WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Boehringer Ingelheim is developing BI-836845, a fully human mAb targeting IGF-1 created using HuCAL technology from Morphosys, for the potential iv infusion treatment of cancer, including solid tumors and breast cancer.
In April 2011, a phase I trial was initiated in the UK . In October 2011, another phase I trial was initiated in Taiwan. In February 2014, recruitment was ongoing. At that time, the trial was expected to be completed in March 2015 In June 2014, the drug was listed as being in phase I development for solid tumors in Japan and for breast cancer
In May 2014, an open-label, randomized, parallel-assigned, phase II trial (NCT02123823; 1280.4; 2013-001110-15) to evaluate the safety and efficacy of BI-836845 and everolimus in combination with exemestane in women with breast cancer (expected n = 198) was planned to be initiated in Belgium, France and the Netherlands. At that time, the trial was expected to complete in December 2017
In June 2014, an open-label, single-group assigned, phase I trial (NCT02145741; 1280.15) to evaluate BI-836845 in Japanese patients (expected n = 18) with advanced solid tumors was planned to be initiated in Japan. At that time, the trial was expected to complete in June 2015
In March 2011, a non-randomized, open-label, phase I study (NCT01317420; 1280.2; 2010-021714-29) was planned to begin later that month in patients with solid tumors (expected n = 70) in the UK, to assess the safety, efficacy, pharmacokinetics, pharmacodynamics and pharmacogenomics of BI-836845. The study began in April 2011; at that time, completion was expected in March 2013 .
In June 2012, preclinical data were presented at the 48th ASCO meeting in Chicago, IL. In the study, the combination of BI-836845 plus rapamycin was more effective than single agent therapy at inhibiting Ewing’s sarcoma cell proliferation in vitro and in a nude mouse xenograft model .
In November 2011, preclinical data were presented at the 23rd AACR-NCI-EORTC International Conference in San Francisco, CA. BI-836845 potently inhibited proliferation of the multiple myeloma cell line LP-1 with an EC50 of 0.4 nM.
BI-836845 is a human monoclonal IgG1 lambda antibody against IGF-1 (insulin growth factor-1) and IGF-2 (insulin growth factor-2). Phase II clinical trials are ongoing at Boehringer Ingelheim for the treatment of patients with breast cancer, and phase I clinical trials are ongoing with patients with advanced solid tumors.
Insulin-like growth factor-1 (IGF-1; a 70 amino-acid polypeptide) and insulin-like growth factor-2 (IGF-2; a 67 amino-acid polypeptide) are 7.5-kD soluble factors present in serum that can potently stimulate the growth of many mammalian cells (reviewed by Pollack et al., 2004). Although IGFs can be detectable in a number of tissues the main source of circulating IGFs is the liver which secretes the IGFs and IGF binding proteins (IGFBPs) in response to a complex signaling pathway that is initiated in the pituitary gland and transduced via growth hormone. On secretion into the bloodstream the IGFs form complexes with the IGFBPs which not only protects them from proteolytic degradation in the serum en route to their target tissues but also prevents their association with the IGF receptors. In addition to this endocrine source of IGFs they are also known to be secreted in an autocrine or paracrine manner in target tissues themselves. This is known to occur during normal fetal development where the IGFs play a key role in the growth of tissues, bone and organs. It is also seen in many cancer tissues where there is thought to be paracrine signaling between tumour cells and stromal cells or autocrine IGF production by the tumour cells themselves (reviewed by LeRoith D, 2003).
30 May 2014
MEDIA ALERT
ASCO 2014: Boehringer Ingelheim to present latest oncology research, including overall survival results
• Highly anticipated new overall survival data for Giotrif® (afatinib*) to be presented on June 2nd (3:00 – 6:00 PM, E Hall D2 [Abstract #8004 scheduled for 4:00 – 4:12 PM])
• 7 total abstracts accepted for Giotrif® (afatinib*), nintedanib** and BI 836845**: 1 for oral presentation and 6 posters
BI 836845 (IGF ligand antibody)**
A Phase I dose escalation study of weekly BI 836845, a fully human, affinity-optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid cancers
Chia-Chi Lin, Kwang-Yu Chang, Dennis Chin-Lun Huang, Vicky Marriott, Ludy van Beijsterveldt, Li-Tzong Chen, Ann-Lii Cheng
Sunday, June 1
8:00 – 11:45 AM
S Hall A2
(Abstract #2617
Poster #80)
Phase I dose escalation study of 3-weekly BI 836845, a fully human, affinity optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid tumours
Rihawi K, Ong M, Michalarea V, Bent L, Buschke S4, Bogenrieder T, Anthoney A, de Bono J, Twelves CJ
Sunday, June 1
8:00 – 11:45 AM
S Hall A2
(Abstract #2622
Poster #85)
The activity of the IGFs is thought to be regulated by a complex and relatively poorly understood interaction involving seven different IGFBPs and other serum proteins. Activation of the IGFs involves their release from this ternary complex after proteolytic release of the serum binding protein and IGFBPs, this is thought to occur in close proximity to cell surfaces where the IGFs are then free to bind to their receptors and transduce intracellular signals that ultimately leads to cellular proliferation and the inhibition of apoptosis. IGF-1 and IGF-2 are able to bind to the IGF-1 receptor (IGF-1R) expressed on many normal tissues, which functionally is a 460 kD heterotetramer consisting of a dimerised alpha- and beta-subunit, with similar affinities (Rubin et al., 1995). IGF-2 can also bind to the IGF-2 receptor (also know as the mannose-6-phosphate receptor) which does not have any known signaling function, rather it is thought to act as a sink for IGF-2 and prevent it from binding and signaling through the IGF-1R. In this respect the IGF-2R has been demonstrated to be a tumour suppressor protein. The IGF-1R is structurally similar to the insulin receptor which exists in two forms, IR-A and IR-B, which differ by an alternatively spliced 12 amino acid exon deletion in the extracellular domain of IR-A. IR-B is the predominant IR isoform expressed in most normal adult tissues where it acts to mediate the effects of insulin on metabolism. IR-A on the other hand is known to be highly expressed in developing fetal tissues but not in adult normal tissues. Recent studies have also shown that IR-A, but not IR-B, is highly expressed in some cancers. The exon deletion in IR-A has no impact on insulin binding but does cause a small conformational change that allows IGF-2 to bind with much higher affinity than for IR-B (Frasca et al., 1999; Pandini et al., 2002). Thus, because of it’s expression in cancer tissues and increase propensity for IGF-2 binding, IR-A may be as important as IGF1-R in mediating the mitogenic effects of IGF-2 in cancer.
Binding of the IGFs to IGF-1R triggers a complex intracellular signaling cascade which results in activation of proteins that stimulate growth and inhibit apoptosis (reviewed by Pollack et al., 2004). In terms of growth, upregulated translation is induced by the activation of p70 S6 kinase, which in turn phosphorylates the S6 ribosomal protein (Dufner and Thomas, 1999). Thus, IGF-stimulated cell growth can be measured by the rapid increase in phosphorylated S6 ribosomal protein.
Unlike the EGFR and Her2neu receptors there is no known amplification of the IGF1-R or IR-A receptors in cancers indicating that receptor activation is controlled by the presence of active ligand. There is a very large body of scientific, epidemiological and clinical literature implicating a role for the IGFs in the development, progression and metastasis of many different cancer types (reviewed by Jerome et al., 2003; and Pollack et al., 2004).
For example, in colorectal cancer the expression of IGF-2 mRNA and protein is elevated in clinical colorectal tumour specimens compared with adjacent normal tissue (Freier et al., 1999; Li et al., 2004). There is also a positive correlation of elevated IGF serum levels with proliferating cell index in patients with colorectal neoplasia (Zhao et al., 2005). In addition, elevated circulating levels of IGF-2 correlate with an increased risk of developing colorectal cancers and adenomas (Renehan et al., 2000a) and b); Hassan et al., 2000). Loss of parental imprinting (LOI) of the IGF-2 gene, an epigenetic alteration that results in elevated IGF-2 expression, is a heritable molecular trait that has recently been identified in patients with colorectal and other tumour types. Loss of IGF-2 imprinting has been shown to be associated with a five-fold risk of colorectal neoplasia (Cui et al., 2003; Cruz-Correa et al., 2004) and adenomas (Woodson et al., 2004). Antibodies targeting the alpha-subunit of the IGF-1R which block IGF binding and internalize the receptor have been shown to delay the growth of the xenografted colon cancer-derived cell lines such as COLO 205 (Burtrum et al., 2003).
Elevated levels of IGFs are associated with a poor prognosis in human pulmonary adenocarcinomas (Takanami et al., 1996) and IGFs are expressed and secreted by many SCLC— and NSCLC-derived cell lines (Quinn et al., 1996). Transgenic over-expression of IGF-2 induces spontaneous lung tumours in a murine model (Moorhead et al., 2003). In terms of hepatocellular carcinoma (HCC), human clinical specimens and animal models of HCC express higher levels of IGF mRNA and protein than corresponding normal tissues and this has been correlated with increased tumour growth (Wang et al., 2003; Ng et al., 1998). IGF-2 has also been shown to be a serological marker of HCC with elevated levels in the serum of HCC patients compared with controls (Tsai et al., 2005). An orthotopic xenograft tumour model of HCC was established using Hep 3B cells, and used to demonstrate that inhibition of IGF-2 expression using a methylated oligonucleotide enhances survival (Yao et al., 2003a) and b).
Many childhood solid tumours such as Ewing sarcoma and rhabdomyosarcoma appear to be particularly dependent on the IGF signaling pathway for their growth (Scotlandi et al., 1996). LOI of the IGF-2 gene has been implicated as a primary genetic event in the development for embryonal rhabdomyosarcoma (Fukuzawa et al., 1999). Autocrine IGF signaling is also thought to strongly influence the growth of Ewing sarcoma in cases where the type-1 EWS-FLI1 chimeric transcription factor is expressed through a chromosomal translocation resulting in elevated expression of target genes including the IGF ligands and IGF-1R, and reduced expression of IGFBP-3. Antibodies and small molecule compounds targeting the IGF-1R have been shown to reduce the growth of xenografted pediatric solid tumour derived cell lines (Kolb et al., 2008; Manara et al., 2007).
Using IGF ligand-specific antibodies it has been demonstrated that the growth of human prostate cancer cells in adult human bone implanted into SCID mice can be inhibited (Goya et al., 2004). In addition, it was demonstrated that the same IGF ligand antibodies could block the paracrine supply of IGF and suppress the liver metastasis of human colorectal cancer cells in a murine xenograft system (Miyamoto et al., 2005).
There is also considerable evidence suggesting that the IGF signaling system reduces the sensitivity of cancers to chemotherapeutic agents and radiation. One of the earliest findings in this respect was the demonstration that IGF-1R knock-out mouse embryos are refractory to transformation by viruses, oncogenes and over-expressed growth factor receptors (Sell et al., 1993; Sell et al., 1994) and that over-expression of IGF-1R protects cells from UV irradiation and gamma radiation-induced apoptosis (Kulik et al., 1997). Furthermore, using liver tumour cell lines that secrete large amounts of IGF-2, it was found that neutralization of IGF-2 significantly increased response to chemotherapeutic agents such as cisplatin and etoposide in vitro, especially at lower, cytostatic doses, suggesting that IGF-2 can reduce the susceptibility to chemotherapeutic agents (Lund et al., 2004). Consistent with these findings it has been demonstrated that antibodies targeting the IGF-1R increase the susceptibility of tumour xenografts to growth inhibition by chemotherapeutic drugs and radiation (Goetsch et al., 2005).
A number of antibodies that show cross-reactive binding to human IGF-1 and human IGF-2 have been reported. Antibody sm1. was raised against human IGF-1 and shows 40% cross-reactivity to human IGF-2 and was shown to inhibit the proliferation of a mouse fibroblast cell line BALB/c3T3 which was stimulated with 20 ng/ml human IGF-1 (Russell et al., 1984). In a study designed to functionally epitope map IGF-1 by raising monoclonal antibodies to whole IGF-1 protein and portions of the protein a number of antibodies where identified that cross reacted with IGF-2 (Manes et al., 1997). The percent cross-reactivity with IGF-2 ranged from 0 to 800% and several antibodies were identified which were equally IGF-1 and IGF-2 reactive. KM1486 is a rat monoclonal antibody that cross-reacts with human IGF-1 and IGF-2 and it was demonstrated that KM1486 can inhibit growth of human prostate cancer cells in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice (Goya et al., 2004). In addition, it was demonstrated that KM1486 suppresses the liver metastasis of human colorectal cancers (Miyamoto et al., 2005). KM1486 has also been described in WO 03/093317, JP 2003-310275, WO 2005/018671, WO 2005/028515, and WO 2005/027970.
For the treatment of human disease an antibody with a fully human sequence is highly desirable in order to minimize the risk of generating a human anti-antibody reaction and neutralizing antibodies that will rapidly eliminate the administered antibody from the body and thereby reduce the therapeutic effect. As such, and given the roles of IGF-1 and IGF-2 dependent signaling in the development and progression of cancers it would be desirable to obtain high affinity fully human antibodies that co-neutralise the mitogenic effects of both ligands.
In addition, to maximize the therapeutic potential of such an antibody, it is important to have a suitably long terminal half life (T1/2). Prior to terminal half life determination in human subjects, the most accurate estimation of an antibody’s human terminal half life can be obtained from administration to non-human primates such as cynomolgus monkeys. For example, bevacizumab, a registered humanized monoclonal antibody against vascular endothelial growth factor (VEGF) used for the treatment of several human cancers, has a terminal half-life in cynomolgus monkeys of 8.57±0.38 days (Lin et al., 1999), which translates to a terminal half life in humans of approximately 20 days allowing for a single administration once every two weeks (Lu et al., 2008).
It was a further object of the invention to obtain an antibody that does not affect binding of insulin to its receptor.
The clinical development of therapeutic agents is supported by pharmacodynamic biomarkers of drug activity. Clinical studies with antibodies targeting the IGF-1R have demonstrated that an increase in total serum IGF-1 levels may be a useful pharmacodynamic marker for these agents (Pollack et al., 2007). The reason for the increase in total serum IGF-1 levels is likely due to a feedback mechanism involving pituitary growth hormone (GH) secretion which releases both IGF-1 and IGFBPs from the liver. Indeed, in humans it has been demonstrated that free or bioactive IGF-1, which represents only around 1% of total IGF-1 levels, determines the feedback response (Chen et al., 2005). The inventors thus sought to confirm whether total serum IGF-1 levels are also a useful pharmacodynamic marker for the activity of a therapeutic anti-IGF antibody. In this case it would be desirable for such antibody to be cross-reactive with IGFs from a suitable animal species, e.g. mouse or rat, such that a pharmacodynamic effect can already be tested pre-clinically.
Boehringer Ingelheim
The Boehringer Ingelheim group is one of the world’s 20 leading pharmaceutical companies. Headquartered in Ingelheim, Germany, Boehringer Ingelheim operates globally with 142 affiliates and a total of more than 47,400 employees. The focus of the family-owned company, founded in 1885, is researching, developing, manufacturing and marketing new medications of high therapeutic value for human and veterinary medicine.
Taking social responsibility is an important element of the corporate culture at Boehringer Ingelheim. This includes worldwide involvement in social projects, such as the initiative “Making more Health” and caring for the employees. Respect, equal opportunities and reconciling career and family form the foundation of the mutual cooperation. In everything it does, the company focuses on environmental protection and sustainability.
In 2013, Boehringer Ingelheim achieved net sales of about 14.1 billion euros. R&D expenditure corresponds to 19.5% of its net sales.
