
Home » Posts tagged 'phase 2' (Page 12)
Tag Archives: phase 2
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CEP 18770, Delanzomib

CEP-18770, Delanzomib
cas 847499-27-8
Chemical Formula: C21H28BN3O5
Exact Mass: 413.21220, UNII-6IF28942WO;
CT-47098
NPH 007098
NPH007098
[(1R)-1-[[(2S,3R)-3-Hydroxy-2-[[(6-phenylpyridin-2-yl)carbonyl]amino]-1-oxobutyl]amino]-3-methylbutyl]boronic acid
[(lR)-l-[[(2S,3R)-3-hydroxy-2- [6-phenyl-pyridine-2-carbonyl)amino]-l-oxobutyl]amino]-3-methylbutylboronic acid,
Boronic acid, ((1R)-1-(((2S,3R)-3-hydroxy-1-oxo-2-(((6-phenyl-2-pyridinyl)carbonyl)amino)butyl)amino)-3-methylbutyl)-
In phase 2, multiple mylenoma, Ethical Oncology Science (EOS), licensee
CEP-18770 was discovered through collaboration between Cephalon and Novuspharma/CTI.
Cephalon, Inc., 145 Brandywine Parkway, West Chester, Pennsylvania 19380, and Cell Therapeutics Europe S.r.l., Via L. Ariosto, 23, I-20091 Bresso, Italy
Cephalon was acquired by Teva in October 2011. In 2013, EOS was acquired by Clovis Oncology.
Chemical Process Research and Development, Teva Branded Pharmaceutical Products R&D Inc., 383 Phoenixville Pike, Malvern, Pennsylvania 19355, United States

CEP-18770 is a reversible P2 threonine boronic acid inhibitor of the chymotrypsin-like activity of the proteasome. Displays anti-multimyeloma (MM) effect.

HPLC………http://www.apexbt.com/downloader/document/A4009/HPLC.pdf
NMR………http://www.apexbt.com/downloader/document/A4009/NMR.pdf


CLICK ON IMAGE FOR CLEAR VIEW
Delanzomib, also known as CEP-18770, is An orally bioavailable synthetic P2 threonine boronic acid inhibitor of the chymotrypsin-like activity of the proteasome, with potential antineoplastic activity. Proteasome inhibitor CEP 18770 represses the proteasomal degradation of a variety of proteins, including inhibitory kappaBalpha (IkappaBalpha), resulting in the cytoplasmic sequestration of the transcription factor NF-kappaB; inhibition of NF-kappaB nuclear translocation and transcriptional up-regulation of a variety of cell growth-promoting factors; and apoptotic cell death in susceptible tumor cell populations. In vitro studies indicate that this agent exhibits a favorable cytotoxicity profile toward normal human epithelial cells, bone marrow progenitors, and bone marrow-derived stromal cells relative to the proteasome inhibitor bortezomib. The intracellular protein IkappaBalpha functions as a primary inhibitor of the proinflammatory transcription factor NF-kappaB
New series of dipeptidyl boronate inhibitors of 20S proteasome were identified to be highly potent drug-like candidates with IC50 values of 1.2 and 1.6 nM, respectively, which showed better activities than the drug bortezomib on the market
ref
The potent, selective, and orally bioavailable threonine-derived 20S human proteasome inhibitor that has been advanced to preclinical development, [(1R)-1-[ [ (2S,3R)- 3-hydroxy-2-[ (6-phenylpyridine- 2-carbonyl) amino]-1 -oxobutyl] amino]- 3-methylbutyl] boronic acid (CEP-18770, has been reported
ref .
Dorsey BD, Iqbal M, Chatterjee S, Menta E, Bernardini R, Bernareggi A, et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J Med Chem. 2008;51:1068–1072. [PubMed]
Further, the anti-multiple myeloma protea-some inhibitor CEP-18770 enhanced the anti-myeloma activity of bortezomib and melphalan. The combination of anti-multiple myeloma proteasome inhibitor CEP-18770 intravenously and bortezomib exhibited complete regression of bortezomib-sensitive tumours. Moreover, this combination markedly delayed progression of bortezomib-resistant tumours compared to treatment with either agent alone
Paper
Development and scale-up of an optimized route to the peptide boronic acid, CEP-18770
Org Process Res Dev 2013, 17(3): 422
http://pubs.acs.org/doi/abs/10.1021/op400010u
 USED AS PRODRUG
USED AS PRODRUGCEP-18770 is an unstable peptide boronic acid and an amorphous solid, making it a challenging synthetic target. Process R&D led to a new process that avoided chromatography through crystalline intermediates, increased atom and volume efficiency, provided a chromophore, and gave higher yields and purity. A stable, crystalline diethanolamine adduct was discovered that has the potential to be used as a prodrug.

Compound 8 proved to be a direct substitute for delanzomib in the formulation process. In the first step of the IV formulation process, delanzomib is dissolved in water along with several excipients. Predictably, the delanzomib degrades during this process. It was found that upon dissolution in the lyophilization medium, 8 hydrolyzes to delanzomib,
N-[(1S,2R)-1-[[[(1R)-1–1[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-methano-1,3,2-benzodioxaborol-2-yl]-3-methylbutyl]amino]carbonyl]-2-hydroxypropyl]-6-phenyl-2-pyridinecarboxamide (5)
PAPER
Discovery of a Potent, Selective, and Orally Active Proteasome Inhibitor for the Treatment of Cancer
http://pubs.acs.org/doi/abs/10.1021/jm7010589

The ubiquitin−proteasome pathway plays a central role in regulation of the production and destruction of cellular proteins. These pathways mediate proliferation and cell survival, particularly in malignant cells. The successful development of the 20S human proteasome inhibitor bortezomib for the treatment of relapsed and refractory multiple myeloma has established this targeted intervention as an effective therapeutic strategy. Herein, the potent, selective, and orally bioavailable threonine-derived 20S human proteasome inhibitor that has been advanced to preclinical development, [(1R)-1-[[(2S,3R)-3-hydroxy-2-[(6-phenylpyridine-2-carbonyl)amino]-1-oxobutyl]amino]-3-methylbutyl]boronic acid 20 (CEP-18770), is disclosed.
[(1R)-1-[[(2S,3R)-3-Hydroxy-2-[(6-phenylpyridine-2-carbonyl)amino]-1-oxobutyl]amino]-3-methylbutyl]boronic Acid (20)
Patent
http://www.google.com/patents/WO2010056733A1?cl=en
Preferred among these compounds is [(lR)-l-[[(2S,3R)-3-hydroxy-2- [6-phenyl-pyridine-2-carbonyl)amino]-l-oxobutyl]amino]-3-methylbutylboronic acid, also known as CEP- 18770, which has the following structure:

PATENT
http://www.google.co.in/patents/WO2005021558A2
NOT SAME BUT SIMILAR
Example E.4 Boronic acid, [(lR)-l-[[(2S,3R)-3-hydroxy-2-[[4-(3-pyridyl)benzoyl]amino]-l- oxobutyI]amino]-3-methyIbutyl].

[00275] A mixture of 4-(pyridin-3-yl)benzamide, N-[(1S,2R)-1-[[[(1R)-1-
[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-methano-l,3,2-benzodioxaborol-2- yl]-3-methylbutyl]amino]carbonyl]-2-hydroxypropyl]- of Example D.8.3 (155 mg, 0.283 mmol), 2-methylpropylboronic acid (81 mg, 0.793 mmol) and 2N aqueous hydrochloric acid (0.3 ml) in a heterogeneous mixture of methanol (3 ml) and hexane (3 ml) was stirred at room temperature for 24 hours. The hexane layer was removed and the methanolic layer was washed with fresh hexane (about 5 ml). Ethyl acetate (10 ml) was added to the methanol layer which was then concentrated. The residue was taken up with ethyl acetate and the mixture was concentrated. This step was repeated (2-3 times) until an amorphous white solid was obtained. The solid was then triturated with diethyl ether (5 ml) and the surnatant was removed by decantation. This step was repeated. The residue (126 mg) was combined with the product of a similar preparation (140 mg) and dissolved in ethyl acetate (about 40 ml) and a small amount of methanol (2-3 ml). The solution was washed with a mixture of NaCl saturated solution (7 ml) and 10% NaHCO3 (2 ml). The layers were separated and the aqueous phase was further washed with ethyl acetate (2 x 20 ml). The combined organic phases were dried over sodium sulfate and concentrated. The residue was taken up with ethyl acetate (about 20 ml) and the minimum amount of methanol, and then concentrated to small volume (about 5 ml). The resulting white was collected by filtration and dried under vacuum at 50°C (160 mg, 65% overall yield).
1H NMR (MeOH-d4): 8.90 (IH, s); 8.49 (IH, d, J=4.0); 8.20 (IH, d, J=8.1); 8.06 (2H, d, J=8.1); 7.85 (2H, d, J=8.1); 7.58 (IH, t br., J=6.0); 4.80 (IH, d, J=3.9); 4.40-4.29 (IH, m); 2.78 (IH, t, J=7.5); 1.73-1.61 (IH, m); 1.38 (2H, t, J=6.9); 1.31 (3H, d, J=6.3); 0.94 (6H, d, J=6.31). [00276] Further compounds prepared according to the above procedure for
Example E.4 are reported in Table E-4. Table E-4

E.4.3 IS THE COMPD
D.8.12 Chemical Name: 6-Phenyl-2-pyridinecarboxamide,N-[(lS,2R)-l-[[[(lR)- l-[(3aS,4S,6S,7aR)-hexahydro-3a,5,5-trimethyl-4,6-
 THIS IS PRECURSOR OF FINAL PDT
THIS IS PRECURSOR OF FINAL PDTmethano-l,3,2-benzodioxaborol-2-yl]-3- methylbutyl]amino]carbonyl]-2-hydroxypropyl]. Analytical Data: Η -NMR (DMSO-d6): 9.20-8.95 (IH, m); 8.76 (IH, d, J=8.55 Hz); 8.26-8.16 (4H, m); 8.12 (IH, t, J= 7.77 Hz); 8.02 (IH, d, J= 7.56 Hz); 7.60-7.47 (4H, m); 5.27 (IH, d, J= 4.97 Hz); 4.50 (IH, dd, J= 4.22 Hz, J= 8.50 Hz); 4.16-4.07 (2H, m); 2.65-2.56 (IH, m); 2.25-2.15 (IH, m); 2.09-1.98 (IH, m); 1.84 (IH, t, J= 5.62 Hz); 1.79- 1.73 (IH, m); 1.73-1.66 (IH, m); 1.66-1.59 (IH, m); 1.40-1.26 (4H, m); 1.23 (7H, d, J= 10.89 Hz); 1.15-1.10 (4H, m); 0.85 (7H, d, J= 6.56 Hz); 0.79 (IH, bs).
|
References |
1. Fuchs, Ota. Proteasome inhibition as a therapeutic strategy in patients with multiple myeloma. Multiple Myeloma (2009), 101-125. CODEN: 69MVM2 AN 2010:737549
2. Genin, E.; Reboud-Ravaux, M.; Vidal, J. Proteasome inhibitors: recent advances and new perspectives in medicinal chemistry. Current Topics in Medicinal Chemistry (Sharjah, United Arab Emirates) (2010), 10(3), 232-256. CODEN: CTMCCL ISSN:1568-0266. CAN 152:516315 AN 2010:423458
3. Sanchez, Eric; Li, Mingjie; Steinberg, Jeffrey A.; Wang, Cathy; Shen, Jing; Bonavida, Benjamin; Li, Zhi-Wei; Chen, Haiming; Berenson, James R. The proteasome inhibitor CEP-18770 enhances the anti-myeloma activity of bortezomib and melphalan. British Journal of Haematology (2010), 148(4), 569-581. CODEN: BJHEAL ISSN:0007-1048. AN 2010:353952
4. Dick, Lawrence R.; Fleming, Paul E. \Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discovery Today (2010), 15(5/6), 243-249. CODEN: DDTOFS ISSN:1359-6446. AN 2010:318415
5. Ruggeri, Bruce; Miknyoczki, Sheila; Dorsey, Bruce; Hui, Ai-Min. The development and pharmacology of proteasome inhibitors for the management and treatment of cancer. Advances in Pharmacology (San Diego, CA, United States) (2009), 57(Contemporary Aspects of Biomedical Research: Drug Discovery), 91-135. CODEN: ADPHEL ISSN:1054-3589. AN 2010:62762
6. Chen-Kiang, Selina; Di Liberto, Maurizio; Huang, Xiangao. Targeting CDK4 and CDK6 kinases or genes thereof in cancer therapy for sensitizing drug-resistant tumors. PCT Int. Appl. (2009), 149pp. CODEN: PIXXD2 WO 2009061345 A2 20090514 CAN 150:531264 AN 2009:586623
7. Rickles, Richard; Lee, Margaret S. Use of adenosine A2A receptor agonists and phosphodiesterase (PDE) inhibitors for the treatment of B-cell proliferative disorders, and combinations with other agents. PCT Int. Appl. (2009), 70 pp. CODEN: PIXXD2 WO 2009011893 A2 20090122 CAN 150:160095 AN 2009:86451
8. Rickles, Richard; Pierce, Laura; Lee, Margaret S. Combinations for the treatment of B-cell proliferative disorders. PCT Int. Appl. (2009), 79pp. CODEN: PIXXD2 WO 2009011897 A1 20090122 CAN 150:160094 AN 2009:83374
9. Hoveyda, Hamid; Fraser, Graeme L.; Benakli, Kamel; Beauchemin, Sophie; Brassard, Martin; Drutz, David; Marsault, Eric; Ouellet, Luc; Peterson, Mark L.; Wang, Zhigang. Preparation and methods of using macrocyclic modulators of the ghrelin receptor. U.S. Pat. Appl. Publ. (2008), 178pp. CODEN: USXXCO US 2008194672 A1 20080814 CAN 149:288945 AN 2008:975261
10. Piva, Roberto; Ruggeri, Bruce; Williams, Michael; Costa, Giulia; Tamagno, Ilaria; Ferrero, Dario; Giai, Valentina; Coscia, Marta; Peola, Silvia; Massaia, Massimo; Pezzoni, Gabriella; Allievi, Cecilia; Pescalli, Nicoletta; Cassin, Mara; di Giovine, Stefano; Nicoli, Paola; de Feudis, Paola; Strepponi, Ivan; Roato, Ilaria; Ferracini, Riccardo; Bussolati, Benedetta; Camussi, Giovanni; Jones-Bolin, Susan; Hunter, Kathryn; Zhao, Hugh; Neri, Antonino; Palumbo, Antonio; Berkers, Celia; Ovaa, Huib; Bernareggi, Alberto; Inghirami, Giorgio. CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood (2008), 111(5), 2765-2775. CODEN: BLOOAW ISSN:0006-4971. CAN 149:486154 AN 2008:292777
11. Dorsey, Bruce D.; Iqbal, Mohamed; Chatterjee, Sankar; Menta, Ernesto; Bernardini, Raffaella; Bernareggi, Alberto; Cassara, Paolo G.; D’Arasmo, Germano; Ferretti, Edmondo; De Munari, Sergio; Oliva, Ambrogio; Pezzoni, Gabriella; Allievi, Cecilia; Strepponi, Ivan; Ruggeri, Bruce; Ator, Mark A.; Williams, Michael; Mallamo, John P. Discovery of a Potent, Selective, and Orally Active Proteasome Inhibitor for the Treatment of Cancer. Journal of Medicinal Chemistry (2008), 51(4), 1068-1072. CODEN: JMCMAR ISSN:0022-2623. CAN 148:345774 AN 2008:146611
12. Dorsey, Bruce D.; Menta, Ernesto; Bernardini, Raffaella; Bernareggi, Alberto; Casara, Paolo G.; D’Arasmo, Germano; Ferretti, Edmondo; De Munari, Sergi; Oliva, Ambrogio; Iqbal, Mohamed; Chatterjee, Sankar; Ruggeri, Bruce; Ator, Mark A.; Williams, Michael; Mallamo, John P. CEP-18770: Discovery of a Potent, Selective and Orally Active Proteasome Inhibitor for the Treatment of Cancer. Frontiers in CNS and Oncology Medicinal Chemistry, ACS-EFMC, Siena, Italy, October 7-9 (2007), COMC-027. CODEN: 69KAR2 AN 2007:1171000
13. Marblestone Jeffrey G Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs : the investigational drugs journal (2009), 12(12), 750-3.
| Patent | Submitted | Granted |
|---|---|---|
| Proteasome inhibitors and methods of using the same [US7576206] | 2005-05-19 | 2009-08-18 |
| PROTEASOME INHIBITORS AND METHODS OF USING THE SAME [US7915236] | 2009-11-26 | 2011-03-29 |
| BORONATE ESTER COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF [US2009325903] | 2009-12-31 |
| US7442830 * | 6 Aug 2007 | 28 Oct 2008 | Millenium Pharmaceuticals, Inc. | Proteasome inhibitors |
| US7687662 * | 2 Jul 2008 | 30 Mar 2010 | Millennium Pharmaceuticals, Inc. | Proteasome inhibitors |
| US8003819 * | 12 Feb 2010 | 23 Aug 2011 | Millennium Pharmaceuticals, Inc. | Proteasome inhibitors |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8962572 | 4 Oct 2011 | 24 Feb 2015 | Fresenius Kabi Usa, Llc | Bortezomib formulations |
| WO2012177835A1 | 21 Jun 2012 | 27 Dec 2012 | Cephalon, Inc. | Proteasome inhibitors and processes for their preparation, purification and use |
/////CEP-18770, delanzomib
B(C(CC(C)C)NC(=O)C(C(C)O)NC(=O)C1=CC=CC(=N1)C2=CC=CC=C2)(O)O
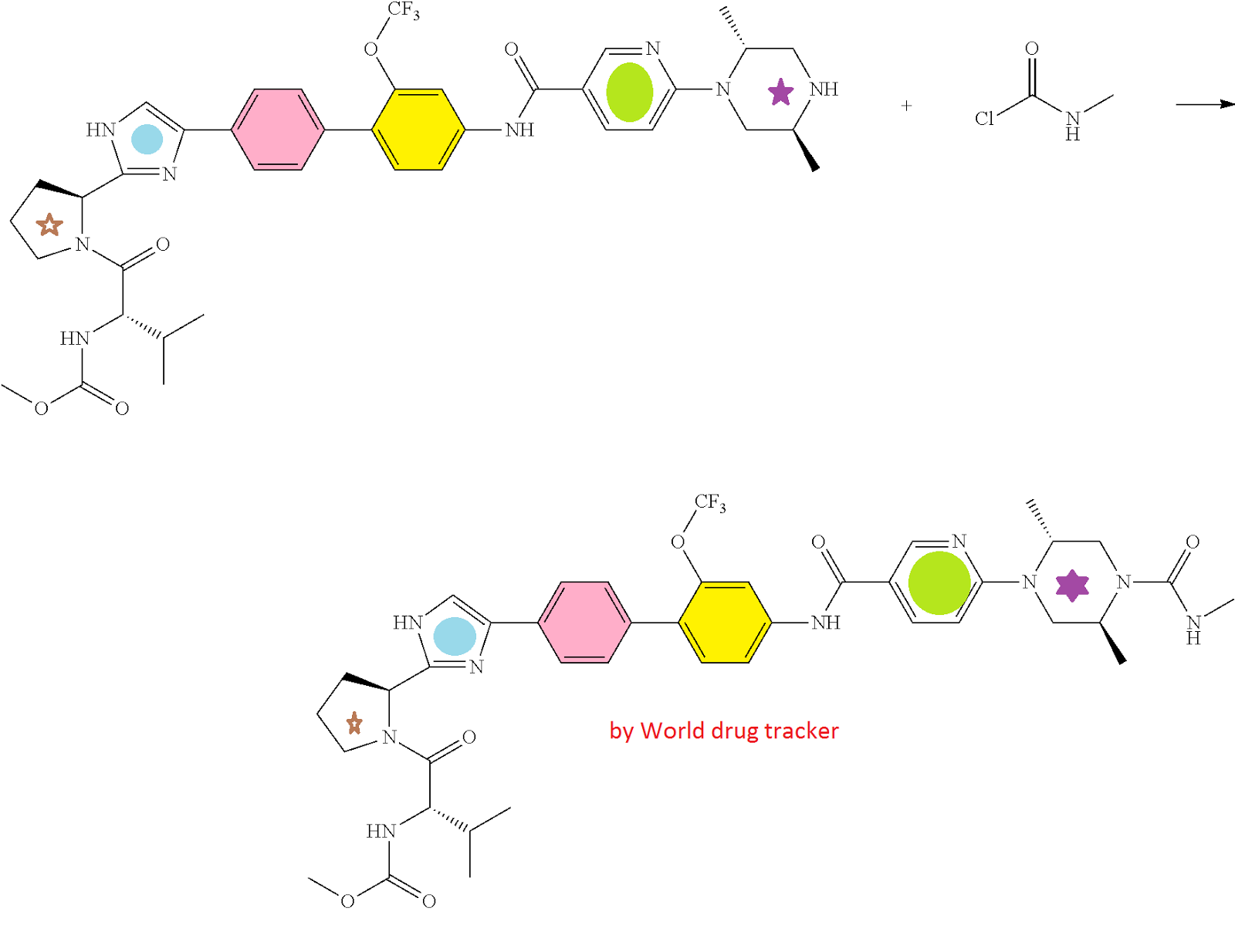
HCV NS5A Inhibitor from Theravance, Inc. to treat hepatitis C virus infection

((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
N-[(1S)-1-[[(2S)-2-[5-[4′-[[[6-[(2R,5S)-2,5-dimethyl-4-[(methylamino)carbonyl]-1-piperazinyl]-3-pyridinyl]carbonyl]amino]-2′-(trifluoromethoxy)[1,1′-biphenyl]-4-yl]-1H-imidazol-2-yl]-1-pyrrolidinyl]carbonyl]-2-methylpropyl]-, Carbamic acid, methyl ester
Carbamic acid, N-[(1S)-1-[[(2S)-2-[5-[4′-[[[6-[(2R,5S)-2,5-dimethyl-4-[(methylamino)carbonyl]-1-piperazinyl]-3-pyridinyl]carbonyl]amino]-2′-(trifluoromethoxy)[1,1′-biphenyl]-4-yl]-1H-imidazol-2-yl]-1-pyrrolidinyl]carbonyl]-2-methylpropyl]-, methyl ester
CAS 1374883-22-3, 819.87, C41 H48 F3 N9 O6
CAS of DIHCl 1480448-59-6
CAS of DIHCl, H2O 1480448-63-2
Theravance, Inc. INNOVATOR
To treat hepatitis C virus infection

-
((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (compound 1):
-
 Recent estimates place the number of people infected with the hepatitis C virus (HCV) worldwide at more than 170 million, including 3 million people in the United States. The infection rate is thought to be roughly 4 to 5 times that of the human immunodeficiency virus (HIV). While in some individuals, the natural immune response is able to overcome the virus, in the majority of cases, a chronic infection is established, leading to increased risk of developing cirrhosis of the liver and hepatocellular carcinomas. Infection with hepatitis C, therefore, presents a serious public health problem.The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.Commonly-assigned U.S. Provisional Application Nos. 61/410,267, filed on Nov. 4, 2010, 61/444,046, filed on Feb. 17, 2011, and 61/492,267, filed on Jun. 1, 2011, and U.S. application Ser. No. 13/288,216, filed on Nov. 3, 2012 disclose pyridyl-piperazinyl compounds
Recent estimates place the number of people infected with the hepatitis C virus (HCV) worldwide at more than 170 million, including 3 million people in the United States. The infection rate is thought to be roughly 4 to 5 times that of the human immunodeficiency virus (HIV). While in some individuals, the natural immune response is able to overcome the virus, in the majority of cases, a chronic infection is established, leading to increased risk of developing cirrhosis of the liver and hepatocellular carcinomas. Infection with hepatitis C, therefore, presents a serious public health problem.The virus responsible for HCV infection has been identified as a positive-strand RNA virus belonging to the family Flaviviridae. The HCV genome encodes a polyprotein that during the viral lifecycle is cleaved into ten individual proteins, including both structural and non-structural proteins. The six non-structural proteins, denoted as NS2, NS3, NS4A, NS4B, NS5A, and NS5B have been shown to be required for RNA replication. In particular, the NS5A protein appears to play a significant role in viral replication, as well as in modulation of the physiology of the host cell. Effects of NS5A on interferon signaling, regulation of cell growth and apoptosis have also been identified. (Macdonald et al., Journal of General Virology (2004), 85, 2485-2502.) Compounds which inhibit the function of the NS5A protein are expected to provide a useful approach to HCV therapy.Commonly-assigned U.S. Provisional Application Nos. 61/410,267, filed on Nov. 4, 2010, 61/444,046, filed on Feb. 17, 2011, and 61/492,267, filed on Jun. 1, 2011, and U.S. application Ser. No. 13/288,216, filed on Nov. 3, 2012 disclose pyridyl-piperazinyl compounds

SYNTHESIS
CLICK ON IMAGES FOR CLEAR VIEW
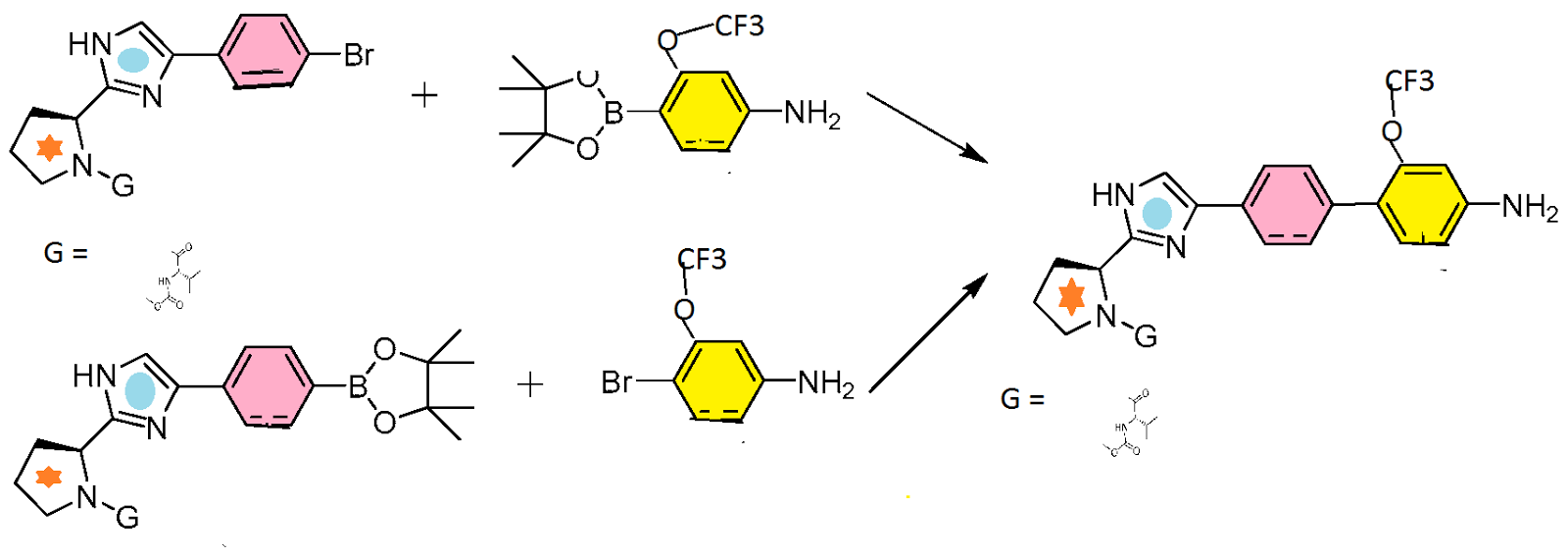
………………………..
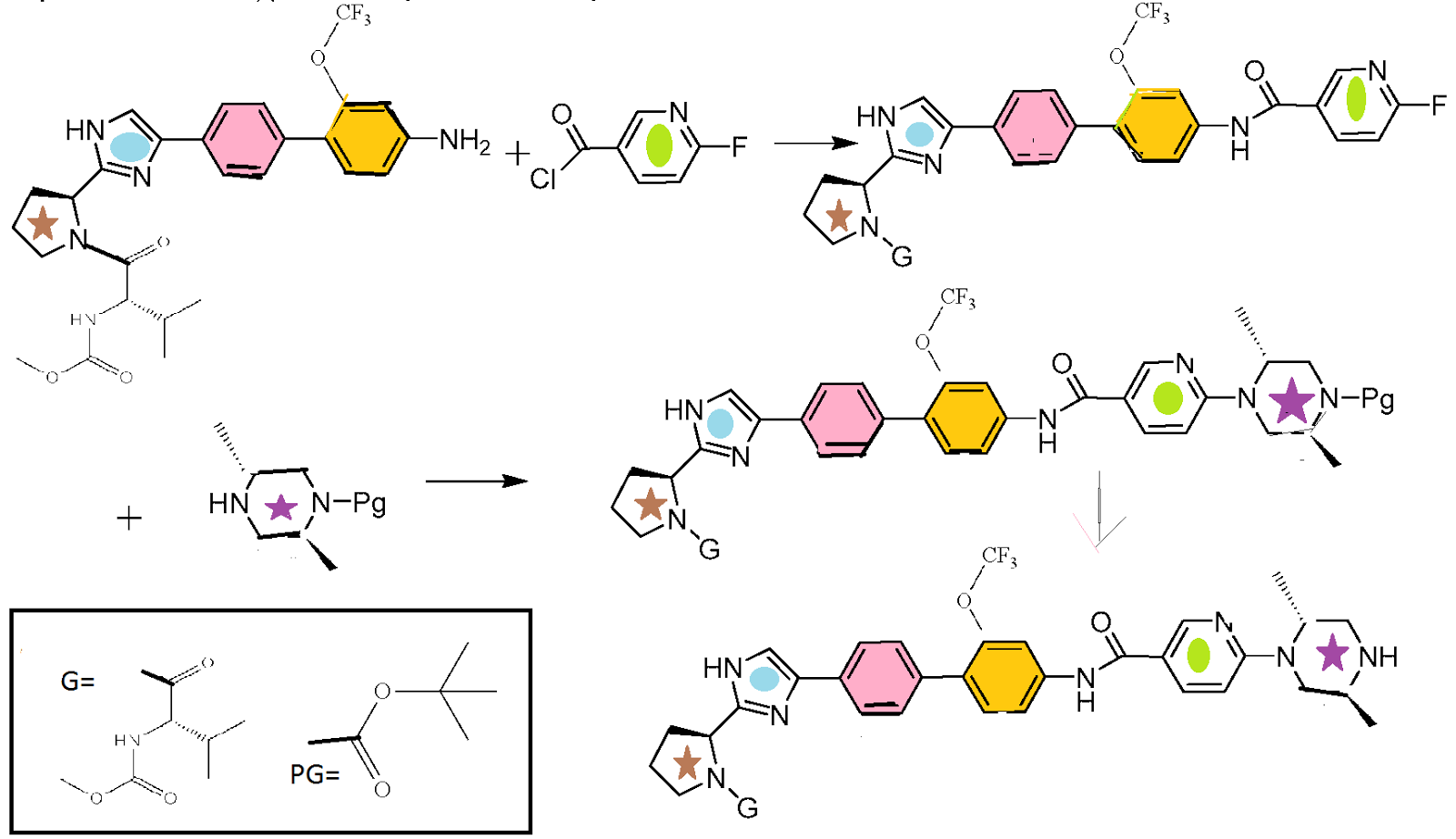
……………………………….
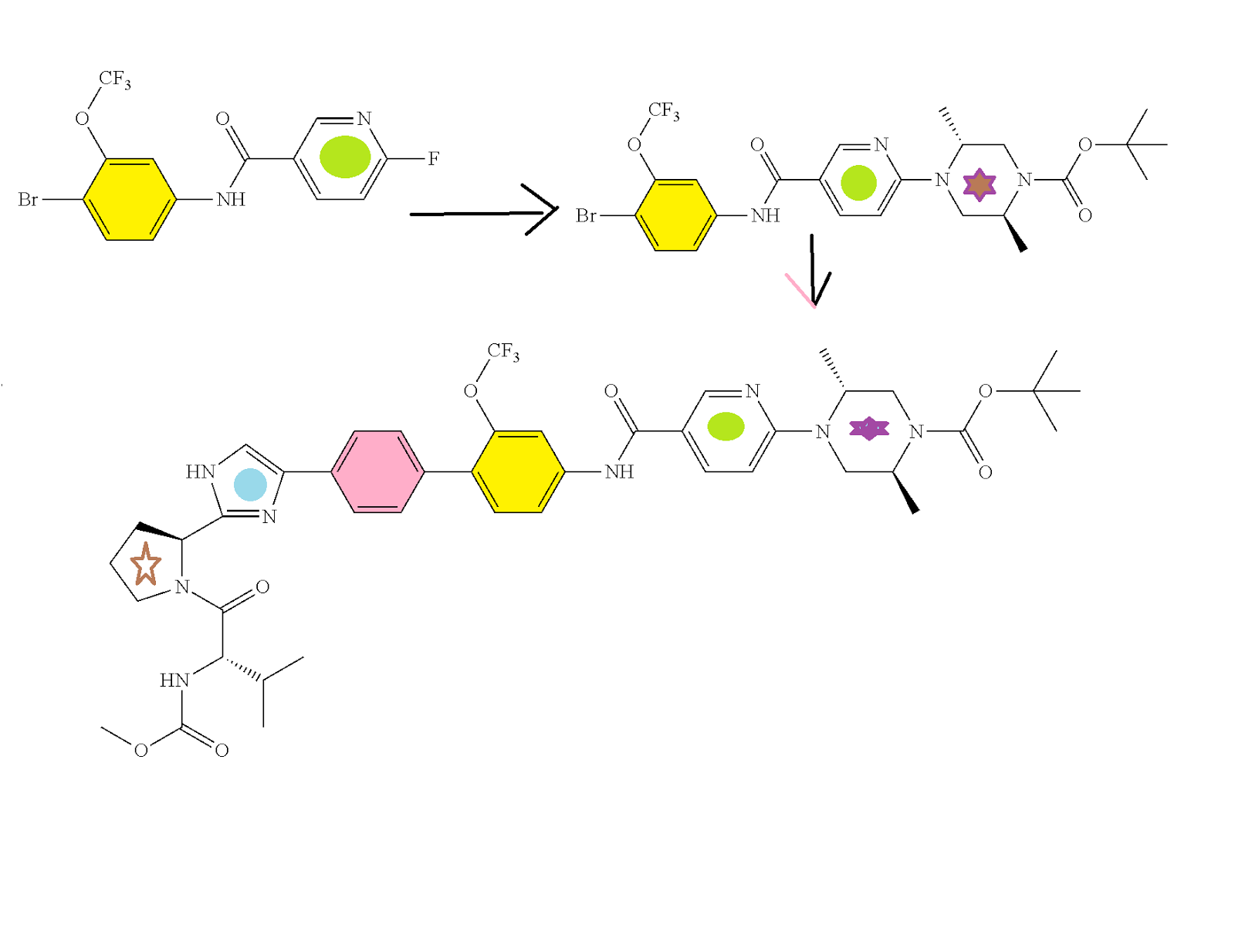
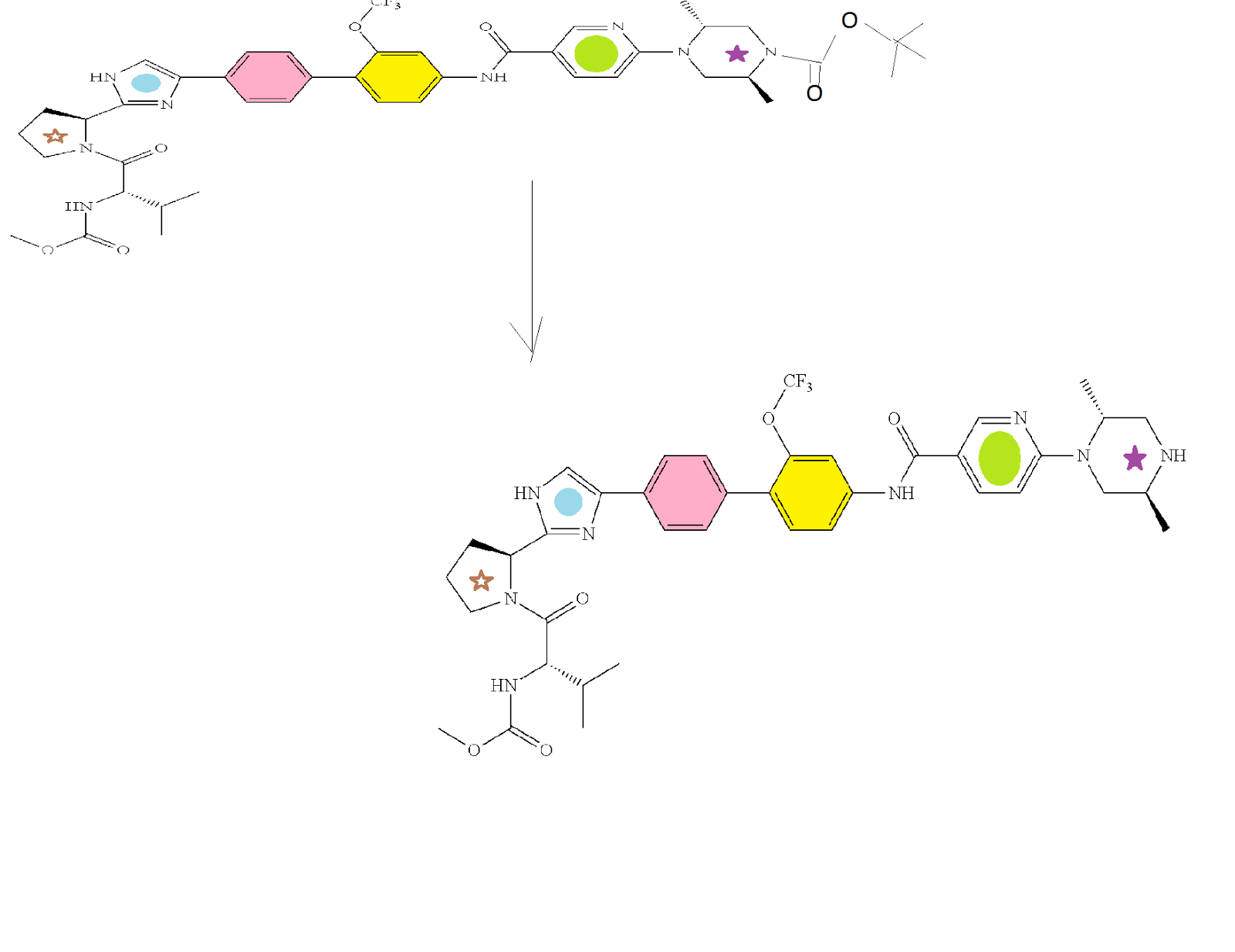
PATENT
WO-2013/165796
https://www.google.co.in/patents/WO2013165796A1?cl=en
PATENT
http://www.google.com/patents/US20130295048
crystalline compound 1 is advantageously prepared directly from the crude product of the final step of the synthesis of compound 1, illustrated in the following scheme, without purification of the amorphous form.

