2CO3 (1.16 g). The mixture was heated at 1000C overnight. The residue obtained after workup (DCM) was stirred in EA, and the solid was collected by filtration and sequentially washed with EA and Hex, affording 2.52 g (94.5% yield) of a beige solid.
1H NMR (DMSOd6; δ ppm): 1.57 (m, 4H), 3.14 (m, 2H), 3.54 (m, IH), 3.64 (m, IH), 3.79 (m, 5 H), 4.03 (t, J = 9.1, 1 H), 4.66 (m, 1 H), 4.78 (s, 1 H), 5.05 (s, 2 H), 5.16 (t,
J = 5.6, 1 H), 7.18 (m, 2 H), 7.32 (m, 5 H), 7.55 (d, J = 12, 1 H).
MS (ESI): 475.0.
1. iii. (R)-3-[3-fluoro-4-(4-hydroxy-piperidin-4-ylmethoxy)-phenyl]-5-hydroxymethyl- oxazolidin-2-one:
A suspension of intermediate l.ii (2.5 g) in EA/MeOH (1 :1; 100 ml) was hydrogenated over Pd/C for 48 h. The suspension was heated at 400C and the catalyst was filtered off.
The filtrate was evaporated under reduced pressure affording 1.61 g (89% yield) of a yellow powder.
1H NMR (DMSOd6; δ ppm): 1.4-1.63 (m, 4H), 2.67 (m, 2H), 2.83 (m, 2H), 3.53 (dd, J = 4.0, 12.0, IH); 3.66 (dd, J = 3.3, 12.0, IH), 3.71 (s, 2H); 3.80 (m, IH), 4.05 (t, J = 9.0,
IH), 4.48 (s, IH), 4.68 (m, IH), 5.20 (s, IH), 7.20 (m, 2H), 7.57 (d, IH).
MS (ESI): 341.5.
l.iv. l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid:
A solution of intermediate l.iii (200 mg), 7-chloro-l-cyclopropyl-6-fiuoro-l,4-dihydro- 4-0X0-3 -quinolinecarboxylic acid boron diacetate complex (241 mg; prepared according to WO 88/07998) and DIPEA (100 μl) in NMP (2 ml) was stirred at 85°C for 5 h. The reaction mixture was evaporated under reduced pressure and the residue was taken up in 5M HCl in MeOH (3 ml) and stirred. The resulting solid was collected by filtration and washed with MeOH to afford 230 mg (67% yield) of a yellow solid.
1H NMR (DMSOd6; δ ppm): 1.66-1.35 (m, 4H), 1.75 (d, J = 12.8, 2H), 1.95 (m, 2H), 3.33 (t broad, J = 11.0, 2H), 3.57 (m, 3H), 3.67 (dd, J = 12.3, 3.3, IH), 3.83 (m, 2H), 3.92 (s, 2H), 4.06 (t, J = 9.0, IH), 4.69 (m, IH), 7.24 (m, 2H), 7.60 (m, 2H), 7.90 (d, J = 13.3, IH), 8.66 (s, IH).
MS (ESI): 585.9.
References
- Boschert, Sherry (19 Sep 2012). “Promising C. difficile Antibiotic in Pipeline”. Internal Medicine News. International Medical News Group. Retrieved 22 May 2013.
- “Cadazolid”. .actelion.com. Retrieved 2013-05-22.
- “Anaerobe – In vitro activity of cadazolid against Clostridium difficile strains isolated from primary and recurrent infections in Stockholm, Sweden”. ScienceDirect.com. 2013-02-26. Retrieved 2013-05-22.
- WO 2008056335
- WO 2009136379
Sonidegib/Erismodegib..Novartis Cancer Drug LDE225 Meets Primary Endpoint in Phase 2


Sonidegib/Erismodegib
CODE DESIGNATION ..LDE225, NVP-LDE-225
Treatment of medulloblastoma PHASE3 2014 FDA FILING
Treatment of advanced basal cell carcinoma PHASE3 2014 FDA FILING
Treatment of SOLID TUMORS..PHASE1 2017 FDA FILING
READMalignant Solid Tumors of Childhood
THERAPEUTIC CLAIM Oncology, Antineoplastics & Adjunctive Therapies

CHEMICAL NAMES
1. [1,1′-Biphenyl]-3-carboxamide, N-[6-[(2R,6S)-2,6-dimethyl-4-morpholinyl]-3-pyridinyl]-2-
methyl-4′-(trifluoromethoxy)-, rel-
2. N-{6-[(2R,6S)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl}-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide
N-[6-[(2S,6R)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl]-2-methyl-3-[4-(trifluoromethoxy)phenyl]benzamide
N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-(trifluoromethoxy)biphenyl-3-carboxamide
MOLECULAR FORMULA C26H26F3N3O3
MOLECULAR WEIGHT 485.5
SPONSOR Novartis Pharma AG
CAS REGISTRY NUMBER 956697-53-3 free form
NOTE… DIPHOSPHATE SALT IS THE DRUG WITH CAS 1218778-77-8
sonidegib – European Medicines Agency READ THIS..
Summary EudraCT Number: 2012-004022-21 Sponsor’s Protocol … READ THIS
About the Study
The Phase II, randomized, double-blind BOLT (Basal cell carcinoma Outcomes in LDE225 Trial) study was designed to assess the safety and efficacy of two oral dose levels of LDE225 (200 mg and 800 mg) in patients with locally advanced or metastatic basal cell carcinoma[4], which are subtypes of advanced basal cell carcinoma.
The primary endpoint was the proportion of patients achieving an objective response rate, defined as a confirmed complete response and partial response as their best overall response per modified RECIST criteria, within six months of starting treatment with LDE225. Key secondary endpoints of the study included assessing the duration of tumor responseand the rate of complete response. Other secondary endpoints included progression-free survival, time to tumor response and overall surviva

Sonidegib (INN) or Erismodegib (USAN), also known as LDE225 is a Hedgehog signalling pathway inhibitor (via smoothened antagonism) being developed as an anticancer agent by Novartis.[1][2] It has been investigated as a potential treatment for:
- Pancreatic cancer[3][4][5][6]
- Breast cancer[7][8]
- Basal cell carcinoma of the skin[9][10][11]
- Small cell lung cancer[12]
- Medulloblastoma[13][14]
- Advanced solid tumours (including ovarian, breast, pancreatic, stomach, oesophageal cancers and glioblastoma multiforme)[15][16][17]
- Acute leukaemia[18]
- Chronic myeloid leukaemia[19]
- Myelofibrosis and Essential thrombocythaemia[20]
NVP-LDE-225, a product candidate developed by Novartis, is in phase III clinical trials for the treatment of medulloblastoma and basal cell carcinoma. Phase II trials are in progress for the treatment of adult patients with relapsed or refractory or untreated elderly patients with acute leukemia.
Early clinical trials are ongoing for the oral treatment of advanced solid tumors, for the treatment of myelofibrosis in combination with ruxolitinib and for the treatment of small cell lung cancer. A phase II clinical trial for the treatment of basal cell carcinomas in Gorlin’s syndrome patients with a cream formulation of NVP-LDE-225 was discontinued in 2011 since the formulation did not demonstrate tumor clearance rate sufficient to support further development.
Dana-Farber Cancer Institute and the Massachusetts General Hospital are conducting phase I clinical trials for the treatment of locally advanced or metastatic pancreatic cancer in combination with chemotherapy. In 2009, orphan drug designation was assigned in the E.U. for the treatment of Gorlin syndrome.
It has demonstrated significant efficacy against melanoma in vitro and in vivo.[21] It also demonstrated efficacy in a mouse model of pancreatic cancer.[22]
NVP-LDE225 Diphosphate salt (Erismodegib, Sonidegib)

- Synonym:Erismodegib, Sonidegib
- CAS Number:1218778-77-8
- Mol. Formula:C26H26F3N3O3 ∙ 2H3PO4
- MW:681.5
- nmr.http://www.chemietek.com/Files/Line2/Chemietek,%20NVP-LDE225%20[02],%20NMR.pdf
- hplc–http://www.chemietek.com/Files/Line3/Chemietek,%20NVP-LDE225%20[02],%20HPLC.pdf
Brief Description:
About LDE225
LDE225 (sonidegib) is an oral, investigational, selective smoothened inhibitor being studied in a variety of cancers. Smoothened (SMO) is a molecule that regulates the hedgehog (Hh) signaling pathway, which plays a critical role in stem cell maintenance and tissue repair. LDE225 is currently in clinical development for a variety of diseases including myelofibrosis, leukemia and solid tumors.
Given that LDE225 is an investigational compound, the safety and efficacy profile has not yet been fully established. Access to this investigational compound is available only through carefully controlled and monitored clinical trials. These trials are designed to better understand the potential benefits and risks of the compound. Given the uncertainty of clinical trials, there is no guarantee that LDE225 will ever be commercially available anywhere in the world.
Possibility (LDE225) is effective in medulloblastoma relapsed or refractory hedgehog pathway inhibitor sonidegib has been revealed. That the anti-tumor effect was observed in some patients and tolerability in 1/2 test phase.
4th Quadrennial Meeting of the World Federation of Neuro-Oncology in conjunction with the 18th Annual Meeting of the Society for Neuro-Oncology, which was held in San Francisco November 21 to 24 in (WFNO-SNO2013), rice Dana-Farber It was announced by Mark Kieran Mr. Children’s Hospital Cancer Center.
The research group, announced the final results of the Phase 1 trial that target advanced solid cancer in children of sonidegib. 1 dose increased multi-test phase, was initiated from 372mg/m2 once-daily dosing to target children under the age of 18 more than 12 months. (233mg/m2 group 11 people, 16 people 372mg/m2 group, 11 people group 425mg/m2, 680mg/m2 group 21 women) who participated 59 people, including medulloblastoma 38 patients. 12 median age was (2-17).
Creatine phosphokinase elevation of grade 4 only were seen at 372mg/m2 as dose-limiting toxicity only, and became two recommended dose phase and 680mg/m2. Nausea muscle pain creatine kinase rise malaise (22.0%) (15.3%) (15.3%), (13.6%), vomiting side effects were many, was (13.6%). Hypersensitivity vomiting creatine kinase increased (3.4%) (1.7%) (1.7%), rhabdomyolysis side effects of grade 3/4 was (1.7%). (One group 372mg/m2, 425mg/m2 group one) complete response was obtained in two people, a strong correlation was found between the activation of the hedgehog pathway and effect.
Phase III clinical trials that target medulloblastoma the activated hedgehog pathway currently are underway.
About Novartis
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, eye care, cost-saving generic pharmaceuticals, preventive vaccines and diagnostic tools, over-the-counter and animal health products. Novartis is the only global company with leading positions in these areas. In 2013, the Group achieved net sales of USD 57.9 billion, while R&D throughout the Group amounted to approximately USD 9.9 billion (USD 9.6 billion excluding impairment and amortization charges). Novartis Group companies employ approximately 136,000 full-time-equivalent associates and operate in more than 140 countries around the world.

The following Examples serve to illustrate the invention without limiting the scope thereof, it is understood that the invention is not limited to the embodiments set forth herein, but embraces ali such forms thereof as come within the scope of the disclosure,

Step 1:
To a solution of 2-chloro-5-nitro-pyridine 1 (5.58 g, 35.2 mmoL) and c/s-2,6- dimethylmorpholine (4.05 g, 35.2 mmoL) in anhydrous DMF (30 mi.) was added K2CO3 (9.71 g, 70.4 mnrtoL). The mixture was heated at 50ºC overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 3 as a yellow solid, after purification by silica gel chromatography, obtained pure product (7.80 g, 93.2%). LC-MS m/z: 238.2 (M+ 1).
Step 2:
The above material 3 (7.3Og. 30.8 mmoL) was hydrogenated in the presence of 10% Pd-C (1.0 g) in MeOH (120 ml) under hydrogen overnight. The suspension was filtered through celite and the filtrate was concentrated to give the crude product 4 (5.92 g) as a dark brown oil which was used directly in the next step without further purification. LC-MS m/z. 208.2 (M+1).
Step 3:
To a solution of 3-bromo-2-methyl benzoic acid (2.71 g, 12.6 mmoL), 6-((2S,6R)-2,6- dimethylmorpholino)pyridin-3-arnine 4 (2.61 g, 12.6 mmoL), and HATU (4.80 g, 12.6 mmoL) in anhydrous DMF (30 mL) was added diisopropylethylamine (6.58 mL, 37.8 mmoL) dropwise. The resulting mixture was stirred overnight at room temperature. The reaction mixture was diluted with water (50 mL), and then extracted with EtOAc (3×120 mL). The organic layer was dried and concentrated to give the crude product. This crude product was then purified by flash column chromatography using 30% EtOAc in hexane as eiuent to give 5 as a white solid (4.23 g, 83.0%). LC-MS m/z: 404.1 (M+1).
Step 4:
A mixture of 4-(trif!uoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo- N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-ylJ-4-methyl-benzamide 5 (250 mg, 0.62mmol), Pd(PPh3)4 (36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL) in a sealed tube was heated at 130ºC overnight. The reaction mixture was diluted with EtOAc and water. The aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine and concentrated to give the crude product which was then purified by preparative mass triggered HPLC (C18 column, etuted with CH3CN-H2O containing 0.05% TFA) to give N-(6-((2S,6R)-2,6-dimethyfmorpholino)pyridin-3-yl)-2-rnethyl- 4′-(trifluoromethoxy)biphenyi-3-carboxamide (183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
The resultant crystalline product (Form A) was converted to the amorphous form by dissolving in 3% w/w aqueous ethanol, and the resultant solution spray dried at about 150ºC.
Form B was prepared by heating the amorphous form in an oven at 110ºC for 2 hours. In a further embodiment, the invention relates to a process step or steps, or an intermediate as described herein.

…………………………..
SYNTHESIS

| LC-MS m/z 486.2 (M + 1) |

Step 1: To a solution of 3-iodo-4-methyl-benzoic acid (10.0 g, 38.2 mmol) in methanol (70 ml) is added concentrated sulfuric acid (0.5 ml). The reaction mixture is heated at 70° C. for 48 hours, cooled to room ambient temperature and then concentrated. After that, ethyl acetate (100 ml) and aqueous NaHCO3 (saturated, 100 ml) solution are added to the residue. The organic layer is separated and washed again with aqueous NaHCO3 (saturated, 100 ml) solution. The organic layer is separated, dried over anhydrous Na2SO4 and concentrated to yield 3-iodo-4-methyl-benzoic acid methyl ester 1. It is used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (s, 1H), 7.87 (d, 1H, J=8.4 Hz), 7.48 (d, 1H, J=8.4 Hz), 3.85 (s, 3H), 3.35 (s, 3H); LC-MS m/z: 277.0 (M+1).
Step 2: To a round-bottom flask containing 3-iodo-4-methyl-benzoic acid methyl ester (1.38 g, 5.00 mmol), 4-cyanophenylboronic acid (1.10 g, 7.48 mmol), palladium acetate (168 mg, 0.748 mmol), 2-(dicyclohexylphosphino)biphenyl (0.526 g, 1.50 mmol) and potassium fluoride (0.870 g, 15.0 mmol) is added anhydrous 1,4-dioxane (15 ml). The flask is purged with argon and sealed. The mixture is stirred at 130° C. for 18 hours, cooled to ambient temperature and then water (20 ml) and ethyl acetate (20 ml) are added. Solid is removed under vacuum filtration. The filtrate is extracted with EtOAc (20 ml×2). The organic layers are combined, washed with aqueous HCl (5%, 20 ml) and saturated NaHCO3 (20 ml). It is dried over MgSO4, and concentrated. The residue is purified by silica gel column chromatography (EtOAc/Hexane, gradient) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2; LC-MS m/z: 252.1 (M+1).
Step 3: To a solution of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2 (2.56 g, 10.3 mmol) in 1,4-dioxane-H2O (1:1 mixture, 20 ml) is added NaOH (1.22 g, 30.2 mmol)). The reaction is stirred at ambient temperature for 24 hours. To this mixture is added aqueous HCl (1 N, 36 ml) and it is then extracted with ethyl acetate (40 ml×3). The organic layers are combined, dried over anhydrous Na2SO4. The solver is removed. The solid obtained is washed with small amount of acetonitrile and air dried to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3: 1H NMR (DMSO-d6) δ 7.94 (d, 2H, J=8.0 Hz), 7.84 (dd, 1H, J1=8.4 Hz, J2=1.2 Hz), 7.75 (d, 1H, J=1.2 Hz), 7.61 (d, 2H, J=8.0 Hz), 7.48 (d, 1H, J=8.4 Hz), 2.29 (s, 3 H); LC-MS m/z 238.1 (M+1).
Step 4: To a suspension of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3 (40 mg, 0.17 mmol) in anhydrous methylene chloride (5 ml) is added 2 drops of DMF. Then oxalyl chloride (32 mg, 22 μl, 0.25 mmol) is added. The mixture is stirred at ambient temperature until it turns clear. After that, it is concentrated, re-dissolved in anhydrous methylene chloride (3 ml), and added to a solution of 4-(morpholine-4-sulfonyl)-phenylamine (61 mg, 0.25 mmol) and triethylamine (34 mg, 47 μl, 0.33 mmol) in methylene chloride (2 ml). The mixture is stirred for 2 hours, concentrated and the residue is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [4-(morpholine-4-sulfonyl)-phenyl]-amide: 1H NMR (DMSO-d6) δ 10.64 (s, 1H), 8.07 (d, 2H, J=8.8 Hz), 7.97 (d, 2H, J=8.4 Hz), 7.95 (d, 1H, J=8.8 Hz), 7.89 (s, 1H), 7.43 (d, 2H, J=8.4 Hz), 7.67 (d, 2H, J=8.8 Hz), 7.53 (d, 2H, J=8.8 Hz), 3.63 (m, 4H), 2.84 (m, 4H) 2.32 (s, 3H); LC-MS m/z: 462.1 (M+1).
Example 2 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide

Step 1: To a solution of 2-chloro-5-nitro-pyridine 4 (2.38 g, 15 mmol.) and cis-2,6-dimethylmorpholine (1.73 g, 15 mmol.) is added K2CO3 (4.14 g, 30 mmol.). The mixture was heated at 50° C. overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 6 as a yellow solid. The crude product is used directly in next step without further purification. LC-MS m/z: 238.1 (M+1).
Step 2: The above crude material 6 is hydrogenated in the presence of Pd—C (0.2 g) in MeOH (100 mL) under hydrogen over 10 h. The suspension is filtered through celite and the filtrate is concentrated to give the crude product 7 as a dark brown oil which is used directly in the next step without further purification. LC-MS m/z: 208.1 (M+1).
Step 3: To a solution of 3-bromo-4-methyl benzoic acid (108 mg, 0.5 mmol.), 6-(2,6-Dimethyl-morpholin-4-yl)-pyridin-3-ylamine 7 (104 mg, 0.5 mmol.), amd HATU (190 mg, 0.5 mmol.) in dry DMF (5 mL) is added triethylamine (139 uL, 1.0 mmol.) dropwise. The resulting mixture is stirred at room temperature for 2 h. After concentration, the residue is partitioned between EtOAc and water. The organic layer is dried and concentrated to give the crude product. The final compound is purified by flash column chromatography using 50% EtOAc in hexane as eluent to give 8 as a white solid. LC-MS m/z: 404.1 (M+1).
Step 4: A mixture of 4-cyanophenyl boronic acid (18 mg, 0.12 mmol), 3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide 8 (40 mg, 0.1 mmol), Pd(PPh3)4 (11 mg, 0.01 mmol), and Na2CO3 (42 mg, 0.4 mmol) in a combined solvent system of toluene (0.2 mL) and water (0.2 mL) and ethanol (0.05 mL) is heated at 140° C. under microwave irradiation for 30 min. The reaction mixture is diluted with EtOAc and water. The aqueous layer is extracted with EtOAc. The combined organic layer is washed with brine and concentrated to give the crude product which is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide. LC-MS m/z: 427.2 (M+1).

