

Home » Posts tagged 'PHASE 1' (Page 3)
Tag Archives: PHASE 1
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
HEC-68498
HEC-68498, CT-365
CAS 1621718-37-3
C20 H13 F2 N5 O3 S
441.41
Benzenesulfonamide, N-[5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxy-3-pyridinyl]-2,4-difluoro-
N-[5-(3-Cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxy-3-pyridinyl]-2,4-difluorobenzenesulfonamide
HEC Pharm , Calitor Sciences Llc; Sunshine Lake Pharma Co Ltd
PHASE 1, idiopathic pulmonary fibrosis and solid tumors
Phosphoinositide 3-kinase inhibitor; mTOR inhibitor

- Originator HEC Pharm
- Developer HEC Pharm; Sunshine Lake Pharma
- Class Anti-inflammatories; Antifibrotics; Isoenzymes
- Mechanism of Action 1 Phosphatidylinositol 3 kinase inhibitors; MTOR protein inhibitors
- Phase I Idiopathic pulmonary fibrosis
- 22 May 2018 Phase-I clinical trials in Idiopathic pulmonary fibrosis in USA (PO) (NCT03502902)
- 24 Apr 2018 Sunshine Lake Pharma in collaboration with Covance plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in China , (NCT03502902)
- 19 Apr 2018 Preclinical trials in Idiopathic pulmonary fibrosis in China (PO)
- US 20140234254
- CN 103965199
CN 103965199

CN 103965199

Sunshine Lake Pharma , a subsidiary of HEC Pharm is developing an oral capsule formulation of HEC-68498 (phase 1, in July 2019) sodium salt, a dual inhibitor of phosphoinositide-3 kinase and the mTOR pathway, for the treatment of idiopathic pulmonary fibrosis and solid tumors
HEC 68498 is an oral inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin in clinical development at HEC Pharm for the treatment of idiopathic pulmonary fibrosis. A phase I trial is under way in healthy volunteers.
The phosphoinositide 3-kinases (PI3 kinases or PI3Ks), a family of lipid kinases, have been found to play key regulatory roles in many cellular processes including cell survival, proliferation and differentiation. The PI3K enzymes consist of three classes with variable primary structure, function and substrate specificity. Class I PI3Ks consist of heterodimers of regulatory and catalytic subunits, and are subdivided into 1A and 1B based on their mode of activation. Class 1A PI3Ks are activated by various cell surface tyrosine kinases, and consist of the catalytic pl lO and regulatory p85 subunits. The three known isoforms of Class 1A pl lO are pl lOot, rΐ ΐqb, and rΐ ΐqd, which all contain an amino terminal regulatory interacting region (which interfaces with p85), a Ras binding domain, and a carboxy terminal catalytic domain. Class IB PI3Ks consist of the catalytic (pl lOy) and regulatory (p 101 ) subunits and are activated by G-protein coupled receptors. (“Small-molecule inhibitors of the PI3K signaling network” Future Med. Chem ., 2011, 3, 5, 549-565).
[0004] As major effectors downstream of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), PI3Ks transduce signals from various growth factors and cytokines into intracellular massages by generating phospholipids, which activate the serine-threonine protein kinase ART (also known as protein kinase B (PKB)) and other downstream effector pathways. The tumor suppressor or PTEN (phosphatase and tensin
homologue) is the most important negative regulator of the PI3K signaling pathway. (“Status of PBK/Akt/mTOR Pathway Inhibitors in Lymphoma.” Clin Lymphoma, Myeloma Leuk , 2014, 14(5), 335-342.)
[0005] The signaling network defined by phosphoinositide 3-kinases (PI3Ks), AKT and mammalian target of rapamycin (mTOR) controls most hallmarks of cancer, including cell cycle, survival, metabolism, motility and genomic instability. The pathway also contributes to cancer promoting aspects of the tumor environment, such as angiogenesis and inflammatory cell recruitment. The lipid second messenger produced by PI3K enzymes, phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3; also known as PIP3), is constitutively elevated in most cancer cells and recruits cytoplasmic proteins to membrane-localized‘onco’ signal osomes.
[0006] Cancer genetic studies suggest that the PI3K pathway is the most frequently altered pathway in human tumors: the PIK3CA gene (encoding the PI3K catalytic isoform pl lOa) is the second most frequently mutated oncogene, and PTEN (encoding phosphatase and tensin homolog, the major PtdIns(3,4,5)P3 phosphatase) is among the most frequently mutated tumor suppressor genes. In accord, a recent genomic study of head and neck cancer found the PI3K pathway to be the most frequently mutated. Indeed, even in cancer cells expressing normal PI3K and PTEN genes, other lesions are present that activate the PI3K signaling network (that is, activated tyrosine kinases, RAS and AKT, etc ). As a net result of these anomalies, the PI3K pathway is activated, mutated or amplified in many malignancies, including in ovarian cancer (Campbell et al., Cancer Res., 2004, 64, 7678-7681; Levine et al., Clin. Cancer Res., 2005, 11, 2875-2878; Wang et al., Hum. Mutat., 2005, 25, 322; Lee et al., Gynecol. Oncol. ,2005, 97, 26-34), cervical cancer, breast cancer (Bachman et al.,· Cancer Biol., Ther, 2004, 3, 772-775; Levine et al., supra; Li et al., Breast Cancer Res. Treat., 2006, 96, 91-95; Saal et al., Cancer Res., 2005, 65, 2554-2559; Samuels and Velculescu, Cell Cycle, 2004, 3, 1221-1224), colorectal cancer (Samuels et al., Science, 2004, 304, 554; Velho et al., Eur. J. Cancer, 2005, 41, 1649-1654), endometrial cancer (Oda et al ., Cancer Res., 2005, 65, 10669-10673), gastric carcinomas (Byun et al., M. J. Cancer, 2003 , 104, 318-327; Li et al., supra; Velho et al., supra; Lee et al., Oncogene, 2005 , 24, 1477-1480), hepatocellular carcinoma (Lee et al., id), small and non-small cell lung cancer (Tang et al., Lung Cancer 2006, 11, 181-191; Massion et al , Am. J. Respir. Crit. Care Med., 2004, 170, 1088-1094), thyroid carcinoma (Wu et al., J. Clin. Endocrinol. Metab., 2005, 90, 4688-4693),
acute myelogenous leukemia (AML) (Sujobert et al., Blood, 1997, 106, 1063-1066), chronic myelogenous leukemia (CML) (Hickey et al., J. Biol. Chem ., 2006, 281, 2441-2450), glioblastomas (Hartmann et al. Jlcta Neuropathol (Bert ), 2005, 109, 639-642; Samuels et al., supra), Hodgkin and non-Hodgkin lymphomas (“PI3K and cancer: lessons, challenges and opportunities”, Nature Reviews Drug Discovery., 2014, 13, 140).
