
Home » Posts tagged 'phase 2' (Page 18)
Tag Archives: phase 2
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
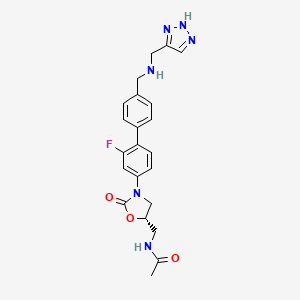
Radezolid in phase 2, Rib-X Pharmaceuticals

Radezolid
869884-78-6 cas no
http://www.ama-assn.org/resources/doc/usan/radezolid.pdf
Rib-X Pharmaceuticals
Phase II completed
N-{[(5S)-3-(2-fluoro-4′-{[(1H-1,2,3-triazol-5-ylmethyl)amino]methyl}biphenyl-4-yl)-2-oxo-1,3-oxazolidin-5-yl]methyl}acetamide
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide
Rib-X Pharmaceuticals has completed two Phase II clinical trials of radezolid for the treatment of pneumonia and uncomplicated skin infections. The trial completion dates were in 2008 and 2009, but to date the Phase III trials have not been initiated [1-6].
Radezolid (INN, codenamed RX-1741) is a novel oxazolidinone antibiotic being developed by Rib-X Pharmaceuticals, Inc. for the treatment of serious multi-drug–resistant infections. Radezolid has completed two phase-II clinical trials. One of these clinical trials was for uncomplicated skin and skin-structure infections (uSSSI) and the other clinical trial was for community acquired pneumonia (CAP).
Oxazolidinone antibiotics are a relatively new class of antibacterial agents with activity against a broad spectrum of gram-positive pathogens. The first member of this new class to be commercialized, linezolid, was approved in 2000. Since that time the development of linezolid resistant organisms has prompted efforts to discover more effective members of the oxazolidinone class.
A new family of biaryl oxazolidinone antibacterials with activity against both linezolid-susceptible and -resistant Gram-positive bacteria, as well as certain Gram-negative bacteria has been reported (see Bioorganic & Medicinal Chemistry Letters, 2008, 18, 6175-6178, and PCT Patent Publication WO 2005/019211).
Among the known biaryloxazolidinones is N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-bipheny- l-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide, more commonly known as radezolid (RX-1741), currently being developed for multi-drug-resistant infections.
Although a monohydrochloride salt of radezolid was disclosed in PCT Patent Publication WO 2006/133397, there is a continuing need for new salts and polymorphs thereof having improved properties such as solubility to optimize bioavailability on therapeutic administration.
Synthesis 1
http://www.google.co.il/patents/WO2005019211A2?hl=iw&cl=en
Scheme A

Scheme B illustrates the synthesis of intermediates 7 and 8 of the present invention using Suzuki coupling chemistry between boronic acids and aryl triflates. Boronic ester 6 is treated with an appropriate aryl triflate to yield the BOC-protected biaryl 7. The BOC group of 7 is removed to provide amine 8, an intermediate useful in the synthesis of certain compounds of the present invention.
Scheme B

8, R = NH2-HCI Scheme C depicts the synthesis of intermediates 9-13, which are useful in producing certain methoxy-substituted biaryl derivatives of the present invention. Suzuki coupling of boronic ester 6 produces biaryl aldehyde 9, which can be reduced to alcohol 10. Mesylation of 10 yields 11 that can be converted to azide 12. Reduction of azide 12 yields amine 13.
Scheme C

Scheme D depicts the synthesis of pyridyl intermediates, which are useful for the synthesis of compounds of the present invention, via similar chemistry to that shown in Scheme C. Coupling of boronic ester 6 to a halopyridine aldehyde produces biaryl aldehyde 14. Aldehyde 14 serves as the precursor to intermediates 15-18 via chemistry described above.
Scheme D

Biaryl aldehyde 19 (Scheme E) can be synthesized from a Suzuki coupling of iodide 1 and 4-formylphenylboronic acid. Scheme E illustrates how intermediate aldehydes of type 19, 9, and 14 can be converted via reductive amination chemistry to other amines, such as amines 20-22, which are useful as intermediates for the synthesis of certain compounds of the invention.
Scheme E

Scheme F depicts the general synthesis of compounds of type la and lb from amines of type 5, 13, 18, and 20-22. Compounds of type la and lb are synthesized via acylation of amines 5, 13 and 18 and 20-22 with the appropriate acids using, for example, l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) as the coupling agent. Compounds 4001-4007 were specifically synthesized from amine 5 and the appropriate carboxylic acids. Scheme F

Scheme G highlights the synthesis of compounds of general structure II from amines of type 5 and 18. The amine can be acylated with carboxylic acids using EDCI (or other commonly employed peptide coupling reagents known in the art) to afford amides II.
Acid chlorides can be purchased or synthesized and allowed to react with amines 5 and 18, in the presence of bases such as triethylamine, to also produce amides II.
Alternatively, carboxylic acids can be pre-loaded onto a solid polymeric support, such as a tetrafluorophenol containing resin (TFP resin), and reacted with amines to yield amide products of general structure II (such as compounds 4008-4015).
Scheme G

Scheme H illustrates the synthesis of compounds of general structure Ilia from amines of type 5, 13, and 18 using reductive amination chemistry. For example, biaryl amine compounds 4016-4028 are synthesized in this manner. Scheme H

Scheme I depicts the synthesis of general structure Illb of the present invention from amine intermediate 8. For example, compounds 4029-4031 are synthesized using this reductive amination chemistry.
Scheme I

Scheme J shows the synthesis of compounds of general structure IVa and IVb. Amines 20, 21, and 22 can be converted to tertiary amines IVa, such as compounds 4032-4034 and 4036, using standard reductive amination chemistry employed earlier for other derivatives.
This reductive amination chemistry can be employed on biaryl aldehyde intermediates such as 19, 9, and 14 to yield optionally substituted amines of general structure IVb, illustrated by compound 4037.
Scheme J

producing compounds of the present invention. Known iodoaryl oxazolidinone intermediate 50 (see U.S. Patent Nos. 5,523,403 and 5,565,571) is coupled to a substituted aryl boronic acid (the Suzuki reaction) to produce biaryl alcohol 51. Mesylate 52, azide 53, and amine 54 are then synthesized using chemistry well known to those skilled in the art. Scheme 1

NaN3, DMF, 70 °C


http://www.google.co.il/patents/WO2005019211A2?hl=iw&cl=en
……………….
NO 2
http://www.google.com/patents/US20100234615
| TABLE 1 | |
| Compound | |
| Number | Structure |
| 1 |
 |
Example 1 Synthesis of Compound 1
Compound 1 and its hydrochloride salt are synthesized according to the following Scheme:


4-Methoxybenzyl Azide
1001.
A solution of 4-methoxybenzyl chloride 1000 (51.8 g, 331.0 mmol) in anhydrous DMF (200 mL) was treated with solid sodium azide (21.5 g, 331.0 mmol, 1.0 equiv) at 25° C., and the resulting mixture was stirred at 25° C. for 24 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was quenched with H2O (400 mL) and ethyl acetate (EtOAc, 400 mL) at room temperature.
The two layers were separated, and the aqueous layer was extracted with EtOAc (200 mL). The combined organic extracts were washed with H2O (2×200 mL) and saturated NaCl aqueous solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude 4-methoxybenzyl azide (51.2 g, 53.95 g theoretical, 94.9% yield) was obtained as colorless oil, which by HPLC and 1H NMR was found to be essentially pure and was directly used in the subsequent reaction without further purifications. For 4-methoxybenzyl azide 1001:
1H NMR (300 MHz, CDCl3) δ 3.84 (s, 3H, ArOCH3), 4.29 (s, 2H, Ar—CH2), 6.96 (d, 2H, J=8.7 Hz), 7.28 (d, 2H, J=7.8 Hz).
C-[1-(4-Methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-Methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine
(1003 and 1004).
A solution of 4-methoxybenzyl azide 1001 (61.2 g, 375.5 mmol) in toluene (188 mL) was heated with propargylamine 1002 (commercially available, 30.97 g, 38.6 mL, 563.0 mmol, 1.5 equiv) at 25° C., and the resulting reaction mixture was warmed up to gentle reflux at 100-110° C. for 21 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was cooled down to room temperature before being concentrated in vacuo to remove the excess amount of propargylamine and solvent.
The oily residue was then treated with 30% ethyl acetate-hexane (v/v, 260 mL), and the resulting mixture was warmed up to reflux and stirred at reflux for 30 min before being cooled down to room temperature for 1 h. The pale-yellow solids were then collected by filtration, washed with 30% ethyl acetate-hexane (v/v, 2×100 mL), and dried in vacuo at 40° C. for overnight to afford the crude, cycloaddition product (78.8 g, 81.75 g theoretical, 96.4%) as a mixture of two regioisomers, C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004), in a ratio of 1.2 to 1 by 1H NMR.
The crude cycloaddition product was found to be essentially pure and the two regioisomers were not separated before being used directly in the subsequent reaction without further purification. For 1003 and 1004:
1H NMR (300 MHz, DMSO-d6) δ 1.82 (br. s, 2H, NH2), 3.72 and 3.73 (two s, 3H, Ar—OCH3), 5.47 and 5.53 (two s, 2H, ArCH2), 6.89 and 6.94 (two d, 2H, J=8.7 Hz, Ar—H), 7.17 and 7.29 (two d, 2H, J=8.7 Hz, Ar—H), 7.58 and 7.87 (two br. s, 1H, triazole-CH); C11H14N4O, LCMS (EI) m/e 219 (M++H) and 241 (M++Na).
4-({tert-Butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-Butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009).
Method A. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.0 g, 91.74 mmol) in 1,2-dichloroethane (DCE, 280 mL) was treated with 4-formylphenylboronic acid 1005 (commercially available, 12.39 g, 82.57 mmol, 0.9 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.2 g, 137.6 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h.
When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture was concentrated in vacuo. The residue, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with tetrahydrofuran (THF, 100 mL) and water (H2O, 100 mL).
The resulting solution was subsequently treated with solid potassium carbonate (K2CO3, 37.98 g, 275.2 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.02 g, 91.74 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL). The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo.
The crude, regioisomeric 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 35.98 g, 37.32 g, 96.4%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo.
This crude material was directly used in the subsequent reaction without further purification. For 1008 and 1009:
1H NMR (300 MHz, DMSO-d6) δ 1.32 and 1.37 (two br. s, 9H, COOC(CH3)3), 3.70, 3.73 and 3.74 (three s, 3H, Ar—OCH3), 4.07-4.39 (m, 4H), 5.49 and 5.52 (two s, 2H), 6.70-8.04 (m, 9H, Ar—H and triazole-CH); C23H29BN4O5, LCMS (EI) m/e 453 (M++H) and 475 (M++Na).
Method B. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.06 g, 92.0 mmol) in tetrahydrofuran (THF, 300 mL) was treated with 4-formylphenylboronic acid (13.11 g, 87.4 mmol, 0.95 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.25 g, 138.0 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h.
When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with water (H2O, 200 mL).
The resulting aqueous solution was subsequently heated with solid potassium carbonate (K2CO3, 38.0 g, 276 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.08 g, 92 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL).
The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude, 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 38.45 g, 39.50 g, 97.3%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo. This crude material was found to be essentially identical in every comparable aspect as the material obtained from Method A and was directly used in the subsequent reaction without further purification.
(5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester
(1011 and 1012).
A suspension of the crude regioisomeric mixture of 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 37.62 g, 83.23 mmol) and N-[3-(3-fluoro-4-iodo-phenyl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1010, 28.32 g, 74.9 mmol, 0.90 equiv) in toluene (150 mL) was treated with powder K2CO3 (34.45 g, 249.7 mol, 3.0 equiv), EtOH (50 mL), and H2O (50 mL) at 25° C.,
and the resulting mixture was degassed three times under a steady stream of Argon at 25° C. Pd(PPh3)4 (866 mg, 0.749 mmol, 0.01 equiv) was subsequently added to the reaction mixture, and the resulting reaction mixture was degassed three times again under a stead stream of Argon at 25° C. before being warmed up to gentle reflux for 18 h. When TLC and HPLC/MS showed the coupling reaction was complete, the reaction mixture was cooled down to room temperature before being treated with H2O (100 mL) and ethyl acetate (100 mL). The two layers were then separated, and the aqueous layer was extracted with EtOAc (100 mL).
The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×150 mL), H2O (100 mL), and the saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The residual oil was solidified upon standing at room temperature in vacuo to afford the crude, (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-y]methyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester (1011) and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1012) as a regioisomeric mixture.
This crude product (43.36 g, 49.28 g theoretical, 88%) was used directly in the subsequent reaction without further purification. For the mixture of 1011 and 1012: 1H NMR (300 MHz, DMSO-d6) δ 1.35 and 1.38 (two br. s, 9H, COO(CH3)3), 1.85 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.73 and 3.76 (two s, 3H, Ar—OCH3), 3.79 (dd, 1H, J=6.6, 9.1 Hz), 4.18 (t, 1H, J=9.1 Hz), 4.35-4.43 (m, 4H), 4.73-4.81 (m, 1H), 5.50 (br. s, 2H), 6.90 and 6.98 (two d, 2H, J=8.7 Hz), 7.28 and 7.32 (two d, 2H, J=8.7 Hz), 7.35 (dd, 2H, J=2.2, 8.6 Hz), 7.42 (dd, 1H, J=2.2, 8.6 Hz), 7.49-7.63 (m, 4H, aromatic-H), 7.90 and 7.99 (two br. s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3); C35H39FN6O6, LCMS (EI) m/e 659 (M++H) and 681 (M++Na).
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1013)
and
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H–[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1014).
A solution of a regioisomeric mixture of (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1011 and 1012, 37.28 g, 56.65 mmol) in ethyl acetate (EtOAc, 150 mL) and methanol (MeOH, 30 mL) was treated with a solution of 4 N hydrogen chloride in 1,4-dioxane (113.3 mL, 453.2 mmol, 8.0 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 12 h. When TLC and HPLC/MS showed that the N-BOC deprotection reaction was complete,
the solvents were removed in vacuo. The residue was then suspended in 250 mL of 5% methanol (MeOH) in acetonitrile (CH3CN), and the resulting slurry was stirred at room temperature for 1 h. The solids were then collected by filtration, washed with toluene (2×100 mL) and 5% methanol in acetonitrile (2×50 mL), and dried in vacuo to afford a regioisomeric mixture of the crude, (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 30.0 g, 33.68 g theoretical, 89.1% yield) as off-white crystals in a ratio of 1.2 to 1.
This material was found by 1H NMR and HPLC/MS to be essentially pure and was directly used in the subsequent reactions without further purification. For the regioisomeric mixture of 1013 and 1014:
1H NMR (300 MHz, DMSO-d6) δ 1.84 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.71 and 3.74 (two s, 3H, Ar—OCH3), 3.80 (dd, 1H, J=6.6, 9.1 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.23-4.30 (m, 4H), 4.73-4.80 (m, 1H), 5.58 and 5.70 (two s, 2H), 6.88 and 6.93 (two d, 2H, J=8.7 Hz), 7.15 and 7.32 (two d, 2H, J=8.7 Hz), 7.43 (dd, 2H, J=2.2, 8.6 Hz), 7.52-7.62 (m, 6H, aromatic-H), 8.28 (s, 1H, triazole-CH), 8.32 (t, 1H, J=5.8 Hz, NHCOCH3), 9.91 and 10.32 (two br. s, 2H, ArCH2N+H2); C30H31FN6O4, LCMS (EI) m/e 559 (M++H) and 581 (M++Na).
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide hydrochloride (1 hydrochloride salt).
A solution of the crude regioisomeric mixture of (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-1H-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 29.17 g, 49.07 mmol) in trifluoroacetic acid(TFA, 150 mL) was warmed up to 65-70° C., and the resulting reaction mixture was stirred at 65-70° C. for 12 h. When TLC and HPLC/MS showed that the deprotection reaction was complete, the solvents were removed in vacuo.
The residual solids were then treated with ethyl acetate (EtOAc, 100 mL) and H2O (150 mL) before being treated with a saturated aqueous solution of sodium carbonate (30 mL) at room temperature. The resulting mixture was then stirred at room temperature for 1 h before the solids were collected by filtration, washed with EtOAc (2×50 mL) and H2O (2×50 mL), and dried in vacuo at 40-45° C. to afford the crude, (5S)-N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl)-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1 as the free base, 18.9 g, 21.49 g theoretical, 87.9%) as off-white powders, which by HPLC/MS and 1H NMR was found to be one pure regioisomer and this regioisomer was found to be identical as the material obtained from deprotection of 1013 alone by the same method.
For 1 as the free base: 1H NMR (300 MHz, DMSO-d6) δ 1.85 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.74 (s, 2H), 3.77 (s, 2H), 3.79 (dd, 1H, J=6.4, 9.2 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.72-4.81 (m, 1H), 7.39-7.62 (m, 7H, aromatic-H), 7.73 (s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH); C22H23FN6O3, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
A suspension of 1 free base (18.0 g, 41.1 mmol) in ethyl acetate (EtOAc, 80 mL), and methanol (MeOH, 20 mL) was treated with a solution of 4.0 N hydrogen chloride in 1,4-dioxane (41.1 mL, 164.4 mmol, 4.0 equiv) at room temperature, and the resulting mixture was stirred at room temperature for 8 h. The solvents were then removed in vacuo, and the residue was further dried in vacuo before being treated with a mixture of 10% methanol in acetonitrile (80 mL). The solids were collected by filtration, washed with 10% MeOH/acetonitrile (2×40 mL), and dried in vacuo to afford 1 hydrochloride salt (18.13 g, 19.50 g theoretical, 93% yield) as off-white crystals.
The crude 1 hydrochloride salt can be recrystallized from acetonitrile and water, if necessary, according to the following procedure: A suspension of the crude 1 hydrochloride salt (50.0 g) in acetonitrile (1250 mL) was warmed up to reflux before the distilled water (H2O, 280 mL) was gradually introduced to the mixture. The resulting clear yellow to light brown solution was then stirred at reflux for 10 min before being cooled down to 45-55° C. The solution was then filtered through a Celite bed at 45-55° C., and the filtrates were gradually cooled down to room temperature before being further cooled down to 0-5° C. in an ice bath for 1 h. The solids were then collected by filtration, washed with acetonitrile (2×50 mL), and dried in vacuo at 40° C. for 24 h to afford the recrystallized 1 hydrochloride salt (42.5 g, 50.0 g theoretical, 85% recovery) as off-white crystals.
For 1: 1H NMR (300 MHz, DMSO-d6) δ 1.86 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.84 (dd, 1H, J=6.4, 9.2 Hz), 4.19 (t, 1H, J=9.1 Hz), 4.24 (br. s, 2H), 4.31 (br. s, 2H), 4.74-4.79 (m, 1H), 7.44 (dd, 1H, J=2.2, 8.6 Hz), 7.57-7.66 (m, 6H, aromatic-H), 8.17 (s, 1H, triazole-CH), 8.30 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH);
13C NMR (75 MHz, DMSO-d6) δ 22.57, 40.69, 41.50, 47.36, 49.23, 71.85, 105.70 (d, J=28.5 Hz), 114.14 (d, J=2.9 Hz), 122.29 (d, J=13.3 Hz), 128.82 (d, J=3.0 Hz), 130.70, 130.94, 131.0, 131.22, 135.30, 137.92 (br. s), 139.66 (d, J=11.2 Hz), 154.11, 159.13 (d, J=243.5 Hz), 170.19;
C22H23FN6O3—HCl, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
……………………………..
http://www.sciencedirect.com/science/article/pii/S0960894X0801192X