As described in Example 3 below, ((S)-1-{(S)-2-[4-(4′-{[6-(2R,5S)-2,5-dimethyl-piperazin-1-yl)-pyridine-3-carbonyl}-amino]-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (2) is reacted with methylaminoformyl chloride to provide a crude product, which is recovered by conventional extraction and drying. The reaction is typically performed in the presence of an excess of base, in an inert diluent such as dichloromethane. Next, methanol is added to the crude product followed by the slow addition of water in a ratio of methanol:water of about 2.5:1 to about 2.7:1 to form a crystallization mixture. Seeds of crystalline compound 1 are added about halfway through the water addition. The crystallization mixture is stirred for a period of several days to form crystalline compound 1. To increase purity, the product can be recrystallized by a similar process: the crystalline compound is dissolved in methanol, water and seeds are added, such that the ratio of methanol to water in the mixture is about 2.5:1, and the mixture is stirred for a period of at least 12 hours to provide crystalline compound 1, which is recovered conventionally
-

-
To a solution of 4-bromo-3-trifluoromethoxy-phenylamine (3.15 g, 12.3 mmol) and triethylamine (3.43 mL, 24.6 mmol) in DCM (25 mL) was slowly added a solution of 2-fluoropyridine-5-carbonyl chloride (2.36 g, 14.8 mmol) in DCM (10 mL). After 2 h at RT, MTBE (90 mL) was added and the reaction mixture was washed with water, brine, and saturated sodium carbonate, dried, and evaporated to give a solid (5.4 g). Ethanol (43 mL) was added to the solid and then water (43 mL) was slowly added. The reaction mixture was stirred for 1.5 h, filtered, and washed with 1:4 ethanol:water (2×25 mL) to give the title intermediate as a white solid (3.87 g). Analytical HPLC: Retention time=21.3 min.
- Preparation 1: (2S,5R)-4-[5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester(a) N-(4-Bromo-3-trifluoromethoxy-phenyl)-6-fluoro-nicotinamide

(b) (2S,5R)-4-[5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester
-

-
The product of the previous step (3.86 g, 10.2 mmol) (2S,5R)-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (2.62 g, 12.2 mmol) and N,N-diisopropylethylamine (5.32 mL, 30.5) was dissolved in DMSO (12 mL). The reaction mixture heated at 120° C. for 3 h, diluted with EtOAc (100 mL), washed with water, and saturated NH4Cl, water, and brine. The reaction mixture was evaporated to about 40% volume and 3 M HCl in cyclopentyl methyl ether (4.24 mL, 12.7 mmol) was added slowly. Seeds from a previous run at smaller scale were added and the reaction mixture was stirred for 2 days and filtered to provide the HCl salt of the title intermediate (5.15 g, 83% yield). Analytical HPLC: Retention time=21.1 min.

Preparation 2: (2S,5R)-4-[5-(4′-{2-[(S)-1-((S)-2-Methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester
-

-
To a solution of ((S)-1-{(S)-2-[4-(4-bromo-phenyl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (3.05 g, 6.8 mmol), bis(pinacolato)diboron (1.81 g, 7.1 mmol) and potassium acetate (1.00 g, 10.2 mmol) was added nitrogen sparged toluene (15 mL). The resulting mixture was sparged with nitrogen and 1,1′-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane (Pd catalyst) (0.17 g, 0.204 mmol) was added. The reaction mixture was stirred at 90° C. overnight.
-
The reaction mixture was cooled to RT and to this mixture was added nitrogen sparged water (7.6 mL), potassium carbonate (5.16 g, 37.3 mmol), and (2S,5R)-4-[5-(4-bromo-3-trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (4.35 g, 7.13 mmol). The reaction mixture was stirred at 95° C. overnight.
-
Another portion of the Pd catalyst used above (0.08 g, 0.10 mmol) was added to the reaction mixture. After 5 h, the reaction mixture was cooled to RT, diluted with EtOAc (150 mL), washed with water (150 mL) and brine (100 mL), dried over sodium sulfate, and evaporated to give a black residue (6.7 g), which was purified by silica gel chromatography (eluted with 50-100% EtOAc/hexane) to provide the title intermediate (5.3 g, 90% yield). Analytical HPLC: Retention time=14.7 min.

Preparation 3: ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
-

-
Acetyl chloride (63.2 mL, 888 mmol) was added to ethanol (360 mL) and stirred at RT for 1 h. To the resulting HCl solution was added a solution of (2S,5R)-4-[5-(4′-{2-[(S)-1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (73 g, 84 mmol) in ethanol (360 mL). The reaction mixture was stirred at RT overnight.
-
The reaction mixture was concentrated to dryness (124 g crude). Water 500 mL) was added and the mixture was extracted with EtOAc (2×500 mL). The aqueous layer was adjusted to pH 4 with 1:1 NaOH:water. Ethyl acetate (400 mL) and sat. aq. Na2CO3 (100 mL) were added and the layers were separated. The organic layer was dried over Na2SO4 and evaporated to give the title intermediate (62.8 g; 88% yield). Analytical HPLC: Retention time=10.0 min.

Example 1Amorphous ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

(a) ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester tri-HCl
-
Acetyl chloride (0.71 mL, 10.0 mmol) was added to ethanol (7 mL) and stirred at RT for 1 h. The resulting HCl solution was added to a solution of (2S,5R)-4-[5-(4′-{2-[(5)-1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-2-trifluoromethoxy-biphenyl-4-ylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-1-carboxylic acid tert-butyl ester (1.55 g, 1.8 mmol) in ethanol (7 mL). The reaction mixture was warmed to 35° C. and stirred overnight. The mixture was concentrated to dryness, and chased with DCM to provide the crude tri-HCl salt of the title intermediate (1.57 g) which was used directly in the next step. HPLC method C: Retention time=10.0 min.
(b) Amorphous ((S)-1-{(S)-2-[4-(4′-{[6-((2R,5S)-2,5-Dimethyl-4-methylcarbamoyl-piperazin-1-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-1H-imidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester
-
To a solution of the product of the previous step (1.57 g crude, ca. 1.80 mmol) and N,N-diisopropylethylamine (3.14 mL, 18.0 mmol) in DCM (24 mL) was slowly added 1 M methylaminoformyl chloride in DMA (1.8 mL). The reaction mixture stirred at RT for 1 h, and then 1 M methylaminoformyl chloride in DMA (1.8 mL) was added. The reaction was quenched with sat. aq. NaHCO3 and the reaction mixture was stirred for 20 min. The layers were separated and the organic layer was dried and evaporated to give a residue. To the residue was added methanol (15 mL) followed by 2 N LiOH/water (3 mL). The reaction mixture was stirred at RT for 1 h, diluted with water, extracted with DCM (80 mL), dried, and evaporated to give a crude product which was purified by silica gel chromatography (40 g silica, 2-8% MeOH/DCM) to provide the title compound (0.93 g, 63% yield). Analytical HPLC: Retention time=11.0 min.
HPLC
- Analytical HPLC Method
-
-
- Column: Zorbax Bonus-RP 3.5 μm. 4.6×150 mm
- Column temperature: 35° C.
- Flow rate: 1.0 mL/min
- Mobile Phases: A=Water/ACN (98:2)+0.1% TFA
- B=Water/ACN (10:90)+0.1% TFA,
- Injection volume: 100-1500 μL
- Detector wavelength: 214 nm
- Sample preparation: Dissolve in 1:1 ACN:water
- Gradient: 29 min total (time (min)/% B): 0.5/10, 24/90, 25/90, 26/10, 29/10
-
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010094977A1 * | 22 Feb 2010 | 26 Aug 2010 | Arrow Therapeutics Limited | Novel biphenyl compounds useful for the treatment of hepatitis c |
| WO2012061552A1 * | 3 Nov 2011 | 10 May 2012 | Theravance, Inc. | Novel inhibitors of hepatitis c virus |
| US201113288216 |
About Theravance Biopharma
The mission of Theravance Biopharma (NASDAQ: TBPH) is to create value from a unique and diverse set of assets: an approved product; a development pipeline of late-stage assets; and a productive research platform designed for long-term growth.
Our pipeline of internally discovered product candidates includes potential best-in-class opportunities in underserved markets in the acute care setting, representing multiple opportunities for value creation. VIBATIV® (telavancin), our first commercial product, is a once-daily dual-mechanism antibiotic approved in the U.S., Europe and certain other countries for certain difficult-to-treat infections. Revefenacin (TD-4208) is an investigational long-acting muscarinic antagonist (LAMA) being developed as a potential once-daily, nebulized treatment for COPD. Axelopran (TD-1211) is an investigational potential once-daily, oral treatment for opioid-induced constipation (OIC). Our earlier-stage clinical assets represent novel approaches for potentially treating diseases of the lung and gastrointestinal tract and infectious disease. In addition, we have an economic interest in future payments that may be made by GlaxoSmithKline plc pursuant to its agreements with Theravance, Inc. relating to certain drug development programs, including the combination of fluticasone furoate, umeclidinium and vilanterol (the “Closed Triple”).
With our successful drug discovery and development track record, commercial infrastructure, experienced management team and efficient corporate structure, we believe that we are well positioned to create value for our shareholders and make a difference in the lives of patients.
For more information, please visit www.theravance.com.
THERAVANCE®, the Cross/Star logo, MEDICINES THAT MAKE A DIFFERENCE® and VIBATIV® are registered trademarks of the Theravance Biopharma group of companies.
Journal of Medicinal Chemistry (2014), 57(5), 1643-1672……….

(e)Thalladi, V. R.; Nzerem, J.; Huang, X.; Zhang, W. Crystalline form of a pyridyl-piperazinyl hepatitis C virus inhibitor. World Patent Application WO-2013/165796, November 7, 2013.
PATENT
WO 2012061552
http://www.google.com.ar/patents/WO2012061552A1?cl=en
Preparation 28: ((S)-l-{(S)-2-[4-(4′-{[6-((2JR,5S)-2,5-Dimethyl-piperazin-l- yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2- yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

A mixture of [(5)-2-methyl-l-((5)-2- {4-[4-(4,4,5,5-tetramethyl- [l,3,2]dioxaborolan-2-yl)-phenyl]-lH-imidazol-2-yl}-pyrrolidine-l-carbonyl)-propyl]- carbamic acid methyl ester (86 mg, 0.17 mmol) and (25′,5R)-4-[5-(4-bromo-3- trifluoromethoxy-phenylcarbamoyl)-pyridin-2-yl]-2,5-dimethyl-piperazine-l-carboxylic acid tert-butyl ester (100 mg, 0.2 mmol, Preparation 27) was dissolved in 1,4-dioxane (1.8 mL, 23 mmol) and water (0.25 mL, 14 mmol). Cesium carbonate (170 mg, 0.52 mmol) was added. The reaction mixture was sparged with nitrogen and then
tetrakis(triphenylphosphine)palladium(0) (12.1 mg, 0.011 mmol) was added. The reaction mixture was sealed under nitrogen and heated at 95 °C overnight. The reaction mixture was extracted with ethyl acetate/water, the organic layer was dried over sodium sulfate and concentrated to produce an orange oil.
The oil from the previous step was treated with 4 M HCl in 1,4-dioxane (2 mL, 7 mmol) and stirred at room temperature for 1 h. The reaction mixture was concentrated and evaporated with ethyl acetate (2 x) to produce the HCl salt of the title compound as a yellow solid which was purified by preparative HPLC to provide the tri-TFA salt of the title compound (150 mg, 30 % overall yield), (m/z): [M+H] calcd for
763.35 found 763.7.
Example 29 ((S)-l-{(S)-2-[4-(4′-{[6-((2JR,5S)-2,5-Dimethyl-4- methylcarbamoyl-piperazin-l-yl)-pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy- biphenyl-4-yl)-lH-imidazol-2-yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)- carbamic a

To a solution of ((5)- l – {(5)-2-[4-(4′- { [6-((2R,55)-2,5-dimethyl-piperazin- l-yl)- pyridine-3-carbonyl]-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl]- pyrrolidine- l-carbonyl} -2-methyl-propyl)-carbamic acid methyl ester tri-TFA (1 1.4 mg, 0.01 1 mmol; Preparation 28) and N,N-diisopropylethylamine (18 uL, 0.1 1 mmol) dissolved in DMA (0.4 mL, 4 mmol) was added 1.0 M methyl isocyanate in toluene (10 uL, 0.01 mmol). The reaction mixture was stirred at RT overnight, concentrated, dissolved in 1 : 1 acetic acid:water (1.5 mL) and purified by preparative HPLC to provide the di-TFA salt of the title compound (7.1 mg). (m/z): [M+H]+ calcd for C41H48F3N906 820.37 found 820.5.
Alternative synthesis of ((5)-1-{(5)-2-[ -(4′-{[6-((2Λ,ί» 2,5- Dimethyl-4-methylcarbamoyl-piperazin-l-yl)-pyridine-3-carbonyl]-amino}-2′- trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl]-pyrrolidine-l-carbonyl}-2- methyl-propyl)-carbamic acid methyl ester
(a) N-(‘4-Bromo-3-trifluoromethoxy-phenyl -6-fluoro-nicotinamide

To a solution of 4-bromo-3-trifluoromethoxy-phenylamine (3.15 g, 12.3 mmol) and triethylamine (3.43 mL, 24.6 mmol) in DCM (25 mL) was slowly added a solution of 2-fluoropyridine-5-carbonyl chloride (2.36 g, 14.8 mmol) in DCM (10 mL). After 2 h at RT, MTBE (90 mL) was added and the reaction mixture was washed with water, brine, and saturated sodium carbonate, dried, and evaporated to give a solid (5.4 g). Ethanol (43 mL) was added to the solid and then water (43 mL) was slowly added. The reaction mixture was stirred for 1.5 h, filtered, and washed with 1 :4 ethanohwater (2 x 25 mL) to give the title intermediate as a white solid (3.87 g). HPLC method C: Retention time = 21.3 min.
(b) (25,,5R)-4-r5-(4-Bromo-3-trifluoromethoxy-phenylcarbamoyl) -pyridin-2-yl1-2,5- dimethyl-piperazin – 1 -carboxylic acid fe/t-butyl ester

The product of the previous step (3.86 g, 10.2 mmol) (2S,5R)-2,5-dimethyl- piperazine-1 -carboxylic acid tert-butyl ester (2.62 g, 12.2 mmol) and N,N- diisopropylethylamine (5.32 mL, 30.5) was dissolved in DMSO (12 mL). The reaction mixture heated at 120 °C for 3 h, diluted with EtOAc (100 mL), washed with water, and saturated NH4C1, water, and brine. The reaction mixture was evaporated to about 40% volume and 3 M HCl in cyclopentyl methyl ether (4.24 mL, 12.7 mmol) was added slowly. Seeds from a previous run at smaller scale were added and the reaction mixture was stirred for 2 days and filtered to provide the HCl salt of the title intermediate (5.15 g, 83 % yield). HPLC method C: Retention time = 21.1 min (c) (2 .5R)-4 5-(4′-{2 ffl -((5f)-2-Methoxycarbonylamino-3-methyl-butyrvn- pyiTolidin-2-yl1-lH-imidazol-4-yl}-2-tri
pyridin-2- -2,5-dimethyl-piperazine-l-carboxylic acid fert-butyl ester

To a solution of ((5′)-l-{(5,)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]- pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester (3.05 g, 6.8 mmol;), bis(pinacolato)diboron (1.81 g, 7.1 mmol) and potassium acetate (1,00 g, 10.2 mmol) was added nitrogen sparged toluene (15 mL). The resulting mixture was sparged with nitrogen and l,l’-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane (Pd catalyst) (0.17 g, 0.204 mmol) was added. The reaction mixture was stirred at 90 °C overnight.
The reaction mixture was cooled to RT and to this mixture was added nitrogen sparged water (7.6 mL), potassium carbonate (5.16 g, 37.3 mmol). The reaction mixture was stirred at 95°C overnight.
Another portion of the Pd catalyst used above (0.08 g, 0.10 mmol) was added to the reaction mixture. After 5 h, the reaction mixture was cooled to RT, diluted with EtOAc (150 mL), washed with water (150 mL) and brine (100 mL), dried over sodium sulfate, and evaporated to give a black residue (6.7 g), which was purified by silica gel chromatography (eluted with 50-100 % EtOAc/hexane) to provide the title intermediate (5.3 g, 90 % yield). HPLC method C: Retention time = 14.7 min.
(d) (ffl ffl-2 4-(4′ r6-((2R,5^-2,5-Dimethyl-piperazin-l-vn-pyridine-3-carbonyl1- amino}-2′ rifluoromethoxy-biphenyl-4-yl -lH-imidazol-2-yl1-pyrrolidine-l- carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

Acetyl chloride (2.57 mL, 36.2 mmol) was added to ethanol (18 mL) and stirred at RT for 1 h. To the resulting HQ solution was added a solution of the product of the previous step (3.90 g, 4.5 mmol) in ethanol (18 mL). The reaction mixture was warmed to 35 °C and stirred overnight. Acetyl chloride (1.28 mL, 18.1 mmol) was added to ethanol (7.8 mL) and stirred for 30 min. The resulting HC1 solution was added to the reaction mixture at 35 °C. The temperature was raised to 40 °C. The mixture was concentrated to dryness chased by dichloromethane to provide the crude tri-HCl salt of the title intermediate (5.4 g) which was used directly in the next step. HPLC method C: Retention time = 10.1 min.
(e) ((^-l- {(^-2-r4-(4′- {r6-((2R.5^-2.5-Dimethyl-4-methylcarbamoyl-piperazin-l-yl)- pyridine-3-carbonyl1-amino}-2′-trifluoromethoxy-biphenyl-4-yl)-lH-imidazol-2-yl1- pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester

To a solution of the product of the previous step (5.4 g crude, ca. 3.96 mmol) and N,N-diisopropylethylamine (6.89 mL, 39.6 mmol) in DCM (52 mL) was slowly added 1 M methylaminoformyl chloride in DMA (4.3 mL). The reaction mixture stirred at room temperature for 1 h, and then water (50 mL) was added. The organic layer was washed with saturated NH4C1 and then brine, dried over Na2S04 and evaporated to give 5.2 g crude product, which was purified by silica gel chromatography (133 g silica, 2 to 8 % methanol/DCM for 15 min then 8 % methanol/DCM for 40 min) to provide the title compound (2.4 g, 74 % yield). HPLC method C: Retention time 1 1.2 min
Synthesis of intermediates
http://www.google.com.ar/patents/WO2012061552A1?cl=en
Preparation 1: 4-(4-bro -phenyl)-2-(S)-pyrrolidin-2-yl-lH-imidazole

(a) 2-Bromo-l-(4-bromo-phenyl)-ethanone
Bromine (80 g, 500 mmol) was added dropwise to a solution of l-(4-bromo- phenyl)-ethanone (100 g, 500 mmol) in dichloromethane (1500 mL) at ambient temperature. The reaction mixture was stirred for 3 h and then concentrated. The residue was washed with dichloromethane (100 mL) to give the crude title compound (120 g, 86
% yield) as a white solid. XH NMR (CDC13, 400 MHz) δ (ppm): 7.78 (d, J=8.4 Hz, 2H), 7.57 (d, J=8.4 Hz, 2H), 4.32 (s, 2H).
(b) (^-pyrrolidine- 1 ,2-dicarboxylic acid 2-r2-(4-bromo-phenyl)-2-oxo-ethyl1 ester \-tert- butyl ester
Diisopropylethylamine (67 g, 518 mmol) was added dropwise to a solution of the product of the previous step (120 g, 432 mmol) and (5)-pyrrolidine-l,2-dicarboxylic acid 1-tert-butyl ester ( -Boc proline) (102 g, 475 mmol) in acetonitrile (2 L) at room temperature. The reaction mixture was stirred overnight and concentrated to dryness. The residue was dissolved in ethyl acetate (2 L) and washed with water (2 L). The organic layer was dried over sodium sulfate and concentrated to give crude title compound (178 g, 100 % yield).
(c) (5f)-2-r4-(4-bromo-phenyl -lH-imidazol-2-yl1-pyrrolidine-l-carboxylic acid fe/t-butyl ester
A solution of the product of the previous step (178 g, 432 mmol) and ammonium acetate (500 g, 6.5 mol) in toluene (2 L) was heated at reflux overnight. The solvent was removed and the residue was dissolved in ethyl acetate (2 L) and washed with water (2 L). The organic layer was dried over sodium sulfate and concentrated. The residue was purified by silica gel column chromatography in 1:3 petroleum ether: ethyl acetate to give the title compound (120 g, 71 % yield) as a yellow solid. ¾ NMR (CDC13, 400 MHz) δ (ppm): 7.56 (s, 1H), 7.39 (d, J=8.0 Hz, 2H), 7.24 (m, 1H), 7.14 (s, 1H), 4.88 (m, 1H), 3.33 (m, 2H), 2.94 (s, 1H), 2.07 (m, 2H), 1.88 (m, 1H), 1.42 (s, 9H).
(d) 4-(4-bromo-phenyl)-2-(5f)-pyrrolidin-2-yl-lH-imidazole
To a solution of (5)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]-pyrrolidine-l- carboxylic acid tert-butyl ester (3 g, 7.6 mmol) in methanol (3 mL) was added 4N HQ in methanol (60 mL) at 0 °C. The reaction mixture was stirred for 2 h and then concentrated to give crude hydrochloride salt of the title compound (2.51 g 100 % yield) as a yellow solid.
Preparation 2: (5)-2-Methoxycarbonylamino-3-methyl-butyric acid

A mixture of (5)-2-amino-3 -methyl-butyric acid (10 g, 85 mmol), NaOH (10.3 g, 255 mmol) in water (100 mL) was treated with methylchloridocarbonate (8 g, 85 mmol) at 0 0 C. The reaction mixture was stirred for 24 h at room temperature and then 5 N aqueous HC1 was added to the reaction mixture to adjust pH to 4. The mixture was filtered through a pad of Celite to give the product (10 g, 67% yield) as a white solid. ¾ NMR (CH3OD, 400 MHz) δ (ppm) 4.05(d, 1H), 3.65(s, 3H), 2.14(m, 1H), 0.95(m, 6H). Preparation 3: ((S)-l-{(S)-2-[4-(4-bromo-phenyl)-lH-imidazol-2-yl]- pyrrolidine-l-carbonyl}-2-met methyl ester