[0007] The PI3K pathway is hyperactivated in most cancers, yet the capacity of PI3K inhibitors to induce tumor cell death is limited. The efficacy of PI3K inhibition can also derive from interference with the cancer cells’ ability to respond to stromal signals, as illustrated by the approved PI3K5 inhibitor idelalisib in B-cell malignancies. Inhibition of the leukocyte-enriched PI3K5 or RI3Kg may unleash antitumor T-cell responses by inhibiting regulatory T cells and immune-suppressive myeloid cells. Moreover, tumor angiogenesis may be targeted by PI3K inhibitors to enhance cancer therapy. (“Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy”, Cancer Discov., 2016, 6(10), 1090-1105.)
[0008] mTOR is a highly conserved serine-threonine kinase with lipid kinase activity and participitates as an effector in the PI3K/AKT pathway. mTOR exists in two distinct complexes, mTORCl and mTORC2, and plays an important role in cell proliferation by monitoring nutrient avaliability and cellular energy levels. The downstream targets of mTORCl are ribosomal protein S6 kinase 1 and eukaryotic translation initiation factor 4E-binding protein 1, both of which are crucial to the regulation of protein synthesis. (“Present and future of PI3K pathway inhibition in cancer: perspectives and limitations”, Current Med. Chem., 2011, 18, 2647-2685).
[0009] Knowledge about consequences of dysregulated mTOR signaling for tumorigenesis comes mostly from studies of pharmacologically disruption of mTOR by repamycin and its analogues such as temsirolimus (CCI-779) and everolimus (RADOOl).Rapamycin was found to inhibit mTOR and thereby induce G1 arrest and apoptosis. The mechanism of rapamycin growth inhibition was found to be related to formation of complexes of rapamycin with FK-binding protein 12 (FKBP-12). These complexes then bound with high affinity to mTOR, preventing activation and resulting in inhibition of protein translation and cell growth. Cellular effects of mTOR inhibition are even more pronounced in cells that have concomitant inactivation of PTEN. Antitumor activity of rapamycin was subsequently identified, and a number of rapamycin analogues such as temsirolimus and everolimus have been approved by the US Food and Drug
Administration for the treatment certain types of cancer.
[0010] Fibrosis is the formation of excess fibrous connective tissue in an organ or tissue in a reparative or reactive process. Examples of fibrosis include, but are not limited to pulmonary fibrosis, liver fibrosis, dermal fibrosis, and renal fibrosis. Pulmonary fibrosis, also called idiopathic pulmonary fibrosis (IPF), interstitial diffuse pulmonary fibrosis, inflammatory pulmonary fibrosis, or fibrosing alveolitis, is a lung disorder and a heterogeneous group of conditions characterized by abnormal formation of fibrous tissue between alveoli caused by alveolitis comprising cellular infiltration into the alveolar septae with resulting fibrosis. The effects of IPF are chronic, progressive, and often fatal.
[0011] The clinical course of IPF is variable and largely unpredictable. IPF is ultimately fatal, with historical data suggesting a median survival time of 2-3 years from diagnosis. A decline in forced vital capacity (FVC) is indicative of disease progression in patients with IPF and change in FVC is the most commonly used endpoint in clinical trials. A decline in FVC of 5% or 10% of the predicted value over 6-12 months has been associated with increased mortality in patients with IPF.
[0012] Our understanding of the pathogenesis of IPF has evolved from that of a predominantly inflammatory disease to one driven by a complex interplay of repeated epithelial cell damage and aberrant wound healing, involving fibroblast recruitment, proliferation and differentiation, and culminating in excess deposition of extracellular matrix. This shift in knowledge prompted a change in the type of compounds being investigated as potential therapies, with those targeted at specific pathways in the development and progression of fibrosis becoming the focus.
[0013] In patients with IPF, the mechanisms whereby PI3K/mTOR inhibitors act may involve inhibition of kinases such as PI3Ks and mTOR. This results in inactivation of cellular receptors for mediators involved in the development of pulmonary fibrosis. As a result, fibroblast proliferation is inhibited and extracellular matrix deposition is reduced. (“Update on diagnosis and treatment of idiopathic pulmonary fibrosis”, J Bras Pneumol. 2015, 41(5), 454-466.)
[0014] Accordingly, small-molecule compounds that specially inhibit, regulate and/or modulate the signal transduction of kinases, particularly including PI3K and mTOR as described above, are desirable as a means to prevent, manage, or treat proliferative disorders and fibrosis, particular idiopathic pulmonary fibrosis in a patient. One such small-molecule is A-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfon-amide, which has the chemical structure as shown in the following:
[0015] WO 2014130375A1 described the synthesis of N-(5 -(‘3 -cyanopyrazol o [l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (Example 3) and also disclosed the therapeutic activity of this molecule in inhibiting, regulating and modulating the signal transduction of protein kinases.
[0016] Different salts and solid state forms of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, improving the dissolution profile, stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also provide improvements to the final dosage form, for example, if they serve to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
Different salts and solid state forms of /V-(5-(3-cyanopyrazolo[l,5- ]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide are described herein.
PATENT
WO2014130375 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014130375
claiming new pyrazolo[1,5-a]pyridine derivatives are PI3K and mTOR inhibitors, useful for treating proliferative diseases
Example 3 N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
Step 1) 5-bromopyrazolo[1,5-a]pyridine
[196] A solution of ethyl 5-bromopyrazolo[1,5-a]pyridine-3-carboxylate (240
mmol) in 40% H2SO4 (12 mL) was stirred at 100 °C for 4 hours, then cooled to rt, and neutralized to pH=7 with aq. NaOH (6 M) in ice bath. The resulted mixture was extracted with DCM (25 mL x 2). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (175 mg, 99.5%).
MS (ESI, pos. ion) m/z: 196.9 [M+H]+.
Step 2) 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde
[197] To a solution of 5-bromopyrazolo[1,5-a]pyridine (175 mg, 0.89 mmol) in DCM (6 mL) was added (chloromethylene)dimethyliminium chloride (632 mg, 3.56 mmol). The reaction was stirred at 44 °C overnight, and concentrated in vacuo. The residue was dissolved in saturated NaHCO3 aqueous solution (25 mL) and the resulted mixture was then extracted with EtOAc (25 mL x 3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (225 mg, 100%).
MS (ESI, pos. ion) m/z: 225.0 [M+H]+.
Step 3) (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime
[198] To a suspension of 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde (225 mg, 1 mmol) in EtOH (10 mL) and H2O (5 mL) was added hydroxylamine hydrochloride (104 mg, 1.5 mmol). The reaction was stirred at 85 °C for 2 hours, then cooled to rt, and concentrated in vacuo. The residue was adjusted to pH=7 with saturated NaHCO3 aqueous solution. The resulted mixture was then filtered and the filter cake was dried in vacuo to give title compound as a yellow solid (240 mg, 99%).
MS (ESI, pos. ion) m/z: 240.0 [M+H]+.
Step 4) 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile
[199] A solution of (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime (240 mg,
1 mmol) in Ac2O (6 mL) was stirred at 140 °C for 18 hours, then cooled to rt, and concentrated in vacuo. The residue was washed with Et2O (1 mL) to give the title compound as a yellow solid (44 mg, 22.5%).