References
- Zhou J, Bhattacharjee A, Chen S, et al. (December 2008). “Design at the atomic level: design of biaryloxazolidinones as potent orally active antibiotics”.Bioorg. Med. Chem. Lett. 18 (23): 6175–8. doi:10.1016/j.bmcl.2008.10.011. PMID 18947996.
- Skripkin E, McConnell TS, DeVito J, et al. (October 2008). “Rχ-01, a new family of oxazolidinones that overcome ribosome-based linezolid resistance”.Antimicrob. Agents Chemother. 52 (10): 3550–7. doi:10.1128/AAC.01193-07. PMC 2565890. PMID 18663023.
- Lawrence L, Danese P, DeVito J, Franceschi F, Sutcliffe J (May 2008). “In vitro activities of the Rχ-01 oxazolidinones against hospital and community pathogens”. Antimicrob. Agents Chemother. 52 (5): 1653–62. doi:10.1128/AAC.01383-07. PMC 2346622. PMID 18316525.
- Hanselmann R, Job G, Johnson G, Lou R, Martynow JG, Reeve MM (2009). “Synthesis of an antibacterial compound containing a 1,4-substituted 1H-1,2,3-triazole- a scaleable alternative to the “click” reaction””. Organic Process Research and Development 14: 152–158. doi:10.1021/op900252a.
- Franceschi F, Duffy EM (March 2006). “Structure-based drug design meets the ribosome”. Biochem. Pharmacol. 71 (7): 1016–25.doi:10.1016/j.bcp.2005.12.026. PMID 16443192.
- Ohlsen K (November 2009). “Novel antibiotics for the treatment of Staphylococcus aureus“. Expert Rev. Clin. Pharmacol. 2 (6): 661–72.doi:10.1586/ecp.09.26.
- Radezolid at Rib-X Pharmaceuticals
- http://www.unil.ch/webdav/site/cnfmi/shared/abstracts_and_lectures/2009/3__F_van_Bambeke.pdf
- Sutcliffe, J.A. Antibiotics in development targeting protein synthesis. Ann. NY Acad. Sci. 2011, 1241, 122–152, doi:10.1111/j.1749-6632.2011.06323.x.
- Rib-X. Radezolid. Available online: http://www.rib-x.com/pipeline/radezolid.php#development (accessed on 14 April 2013).
- Rib-X Pharmaceuticals, Inc. Safety and efficacy study of oxazolidinone to treat pneumonia. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00640926 (accessed on 14 April 2013).
- Rib-X Pharmaceuticals, Inc. Safety and efficacy study of oxazolidinones to treat uncomplicated skin infections. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00646958 (accessed on 14 April 2013).
- Shaw, K.J.; Barbachyn, M.R. The oxazolidinones: Past, present, and future. Ann. NY Acad. Sci. 2011, 1241, 48–70, doi:10.1111/j.1749-6632.2011.06330.x.
- Skripkin, E.; McConnell, T.S.; DeVito, J.; Lawrence, L.; Ippolito, J.A.; Duffy, E.M.; Sutcliffe, J.; Franceschi, F. Rχ-01, a new family of oxazolidinones that overcome ribosome-based linezolid resistance.Antimicrob. Agents Chemother. 2008, 52, 3550–3557, doi:10.1128/AAC.01193-07.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6969726 * | Jun 2, 2004 | Nov 29, 2005 | Rib X Pharmaceuticals Inc | Biaryl heterocyclic compounds and methods of making and using the same |
| US20050043317 * | Jun 2, 2004 | Feb 24, 2005 | Jiacheng Zhou | Biaryl heterocyclic compounds and methods of making and using the same |
|
9-17-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
|
|
|
9-17-2010
|
Process for the synthesis of triazoles
|
|
|
4-28-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
|
|
|
11-26-2008
|
Biaryl heterocyclic compounds and methods of making and using the same
|
|
|
10-26-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
|
|
|
12-13-2006
|
Biaryl heterocyclic compounds and methods of making and using the same
|
|
|
11-30-2005
|
Biaryl heterocyclic compounds and methods of making and using the same
|

October 10, 2012
QIDP Designation for Radezolid for Acute Bacterial Skin and Skin Structure Infections, Community-acquired Bacterial Pneumonia
Rib-X Pharmaceuticals announced that the FDA designated radezolid as a Qualified Infectious Disease Product (QIDP) for the indications of acute bacterial skin and skin structure infections (ABSSSI) and community-acquired bacterial pneumonia (CABP).
The QIDP designation will enable Rib-X to benefit from certain incentives for the development of new antibiotics, including an additional five years of market exclusivity, priority review and eligibility for fast-track status, provided under the new Generating Antibiotic Incentives Now (GAIN) program. GAIN was included in the FDA Safety and Innovation Act (FDASIA), formerly known as PDUFA V, which received bipartisan Congressional support and was signed into law by President Obama in July 2012.
Radezolid has completed two Phase 2 clinical trials with an oral formulation in uncomplicated skin and skin structure infections (uSSSI) and in CABP. A Phase 1 study with an IV formulation was recently completed in healthy subjects. Rib-X recently announced data from a positive Phase 1 IV dosing study conducted in healthy subjects and an in vivo long-term safety study vs. linezolid (Zyvox; Pfizer).
Radezolid is a next-generation oxazolidinone with a safety profile permitting long-term treatment of resistant infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
For more information call (203) 624-5606 or visit www.rib-x.com


Sonidegib/Erismodegib..Novartis Cancer Drug LDE225 Meets Primary Endpoint in Phase 2


Sonidegib/Erismodegib
CODE DESIGNATION ..LDE225, NVP-LDE-225
Treatment of medulloblastoma PHASE3 2014 FDA FILING
Treatment of advanced basal cell carcinoma PHASE3 2014 FDA FILING
Treatment of SOLID TUMORS..PHASE1 2017 FDA FILING
READMalignant Solid Tumors of Childhood
THERAPEUTIC CLAIM Oncology, Antineoplastics & Adjunctive Therapies

CHEMICAL NAMES
1. [1,1′-Biphenyl]-3-carboxamide, N-[6-[(2R,6S)-2,6-dimethyl-4-morpholinyl]-3-pyridinyl]-2-
methyl-4′-(trifluoromethoxy)-, rel-
2. N-{6-[(2R,6S)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl}-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide
N-[6-[(2S,6R)-2,6-dimethylmorpholin-4-yl]pyridin-3-yl]-2-methyl-3-[4-(trifluoromethoxy)phenyl]benzamide
N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-(trifluoromethoxy)biphenyl-3-carboxamide
MOLECULAR FORMULA C26H26F3N3O3
MOLECULAR WEIGHT 485.5
SPONSOR Novartis Pharma AG
CAS REGISTRY NUMBER 956697-53-3 free form
NOTE… DIPHOSPHATE SALT IS THE DRUG WITH CAS 1218778-77-8
sonidegib – European Medicines Agency READ THIS..
Summary EudraCT Number: 2012-004022-21 Sponsor’s Protocol … READ THIS
About the Study
The Phase II, randomized, double-blind BOLT (Basal cell carcinoma Outcomes in LDE225 Trial) study was designed to assess the safety and efficacy of two oral dose levels of LDE225 (200 mg and 800 mg) in patients with locally advanced or metastatic basal cell carcinoma[4], which are subtypes of advanced basal cell carcinoma.
The primary endpoint was the proportion of patients achieving an objective response rate, defined as a confirmed complete response and partial response as their best overall response per modified RECIST criteria, within six months of starting treatment with LDE225. Key secondary endpoints of the study included assessing the duration of tumor responseand the rate of complete response. Other secondary endpoints included progression-free survival, time to tumor response and overall surviva

Sonidegib (INN) or Erismodegib (USAN), also known as LDE225 is a Hedgehog signalling pathway inhibitor (via smoothened antagonism) being developed as an anticancer agent by Novartis.[1][2] It has been investigated as a potential treatment for:
- Pancreatic cancer[3][4][5][6]
- Breast cancer[7][8]
- Basal cell carcinoma of the skin[9][10][11]
- Small cell lung cancer[12]
- Medulloblastoma[13][14]
- Advanced solid tumours (including ovarian, breast, pancreatic, stomach, oesophageal cancers and glioblastoma multiforme)[15][16][17]
- Acute leukaemia[18]
- Chronic myeloid leukaemia[19]
- Myelofibrosis and Essential thrombocythaemia[20]
NVP-LDE-225, a product candidate developed by Novartis, is in phase III clinical trials for the treatment of medulloblastoma and basal cell carcinoma. Phase II trials are in progress for the treatment of adult patients with relapsed or refractory or untreated elderly patients with acute leukemia.
Early clinical trials are ongoing for the oral treatment of advanced solid tumors, for the treatment of myelofibrosis in combination with ruxolitinib and for the treatment of small cell lung cancer. A phase II clinical trial for the treatment of basal cell carcinomas in Gorlin’s syndrome patients with a cream formulation of NVP-LDE-225 was discontinued in 2011 since the formulation did not demonstrate tumor clearance rate sufficient to support further development.
Dana-Farber Cancer Institute and the Massachusetts General Hospital are conducting phase I clinical trials for the treatment of locally advanced or metastatic pancreatic cancer in combination with chemotherapy. In 2009, orphan drug designation was assigned in the E.U. for the treatment of Gorlin syndrome.
It has demonstrated significant efficacy against melanoma in vitro and in vivo.[21] It also demonstrated efficacy in a mouse model of pancreatic cancer.[22]
NVP-LDE225 Diphosphate salt (Erismodegib, Sonidegib)

- Synonym:Erismodegib, Sonidegib
- CAS Number:1218778-77-8
- Mol. Formula:C26H26F3N3O3 ∙ 2H3PO4
- MW:681.5
- nmr.http://www.chemietek.com/Files/Line2/Chemietek,%20NVP-LDE225%20[02],%20NMR.pdf
- hplc–http://www.chemietek.com/Files/Line3/Chemietek,%20NVP-LDE225%20[02],%20HPLC.pdf
Brief Description:
About LDE225
LDE225 (sonidegib) is an oral, investigational, selective smoothened inhibitor being studied in a variety of cancers. Smoothened (SMO) is a molecule that regulates the hedgehog (Hh) signaling pathway, which plays a critical role in stem cell maintenance and tissue repair. LDE225 is currently in clinical development for a variety of diseases including myelofibrosis, leukemia and solid tumors.
Given that LDE225 is an investigational compound, the safety and efficacy profile has not yet been fully established. Access to this investigational compound is available only through carefully controlled and monitored clinical trials. These trials are designed to better understand the potential benefits and risks of the compound. Given the uncertainty of clinical trials, there is no guarantee that LDE225 will ever be commercially available anywhere in the world.
Possibility (LDE225) is effective in medulloblastoma relapsed or refractory hedgehog pathway inhibitor sonidegib has been revealed. That the anti-tumor effect was observed in some patients and tolerability in 1/2 test phase.
4th Quadrennial Meeting of the World Federation of Neuro-Oncology in conjunction with the 18th Annual Meeting of the Society for Neuro-Oncology, which was held in San Francisco November 21 to 24 in (WFNO-SNO2013), rice Dana-Farber It was announced by Mark Kieran Mr. Children’s Hospital Cancer Center.
The research group, announced the final results of the Phase 1 trial that target advanced solid cancer in children of sonidegib. 1 dose increased multi-test phase, was initiated from 372mg/m2 once-daily dosing to target children under the age of 18 more than 12 months. (233mg/m2 group 11 people, 16 people 372mg/m2 group, 11 people group 425mg/m2, 680mg/m2 group 21 women) who participated 59 people, including medulloblastoma 38 patients. 12 median age was (2-17).
Creatine phosphokinase elevation of grade 4 only were seen at 372mg/m2 as dose-limiting toxicity only, and became two recommended dose phase and 680mg/m2. Nausea muscle pain creatine kinase rise malaise (22.0%) (15.3%) (15.3%), (13.6%), vomiting side effects were many, was (13.6%). Hypersensitivity vomiting creatine kinase increased (3.4%) (1.7%) (1.7%), rhabdomyolysis side effects of grade 3/4 was (1.7%). (One group 372mg/m2, 425mg/m2 group one) complete response was obtained in two people, a strong correlation was found between the activation of the hedgehog pathway and effect.
Phase III clinical trials that target medulloblastoma the activated hedgehog pathway currently are underway.
About Novartis
Novartis provides innovative healthcare solutions that address the evolving needs of patients and societies. Headquartered in Basel, Switzerland, Novartis offers a diversified portfolio to best meet these needs: innovative medicines, eye care, cost-saving generic pharmaceuticals, preventive vaccines and diagnostic tools, over-the-counter and animal health products. Novartis is the only global company with leading positions in these areas. In 2013, the Group achieved net sales of USD 57.9 billion, while R&D throughout the Group amounted to approximately USD 9.9 billion (USD 9.6 billion excluding impairment and amortization charges). Novartis Group companies employ approximately 136,000 full-time-equivalent associates and operate in more than 140 countries around the world.

The following Examples serve to illustrate the invention without limiting the scope thereof, it is understood that the invention is not limited to the embodiments set forth herein, but embraces ali such forms thereof as come within the scope of the disclosure,

Step 1:
To a solution of 2-chloro-5-nitro-pyridine 1 (5.58 g, 35.2 mmoL) and c/s-2,6- dimethylmorpholine (4.05 g, 35.2 mmoL) in anhydrous DMF (30 mi.) was added K2CO3 (9.71 g, 70.4 mnrtoL). The mixture was heated at 50ºC overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 3 as a yellow solid, after purification by silica gel chromatography, obtained pure product (7.80 g, 93.2%). LC-MS m/z: 238.2 (M+ 1).
Step 2:
The above material 3 (7.3Og. 30.8 mmoL) was hydrogenated in the presence of 10% Pd-C (1.0 g) in MeOH (120 ml) under hydrogen overnight. The suspension was filtered through celite and the filtrate was concentrated to give the crude product 4 (5.92 g) as a dark brown oil which was used directly in the next step without further purification. LC-MS m/z. 208.2 (M+1).
Step 3:
To a solution of 3-bromo-2-methyl benzoic acid (2.71 g, 12.6 mmoL), 6-((2S,6R)-2,6- dimethylmorpholino)pyridin-3-arnine 4 (2.61 g, 12.6 mmoL), and HATU (4.80 g, 12.6 mmoL) in anhydrous DMF (30 mL) was added diisopropylethylamine (6.58 mL, 37.8 mmoL) dropwise. The resulting mixture was stirred overnight at room temperature. The reaction mixture was diluted with water (50 mL), and then extracted with EtOAc (3×120 mL). The organic layer was dried and concentrated to give the crude product. This crude product was then purified by flash column chromatography using 30% EtOAc in hexane as eiuent to give 5 as a white solid (4.23 g, 83.0%). LC-MS m/z: 404.1 (M+1).
Step 4:
A mixture of 4-(trif!uoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo- N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-ylJ-4-methyl-benzamide 5 (250 mg, 0.62mmol), Pd(PPh3)4 (36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL) in a sealed tube was heated at 130ºC overnight. The reaction mixture was diluted with EtOAc and water. The aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine and concentrated to give the crude product which was then purified by preparative mass triggered HPLC (C18 column, etuted with CH3CN-H2O containing 0.05% TFA) to give N-(6-((2S,6R)-2,6-dimethyfmorpholino)pyridin-3-yl)-2-rnethyl- 4′-(trifluoromethoxy)biphenyi-3-carboxamide (183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
The resultant crystalline product (Form A) was converted to the amorphous form by dissolving in 3% w/w aqueous ethanol, and the resultant solution spray dried at about 150ºC.
Form B was prepared by heating the amorphous form in an oven at 110ºC for 2 hours. In a further embodiment, the invention relates to a process step or steps, or an intermediate as described herein.