Fig.1 Production of MAb
Adam, P.J.; Friedbichler, K.; Hofmann, M.H.; Bogenrieder, T.; Borges, E.; Adolf, G.R. BI 836845, a fully human IGF ligand neutralizing antibody, to improve the efficacy of rapamycin by blocking rapamycin-induced AKT activation
48th Annu Meet Am Soc Clin Oncol (ASCO) (June 1-5, Chicago) 2012, Abst 3092
Lin, C.-C.; Chang, K.-Y.; Huang, D.C.; Marriott, V.; Van Beijsterveldt, L.; Chen, L.-T.; Cheng, A.-L. A phase I dose escalation study of weekly BI 836845, a fully human, affinity-optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid cancers
50th Annu Meet Am Soc Clin Oncol (ASCO) (May 30-June 3, Chicago) 2014, Abst 2617
Adam, P.J.; Ostermann, E.; Lamche, H.R.; Hofmann, M.H.; Kroez, M.; Borges, E.; Adolf, G.R. Pharmacodynamic properties and anti-tumor efficacy of BI 836845, a fully human IGF ligand neutralizing antibody
AACR-NCI-EORTC Int Conf Mol Targets Cancer Ther (November 12-16, San Francisco) 2011, Abst A208
Rihawi, K.; Ong, M.; Michalarea, V.; et al. Phase I dose escalation study of 3-weekly BI 836845, a fully human, affinity optimized, insulin-like growth factor (IGF) ligand neutralizing antibody, in patients with advanced solid tumors
50th Annu Meet Am Soc Clin Oncol (ASCO) (May 30-June 3, Chicago) 2014, Abst 2622
“These patients were treated in our Phase 1 clinical trial of Selinexor in … Additional Phase 1 and Phase 2 studies are ongoing or currently planned and … the discovery and development of novel first-in-class drugs directed against …
600 MG TABLET ORAL, DRUGS FOR NEGLECTED DISEASES INITIATIVE
US FDA approves fexinidazole as the first all-oral treatment for sleeping sickness
POSTED ON JULY 19
The US Food and Drug Administration (FDA) has approved fexinidazole as the first all-oral treatment for both stages of the Trypanosoma brucei gambiense form of sleeping sickness (Human African trypanosomiasis) in patients 6 years of age and older and weighing at least 20 kg.
Fexinidazole was developed as part of an innovative partnership between the non-profit research and development organization Drugs for Neglected Diseases initiative (DNDi), which conducted the pivotal clinical trials for this treatment, in partnership with the National Sleeping Sickness Programs of the Democratic Republic of Congo (DRC) and Central African Republic (CAR), and Sanofi.
Sleeping sickness is a parasitic disease transmitted by the bite of an infected tse-tse fly. It affects mostly populations living in remote rural areas of sub-Saharan Africa, where about 65 million people are at risk of infection. Left untreated, sleeping sickness is almost always fatal. Through Sanofi’s collaboration the number of sleeping sickness cases reported to the WHO has been reduced by ~97% between 2001 and 2020. DNDi, Sanofi and partners are deeply committed to ensuring access to fexinidazole in all sleeping sickness-endemic countries.
Current treatment options for the disease are effective, but burdensome for patients and health workers due to the need for infusion or injection, requiring hospitalization, especially challenging for people living in remote areas.
“Having a simple, all-oral treatment for sleeping sickness is a dream come true for frontline clinicians,” said Dr Bernard Pécoul, DNDi Executive Director. “We are proud of this latest milestone in our long-term partnership with Sanofi, developed in close collaboration with researchers in countries hard-hit by sleeping sickness.”
Fexinidazole is indicated as a 10-day once-a-day treatment for Trypanosoma brucei gambiense sleeping sickness, the most common form of the disease found in West and Central Africa. Fexinidazole is the first all-oral treatment that works both for the first stage of the disease, as well as the second stage of the disease in which the parasites have crossed the blood-brain barrier, causing patients to suffer from neuropsychiatric symptoms.
“This FDA approval is a key milestone in Sanofi’s long-term commitment to fight sleeping sickness, started 20 years ago alongside the WHO through an ambitious partnership to combat Neglected Tropical Diseases” said Luc Kuykens, Senior Vice President, Sanofi Global Health unit. “Following the positive scientific opinion granted by the European Medicines Agency end 2018, the FDA approval is an important step to revitalize efforts to support the sustainable elimination of the disease”.
As a result of FDA approval, a Tropical Disease Priority Review Voucher (PRV) has been awarded to DNDi. The FDA Tropical Disease PRV Program was established in 2007 to incentivize development of new treatments for neglected tropical diseases, including sleeping sickness. Any benefits from the PRV will be shared between Sanofi and DNDi, which will enable continued investments in innovating for and ensuring access to new health tools for sleeping sickness and other neglected diseases. Sanofi commits to continue to provide the drug free-of-charge to the World Health Organization for distribution to affected countries, as part of a long-term collaboration with WHO.
About Sleeping sickness
Sleeping sickness, or human African trypanosomiasis (HAT), is usually fatal without treatment. Transmitted by the bite of an infected tse-tse fly, following a period with nonspecific symptoms, it evolves to cause neuropsychiatric symptoms, including abnormal behaviour, and a debilitating disruption of sleep patterns that have given this neglected disease its name. About 65 million people in sub-Saharan Africa are at moderate to very high risk of infection.
About DNDi
The Drugs for Neglected Diseases initiative (DNDi) is a collaborative, patient needs-driven, not-for-profit research and development (R&D) organization that develops safe, effective, and affordable treatments for sleeping sickness, leishmaniasis, Chagas disease, filarial infections, mycetoma, paediatric HIV, hepatitis C, and covid-19. Since its inception in 2003, DNDi has delivered eight new treatments, including nifurtimox-eflornithine combination therapy (NECT) for late-stage sleeping sickness, and fexinidazole, the first all-oral drug for sleeping sickness.
Fexinidazole was discovered by the German pharmaceutical company Hoechst AG, but its development as a pharmaceutical was halted in the 1980s.[5] Fexinidazole is now being studied through a collaboration between Sanofi and the Drugs for Neglected Diseases Initiative for the treatment of Chagas disease and human African trypanosomiasis (sleeping sickness).[6][7] Fexinidazole is the first drug candidate for the treatment of advanced-stage sleeping sickness in thirty years.[8]
Fexinidazole is currently in phase II/III clinical development at Drugs for Neglected Diseases Initiative for the oral treatment of African trypanosomiasis (sleeping sickness). In May 2009, Sanofi (formerly known as sanofi-aventis) licensed the drug candidate to Drugs for Neglected Diseases Initiative for the development, manufacturing and distribution as a treatment of human African trypanosomiasis. Once approved, the companies plan to make the drug available on a nonprofit basis.
Fexinidazole was originally developed by a German pharmaceutical company called Hoechst, now part of Sanofi; however, its development was abandoned in the 1980s when the company gave up its tropical disease programs. Fexinidazole is one of a class of drugs known as azoles, like fluconazole, that work against fungi and may work against cancer.
Onset of trypanosomiasis is caused by Trypanosoma protozoa and it is said that every year 200,000 to 300,000 of new patients of African sleeping sickness fall sick. At present the number of patients of African sleeping sickness cannot be confirmed due to the low reliability of the investigative data. According to the WHO, at least 150,000 people died of African sleeping sickness in 1996 and it is said that its aftereffect remains in not less than 100,000 people. Beyond that, enormous is the damage to domestic animals caused by a disease called as nagana, and several hundred thousands of cattle which are to be protein sources for people die every year. Further, in the area of about 10,000,000 km2of savanna equal to the United States of America, cattle-breeding is impossible due to Trypanosoma. Thus, African sleeping sickness remarkably damages the health and the economical development of African people, and this is the reason why the WHO adopts the trypanosomiasis as one of the infectious diseases that should be controlled.
African sleeping sickness is a protozoal infectious disease by Trypanosoma transmitted through tsetse flies and the protozoa appear in the blood stream in about 10 days after infection. In the initial period of infection the protozoa multiply in the blood stream and give fever, physical weakness, headache, a pain of muscles and joints and a feeling of itching to proceed. On entering the chromic period, the central nerve is affected to show symptoms such as mental confusion and systemic convulsion, and finally the patients lapse into lethargy and are led to death.
The trypanosomiasis of domestic animals has Trypanosoma brucei brucei, Trypanosoma evansi, Trypanosoma congolense and Trypanosoma vivax as pathogens and is a communicable disease which affects domestic animals such as horses, cattle, pigs and dogs and, in addition, mice, guinea pigs, rabbits and the like. Particularly, the loss of cattle and horses is greatest and almost fetal, and they are led to anemia, edema, weakening and the like and fall dead in one month after infection.
In treating trypanosomiasis, pentamidine, melarsoprol, eflornithine and the like are used and there was a feeling in the 1960s that its eradication might be possible. However, these drugs are old and are gradually losing their efficacy. Particularly, the resistance to melarsoprol of an arsenic agent causes a big problem and the situation is so dire that patients with no efficacy only await death and the development of novel antitrypanosoma agents are strongly desired.
Trypanosoma mainly lives in the blood stream of the human body. This bloodstream energy metabolism depends on the glycolytic pathway localized in the organelle characteristic of the protozoa which is called as glycosome and the so-called oxidative phosphorylation does not function. However, in order to efficiently drive this glycolytic pathway, the produced NADH has to be reoxidized, and the glycerol-3-phosphate oxidation system of mitochondria plays an important role in this reoxidation. The terminal oxidase of this oxidation system functions as a quinol oxidase having a reduced ubiquinone as an electron donor and has properties greatly different from those of cytochrome oxidase of an aerobic respiration system which the host has. Particularly, a remarkable point is that the terminal oxidase of the oxidation system is non-sensitive to the cyanide which quickly inhibits the cytochrome oxidase of the host. Then, many researchers centered around Western countries have tried to develop drugs targeting this cyanide resistant oxidase but effective drugs having a selective toxicity have not been obtained.
Under these circumstances the present inventors et al. found that isoprenoid based physiologically active substances of ascochlorin, ascofuranone and derivatives thereof, particularly ascofuranone specifically inhibits the glycerol-3-phosphate oxidation system of trypanosome at a very low concentration of the order of nM and filed a patent application (Japanese Patent Publication A No. : H09-165332). They also clarified that acofuranone exhibits a very strong multiplication inhibition effect in the copresence of glycerin (Molecular and Biochemical Parasitology, 81: 127-136, 1996).
In consideration of practical use of ascofuranone, it was found essential to discover agents which replace glycerin and exhibit an effect of the combined use in a small amount, and by using an alkaloid compound having an indole skeleton existing in a plant of the family Simaroubaceae together with ascofuranone, the prolongation of life and recovery effect in African seeping sickness was found and a patent application was filed (Japanese Patent Application No.: 2003-24643, Japanese Patent Publication A No.: 2004-23601).
Method for the preparation of fexinidazole, useful for the treatment of parasitic diseases, visceral leishmaniasis, chagas disease and human African trypanosomiasis. Family members of the product patent, WO2005037759, are expected to expire from October 2024. This to be the first application from Drugs for Neglected Diseases Initiative (DNDi) on this API. DNDi in collaboration with Sanofi, the Swiss Tropical & Public Health Institute and the University of Dundee, is developing fexinidazole, an antiparasitic agent, for treating human African trypanosomiasis (HAT) and visceral Leishmaniasis (VL). By June 2013, phase I clinical studies had been completed and at that time, DNDi was planning to initiate a phase II proof-of-concept study in VL patients in early 2013.
Chemotherapeutically active nitro compounds. 4,5-Nitroimidazoles. Part III
Synthesis
By condensation of 4 – (methylmercapto) phenol (II) with 1-mehtyl-2-chloromethyl-5-nitroimidazole (I) by means of K2CO3 in DMF (1,2) Description:. Crystals, mp 116 C. References: 1) Raether, W., Winkelman, E.; Chemotherapeutically active nitro compounds 4,5-Nitroimidazoles Part III Arzneim-Forsch 1978, 28 (5):. 739 2) Winkelmann,… E., Raether, W. (Hoechst AG); DE 2531303.
Winkelman, E.; Raether, W.;… Chemotherapeutically active nitro compounds 4,5-Nitroimidazoles Part III Arzneim-Forsch 1978, 28, 5, 739
Process for preparing fexinidazole – comprising the reaction of 1-methyl-2-hydroxymethyl-5-nitro-imidazole with methanesulfonyl chloride, followed by reaction with 4-methylmercapto-phenol, and further manipulative steps.
1-Methyl-2-hydroxymethyl-5-nitro-imidazole is (I) and 1-methyl-2-(4-methylmercapto-phenyloxymethyl)-5-nitro-imidazole (fexinidazole) is (II) (claim 1, page 12).
The synthesis of (II) via intermediate (I) is described (example 1, pages 6-8).
A process for preparing fexinidazole comprising the reaction of 1-methyl-2-hydroxymethyl-5-nitro-imidazole with methanesulfonyl chloride in the presence of a suspension of powdered alkaline carbonate (eg potassium carbonate) in an anhydrous organic solvent (eg acetone), followed by reaction with 4-methylmercapto-phenol, removal of hydrochloride salt, and isolation and purification is claimed. Also claimed is their use for treating parasitic diseases, visceral leishmaniasis, chagas disease, and human African trypanosomiasis. Fexinidazole is known to be an antiparasitic agent.
Topical chemotherapy for experimental murine African CNS-trypanosomiasis: the successful use of the arsenical, melarsoprol, combined with the 5-nitroimidazoles, fexinidazole or MK-436.
Tropical medicine & international health : TM & IH
Raether, W; Seidenath, H (1983). “The activity of fexinidazole (HOE 239) against experimental infections with Trypanosoma cruzi, trichomonads and Entamoeba histolytica”. Annals of Tropical Medicine and Parasitology77 (1): 13–26. PMID6411009.
Jennings, FW; Urquhart, GM (1983). “The use of the 2 substituted 5-nitroimidazole, Fexinidazole (Hoe 239) in the treatment of chronic T. brucei infections in mice”. Zeitschrift für Parasitenkunde69 (5): 577–581. doi:10.1007/bf00926669. PMID6636983.
(RTTNews.com) – BioCryst Pharmaceuticals Inc. ( BCRX ) will be reporting results from OPuS-1, a phase IIa trial of orally-administered BCX4161 in patients with hereditary angioedema, on Tuesday, May 27, 2014 at 8:30 a.m. Eastern Time.
BCX-4161 is a novel, selective inhibitor of plasma kallikrein in development for prevention of attacks in patients with hereditary angioedema (HAE). By inhibiting plasma kallikrein, BCX-4161 suppresses bradykinin production. Bradykinin is the mediator of acute swelling attacks in HAE patients.
……………………………….
old article
Jul 29, 2013
BCRX – BioCryst – Entering The HAE Market
BioCryst announced on Monday July 22 the successful completion of a Phase I study on the safety and PK of BCX4161, a candidate for the treatment of Hereditary angioedema (HAE). HAE is a genetic disorder resulting from the loss or dysfunction of complement C1 Inhibitor (C1INH).
Among the functions performed by C1INH is regulation of the hormone bradykinin, which when activated, leads to the dilation of blood vessels. Left unchecked, excess bradykinin can cause painful attacks of swelling, or angioedemas, in any part of the body, including the face, abdomen, hands, and larynx. Death can occur from asphyxiation, particularly in children.
The mechanics involved in HAE are fairly well understood today. There are several approved drugs available today that work at three major points in the pathway. Ultimately, each prevents bradykinin from activating its receptor on endothelial cells.
C1 Inhibitors, of which four have been approved, prevent Factor XIIa activation of Plasma Kallikrein and inhibit Kallikrein itself. The single specific Kallikrein inhibitor is Kalbitor from Dyax. C1INHs and kallikrein inhibitors prevent the formation of bradykinin (labeled “BK” in this diagram). Then there is Firazyr from Shire, a B2 bradykinin receptor antagonist; while not preventing overproduction of the hormone, activation of downstream activity is suppressed.
Interestingly, of all the available therapies, only C1INH Cinryze from Viropharma is approved for prophylactic use- all others are designated strictly for treatment of acute attacks. A key reason for this is Cinryze’s long half-life, allowing sustained activity over longer intervals. As each of these drugs are given by injection, frequent treatment is not practical. Consider, for instance, Kalbitor has a half life of just two hours.
This is where BioCryst comes in. The company is pursuing the less crowded prophylaxis indication. It has the only orally available (although just barely) plasma kallikrein inhibitor. And while PK is not great, requiring three-times daily dosing to ensure adequate drug levels, pills make this a feasible option. As you can see, 800 mg appears optimal, however, 400 mg was selected as the Phase IIa dose due to 3 cases of moderate AEs seen at 800. This study was in healthy volunteers and the drug was otherwise well tolerated [ref].
(From Company Presentation)
BCX4161 is an interesting compound. Based on patent literature, we believe the molecule has a similar structure to the one illustrated below:
BCX4161 is not a specific inhibitor of kallikrein, and in fact has near equal potency against Factor XIIa. This dual-activity is also seen with C1INH, setting the compound apart from Kalbitor and Firazyr.
The different profile may improve efficacy, but that is unknown at this point. Along with Factor XIIa, BCX4161 inhibits additional factors involved in coagulation. Bleeding issues has been something the company has been testing and will be certain to monitor. As a drug designed for chronic use, safety will be a major concern.
A 25 patient Phase IIa study set for Q4 will be placebo-controlled double-blind crossover of the following design:
(From Company Presentation)
Individuals with a high frequency of attacks(~1/week) will be enrolled, the primary endpoint is attack frequency. Viropharma conducted a pivotal trial of similar design (but two twelve week dosing periods), reporting ~50% reduction in attacks vs. placebo. We imagine BioCryst would need to achieve results in this range for the drug to be competitive.