Triethylamine (2.3 g, 11.4 mmol) was added to a solution of 4-(4-bromo-phenyl)- 2-(5)-pyrrolidin-2-yl-lH- imidazole hydrochloride (2 g, 11.4 mol), (5)-2- methoxycarbonylamino-3-methyl-butyric acid (2.5 g, 7.6 mmol), and HATU (4.3 g, 11.4 mmol) in dimethylformamide (50 mL) at 0 °C under nitrogen. The reaction mixture was stirred at room temperature overnight and treated with ethyl acetate (100 mL) and water (1000 mL). The organic layer was washed with water (2 x 100 mL) and brine (100 mL), dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel column chromatography in 1 : 1 petroleum ether: ethyl acetate to give the title compound (2.5 g 74 % yield) as a yellow solid. XH NMR (i¾-DMSO, 400 MHz) δ (ppm) 7.63 (d, J=8.8 Hz, 2H), 7.54 (m, IH), 7.47 (m, 2H), 7.26 (d, J=8.4 Hz, IH), 5.03 (m, IH), 4.02 (t, J =8.4 Hz, IH), 3.76 (m, 2H), 3.51 (s, 3H), 2.10 (m, 2H), 1.93 (m, 3H), 0.85 (d, J=6.8 Hz, 3H), 0.81 (d, J=6.8 Hz, 3H).
I AM NOT SURE OF BELOW DATA, It is a cut paste for TD 6450 , NOT ABLE TO CONNECT CAS 1374883-22-3 WITH TD 6450
IF YOU HAVE A STRUCTURE PIC FOR THE SAME MAIL ME amcrasto@gmail.com, call +919323115463
POSTER
50th Annu Meet Eur Assoc Study Liver (EASL) (April 22-26, Vienna) 2015, Abst P0898
http://ilc-congress.eu/abstract_25_04/ILC2015-abstract-book-25-04-Saturday.pdf
P0898
TD-6450,
A NEXT GENERATION ONCE-DAILY NS5A INHIBITOR, HAS POTENT ANTIVIRAL ACTIVITY FOLLOWING A 3-DAY MONOTHERAPY STUDY IN GENOTYPE 1 HCV INFECTION
E. Lawitz1, M. Rodriguez-Torres2, R. Kohler3, A. Amrite3, C. Barnes3, M.L.C. Pecoraro3, J. Budman3, M. McKinnell3, C.B. Washington3. 1Texas Liver Institute, University of Texas Health Science Center, San Antonio, TX, United States; 2Fundacion de Investigacion, San Juan, Puerto Rico; 3Theravance Biopharma, South San Francisco, CA, United States
E-mail: cwashington@theravance.com
Background and Aims: TD-6450 is a next generation HCV NS5A inhibitor with superior in vitro potency against resistanceassociated variants (RAVs) encountered with first-generation NS5A inhibitors. This study evaluated the safety, pharmacokinetics (PK) and antiviral activity of TD-6450 following multiple oral doses in HCV patients
Theravance Biopharma Announces Positive Results From Phase 1 Proof-of-Concept Study of TD-6450, an NS5A Inhibitor to Treat Hepatitis C
240 mg Achieved a Median Maximal Viral Load Decline of 4.9 Log10 IU/mL Following Three Daily Doses in Genotype 1a Patients
SOUTH SAN FRANCISCO, CA — (Marketwired) — 11/03/14 — Theravance Biopharma (NASDAQ: TBPH), through its U.S. operating subsidiary, Theravance Biopharma US, Inc., today announced positive results from the first three cohorts of Study 0110, a Phase 1 proof-of-concept study of TD-6450, a next-generation investigational NS5A inhibitor in development to treat patients with hepatitis C virus infection (HCV).
TD-6450 was evaluated in three cohorts of eight genotype 1a (GT-1a) patients each at doses of 60, 120 and 240 mg, administered once-daily for three days. TD-6450 demonstrated dose-dependent antiviral activity with median maximal declines of HCV RNA of 3.87, 4.63 and 4.89 log10 IU/mL for doses of 60, 120 and 240 mg, respectively.
In the 120 and 240 mg dose groups, three days of once-daily oral treatment resulted in levels of serum HCV RNA below the limit of detection (LOD) in 43% (3/7) and 57% (4/7) of patients treated with TD-6450, respectively. Three of the seven LOD patients went on to show no measurable virus at Day 14, and two of these patients still had no measurable virus at Day 28. At a two-month time point in a long-term follow-up study, the viral load in these two patients was measurable, but both remained more than three logs below their baseline.
None of the patients in the three dose groups had virologic breakthrough during their three-day treatment course, and 100% of the treated GT-1a patients in the study achieved at least a three log10 IU/mL reduction of HCV RNA. At the 120 and 240 mg doses, 71% (5/7) and 86% (6/7) of treated patients achieved at least a four log10 IU/mL reduction in HCV RNA, respectively.
All doses of TD-6450 were generally well tolerated after three doses and for the 28-day observation period. There were no serious adverse events and no patient discontinuations. There was no pattern of clinical adverse events or laboratory abnormalities related to treatment.
“We see diverse responses to direct antivirals in genotype 1 populations. Despite recent advances in HCV therapy, significant treatment challenges remain, including the required length of drug therapy. The robust activity of TD-6450 in genotype 1a patients suggests that this potentially best-in-class NS5A inhibitor could be a component of short and highly active combination therapy regimens,” said Eric Lawitz, MD, Vice President of Scientific and Research Development at the Texas Liver Institute and Clinical Professor of Medicine, The University of Texas Health Science Center San Antonio, and one of the principal investigators on the Phase 1 study.
“TD-6450, created using the principles of multivalent design, has a heterodimeric structure distinct from other NS5A inhibitors. We believe this unique structure allows it to bind asymmetrically across the NS5A protein interface, providing high in vitro potency against clinically encountered resistance-associated variants. We believe the potency of TD-6450 against both wild type virus and these resistance-associated variants enables the robust antiviral activity that we reported today,” said Mathai Mammen, MD, Senior Vice President, Research and Development, Theravance Biopharma. “We look forward to analyzing the full set of results from this Phase 1 study and evaluating the next steps in the development strategy for TD-6450.”
About the Phase 1 Proof-of-Concept Study (Study 0110)
This Phase 1 study is a double-blind, randomized, placebo-controlled, multiple-dose study to evaluate the safety, tolerability, pharmacokinetics and antiviral activity of orally administered TD-6450 in non-cirrhotic, treatment-naive patients with GT-1, 2, or 3 chronic HCV infection. The study includes seven cohorts. The first three cohorts enrolled eight GT-1a patients each (7 active; 1 placebo) and tested once-daily oral doses of 60, 120 or 240 mg, respectively. Patients were dosed for three days and followed for up to 28 days for viral load quantification. The limit of detection for the viral load quantification assay is 15 IU/mL.
Safety evaluations include monitoring for adverse events, routine laboratory assessments, vital signs and 12-lead ECG tracings.
In cohorts 4 through 6, patients with GT-1b, GT-2 and GT-3 are dosed once-daily at 240 mg. An additional cohort (cohort 7) of GT-1a patients is dosed twice daily with 240 mg. Data generation and analysis of results for cohorts 4 through 7 is ongoing. An interim analysis of those cohorts showed antiviral activity for GT-1b similar to that for GT-1a, but minimal antiviral activity for GT-2 and GT-3.
The Company anticipates presenting further data on all cohorts at a future scientific conference.
About TD-6450
TD-6450 is an internally discovered multivalent NS5A inhibitor designed to have improved antiviral activity against GT-1 resistance-associated variants (RAV) resistant to first generation NS5A inhibitors. TD-6450’s heterodimeric structure permits an asymmetric binding mode to NS5A relative to structurally symmetric inhibitors. TD-6450 has demonstrated additive activity with other classes of anti-HCV agents in replicon assays, and no cross-resistance with RAVs that confer resistance to other anti-HCV agents. The Company believes that the antiviral activity of TD-6450, in combination with other antivirals, may help improve cure rates and/or reduce treatment times for appropriate patients.
TD-6450 was previously evaluated in a single-ascending dose and a 14-day multiple-ascending dose study in healthy subjects (study 0094). This randomized, double-blind, placebo-controlled study evaluated the safety, tolerability and pharmacokinetics of TD-6450. Single doses (up to 500 mg) and multiple doses of TD-6450 (up to 240 mg daily for 14 days) were evaluated in healthy subjects. Following single and multiple doses, TD-6450 was generally well-tolerated and no subjects discontinued due to adverse events. Headache was the most commonly reported adverse event following multiple doses (n=4). TD-6450 pharmacokinetics were linear up to 240 mg following single and multiple doses and its long half-life supports once-daily dosing.
About Hepatitis C and the NS5A Inhibitor Class
Hepatitis C is an infectious disease of the liver. Worldwide, health experts estimate that 130 – 150 million people have chronic hepatitis C, with as many as four million of those cases in the United States. Hepatitis C, like all forms of hepatitis, can damage the liver. Of people infected, 55 to 85 percent will develop chronic infection, and 75 percent of those with chronic infection will develop chronic liver disease.
The hepatitis C non-structural 5A (NS5A) protein of HCV has emerged as an attractive drug target and inhibitors of NS5A have a central role in all-oral HCV therapy. The multi-functional NS5A protein is required for ribonucleic acid (RNA) replication and virion assembly, and a number of investigational and approved NS5A inhibitors have shown antiviral efficacy in HCV-infected patients.
Theravance Biopharma and Trek Therapeutics Announce Initiation of Phase 2a Trial of TD-6450, an NS5A Inhibitor to Treat Hepatitis C
Study Being Conducted by Trek Therapeutics Following Licensing of Worldwide Rights to Drug Candidate From Theravance Biopharma
DUBLIN, IRELAND and CAMBRIDGE, MA — (Marketwired) — 10/27/15 — Theravance Biopharma, Inc. (NASDAQ: TBPH) (“Theravance Biopharma”) and Trek Therapeutics (“TREKtx”) today announced that TREKtx has initiated a Phase 2a clinical trial of TD-6450, a next-generation investigational NS5A inhibitor in development to treat patients with hepatitis C virus (HCV). Theravance Biopharma recently granted TREKtx an exclusive worldwide license for the development, manufacturing, use, marketing and sale of TD-6450 as a component in combination HCV products. Other terms of the transaction have not been disclosed.
The Phase 2a clinical trial will evaluate faldaprevir (FDV), an HCV protease inhibitor, combined with TD-6450 and ribavirin (RBV) in patients infected with HCV genotype 4. The trial is being conducted in the United States.
Mathai Mammen, M.D., Ph.D., Senior Vice President of Research and Development at Theravance Biopharma commented, “We are pleased to see the initiation of this Phase 2a clinical trial with TD-6450. This NS5A inhibitor has shown robust antiviral activity in a Phase 1 trial in patients with HCV genotype 1, as well as preclinical potency against both wild type HCV and resistance-associated variants. We believe that its antiviral activity, in combination with other antivirals, may help improve cure rates and/or reduce treatment times for appropriate patients. We are especially pleased to collaborate with TREKtx and support their commitment to delivering novel and accessible combination HCV treatments to patients worldwide.”
“We are very excited about dosing our first genotype 4 patients in this combination study. If safety and efficacy are demonstrated, the goal is to initiate clinical trials in Egyptnext year, where the need is enormous,” said Dr. Robert Hindes, Chief Medical Officer of Trek Therapeutics.
About TD-6450
Theravance Biopharma discovered TD-6450, a multivalent NS5A inhibitor designed to have improved antiviral activity against genotype 1 resistance-associated variants (RAV) resistant to first generation NS5A inhibitors. TD-6450 has successfully completed Phase 1 studies in both healthy volunteers and HCV patients.
About Faldaprevir
Faldaprevir is a protease inhibitor that TREKtx acquired from Boehringer Ingelheim. FDV has completed Phase 3 studies in combination with pegylated interferon and RBV.
About HCV
Hepatitis C is an infectious disease of the liver. Of people infected, 55 to 85 percent will develop chronic infection, and 75 percent of those with chronic infection will develop chronic liver disease.
The U.S. Centers for Disease Control and Prevention estimates 2.7 million individuals in the United States have active hepatitis C virus (HCV) infection, most of whom are “baby boomers.” In the United States, chronic HCV infection is the leading cause of cirrhosis and liver cancer and the most common reason for liver transplantation. Worldwide, more than 135 million people have chronic HCV infection and most are undiagnosed.
About Trek Therapeutics
TREKtx is a private, clinical stage public benefit corporation developing treatments for serious infections. Its mission is to profitably develop affordable and accessible medicines to treat infectious diseases and to commercialize them for global populations. The company’s founders collectively participated in the development of seven approved antiviral drugs.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008021927A2 * | 9 Aug 2007 | 21 Feb 2008 | Squibb Bristol Myers Co | Hepatitis c virus inhibitors |
| WO2010094977A1 * | 22 Feb 2010 | 26 Aug 2010 | Arrow Therapeutics Limited | Novel biphenyl compounds useful for the treatment of hepatitis c |
| US201161492267 |
| WO2013067267A1 * | 2 Nov 2012 | 10 May 2013 | Theravance, Inc. | Rod -like hepatitis c virus inhibitors containing the fragement {2- [4- (bi phenyl – 4 – yl) – 1h – imidazo – 2 – yl] pyrrolidine – 1 – carbonlymethyl} amine |
| WO2013163270A1 * | 24 Apr 2013 | 31 Oct 2013 | Theravance, Inc. | Hepatitis c virus inhibitors |
| WO2013165796A1 * | 25 Apr 2013 | 7 Nov 2013 | Theravance, Inc. | Crystalline form of a pyridyl-piperazinyl hepatitis c virus inhibitor |
//////////////
AN-2718, a New Borole in the pipeline
AN-2718,
2,1-Benzoxaborole, 5-chloro-1,3-dihydro-1-hydroxy-
5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole
5-Chloro-2,1-benzoxaborol-1(3H)-ol
CAS 174672-06-1
UNII: 810U6C2DGG
MW 168.3864, MF C7 H6 B Cl O2
MP…150-154 °C [WO 9533754]
MP 142-144 °C [ jmc 2006, 49(15) 447]
M.p. 147- 149 °C. WO2013050591
Anacor Pharmaceuticals Inc, INNOVATOR
Onychomycosis is a disease of the nail caused by yeast, dermatophytes, or other molds, and represents approximately 50% of all nail disorders. Toenail infection accounts for approximately 80% of onychomycosis incidence, while fingernails are affected in about 20% of the cases. Dermatophytes are the most frequent cause of nail plate invasion, particularly in toenail onychomycosis. Onychomycosis caused by a dermatophyte is termed Tinea unguium. Trichophyton rubrum is by far the most frequently isolated dermatophyte, followed by T. mentagrophytes. Distal subungual onychomycosis is the most common presentation of tinea unguium, with the main site of entry through the hyponychium (the thickened epidermis underneath the free distal end of a nail) progressing in time to involve the nail bed and the nail plate. Discoloration, onycholysis, and accumulation of subungual debris and nail plate dystrophy characterize the disease. The disease adversely affects the quality of life of its victims, with subject complaints ranging from unsightly nails and discomfort with footwear, to more serious complications including secondary bacterial infections.
Many methods are known for the treatment of fungal infections, including the oral and topical use of antibiotics (e.g., nystatin and amphotericin B), imidazole anti-fungal agents such as miconazole, clotrimazole, fluconazole, econazole and sulconazole, and non-imidazole fungal agents such as the allylamine derivatives terbinafme and naftifϊne, and the benzylamine butenafine. However, onychomycosis has proven to be resistant to most treatments. Nail fungal infections reside in an area difficult to access by conventional topical treatment and anti-fungal drugs cannot readily penetrate the nail plate to reach the infection sites under the nail. Therefore, onychomycosis has traditionally been treated by oral administration of anti-fungal drugs; however, clearly this is undesirable due to the potential for side effects of such drugs, in particular those caused by the more potent anti-fungal drugs such as itraconazole and ketoconazole. An alternative method of treatment of onychomycosis is by removal of the nail before treating with a topically active anti-fungal agent; such a method of treatment is equally undesirable. Systemic antimycotic agents require prolonged use and have the potential for significant side effects. Topical agents have usually been of little benefit, primarily because of poor penetration of the anti-fungal agents into and through the nail mass.
- 51. Hui X, Baker SJ, Wester RC, In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci 2007;96(10):2622-31 [CrossRef], [PubMed], [Web of Science ®]
- 55. Mao W, Seiradake E, Crepin T, AN2718 has Broad Spectrum Antifungal Activity Necessary for the Topical Treatment of Skin and Nail Fungal Infections. J Am Acad Dermatol 2007:56(2 Suppl):AB124
- 56. ClinicalTrials.gov. Cumulative Irritation Test. Available from: http://clinicaltrials.gov/ct2/show/NCT00781664 [Cited 11 December 2011]
SYNTHESIS

Reduction of 2-bromo-5-chlorobenzoic acid with BH3/THF in THF gives 2-bromo-5-chlorobenzyl alcohol , which is protected as the methoxymethyl derivative by treatment with MOM-Cl in the presence of DIEA in CH2Cl2. Metalation and boronylation of aryl bromide either with t-BuLi or BuLi (1,2,3,4) and (i-PrO)3B (4) or B(OMe)3 in THF affords the title compound .
Alternatively, reaction of 3-chlorobenzaldehyde with p-toluenesulfonylhydrazide in MeOH provides tosyl hydrazone which undergoes thermal decomposition in the presence of BBr3 and catalytic FeCl3 in refluxing CH2Cl2, followed by heating with aqueous NaOH to produce the title oxaborole compound .
You can construct from this…………….
 OH A
OH A
(2-bromo-5-chloro-phenyl)methanol (A)
 B
B
l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
 1
1
5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
PATENT
WO 9533754
https://www.google.com/patents/WO1995033754A1?cl=en
Example 1 Preparation of 5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole (Method B).
a)
Preparation of 3-chlorobenzaldehyde tosyl hydrazide
A solution of 3-chlorobenzaldehyde (15.56 parts; 0.109M;
Aldrich) in methylated spirits (40 ml) was added slowly at below 10°C to a stirred suspension of p-toluene-sulphonylhydrazide (20.7 parts;
0.108M) in methylated spirits (150 ml). The reaction mass was then stirred at 20 to 25°C for 1 hour and then heated at 60-70°C for 1M hours when the reactants and products dissolved. The solvent was then removed by rotary evaporation and the product was obtained as a solid which was slurried with ether and washed with n-hexane. Yield = 27.2 parts (81.5% theory) mpt 122-3°C. Elemental analysis
Theory 54.5%C; 4.2%H; 9.1%N
Found 54.5%C; 4.3%H; 9.1%N
Proton NMR (CDCl3:ppm)
8.5, s, 1H(-NH-); 7.9, d, 2H(Tosyl aromatic); 7.7,s, 1H(CH=N); 7.5, s, 1H (aromatic); 7.2-7.4, m, 5H(Tosyl aromatic); 2.3, s, 3H(-CH3) b) Preparation of title compound
A suspension of anhydrous ferric chloride catalyst (0.75 parts, Fisons) in dry dichloromethane (20 ml) was added at 20 to 25°C simultaneously with boron tribromide (25 parts, 0.1M, Aldrich) in dry dichloromethane (100 mis) to a stirred suspension of the hydrazide from a) above (10.18 part, 0.033M) in dry dichloromethane (160 mis) under a nitrogen blanket. The reactants were then stirred under reflux and the evolved hydrogen bromide trapped under aqueous sodium hydroxide. After 3 hours stirring at reflux, the reactants were allowed to stand at 20- 25°C for 48 hours and then stirred under reflux for a further 4 hours. The reaction mass was then cooled and the solvent removed by rotary evaporation. The solid obtained was then stirred under reflux with 2N sodium hydroxide solution (160 ml) for 3 hours. The brown aqueous suspension was extracted with dichloromethane (50 ml), screened and then acidified to about pH 2 by addition of 2N hydrochloric acid. The solid was filtered, slurried with dichloromethane (400 ml) and then washed with a saturated solution of sodium bicarbonate followed by water.
Yield = 24 parts (43% theory). The solid was slurried in hot
dichloromethane and filtered to give 0.36 parts oxaborole mp 140-45°C. The dichloromethane solution was cooled and the solid filtered giving a further 0.35 parts oxaborole mp 146-8°C. The solids were combined and recrystallised from methylated spirits.
Yield = 0.51 parts (9.2% theory) mp 150-4°C.
Elemental Analysis
Theory 49.8%C, 3.5%H, 21.06%C1
Found 49.5%C, 3.5%H, 21.0%C1
Proton NMR (CDCl3) ppm
9.3, s, 1H(-OH); 7.5, d, s, d, 3H(aromatic);
5.0, s, 2H(-CH2-O).
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en

Analytical data for exemplary compounds of structure I are provided below.
4.2. a 5-Chloro-1.3-dihydro-l -hvdroxy-2J-benzoxaborole (Cl) M.ρ. 142-15O0C. MS (ESI): m/z = 169 (M+l, positive) and 167 (M-I, negative). HPLC (220 nm): 99% purity. 1H NMR (300 MHz, DMSO-d6): δ 9.30 (s, IH), 7.71 (d, J = 7.8 Hz, IH), 7.49 (s, IH), 7.38 (d, J = 7.8 Hz, IH) and 4.96 (s, 2H) ppm.
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm0603724?source=chemport
COMPD IS 19d
5-Chloro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (19d) This compound was made from 18d in the same manner as compound 19b (triturated with hexane, 75% yield):: mp 142-144°C. 1 H NMR (300MHz, DMSO-d6) δ (ppm) 4.96 (s, 2H), 7.38 (d, J = 7.8 Hz, 1H), 7.49 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 9.30 (s, 1H); ESI-MS m/z 167 (M-H)- ; HPLC purity 99.0%; Anal (C7H6BClO2 ⋅ 0.1H2O) C, H
precursor 18d
2-Bromo-5-chloro-1-(methoxymethoxymethyl)benzene (18d) To a solution of 2-bromo-5-chlorobenzoic acid (5.49 g, 23.3 mmol) in anhydrous THF (70 mL) under nitrogen was added dropwise a BH3 THF solution (1.0 M, 55 mL) at 0o C and the reaction mixture was stirred overnight at room temperature. Then the mixture was cooled on an ice bath and MeOH (20 mL) was added dropwise to decompose excess BH3. The resulting mixture was stirred until no bubble was released and then 10% NaOH (10 mL) was added. The mixture was concentrated and the residue was S6 mixed with water (200 mL) and extracted with EtOAc. The residue from rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 2-bromo-5-chlorobenzyl alcohol as a white solid (4.58 g, 88%): 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 7.57 (d, J = 8.7 Hz, 1H), 7.50-7.49 (m, 1H), 7.28-7.24 (m, 1H), 5.59 (t, J = 6.0 Hz, 1H), 4.46 (d, J = 6.0 Hz, 2H). 2-Bromo-5-chlorobenzyl alcohol obtained above was dissolved in CH2Cl2 (150 mL) and cooled to 0o C on an ice bath. To this solution under nitrogen were added in sequence i-Pr2NEt (5.4 mL, 31 mmol) and chloromethyl methyl ether (2.0 mL, 26 mmol). The reaction mixture was stirred overnight at room temperature and washed with NaHCO3-saturated water and then brine. The residue after rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 18d (4.67 g, 85%) as a colorless oil: 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 3.30 (s, 3H), 4.53 (s, 2H), 4.71 (s, 2H), 7.32 (dd, J = 8.4, 2.4 Hz, 1H), 7.50 (dd, J = 2.4, 0.6 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H).
PATENT
http://www.google.com/patents/WO2013050591A2?cl=en
EXAMPLES Example 1 : Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
Step 1 : Preparation of (2-bromo-5-chloro-phenyl)methanol (A)
A solution of borane-tetrahydrofuran complex in THF (0.15 L, 1.5 eq) was added dropwise to a solution of 2-bromo-5-chlorobenzoic acid (24 g) in anhydrous tetrahydrofuran (0.24 L) at 0°C and under argon atmosphere. The reaction mixture was stirred at room temperature for 16 h, before being slowly poured onto 0.10 L of a 2N aqueous solution of hydrogen chloride at 0°C. The mixture was stirred for 15 minutes and the volatiles were removed under reduced pressure. The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed with a IN aqueous solution of sodium hydroxide and then water. After drying over sodium sulfate, filtration and concentration under reduced pressure, the crude product was purified by column chromatography; 23.2 g; M.p. 79-80 °C.
Step 2: Preparation of l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
(2-bromo-5-chloro-phenyl)methanol (A, 12 g) was dissolved in dichloromethane (0.35 mL) and cooled to 0 °C. Under argon atmosphere, diisopropylethylamine (14 mL, 1.5 eq) and chloromethyl methyl ether (5.0 mL, 1.2 eq) were added. After 1 night of stirring at room temperature, the crude reaction mixture was washed with a saturated solution of sodium hydrogen carbonate, dried over sodium sulfate and evaporated under reduced pressure. Purification by column chromatography afforded 10.5 g of l-bromo-4-chloro-2- (methoxymethoxymethyl)benzene (B) as an oil.
Step 3: Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
To a solution of (B) (6.0 g) in anhydrous tetrahydrofuran (120 mL) at -78°C was added dropwise a solution of butyllithium in hexane (15.6 mL, 1.1 eq). To the resulting yellow- brown solution trimethyl borate (2.5 mL, 1.0 eq) was injected in one portion and the cooling bath was removed. The mixture was warmed gradually for 30 minutes. After stirring at room temperature for 2 hours, 8.0 ml of a 6N aqueous solution of hydrogen chloride were added and the reaction mixture was left stirring overnight at room temperature. Evaporation of the volatiles gave a residue which was taken up in ethyl acetate, washed with water, brine, dried over sodium sulfate and then evaporated. The crude product was crystallized from ethyl acetate to give 1.4 g of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1) as a white powder. Purification of the filtrate by column chromatography afforded 1.2 g more of 1. M.p. 147- 149 °C.
PATENT
http://www.google.fr/patents/WO2010110400A1?cl=en&hl=fr
Reference Example 187
5-chloro-2,1-benzoxazine ball roll -1 (3H) – All

5-chloro-2- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzaldehyde 8.50g synthesized in Reference Example 185 was dissolved in methanol 100ml, borohydride Sodium 2.40g was added, and after stirring for 30 minutes at room temperature and stirred for 2 hours at 60 ℃. The reaction solution was concentrated, and the organic layer after the layers were separated with ethyl acetate and saturated aqueous ammonium chloride and then concentrated under reduced pressure. The residue was added 100ml of tetrahydrofuran, and 6N hydrochloric acid 60ml, and stirred for 8 hours at room temperature.After the reaction mixture was dried and the organic layer was extracted with ethyl acetate, and concentrated. The residue was purified by silica gel column chromatography to give the title compound 9.6g. 1 H-NMR (400MHz, DMSO-d 6) δ: 4.95 (2H, s), 7.36 (1H, dd, J = 8.0,1.6Hz), 7.47 (1H, s) , 7.70 (1H, d, J = 8.0Hz), 9.28 (1H, s).
PATENT
http://www.google.com/patents/WO2008070257A2?cl=en
5-Fluoro-l,3-dihvdro-l-hvdroxy-2, 1-benzoxaborole (Cl)
1-Hydroxy-dihydrobenzoxaboroles, such as Cl, were synthesized as shown in Scheme 1. The protected o-bromobenzyl alcohol derivative (18), prepared from 16 or 17, was converted into the corresponding phenyl boronic acid. Deprotection of the methoxymethyl ether using hydrochloric acid followed by spontaneous cyclization gave the target compounds.
Scheme 7

Conditions (a) NaBH4, MeOH, rt (when X = H ), or BH3-THF, THF, rt (when X = OH), (b) MeOCH2CI, /-Pr2NEt, CH2CI2, rt, (c) MeMgBr, THF, -78 0C to rt , (d) NBS, AIBN, CCI4, reflux, (e) NaOAc, DM F, 70 0C, (f) NaOH, MeOH, reflux, (g) n-BuLι, (/-PrO)3B, THF, -780C to rt, (h) 6N HCI, THF, rt
References
- Hui, Xiaoying; Journal of Pharmaceutical Sciences 2007, 96(10), Pg2622-2631
- Baker, Stephen J.; Journal of Medicinal Chemistry 2006, 49(15), Pg4447-4450
- Austin, Peter William; WO 9533754 A1 1995 CAPLUS
| US5880188 * | 26 May 1995 | 9 Mar 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US6083903 * | 16 May 1995 | 4 Jul 2000 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| WO2005013892A2 | 15 Jun 2004 | 17 Feb 2005 | Tsutomu Akama | Hydrolytically-resistant boron-containing therapeutics and methods of use |
| Reference | ||
|---|---|---|
| 1 | * | Austin et al., 1996, CAS: 124:234024. |
| 2 | * | fungicide: definition from Answre.com, 1998. |
| 3 | S. J. Baker, et al., “Progress on New Therapeutics for Fungal Nail Infections,“Annual Reports in Medicinal Chemistry, 40:323-335 (2005). | |
| 4 | Sudaxshina Murdan, “Drug Delivery to the Nail Following Topical Application,” International Journal of Pharmaceutics, 236:1-26 (2002). | |
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
//////////AN-2718, Borole, PHASE 2
B1(c2ccc(cc2CO1)Cl)O
Voxtalisib, SAR-245409, XL-765

Voxtalisib
SAR-245409, XL-765
2-amino-8-ethyl-4-methyl-6-(1H-pyrazol-3-yl)pyrido[2,3-d]pyrimidin-7(8H)-one
2-Amino-8-ethyl-4-methyl-6-(1H-pyrazol-5-yl)pyrido[2,3-d]pyrimidin-7(8H)-one hydrochloride
C13 H14 N6 O . Cl H, 306.751
934493-76-2
INNOVATOR Exelixis Inc,, LICENSE SANOFI
PHASE 2, Malignant neoplasms
0.2H2O
- Mol. Formula:C13H14N6O∙0.2H2O, MW:273.9
- NMR………http://www.chemietek.com/Files/Line2/CHEMIETEK,%20XL765,%20Lot%2001,%20NMR%20in%20CD3OD.pdf
- Mechanism of Action:selective oral inhibitor of PI3K and mTOR
Indication:Cancer Treatment
Stage of Development: phase ll study in chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma (NHL). A phase I/II trial is assessing SAR245409 in combination with letrozole in ER/PR+ HER2- breast cancer. 
SAR245409 (XL765)
SAR245409 (XL765) is an orally available inhibitor of PI3K and the mammalian target of rapamycin (mTOR), which are frequently activated in human tumors and play central roles in tumor cell proliferation. Exelixis discovered SAR245409 internally and out-licensed the compound to Sanofi. SAR245409 is being evaluated by Sanofi as a single agent and in multiple combination regimens in a variety of cancer indications. Clinical trials have included a single agent phase 2 trial in Non-Hodgkin’s lymphoma, combination phase 1b/2 trials with temozolomide in patients with glioblastoma, with letrozole in hormone receptor positive breast cancer, with bendamustine and/or rituximab in lymphoma or leukemia, and a phase 1 trial in combination with a MEK inhibitor.
SAR-245409 is an investigational drug originated by Exelixis that dually inhibits mammalian target of rapamycin (mTOR) and phosphatidylinositol 3-kinase (PI3K).

Sanofi is also evaluating the compound in phase I/II clinical trials for the treatment of malignant neoplasm as monotherpay or in combination regimen. It has also completed phase I clinical trials as an oral treatment for brain cancer.
In 2009, the drug candidate was licensed to Sanofi (formerly known as sanofi-aventis) by Exelixis worldwide for the treatment of solid tumors.
XL765 (Voxtalisib, SAR245409, Sanofi)*, a PYRIDOPYRIMIDINONE-derivative, is a highly selective, potent and reversible ATP-competitive inhibitor of pan-Class I PI3K (α, β, γ, and δ) and mTORC1/mTORC2. It is orally active, highly selective over 130 other protein kinases. In cellular assays, XL765 inhibits the formation of PIP3 in the membrane, and inhibits phosphorylation of AKT, p70S6K, and S6 phosphorylation in multiple tumor cell lines with different genetic alterations affecting the PI3K pathway.

In mouse xenograft models, oral administration of XL-765 results in dose-dependent inhibition of phosphorylation of AKT, p70S6K, and S6 with a duration of action of approximately 24 hours. Repeat dose administration of XL765 results in significant tumor growth inhibition in multiple human xenograft models in nude mice that is associated with antiproliferative, antiangiogenic, and proapoptotic effects
PATENT
WO 2014058947
http://www.google.co.in/patents/WO2014058947A1?cl=en
Example 1. Synthesis of Compound (1)
Compound (1) can be synthesized as described in WO 07/044813, which is hereby incorporated in its entirety.

Briefly, a base and an intermediate, compound (a), are added to solution of commercially available 2-metfiyl-2-thiopseudourea sulfate in a solvent such as water and stirred overnight at room temperature. After neutralization, compound (b) is collected by filtration and dried under vacuum. Treatment of compound (b) with POCI3 and heating at reflux for 2 hours yields compound (c) which can be concentrated under vacuum to dryness. Compound (c) can be used directly in the following reaction with ethylamine carried out in a solvent such as water with heating to give compound (d). Compound (d) is then treated with iodine monochloride in a solvent such as methanol to form compound (e). Compound (e) is then dissolved in DMA, to which ethyl acrylate, Pd(OAc)2 and a base are added. This reaction mixture is heated and reacted overnight until completion of the reaction to give compound (f), which can be purified via column chromatography.
Compound (f) is then be treated with DBU in the presence of a base, such as DIEA, and heated at reflux for 15 hours. Upon completion of the reaction, the solvent is evaporated and the residue triturated with acetone to yield compound (g). Bromination of compound (g) can be achieved through drop-wise addition of Br2 to compound (g) in CH2C12, followed by stirring overnight at room temperature. Next, filtration is carried out, and triethylamine is added so that, upon washing and drying, the product, compound (h) is obtained. A Suzuki coupling between compound (h) and lH-pyrazol-5-yl boronic acid is carried out using a Pd- catalyst such as [1,1 -bis(diphenylphosphino)ferrocene]dichloropalladium(II) in the presence of a base to yield compound (i). Finally, compound (i) can be converted to compound (1) of the instant invention through 1) oxidation of the methylthio group with m-CPBA, carried out at room temperature with stirring and 2) treatment of the resulting product dissolved in dioxane, with liquid ammonia. Stirring at room temperature overnight followed by purification by column chromatography gives the desired product, 2-amino-8-ethyl-4-methyl- 6-(lH-pyrazol-5-yl)pyrido[2,3-d]pyrimidin-7(8H)-one, compound (1).
PATENT
WO 2007044813
http://www.google.co.in/patents/WO2007044813A1?cl=en
Example 1 2-amino-8-ethyl-4-methyl-6-(lJΪ-pyrazol-5-yl)pyrido[2,3-</]pyrimidin-7(8J?)-one

To a solution of 2-methyl-2-thiopseudourea sulfate (Aldrich, 58.74 g, 0.422 mol) in water (1000 mL) were added sodium carbonate (81.44 g, 0.768 mol) and ethyl acetoacetate (50 g, 0.384 mol) at room temperature. The reaction mixture was stirred overnight. After neutralizing to pH = 8, the solid was collected through filtration followed by drying under vacuum overnight to afford 6-methyl-2-(methylthio)pyrimidin-4(3H)-one (57.2 g, 95% yield) of product. 1H NMR (400 MHz, DMSO-d6): δ 12.47 (bs, IH), 5.96 (bs, lH), 2.47(s, 3H), 2.17 (s, 3H).

To the round bottom flask containing 6-methyl-2-(methylthio)pyrimidin-4(3H)- one (19 g, 121.6 mmol) was added POCl3 (30 mL). The reaction mixture was heated to reflux for 2 h and then concentrated on a rotary evaporator to dryness. The crude 4-chloro- 6-methyl-2-(methylthio)pyrimidine was used directly in the next reaction without further purification.

To the 4-chloro-6-methyl-2-(methylthio)pyrimidine from above was added 30 mL of a solution of 70% ethylamine in water. The reaction mixture was heated to 50 0C for 3 h. After completion, excess ethylamine was evaporated on rotary evaporator under vacuum. The solid was filtered and dried under vacuum to afford 7V-ethyl-6-methyl-2- (methylthio)pyrimidin-4-amine (20 g, 90% yield).

To the solution of N-emyl-6-methyl-2-(methylthio)pyrimidin-4-amine (20 g, 121.6 mmol) in methanol was added iodine monochloride (26.58 g, 163.7 mmol) in small portions at 0 °C. Then the reaction mixture was stirred overnight. After evaporation of solvent, the residue was triturated with acetone. The product iV-ethyl-5-iodo-6-methyl-2- (methylthio)pyrimin-4-amine (25.2 g, 75% yield) was collected by filtration. 1H NMR (400 MHz, CDCl3): δ 5.37 (bs, IH), 3.52 (q, J = 7.2 Hz, IH), 2.50 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H).

To the solution of N-ethyl-5-iodo-6-methyl-2-(methylthio)pyrimin-4-amine (25.2 g, 81.48 mmol) in DMA (260 mL) were added ethyl acrylate (12.23 g, 122.2 mmol), Pd(OAc)2 (3.65 g, 16.25 mmol), (+)BINAP and triethyl amine (24.68 g, 244.4 mmol). Then the reaction mixture was heated to 100 0C and reacted overnight. After evaporation of solvent, the residue was diluted with water and the aqueous layer was extracted with ethyl acetate. The product (E)-ethyl-3-(4-(ethylamino)-6-methyl-2-(methylthio)pyrimidin-5- yl)acrylate (16.8 g, 73% yield) was isolated by silica gel column chromatography with 6-8% ethyl acetate in hexane as eluent. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 16.4Hz, IH), 6.20 (d, J = 16.4Hz, IH), 5.15 (bs, IH), 4.28(q, J = 7.2 Hz, 2H), 3.54 (q, J = 7.2 Hz, 2H), 2.53 (s, 3H), 2.37 (s, 3H), 1.35 (t, J = 7.2 Hz, 3H), 1.24 (t, J = 7.2 Hz, 3H).

To a solution of (E)-ethyl-3-(4-(ethylamino)-6-methyl-2-(methylthio)pyrimidin- 5-yl)acrylate (16.8 g, 59.8 mmol) in DIPEA was added l,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 18.21 g, 119.6 mmol) at room temperature. Then the reaction mixture was heated to reflux and reacted for 15 h. After evaporation of solvent, the residue was triturated with acetone. The product 8-ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (10.77 g, 77% yield) was collected by filtration. 1H NMR (400 MHz, CDCl3): δ 7.78 (d, J = 9.6 Hz, IH), 6.63 (d, J = 9.6 Hz5 IH), 4.5(q, J = 7.2 Hz, 2H), 2.67 (s, 3H), 2.62 (s, 3H), 1.33 (t, J = 7.2 Hz, 3H).