4-(Trifluoromethoxy)phenylboronic acid
- CAS Number 139301-27-2
- Linear Formula CF3OC6H4B(OH)2
- Molecular Weight 205.93
CONDENSE WITH …3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamideACS Medicinal Chemistry Letters, 2010 , vol. 1, 3 p. 130 – 134

A mixture of 4-(trifluoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo-N-[6-(2,6-
dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide E (250 mg, 0.62mmol), Pd(PPh3)4
(36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL)
in a sealed tube was heated at 1300C overnight. The reaction mixture was diluted with EtOAc
and water. The aqueous layer was extracted with EtOAc. The combined organic layer was
washed with brine and concentrated to give the crude product which was then purified by
preparative mass triggered HPLC (C18 column, eluted with CH3CN-H2O containing 0.05% TFA)
to give N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide (5m, 183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
HRMS (m/z): [M+H]+
calcd for C26H27N3O3F3 486.2005; found 486.1986,
1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.15 (s, 1H), 8.43 (d, 1H), 7.94 (dd, 1H), 7.52-7.43
(m, 5H), 7.38 (m, 1H), 7.33 (m, 1H), 6.86 (d, 1H), 4.06 (d, 2H), 3.62 (m, 2H), 2,34 (m, 2H), 2.22
(s, 3H), 1.16 (d, 6H).
http://pubs.acs.org/doi/suppl/10.1021/ml1000307/suppl_file/ml1000307_si_001.pdf
Reference
- “LDE225 – PubChem”. PubChem. National Institutes of Health. Retrieved 16 February 2014.
- Pan, S; Wu, X; Jiang, J; Gao, W; Wan, Y; Cheng, D; Han, D; Liu, J; Englund, NP; Wang, Y; Peukert, S; Miller-Moslin, K; Yuan, J; Guo, R; Matsumoto, M; Vattay, A; Jiang, Y; Tsao, J; Sun, F; Pferdekamper, AC; Dodd, S; Tuntland, T; Maniara, W; Kelleher, JF; Yao, Y; Warmuth, M; Williams, J; Dorsch, M (10 June 2010). “Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist”. ACS Medicinal Chemistry Letters 1 (3): 130–134. doi:10.1021/ml1000307.
- “A Biomarker Study to Identify Predictive Signatures of Response to LDE225 (Hedgehog Inhibitor) In Patients With Resectable Pancreatic Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Gemcitabine + Nab-paclitaxel With LDE-225 (Hedgehog Inhibitor) as Neoadjuvant Therapy for Pancreatic Adenocarcinoma”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Dose-escalation, and Safety Study of LDE225 and Gemcitabine in Locally Advanced or Metastatic Pancreatic Cancer Patients”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Pilot Study of a Hedgehog Pathway Inhibitor (LDE-225) in Surgically Resectable Pancreas Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Study With LDE225 in Combination With Docetaxel in Triple Negative (TN) Advanced Breast Cancer (ABC) Patients (EDALINE)”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014.
- “LDE225 in Treating Patients With Stage II-III Estrogen Receptor- and HER2-Negative Breast Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “To Evaluate the Safety, Local Tolerability, PK and PD of LDE225 on Sporadic Superficial and Nodular Skin Basal Cell Carcinomas(sBCC)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Trial to Evaluate the Safety, Local Tolerability, Pharmacokinetics and Pharmacodynamics of LDE225 on Skin Basal Cell Carcinomas in Gorlin Syndrome Patients”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Combination of the Hedgehog Inhibitor, LDE225, With Etoposide and Cisplatin in the First-Line Treatment of Patients With Extensive Stage Small Cell Lung Cancer (ES-SCLC)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase III Study of Oral LDE225 Versus (vs) Temozolomide (TMZ) in Patients With Hedge-Hog (Hh)-Pathway Activated Relapsed Medulloblastoma (MB)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase I Dose Finding and Safety Study of Oral LDE225 in Children and a Phase II Portion to Assess Preliminary Efficacy in Recurrent or Refractory MB”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Phase Ib, Dose Escalation Study of Oral LDE225 in Combination With BKM120 in Patients With Advanced Solid Tumors”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Dose Finding and Safety of Oral LDE225 in Patients With Advanced Solid Tumors”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “LDE225 and Paclitaxel in Solid Tumors”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Study of Efficacy and Safety of LDE225 in Adult Patients With Relapsed/Refractory Acute Leukemia”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Nilotinib and LDE225 in the Treatment of Chronic or Accelerated Phase Myeloid Leukemia in Patients Who Developed Resistance to Prior Therapy”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase Ib/II Dose-finding Study to Assess the Safety and Efficacy of LDE225 + INC424 in Patients With MF”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- Jalili, A; Mertz, KD; Romanov, J; Wagner, C; Kalthoff, F; Stuetz, A; Pathria, G; Gschaider, M; Stingl, G; Wagner, SN (30 July 2013). “NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo.” (PDF). PloS one 8 (7): e69064. doi:10.1371/journal.pone.0069064. PMC 3728309.PMID 23935925.
- Fendrich, V; Wiese, D; Waldmann, J; Lauth, M; Heverhagen, AE; Rehm, J; Bartsch, DK (November 2011). “Hedgehog inhibition with the orally bioavailable Smo antagonist LDE225 represses tumor growth and prolongs survival in a transgenic mouse model of islet cell neoplasms.”. Annals of Surgery 254 (5): 818–23.doi:10.1097/SLA.0b013e318236bc0f. PMID 22042473.
- ChemMedChem, 2013 , vol. 8, 8 p. 1261 – 1265
- ACS Med. Chem. Lett., 2010, 1 (3), pp 130–134.
- MORE REF
sonidegib
Skin Cancer Foundation. “Skin Cancer Facts.” Available at:http://www.skincancer.org/skin-cancer-information/skin-cancer-facts . Accessed on February 14, 2014.
Rubin AI, Chen EH, Ratner D (2005). Current Concepts: Basal-Cell Carcinoma. N Engl J Med; 353:2262-9.
ClinicalTrials.gov. “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)” Available at:http://clinicaltrials.gov/ct2/show/NCT01327053?term=%22LDE225%22+and+%22BOLT%22&rank=1. Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Complete Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45652 . Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Partial Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45819 . Accessed on February 14, 2014.
Wong C S M, Strange R C, Lear J T (2003). Basal cell carcinoma. BMJ; 327:794-798.
Copcu E, Aktas A. Simultaneous two organ metastases of the giant basal cell carcinoma of the skin. Int Semin Surg Oncol. 2005;2:1-6. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC544837/ . Accessed on February 14, 2014.
Skin Cancer Foundation. “Basal Cell Carcinoma Treatment Options.” Available athttp://www.skincancer.org/skin-cancer-information/basal-cell-carcinoma/bcc-treatment-options . Accessed on February 14, 2014.
Stuetz A, et al. LDE225, a specific smoothened inhibitor, for the topical treatment of nevoid basal cell carcinoma syndrome (Gorlin’s syndrome). Melanoma Research. 2010; 20:e40. Available at:http://journals.lww.com/melanomaresearch/Fulltext/2010/06001/FC24_LDE225,_a_specific_smoothened_inhibitor,_for.87.aspx#FC24_LDE225%2C_a_specific_smoothened_inhibitor%2C_for.87.aspx?s=2&_suid=139234380607909969110518506816.
Novartis.com. “The Pipeline of Novartis Oncology: LDE225.” Available at:http://www.novartisoncology.com/research-innovation/pipeline.jsp #. Accessed on February 14, 2014.
Children’s Medical Research Center, Children’s Memorial Hospital/Northwestern University Feinberg School of Medicine. “The Sonic hedgehog/patched/gli signal transduction pathway.” Available at http://www.childrensmrc.org/iannaccone/gli/ . Accessed on February 14, 2014.
Gupta S, Takebe N, LoRusso P. Targeting the Hedgehog pathway in cancer. Ther Adv Med Oncol. 2010 July; 2(4): 237-250. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3126020/ . Accessed on February 14, 2014.
| WO2004078163A2 | Feb 26, 2004 | Sep 16, 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2007113120A1 | Mar 22, 2007 | Oct 11, 2007 | Frank Hoffmann | Stamping apparatus with feed device |
| WO2007131201A2 * | May 4, 2007 | Nov 15, 2007 | Irm Llc | Compounds and compositions as hedgehog pathway modulators |
| WO2008154259A1 | Jun 4, 2008 | Dec 18, 2008 | Irm Llc | Biphenylcarboxamide derivatives as hedgehog pathway modulators |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link
blogs are

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ


‘Female Viagra’ Flibanserin now on track for Q3 filing in USA

Flibanserin, girosa
167933-07-5 cas no
147359-76-0 (monoHCl)
- Bimt 17
- BIMT 17 BS
- Bimt-17
- Flibanserin
- Girosa
- UNII-37JK4STR6Z
Women with low libido in the US will have to wait even longer for approval of the first ever treatment for the condition after regulators requested more data on the forerunner flibanserin, delaying its submission until later this year.
The US Food and Drug Administration has asked manufacturer Sprout Pharmaceuticals for data on how flibanserin interacts with other medicines and also how it affects driving ability, after around 10% of patients experienced sleepiness while on the drug
Read more at: http://www.pharmatimes.com/Article/14-02-11/Female_Viagra_now_on_track_for_Q3_filing_in_USA.aspx#ixzz2tAWxwzRD
December 11, 2013 – Sprout Pharmaceuticals today announced that it has received and appealed the Food and Drug Administration’s (FDA) Complete Response Letter (CRL) for flibanserin through the Formal Dispute Resolution process.
Flibanserin is an investigational, once-daily treatment for Hypoactive Sexual Desire Disorder, or HSDD, in premenopausal women. HSDD is the most commonly reported form of female sexual dysfunction
read all here
A new drug being developed by Boehringer Ingelheim could give a boost to the sex drive of women with low libido. The drug, known as flibanserin, has been shown in clinical trials to increase their sexual desire when taken once a day at bedtime.
The results from four pivotal Phase III clinical trials on women with hypoactive sexual desire disorder (HSDD) were presented this week at the European Society for Sexual Medicine’s congress in Lyon, France. The trials showed that participants taking flibanserin had a significant improvement in their sexual desire compared to those given a placebo. They also experienced less of the distress associated with sexual dysfunction.
The drug was initially being investigated as a treatment for depression, and acts on the serotonin receptors in the brain – it is both a 5-HT1A receptor agonist and a 5-HT2A receptor antagonist. It is also a partial agonist at the dopamine D4 receptor.

Neurotransmitters such as serotonin are believed to be involved in sexual function, and antidepressants are commonly associated with a loss of libido, so this was an obvious side-effect to look out for during clinical trials in depression. But far from suppressing the libido in women, it appeared to have the opposite effect, so trials in women with HSDD were initiated.
Hormone replacement can improve the libido of women who have had their ovaries removed, but there is no available drug to treat those who have not. There have been accusations that pharma companies invent new diseases like HSDD in order to sell more medicines, but according to Kathleen Segraves, an assistant professor at Case Western Reserve University in the US who has worked in the field of sexual functioning for many years, this is not the case here. HSDD is a very real disorder, she says, and the potential for a treatment for these women is very exciting.

Flibanserin (code name BIMT-17; proposed trade name Girosa) is a drug that was investigated by Boehringer Ingelheim as a novel, non-hormonal treatment for pre-menopausal women with Hypoactive Sexual Desire Disorder (HSDD).[1][2] Development was terminated in October 2010 following a negative report by the U.S. Food and Drug Administration.[3]
HSDD is the most commonly reported female sexual complaint and characterized by a decrease in sexual desire that causes marked personal distress and/or personal difficulties. According to prevalence studies about 1 in 10 women reported low sexual desire with associated distress, which may be HSDD.[4] The neurobiological pathway of female sexual desire involves interactions among multiple neurotransmitters, sex hormones and various psychosocial factors. Sexual desire is modulated in distinct brain areas by a balance between inhibitory and excitatory neurotransmitters, serotonin acting as an inhibitor while dopamine and norepinephrine act as a stimulator of sexual desire.[5][6]Flibanserin is a 5-HT1A receptor agonist and 5-HT2A receptor antagonist that had initially been investigated as an antidepressant. Preclinical evidence suggested that flibanserin targets these receptors preferentially in selective brain areas and helps to restore a balance between these inhibitory and excitatory effects.[6] HSDD has been recognized as a distinct sexual function disorder for more than 30 years.
The proposed mechanism of action refers back to the Kinsey dual control model. Several sex steroids, neurotransmitters, and hormones have important excitatory or inhibitory effects on the sexual response. Among the neurotransmitters, the excitatory activity is driven by dopamine and norepinephrine, while the inhibitory activity is driven by serotonin. The balance between these systems is relevant for a healthy sexual response. By modulating these neurotransmitters in selective brain areas, flibanserin, a 5-HT1A receptoragonist and 5-HT2A receptor antagonist, is likely to restore the balance between these neurotransmitter systems.[6]
Several large pivotal Phase III studies with Flibanserin were conducted in the USA, Canada and Europe. They involved more than 5,000 pre-menopausal women with generalized acquired Hypoactive Sexual Desire Disorder (HSDD). The results of the Phase III North American Trials demonstrated that
Although the two North American trials that used the flibanserin 100 mg qhs dose showed a statistically significant difference between flibanserin and placebo for the endpoint of [satisfying sexual events], they both failed to demonstrate a statistically significant improvement on the co-primary endpoint of sexual desire. Therefore, neither study met the agreed-upon criteria for success in establishing the efficacy of flibanserin for the treatment of [Hypoactive Sexual Desire Disorder].
These data were first presented on November 16, 2009 at the congress of the European Society for Sexual Medicine in Lyon, France. The women receiving Flibanserin reported that the average number of times they had “satisfying sexual events” rose from 2.8 to 4.5 times a month. However, women receiving placebo reported also an increase of “satisfying sexual events” from 2.7 to 3.7 times a month.
Evaluation of the overall improvement of their condition and whether the benefit was meaningful to the women, showed a significantly higher rate of a meaningful benefit in the flibanserin-treated patient group versus the placebo group.The onset of the Flibanserin effect was seen from the first timepoint measured after 4 weeks of treatment and maintained throughout the treatment period.
The overall incidence of adverse events among women taking flibanserin was low, the majority of adverse events being mild to moderate and resolved during the treatment. The most commonly reported adverse events included dizziness, nausea, fatigue, somnolence and insomnia.

On June 18, 2010, a federal advisory panel to the U.S. Food and Drug Administration (FDA) unanimously voted against recommending approval of Flibanserin.
Earlier in the week, a FDA staff report also recommended non-approval of the drug. While the FDA still might approve Flibanserin, in the past, negative panel votes tended to cause the FDA not to approve.
On October 8, 2010, Boehringer Ingelheim announced that it would discontinue its development of flibanserin in light of the FDA advisory panel’s recommendation.
On June 27, 2013, Sprout Pharmaceuticals confirmed they had resubmitted flibanserin for FDA approval.
Flibanserin, chemically 1 -[2-(4-(3-trifluoromethylphenyl)piperazin-1 – yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one was disclosed in form of its hydrochloride in European Patent No. 526,434 (‘434) and has the following chemical structure:

Process for preparation of flibanserin were disclosed in European Patent No. ‘434, U.S. Application Publication No. 2007/0032655 and Drugs of the future 1998, 23(1): 9-16.
According to European Patent No. ‘434 flibanserin is prepared by condensing 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethyl phenyl piperazine. According to U.S. Application Publication No. 2007/0032655 flibanserin is prepared by condensing 1-[(3-trifluoromethyl)phenyl]-4-(2- chloroethyl)piperazine with 1 -(2-propenyl)-1 ,3-dihydro-benzimidazol-2H-one.
According to Drugs of the future 1998, 23(1): 9-16 flibanserin is prepared by reacting 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethylphenylpiperazine.
PATENT
1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1H-benzimidazol-2-one
Compound 3
Hydrochloride salt (isopropanol) M.p. 230-231°C
Analysis

¹H NMR (DMSO-d₆/CDCL₃ 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (8H), 4.36 (t, 2H), 4.1-3.1 (10H)
CLIP

The compound 1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazol-2-one (flibanserin) is disclosed in form of its hydrochlorid in European Patent Application EP-A-526434 and has the following chemical structure:

Flibanserin shows affinity for the 5-HTιA and 5-HT2-receptor. It is therefore a promising therapeutic agent for the treatment of a variety of diseases, for instance depression, schizophrenia, Parkinson, anxiety, sleep disturbances, sexual and mental disorders and age associated memory impairment.

EXAMPLE……… EP1518858A1
375 kg of 1-[(3-trifluoromethyl)phenyl]-4-(2-cloroethyl)piperazin are charged in a reactor with 2500 kg of water and 200 kg of aqueous Sodium Hydroxide 45%. Under stirring 169.2 kg of 1-(2-propenyl)-1,3-dihydro-benzimidazol-2H-one, 780 kg of isopropanol, 2000 kg of water and 220 kg of aqueous Sodium Hydroxide 45% are added. The reaction mixture is heated to 75-85° C. and 160 kg of concentrated hydrochloric acid and 200 kg of water are added.
The reaction mixture is stirred at constant temperature for about 45 minutes. After distillation of a mixture of water and Isopropanol (about 3000 kg) the remaining residue is cooled to about 65-75° C. and the pH is adjusted to 6.5-7.5 by addition of 125 kg of aqueous Sodium Hydroxide 45%. After cooling to a temperature of 45-50° C., the pH value is adjusted to 8-9 by addition of about 4 kg of aqueous Sodium Hydroxide 45%. Subsequently the mixture is cooled to 30-35° C. and centrifuged. The residue thus obtained is washed with 340 l of water and 126 l of isopropanol and then with water until chlorides elimination.
The wet product is dried under vacuum at a temperature of about 45-55° C. which leads to 358 kg of crude flibanserin polymorph A. The crude product thus obtained is loaded in a reactor with 1750 kg of Acetone and the resulting mixture is heated under stirring until reflux. The obtained solution is filtered and the filtrate is concentrated by distillation. The temperature is maintained for about 1 hour 0-5° C., then the precipitate solid is isolated by filtration and dried at 55° C. for at least 12 hours.
The final yield is 280 kg of pure flibanserin polymorph A.
CLIP
Flibanserin may be prepared by reacting 1-(phenylvinyl)-2,3-dihydro-1H-benzimidazol-2-one (I) with 1,2-dichloroethane (II) in the presence of NaH in warm dimethylformamide. The resulting 1-(2-chloroethyl)-2,3-dihydro-1H-benzimidazol-one (III) is in turn coupled with commercially available m-trifluoromethylphenylpiperazine hydrochloride (IV) in the presence of sodium carbonate and catalytic potassium iodide in refluxing ethanol. The crude flibanserin hydrochloride (V) is then dissolved in aqueous ethanol and the pure base is precipitated upon addition of sodium hydroxide.
PICK UP INTERMEDIATES FROM CHEM24H.COM
1-(1-phenylvinyl)-1,3-dihydro-2H-benzimidazol-2-one (I)
1,2-dichloroethane (II)
1-(2-chloroethyl)-1,3-dihydro-2H-benzimidazol-2-one (III)
1-[3-(trifluoromethyl)phenyl]piperazine; N-[3-(trifluoromethyl)phenyl]piperazine (IV)
1-(2-[4-[3-(trifluoromethyl)phenyl]piperazino]ethyl)-1,3-dihydro-2H-benzimidazol-2-one (V)
PATENT


wherein R2 is as defined in formula X; with a compound of formula Xl:

According to another aspect of the present invention there is provided a novel compound or a salt thereof selected from the compounds of formula I, IV and VII:


Wherein R is hydrogen or an amino protecting group.
Preferable the amino protecting groups are selected from butyl, 1 ,1- diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl.
R1 is independently selected from chlorine, bromine, iodine, methanesulphonate, trifluoromethanesulphonate, paratoluenesulphonate or benzenesulphonate. Preferable R1 is independently selected from chlorine, bromine or iodine and more preferable R1 is chlorine.
Wherein R2 is hydrogen or an amino protecting group.
The amino protecting group may be any of the groups commonly used to protect the amino function such as alkyl, substituted alkyl, hetero substituted alkyl, substituted or unsubstituted unsaturated alkyl, alkyl substituted hetero atoms, substituted or unsubstituted phenyl, substituted or unsubstituted benzyl, alkyoxy carbonyl groups and aryloxy carbonyl groups.
Preferable the amino protecting groups are selected from butyl, 1 ,1 – diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl. The following examples are given for the purpose of illustrating the present invention and should not be considered as limitations on the scope and spirit of the invention.
EXAMPLES Example 1
A mixture of sodium hydroxide (47 gm) and i-(α-methylvinyl) benzimidazol-2-one (100 gm) in dimethylformamide (400 ml) was .stirred for 1 hour at room temperature. Dibromoethane (217 gm) was slowly added to the mixture and stirred at 1 hour 30 minutes. The resulting solution after addition water (500 ml) was extracted with ethyl acetate. The combined ethyl acetate extract washed with water. After drying the solvent was removed under vacuum to yield 132 gm of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H-benzimidazol- 2-one as a yellow oily liquid.
Example 2 A mixture of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H- benzimidazol-2-one (100 gm), diethanolamine (175 ml), sodium carbonate (40 gm) and potassium iodide (10 gm) was heated to 90 to 95 deg C and stirred for 2 hours. The reaction mass was cooled to room temperature and added water (500 ml). The resulting mixture extracted into ethyl acetate and the organic layer washed with water. After drying the solvent was removed under vacuum to yield 105 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3-isopropenyl- 2H-benzimidazol-2-one as a thick yellow oily liquid.
Example 3
To the mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 2 and chloroform (300 ml), thionyl chloride (95 ml) was slowly added. The mixture was heated to reflux and stirred for 2 hours. The excess thionyl chloride and chloroform was distilled off to yield 98 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2- chloroethyl)amino]ethyl]-3-isopropenyl-2H-benzimidazol-2-one as a brown coloured sticky residue.
Example 4
1 ,3-dihydro-1-[2-[N-[bis-(2-chloroethyl)amino]ethyl]-3-isopropenyl-2H- benzimidazol-2-one (98 gm) obtained as in example 3 was added to water (500 ml) and concentrated hydrochloric acid (200 ml) mixture. The mixture was heated to 60 to 65 deg C and stirred for 1 hour. The contents of the flask cooled to room temperature and pH of the solution adjusted to 9 – 10 with 10% sodium hydroxide solution. The resulting solution extracted with ethyl acetate and washed the organic layer with water. Evaporate the solvent under reduced pressure to yield 82 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]- 2H-benzimidazol-2-one as a dark brown coloured oily liquid
Example 5
A mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]-1,2-H- benzimidazol-2-one (82 gm) obtained as in example 4, xylene (300 ml) and m- trifluoromethyl aniline (58 gm) was refluxed for 64 hours. The reaction mass was cooled to room temperature and filtered to obtain 1-[2-(4-(3- thfluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one hydrochloride (Flibanserin hydrochloride) as a light brown coloured solid.
The crude flibanserin hydrochloride was purified in isopropyl alcohol to give 85 gm of pure flibanserin hydrochloride as off white solid.
Example 6
Piperazine (12 gm), toluene(60 ml) and tetra butyl ammonium bromide (1 gm) mixture was heated to 60 deg C, added 1 ,3-dihydro-1-(2-bromoethyl)-3- isopropenyl-2H-benzimidazol-2-one (10 gm) and stirred for 4 hours at 90 to 95 deg C. The mixture was cooled to 60 deg C and added water (50 ml). The separated toluene layer distilled under vacuum to give 8.5 gm of 1 ,3-dihydro-1- (2-piperazinyl)ethyl-3-isopropenyl-2H-benzimidazol-2-one as a white solid.
Example 7
To the mixture of concentrated hydrochloric acid (20 ml) and water (100 ml) was added 1 ,3-dihydro-1-(2-piperazinylethyl)-3-isopropenyl-2H- benzimidazol-2-one (10 gm) obtained as in example 6 and heated to 60 to 65 deg C 1 hour. The mixture was cooled to room temperature and pH of the solution was adjusted to 9 – 10 with 10% sodium hydroxide solution, extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield 8.5 gm of 1 ,3-dihydro-1-(2- piperazinyl ethyl)-2H-benzimidazol-2-one as a white solid.
Example 8
3-trifluoromethylaniline (40 gm) and hydrobromic acid (85 ml; 48- 50%w/w) mixture was cooled to 0 to 5 deg C. To this mixture added sodium nitrite solution (18.5 gm in 25 ml of water) at 5 to 10 deg C and copper powder (1 gm). The temperature was slowly raised to 50 to 55 deg C and stirred for 30 minutes. Added water (200 ml) to reaction mass and applied steam distillation, collected m-trifluoromethylbromobenzene as oily liquid. The oily liquid washed with sulfuric acid for two times (2 X 10 ml) followed by washed with water (2 X 20 ml) and dried the liquid with sodium sulphate to give 22 gm of m- trifluoromethylbromobenzene.
Example 9
To a mixture of 1 ,3-dihydro-1-(2-piperazinyl ethyl)-2H-benzimidazol-2- one (10 gm) obtained as in example 7, m-trifluoromethylbromobenzene (9 gm) obtained as in example 8, sodium tert-butoxide (5.5 gm), palladium acetate (4.5 mg) and xylene (80 ml) was added tri-tert.-butylphosphine (0.2 ml). The mixture was heated to 120 deg C and stirred for 3 hours. The reaction mass was cooled, added water (100 ml) and extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield
10 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazole-2-one (Flibanserin).
Example 10
To a mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 3, cyclohexane (400 ml) and sodium carbonate (35 gm) was added benzene sulfonyl chloride (116 gm) at room temperature. The mixture was heated to 80 to
85 deg C and stirred for 8 hours . The contents were cooled to room temperature and added water (500 ml). Distilled the organic layer to give 182 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one.
Example 11
1 ,3-dihydro-1 -[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzitηidazol-2-one (100 gm) obtained as in example 10, dimethylformamide (500 ml) and sodium corbonate (18 gm) was mixed and heated to 70 deg C. To the mixture was added m-trifluoromethyl aniline (27 gm) and heated to 80 to 85 deg C, stirred for 5 hours. The reaction mass was cooled and added water (2000 ml), filtered the solid to yield 1 ,3-dihydro-1-[2-[4-(3- trifluoromethylphenyl)piperazinyl]ethyl]-3-isopropenyl-2H benzimidazol-2-one. Example 12
1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one (100 gm) obtained as in example 11 added to the mixture of water (500 ml) and concentrated hydrochloric acid (200 ml), heated to 65 deg C and stirred for 1 hour. The reaction mass was cooled to room temperature and pH adjusted to 10 to 10-5 with 10% sodium hydroxide solution. The resulting mixture was extracted with ethyl acetate and the organic
■ layer was washed with water. After drying the solvent was removed under vacuum to yield 87 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]- 2,3-dihydro-1 H-benzimidazole -2-one (Flibanserin).
Paper
Journal of Pharmaceutical and Biomedical Analysis, v.57, 2012 Jan 5, p.104(5)
Isolation and structural elucidation of flibanserin as an adulterant in a health supplement used for female sexual performance enhancement
Low, Min-Yong et al
http://www.sciencedirect.com/science/article/pii/S0731708511004833