A major impedance toward these efficacy goals will likely be individual adherence to dosing every eight hours schedule. Missed doses will mean severe drops in drug levels, potentially putting the patient at risk for an attack. The company noted patients on Cinryze occasionally miss doses with no apparent adverse effect. We will see if this holds true for their own compound.
The Phase IIa is being run in Germany, ostensibly because of the country’s well organized HAE medical treatment system. The study is expected to initiate in 4Q 2013. BioCryst aims to market the drug in the U.S. on their own, likely partnering in the EU.
Handicapping this Phase II is rather difficult with the lack of any prior efficacy results. BioCryst has selected a well-validated target in a fairly well understood disease. The data suggests BCX4161 is an active drug. What we will soon find out is whether the compound is active enough and has a sufficiently clean profile. As attractive as oral dosing is- it has an achilles heel. Regardless of the medication, patients continue to have attacks, only of less frequency and severity. If a patient should suffer major laryngeal swelling, pills may not be an option as a rescue medicine. Cinryze on the hand can serve as both prophylaxis and acute treatment.
Commercially, we believe the compound will have a difficult time competing with Cinryze. True, Cinryze has its own issues, namely a requirement for infusions every 3 to 4 days, but it is difficult to see how a 3-times/day treatment is much of an improvement. In any case, by the time BCX4161 reaches the market, Viropharma should have a much simpler subcutaneous version of its C1INH available, allowing it to maintain a strong monopoly in prophylaxis HAE treatments. Additional competition may come in the form of a follow-up kallikrein inhibitor in development at Dyax; the long acting antibody is designed specifically for the prophylaxis market and is expected to enter the clinic 2H 2013.
GSK-1292263 is a novel GPR119 receptor agonist that is currently under development for the treatment of type 2 diabetes. Treatment of male Sprague-Dawley rats with a single dose of GSK-1292263 (3-30 mg/kg) in the absence of nutrients correlated with increased levels of circulating gastrointestinal peptides; glucagon-like peptide 1 (GLP-1), gastric inhibitory polypeptide (GIP), peptide YY (PYY) and glucagon.
GSK-1292263 had been evaluated in phase II clinical studies at GlaxoSmithKline for the oral treatment of type 2 diabetes and as monotherapy or in combination with sitagliptin for the treatment of dyslipidemia; however no recent development has been reported for this research.
Following administration of glucose in the oral glucose tolerance test (OGTT), greater increases in total GLP-1, GIP and PYY were seen in GSK-1292263-treated rats than in control animals. Despite significant decreases in the glucose AUC, no statistically significant differences in insulin responses and insulin AUC were observed between rats administered GSK-1292263 and those receiving vehicle control.
In the intravenous glucose tolerance test, significant increases in the peak insulin response and insulin AUC(0-15 min) of 30-60% were reported in the GSK-1292263 treatment group, compared with values in the vehicle control cohort. This insulin upregulation correlated with a significant increase in the glucose disposal rate (Brown, K.K. et al. Diabetes [70th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 25-29, Orlando) 2010] 2010, 59(Suppl. 1): Abst 407).
The safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple oral doses of GSK-1292263 were evaluated in a recently completed randomized, placebo-controlled clinical trial in healthy volunteers (ClinicalTrials.gov Identifier NCT00783549).
A total of 69 subjects received single escalating doses of GSK-1292263 (10-400 mg) prior to administration of a 250-mg dose given once daily for 2 and 5 days, which was also evaluated in combination with sitagliptin (100 mg). Treatment with GSK-1292263 at all doses was described as well tolerated, with the most common drug-related effects being mild headache, dizziness, hyperhidrosis, flushing and post-OGTT hypoglycemia.
HRMS calcd for C23H29N4O4S (M + H)+ 457.1904, found, 457.1900.
Anal. Calcd for C23H28N4O4S: C, 60.51; H, 6.18; N, 12.27. Found: C, 60.64; H, 6.16; N, 12.24.
Hypoglycemia was not reported with the 5-day dosing schedule. Pharmacokinetic profiling revealed dose-proportional AUC and Cmax at single lower doses, but not at single higher ones. Following repeated once-daily dosing (5 days), drug accumulation was observed consistent with a mean half-life of 12-18 hours. A dose-dependent increase in glucose AUC(0-3 h) during OGTT was seen in GSK-1292263-treated subjects. The treatment was also associated with an increase in PYY during the prandial periods.
Coadministration with sitagliptin led to increases in the plasma concentrations of active GLP-1 but reduced the levels of total GLP-1, GIP and PYY. Sitagliptin affected the exposure to GSK-1292263 (50% increase) but GSK-1292263 did not affect sitagliptin exposure. The data support further evaluation of GSK-1292263 for the treatment of type 2 diabetes (Source: Nunez, D.J. et al. Diabetes [70th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 25-29, Orlando) 2010] 2010, 59(Suppl. 1): Abst 80-OR).
Example 169: 5-[({1 -[3-(1 -Methylethyl)-1,2,4-oxadiazol-5-yl]-4- piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine hydrochloride
Step 1 : A mixture of 6-bromo-3-pyridinol (7 g, 40 mmol), [4-(methylsulfonyl)phenyl]boronic acid (8 g, 40 mmol), 2M Na2CO3 (30 ml_), PdCI2(PPh3)2 (1 g) and DME (60 ml.) under N2 was heated at 80 0C overnight. The reaction was allowed to cool to room temperature and was diluted with EtOAc and water. The resulting precipitate was filtered off and the aqueous layer was extracted with EtOAc. The combined organic extracts were dried over MgSO4, filtered and concentrated. The aqueous phase was also concentrated. Each of the residues was recrystallized from MeOH. The solid material from the organic phase recrystallization and the mother liquors from both aqueous and organic recrystallizations were combined, concentrated and purified by chromatography on a silica gel column using 0 to 10% MeOH/CH2CI2 to give 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (2.9 g, 29%) as a tan solid. Step 2: Diisopropyl azodicarboxylate (0.175 ml_, 0.89 mmol) was added dropwise to a solution of 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (150 mg, 0.59 mmol), {1-[3-(1- methylethyl)-1 ,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Example 20, Steps 1-3, 200 mg, 0.89 mmol), PPh3 (233 mg, 0.89 mmol), and THF (10 ml.) at ambient temperature. The mixture was stirred at ambient temperature for 4 h. The mixture was concentrated, and the resulting crude was purified by reverse-phase preparative HPLC using a CH3CN:H2O gradient (10:90 to 100:0) with 0.05% TFA as a modifier, then taken up in CH2CI2 and free-based with saturated NaHCO3 (aq) to give 5-[({1-[3-(1-methylethyl)-1 ,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4- (methylsulfonyl)phenyl]pyridine (220 mg) as a white solid. Step 3: A mixture of the resulting white solid (50 mg, 0.1 1 mmol) in THF (3 ml.) was stirred at ambient temperature as 4Λ/ HCI in dioxane (28 μl_) was added dropwise. The resulting white precipitate was filtered, air-dried, then triturated with diethyl ether to give 35 mg (65%) of the title compound as a white solid. 1H NMR (400 MHz, CDCI3): δ 8.46 (d, 1 H, J = 0.7 Hz), 8.18 (bs, 2H), 8.05 (bs, 2H), 7.83 (bs, 1 H), 7.61- 7.45 (m, 1 H), 4.24 (d, 2H, J = 10.4 Hz), 4.00 (d, 2H, J = 0.6 Hz), 3.21-3.03 (m, 5H), 2.89 (m, 1 H), 2.15 (d, 1 H, J = 1.1 Hz), 1.96 (bs, 2H), 1.50 (bs, 2H), 1.28 (d, 6H, J = 6.9 Hz); LRMS (ESI), m/z 457 (M+H).
Step 1: A mixture of 2-methylpropanenitrile (100 g, 1.45 mol), hydroxylamine hydrochloride (111 g, 1.59 mol) and NaOH (64 g, 1.59 mol) in EtOH (2 L) and water (500 mL) was stirred at reflux overnight. The mixture was evaporated to dryness and extracted with dichloromethane. The organic extract was dried over Na2SO4 and concentrated to afford the desired N-hydroxy-2-methylpropanimidamide (50 g, 34%).
Step 2: A solution of 4-piperidinemethanol (140 g, 1.22 mol) in CH2Cl2 (1 L) was treated with a slurry of NaHCO3(205 g, 2.44 mol) in water (1.4 L) at 0° C. The mixture was stirred at 0° C. for 15 min, and then charged with a solution of cyanogen bromide in CH2Cl2, (1.34 mol) at 0° C. The reaction mixture was stirred and allowed to warm to ambient temperature, and stirred overnight. The aqueous layer was separated and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4, filtered, and the filtrate was concentrated. The crude product was combined with other batches made similarly and purified by chromatography on a silica gel column to give 300 g of 4-(hydroxymethyl)-1-piperidinecarbonitrile. Step 3: A solution of 1N ZnCl2 in Et2O (182 mL, 182 mmol) was added to a solution of 4-(hydroxymethyl)-1-piperidinecarbonitrile (21.3 g, 152 mmol) and N-hydroxy-2-methylpropanimidamide (18.6 g, 182 mmol) in EtOAc (50 mL) at ambient temperature. The reaction mixture was left at ambient temperature for 30 min, decanted, and was treated with concentrated HCl (45 mL) and ethanol 20 mL). The mixture was heated at reflux for 2 h. The mixture was evaporated to dryness, and the resulting residue was charged with water and the pH was adjusted to basic with K2CO3. The mixture was extracted with EtOAc and the material obtained was combined with 9 other batches prepared similarly and purified by silica gel chromatography to give 150 g of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol.
Step 4: A solution of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Step 3, 174 g, 0.77 mol) and triethylamine (140 mL, 1.0 mol) in dichloromethane (1 L) at 5° C. was treated with a solution of methanesulfonyl chloride (69 mL, 0.89 mol) in dichloromethane (150 mL) over a 1 h period. The mixture was stirred at 5° C. for 30 min, and then was quenched by the addition of water (400 mL). The mixture was stirred for 30 min, and then the organic extract was washed with water (2×400 mL), dried (MgSO4) and concentrated. The residue was treated with heptane (1 L), stirred for 3 h, and the resulting solid was collected by filtration (heptane wash) and air-dried to afford {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl methanesulfonate (219.7 g, 94%) as an off-white solid. 1NMR (400 MHz, CDCl3): δ 4.21-4.15 (m, 2H), 4.08 (d, 2H, J=6.6 Hz), 3.04 (m, 2H), 3.01 (s, 3H), 2.86 (septet, 1H, J=6.9 Hz), 2.05-1.93 (m, 1H), 1.88-1.81 (m, 2H), 1.43-1.31 (m, 2H), 1.26 (d, 6H, J=6.8 Hz); LRMS (ESI), m/z 304 (M+H).
Step 5: A mixture of 6-bromo-3-pyridinol (36 g, 207 mmol), [4-(methylsulfonyl)phenyl]boronic acid (50 g, 250 mmol), 2M Na2CO3 (315 mL) and DME (500 mL) was degassed with N2 for 30 min, and then Pd(PPh3)4 (12 g, 10 mmol) was added and the mixture was heated at 80° C. for 18 h. The reaction was allowed to cool to room temperature and was diluted with dichloromethane (500 mL) and water (500 mL) and stirred for 30 min. The reaction was filtered and the solids were rinsed with dichloromethane and the aqueous layer was extracted with dichloromethane. The combined organic extracts were extracted with 1N NaOH (2×600 mL), and then cooled to 5° C. and the pH was adjusted to ˜8 with 6N HCl. The resulting precipitate was collected by filtration (water wash) and air-dried to afford a yellow solid. This procedure was repeated and the solids were combined to provide (71.2 g, 68%) of 6-[4-(methylsulfonyl)phenyl]-3-pyridinol. 1H NMR (400 MHz, DMSO-d6): δ 10.27 (s, 1H), 8.25 (d, 1H, J=2.7 Hz), 8.21 (d, 2H, J=8.5 Hz), 8.00-7.90 (m, 3H), 7.27 (dd, 1H, Ja=8.7 Hz, Jb=2.8 Hz), 3.21 (s, 3H); LRMS (ESI), m/z 250 (M+H).
Step 6: A mixture of {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl methanesulfonate (82.3 g, 271 mmol), 6-[4-(methylsulfonyl)phenyl]-3-pyridinol (71.0 g, 285 mmol), powdered potassium carbonate (118 g, 855 mmol) and N,N-dimethylformamide (750 mL) was mechanically stirred and heated at 80° C. under nitrogen for 20 h. The reaction was cooled to ambient temperature, poured onto ice water (3 L) and allowed to stand for 1 h. The resulting solid was filtered, rinsed with water (2×500 mL) and air-dried. The solid was taken up in dichloromethane (300 mL) and methanol (500 mL). The dichloromethane was slowly removed via rotovap at 55° C. The methanol solution was allowed to stand at ambient temperature for 16 h. The resulting crystalline solid was filtered, rinsed with cold methanol and dried under vacuum at 60° C. for 18 h to afford the desired product (105.7 g, 84%) as a light tan solid. 1H NMR (400 MHz, CDCl3): δ 8.41 (d, 1H, J=2.8 Hz), 8.13 (d, 2H, J=8.6 Hz), 8.01 (d, 2H, J=8.6 Hz), 7.74 (d, 1H, J=8.7 Hz), 7.29 (dd, 1H, Ja=8.7 Hz, Jb=3.0 Hz), 4.24 (d, 2H, J=13.1 Hz), 3.95 (d, 2H, J=6.2 Hz), 3.17-3.04 (m, 5H), 2.94-2.84 (m, 1H), 2.11 (bs, 1H), 1.97 (d, 2H, J=12.6 Hz), 1.54-1.42 (m, 2H), 1.29 (d, 6H, J=7.0 Hz); LRMS (ESI), m/z 457 (M+H).
Alternative preparation: Step 1: 2-Bromo-5-[({1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]pyridine (220 mg, 29%) was prepared as a white solid from {1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methanol (prepared as in Example 20, Steps 1-3, 348 mg, 2.0 mmol), 6-bromo-3-pyridinol (348 mg, 2.0 mmol) and Ph3P (629 mg, 2.4 mmol) in THF (5 mL) followed by diisopropyl azodicarboxylate (0.51 mL, 2.6 mmol) in a manner similar to Example 1, Step 2. 1H NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 7.37 (d, 1H, J=8.8 Hz), 7.08 (d, 1H, J=8.8 Hz), 4.26-4.16 (m, 2H), 3.85 (d, 2H, J=6.2 Hz), 3.14-3.04 (m, 2H), 2.95-2.76 (m, 1H), 2.11-1.96 (m, 1H), 1.98-1.88 (m, 2H), 1.52-1.36 (m, 2H), 1.28 (d, 6H, J=6.9 Hz); LRMS (ESI), m/z 381/383 (M+H).
Step 2: 5-[({1-[3-(1-Methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]-2-[4-(methylsulfonyl)phenyl]pyridine (51 mg, 21%) was prepared from 2-bromo-5-[({1-[3-(1-methylethyl)-1,2,4-oxadiazol-5-yl]-4-piperidinyl}methyl)oxy]pyridine (220 mg, 0.52 mmol), [4-(methylsulfonyl)phenyl]boronic acid (105 mg, 0.52 mmol), 2M Na2CO3 (5 mL), Pd(PPh3)4 (50 mg, 0.04 mmol) and DME (5 mL) in a manner similar to Example 21, Step 3.
Paper
Development of Large-Scale Routes to Potent GPR119 Receptor Agonists
†API Chemistry Department, ‡Analytical Science & Development Department, #Medicinal Chemistry Department, and§Particle Sciences and Engineering Department, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
Practical and scalable syntheses were developed that were used to prepare multikilogram batches of GSK1292263A (1) and GSK2041706A (15), two potent G protein-coupled receptor 119 (GPR119) agonists. Both syntheses employed relatively cheap and readily available starting materials, and both took advantage of an SNAr synthetic strategy.
Enzyme acyl-CoA:diacylglycerol acyltransferase-1 (DGAT-1) catalyzes the rate-limiting step in triglyceride synthesis. It has recently emerged as an attractive target for therapeutic intervention in the treatment of Type II diabetes and obesity.