[00187] To a solution of 8-ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)- one (6.31 g, 26.84 mmol) in DCM was added Br2 (4.79 g, 29.52 mmol) dropwise at room temperature. Then the reaction mixture was stirred at room temperature overnight. After filtration the solid was suspended in DCM (100 mL), and triethylamine (20 mL) was added. The mixture was washed with water and dried with Na2SO4, and the product 6-bromo-8- ethyl-4-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (6.96 g, 83 % yield) was obtained after evaporation of DCM. 1H NMR (400 MHz, CDCl3): δ 8.22 (s, IH), 4.56 (q, J = 7.2 Hz, 2H), 2.68 (s, 3H), 2.62 (s, 3H), 1.34 (t, J = 7.2Hz, 3H).

To a solution of 6-bromo-8-ethyl-4-methyl-2-(methylthio)ρyrido[2,3- d]pyrimidin-7(8H)-one (0.765 g, 2.43 mmol) in DME-H2O (10:1 11 mL) was added IH- pyrazol-5-ylboronic acid (Frontier, 0.408 g, 3.65 mmol), [1,1′- bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with CH2Cl2 (Pd(dρρρf),0.198 g, 0.243 mmol) and triethylamine (0.736 g, 7.29 mmol) at room temperature. Then the reaction mixture was heated to reflux and reacted for 4 h. After cooling down to room temperature, the reaction mixture was partitioned with water and ethyl acetate. After separation, the. organic layer was dried with Na2SO4, and the product 8- ethyl-4-methyl-2-(methylthio)-6-(lH-pyrazol-5-yl)pyrido[2,3-d]pyrimidin-7(8H)-one (0.567 g, 77% yield) was obtained by silica gel column chromatography. 1H NMR (400 MHz, CDCl3): δ 13.3 (bs, IH), 8.54 (s, IH), 7.82-7.07 (m, 2H), 4.45 (q, J = 7.2 Hz, 2H), 2.71 (s, 3H), 2.60 (s, 3H), 1.26 (t, J = 7.2Hz, 3H).

To the solution of 8-ethyl-4-methyl-2-(methylthio)-6-(lH-pyrazol-5- yl)pyrido[2,3-d]pyrimidin-7(8H)-one (0.123 g, 0.41mmol) in DCM (2 mL) was added MCPBA (0.176 g, 77%, 0.785 mmol) in a small portion at room temperature. Then the reaction mixture was stirred for 4 h. After evaporation of DCM, dioxane (1 mL) and liquid ammonia (1 mL) were introduced. The reaction was stirred at room temperature overnight. The product 2-amino-8-ethyl-4-methyl-6-(lH-pyrazol-5-yl)pyrido[2,3-(/lpyrimidin-7(8H)- one (50.4 mg) was obtained by silica gel column chromatography. 1H NMR (400 MHz, CD3OD): δ 8.41 (s, IH), 7.62 (d, J – 2.0 Hz, IH), 6.96 (d, J = 2.0Hz5 IH), 4.51 (q, J = 7.2Hz, 2H), 2.64 (s, 3H), 1.29 (t, J = 7.2Hz, 3H); MS (EI) for C13H14N6O: 271.3 (MH+)
References:
| WO2007044813A1 | 9 Oct 2006 | 19 Apr 2007 | Exelixis Inc | PYRIDOPYRIMIDINONE INHIBITORS OF PI3Kα |
| WO2012054748A2 * | 20 Oct 2011 | 26 Apr 2012 | Seattle Genetics, Inc. | Synergistic effects between auristatin-based antibody drug conjugates and inhibitors of the pi3k-akt mtor pathway |
| WO2012065019A2 * | 11 Nov 2011 | 18 May 2012 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of p13k alpha |
| US7811572 | 14 Aug 2006 | 12 Oct 2010 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| US20040235840 | 20 May 2004 | 25 Nov 2004 | Immunogen, Inc. | Cytotoxic agents comprising new maytansinoids |
Exelixis, Inc.
210 East Grand Avenue
So. San Francisco, CA 94080
(650) 837-7000 phone
(650) 837-8300 fax

////////////Voxtalisib hydrochloride, Exelixis, SANOFI, PHASE 2, Malignant neoplasms, SAR-245409, XL-765


TAK 272, For Hypertension, Takeda’s Next Sartan
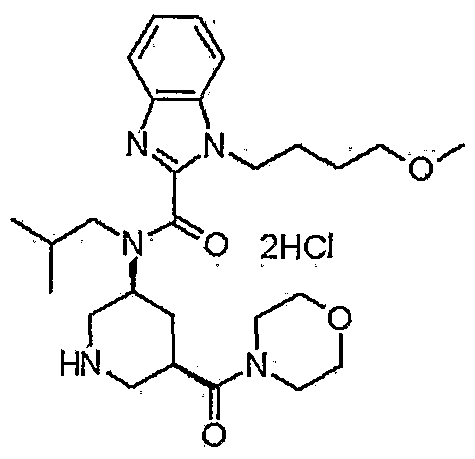
TAK 272
C27 H41 N5 O4 . Cl H, 536.106
CAS.1202269-24-6. MonoHCl
1202265-90-4 DIHCL
Base cas…1202265-63-1
Metanesulfonate…1202266-34-9
Takeda Pharmaceutical Company Limited, INNOVATOR
see……….http://www.allfordrugs.com/2015/10/21/tak-272-for-hypertension-takedas-next-sartan/
1-(4-methoxybutyl)-N-(2-methylpropyl)-N-[(3S,5R)-5-(morpholin-4-ylcarbonyl)-piperidin-3-yl]-1H-benzimidazole-2-carboxamide
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
N-Isobutyl-1-(4-methoxybutyl)-N-[5(R)-(morpholin-4-ylcarbonyl)piperidin-3(S)-yl]-1H-benzimidazole-2-carboxamide hydrochloride
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidine-3 – yl] -1H- benzimidazole-2-carboxamide hydrochloride,
![]()
The compound is used as renin inhibitor for treating diabetic nephropathy and hypertension
Takeda’s TAK-272, was reported to be in phase II in October 2015), an oral renin inhibitor, for treating diabetic nephropathy and hypertension
- 01 Apr 2015Takeda completes a phase I drug-drug interaction trial in Healthy volunteers in Japan (NCT02370615)
- 18 Feb 2015Takeda plans a phase I drug-drug interaction trial in Healthy volunteers in Japan (NCT02370615)
- 13 Feb 2015Takeda plans a phase I pharmacokinetics trial in Renal or Hepatic impairment patients in Japan (NCT02367872)

In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.
(Wherein each symbol is as described in Patent Document 2.)
Patent literature
Patent Document 2: International Publication No. 2007/077005
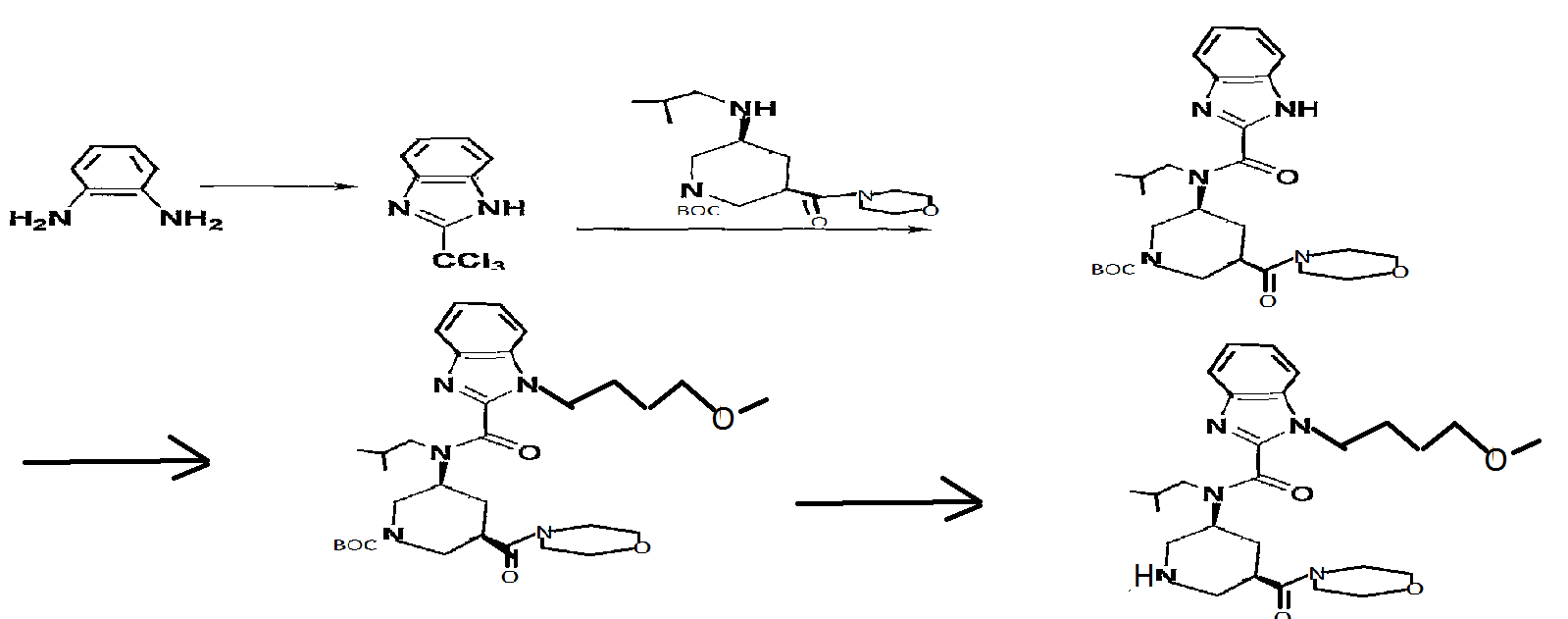
WO2009154300
https://www.google.co.in/patents/WO2009154300A2?cl=en
INTERMEDIATES FOR CONSTRUCTION

USE THIS ONE




Reference Example 31 tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate and 1- (4-methoxybutyl) -N-
(2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4- ylcarbonyl)piperidin-3-yl]-lH-benzimidazole-2-carboxamide
tert-Butyl (3S, 5R) -3-{ [ ( {2- [ (4- methoxybutyl) amino] phenyl}amino) (oxo) acetyl] (2- methylpropyl) amino} -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.11 g) was dissolved in acetic acid (50 ml), and the mixture was stirred at 😯0C for 15 hr. The reaction mixture was cooled to room temperature and concentrated under reduced pressure, the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give tert- butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl } (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (5.85 g) , and a fraction eluted with ethyl acetate-methanol (85:15) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2- methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin- 3-yl] -lH-benzimidazole-2-carboxamide (580 mg) . [0424] tert-butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl ) piperidine-1-carboxylate 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3. 46 ( 6H, m) , 3.49-3. 91 (1OH, m) , 3. 95-4 . 47 (5H, m) , 7 . 18-7 . 51 (3H, m) , 7. 56-7 . 84 ( IH, m) .
MS (ESI+, m/e) 600 (M+l )
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl)piperidin-3-yl] -lH-benzimidazole-2-carboxamide BASE
1H-NMR (CDCl3) δ 0.64-0.74 (2H, m) , 0.95-1.07 (4H, m) , 1.43-
1.74 (3H, m) , 1.84-2.41 (4H, m) , 2.48-2.67 (IH, m) , 2.67-3.01
(3H, m), 3.03-3.44 (8H, m) , 3.47-3.78 (9H, m) , 4.06-4.46 (3H, m) , 7.28-7.47 (3H, m) , 7.62-7.81 (IH, m) . MS (ESI+, m/e) 500 (M+l)
Example 10
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-
4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide dihydrochloride
tert-Butyl (3S,5R)-3-[{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5-
(morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, and the residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give 1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide (4.40 g) . The obtained 1- (4-methoxybutyl) -N- (2-methylpropyl) – N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -IH- benzimidazole-2-carboxamide (2.20 g) was dissolved in ethyl acetate (20 ml) , 4M hydrogen chloride-ethyl acetate (5 ml) and methanol (20 ml) were added, and the mixture was stirred at room temperature for 5 min. The reaction mixture was concentrated under reduced pressure to give the object product (2.52 g).
dihydrochloride
1H-NMR (DMSO-d6) δ 0.63-0.76 (2H, m) , 0.85-1.00 (4H, m) , 1.40-
1.60 (2H, m) , 1.68-1.89 (2H, m) , 1.93-2.17 (2H, m) , 2.20-2.44
(2H, m) , 2.81-3.81 (2OH, m) , 4.19-4.39 (3H, m) , 7.23-7.46 (2H, m) , 7.57-7.81 (2H, m) , 8.38-9.77 (2H, m) .
MS (ESI+, m/e) 500 (M+l)
Example 252
1- ( 4-methoxybutyl ) -N- ( 2-methylpropyl ) -N- [ ( 3S 1. 5R) -5- (morpholin- 4-ylcarbonyl ) piperidin-3-yl ] -lH-benzimidazole-2-carboxamide methanesulfonate

l-(4-Methoxybutyl) -N- (2-methylpropyl) -N- [ (3S,5R)-5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide (208 mg) was dissolved in ethyl acetate (2 ml) , a solution of methanesulfonic acid (40 μl) in ethyl acetate (1 ml) was added at 75°C, hexane (1 ml) was added, and the mixture was heated under reflux and stood at room temperature overnight. The precipitated crystals were collected by filtration, and dried at 7O0C for 3 hr to give the object product (158 mg) . MS (ESI+, m/e) 500 (M+l) melting point : 144.40C
EXTRAS IF REQD .………….
Example 32
methyl (3R, 5S)-5-[{ [1- (4-methoxybutyl) -lH-benzimidazol-2- yl] carbonyl} (2-methylpropyl) amino] piperidine-3-carboxylate dihydrochloride [0675]

MS (ESI+, m/e) 445 (M+l)
Example 33
(3R, 5S) -5- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yljcarbonyl} (2-methylpropyl) amino] piperidine-3-carboxylic acid dihydrochloride

MS (ESI+, m/e) 431 (M+l)
Reference Example 29
{ [ ( 3S , 5R) -1- (tert-butoxycarbonyl ) -5- (morpholin-4- ylcarbonyl ) piperidin-3~yl ] ( 2-itιethylpropyl ) amino } (oxo ) acetic acid
To a solution of tert-butyl (3S,5R)~3-{ [ethoxy (oxo) acetyl] (2-methylpropyl) amino}-5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate (10.3 g) in ethanol (40 ml) was added 2M aqueous sodium hydroxide solution (22 ml) , and the mixture was stirred at room temperature for 6 hr. The reaction mixture was adjusted to pH 7 with IM hydrochloric acid, and extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure to give the object product (10.3 g) .
1H-NMR (CDCl3) δ 0.78-0.99 (6H, m) , 1.37-1.52 (9H, m) , 1.79- 2.16 (3H, m) , 2.38-3.86 (14H, m) , 3.93-4.43 (2H, m) . MS (ESI+, m/e) 442 (M+l)
Reference Example 28
tert-butyl (3S, 5R) -3-{ [ethoxy (oxo) acetyl] (2- methylpropyl ) amino } -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate
To a solution of tert-butyl (3S, 5R) -3- [ (2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine-1- carboxylate (9.24 g) and diisopropylethylamine (10.5 ml) in DMA (100 ml) was added dropwise ethyl chloroglyoxylate (3.4 ml) at 0°C. The reaction mixture was stirred at room temperature for 15 hr, and the reaction mixture was concentrated. An aqueous sodium bicarbonate solution was added to the residue, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (10.3 g) . 1H-NMR (CDCl3) δ 0.84-1.00 (6H, m) , 1.37 (3H, q) , 1.42-1.53 (9H, m) , 1.80-2.19 (3H, m) , 2.26-2.42 (IH, m) , 2.59-2.96 (IH, in) , 2.97-3.30 (3H, m) , 3.37-3.92 (9H, m) , 4.01-4.26 (2H, m) , 4.26- 4.40 (2H, m) . MS (ESI4-, m/e) 470 (M+l) “
Reference Example 22 tert-butyl (3S, 5R) -3- [ (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate
[0369] tert-Butyl (3S,5R)-3-{ [ (benzyloxy) carbonyl] aminoJ-5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (58 g) and palladium (II) hydroxide-carbon (5 g) were suspended in methanol (400 ml) and the mixture was stirred under a hydrogen atmosphere (1 atom) at room temperature for 16 hr. The palladium catalyst was filtered off, and the filtrate was concentrated under reduced pressure. The obtained residue and acetic acid (8.8 ml) were dissolved in methanol (400 ml), 2- methylpropanal (14.0 ml) was added, and the mixture was stirred at room temperature for 1 hr. Sodium triacetoxyborohydride (40.4 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 2 hr. The reaction mixture was concentrated under reduced pressure, and the concentrate was basified with 3.5M aqueous potassium carbonate solution, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:5) – ethyl acetate-hexane (1:1) was concentrated under reduced pressure to give the object product (33.3 g) .
1H-NMR (CDCl3) δ: 0.90 (6H, d) , 1.46 (9H, s) , 1.54 (IH, d) , 1.69 (IH, dt), 1.96-2.12 (2H, m) , 2.23-2.37 (IH, m) , 2.47 (3H, d) , 2.66 (IH, d) , 3.61 (IH, br s) , 3.55 (2H, d) , 3.69 (5H, ddd) , 4.01-4.46 (2H, m) .
Example 6 1-tert-butyl 3-methyl (3R, 5S) -5-aminopiperidine-l, 3- dicarboxylate [0318]

(3S, 5R) -1- (tert-Butoxycarbonyl) -5-(methoxycarbonyl)piperidine-3-carboxylic acid (2.83 g) was suspended in toluene (36 ml), diphenylphosphoryl azide (2.60 ml) and triethylamine (1.70 ml) were added, and the mixture was stirred at 100°C for 1 hr. The reaction mixture was cooled to room temperature, benzyl alcohol (1.53 ml) and triethylamine (7.00 ml) were added and the mixture was stirred at 80°C for 3 hr. The reaction mixture was concentrated, the residue was dissolved in ethyl acetate, and the solution was washed with water, 0.5M hydrochloric acid, saturated aqueous sodium hydrogen carbonate and saturated brine in this order, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:3 – 3:1) was concentrated under reduced pressure. The obtained residue was dissolved in methanol (60 ml), 10% palladium carbon (50% in water) (150 mg) was added and the mixture was stirred under a hydrogen pressurization (5 atom) at ambient temperature and normal pressure for 5 hr. The catalyst was filtered off, and the filtrate was concentrated under reduced pressure to give the object product (1.83 g) as an oil.
1H-NMR (CDCl3) δ 1.22-1.43 (4H, m) , 1.46 (9H, s), 2.27-2.79 (4H, m) , 3.70 (3H, s) , 4.13 (2H, br s) [0320] In the same manner as in the method shown in Reference Example 6, the following compound (Reference Example 7) was obtained.
Reference Example 8
1-tert-butyl 3-methyl (3R, 5S) -5- [ (2- methylpropyl) amino] piperidine-1, 3-dicarboxylate [0325]

1-tert-Butyl 3-methyl (3R, 5S) -5-aminopiperidine-l, 3- dicarboxylate (1.83 g) , isobutyraldehyde (0.78 ml) and acetic acid (0.49 ml) were dissolved in methanol (50 ml), and the mixture was stirred at room temperature for 30 min. Sodium triacetoxyborohydride (3.80 g) was added to the reaction mixture, and the mixture was stirred at room temperature for 7 hr. The reaction mixture was concentrated under reduced pressure, the concentrate was basified with aqueous sodium bicarbonate, and extracted with ethyl acetate. The extract was washed with water and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:1) – ethyl acetate 100% – ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give the object product (1.42 g) as an oil.
1H-NMR (CDCl3) δ 0.90 (6H, d) , 1.22-1.38 (3H, m) , 1.46 (9H, s) , 1.69 (IH, dt), 2.23-2.39 (2H, m) , 2.44-2.59 (IH, m) , 2.47 (2H, d) , 2.74 (IH, br s) , 3.69 (3H, s) , 4.18-4.34 (2H, m)
Reference Example 27
N- (4-methoxybutyl) benzene-1, 2-diamine

To a solution of phenylenediamine (10.8 g) and 4- methoxybutyl methanesulfonate (9.11 g) in acetonitrile (100 ml) was added potassium carbonate (20.7 g) , and the mixture was stirred heated under reflux for 15 hr. Water was added to the reaction mixture, and the mixture was extracted twice with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (35:65) was concentrated under reduced pressure to give the object product (5.44 g) . 1H-NMR (CDCl3) δ 1.67-1.82 (4H, m) , 3.13 (2H, t) , 3.24-3.39 (6H, m) , 3 . 38 -3 . 50 ( 2H, m) , 6 . 62 – 6 . 74 ( 3H, m) , 6 . 81 ( IH, in) . MS ( ESI+ , m/e ) 195 (M+l )
Reference Example 146 tert-butyl (3S, 5R) -3- [ { [1- (4-methoxybutyl) -lH-benzimidazol-2- yl]carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate

A solution of tert-butyl (3S, 5R) -3- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl)piperidine-l-carboxylate (200 mg) , 4-itιethoxybutyl methanesulfonate (107 mg) and cesium carbonate (254 mg) in N,N-dimethylacetamide (5 ml) was stirred at 60°C for 15 hr. After cooling to room temperature, the reaction mixture was diluted with water and extracted with ethyl acetate (10 ml*2) . The extract was washed with saturated brine, and dried over anhydrous magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (5:95 – 3:7) was concentrated under reduced pressure to give the object product (190 mg) . 1H-NMR (CDCl3) δ 0.63-0.80 (2H, m) , 0.89-1.07 (4H, m) , 1.41- 1.59 (9H, m) , 1.59-1.80 (2H, m) , 1.87-2.23 (4H, m) , 2.30-2.98 (3H, m) , 3.21-3.46 (6H, m) , 3.49-3.91 (1OH, m) , 3.95-4.47 (5H, m) , 7.18-7.51 (3H, m) , 7.56-7.84 (IH, m) . MS (ESI+, m/e) 600 (M+l)
ALTERNATE METHOD IN THIS PATENT



Reference Example 61
2- (trichloromethyl) -lH-benzimidazole

O-Phenylenediamine (25 g) was dissolved in acetic acid (750 ml), and methyl 2, 2, 2-trichloroacetimidate (28.5 ml) was added dropwise over 15 min. After stirring at room temperature for 1 hr, the reaction mixture was concentrated to about 150 ml, and poured into water (1500 ml) . The precipitated crystals were collected by filtration, washed with water (1000 ml) and suspended in toluene (500 ml) . The solvent was evaporated under reduced pressure. The residue was again suspended in toluene (500 ml) and the solvent was evaporated under reduced pressure. The residue was dried under reduced pressure to give the object product (51.8 g) . 1H-NMR (CDCl3) δ 7.31-7.45 (2H, m) , 7.49-7.55 (IH, m) , 7.89 (IH, d) , 9 . 74 ( IH, br s )
Reference Example 64
1-tert-butyl 3-methyl (3R, 5S) -5- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] piperidine-1, 3-dicarboxylate

2- (Trichloromethyl) -lH-benzimidazole (19 g) and 1-tert- butyl 3-methyl (3R, 5S) -5- [ (2-methylpropyl) amino] piperidine- 1,3-dicarboxylate (25 g) were dissolved in THF (1200 ml), sodium hydrogen carbonate (67 g) and water (600 ml) were added, and the mixture was stirred at room temperature for 1 hr and at 5O0C for 1 hr. After evaporation of the solvent, the residue was extracted 3 times with ethyl acetate (700 ml) . The extract was washed successively with 10%-aqueous citric acid solution (500 ml) and brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure.
The residue was dissolved in ethyl acetate (1000 ml), subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate was concentrated under reduced pressure to give the object product (30.6 g) .
1H-NMR (CDCl3) δ 0.78-1.09 (6 H, m) , 1.17-1.55 (9 H, m) , 1.77-2.95 (5 H, m) , 3.11-3.79 (6 H, m) , 3.99-4.73 (4 H, m) , 7.24- 7.41 (2 H, m) , 7.45-7.59 (1 H, m) , 7.72-7.88 (1 H, m) , 10.66-10.98 (1 H, m)MS (ESI+, m/e) 459 (M+l)
Reference Example 69
1-tert-butyl 3-methyl (3R, 5S) -5- [ { [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] piperidine-1 , 3-dicarboxylate

1-tert-Butyl 3-methyl (3R, 5S) -5- [ (lH-benzimidazol-2- ylcarbonyl) (2-methylpropyl) amino] piperidine-1, 3-dicarboxylate (30 g) and 4-methoxybutyl methanesulfonate (12.5 g) were dissolved in DMA (600 ml), cesium carbonate (32 g) was added, and the mixture was stirred at 70°C for 12 hr. The reaction mixture was poured into ice water (1000 ml), and the mixture was extracted twice with ethyl acetate (1000 ml) . The extract was washed with brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to silica gel column chromatography, and a fraction eluted with ethyl acetate-hexane (1:4 – 1:1) was concentrated under reduced pressure to give the object product (28.7 g) .
1H-NMR (CDCl3) δ 0.76 (4H, d) , 1.01 (2H, d) , 1.30-1.52 (9H, m) , 1.58-2.07 (4H, m) , 2.10-2.93 (4H, m) , 3.27-3.75 (12H, m) , 4.06-4.57 (5H, m) , 7.26-7.48 (3H, m) , 7.79 (IH, d) MS (ESI+, m/e) 545 (M+l)
Example 71
1- (4-methoxybutyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin- 4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2-carboxamide

tert-Butyl (3S, 5R) -3- [{ [1- (4-methoxybutyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (5.85 g) was dissolved in methanol (20 ml) , 4M hydrogen chloride-ethyl acetate (20 ml) was added, and the mixture was stirred at room temperature for 15 hr. The reaction mixture was concentrated, the residue was diluted with aqueous sodium bicarbonate,…and, the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure. The residue was subjected to basic silica gel column chromatography, and a fraction eluted with ethyl acetate- methanol (9:1) was concentrated under reduced pressure to give the object product (4.40 g) . MS (ESI+, m/e) 500 (M+l)
Example 101
1- (5-methoxypentyl) -N- (2-methylpropyl) -N- [ (3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -lH-benzimidazole-2- carboxamide dihydrochloride