This proposed formula and structure was further confirmed by 1H and 13C NMR data which indicated the presence of 20 carbon atoms and 21 protons.
1H NMR
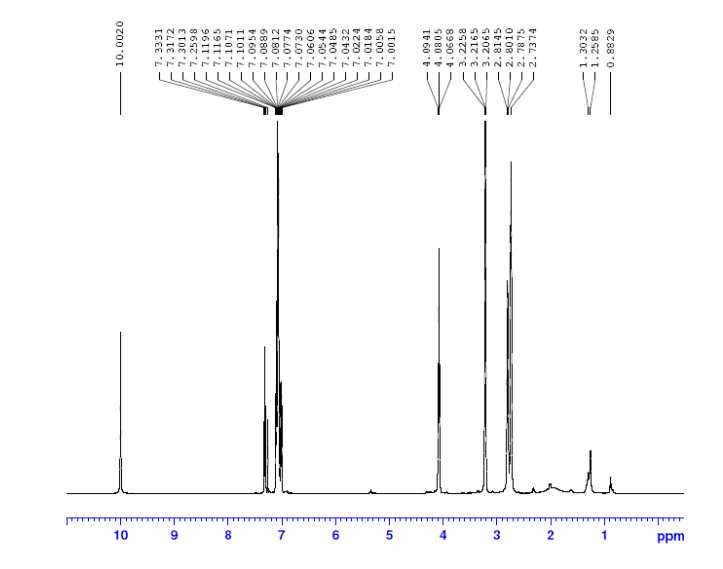
13C NMR
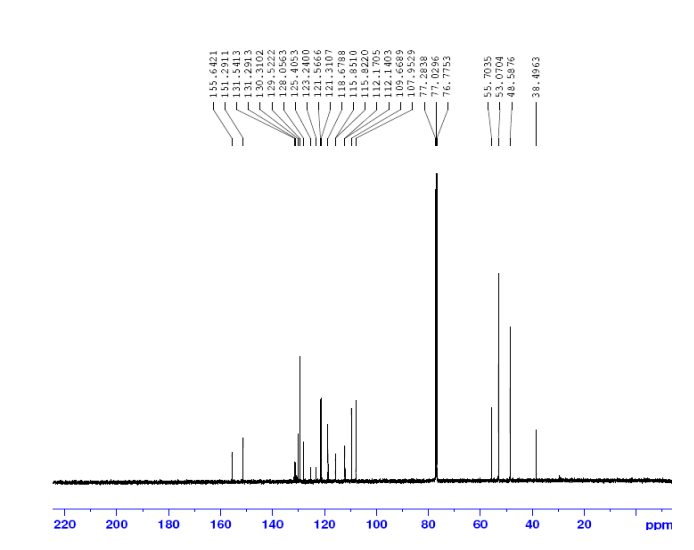
1D and 2DNMR data were used to assign the protons and carbon atoms.
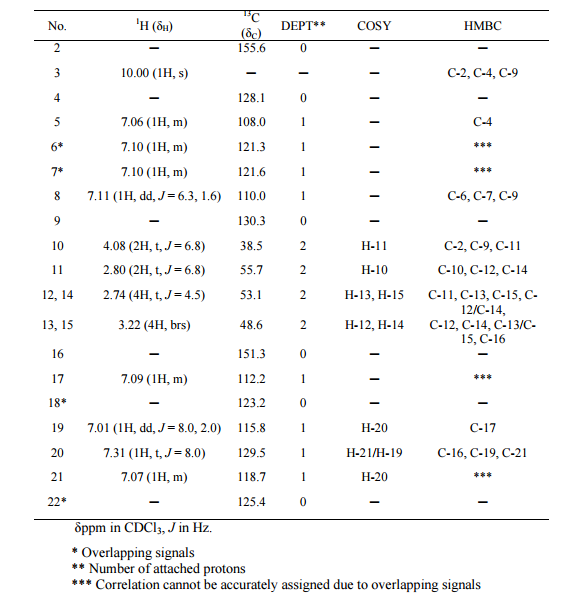
In the1H NMR spectrum , a sharp singlet at 10.00 ppm integrating for one
proton is a typical proton attached to nitrogen. HMBC correlated this proton to C-2, C-4, and C-9 suggesting that it was H-3.
Complex signals were observedbetween 7.00 to 7.31 ppm, integrating for eight protons. A triplet at 7.31 ppm,integrating for a proton has a coupling constant of 8.0 Hz. HMBC correlated thisproton with C-16, C-19, and C-21 suggesting that it was H-20.
A double-doubletsplitting pattern at chemical shift 7.11 ppm, integrating for a proton, has couplingconstants of 6.3 Hz and 1.6 Hz.
HMBC correlated this proton to C-6, C-7, and C-9 showing that it was H-8. Overlapped signals were observed from 7.04 ppm to7.10 ppm, integrating for five protons. A double-doublet splitting pattern at 7.01ppm with coupling constant 8.0 Hz and 2.0 Hz, integrating for a proton was
observed.
HMBC correlated this proton to C-17 suggesting that it was either H-19or H-21. Four triplet signals were also observed from 2.73 ppm to 4.08 ppm,integrating for a total of twelve protons.
Two of these triplet signals at 2.74 ppmand 3.22 ppm integrated for four protons each, suggesting overlapping signals ofmethylene protons. This was further confirmed by 13C and DEPT NMR.
13C and DEPT NMR data showed the signals of four methylene, eight methineand six quaternary carbon atoms. The DEPT signals at 53.1 ppm and 48.6 ppmhave intensities which were double of those from the rest of the methylene carbonsignals, suggesting two methylene carbon atoms each contributing to the signal at 53.1 ppm and 48.6 ppm.
DEPT

HMQC results further indicated that these two methylene carbon signals at 53.1 ppm and 48.6 ppm were correlated to the protons signal at 2.73 ppm and 4.08 ppm respectively, which corresponded to four protons each. The finding confirmed overlapping methylene carbon signals (at 53.1 ppm and 48.6 ppm) and methylene proton signals (at 2.73 ppm and 4.08 ppm). Hence, the unknown compound has six methylene carbon atoms with a total of twelve methylene protons.
The chemical shifts of the twelve methylene protons suggested that they were attached to relatively electronegative atoms. It was speculated that the six methylene groups were attached to the nitrogen atoms and the electron withdrawing effect of these electronegative nitrogen atoms resulted in the deshielding of the protons. HMBC and COSY correlations were used to assign the rest of the protons
HMBC

HMQC
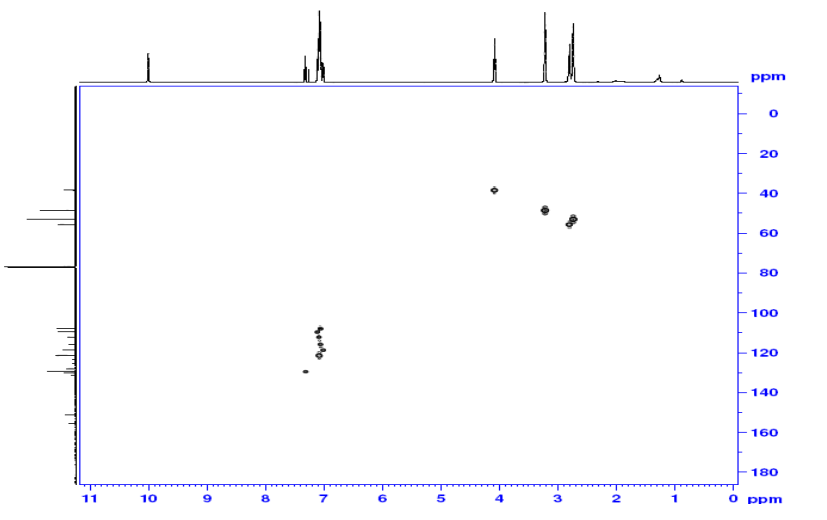
COSY
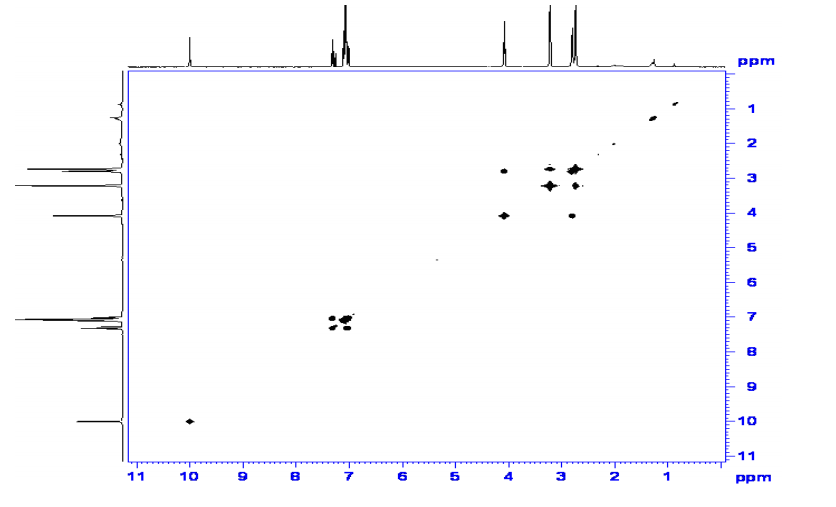
The 13C NMR data showed that there were two quaternary carbon at
155.6 ppm and 151.3 ppm. The carbon with chemical shift 155.6 ppm was C-2. Inthe structure of imidazolone, carbonyl carbon C-2 was attached to two nitrogenatoms which helped to withdraw electrons from oxygen to C-2. Hence, C-2 wasless deshielded as compared to a normal carbonyl carbon which has chemical shiftabove 170 ppm.
Eight methine carbons and two quaternary carbons with chemicalshifts above 108 ppm suggested the presence of two aromatic rings. Thequaternary carbon with chemical shift 125.4 ppm was C-22 which was attached tothree fluorine atoms. Due to the strong electron withdrawing effect of the fluorineatoms, C-22 was highly deshielded and had a high chemical shift.
The IR spectrum of the isolated compound showed absorption bands of amide (νC=O 1685 cm-1, νN-H (stretch) 3180 cm-1, νN-H (bending) 1610 cm-1), alkyl fluoride (νC-F1077 cm-1, 1112 cm-1, 1158 cm-1), aromatic ring (ν Ar-H 3028 cm-1, 3078 cm-1 andνC=C 1401 cm-1, 1446 cm-1, 1453 cm-1, 1468 cm-1, 1487 cm-1) and alkane (νC-H2891 cm-1, 2930 cm-1 2948 cm-).
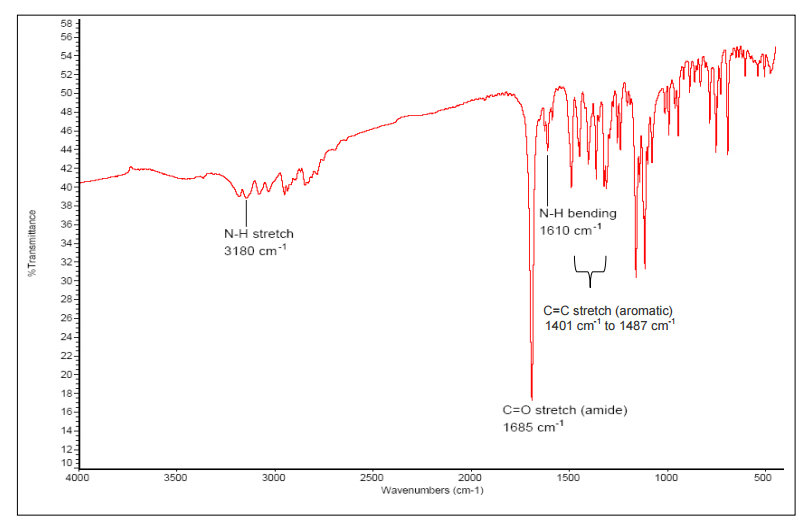
FOR MASS, HMBC ETC SEE………http://orgspectroscopyint.blogspot.in/2015/06/flibanserin.html
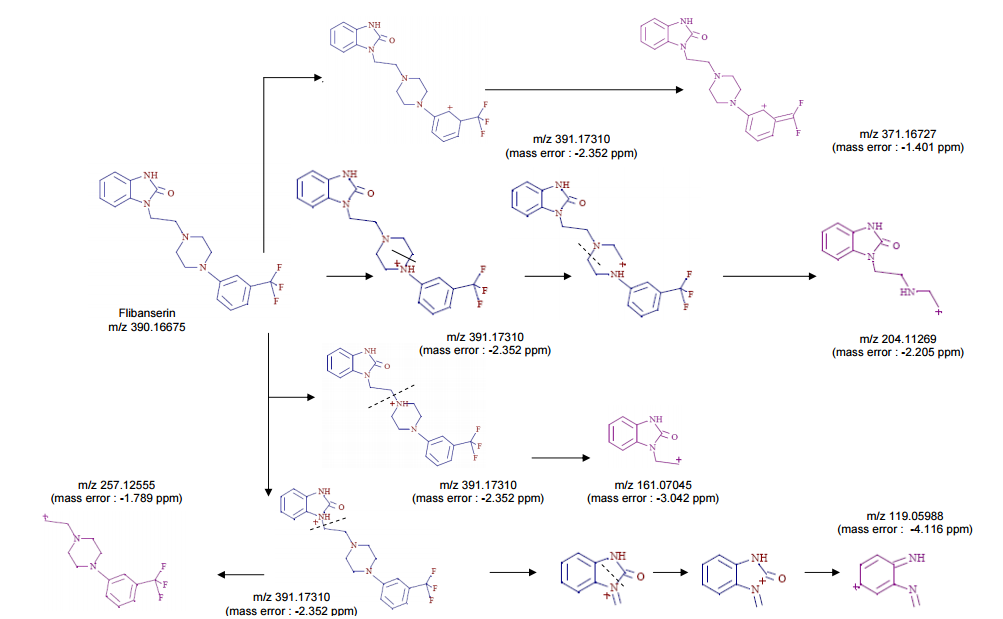
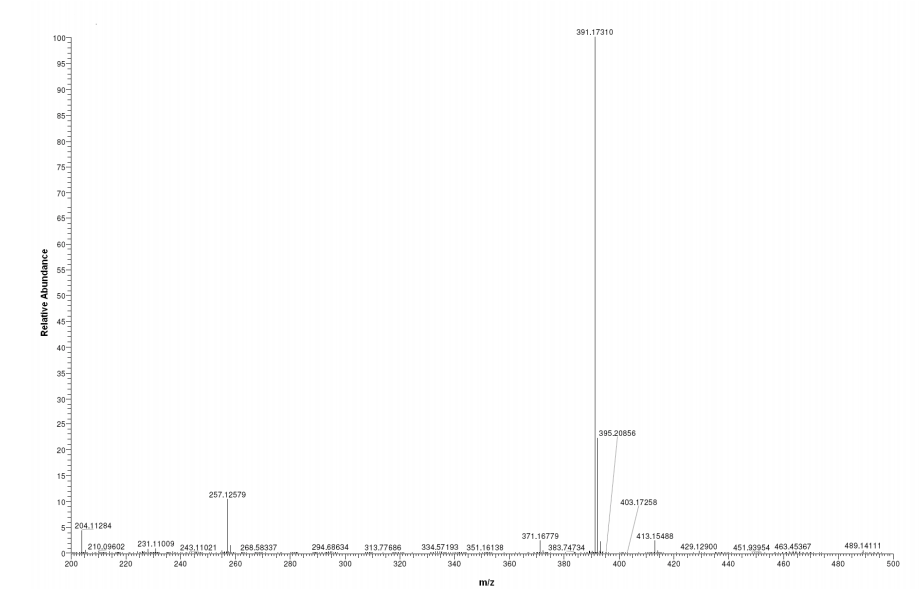
NMR PREDICT
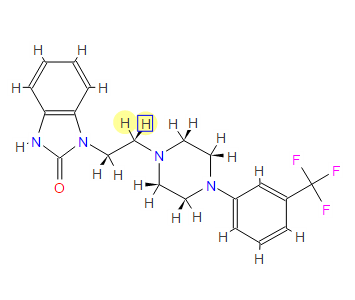
1H NMR PREDICT
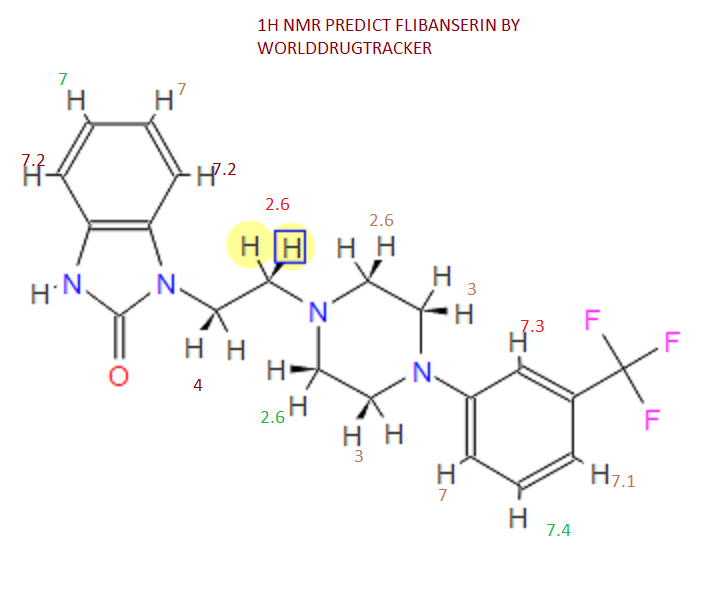
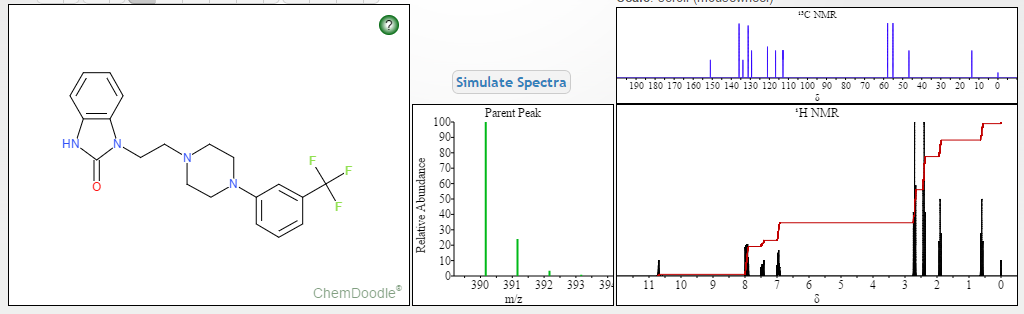
13C NMR PREDICT
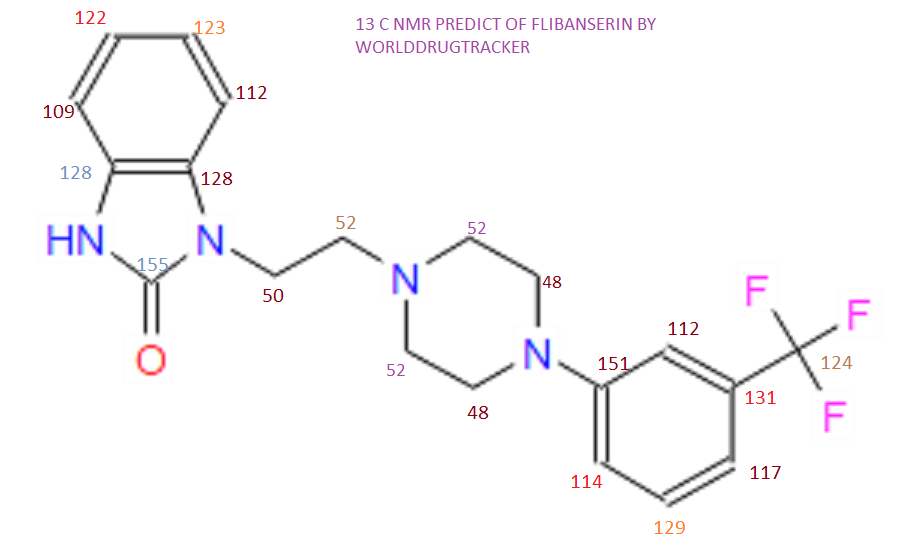
COSY PREDICT
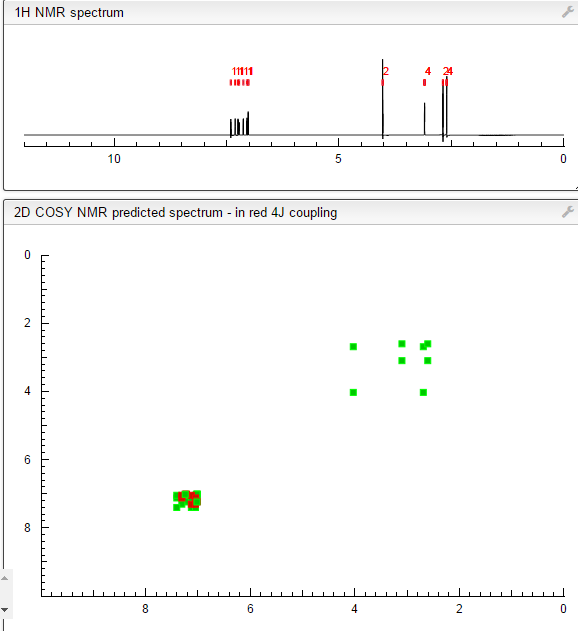
NMR PREDICT FROM MOLBASE
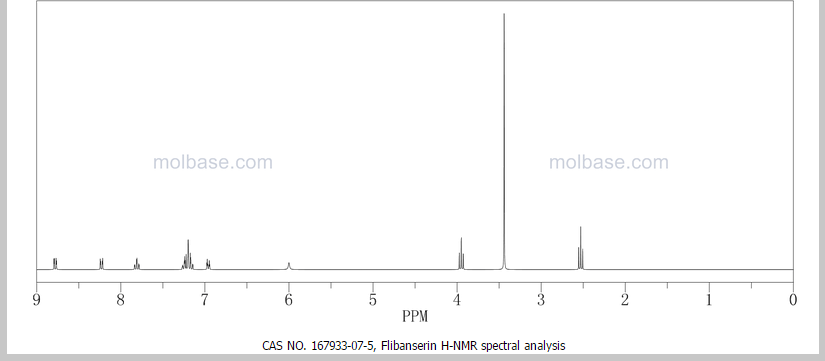
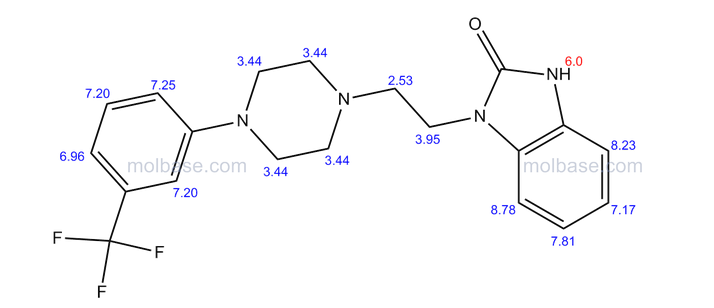
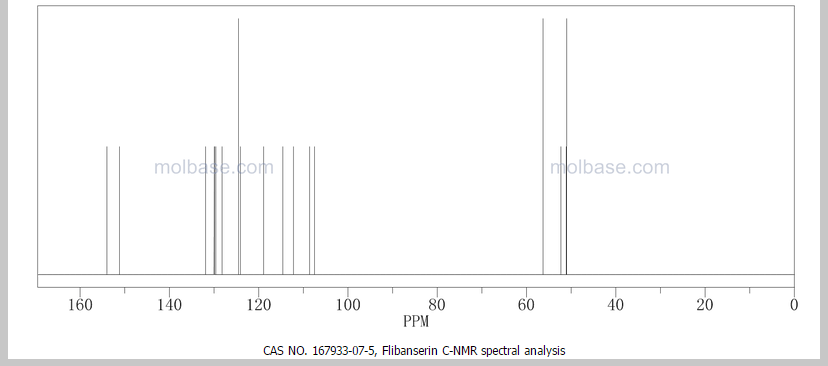