It is estimated that somewhere between 34 and 61 million people in the US are obese and, in much of the developing world, incidence is increasing by about 1% per year. Obesity increases the likelihood of death from all causes by 20%, and more specifically, death from coronary artery disease and stroke are increased by 25% and 10%, respectively. Key priorities of anti-obesity treatments are to reduce food intake and/or hyperlipidemia. Since the latter has been suggested to provoke insulin resistance, molecules developed to prevent the accumulation of triglyceride would not only reduce obesity but they would also have the additional effect of reducing insulin resistance, a primary factor contributing to the development of diabetes. The therapeutic activity of leptin agonists has come under scrutiny through their potential to reduce food intake and, also, to reverse insulin resistance; however, their potential may be compromised by leptin-resistance, a characteristic of obesity. Acyl coenzyme A:diacylglycerol acyltransferase 1 (DGAT-1) is one of two known DGAT enzymes that catalyze the final step in mammalian triglyceride synthesis and an enzyme that is tightly implicated in both the development of obesity and insulin resistance. DGAT-1 deficient mice are resistant to diet-induced obesity through a mechanism involving increased energy expenditure. US researchers have now shown that these mice have decreased levels of tissue triglycerides, as well as increased sensitivity to insulin and to leptin. Importantly, DGAT-1 deficiency protects against insulin resistance and obesity in agouti yellow mice, a model of severe leptin resistance. Thus, DGAT-1 may represent a useful target for the treatment of insulin and leptin resistance and hence human obesity and diabetes. Chen, H. C., et al., J Clin Invest, 109(8), 1049-55 (2002).
Although studies show that DGAT-1 inhibition is useful for treating obesity and diabetes, there remains a need for DGAT-1 inhibitors that have efficacy for the treatment of metabolic disorders (e.g., obesity, Type 2 diabetes, and insulin resistance syndrome (also referred to as “metabolic syndrome”)).
………………………………..
US 20100197591
Scheme II outlines the general procedures one could use to provide compounds of the general Formula (II).
Scheme IV outlines a general procedure for the preparation of compounds of the general Formula VI.
1-[4-(4-amino-2-methoxy-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl)phenyl]cyclobutanecarbonitrilePotassium nitrate (7.88 g, 77.0 mmol) was suspended in sulfuric acid (45 mL) at 0° C. and stirred for 30 minutes until a clear and colorless solution was obtained (NOTE—a blast shield is highly recommended). An addition funnel was charged with 1-phenylcyclobutanecarbonitrile (11.40 g, 72.5 mmol), and this neat starting material was added drop wise at such a rate that the internal reaction temperature did not exceed 10° C. Upon completion of the addition (which required 90 min), the mixture was poured onto 300 g of ice and stirred vigorously for 30 minutes. The resulting suspension was filtered, and the solid was washed with water and dried under vacuum to afford give 1-(4-nitrophenyl)cyclobutanecarbonitrile (13.53 g, 92%) as a light tan powder.
A steel hydrogenation vessel was loaded with 1-(4-nitrophenyl)cyclobutanecarbonitrile (103.6 g, 0.51 mol), 10% palladium on activated carbon (10.3 g; contains ˜50% of water), and 2-methyltetrahydrofuran (1.3 L). The mixture was stirred under 30 psi of hydrogen gas at 45° C. for 4 h. The mixture was filtered through a pad of celite and filtrate concentrated. Heptane (1 L) was added to the obtained oil and the heterogeneous mixture was stirred while slowly cooled to room temperature, causing the product aniline to solidify. The solid was filtered off and dried in vacuum to give 1-(4-aminophenyl)cyclobutanecarbonitrile (86.6 g, 98%).
A mixture of 1-(4-aminophenyl)cyclobutanecarbonitrile (42.2 g, 245 mmol), triethylamine (27.1 mL, 394 mmol), and ethyl acrylate (28.0 mL, 258 mmol) were combined in ethanol (27 mL) and heated to reflux for 24 hours. The mixture was concentrated to dryness and toluene (600 mL) added and concentrated to dryness to give ethyl N-[4-(1-cyanocyclobutyl)phenyl]beta-alaninate as brown oil, which was used without further purification.
Ethyl N-[4-(1-cyanocyclobutyl)phenyl]-beta-alaninate was combined with cyanoacetic acid (22.9 g, 270 mmol) and 4-dimethylaminopyridine (2.30 g, 18.8 mmol) in N,N-dimethylformamide (400 mL) and cooled to 0° C. Diisopropylcarbodiimide (41.7 mL, 270 mmol) was then added drop wise over 30 minutes. Once addition was complete, the reaction was slowly warmed up to room temperature and stirred for 16 hours. Reaction was then poured into saturated aqueous sodium bicarbonate (600 mL) and stirred for 30 mintues. Ethyl acetate (1 L) was added and the mixture was filtered to remove the insoluble diisopropylurea. The phases of the filtrate were separated, and the organic phase was washed with brine and dried over sodium sulfate and concentrated to give ethyl N-(cyanoacetyl)-N-[4-(1-cyanocyclobutyl)phenyl]-beta-alaninate as yellow oil that was used with out further purification in the following step.
ethyl N-(cyanoacetyl)-N-[4-(1-cyanocyclobutyl)phenyl]-beta-alaninate and 1,8-diazabicyclo[5.4.0]undec-7-ene (350 mmol) were combined in methanol (400 mL) and heated to 70° C. for 30 minutes. The mixture was concentrated to dryness then partitioned between water (400 mL) and 2:1 ethyl acetate:heptane (400 mL). The aqueous phase was separated and acidified to pH 2 by the addition of 1M hydrochloric acid (400 mL). The precipitate was filtered off and washed with water (300 mL) and 2:1 ethyl acetate:heptane (300 mL) give 1-(4-(1-cyanocyclobutyl)phenyl)-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (31.7 g, 44% over 3 steps) as an off-white solid.
1-(4-(1-Cyanocyclobutyl)phenyl)-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (50.0 g, 170 mmol) and N,N-dimethylformamide (0.66 mL, 8.5 mmol) in dichloromethane (350 mL) was cooled to 0° C. Oxalyl chloride (18.0 mL, 203 mmol) was added over 15 minutes. The mixture was warmed to room temperature over 2 hours. Methanol (300 mL) was then added as a steady stream, and the mixture was heated at 45° C. for 16 hours. The mixture was cooled to room temperature and concentrated to get rid of most of the dichloromethane. Methanol (200 mL) was added and the thick slurry was stirred for 2 hours. The solid was filtered and dried under vacuum to give 1-(4-(1-cyanocyclobutyl)phenyl)-4-methoxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (48.3 g, 92%) as an off-white powder.
1-(4-(1-Cyanocyclobutyl)phenyl)-4-methoxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (12.04 g, 37.9 mmol) and cyanamide (1.64 g, 41.0 mmol) were suspended in methanol (200 mL) at room temperature. A solution of 25% sodium methoxide in methanol (45.0 mmol) was then added drop wise over 10 minutes to obtain a clear homogeneous solution of the intermediate cyanamide adduct. In one portion, sulfuric acid (5.06 mL, 94.9 mmol) was added, and the mixture was heated to 50° C. for 16 hours. The mixture was then cooled to room temperature and basified to pH 10-11 by the addition of 1N sodium hydroxide, and the thick suspension was stirred for 20 minutes. The solid was filtered, washed with cold methanol and water, and dried under vacuum to obtain the crude product as a mixture contaminated with the vinylogous amide (4-amino-1-[4-(1-cyanocyclobutyl)phenyl]-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile). This solid mixture was heated to reflux in methanol (150 mL) for 3 hours then cooled to room temperature and filtered. The solid collected was then dissolved in a minimal amount of acetic acid (30 mL) at 60° C. to obtain a clear yellow solution. Water was then added drop wise at 60° C. until the cloudiness persisted, and the mixture was allowed to return to room temperature. Another 50 mL of water was added and the fine suspension was filtered, washed with water, and dried under vacuum to afford the title compound (4A) (6.80 g, 51%) as a light yellow solid.
Org. Process Res. Dev., 2013, 17 (12), pp 1510–1516
DOI: 10.1021/op400215h
A practical large-scale synthesis was developed for 1, a DGAT-1 inhibitor, involving an aza-Michael reaction, amidation, Dieckman cyclization, and conjugate addition of cyanamide followed by cyclization, to form the fused 4-amino-7,8-dihydropyrido[4,3-d]pyrimidin-5-one scaffold. The enabled process presented here substantially improved safety (in particular, due to eliminating a nitration step and optimizing a high-energy intermediate step), reproducibility, and scalability, resulting in delivery of a multikilogram quantity of the API with high purity. The controls of API quality and particle size were also discussed.
Purification of Crude 1-(4-(4-Amino-2-methoxy-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl)phenyl)cyclobutanecarbonitrile (1)
compound 1 as a white powder (2.61 kg, 51.8%). HPLC purity was 99.63%, associated with 0.16% of 14 and 0.13% of 15. Particle Size: D[4, 3] = 25 μm, D[v, 0.95] = 58 μm. Residual Solvents: acetic acid 0.4 wt %, water 0.1 wt % and DMF <0.1 wt %.
HRMS (m/z): calculated for C19H19N5O2, [M + H]+ 350.1612; found 350.1620.
Elemental analysis: calculated for C19H19N5O2: C 65.32, H 5.48, N 20.04; found: C 65.40, H 5.45, N 20.16.
hplc
Liquid chromatography mass spectrometry (LCMS) was performed on an Agilent 1100 Series (Waters Atlantis C18 column, 4.6 mm × 50 mm, 5 μm; 95% water/acetonitrile linear gradient to 5% water/acetonitrile over 4 min, hold at 5% water/acetonitrile to 5 min, trifluoroacetic acid modifier (0.05%); flow rate = 2.0 mL/min). Reaction monitoring and purity of intermediates and the final compound were checked by HPLC in the following conditions: Column: Zorbax SB-CN, 5 μm, 4.6 mm × 150 mm; Column Temperature: 30 °C; Flow Rate: 2 mL/min; Detection: UV @ 210 nm; Mobile phase: A: 0.2% phosphoric acid in water, B: Acetonitrile; Linear Gradient: from 95% of A to 5% of A within 15 min. HPLC purity was reported at 210 nm wavelength.
(a) Birch, A. M.; Buckett, L. K.; Turnbull, A. V. Opin. Drug Discovery Dev.2010, 13,489
(b) Zammit, V. A.; Buckett, L. K.; Turnbull, A. V.; Wure, H. Pharmacol. Ther.2008, 118, 295
(a) Dow, R. L.; Munchhof, M. J. U.S. Patent Appl.2010/0197590.
(b) Aspnes, G. E.; Dow, R. L.; Munchhof, M. J. U.S. Patent Appl. 2010/0197591.
(c) Bahnck, K. B.; Shavnya, A.; Tao,Y.; Lilley, S. C.; Andrews, M. P.; Aspnes, G. E.; Bernhardson, D. J.; Bill, D. R.; Bundesmann, M. W.; Dow, R. L.; Karki, K.; Le, T.; Li, Q.; Munchhof, M. J.; Nematalla, A.; Nihlawi, M.; Patel, L.; Perreault, C.; Waldo, M. Synthesis2012, 44, 3152
(a) Yendapally, R.; Hurdle, J. G.; Carson, E. I.; Lee, R. B.; Lee, R. E. J. Med. Chem.2008,51, 1487
(b) Kulkarni, B. A.; Ganesan, A. Angew. Chem., Int. Ed.1997, 109, 2565
anto obesity agents
Obesity and overweight are generally defined by body mass index (BMI), which is correlated with total body fat and estimates the relative risk of disease. BMI is calculated by weight in kilograms divided by height in meters squared (kg/m2). Overweight is typically defined as a BMI of 25-29.9 kg/m2, and obesity is typically defined as a BMI of 30 kg/m2. See, e.g., National Heart, Lung, and Blood Institute, Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults, The Evidence Report, Washington, D.C.: U.S. Department of Health and Human Services, NIH publication no. 98-4083 (1998).
Another aspect of the invention is for the treatment or delaying the progression or onset of diabetes or diabetes-related disorders including Type 1 (insulin-dependent diabetes mellitus, also referred to as “IDDM”) and Type 2 (noninsulin-dependent diabetes mellitus, also referred to as “NIDDM”) diabetes, impaired glucose tolerance, insulin resistance, hyperglycemia, and diabetic complications (such as atherosclerosis, coronary heart disease, stroke, peripheral vascular disease, nephropathy, hypertension, neuropathy, and retinopathy).
Yet another aspect of the invention is the treatment of diabetes- or obesity-related co-morbidities, such as metabolic syndrome. Metabolic syndrome includes diseases, conditions or disorders such as dyslipidemia, hypertension, insulin resistance, diabetes (e.g., Type 2 diabetes), weight gain, coronary artery disease and heart failure. For more detailed information on Metabolic Syndrome, see, e.g., Zimmet, P.Z., et al., “The Metabolic Syndrome: Perhaps an Etiologic Mystery but Far From a Myth —Where Does the International Diabetes Federation Stand?,” Diabetes & Endocrinology, 7(2), (2005); and Alberti, K. G., et al., “The Metabolic Syndrome —A New Worldwide Definition,” Lancet, 366, 1059-62 (2005). Administration of the compounds of the invention may provide a statistically significant (p<0.05) reduction in at least one cardiovascular disease risk factor, such as lowering of plasma leptin, C-reactive protein (CRP) and/or cholesterol, as compared to a vehicle control containing no drug. The administration of compounds of the invention may also provide a statistically significant (p<0.05) reduction in glucose serum levels.
In yet another aspect of the invention, the condition treated is impaired glucose tolerance, hyperglycemia, diabetic complications such as sugar cataracts, diabetic neuropathy, diabetic nephropathy, diabetic retinopathy and diabetic cardiomyopathy, anorexia nervosa, bulimia, cachexia, hyperuricemia, hyperinsulinemia, hypercholesterolemia, hyperlipidemia, dyslipidemia, mixed dyslipidemia, hypertriglyceridemia, nonalcoholic fatty liver disease, atherosclerosis, arteriosclerosis, acute heart failure, congestive heart failure, coronary artery disease, cardiomyopathy, myocardial infarction, angina pectoris, hypertension, hypotension, stroke, ischemia, ischemic reperfusion injury, aneurysm, restenosis, vascular stenosis, solid tumors, skin cancer, melanoma, lymphoma, breast cancer, lung cancer, colorectal cancer, stomach cancer, esophageal cancer, pancreatic cancer, prostate cancer, kidney cancer, liver cancer, bladder cancer, cervical cancer, uterine cancer, testicular cancer and ovarian cancer.
anti-obesity agent is selected from the group consisting of dirlotapide, mitratapide, implitapide, R56918 (CAS No. 403987), CAS No. 913541-47-6, lorcaserin, cetilistat, PYY3-36, naltrexone, oleoyl-estrone, obinepitide, pramlintide, tesofensine, leptin, liraglutide, bromocriptine, orlistat, exenatide, AOD-9604 (CAS No. 221231-10-3) and sibutramine; and said anti-diabetic agent is selected from the group consisting of metformin, acetohexamide, chlorpropamide, diabinese, glibenclamide, glipizide, glyburide, glimepiride, gliclazide, glipentide, gliquidone, glisolamide, tolazamide, tolbutamide, tendamistat, trestatin, acarbose, adiposine, camiglibose, emiglitate, miglitol, voglibose, pradimicin-Q, salbostatin, balaglitazone, ciglitazone, darglitazone, englitazone, isaglitazone, pioglitazone, rosiglitazone, troglitazone, exendin-3, exendin-4, trodusquemine, reservatrol, hyrtiosal extract, sitagliptin, vildagliptin, alogliptin and saxagliptin.
Exemplary anti-obesity agents for use in the combination aspects of the invention include gut-selective MTP inhibitors (e.g., dirlotapide, mitratapide and implitapide, R56918 (CAS No. 403987) and CAS No. 913541-47-6), CCKa agonists (e.g., N-benzyl-2-[4-(1H-indol-3-ylmethyl)-5-oxo-1-phenyl-4,5-dihydro-2,3,6,10 b-tetraaza-benzo[e]azulen-6-yl]-N-isopropyl-acetamide described in PCT Publication No. WO 2005/116034 or US Publication No. 2005-0267100 A1), 5HT2c agonists (e.g., lorcaserin), MCR4 agonist (e.g., compounds described in U.S. Pat. No. 6,818,658), lipase inhibitor (e.g., Cetilistat), PYY3-36(as used herein “PYY3-36” includes analogs, such as peglated PYY3-36 e.g., those described in US Publication 2006/0178501), opioid antagonists (e.g., naltrexone), oleoyl-estrone (CAS No. 180003-17-2), obinepitide (TM30338), pramlintide (Symlin®), tesofensine (NS2330), leptin, liraglutide, bromocriptine, orlistat, exenatide (Byetta®), AOD-9604 (CAS No. 221231-10-3) and sibutramine
LX-4211 is a dual SGLT2/1 inhibitor; Antidiabetic agents.