[1144] tert-Butyl (3S, 5R) -3- [ { [1- (5-methoxypentyl) -IH- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4-ylcarbonyl)piperidine-l-carboxylate (123 mg) was dissolved in 4M hydrogen chloride-ethyl acetate (5 ml) , and the mixture was stirred at room temperature for 3 hr. The reaction mixture was concentrated, and the residue was subjected to reversed-phase preparative HPLC and the eluted fraction was concentrated under reduced pressure. The residue was diluted with aqueous sodium bicarbonate, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, and dried over anhydrous sodium sulfate. 4M Hydrogen chloride-ethyl acetate (1 ml) was added and the mixture was stirred for 5 min. The solvent was evaporated under reduced pressure to give the object product (76 mg) . MS (ESI+, m/e) 514 (M+l)
PATENT
WO2013122260
http://www.google.co.in/patents/WO2013122260A1?cl=en
PATENT
WO 2011158880
http://www.google.co.in/patents/WO2011158880A1?cl=en
Reference Example 1
1- (4-methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-ylcarbonyl) piperidin-3-yl] -1H- benzimidazole -2 – carboxamide hydrochloride (A-type crystal)
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) was suspended dissolved piperidine-1-carboxylate The (300g) in 3N- hydrochloric acid water (1200mL) and Ethyl acetate (60mL), and stirred over 3 h at 25 ~ 35 ℃. After completion of the reaction, it was added ethyl acetate (2400mL) in the same temperature. After the addition, it was added 25% aqueous ammonia (600mL) with cooling. After the addition stirring and extracted the organic layer of 5% aqueous ammonia (600mL) was added and stirred. After stirring, the resulting organic layer it was concentrated until the solvent no longer distilled off. After concentrated, dissolved with ethyl acetate (1500mL), and transferred to solution to the crystallizer vessel, and washed with ethyl acetate (750mL). After washing, it was raised in stirring under 45 ~ 55 ℃. After raising the temperature, at the same temperature 4N- hydrogen chloride – it was dropped ethyl acetate (131.3mL). After dropping, it was to dissolve the precipitate at the same temperature. After dissolution confirmation, it was added heptane (750mL) at 40 ~ 50 ℃, after the addition, then cooled to 25 ~ 35 ℃. After cooling, the addition of A-type crystals of the seed crystals (300mg) which was obtained according to the method described in Example 265 of WO2009 / 154300, and stirred for 30 minutes or more. After stirring, the temperature was raised to 40 ~ 45 ℃, it was dropped heptane (1500mL). After the completion of the dropping, it was stirred at the same temperature. Then gradually cooled to 5 ℃ below, followed by stirring at the same temperature for 1 hour. After stirring, ethyl acetate and filtered crystals – heptane: washed with (1 1,600mL), to obtain a wet crystal. The obtained wet crystals dried under reduced pressure at 50 ℃, 1- (4- methoxybutyl) -N- (2- methylpropyl) -N – [(3S, 5R) -5- (morpholin-4-yl carbonyl) piperidin-3-yl] -1H- obtained a crystalline powder of benzimidazole-2-carboxamide hydrochloride (A-type crystal, 198.82g, 74.1% yield). FINAL PRODUCT
TERT BUTYL DERIVATIVE, N-1
Reference Example 4
tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5- (morpholin-4- ylcarbonyl) piperidine-1-carboxylate 1)
o- nitro aniline (50.0g, 0.362mol), tetrabutylammonium bromide (58.3g, 0.181mol), potassium bromide (43.1g, 0.362mol) in toluene (500mL ) and it was added. At a temperature of 20 ~ 30 ℃ 1- chloro-4-methoxy-butane (66.6g, 0.543mol) and, I was added to 50w / v% sodium hydroxide solution (145mL, 1.81mol). The reaction was heated to a temperature 85 ~ 95 ℃, and stirred for 6 hours. After cooling to a temperature 20 ~ 30 ℃, the reaction mixture water (250mL), 1N- aqueous hydrochloric acid (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), it was washed successively with water (250mL). After concentration under reduced pressure the organic layer to Contents (250mL), was added toluene (100mL), was obtained
N- (4- methoxy-butyl) -2-nitroaniline in toluene (350mL, 100% yield).
1 H-NMR (300MHz, CDCl 3) δ 1.64-1.89 (m, 4H), 3.25-3.39 (m, 2H), 3.35 (s, 3H), 3.44 (t, J = 6.1 Hz, 2H), 6.63 ( ddd, J = 8.5, 6.9, 1.2 Hz, 1H), 6.86 (dd, J = 8.5, 1.2 Hz, 1H), 7.43 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 8.07 (br s, 1H ), 8.17 (dd, J = 8.5, 1.5 Hz, 1H).
2) N- (4-methoxy-butyl) -2-10 percent in nitroaniline of toluene solution (350mL) Pd / C (K-type, 50% water-containing product) (10.0g) and toluene (100mL) it was added. Hydrogen pressure of 0.1MPa, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. A stream of nitrogen, the catalyst was filtered, I was washed with toluene (100mL). After the water in the filtrate was separated off and adding magnesium sulfate (25.0g) at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered over magnesium sulfate, washed with toluene (100mL), was obtained N- (4- methoxybutyl) -o- toluene solution of phenylenediamine (100% yield).
1 H NMR (500 MHz, CDCl 3) δ1.67-1.78 (m, 4H), 3.12-3.14 (m, 2H), 3.32 (br, 3H), 3.35 (s, 3H), 3.41-3.47 (m, 2H), 6.63-6.69 (m, 2H), 6.69-6.74 (m, 1H), 6.82 (td, J = 7.57, 1.58 Hz, 1H).
3) N- (4- methoxy-butyl) -o- After the toluene solution of phenylenediamine cooled to a temperature 0 ~ 10 ℃, acetic acid (65.2g, 1.09mol) and 2,2,2 trichloroacetimide acid methyl ( 70.3g, 0.398mol) and I were added. After stirring for 30 minutes at a temperature 0 ~ 10 ℃, it was stirred for 3 hours at a temperature of 20 ~ 30 ℃. The reaction was 5w / v% saline (250mL), 2N- aqueous hydrochloric acid / 5w / v% sodium chloride solution: a mixture of (1 1) (250mL × 2), 5w / v% aqueous solution of sodium bicarbonate (250mL), 5w / v It was washed successively with% saline solution (250mL). A stream of nitrogen, was added magnesium sulfate (25.0g) to the organic layer at a temperature 20 ~ 30 ℃, and stirred at the same temperature for 30 minutes. Filtered magnesium sulfate, and washed with toluene (100mL). The filtrate was concentrated under reduced pressure and the amount of contents (150mL). Stir the concentrated solution at a temperature 20 ~ 30 ℃, was allowed to precipitate crystals, was added dropwise heptane (750mL). The crystals bleeding is heated to a temperature 40 ~ 50 ℃, after stirring for 30 min, cooled to a temperature 0 ~ 10 ℃, and the mixture was stirred at the same temperature for 2 hours.The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,150 mL). And dried under reduced pressure at 40 ℃, it was obtained 1- (4-methoxy-butyl) -2-fine brown crystals of trichloromethyl -1H- benzimidazole (96.5g, 82.9% yield from o- nitroaniline).
1 H-NMR (300MHz, CDCl 3) δ: 1.68-1.85 (m, 2H), 1.99-2.17 (m, 2H), 3.37 (s, 3H), 3.48 (t, J = 6.1 Hz, 2H), 4.50 -4.65 (m, 2H), 7.27-7.49 (m, 4H), 7.82-7.93 (m, 1H).
. Anal Calcd for C 13 H 15 Cl 3 N 2 O:. C, 48.55; H, 4.70; N, 8.71; Cl, 33.07 Found: C, 48.30; H, 4.61; N, 8.74; Cl, 33.30.
4) pyridine-3,5-dicarboxylic acid (110g, 0.66mol), it was dropped methanol (660 mL) mixture of concentrated sulfuric acid at a temperature of 50 ℃ or less of (226.0g, 2.30mol). Thereafter, the mixture was stirred and heated to a temperature 55 ~ 65 ℃ 7 hours. The reaction was the temperature 40 ~ 50 ℃, was added water (220mL). And further dropping temperature 40-50 5% aqueous ammonia at ℃ (about 1.10L) was adjusted to pH8.0 ~ 8.5. After stirring at a temperature 40 ~ 50 ℃ 30 minutes and stirred for 1 hour and cooled to a temperature 0 ~ 10 ℃. Was collected by filtration precipitated crystals, methanol – water (1: 3,165mL), and washed successively with water (440mL). To obtain a white crystalline powder pyridine-3,5-dicarboxylic acid dimethyl and dried under reduced pressure at 50 ℃ (105.0g, 82.0% yield).
1 H-NMR (300 MHz, CDCl 3) δ 4.00 (s, 6H), 8.87 (s, 1H), 9.37 (s, 2H).
. Anal Calcd for C 9 H 9 NO 4:. C, 55.39; H, 4.65; N, 7.18; O, 32.79 Found: C, 55.42; H, 4.65; N, 7.16.
5) 1 L autoclave pyridine-3,5-dicarboxylic acid dimethyl (100g, 0.51mol) and was charged with dimethylacetamide (400mL), temperature 30 ℃ below with trifluoroacetic acid (59.2mL, after dropping the 0.77mol), 10% Pd-C (PE-type) the (20.0g) it was added. Hydrogen pressure of 0.5 ~ 0.7MPa, it was stirred for 12 hours at a temperature of 55 ~ 65 ℃. The catalyst was filtered off, it was washed with dimethylacetamide (50mL × 2). Triethylamine and the combined filtrates at a temperature 20 ~ 30 ℃ (77.8g, 0.77mol) was added dropwise, and adjusted to pH9.0 ~ 10.0. Temperature 30 ~ 40 ℃ by di -tert- butyl (134g, 0.614mol) was added dropwise and stirred at the same temperature for 2 hours. After the reaction mixture as a 20 ~ 30 ℃, it was added ethyl acetate (600mL), washed with water (900mL). The aqueous layer it was re-extracted with ethyl acetate (400mL). The combined organic layers 5w / v% citric acid -10w / v% sodium chloride solution (600mL), 3% aqueous sodium bicarbonate (600mL), and washed successively with water (600mL). Contents The organic layer (200mL) until it was concentrated under reduced pressure, methanol (250mL) was added to the concentrated solution, and then concentrated under reduced pressure until Contents (200mL). The addition of methanol (250mL) again concentrate, After concentration under reduced pressure until Contents (200mL), was added methanol (2.40L). The solution in water (18.5g, 1.03mol), cesium carbonate (417g, 1.28mol) was added and stirred for about 24 hours at a temperature 55 ~ 65 ℃. The reaction solution was the temperature 20 ~ 30 ℃, concentrated to Contents (700mL), it was added tetrahydrofuran (500mL). The solution temperature at 15 ~ 35 ℃ 2N- hydrochloric acid solution (1.28L, 2.56mol) was added dropwise and adjusted to pH3.0 ~ 3.5, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Extracted with ethyl acetate (750mL × 2), and the organic layer was washed with 10w / v% aqueous sodium chloride solution (500mL × 3). Contents The organic layer (300mL) until it was concentrated under reduced pressure, to obtain a weight content by adding ethyl acetate (650mL).Heating the concentrate to a temperature of 55 ~ 65 ℃, it was added dropwise heptane (500mL). It cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. The precipitated crystals were collected by filtration, ethyl acetate – heptane: was washed with (1 1,120mL). Dried under reduced pressure at 50 ℃ 1- (tert- butoxycarbonyl) to give a white crystalline powder of piperidine-3,5-dicarboxylic acid (113.3g, 80.9% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.40 (s, 9H), 1.44-1.61 (m, 1H), 2.21-2.26 (m, 1H), 2.31-2.41 (m, 2H), 4.10- 4.12 (m, 2H).
. Anal Calcd for C 12 H 19 NO 6:. C, 52.74; H, 7.01; N, 5.13; O, 35.13 Found: C, 52.96; H, 6.99; N, 5.39.
6) Under a nitrogen stream, 1- (tert- butoxycarbonyl) piperidine-3,5-dicarboxylic acid (5.00g, 18.3mmol) was suspended in tetrahydrofuran (10.0mL), trifluoroacetic acid anhydride at a temperature 20 ~ 30 ℃ It was dropping things (3.80mL, 27.5mmol). After the completion of the dropping, it was stirred for 1 hour at a temperature of 20 ~ 30 ℃. It was added dropwise heptane (20.0mL) at a temperature 20 ~ 30 ℃ the reaction solution, and stirred for 3 hours then cooled to a temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with heptane (3.00mL). Dried under reduced pressure at 40 ℃ 2,4- dioxo-3-oxa-7-azabicyclo [3,3,1] white crystalline powder of nonane-7-carboxylic acid tert- butyl was obtained (4.03g, yield 86.1%).
1 H-NMR (300 MHz, CDCl 3) δ 1.43 (s, 9H), 1.93-1.99 (m, 1H), 2.40-2.46 (m, 1H), 3.06-3.11 (m, 4H), 4.50-4.54 ( m, 2H).
. Anal Calcd for C 12 H 17 NO 5:. C, 56.46; H, 6.71; N, 5.49; O, 31.34 Found: C, 56.51; H, 6.63; N, 5.69.
7) Under a nitrogen stream, quinidine (69.9g, 0.215mol) and was charged with tetrahydrofuran (200mL), and cooled to a temperature -5 ~ 5 ℃. At the same temperature 2,4-dioxo-3-oxa-7-azabicyclo [3,3,1] nonane-7-carboxylic acid tert- butyl (50.0g, 0.196mol) was added and washed with tetrahydrofuran (50.0mL) crowded. Temperature -5 ~ 5 methanol at ℃ (9.41g, 0.29 4mol) was added dropwise, and the mixture was stirred for 2 hours at a temperature -5 ~ 5 ℃. Ethyl acetate (350mL) to the reaction mixture, was by adding minute solution 20w / v% citric acid aqueous solution (250mL). The aqueous layer it was re-extracted with ethyl acetate (125mL × 2). The organic layers were combined 20w / v% aqueous solution of citric acid (250mL), I was washed successively with water (250mL × 2). The organic layer it was concentrated under reduced pressure. To the residue ethanol (100mL) was added ethyl acetate (450mL) was heated to a temperature 60 ~ 70 ℃, (R) – was added phenethylamine (23.7g, 0.196mol). Temperature 50-60 for one hour at ℃, 1 hour at a temperature of 20 ~ 30 ℃, it was stirred for 1 hour at a temperature of -5 ~ 5 ℃. The precipitated crystals were collected by filtration, ethanol – ethyl acetate: and washed with (2 9,100mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidin-3 to give a white crystalline powder of the carboxylic acid (1R) -1- phenylethylamine salt It was (55.7g, 69.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.42 (s, 9H), 1.43-1.51 (m, 3H), 2.06-2.14 (m, 1H), 2.21-2.26 (m, 1H), 2.39- 2.44 (m, 1H), 2.52-2.53 (m, 1H), 2.57 (br s, 2H), 3.64 (s, 3H), 4.12 (br s, 2H), 4.19-4.26 (m, 1H), 7.30- 7.40 (m, 3H), 7.45-7.48 (m, 2H).
. Anal Calcd for C 21 H 32 N 2 O 6:. C, 61.75; H, 7.90; N, 6.86; O, 23.50 Found: C, 61.54; H, 7.77; N, 6.86.
8) (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (1R) -1- phenylethylamine salt (20.0g, 49.0mmol), methanol (20mL) and it was charged with water (80mL). Temperature 20-30 citric acid at ℃ (11.3g, 58.8mmol) was added dropwise a solution prepared by dissolving in water (20.0mL), and the mixture was stirred 1.5 hours at the same temperature. The precipitated crystals were collected by filtration and washed with water (60mL). And dried under reduced pressure at 50 ℃ (3S, 5R) -1- (tert- butoxycarbonyl) -5- give a white crystalline powder (methoxycarbonyl) piperidine-3-carboxylic acid (13.5g, 96.1% yield ).
1 H-NMR (300 MHz, CDCl 3) δ 1.40 (s, 9H), 1.46-1.59 (m, 1H), 2.22-2.27 (m, 1H), 2.37-2.45 (m, 2H), 2.63-2.73 ( m, 2H), 3.63 (s, 3H), 4.14 (br s, 2H), 12.51 (br s, 1H).
. Anal Calcd for C 13 H 21 NO 6:. C, 54.35; H, 7.37; N, 4.88; O, 33.41 Found: C, 54.14; H, 7.28; N, 4.85.
9) Under a nitrogen stream, (3S, 5R) -1- (tert- butoxycarbonyl) -5- (methoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 104mmol), triethylamine (31.7g, 313mmol) and toluene ( It was charged with 180mL). Diphenylphosphorylazide at a temperature of 15 ~ 35 ℃ (28.7g, 313mmol) I was dropped a toluene (30.0mL) solution. After stirring at a temperature 30 ± 5 ℃ 30 minutes, and the mixture was stirred and heated to a temperature 65 ~ 75 ℃ 30 minutes. Temperature 60 ~ 70 ℃ in the benzyl alcohol (12.4g, 115mmol) it was dropped. To a temperature 80 ~ 90 ℃ was stirred and heated for 3 hours. The reaction mixture was cooled to a temperature 20 ~ 30 ℃, sodium nitrite (7.20g, 104mmol) and after stirring was added a solution prepared by dissolving in water (150mL) 1 hour, the aqueous layer was separated. The organic layer 5w / v% aqueous sodium bicarbonate solution (150mL), 20w / v% aqueous citric acid solution (150mL), washed successively with 5w / v% aqueous sodium chloride solution (150mL), the organic layer was concentrated under reduced pressure. The residue methanol (60.0mL) was added and concentrated under reduced pressure to. The more we went once in the same manner.To the residue was added methanol and the content amount of the (90.0g). Temperature 15 ~ 35 ℃ 2N- aqueous sodium hydroxide (62.6mL, 125mmol) was added and stirred for 1 hour at a temperature 30 ± 5 ℃. Temperature 20 ~ 30 ℃ in methanol (120mL), was added to 20w / v% aqueous citric acid solution (300mL), it was a pH3.0 ~ 3.5. After stirring for 30 minutes at a temperature 50 ~ 60 ℃, cooled to a temperature 20 ~ 30 ℃ and stirred for 1 hour. It was stirred for 1 hour at the temperature 0 ~ 10 ℃. The precipitated crystals were collected by filtration, and washed with water (90.0mL). And dried under reduced pressure at 50 ℃ (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) to yield a white crystalline powder piperidine-3-carboxylic acid (35.0 g, 88.6% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 2.11 (d, J = 12.4 Hz, 1H), 2.40-2.48 (m, 4H), 2.62 (br s, 1H), 4.08 (t, J = 14.4 Hz, 2H), 5.04 (s, 2H), 7.31-7.41 (m, 5H), 12.53 (br s, 1H).
. Anal Calcd for C 19 H 26 N 2 O 6:. C, 60.30; H, 6.93; N, 7.40; O, 25.37 Found: C, 60.03; H, 6.99; N, 7.41.
10) Under a nitrogen stream, (3R, 5S) -5 – {[(benzyloxy) carbonyl] amino} -1- (tert- butoxycarbonyl) piperidine-3-carboxylic acid (30.0g, 79.3mmol), morpholine (7.60 g, 87.2mmol), 1- hydroxybenzotriazole monohydrate (2.43g, it was charged with 15.9mmol) and dimethylacetamide (90.0mL). Hydrochloride 1-ethyl at a temperature 20 ~ 30 ℃ -3- (3- dimethylaminopropyl) carbodiimide (16.7g, 87.1mmol) after addition and stirred for 1 hour at a temperature 45 ~ 55 ℃. Temperature 45 ~ 55 ℃ with tetrahydrofuran (90.0mL), sequentially dropwise addition of water (210mL), and stirred for 1 hour. After stirring for 1 hour and cooled to a temperature 20 ~ 30 ℃, were collected by filtration the precipitated crystals, tetrahydrofuran – water: washing with (1 3,120mL). And dried under reduced pressure at 50 ℃ tert- butyl piperidine -1- (3S, 5R) -3 – a white crystalline powder of {[(benzyloxy) carbonyl] amino} -5 (morpholin-4-yl-carbonyl) carboxylate It was obtained (32.7g, 92.3% yield).
1 H-NMR (300 MHz, DMSO-d 6) δ 1.41 (s, 9H), 1.49-1.57 (m, 1H), 1.87 (d, J = 12.3 Hz, 1H), 2.43 (br s, 1H), 2.63-2.71 (m, 1H), 2.79-2.83 (m, 1H), 3.37-3.54 (m, 9H), 3.89 (d, J = 11.5 Hz, 1H), 4.06 (br s, 1H), 5.03 (s , 2H), 7.30-7.38 (m, 5H).
. Anal Calcd for C 23 H 33 N 3 O 6:. C, 61.73; H, 7.43; N, 9.39; O, 21.45 Found: C, 61.59; H, 7.50; N, 9.43.
11) tert- Butyl piperidin -1- (3S, 5R) -3 – {[(benzyloxy) carbonyl] amino} -5- (morpholin-4-ylcarbonyl) carboxylate (30.0g, 67.0mmol), isobutyraldehyde (7.25g, 101mmol), it was charged with 10% Pd-C (PE type) (1.50g) and methanol (240mL).Hydrogen pressure of 0.2 ~ 0.3MPa, it was stirred for 4 hours at a temperature of 20 ~ 30 ℃. The catalyst is filtered off and washed with methanol (60.0mL). The filtrate was concentrated under reduced pressure, ethyl acetate was added (60.0mL), and concentrated under reduced pressure again. The residue ethyl acetate was added, followed by the amount of contents (360mL). Temperature 45-55 succinate by heating to ℃ (7.90g, 67.0mmol) was added. After stirring for 1 hour at a temperature 45 ~ 55 ℃, cooled to a temperature 20 ~ 30 ℃, and stirred for 1 hour. The precipitated crystals were collected by filtration, and washed with ethyl acetate (90.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [(2- methyl-propyl) amino] -5- (morpholin-4-yl-carbonyl) piperidine – 1-carboxylate white crystals of alert succinate got sex powder (30.2g, 92.5% yield).
1 H-NMR (300 MHz, D 2 O) δ 1.02 (s, 3H), 1.04 (s, 3H), 1.47 (s, 9H), 1.97-2.09 (m, 2H), 2.26-2.30 (m, 1H ), 2.55 (s, 4H), 2.99 (d, J = 7.0 Hz, 2H), 3.23 (br s, 1H), 3.39-3.45 (m, 2H), 3.53-3.80 (m, 10H), 3.82-3.93 (br s, 1H).
. Anal Calcd for C 23 H 41 N 3 O 8:. C, 56.66; H, 8.48; N, 8.62; O, 26.25 Found: C, 56.48; H, 8.46; N, 8.39.
12) tert- Butyl (3S, 5R) -3 – [(2- methylpropyl) amino] -5- (morpholin-4-ylcarbonyl) piperidine – 1 – carboxylate succinate (30.3g, 62.2mmol), acetonitrile (60.0mL) and, it was charged with water (40.0mL). Then after stirring was added potassium carbonate (34.4g, 0.249mmol) 10 minutes, 1- (4-methoxybutyl) -2-trichloromethyl -1H- benzimidazole (20.0g, 62.2mmol) was added. After stirring for 2 hours at a temperature of 70 ~ 80 ℃, it was added dimethyl sulfoxide (15.0mL), and the mixture was stirred for 6 hours at a temperature 70 ~ 80 ℃. After cooling the reaction mixture to a temperature 20 ~ 30 ℃, water (120mL), it was separated and by adding toluene (240mL). The organic layer 10w / v% sodium chloride solution (100mL), 10w / v% aqueous solution of citric acid (100mL), it was washed sequentially with 10w / v% sodium chloride solution (100mL). The organic layer of activated carbon Shirasagi A a (1.0g) was added, and the mixture was stirred for 30 minutes at a temperature 20 ~ 30 ℃. Activated carbon was filtered, washed with toluene (40.0mL), and concentrated under reduced pressure of the filtrate to 110 mL. By heating to a temperature 35 ~ 45 ℃ was added dropwise heptane (280mL). At a temperature 35 ~ 45 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] -5 – and the mixture was stirred for 1 hour at (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was added to the same temperature the crystals (10mg) of the acrylate. Heptane (140mL) was stirred and added dropwise to 30 minutes at a temperature 35 ~ 45 ℃. It was cooled to a temperature 20 ~ 30 ℃ and stirred for 2 hours. The precipitated crystals were collected by filtration, toluene – heptane: was washed with (1 5,40.0mL). And dried under reduced pressure at 50 ℃ tert- butyl (3S, 5R) -3 – [{[1- (4- methoxy-butyl) -1H- benzoimidazol-2-yl] carbonyl} (2-methylpropyl) amino] – 5- (morpholin-4-ylcarbonyl) piperidine-1-carboxylate was obtained a pale yellowish crystalline powder of alert (27.7g, 74.2% yield).
1 H-NMR (300 MHz, CDCl 3) δ 0.68-0.80 (m, 3H), 0.96-1.08 (m, 3H), 1.31 (br s, 5H), 1.49 (s, 4H), 1.61-1.71 (m , 2H), 1.71 (br s, 0.5H), 1.92-2.05 (m, 3H), 2.05-2.24 (m, 2H), 2.45 (br s, 1H), 2.60 (br s, 1H), 2.72-2.96 (m, 2H), 3.26-3.35 (m, 3H), 3.35-3.47 (m, 2H), 3.47-3.73 (m, 10H), 4.02-4.26 (m, 2H), 4.26-4.34 (m, 1H) , 4.34-4.47 (m, 0.5H), 7.25-7.29 (m, 1H), 7.29-7.41 (m, 1H), 7.41-7.53 (m, 1H), 7.64 (br s, 0.5H), 7.79 (d , J = 8.2 Hz, 0.5H).
. Anal Calcd for C 32 H 49 N 5 O 6:. C, 64.08; H, 8.23; N, 11.68; O, 16.01 Found: C, 63.82; H, 8.12; N, 11.64.
PATENT
TAKEDA PHARMACEUTICAL COMPANY LIMITED [JP/JP]; 1-1, Doshomachi 4-chome, Chuo-ku, Osaka-shi, Osaka 5410045 (JP)
Provided is a method for producing a synthetic intermediate of a heterocyclic compound having a renin inhibitory activity and effective as a prophylactic or therapeutic drug against diabetic renal disease, hypertension, and the like. A method for producing a compound represented by formula (III-1a), (III-1b), (III-1c), and/or (III-1d) [where the symbols in the formulas are as defined in the description], or a salt thereof, said method characterized in that a compound represented by formula (Ia) or (Ib) [where the symbols in the formulas are as defined in the description] or a salt thereof is reacted with a compound represented by formula (II) [where the symbols in the formula are as defined in the description] or a salt thereof in the presence of an aluminum compound and a chiral amine compound.
In the above method, the acid anhydride (BANC) from chiral dicarboxylic acid monoester ((-) – BMPA) were synthesized and then the carboxylic acid after conversion and hydrolysis reaction of the Z amine by the Curtius rearrangement of the carboxylic acid (BAPC) and it was then performs amidation by the condensation reaction with the amine (morpholine), is synthesized heterocyclic amide compound (BMPC). Further, Patent Document 2, the preparation of compounds useful as synthetic intermediates of the above heterocyclic compounds are disclosed.[Formula 3]

(Wherein each symbol is as described in Patent Document 2.)
Prior art documents
Patent literaturePatent Document 1: Patent No. 4,800,445 Patent
reaction vessel (1R, 3S) – was added to cyclohexane-1,3-dicarboxylic acid (10g) and THF (20mL), 5 It was cooled to ℃. It was added dropwise trifluoroacetic anhydride (8.19mL), and the mixture was stirred for about 1 hour. The reaction mixture was allowed to warm to room temperature, heptane (20mL) was added, up to 5 ℃ was cooled and stirred for about 30 minutes. The precipitate was filtered off, washed with heptane to give the title compound. Yield (6.7g)
reactor in THF (240ml), (3S, 5R) -1- (tert – butoxycarbonyl) -5- (morpholine-4-carbonyl) piperidine-3-carboxylic acid (20.0g), triethylamine (12.2mL) and diphenylphosphoryl azide (15.1mL) They were charged and allowed to react for 1 hour at 60 ℃, cooled to 25 ℃. After cooling the THF (60ml) and sodium trimethyl silanolate (19.7g) to charged 0 ℃ separately reaction vessel, was added dropwise to this was allowed to react before the reaction solution over about 1 hour, 0 at 0 ℃. 5 hours it was allowed to react. 0 slowly added dropwise acetic acid (40mL) at ℃, After stirring for 10 minutes, was added ethanol (60ml) and isobutyraldehyde (5.3mL) at 25 ℃, and stirred for 10 minutes. Then added sodium borohydride (1.88g), and the mixture was stirred for 30 minutes, and further addition of sodium borohydride (1.88g) at 25 ℃, and the mixture was stirred for 30 minutes. After completion of the reaction, water (100mL) was added and stirred for 10 minutes at room temperature. The organic layer was concentrated, then added dropwise slowly toluene (140ml) and 5N aqueous sodium hydroxide solution (120ml), the layers were separated. After washing and addition of aqueous 1N sodium hydroxide (100ml) the organic layer was washed 1N aqueous sodium hydroxide (100ml) was added again organic layer. The aqueous layers were combined and extracted by addition of toluene (100ml). The organic layers were combined, washed with 10w / v% aqueous sodium chloride solution (100ml), and the organic layer was concentrated. It was added ethanol (100ml), after it was concentrated under reduced pressure until about 60ml, warmed to 60 ℃ by the addition of ethyl acetate (40ml). Was added succinic acid (6.9g), After stirring for 30 minutes, it was added dropwise ethyl acetate (200ml) at 60 ℃, and stirred for 30 minutes. After stirring for 1 hour at room temperature, and the mixture was stirred for 1 hour at 0 ℃. The crystals were collected by filtration and washed with a mixture of ethyl acetate / n-heptane (6/1) (60mL). The obtained crystals at an external temperature of 50 ℃ to constant weight and then dried under reduced pressure to give the title compound as almost white crystals. Yield (22.8g)
the reaction vessel in chlorobenzene (7.5mL) and quinine (0.70g ) is added and stirred, it was added dropwise DIBAL1.0M hexane solution (2.16mL). The reaction mixture was cooled to -40 ℃, tert – butyl 2,4-dioxo-3-oxa-7-azabicyclo [3.3.1] was added nonane-7-carboxylic acid ester (0.50g), about 1 hour stirring. Was added chlorobenzene to another reaction vessel (2.5mL) and morpholine (0.17mL), the resulting solution was cooled to -40 ℃ was added dropwise to the previous reaction solution. After completion of the reaction, the mixture was separated with ethyl acetate and 10w / w% aqueous citric acid solution, and the resulting aqueous layer was re-extracted with ethyl acetate. The organic layers were combined, washed with 10w / w% saline, and concentrated to give the title compound. 1 H NMR (500 MHz, DMSO-D 6 ) delta ppm 1.41 (s, 9 H), 1.47 – 1.72 (M, 1 H), 1.89 – 2.10 (M, 1 H), 2.36 – 2.49 (M, 1 H ), 2.55 – 2.83 (m, 3 H), 3.40 – 3.50 (m, 2 H), 3.51 -.. 3.57 (m, 4 H), 3.59 (br s, 2 H), 3.83 – 4.04 (m, 1 H), 4.05 – 4.29 (m, 1 H), 12.52 (s, 1 H) optical purity of 94.3% EE <HPLC analytical conditions> column: CHIRALPAK IC (Co., Ltd. Daicel) column temperature: constant around 15 ℃ Temperature Mobile phase: A solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 70: 30 B solution) 0.02 mol / L KH 2 PO 4 buffer solution (pH3.0): acetonitrile = 50 : 50 gradient program
| WO2010150840A1 | 24 Jun 2010 | 29 Dec 2010 | Dainippon Sumitomo Pharma Co., Ltd. | N-substituted-cyclic amino derivative |
| WO2011158880A1 | 15 Jun 2011 | 22 Dec 2011 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| WO2012062687A1 * | 7 Nov 2011 | 18 May 2012 | F. Hoffmann-La Roche Ag | Triazole derivatives and their use for neurological disorders |
| WO2013122260A1 | 14 Feb 2013 | 22 Aug 2013 | Takeda Pharmaceutical Company Limited | Tablet |
| CN103221402B * | 7 Nov 2011 | 17 Jun 2015 | 霍夫曼-拉罗奇有限公司 | 三唑衍生物及其用于神经障碍的用途 |
| US8329691 | 14 Oct 2008 | 11 Dec 2012 | Takeda Pharmaceutical Company Limited | Amide compounds and use of the same |
| US8389511 | 19 Dec 2008 | 5 Mar 2013 | Dainippon Sumitomo Pharma Co., Ltd. | Bicyclic heterocyclic derivative |
| US8658639 | 24 Jun 2010 | 25 Feb 2014 | Dainippon Sumitomo Pharma Co., Ltd | N-substituted-cyclic amino derivative |
| US8742097 | 2 Nov 2011 | 3 Jun 2014 | Hoffmann-La Roche Inc. | Triazole compounds I |
| US9018374 | 15 Jun 2011 | 28 Apr 2015 | Takeda Pharmaceutical Company Limited | Crystal of amide compound |
| US9090601 | 28 Jan 2010 | 28 Jul 2015 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
///////////TAK 272, Hypertension
Daprodustat, ダプロデュスタット

Daprodustat, GSK1278863
ダプロデュスタット
CAS 960539-70-2
GSK1278863; GSK 1278863; GSK-1278863; Daprodustat
C19H27N3O6
Exact Mass: 393.18999
(1,3-dicyclohexyl-2,4,6-trioxohexahydropyrimidine-5-carbonyl)glycine
N-[(l,3-dicyclohexyl-6-hydroxy-2,4-dioxo-l,2,3,4- tetrahydro-5-pyrimidinyl)carbonyl]glycine
2-(1,3-dicyclohexyl-2,4,6-triohexahydropyrimidine-5-carboxamide acetic acid
Mechanism of Action: HIF-prolyl hydroxylase inhibitor
Indication: anemia, diabetic wounds, and reduction of ischemic complications
Development Stage: Phase II
Developer:GlaxoSmithKline
UNII:JVR38ZM64B
ダプロデュスタット
Daprodustat

C19H27N3O6 : 393.43
[960539-70-2]
Daprodustat , also known as GSK1278863, is a novel HIF-prolyl hydroxylase inhibitor. Hypoxia inducible factor (HIF) stabilization by HIF-prolyl hydroxylase (PHD) inhibitors may improve ischemic conditions such as peripheral artery disease (PAD). Short-term treatment with a novel HIF-prolyl hydroxylase inhibitor (GSK1278863) failed to improve measures of performance in subjects with claudication-limited peripheral artery disease
- Originator GlaxoSmithKline
- Class Antianaemics; Pyrimidines; Small molecules
- Mechanism of ActionErythropoiesis stimulants; Prolyl hydroxylase inhibitors
- Phase II Anaemia; Perioperative ischaemia
- Phase I Diabetic foot ulcer; Tendon injuries
- DiscontinuedPeripheral arterial disorders
Most Recent Events
- 27 Jul 2015No recent reports of development identified – Phase-II for Anaemia in India and New Zealand (PO)
- 27 Jul 2015Daprodustat is still in phase II trials for Anaemia in the USA, Australia, Canada, Czech Republic, Denmark, France, Germany, Hungary, Japan, Poland, Russia, Spain, South Korea, and United Kingdom
- 01 Jun 2015GlaxoSmithKline completes a phase I trial in Tendon injuries (In volunteers) in USA (PO) (NCT02231190)
| WHO ATC code: | B03 (Antianemic Preparations)C (Cardiovascular System)
C01 (Cardiac Therapy) D03 (Preparations for Treatment of Wounds and Ulcers) M09A-X (Other drugs for disorders of the musculo-skeletal system) |
| EPhMRA code: | B3 (Anti-Anaemic Preparations)C1 (Cardiac Therapy)
C6A (Other Cardiovascular Products) D3A (Wound Healing Agents) M5X (All Other Musculoskeletal Products) |
Daprodustat (INN) (GSK1278863) is a drug which acts as a HIF prolyl-hydroxylase inhibitor and thereby increases endogenous production of erythropoietin, which stimulates production of hemoglobin and red blood cells. It is in Phase III clinical trials for the treatment of anemia secondary to chronic kidney disease.[1][2] Due to its potential applications in athletic doping, it has also been incorporated into screens for performance-enhancing drugs.[3]
SYN 1

SYN 2

PATENT
WO 2007150011
https://www.google.com.ar/patents/WO2007150011A2
Illustrated Methods of preparation
Scheme 1

a) 1. NaH, THF, rt 2. R1NCO, 60 0C; b) 1. NaH, THF or dioxane, rt 2. R4NCX, heat; c) H2NCH2CO2H, DBU, EtOH, 1600C, microwave.
Scheme 2

a) R1NH2, CH2Cl2 or R1NH2-HCl, base, CH2Cl2; b) CH2(C(O)Cl)2, CH2Cl2, reflux or CH2(CO2Et)2, NaOEt, MeO(CH2)2OH, reflux or 1. EtO2CCH2COCl, CHCl3, 70 0C 2.
DBU, CHCl3, 70 0C; c) 1. YCNCH2CO2Et,, EtPr’2N, CHCl3 or CH2Cl2 2. aq NaOH, EtOH, rt. Scheme 3 (for R1 = R4)
a) CDI,