…………………………..
SYNTHESIS

| LC-MS m/z 486.2 (M + 1) |

Step 1: To a solution of 3-iodo-4-methyl-benzoic acid (10.0 g, 38.2 mmol) in methanol (70 ml) is added concentrated sulfuric acid (0.5 ml). The reaction mixture is heated at 70° C. for 48 hours, cooled to room ambient temperature and then concentrated. After that, ethyl acetate (100 ml) and aqueous NaHCO3 (saturated, 100 ml) solution are added to the residue. The organic layer is separated and washed again with aqueous NaHCO3 (saturated, 100 ml) solution. The organic layer is separated, dried over anhydrous Na2SO4 and concentrated to yield 3-iodo-4-methyl-benzoic acid methyl ester 1. It is used without further purification in the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (s, 1H), 7.87 (d, 1H, J=8.4 Hz), 7.48 (d, 1H, J=8.4 Hz), 3.85 (s, 3H), 3.35 (s, 3H); LC-MS m/z: 277.0 (M+1).
Step 2: To a round-bottom flask containing 3-iodo-4-methyl-benzoic acid methyl ester (1.38 g, 5.00 mmol), 4-cyanophenylboronic acid (1.10 g, 7.48 mmol), palladium acetate (168 mg, 0.748 mmol), 2-(dicyclohexylphosphino)biphenyl (0.526 g, 1.50 mmol) and potassium fluoride (0.870 g, 15.0 mmol) is added anhydrous 1,4-dioxane (15 ml). The flask is purged with argon and sealed. The mixture is stirred at 130° C. for 18 hours, cooled to ambient temperature and then water (20 ml) and ethyl acetate (20 ml) are added. Solid is removed under vacuum filtration. The filtrate is extracted with EtOAc (20 ml×2). The organic layers are combined, washed with aqueous HCl (5%, 20 ml) and saturated NaHCO3 (20 ml). It is dried over MgSO4, and concentrated. The residue is purified by silica gel column chromatography (EtOAc/Hexane, gradient) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2; LC-MS m/z: 252.1 (M+1).
Step 3: To a solution of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid methyl ester 2 (2.56 g, 10.3 mmol) in 1,4-dioxane-H2O (1:1 mixture, 20 ml) is added NaOH (1.22 g, 30.2 mmol)). The reaction is stirred at ambient temperature for 24 hours. To this mixture is added aqueous HCl (1 N, 36 ml) and it is then extracted with ethyl acetate (40 ml×3). The organic layers are combined, dried over anhydrous Na2SO4. The solver is removed. The solid obtained is washed with small amount of acetonitrile and air dried to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3: 1H NMR (DMSO-d6) δ 7.94 (d, 2H, J=8.0 Hz), 7.84 (dd, 1H, J1=8.4 Hz, J2=1.2 Hz), 7.75 (d, 1H, J=1.2 Hz), 7.61 (d, 2H, J=8.0 Hz), 7.48 (d, 1H, J=8.4 Hz), 2.29 (s, 3 H); LC-MS m/z 238.1 (M+1).
Step 4: To a suspension of 4′-cyano-6-methyl-biphenyl-3-carboxylic acid 3 (40 mg, 0.17 mmol) in anhydrous methylene chloride (5 ml) is added 2 drops of DMF. Then oxalyl chloride (32 mg, 22 μl, 0.25 mmol) is added. The mixture is stirred at ambient temperature until it turns clear. After that, it is concentrated, re-dissolved in anhydrous methylene chloride (3 ml), and added to a solution of 4-(morpholine-4-sulfonyl)-phenylamine (61 mg, 0.25 mmol) and triethylamine (34 mg, 47 μl, 0.33 mmol) in methylene chloride (2 ml). The mixture is stirred for 2 hours, concentrated and the residue is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [4-(morpholine-4-sulfonyl)-phenyl]-amide: 1H NMR (DMSO-d6) δ 10.64 (s, 1H), 8.07 (d, 2H, J=8.8 Hz), 7.97 (d, 2H, J=8.4 Hz), 7.95 (d, 1H, J=8.8 Hz), 7.89 (s, 1H), 7.43 (d, 2H, J=8.4 Hz), 7.67 (d, 2H, J=8.8 Hz), 7.53 (d, 2H, J=8.8 Hz), 3.63 (m, 4H), 2.84 (m, 4H) 2.32 (s, 3H); LC-MS m/z: 462.1 (M+1).
Example 2 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide

Step 1: To a solution of 2-chloro-5-nitro-pyridine 4 (2.38 g, 15 mmol.) and cis-2,6-dimethylmorpholine (1.73 g, 15 mmol.) is added K2CO3 (4.14 g, 30 mmol.). The mixture was heated at 50° C. overnight. After concentration, the residue is partitioned between EtOAc and water. The EtOAc layer is dried over anhydrous Na2SO4 and concentrated to give crude product 6 as a yellow solid. The crude product is used directly in next step without further purification. LC-MS m/z: 238.1 (M+1).
Step 2: The above crude material 6 is hydrogenated in the presence of Pd—C (0.2 g) in MeOH (100 mL) under hydrogen over 10 h. The suspension is filtered through celite and the filtrate is concentrated to give the crude product 7 as a dark brown oil which is used directly in the next step without further purification. LC-MS m/z: 208.1 (M+1).
Step 3: To a solution of 3-bromo-4-methyl benzoic acid (108 mg, 0.5 mmol.), 6-(2,6-Dimethyl-morpholin-4-yl)-pyridin-3-ylamine 7 (104 mg, 0.5 mmol.), amd HATU (190 mg, 0.5 mmol.) in dry DMF (5 mL) is added triethylamine (139 uL, 1.0 mmol.) dropwise. The resulting mixture is stirred at room temperature for 2 h. After concentration, the residue is partitioned between EtOAc and water. The organic layer is dried and concentrated to give the crude product. The final compound is purified by flash column chromatography using 50% EtOAc in hexane as eluent to give 8 as a white solid. LC-MS m/z: 404.1 (M+1).
Step 4: A mixture of 4-cyanophenyl boronic acid (18 mg, 0.12 mmol), 3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide 8 (40 mg, 0.1 mmol), Pd(PPh3)4 (11 mg, 0.01 mmol), and Na2CO3 (42 mg, 0.4 mmol) in a combined solvent system of toluene (0.2 mL) and water (0.2 mL) and ethanol (0.05 mL) is heated at 140° C. under microwave irradiation for 30 min. The reaction mixture is diluted with EtOAc and water. The aqueous layer is extracted with EtOAc. The combined organic layer is washed with brine and concentrated to give the crude product which is purified by preparative mass triggered HPLC (C18 column, eluted with CH3CN—H2O containing 0.05% TFA) to give 4′-cyano-6-methyl-biphenyl-3-carboxylic acid [6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-amide. LC-MS m/z: 427.2 (M+1).

4-(Trifluoromethoxy)phenylboronic acid
- CAS Number 139301-27-2
- Linear Formula CF3OC6H4B(OH)2
- Molecular Weight 205.93
CONDENSE WITH …3-bromo-N-[6-(2,6-dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamideACS Medicinal Chemistry Letters, 2010 , vol. 1, 3 p. 130 – 134

A mixture of 4-(trifluoromethoxy)phenylboronic acid (254 mg, 1.24 mmol), 3-bromo-N-[6-(2,6-
dimethyl-morpholin-4-yl)-pyridin-3-yl]-4-methyl-benzamide E (250 mg, 0.62mmol), Pd(PPh3)4
(36 mg, 0.03 mmol), Na2CO3 (2.0M aqueous solution, 1.23 mL, 2.4 mmol) and DME (4.5 mL)
in a sealed tube was heated at 1300C overnight. The reaction mixture was diluted with EtOAc
and water. The aqueous layer was extracted with EtOAc. The combined organic layer was
washed with brine and concentrated to give the crude product which was then purified by
preparative mass triggered HPLC (C18 column, eluted with CH3CN-H2O containing 0.05% TFA)
to give N-(6-((2S,6R)-2,6-dimethylmorpholino)pyridin-3-yl)-2-methyl-4′-
(trifluoromethoxy)biphenyl-3-carboxamide (5m, 183.5 mg, 61.1% yield). LC-MS m/z: 486.2 (M+1).
HRMS (m/z): [M+H]+
calcd for C26H27N3O3F3 486.2005; found 486.1986,
1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.15 (s, 1H), 8.43 (d, 1H), 7.94 (dd, 1H), 7.52-7.43
(m, 5H), 7.38 (m, 1H), 7.33 (m, 1H), 6.86 (d, 1H), 4.06 (d, 2H), 3.62 (m, 2H), 2,34 (m, 2H), 2.22
(s, 3H), 1.16 (d, 6H).
http://pubs.acs.org/doi/suppl/10.1021/ml1000307/suppl_file/ml1000307_si_001.pdf
Reference
- “LDE225 – PubChem”. PubChem. National Institutes of Health. Retrieved 16 February 2014.
- Pan, S; Wu, X; Jiang, J; Gao, W; Wan, Y; Cheng, D; Han, D; Liu, J; Englund, NP; Wang, Y; Peukert, S; Miller-Moslin, K; Yuan, J; Guo, R; Matsumoto, M; Vattay, A; Jiang, Y; Tsao, J; Sun, F; Pferdekamper, AC; Dodd, S; Tuntland, T; Maniara, W; Kelleher, JF; Yao, Y; Warmuth, M; Williams, J; Dorsch, M (10 June 2010). “Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist”. ACS Medicinal Chemistry Letters 1 (3): 130–134. doi:10.1021/ml1000307.
- “A Biomarker Study to Identify Predictive Signatures of Response to LDE225 (Hedgehog Inhibitor) In Patients With Resectable Pancreatic Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Gemcitabine + Nab-paclitaxel With LDE-225 (Hedgehog Inhibitor) as Neoadjuvant Therapy for Pancreatic Adenocarcinoma”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Dose-escalation, and Safety Study of LDE225 and Gemcitabine in Locally Advanced or Metastatic Pancreatic Cancer Patients”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Pilot Study of a Hedgehog Pathway Inhibitor (LDE-225) in Surgically Resectable Pancreas Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Study With LDE225 in Combination With Docetaxel in Triple Negative (TN) Advanced Breast Cancer (ABC) Patients (EDALINE)”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014.
- “LDE225 in Treating Patients With Stage II-III Estrogen Receptor- and HER2-Negative Breast Cancer”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “To Evaluate the Safety, Local Tolerability, PK and PD of LDE225 on Sporadic Superficial and Nodular Skin Basal Cell Carcinomas(sBCC)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Trial to Evaluate the Safety, Local Tolerability, Pharmacokinetics and Pharmacodynamics of LDE225 on Skin Basal Cell Carcinomas in Gorlin Syndrome Patients”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Combination of the Hedgehog Inhibitor, LDE225, With Etoposide and Cisplatin in the First-Line Treatment of Patients With Extensive Stage Small Cell Lung Cancer (ES-SCLC)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase III Study of Oral LDE225 Versus (vs) Temozolomide (TMZ) in Patients With Hedge-Hog (Hh)-Pathway Activated Relapsed Medulloblastoma (MB)”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase I Dose Finding and Safety Study of Oral LDE225 in Children and a Phase II Portion to Assess Preliminary Efficacy in Recurrent or Refractory MB”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Phase Ib, Dose Escalation Study of Oral LDE225 in Combination With BKM120 in Patients With Advanced Solid Tumors”.ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Dose Finding and Safety of Oral LDE225 in Patients With Advanced Solid Tumors”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “LDE225 and Paclitaxel in Solid Tumors”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Study of Efficacy and Safety of LDE225 in Adult Patients With Relapsed/Refractory Acute Leukemia”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “Nilotinib and LDE225 in the Treatment of Chronic or Accelerated Phase Myeloid Leukemia in Patients Who Developed Resistance to Prior Therapy”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- “A Phase Ib/II Dose-finding Study to Assess the Safety and Efficacy of LDE225 + INC424 in Patients With MF”. ClinicalTrials.gov. National Institutes of Health. 13 February 2014. Retrieved 16 February 2014.
- Jalili, A; Mertz, KD; Romanov, J; Wagner, C; Kalthoff, F; Stuetz, A; Pathria, G; Gschaider, M; Stingl, G; Wagner, SN (30 July 2013). “NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo.” (PDF). PloS one 8 (7): e69064. doi:10.1371/journal.pone.0069064. PMC 3728309.PMID 23935925.
- Fendrich, V; Wiese, D; Waldmann, J; Lauth, M; Heverhagen, AE; Rehm, J; Bartsch, DK (November 2011). “Hedgehog inhibition with the orally bioavailable Smo antagonist LDE225 represses tumor growth and prolongs survival in a transgenic mouse model of islet cell neoplasms.”. Annals of Surgery 254 (5): 818–23.doi:10.1097/SLA.0b013e318236bc0f. PMID 22042473.
- ChemMedChem, 2013 , vol. 8, 8 p. 1261 – 1265
- ACS Med. Chem. Lett., 2010, 1 (3), pp 130–134.
- MORE REF
sonidegib
Skin Cancer Foundation. “Skin Cancer Facts.” Available at:http://www.skincancer.org/skin-cancer-information/skin-cancer-facts . Accessed on February 14, 2014.
Rubin AI, Chen EH, Ratner D (2005). Current Concepts: Basal-Cell Carcinoma. N Engl J Med; 353:2262-9.
ClinicalTrials.gov. “A Phase II Study of Efficacy and Safety in Patients With Locally Advanced or Metastatic Basal Cell Carcinoma (BOLT)” Available at:http://clinicaltrials.gov/ct2/show/NCT01327053?term=%22LDE225%22+and+%22BOLT%22&rank=1. Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Complete Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45652 . Accessed on February 14, 2014.
National Cancer Institute Dictionary of Cancer Terms. “Partial Response.” Available at: http://www.cancer.gov/dictionary?CdrID=45819 . Accessed on February 14, 2014.
Wong C S M, Strange R C, Lear J T (2003). Basal cell carcinoma. BMJ; 327:794-798.
Copcu E, Aktas A. Simultaneous two organ metastases of the giant basal cell carcinoma of the skin. Int Semin Surg Oncol. 2005;2:1-6. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC544837/ . Accessed on February 14, 2014.
Skin Cancer Foundation. “Basal Cell Carcinoma Treatment Options.” Available athttp://www.skincancer.org/skin-cancer-information/basal-cell-carcinoma/bcc-treatment-options . Accessed on February 14, 2014.
Stuetz A, et al. LDE225, a specific smoothened inhibitor, for the topical treatment of nevoid basal cell carcinoma syndrome (Gorlin’s syndrome). Melanoma Research. 2010; 20:e40. Available at:http://journals.lww.com/melanomaresearch/Fulltext/2010/06001/FC24_LDE225,_a_specific_smoothened_inhibitor,_for.87.aspx#FC24_LDE225%2C_a_specific_smoothened_inhibitor%2C_for.87.aspx?s=2&_suid=139234380607909969110518506816.
Novartis.com. “The Pipeline of Novartis Oncology: LDE225.” Available at:http://www.novartisoncology.com/research-innovation/pipeline.jsp #. Accessed on February 14, 2014.
Children’s Medical Research Center, Children’s Memorial Hospital/Northwestern University Feinberg School of Medicine. “The Sonic hedgehog/patched/gli signal transduction pathway.” Available at http://www.childrensmrc.org/iannaccone/gli/ . Accessed on February 14, 2014.
Gupta S, Takebe N, LoRusso P. Targeting the Hedgehog pathway in cancer. Ther Adv Med Oncol. 2010 July; 2(4): 237-250. Available at:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3126020/ . Accessed on February 14, 2014.
| WO2004078163A2 | Feb 26, 2004 | Sep 16, 2004 | Oern Almarsson | Pharmaceutical co-crystal compositions of drugs such as carbamazepine, celecoxib, olanzapine, itraconazole, topiramate, modafinil, 5-fluorouracil, hydrochlorothiazide, acetaminophen, aspirin, flurbiprofen, phenytoin and ibuprofen |
| WO2007113120A1 | Mar 22, 2007 | Oct 11, 2007 | Frank Hoffmann | Stamping apparatus with feed device |
| WO2007131201A2 * | May 4, 2007 | Nov 15, 2007 | Irm Llc | Compounds and compositions as hedgehog pathway modulators |
| WO2008154259A1 | Jun 4, 2008 | Dec 18, 2008 | Irm Llc | Biphenylcarboxamide derivatives as hedgehog pathway modulators |

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
web link
blogs are

MY BLOG ON MED CHEM
ALL FOR DRUGS ON WEB
http://scholar.google.co.uk/citations?user=bxm3kYkAAAAJ


Fiduxosin ….An α1-Adrenoceptor antagonist
Fiduxosin hydrochloride, 208992-74-9, NCGC00162178-02, AC1L58WW,
208993-54-8 (free base)
 Fiduxosin
Fiduxosin-

-

-

-

-

-

-

-

-
Example 108
- 3-[4-((3aR,9bR)-cis -9-Methoxy-1,2,3,3a,4,9b-hexahydro-[1]-benzopyrano[3,4-c]pyrrol-2-yl)butyl]-8-(4-hydroxyphenyl)-pyrazino[2′,3′:4,5]thieno[3,2-d]pyrimidine-2,4(1H,3H)-dione
-
The product of Example 16 (0.07 g,0.105 mmol) and 4-(methoxymethyloxy) phenyl boronic acid (0.02 g, 0.11 mmol) prepared by the procedure described in Tetr.Lett., 31, 27, (1990) were treated as described in Example 106 to yield 0.029g(45%) of MOM-protected product. To the solution of this product (0.11g, 0.17 mmol) in CH3OH/THF was added 2N HCl (0.2ml) and the reaction mixture was refluxed for 1 hour. The reaction was evaporated and partitioned in NaHCO3 sol. and CH2Cl2/CH3OH to yield 0.005 g (51%) of the title compound.
-
1H NMR (500 MHz, CDCl3) d 1.81 (m, 2H), 1.98 (m, 2H), 2.25 (m, 1H), 2.65 (m, 1H), 2.88 (m, 1H), 3.08 (m, 2H), 3.22(m, 2H), 3.65 (m, 1H), 3.73 (m, 1H), 3.82 (s, 3H), 3.9 (m, 1H), 4.25 (m, 1H), 4.42 (m, 1H), 6.52 (m, 2H), 7.38 (m, 2H),7.49(m, 1H), 7.9 (t, 1H), 8.09 (d, 1H),9.12 (s, 1H);
-
MS(ESI)m/e 572 (M+H)+.
-








3-[4-((3aR,9bR)- cis -9-Methoxy-1,2,3,3a,4,9b-hexahydro-[1]-benzopyrano[3,4-c]pyrrol-2-yl)butyl]-8-chloro-pyrazino[2′,3′:4,5]thieno[3,2-d]pyrimidine-2,4(1H,3H)-dione hydrochloride
-
The product from Example 10 C (0.27 g, 1.0 mmol) and the product from Example 1E (0.20 g, 0.73 mmol) were treated as described in Example 1F to yield 0.29 g (77%) of the title compound: m.p. 220-222°;
-
1H NMR (300 MHz, CDCl3(free base)) δ 8.68 (s, 1H), 7.0 (t, 1H), 6.48 (d, 1H), 6.45 (d, 1H), 4.28 (m, 1H), 4.12 (m, 3H), 4.0 (m, 2H), 3.75 (s, 3H), 3.6 (m, 1H), 3.08 (m, 3H), 2.9 (m, 2H), 1.75 (m, 4H); MS (DCI/NH3) m/e 514(M+H)+;
-
Analysis calc’d for C24H24ClN5O4S·HCl·0.75H2O: C, 51.11; H, 4.74; N, 12.42; found: C, 51.09; H, 4.75; N, 12.43.