MS (ESI, pos. ion) m/z: 222.0 [M+H]+.
Step 5) N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
[200] 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (612 mg, 1.5 mmol), 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile (222 mg, 1 mmol), Pd(dppf)Cl2·CH2Cl2 (16 mg, 0.02 mmol) and Na2CO3 (85 mg, 0.8 mmol) were placed into a two-neck flask, then degassed with N2 for 3 times, and followed by adding 1,4-dioxane (5 mL) and water (1 mL). The resulted mixture was degassed with N2 for 3 times, then heated to 90 °C and stirred further for 5 hours. The mixture was cooled to rt and filtered. The filtrate was concentrated in vacuo and the residue was purified by a silica gel column chromatography (PE/EtOAc (v/v) = 1/2) to give the title compound as a light yellow solid (400 mg, 81.6%).
MS (ESI, pos. ion) m/z: 442.0 [M+H]+;
1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.37 (s, 1H), 9.02 (d, J = 7.2 Hz, 1H), 8.67 (s, 1H), 8.60 (d, J = 2.2 Hz, 1H), 8.26-8.16 (m, 2H), 7.82-7.72 (m, 1H), 7.57 (dd, J = 13.2, 5.8 Hz, 2H), 7.21 (t, J= 8.5 Hz, 1H), 3.67 (s, 3H).
PATENT
WO-2019125967
The invention relates to salts of pyrazolo[l,5-a]pyridine derivatives and use thereof, specifically relates to salt of /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl) -2,4-difluorobenzenesulfonamide (compound of formula (I)) and use thereof, further relates to composition containing said salts above. The salts or the composition can be used to inhibit/modulate protein kinases, further prevent, manage or treat proliferative disorders or pulmonary fibrosis in a patient.
Amorphous form of mono-sodium salt of HEC-68498 , useful for treating a proliferative disorder or pulmonary fibrosis.
The invention is further illustrated by the following examples, which are not be construed as limiting the invention in scope.
[00108] /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluoroben zenesulfonamide can be prepared according to the synthetic method of example 3 disclosed in WO2014130375 Al.
//////////////HEC-68498, HEC 68498, HEC68498, HEC Pharm , Calitor Sciences, Sunshine Lake Pharma, PHASE 1, proliferative disorder, pulmonary fibrosis, idiopathic pulmonary fibrosis, solid tumors, CT-365 , CT 365 , CT365
Fc1ccc(c(F)c1)S(=O)(=O)Nc2cc(cnc2OC)c3ccn4ncc(C#N)c4c3
GNE-0877

GNE-0877
Maybe DNL-151 ?
CAS 1374828-69-9
Chemical Formula: C14H16F3N7
Molecular Weight: 339.31895
2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-1H-pyrazol-1-yl)propanenitrile
Denali Therapeutics Inc, useful for treating Alzheimer’s disease, breast tumor, type I diabetes mellitus and Crohn’s disease
GNE-0877 is a highly potent and selective LRRK2 inhibitor. Leucine-rich repeat kinase 2 (LRRK2) has drawn significant interest in the neuroscience research community because it is one of the most compelling targets for a potential disease-modifying Parkinson’s disease therapy.
- Developer Denali Therapeutics Inc
- Class Antiparkinsonians; Small molecules
- Mechanism of Action LRRK2 protein inhibitors
- Phase I Parkinson’s disease
- 20 Dec 2017 Denali Therapeutics plans clinical studies for Parkinson’s disease
- 13 Nov 2017 Phase-I clinical trials in Parkinson’s disease (In volunteers) in Netherlands (unspecified route)
- 13 Nov 2017 Preclinical trials in Parkinson’s disease in USA (unspecified route) before November 2017
Denali Therapeutics is developing DNL-151 (phase 1, in July 2019), a lead from a program of small-molecule inhibitors of LRRK2 originally licensed from Genentech, for the treatment of Parkinson’s disease.
Leucine-rich repeat kinase 2 (LRRK2) is a complex signaling protein that is a key therapeutic target, particularly in Parkinson’s disease (PD). Combined genetic and biochemical evidence supports a hypothesis in which the LRRK2 kinase function is causally involved in the pathogenesis of sporadic and familial forms of PD, and therefore that LRRK2 kinase inhibitors could be useful for treatment (Christensen, K.V. (2017) Progress in medicinal chemistry 56:37-80). Inhibition of the kinase activity of LRRK2 is under investigation as a possible treatment for Parkinson’s disease (Fuji, R.N. et al (2015) Science Translational Medicine 7(273):ral5;
Taymans, J.M. et al (2016) Current Neuropharmacology 14(3):214-225). A group of LRRK2 kinase inhibitors have been studied (Estrada, A.A. et al (2015) Jour. Med. Chem. 58(17): 6733-6746; Estrada, A.A. et al (2013) Jour. Med. Chem. 57:921-936; Chen, H. et al (2012) Jour. Med. Chem. 55:5536-5545; Estrada, A.A. et al (2015) Jour. Med. Chem. 58:6733-6746; US 8354420; US 8569281; US8791130; US 8796296; US 8802674; US 8809331; US 8815882; US 9145402; US 9212173; US 9212186; WO 2011/151360; WO 2012/062783; and WO 2013/079493.
PATENT
WO2012062783 , assigned to Hoffmann-La Roche , but naming inventors specifically associated with both Genentech and BioFocus (which had an agreement with Genentech for drug discovery programs); the compound was also later identified in J.Med.Chem (57(3), 921-936, 2014) in an article from these two companies, with the lab code GNE-0877. So while this represents the first application in the name of Denali Therapeutics Inc that focuses on this compound, it is likely that it provides the structure of DNL-151 , a lead from a program of small-molecule inhibitors of leucine-rich repeat kinase 2 (LRRK2) originally licensed from Genentech, being developed for the oral treatment of Parkinson’s disease, and which had begun phase I trials by December 2017 (when this application was lodged).
PATENT
WO2019104086 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019104086
claiming novel crystalline and amorphous forms of pyrimidinylamino-pyrazole compound, useful for treating Alzheimer’s disease, breast tumor, type I diabetes mellitus and Crohn’s disease.
Novel crystalline and amorphous forms of 2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-1H-pyrazol-1-yl)propanenitrile (which is substantially pure form) and their anhydrous and solvates such as cyclohexanol solvate (designated as Forms B-D), processes for their preparation and compositions comprising them are claimed. The compound is disclosed to be leucine rich serine threonine kinase 2 inhibitor, useful for treating Gaucher disease, Alzheimer’s disease, motor neurone disease, Parkinson’s disease, prostate tumor, Lewy body dementia, mild cognitive impairment, breast tumor, type I diabetes mellitus and Crohn’s disease.