LX-4211 is a SGLT-2 inhibitor being evaluated in phase II clinical studies at Lexicon Pharmaceuticals for the oral treatment of type 2 diabetes.
More Positive News For Lexicon, But Not The Big Announcement
Apr. 14, 2014 1:54 PM ET |
Summary
Co-administration of LX4211 led to a nearly one-third reduction in mealtime insulin for Type 1 diabetics.
Although there was no reduction in basal insulin use, the LX4211 group saw better glucose control, lower HbA1c, and weight loss.
Partnering LX4211 is still management’s top priority but independent development in Type 1 diabetes is at least an option.
Lexicon Pharmaceuticals (LXRX) continues to generate data on its SGLT-1/2 inhibitor LX4211 that suggest this is an effective and promising medication for treating not only Type 2 diabetes (the common target for non-insulin medications for diabetes), but also Type 1 as well. Lexicon’s most recent update, a small short-term Phase II study in Type 1 diabetics is certainly a positive update, but it’s not what investors really want to see. Lexicon still needs to find a development partner for LX4211 and the ongoing delays don’t help sentiment or the long-term prospects for the drug.
A Potentially Meaningful Addition To Type 1 Care
On Monday morning, Lexicon released top-line data from a small (33-patient) Phase II study of LX4211 in Type 1 diabetics on insulin. The results support the notion that SGLT inhibition can play a valuable role in improving glucose control for Type 1 diabetics.
This small study enrolled generally well-controlled patients (HbA1c levels ranging from 7 to 9, with an average of 7.9) and the addition of LX4211 led to 32% reduction in bolus (mealtime) insulin versus a 6% reduction in the placebo group. Even with the lower bolus insulin, patients in the LX4211 group showed a 0.55% reduction in HbA1c versus a 0.06% reduction in the placebo group. Patients taking LX4211 demonstrated better glucose control (more time spent in the target range of 70-180 mg/dL) and saw a 1.7kg weight loss versus a 0.5kg weight gain in the placebo group
Aspects of this invention can be understood from the following examples.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2.3-d][13]dioxol-5-yl)(morpholino)methanone
To a 12L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(-)-xylose (504.40 g, 3.360 mol), acetone (5L, reagent grade) and anhydrous MgSO4 powder (811.23g, 6.740 mol / 2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol / 0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24°C over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH = 8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt% water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23g, 86% yield, 96.7 area% pure by GC). 1H NMR (400 MHz, DMSO-d6)δ1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93 – 4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 13C NMR (101MHz, DMSO-d6) δ26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73.
To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0g, 131 mmol) in acetone (375 mL, 15X) and H2O (125 mL, 5X) was added NaHC03 (33.0g, 3.0 equiv), NaBr (2.8g, 20 mol%) and TEMPO (0.40g, 2 mol%) at 20°C. The mixture was cooled to 0-5°C and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20°C for 24h. Methanol (20 mL) was added and the mixture was stirred at 20°C for 1h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2X). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12X x3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5X) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0g, 79%). 1H NMR (methanol-d4), δ 6.00 (d, J= 3.2 Hz, 1H), 4.72 d, J= 3.2 Hz, 1H), 4.53 (d, J= 3.2 Hz, 1H), 4.38 (d, J= 3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H).
To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0g, 24.5 mmol) in THF (100 mL, 20X) was added TBTU (11.8g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20°C for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20°C for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2X x2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4:1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), 8 6.02 (d, J= 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J= 3.2 Hz, 1H), 4.58 (d, J= 3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahvdrofuro[2.3-d][1,3]dioxol-5-yl)(morpholino)methanone
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2 solution (1.6 L) and bleach/K2HPO4 solution (400 mL),∼1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25°C. Additional charges of TEMPO (14.3g, 0.5 mol%) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to < 10°C and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15°C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L x 3). The combined organic layer was concentrated under vacuo (∼100-120 torr) at < 35°C (28-32°C vapor, 45-50°C bath) to low volume (- 6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached < 1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5°C and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ5.96 (d, J = 3.6 Hz, 1H), 4.5 8 (d, J = 3.6 Hz, 1H), 4.53 (d, J =3.2Hz,1H), 4.30 (d, J= 3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) 8 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25. 1.
The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (-80 L) to remove excess morpholine. When final volume reached -12 L, heptane (14 L) was added slowly at 60-70°C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136°C (DSC). 1H NMR (CDCl3), δ 6.02 (d, J = 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene:
6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene
A 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF = 0.003 wt% water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step.
A jacketed 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF = 0.003 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 6°C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9°C. The resulting orange solution was diluted with dichloromethane (75mL) and was cooled to -7°C. Then a solution of 2-chloro-5-iodobenzoyl chloride (≤ 0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3°C. The reaction mixture was warmed slightly and held at +5°C for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450mL pre-cooled (∼5°C) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10min with internal temperature remaining below 28°C. The quenched biphasic mixture was stirred at 20°C for 45min and the lower organic phase was washed with 1N aq. HCl (200mL), twice with saturated aq sodium bicarbonate (200mL per wash), and with saturated aq sodium chloride (200mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93g, 99.0 area% by HPLC at 220nm, 1.0 area% regioisomer at 200nm, 98.5 % “as-is” yield).
A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300mL, KF = 0.004 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 5°C.Then triethylsilane (28mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24mL, 194.46mmo1,2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150mL) followed by saturated aq sodium bicarbonate (150mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq sodium bicarbonate (100 mL), and with saturated aq sodium chloride (50mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45°C was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45°C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone
To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500mag, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5°C, and the mixture was stirred for 1.5 h at 0-5°C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5°C and the mixture was kept stirring for 1h, warmed to 20°C and stirred at 20°C for 2 hours. The reaction was quenched with saturated aq NH4CI, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7. 88 (dd, J= 8.4, 2.0 Hz, 1H), 7.82 (d, J= 2.0 Hz, 1H), 7.50 (d, J= 8.4 Hz, 1H), 7.12 (d, J= 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.07 (d, J = 3.2 Hz, 1H), 5.21 (d, J = 3.2 Hz, 1H), 4.58 (d, J = 3.2 Hz, 1H), 4.56 (d, J = 3.2 Hz, 1H), 4.16 (d, J = 7.2 Hz, 2H), 4.03 (q, J = 7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4X to the morpholinoamide) at room temperature and cooled to -5°C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at -5°C over 3 hours. This Grignard solution was used in the ketone formation below.
[0055]
To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity = 97 wt%, 2.01 kg, 7.34 mol) and THF (11 L, 5.5X) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30°C. The homogeneous solution was then cooled to – 25°C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at -25°C over 3 hours. Then the above Grignard solution was added to this solution at -20 over 41 minutes. The resulting solution was further stirred at -20°C before quench. The reaction mixture was added to 10 wt% aqueous NH4Cl (10 L, 5X) at 0°C with vigorous stirring, and stirred for 30 minutes at 0°C. To this mixture was added slowly 6 N HCl (4 L, 2X) at 0°C to obtain a clear solution and stirred for 30 minutes at 10°C. After phase split, the organic layer was washed with 25 wt% aq NaCl (5 L, 2.5X). Then the organic layer was concentrated to a 3X solution under the conditions (200 mbar, bath temp 50°C). EtOAc (24 L, 12X) was added, and evaporated to a 3X solution under the conditions (150 mbar, bath temp 50°C). After removed solids by a polish filtration, EtOAc (4 L, 2X) was added and concentrated to dryness (150 mbar, bath temp 50°C). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70°C to obtain a 2.5X homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5X) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5X) was added slowly to a little cloudy solution at 70°C. After stirred for 0.5 h at 70°C, the suspension was slowly cooled to 60°C and stirred for 1 h at 60°C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4X), dried under vacuum at 45°C to give the desired ketone as fluffy solids (2.57 kg, 100 wt% by HPLC, purity-adjusted yield: 81%).
6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)-((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17X) was added CeCl3.7H2O (118.5g, 1.2 equiv) and the mixture was stirred at 20°C until all solids were dissolved. The mixture was then cooled to -78°C and NaBH4 (12.03g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed -70°C. The mixture was stirred at – 78°C for 1 hour, slowly warmed to 0°C and quenched with saturated aq NH4Cl (550 mL, 5X). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1L, 10X x2) and washed with brine (550 mL, 5X). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100°C and stirred for 15 hours. The mixture was then cooled to room temperature (20°C) and concentrated under vacuum to give a yellow oil (crude, ∼118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0°C. Then, Ac2O (195 mL, -8.0 equiv) was added and the mixture was warmed to 20°C and stirred at 20°C for 2h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, x2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (-133g).
To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4X) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80°C for 3.5 hours. The mixture was cooled to 20°C and Mel (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20°C for 3h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1.3 L, 10X) and washed with H2O (650 mL, 5X x2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5X) and the mixture was reslurried at 60°C for 2h and then cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 70 mL, x3). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) δ 7.37 (d, J= 8.0 Hz, 1H), 7.20 (dd, J= 8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J = 9.6 Hz, 1H), 5.20 (t, J = 9.6 Hz, 1H), 5.05 (t, J= 9.6 Hz, 1H), 4.51 (d, J=9.6Hz, 1H), 4.38 (d, J= 9.6Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2.11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19X). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25°C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt% aq NH4Cl (2.5 L, 1X) and the mixture was concentrated under vacuum to 5X, diluted with water (10 L, 4X) and MTBE (12.5L, 5X). The mixture was cooled to 10°C and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2X). The combined aqueous layer was extracted with MTBE (12.5 L, 5X). The combined organic layers were washed with brine (2.5 L, 1X) and concentrated under vacuum to 3X. MeCN (15 L, 6X) was added. The mixture was concentrated again to 10 L (4X) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN.
The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol% aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80°C for 2 hours and then cooled to 20°C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2X) and diluted with MTBE (15 L, 6X). The organic layer was separated, washed with brine (5 L, 2X) and concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the mixture was concentrated to 7.5 L (3X).
The above MeCN solution of (3S,4R,SR,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10°C, added with dimethylaminopyridine (17.53 g, 2.5 mol%), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2X, 6.0 equiv) so that the temperature of the mixture was kept below 20°C. The reaction was then warmed to 20°C and stirred for 1 hour and diluted with MTBE (15 L, 6X). The mixture was slowly quenched with water (7.5 L, 3X). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2X), 1 N NaHSO4 (5 L, 2X), and brine (5 L, 2X) in sequence.
The organic layer was then concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the solution was concentrated to 7.5 L (3X) (KF = 0.08%). Dioxane (12.5 L, 5X) was added and the solution was concentrated to 7.50 L (3X) (KF = 0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL).
To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80°C for 3 hours (>97% conversion). The mixture was cooled to 20°C and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20°C for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20°C for 1 hours. The mixture was then diluted with MTBE (25 L, 10X) and washed with water (12.5 L, 5X x2). The organic layer was separated and concentrated under vacuum to -5 L (2X). MeOH (12.5 L, 5X) was added and the mixture was concentrated to 5X to afford a slurry. The mixture was then heated at 60°C for 1 hour and cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 2.5 L, 1X x2, 1.0 L, 0.4X). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol
To a slurry of (2S,3S,4R,SS,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mo1) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 20°C and the mixture was stirred at 20°C for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.0g, 95%). 1H NMR (CDCl3) δ 7.38 (d, J = 8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, 1H), 4.15 (d, J = 9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, 1H), 3.50 (m, 2H), 3.42 (br s, 1H), 2.95 (br s, 1H), 2.57 (br s, 1H), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
LEX-1287 The compound is an inhibitor of the sodium glucose co-transporter 2, and may be useful in the treatment of diabetes and a variety of other diseases and conditions. See U.S. patent application no. 11/862,690, filed September 28, 2007.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran-3,4,5-triol To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-
(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mol) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. The mixture was then
18
LEX-1287 concentrated to 300 mL, added to H2O (IL) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.Og, 95%). IH NMR (CDC13) δ 7.38 (d, J = 8.4 Hz, IH), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, IH), 4.15 (d, J = 9.6 Hz, IH), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, IH), 3.50 (m, 2H), 3.42 (br s, IH), 2.95 (br s, IH), 2.57 (br s, IH), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
6.9. Preparation of Crystalline Anhydrous (2S,3R,4R,5S,6R)-2-(4-chloro-
3-f4-ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran- 3,4,5-triol Form 1
Under slightly positive nitrogen pressure, to a 50 L reactor was charged MeOH (12 L) and the triacetate (1.70 Kg, 3.09 mol). Methanol (5L) was added as a rinse. The slurry was then added NaOMe in MeOH (25 wt%, 340 mL, 0.2X) in 15 minutes at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. To the mixture was added slowly water (25.5 L, 15X) in 45 minutes with 5 g seeding (DSC123°C). Solids crashed out and the mixture was stirred at 200C for 1 hour, cooled to 00C and stirred for 30 minutes. The solid was filtered and washed with water (1.7 L, IX, x2) and the cake was dried under vacuum at 45°C overnight to afford the title compound (m.p. ~ 123 0C by DSC peak; 1.28 Kg, 97.7% yield).
Aspects of this invention can be understood from the following examples, which do not limit its scope.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone
To a 12 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(−)-xylose (504.40 g, 3.360 mol), acetone (5 L, reagent grade) and anhydrous MgSO4 powder (811.23 g, 6.740 mol/2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol/0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24° C. over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH =8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142 mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5 S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25 L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt % water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23 g, 86% yield, 96.7 area % pure by GC). 1H NMR (400 MHz, DMSO-d6) δ 1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93-4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 3C NMR (101 MHz, DMSO-d6) δ 26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73. To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0 g, 131 mmol) in acetone (375 mL, 15×) and H2O (125 mL, 5×) was added NaHCO3 (33.0 g, 3.0 equiv), NaBr (2.8 g, 20 mol %) and TEMPO (0.40 g, 2 mol %) at 20° C. The mixture was cooled to 0-5° C. and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20° C. for 24h. Methanol (20 mL) was added and the mixture was stirred at 20° C. for 1 h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2×). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12× ×3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5×) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0 g, 79%).1H NMR (methanol-d4), δ 6.00 (d, J=3.2 Hz, 1H), 4.72 d, J=3.2 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.38 (d, J=3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H). To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0 g, 24.5 mmol) in THF (100 ML, 20×) was added TBTU (11.8 g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20° C. for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20° C. for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2× ×2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4: 1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0 L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2solution (1.6 L) and bleach/K2HPO4 solution (400 mL, 1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25° C. Additional charges of TEMPO (14.3 g, 0.5 mol %) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to <10° C. and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15° C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L ×3). The combined organic layer was concentrated under vacuo (˜100-120 torr) at <35° C. (28-32° C. vapor, 45-50° C. bath) to low volume (˜6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached <1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5° C. and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ 5.96 (d, J=3.6 Hz, 1H), 4.58 (d, J=3.6 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.30 (d, J=3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) δ 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25.1. The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (˜80 L) to remove excess morpholine. When final volume reached 12 L, heptane (14 L) was added slowly at 60-70° C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136° C. (DSC). 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene
A 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF=0.003 wt % water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step. A jacketed 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF=0.003 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 6° C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9° C. The resulting orange solution was diluted with dichloromethane (75 mL) and was cooled to −7° C. Then a solution of 2-chloro-5-iodobenzoyl chloride (<0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3° C. The reaction mixture was warmed slightly and held at +5° C. for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450 mL pre-cooled (˜5° C.) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10 min with internal temperature remaining below 28° C. The quenched biphasic mixture was stirred at 20° C. for 45 min and the lower organic phase was washed with 1N aq. HCl (200 mL), twice with saturated aq. sodium bicarbonate (200 mL per wash), and with saturated aq. sodium chloride (200 mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93 g, 99.0 area % by HPLC at 220 nm, 1.0 area % regioisomer at 200 nm, 98.5 % “as-is” yield). A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300 mL, KF=0.004 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 5° C. Then triethylsilane (28 mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24 mL, 194.46 mmol, 2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30 min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150 mL) followed by saturated aq sodium bicarbonate (150 mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq. sodium bicarbonate (100 mL), and with saturated aq. sodium chloride (50 mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45° C. was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45° C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220 nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone
To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500 mg, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5° C., and the mixture was stirred for 1.5 h at 0-5° C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5° C. and the mixture was kept stirring for 1 h, warmed to 20° C. and stirred at 20° C. for 2 hours. The reaction was quenched with saturated aq NH4Cl, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7.88 (dd, J=8.4, 2.0 Hz, 1H), 7.82 (d, J=2.0 Hz, 1H), 7.50 (d, J=8.4 Hz, 1H), 7.12 (d, J=8.4 Hz, 2H), 6.86 (d, J=8.4 Hz, 2H), 6.07 (d, J=3.2 Hz, 1H), 5.21 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 4.56 (d, J=3.2 Hz, 1H), 4.16 (d, J=7.2 Hz, 2H), 4.03 (q, J=7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J=7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d]1,3]dioxol-5-yl)methanone
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4× to the morpholinoamide) at room temperature and cooled to −5° C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at −5° C. over 3 hours. This Grignard solution was used in the ketone formation below. To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity=97 wt %, 2.01 kg, 7.34 mol) and THF (11 L, 5.5×) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30° C. The homogeneous solution was then cooled to −25° C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at −25° C. over 3 hours. Then the above Grignard solution was added to this solution at −20 over 41 minutes. The resulting solution was further stirred at −20° C. before quench. The reaction mixture was added to 10 wt % aqueous NH4Cl (10 L, 5×) at 0° C. with vigorous stirring, and stirred for 30 minutes at 0° C. To this mixture was added slowly 6 N HCl (4 L, 2×) at 0° C. to obtain a clear solution and stirred for 30 minutes at 10° C. After phase split, the organic layer was washed with 25 wt % aq NaCl (5 L, 2.5×). Then the organic layer was concentrated to a 3× solution under the conditions (200 mbar, bath temp 50° C.). EtOAc (24 L, 12×) was added, and evaporated to a 3× solution under the conditions (150 mbar, bath temp 50° C.). After removed solids by a polish filtration, EtOAc (4 L, 2×) was added and concentrated to dryness (150 mbar, bath temp 50° C.). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70° C. to obtain a 2.5× homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5×) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5×) was added slowly to a little cloudy solution at 70° C. After stirred for 0.5 h at 70° C., the suspension was slowly cooled to 60° C. and stirred for 1 h at 60° C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4×), dried under vacuum at 45° C. to give the desired ketone as fluffy solids (2.57 kg, 100 wt % by HPLC, purity-adjusted yield: 81%).