Fiduxosin (ABT-980), α1a-adrenoreceptor antagonist, a development compound at Abbot for the treatment of benign prostate hyperplasia, is disclosed in Organic Process Research & Development 2004, 8, 897-902 and references cited therein.
The synthetic route for preparation of Fiduxosin is as follows:


Fiduxosin (1) has been under development at Abbott Laboratories for the treatment of benign prostatic hyperplasia. A convergent strategy required methodologies for preparation of an enantiomerically pure 3,4-cis-disubstituted pyrrolidine and a 2,3,5-trisubstituted thienopyrazine in a regiospecific manner.
A [3+2] cycloaddition of an enantiopure azomethine ylide followed by a diastereoselective crystallization was employed to prepare the benzopyranopyrrolidine in high diastereomeric and enantiomeric purity. Conditions for reduction of an O-aryl lactone susceptible to epimerization were developed, and cyclization of the alcohol/phenol to the ether was accomplished in high yield.
The thienopyrazine was prepared by condensation of methyl thioglycolate and a regiospecifically prepared 2-bromo-3-cyano-5-phenylpyrazine. Conditions for effective halogen substitutive deamination to prepare regiospecific trisubstituted pyrazines will be described.
The mixture of 5 – and 6-phenyl regioisomers of 2-hydroxy-3-carboxamidopyrazine (IX) and (X), prepared by a known method, was treated with POCl3 and Et3N to produce the corresponding chloro nitriles (XI) and (XII ). Condensation of this mixture with methyl thioglycolate in the presence of NaOMe, followed by chromatographic separation of isomers furnished the desired thienopyrazine intermediate (XIII).
http://pubs.acs.org/doi/suppl/10.1021%2Fop049889k
…………………………………………………..
 Fiduxosin
Fiduxosin
……………………………………………………….
SYNTHESIS

Cycloaddition of the azomethine ylide resulting from N-trimethylsilylmethyl-N-methoxymethyl-(R)-alpha-methylbenzylamine (II) to 5-methoxycoumarin (I) produced the chiral cis-benzopyranopyrrole system (III). Lactone reduction by means of LiAlH4 or LiBH4 afforded diol (IV). After conversion of the primary alcohol of (IV) to either the corresponding chloride or the mesylate, cyclization in the presence of potassium tert-butoxide generated the tricyclic compound (V).
The alpha-methylbenzyl group of ( V) was removed by catalytic hydrogenation to give amine (VI), which was alkylated with 4-bromobutyronitrile yielding (VII). Reduction of the cyano group of (VII) using LiAlH4 in the presence AlCl3 or by catalytic hydrogenation in the presence of Raney -Ni produced the primary amine (VIII).
…………………………………………………

The mixture of 5 – and 6-phenyl regioisomers of 2-hydroxy-3-carboxamidopyrazine (IX) and (X), prepared by a known method, was treated with POCl3 and Et3N to produce the corresponding chloro nitriles (XI) and (XII ). Condensation of this mixture with methyl thioglycolate in the presence of NaOMe, followed by chromatographic separation of isomers furnished the desired thienopyrazine intermediate (XIII).
………………………………………………………….

In a regioselective synthetic method, phenyl glyoxime (XIV) was condensed with aminomalononitrile to produce the pyrazine N-oxide (XV). Reduction of the N-oxide of (XV) with triethyl phosphite yielded (XVI). Diazotization of the amino group of (XVI), followed by diazo displacement with CuBr2, furnished bromo pyrazine (XVII). This was then cyclized with methyl thioglycolate as above to yield the desired thienopyrazine intermediate (XIII).
………………………………………………….

In an alternative synthesis, phenylacetaldehyde (XVIII) was condensed with pyrrolidine (XIX) to give enamine (XX). Nitrosation of malononitrile (XXI), followed by treatment with tosyl chloride, produced the O-tosyl oxime (XXII). This was condensed with enamine (XX), and to the intermediate adduct (XXIII) was added thiophenol producing the phenylthiopyrazine (XXIV). Subsequent oxidation of the sulfide group of (XXIV) to sulfone (XXV), followed by condensation with methyl thioglycolate, gave the desired thienopyrazine (XIII).
……………………………………………………………..

The amino ester intermediate (XIII) was treated with phosgene and Et3N, and to the resulting isocyanate (XXVI) was added the primary amine (VIII), producing urea (XXVII). Then, cyclization of (XXVII) in refluxing toluene generated the desired compound,
fiduxosin
|
2-1-2002
|
Effect of fiduxosin, an antagonist selective for alpha(1A)- and alpha(1D)-adrenoceptors, on intraurethral and arterial pressure responses in conscious dogs.
|
The Journal of pharmacology and experimental therapeutics
|
|
|
2-1-2002
|
Modeling of relationships between pharmacokinetics and blockade of agonist-induced elevation of intraurethral pressure and mean arterial pressure in conscious dogs treated with alpha(1)-adrenoceptor antagonists.
|
The Journal of pharmacology and experimental therapeutics
|
|
|
1-1-2002
|
Effect of food on the pharmacokinetics of fiduxosin in healthy male subjects.
|
European journal of drug metabolism and pharmacokinetics
|
|
9-1-2012
|
Identification and analysis of hepatitis C virus NS3 helicase inhibitors using nucleic acid binding assays.
|
Nucleic acids research
|
|
|
3-1-2012
|
Small molecule screening identifies targetable zebrafish pigmentation pathways.
|
Pigment cell & melanoma research
|
|
|
7-1-2010
|
A small molecule inverse agonist for the human thyroid-stimulating hormone receptor.
|
Endocrinology
|
|
|
11-1-2009
|
A new homogeneous high-throughput screening assay for profiling compound activity on the human ether-a-go-go-related gene channel.
|
Analytical biochemistry
|
|
|
10-1-2009
|
Genetic mapping of targets mediating differential chemical phenotypes in Plasmodium falciparum.
|
Nature chemical biology
|
|
|
5-1-2007
|
Chemical genetics reveals a complex functional ground state of neural stem cells.
|
Nature chemical biology
|
|
|
5-1-2006
|
Microsphere-based protease assays and screening application for lethal factor and factor Xa.
|
Cytometry. Part A : the journal of the International Society for Analytical Cytology
|
|
|
5-1-2002
|
Single- and multiple-dose pharmacokinetics of fiduxosin under nonfasting conditions in healthy male subjects.
|
Journal of clinical pharmacology
|
|
|
5-1-2002
|
Multiple dose pharmacokinetics of fiduxosin under fasting conditions in healthy elderly male subjects.
|
The Journal of pharmacy and pharmacology
|
|
|
2-1-2002
|
Preclinical pharmacology of fiduxosin, a novel alpha(1)-adrenoceptor antagonist with uroselective properties.
|
The Journal of pharmacology and experimental therapeutics
|
DASANTAFIL
 Dasantafil
Dasantafil

569351-91-3 CAS NO
405214-79-1 (racemate)
THERAPEUTIC CLAIM treatment of erectile dysfunction (phosphodiesterase (PDE) 5 isoenzyme inhibitor)
read all at
ALL ABOUT DRUGS
CLICK BELOW
http://www.allfordrugs.com/2014/01/29/dasantafil-for-treatment-of-erectile-dysfunction/

TEGOBUVIR ..IN PHASE II FOR HEPATITIS C

TEGOBUVIR
A non-structural protein 5B polymerase inhibitor
for Treatment of chronic hepatitis C
5-[6-[2,4-Bis(trifluoromethyl)phenyl]pyridazin-3-ylmethyl]-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine
CHEMICAL NAMES
1. 5H-Imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis(trifluoromethyl)phenyl]-3-pyridazinyl]methyl]-
2-(2-fluorophenyl)-
2. 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-
imidazo[4,5-c]pyridine
MOLECULAR FORMULA C25H14F7N5
MOLECULAR WEIGHT 517.4
MANUFACTURER Gilead Sciences, Inc.
CODE DESIGNATION
- GS 333126
- GS 9190
- GS-333126
- GS-9190
- Tegobuvir
- UNII-5NOK5X389M
CAS REGISTRY NUMBER 1000787-75-6
GS-9190, an RNA-directed RNA polymerase (NS5B) inhibitor, is in phase II clinical evaluation at Gilead for the treatment of hepatitis C virus (HCV) infection. A clinical trial with GS-9190 in combination with peginterferon alfa-2a and ribavirin and with GS-9451 or with GS-9256 in treatment-naive subjects with chronic genotype 1 HCV infection was discontinued due to serious adverse events.
Gilead (Originator)
Katholieke Universiteit Leuven (Originator)
……………………………………….
tegobuvir
PATENTS
WO 2005063744
WO 2008005519
WO 2009009001
WO 2010151488
WO 2010151487
WO 2010151472
WO 2011072370
WO 2011156757
WO 2012087596
WO 2013101550
Hebner CM, Han B, Brendza KM, Nash M, Sulfab M, Tian Y, Hung M, Fung W, Vivian RW, Trenkle J, Taylor J, Bjornson K, Bondy S, Liu X, Link J, Neyts J, Sakowicz R, Zhong W, Tang H, Schmitz U.
PLoS One. 2012;7(6):e39163. doi: 10.1371/journal.pone.0039163. Epub 2012 Jun 13.
Wong KA, Xu S, Martin R, Miller MD, Mo H.
Virology. 2012 Jul 20;429(1):57-62. doi: 10.1016/j.virol.2012.03.025. Epub 2012 Apr 28.
Zeuzem S, Buggisch P, Agarwal K, Marcellin P, Sereni D, Klinker H, Moreno C, Zarski JP, Horsmans Y, Mo H, Arterburn S, Knox S, Oldach D, McHutchison JG, Manns MP, Foster GR.
Hepatology. 2012 Mar;55(3):749-58. doi: 10.1002/hep.24744.
Shih IH, Vliegen I, Peng B, Yang H, Hebner C, Paeshuyse J, Pürstinger G, Fenaux M, Tian Y, Mabery E, Qi X, Bahador G, Paulson M, Lehman LS, Bondy S, Tse W, Reiser H, Lee WA, Schmitz U, Neyts J, Zhong W.
Antimicrob Agents Chemother. 2011 Sep;55(9):4196-203. doi: 10.1128/AAC.00307-11. Epub 2011 Jul 11.
- ……………………..
- http://www.google.com/patents/WO2013040492A2
- ompound 1 can be prepared using synthetic methods and intermediates like those described in US 7,754,720. Compound 1 can also be prepared as described in the following Example.
- Compound 1 is:

Compound 1 may also be referred to as 5-((6-(2,4-bis(trifluoromethyl)phenyl)pyridazin-3-yl)methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridine, 5-[[6-[2,4-bis (trifluoromethyl)phenyl]pyridazin=3-yl]methyl]-2-(2-fluorophenyl).
- Example 1 : 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H- imidazo[4,5-c]pyridi


Compound 103 was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid 105 and a 2N aq. Na2C03 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80°C for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6g Si02 using MeOH/CH2CI2 to elute compound. The compound thus obtained was contaminated with PPh3(0). The product was repurified on a 1 mm Chromatotron plate with 0 to 5%
MeOH/CH2CI2 in 1 % steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound 1 was obtained with no PPh3 contamination. 1H NMR (300MHz,CD3OD) δ 6.20 (s, 2), 7.32 (m, 3), 7.52 (m, 1 ), 7.78 (d, 1), 7.89 (d, 1), 7.95 (s, 2), 8.15 (m, 3), 8.35 (d, 1), 9.12 (s, 1); LC/MS M+H = 518.
The intermediate compound 104 was prepared as follows, a. Preparation of Compound 10

101 102

To a solution of the commercially available starting material 101 in CHCI3, trichloroisocyanuric acid (TCCA) was added at 60°C. Then the solution was stirred for 1.5 hrs, cooled, and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g of compound 102. b. Preparation of Compound 104.

102 104

To a solution of compound 103 in DMF (dimethylformamide), NaOH was added.
Compound 102 was dissolved in DMF (20 mL) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2S0 . The solvent was removed and the product recrystallized with
dichloromethane. The yield was 5.7 g of compound 103.
- ……………………………
- US7754720
- Example 1a Synthesis of 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridineIn this method, dimethoxyethane or its related solvents, all having the general formula R1OR2O(R4O)aR3 wherein each of R1, R2, R3 and R4 are independently selected from C1-C6 alkyl and a is 0 or 1, have been found to be particularly advantageous over the conventional solvent DMF. Typically, each of R1, R2, R3 and R4 are independently C1-C2 alkyl and usually a is 0. C1-C6 alkyl includes fully saturated primary, secondary or tertiary hydrocarbon groups with 1 to 6 carbon atoms and thereby includes, but is not limited to methyl, ethyl, propyl, butyl, etc.Step 1



Compound MW Amount mmoles Equivalents SM 128.56 5 g 38.9 1 TCCA 232.41 3.62 g 15.6 0.4 CHCl3 130 ml To a solution of the commercially available starting material (SM) in CHCl3, trichloroisocyanuric acid (TCCA) was added at 60° C. Then the solution was stirred for 1.5 hrs., cooled down and filtered with HiFlo-Celite. The filtrate was concentrated and dried with vacuum. The yield was 5.037 g.
Step 2



Compound MW Amount mmoles Equivalents S.M. 163 5.073 g 31.12 1 Core 213.2 6.635 g 31.12 1 NaOH (10%) 40 1.245 g 31.12 1 DMF 320 ml To a solution of core (obtained as described in literature in DMF (dimethylformamide), NaOH was added. Then SM for this step (obtained from step 1) was dissolved in DMF (20 ml) and added to the solution slowly. The reaction was stirred for 3 hrs, was diluted with water and extracted with EtOAc. The organic layer was dried with Na2SO4. The solvent was removed and the product recrystallized with DCM (dichloromethane). The yield was 5.7 g.
Step 3



Compound MW Amount Moles Equivalents A 453.79 95 mg 0.209 1 DME 500 ul 2 N aq. Na2CO3 313ul 0.626 3 2,4-bisCF3– 257.93 80.9 mg 0.313 1.5 phenylboronic acid Pd(PPh3)4 1155 12 mg 0.0104 0.05 Compound A was dissolved in dimethoxyethane (DME). To this solution was added 2,4-bis(trifluromethyl)phenylboronic acid and a 2N aq. Na2CO3 solution. To the resulting biphasic mixture was added Pd(PPh3)4 and the reaction was then heated at 80° C. for 72 hrs. The reaction was cooled to room temperature and filtered through Celite and the Celite washed with EtOAc. The filtrate was concentrated in vacuo. The residue was purified on 6 g SiO2 using MeOH/CH2Cl2 to elute compound. The compound thus obtained was contaminated with PPh3(O). The product was repurified on a 1 mm Chromatotron plate with 0 to 5% MeOH/CH2Cl2 in 1% steps. The pure fractions were combined and concentrated in vacuo, then dried on high vacuum for 12 hrs. 11.8 mg of the free base of compound (1) was obtained with no PPh3 contamination.
1H NMR (300 MHz, CD3OD)
6.20 (s, 2)
7.32 (m, 3)
7.52 (m, 1)
7.78 (d, 1)
7.89 (d, 1)
7.95 (s, 2)
8.15 (m, 3)
8.35 (d, 1)
9.12 (s, 1)
LC/MS M+H=518
Example 1b Synthesis of 5-({6-[2,4-bis(trifluoromethyl)phenyl]pyridazin-3-yl}methyl)-2-(2-fluorophenyl)-5H-imidazo[4,5-c]pyridineThis example is directed to an additional method for making compound (1), employing the following schemes.