The present disclosure relates to crystalline polymorph or amorphous forms of a pyrimidinylamino-pyrazole kinase inhibitor, referred to herein as the Formula I compound and having the structure:
FORMULA I COMPOUND
The present disclosure includes polymorphs and amorphous forms of Formula I compound, (CAS Registry Number 1374828-69-9), having the structure:
and named as: 2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-lH-pyrazol-l-yl)propanenitrile (WO 2012/062783; US 8815882; US 2012/0157427, each of which are incorporated by reference). As used herein, the Formula I compound includes tautomers, and pharmaceutically acceptable salts or cocrystals thereof. The Formula I compound is the API (Active Pharmaceutical Ingredient) in formulations for use in the treatment of neurodegenerative and other disorders, with pKa when protonated calculated at 6.7 and 2.1.
CRYSTALLIZATION
Initial polymorph screening experiments were performed using a variety of
crystallization or solid transition methods, including: anti-solvent addition, reverse anti-solvent addition, slow evaporation, slow cooling, slurry at room temperature (RT), slurry at 50 °C, solid vapor diffusion, liquid vapor diffusion, and polymer induced crystallization. By all these methods, the Form A crystal type was identified. Polarized light microscopy (PLM) images of Form A obtained from various polymorph screening methods were collected (Example 5).
Particles obtained via anti-solvent addition showed small size of about 20 to 50 microns (pm) diameter while slow evaporation, slow cooling (except for THF/isooctane), liquid vapor diffusion and polymer-induced crystallization resulted in particles with larger size. Adding isooctane into a dichloromethane (DCM) solution of the Formula I compound produced particles with the most uniform size. Crude Formula I compound crystallized from THF///-heptane and then was micronized. A crystallization procedure was developed to control particle size.
A total of four crystal forms (Forms A, B, C, and D) and an amorphous form E of Formula I compound were prepared, including 3 anhydrates (Form A, C, and D) and one solvate (Form B). Slurry competition experiments indicated that Form D was thermodynamically more stable when the water activity aw< 0.2 at RT, while Form C was more stable when aw> 0.5 at RT. The 24 hrs solubility evaluation showed the solubility of Form A, C and D in FLO at RT was 0.18, 0.14 and 0.11 mg/mL, respectively. DVS (dynamic vapor sorption) results indicated that Form A and D were non-hygroscopic as defined by less than 0.1% reversible water intake in DVS, while Form C was slightly hygroscopic. Certain characterization data and observations of the crystal forms are shown in Table 1.
Table 1 Characterization summary for crystal forms of Formula I compound
Differential Scanning Calorimetry (DSC) analysis of Forms A and C showed that Form C had higher melting point and higher heat of fusion (Table 1), suggesting that the two forms are monotropic with Form C being the more stable form. Competitive slurry experiments with 1 : 1 Form A and C in a variety of solvents always produced Form C confirming that Form C was
more stable than Form A. In accordance with this, Form C was produced even when the crystallization batch was seeded with seeds of Form A.
PATENT
WO-2019126383
Methods of making leucine-rich repeat kinase 2 (LRRK2)-inhibiting, pyrimidinyl-4-aminopyrazole compounds (eg 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol-1-yl)propanenitrile), useful for treating LRRK2 mediated diseases such as Parkinson’s disease.
Example 1 Preparation of 2-(4-amino-3 -methyl- liT-pyrazol-l -yl)-2-methylpropanamide 5a
4a 5a
To a 20-L reactor containing dimethyl formamide (4.5 L) was charged 5-methyl-4-nitro-lH-pyrazole la (1.5 kg, 1.0 equiv). The solution was cooled to 0 °C and charged with finely ground K2CO3 (2.45 kg, 1.5 equiv) in three portions over ~l h. Methyl 2-bromo-2-methylpropanoate (3.2 kg, 1.5 equiv) was added dropwise to the mixture and then was allowed to warm to ~25 °C. The reaction mixture was maintained for 16 h and then quenched with water (15 L) and product was extracted with ethyl acetate. The combined organic layer was washed with water, and then with a brine. The organic layer was dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure to give a light yellow solid. The crude product was purified by crystallization with petroleum ether (15 L), filtered, and dried to give methyl 2-m ethyl -2-(3 -methyl -4-nitro- l//-pyrazol- l -yl)propanoate 3a (2.25 kg, >99% purity by HPLC, 84 % yield) as an off-white solid. ¾ NMR (400 MHz, CDCb) 8.28 (s, 1H), 3.74 (s, 3H), 2.53 (s, 3H), 1.85 (s, 6H).
Methanol (23 L) and 2-methyl-2-(3-methyl-4-nitro-lif-pyrazol-l-yl)propanoate 3a (2.25 kg, 1.0 equiv) were charged into a 50-L reactor and cooled to approximately -20 °C. Ammonia gas was purged over a period of 5 h and then the reaction mixture warmed to 25 °C. After 16 h, the reaction mixture was concentrated under reduced pressure (~50 °C) to give the crude product. Ethyl acetate (23 L) was charged and the solution agitated in the presence of charcoal (0.1 w/w) and Celite® (0.1 w/w) at 45 °C. The mixture was filtered and concentrated under reduced pressure, and then the solid was slurried in methyl tert-butyl ether (MTBE, 11.3 L) at RT for 2 h. Filtration and drying at ~45 °C gave 2-m ethyl -2-(3 -m ethyl -4-ni tro- 1 //-pyrazol – 1 -yl)propanamide 4a (1.94 kg, >99% purity by HPLC, 92% yield).
Methanol (5 L) and 2-m ethyl-2-(3 -methyl -4-nitro-lif-pyrazol-l-yl)propanamide 4a (0.5 kg) were charged into a 10-L autoclave under nitrogen atmosphere, followed by slow addition of 10 % (50% wet) Pd/C (50 g). Hydrogen was charged (8.0 kg pressure/l 13 psi) and the reaction mixture agitated at 25 °C until complete. The mixture was filtered, concentrated under reduced
pressure and then slurried in MTBE (2.5 L) for 2 h at 25 °C. Filtration and drying under reduced pressure (45 °C) gave 2-(4-amino-3-methyl- l//-pyrazol- l -yl)-2-methyl propanamide 5a (0.43 kg, >99% purity by HPLC, 99% yield).
Example 2 Preparation of 2-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-3-methyl-lH-pyrazol-l-yl)-2-methylpropanamide 7a
DCM
Into a first reactor was charged /-BuOH (or alternatively 2-propanol) (15.5 vol) and 2-(4-amino-3 -methyl- li7-pyrazol-l-yl)-2-methylpropanamide 5a (15 kg), followed by zinc chloride (13.5 kg, 1.2 equiv) at room temperature and the suspension agitated ~2 h. Into a second reactor was charged dichloromethane (DCM, 26.6 vol) and 2,4-dichloro-5-trifluoromethyl pyrimidine 6a (19.6 kg, 1.1 equiv) and then cooled to 0 °C. The contents from first reactor were added portion-wise to the second reactor. After addition, the reaction mixture was agitated at 0 °C for ~l h and then Et3N (9.2 kg, 1.1 equiv) was slowly charged. After agitation for 1 h, the temperature was increased to 25 °C and monitored for consumption of starting material. The reaction mixture was quenched with 5% aqueous NaHCO, and then filtered over Celite®. The DCM layer was removed and the aqueous layer was back-extracted with DCM (3x). The combined organics were washed with water, dried (Na2S04), and concentrated. Methanol (2.5 vol) was charged and the solution was heated to reflux for 1 h, then cooled to 0 °C. After 1 h, the solids were filtered and dried under reduced pressure to give 2-(4-((4-chloro-5-(tri fluoromethyl)pyri mi din-2-yl)amino)-3 -methyl – l//-pyrazol- l -yl)-2-methyl propanamide 7a
(31.2 kg (wet weight)). 1H NMR (600 MHz, DMSO-de) 10.05 (br. s., 1H), 8.71 (d, J= 11 Hz, 1H), 7.95 (app. d, 1H), 7.18 (br. s., 1H), 6.78 (br. s., 1H), 2.14 (s, 3H), 1.67 (s, 6H).