6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17×) was added CeCl3.7H2O (118.5 g, 1.2 equiv) and the mixture was stirred at 20° C. until all solids were dissolved. The mixture was then cooled to −78° C. and NaBH4 (12.03 g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed −70° C. The mixture was stirred at −78° C. for 1 hour, slowly warmed to 0° C. and quenched with saturated aq NH4Cl (550 mL, 5×). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1 L, 10× ×2) and washed with brine (550 mL, 5×). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115 g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100° C. and stirred for 15 hours. The mixture was then cooled to room temperature (20° C.) and concentrated under vacuum to give a yellow oil (crude, 118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0° C. Then, Ac2O (195 mL, ˜8.0 equiv) was added and the mixture was warmed to 20° C. and stirred at 20° C. for 2 h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, ×2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (˜133 g). To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4×) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80° C. for 3.5 hours. The mixture was cooled to 20° C. and MeI (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20° C. for 3 h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1. 3 L, 10×) and washed with H2O (650 mL, 5× ×2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5×) and the mixture was reslurried at 60° C. for 2 h and then cooled to 0C and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 70 mL, ×3). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) 6 7.37 (d, J=8.0 Hz, 1H), 7.20 (dd, J=8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J=9.6 Hz, 1H), 5.20 (t, J=9.6 Hz, 1H), 5.05 (t, J =9.6 Hz, 1H), 4.51 (d, J =9.6 Hz, 1H), 4.38 (d, J=9.6 Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2. 11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J=7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19×). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25° C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt % aq NH4Cl (2.5 L, 1×) and the mixture was concentrated under vacuum to 5×, diluted with water (10 L, 4×) and MTBE (12.5 L, 5×). The mixture was cooled to 10° C. and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2×). The combined aqueous layer was extracted with MTBE (12.5 L, 5×). The combined organic layers were washed with brine (2.5 L, 1×) and concentrated under vacuum to 3×. MeCN (15 L, 6×) was added. The mixture was concentrated again to 10 L (4×) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN. The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol % aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80° C. for 2 hours and then cooled to 20° C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2×) and diluted with MTBE (15 L, 6×). The organic layer was separated, washed with brine (5 L, 2×) and concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the mixture was concentrated to 7.5 L (3×). The above MeCN solution of (3S,4R,5R,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10° C., added with dimethylaminopyridine (17.53 g, 2.5 mol %), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2×, 6.0 equiv) so that the temperature of the mixture was kept below 20° C. The reaction was then warmed to 20° C. and stirred for 1 hour and diluted with MTBE (15 L, 6×). The mixture was slowly quenched with water (7.5 L, 3×). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2×), 1 N NaHSO4 (5 L, 2×), and brine (5 L, 2×) in sequence. The organic layer was then concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the solution was concentrated to 7.5 L (3×) (KF=0.08%). Dioxane (12.5 L, 5×) was added and the solution was concentrated to 7.50 L (3×) (KF=0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL). To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80° C. for 3 hours (>97% conversion). The mixture was cooled to 20° C. and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20° C. for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20° C. for 1 hours. The mixture was then diluted with MTBE (25 L, 10×) and washed with water (12.5 L, 5× ×2). The organic layer was separated and concentrated under vacuum to ˜5 L (2×). MeOH (12.5 L, 5×) was added and the mixture was concentrated to 5× to afford a slurry. The mixture was then heated at 60° C. for 1 hour and cooled to 0° C. and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 2.5 L, 1× ×2, 1.0 L, 0.4×). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol
To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0. 164 mol) in MeOH (900 mL, 10×) was added NaOMe in MeOH (25 wt %, 18 mL, 0.2×) at 20° C. and the mixture was stirred at 20° C. for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1 L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, ×3) and the cake was dried under vacuum at 45° C. overnight to afford the desired methyl thiolate (67.0 g, 95%). 1H NMR (CDCl3) 6 7.38 (d, J=8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J=8.8 Hz, 2H), 6.83 (d, J=8.8 Hz, 2H), 4.35 (d, J=9.6 Hz, 1H), 4.15 (d, J=9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J=8.8 Hz, 1H), 3.50 (m, 2H), 2.73 (br s, 3H), 2.17 (s, 3H), 1.40 (t, J=7.2 Hz, 3H).
MK-8742 is in phase II clinical development at Merck & Co. for the oral treatment of chronic hepatitis C infection in combination with MK-5172 and ribavirin. Phase I clinical trials are uongoing for the treatment of hepatitis C infected males. In 2013, breakthrough therapy designation was assigned to the compound.
MK-8742 is an inhibitor of Hepatitis C Virus (HCV) non-structural protein 5A (NS5A) that is being developed for the treatment of HCV infection. MK-8742 has broad, potent HCV genotypic activity in vitro against viral variants that are resistant to other NS5A inhibitors. MK-8742 exhibits potent antiviral activity during 5 days of monotherapy in patients with GT1 and GT3 chronic HCV infection. MK-8742 is currently in Phase IIB development.
ELBASVIR
MK-8742 is an inhibitor of Hepatitis C Virus (HCV) non-structural protein 5A (NS5A) that is being developed for the treatment of HCV infection. MK-8742 has broad, potent HCV genotypic activity in vitro against viral variants that are resistant to other NS5A inhibitors. MK-8742 exhibits potent antiviral activity during 5 days of monotherapy in patients with GT1 and GT3 chronic HCV infection. MK-8742 is currently in Phase IIB development.
A mixture of Compound Int-19b (1.1 g, 3 mmol), (dibromomethyl)benzene (2.25 g, 9 mmol) and K2C03 (1.2 g, 9 mmol) in 15 mL of DMF was heated to 100 °C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature, concentrated in vacuo and the residue obtained was dissolved with
dichloromethane and water. The aqueous phase was extracted with dichloromethane. The combined organic extracts were washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using flash column
chromatography on silica gel to provide Compound Int-23a (380 mg, 28 %) as a white solid. 1H MR (CDCI3): δ 7.72 (bs, 1 H), 7.44 – 7.46 (d, J= 8.4 Hz, 1 H), 7.21 – 7.28 (m, 3 H), 7.09 – 7.12 (m, 3 H), 7.04 (s, 1 H), 6.99 – 7.01 (bs, J= 6.8 Hz, 2 H), 6.78 (s, 1 H), 6.63 – 6.65 (d, J = 8.4 Hz, 1 H). MS (ESI)
m/e (M+H+): 456. Step B – Pre aration of Compound Int-23b
lnt-23a lnt-23b
To a solution of Int-23a (456 mg, 1.0 mmol) in 1,4-dioxane was added bis pinacol borate (2.2 mmol) , Pd(dppf)Cl2 (0.04 mmol) and KOAc (4 mmol). The reaction mixture was put under N¾ heated to 110°C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature, concentrated in vacuo, and the residue obtained was purified using column chromatography on silica gel to provide Compound Int- 23b (590 mg, 87 % yield). 1H MR (CDC13): δ 8.13 (s, 1 H), 7.60 (d, J= 7.6 Hz, 1 H), 7.52 (d, J= 8.0 Hz, 1H), 7.36 – 7.39 (m, 1 H), 7.14 -7.19 (m, 4 H), 6.93 – 6.95 (m, 3 H), 6.90 (s, 1 H), 1.26 – 1.29 (s, 24 H). MS (ESI) m / e (M+H+): 550.
– Pre aration of Compound Int-23c
lnt-23b lnt-23c
A suspension of Int-23b (550 mg, 1.0 mmol), tert-butyl 2-(2-bromo-lH- imidazol-5-yl) pyrrolidine- 1-carboxylate (2.4 mmol), Pd(dppf) Cl2 (200 mg), Na2C03 (3 mmol) and in THF/H20 (10: 1, 33 mL) was allowed to stir at reflux for about 15 hours under N2. The reaction mixture was cooled to room temperature and filtered, and the filtrate was washed with water (50 mL) and extracted with EtOAc (100 mL). The organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography on silica gel to provide Compound Int-23c (160 mg). MS (ESI) m / e (M+H+): 768.
Preparation of Compound Int-23d
Int-23c (0.10 g, 0.13 mmol) was added to HCl/CH3OH (5 mL, 3M) and the resulting reaction was allowed to stir at room temperature for about 3 hours. The reaction mixture was then concentrated in vacuo to provide Compound Int-23d, which was used without further purification. MS (ESI) m / e (M+H+): 568.
– Preparation of Compound A
To a solution of Int-23d (56.8 mg, 0.10 mmol), (S)-2- (methoxycarbonylamino)-3-methylbutanoic acid (35.0 mg, 0.20 mmol) and DIPEA (0.8 mmol) in CH3CN (1 mL) was added BOP (98 mg, 0.22 mmol). The resulting reaction was allowed to stir at room temperature and monitored using LCMS. After LCMS showed the starting material to be consumed, the reactionmixture was filtered, and the filtrate was purified using HPLC to provide Compound A as a white solid. 1H MR (MeOD): δ 7.94 (s,
The NS5A protein plays a critical role in the replication of HCV and has been the focus of numerous research efforts over the past few years. NS5A inhibitors have shown impressive in vitro potency profiles in HCV replicon assays, making them attractive components for inclusion in all oral combination regimens. Early work in the NS5A arena led to the discovery of our first clinical candidate, MK-4882 [2-((S)-pyrrolidin-2-yl)-5-(2-(4-(5-((S)-pyrrolidin-2-yl)-1H-imidazol-2-yl)phenyl)benzofuran-5-yl)-1H-imidazole]. While preclinical proof-of-concept studies in HCV-infected chimpanzees harboring chronic genotype 1 infections resulted in significant decreases in viral load after both single- and multiple-dose treatments, viral breakthrough proved to be a concern, thus necessitating the development of compounds with increased potency against a number of genotypes and NS5A resistance mutations. Modification of the MK-4882 core scaffold by introduction of a cyclic constraint afforded a series of tetracyclic inhibitors, which showed improved virologic profiles. Herein we describe the research efforts that led to the discovery of MK-8742, a tetracyclic indole-based NS5A inhibitor, which is currently in phase 2b clinical trials as part of an all-oral, interferon-free regimen for the treatment of HCV infection.
see
Journal of Medicinal Chemistry (2014), 57(5), 1643-1672.
In December 2021, the U.S. Food and Drug Administration approved cabotegravir for pre-exposure prophylaxis (PrEP) in at-risk people under the brand name Apretude.[11]
GSK744 (also known as S/GSK1265744) is an investigational new drug under development for the treatment of HIV infection. It is anintegrase inhibitor, with a carbamoyl pyridone structure similar to dolutegravir. In investigational studies, the agent has been packaged into nanoparticles (GSK744LAP) conferring an exceptionally long half-life of 21–50 days following a single dose. In theory, this would make possible suppression of HIV with dosing as infrequently as once every three months.[1]
S-265744 LAP is in phase II clinical development at Shionogi-GlaxoSmithKline for the treatment of HIV infection. Phase III clinical trials had been ongoing for this indication; however, no recent development has been reported for this study.
Cabotegravir, or GSK1265744, is an HIV-1 integrase inhibitor that is prescribed with the non-nucleoside reverse transcriptase inhibitor, rilpivirine.4,6,7 Early research into cabotegravir showed it had lower oral bioavailability than dolutegravir,4 which resulted in the development of long acting monthly intramuscular injection formulation for cabotegravir.4,7
Cabotegravir was granted FDA approval on 21 January 2021 in combination with rilpivirine to treat HIV-1 infection in virologically suppressed individuals.8 While previously administered once monthly only, this combination product was granted FDA approval for dosing every two months on February 01, 2022 11 and without the need for an oral lead-in period prior.7
The human immunodeficiency virus (“HIV”) is the causative agent for acquired immunodeficiency syndrome (“AIDS”), a disease characterized by the destruction of the immune system, particularly of CD4+ T-cells, with attendant susceptibility to opportunistic infections, and its precursor Al DS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. HIV is a retrovirus; the conversion of its RNA to DNA is accomplished through the action of the enzyme reverse transcriptase. Compounds that inhibit the function of reverse transcriptase inhibit replication of HIV in infected cells. Such compounds are useful in the prevention or treatment of HIV infection in humans.
A required step in HIV replication in human T-cells is the insertion by virally-encoded integrase of proviral DNA into the host cell genome. Integration is believed to be mediated by integrase in a process involving assembly of a stable nucleoprotein complex with viral DNA sequences, cleavage of two nucleotides from the 3′ termini of the linear proviral DNA and covalent joining of the recessed 3′ OH termini of the proviral DNA at a staggered cut made at the host target site. The repair synthesis of the resultant gap may be accomplished by cellular enzymes. There is continued need to find new therapeutic agents to treat human diseases. HIV integrase is an attractive target for the discovery of new therapeutics due to its important role in viral infections, particularly HIV infections. Integrase inhibitors are disclosed in WO2006/116724.
(3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, a compound of formula (I), also referred to as compound (I), has proven antiviral activity against human immunodeficiency virus (HIV).
The present invention features pharmaceutical compositions comprising the active ingredient (3S, 1 1 aR)- N-[(2,4-difluorophenyl)methyl]-2,3,5,7, 1 1 , 1 1 a-hexahydro-6-hydroxy-3- methyl-5,7- dioxo-oxazolo[3,2-a]pyrido[1 ,2-d]pyrazine-8-carboxamide, or a pharmaceutically acceptable salt thereof, suitable for administration once monthly or longer.