PATENT
US5576318, 1996
1 H NMR (DMSO-d6 /CDCL3 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (SH), 4.36 (t, 2H), 4.1-3.1 (10 H)
UPDATES………..
A Facile Route of Synthesis for Making Flibanserin

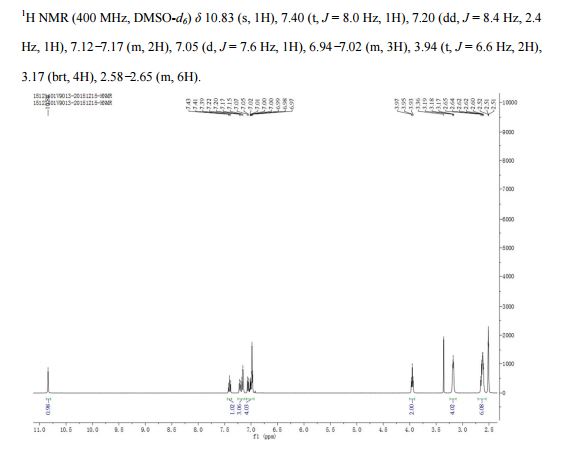
REFERENCES
- Borsini F, Evans K, Jason K, Rohde F, Alexander B, Pollentier S (summer 2002). “Pharmacology of flibanserin”. CNS Drug Rev. 8 (2): 117–142. doi:10.1111/j.1527-3458.2002.tb00219.x. PMID 12177684.
- Jolly E, Clayton A, Thorp J, Lewis-D’Agostino D, Wunderlich G, Lesko L (April 2008). “Design of Phase III pivotal trials of flibanserin in female Hypoactive Sexual Desire Disorder (HSDD)”. Sexologies 17 (Suppl 1): S133–4. doi:10.1016/S1158-1360(08)72886-X.
- Spiegel online: Pharmakonzern stoppt Lustpille für die Frau, 8 October 2010 (in German)
- Nygaard I (November 2008). “Sexual dysfunction prevalence rates: marketing or real?”. Obstet Gynecol 112 (5): 968–9.doi:10.1097/01.AOG.0000335775.68187.b2. PMID 18978094.
- Clayton AH (July 2010). “The pathophysiology of hypoactive sexual desire disorder in women”. Int J Gynaecol Obstet 110 (1): 7–11.doi:10.1016/j.ijgo.2010.02.014. PMID 20434725.
- Pfaus JG (June 2009). “Pathways of sexual desire”. J Sex Med 6 (6): 1506–33. doi:10.1111/j.1743-6109.2009.01309.x.PMID 19453889.
- Yves Aubert, Thesis, Leiden University. (Dec 11, 2012) Sex, aggression and pair-bond: a study on the serotonergic regulation of female sexual function in the marmoset monkey
- Viagra for women?
- Marazziti D, Palego L, Giromella A, et al. (June 2002). “Region-dependent effects of flibanserin and buspirone on adenylyl cyclase activity in the human brain”. Int. J. Neuropsychopharmacol. 5 (2): 131–40. doi:10.1017/S1461145702002869.PMID 12135537.
- Podhorna J, Brown RE (June 2000). “Flibanserin has anxiolytic effects without locomotor side effects in the infant rat ultrasonic vocalization model of anxiety”. Br J Pharmacol 130 (4): 739–746. doi:10.1038/sj.bjp.0703364. PMC 1572126.PMID 10864879.
- Brambilla A, Baschirotto A, Grippa N, Borsini F (December 1999). “Effect of flibanserin (BIMT 17), fluoxetine, 8-OH-DPAT and buspirone on serotonin synthesis in rat brain”. Eur Neuropsychopharmacol 10 (1): 63–7. doi:10.1016/S0924-977X(99)00056-5.PMID 10647099.
| EP0200322A1 * | Mar 18, 1986 | Nov 5, 1986 | H. Lundbeck A/S | Heterocyclic compounds |
| BE904945A1 * | Title not available | |||
| GB2023594A * | Title not available | |||
| US3472854 * | May 29, 1967 | Oct 14, 1969 | Sterling Drug Inc | 1-((benzimidazolyl)-lower-alkyl)-4-substituted-piperazines |
| US4954503 * | Sep 11, 1989 | Sep 4, 1990 | Hoechst-Roussel Pharmaceuticals, Inc. | 3-(1-substituted-4-piperazinyl)-1H-indazoles |
update………..
1-(2-(4-(3-(Trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1H-benzo[d]imidazol-2(3H)-one (1)

A novel and efficient route of synthesis for making flibanserin via 2-ethoxy-1H-benzo[d]imidazole (12) was described with excellent yield. This protocol provided a more facile approach toflibanserin.
A Facile Route of Synthesis for Making Flibanserin
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00108

aReagents and conditions: (a) ethyl benzoylacetate, 200 °C; (b) dichloroethane, NaH, DMF; (c) conc HCl (aq); (d) 1-(3-(trifluoromethyl)phenyl)piperazine hydrochloride, Na2CO3, KI, EtOH; (e)
- 3.Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignotti, M. For treatment of central nervous system disorders. U.S. Patent 5,576,318, 1996.
- 4.Mohan Rao, D.; Krishna Reddy, P.; Venkat Reddy, B. Preparing benzoimidazol-2-one compound, useful to prepare flibanserin, comprises reacting benzoimidazol-2-one compound with 2-(2-hydroxy-ethylamino)-ethanol to give (bis-(hydroxy-ethyl)-amino)-ethyl-benzoimidazol-2-one compound. PCT. Int.WO2,010,128,516, 2010.5.
- 5.Vernin, G.; Domlog, H.; Siv, C.; Metzger, J.; El-Shafei, A. K.Synthesis of 1-alkyl and 1, 3-dialkyl-2-benzimidazolones from 1-alkenyl-2-benzimidazolones using phase-transfer catalysis technique J. Heterocycl. Chem. 1981, 18, 85– 89, DOI: 10.1002/jhet.5570180118
-
A patent application for the new synthetic route has been filed in China (CN201610527244.4).

aReagents and conditions: (a) ethyl acetoacetate, KOH, EtOH, xylene, reflux, 56%; (b) 1,2-dibromoethane, K2CO3, DMF, 50 °C, 50%; (c) K2CO3, CH3CN, 70 °C, 80%; (d) conc. HCl (aq), isopropanol, 70 °C; (e) NaOH (aq), rt, 72% over two steps.

aReagents and conditions: (a) tetraethyl orthocarbonate, AcOH, 70 °C, 94%; (b) 1-bromo-2-chloroethane, K2CO3, acetone, reflux, 75%; (c) K2CO3, NaI, H2O, reflux, 92%; (d) conc. HCl (aq), isopropanol, 70 °C; (e) NaOH (aq), 68% over two steps.
//////////////
Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue
![]()


Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue

INTRODUCTION
Benzimidazol-2-ones 1 are an important class of heterocycles and a privileged scaffold in medicinal chemistry. They consist of cyclic urea fused with the aromatic backbone, which can potentially interact in a biological system by various noncovalent interactions such as hydrogen bonding and π stacking. Benzimidazolone derivatives exhibit a wide range of biological activities, and they are useful in treating various diseases including cancer, type II diabetes, central nervous system disorders, pain management, and infectious disease.1 Selected compounds embedded with a benzimidazol-2-one moiety along with their use are captured in Figure 1. It is worth mentioning that oxatomide drug with a benzimidazol-2-one core was approved for marketing a few years ago.2a Very recently, US Food and Drug Administration approved a new drug called flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in females, which contains benzimidazol-2- one motif.2b
CONCLUSIONS
We have developed a mild and new protocol for the synthesis of benzimidazol-2-ones from quinoxalinediones through decarbonylation. The present methodology can be an addition to the toolbox to prepare benzimidazolones, and it will be useful in medicinal chemistry, particularly, late-stage functionalization of natural products, drug scaffolds, or an intermediate containing quinoxaline-2,3-diones. As direct application of this method, we have successfully developed a new route for the synthesis of recently approved drug flibanserin and a urea analogue of antibiotic natural product hunanamycin A. Later application demonstrates the utility of the present method in late-stage functionalization
Synthesis of 1-(2-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1,3-dihydro-2Hbenzo[d]imidazol-2-one (Flibanserin)
Flibanserin hydrochloride as white solid.
1H NMR (400MHz ,DMSO-d6) 11.06 (s, 1 H), 10.93 (br. s., 1 H), 7.54 – 7.41 (t, J = 7.9 Hz, 1 H), 7.36 – 7.22 (m, 3 H), 7.15 (d, J = 7.6 Hz, 1 H), 7.09 – 7.01 (m, 3 H), 4.30 (t, J = 6.7 Hz, 2 H), 4.01 (d, J = 11.6 Hz, 2 H), 3.75 (d, J = 10.4 Hz, 2 H), 3.54 – 3.43 (d, J = 4.2 Hz 2 H), 3.31 – 3.10 (m, 4 H);
HRMS (ESI): m/z calculated for C20H22ON4F3[M+H]+ 391.1740 found 391.1743;
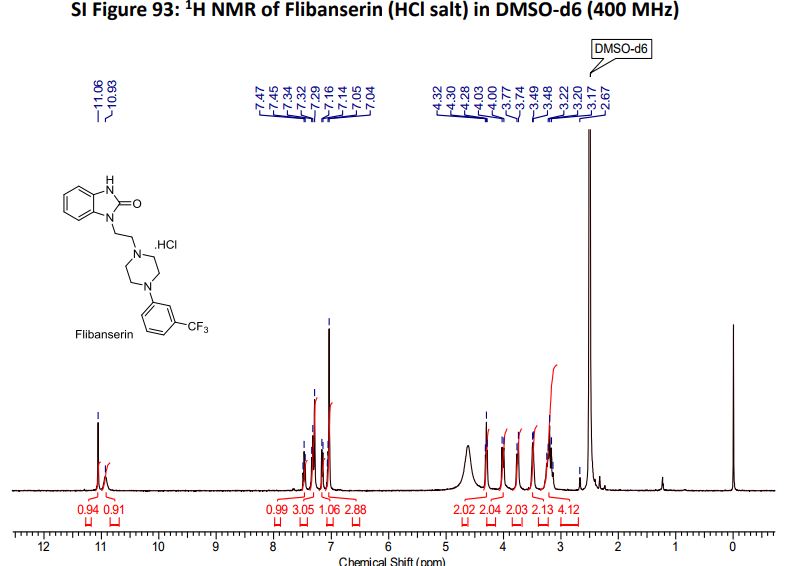
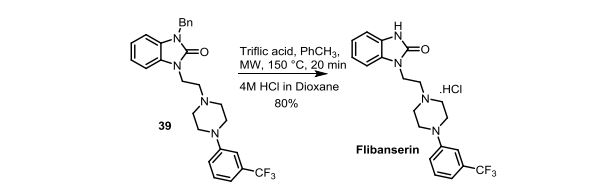

Scheme 4. Synthesis of Flibanserin through Ring Contraction
The same methodology was applied for the synthesis of flibanserin, also known as “female viagra”, which is the first approved medication for treating HSDD in women and is classified as a multifunctional serotonin agonist antagonist.(14, 15) Our synthesis of flibanserin commenced with 1-benzyl-1,4-dihydroquinoxaline-2,3-dione 36,(16) which was reacted with known chloride 37(17) under the basic condition in DMF to give the desired product 38 in good yield. Compound 38 was subjected for the decarbonylative cyclization under the optimized condition to afford the product 39 in 59% yield. Finally, the benzyl group was deprotected using trifluoromethanesulfonic acid in toluene under microwave irradiation,(8b, 18) which gave flibanserin in excellent yield (Scheme 4). The final product was isolated as HCl salt, and all of the spectral data are in agreement with the published data.(15c)

Rahul D. Shingare completed his M.Sc (Chemistry) from Fergusson College, Pune in 2008. He worked as a research associate in Ranbaxy and Lupin New drug discovery center, Gurgaon and Pune respectively until 2012 and currently pursuing his doctoral research in NCL – Pune from 2012.
Current Research Interests: Antibacterial Natural Product Hunanamycin A: Total Synthesis, SAR and Related Chemistry.
e-mail: rd.shingare@ncl.res.in

Akshay Kulkarni completed his M.Sc. from Ferguson College, Pune University in the year 2015 and joined our group as a Project Assistant in the month of October, 2015.
Current research interest: Synthesis of silicon incorporated biologically active antimalerial compounds.
e-mail : as.kulkarni@ncl.res.in
Dr.D. Srinivasa Reddy
Organic Chemistry Division
CSIR-National Chemical Laboratory
-
See, previous synthesis of Flibanserin:
(a) Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignotti, M. For treatment of central nervous system disorders. U.S. Patent 5,576,318, 1996.
(b) Mohan, R. D.; Reddy, P. K.;Reddy, B. V. Process for the preparation of Flibanserin involving novel intermediates. WO2010128516 A2,2010.
(c) Yang, F.; Wu, C.; Li, Z.; Tian, G.; Wu, J.; Zhu, F.; Zhang, J.; He, Y.; Shen, J. A Facile route of synthesis for making Flibanserin Org. Process Res. Dev. 2016, 20, 1576 DOI: 10.1021/acs.oprd.6b00108
[ACS Full Text ], [CAS]
], [CAS] -
Xueong, X. Preparation method of Flibanserin. CN104926734 A, 2015.
//////////
Eluxadoline …Diarrhea-predominant irritable bowel syndrome
Eluxadoline
5 JAN 2014
Furiex Pharmaceuticals Inc. more than doubled in its best day of trading after its experimental drug alleviated diarrhea and abdominal pain caused by irritable bowel syndrome in two studies.
The drug eluxadoline met targets for improvements in stool consistency and abdominal pain that were developed in conjunction with U.S. and European regulators, the company said today. Furiex will apply for approval in June, Chairman Fred Eshelman said in an investor call today. He estimated annual sales of $750 million to $1 billion.
“By our math, it looks like a pretty doggone good market,” Eshelman said on the call, noting that there is only one currently approved drug available in the U.S. for the condition.
Diarrhea-predominant irritable bowel syndrome is a chronic disorder that affects about 28 million patients in the U.S. and Europe, Furiex said in the statement.Furiex said it would apply by mid-year for U.S. approval of the drug, eluxadoline, to treat diarrhea-predominant irritable bowel syndrome (IBS-d), a debilitating bowel disorder that affects about 28 million people in the United States and major European markets.
Furiex said it expected to seek European approval in early 2015.
“We believe that there are a lot of patients out there who need this drug. There is a huge unmet need,” Furiex Chief Medical Officer June Almenoff said in a telephone interview.
Currently approved drugs for IBS address constipation associated with the disorder, but there are few options for diarrhea predominant IBS.
Furiex founder and chairman Fred Eshelman said he believes the drug has the potential for blockbuster sales, which he defined as annual sales of between $750 million and $1 billion.
Eluxadoline was tested at two doses against a placebo over the course of 12 weeks to meet requirements by the U.S. Food and Drug Administration, and for 26 weeks for European health regulators, in Phase III studies involving 2,428 patients, Furiex said.
For the combined goal of improvement in abdominal pain and stool consistency for at least half the days in the study, eluxadoline achieved a statistically significant improvement at the 100 milligram and 75 mg doses through 12 weeks in both studies.
On the 26-week measure, the higher dose succeeded in both studies but the lower dose missed statistical significance in one of the two trials, according to initial results released by the company.
The success appeared to be driven by the percentage of patients reporting improvements in diarrhea, which ranged from 30 percent to 37 percent versus 22 percent and 20.9 percent for the placebo groups.
When the composite goal was broken into its two components, researchers found a numerical improvement in pain response rates that did not achieve statistical significance.
The drug appeared to be safe and well-tolerated in both studies, Furiex said. The most commonly reported side effects were constipation and nausea.
The company plans to present a far more detailed analysis of the late stage studies at an upcoming medical meeting.
“We’re very excited about the path ahead and about how this can transform patients’ lives,” Almenoff said.
Eluxadoline
5-({[(2S)-2-amino-3-(4-carbamoyl-2,6-dimethylphenyl)propanoyl][(1S)-1-(4-phenyl-1H-imidazol-2-yl)ethyl]amino}methyl)-2-methoxybenzoic acid
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
864821-90-9 CAS
JNJ-27018966
Molecular Formula: C32H35N5O5
Molecular Weight: 569.6508
Agents for Irritable Bowel Syndrome, mu-Opioid Agonists, delta-Opioid Antagonists
Mu Delta is a locally active mu opioid receptor agonist and delta opioid receptor antagonist in phase III clinical evaluation at Furiex Pharmaceuticals for the oral treatment of diarrheal predominant irritable bowel syndrome (d-IBS).
The product candidate holds an advantage over currently marketed products for this indication because it acts locally on the enteric nervous system, possibly decreasing adverse effects on the central nervous system. In 2011, fast track designation was assigned in the U.S. for the treatment of d-IBS. In 2011, Mu Delta was licensed to Furiex Pharmaceuticals by Janssen for the treatment of d-IBS, granting an option to Furiex to continue development and commercialization following phase II proof of concept studies.
The opioid receptors were identified in the mid-1970’s, and were quickly categorized into three sub-sets of receptors (mu, delta and kappa). More recently the original three types of receptors have been further divided into sub-types. Also known is that the family of opioid receptors are members of the G-protein coupled receptor (GPCR) super-family. More physiologically pertinent are the well established facts that opioid receptors are found throughout the central and peripheral nervous system of many mammalian species, including humans, and that modulation of the respective receptors can elicit numerous, albeit different, biological effects, both desirable and undesirable (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye, T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269; J. V. Aldrich, “Analgesics”, Burger’s Medicinal Chemistry and Drug Discovery, 5thEdition, Volume 3: Therapeutic Agents, John Wiley & Sons, Inc., 1996, pp. 321-441). In the most current literature, the likelihood of heterodimerization of the sub-classes of opioid receptors has been reported, with respective physiological responses yet undetermined (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development”, Drug Development 2000, pp. 203-238).
A couple biological effects identified for opioid modulators have led to many useful medicinal agents. Most significant are the many centrally acting mu opioid agonist modulators marketed as analgesic agents to attenuate pain (e.g., morphine), as well as peripherally acting mu agonists to regulate motility (e.g., loperamide). Currently, clinical studies are continuing to evaluate medicinal utility of selective delta, mu, and kappa modulators, as well as compounds possessing combined sub-type modulation. It is envisioned such explorations may lead to agents with new utilities, or agents with minimized adverse side effects relative to currently available agents (examples of side effects for morphine includes constipation, respiratory depression, and addiction potential). Some new GI areas where selective or mixed opioid modulators are currently being evaluated includes potential treatment for various diarrheic syndromes, motility disorders (post-operative ileus, constipation), and visceral pain (post operative pain, irritable bowel syndrome, and inflammatory bowel disorders) (Pierre J. M. Riviere and Jean-Louis Junien, “Opioid receptors: Targets for new gastrointestinal drug development” Drug Development, 2000, pp. 203-238).
Around the same time the opioid receptors were identified, the enkephalins were identified as a set of endogenous opioid ligands (D. S. Fries, “Analgesics”, inPrinciples of Medicinal Chemistry, 4th ed.; W. O. Foye; T. L. Lemke, and D. A. Williams, Eds.; Williams and Wilkins: Baltimore, Md., 1995; pp. 247-269). Schiller discovered that truncating the original pentapeptide enkephalins to simplified dipeptides yielded a series of compounds that maintained opioid activity (Schiller, P. WO 96/06855). However one potential drawback cited for such compounds is the likelihood of their inherent instability (P. W. Schiller et al., Int. J. Pept. Protein Res. 1993, 41 (3), pp. 313-316).
More recently, a series of opioid pseudopeptides containing heteroaromatic or heteroaliphatic nuclei were disclosed, however this series is reported showing a different functional profile than that described in the Schiller works. (L. H. Lazarus et al., Peptides 2000, 21, pp. 1663-1671).
Most recently, works around morphine related structures were reported by Wentland, et al, where carboxamido morphine derivatives and it’s analogs were prepared (M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 1717-1721; M. P. Wentland et al., Biorg. Med. Chem. Letters 2001, 11, pp. 623-626). Wentland found that substitution for the phenol moiety of the morphine related structures with a primary carboxamide led anywhere from equal activities up to 40 fold reduced activities, depending on the opioid receptor and the carboxamide. It was also revealed that any additional N-substitutions on the carboxamide significantly diminished the desired binding activity.
Compounds of the present invention have not been previously disclosed and are believed to provide advantages over related compounds by providing improved pharmacological profiles.
Opioid receptor modulators, agonists or antagonists are useful in the treatment and prevention of various mammalian disease states, for example pain and gastrointestinal disorders such as diarrheic syndromes, motility disorders including post-operative ileus and constipation, and visceral pain including post-operative pain, irritable bowel syndrome and inflammatory bowel disorders.
It is an object of the present invention to provide opioid receptor modulators. It is a further object of the invention to provide opioid receptor agonists and opioid receptor antagonists. It is an object of the present invention to provide opioid receptor ligands that are selective for each type of opioid receptor, mu, delta and kappa. It is a further object of the present invention to provide opioid receptor ligands that modulate two or three opioid receptor types, mu, delta and kappa, simultaneously.
It is an object of the invention to provide certain instant compounds that are also useful as intermediates in preparing new opioid receptor modulators. It is also an object of the invention to provide a method of treating or ameliorating a condition mediated by an opioid receptor. And, it is an object of the invention to provide a useful pharmaceutical composition comprising a compound of the present invention useful as an opioid receptor modulator.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1 h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid is an opoid receptor modulator (mu receptor agonist and delta receptor antagonist) and may be useful for treating irritable bowel syndrome, pain or other opioid receptor disorders.
5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and methods of making this molecule are disclosed in
US application 2005/02033143. Example 9 of US application 2005/02033143 makes the hydrochloride salt of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
Applicants have discovered a process of making the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid and two novel crystals of this zwitterion. In Applicant’s hands, these novel crystals provide improved properties and can be purified at higher purity. Applicant’s new process results in improved and less costly process manufacturing conditions than the procedure disclosed in US application 2005/02033143.
………………..
FIG. 6 is the molecular structure of the zwitterion 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1h-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.