Example 3 Preparation of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol- 1 -yl)propanamide 8a
A reactor was charged with anhydrous tetrahydrofuran (THF, 10 vol) and 2-(4-((4-chloro-5-(trifl uoromethyl )pyrimi din-2-yl)amino)-3 -methyl – l //-pyrazol- l -yl)-2-methylpropanamide 7a (21 kg) at room temperature with agitation. A solution of 2M
methylamine in THF (3.6 vol) was slowly charged to the reactor at 25 °C and maintained for ~3 h. The reaction mixture was diluted with 0.5 w/w aqueous sodium bicarbonate solution (10 w/w), and extracted with ethyl acetate (EtOAc, 4.5 w/w). The aqueous layer was extracted with EtOAc (4x), the organics were combined and then washed with H20 (7 w/w). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. «-Heptane (3 w/v) was added to the residue, agitated, filtered and dried under reduced pressure to give 2-m ethyl -2-(3 -methyl -4-((4-(methyl ami no)-5-(trifl uoromethyl )pyri mi din-2-yl)amino)- l //-pyrazol-1 -yl)propanamide 8a (19.15 kg, 93% yield). ¾ NMR (600 MHz, DMSO-d6) 8.85 (m, 1H), 8.10 (s, 1H), 8.00 (m, 1H), 7.16 (br. s., 1H), 6.94 (m, 1H), 6.61 (br. s., 1H), 2.90 (d, J = 4.3 Hz, 3H), 2.18 (br. s., 3H), 1.65 (s, 6H).
Example 4 Preparation of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol- 1 -yl)propanenitrile 9a
To a reactor was charged 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifl uoromethyl )pyri mi din-2-yl)amino)- l //-pyrazol- l -yl)propan amide 8a (15 kg, 1 equiv) at room temperature followed by EtOAc (2 vol) and 6.7 vol T3P (50% w/w in EtOAc). The reaction mixture was heated to 75 °C over 1 h and then agitated for 16 h until consumption of starting material. The reaction mixture was cooled between -10 to -15 °C then added drop-wise 5N aqueous NaOH (7 vol) resulting in pH 8-9. The layers were separated and the aqueous layer back-extracted with EtOAc (2 x 4 vol). The combined organic extracts were washed with 5 %
aqueous NaHCO, solution, and then distilled to azeotropically remove water. The organics were further concentrated, charged with «-heptane (2 vol) and agitated for 1 h at room temperature. The solids were filtered, rinsed with «-heptane (0.5 vol) and then dried under vacuum (<50 °C). The dried solids were dissolved in EtOAc (1.5 vol) at 55 °C, and then «-heptane (3 vol) was slowly added followed by 5-10% of 9a seeds. To the mixture was slowly added «-heptane (7 vol) at 55 °C, agitated for 1 h, cooled to room temperature and then maintained for 16 h. The suspension was further cooled between 0-5 °C, agitated for 1 hour, filtered, and then rinsed the filter with chilled 1 :6.5 EtOAc/«-heptane (1 vol). The product was dried under vacuum at 50 °C to give 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1 //-pyrazol – 1 -yl )propaneni tri 1 e 9a (9.5 kg, first crop), 67% yield). ‘H NMR (600 MHz, DMSO-d6) 8.14 (s, 1H), 8.13 (br. s., 1H), 7.12 (br. s., 1H), 5.72 (br. s, 1H), 3.00 (d, J= 4.6 Hz, 3H),
2.23 (s, 3H), 1.96 (s, 3H).
Example 5 Preparation of methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a
Following the procedure of Example 1, a mixture of methanol and methyl 2-methyl-2-(3-methyl-4-nitro-lH-pyrazol-l-yl)propanoate 3a (0.5 kg) was charged into an autoclave under nitrogen atmosphere, followed by slow addition of 10 % (50% wet) Pd/C. Hydrogen was charged under pressure and the reaction mixture agitated at 25 °C until complete. The mixture was filtered, concentrated under reduced pressure and then slurried in MTBE for 2 h at 25 °C. Filtration and drying under reduced pressure gave methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a (LC-MS, M+l=l98).
Example 6 Preparation of methyl 2-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-3 -methyl- lH-pyrazol- 1 -yl)-2-methylpropanoate 11a
Following the procedure of Example 2, a mixture of methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a and DIPEA (1.2 equiv) in /-BuOH was warmed to 80 °C. Then a solution of 2,4-dichloro-5-trifluoromethyl pyrimidine 6a in /-BuOH was added slowly drop wise at 80 °C. After 15 minutes, LCMS showed the reaction was complete, including later eluting 59.9% of product ester 11a, earlier eluting 31.8% of undesired regioisomer (ester), and no starting material 10a. After completion of reaction, the mixture was cooled to room temperature and a solid was precipitated. The solid precipitate was filtered and dried to give methyl 2-(4-((4-chloro-5-(trifluoromethyl)pyrimi din-2 -yl)amino)-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 11a (LC-MS, M+l=378).
PAPER
J.Med.Chem (57(3), 921-936, 2014
https://pubs.acs.org/doi/full/10.1021/jm401654j
2-Methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1H-pyrazol-1-yl)propanenitrile (11)
A solution of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1H-pyrazol-1-yl)propanamide (34, 250 mg, 0.7 mmol) in POCl3 (5 mL) was stirred at 90 °C for 1 h. The POCl3 was removed by evaporation. The mixture was then slowly poured onto ice (10 mL). The pH of the solution was adjusted to 8 with saturated sodium carbonate. The aqueous phase was extracted with EtOAc (3×). The combined organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated to give a residue that was purified by recrystallization to give 11 (100 mg, 42% yield) as a white solid. 1H NMR (300 MHz, DMSO) δ 9.18 (s, 1H), 8.29 (s, 1H), 8.14 (s, 1H), 7.10 (s, 1H), 2.91 (d, 3H), 2.22 (s, 3H), 1.94 (s, 6H). HRMS (ES) m/z: [M + H]+ calcd for C14H16F3N7H+, 340.1492; found, 340.1484.