Methanesulfonic acid was added to 2-fluorobenzoic acid in a reactor with active cooling keeping T≦50° C. 3,4-Diaminopyridine was then added portionwise to this cooled slurry, keeping T≦35° C. The contents of the reactor were then heated to 50° C. Phosphorus pentoxide was added in a single charge. The reaction was then heated at 90-110° C. for at least 3 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature and water was added portionwise slowly to quench the reaction. The reaction was then diluted with water. In solubles were removed by filtration. The pH of the filtrate was adjusted to 5.5-5.8 with ammonium hydroxide. The reaction was allowed to self-seed and granulate for ˜4 hours at ambient temperature. The pH was then adjusted to 8.0-9.3 with ammonium hydroxide. The slurry was held at ambient temperature for at least 2 hours. The solids were isolated by filtration and washed with water, followed by IPE. The wet cake was dried in vacuo at not more than 60° C. until ≦1% water remains. The dry product is core (2).
Summary of Materials M.W. Wt. Ratio Mole ratio 3,4-Diaminopyridine 109.13 1.0 1.0 2-Fluorobenzoic acid 140.11 1.4 1.1 Methanesulfonic acid 96.1 7.0 8.0 Phosphorus pentoxide 141.94 1.3 1.0 Water 18.02 40 — Isopropyl ether 102.17 5.0 — Ammonium hydroxide 35.09 ~10 — 
A solution of compound (2a) in 1,2-dichloroethane was heated to 40-45° C. Trichloroisocyanuric acid was added and the mixture was heated at 60-70° C. for at least 2 hours. The reaction was sampled for completion by HPLC analysis. The reaction was cooled to ambient temperature. Celite was added to absorb insolubles, then solids were removed by filtration. The filtrate was washed with 0.5 N sodium hydroxide solution. The organic layer was concentrated to lowest stirrable volume and displaced with DMF. Core (2) and 10% aqueous sodium hydroxide solution were added. The reaction was stirred at ambient temperature for at least 8 hours. The reaction was sampled for completion by HPLC analysis. An additional 10% charge of 10% sodium hydroxide solution was added to the reaction. The reaction was then charged into water to isolate the crude product. After granulating for at least 1 hour, the solids were isolated and washed with water and isopropyl ether. Ethyl acetate was added and refluxed (internal T=70-77° C.) for 1-5 hours to dissolve product, then cooled to 18-23° C. slowly over 4-8 hours. The reactor contents were agitated at 18-23° C. for 8-20 hours and solids collected by filtration and rinsed with ethyl acetate. Low melt (i.e., DSC about 220 degrees C.) amorphous compound (1) was discharged. Amorphous compound (1) was dissolved in ethyl acetate by heating at reflux (internal T=70-77° C.) for 1-5 hours. Water content is controlled to about 0.2% by azeotropically removing water (with ethyl acetate the upper limit on water content is about 0.6% by weight; at about 0.9% by weight water the amorphous material will reprecipitate and crystals will not be obtained). The reactor contents are cooled slowly to 18-23° C. over 4-8 hours, then agitated at 18-23° C. for 8-20 hours and solids collected by filtration. The solids were rinsed with ethyl acetate and dried in vacuo at not more than 60° C. to obtain the dry crystalline compound (1).
Summary of Materials M.W. Wt. Ratio Mole ratio 3-chloro-6-methylpyridazine 128.56 1.0 1.0 2,4bis(trifluromethyl)phenylboronic 257.93 4.0 2.0 acid X-Phos 476.72 0.18 0.05 Palladium acetate 224.49 0.04 0.025 1,2-Dimethoxyethane 90.12 16.7 — Potassium carbonate 138.21 2.15 2.0 Water 18.02 7.8 — Copper iodide 190.45 0.037 0.025 Celite — 0.25 — Heptane 100.2 22.4 — Nuclear Magnetic Resonance (1H-, 13C-, and 19F-NMR) SpectraNuclear magnetic resonance (NMR) spectra of compound (1) is consistent with the proposed structure. The 13C, 19F, and 1H-NMR spectra of compound (1) in DMSO-d6 were measured using a Varian UnityInova-400 FT-NMR spectrometer. Spectra are shown in the table below. The NMR chemical shift assignments were established using 2D correlation experiments (COSY, HSQC, HMBC and HSQCTOCSY).
1H- and 13C-NMR Chemical Shift Assignments for Compound (1) Reference Standard
Atom δC/ppm (DMSO-d6) δF/ppm (DMSO-d6) δH/ppm (DMSO-d6) 1A 140.16 2A 128.32 (qa, JCF = 32 Hz) 3A 123.61, m 8.24 (m, 1 H) 4A 130.27 (q, JCF = 34 Hz) 5A 129.54 (q, JCF = 3 Hz) 8.22 (m, 1 H) 6A 133.36 7.88 (m, 1 H) 7A 123.20 (q, JCF = 273 Hz) −56.4b 8A 123.02 (q, JCF = 275 Hz) −62.0b 1B 158.76 2B 128.16 8.01 (d, 1 H, J = 8.4 Hz) 3B 126.20 7.95 (d, 1 H, J = 8.8 Hz) 4B 157.70 5B 60.49 6.17 (s, 2 H) 2C 131.86 8.31 (m, 1 H) 3C 112.63 7.86 (m, 1 H) 4C 155.44 6C 168.11 (d, JCF = 6 Hz) 8C 145.08 9C 133.06 9.25 (s, 1 H) 1D 123.11 (d, JCF = 10 Hz) 2D 160.46 (d, JCF = 254 Hz) −111.7 3D 116.59 (d, JCF = 22 Hz) 7.29 (m, 1 H) 4D 130.84 (d, JCF = 8 Hz) 7.46 (m, 1 H) 5D 124.13 (d, JCF = 4 Hz) 7.31 (m, 1 H) 6D 131.72 (d, JCF = 2 Hz) 8.35 (m, 1 H) amultiplicity, s: singlet, d: doublet, q: quartet, m: multiplet binterchangeable signals 
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html







DANOPREVIR (ITMN-191) …..a peptidomimetic inhibitor of the NS3/4A protease of hepatitis C virus (HCV)

Danoprevir
Danoprevir(ITMN-191) is a peptidomimetic inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.
Array BioPharma (Originator)
| RG7227 |
| ITMN-191 |
| RO5190591 |
2H-Isoindole-2-carboxylic acid, 4-fluoro-1,3-dihydro-, (2R,6S,12Z,13aS,14aR,16aS)-
14a-[[(cyclopropylsulfonyl)amino]carbonyl]-6-[[(1,1-dimethylethoxy)carbonyl]amino]-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydro-5,16-
dioxocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl ester
2. (2R,6S,12Z,13aS,14aR,16aS)-14a-[(cyclopropylsulfonyl)carbamoyl]-6-{[(1,1-
dimethylethoxy)carbonyl]amino}-5,16-dioxo-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-
a][1,4]diazacyclopentadecin-2-yl 4-fluoro-1,3-dihydro-2H-isoindole-2-carboxylate
Treatment of hepatitis C
MOLECULAR FORMULA C35H46FN5O9S
MOLECULAR WEIGHT 731.8
MANUFACTURER Genentech
CODE DESIGNATION R05190591
CAS REGISTRY NUMBER 850876-88-9, 916881-67-9
Danoprevir(ITMN-191) is a peptidomimetic
ITMN-191 (R-7227), a macrocyclic protease inhibitor, is in phase II clinical evaluation for the treatment of chronic hepatitis C virus (HCV) infection as monotherapy and in combination with Pegasys(R) (pegylated interferon alpha-2a) and Copegus(R) (ribavirin). The product candidate is also being evaluated in combination with R-7128 in treatment-naive patients infected with HCV genotype 1.
Danoprevir (ITMN-191; RG-7227), under development by InterMune Inc and Roche Holding AG, is a promising, potent NS3/4A protease inhibitor for the oral treatment of HCV infection. Preclinical data demonstrated that danoprevir binds with high affinity and dissociates slowly from the HCV NS3 protease, allowing high liver drug exposure with only modest plasma drug exposure.
In 2006, originator InterMune and licensee Roche entered into an exclusive worldwide collaboration agreement to develop and commercialize products from InterMune’s hepatitis C (HCV) protease inhibitor program, including ITMN-191. In 2010, the licensing agreement was terminated. Also in 2010, Roche acquired worldwide development and commercialization rights to R-7227 from InterMune. Preclinical pharmacokinetic results support the exploration of twice-daily oral dosing in HCV.
A phase Ib, ‘IFN-free’ clinical trial demonstrated that danoprevir, combined with the HCV polymerase inhibitor RG-7128 (Pharmasset Inc/Roche Holding AG), was effective in reducing HCV-RNA levels in a large proportion of treatment-naïve patients with HCV infection and in approximately half of previously non-responsive patients with HCV-1 infection, without resistance or safety concerns. In a phase IIb trial in treatment-naïve patients with HCV-1 infection, danoprevir plus pegylated IFNalpha2a and ribavirin resulted in undetectable levels of HCV-RNA in the majority of patients, without any evidence of viral resistance; however, the high-dose danoprevir arm was prematurely terminated because of grade 4 ALT elevations. Phase I trials have also demonstrated that ritonavir boosting improved the pharmacokinetic profile of danoprevir; therefore, at the time of publication, a phase IIb trial to evaluate ritonavir-boosted, low-dose danoprevir in combination with RG-7128 was planned. (source:
inhibitor of the NS3/4A protease of hepatitis C virus (HCV) with IC50 of 0.2-3.5 nM, inhibition effect for HCV genotypes 1A/1B/4/5/6 is ~10-fold higher than 2B/3A. Phase 2.
SODIUM SALT
HERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES
1. 2H-Isoindole-2-carboxylic acid, 4-fluoro-1,3-dihydro-, (2R,6S,12Z,13aS,14aR,16aS)-
14a-[[(cyclopropylsulfonyl)amino]carbonyl]-6-[[(1,1-dimethylethoxy)carbonyl]amino]-
1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydro-5,16-dioxocyclopropa
[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl ester, sodium salt (1:1)
2. sodium (cyclopropylsulfonyl){[(2R,6S,12Z,13aS,14aR,16aS)-6-{[(1,1-dimethylethoxy)
carbonyl]amino}-2-{[(4-fluoro-1,3-dihydro-2H-isoindol-2-yl)carbonyl]oxy}-5,16-dioxo-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydrocyclopropa[e]pyrrolo[1,2-
a][1,4]diazacyclopentadecine-14a(5H)-yl]formyl}azanide
MOLECULAR FORMULA C35H45FN5NaO9S
MOLECULAR WEIGHT 753.8
SPONSOR Genentech
CODE DESIGNATION
- Danoprevir sodium
- ITMN-191
- R 7227 sodium
- R7227
- RO 5190591-001
- RO5190591-001
- UNII-217RJI972K
CAS REGISTRY NUMBER 916826-48-7
DANOPREVIR SODIUM
The HCV protease mediates the cleavage of the HCV polyprotein to release the functional proteins that are essential for viral propagation. The inhibition of the HCV protease activity is expected to block HCV replication in infected host cells. Numberous HCV protease inhibitors have been identified. Non- limiting examples of HCV protease inhibitors are described in U.S. Patent Application Pub. Nos. 20040106559, 20040180815, 20040266668, 2004038872, 20050090432, 20050267018, 20070054842, 20070281885, 2007299078, 20080032936, 20080125444, 20080279821, 20090111757, 20090148407, 20090202480, 20090269305, 20090285773, 20090285774, 20100081700, 20100144608, 2010018355, 20100183551, 20100221217, 20100260710, 20100286185 and 20110135604, and U.S. Patent Nos. 6608027, 6767991, 7091184, 7119072, 7544798, 7642235 and 7829665, as well as WO2007014919, WO2007014926, WO2008046860, WO2008095058,
………………………………
danoprevir
patents and journal ref
1. WO 2005037214..
2. WO 2005095403
3. WO 2007015824..
4. WO 2008128921
5. WO 2009080542
6. WO 2009142842
7. WO 2010015545
8. WO 2013079424
9. WO 2012062685
10.WO 2013106631
11. Concise asymmetric synthesis of a (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid-derived sulfonamide and ethyl ester
Org Biomol Chem 2013, 11(39): 6796http://pubs.rsc.org/en/content/articlelanding/2013/ob/c3ob41394b/unauth#!divAbstract
12.J. Med. Chem., Article ASAP,DOI: 10.1021/jm400164c
Kazmierski WM, Hamatake R, Duan M, Wright LL, Smith GK, Jarvest RL, Ji JJ, Cooper JP, Tallant MD, Crosby RM, Creech K, Wang A, Li X, Zhang S, Zhang YK, Liu Y, Ding CZ, Zhou Y, Plattner JJ, Baker SJ, Bu W, Liu L.
J Med Chem. 2012 Apr 12;55(7):3021-6. doi: 10.1021/jm201278q. Epub 2012 Apr 3.
14 . Discovery of novel P3-oxo inhibitor of hepatitis C virus NS3/4A serine protease.
Duan M, Kazmierski W, Crosby R, Gartland M, Ji J, Tallant M, Wang A, Hamatake R, Wright L, Wu M, Zhang YK, Ding CZ, Li X, Liu Y, Zhang S, Zhou Y, Plattner JJ, Baker SJ.
Bioorg Med Chem Lett. 2012 Apr 15;22(8):2993-6. doi: 10.1016/j.bmcl.2012.02.039. Epub 2012 Feb 22.
……………….

(2R,6S,13aS,14aR,16aS,Z)-6-(tert-Butoxycarbonylamino)-14a-(cyclopropylsulfonylcarbamoyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl 4-fluoroisoindoline-2-carboxylate (49)



For certain NS3 inhibitors shown in this section, additional chemical transformations are utilized to obtain the final products. The preparations of two such examples are described for compounds 153 and 154 below:

(2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-2-(4-fluoroisoindoline-2-carbonyloxy)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecine-14a-carboxylic acid (0.10 g, 0.16 mmol) and TEA (0.024 mL, 0.18 mmol) in THF (5 mL) was added ethyl carbonochlridate (0.016 mL, 0.17 mmol) at 0° C. The reaction was stirred at 0° C. for 2 hrs. Sodium boronhydride (0.012 g, 0.32 mmol) was added and the reaction was stirred at rt for 3 days. Water (5 mL) and ethyl acetate (10 mL) were added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was purified by column chromatography (ethyl acetate) to give the product (0.060 g, 61.4%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.47 (b, 1H), 7.35 (m, 1H), 7.10-7.20 (m, 2H), 7.03 (m, 1H), 5.47 (m, 1H), 5.28 (b, 1H), 4.98 (m, 1H), 4.67 (b, 4H), 4.56 (m, 1H), 4.46 (m, 1H), 4.26 (m, 1H), 3.92 (m, 1H), 3.66 (m, 2H), 3.16 (m, 1H), 2.67 (m, 1H), 2.21 (m, 2H), 1.80 (m, 1H), 1.68 (m, 1H), 1.30 (m, 8H), 1.11-1.20 (m, 9H), 0.85 (m, 1H), 0.77 (m, 1H).

A solution of oxalyl chloride 90.045 mL, 0.089 mmol) in DCM (5 mL) at −78° C. was added a solution of DMSO (0.015 g, 0.020 mmol) in DCM (2 mL) dropwise over 2 ninytes. The reaction was stirred at −78° C. for 10 minutes and the a solution of (2R,6S,13aS,14aR,16aS,Z)-6-(tert-botoxycarbonylamino)-14a-(hydroxymethyl)-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-α][1,4]dizacyclopentadecin-2-yl-4-fluoroisoindoline-2-carboxylate (0.050 g, 0.081 mmol) in DCM (2 mL) was added. After stirred at −78° C. for 40 min, TEA (0.051 mL, 0.37 mmol) was added. The reaction was warmed to rt, water (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in MeOH (5 mL) and ammonium hydroxide (0.085 g, 2.45 mmol) and acetic acid (0.014 mL, 0.25 mmol) were added. The reaction stirred at rt for 3 minutes. NaCNBH3 90.015 g, 0.245 mmol) was added and stirred at rt for 30 minutes. The MeOH was removed. DCM (20 mL) and saturated sodium bicarbonate (5 mL) was added. The organic layer was separated, washed with brine and dried over sodium sulfate. After removal of solvent, the residue was dissolved in DCM (5 mL). TEA (0.017 mL, 0.122 mmol) was added and followed by the cyclopropanesulfonyl chloride (0.015 g, 0.098 mmol). The reaction was stirred at rt for 5 hrs. The solvent was removed. The residue was purified by column chromatography (ethyl acetate) to give the product (0.017 g, 28.2%) as white solid. 1H NMR (400 MHz, d6-DMSO) δ 8.52 (m, 1H), 7.35 (m, 1H), 7.02-7.20 (m, 4H), 5.56 (m, 1H), 4.99 (m, 1H), 4.97 (m, 1H), 4.67 (m, 2H), 4.66 (s, 2H), 4.46 (m, 1H), 4.24 (m, 1H), 3.92 (m, 1H), 3.67 (m, 1H), 3.46 (m, 1H), 2.74 (m, 1 h), 2.67 (m, 1H), 2.22 (m, 2H), 1.84 (m, 1H), 1.68 (m, 1H), 1.08-1.36 (m, 20H), 0.89 (m, 2H), 0.81 (m, 2H).