…………………..
SYNTHESIS OF 5-formyl-2- methoxy-benzoic acid methyl ester
Example 8: 2-Methoxy-5-formylbenzoic acid

Lithium hydroxide (1.04g, 0.043mol, 3eq) in water (lOmL) was added to a stirred solution of methyl 2-methoxy-5-formylbenzoate (2.8g, 0.014mol, leq) in a mixture of tetrahydrofuran (30mL) and methanol (20mL). The solution was stirred overnight, acidified to pH 1 with 10% HCl and the organic solvents removed in vacuo. The aqueous solution was extracted with ethyl acetate (lOOmL) and the organic solution washed with brine (lOOmL), then extracted with saturated aqueous sodium bicarbonate (3 x lOOmL). The basic solution was washed with ethyl acetate (lOOmL), then acidified to pH 1 with 10% HCl and back extracted with dichloromethane (3 x lOOmL). The organic solution was dried over sodium sulfate and evaporated in vacuo to give a cream coloured powder (2.01g, 77%). 1H NMR (CDC13) δ 9.99 (s, IH, O=C- H), 4.14 (s, 3H, CH3).
………………
ANALOGOUS METHOD TO PREPARE..2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester

USE 5-formyl-2- methoxy-benzoic acid methyl ester for 3,4- dimethoxybenzaldehyde, TO GET 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester
Example 4
(3,4-Dimethoxy-benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine

A solution of 1-(4-phenyl-1 W-imidazol-2-yl)-ethylamine (0.061 g, 0.33 mmol) of Example 3, and 0.55 g (0.33 mmol) of 3,4-dimethoxybenzaldehyde in 5 ml_ of anhydrous methanol was stirred at room temperature for 1 h and then cooled to about 0-100C in an ice bath for 1 h. The reaction was treated carefully with 0.019 g (0.49 mmol) of sodium borohydride in one portion and maintained at about 0-100C for 21 h. Cold 2M aqueous HCI was added dropwise (30 drops), the mixture was stirred for 5 min, and then partially concentrated in vacuo unheated. The residual material was taken up in EtOAc to yield a suspension that was treated with 5 ml_ of cold 3M aqueous NaOH and stirred vigorously until clear. The phases were separated and the aqueous layer was extracted three times additional with EtOAc. The combined extracts were dried over MgSO4, filtered, and concentrated to yield (3,4-dimethoxy- benzyl)-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amine as a light yellow oil (HPLC: 87% @ 254nm and 66% @ 214 nm).
MS (ES+) (relative intensity): 338.1 (100) (M+1)
This sample was of sufficient quality to use in the next reaction without further purification.
…………………..
SYNTHESIS
In an embodiment, the present invention is directed to processes for the preparation of the compound of formula (IV)

also known as, 5-({[2-amino-3-(4-carbamoyl-2,5-dimethyl-phenyl)- propionyl]-[1 -(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid
Example 1
(S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2.6-dimethyl-phenyl)- propionic acid


STEP A: Trifluoromethanesulfonic acid 4-bromo-3,5-dimethyl-phenyl ester
To a cooled (0°C) solution of 4-bromo-3,5-dimethylphenol (3.05 g, 15.2 mmol) in pyridine (8 ml_) was added trifluoromethanesulfonic anhydride (5.0 g, 17.7 mmol) dropwise. After completion of addition, the resulting mixture was stirred at 0°C for 15 min, and then at room temperature overnight. The reaction was quenched by addition of water, and then extracted with EtOAc. The organic extracts were washed sequentially with water, 2N HCI (2x ), brine, and then dried over MgSO4. Filtration and evaporation to dryness yielded compound 1 b as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 2.45 (6H, s), 7.00 (2H, s).
Step B: 4-Bromo-3,5-dimethylbenzoic acid
Into a solution of compound 1 b (6.57 g, 19.7 mmol) in DMF (65 ml_) were added K2CO3 (13.1 g, 94.7 mmol), Pd(OAc)2 (0.44 g, 1.97 mmol) and 1 ,1′-bis(diphenylphosphino)ferrocene (2.29 g, 4.14 mmol). The resulting mixture was bubbled in gaseous CO for 10 min and was heated to 60°C for 7.5h with a CO(9) balloon. The cooled mixture was partitioned between aqueous NaHCO3 and EtOAc, and filtered. The aqueous phase was separated, acidified with aqueous 6N HCI, extracted with EtOAc, and then dried over Na2SO4. Filtration and concentration of the filtrate yielded crude compound 1c as a brown residue, which was used in the next step without further purification. STEP C: Method A: 4-Bromo-3,5-dimethyl-benzamide
Into a suspension of compound 1c in DCM (40 ml_) was added SOCI2 (3.1 rnL, 42 mmol) and the mixture was heated at reflux for 2 h. Upon removal of the solvent by evaporation, the residue was dissolved in DCM (40 ml_) and then ammonium hydroxide (28% NH3 in water, 2.8 ml_) was added. The reaction mixture was heated at 5O0C for 2 h and concentrated. The residue was diluted with H2O, extracted with EtOAc, and the organic portion was dried over Na2SO4. After filtration and evaporation, the residue was purified by flash column chramotagraphy (eluent: EtOAc) to yield compound 1 d as an off-white solid.
1H NMR (300 MHz, CD3CN): δ 2.45 (6H, s), 5.94 (1 H, br s), 6.71 (1 H, br s), 7.57 (2H, s)
MS(ES+)(relative intensity): 228.0 (100%) (M+1).
Step C: Method B: 4-Bromo-3,5-dimethyl-benzamide
A mixture of compound 1 b (3.33 g, 10 mmol), PdCI2 (0.053 g, 0.3 mmol), hexamethyldisilazane (HMDS, 8.4 ml_, 40 mmol), and DPPP (0.12 g, 0.3 mmol) was bubbled with a gaseous CO for 5 min and then stirred in a CO balloon at 80°C for 4 h. To the reaction mixture was added MeOH (5 ml_). The reaction mixture was stirred for 10 min, diluted with 2N H2SO4 (200 ml_), and then extracted with EtOAc. The EtOAc extract was washed with saturated aqueous NaHCO3, brine, and then dried over Na2SO4. Filtration and evaporation of the resultant filtrate yielded a residue, which was purified by flash column chromatography (eluent: EtOAc) to yield compound 1d as a white solid.
Step D: 2-terf-Butoxycarbonylaminoacrylic acid methyl ester
To a suspension of /V-Boc-serine methyl ester (Compound 1e, 2.19 g, 10 mmol) and EDCI (2.01 g, 10.5 mmol) in DCM (70 ml_) was added CuCI (1.04 g, 10.5 mmol). The reaction mixture was stirred at room temperature for 72 h. Upon removal of the solvent, the residue was diluted with EtOAc, washed sequentially with water and brine and then dried over MgSO4. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :4) to yield compound 1f as a colorless oil.
1H NMR (300 MHz, CDCI3): δ 1.49 (9H, s), 3.83 (3H, s), 5.73 (1 H, d, J = 1.5 Hz), 6.16 (1 H1 S), 7.02 (1 H, s).
STEP E: (2)-2-fert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)acrylic acid methyl ester
A flask charged with compound 1d (0.46 g, 2.0 mmol), compound 1f (0.80 g, 4.0 mmol), tri-o-tolylphosphine (0.098 g, 0.32 mmol) and DMF (8 ml_) was purged with N2(g) 3 times. After the addition of tris(dibenzylideneacetone)dipalladium (0) (0.074 g, 0.08 mmol) and TEA (0.31 ml_, 2.2 mol), the reaction mixture was heated at 110°C for 24 h. At that time, the reaction was quenched by addition of water, and then extracted with EtOAc. The organic phase was washed with 1 N HCI, saturated aqueous NaHCO3, brine, and dried over MgSO4. The mixture was concentrated to a residue, which was purified by flash column chromatography (eluent: EtOAc:hexane~1 :1 to EtOAc only) to yield compound 1g as a white solid.
1H NMR (300 MHz, CD3OD): δ 1.36 (9H, s), 2.26 (6H, s), 3.83 (3H, s), 7.10 (1 H, s), 7.56 (2H, s); 13C NMR (75 MHz, DMSO-d6): δ 17.6, 25.7, 50.2, 78.7, 124.9, 126.4,
128.3, 131.2, 135.2, 135.5, 152.8, 164.3, 169.6;
MS (ES+) (relative intensity): 349.1 (38%)(M+1).
STEP F: (S)-2-ferf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid methyl ester
Into a reactor charged with a solution of compound 1g (0.56 g, 1.6 mmol) in degassed MeOH (80 mL) was added [Rh(COd)(H1R-DIPAMP)J+BF4 “ under a stream of argon. The reactor was sealed and flushed with H2, stirred at 6O0C under 1000 psi of H2 for 14 days. The crude product was purified by flash column chromatography (eluent: EtOAc:hexane ~1 :1) to yield compound 1 h as a white solid. ee: >99%; 1H NMR (300 MHz, CDCI3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J = 7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1 H, m), 5.12 (1 H, d, J = 8.7 Hz), 5.65 (1 H, br s), 6.09 (1 H, br s), 7.46 (2H, s);
MS(ES+) (relative intensity): 250.9 (100) (M-BoC)+.
STEP G: (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid
Into an ice-cooled solution of compound “I h (0.22 g, 0.63 mmol) in THF (3.5 ml_) was added an aqueous LiOH solution (1 N, 3.5 ml_) and the reaction mixture stirred at 0°C. Upon completion of the reaction, the reaction mixture was concentrated and the aqueous phase was neutralized with cooled aqueous 1 N HCI at 0°C, and then extracted with EtOAc. The combined extracts were dried over Na2SO4 overnight. Filtration and evaporation of the filtrate to dryness yielded compound 1j as a white solid. 1H NMR (300 MHz, DMSO-cfe): δ 1.30 (9H, s), 2.32 (6H, s), 2.95(1 H, dd,
J= 8.8, 13.9 Hz), 3.10 (1 H, dd, J= 6.2, 14.0 Hz), 4.02-4.12 (1 H, m), 7.18-7.23 (2H, m), 7.48 (2H1 s), 7.80 (1 H, s);
MS(ES+) (relative intensity): 236.9 (6) (M-BoC)+.
Example 5
5-((r2-Amino-3-(4-carbamoyl-2.6-dimethyl-phenyl)-propionvn-n-(4-phenyl- 1 H-imidazol-2-yl)-ethvπ-aminol-methyl)-2-methoxy-benzoic acid


STEP A. 2-Methoxy-5-{[1-(4-phenyl-1 W-imidazol-2-yl)-ethylamino]-methyl}- benzoic acid methyl ester
Using the procedures described for Example 4, substituting 5-formyl-2- methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4- dimethoxybenzaldehyde, 2-methoxy-5-{[1 -(4-phenyl-1 H-imidazol-2-yl)- ethylamino]-methyl}-benzoic acid methyl ester was prepared.
STEP B. 5-({[2-ferf-ButoxycarbonylmethyI-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 H-imidazoI-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid methyl ester
Using the procedure of Example 3 for the conversion of Cpd 3d to Cpd 3e, substituting 2-methoxy-5-{[1-(4-phenyl-1 /-/-imidazol-2-yl)-ethylamino]- methylj-benzoic acid methyl ester for Cpd 3d and substituting 2-tert- Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionic acid for 2- tø/t-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 5a was prepared.
STEP C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)-propionyl]-[1 -(4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2- methoxy-benzoic acid
5-({[2-tørf-Butoxycarbonylmethyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)- propionyl]-[1-(4-phenyl-1 H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy- benzoic acid methyl ester was dissolved in an ice-chilled (0-10°C), mixed solvent system of THF (10 ml_) and MeOH (5 ml_). A LiOH H2O/water suspension (2.48 M; 3.77 ml_) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3 x 26 ml_). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to yield a pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2CI2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOHZCH2CI2 system as follows: Initial 100% CH2CI2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 5-({[2-terf- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4- phenyl-1 /-/-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 5b, as a white solid.
STEP D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1 – (4-phenyl-1 W-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A portion of Cpd 5b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 ml_)/THF (5 ml_), filtered, and subsequently treated with gaseous HCI for 15 min. After completion of the HCI addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield Cpd 5c as a white solid di-HCI salt.
Example 2
Racemic 2-terf-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethvl- phenvD-propionic acid

STEP A: Racemic 2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid methyl ester
To a reactor charged with a solution of compound 1g (0.68 g, 1.95 mmol) in MeOH (80 mL) was added 10% Pd-C (0.5 g). The reactor was connected to a hydrogenator and shaken under 51 psi of H2 overnight. The mixture was filtered through a pad of Celite and the filtrate was concentrated to dryness to yield compound 2a as a white solid.
The 1H NMR spectrum was identical to that of (S)-2-tert- butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)propionic acid methyl ester, compound 1 h.
STEP B: Racemic 2-terf-butoxycarbonylamino-3-(4-carbamoyl-2,6- dimethyl-phenyl)propionic acid
Following the procedure described for Example 1 , STEP G (preparation of (S)-2-teAt-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl- phenyl)propionic acid), compound 2b – racemic 2-te/?-butoxycarbonylamino-3- (4-carbamoyl-2,6-dimethyl-phenyl)propionic acid – was prepared.
…………….
POLYMORPHS
Example 1 Preparation of the zwitterion of 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid
A 1 L three-necked round-bottomed flask equipped with a mechanical stirrer, addition funnel and a thermocouple was charged without agitation. 34.2 g of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid (see Example 9 of US 2005/0203143), 340 ml of acetone, and 17 ml of 204 mmolar concentrated HCl were combined in the flask. The stirring was started and the resulting slurry formed a clear solution. This solution was heated to 45° C. under vigorous stirring and aged at this temperature for a period of two hours. After the completion, the reaction mass was cooled to ambient temperature and the supernatant was removed by suction. The vessel along with the residue was rinsed with 20 ml of acetone and then removed as previously. 170 ml of water was added and the reaction mass and was aged under stirring until a homogeneus solution resulted. This solution was then added over a period of ˜½ hr to a solution of 90 ml of 1N NaOH and water. The pH was adjusted to 6.5-7.0 accordingly. The resulting slurry was aged for about 2 hrs at ambient temperature, cooled to 10-15° C., aged at that temperature for about 1 hr, and then filtered. The solid was washed with 10 ml water, air-dried for a period of 4 to 5 hrs, and then placed in a vacuum oven at 50-55° C. until the water content was less than 3%.
Example 2 Preparation of the Form α Crystal
The Form α crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at 0-25% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1-3 respectively.
Example 3 Preparation of the Form β crystal
The Form β crystal can be prepared by storing the zwitterion of 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid at greater than 60% relative humidity for 3 days. Representative PXRD, TGA, and DSC data are shown in FIGS. 1, 4, and 5 respectively.
…………….
SYNTHESIS
Example 9 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid

A. 2-Methoxy-5{[1-(4-phenyl-1 H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester.
Using the procedures described for Example 3, substituting 5-formyl-2-methoxy-benzoic acid methyl ester (WO 02/22612) for 3,4-dimethoxybenzaldehyde, 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester was prepared.
B. 5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester.
Using the procedure of Example 1 for the conversion of Cpd 1d to Cpd 1e, substituting 2-methoxy-5-{[1-(4-phenyl-1H-imidazol-2-yl)-ethylamino]-methyl}-benzoic acid methyl ester for Cpd 1 d and substituting 2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl-propionic acid of Example 8 for 2-tert-Butoxycarbonylamino-3-(4-hydroxy-2,6-dimethyl-phenyl)-propionic acid, Cpd 9a was prepared.
C. 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[11-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
5-({[2-tert-Butoxycarbonyl methyl-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid methyl ester was dissolved in an ice-chilled (0-10° C.), mixed solvent system of THF (10 mL) and MeOH (5 mL). A LiOH.H2O/water suspension (2.48 M; 3.77 mL) was added dropwise, then the reaction was allowed to warm to room temperature and stirred overnight. The resulting mixture was cooled in an ice bath and the basic solution was neutralized with 2N citric acid until slightly acidic. The mixture was concentrated under reduced pressure to remove the volatile materials, after which time the remaining aqueous phase was extracted with EtOAc (3×26 mL). These combined organic phases were dried over MgSO4, filtered, and concentrated under reduced pressure to give 2.26 g (146% of theory) of pale yellowish white solid. This crude material was dissolved in a 10% MeOH/CH2Cl2 solution and adsorbed onto 30 g of silica. The adsorbed material was divided and chromatographed on an ISCO normal phase column over two runs, using a 40 g Redi-Sep column for both runs. The solvent system was a gradient MeOH/CH2Cl2 system as follows: Initial 100% CH2Cl2, 98%-92% over 40 min; 90% over 12 min, and then 88% over 13 min. The desired product eluted cleanly between 44-61 min. The desired fractions were combined and concentrated under reduced pressure to yield 1.74 g (113% of theory) of 5-({[2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid, Cpd 9b, as a white solid.
D. 5-({[2-Amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid.
A portion of Cpd 9b (0.27g, 0.41 mmol) was dissolved in EtOAc (39 mL)/THF (5 mL), filtered, and subsequently treated with gaseous HCl for 15 min. After completion of the HCl addition, the reaction was slowly warmed to room temperature and a solid precipitate formed. After 5 h the reaction appeared >97% complete by LC (@214 nm; 2.56 min.). The stirring was continued over 3 d, then the solid was collected and rinsed with a small amount of EtOAc. The resulting solid was dried under high vacuum under refluxing toluene for 2.5 h to yield 0.19 g (71%) of desired Cpd 9c as a white solid di-HCl salt.
Example 8 (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester

A. (S)-2-tert-Butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester. Into a cool solution of Boc-L-(2,6-diMe)Tyr-OMe (7.0 g, 21.6 mmol; Sources: Chiramer or RSP AminoAcidAnalogues) and N-phenyltrifluoromethanesulfonimide (7.9 g, 22.0 mmol) in dichloromethane (60 mL) was added triethylamine (3.25 mL, 23.3 mmol). The resulting solution was stirred at 0° C. for 1 h and slowly warmed to rt. Upon completion, the reaction was quenched by addition of water. The separated organic phase was washed with 1 N NaOH aqueous solution, water and dried over Na2SO4 overnight. After filtration and evaporation, the residue was purified by flash column chromatography (eluent: EtOAc-hexane: 3:7) to give the desired product (9.74 g, 99%) as a clear oil; 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.06 (2H, d, J=7.7 Hz), 3.64 (3H, s), 4.51-4.59 (1H, m), 5.12 (1H, d, J=8.5 Hz), 6.92 (2H, s); MS (ES+) (relative intensity): 355.8 (100) (M−Boc)+.
B. (S)4-(2-tert-Butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethylbenzoic acid. To a suspension of (S)-2-tert-butoxycarbonylamino-3-(2,6-dimethyl-4-trifluoromethanesulfonylphenyl)-propionic acid methyl ester (9.68 g, 21.3 mmol), K2CO3 (14.1 g, 0.102 mol), Pd(OAc)2 (0.48 g, 2.13 mmol) and 1,1′-bis(diphenylphosphino)ferrocene (2.56 g, 4.47 mmol) in DMF (48 mL) was bubbled in gaseous CO for 15 min. The mixture was heated to 60° C. for 8 h with a CO balloon. The cool mixture was partitioned between NaHCO3 and EtOAc, and filtered. The aqueous layer was separated, acidified with 10% citric acid aqueous solution, extracted with EtOAc, and finally dried over Na2SO4. Filtration and concentration of the filtrate resulted in a residue. The residue was recrystallized from EtOAc-hexanes to afford the desired product (7.05 g, 94%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.42 (6H, s), 3.14 (2H, J=7.4 Hz), 3.65 (3H, s), 4.57-4.59 (1H, m), 5.14 (1H, d, J=8.6 Hz), 7.75 (2H, s); MS(ES+) (relative intensity): 251.9 (100) (M−Boc)+.
C. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid methyl ester. Into a stirring solution of (S)-4-(2-tert-butoxycarbonylamino-2-methoxycarbonylethyl)-3,5-dimethyl benzoic acid (3.00 g, 8.54 mmol), PyBOP (6.68 g, 12.8 mmol) and HOBt (1.74 g, 12.8 mmol) in DMF (36 mL) was added DIPEA (5.96 mL, 34.2 mmol) and NH4Cl (0.92 g, 17.1 mmol). The resulting mixture was stirred at rt for 40 min before being partitioned between aqueous NH4Cl solution and EtOAc. The separated organic phase was washed sequentially with 2N citric acid aqueous solution, saturated aqueous NaHCO3 solution, and brine, then dried over Na2SO4 overnight. After filtration and concentration, the residue was purified by flash column chromatography (eluent: EtOAc) to give the product. (3.00 g, 100%); 1H NMR (300 MHz, CDCl3): δ 1.36 (9H, s), 2.39 (6H, s), 3.11 (2H, J=7.2 Hz), 3.65 (3H, s), 4.53-4.56 (1H, m), 5.12 (1H, d, J=8.7 Hz), 5.65 (1H, brs), 6.09 (1H, br s), 7.46 (2H, s); MS(ES+) (relative intensity): 250.9 (100) (M−Boc)+.
D. (S)-2-tert-Butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid. Into an ice-cooled solution of methyl ester from Step C (2.99 g, 8.54 mmol) in THF (50 mL) was added an aqueous LiOH solution (1N, 50 mL) and stirred at 0° C. Upon consumption of the starting materials, the organic solvents were removed and the aqueous phase was neutralized with cooled 1N HCl at 0° C., and extracted with EtOAc, and dried over Na2SO4 overnight. Filtration and evaporation to dryness led to the title acid (S)-2-tert-butoxycarbonylamino-3-(4-carbamoyl-2,6-dimethylphenyl)propionic acid (2.51 g, 87%); 1H NMR (300 MHz, DMSO-d6): δ 1.30 (9H, s), 2.32 (6H, s), 2.95 (1H, dd, J=8.8, 13.9 Hz), 3.10 (1H, dd, J=6.2, 14.0 Hz), 4.02-4.12 (1H, m), 7.18-7.23 (2H, m), 7.48 (2H, s), 7.80 (1H, s); MS(ES+) (relative intensity): 236.9 (6) (M−Boc)+.
…………………..
PATENTS
1.WO 2005090315
2..WO 2006099060
3.WO 2009009480
4. WO 2010062590
5.US 2011263868 *
|
12-24-2010
|
NOVEL COMPOUNDS AS OPIOID RECEPTOR MODULATORS
|
|
|
8-32-2010
|
Compounds as opioid receptor modulators
|
|
|
6-23-2010
|
Compounds as opioid receptor modulators
|
|
|
2-12-2010
|
PROCESS FOR THE PREPARATION OF OPIOD MODULATORS
|
|
|
12-9-2009
|
Process for the preparation of opioid modulators
|
| US7629488 * | Mar 6, 2006 | Dec 8, 2009 | Janssen Pharmaceutica N.V. | Process for the preparation of opioid modulators |
| US7741356 * | Mar 14, 2005 | Jun 22, 2010 | Janssen Pharmaceutica N.V. | Compounds as opioid receptor modulators |
| US7786158 * | Oct 24, 2007 | Aug 31, 2010 | Janssen Pharmaceutica N.V. | Compounds as opioid receptor modulators |
| US7994206 | Jul 7, 2008 | Aug 9, 2011 | Janssen Pharmaceutica, N.V. | Crystals and process of making 5-({[2-amino-3-(4-carbamoyl-2,6-dimethyl-phenyl)-propionyl]-[1-(4-phenyl-1H-imidazol-2-yl)-ethyl]-amino}-methyl)-2-methoxy-benzoic acid |
| CN1950342A | Mar 14, 2005 | Apr 18, 2007 | 詹森药业有限公司 | Novel compounds as opioid receptor modulators |
Update july 2015
Eluxadoline
Trade Name: Viberzi®
Research Code: JNJ-27018966, JNJ27018966, JNJ 27018966
Chemical Name: 5 – [[[(2S) -2-amino-3- [4- (aminocarbonyl) -2,6-dimethylphenyl ] -1- oxopropyl] [(1S) -1- (4-phenyl-1H-imidazol-2-yl) ethyl] amino] methyl] -2-methoxybenzoic acid
CAS No: 864821-90-9
MOA: mu opioid receptor agonist
Indication: Irritable bowel syndrome with diarrhea (IBS-D)
Approval Date: May 27, 2015 (US)
Originator: Furiex Pharmaceuticals Inc ( Furiex acquired Eluxadoline from Janssen in 2011 )
Developer: Forest Laboratories Inc. (acquired by Actavis PLC in 2014 )

Lodenafil Carbonate … an Erectile Dysfunction Drug in Phase III

Lodenafil carbonate
UNII-29X84F932D, CRIS-031
bis-(2-{4-[4-ethoxy-3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-benzenesulfonyl]piperazin-1-yl}-ethyl)carbonate
5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one. IS THE NAME OF MONOMER
398507-55-6 CAS
Cristalia (Originator)
| C47 H62 N12 O11 S2= MF | |
| Molecular Weight | 1035.199 |
Lodenafil is a drug belonging to a class of drugs called PDE5 inhibitor, which many other erectile dysfunction drugs such as sildenafil, tadalafil, and vardenafil also belong to. Like udenafil and avanafil it belongs to a new generation of PDE5 inhibitors.
- Sildenafil, tadalafil, vardenafil, and the newer udenafil and avanafil selectively inhibit PDE5, which is cGMP-specific and responsible for the degradation of cGMP in the corpus cavernosum. These phosphodiesterase inhibitors are used primarily as remedies for erectile dysfunction, as well as having some other medical applications such as treatment of pulmonary hypertension.
Lodenafil is formulated as a dimer, lodenafil carbonate, which breaks down in the body to form two molecules of the active drug lodenafil. This formulation has higher oral bioavailability than the parent drug.[1]
It is manufactured by Cristália Produtos Químicos e Farmacêuticos in Brazil and sold there under the brand-name Helleva.[2]

Helleva (Lodenafil Carbonate) is an oral PDE5 inhibitor prescribed to treat men suffering from erectile dysfunction. It operates by relaxing muscles and dilating blood vessels in the penis to increase circulation making it easier to attain and maintain an erection.
It has undergone Phase III clinical trials,[3][4][5] but is not yet approved for use in the United States by the U.S. Food and Drug Administration.

………..
SYNTHESIS
WO 2002012241 OR US7148350
MONOMER synthesis
 PIPERAZINE
PIPERAZINE
AND
 ETHYL CHLORO ACETATE
ETHYL CHLORO ACETATE
WILL GIVE
Ethyl 1-piperazinylacetate
SEE RXN 1 BELOW
Reaction 1:
Synthesis of Piperazine Ethyl Acetate
To a reaction blend containing 100 g (3 Eq, 0.515 mol, MW=194) of piperazine, 26.3 mL (1.1 Eq, 0.189 mol, MW=101, d=0.726) of triethylamine in 200 mL of isopropanol, add to a solution previously prepared of 18.4 mL (1 Eq., 0.172 mol, MW=122.55, d=1.15) of chloroacetate of ethyl in 140 mL of isopropanol under stirring, at room temperature. Keep the reaction medium under stirring, monitoring the reaction termination by means of a chromatography of the thin layer (about 2–3 hours). Add a solution of 40.6 g (0.344 mol) of succinic acid in 140 mL of isopropanol. Keep the system under stirring for about 30 minutes to assure total precipitation of the succinate salt of piperazine formed. Filter this salt and concentrate the filtrate containing the mono and dialkyled derivatives. We obtain a slightly yellowish oil, which is used in later phases without purification.
Mass obtained=33 g
GC/MS: Monoalkylated derivative 72%, and dialkylated 22%.
NEXT
Piperazine Ethyl Acetate
AND
![5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one Structure](https://i0.wp.com/www.chemicalbook.com/CAS/GIF/139756-22-2.gif)
5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one
WILL REACT TO GIVE… 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one AS IN RXN 4 BELOW
Reaction 4:
Synthesis of 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one.
Suspend 24.6 g (60 mmol, MW=410.9) of 5-(5-chlorosulfonyl-2-etoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one in 900 mL of ethanol absolute. Under stirring and at room temperature, add at only one time, a solution containing 31.0 g (3 Eq., 180 mmol MW=172) of N-piperazine ethyl acetate (Reaction 1) dissolved in 150 mL of ethanol absolute. In an interval of 2–10 minutes, all solid is consumed, forming a clean and homogeneous solution, and after that starts the precipitation of the expected product. At the end of the reaction, which lasts 2–3 hours (monitored by chromatography of thin layer), the product is vacuum filtered and the solid is washed with two portions of 50 mL of iced absolute ethanol. 29 g are obtained (yielding=89%) from the product as a white solid of MP=165.5–166.5° C.
Reaction 7:
Intermediate 1
5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one. IS MONOMER
please note during LAH redn …………. the PIP CH2-C=O-O CH2 CH3 BECOMES PIP-CH2CH2-OH
To a suspension of lithium aluminum hydride (0.74 g 2.2 Eq. MW=37.9) in 25 mL of THF, slowly add, under stirring and at room temperature, a suspension of 5.0 g (9.1 mmol, MW=546.6) of 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one in 50 mL of THF. The system is maintained under stirring, monitoring the consumption of the product by chromatography of thin layer, until the complete consumption of the starting reagent (about 5–6 hours). Slowly add water to the reaction medium and, when there is no longer release of H2, add HCl 1M regulating pH for 7. Extract the product with 3 200 mL-portions of chloroform, dry with anhydrous sodium sulfate and vacuum concentrate the product. It is obtained 3.8 g of the product as a cream solid MP=183–187° C. yielding 83%. The same was crystallized from methanol and DMF yielding a slightly yellowish solid with melting point at 189–192° C.

note …………. the PIP CH2-C=O-O CH2 CH3 BECOMES PIP-CH2CH2-OH
HOMODIMER CARBONATE
EXAMPLE 1B
Homodimer Carbonate of Intermediate 1—Alternative Method
A phosgene solution (3.5 g, 35 mmol) dissolved in 20 mL of toluene was added dropwise to a solution of 2.02 g (4 mmol) of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, suspended in 44 mL of toluene. The reaction mixture resulting is stirred and followed by chromatography analysis of thin layer every hour until the reagent conversion in its chloroformate was completed. When the analysis indicates the complete consumption of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, the volatile compounds of the reaction are vacuum removed (solvents and phosgene), yielding the esther chloroformate raw derivative of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one.
The raw chloroformate obtained above (4.0 mmol, 2.27 g) is dissolved in about 30 mL of dichloromethane, to which is added 2.07 g (4.1 mmol) of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, followed by the addition of 4 mL of dichloromethane containing 450 mg of triethylamine. The reaction mixture is maintained under stirring, being followed by chromatography of thin layer every hour until this indicates the end of the reaction (disappearing of chloroformate derivative). The reaction mixture is then diluted with 60 mL of dichloromethane, washed with NaCl saturated solution, after with sodium bicarbonate saturated solution and again with NaCl saturated solution. Organic phase is separated and dry with anhydrous sodium sulfate. The solvent is then evaporated to dry, yielding the dimer carbonate as a slightly yellowish solid.
This compound is re-crystallized from ethanol:DMF, yielding a pale white solid. Yielding m=3.2 g (76%)
Microanalysis: Theoretical C, (54.53%); H, (6.04%); N, (16.24%);
Obtained C, (54.45%); H, (6.02%); N, (16.17%).
INFO ABOUT INTERMEDIATE
5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one
| CAS No. | 139756-22-2 |
| Chemical Name: | 5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one |
| Synonyms: | Sildenafil Chlorosulfone IMpurity;Sildenafil Chlorosulfonyl IMpurity;5-(5-CHLOROSULFONYL-2-ETHOXY PHENYL)-1-METHYL-3-N-PROPYL-1;3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1 H-pyrazolo-(4-3-d)-pyrimidine-5;5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one;3-(4,7-Dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxy-benzenesulfonyl Chloride;4-Ethoxy-3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyriMidin-5-yl)benzene-1-sulfonyl chloride |
| CBNumber: | CB11175931 |
| Molecular Formula: | C17H19ClN4O4S |
http://www.chemicalbook.com/ChemicalProductProperty_EN_CB11175931.htm
…………..
SYNTHESIS OF

http://www.google.co.in/patents/US6362178

22.27 g (250 mmol) of D,L-alanine and 55.66 g (550 mmol) of triethylamine are dissolved in 250 ml of dichloromethane, and the solution is cooled to 0° C. 59.75 g (550 mmol) of trimethylsilyl chloride are added dropwise, and the solution is stirred for 1 hour at room temperature and for 1 hour at 40° C. After cooling to −10° C., 26.64 g (250 mmol) of butyryl chloride are added dropwise, and the resulting mixture is stirred for 2 hours at −10° C. and for one hour at room temperature.
With ice-cooling, 125 ml of water are added dropwise and the reaction mixture is stirred at room temperature for 15 minutes. The aqueous phase is evaporated to dryness, the residue is titrated with acetone and the mother liquor is filtered off with suction. The solvent is removed and the residue is chromatographed. The resulting product is dissolved in 3N aqueous sodium hydroxide solution and the resulting solution is evaporated to dryness. The residue is taken up in conc. HCl and once more evaporated to dryness. The residue is stirred with acetone, precipitated solid is filtered off with suction and the solvent is removed under reduced pressure. This gives 28.2 g (71%) of a viscous oil which crystallizes after some time.
200 MHz 1H-NMR (DMSO-d6): 0.84, t, 3H; 1.22, d, 3H; 1.50, hex, 2H; 2.07, t, 2H; 4.20, quin., 1H; 8.09, d, 1H.

25 g (210 mmol) of 2-hydroxybenzonitrile are refluxed with 87 g of potassium carbonate and 34.3 g (314.8 mmol) of ethyl bromide in 500 ml of acetone overnight. The solid is filtered off, the solvent is removed under reduced pressure and the residue is distilled under reduced pressure. This gives 30.0 g (97%) of a colourless liquid.
200 MHz 1H-NMR (DMSO-d6): 1.48, t, 3H; 4.15, quart., 2H; 6.99, dt, 2H; 7.51, dt, 2H.

21.4 g (400 mmol) of ammonium chloride are suspended in 375 ml of toluene, and the suspension is cooled to 0° C. 200 ml of a 2M solution of trimethylaluminium in hexane are added dropwise, and the mixture is stirred at room temperature until the evolution of gas has ceased. After addition of 29.44 g (200 mmol) of 2-ethoxybenzonitrile, the reaction mixture is stirred at 80° C. (bath) overnight.
With ice-cooling, the cooled reaction mixture is added to a suspension of 100 g of silica gel and 950 ml of chloroform, and the mixture is stirred at room temperature for 30 minutes. The mixture is filtered off with suction, and the filter residue is washed with the same amount of methanol. The mother liquor is concentrated, the resulting residue is stirred with a mixture of dichloromethane and methanol (9:1), the solid is filtered off with suction and the mother liquor is concentrated. This gives 30.4 g (76%) of a colourless solid.
200 MHz 1H-NMR (DMSO-d6): 1.36, t, 3H; 4.12, quart., 2H; 7.10, t, 1H; 7.21, d, 1H; 7.52, m, 2H; 9.30, s, broad, 4H.
EXAMPLE 10A 2-(2-Ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

7.16 g (45 mmol) of 2-butyrylamino-propionic acid and 10.67 g of pyridine are dissolved in 45 ml of THF and, after addition of a spatula tip of DMAP, heated to reflux. 12.29 g (90 mmol) of ethyl oxalyl chloride are slowly added dropwise, and the reaction mixture is refluxed for 3 hours. The mixture is poured into ice-water and extracted three times with ethyl acetate and the organic phase is dried over sodium sulphate and concentrated using a rotary evaporator. The residue is taken up in 15 ml of ethanol and refluxed with 2.15 g of sodium bicarbonate for 2.5 hours. The cooled solution is filtered.
With ice-cooling, 2.25 g (45 mmol) of hydrazine hydrate are added dropwise to a solution of 9.03 g (45 mmol) of 2-ethoxybenzamidine hydrochloride in 45 ml of ethanol, and the resulting suspension is stirred at room temperature for another 10 minutes. The ethanolic solution described above is added to this reaction mixture, and the mixture is stirred at a bath temperature of 70° C. for 4 hours. After filtration, the mixture is concentrated, the residue is partitioned between dichloromethane and water, the organic phase is dried over sodium sulphate and the solvent is removed under reduced pressure.
This residue is dissolved in 60 ml of 1,2-dichloroethane and, after addition of 7.5 ml of phosphorus oxychloride, refluxed for 2 hours. The mixture is diluted with dichloromethane and neutralized by addition of sodium bicarbonate solution and solid sodium bicarbonate. The organic phase is dried and the solvent is removed under reduced pressure. Chromatography using ethyl acetate and crystallization afford 4.00 g (28%) of a colourless solid, Rf=0.42 (dichloromethane/methanol=95:5)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.56, t, 3H; 1.89, hex, 2H; 2.67, s, 3H; 3.00, t, 2H; 4.26, quart., 2H; 7.05, m, 2H; 7.50, dt, 1H; 8.17, dd, 1H; 10.00, s, 1H.
EXAMPLE 15A 4-Ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride

At 0° C., 2.00 g (6.4 mmol) of 2-(2-ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are slowly added to 3.83 ml of chlorosulphonic acid. At room temperature, the reaction mixture is stirred ovemight, and then poured into ice-water and extracted with dichloromethane. This gives 2.40 g (91%) of a colourless foam.
200 MHz 1H-NMR (CDCl3): 1.03, t, 3H; 1.61, t, 2H; 1.92, hex, 2H; 2.67, s, 3H; 3.10, t, 2H; 4.42, quart., 2H; 7.27, t, 1H; 8.20, dd, 1H; 8.67, d, 1H; 10.18, s, 1H.
Example 22 2-[2-Ethoxy-5-(4-hydroxyethyl-1-amino-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one
By the same method, starting with 0.04 g (0.097 mmol) of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride and 0.04 g (0.29 mmol) of 1-amino-4-hydroxyethylpiperazine, 46 mg (91%) of 2-[2-ethoxy-5-(4-hydroxyethyl-1-amino-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are obtained.
Rf=0.08 (dichloromethane/methanol=19:1)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.59, t, 3H; 1.90, sex., 2H; 2.49, m, 6H; 2.62, s, 3H; 2.71, m, 4H; 3.00, t, 2H; 3.55, t, 2H; 4.31, quart., 2H; 7.14, d, 1H; 8.05, dd, 1H; 8.60, d, 1H.
…………..
Methods of analysis
The development of lodenafil carbonate was reported by Toque et al. (2008). They observed the effects of lodenafil carbonate on rabbit and human corpus cavernosum relaxation, activity of PDE5 in human platelets, stability and metabolic studies in comparison with sildenafil and lodenafil, as well as the pharmacological evaluation of lodenafil carbonate after intravenous and oral administration in male beagles.
The determination of PDE activity, stability of lodenafil carbonate in human, dog and rat plasma and the pharmacokinetic parameters after a single intravenous or oral dose was carried out by LC-MS/MS analysis
Codevilla et al. (2011a) developed a stability-indicating reversed-phase liquid chromatography method using ultraviolet (UV) detection for the quantitative determination of lodenafil carbonate in tablets. The method can be useful for routine quality control assay and stability studies.
Another study for the determination of lodenafil carbonate in tablets was developed by Codevilla et al. (2011b). As an alternative to the LC method the authors suggested a UV-spectrophotometric method for the analysis of lodenafil carbonate in pharmaceutical form. The UV method offers advantages over other analytical methods due to its rapidity, simplicity, and lower cost. Recently, Codevilla et al. (2012) developed and validated a capillary zone electrophoresis (CZE) method for determination of lodenafil carbonate in drug products. There are some advantages to use the CZE method, such as rapid analysis, small sample and reagent consumption, high separation efficiency (Furlanetto et al., 2001; Yang et al., 2010). The results obtained from the UV-spectrophotometric method and CZE method were compared statistically with the LC method (Codevilla et al., 2011a) and the results showed no significant difference between these methods.
References
- Toque HA, Teixeira CE, Lorenzetti R, Okuyama CE, Antunes E, De Nucci G (September 2008). “Pharmacological characterization of a novel phosphodiesterase type 5 (PDE5) inhibitor lodenafil carbonate on human and rabbit corpus cavernosum”. European Journal of Pharmacology 591 (1–3): 189–95. doi:10.1016/j.ejphar.2008.06.055. PMID 18593576.
- Cristália Product page. Retrieved on September 16, 2009.
- ukmedix Lodenafil article. Retrieved on September 16, 2009.
- Glina S, Toscano I, Gomatzky C, de Góes PM, Júnior AN, Claro JF, Pagani E (February 2009). “Efficacy and tolerability of lodenafil carbonate for oral therapy in erectile dysfunction: a phase II clinical trial”. The Journal of Sexual Medicine 6 (2): 553–7. doi:10.1111/j.1743-6109.2008.01079.x.PMID 19040623.
- Glina S, Fonseca GN, Bertero EB, Damião R, Rocha LC, Jardim CR, Cairoli CE, Teloken C, Torres LO, Faria GE, da Silva MB, Pagani E (February 2010). “Efficacy and Tolerability of Lodenafil Carbonate for Oral Therapy of Erectile Dysfunction: A Phase III Clinical Trial”. The Journal of Sexual Medicine 7 (5): 1928–1936. doi:10.1111/j.1743-6109.2010.01711.x. PMID 20214718.
- Toque H A et al., (2008) European Journal of Pharmacology, 591(1-3):189-95.
- Exploring the role of PDE5 inhibition in the treatment of muscular dystrophy
Drugs Fut 2011, 36(4): 321
PANOBINOSTAT

Panobinostat
HDAC inhibitors, orphan drug
cas 404950-80-7
2E)-N-hydroxy-3-[4-({[2-(2-methyl-1H-indol-3-yl)ethyl]amino}methyl)phenyl]acrylamide
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide)
Molecular Formula: C21H23N3O2 Molecular Weight: 349.42622
- Faridak
- LBH 589
- LBH589
- Panobinostat
- UNII-9647FM7Y3Z
A hydroxamic acid analog histone deacetylase inhibitor from Novartis.
NOVARTIS, innovator
Histone deacetylase inhibitors
Is currently being examined in cutaneous T-cell lymphoma, CML and breast cancer.
clinical trials click here phase 3
DRUG SUBSTANCE–LACTATE AS IN http://www.google.com/patents/US7989639 SEE EG 31