Medical uses
Cabotegravir in combination with rilpivirine is indicated for the treatment of human immunodeficiency virus type-1 (HIV-1) in adults.[1][5] The combination injection is intended for maintenance treatment of adults who have undetectable HIV levels in the blood (viral load less than 50 copies/mL) with their current antiretroviral treatment, and when the virus has not developed resistance to non-nucleoside reverse transcriptase inhibitors (NNRTIs) and integrase strand transfer inhibitors.[5] The tablets are used to check whether a person tolerates the treatment before the injection therapy is started.[12][5]
The two medicines are the first antiretroviral drugs that come in a long-acting injectable formulation.[12]
Cabotegravir (Apretude) is indicated for use in at-risk people weighing at least 35 kilograms (77 lb) for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV.[11]
Contraindications and interactions
Cabotegravir must not be combined with the drugs rifampicin, rifapentine, carbamazepine, oxcarbazepine, phenytoin or phenobarbital, which induce the enzyme UGT1A1.[5] These drugs significantly decrease cabotegravir concentrations in the body and thus may reduce its effectiveness.[9][5] Additionally, they induce the enzyme CYP3A4, which leads to reduced rilpivirine concentrations in the body.[5][13][14][15] Additionally, patients who are breastfeeding or plan to breastfeed should not take Cabotegravir because it is not known if it will pass within the breast milk.[16]
Adverse effects
The most common side effects of the injectable combination therapy with rilpivirine are reactions at the injection site (in up to 84% of patients) such as pain and swelling, as well as headache (up to 12%) and fever or feeling hot (in 10%). For the tablets, headache and a hot feeling were slightly less frequent. Less common side effects (under 10%) for both formulations are depressive disorders, insomnia, and rashes.[9]
Pharmacology
Mechanism of action
Cabotegravir is an integrase strand transfer inhibitor. This means it blocks the HIV’s enzyme integrase, thereby preventing its genome from being integrated into the human cells’ DNA.[9] As this is a necessary step for the virus to replicate, its further spread is hampered.[9]
When taken by mouth, cabotegravir reaches highest blood plasma levels after three hours. Taking the drug together with food slightly increases its concentrations in the blood, but this is not clinically relevant. After injection into the muscle, cabotegravir is slowly absorbed into the bloodstream, reaching its highest blood plasma levels after about seven days.[9]
Over 99% of the substance are bound to plasma proteins. The drug is inactivated in the body by glucuronidation, mainly by the enzyme UGT1A1, and to a much lesser extent by UGT1A9. More than 90% of the circulating substance are the unchanged cabotegravir, however. The biological half-life is 41 hours for the tablets and 5.6 to 11.5 weeks for the injection.[9]
Elimination has only been studied for oral administration: Most of the drug is eliminated via the faeces in unchanged form (47%). It is not known how much of this amount comes from the bile, and how much was not absorbed in the first place. (The bile actually contains the glucuronide, but this could be broken up again in the gut lumen to give the parent substance that is observed in the faeces.) To a lesser extent it is excreted via the urine (27%), almost exclusively as the glucuronide.[9]
Pharmacogenomics
UGT1A1 poor metabolizers have 1.3- to 1.5-fold increased cabotegravir concentrations in the body. This is not considered clinically significant.[9]
Chemistry
Cabotegravir is a white to off-white, crystalline powder that is practically insoluble in aqueous solutions under pH 9, and slightly soluble above pH 10. It is slightly acidic with a pKa of 7.7 for the enolic acid and 1.1 (calculated) for the carboxamide. The molecule has two asymmetric carbon atoms; only one of the four possible configurations is present in the medication.[18]
Formulation
In studies, the agent was packaged into nanoparticles (GSK744LAP) conferring a biological half-life of 21 to 50 days[citation needed] following a single dose. The marketed injection achieves its long half-life not via nanoparticles but with a suspension of the free cabotegravir acid. The tablets contain cabotegravir sodium salt.[18]
History
Cabotegravir was examined in the clinical trials HPTN 083 and HPTN 084.[19][20] In 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vocabria intended for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with rilpivirine injection.[21] The EMA also recommended marketing authorization be given for rilpivirine and cabotegravir injections to be used together for the treatment of people with HIV-1 infection.[12] Cabotegravir was approved for medical use in the European Union in December 2020.[8]
In 2020, results for some studies were released showing success in using injectable cabotegravir for long-acting pre-exposure prophylaxis (PrEP) with greater efficacy than the emtricitabine/tenofovir combination being widely used for PrEP at the time.[24][25]
The safety and efficacy of cabotegravir to reduce the risk of acquiring HIV were evaluated in two randomized, double-blind trials that compared cabotegravir to emtricitabine/tenofovir, a once daily oral medication for HIV PrEP.[11] Trial 1 included HIV-uninfected men and transgender women who have sex with men and have high-risk behavior for HIV infection.[11] Trial 2 included uninfected cisgender women at risk of acquiring HIV.[11]
In Trial 1, 4,566 cisgender men and transgender women who have sex with men received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections among trial participants taking daily cabotegravir followed by cabotegravir injections every two months compared to daily oral emtricitabine/tenofovir.[11] The trial showed participants who took cabotegravir had 69% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In Trial 2, 3,224 cisgender women received either cabotegravir or emtricitabine/tenofovir.[11] The trial measured the rate of HIV infections in participants who took oral cabotegravir and injections of cabotegravir compared to those who took emtricitabine/tenofovir orally.[11] The trial showed participants who took cabotegravir had 90% less risk of getting infected with HIV when compared to participants who took emtricitabine/tenofovir.[11]
In December 2021, the U.S. Food and Drug Administration (FDA) approved cabotegravir for pre-exposure prophylaxis.[11] The FDA granted the approval of Apretude to Viiv.[11]
Methods for the preparation of a compound of formula (I) are described in WO 2006/1 16764, WO2010/01 1814, WO2010/068262, and WO2010/068253
3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0,14 mL, 1.74 mmol) and 10 drops of glacial acetic acid.
The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added
to the mixture and the solvents removed in vacuo and the material was purified via
silica gel chromatography (2% CH3OH/CH2CI2 gradient elution) to give
The title compound was made in two steps using a similar process to that described
in example Z-I. 16a (510 mg, 1.08 mmol) and (2«5)-2-amino-1-propanol (0.17 mL, 2,17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give
The starting material of Example A is compound 8, which is identical to formula (Ia). Thus, Example A depicts a process in providing an intermediate for the compound of formula 17 below which is isomeric to the compound ZZ-2 at page 237 of WO 2006/116764 to Brian Johns et al.
14
Example Aa After dissolution of mixture of 320 g of compound 8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 mL of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 °C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 mL of cold 2-propanol and drying provided 167 g of compound 14 (52% yield) as a crystal. 1H NMR(300 MHz1 CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1H), 5.46 (d, J = 10.5 Hz, 1H), 5.23 (d, J = 10.2 Hz, 1H), 5.20 (dd, J = 3.9, 9.6 Hz, 1H), 4.46- 4.34 (m, 1H)1 4.31 (dd, J = 6.6, 8.7 Hz, 1H)1 4.14 (dd, J = 3.9, 12.3 Hz1 1H)1 3.79 (dd, J = 9.9, 12.3 Hz1 1 H), 3.62 (dd, J = 6.9, 8.7 Hz1 1 H), 1.38 (d, J = 6.3 Hz1 3H).
Example Ab
To slurry of 156 g of compound 14 (1.0 eq.) in 780 ml_ of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound 15 (84% yield) as a crystal.
Under carbon mono-oxide atmosphere, a mixture of 163 g of compound 15 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 ml_ of 2,4-difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 11.3 g of
Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O1 the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt. The organic layers were combined and concentrated. Silica gel column chromatography of the residue provided 184 g of compound 16 (96% yield) as foam.
Under hydrogen atmosphere, a mixture of 184 g of compound 16 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1), the filtrate was concentrated. After 200 ml_ of AcOEt was added to the residue, filtration afforded crude solid of compound 17. The precipitates were combined and extracted with 4.0 L of CHCl3/MeOH(5/1). After concentration of the CHCI3ZMeOH solution and addition of 250 ml_ of AcOEt to the residue, filtration afforded crude solid of compound 17. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated (three times). After cooling of the residue, filtration and drying provided 132 g of compound 17 (88% yield) as a crystal. 1H NMR(300 MHz, DMSO-cfe) δ 11.47 (brs, 1H), 10.31 (t, J = 6.0 Hz, 1H), 8.46 (s, 1H), 7.40 (td, J = 8.6, 6.9 Hz, 1H), 7.24 (ddd, J = 2.6, 9.4, 10.6, 1H), 7.11-7.01 (m, 1H), 5.39 (dd, J = 4.1, 10.4 Hz, 1H), 4.89 (dd, J = 4.2, 12.3 Hz, 1H), 4.55 (d, J = 6.0 Hz, 2H), 4.40 (dd, J = 6.8, 8.6 Hz, 1H), 4.36-^.22 (m, 1H)1 4.00 (dd, J = 10.2, 12.3 Hz, 1H), 3.67 (dd, J = 6.7, 8.6 Hz, 1H), 1.34 (d, J = 6.3 Hz, 3H).
Example Ae
After dissolution of 16.0 g of compound 17 (1.0 eq.) in 2.56 L of EtOH and 0.64 L of H2O by heating, followed by filtration, 39 ml_ of 1N NaOHaq.(1.0 eq.) was added to the solution at 75 0C. The solution was gradually cooled to room temperature. Filtration, washing with 80 ml_ of EtOH and drying provided 13.5 g of compound 18 (80% yield) as a crystal.
The following examples are intended for illustratation only and are not intended to limit the scope of the invention in any way. Preparation 1 : (3S.11 af?VΛ/-r(2.4-DifluoroDhenvnmethyll-6-hvdroxy-3-methyl-5.7-dioxo- 2,3,5,7, 11 ,11 a-hexahydroM ,31oxazolor3,2-alpyridori ,2-c/1pyrazine-8-carboxamide sodium salt (compound 1 b, scheme 2).
I) MsCI, Et3N
2) DBU
P-1 P-2 P-3
a) Synthesis of 2-methyl-3-[(phenylmethvl)oxvl-4/-/-pvran-4-one (compound P-2). To a slurry of 2000 g of compound P-1(1.0 eq.) in 14.0 L of MeCN were added 2848 g of benzyl bromide(1.05 eq.) and 2630 g of K2CO3(1.2 eq.). The mixture was stirred at 80 0C for 5 h and cooled to 13°C. Precipitate was filtered and washed with 5.0 L of MeCN. The filtrate was concentrated and 3.0 L of THF was added to the residue. The THF solution was concentrated to give 3585 g of crude compound P-2 as oil. Without further purification, compound P-2 was used in the next step. 1H NMR(300 MHz, CDCI3) δ 7.60 (d, J = 5.7 Hz, 1 H), 7.4-7.3 (m, 5H), 6.37 (d, J = 5.7 Hz, 1 H), 5.17 (s, 2H), 2.09 (s, 3H).
b) Synthesis of 2-(2-hydroxy-2-phenylethyl)-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound P-3). To 904 g of the crude compound P-2 was added 5.88 L of THF and the solution was cooled to -60 0C. 5.00 L of 1.0 M of Lithium bis(trimethylsilylamide) in THF(1.25 eq.) was added dropwise for 2 h to the solution of compound 2 at -60 0C. Then, a solution of 509 g of benzaldehyde(1.2 eq.) in 800 ml. of THF was added at -60 0C and the reaction mixture was aged at -60 0C for 1 h. The THF solution was poured into a mixture of 1.21 L of conc.HCI, 8.14 L of ice water and 4.52 L of EtOAc at less than 2 0C.
The organic layer was washed with 2.71 L of brine (twice) and the aqueous layer was extracted with 3.98 L of EtOAc. The combined organic layers were concentrated. To the mixture, 1.63 L of toluene was added and concentrated (twice) to provide toluene slurry of compound P-3. Filtration, washing with 0.90 L of cold toluene and drying afforded 955 g of compound P-3 (74% yield from compound P-1 ) as a solid. 1H NMR(300 MHz, CDCI3) δ
c) Synthesis of 2-[(£)-2-phenylethenyl]-3-[(phenylmethyl)oxy]-4H-pyran-4-one (compound
P-4). To a solution of 882 g of compound P-3 (1.0 eq.) in 8.82 L of THF were added 416 g of Et3N(1.5 eq.) and 408 g of methanesulfonyl chloride(1.3 eq.) at less than 30 0C. After confirmation of disappearance of compound P-3, 440 ml. of NMP and 1167 g of DBU(2.8 eq.) were added to the reaction mixture at less than 30 0C and the reaction mixture was aged for 30 min. The mixture was neutralized with 1.76 L of 16% sulfuric acid and the organic layer was washed with 1.76 L of 2% Na2S03aq. After concentration of the organic layer, 4.41 L of toluene was added and the mixture was concentrated (tree times). After addition of 4.67 L of hexane, the mixture was cooled with ice bath. Filtration, washing with 1.77 L of hexane and drying provided 780 g of compound P-4 (94% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.69 (d, J = 5.7 Hz, 1 H), 7.50-7.25 (m, 10H), 7.22 (d, J = 16.2
Hz, 1 H), 7.03 (d, J = 16.2 Hz, 1 H), 6.41 (d, J = 5.7 Hz, 1 H), 5.27 (s, 2H). d) Synthesis of 4-oxo-3-[(phenylmethyl)oxy]-4H-pyran-2-carboxylic acid (compound P-5). To a mixture of 822 g of compound P-4 (1.0 eq.) and 1 1.2 g of RuCI3-nH2O(0.02 eq.) in 2.47 L of MeCN, 2.47 L of EtOAc and 2.47 L of H2O was added 2310 g of NalO4(4.0 eq.) at less than 25 0C. After aging for 1 h, 733 g of NaCIO2(S-O eq.) was added to the mixture at less than 25 0C. After aging for 1 h, precipitate was filtered and washed with 8.22 L of
EtOAc. To the filtrate, 1.64 L of 50% Na2S203aq, 822 ml. of H2O and 630 ml. of coc.HCI were added. The aqueous layer was extracted with 4.11 L of EtOAc and the organic layers were combined and concentrated. To the residue, 4 L of toluene was added and the mixture was concentrated and cooled with ice bath. Filtration, washing with 1 L of toluene and drying provided 372 g of compound P-5 (56% yield) as a solid. 1H NMR(300 MHz,
e) Synthesis of 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylic acid (compound P-6). A mixture of 509 g of compound P-5 (1.0 eq.) and
407 g of 3-amino-propane-1 ,2-diol(2.5 eq.) in 1.53 L of EtOH was stirred at 65 0C for 1 h and at 80 0C for 6 h. After addition of 18.8 g of 3-Amino-propane-1 ,2-diol(0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 1 h. After addition of 18.8 g of 3-amino- propane-1 ,2-diol (0.1 eq.) in 200 ml. of EtOH, the mixture was stirred at 80 0C for 30 min. After cooling and addition of 509 ml. of H2O, the mixture was concentrated. To the residue,
2.54 L of H2O and 2.54 L of AcOEt were added. After separation, the aqueous layer was washed with 1.02 L of EtOAc. To the aqueous layer, 2.03 L of 12% sulfuric acid was added at less than 12 0C to give crystal of compound P-6. Filtration, washing with 1.53 L of cold H2O and drying provided 576 g of compound P-6 (83% yield) as a solid. 1H NMR(300 MHz, DMSO-de) δ 7.67 (d, J = 7.5 Hz, 1 H), 7.5-7.2 (m, 5H), 6.40 (d, J = 7.5 Hz, 1 H), 5.07
f) Synthesis of methyl 1-(2,3-dihydroxypropyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-7). To a slurry of 576 g of compound P-6 (1.0 eq.: 5.8% of H2O was contained) in 2.88 L of NMP were added 431 g of NaHCO3(3.0 eq.) and 160 ml. of methyl iodide(1.5 eq.) and the mixture was stirred at room temperature for 4 h. After cooling to 5 0C, 1.71 L of 2N HCI and 1.15 L of 20% NaClaq were added to the mixture at less than 10 0C to give crystal of compound 7. Filtration, washing with 1.73 L of H2O and drying provided 507 g of compound P-7 (89% yield) as a solid. 1H NMR(300 MHz, DMSO- cfe) δ 7.59 (d, J = 7.5 Hz, 1 H), 7.40-7.28 (m, 5H), 6.28 (d, J = 7.5 Hz, 1 H), 5.21 (d, J = 5.4 Hz, 1 H), 5.12 (d, J = 10.8 Hz, 1 H), 5.07 (d, J = 10.8 Hz, 1 H), 4.83 (t, J = 5.7 Hz, 1 H), 3.97 (dd, J = 2.4, 14.1 Hz, 1 H), 3.79 (s, 3H), 3.70 (dd, J = 9.0, 14.4 Hz, 1 H), 3.65-3.50 (m, 1 H), 3.40-3.28 (m, 1 H), 3.26-3.14 (m, 1 H).
g) Synthesis of methyl 1-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-1 ,4-dihydro-2- pyridinecarboxylate (compound P-8). To a mixture of 507 g of compound P -7 (1.0 eq.) in
5.07 L of MeCN, 5.07 L of H2O and 9.13 g of AcOH(0.1 eq.) was added 390 g of NaIO4(1.2 eq.) and the mixture was stirred at room temperature for 2 h. After addition of 1.52 L of 10% Na2S2OsBq., the mixture was concentrated and cooled to 10 0C. Filtration, washing with H2O and drying provided 386 g of compound P-8 (80% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 7.62 (d, J = 7.5 Hz, 1 H), 7.42-7.30 (m, 5H), 6.33 (d, J = 6.0 Hz, 2H),
h) Synthesis of (3S, 11 aR)-3-methyl-6-[(phenylmethyl)oxy]-2,3, 1 1 ,1 1a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-9). After dissolution of mixture of 320 g of compound P-8 (1.0 eq.) in 3.20 L of MeOH by heating, the solution was concentrated. To the residue, 1.66 L of MeCN, 5.72 ml. of AcOH(0.1 eq.) and 82.6 g of (S)-2-Amino-propan-1-ol(1.1 eq.) were added and the mixture was heated to 70 0C, stirred at 70 0C for 4 h and concentrated. To the residue, 1.67 L of 2-propanol was added and the mixture was concentrated (twice). After cooling of the residue, filtration, washing with 500 ml. of cold 2-propanol and drying provided 167 g of compound P-9 (52% yield) as a solid. 1H NMR(300 MHz, CDCI3) δ 7.61-7.55 (m, 2H), 7.40-7.20 (m, 4H), 6.53 (d, J = 7.2, 1 H), 5.46 (d, J = 10.5 Hz, 1 H), 5.23 (d, J = 10.2 Hz, 1 H), 5.20 (dd, J = 3.9, 9.6 Hz, 1 H), 4.46-4.34 (m, 1 H), 4.31 (dd, J = 6.6, 8.7 Hz, 1 H), 4.14 (dd, J = 3.9, 12.3 Hz, 1 H), 3.79 (dd, J = 9.9, 12.3 Hz, 1 H), 3.62 (dd, J = 6.9, 8.7 Hz, 1 H), 1.38 (d, J = 6.3 Hz, 3H).