DMF, 70 0C or , EtOAc, rt
Scheme 4

a) OCNCH2CO2Et, EtPr’2N, CHCl3 or CH2Cl2; b) 1. R1HaI, Na/K2CO3, DMF or DMA, 100 0C or R1HaI, pol-BEMP, DMF, 120 0C, microwave 2. aq NaOH, MeOH or EtOH, rt.
Scheme 5

a) 1. CH2(CO2H)2, THF, O 0C – rt 2. EtOH, reflux; b) 1. OCNCH2CO2Et, EtPr’2N, CH2Cl2 2. aq NaOH, EtOH, rt.
Scheme 6

a) 1. Phthalimide, DIAD, PPh3, THF 2. (NH2)2, EtOH, reflux.
Scheme 7

a) Ac2O, AcOH, 130 0C.
Example 18
N-T(1 ,3-Dicvclohexyl-6-hydroxy-2,4-dioxo- 1 ,2,3,4-tetrahvdro-5-pyrimidinyl)carbonyl1grycine Method 1
18.1a) h3-Dicvclohexyl-2A6(lH,3H,5H)-pyrimidinetrione. Dicyclohexylurea (3.0 g, 13.39 mmoles) was stirred in chloroform (80 mL) and treated with a solution of malonyl dichloride (1.3 mL, 13.39 mmoles) in chloroform (20 mL), added dropwise under argon. The mixture was heated at 500C for 4 hours, wasahed with 1 molar hydrochloric acid and evaporated onto silica gel. Flash chromatography (10-30% ethyl acetate in hexane) to give the title compound (2.13 g, 55%). 1Η NMR (400 MHz, OMSO-d6) δ ppm 4.46 (tt, J=12.13, 3.54 Hz, 2 H), 3.69 (s, 2 H), 2.15 (qd, J=12.46, 3.28 Hz, 4 H), 1.77 (d, J=13.14 Hz, 4 H), 1.59 (t, J=12.76 Hz, 6 H), 1.26 (q, J=12.97 Hz, 4 H), 1.04 – 1.16 (m, 2 H)
18.1b) N-r(1.3-Dicvclohexyl-6-hvdroxy-2.4-dioxo-1.2.3.4-tetrahvdro-5- pyrimidinvDcarbonyll glycine. Ethyl isocyanatoacetate (802 uL, 7.15 mmoles) was added to a mixture of l,3-dicyclohexyl-2,4,6(lH,3H,5H)-pyrimidinetrione (2.1 g, 7.15 mmoles) and diisopropylethylamine (2.47 mL, 14.3 mmoles) in dichloromethane (100 mL) and stirred overnight. The reaction mixture was washed with 1 molar hydrochloric acid (x2) and evaporated. The residue was dissolved in ethanol (10 mL) and treated with 1.0 molar sodium hydroxide (5 mL). The mixture was stirred for 72 hours, acidified and extracted into ethyl acetate. Some ester remained, therefore the solution was evaporated and ther residue was dissolved in 1 molar soldium hydroxide solution with warming and strred for 2 hours. The mixture was acidified with IM HCl and extracted with ethyl acetate (x2). The combined extracts were washed with 1 molar hydrochloric acid , dried and evaporated to a solid which was slurried in a mixture of diethyl ether and hexane, collected, washed with the same solvent mixture and dried to give the title compound (1.86 g, 66%). IH NMR (400 MHz, DMSO-^6) δ ppm 13.07 (br. s., 1 H), 10.19 (t, J=5.31 Hz, 1 H), 4.63 (t, J=10.99 Hz, 2 H), 4.12 (d, J=5.56 Hz, 2 H), 2.27 (q, J=I 1.71 Hz, 4 H), 1.79 (d, J=12.88 Hz, 4 H), 1.50 – 1.69 (m, 6 H), 1.28 (q, J=12.97 Hz, 4 H), 1.12 (q, J=12.72 Hz, 2 H)
Method 2
18.2a) 1.3-Dicvclohexyl-2.4.6πH.3H.5H)-pyrimidinetrione. A solution of N5N- dicyclohexylcarbodiimide (254 g; 1.23 mol.) in anhydrous TΗF (700 mL) was added dropwise to a cold (0 0C) solution of malonic acid (64.1 g; 0.616 mol.) in anhydrous TΗF (300 mL) over a period of- 30 minutes. The mixture was stirred and allowed to warm to room temperature over 2 h. (After 1 h, the mixture became very thick with precipitate so further anhydrous TΗF (500 mL) was added to facilitate agitation.). The mixture was filtered and the filtrate evaporated to afford a yellow solid which was immediately slurried in ethanol (1 L) and heated to reflux temperature. The mixture was then allowed to cool to room temperature then filtered and the solid washed with cold ethanol (250 mL) to afford the title compound (129.4 g; 72%) as a colorless solid. 1Η NMR (400 MHz, DMSO-(Z6) δ ppm 1.03 – 1.18 (m, 2 H) 1.18 – 1.34 (m, 4 H) 1.59 (t, J=13.14 Hz, 6 H) 1.76 (d, J=12.88 Hz, 4 H) 2.04 – 2.24 (m, 4 H) 3.69 (s, 2 H) 4.35 – 4.54 (m, 2 H).
18.2b) Ethyl N-[(l .3-dicvclohexyl-6-hvdroxy-2.4-dioxo- 1.2.3.4-tetrahydro-5- pyrimidinyPcarbonyll glycinate. A solution of l,3-dicyclohexyl-2,4,6(lH,3H,5H)-pyrimidinetrione (120.0 g; 0.41 mol.) and diisopropylethylamine (105.8 g; 0.82 mol.) in dichloromethane (1 L) was stirred and treated dropwise with a solution of ethyl isocyanatoacetate (53.0 g; 0.41 mol.) in dichloromethane (500 mL) and the mixture was then stirred at room temperature overnight. The mixture was then treated dropwise with 6M aq. hydrochloric acid (500 mL) and the separated organic layer was dried and evaporated. The resulting solid was slurried in hexanes (500 mL) and heated to reflux temperature. The mixture was then allowed to cool and filtered to afford ethyl N- [(1 ,3-dicyclohexyl-6-hydroxy-2,4-dioxo- 1 ,2,3,4-tetrahydro-5-pyrimidinyl)carbonyl]glycinate (159.1 g; 92%) as a cream powder. IH NMR (400 MHz, CHLOROFORM-,/) δ ppm 1.24 (s, 2 H) 1.37 (s, 7 H) 1.52 – 1.76 (m, 6 H) 1.78 – 1.94 (m, 4 H) 2.25 – 2.48 (m, 4 H) 4.17 (d, J=5.81 Hz, 2 H) 4.28 (q, J=7.24 Hz, 2 H) 4.74 (s, 2 H) 10.37 (t, J=4.67 Hz, 1 H). 18.2c)
N-rπ^-Dicyclohexyl-ό-hydroxy^^-dioxo-l^J^-tetralivdro-S- pyrimidinyDcarbonyll glycine. A stirred suspension of ethyl Ν-[(l,3-dicyclohexyl-6-hydroxy-2,4- dioxo-l,2,3,4-tetrahydro-5-pyrimidinyl)carbonyl]glycinate (159.0 g; 0.377 mol.) in ethanol (1.5 L) was treated dropwise with 6M aq. Sodium hydroxide (250 mL) and stirred at room temperature for 3 h. The solution was then acidified by the dropwise addition of 6M aq. hydrochloric acid (300 mL), diluted with water (IL) and then filtered. The crude solid was slurried in water (2 L) then stirred vigorously and heated at 35 0C for 1 h and filtered and dried. The solid material (~ 138 g) was then crystallized from glacial acetic acid (1.5 L) (with hot filtration to remove a small amount of insoluble material). The solid, which crystallized upon cooling, was collected and washed with cold glacial acetic acid (3 x 100 mL) to afford N-[(l,3-dicyclohexyl-6-hydroxy-2,4-dioxo-l,2,3,4- tetrahydro-5-pyrimidinyl)carbonyl]glycine (116.2 g; 78%) as a colorless solid.
IH NMR (400 MHz, DMSO-(Z6) δ ppm 1.11 (d, J=12.88 Hz, 2 H) 1.27 (q, J=12.80 Hz, 4 H) 1.62 (s, 6 H) 1.70 – 1.90 (m, J=12.88 Hz, 4 H) 2.11 – 2.44 (m, 4 H) 4.11 (d, J=5.81 Hz, 2 H) 4.45 – 4.77 (m, 2 H) 10.19 (t, J=5.81 Hz, 1 H) 13.08 (s, 1 H).
References
- Jump up^ Schmid H, Jelkmann W. Investigational therapies for renal disease-induced anemia. Expert Opin Investig Drugs. 2016 Aug;25(8):901-16. . doi:10.1080/13543784.2016.1182981. PMID 27122198. Missing or empty
|title=(help) - Jump up^ Ariazi JL, Duffy KJ, Adams DF, Fitch DM, Luo L, Pappalardi M, Biju M, DiFilippo EH, Shaw T, Wiggall K, Erickson-Miller C. Discovery and Preclinical Characterization of GSK1278863 (Daprodustat), a Small Molecule Hypoxia Inducible Factor-Prolyl Hydroxylase Inhibitor for Anemia. J Pharmacol Exp Ther. 2017 Dec;363(3):336-347. . doi:10.1124/jpet.117.242503. PMID 28928122. Missing or empty
|title=(help) - Jump up^ Thevis M, Milosovich S, Licea-Perez H, Knecht D, Cavalier T, Schänzer W. Mass spectrometric characterization of a prolyl hydroxylase inhibitor GSK1278863, its bishydroxylated metabolite, and its implementation into routine doping controls. Drug Test Anal. 2016 Aug;8(8):858-63. . doi:10.1002/dta.1870. PMID 26361079. Missing or empty
|title=(help)
 |
|
| Clinical data | |
|---|---|
| Synonyms | GSK1278863 |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| Chemical and physical data | |
| Formula | C19H27N3O6 |
| Molar mass | 393.44 g/mol |
| 3D model (JSmol) | |
//////////////Daprodustat, GSK1278863, ダプロデュスタット , HIF-prolyl hydroxylase inhibitor, anemia, diabetic wounds, reduction of ischemic complications, Phase II, GlaxoSmithKline
- Daprodustat
- 960539-70-2
- GSK1278863
- UNII-JVR38ZM64B
- GSK-1278863
- JVR38ZM64B
- N-((1,3-Dicyclohexylhexahydro-2,4,6-trioxopyrimidin-5-yl)carbonyl)glycine
- Daprodustat [USAN:INN]
- GSK 1278863
- D0F6JC
- Daprodustat(GSK1278863)
- Daprodustat; GSK1278863
- Daprodustat (JAN/USAN/INN)
- GTPL8455
- Daprodustat (GSK1278863)
- CHEMBL3544988
- BCP16766
- EX-A1121
- KS-00000M8Z
- s8171
C1CCC(CC1)N2C(=O)C(C(=O)N(C2=O)C3CCCCC3)C(=O)NCC(=O)O
Sparsentan, PS433540, RE-021

Sparsentan (PS433540, RE-021)
- C32H40N4O5S
- Average mass592.749
FDA APPROVED 2023/2/17, Filspari
4′-((2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl)-N-(4,5-dimethylisoxazol-3-yl)-2′-(ethoxymethyl)-[1,1′-biphenyl]-2-sulfonamide
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide
Sparsentan
PS433540; RE-021, formerly known as DARA
CAS :254740-64-2
4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(4,5- dimethylisoxazol-3-yl)-2-(ethoxymethyl)biphenyl-2-sulfonamide
Mechanism of Action:acting as both an Endothelin Receptor Antagonist (ERA) and Angiotensin Receptor Blocker (ARB).
Indication: Focal Segmental Glomerulosclerosis (FSGS).Focal Segmental Glomerulosclerosis (FSGS) is a rare and severe nephropathy which affects approximately 50,000 patients in the United States. Most cases of FSGS are pediatric.
Development Stage: Phase II
Developer:Retrophin, Inc
- OriginatorBristol-Myers Squibb
- DeveloperRetrophin
- ClassAntihypertensives; Isoxazoles; Small molecules; Spiro compounds; Sulfonamides
- Mechanism of ActionAngiotensin type 1 receptor antagonists; Endothelin A receptor antagonists
- Orphan Drug Status Yes – Focal segmental glomerulosclerosis
-
- 09 Jan 2015 Sparsentan receives Orphan Drug status for Focal segmental glomerulosclerosis in USA
- 31 Dec 2013 Phase-II/III clinical trials in Focal segmental glomerulosclerosis in USA (PO)
- 07 May 2012I nvestigation in Focal segmental glomerulosclerosis in USA (PO)
Sparsentan is an investigational therapeutic agent which acts as both a selective endothelin receptor antagonist and an angiotensin receptor blocker. Retrophin is conducting the Phase 2 DUET trial of Sparsentan for the treatment of FSGS, a rare and severe nephropathy that is a leading cause of end-stage renal disease. There are currently no therapies approved for the treatment of FSGS in the United States. Ligand licensed worldwide rights of Sparsentan (RE-021) to Retrophin in 2012 .The Food and Drug Administration (FDA) has granted orphan drug designation for Retrophins sparsentan for the treatment of focal segmental glomerulosclerosis (FSGS) in January 2015.
In 2006, the drug candidate was licensed to Pharmacopeia by Bristol-Myers Squibb for worldwide development and commercialization. In 2012, a license was obtained by Retrophin from Ligand. In 2015, Orphan Drug Designation was assigned by the FDA for the treatment of focal segmental glomerulosclerosis.
Sparsentan, also known as RE-021, BMS346567, PS433540 and DARA-a, is a Dual angiotensin II and endothelin A receptor antagonist. Retrophin intends to develop RE-021 for orphan indications of severe kidney diseases including Focal Segmental Glomerulosclerosis (FSGS) as well as conduct proof-of-concept studies in resistant hypertension and diabetic nephropathy. RE-021, with its unique dual blockade of angiotensin and endothelin receptors, is expected to provide meaningful clinical benefits in mitigating proteinuria in indications where there are no approved therapies
Sparsentan, sold under the brand name Filspari, is a medication used for the treatment of primary immunoglobulin A nephropathy.[1] Sparsentan is an endothelin and angiotensin II receptor antagonist.[1][4] It is taken by mouth.[1]
The most common side effects include swelling of the extremities, low blood pressure, dizziness, high blood potassium, anemia, injury to the kidney, and increased liver enzymes in the blood.[5]
It was approved for medical use in the United States in February 2023.[5][6][7] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[8]
PATENT
WO 2000001389
https://www.google.co.in/patents/WO2000001389A1?cl=en


Example 41
4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.4! non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l, l’-biphenyl! -2-sulfonamide

A. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl)-N-[(2-trimethylsilylethoxy)methyl]-2′- hydroxym ethyl [1, l’-biphenyl] -2-sulfonamide P14 (243 mg, 0.41 mmol) was used to alkylate 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene hydrochloride according to General Method 4. 41A (100 mg, 35% yield) was isolated as a slightly yellow oil after silica gel chromatography using 1:1 hexanes/ethyl acetate as eluant. B. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methvn -N-0.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l,l’-biphenyn-2- sulfonamide
Deprotection of 41A (100 mg, 0.14 mmol) according to General Method 8 (ethanol) gave the title compound as white solid in 46% yield following silica gel chromatography (96:4 methanol/chloroform eluant):
MS m/e 565 (ESI+ mode); HPLC retention time 3.21 min (Method A);
HPLC purity >98%.
Example 42
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide

A. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3 ,4- dimethyl-5-isoxazolyl)-N-[(2-methoxyethoxy)methyll-2′- hvdroxym ethyl [1 , l’-biphenyl] -2-sulfonamide
Triethylsilane (6 ml) and TFA (6 ml) were added to a solution of 5F (960 mg, 1.5 mmol) in 15 ml dichloromethane at RT. The mixture was stirred at RT for 2 h and was then concentrated. The residue was taken up in ethyl acetate and was washed successively with aqueous sodium bicarbonate, water, and brine. The organic layer was dried over sodium sulfate and concentrated. The residue was chromatographed on silica gel using 100:2 dichloromethane/methanol to afford 42A (740 mg, 77%) as a colorless gum. Rf=0.13, silica gel, 100:5 dichloromethane/methanol. B. 4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-N-r(2-methoxyethoxy)methyll-2′- ethoxymethyl[l.l’-biphenyll-2-sulfonamide A mixture of 42A (100 mg, 0.15 mmol), iodoethane (960 mg, 6.1 mmol) and silver (I) oxide (180 mg, 0.77 mmol) in 0.7 ml DMF was heated at 40 ° C for 16 h.. Additional iodoethane (190 mg, 1.2 mmol) and silver (I) oxide (71 mg, 0.31 mmol) were added and the reaction mixture was heated at 40 ° C for an additional 4 h. The mixture was diluted with 1:4 hexanes/ethylacetate and was then washed with water and brine. The organic layer was dried over sodium sulfate and was then concentrated. The residue was chromatographed on silica gel using 200:3 dichloromethane/methanol as eluant to afford 42B (51mg, 49%) as a colorless gum. Rf=0.35, silica gel, 100:5 dichloromethane/methanol.
C. 4,-[(2-Butyl-4-oxo-1.3-diazaspirof4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl )-2′-ethoxym ethyl [ 1. l’-biphenyll -2-sulfonamide
42B (51 mg) was deprotected according to General Method 7 to afford the title compound in 80% yield following preparative reverse-phase HPLC purification: white solid; m.p. 74-80 ° C (amorphous); IH NMR (CDCL, )δ0.87(tr, J=7Hz, 3H), 0.99(tr, J=7Hz, 3H), 1.32(m, 2H), 1.59(m, 2H), 1.75-2.02(m, 11H), 2.16(s, 3H), 2.35(m, 2H), 3.38 (m, 2H), 4.23(m, 2H), 4.73(s, 2H), 7.11-7.85 (m, 7H); MS m/e 593 (ESI+ mode); HPLC retention time 18.22 min. (Method E); HPLC purity >97%.
PATENT
WO 2001044239
http://www.google.co.in/patents/WO2001044239A2?cl=en
……………………
Dual angiotensin II and endothelin A receptor antagonists: Synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides with improved potency and pharmacokinetics
J Med Chem 2005, 48(1): 171
 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPDThe ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a DIFFERENT COMPD
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
PATENT
WO 2010135350
http://www.google.com/patents/WO2010135350A2?cl=en
Compound 1 :

Scheme IV

Scheme V

Formula IV 1
Scheme VII

Formula Vl

A solution of 2-(2,4-dimethylphenyl)benzenesulfonic acid (Compound 12) (0.5 g, 1.9 mmol) in 50 mL of anhydrous acetonitrile was prepared and transferred to a round-bottom flask. After flushing with nitrogen gas, N-bromosuccinimide (0.75 g, 4.2 mmol) was added followed by 50 mg (0.2 mmol) of benzoyl peroxide. The solution was heated at reflux for 3 hours. The solvent was removed in-vacuo and the resulting syrup purified by silica gel chromatography (1 :1 hexanes/EtOAc) to yield Compound 13 as a white solid. 1H NMR (500 MHz, CD3CN) 8.12 (d, J = 7.5 Hz, IH), 7.92 (t, J = 7.5 Hz, IH), 7.78 (d, J= 7.5 Hz, IH), 7.74-7.71 (m, 2H), 7.68-7.65 (m, 2H), 5.12 (s, 2H), 4.70 (s, 2H). Example 4 2-(4-Bromomethyl-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 14)

A solution of 20 mg (0.058 mmol) of (l-bromomethylbenzo[3,4- d])benzo[l,2-f]-2-oxa-l,l-dioxo-l-thiocycloheptane (Compound 13) in ethanol was stirred at elevated temperature until the starting material was consumed to give crude product (compound 14) that was used directly in the next step without isolation or purification.
Example 5
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonic acid (Compound 15)

To the above ethanol solution of crude 2-(4-bromomethyl-2- ethoxymethylphenyl)benzenesulfonic acid (Compound 14) described in Example 4 was added approximately 25 mL of anhydrous DMF. The ethanol was removed from the system under reduced pressure. Approximately 15 mg (0.065 mmol) of 2-butyl-l,3- diazaspiro[4.4]non-l-en-4-one (compound 7 in Scheme IV) was added followed by 300 μL of a IM solution of lithium bis-trimethylsilylamide in THF. The solution was allowed to stir at room temperature for 3 hours. The solvents were removed under reduced pressure and the remaining residue purified by preparative RP-HPLC employing a Cl 8 column and gradient elution (H2O:MeCN) affording the title compound as a white solid; [M+H]+ calcd for C27H34N2O5S 499.21, found, 499.31 ; 1H NMR (500 MHz, CD3CN) 8.04 (t, J= 5.5 Hz, IH), 7.44-7.10 (m, 2H), 7.28 (s, IH), 7.22 (d, J= 8.0 Hz, 2H), 7.08- 7.04 (m, 2H), 4.74 (br s, 2H), 4.32 (d, J= 13.0 Hz IH), 4.13 (d, J= 13.0 Hz IH), 3.40- 3.31 (m, 2H), 2.66 (t, J= 8 Hz, 2H), 2.18-2.13 (m, 5H), 1.96-1.90 (m, 2H obscured by solvent), 1.48 (m, 2H), 1.27 (s, J= 7 Hz, 2H), 1.16 (t, J= 7 Hz, 3H), 0.78 (t, J= 7.5 Hz, 3H).
Example 6
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16)

To a solution of DMF (155 μL, 2 mmol, 2 equiv.) in dichloromethane (5 mL) at 0 0C was added dropwise oxalyl chloride (175 μL, 2 mmol, 2 equiv.) followed by a dichloromethane (5 mL) solution of 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l- en-3-yl)methyl)-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 15) (0.50 g, 1.0 mmol). The resulting mixture was stirred at 0 0C for ~2 hours, diluted with additional dichloromethane (25 mL), washed with saturated sodium bicarbonate solution (10 mL), water (10 mL), and brine (10 mL), dried over sodium sulfate, and then concentrated to give crude sulfonyl chloride (compound 16) that was used without purification.
Example 7
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

[0062] To a solution of 5-amino-3,4-dimethylisoxazole (60 mg, 0.54 mmol) in THF at -60 °C was added dropwise potassium tert-butoxide (1 mL of 1 M solution) followed by a solution of crude 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3- yl)methyl)-2-ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16) (0.28 g, 0.54 mmol) in THF (4 mL). The resulting mixture was stirred at about -60 °C for 1 hour, allowed to warm to room temperature overnight, and then quenched with IN HCl solution to about pH 4. Standard workup of extraction with ethyl acetate, washing with water, drying, and concentration provided the final compounds as a white solid. 1H NMR (400 MHz, CDCl3) 8.03 (dd, J = 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J = 7.5, 2H), 1.37 (sextet, J = 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
Example 8 l-Bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17)

To a solution of ethyl 4-bromo-3-ethoxymethylbenzoate (9.4 g, 33 mmol) in toluene (56 mL) at about -10 0C was added 51 g of a 20% diisobutylaluminum hydride solution in toluene (ca. 70 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy 5% NaOH solution to keep the temperature below about 10 °C. Organic phase of the resulting mixture was separated and the aqueous phase was extracted with toluene. The combined organic phase was concentrated in vacuo to a final volume of ~60 mL toluene solution of l-bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) that was used in next step without purification.
Example 9 l-Bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18)

To a solution of 1 -bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) (8.4 g, 33 mmol) in toluene (60 mL) prepared in Example 8 at about -10 °C was added methanesulfonyl chloride (7.9 g, 68 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy water to keep the temperature at about 0 °C. The organic layer was separated and washed again with icy water to provide a crude product solution of 1 – bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18) that was used without purification.
Example 10
1 -Bromo-4-((2-butyl-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 -yl)methy l)-2- ethoxymethylbenzene bisoxalic acid salt (Compound 19)

To the crude solution of 1 -bromo-2-ethoxymethyl-4- methanesulfonyloxymethylbenzene (Compound 18) (1 1 g, 33 mmol) in toluene (80 mL) prepared in Example 9 was added a 75% solution of methyltributylammonium chloride in water (0.47 mL). The resulting mixture was added to a solution of 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene (compound 7 in Scheme VI) (7.5 g, 32 mmol) in dichloromethane (33 mL) pretreated with a 10 M NaOH solution (23 mL). The reaction mixture was stirred at room temperature for 2 hours until compound 18 was not longer detectable by HPLC analysis and then was quenched with water (40 mL). After stirring about 10 minutes, the organic layer was separated and aqueous layer was extracted with toluene. The combined organic phase was washed with water and concentrated to a small volume. Filtration through a silica gel pad using ethyl acetate as solvent followed by concentration yielded 1 -bromo-4-((2-buty 1-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 – yl)methyl)-2-ethoxymethylbenzene as a crude oil product.
The crude oil was dissolved in ethyl acetate (22 mL) and warmed to around 50 °C. Anhydrous oxalic acid (4.6 g) was added to the warm solution at once and the resulting mixture was stirred until a solution was obtained. The mixture was cooled gradually and the bisoxalic acid salt (compound 19) was crystallized. Filtration and drying provided pure product (compound 19) in 50-60% yield from ethyl 4-bromo-3- ethoxymethylbenzoate in 3 steps. 1H NMR (400 MHz, CDCl3) 12.32 (bs, 4H), 7.58 (d, J = 7.8, IH), 7.36 (s, IH), 7.12 (d, J= 7.8, IH), 4.90 (s, 2H), 4.56 (s, 2H), 3.68 (q, J= 7.5, 2H), 2.87-2.77 (m, 2H), 2.40-1.95 (m, 8H), 1.62-1.53 (m, 2H), 1.38-1.28 (m, 4H), and 1.82 (t, J= 7.5, 3H).
Example 11
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

To a suspension of l-bromo-4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non- l-en-3-yl)methyl)-2-ethoxymethylbenzene bisoxalic acid salt (Compound 19) (5.0 g, 8.3 mmol) in toluene (20 niL) under nitrogen was added water (30 mL) and pH was adjusted to 8-9 by addition of a 2 M NaOH solution at room temperature. The organic phase was separated and mixed with 2-(N-(3,4-dimethyl-5-isoxazolyl)-N- methoxymethylamino)sulfonylphenylboronic acid pinacol ester (Scheme VII, Formula IX, where R8is methoxymethyl and M = boronic acid pinacol ester) (3.6 g, 8.5 mmol), bis(dibenzylideneacetone)palladium(0) (Pd(dba)2) (0.12 g), and a standard phosphine ligand. After a 2 M sodium carbonate solution was added, the reaction mixture was warmed to 70 0C and stirred until the reaction was complete by HPLC analysis. The reaction was cooled to room temperature and quenched with water, and then separated in phases. The organic phase was treated with activated carbon, filtered through a pad of silica gel, and was concentrated to afford a crude mixture.
The crude reaction mixture was dissolved in ethanol (40 mL) after palladium catalyst was removed and was treated with 6 M HCl solution (ca. 40 mL). The mixture was warmed to 75-80 °C and stirred for about 2 hours until the reaction was completed by HPLC analysis. After the mixture was cooled to room temperature, the pH of the mixture was adjusted to 8 by addition of 10 M NaOH solution. The mixture was stirred for 2 more hours and the pH was adjusted to 6 by adding 2 M HCl and the crystal seeds. Filtration of the crystalline solid followed by drying provided N-(3,4-dimethyl-5- isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en-3yl)methyl-2- ethoxymethylphenyl)phenylsulfonamide (Compound 1) as a white solid.1H NMR (400 MHz, CDCIa) 8.03 (dd, J= 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J= 7.5, 2H), 1.37 (sextet, J= 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
| US20040002493 * | Aug 20, 2001 | Jan 1, 2004 | Kousuke Tani | Benzoic acid derivatives and pharmaceutical agents comprising the same as active ingredient |
| US20070054806 * | Sep 6, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Novel sulfonamide-comprising solid formulations |
| US20070054807 * | Sep 8, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Storage-stable formulations of sulfonamides |
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Filspari |
| Other names | RE-021, PS433540 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623018 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.275.317 |
| Chemical and physical data | |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
References
- ^ Jump up to:a b c d e f “Filspari- sparsentan tablet, film coated”. DailyMed. 17 February 2023. Retrieved 6 March 2023.
- ^ Jump up to:a b c d “Filspari EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Filspari Product information”. Union Register of medicinal products. 23 April 2024. Retrieved 7 September 2024.
- ^ Chiu AW, Bredenkamp N (September 2023). “Sparsentan: A First-in-Class Dual Endothelin and Angiotensin II Receptor Antagonist”. The Annals of Pharmacotherapy. 58 (6): 645–656. doi:10.1177/10600280231198925. PMID 37706310. S2CID 261743204.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Drug Trials Snapshots: Filspari”. U.S. Food and Drug Administration (FDA). 17 February 2023. Retrieved 7 September 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Travere Therapeutics Announces FDA Accelerated Approval of Filspari (sparsentan), the First and Only Non-immunosuppressive Therapy for the Reduction of Proteinuria in IgA Nephropathy” (Press release). Travere Therapeutics. 17 February 2023. Retrieved 17 February 2023 – via GlobeNewswire.
- ^ Syed YY (April 2023). “Sparsentan: First Approval”. Drugs. 83 (6): 563–568. doi:10.1007/s40265-023-01864-x. PMC 10232600. PMID 37022667.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ “PHARMACOPEIA LAUNCHES STUDY OF DARA COMPOUND | FDAnews”. http://www.fdanews.com.
- ^ “Ligand Licenses DARA Program to Retrophin”. investor.ligand.com. 21 February 2012.
- ^ https://www.fiercebiotech.com/biotech/retrophin-sheds-shkreli-connection-new-name-travere-therapeutics.
{{cite news}}: Missing or empty|title=(help) - ^ “Ongoing Non-malignant Hematological, Neurological, and Other Disorder Indications Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 21 August 2024. Retrieved 7 September 2024.
- ^ “Travere Therapeutics Announces Full FDA Approval of Filspari (sparsentan), the Only Non-Immunosuppressive Treatment that Significantly Slows Kidney Function Decline in IgA Nephropathy” (Press release). Travere Therapeutics. 5 September 2024. Retrieved 7 September 2024 – via GlobeNewswire.
- ^ “Despite trial scare, Travere’s Filspari gains full FDA nod in kidney disease showdown with Novartis”. fiercepharma.com.
External links
- Clinical trial number NCT03762850 for “A Study of the Effect and Safety of Sparsentan in the Treatment of Patients With IgA Nephropathy (PROTECT)” at ClinicalTrials.gov
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Sparsentan (Filspari). Sparsentan (27), marketed by Travere Therapeutics, is an oral, dual endothelin angiotensin receptor antagonist that received accelerated USFDA approval in February 2023 for reducing proteinuria in adults with primary immunoglobulin A (IgA) nephropathy who are at risk of rapid
disease progression.205206,207 Also known as Berger’s disease, IgAnephropathy is an immune-complex mediated disease characterized by deposits of IgA in the kidneys, resulting in inflammation and damage which can eventually lead to kidney failure. Typical treatment of IgA nephropathy has focused
on supportive care to slow kidney decline, for example, lowering blood pressure, reducing proteinuria, and minimizing lifestyle risk factors; immunosuppressive therapy has also been utilized, though it is controversial and carries risks.208 Sparsentan is the first nonimmunosuppressive treatment for IgA nephropathy and has received first-in-class and orphan drug designations. Accelerated approval was based on reduction of proteinuria (which is a risk factor for disease progression) during interim
analysis in phase III clinical trials. 209 endothelin type A (ETASparsentan blocks ) and angiotensin II type 1 receptors(AT1), interrupting the signaling pathway that contributes to disease progression. 210
The structure of the drug combines 211,212 elements that target both of these receptor types.
213 Thesynthesis of sparsentan (27), as shown in Scheme 50 and Scheme 51, was disclosed by Retrophin Pharmaceuticals (now Travere Therapeutics). Its telescoped sequences and isolation of intermediates as salts suggest that this route may be suitable for large-scale manufacturing.
The synthesis of the spirocyclic imidazolinone intermediate 27.7 is shown in Scheme 50.
Displacement of the benzylic bromide in 27.1 with sodium ethoxide produced ether 27.2. Reduction of the ester with sodium borohydride and zinc chloride yielded alcohol 27.3 which was then converted to mesylate 27.4. Reaction with spirocyclic imidazolinone 27.5 under phase transfer conditions
yielded 27.6 whichwasisolatedasthebisoxalatesalt (27.7).The sequence from 27.1 to 27.7 is telescoped, and no yields were given in the patent.
The construction of the biphenyl framework is shown in Scheme 51. Treatment of aryl bromide 27.8 with n-BuLi and triisopropyl borate followed by reaction with pinacol yielded boronic ester 27.9. Intermediates 27.7 and 27.9 were coupled via a Suzuki reaction to form the biphenyl which was isolated as
the camphorsulfonate salt (27.10). The synthesis was finished with deprotection of the methoxymethyl group under acidic conditions followed by recrystallization from isopropanol and heptane to yield sparsentan (27).
(206) Donadio, J. V.; Grande, J. P. IgA nephropathy. N. Engl. J. Med.2002, 347, 738−748.
(207) Fabiano, R. C. G.; Pinheiro, S. V. B.; Simões e Silva, A. C.Immunoglobulin A nephropathy: a pathophysiology view. Inflammation Res. 2016, 65, 757−770.
(208) Floege, J.; Rauen, T.; Tang, S. C. W. Current treatment of IgAnephropathy. Springer Semin. Immunopathol. 2021, 43, 717−728.
(209) Rovin, B.H.; Barratt, J.; Heerspink, H. J. L.; Alpers, C. E.; Bieler,S.; Chae, D.-W.; Diva, U. A.; Floege, J.; Gesualdo, L.; Inrig, J. K.; et al.Efficacy and safety of sparsentan versus irbesartan in patients with IgA
nephropathy (PROTECT): 2-year results from a randomised, active controlled, phase 3 trial. Lancet 2023, 402, 2077−2090.
(210) Komers, R.; Plotkin, H. Dual inhibition of renin-angiotensin aldosterone system and endothelin-1 in treatment of chronic kidney disease. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 2016, 310, R877−
R884.
(211) Murugesan, N.; Tellew, J. E.; Gu, Z.; Kunst, B. L.; Fadnis, L.;Cornelius, L. A.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; et al. Discovery of N-isoxazolyl biphenylsulfonamides as potent dual
angiotensin II and endothelin A receptor antagonists. J. Med. Chem.2002, 45, 3829−3835.
(212) Murugesan, N.; Gu, Z.; Fadnis, L.; Tellew, J. E.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; Dickinson, K. E.; Valentine,M.T.; et al. Dual angiotensin II and endothelin A receptor antagonists:
synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides withimproved potencyandpharmacokinetics. J. Med. Chem. 2005, 48, 171−179.
(213) Komers, R.; Shih, A. Biphenyl sulfonamide compounds for the treatment of kidney diseases or disorders. WO 2018071784, 2018.
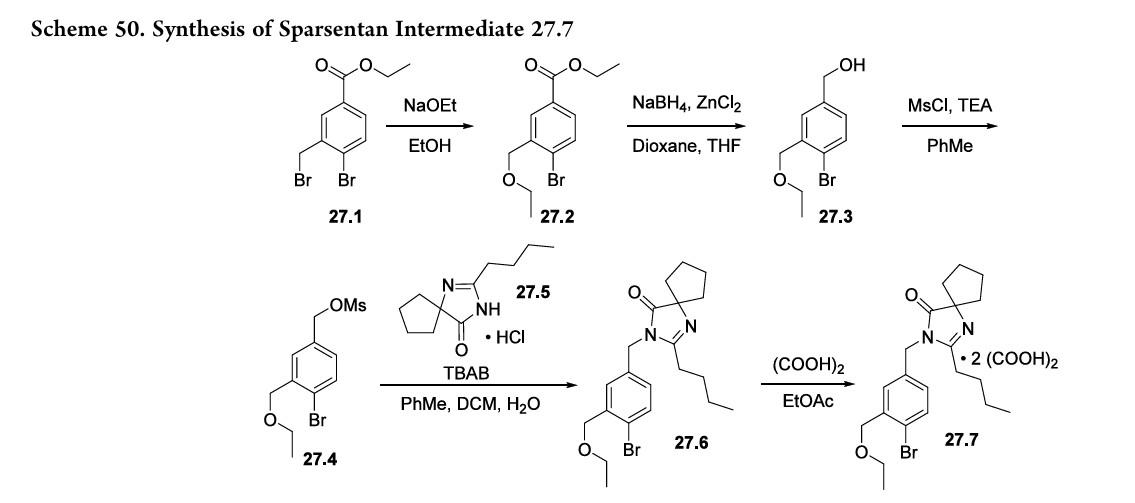