Panobinostat (LBH-589) is an experimental drug developed by Novartis for the treatment of various cancers. It is a hydroxamic acid[1] and acts as a non-selective histone deacetylase inhibitor (HDAC inhibitor).[2]
panobinostat
Panobinostat is a cinnamic hydroxamic acid analogue with potential antineoplastic activity. Panobinostat selectively inhibits histone deacetylase (HDAC), inducing hyperacetylation of core histone proteins, which may result in modulation of cell cycle protein expression, cell cycle arrest in the G2/M phase and apoptosis. In addition, this agent appears to modulate the expression of angiogenesis-related genes, such as hypoxia-inducible factor-1alpha (HIF-1a) and vascular endothelial growth factor (VEGF), thus impairing endothelial cell chemotaxis and invasion. HDAC is an enzyme that deacetylates chromatin histone proteins. Check for
As of August 2012, it is being tested against Hodgkin’s Lymphoma, cutaneous T cell lymphoma (CTCL)[3] and other types of malignant disease in Phase III clinical trials, against myelodysplastic syndromes, breast cancer and prostate cancer in Phase II trials, and against chronic myelomonocytic leukemia (CMML) in a Phase I trial.[4][5]
Panobinostat is a histone deacetylase (HDAC) inhibitor which was filed for approval in the U.S. in 2010 for the oral treatment of relapsed/refractory classical Hodgkin’s lymphoma in adult patients. The company is conducting phase II/III clinical trials for the oral treatment of multiple myeloma, chronic myeloid leukemia and myelodysplasia. Phase II trials are also in progress for the treatment of primary myelofibrosis, post-polycythemia Vera, post-essential thrombocytopenia, Waldenstrom’s macroglobulinemia, recurrent glioblastoma (GBM) and for the treatment of pancreatic cancer progressing on gemcitabine therapy. Additional trials are under way for the treatment of hematological neoplasms, prostate cancer, colorectal cancer, renal cell carcinoma, non-small cell lung cancer (NSCLC), malignant mesothelioma, acute lymphoblastic leukemia, acute myeloid leukemia, head and neck cancer and gastrointestinal neuroendocrine tumors. Early clinical studies are also ongoing for the treatment of HER2 positive metastatic breast cancer. Additionally, phase II clinical trials are ongoing at Novartis as well as Neurological Surgery for the treatment of recurrent malignant gliomas as are phase I/II initiated for the treatment of acute graft versus host disease. The National Cancer Institute had been conducting early clinical trials for the treatment of metastatic hepatocellular carcinoma; however, these trials were terminated due to observed dose-limiting toxicity. In 2009, Novartis terminated its program to develop panobinostat for the treatment of cutaneous T-cell lymphoma. A program for the treatment of small cell lung cancer was terminated in 2012. Phase I clinical trials are ongoing for the treatment of metastatic and/or malignant melanoma and for the treatment of sickle cell anemia. The University of Virginia is conducting phase I clinical trials for the treatment of newly diagnosed and recurrent chordoma in combination with imatinib. Novartis is evaluating panobinostat for its potential to re-activate HIV transcription in latently infected CD4+ T-cells among HIV-infected patients on stable antiretroviral therapy.
Mechanistic evaluations revealed that panobinostat-mediated tumor suppression involved blocking cell-cycle progression and gene transcription induced by the interleukin IL-2 promoter, accompanied by an upregulation of p21, p53 and p57, and subsequent cell death resulted from the stimulation of caspase-dependent and -independent apoptotic pathways and an increase in the mitochondrial outer membrane permeability. In 2007, the compound received orphan drug designation in the U.S. for the treatment of cutaneous T-cell lymphoma and in 2009 and 2010, orphan drug designation was received in the U.S. and the E.U., respectively, for the treatment of Hodgkin’s lymphoma. This designation was also assigned in 2012 in the U.S. and the E.U. for the treatment of multiple myeloma.
Cardiovascular disease is the leading cause of morbidity and mortality in the western world and during the last decades it has also become a rapidly increasing problem in developing countries. An estimated 80 million American adults (one in three) have one or more expressions of cardiovascular disease (CVD) such as hypertension, coronary heart disease, heart failure, or stroke. Mortality data show that CVD was the underlying cause of death in 35% of all deaths in 2005 in the United States, with the majority related to myocardial infarction, stroke, or complications thereof. The vast majority of patients suffering acute cardiovascular events have prior exposure to at least one major risk factor such as cigarette smoking, abnormal blood lipid levels, hypertension, diabetes, abdominal obesity, and low-grade inflammation.
Pathophysiologically, the major events of myocardial infarction and ischemic stroke are caused by a sudden arrest of nutritive blood supply due to a blood clot formation within the lumen of the arterial blood vessel. In most cases, formation of the thrombus is precipitated by rupture of a vulnerable atherosclerotic plaque, which exposes chemical agents that activate platelets and the plasma coagulation system. The activated platelets form a platelet plug that is armed by coagulation-generated fibrin to form a biood clot that expands within the vessel lumen until it obstructs or blocks blood flow, which results in hypoxic tissue damage (so-called infarction). Thus, thrombotic cardiovascular events occur as a result of two distinct processes, i.e. a slowly progressing long-term vascular atherosclerosis of the vessel wall, on the one hand, and a sudden acute clot formation that rapidly causes flow arrest, on the other. This invention solely relates to the latter process.
Recently, inflammation has been recognized as an important risk factor for thrombotic events. Vascular inflammation is a characteristic feature of the atherosclerotic vessel wall, and inflammatory activity is a strong determinant of the susceptibility of the atherosclerotic plaque to rupture and initiate intravascular clotting. Also, autoimmune conditions with systemic inflammation, such as rheumatoid arthritis, systemic lupus erythematosus and different forms of vasculitides, markedly increase the risk of myocardial infarction and stroke.
Traditional approaches to prevent and treat cardiovascular events are either targeted 1) to slow down the progression of the underlying atherosclerotic process, 2) to prevent clot formation in case of a plaque rupture, or 3) to direct removal of an acute thrombotic flow obstruction. In brief, antiatherosclerotic treatment aims at modulating the impact of general risk factors and includes dietary recommendations, weight loss, physical exercise, smoking cessation, cholesterol- and blood pressure treatment etc. Prevention of clot formation mainly relies on the use of antiplatelet drugs that inhibit platelet activation and/or aggregation, but also in some cases includes thromboembolic prevention with oral anticoagulants such as warfarin. Post-hoc treatment of acute atherothrombotic events requires either direct pharmacological lysis of the clot by thrombolytic agents such as recombinant tissue-type plasminogen activator or percutaneous mechanical dilation of the obstructed vessel.
Despite the fact that multiple-target antiatherosclerotic therapy and clot prevention by antiplatelet agents have lowered the incidence of myocardial infarction and ischemic stroke, such events still remain a major population health problem. This shows that in patients with cardiovascular risk factors these prophylactic measures are insufficient to completely prevent the occurrence of atherothrombotic events.
Likewise, thrombotic conditions on the venous side of the circulation, as well as embolic complications thereof such as pulmonary embolism, still cause substantial morbidity and mortality. Venous thrombosis has a different clinical presentation and the relative importance of platelet activation versus plasma coagulation are somewhat different with an preponderance for the latter in venous thrombosis, However, despite these differences, the major underlying mechanisms that cause thrombotic vessel occlusions are similar to those operating on the arterial circulation. Although unrelated to atherosclerosis as such, the risk of venous thrombosis is related to general cardiovascular risk factors such as inflammation and metabolic aberrations.

Panobinostat can be synthesized as follows: Reduction of 2-methylindole-3-glyoxylamide (I) with LiAlH4 affords 2-methyltryptamine (II). 4-Formylcinnamic acid (III) is esterified with methanolic HCl, and the resulting aldehyde ester (IV) is reductively aminated with 2-methyltryptamine (II) in the presence of NaBH3CN (1) or NaBH4 (2) to give (V). The title hydroxamic acid is then obtained by treatment of ester (V) with aqueous hydroxylamine under basic conditions.
Panobinostat is currently being used in a Phase I/II clinical trial that aims at curing AIDS in patients on highly active antiretroviral therapy (HAART). In this technique panobinostat is used to drive the HI virus’s DNA out of the patient’s DNA, in the expectation that the patient’s immune system in combination with HAART will destroy it.[6][7]
 panobinostat
panobinostat
Panobinostat has been found to synergistically act with sirolimus to kill pancreatic cancer cells in the laboratory in a Mayo Clinic study. In the study, investigators found that this combination destroyed up to 65 percent of cultured pancreatic tumor cells. The finding is significant because the three cell lines studied were all resistant to the effects of chemotherapy – as are many pancreatic tumors.[8]
Panobinostat has also been found to significantly increase in vitro the survival of motor neuron (SMN) protein levels in cells of patients suffering fromspinal muscular atrophy.[9]
Panobinostat was able to selectively target triple negative breast cancer (TNBC) cells by inducing hyperacetylation and cell cycle arrest at the G2-M DNA damage checkpoint; partially reversing the morphological changes characteristic of breast cancer cells.[10]
Panobinostat, along with other HDAC inhibitors, is also being studied for potential to induce virus HIV-1 expression in latently infected cells and disrupt latency. These resting cells are not recognized by the immune system as harboring the virus and do not respond to antiretroviral drugs.[11]
Panobinostat inhibits multiple histone deacetylase enzymes, a mechanism leading to apoptosis of malignant cells via multiple pathways.[1]
The compound N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (alternatively, N-hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide) has the formula

as described in WO 02/22577. Valuable pharmacological properties are attributed to this compound; thus, it can be used, for example, as a histone deacetylase inhibitor useful in therapy for diseases which respond to inhibition of histone deacetylase activity. WO 02/22577 does not disclose any specific salts or salt hydrates or solvates of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide.
The compounds described above are often used in the form of a pharmaceutically acceptable salt. Pharmaceutically acceptable salts include, when appropriate, pharmaceutically acceptable base addition salts and acid addition salts, for example, metal salts, such as alkali and alkaline earth metal salts, ammonium salts, organic amine addition salts, and amino acid addition salts, and sulfonate salts. Acid addition salts include inorganic acid addition salts such as hydrochloride, sulfate and phosphate, and organic acid addition salts such as alkyl sulfonate, arylsulfonate, acetate, maleate, fumarate, tartrate, citrate and lactate. Examples of metal salts are alkali metal salts, such as lithium salt, sodium salt and potassium salt, alkaline earth metal salts such as magnesium salt and calcium salt, aluminum salt, and zinc salt. Examples of ammonium salts are ammonium salt and tetramethylammonium salt. Examples of organic amine addition salts are salts with morpholine and piperidine. Examples of amino acid addition salts are salts with glycine, phenylalanine, glutamic acid and lysine. Sulfonate salts include mesylate, tosylate and benzene sulfonic acid salts.
……………………………..
GENERAL METHOD OF SYNTHESIS
ADD YOUR METHYL AT RIGHT PLACE
As is evident to those skilled in the art, the many of the deacetylase inhibitor compounds of the present invention contain asymmetric carbon atoms. It should be understood, therefore, that the individual stereoisomers are contemplated as being included within the scope of this invention.
The hydroxamate compounds of the present invention can be produced by known organic synthesis methods. For example, the hydroxamate compounds can be produced by reacting methyl 4-formyl cinnamate with tryptamine and then converting the reactant to the hydroxamate compounds. As an example, methyl 4-formyl cinnamate 2, is prepared by acid catalyzed esterification of 4-formylcinnamic acid 3 (Bull. Chem. Soc. Jpn. 1995; 68:2355-2362). An alternate preparation of methyl 4-formyl cinnamate 2 is by a Pd- catalyzed coupling of methyl acrylate 4 with 4-bromobenzaldehyde 5.
CHO

Additional starting materials can be prepared from 4-carboxybenzaldehyde 6, and an exemplary method is illustrated for the preparation of aldehyde 9, shown below. The carboxylic acid in 4-carboxybenzaldehyde 6 can be protected as a silyl ester (e.g., the t- butyldimethylsilyl ester) by treatment with a silyl chloride (e.g., f-butyldimethylsilyl chloride) and a base (e.g. triethylamine) in an appropriate solvent (e.g., dichloromethane). The resulting silyl ester 7 can undergo an olefination reaction (e.g., a Horner-Emmons olefination) with a phosphonate ester (e.g., triethyl 2-phosphonopropionate) in the presence of a base (e.g., sodium hydride) in an appropriate solvent (e.g., tetrahydrofuran (THF)). Treatment of the resulting diester with acid (e.g., aqueous hydrochloric acid) results in the hydrolysis of the silyl ester providing acid 8. Selective reduction of the carboxylic acid of 8 using, for example, borane-dimethylsuflide complex in a solvent (e.g., THF) provides an intermediate alcohol. This intermediate alcohol could be oxidized to aldehyde 9 by a number of known methods, including, but not limited to, Swern oxidation, Dess-Martin periodinane oxidation, Moffatt oxidation and the like.

The aldehyde starting materials 2 or 9 can be reductively aminated to provide secondary or tertiary amines. This is illustrated by the reaction of methyl 4-formyl cinnamate 2 with tryptamine 10 using sodium triacetoxyborohydride (NaBH(OAc)3) as the reducing agent in dichloroethane (DCE) as solvent to provide amine 11. Other reducing agents can be used, e.g., sodium borohydride (NaBH ) and sodium cyanoborohydride (NaBH3CN), in other solvents or solvent mixtures in the presence or absence of acid catalysts (e.g., acetic acid and trifluoroacetic acid). Amine 11 can be converted directly to hydroxamic acid 12 by treatment with 50% aqueous hydroxylamine in a suitable solvent (e.g., THF in the presence of a base, e.g., NaOH). Other methods of hydroxamate formation are known and include reaction of an ester with hydroxylamine hydrochloride and a base (e.g., sodium hydroxide or sodium methoxide) in a suitable solvent or solvent mixture (e.g., methanol, ethanol or methanol/THF).

NOTE ….METHYL SUBSTITUENT ON 10 WILL GIVE YOU PANOBINOSTAT

……………………………….
Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
(E)-N-Hydroxy-3-(4-{[2-(2-methyl-1H-indol-3-yl)-ethylamino]-methyl}-phenyl)-acrylamide
lactate
(34, panobinostat, LBH589)
http://pubs.acs.org/doi/full/10.1021/jm2003552
http://pubs.acs.org/doi/suppl/10.1021/jm2003552/suppl_file/jm2003552_si_001.pdf
for str see above link
α-methyl-β-(β-bromoethyl)indole (29) was made according to method reported by Grandberg et al.(2. Grandberg, I. I.; Kost, A. N.; Terent’ev, A. P. Reactions of hydrazine derivatives. XVII. New synthesis of α-methyltryptophol. Zhurnal Obshchei Khimii 1957, 27, 3342–3345. )
The bromide 29 was converted to amine 30 by using similar method used by Sletzinger et al.(3. Sletzinger, M.; Ruyle, W. V.; Waiter, A. G. (Merck & Co., Inc.). Preparation of tryptamine
derivatives. U.S. Patent US 2,995,566, Aug 8, 1961.)
To a 500 mL flask, crude 2-methyltryptamine 30 (HPLC purity 75%, 1.74 g, 7.29 mmol) and 3-(4-
formyl-phenyl)-acrylic acid methyl ester 31 (HPLC purity 84%, 1.65 g, 7.28 mmol) were added,
followed by DCM (100 mL) and MeOH (30 mL). The clear solution was stirred at room temp for 30
min, then NaBH3CN (0.439 g, 6.99 mmol) was added in small portions. The reaction mixture was
stirred at room temp overnight. After removal of the solvents, the residue was diluted with DCM and
added saturated NaHCO3 aqueous solution, extracted with DCM twice. The DCM layer was dried
and concentrated, and the resulting residue was purified by flash chromatography (silica, 0–10%
MeOH in DCM) to afford 33 as orange solid (1.52 g, 60%). LC–MS m/z 349.2 ([M + H]+). 33 was
converted to hydroxamic acid 34 according to procedure D (Experimental Section), and the freebase
34 was treated with 1 equiv of lactic acid in MeOH–water (7:3) to form lactic acid salt which was
further recrystallized in MeOH–EtOAc to afford the lactic acid salt of 34as pale yellow solid. LC–MS m/z 350.2 ([M + H − lactate]+).
= DELTA
1H NMR (DMSO-d6) 10.72 (s, 1H, NH), 7.54 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 16 Hz, 1H), 7.43 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 7.8 Hz, 1H), 6.97 (td, J = 7.8 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8Hz, 2H), 7.01 (t, J = 7.4, 0.9 Hz, 1H), 6.91 (td, J = 7.4, 0.9 Hz, 1H), 6.47 (d, J = 15.2 Hz, 1H), 3.94(q, J = 6.8 Hz, 1H, lactate CH), 3.92 (s, 2H), 2.88 and 2.81 (m, each, 4H, AB system, CH2CH2),2.31 (s, 3H), 1.21 (d, J = 6.8 Hz, 3H).;
13C NMR (DMSO-d6) 176.7 (lactate C=O), 162.7, 139.0,
137.9, 135.2, 134.0, 132.1, 129.1, 128.1, 127.4, 119.9, 119.0, 118.1, 117.2, 110.4, 107.0, 66.0, 51.3,
48.5, 22.9, 20.7, 11.2.
…………………………………………..
PANOBINOSTAT DRUG SUBSTANCE SYNTHESIS AND DATA
http://www.google.com/patents/US7989639