Scheme 2

Scheme 2. Synthesis of N-Alkyl Pyrazole Analoguesa
aReagents and conditions: (a) NaH, methyl 2-bromo-2-methylpropanoate, DMF, 70%; (b) LiOH, THF-H2O, 90%; (c) (i) (COCl)2, CH2Cl2, (ii) R-NH2, THF; (d) Pd/C, H2, MeOH; (e) 26, Et3N, n-BuOH, 120 °C; (f) 26, TFA, 2-methoxyethanol, 70 °C; (g) POCl3, 90 °C, 42%.
GNE-9605
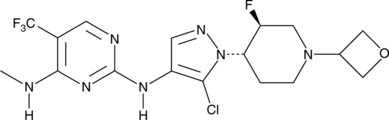
CAS № 1536200-31-3
Molecular Formula
C17H20ClF4N7O
Formula Weight
449.8
GNE-9065 is an orally bioavailable and potent inhibitor of leucine-rich repeat kinase 2 (LRRK2; IC50 = 18.7 nM).1 It is selective for LRRK2 over 178 kinases, inhibiting only TAK1-TAB1 >50% at a concentration of 0.1 μM. GNE-9065 (10 and 50 mg/kg) inhibits LRRK2 Ser1292 autophosphorylation in BAC transgenic mice expressing human LRRK2 protein with the G2019S mutation found in families with autosomal Parkinson’s disease.
CNC1=C(C(F)(F)F)C=NC(NC2=C(Cl)N([C@H]3CCN(C4COC4)C[C@@H]3F)N=C2)=N1
N2-(5-Chloro-1-((trans)-3-fluoro-1-(oxetan-3-yl)piperidin-4-yl)-1H-pyrazol-4-yl)-N4-methyl-5-(trifluoromethyl)pyrimidine-2,4-diamine (20)
A mixture of (±)-(trans)-4-(5-chloro-4-nitro-1H-pyrazol-1-yl)-3-fluoro-1-(oxetan-3-yl)piperidine (53, 2.2 g, 3.9 mmol), iron dust (1.6 g, 29 mmol), and ammonium chloride (1.5 g, 29 mmol) in ethanol (20 mL) was stirred at 90 °C for 30 min. The reaction was filtered and concentrated. The residue was sonicated with 100 mL of EtOAc for 5 min. The mixture was filtered to remove all insoluble solids. The filtrate was then concentrated to give crude (±)- (trans)-4-(5-chloro-4-amino-pyrazol-1-yl)-3-fluoro-1-(oxetan-3-yl)piperidine (1.9 g).
To a mixture of the crude (±)-(trans)-4-(5-chloro-4-amino-pyrazol-1-yl)-3-fluoro-1-(oxetan-3-yl)piperidine (1.9 g) and 2-chloro-N-methyl-5-(trifluoromethyl)pyrimidin-4-amine (26, 1.5 g, 6.9 mmol) in 2-methoxyethanol (25 mL) was added TFA (0.60 mL, 7.7 mmol). The reaction was stirred at 90 °C for 15 min. The mixture was then diluted with saturated sodium bicarbonate and extracted with EtOAc (3×). The combined extracts were washed with brine, dried over sodium sulfate, filtered, and concentrated. The crude product was purified by preparative HPLC, chiral SFC, and recrystallized in isopropanol to give 20 (0.70 g, 40% yield). 1H NMR (400 MHz, DMSO) δ 8.91 (s, 1H), 8.08 (s, 1H), 7.87 (s, 1H), 7.00 (s, 1H), 5.03 4.79 (m, 1H), 4.56 (m, 1H), 4.46 (m, 2H), 3.68–3.51 (m, 1H), 3.26–3.12 (m, 1H), 2.92–2.73 (m, 3H), 2.54 (s, 2H), 2.20–1.88 (m, 3H). HRMS (ES) m/z: [M + H]+ calcd for C17H20ClF4N7OH+, 450.1427; found, 450.1418.
Scheme 8

Scheme 8. Synthesis of Inhibitor 20a
aReagents and conditions: (a) (±)-(cis)-tert-butyl 3-fluoro-4-hydroxypiperidine-1-carboxylate, PPh3, diisopropyl azodicarboxylate, THF; (b) TFA, DCM, 58% over two steps; (c) oxetan-3-one, DIPEA, NaBH(OAc)3, acetic acid, DCE, 85%; (d) LiHMDS then C2Cl6, THF, −78 °C, 65%; (e) iron dust, NH4Cl, EtOH, 90 °C; (f) 26, TFA, 2-methoxyethanol, 90 °C, 40%, two steps.
REFERENCES
1: Estrada AA, Chan BK, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, Chen H, Dominguez SL, Dotson J, Drummond J, Flagella M, Fuji R, Gill A, Halladay J, Harris SF, Heffron TP, Kleinheinz T, Lee DW, Pichon CE, Liu X, Lyssikatos JP, Medhurst AD, Moffat JG, Nash K, Scearce-Levie K, Sheng Z, Shore DG, Wong S, Zhang S, Zhang X, Zhu H, Sweeney ZK. Discovery of Highly Potent, Selective, and Brain-Penetrant Aminopyrazole Leucine-Rich Repeat Kinase 2 (LRRK2) Small Molecule Inhibitors. J Med Chem. 2014 Jan 15. [Epub ahead of print] PubMed PMID: 24354345.
/////////////DNL-151, DNL 151, DNL151, Alzheimer’s disease, breast tumor, type I diabetes mellitus, Crohn’s disease, phase 1, Parkinson’s disease, GNE0877, GNE 0877, GNE-0877, GNE-9605, GNE 9605, GNE9605, Genentech
CC(N1N=C(C)C(NC2=NC=C(C(F)(F)F)C(NC)=N2)=C1)(C)C#N
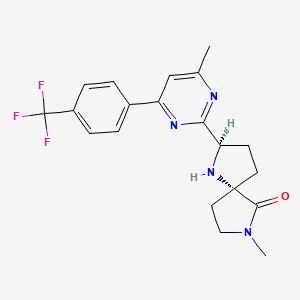
BIIB-095

BIIB-095
ROTATION (+)
1493790-64-9 CAS free form,
1493772-48-7 cas Hcl salt
cas 1493790-65-0, 1496563-32-6 ,SULPHATE ???
cas 1496563-31-5 SULFATE 1;1
cas 1496563-32-6 SULFATE HYDRATE 1;1;1
(2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one
1,7-Diazaspiro[4.4]nonan-6-one, 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)phenyl]-2-pyrimidinyl]-, (2R,5S)-
C20 H21 F3 N4 O, 390.40
- Originator Biogen
- Class Analgesics
- Mechanism of Action Nav1.7 voltage-gated sodium channel inhibitors
- Phase I Neuropathic pain
- 29 Mar 2018 Phase-I clinical trials in Neuropathic pain (In volunteers) in United Kingdom (PO) (NCT03454126)
- 05 Mar 2018 Biogen plans a phase I trial for Pain, including Neuropathic pain (In volunteers) in USA (PO) (NCT03454126)
- 05 Mar 2018 Preclinical trials in Neuropathic Pain in USA (PO), before March 2018
In March 2018, a randomized, double blind, placebo controlled, single and multiple-ascending dose, dose-escalation phase I study ( NCT03454126; 255HV101; 2017-003982-90) was initiated in the UK in healthy subjects (expected n = 80) to evaluate the safety, tolerability and pharmacokinetics of BIIB-095. At that time, the trial was expected to complete in December 2018
Biogen is developing BIIB-095, a voltage-gated sodium channel 1.7 inhibitor, for the potential oral treatment of neuropathic pain [2027279], [2027426]. In March 2018, a phase I trial was initiated in healthy subjects
Biogen is developing oral agent BIIB-095 for the treatment of chronic pain, including neuropathic pain. A phase I clinical trial is under way in healthy volunteers.