Hoffmann-La Roche and Genentech’s danoprevir/r (RG7227) is a twice-daily, ritonavir-boosted HCV protease inhibitor with activity against HCV genotypes 1, 4 and 6. DAUPHINE, an ongoing phase II trial in 421 treatment-naive people with HCV genotypes 1 and 4, is comparing doses (200, 100, and 50 mg danoprevir, boosted with 100 mg ritonavir, twice-daily) and response-guided therapy with danoprevir/r plus PEG-IFN/RBV. At 12 weeks after treatment completion, HCV RNA was undetectable in 86% of the highest-dosing arm, 77% of the 100 mg arm, and 65% of the 50 mg arm.
Response to treatment in the 200 mg dosing arm did not differ according to HCV subtype or IL28B genotype; at 12 weeks after treatment completion, 88% of people with HCV subtype 1a and an IL28B non-CC genotype had undetectable HCV RNA. Across all dosing arms, HCV RNA remained undetectable 12 weeks after treatment completion in 100% of people with HCV genotype 4.
In the response-guided therapy arm, 76% of early responders (who were treated for 12 weeks) and 67% of late responders (treated for 24 weeks) maintained undetectable HCV RNA 12 weeks after treatment completion, bringing the overall total to 72%.
One death occurred during the trial—from sudden heart attack, in a participant with preexisting diabetes and hypertension—it was considered unrelated to study drugs. Adverse events were reported in virtually all study participants. Side effects from ritonavir, which is used to boost danoprevir levels, increased the likelihood of more than one serious adverse event among people in the danoprevir/r arms (range 4–9% vs. 1% for placebo). The rate of danoprevir/r-related treatment discontinuations was similar to the rate of PEG-IFN/RBV-associated discontinuations (3–7%, and 3–8%, respectively).
Common side effects (experienced by more than 15% of study participants) included fatigue, fever, chills, weakness, nausea, diarrhea, itching, rash, hair loss, headache, aching muscles and joints, insomnia, cough, and appetite loss. Diarrhea was the only side effect associated with danoprevir/r. Adding danoprevir/r did not increase rates of rash or anemia (known side effects of other HCV protease inhibitors). Most grade 3 and grade 4 lab abnormalities were neutropenia, reported in 22% to 38% of study participants.
Interferon-free DAA Combinations
Danoprevir/r and Mericitabine, plus Ribavirin (HCV Genotypes 1 and 4)
Roche’s phase IIb study, INFORM-SVR, is combining response-guided therapy with danoprevir/r, a twice-daily ritonavir-boosted HCV protease inhibitor, and mericitabine, a twice-daily nucleoside polymerase inhibitor, with or without ribavirin for 12 to 24 weeks in non-cirrhotic people with HCV genotype 1. The original study design was modified after high relapse rates were observed in the 12-week treatment and ribavirin-free arms. Treatment was extended to 24 weeks, and ribavirin was given to all participants.
The majority of INFORM-SVR participants were male, had HCV genotype 1a, and non-CC genotypes. Of the 64 people treated for 24 weeks with all three drugs, 41% experienced SVR-12. People with HCV genotype 1b were more likely to achieve SVR-12 (71% versus 26% in HCV genotype 1a). In contrast, SVR-12 was more likely among people with non-CC genotypes (32% for CC versus 44% for non-CC), although only 4 people had HCV genotype 1b and CC genotype. Breakthrough rates were higher in people who did not receive ribavirin, and in HCV genotype 1a versus 1b. Resistance to danoprevir/r was observed in all patients who experienced viral breakthrough; mericitabine resistance was found in one person.
Almost all participants had more than one adverse event; a total of 567 mild-to-moderate events were reported among 83 people. The most common side effects, occurring in >10% of people were headache, fatigue, nausea, diarrhea, colds, insomnia, itching, weakness, dizziness, irritability, shortness of breath, cough, upset stomach, painful joints, and vomiting. As for laboratory abnormalities, one person experienced grade 3 anemia, four people had grade 3 lipid elevations, and one case each of grade 3 elevations in phosphate and lipase were observed.
A single serious adverse event, multiple myeloma, occurred 53 days after treatment completion and one person discontinued due to pain in the back of the throat (it was not specified whether or not this was a treatment-related adverse event).
- Everson G, Cooper C, Shiffman ML, et al. Rapid and sustained achievement of undetectable HCV RNA during treatment with ritonavir-boosted danoprevir/PEG-IFNa-2A/RBV in HCV genotype 1 or 4 patients: Dauphine week 36 interim analysis (Abstract 1177). Paper presented at: 47th Annual Meeting of the European Association for the Study of the Liver; 2012 April 18–22; Barcelona, Spain. Available from: http://mobile.ilcapp.eu/EASL_161/poster_24544/program.aspx. (Accessed 2012 June 25)
- Gane EJ, Pockros P, Zeuzem S, et al. Interferon-free treatment with combination of mericitabine and danoprevir/r with or without ribavirin in treatment-naïve HCV genotype-1 infected patients (Abstract 1412). 47th Annual Meeting of the European Association for the Study of the Liver; 2012 April 18–22; Barcelona, Spain. Available from:http://mobile.ilcapp.eu/EASL_161/poster_24848/program.aspx. (Accessed 2012 June 25)
Non- limiting examples of suitable HCV protease inhibitors include ACH-1095
(Achillion), ACH-1625 (Achillion), ACH-2684 (Achillion), AVL-181 (Avila), AVL-192 (Avila), BI-201335 (Boehringer Ingelheim), BMS-650032 (BMS), boceprevir, danoprevir, GS- 9132 (Gilead), GS-9256 (Gilead), GS-9451 (Gilead), IDX-136 (Idenix), IDX-316 (Idenix), IDX- 320 (Idenix), MK-5172 (Merck), narlaprevir, PHX-1766 (Phenomix), telaprevir, TMC-435 (Tibotec), vaniprevir, VBY708 (Virobay), VX-500 (Vertex), VX-813 (Vertex), VX-985 (Vertex), or a combination thereof. Non-limiting examples of suitable HCV polymerase inhibitors include ANA-598 (Anadys), BI-207127 (Boehringer Ingelheim), BILB-1941 (Boehringer Ingelheim), BMS-791325 (BMS), filibuvir, GL59728 (Glaxo), GL60667 (Glaxo), GS-9669 (Gilead), IDX-375 (Idenix), MK-3281 (Merck), tegobuvir, TMC-647055 (Tibotec), VCH-759 (Vertex & ViraChem), VCH-916 (ViraChem), VX-222 (VCH-222) (Vertex & ViraChem), VX-759 (Vertex), GS-6620 (Gilead), IDX-102 (Idenix), IDX-184 (Idenix), INX-189 (Inhibitex), MK-0608 (Merck), PSI-938 (Pharmasset), RG7128 (Roche), TMC64912 (Medivir), GSK625433 (Glaxo SmithKline), BCX-4678 (BioCryst), ALS-2200 (Alios BioPharma/Vertex), ALS-2158 (Alios BioPharma/Vertex), or a combination thereof. A polymerase inhibitor may be a nucleotide polymerase inhibitor, such as GS-6620 (Gilead), IDX-102 (Idenix), IDX-184 (Idenix), INX-189 (Inhibitex), MK-0608 (Merck), PSI-938 (Pharmasset), RG7128 (Roche), TMC64912 (Medivir), ALS-2200 (Alios BioPharma/Vertex), ALS-2158 (Alios BioPharma/Vertex), or a combination therefore. A polymerase inhibitor may also be a non- nucleoside polymerase inhibitor, such as ANA-598 (Anadys), BI-207127 (Boehringer Ingelheim), BILB-1941 (Boehringer Ingelheim), BMS-791325 (BMS), filibuvir, GL59728 (Glaxo), GL60667 (Glaxo), GS-9669 (Gilead), IDX-375 (Idenix), MK-3281 (Merck), tegobuvir, TMC-647055 (Tibotec), VCH-759 (Vertex & ViraChem), VCH-916 (ViraChem), VX-222 (VCH-222) (Vertex & ViraChem), VX-759 (Vertex), or a combination thereof. Non-limiting examples of suitable NS5A inhibitors include GSK62336805 (Glaxo SmithKline), ACH-2928 (Achillion), AZD2836 (Astra-Zeneca), AZD7295 (Astra-Zeneca), BMS-790052 (BMS), BMS- 824393 (BMS), GS-5885 (Gilead), PPI-1301 (Presidio), PPI-461 (Presidio), or a combination thereof. Non-limiting examples of suitable cyclophilin inhibitors include alisporovir (Novartis & Debiopharm), NM-811 (Novartis), SCY-635 (Scynexis), or a combination thereof. Non-limiting examples of suitable HCV entry inhibitors include ITX-4520 (iTherx), ITX-5061 (iTherx), or a combination thereof.
WO 2007015824WO 2003053349WO 2005095403WO 2005037214WO 2005095403WO 2005037214WO 2003053349WO 2007015824WO 2008128921
| US8048862 | 14 Apr 2009 | 1 Nov 2011 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US8119592 | 10 Oct 2006 | 21 Feb 2012 | Intermune, Inc. | Compounds and methods for inhibiting hepatitis C viral replication |
| US8232246 | 30 Jun 2009 | 31 Jul 2012 | Abbott Laboratories | Anti-viral compounds |
| US8299021 | 19 Apr 2012 | 30 Oct 2012 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US8420596 | 10 Sep 2009 | 16 Apr 2013 | Abbott Laboratories | Macrocyclic hepatitis C serine protease inhibitors |
| WO2013106631A1 | 11 Jan 2013 | 18 Jul 2013 | Abbvie Inc. | Processes for making hcv protease inhibitors |
Danoprevir Clinical Trial Information( data from http://clinicaltrials.gov)
| NCT Number | Recruitment | Conditions | Sponsor /Collaborators |
Start Date | Phases |
|---|---|---|---|---|---|
| NCT01331850 | Completed | Hepatitis C, Chronic | Hoffmann-La Roche | 2011-05 | Phase 2 |
| NCT01531647 | Completed | Healthy Volunteer | Hoffmann-La Roche | 2012-01 | Phase 1 |
| NCT01588002 | Completed | Healthy Volunteer | Hoffmann-La Roche | 2012-04 | Phase 1 |
| NCT01592318 | Active, not recruiting | Healthy Volunteer | Hoffmann-La Roche | 2012-05 | Phase 1 |
| NCT01749150 | Recruiting | Hepatitis C, Chronic | Hoffmann-La Roche | 2013-04 | Phase 2 |

WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
TOSEDOSTAT ….An aminopeptidase inhibitor with antineoplastic activity.

TOSEDOSTAT
An aminopeptidase inhibitor with antineoplastic activity.
- CHR 2797
- CHR-2797
- Tosedostat
- UNII-KZK563J2UW
- BB-76163Vernalis (Originator)
| CAS No. | 238750-77-1 |
| Chemical Name: | Tosedostat |
| Synonyms: | BB-76163;Chr-2797;tosedostat;CHR2797 (Tosedostat);Tosedostat (CHR2797);α-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]-benzeneaceticacidcyclopentlyester;alpha-[[(2R)-2-[(1S)-1-Hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]benzeneacetic acid cyclopentyl ester;Benzeneacetic acid, alpha-(((2R)-2-((1S)-1-hydroxy-2-(hydroxyamino)-2-oxoethyl)-4-methyl-1-oxopentyl)amino)-, cyclopentyl ester, (alphas)- |
| Molecular Formula: | C21H30N2O6 |
| Formula Weight: | 406.47 |
CHR-2797 is an oral, once-daily experimental cancer therapy in phase II clinical development at Chroma Therapeutics for the oral treatment of refractory acute myeloid leukemia in elderly patients. It is also in early clinical development for the treatment of refractory solid tumors alone or in combination with chemotherapy.
No recent development has been reported for phase I/II studies evaluating CHR-2797 as monotherapy in hematologic/blood cancer. A phase I/II clinical trial of the compound in combination with erlotinib for non-small cell lung cancer was terminated in 2010 due to very poor recruitment of patients to the study.
Cell Therapeutics is also conducting phase II clinical trials of the compound for the treatment of myelodysplasia and acute myeloid leukemia.
CHR- 2797 is an inhibitor of aminopeptidases and has demonstrated strong preclinical efficacy as monotherapy in addition to demonstrating strong synergy with a number of leading cancer therapies in a range of cancer cells. It was originally licensed from Vernalis, where it was being evaluated for its potential in treating multiple sclerosis; however development in this indication has been discontinued.
In 2008, orphan drug designation was assigned to CHR-2797 in the U.S. for the treatment of acute myeloid leukemia. In 2011, the compound was licensed to Cell Therapeutics by Chroma Therapeutics in Central America, North America and South America for exclusive marketing and codevelopment for the oral treatment of blood-related cancers and other cancers.
In corporate news, biopharmaceutical company Cell Therapeutics, Inc. (CTIC) was up more than 6% and near 52 week highs after saying Thursday that the U.S. FDA has removed the partial clinical hold on tosedostat and all studies underway have been allowed to continue. Tosedostat is under development for the treatment of blood-related cancers. It is currently being studied in Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia and high-risk myelodysplastic syndromes.

Tosedostat is a proprietary orally bioavailable inhibitor of the M1 family of aminopeptidases with potential antineoplastic activity.
Tosedostat is converted intracellularly into a poorly membrane-permeable active metabolite (CHR-79888) which inhibits the M1 family of aminopeptidases, particularly puromycin-sensitive aminopeptidase (PuSA), and leukotriene A4 (LTA4) hydrolase; inhibition of these aminopeptidases in tumor cells may result in amino acid deprivation, inhibition of protein synthesis due to a decrease in the intracellular free amino acid pool, an increase in the level of the proapoptotic protein Noxa, and cell death.
Noxa is a member of the BH3 (Bcl-2 homology 3)-only subgroup of the proapoptotic Bcl-2 (B-cell CLL/lymphoma 2) protein family
Cell Therapeutics announced that it has received notification from the U.S. Food and Drug Administration (FDA) that the partial clinical hold on tosedostat (IND 075503) has been removed and all studies underway may continue. Tosedostat is a first-in-class selective inhibitor of aminopeptidases, which are required by tumor cells to provide amino acids necessary for growth and tumor cell survival, and is under development for the treatment of blood-related cancers.
Tosedostat is currently being studied in the United States and European Union in investigator-sponsored and cooperative group-sponsored Phase 2 trials in elderly patients with newly diagnosed and relapsed acute myeloid leukemia (AML) and high-risk myelodysplastic syndromes (MDS).
“We are pleased that the FDA has responded favorably to the tosedostat clinical trial data provided and removed the partial clinical hold to allow further development of tosedostat in ongoing and future studies,” said John Pagel, MD, PhD, Associate Member, Clinical Research Division, Fred Hutchinson Cancer Research Center; Associate Professor, Medical Oncology Division, University of Washington School of Medicine; and Principal Investigator in the tosedostat first-line AML/MDS trial.
Recently, WO 93/20047 disclosed a class of hydroxamic acid based MMP inhibitors which also are active in inhibiting TNF production.
As mentioned above, MMP inhibitors have been proposed with hydroxamic acid or carboxylic acid zinc binding groups. The following patent publications disclose hydroxamic acid-based MMP inhibitors:
US 4599361 (Searle) EP-A-0236872 (Roche) EP-A-0274453 (Bellon) WO 90/05716 (British Bio-technology) WO 90/05719 (British Bio-technology) WO 91/02716 (British Bio-technology) EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) EP-A-0497192 (Roche) WO 92/13831 (British Bio-technology) WO 92/17460 (SmithKline Beecham) WO 92/22523 – (Research Corporation Technologies) WO 93/09090 (Yamanouchi) WO 93/09097 (Sankyo) WO 93/20047 (British Bio-technology) WO 93/24449 (Celltech) WO 93/24475 (Celltech) EP-A-0574758 (Roche) The following patent publications disclose carboxylic acid-based MMP inhibitors:
EP-A-0489577 (Celltech) EP-A-0489579 (Celltech) WO 93/24449 (Celltech) WO 93/24475 (Celltech)
|
|
|
TOSEDOSTAT
| WO1996033166A1 * | 17 Apr 1996 | 24 Oct 1996 | Du Pont Merck Pharma | Hydroxamic and carboxylic acids as metalloprotease inhibitors |
| WO1998011063A1 * | 8 Sep 1997 | 19 Mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| GB2268934A * | Title not available |
| US5652262 * | 14 mar 1994 | 29 lug 1997 | British Biotech Pharmaceutical, Ltd. | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| US5821262 * | 4 ott 1994 | 13 ott 1998 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as inhibitors of cytokine production |
| US5861436 * | 29 apr 1997 | 19 gen 1999 | British Biotech Pharmaceuticals Limited | Hydroxamic acid derivatives as metalloproteinase inhibitors |
| EP0423943A2 | 19 set 1990 | 24 apr 1991 | Beecham Group p.l.c. | Use of collagenase inhibitors in the treatment of demyelinating diseases, in particular multiple sclerosis |
| JPH03157372A | Titolo non disponibile | |||
| WO1997049674A1 | 20 giu 1997 | 31 dic 1997 | Francesca Abrate | Matrix metalloproteinase inhibitors |
| WO1998011063A1 | 8 set 1997 | 19 mar 1998 | British Biotech Pharm | Cytostatic hydroxamic acid derivatives |
| WO1999040910A1 | 27 gen 1999 | 19 ago 1999 | Andrew Paul Ayscough | Anti-inflammatory agents |
| WO1999044602A1 | 5 mar 1999 | 10 set 1999 | British Biotech Pharm | Inflammatory cell inhibitors |
| WO1999046241A1 | 12 mar 1998 | 16 set 1999 | British Biotech Pharm | Cytostatic agents |
| WO2000044373A1 * | Jan 27, 2000 | Aug 3, 2000 | Raymond Paul Beckett | Antibacterial hydroxamic acid derivatives |
| US6545051 | Jan 27, 2000 | Apr 8, 2003 | British Biotech Pharmaceuticals, Ltd. | Antibacterial hydroxamic acid derivatives |
Drugs Fut 2009, 34(2): 115
PLoS One (2013), 8(2), e57641.
WO 1999046241
WO 1995019956
WO 1998011063
US 6462023
US 20100260674
WO 2000044373
WO 9940910
NMR
http://file.selleckchem.com/downloads/nmr/S152202-CHR-2797-NMR-Selleck.pdf
Anti-Metastatic and Anti-Invasive Agents Compounds which have the property of inhibiting the action of the metalioproteinase enzymes involved in connective tissue breakdown and remodelling, such as fibroblast collagenase (Type 1 ), PMN-collagenase, 72 kDa-gelatinase, 92 kDa- gelatinase, stromelysin, stromelysin-2 and PUMP-1 (known as “matrix metalloproteinases”, and herein referred to as MMPs) have been proposed and are being tested in the clinic for the treatment of solid tumours. Cancer cells are particularly adept at utilising the MMPs to achieve rapid remodelling of the extracellular matrix, thereby providing space for tumour expansion and permitting metastasis. MMP inhibitors should minimise these processes and thus slow or prevent cancer progression.
In view of the rapid emergence of multidrug-resistant bacteria, the development of antibacterial agents with novel modes of action that are effective against the growing number of resistant bacteria, particularly the vancomycin resistant enterococci and β-lactam antibiotic-resistant bacteria, such as methicillin-resistant Staphylocccus aureus, is of utmost importance.
The natural antibiotic actinonin (see for example J. C. S Perkin I, 1975, 819) is a hydroxamic acid derivative of Structure (A):