//////////////Sparsentan, PS433540, RE-021, Bristol-Myers Squibb, ORPHAN DRUG, Retrophin, FDA 2023, APPROVALS 2023
O=S(C1=CC=CC=C1C2=CC=C(CN3C(CCCC)=NC4(CCCC4)C3=O)C=C2COCC)(NC5=NOC(C)=C5C)=O,
Ciraparantag, Aripazine
Ciraparantag
PER977, Aripazine
CAS Number:1438492-26-2
Chemical Name:N1,N1-[piperazine-1,4-diylbis(propane-1,3-diyl)]bis-L-argininamide
(2S,2’S)-N,N’-(Piperazine-1,4-diyldipropane-3,1-diyl)bis(2-amino-5-carbamimidamidopentanamide)
2S,2’S)-N,N’-(piperazine-1,4-diylbis(propane-3,1-diyl))bis(2-amino-5-guanidinopentanamide)
C22H48N12O2
Mw: 512.40232
Mechanism of Action: an intravenously administered anticoagulant Reversal Agent
Blood coagulation factor modulators; Factor Xa inhibitors
Indication: Anticoagulant Reversal
Development Stage: Phase II
Developer:Perosphere, Inc..Perosphere Inc.
Highest Development Phases
- Phase IIHaemorrhage
Most Recent Events
- 02 Apr 2015Ciraparantag receives Fast Track designation for Haemorrhage [IV] (In volunteers) in USA
- 05 Nov 2014Efficacy and adverse events data from a phase I/II trial in Haemorrhage released by Perosphere
- 06 Oct 2014Aripazine is available for licensing as of 06 Oct 2014. http://www.perosphere.com/
Aripazine(PER977, ciraparantag)

Ciraparantag, also known as PER977, is a A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. PER977 is water-soluble, cationic molecule that is designed to bind specifically to unfractionated heparin and low-molecular-weight heparin through noncovalent hydrogen bonding and charge–charge interactions.
PER-977 is an intravenous heparin neutralizer in phase II clinical trials at Perosphere to reverse edoxaban’s induced anticoagulation.
In April 2015, fast track designation was assigned in the U.S. as an investigational anticoagulant reversal agent.
WO 2013082210
http://www.google.com/patents/WO2013082210A1?cl=en
In one scheme, the compound of Formula V (DAP)

is synthesized by reacting excess equivalents (e.g., at least about two equivalents) of compound 1

with one equivalent of compound 2

in the presence of a peptide coupling reagent, to obtain a compound 3

wherein PI is a protecting group and P2 is a protecting group or is a hydrogen.
the coupling involved reacting compound 1, wherein PI was Boc and P2 was a hydrogen (depicted as Boc-Arg-OH HCl below), with compound 2 as depicted below:

The resultant crude product was more than 95% pure by thin layer
chromatography (TLC).
Subsequently, the deprotection step was carried out as depicted below:

The deprotected product was purified by preparative HPLC using 1% acetic acid buffer. Product purity of >98% was observed. Residual TFA was removed by low quantity of DOWEX resin. The molecular weight of DAP (the compound of Formula V) is 512.4, and the compound synthesized according to the above scheme exhibited the following primary peak by mass spectroscopy: [M+H]+=513.4.
|
References |
1: Dzik WH. Reversal of oral factor Xa inhibitors by prothrombin complex concentrates: a re-appraisal. J Thromb Haemost. 2015 Jun;13 Suppl 1:S187-94. doi: 10.1111/jth.12949. PubMed PMID: 26149022.
2: Crowther M, Crowther MA. Antidotes for Novel Oral Anticoagulants: Current Status and Future Potential. Arterioscler Thromb Vasc Biol. 2015 Aug;35(8):1736-45. doi: 10.1161/ATVBAHA.114.303402. Epub 2015 Jun 18. PubMed PMID: 26088576.
3: Sullivan DW Jr, Gad SC, Laulicht B, Bakhru S, Steiner S. Nonclinical Safety Assessment of PER977: A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. Int J Toxicol. 2015 Jun 15. pii: 1091581815590667. [Epub ahead of print] PubMed PMID: 26079256.
4: Mo Y, Yam FK. Recent advances in the development of specific antidotes for target-specific oral anticoagulants. Pharmacotherapy. 2015 Feb;35(2):198-207. doi: 10.1002/phar.1532. Epub 2015 Feb 3. PubMed PMID: 25644580.
5: Yates SW. Interrupting anticoagulation in patients with nonvalvular atrial fibrillation. P T. 2014 Dec;39(12):858-80. PubMed PMID: 25516695; PubMed Central PMCID: PMC4264672.
6: Vanden Daelen S, Peetermans M, Vanassche T, Verhamme P, Vandermeulen E. Monitoring and reversal strategies for new oral anticoagulants. Expert Rev Cardiovasc Ther. 2015 Jan;13(1):95-103. doi: 10.1586/14779072.2015.987126. Epub 2014 Nov 28. PubMed PMID: 25431993.
7: Costin J, Ansell J, Laulicht B, Bakhru S, Steiner S. Reversal agents in development for the new oral anticoagulants. Postgrad Med. 2014 Nov;126(7):19-24. doi: 10.3810/pgm.2014.11.2829. Review. PubMed PMID: 25387210.
8: Ansell JE, Bakhru SH, Laulicht BE, Steiner SS, Grosso M, Brown K, Dishy V, Noveck RJ, Costin JC. Use of PER977 to reverse the anticoagulant effect of edoxaban. N Engl J Med. 2014 Nov 27;371(22):2141-2. doi: 10.1056/NEJMc1411800. Epub 2014 Nov 5. PubMed PMID: 25371966.
9: Hankey GJ. Intracranial hemorrhage and novel anticoagulants for atrial fibrillation: what have we learned? Curr Cardiol Rep. 2014 May;16(5):480. doi: 10.1007/s11886-014-0480-9. Review. PubMed PMID: 24643903.
///////
Vintafolide

Vintafolide, EC-145 , MK-8109
mw 1917.041, cas 742092-03-1, mf C86 H109 N21 O26 S2
(2S)-2-[(4-{[(2-amino-4-oxo-3H-pteridin-6-yl)methyl]amino}phenyl)formamido]-4-{[(1S)-1-{[(1S)-4-carbamimidamido-1-{[(1S)-2-carboxy-1-{[(1S)-2-carboxy-1-{[(1R)-1-carboxy-2-({2-[({[(1R,9R,10S,11R,12R,19R)-12-ethyl-4-[(13S,15R,17S)-17-ethyl-17-hydroxy-13-(methoxycarbonyl)-1,11-diazatetracyclo[13.3.1.04,12.05,10]nonadeca-4(12),5,7,9-tetraen-13-yl]-10,11-dihydroxy-5-methoxy-8-methyl-8,16-diazapentacyclo[10.6.1.01,9.02,7.016,19]nonadeca-2,4,6,13-tetraen-10-yl]formohydrazido}carbonyl)oxy]ethyl}disulfanyl)ethyl]carbamoyl}ethyl]carbamoyl}ethyl]carbamoyl}butyl]carbamoyl}-2-carboxyethyl]carbamoyl}butanoic acid
Vincaleukoblastin-23-oic acid, O4-deacetyl-, 2-[(2-mercaptoethoxy)carbonyl]hydrazide, disulfide with N-[4-[[(2-amino-1,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine
Endocyte innovator
Vintafolide is an investigational targeted cancer therapeutic currently under development by Endocyte and Merck & Co.[1] It is a small molecule drug conjugate consisting of a small molecule targeting the folate receptor, which is overexpressed on certain cancers, such as ovarian cancer, and a potent chemotherapy drug, vinblastine.[2] It is being developed with a companion imaging agent, etarfolatide, that identifies patients that express the folate receptor and thus would likely respond to the treatment with vintafolide.[3] A Phase 3 study evaluating vintafolide for the treatment of platinum-resistant ovarian cancer (PROCEED trial) and a Phase 2b study(TARGET trial) in non-small-cell lung carcinoma (NSCLC) are ongoing.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
A Marketing Authorization Application (MAA) filing for vintafolide and etarfolatide for the treatment of patients withfolate receptor-positive platinum-resistant ovarian cancer in combination with doxorubicin, pegylated liposomal doxorubicin (PLD), has been accepted by the European Medicines Agency.[6] The drug received an orphan drug status in Europe in March 2012.[1] Merck & Co. acquired the development and marketing rights to this experimental cancer drug from Endocyte in April 2012.[1] The drug received orphan drug status in Europe in March 2012.[3]Endocyte remains responsible for the development and commercialization of etarfolatide, a non-invasive companion imaging agent used to identify patients expressing the folate receptor that will likely respond to treatment with vintafolide.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
In 2014 Merck and Endocyte stopped a late-stage study of vintafolide in treating ovarian cancer on the recommendation of a data safety monitoring board, saying that the drug failed to improve progression-free survival.[7]
Vintafolide is folate-conjugated with DAVBLH, which is a derivative of the vinca alkaloid vinblastine.Vinblastine is a microtubule-destabilizing agent that binds tubulin and causes M phase-specific cell cycle arrest and apoptosis of mitotically active cells. Vinblastine is an extremely potent chemotherapeutic agent but has significant toxicities including bone marrow suppression, neurotoxicity, gastrointestinal toxicity and vesicant injury.
Endocyte’s desacetylvinblastinehydrazide/folate conjugate (EC-145) is a folate-targeted cytotoxic anticancer drug in early development for the treatment of non-small cell lung cancer (NSCLC) and breast cancer. The compound had been pre-registered in the E.U. by Merck for the treatment of ovarian cancer, but the application was withdrawn due to lack of efficacy.
In 2012, the product was licensed to Merck & Co. by Endocyte for worldwide exclusive development and commercialization. In 2014, however, this license agreement was terminated and Endocyte regained all rights.
Folates can serve as one-carbon donors in reactions that are critical in the de novo biosynthesis of purines and thymidylate, amino acid metabolism and methylation reactions. Folate can enter a cell by two routes: RFC or by membrane-bound FRs. RFC is a bidirectional anion transporter that is the normal entry method for reduced folates in most cells. By contrast, FRs are expressed in a limited distribution in normal tissues but are overexpressed in multiple cancers including ovarian, lung, breast and colorectal cancer. FRs bind folate derivatives with high affinity and mediate their internalization by endocytosis. Given that FRs are not typically expressed on the luminal surface of epithelial cells, making them inaccessible to normal circulation, they are attractive therapeutic targets with limited toxicity. In addition to the therapeutic agent vintafolide, a radiodiagnostic agent (99mTc-etarfolatide [EC20]) has been developed to allow single-photon emission computed tomography (SPECT) imaging to identify FR-expressing tissues (tumors).
In 2012, orphan drug designations were assigned in the E.U. for the treatment of ovarian cancer and to be used with folic acid for the diagnosis of positive folate-receptor status in ovarian cancer. In 2013, orphan drug designation was assigned in the U.S. for the treatment of ovarian cancer.
Vintafolide is a water-soluble derivative of folic acid and the vinca alkaloid DAVLBH. The molecules are connected through a hydrophilic L-peptide spacer and a disulfide linker (Figure 1). The disulfide linker serves as a cleavable bond that is necessary for drug release following receptor mediated endocytosis. The disulfide bond is reduced in the acidic environment of the endosome, leading to efficient release of vinblastine.

Vintafolide.
DAVBLH: Desacetylvinblastine hydrazide

Structure of vintafolide and mechanism of release of the payload in the endosome.

Mechanism of action
Folate is required for cell division, and rapidly dividing cancer cells often express folate receptors in order to capture enough folate to support rapid cell growth. Elevated expression of the folate receptor occurs in many diseases, including other aggressively growing cancers and inflammatory disorders.[8] Vintafolide binds to the folate receptor and is subsequently taken up by the cell through a natural internalization process called endocytosis. Once inside the cell, vintafolide’s linker releases the chemotherapy drug which kills the cell.[3]
……………
Bioorganic & Medicinal Chemistry Letters (2006), 16(19), 5093-5096
http://www.sciencedirect.com/science/article/pii/S0960894X06008079
An efficient synthesis of the folate receptor (FR) targeting conjugate EC145 is described. EC145 is a water soluble derivative of the vitamin folic acid and the potent cytotoxic agent, desacetylvinblastine monohydrazide. Both molecules are connected in regioselective manner via a hydrophilic peptide spacer and a reductively labile disulfide linker.

………approach for the design and regioselective synthesis of a FA-vinca alkaloid conjugate 1 (EC145,BELOW). As indicated in the retrosynthetic scheme, 1 can be assembled by tethering a FA-Spacer unit 2 to the highly potent cytotoxic molecule, desacetylvinblastine monohydrazide 3, via a linker containing a reducible disulfide bond. The latter is important for drug delivery applications since real-time imaging using a fluorescence resonance energy transfer technique has recently demonstrated that reduction-mediated release of the drug cargo from a disulfide linked FA-conjugate efficiently occurs within the endosomes of cancer cells.

-
Scheme 1.
Reagents and conditions: (i) a—Fmoc-Asp(OtBu)-OH, PyBOP, DIPEA, RT, 1 h; b—20% piperidine/DMF, rt, 10 min; (ii) a—Fmoc-Arg(Pbf)-OH, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iii) a—Fmoc-Glu-OtBu, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iv) N10-TFA-pteroic acid, PyBOP, DIPEA, rt, 1.5 h; (v) TFA/H2O/TIPS/EDT (92.5:2.5:2.5:2.5), rt, 1 h; (vi) aq NH4OH, pH 9.3, rt, 1 h.
- Selected 1H NMR data for 2 (D2O, 300 MHz): δ 8.68 (s, 1H, FA H-7), 7.57 (d, 2H,J = 8.4 Hz, FA H-12 & 16), 6.67 (d, 2H, J = 9 Hz, FA H-13 & 15), 4.40–4.75 (series of m, 5H), 4.35 (m, 2H), 4.16 (m, 1H), 3.02 (m, 2H), 2.55–2.95 (series of m, 8H), 2.42 (m, 2H), 2.00–2.30 (m, 2H), 1.55–1.90 (m, 2H), 1.48 (m, 2H).
- 1H NMR for compound 6 (DMSO-d6, 300 MHz): δ 8.38 (m, 1H), 8.16 (dt, 1H, J = 8 Hz, 1 Hz), 8.02 (dt, 1H, J = 8 Hz, 1 Hz), 7.88 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz), 7.7 (m, 2H), 7.63 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz,), 7.4–7.2 (br, 1H), 7.2 (m, 1H), 4.72 (t, 2H,J = 6 Hz), 3.36 (t, 2H, J = 6 Hz).
- Selected 1H NMR data for
EC145 (D2O, 300 MHz): δ 8.67 (s, 1H, FA H-7), 7.50 (br s, 1H, VLB H-11′), 7.30–7.40 (br s, 1H, VLB H-14′), 7.35 (d, 2H, J = 7.8 Hz, FA H-12 & 16), 7.25 (m, 1H, VLB H-13′), 7.05 (br s, 1H, VLB H-12′), 6.51 (d, 2H, J = 8.7 Hz, FA H-13 & 15), 6.4 (s, 2H, VLB H-14 & 17), 5.65 (m, 1H, VLB H-7), 5.5 (m, 1H, VLB H-6), 4.15 (m,1H, VLB H-8′), 3.82 (s, 3H, VLB C18′ –CO2CH3), 3.69 (s, 3H, VLB C16 –OCH3), 2.8 (s, 3H, VLB N–CH3), 1.35 (br s, 1H, VLB H-3′), 1.15 (m, 1H, VLB H-2′), 0.9 (t, 3H, J = 7 Hz, VLB H-21′), 0.55 (t, 3H, J = 6.9 Hz, VLB H-21).
 CLICK ON IMAGE FOR CLEAR VIEW
CLICK ON IMAGE FOR CLEAR VIEW
…………
WO 2004069159
http://www.google.com/patents/WO2004069159A2?cl=en
EXAMPLE 16b

The compounds of Examples 16a and 16b were prepared from the peptidyl fragment Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH , prepared according to the general procedure described in Scheme 12. The Michael addition of this peptidyl fragment to the maleimido derivative of seco-CBI-bis-indole resulted in the folate conjugates Example 16a. The peptidyl fragment also reacted with either the thiosulfonate or pyridyldithio-activated vinblastine to form Example 16b. The maleimido derivative of seco-CBI-bis-indole, and the thiosulfonate and pyridyldithio- activated vinblastine intermediates were prepared using the procedures described herein for other examples.
……………..
https://www.google.com/patents/WO2012142281A1?cl=en
Folate-targeted drugs have been developed and are being tested in clinical trials as cancer therapeutics. EC145, also known as vintafolide, comprises a highly potent vinca alkaloid cytotoxic compound, desacetylvinblastine hydrazide (DAVLBH), conjugated to folate. The EC 145 molecule targets the folate receptor found at high levels on the surface of epithelial tumors, including non-small cell lung carcinomas (NSCLC), ovarian, endometrial and renal cancers, and others, including fallopian tube and primary peritoneal carcinomas. It is believed that EC 145 binds to tumors that express the folate receptor delivering the vinca moiety directly to cancer cells while avoiding normal tissue. Thus, upon binding, EC 145 enters the cancer cell via endocytosis, releases DAVLBH and causes cell death or inhibits cell function. EC 145 has the following formula

EC145
and has been accorded the Chemical Abstracts Registry Number 742092-03-1. As used herein, according to the context, the term EC 145 means the compound, or a pharmaceutically acceptable salt thereof; and the compound may be present in a solid, solution or suspension in an ionized form, including a protonated form. EC145 is disclosed in U.S. Patent No. 7,601,332; and particular uses and an aqueous liquid pH 7.4, phosphate-buffered formulation for intravenous administration are disclosed in WO 2011/014821. As described in WO 2011/014821, it is necessary to store the aqueous liquid formulation in the frozen state to ensure its stability. To avoid this necessity, a formulation is needed which has adequate stability at ambient temperature.
As one aspect of the invention described herein, there is provided a pharmaceutical composition of EC145 which is a lyophilized solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
In another aspect of the invention, there is provided a pharmaceutical composition of EC 145 which is an X-ray amorphous solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
| Systematic (IUPAC) name | |
|---|---|
|
N-(4-{[(2-Amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoyl)-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine disulfide with methyl (5S,7R,9S)-5-ethyl-9-[(3aR,4R,5S,5aR,10bR,13aR)-3a-ethyl-4,5-dihydroxy-8-methoxy-6-methyl-5-({2-[(2-sulfanylethoxy)carbonyl]hydrazinyl}carbonyl)-3a,4,5,5a,6,11,12,13a-octahydro-1H-indolizino[8,1-cd]carbazol-9-yl]-5-hydroxy-1,4,5,6,7,8,9,10-octahydro-2H-3,7-methanoazacycloundecino[5,4-b]indol-9-carboxylate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 742092-03-1 |
| ATC code | L01CA06 |
| ChemSpider | 27444385 |
| Synonyms | EC-145 |
| Chemical data | |
| Formula | C86H109N21O26S2 |
| Molecular mass | 1917 g/mol |
References
- Sridharan, Balaji (Apr 16, 2012). “Endocyte soars on cancer drug deal with Merck”. Reuters.
- Statement on a nonproprietary name adopted by the USAN Council, United States Adopted Names (USAN) Council, 6 April 2012
- Kuo, Phillip H. (February 2013). “Companion Imaging Diagnostics for Targeted Therapies”. Radiology Today 14 (2): 32.
- “Merck, Endocyte in Development Deal”. Drug Development & Discovery magazine. 2012-04-25.
- Dosio, F.; Milla, P.; Cattel, L. (2010). “EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers”. Current opinion in investigational drugs (London, England : 2000) 11 (12): 1424–1433. PMID 21154124.
- “EMA Accepts For Review MAA Filings For Vintafolide And Etarfolatide”. rttnews.com. 2012-11-27.
- Garde, Damian (2014-05-02). “Merck halts study of the billion-dollar cancer drug vintafolide”. Fierce Biotech. Retrieved 21 April 2015.
- “Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay” 338 (2). March 2005. pp. 284–93. doi:10.1016/j.ab.2004.12.026.PMID 15745749.
-
WO2008098970A1 * Feb 13, 2008 Aug 21, 2008 Pf Medicament Anhydrous crystalline vinflunine salts, method of preparation and use thereof as a drug and means of vinflunine purification WO2010150100A1 * Jun 23, 2010 Dec 29, 2010 Entarco Sa The use of spinosyns and spinosyn compositions against diseases caused by protozoans, viral infections and cancer WO2011014821A1 * Jul 30, 2010 Feb 3, 2011 Endocyte, Inc. Folate-targeted diagnostics and treatment US20100247669 * Sep 30, 2010 Cerulean Pharma Inc. Polymer-agent conjugates, particles, compositions, and related methods of use
////////Vintafolide, BMS-753493, DAVBLH, Desacetylvinblastine hydrazide, EC-145 , MK-8109 , phase 2
ASLAN Pharmaceuticals Gains Orphan Designation for Rare Cancer Drug ASLAN001 (varlitinib)

(R)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine

ASLAN001 , Varlitinib
C22H19ClN6O2S
Molecular Weight: 466.94
Elemental Analysis: C, 56.59; H, 4.10; Cl, 7.59; N, 18.00; O, 6.85; S, 6.87
CAS: 845272-21-1 (Varlitinib); 1146629-86-8 (Varlitinib tosylate).
ASLAN001; ASLAN-001; ASLAN 001; AR 00334543; ARRY-334543; ARRY334543; ARRY-543; ARRY543; ARRY 543.
(R)-N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)-N6-(4-methyl-4,5-dihydrooxazol-2-yl)quinazoline-4,6-diamine.
(R)-4-[[3-Chloro-4-[(thiazol-2-yl)methoxy]phenyl]amino]-6-[(4-methyl-4,5-dihydrooxazol-2-yl)amino]quinazoline
4,6-Quinazolinediamine, N4-[3-chloro-4-(2-thiazolylmethoxy)phenyl]-N6-[(4R)-4,5-dihydro-4-methyl-2-oxazolyl]-
ASLAN Pharmaceuticals, a Singapore-based drugmaker, announced The Food and Drug Administration (FDA) gave an orphan drug designation on August 13 to its pan-HER inhibitor ASLAN001 (varlitinib), a drug candidate created to treat a destructive form of bile duct cancer called cholangiocarcinoma that has no known cure. ………http://www.dddmag.com/news/2015/08/aslan-pharmaceuticals-gains-orphan-designation-rare-cancer-drug
Current developer: Array Biopharma Inc,
![]()
Varlitinib, also known as ARRY-543 and ASLAN001, is an orally bioavailable inhibitor of the epidermal growth factor receptor family with potential antineoplastic activity.
Varlitinib (ASLAN-001) is an oncolytic drug in phase II clinical trials at ASLAN Pharmaceuticals for the treatment of gastric cancer and for the treatment of metastatic breast cancer in combination with capecitabine. Clinical development is also ongoing for the treatment of solid tumors in combination with cisplatin/FU and cisplatin/capecitabine. The product had been in phase I/II clinical trials at Array BioPharma for the treatment of patients with advanced pancreatic cancer. Phase II clinical trials had also been ongoing for the treatment of solid tumors. No recent development has been reported for this research
Varlitinib selectively and reversibly binds to both EGFR (ErbB-1) and Her-2/neu (ErbB-2) and prevents their phosphorylation and activation, which may result in inhibition of the associated signal transduction pathways, inhibition of cellular proliferation and cell death. EGFR and Her-2 play important roles in cell proliferation and differentiation and are upregulated in various human tumor cell types. Due to the dual inhibition of both EGFR and Her-2, this agent may be therapeutically more effective than agents that inhibit EGFR or Her-2 alone.
The drug is a dual inhibitor of the ErB-2 and EGFR receptor kinases, both of which have been shown to stimulate aberrant growth, prolong survival and promote differentiation of many tumor types. The compound behaves as a reversible ATP-competitive inhibitor with nanomolar potency both in vitro and in cell-based proliferation assays.
In 2011, the compound was licensed to Aslan Pharmaceuticals by Array BioPharma worldwide for the treatment of solid tumors, initially targeting patients with gastric cancer through a development program conducted in Asia.
In 2015, orphan drug designation was assigned to the compound in the U.S. for the treatment of cholangiocarcinoma.
SEE NMR ………….http://www.medkoo.com/Product-Data/Varlitinib/Varlitinib-QC-KB20121128web.pdf
……………..
https://www.google.co.in/patents/US20050043334
Example 52
(R)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine
Prepared using (R)-2-aminopropan-1-o1. MS APCI (+) m/z 467, 469 (M+1, Cl pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, 1H), 8.47 (s, 1H), 8.09 (s, 1H), 7.86 (d, 1H), 7.81 (d, 1H), 7.77 (d, 1H), 7.69 (m, 3H), 7.32 (d, 1H), 7.02 (s, 1H), 5.54 (s, 2H), 4.47 (m, 1H), 3.99 (m, 1H), 3.90 (m, 1H), 1.18 (d, 3H).
Example 53

(S)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine
Prepared using (S)-2-amino-propan-1-o1. MS APCI (+) m/z 467, 469 (M+1, Cl pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, 1H), 8.47 (s, 1H), 8.09 (s, 1H), 7.86 (d, 1H), 7.81 (d, 1H), 7.77 (d, 1H), 7.69 (m, 3H), 7.32 (d, 1H), 7.02 (s, 1H), 5.54 (s, 2H), 4.47 (m, 1H), 3.99 (m, 1H), 3.90 (m, 1H), 1.18 (d, 3H).
………………
PATENT
http://www.google.co.in/patents/WO2005016346A1?cl=en
Example 52

R VN4-r3-Chloro-4-(‘thiazol-2-v-metho-xy)-phenyll-N6-(4-methyl-4,5-dihvdro-oxazol- 2-yl)-quinazoUne-4,6-diamine
[00194] Prepared using (R)-2-aminopropan- 1 -ol. MS APCI (+) m/z 467, 469
(M+l, CI pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, IH), 8.47 (s, IH), 8.09 (s, IH), 7.86 (d, IH), 7.81 (d, IH), 7.77 (d, IH), 7.69 (m, 3H), 7.32 (d, IH), 7.02 (s, IH), 5.54 (s, 2H), 4.47 (m, IH), 3.99 (m, IH), 3.90 (m, IH), 1.18 (d, 3H). Example 53