The compound was first claimed in WO2013175205 , for treating schizophrenia, assigned to subsidiary Convergence Pharmaceuticals Limited , naming some of the inventors. This might present the structure of BIIB-095 , a voltage-gated sodium channel 1.7 inhibitor, being developed by Biogen for the oral treatment of neuropathic pain; in March 2018, a phase I trial was initiated in healthy subjects.
PATENT
WO2013175205

CONTD………………

INTERMEDIATE

WO 2013175206
US 20150119404
https://patents.google.com/patent/US20150119404
Patent
WO-2019067961
Novel salts (citrate, mesylate, hydrosulfate, saccharinate and oxalate) forms of 7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one, processes for their preparation and compositions comprising them are claimed. Also claimed are their use for treating diseases and conditions mediated by modulation of voltage-gated sodium channels.
Voltage-gated sodium channels are responsible for the initial phase of the action potential, which is a wave of electrical depolarisation usually initiated at the soma of the neuron and propagated along the axon to the terminals. At the terminals, the action potential triggers the influx of calcium and the release of neurotransmitter. Drugs, such as lidocaine, that block voltage-gated sodium channels are used as local anaesthetics. Other sodium channel blockers, such as lamotrigine and carbamazepine are used to treat epilepsy. In the latter case, partial inhibition of voltage-gated sodium channels reduces neuronal excitability and reduces seizure propagation. In the case of local anaesthetics, regional block of sodium channels on sensory neurons prevents the conduction of painful stimuli. A key feature of these drugs is their state-dependent mechanism of action. The drugs are thought to stabilise an inactivated conformation of the channel that is adopted rapidly after the channel opens. This inactivated state provides a refractory period before the channel returns to its resting (closed) state ready to be reactivated. As a result, state-dependent sodium channel blockers inhibit the firing of neurons at high frequency, for example in response to painful stimuli, and will help to prevent repetitive firing during periods of prolonged neuronal depolarisation that might occur, for example, during a seizure. Action potentials triggered at lower frequencies, for example in the heart, will not be significantly affected by these drugs, although the safety margin differs in each case, since at high enough concentrations each of these drugs is capable of blocking the resting or open states of the channels.
The voltage-gated sodium channel family is made up of 9 subtypes, four of which are found in the brain, NaV1.1 , 1.2, 1.3 and 1.6. Of the other subtypes, NaV1.4 is found only in skeletal muscle, NaV1.5 is specific to cardiac muscle, and NaV1.7, 1.8, and 1.9 are found
predominantly in sensory neurons. The hypothesised binding site for state-dependent sodium channel blockers is the local anaesthetic (LA) binding site in the inner vestibule of the pore on transmembrane S6 of domain IV. Critical residues are located in a highly conserved region among the different subtypes, thus presenting a challenge for the design of new subtype selective drugs. Drugs such as lidocaine, lamotrigine and carbamazepine do not distinguish between the subtypes. However, selectivity can be achieved, and can be further enhanced functionally, as a result of the different frequencies at which the channels operate.
Drugs that block voltage-gated sodium channels in a state-dependent manner are also used in the treatment of bipolar disorder, either to reduce symptoms of mania or depression, or as mood stabilisers to prevent the emergence of mood episodes. Clinical and preclinical evidence also suggests that state-dependent sodium channel blockers may help to reduce the symptoms of schizophrenia. For example, lamotrigine has been shown to reduce symptoms of psychosis induced by ketamine in healthy human volunteers, and furthermore, studies in patients suggest that the drug can augment the antipsychotic efficacy of some atypical antipsychotic drugs, such as clozapine or olanzapine. It is hypothesised that efficacy in these psychiatric disorders may result in part from a reduction of excessive glutamate release. The reduction in glutamate release is thought to be a consequence of sodium channel inhibition in key brain areas, such as the frontal cortex. However, interaction with voltage-gated calcium channels may also contribute to the efficacy of these drugs.
WO 2013/175205 (Convergence Pharmaceuticals Limited) describes (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one hydrochloride, sulfuric acid salt and sulfuric acid salt hydrate which are claimed to be modulators of voltage-gated sodium channels. The object of the invention is to identify alternative salts of said compound which have advantageous properties.
Example 1
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of citric acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 30 minutes a seed of citrate salt (0.1 wt%) was added and the mixture allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a very thick suspension (white) that required mobilisation with 20 ml additional ethanol and a further maturation period of 2 hours at ambient temperature. Filtration was carried out under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 6.0 g of crystalline white solid (91 % yield).
H NMR (400MHz, DMSO-D6): δΗ 1.90-2.05 (2H, m), 2.10-2.20 (2H, m,), 2.20-2.30 (1 H, m), -2.50 (1 H, m, partially masked by solvent)), 2.55-2.68 (4H, m), 2.56 (3H, s), 2.79 (3H, s),
3.28-3.40 (2H, m), 4.79 (1 H, t, J= 8.0 Hz), 7.92 (2H, d, J = 8.4 Hz), 8.03 (1 H, s), 8.45 (2H, d, J= 8.8Hz) ppm, (exchangeables not reported)
Characterisation of Example 1
The XRPD of Example 1 is presented in FIG. 1 and the DSC/TGA of Example 1 is presented in FIG. 2. The citrate salt of Example 1 displayed the following characteristics:
1 endotherm onset: 171.82°C
peak maximum: 174.55°C
There was an endotherm post the main endotherm.
There was no weight reduction until ca 168°C had been reached. The weight reduction commenced with the start of the main endotherm and coincided with the endotherm post the main endotherm which indicated that this thermal event was the onset of compound decomposition and loss of citric acid. Thermal events >220°C were due to compound decomposition.
The XPRD data in FIG. 1 demonstrated that under different extremes of humidity indicate a stable crystalline form of the citrate salt of Example 1 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 2 which show clear transitions and no evidence of solvates.
Aqueous solubility of the citrate salt (Example 1) = 22mg/ml (25°C).