In ddition to actinonin, various structural analogues of actinonin have also been shown to have antibacterial activity (see for example Broughton et al. (Devlin et al. Journal of the Chemical Society. Perkin Transactions 1 (9):830-841, 1975; Broughton et al. Journal of the Chemical Society. Perkin Transactions 1 (9):857-860, 1975).
The matlystatin group of compounds, share a number of structural similarities with actinonin. Both are peptidic molecules with functional hydroxamic acid metal binding groups (Ogita et al., J. Antibiotics. 45(11):1723-1732; Tanzawa et al., J. Antibiotics. 45(11):1733-1737; Haruyama et al., J. Antibiotics. 47(12):1473-1480; Tamaki et al., J. Antibiotics. 47(12):1481-1492).
………………………………………………………….
EXAMPLE 44 2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl-pentanoylamine]-2-phenyl-ethanoic acid cyclopentyl ester

The above compound was prepared using procedures similar to those described in example 8 using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1H, s), 5.2-5.14 (1H, m), 4.02 (1H, d, J=6.9 Hz), 2.94-2.85 (1H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1H, m) and 0.86 (6H, dd, J=6.5, 11 5 Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1, 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; 8 (MeOD), 7.33-7.19 (5H, m), 5.3 (1H, s), 5.11-5.06 (1H, m), 3.81 (1H, d, J=7.3 Hz), 2.83-2.74 (lH, m), 1.83-1.45 (10H, bm), 1.12-1.03 (lH, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3 Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2
Example 1
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

HO Ξ CONHOH
Prepared using procedures similar to those described in Preparative Example A using phenylglycine cyclopentyl ester.
Diastereoisomer A
Η-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d,
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, 14 dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2,
58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
Η-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d, J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and 0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3, 129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.

tosedostat
http://www.google.it/patents/US6545051

42
| WO98/11063 | WO99/46241 ex 1b | WO 98/11063 analogy ex 8 |

43
| WO98/11063 | WO99/46241 ex 1a | WO 98/11063 analogy ex 8 |
……………………………………………………………………
entry 65 in http://www.google.com/patents/WO2000044373A1
……………………………………………………………………………………………………….
http://www.google.com/patents/WO1999044602A1
Example 43
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester
TC

HO Ξ CONHOH
Prepared using procedures similar to those described in example 8 of WO 98/11063, using phenylglycine cyclopentyl ester.
Diastereoisomer A
1H-NMR; δ (MeOD), 7.4-7.29 (5H, m), 5.43 (1 H, s), 5.2-5.14 (1 H, m), 4.02 (1 H, d, 34
J=6.9Hz), 2.94-2.85 (1 H, m), 1.91-1.34 (10H, bm), 1.25-1.14 (1 H, m) and 0.86 (6H, dd, J=6.5, 11.5Hz).
13C-NMR; δ (MeOD), 175.6, 171.8, 171.4, 137.8, 129.8, 129.4, 128.6, 80.0, 73.2, 58.5, 49.2, 39.1 , 33.3, 33.3, 26.8, 24.5, 24.4, 23.7 and 22.1.
Diastereoisomer B
1H-NMR; δ (MeOD), 7.33-7.19 (5H, m), 5.3 (1 H, s), 5.11-5.06 (1 H, m), 3.81 (1 H, d,
J=7.3Hz), 2.83-2.74 (1 H, m), 1.83-1.45 (10H, bm), 1.12-1.03 (1 H, m) and
0.88-0.81 (6H, dd, J=6.4, 12.3Hz). 13C-NMR; δ (MeOD), 175.8, 171.8, 171.5, 137.3,
129.8, 129.5, 128.8, 79.9, 73.3, 58.7, 48.9, 39.2, 33.3, 33.3, 26.7, 24.5, 24.5, 24.0 and 22.2.
……………………………..
3R-isobutyl-4S-methoxy-dihydrofuran-2,5-dione (WO 97/02239)
…………………………………………………………………………..
2(S)-Amino(phenyl)ethanoic acid cyclopentyl ester

…………………………………………………………………..
2(R)-[2,2-Dimethyl-5-oxo-1,3-dioxolan-4(S)-yl]-4-methylpentanoic acid pentafluorophenyl ester

…………………………………………………………..
intermediates
238750-91-9
α-amino-, cyclopentyl ester Benzeneacetic acid,
……………….
cas 240489-34-3
2-[2R-(S-Hydroxy-hydroxycarbamoyl-methyl)-4-methyl-pentanoylamine]-2-phenyl- ethanoic acid cyclopentyl ester

…………………..
will be updated very soon… keep watching

THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock
need help, email or call me
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family

Vedroprevir
GS 9451, GS-9451, 1098189-15-1 USAN ZZ-81
VEDROPREVIR THERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES 1. Cyclopropanecarboxylic acid, N-[[(1α,3β,5α)-bicyclo[3.1.0]hex-3- yloxy]carbonyl]-3-methyl-L-valyl-(4R)-4-[[8-chloro-2-[2-[(1-methylethyl)amino]- 4-thiazolyl]-7-[2-(4-morpholinyl)ethoxy]-4-quinolinyl]oxy]-L-prolyl-1-amino-2- ethyl-, (1R,2R)-
2. N-{[(1R,3r,5S)-bicyclo[3.1.0]hex-3-yloxy]carbonyl}-3-methyl-L-valyl-(4R)-4-[(8- chloro-2-{2-[(1-methylethyl)amino]thiazol-4-yl}-7-[2-(morpholin-4- yl)ethoxy]quinolin-4-yl)oxy]-L-prolyl-(1R,2R)-1-amino-2- ethylcyclopropanecarboxylic acid
MOLECULAR FORMULA C45H60ClN7O9S
MOLECULAR WEIGHT 910.5 daltons
SPONSOR Gilead Sciences, Inc.
CODE DESIGNATION GS-9451
CAS REGISTRY NUMBER1098189-15-1
WHO NUMBER9745
GS-9451 is a NS3 protease inhibitor in phase II clinical trials at Gilead for the oral treatment of hepatitis C.
…………………………………………………………………………
Discovery of GS-9451: An acid inhibitor of the hepatitis C virus NS3/4A protease Bioorg Med Chem Lett 2012, 22(7): 2629 ……………………………………………………………
PATENTS WO 2012087596 WO 2009005676 WO 2013106631 WO2013101550 ……………………….
WO2012087596A1 Compound 3 can be prepared using synthetic methods and intermediates like those described in USSN 12/215,605 (US 20090257978 A1). Compound 3 can also be prepared described in the following Example. Example 3: Preparation of Compound 3

Compound 315 (12 g, 13 mmol) was dissolved in THF (200 ml), LiOH (11g, 260 mmol) in H20 (200 ml) was added, followed by MeOH (200 ml). The mixture was kept stirring at room temperature for 20 hours. Upon completion of the reaction, 4 N HCI in H20 was added to adjust pH to 7 at 0 °C. The mixture was extracted with EtOAc (2 x 400 ml). The combined organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo to give compound 3 as a yellow solid (11 g, 93%). LC/MS = 911.52(M++1 ). 1H NMR (300MHz, CD3OD)57.95 (d, 1H), 7.90 (s, 1H), 7.48 (s, 1H), 7.31 (d, 1H), 5.42 (s, 1H), 4.37 (dd, 1H), 4.20 (m, 2H), 3.83-3.56 (m, 7H), 3.50 (m, 2H), 3.39 (m, 2H), 2.45 (m, 1H), 2.27(m, 1H), 1.62 (m, 2H), 1.50 (m, 1H), 1.33 (m, 2H), 1.18 (m, 1H), 1.05 (m, 8H), 0.90 (m, 3H), 0.76 (m, 11H), 0.14-0.04 (m, 2H) The intermediate compound 315 was prepared as follows.

301 302 a. Preparation of compound 301. To a dry, argon purged three-neck round bottom flask (1000 mL) were added anhydrous dichloromethane (100 mL) and Et2Zn (28 mL, 273 mmol) at 0 °C. (CAUTION: Source of argon can not be from needle. Use appropriate glass adapter only. A second bubbler can also be attached to the flask to prevent excessive pressure build up.) Cyclopenten-3-ol (10.0 mL, 119 mmol) was then added dropwise (large quantity of ethane gas was produced) to the flask and the reaction mixture was allowed to stir until the evolution of gas had ceased. Diiodomethane (22 mL, 242 mmol) was then added dropwise over a period of 30 minutes. The reaction was allowed to warm to room temperature and continued to stir overnight under a positive flow of argon, at which point TLC analysis had indicated complete disappearance of the starting alcohol. The reaction was then diluted with CH2CI2 and quenched with 2M HCI (white precipitate should be completely dissolved). The biphasic mixture was poured into a separatory funnel and the organic layer was collected. The solvent was removed under reduced pressure until 100 mL of material containing compound 301 remained. b. Preparation of compound 302. Anhydrous dichloromethane (525 mL) was added to the flask followed by the dropwise addition of triethylamine (34 mL, 245 mmol). The reaction continued to stir at room temperature under a positive flow of nitrogen at which point, disuccinimidylcarbonate (40.7 g, 159 mmol) was added to the flask portion wise. The reaction was allowed to stir until TLC analysis indicated complete disappearance of the starting material (2-3 days). Upon completion, the reaction mixture was quenched with 1 M HCI (200 mL x 2) and washed with H20 (200 mL x 2). The desired material was extracted using CH2CI2and the combined organic layers were dried using anhydrous MgS0 and passed through a silica plug. The solvent was removed under reduced pressure and the crude material was purified using flash chromatography (Rf = 0.33, 1 :1 Hex/EtOAc) to provide compound 302 (22 g, 75%): 1H NMR (300 MHz, CDCI3): δ 5. 24 (t, 1 H), 3.82 (s, 4H), 2.24 (m, 2H), 2.03 (d, 2H), 1.38 (m, 2H), 0.48 (m, 1 H), 0.40 (m, 1 H).


c. Preparation of compound 304. N-i-Boc-cis-4-Hydroxy-L-Proline methyl ester 303 (100.0 g, 407.7 mmol) and DABCO (1.5eq, 68.6g, 61 1.6 mmol) were dissolved in anhydrous toluene (200 mL) in a 2 L three necked round bottom flask with a mechanical stirrer and an addition funnel. After cooling the solution to 0 °C under N2, A solution of 4-Bromo-benzenesulfonyl chloride (1.3eq, 135.6g, 530.0 mmol) in 300 mL of toluene was added through addition funnel over 60 minutes. The reaction mixture was stirred and warmed to room temperature overnight (16 hours). The mixture was slowly poured into 2L 1 M Na2C03 (aq.), and the product was extracted with EtOAc (2L). After the organic phase was washed by 0.5 N HCI (2L), H20 (1 L), and brine (1 L), it was dried (MgS04), concentrated to give 195.45 g of a yellow oily brosylate product. To a solution of the above brosylate (407.7 mmol) in dichloromethane (300 mL) was slowly added 4.0 M HCI in dioxane (500 mL, 5eq) and the resulting solution was allowed to stir at room temperature for 2 hours. After ether (500mL) was added to the reaction mixture, the mixture was stirred for 15 minutes and the white precipitate was collected by filtration. The solid was washed with ether and hexane and then dried under vacuum overnight to obtain 153.0 g of the HCI amine salt of compound 304, 381.8 mmol, in 94% yield for two steps. d. Preparation of compound 305. To a solution of Boc-fert-butyl-glycine (97.0g, 420.0 mmol) in DMF (200mL) and DCM (200mL) were added HATU (217.76g, 572.7 mmol) and Hunig’s base (126 mL, 1 145.4 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of the previous HCI salt (153.0 g, 381.8 mmol) and Hunig’s base (126 mL, 1 145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3h, with monitoring by LCMS. The reaction mixture was concentrated to remove dichloromethane under reduced pressure and the white solid that formed was filtered off. The remaining DMF solution was diluted with ethyl acetate (1 L), washed successively with 3% LiCI (aq) (3x650mL), sat’d NH4CI (2x500mL), 0.5N HCI (aq) (2x600ml_), brine (500ml_), sat’d NaHC03 (3x500mL), and brine (500mL). The resulting organic fraction was dried (MgS04) and concentrated to afford compound 305 (111g). e. Preparation of compound 306. To a solution of the methyl ester 305 (120 g, 207.8 mmol) in THF (300 ml_), MeOH (75 mL) was added a solution of LiOH (26.18 g, 623.4 mmol) in H20 (150 ml_). The solution was allowed to stir at room temperature for 4 hours. The mixture was cooled in an ice-bath while acidifying with 3N HCI to pH about 5.5, stirred for 10minut.es, and the resulting white solids were collected by filtration. The solids were washed with more water, ether and hexane. The solids were dried under vacuum at 40°C overnight to give 95.78g (82%) of the acid 306. f. Preparation of compound 307. To a solution of the carboxylic acid 306 (81.4 g, 144.27 mmol) in DMF (200ml_) and dichloromethane (200mL) was added HATU (82.3g, 216.4 mmol) and Hunig’s base (47.5 mL, 432.8 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of amine (158.7 mmol) and Hunig’s base (47.5 mL, 1145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3 hours and monitored by LCMS. After the mixture was concentrated under reduced pressure to remove dichloromethane, the white solids that formed were filtered off. The remaining DMF solution was diluted with ethyl acetate (600mL) and successively washed with 3% LiCI (aq) (2x550mL), sat’d NH4CI (500mL), 1 N HCI (aq) (500mL), sat’d NaHC03(500mL), and brine (300mL). The resulting organic fraction was dried (Na2S04) and concentrated to afford compound 307 (111 g). g. Preparation of compound 308. Compound 307 was dissolved in 4N HCI in dioxane (300 mL) at room temperature and stirred for 2 hours. It was then concentrated under vacuum, and co-evaporated with dichloromethane (2 x 200mL) to dryness. The residue was dissolved in EtOAc (600mL) and sat’d aq. NaHC03 (1 L). It was stirred vigorously. After 10 minutes, carbonic acid bicyclo[3.1.0]hex-3-yl ester 2,5-dioxo-pyrrolidin-1-yl ester 302 (41.4 g, 173.1 mmol) was added in one portion. After the resulting mixture was stirred for another 30 minutes, the organic layer was collected and washed with brine (500mL), dried (Na2S04), and concentrated. The crude product was purified by flash chromatography on silica gel with ethyl acetate/hexane to afford 94.44 g (92%) of compound 308.



h. Preparation of compound 310.1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 309 (70.7 g, 354 mmol) was stirred in 48% aq. HBr (500 mL) at 110 °C for 72 hours. After the mixture was cooled to 0 °C with stirring, the solids were filtered and washed with water. The resulting solids were triturated with a saturated NaHC03 solution (-350 mL), filtered, washed with water, and dried under vacuum to give – 40 g (61%) of crude 310 as a dark brown solid. LC/MS = 186 (M++1). i. Preparation of compound 311. 1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 310 (40 g, 215 mmol) was dissolved in DMF (360 ml). Cesium carbonate (140 g, 430 mmol) was added, followed by bromoacetaldehyde dimethyl acetal (54.5 g, 323 mmol). The mixture was then vigorously stirred at 65 °C for 24 hours. Upon cooling to room temperature, EtOAc (1 L) and H20 (1 L) were added to the mixture. The organic layer was extracted with EtOAc (1 x 400 ml). The combined organic layer was washed with aqueous 3% LiCI solution (2 x 1 L), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give compound 311 as a white solid (39 g, 67%). j. Preparation of compound 312. To a mixture of 1-[2-Amino-3-chloro-4-(2,2-dimethoxy-ethoxy)-phenyl]-ethanone 311 ( 13 g, 47.5 mmol) and isopropylaminothiazole-4-carboxylic acid hydrobromide (12.64 g, 47.5 mmol) in pyridine (150 ml) was slowly added phosphorus oxychloride (9.47 g, 61.8 mmol) at -40 °C. The mixture was then stirred at 0 °C for 4 hours. Upon completion of the reaction, H20 (30 ml) was added dropwise to the mixture. The mixture was then stirred at 0 °C for another 15 minutes. The mixture was concentrated in vacuo. The residue was diluted with EtOAc, washed with a sat. NaHC03 aqueous solution. The organic layer was dried (Na2S04) and concentrated in vacuo. The residue was dissolved in CH2CI2, hexanes were added slowly to the solution, and a yellow solid started to crash out. More hexanes were added until not much product was left in the mother liquid to provide compound 312 (18 g, 85%). k. Preparation of compound 313. 2-lsopropylamino-thiazole-4-carboxylic acid [6-acetyl-2-chloro-3-(2,2-dimethoxy-ethoxy)- phenyl]-amide 312 (18 g, 40.7 mmol) was suspended in toluene (400 ml). NaH (2.4 g, 61 mmol) was added to the vigorously stirred mixture while monitoring H2evolution. The mixture became a clear solution during heating to reflux. The reaction was complete after refluxing for 3 hours. The mixture was cooled to room temperature. A solution of AcOH (69.2 mmol) in H20 (3 vol) was added to the mixture. After vigorous agitation for 1 hour at 0 °C, the solids were collected by filtration, rinsed forward with H20. The wet cake was dried under high vacuum to a constant weight to provide compound 313 ( 5 g, 86%). I. Preparation of compound 314. To a mixture of brosylate intermediate 303 (15 g, 35 mmol) and compound 313 (27.5 g, 38.5 mmol) in NMP (200 ml) was added cesium carbonate (25.1 g, 77 mmol). The mixture was stirred at 65 °C for 5 hours. The reaction was cooled to room temperature and EtOAc (600 ml) and an aqueous solution of 3% LiCI (600 ml) were added to the mixture. The organic layer was washed with aqueous 3% LiCI (1 x 600 ml), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired methyl ester as a yellow solid (23.6 g, 75%). LC/MS = 900. 1 3(M++ 1 ) . m. Preparation of compound 315. Methyl ester 314 (23.6 g, 26 mmol) was dissolved in glacial acetic acid (200 ml), 1.4 N HCI in H20 (75 ml) was added to the solution. The mixture was stirred at 60 °C for 1 hour. Upon completion of the reaction, the mixture was concentrated to remove the solvents, coevaporated with toluene (x 2) to remove residual acetic acid. The residue was then dissolved in EtOAc (500 ml) and sat. NaHC03 aqueous solution (enough to neutralize the mixture) while monitoring C02 evolution. The organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was further dried under high vacuum for 1 h and used as is for the next step. The crude was dissolved in CH2CI2 (360 ml), morpholine (3.4 g, 39 mmol) and sodium triacetoxyborohydride (7.2 g, 34 mmol) were added to the mixture at 0 °C. Then glacial acetic acid (0.47 g, 7.8 mmol) was added dropwise to the mixture. The reaction was complete in 10 minutes at 0 °C. Sat. NaHC03 aqueous solution was added to quench the reaction. After stirring for another 20 minutes, the organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired amine product 315 as a yellow solid (12 g, 50%). LC/MS = 924.63(M++ 1 )
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
GRAZOPREVIR, MK 5172
- Grazoprevir hydrate
- UNII-4O2AB118LA
- MK 5172
MW804.99
quinoxalinyl)pentyl]cyclopropyl]oxy]carbonyl]-3-methyl-L-valyl-(4R)-4-hydroxy-L-prolyl-1-
amino-N-(cyclopropylsulfonyl)-2-ethenyl-, cyclic (1→2)-ether, hydrate (1 :1) (1R,2S)-
ethenylcyclopropyl}-5-(1,1-dimethylethyl)-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide hydrate
MOLECULAR WEIGHT 784.92
9857
ACS Med Chem Lett 2012, 3(4): 332DOI: 10.1021/ml300017p
Org Lett 2013, 15(16): 4174
[1]. Steven Harper , John A. McCauley , Michael T. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med. Chem. Lett., 2012, 3 (4), pp 332-336[2]. Summa V, Ludmerer SW, McCauley JA, MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012 Aug;56(8):4161-7.
IC50 Value: 7.4nM and 7nM for genotype1b and 1a respectively, in replicon system [1]
MK-5172 is a novel P2-P4 quinoxaline macrocyclic HCV NS3/4a protease inhibitor currently in clinical development.
in vitro: In biochemical assays, MK-5172 was effective against a panel of major genotypes and variants engineered with common resistant mutations observed in clinical studies with other NS3/4a protease inhibitors. In the replicon assay, MK-5172 demonstrated subnanomolar to low-nanomolar EC50s against genotypes 1a, 1b, and 2a [2].
in vivo: In rats, MK-5172 showed a plasma clearance of 28 ml/min/kg and plasma half-life of 1.4 hr. When dosed p.o. at 5 mg/kg, the plasma exposure of MK-5172 was good with an AUC of 0.7 uM.hr. The liver exposure of the compound was quite good (23 uM at 4 hr), and MK-5172 remained in liver 24 hr after a single p.o. 5 mg/kg dose. At 24 hr, the liver concentration of MK-5172 was 0.2 uM, which was over 25-fold higher than the IC50 in the replicon assay with 50% NHS. When dosed to dogs, MK-5172 showed low clearance of 5 ml/min/kg and a 3 hr half-life after i.v. 2 mg/kg dosing and had good plasma exposure (AUC=0.4 uM.hr) after a p.o. 1 mg/kg dose [1].
Clinical trial: Evaluation of Hepatic Pharmacokinetics for MK-5172 in Participants With Chronic Hepatitis C . Phase1