i) Synthesis of (3 S, 1 1 aR)-8-bromo-3-methyl-6-[(phenylmethyl)oxy]-2,3, 11 ,11a- tetrahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-5,7-dione (compound P-10). To slurry of 156 g of compound P-9 (1.0 eq.) in 780 ml. of NMP was added 93.6 g of NBS(1.1 eq.) and the mixture was stirred at room temperature for 2.5 h. The reaction mixture was added to 3.12 L of H2O. Filtration, washing with 8.0 L of H2O and drying provided 163 g of compound P-10 (84% yield) as a solid. 1H NMR(300 MHz, DMSO-d6) δ 8.37 (s, 1 H), 7.55- 7.50 (m, 2H), 7.42-7.25 (m, 3H), 5.34 (dd, J = 3.6, 9.9 Hz, 1 H), 5.18 (d, J = 10.8 Hz, 1 H), 5.03 (d, J = 10.5 Hz, 1 H), 4.53 (dd, J = 3.6, 12.0 Hz, 1 H), 4.40-4.20 (m, 2H), 3.99 (dd, J = 9.9, 1 1.7 Hz, 1 H), 3.64 (dd, J = 5.7, 8.1 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3H). j) Synthesis of (3S,1 1aS)-Λ/-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6- [(phenylmethyl)oxy]-2,3,5,7, 11 ,1 1 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8- carboxamide (compound P-11). Under carbon mono-oxide atmosphere, a mixture of 163 g of compound P-10 (1.0 eq.), 163 mL of /-Pr2NEt(2.5 eq.), 68.4 mL of 2,4- difluorobenzylamine(1.5 eq.) and 22.5 g of Pd(PPh3)4(0.05 eq.) in 816 mL of DMSO was stirred at 90 0C for 7 h. After cooling, removal of precipitate, washing with 50 mL of DMSO and addition of 1 1.3 g of Pd(PPh3)4(0.025 eq.), the reaction mixture was stirred at 90 0C for 2 h under carbon mono-oxide atmosphere again. After cooling, removal of precipitate and addition of 2.0 L of AcOEt and 2.0 L of H2O, the organic layer was washed with 1.0 L of 1 N HCIaq. and 1.0 L of H2O (twice) and the aqueous layer was extracted with 1.0 L of AcOEt.
k) Synthesis of (3S,1 1aR)-Λ/-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo- 2,3,5,7, 11 ,11 a-hexahydro[1 ,3]oxazolo[3,2-a]pyrido[1 ,2-c/]pyrazine-8-carboxamide (compound 1a). Under hydrogen atmosphere, a mixture of 184 g of compound P-11 (1.0 eq.) and 36.8 g of 10%Pd-C in 3.31 L of THF and 0.37 L of MeOH was stirred for 3 h. After filtration of precipitate(Pd-C), washing with THF/MeOH(9/1 ) and addition of 36.8 g of 10% Pd-C, the mixture was stirred for 20 min under hydrogen atmosphere. After filtration of precipitate(Pd-C) and washing with THF/MeOH(9/1 ), the filtrate was concentrated. After 200 mL of AcOEt was added to the residue, filtration afforded crude solid of compound 1 a.
The precipitates were combined and extracted with 4.0 L of CHCI3/Me0H(5/1 ). After concentration of the CHCI3/MeOH solution and addition of 250 mL of AcOEt to the residue, filtration afforded crude solid of compound 1a. The crude solids were combined and dissolved in 8.2 L of MeCN/H2O(9/1 ) by heating. After filtration, the filtrate was concentrated. To the residue, 1.5 L of EtOH was added and the mixture was concentrated
†Global API Chemistry, ‡MDR Chemical Science,§Analytical Sciences, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
A novel synthesis of GSK1265744, a potent HIV integrase inhibitor, is described. The synthesis is highlighted by an efficient construction of the densely functionalized pyridinone core as well as a highly diastereoselective formation of the acyl oxazolidine moiety. The latter exploits the target molecule’s ability to chelate to Mg2+, a key feature in the integrase inhibitor’s mechanism of action.
Bictegravir and dolutegravir are two recently approved integrase inhibitors for the treatment of HIV. A third inhibitor, cabotegravir, is in Phase 3 development. As a continuation of a series of articles on synthetic routes to newly approved drugs, the current article reviews the patent and journal literature regarding synthetic routes and final forms of these drug
^“Adopted USANs”(PDF). American Medical Association. Retrieved 19 September 2014.
^World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 73”. WHO Drug Information. 29 (1): 70–1. hdl:10665/331088.
Ziegler, Robert E.; Desai, Bimbisar K.; Jee, Jo-Ann; Gupton, B. Frank; Roper, Thomas D.; Jamison, Timothy F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angewandte Chemie, International Edition. Volume 57. Issue 24. Pages 7181-7185. Journal; Online Computer File. (2018).
SYN 4
Synthetic Reference
Rajan, Srinivasan Thirumalai; Eswaraiah, Sajja; Reddy, Ghojala Venkat; Reddy, Sagyam Rajeshwar; Markandeya, Bekkam; Rajesham, Boge. Novel crystalline polymorph of sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate and process for preparation thereof. Assignee MSN Research & Development Center, India. IN 201641037221. (2018).
Synthetic Reference 5
Sharma, Pramodkumar; Rao, Bhatraju Srinivasa; Deo, Keshav. A process for the preparation of Dolutegravir or its pharmaceutical acceptable salts thereof. Assignee Wockhardt Limited, India. IN 2015MU01007. (2016).
Synthetic Reference 6
Weaver, Jimmie Dean. Preparation of fluoroarenes via hydrogen bond directed photocatalytic hydrodefluorination of perfluoroarenes. Assignee The Board of Regents for Oklahoma State University, USA. WO 2018187336. (2018).
Vellanki, Sivaram Prasad; Nadella, Madumurthy; Bhalme, Mitali; Ramabhotla, Revathi Srinivas. Process for the preparation of dolutegravir, an integrase inhibitor for HIV-1 infection therapy. Assignee Mylan Laboratories Ltd., India. IN 2015CH00588. (2016).
SYN 9
Synthetic Reference
Sankareswaran, Srimurugan; Mannam, Madhavarao; Chakka, Veerababu; Mandapati, Srirami Reddy; Kumar, Pramod. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Organic Process Research & Development. Volume 20. Issue 8. Pages 1461-1468. Journal; Online Computer File. (2016).
Condensation of ethyl 2,4,5-trifluorobenzoylacetate (I) with triethyl orthoformate in refluxing Ac2O produced the benzoyl ethoxyacrylate (II), which was further condensed with 2-amino-5-fluoropyridine (III) to afford enamine (IV). Cyclization of (IV) in the presence of K2CO3 gave rise to the quinolone (V). The 7-fluoride group of (V) was then displaced by N-methylpiperazine (VI) in cold pyridine to furnish the piperazinyl quinolone (VII). Finally, ester hydrolysis in (VII) under acidic conditions yielded the target compound. In a closely related procedure, ester (V) was hydrolyzed to acid (VIII) using HCl. Subsequent displacement of the 7-fluoride of (VIII) with N-methylpiperazine (VI) provided the desired piperazinyl quinolone.
synthesis 2
Condensation of ethyl 2,4,5-trifluorobenzoylacetate (I) with triethyl orthoformate in refluxing Ac2O produced the benzoyl ethoxyacrylate (II), which was further condensed with 2-amino-5-fluoropyridine (III) to afford enamine (IV). Cyclization of (IV) in the presence of K2CO3 gave rise to the quinolone (V). The 7-fluoride group of (V) was then displaced by N-methylpiperazine (VI) in cold pyridine to furnish the piperazinyl quinolone (VII). Finally, ester hydrolysis in (VII) under acidic conditions yielded the target compound. In a closely related procedure, ester (V) was hydrolyzed to acid (VIII) using HCl. Subsequent displacement of the 7-fluoride of (VIII) with N-methylpiperazine (VI) provided the desired piperazinyl quinolone.
Synthesis, pharmacokinetics, and biological activity of a series of new pyridonecarboxylic acid antibacterial agents bearing a 5-fluoro-2-pyridyl group or a 3-fluoro-4-pyridyl group at N-1
J Heterocycl Chem 1997, 34(3): 1021

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....


























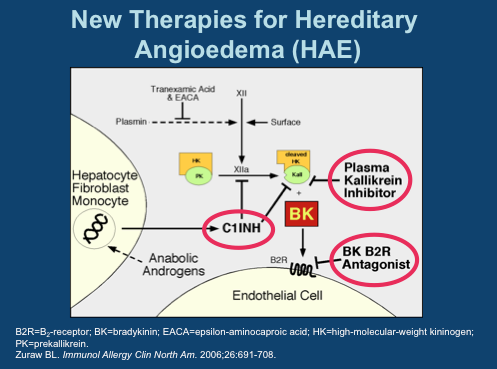
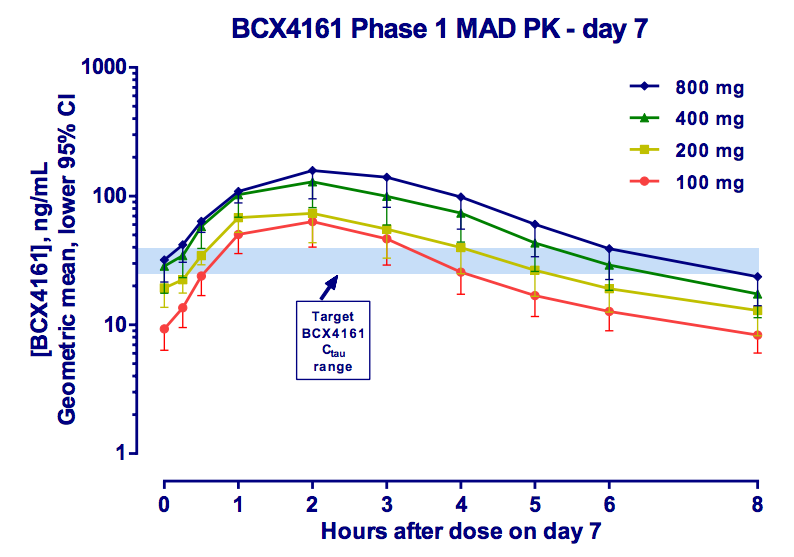
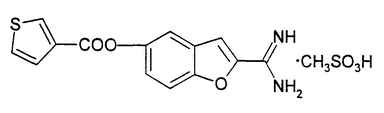
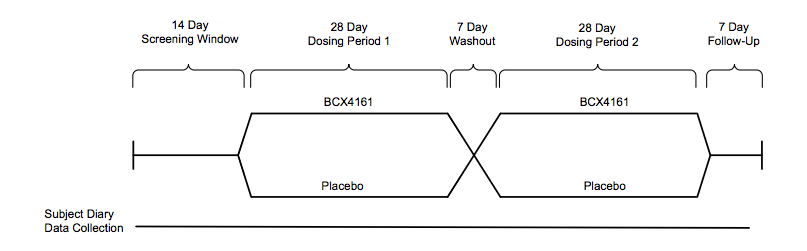
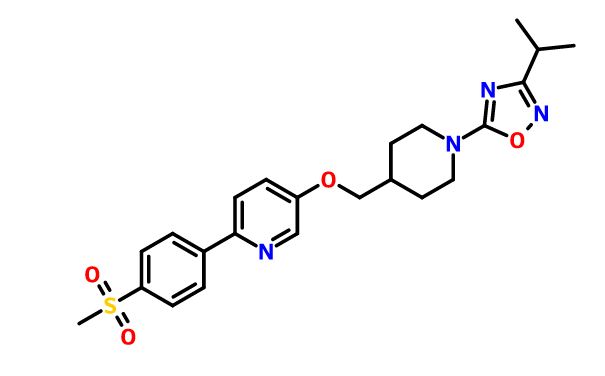




































 ELBASVIR
ELBASVIR


































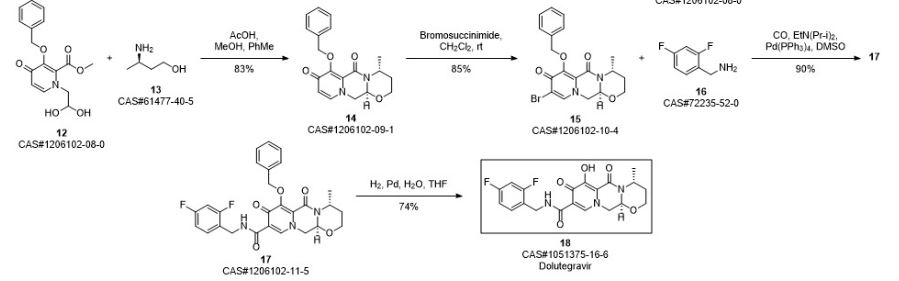







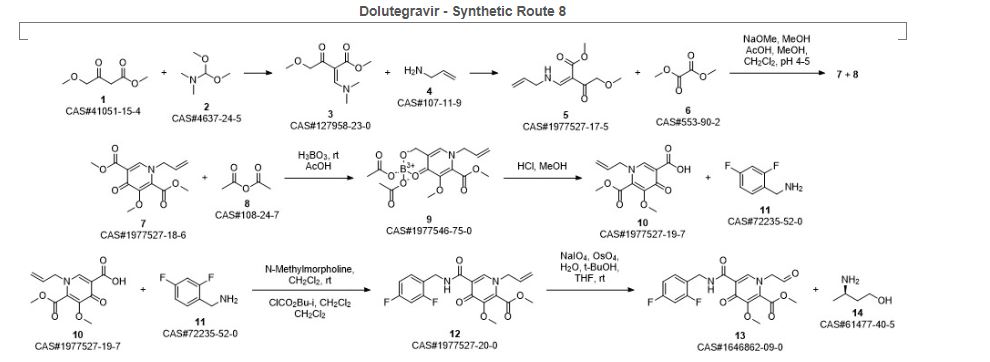

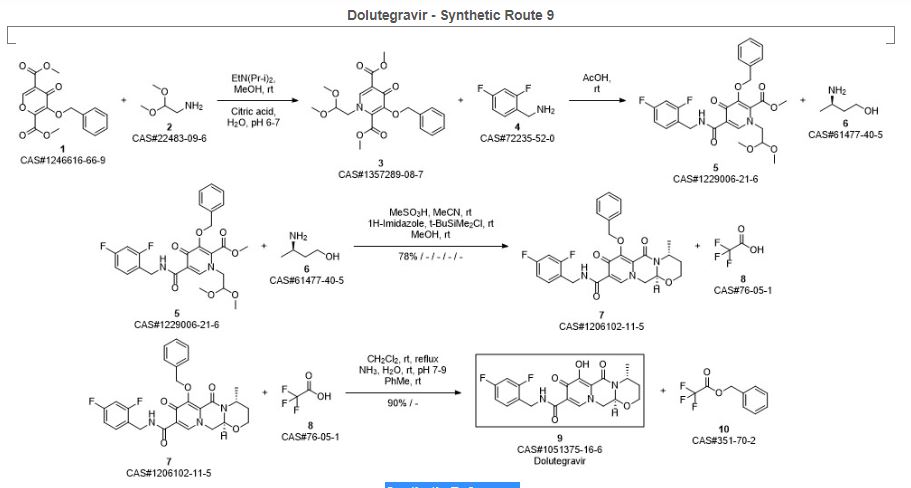



.svg){kind=link}
{kind=link}