(S)-N4-|“3-Chloro-4- thiazol-2-ylmethoxy)-phenyll-N6-(‘4-methyl-4,5-dihvdro-oxazol- 2-yl)-quinazoline-4,6-diamine [00195] Prepared using (S)-2-amino-propan- 1 -ol. MS APCI (+) m z 467, 469
(M+l, CI pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, IH), 8.47 (s, IH), 8.09 (s, IH), 7.86 (d, IH), 7.81 (d, IH), 7.77 (d, IH), 7.69 (m, 3H), 7.32 (d, IH), 7.02 (s, IH), 5.54 (s, 2H), 4.47 (m, IH), 3.99 (m, IH), 3.90 (m, IH), 1.18 (d, 3H).
………
CAUTION a very similar molecule but not same
 NOTE……..METHYL NEXT TO OXYGEN ATOM
NOTE……..METHYL NEXT TO OXYGEN ATOM
Design, Synthesis and Bioactivities Evaluation of Novel Quinazoline Analogs Containing Oxazole Units
DOI: 10.1002/cjoc.201400271,…………http://onlinelibrary.wiley.com/doi/10.1002/cjoc.201400271/abstract;jsessionid=04211D7526D97240F44E4355C6C57F50.f01t01
CAUTION …THIS IS NOT SAME
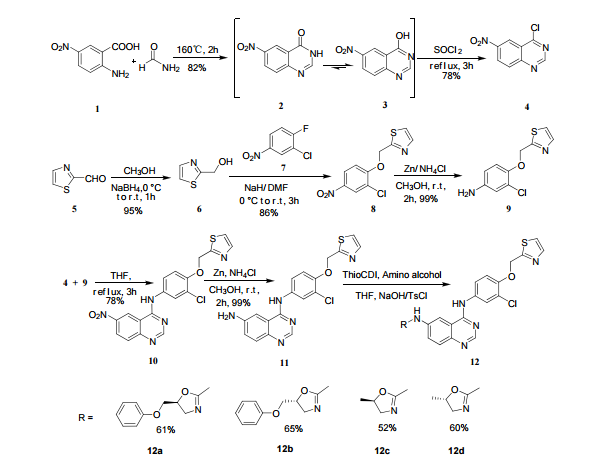
A novel type of quinazoline derivatives, which were designed by the combination of quinazoline as the backbone and oxazole scaffold as the substituent, have been synthesized and their biological activities were evaluated for anti-proliferative activities and EGFR inhibitory potency. Compound 12b demonstrated the most potent inhibitory activity (IC50=0.95 µmol/L for EGFR), which could be optimized as a potential EGFR inhibitor in the further study. The structures of the synthesized quinazoline analogs and all intermediates were comfirmed by 1H and 13C NMR, 2D NMR spectra, IR spectra and MS spectra.
12c: Employing the same method as above, compound 12c was prepared and the amino alcohol was (S)-2-amino-propan-1-ol. Yellow solid, yield 52 %. m.p. 243-244 °C; [α] 20D =﹢22.5 ° (c 1.0, CH3CN); 1 H NMR (DMSO-D6): δ 9.54 (s, 1 H), 8.46 (s, 1 H), 8.06 (s, 2 H), 7.85 (d, 2 H, J=3.3 Hz), 7.79 (d, 2 H, J=3.3 Hz), 7.75 (d, 1 H, J=8.9 Hz), 7.64 (d, 1 H, J=8.3 Hz), 7.30 (d, 1 H, J=9.0 Hz), 5.54 (s, 2 H), 4.76 (m, 1 H), 3.72 (s, 1 H), 3.19 (s, 1 H), 1.34 (d, 3 H, J=6.15 Hz). 13C NMR (DMSO-D6) δ: 165.8, 156.9, 152.0, 148.8, 145.3, 142.6, 134.3, 128.7, 128.0, 123.5, 121.7, 121.3, 121.0, 115.6, 114.6, 72.5, 67.7, 63.0, 29.8, 29.0, 20.0, 13.9. IR (KBr) ν: 3439, 3278, 3101, 2925, 1660, 1631, 1601, 1557, 1500, 1428, 1404, 1384, 1329, 1291, 1257, 1225, 1052 cm-1. Anal. calcd for C22H19N6O2SCl: C 55.59, H 4.10, N 18.00, O 6.85; found C 55.55, H 4.13, N 18.02, O 6.78; MS (ESI) m/z: 467.2 (M+H).
12d: Employing the same method as above, compound 12d was prepared and the amino alcohol was (R)-2-amino-propan-1-ol. Yellow solid, yield 60%. m.p. 242-243 °C; [α] 20D = ﹣22.3 ° (c 1.0, CH3CN); 1 H NMR (DMSO-D6): δ 9.52 (s, 1 H), 8.80 (s, 1 H), 8.52 (dd, 1 H, J=2.7 Hz, J=8.9 Hz), 8.45 (s, 1 H), 8.30 (s, 1 H), 8.07 (s, 1 H), 7.85 (d, 1 H, J=3.2 Hz), 7.79 (d, 1 H, J=3.2 Hz), 7.75 (s, 1 H), 7.63 (d, 1 H, J=8.2 Hz), 7.31 (d, 1 H, J=9.0 Hz), 5.53 (s, 2 H), 4.76 (m, 1 H), 3.81 (s, 1 H), 3.19 (s, 1 H), 1.34 (d, 3 H, J=6.2 Hz). 13C NMR (DMSO-D6) δ: 165.8, 156.9, 152.0, 148.8, 145.3, 142.6, 134.3, 128.7, 128.0, 123.5, 121.7, 121.3, 121.0, 115.6, 114.6, 72.5, 67.7, 63.0, 29.8, 29.0, 20.0, 13.9. IR (KBr) ν: 3439, 3278, 3101, 2925, 1660, 1631, 1601, 1557, 1500, 1428, 1404, 1384, 1329, 1291, 1257, 1225, 1052 cm-1. Anal. calcd for C22H19N6O2SCl: C 55.59, H 4.10, N 18.00, O 6.85; found C 55.55, H 4.13, N 18.02, O 6.78; MS (ESI) m/z: 467.20 (M+H).
The above paper allows you to synthesize the key amino int 11 ………N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)quinazoline-4,6-diamine (11)
this can be applied to varlitinib till int 11
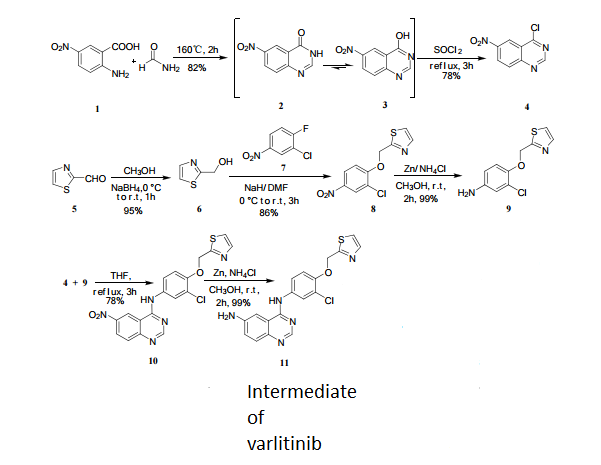
6-Nitro-4-hydroxyquinazoline (3)
2-amino-5-nitrobenzoic acid (5.46 g, 30 mmol) was added to a 250 mL flask equipped with a reflux condenser. Then 50 mL formamide was added. The mixture was heated with vigorous stirring at 160 °C for 3 h. After cooling the solution was poured in ice-water to give 3 in almost pure form (Yellow solid 4.70 g, yield 82.0%). m.p. 317-318 °C; 1 H NMR (DMSO-d6): δ 12.74 (1 H, s, OH, exchangeable), 8.78 (1 H, d, J=2.4 Hz), 8.53 (1 H, dd, J=2.6 Hz, 9.0 Hz), 8.30 (s, 1 H), 7.84 (1 H, d, J=9.0 Hz); 13C NMR (DMSO-d6) δ: 160.1, 152.9, 148.9, 145.0, 129.1, 128.3, 122.7, 121.9. IR (KBr) ν: 3172, 3046, 2879, 1674, 1615, 1577, 1514, 1491, 1469, 1343, 1289, 1242, 1167, 1112, 928, 920, 901, 803, 753, 630, 574, 531 cm-1. Anal. calcd for C8H5N3O3: C 50.27, H 2.64, N 21.98; found C 50.30, H 2.65, N 21.96; MS (ESI) m/z: 189.97 (M-H).
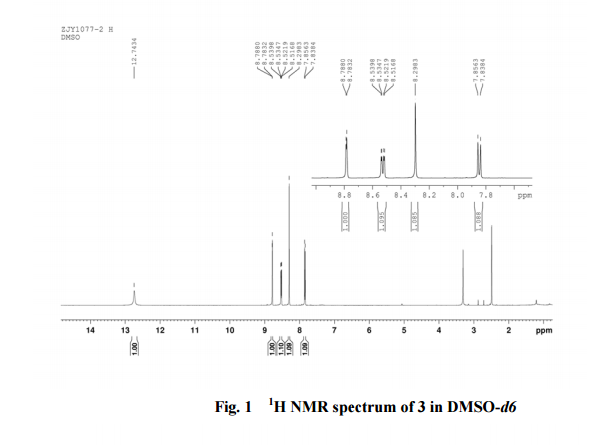
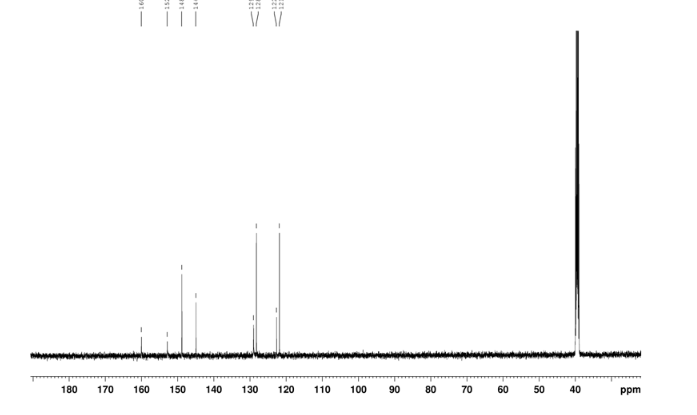 13C NMR OF 3 IN DMSOD6
13C NMR OF 3 IN DMSOD6
IR
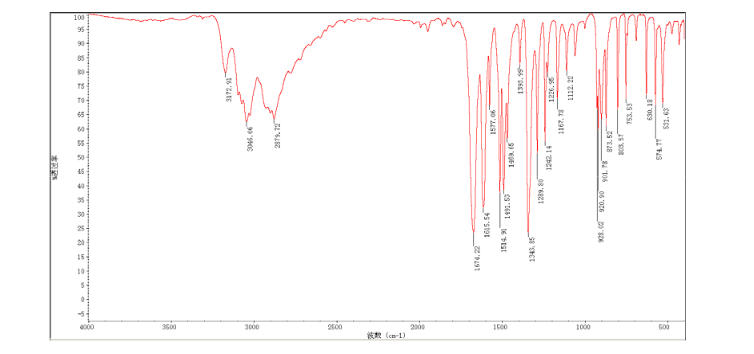
4-chloro-6-Nitroquinazoline (4)
In a 100 mL flask equipped with a reflux condenser, 6-nitroquinazolin-4-one (2.86 g, 15 mmol) and thionyl chloride (SOCl2) 25 mL were added. The mixture was heated under reflux with vigorous stirring for 2 h. After the solution was clear, the reaction mixture was heated for another 2 h. Then, 150 mL of ice MeOH was dropped into it carefully, the mixture was extracted with CH2Cl2. The organic layer was S3 dried under MgSO4, filtered and the solvent removed to give 4-chloro-6-nitroquinazoline (4). Yellow solid 2.45 g, yield 78%. m.p. 134-135 °C; 1 H NMR (DMSO-d6): δ 8.80 (1 H, d, J=3.0 Hz), 8.54(1 H, dd, J=2.7 Hz, 9.0 Hz), 8.35(s, 1 H), 7.87 (1 H, d, J= 9.0 Hz); 13C NMR (DMSO-d6) δ: 160.0, 152.5, 149.1, 145.1, 128.7, 128.4, 122.7, 122.0. IR (KBr) ν: 3431, 3082, 3038, 2664, 2613, 2567, 1724, 1685, 1676, 1646, 1617, 1578, 1526, 1468, 1359, 1346, 1269 cm-1. Anal. calcd for C8H4N3O2Cl: C 45.84, H 1.92, N 20.05, O 15.27; found C 45.81, H 1.97, N 20.02, O 15.21; MS (ESI) m/z: 207.96 (M-H).
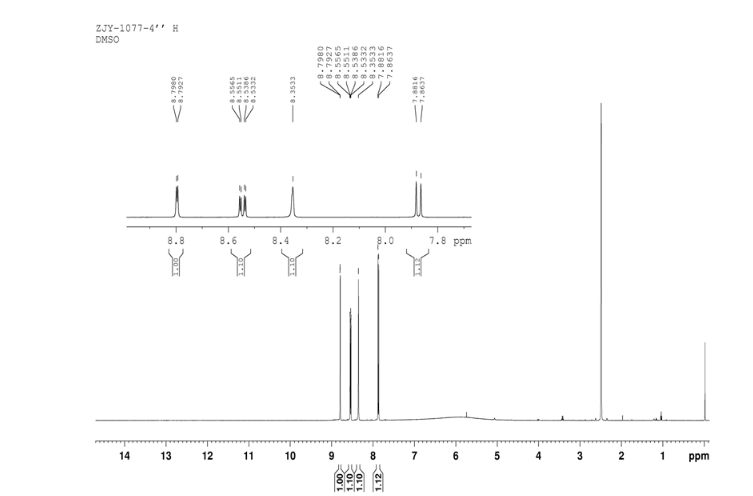 4 nmr dmsod6
4 nmr dmsod6
13C NMR OF4 IN DMSOD6
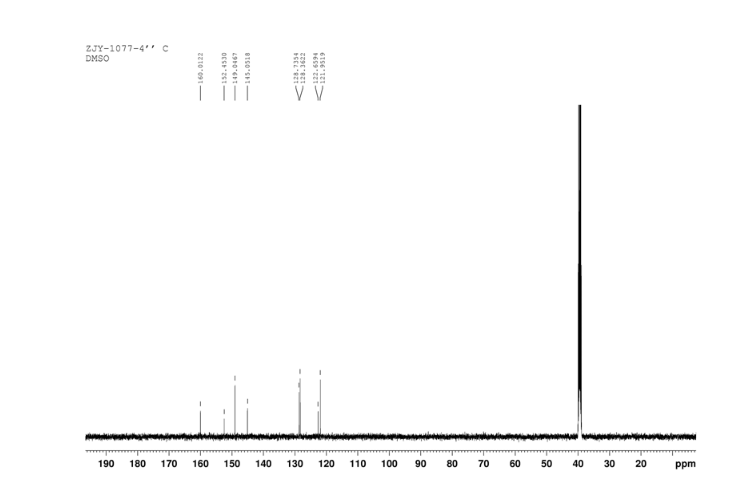
IR
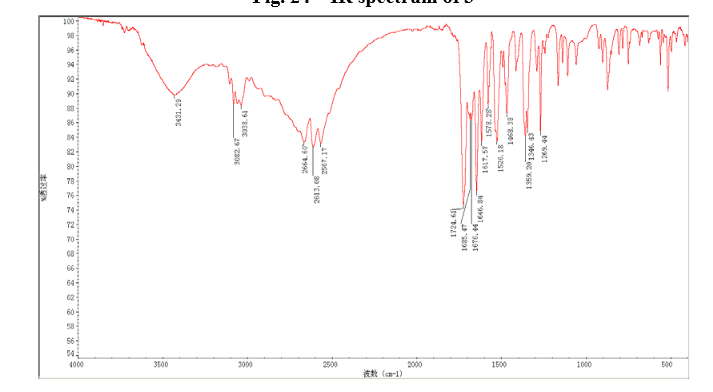
Thiazol-2-yl-methano1 (6)
Sodium borohydride (16.0 g, 140 mmol) was added to a stirred solution of thiazole-2-carbaldehyde (24.2 g, 214 mmol) in MeOH (400 mL) at 0 °C . The reaction mixture was warmed to room temperature. After 1 hour, the reaction mixture was quenched by the addition of water and the organics were removed by concentration. The resulting aqueous mixture was extracted with EtOAc. The combined organic extracts were dried under Na2SO4 and concentrated to give thiazol-2-yl-methano1 (23.39 g, 95%). bp:75-76 °C (0.2 mmHg) [lit.[19] bp:70-80 °C (0.2 mmHg)]; m. p. 63-64 °C. 1 H NMR (CDCl3) δ 4.91 (s, 2 H), 5.1(br, l H), 7.28(d, 1 H, J=3.2 Hz), 7.68 (d, 1 H, J=2.9 Hz). IR (KBr) ν: 3135, 3099, 3082, 2814, 1509, 1446, 1351, 1189, 1149, 1073, 1050, 977, 775, 745, 613, 603 cm-1. Anal. calcd for C4H5NOS: C 41.72, H 4.38, N 12.16; found C 41.74, H 4.33, N 12.18; MS (ESI) m/z: 116.11 (M+H).
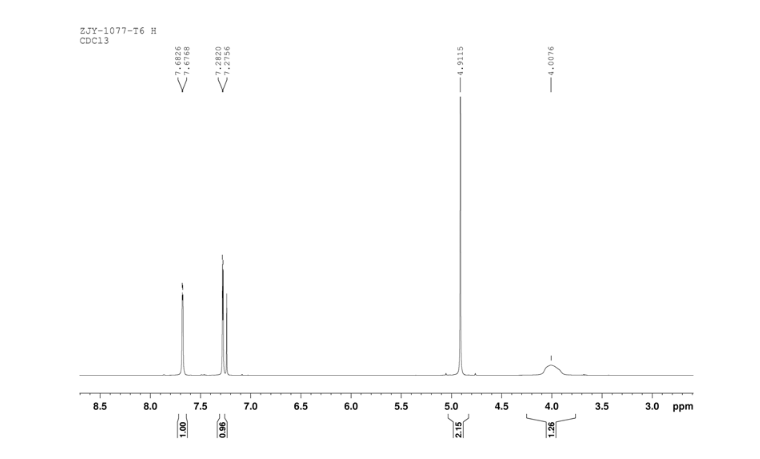 6 in dmsod6 1H NMR
6 in dmsod6 1H NMR
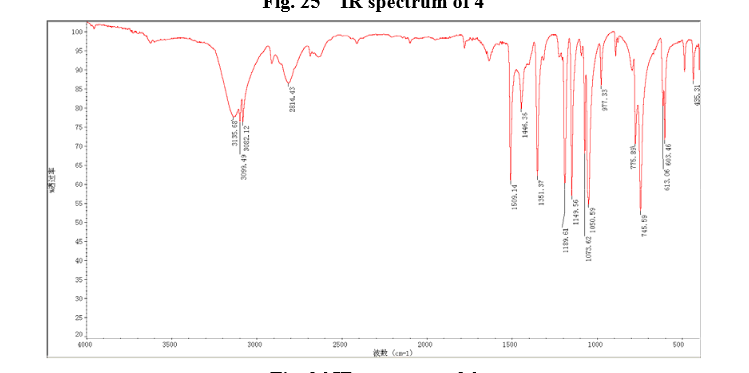
2-((2-Chloro-4-nitrophenoxy)methyl)thiazole (8)
2-(2-chloro-4-nitro-phenoxymethy1)-thiazole was prepared by adding thiazol-2-yl-methanol (5.48 g, 47.65 mmol) to a slurry of sodium hydride (2.42 g of a 60% dispersion in oil, 60.5 mmol) in THF (50 ml) at 0 °C After several minutes, 2-chloro-1-fluoro- 4-nitro-benzene (7.58 g, 43.60 mmol) was added and the reaction mixture warmed to room temperature. The reaction mixture was stirred at room temperature for 3 h, and 60 °C for 16 h. After cooling to room temperature, the reaction mixture was poured into 300 mL water. The resulting precipitate was collected by filtration, washed with water, and dried in vacuo to give 2-(2- chloro-4-nitrophenoxymethy1)-thiazole (11.06 g, 86%) which was used in next step without further purification. m.p. 170-171 °C; 1 H NMR (DMSO-d6): δ 8.35 (1 H, d, J=2.8 Hz), 8.25 (1 H, dd, J=2.8 Hz, 9.15 Hz), 7.87 (1 H, d, J=3.3 Hz), 7.83(1 H, d, J=3.3 Hz), 7.54 (1 H, d, J=9.2 Hz), 5.73(s, 1 H); 13C NMR (DMSO-d6) δ: 164.2, 158.5, 143.2, 141.7, 125.9, 124.9, 122.4, 122.2, 114.6, 68.4; IR (KBr) ν: 3112, 3009, 1587, 1509, 1500, 1354, 1319, 1284, 1255, 1154, 1125, 1054, 1006, 894, 780, 746, 728 cm-1. Anal. calcd for C10H7N2O3SCl: C 44.37, H 2.61, N 10.35, O 17.73; found C 44.31, H 2.67, N 10.29; MS (ESI) m/z: 268.89 (M-H).
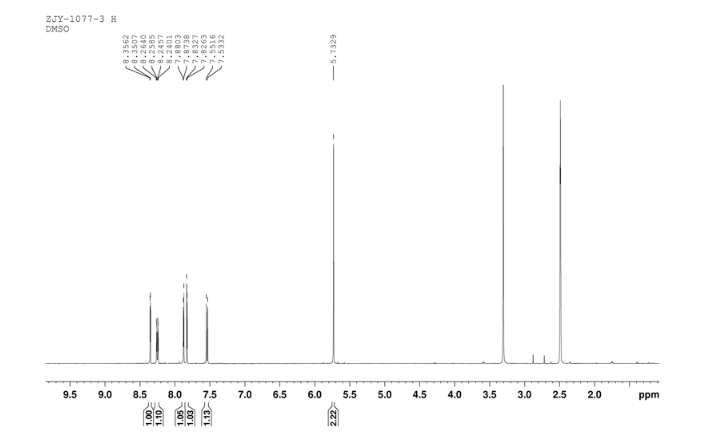 1H NMR 8 DMSOD6
1H NMR 8 DMSOD6
13C NMR OF 8 IN DMSOD6
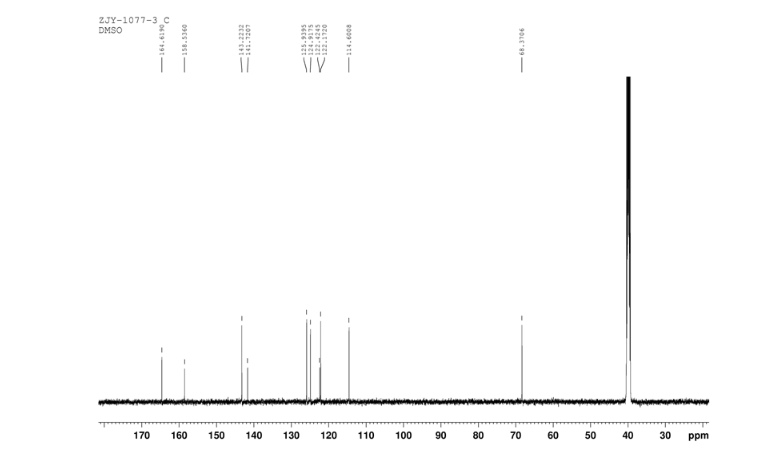
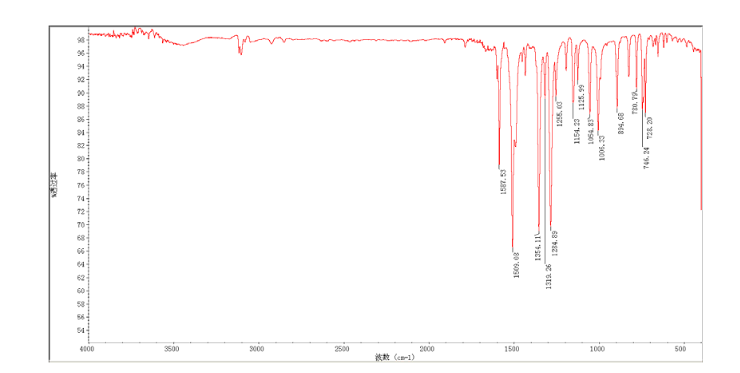
3-Chloro-4-(thiazol-2-ylmethoxy)aniline (9)
In a flask equipped with a reflux condenser, the compound 8 15.00 g (55.6 mmol), reduced zinc powder 14.44 g (222.0 mmo1, 4 eq), saturated ammonia chloride (5 mL) and methanol (100 mL) were mixed. The mixture was stirred at a temperature of 40 °C for 1.5 h. Then the zinc powder was filtered off, the filtrate was concentrated to obtain yellow solid 13.21 g, yield 99%. m.p. 60-61 °C; 1 H NMR (DMSO-d6): δ 7.80 (1 H, d, J=3.3 Hz), 7.75 (1 H, d, J=3.3 Hz), 6.96 (1 H, d, J=8.8 Hz), 6.64(1 H, d, J=2.7 Hz), 6.46 (1 H, dd, J=2.7 Hz, J=8.7 Hz), 5.30 (s, 2 H), 5.04 (s, 2 H, NH2, exchangeable); 13C NMR (DMSO-d6) δ: 166.8, 145.1, 144.1, 142.80, 123.1, 121.5, 117.7, 115.2, 113.6, 69.1. IR (KBr) ν: 3322, 3192, 3112, 1607, 1499, 1457, 1436, 1291, 1274, 1221, 1191, 1144, 1057, 1027, 857, 797, 767, 733, 584 cm-1. Anal. calcd for C10H9N2OSCl: C 49.90, H 3.77, N 11.64, O 6.65; found C 49.95, H 3.76, N 11.66, O 6.60; MS (ESI) m/z: 239.01 (M-H).
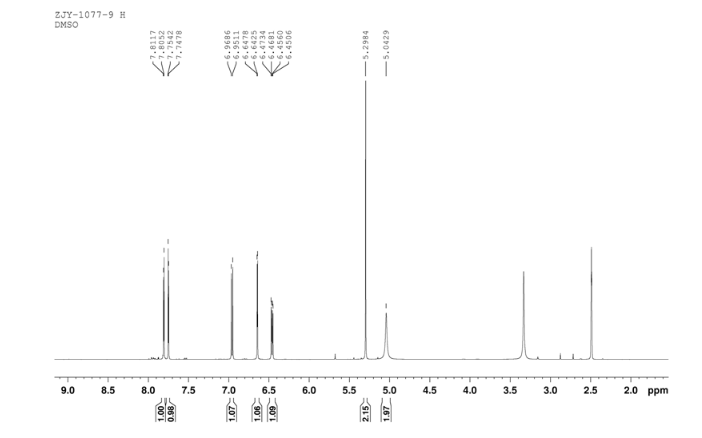 1H NMR DMSOD6 OF 9
1H NMR DMSOD6 OF 9
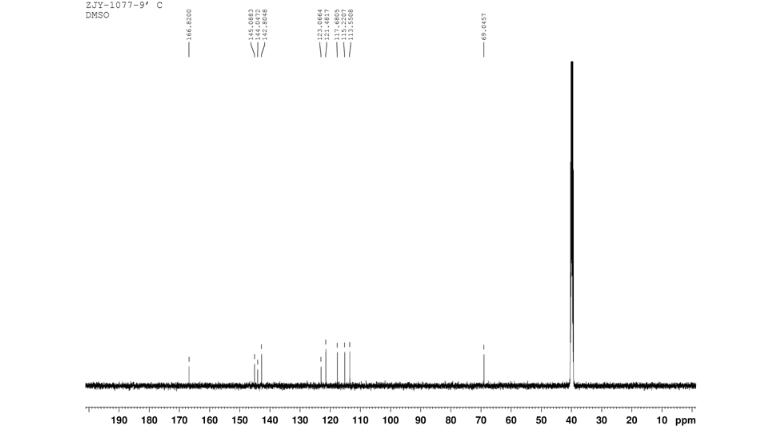 13C NMR OF 9 IN DMSOD6
13C NMR OF 9 IN DMSOD6
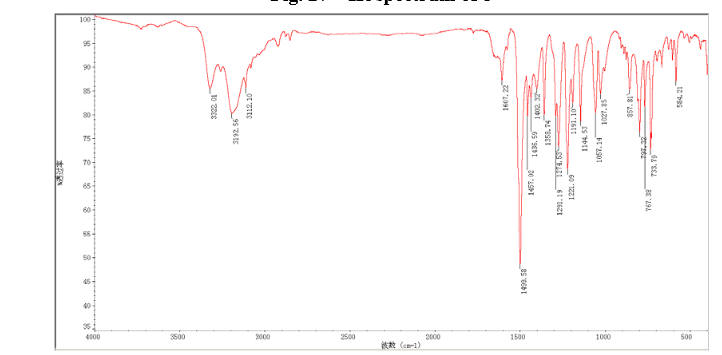
N-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)-6-nitro- quinazolin-4-amine(10)
In a flask equipped with a reflux condenser, 6-nitro-4-chloro- quinazoline 8.0 g (38.3 mmol) and 3-Chloro-4-(thiazol-2-ylmethoxy)aniline 8.9 g (37.0 mmol) were dissolved into 150 mL of THF, and the solution was refluxed for 3 h.Then a lot of yellow solid was deposited. Then it was filtered affording to yellow solid 12.8 g, yield 81%. m.p. 183-184 °C (decompose); 1 H NMR (DMSO-d6): δ 11.97(s, 1 H, exchangeable), 9.84 (s, 1 H), 9.00 (s, 1 H), 8.76 (1 H, d, J=9.1 Hz), 8.12-8.14 (m, 1 H), 7.94 (1 H, d, J=2.3 Hz), 7.87 (1 H, d, J=3.2 Hz), 7.81 (1 H, d, J=3.2 Hz), 7.44 (1 H, d, J=9.0 Hz), 7.69 (1 H, dd, J=2.5 Hz, J=8.9 Hz), 5.61 (s, 2 H); 13C NMR (DMSO-d6) δ: 166.8, 145.1, 144.1, 142.8, 123.1, 121.5, 117.7, 115.2, 113.7, 69.1. IR (KBr) ν: 3442, 3100, 1636, 1618, 1570, 1552, 1523, 1492, 1442, 1400, 1377, 1344, 1301, 1267, 1069, 805 cm-1. Anal. calcd for C18H12N5O3SCl: C 52.24, H 2.92, N 16.92, O 11.60; found C 52.26, H 2.93, N 16.96, O 11.58; MS (ESI) m/z: 412.84 (M-H).
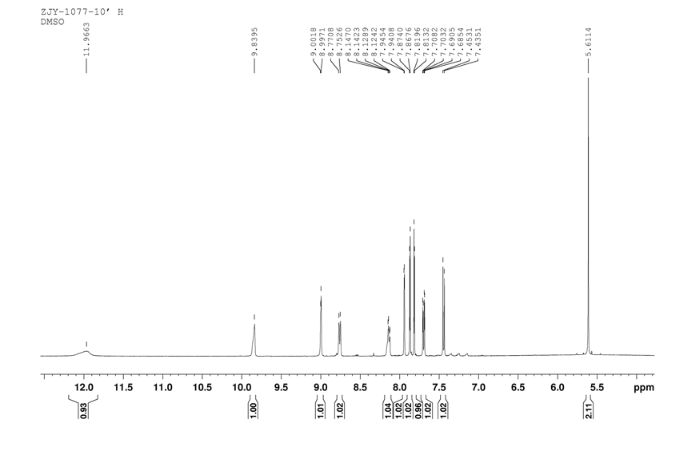 1H NMR DMSOD6 OF 10
1H NMR DMSOD6 OF 10
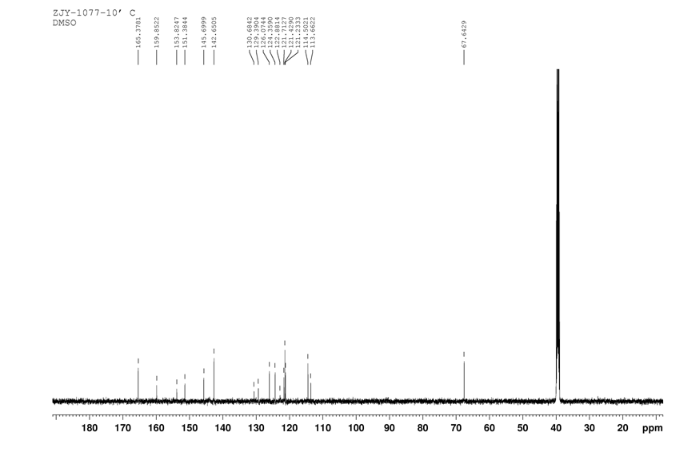 13C NMR OF 10 IN DMSOD6
13C NMR OF 10 IN DMSOD6
N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)quinazoline-4,6-diamine (11)
In a flask equipped with a reflux condenser, the compound 10 5.00 g (12.1 mmol), reduced zinc powder 3.2 g (48.5 mmo1, 4 eq), saturated ammonia chloride (3 mL) and methanol (60 mL) were mixed. The mixture was stirred at room temperature for 30 min. Then the zinc powder was filtered off, the filtrate was concentrated to obtain yellow solid 4.58 g, yield 98%. m.p. 197-198 °C (decompose); 1 H S4 NMR (DMSO-d6): δ 9.33(s, 1 H, exchangeable), 8.31 (s, 1 H), 8.05 (d, 1 H, J=2.6 Hz), 7.85 (d, 1 H, J=3.3 Hz), 7.79 (1 H, d, J=3.3 Hz), 7.73 (1 H, dd, J=2.5 Hz, J=9.0 Hz), 7.51 (1 H, d, J=8.9 Hz), 7.30 (1 H, d, J=2.4 Hz), 7.29 (1 H, d, J=4.7 Hz), 7.23 (1 H, dd, J=2.3 Hz, J=8.9 Hz), 5.57 (s, 2 H, exchangeable), 5.52 (s, 2 H); 13C NMR (DMSO-d6) δ: 165.9, 155.8, 149.7, 148.5, 147.3, 142.6, 142.5, 134.6, 128.7, 123.6, 123.2, 121.4, 121.3, 121.1, 116.5, 114. 7, 100.9, 67.8. IR (KBr) ν: 3443, 3358, 3211, 3100, 1631, 1596, 1577, 1560, 1530, 1494, 1431, 1383, 1217, 910 cm-1. Anal. calcd for C18H14N5OSCl: C 56.32, H 3.68, N 18.24, O 4.17; found C 56.34, H 3.70, N 18.22, O 4.14; MS (ESI) m/z: 382.66 (M-H).
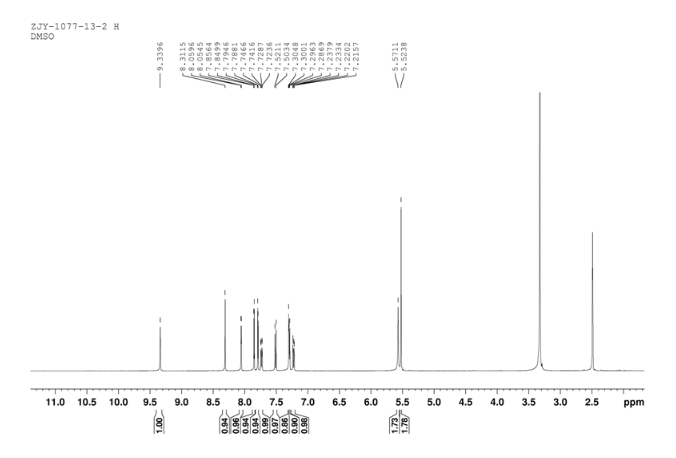 11 1HNMR DMSOD6
11 1HNMR DMSOD6
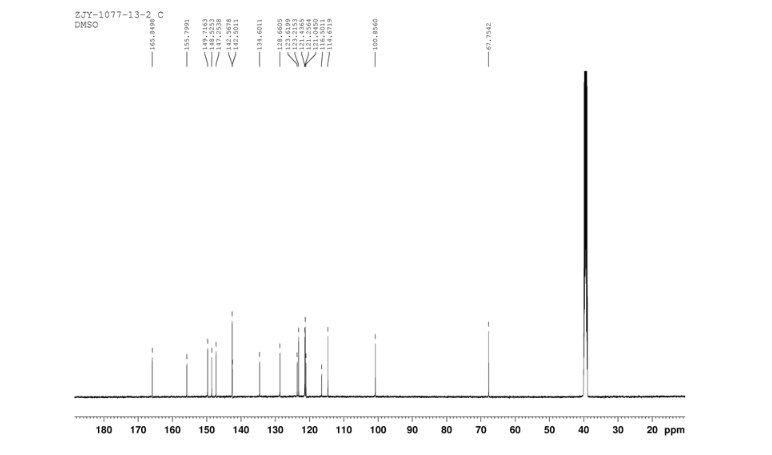 13C NMR OF 11 IN DMSOD6
13C NMR OF 11 IN DMSOD6
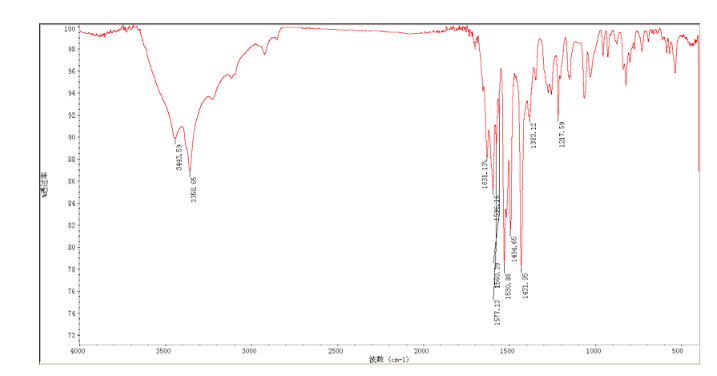
Construction finally as per patent ……….US20050043334
Treatment of N4-[3-chloro-4-(thiazol-2-ylmethoxy)phenyl]quinazoline-4,6-diamine (11) with 1,1′-thiocarbonyldiimidazole , followed by condensation with 2(R)-amino-1-propanol in THF/CH2Cl2 affords thiourea derivative , which finally undergoes cyclization in the presence of TsCl and NaOH in THF/H2O to furnish varlitinib .
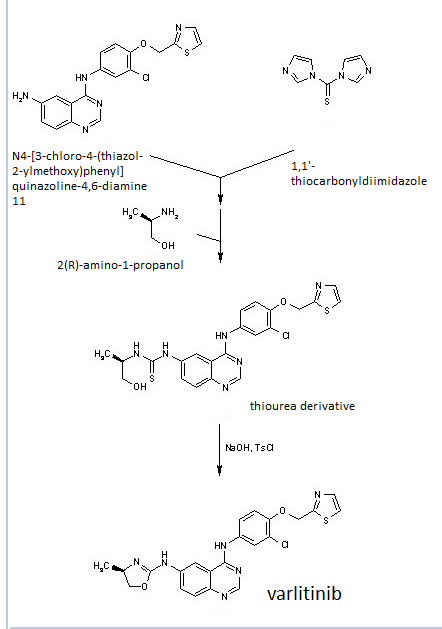
-
ASLAN Pharmaceuticals
-
Address: 10 Bukit Pasoh Rd, Singapore 089824Phone:+65 6222 4235


 carl fith
carl fith
Mr Carl Firth, CEO, Aslan Pharmaceuticals, Singapore (left) and Mr Dan Devine, CEO, Patrys, Australia (right)
///////ASLAN001, varlitinib, ASLAN Pharmaceuticals, Orphan Designation, ARRY-534, ARRY-334543 , PHASE 2, ORPHAN DRUG DESIGNATION, array