1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-
yl]carbonyl}amino)-2-ethenylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide (15 K-salt).
8.3 Hz, 1 H), 7.15 (m, 1 H), 7.04 (m, 1 H), 5.97 (m, 1 H), 5.73 (br s, 1 H), 4.96 (m, 1 H), 4.79 (apparent q, J = 9.3 Hz, 1 H), 4.26 (dd, J = 9.7, 7.7 Hz, 1 H), 4.20 (d, J = 11.3 Hz, 1 H), 4.14 (d, J = 8.8 Hz, 1 H), 3.90 (dd, J = 11.1, 3.2 Hz, 1 H), 3.86 (s, 3 H), 3.62 (m, 1 H), 2.86-2.60 (m, 3 H), 2.38 (m, 1 H), 2.21 (m, 1 H), 1.80-1.48 (m, 6 H), 1.42 (m, 5 H), 1.14 (m, 1 H), 0.95 (m, 10 H), 0.81 (m, 2 H), 0.72-0.50 (m, 3 H), 0.41 (m, 1 H) ppm.http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-
7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide (MK-5172, 15).
OD) δ 7.79 (dd, J = 9.6, 1.8 Hz, 1 H), 7.23 (s, 1 H), 7.22 (m, 1 H), 7.10 (d, J = 9.6 Hz, 1 H), 6.01 (apparent t, J = 3.6 Hz, 1 H), 5.74 (m, 1 H), 5.24 (dd, J = 17.0 Hz, 1.6 Hz, 1 H), 5.11 (dd, J = 10.4 Hz, 1.6 Hz, 1 H), 4.49 (d, J = 11.2 Hz, 1 H), 4.40 (m, 2 H), 4.13 (dd, J = 12.0 Hz, 4.0 Hz, 1 H), 3.92 (s, 3 H), 3.76 (m, 1 H), 2.92 (m, 2 H), 2.85 (m, 1 H), 2.55 (dd, J = 13.6 Hz, 6.4 Hz, 1 H), 2.28 (m, 1 H), 2.18 (apparent q, J =8.8 Hz, 1 H), 1.85 (dd, J = 8.0 Hz, 5.6 Hz, 1 H), 1.73 (m, 2 H), 1.5 (m, 2 H), 1.40 (dd, J = 9.6 Hz, 5.6 Hz, 1 H), 1.3 (m, 2 H), 1.23 (m, 4 H), 1.08 (s, 9 H), 0.99 (m, 2 H), 0.89 (m, 3 H), 0.73 (m, 1 H), 0.49 (m, 1 H) ppm; HRMS (ESI) m/z 767.3411 [(M+H)+; calcd for C38H51N6O9S: 767.3433].http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
| Intermediate # | Structure | Name | Lit. Reference |
| A1 |  |
(1R,2S)-1-Amino-N- (cyclopropylsulfonyl)-2- vinylcyclopropanecarboxamide hydrochloride | Wang et al., U.S. Pat. No. 6,995,174 |

















Step 1: Quinoxaline Hydroxyproline Methyl Ester HCl Salt

A 250-ml RB, equipped with magnetic stirrer and N2 bubbler, was charged with chloroquinoxaline BOC hydroxyproline adduct in MeOH (100 ml), and the mixture was cooled in an ice bath. Acetyl chloride (17.9 g) was then added, and the mixture was stirred at RT for 2 h. The batch was diluted with IP Ac (80 ml). Solids were filtered off and washed with IPAc (20 ml). The washed solids were dried under vacuum for 3 d, to provide 48.9 g (100% yield ). Part of this material was used in the next step.
Example 17: Preparation of Compound A, Method A

Macrocyclic acid hemihydrate, the product of Example 15 (10.16 g, 18.03 mmol) was dissolved in THF (50 mL to 90 mL). The solution was azetropically dried at a final volume of 100 mL. Sulfonamide pTSA salt (7.98 g, 1.983 mmol) followed by DMAc (15 mL) was added at RT. The batch was cooled to 0°C to 10°C, and pyridine (10 mL) was added dropwise. Then, EDC HCl (4.49 g, 23.44 mmol) was added in portions or one portion at 0°C to 10°C. The reaction mixture was aged at 0°C to 10°C for 1 h, and then warmed to 15°C to 20°C for 2 h to 4 h. MeOAc (100 mL) followed by 15 wt% citric acid in 5% NaCl in water (50 mL) was added, while the internal temperature was maintained to < 25°C with external cooling. The separated organic phase was washed with 15 wt% citric acid in 5% NaCl in water (50 mL) followed by 5% NaCl (50 mL). The organic phase was solvent-switched to acetone at a final volume of ~80 mL. Water (10 mL) was added dropwise at 35°C to 40°C. The batch was seeded with Compound A monohydrate form III (~10 mg) and aged for 0.5 h tol h at 35°C to 40°C. Additional water (22 mL) was added dropwise over 2 h to 4 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 60% acetone in water (2x 40 mL). Suction drying at RT gave Compound A monohydrate form III as a white solid.
XH NMR (400 MHz, CDC13) δ 9.95 (s, br, 1 H), 7.81 (d, J = 9.1 Hz, 1 H), 7.18 (dd, J = 9.1, 2.7 Hz, 1 H), 7.16 (s, br, 1 H), 7.13 (d, J = 2.7 Hz, 1 H), 5.96 (t, J = 3.8 Hz, 1 H), 5.72 (m, 1 H), 5.68 (d, J = 10.1 Hz, 1 H), 5.19 (d, J = 17.1 Hz, 1 H), 5.07 (d, J = 10.1 Hz, 1 H), 4.52 (d, J = 11.4 Hz, 1 H), 4.45 (d, J = 9.8 Hz, 1 H), 4.36 (d, J = 10.5, 6.9 Hz, 1 H), 4.05 (dd, J = 11.5, 3.9 Hz, 1 H), 3.93 (s, 3 H), 3.78 (m, 1 H), 2.90 (m, 1 H), 2.82 (tt, J = 8.0, 4.8 Hz, 1 H), 2.74 (dt, J = 13.2, 4.8 Hz, 1 H), 2.59 (dd, J = 14.0, 6.7 Hz, 1 H), 2.40 (ddd, J = 14.0, 10.6, 4.0 Hz, 1 H), 2.10 (dd, J = 17.7, 8.7 Hz, 1 H), 1.98 (2 H, mono hydrate H20), 1.88 (dd, J 8.2, 5.9 Hz, 1 HO, 1.74 (m, 3 H), 1.61 (m, 1 H), 1.50 (m, 3 H), 1.42 (dd, J = 9.6, 5.8 Hz, 1 H), 1.22 (m, 2 H), 1.07 (s, 9 H), 0.95 (m, 4 H), 0.69 (m, 1 H), 0.47 (m, 1 H).
1 C NMR (100 MHz, CDC13) δ 173.5, 172.1, 169.1, 160.4, 157.7, 154.9, 148.4, 141.0, 134.3, 132.7, 129.1, 118.8, 118.7, 106.5, 74.4, 59.6, 59.4, 55.8, 55.5, 54.9, 41.8, 35.4, 35.3, 35.2, 34.3,. 31.2, 30.7, 29.5, 28.6, 28.2, 26.6, 22.6, 18.7, 11.2, 6.31, 6.17.
HPLC conditions: Ascentis Express Column, 10 cm x 4.6 mm, 2.7 μηι; Column temperature of 40°C; Flow rate of 1.8 mL/min; and Wavelength of 215 nm.
Gradiant: mm 0.1% ¾PO4
0 20 80
5 55 45
15 55 45
25 95 5
27 95 5
27.1 20 80
32 20 80
Retention time: mm.
Compound A 14.50
Example 18: Preparation of Compound A, Method B

To a 50-L flask equipped with overhead stirring was added macrocyclic acid (1.06 kg crude, 1.00 eq), amine-pTSA (862 g crude, 1.12 eq) and MeCN (7.42 L) at 19°C. The slurry was cooled in a water bath, pyridine (2.12 L, 13.8 eq) was added, aged 15 min, and then added EDC (586 g, 1.60 eq) in one portion, aged 1.5 h, while it turned into a clear homogeneous solution.
The solution cooled in a water bath, then quenched with 2 N HC1 (1.7 L), and seeded (9.2 g), aged 15 min, and the rest of the aqueous HC1 was added over 2.5 h. A yellow slurry was formed. The slurry was aged overnight at RT, filtered, washed with MeCN/water (1 : 1 v/v, 8 L), to obtain Compound A (Hydrate II).
Compound A was dissolved in acetone (4 L) at RT, filtered and transferred to a
12-L round-bottom flask with overhead stirring, rinsed with extra acetone (1 L), heated to 50°C, water (0.9 L) was added, seeded with Compound A monohydrate form III (-10 mg), and aged 15 min, and then added water (0.8 L) over 2.5 h, extra water 3.3 v over 2.5 h was added, stopped heating, cooled to RT, aged at RT overnight, filtered, washed with water/acetone (1 : 1 v/v, 4 L), and dried in air under vacuum. Compound A Hydrate III, 670 g, was obtained as an off-white solid.
Example 19: Preparation of Compound A, Method C

Macrocyclic acid hemihydrate from Example 15 (10.16 g, 18.03 mmol) was dissolved in THF (50 ml to 90 mL). The solution was azetropically dried at a final volume of 100 mL. Sulfonamide pTSA salt (7.98 g, 19.83 mmol) was added, followed by DMAc (15 mL), at RT. The batch was cooled to 0° to 10°C, and pyridine (10 mL) was added dropwise. Then, EDC HC1 (4.49 g, 23.44 mmol) was added (in portions or one portion) at 0°C to 10°C. The reaction mixture was aged at 0°C to 10°C for 1 h, and then warmed to 15°C to 20°C for 2 h to 4 h. THF (50 mL) was added, followed by 15 wt% citric acid in 15 wt% aq. NaCl (50 mL), while the internal temperature was maintained at < 25°C with external cooling. The separated organic phase was washed with 15 wt% citric acid in 1 % aq. NaCl (40 mL), followed by 15% NaCl (40 mL). The organic phase was solvent-switched to acetone at a final volume of ~75 mL Water (1 1 mL to 12 mL) was added dropwise at 35°C to 40°C. The batch was seeded with Compound A monohydrate form III (~20 mg) and aged for 0.5 h to 1 h at 35°C to 40°C.
Additional water (22 mL) was added dropwise over 2 h to 4 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 60% acetone in water (40 mL x 2). Suction drying at RT or vacuum-oven drying at 45°C gave Compound A monohydrate form III as a white solid.
Example 20: Preparation of Compound A, Method D

Macrocyclic acid hemihydrate from Example 12 (10 g, 98.4wt%, 17.74 mmol) was dissolved in THF (70 mL). The solution was azetropically dried at a final volume of ~60 mL. Sulfonamide pTSA salt (7.53 g, 18.7 mmol) was added at RT. The batch was cooled to 0°C to 5°C, and pyridine (1 1.4 mL) was added dropwise. Then, EDC HC1 (4.26 g, 22.2 mmol) was added in portions at 0°C to 15°C. The reaction mixture was aged at 10°C to 15°C for 2 h to 4 h. 35 wt% Citric acid in 10 wt% aq. NaCl (80 mL) was added, while the internal temperature was maintained at < 25°C with external cooling. The separated organic phase was solvent-switched to acetone at a final volume of ~75 mL. Water (12 mL) was added dropwise at 50°C. The batch was seeded with Compound A monohydrate form III (-300 mg) and aged for 0.5 h to 1 h at 50°C. Additional water (25 mL) was added dropwise over 6 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 65%) acetone in water (40 mL). Suction drying at RT or vacuum-oven drying at 45°C gave Compound A monohydrate form III as a white solid.
Example 24: Ring Closing Metathesis

To a 50 mL 2-neck RB flask with reflux condenser and needle for N2 bubbling was charged the product of Example 20 (1.034 g, 0.869 mmol, 1.0 eq), toluene (20.68 ml, 20X), and the resulting solution was degassed with N2. Hoveyda-Grubbs 2nd generation catalyst (10.90 mg, 0.017 mmol) was charged to the pot, and the system was heated to 80°C with constant sparge of N2, with color change from green to reddish. The reaction was sampled (5 h) and assay by HPLC to be approximately 80% converted. The system was removed from the heat and allowed to stir at RT overnight under N2. The reaction was again assayed and deemed complete by HPLC. Toluene was removed by concentration and the resulting red oil was purified by gradient silica gel chromatography (50 g BlOTAGE SNAP Si gel column; loaded with DCM; eluted with 0 to 10% EtOAc in DCM over 10 column volumes; then 10 to 20% EtOAc in DCM over 3 column volumes; then hold; detect by TLC-UV) to yield a yellow solid, which was further slurried in EtOAc (3 mL) and hexanes (6 mL). The resulting slurry was filtered and washed with 25% EtOAc in hexanes (6 mL) to yield the product (445 mg, 0.754 mmol, 87% yield) as a white solid.


Merck reported interim data from the Phase 2 C-WORTHY study in April 2014 at the International Liver Congress (ILC) in London that evaluated the efficacy and safety of its two-drug regimen based on NS3/4A protease inhibitor MK-5172 and NS5A replication complex inhibitor MK-8742, given with or without ribavirin, in GT1 HCV patients with cirrhosis. The once-daily single pill (without ribavirin) showed a 98% SVR12 (12-week sustained virologic response) in genotype-1, treatment-naive patients. Merck will start the phase III clinical trials (NCT02105688, NCT02105662, NCT02105467 andNCT02105701) for the combination in June 2014.
Development of a Practical, Asymmetric Synthesis of the Hepatitis C Virus Protease Inhibitor MK-5172.Org. Lett. 2013;
15: 4174-4177

MK-5172 is a hepatitis C virus protease inhibitor. Key steps in the synthesis depicted are (1) the regioselective SNAr reaction of dichloroquinoxaline A with prolinol derivative B and (2) construction of the 18-membered macrocycle using a macrolactamization (F → G).
Comment
The medicinal chemistry route to MK-5172 is based on a ring-closing metathesis strategy (S. Harper et al.ACS Med. Chem. Lett. 2012, 3, 332). The best regioselectivity (20:1) and minimization of double substitution in the SNAr reaction of A with B was achieved using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base in polar solvents such as DMSO, NMP, or DMAc.
SYNTHESIS, THESIS PROCEDURES, NMR see………..http://www.allfordrugs.com/2015/07/31/mk-5172-grazoprevir/
/////////////http://www.allfordrugs.com/2015/07/31/mk-5172-grazoprevir/
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