A flow diagram for the synthesis of LBH589 lactate is provided in FIG. A. A nomenclature reference index of the intermediates is provided below in the Nomenclature Reference Index:
| Nomenclature reference index | |
| Compound | Chemical name |
| 1 | 4-Bromo-benzaldehyde |
| 2 | Methyl acrylate |
| 3 | (2E)-3-(formylphenyl)-2-propenoic acid, methyl ester |
| 4 | 3-[4-[[[2-(2-Methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2- | |
| propenoic acid, methyl ester, monohydrochloride | |
| 5 | (2E)-N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3- |
| yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| 6 | 2-hydroxypropanoic acid, compd. with 2(E)-N- |
| hydroxy-3-[4-[[[2-(2-methyl-1H- | |
| indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide | |
| Z3a | 2-Methyl-1H-indole-3-ethanamine |
| Z3b | 5-Chloro-2-pentanone |
| Z3c | Phenylhydrazine |
The manufacture of LBH589 lactate (6) drug substance is via a convergent synthesis; the point of convergence is the condensation of indole-amine Z3a with aldehyde 3.
The synthesis of indole-amine Z3a involves reaction of 5-chloro-2 pentanone (Z3b) with phenylhydrazine (Z3c) in ethanol at reflux (variation of Fischer indole synthesis).
Product isolation is by an extractive work-up followed by crystallization. Preparation of aldehyde 3 is by palladium catalyzed vinylation (Heck-type reaction; Pd(OAc)2/P(o-Tol)3/Bu3N in refluxing CH3CN) of 4-bromo-benzyladehyde (1) with methyl acrylate (2) with product isolation via precipitation from dilute HCl solution. Intermediates Z3a and 3 are then condensed to an imine intermediate, which is reduced using sodium borohydride in methanol below 0° C. (reductive amination). The product indole-ester 4, isolated by precipitation from dilute HCl, is recrystallized from methanol/water, if necessary. The indole ester 4 is converted to crude LBH589 free base 5 via reaction with hydroxylamine and sodium hydroxide in water/methanol below 0° C. The crude LBH589 free base 5 is then purified by recrystallization from hot ethanol/water, if necessary. LBH589 free base 5 is treated with 85% aqueous racemic lactic acid and water at ambient temperature. After seeding, the mixture is heated to approximately 65° C., stirred at this temperature and slowly cooled to 45-50° C. The resulting slurry is filtered and washed with water and dried to afford LBH589 lactate (6).
If necessary the LBH589 lactate 6 may be recrystallised once again from water in the presence of 30 mol % racemic lactic acid. Finally the LBH589 lactate is delumped to give the drug substance. If a rework of the LBH589 lactate drug substance 6 is required, the LBH589 lactate salt is treated with sodium hydroxide in ethanol/water to liberate the LBH589 free base 5 followed by lactate salt formation and delumping as described above.
All starting materials, reagents and solvents used in the synthesis of LBH589 lactate are tested according to internal specifications or are purchased from established suppliers against a certificate of analysis.
EXAMPLE 7 Formation of Monohydrate Lactate Salt
About 40 to 50 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base was suspended in 1 ml of a solvent as listed in Table 7. A stoichiometric amount of lactic acid was subsequently added to the suspension. The mixture was stirred at ambient temperature and when a clear solution formed, stirring continued at 4° C. Solids were collected by filtration and analyzed by XRPD, TGA and 1H-NMR.
| TABLE 7 | |||||
| LOD, % | |||||
| Physical | Crystallinity | (Tdesolvation) | |||
| Solvent | T, ° C. | Appear. | and Form | Tdecomposit. | 1H-NMR |
| IPA | 4 | FFP | excellent | 4.3 (79.3) | — |
| HA | 156.3 | ||||
| Acetone | 4 | FFP | excellent | 4.5 (77.8) | 4.18 (Hbz) |
| HA | 149.5 | ||||
The salt forming reaction in isopropyl alcohol and acetone at 4° C. produced a stoichiometric (1:1) lactate salt, a monohydrate. The salt is crystalline, begins to dehydrate above 77° C., and decomposes above 150° C.
EXAMPLE 18 Formation of Anhydrous Lactate Salt
DL-lactic acid (4.0 g, 85% solution in water, corresponding to 3.4 g pure DL-lactic acid) is diluted with water (27.2 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (10.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (110.5 g) is added, and the suspension is heated to 65° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 min at 65° C. During the addition of the lactate salt solution, the suspension converted into a solution. The addition funnel is rinsed with demineralized water (9.1 g), and the solution is stirred at 65° C. for an additional 30 minutes. The solution is cooled down to 45° C. (inner temperature) and seed crystals (10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate monohydrate) are added at this temperature. The suspension is cooled down to 33° C. and is stirred for additional 20 hours at this temperature. The suspension is re-heated to 65° C., stirred for 1 hour at this temperature and is cooled to 33° C. within 1 hour. After additional stirring for 3 hours at 33° C., the product is isolated by filtration, and the filter cake is washed with demineralized water (2×20 g). The wet filter-cake is dried in vacuo at 50° C. to obtain the anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt as a crystalline product. The product is identical to the monohydrate salt (form HA) in HPLC and in 1H-NMR, with the exception of the integrals of water signals in the 1H-NMR spectra.
In additional salt formation experiments carried out according to the procedure described above, the product solution was filtered at 65° C. before cooling to 45° C., seeding and crystallization. In all cases, form A (anhydrate form) was obtained as product.
EXAMPLE 19 Formation of Anhydrous Lactate Salt
DL-lactic acid (2.0 g, 85% solution in water, corresponding to 1.7 g pure DL-lactic acid) is diluted with water (13.6 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide free base (5.0 g) is placed in a 4-necked reaction flask with mechanical stirrer. Demineralized water (54.85 g) is added, and the suspension is heated to 48° C. (inner temperature) within 30 minutes. The DL-lactic acid solution is added to this suspension during 30 minutes at 48° C. A solution is formed. Seed crystals are added (as a suspension of 5 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form A, in 0.25 g of water) and stirring is continued for 2 additional hours at 48° C. The temperature is raised to 65° C. (inner temperature) within 30 minutes, and the suspension is stirred for additional 2.5 hours at this temperature. Then the temperature is cooled down to 48° C. within 2 hours, and stirring is continued at this temperature for additional 22 hours. The product is isolated by filtration and the filter cake is washed with demineralized water (2×10 g). The wet filter-cake is dried in vacuo at 50° C. to obtain anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A) as a crystalline product.
EXAMPLE 20 Conversion of Monohydrate Lactate Salt to Anhydrous Lactate Salt
DL-lactic acid (0.59 g, 85% solution in water, corresponding to 0.5 g pure DL-lactic acid) is diluted with water (4.1 g), and the solution is heated to 90° C. (inner temperature) for 15 hours. The solution is allowed to cool down to room temperature and is used as lactic acid solution for the following salt formation step.
10 g of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt monohydrate is placed in a 4-necked reaction flask. Water (110.9 g) is added, followed by the addition of the lactic acid solution. The addition funnel of the lactic acid is rinsed with water (15.65 g). The suspension is heated to 82° C. (inner temperature) to obtain a solution. The solution is stirred for 15 minutes at 82° C. and is hot filtered into another reaction flask to obtain a clear solution. The temperature is cooled down to 50° C., and seed crystals are added (as a suspension of 10 mg N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form, in 0.5 g of water). The temperature is cooled down to 33° C. and stirring is continued for additional 19 hours at this temperature. The formed suspension is heated again to 65° C. (inner temperature) within 45 minutes, stirred at 65° C. for 1 hour and cooled down to 33° C. within 1 hour. After stirring at 33° C. for additional 3 hours, the product is isolated by filtration and the wet filter cake is washed with water (50 g). The product is dried in vacuo at 50° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 21 Formation of Anhydrous Lactate Salt
DL-lactic acid (8.0 g, 85% solution in water, corresponding to 6.8 g pure DL-lactic acid) was diluted with water (54.4 g), and the solution was heated to 90° C. (inner temperature) for 15 hours. The solution was allowed to cool down to room temperature and was used as lactic acid solution for the following salt formation step.
N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (20 g) is placed in a 1 L glass reactor, and ethanol/water (209.4 g of a 1:1 w/w mixture) is added. The light yellow suspension is heated to 60° C. (inner temperature) within 30 minutes, and the lactic acid solution is added during 30 minutes at this temperature. The addition funnel is rinsed with water (10 g). The solution is cooled to 38° C. within 2 hours, and seed crystals (20 mg of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt, anhydrate form) are added at 38° C. After stirring at 38° C. for additional 2 hours, the mixture is cooled down to 25° C. within 6 hours. Cooling is continued from 25° C. to 10° C. within 5 hours, from 10° C. to 5° C. within 4 hours and from 5° C. to 2° C. within 1 hour. The suspension is stirred for additional 2 hours at 2° C., and the product is isolated by filtration. The wet filter cake is washed with water (2×30 g), and the product is dried in vacuo at 45° C. to obtain crystalline anhydrous N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide lactate salt (form A).
EXAMPLE 28 Formation of Lactate Monohydrate Salt
3.67 g (10 mmol) of the free base monohydrate (N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl) ethyl]amino]methyl]phenyl]-2E-2-propenamide) and 75 ml of acetone were charged in a 250 ml 3-neck flask equipped with a magnetic stirrer and an addition funnel. To the stirred suspension were added dropwise 10 ml of 1 M lactic acid in water (10 mmol) dissolved in 20 ml acetone, affording a clear solution. Stirring continued at ambient and a white solid precipitated out after approximately 1 hour. The mixture was cooled in an ice bath and stirred for an additional hour. The white solid was recovered by filtration and washed once with cold acetone (15 ml). It was subsequently dried under vacuum to yield 3.94 g of the lactate monohydrate salt of N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2E-2-propenamide (86.2%).
References
- Revill, P; Mealy, N; Serradell, N; Bolos, J; Rosa, E (2007). “Panobinostat”. Drugs of the Future 32 (4): 315. doi:10.1358/dof.2007.032.04.1094476. ISSN 0377-8282.
- Table 3: Select epigenetic inhibitors in various stages of development from Mack, G. S. (2010). “To selectivity and beyond”. Nature Biotechnology 28 (12): 1259–1266.doi:10.1038/nbt.1724. PMID 21139608. edit
- ClinicalTrials.gov NCT00425555 Study of Oral LBH589 in Adult Patients With Refractory Cutaneous T-Cell Lymphoma
- ClinicalTrials.gov: LBH-589
- Prince, HM; M Bishton (2009). “Panobinostat (LBH589): a novel pan-deacetylase inhibitor with activity in T cell lymphoma”. Hematology Meeting Reports (Parkville, Australia: Peter MacCallum Cancer Centre and University of Melbourne) 3 (1): 33–38.
- Simons, J (27 April 2013). “Scientists on brink of HIV cure”. The Telegraph.
- ClinicalTrials.gov NCT01680094 Safety and Effect of The HDAC Inhibitor Panobinostat on HIV-1 Expression in Patients on Suppressive HAART (CLEAR)
- Mayo Clinic Researchers Formulate Treatment Combination Lethal To Pancreatic Cancer Cells
- Garbes, L; Riessland, M; Hölker, I; Heller, R; Hauke, J; Tränkle, Ch; Coras, R; Blümcke, I; Hahnen, E; Wirth, B (2009). “LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate”. Human Molecular Genetics 18 (19): 3645–3658. doi:10.1093/hmg/ddp313.PMID 19584083.
- Tate, CR; Rhodes, LV; Segar, HC; Driver, JL; Pounder, FN; Burow, ME; and Collins-Burow, BM (2012). “Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat”. Breast Cancer Research 14 (3).
- TA Rasmussen, et al. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Human Vaccines & Immunotherapeutics 9:5, 1-9, May 2013.
- Drugs of the Future 32(4): 315-322 (2007)
- WO 2002022577…
- WO 2007146718
- WO 2013110280
- WO 2010009285
- WO 2010009280
- WO 2005013958
- WO 2004103358
- WO 2003048774…
- Journal of Medicinal Chemistry, 2011 , vol. 54, 13 pg. 4694 – 4720
-
23009203 11-26-2012 Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. Journal of medicinal chemistry -
21634430 7-14-2011 Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. Journal of medicinal chemistry -
21417419 4-28-2011 Discovery, synthesis, and pharmacological evaluation of spiropiperidine hydroxamic acid based derivatives as structurally novel histone deacetylase (HDAC) inhibitors. Journal of medicinal chemistry -
19317450 4-23-2009 Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. Journal of medicinal chemistry -
15650931 1-1-2005 The American Society of Hematology–46th Annual Meeting and Exposition. HDAC, Flt and farnesyl transferase inhibitors. IDrugs : the investigational drugs journal -
US7989639 8-3-2011 PROCESS FOR MAKING SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2010286409 11-12-2010 SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2010179208 7-16-2010 Use of HDAC Inhibitors for the Treatment of Bone Destruction US2010160257 6-25-2010 USE OF HDAC INHIBITORS FOR THE TREATMENT OF MYELOMA US2010137398 6-4-2010 USE OF HDAC INHIBITORS FOR THE TREATMENT OF GASTROINTESTINAL CANCERS US2009306405 12-11-2009 PROCESS FOR MAKING N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE AND STARTING MATERIALS THEREFOR US2009281159 11-13-2009 USE OF HDAC INHIBITORS FOR THE TREATMENT OF LYMPHOMAS US2009264439 10-23-2009 Combination of a) N–4-(3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia US2009197936 8-7-2009 SALTS OF N-HYDROXY-3-[4-[[[2-(2-METHYL-1H-INDOL-3-YL)ETHYL]AMINO]METHYL]PHENYL]-2E-2-PROPENAMIDE US2009012066 1-9-2009 Method of Use of Deacetylase Inhibitors
| US2008319045 | 12-26-2008 | Combination of Histone Deacetylase Inhibitors and Radiation |
| US2008221126 | 9-12-2008 | Use of Hdac Inhibitors for the Treatment of Myeloma |
| US2008176849 | 7-25-2008 | DEACETYLASE INHIBITORS |
| US2006189674 | 8-25-2006 | Deacetylase inhibitors |
| US7067551 | 6-28-2006 | Deacetylase inhibitors |
| US2006100140 | 5-12-2006 | Combination of a) n-{5-[4-(4-methyl-piperazino-methyl)-benzoylamido]2-methylphenyl}-4- (3-pyridyl)-2-pyrimidine-amine and b) a histone deacetylase inhibitor for the treatment of leukemia |
| US6833384 | 12-22-2004 | Deacetylase inhibitors |
| US6552065 | 4-23-2003 | Deacetylase inhibitors |
| GB776693A | Title not available | |||
| GB891413A | Title not available | |||
| GB2185020A | Title not available | |||
| WO2002022577A2 | Aug 30, 2001 | Mar 21, 2002 | Kenneth Walter Bair | Hydroxamate derivatives useful as deacetylase inhibitors |
| WO2003016307A1 | Aug 6, 2002 | Aug 19, 1993 | Jolie Anne Bastian | β3 ADRENERGIC AGONISTS |
| WO2003039599A1 | Nov 5, 2002 | May 15, 2003 | Ying-Nan Pan Chen | Cyclooxygenase-2 inhibitor/histone deacetylase inhibitor combination |
| WO2005105740A2 | Apr 26, 2005 | Nov 10, 2005 | Serguei Fine | Preparation of tegaserod and tegaserod maleate |
| WO2006021397A1 | Aug 22, 2005 | Mar 2, 2006 | Recordati Ireland Ltd | Lercanidipine salts |
…………………………………..
extras
5. Mocetinostat (MGCD0103), including pharmaceutically acceptable salts thereof. Balasubramanian et al., Cancer Letters 280: 211-221 (2009).
Mocetinostat, has the following chemical structure and name:

Vorinostat, including pharmaceutically acceptable salts thereof. Marks et al., Nature Biotechnology 25, 84 to 90 (2007); Stenger, Community Oncology 4, 384-386 (2007).
Vorinostat has the following chemical structure and name:

Belinostat (PXD-101 , PX-105684)
(2E)-3-[3-(anilinosulfonyl)phenyl]-N-hydroxyacrylamide

……………………………………………….
Dacinostat (LAQ-824, NVP-LAQ824,)
((E)-N-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1 H-indol-3-yl)ethyl]amino]methyl]phenyl]prop-2-enamide

Entinostat (MS-275, SNDX-275, MS-27-275)
4-(2-aminophenylcarbamoyl)benzylcarbamate

(a) The HDAC inhibitor Vorinostat™ or a salt, hydrate, or solvate thereof.

Vorinostat………………..
(b) The HDAC inhibitor Givinostat or a salt, hydrate, or solvate thereof.

Givinostat or a salt, hydrate, or solvate thereof.
PSC 833 ( Valspodar )

Valspodar, SDZ-PSC-833, PSC-833, Amdray
P-Glycoprotein (MDR-1; ABCB1) Inhibitors , Multidrug Resistance Modulators
Valspodar is a cyclosporine derivative and a P-glycoprotein inhibitor currently in phase III clinical trials at the National Cancer Institute (NCI) in combination with chemotherapy for the treatment of leukemia. The drug was also being developed in combination with chemotherapy for the treatment of various other types of cancers, however, no recent developments on these trials have been reported.
P-glycoprotein is an ABC-transporter protein that has been implicated in conferring multidrug resistance to tumor cells. In previous trials, valspodar was associated with greater disease-free and overall survival in younger patients (45 years or below), and was shown to significantly increase the cellular uptake of daunorubicin in leukemic blast cells in vivo. However, in a phase III trial examining the drug candidate’s effects on AML in patients at least 60 years of age, valspodar was associated with excessive mortality and complete remission rates were higher in groups not treated with the compound.
Nonimmunosuppressive cyclosporin analog which is a potent multidrug resistance modifier; 7-10 fold more potent than cyclosporin A; a potent P glycoprotein inhibitor; MW 1215.
M.Wt: 1214.62
Formula: C63H111N11O12
CAS : 121584-18-7
IUPAC/Chemical name:
(3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-6,9,18,24-tetraisobutyl-3,21,30-triisopropyl-1,4,7,10,12,15,19,25,28-nonamethyl-33-((R,E)-2-methylhex-4-enoyl)-1,4,7,10,13,16,19,22,25,28,31-undecaazacyclotritriacontan-2,5,8,11,14,17,20,23,26,29,32-undecaone
6 – [(2S, 4R, 6E)-4-Methyl-2-(methylamino)-3-oxo-6-octenoic acid]-7-L-valine-cyclosporin A; Cyclo [[(2S, 4R, 6E) -4-methyl-2-(methylamino)-3-oxo-6-octenoyl]-L-valyl-N-methylglycyl-N-methyl-L-leucyl-L-valyl-N-methyl-L-leucyl-L- alanyl-D-alanyl-N-methyl-L-leucyl-Nm
[3′-oxo-4-butenyl-4-methyl-Thr1]-[Val2]-cyclosporine
Clinical trials
http://clinicaltrials.gov/search/intervention=psc+833
Synonyms
- 3′-Keto-bmt(1)-val(2)-cyclosporin A
- Amdray
- Psc 833
- PSC-833
- PSC833
- SDZ PSC 833
- Sdz-psc-833
- UNII-Q7ZP55KF3X
- Valspodar
Valspodar or PSC833 is an experimental cancer treatment and chemosensitizer drug.[1] It is a derivative of ciclosporin D.
Its primary use is that of a p-glycoprotein inhibitor. Previous studies in animal models have found it to be effective at preventing cancer cell resistance to chemotherapeutics, but these findings did not translate to clinical success.[2]
Valspodar, also known as PSC-833 is an analogue of cyclosporin-A. Valspodar inhibits p-glycoprotein, the multidrug resistance efflux pump, thereby restoring the retention and activity of some drugs in some drug-resistant tumor cells. This agent also induces caspase-mediated apoptosis.
PSC-833 is a non-immunosuppressive cyclosporin derivative that potently and specifically inhibits P-gp. In vitro experiments indicate that PSC-833interacts directly with P-gp with high affinity and probably interferes with the ATPase activity of P-gp. Studies in multidrug resistant tumor models confirm P-gp as the in vivo target of PSC-833 and demonstrate the ability of PSC-833 to reverse MDR leukemias and solid tumors in mice. Presently,PSC-833 is being evaluated in the clinic.
Valspodar can cause nerve damage.[1]
Valspodar

Synthesis By oxidation of cyclosporin D (I) with N-chlorosuccinimide and dimethylsulfide in toluene (1) Scheme 1 Description alpha (20, D) -..?. 255.1 (c 0.5, CHCl3) Manufacturer Sandoz Pharmaceuticals Corp (US).. . References 1 Bollinger, P., B flounder sterli, JJ, Borel, J.-F., Krieger, M., Payne, TG, Traber, RP, Wenger, R. (Sandoz AG; Sandoz Patent GmbH; Sandoz Erfindungen VmbH ). Cyclosporins and their use as pharmaceuticals.
AU 8817679, EP 296122, JP 89045396. AU 8817679; EP 0296122; JP 1989045396; JP 1996048696; US 5525590
……………………………..
- The cyclosporins comprise a class of structurally distinctive, cyclic, poly-N-methylated undecapeptides, generally possessing pharmacological, in particular immunosuppressive, anti-inflammatory and/or anti-parasitic activity, each to a greater or lesser degree. The first of the cyclosproins to be isolated was the naturally occurring fungal metabolite Ciclosporin or Cyclosporine, also known as cyclosporin A and now commercially available under the Registered Trade Mark SANDIMMUN®. Ciclosporin is the cyclosporin of formula A

wherein -MeBmt- represents the N-methyl-(4R)-4-but-2E-en-1-yl-4-methyl-(L)threonyl residue of formula B

in which -x-y- is trans -CH=CH- and the positive 2′, 3′ and 4′ have the configuration S, R and R respectively.
-
Since the original discovery of Ciclosporin, a wide variety of naturally occurring cyclosporins have been isolated and identified and many further non-natural cyclosporins have been prepared by total- or semi-synthetic means or by the application of modified culture techniques. The class comprised by the cyclosporins is thus now substantial and includes, for example, the naturally occurring cyclosporins A through Z [c.f. Traber et al. 1, Helv. Chim. Acta, 60, 1247-1255 (1977); Traber et al. 2, Helv. Chim. Acta, 65, 1655-1667 (1982); Kobel et al., Europ. J. Applied Microbiology and Biotechnology 14, 273-240 (1982); and von Wartburg et al. Progress in Allergy, 38, 28-45 (1986)], as well as various non-natural cyclosporin derivatives and artificial or synthetic cyclosporins including the dihydro- and iso-cyclosporins [in which the moiety -x-y- of the -MeBmt- residue (Formula B above) is saturated to give -x-y- = -CH₂-CH₂- / the linkage of the residue -MeBmt- to the residue at the 11-position of the cyclosporin molecule (Formula A above) is via the 3′-O-atom rather than the α-N-atom]; derivatised cyclosporins (e.g. in which the 3′-O-atom of the -MeBmt- residue is acylated or a further substituent is introduced at the α-carbon atom of the sarcosyl residue at the 3-position); cyclosporins in which the -MeBmt- residue is present in isomeric form (e.g. in which the configuration across positions 6′ and 7′ of the -MeBmt- residue is cis rather than trans); and cyclosporins wherein variant amino acids are incorporated at specific positions within the peptide sequence employing e.g. the total synthetic method for the production of cyclosporins developed by R. Wenger – see e.g. Traber et al. 1, Traber et al. 2 and Kobel et al. loc. cit.; U.S. Patents Nos 4 108 985, 4 210 581, 4 220 641, 4 288 431, 4 554 351 and 4 396 542; European Patent Publications Nos. 0 034 567 and 0 056 782; International Patent Publication No. WO 86/02080; Wenger 1, Transpl. Proc. 15, Suppl. 1:2230 (1983); Wenger 2, Angew. Chem. Int. Ed., 24, 77 (1985); and Wenger 3, Progress in the Chemistry of Organic Natural Products 50, 123 (1986).
-
The class comprised by the cyclosporins is thus now very large indeed and includes, for example [Thr]²-, [Val]²-, [Nva]²- and [Nva]²-[Nva]⁵-Ciclosporin (also known as cyclosporins C, D, G and M respectively), [3-O-acetyl-MeBmt]¹-Ciclosporin (also known as cyclosporin A acetate), [Dihydro-MeBmt]¹-[Val]²-Ciclosporin (also known as dihydro-cyclosporin D), [Iso-MeBmt]¹-[Nva]²-Ciclosporin (also known as isocyclosporin G), [(D)Ser]⁸-Ciclosporin, [MeIle]¹¹-Ciclosporin, [(D)MeVal]¹¹-Ciclosporin (also known as cyclosporin H), [MeAla]⁶-Ciclosporin, [(D)Pro]³-Ciclosporin and so on.
-
[In accordance with conventional nomenclature for cyclosporins, these are defined throughout the present specification and claims by reference to the structure of Ciclosporin (i.e. Cyclosporin A). This is done by first indicating the amino acid residues present which differ from those present in Ciclosporin (e.g. “[(D)Pro]³” to indicate that the cyclosporin in question has a -(D)Pro- rather than -Sar- residue at the 3-position) and then applying the term “Ciclosporin” to characterise remaining residues which are identical to those present in Ciclosporin.
-
The residue -MeBmt- at position 1 in Ciclosporin was unknown before the discovery of the cyclosporins. This residue and variants or modifications of it, e.g. as described below, are thus generally characteristic of the cyclosporins. In general, variants or alternatives to [MeBmt]¹ are defined by reference to the -MeBmt- structure. Thus for dihydrocyclosporins in which the moiety -x-y- (see formula B above) is reduced to -CH₂-CH₂-, the residue at the 1-position is defined as “-dihydro-MeBmt-“. Where the configuration across the moiety -x-y- is cis rather than trans, the resulting residue is defined as “-cis-MeBmt-“.
-
Where portions of the -MeBmt- residue are deleted, this is indicated by defining the position of the deletion, employing the qualifier “des” to indicate deletion, and then defining the group or atom omitted, prior to the determinant “-MeBmt-“, “-dihydro-MeBmt-“, “-cis-MeBmt-” etc.. Thus “-N-desmethyl-MeBmt-“, “-3′-desoxy-MeBmt-“, and “-3′-desoxy-4′-desmethyl-MeBmt-” are the residues of Formula B¹, B² and B³ respectively:

B¹ – X = CH₃, Y = OH, Z = H.
B² – X = CH₃, Y = H, Z = CH₃.
B³ – X = H, Y = H, Z = CH₃. -
Where positions or groups, e.g. in -MeBmt-, are substituted this is represented in conventional manner by defining the position and nature of the substitution. Thus -3′-O-acetyl-MeBmt- is the residue of formula B in which the 3′-OH group is acetylated (3′-O-COCH₃). Where substituents of groups, in e.g. -MeBmt-, are replaced, this is done by i) indicating the position of the replaced group by “des-terminology” as described above and ii) defining the replacing group. Thus -7′-desmethyl-7′-phenyl-MeBmt- is the residue of formula B above in which the terminal (8′) methyl group is replaced by phenyl. 3′-Desoxy-3′-oxo-MeBmt- is the residue of formula B above in which the 3′-OH group is replaced by =O.
-
In addition, amino acid residues referred to by abbreviation, e.g. -Ala-, -MeVal-, -αAbu- etc… are, in accordance with conventional practice, to be understood as having the (L)-configuration unless otherwise indicated, e.g. as in the case of “-(D)Ala-“. Residue abbreviations preceded by “Me” as in the case of “-MeLeu-“, represent α-N-methylated residues. Individual residues of the cyclosporin molecule are numbered, as in the art, clockwise and starting with the residue -MeBmt-, -dihydro-MeBmt- etc. … in position 1. The same numerical sequence is employed throughout the present specification and claims.]
-
[0010]Because of their unique pharmaceutical potential, the cyclosporins have attracted very considerable attention, not only in medical and academic circles, but also in the lay press. Cyclosporin itself is now commonly employed in the prevention of rejection following allogenic organ, e.g. heart, heart-lung, kidney and bone-marrow transplant, as well as, more recently, in the treatment of various auto-immune and related diseases and conditions. Extensive work has also been performed to investigate potential utility in the treatment of various parasitic diseases and infections, for example coccidiomycosis, malaria and schistosomiasis. Reports of investigative work into the potential utility of the very many other known cyclosporins in these or related indications now abound in the literature.



………………………………
References
- Wilkes, Gail; Ades, Terri B. (2004). Consumers Guide to Cancer Drugs. Jones & Bartlett Learning. p. 226. ISBN 9780763722548. Retrieved 29 May 2013.
- Tao, Jian’guo; Sotomayor, Eduardo. (2012). Hematologic Cancers: From Molecular Pathobiology to Targeted Therapeutics. Springer. p. 335. ISBN 9789400750289.
- PSC-833Drugs Fut 1995, 20(10): 1010
- US 5525590
- Synthesis of [S-[1-14C]Val(7)]VALSPODAR application of (+)/(-)-[13,14Cn]BABS and (+)/(-)-[13,14Cn]DPMGBS, part 4J Label Compd Radiopharm 2000, 43(3): 205
- WO 2006013094
- WO 2005013947
- WO 2002098418
- WO 1999017757
- Pharmaceutical Research, 2001 , vol. 18, 2 pg. 183 – 190
- US2003/158097 A1
- Valspodar; EP-B1 0 296 122:
- WO 94/07858