Example 2
(2R,5S)-7-Methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1,7-diazaspiro[4.4]nonan-6-one ) salt (E2)
To a solution of (2R,5S)-7-methyl-2-[4-methyl-6-[4-(trifluoromethyl)-phenyl]pyrimidin-2-yl]-1 ,7-diazaspiro[4.4]nonan-6-one (which may be prepared in accordance with the procedure described in Example 1 of WO 2013/175205) (4.45g, 0.0114 mol) dissolved in absolute ethanol (66.82 ml, 15 vol) at 45 °C was added a solution of methanesulfonic acid in ethanol (1 M, 1.05 equiv. 12 ml) over a period of 2-3 minutes. The solution was aged at 45 °C for a period of 1 hour. After 10 minutes nucleation and gradual crystallisation was noted to afford a thick mixture. Additional ethanol was added (10 ml) to mobilise the suspension that was then allowed to cool over approximately 2 hours and mature for 18 hours at ambient temperature (approximately 10-15 °C). Following maturation the salt was noted to be a thin, mobile suspension (white) that was filtered under vacuum and the vessel and cake rinsed with 15 ml ethanol. The de-liquored cake was dried further in a vacuum oven at 50 °C to provide 4.0 g of crystalline white solid (72% yield).
H NMR (400MHz, DMSO-D6): δΗ 2.1-2.45 (4H, m), 2.27 (3H, s), 2.50-2.75 (2H, m), 2.61 (3H, s), 2.86 (3H, s), 3.35-3.50 (2H, m), 5.20 (1 H, t, J = 8 Hz), 7.96 (2H, d, J = 8.8 Hz), 8.17 (1 H, s), 8.51 (2H, d, J = 8.4Hz), 9.45 (1 H, br), 10.16 (1 H, br) ppm.
Characterisation of Example 2
The XRPD of Example 2 is presented in FIG. 3 and the DSC/TGA of Example 2 is presented in FIG. 4. The DSC thermograph of the methanesulfonate (mesylate) (Example 2) displayed the following characteristics:
One distinct endotherm onset: 247.34°C
peak maximum: 250.34°C
The TGA thermograph showed no weight reduction until ca 250°C had been reached. The weight reduction commenced with the start of the main endotherm and indicated that this thermal event was the onset of compound decomposition. There is no evidence of entrapped solvents or water.
The XPRD data in FIG. 3 demonstrated that under different extremes of humidity indicate a stable crystalline form of the mesylate salt of Example 2 with no tendency to form hydrates. This is supported by DSC/TGA data in FIG. 4 which show clear transitions and no evidence of solvates.
Aqueous solubility of the mesylate salt (Example 2) = 65mg/ml (25°C).
Example 3
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one hydrosulfate single crystals: 25.0 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluorome
one hydrosulfate was added to 4 mL vial. 1.000 mL of anhydrous EtOH was added, and the sample was filtered. Anhydrous hexanes were added dropwise until the solution neared the precipitation point. The vial was sealed and left undisturbed for 24 hr, after which time a crop of single crystals was evident. The sample was sent for single crystal analysis and confirmed as the anhydrous (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate form (FIGs. 5A-5B).
Example 4
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one freebase: 8.00 g of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate (JM Lot R-2017-4323 D 301) was added to a 1 L Erlenmeyer flask and suspended and stirred vigorously in 400 mL of THF. 20% K2C03 (250 mL) was added and dissolved. The mixture was transferred to 1 L sep. funnel. 100 mL EtOAc was added and the aqueous and organic layers were separated. The aqueous layer was re-extracted with 50 mL of EtOAc and the combined organics were back-extracted with brine (100 mL) and water (100 mL). Due to fairly poor separation, a significant quantity of MgSCU was required to dry the solution. The solution was reduced via Rotavap (45 °C) to -50 mL, transferred to a 100 mL RB flask, reduced down to -10 mL, transferred to 20 mL scintillation vial and continued to be reduced to a thick oil. The oil was left on the Rotavap for another hour and a “wet” solid was obtained. Loosened solids on the bottom of the vial were left on the Rotavap for 1 hr with no heat applied to obtain a chunky solid. The contents was transferred to a mortar and pestle, ground to powder and fine granules, placed back in a 20 mL scintillation vial and left on a Rotavap overnight to obtain a dry solid (5.1 g). The XRPD pattern of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase is shown in FIG. 6.
Example 5
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one saccharinate: 199.7 mg of (2R,5S)-7-Methyl-2-(4-
methyl-6-(4-(trifluoromethyl)phenyl)pyrirnidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one free base (0.5115 mmol) was dissolved in 4.2 mL of 2-Me-THF. 98.1 mg of saccharin (0.5106 mmol) was dissolved in 4.2 mL of 2-Me-THF. Saccharin was added to the freebase, and after 15 seconds the mixture began to precipitate and solidify. 10 mL of 2-Me-THF was added and stirred at max rpm as to provide a thick white suspension in 10 min. The suspension was filtered, air dried under vacuum for 10 min on frit, then dried under a stream of nitrogen for 30 min resulting in 215 mg of white solid product. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one saccharinate is shown in FIG. 7.
Example 6
Preparation of (2R,5S)-7-methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1,7-diazaspiro[4.4]nonan-6-one oxalate: 403 mg of (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase was dissolved in 4.03 mL EtOH. 1.000 mL of this solution was added to a 4 mL vial. 23.8 mg of oxalic acid was dissolved in 1.000 mL of EtOH and added dropwise to the stirring (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one freebase solution. After 5 min, a white precipitate was evident and 2.000 mL of EtOH was added to the slurry to aid stirring. The resulting suspension was stirred overnight. The following day the suspension was filtered and dried on a frit under vacuum for 10 min yielding 106 mg of white solid. The XRPD pattern for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one oxalate is shown in FIG. 8.
Example 7
The single crystal structural information and refinement parameters for (2R,5S)-7-Methyl-2-(4-methyl-6-(4-(trifluoromethyl)phenyl)pyrimidin-2-yl)-1 ,7-diazaspiro[4.4]nonan-6-one hydrosulfate are shown in Table 1.
Table 1.
Largest peak, hole / e A-3 0.363, -0.264
The most prominent XRPD diffraction peaks were (2Θ): 7.8±0.2°, 8.1±0.2°, 12.6±0.2°, 14.3±0.2°, 16.5±0.2°, 18.5±0.2°, 19.6±0.2°, 24.8±0.2° and 25.3±0.2°.
PATENTS
US2018360833NOVEL PYRIMIDINYL-DIAZOSPIRO COMPOUNDS2018-06-27
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017304303 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2017-07-11 | |
| US9737536 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-05-25 | 2016-09-15 |
| US2016184306 | Novel Pyrimidinyl-DiazoSpiro Compounds | 2016-02-15 | 2016-06-30 |
| US9309254 | NOVEL COMPOUNDS | 2013-05-22 | 2015-04-30 |
| US9376445 | NOVEL COMPOUNDS | 2013-05-22 | 2015-06-18 |
////////////////BIIB-095, BIIB095, BIIB 095, PHASE 1
CC1=NC(=NC(=C1)C2=CC=C(C=C2)C(F)(F)F)C3CCC4(N3)CCN(C4=O)C
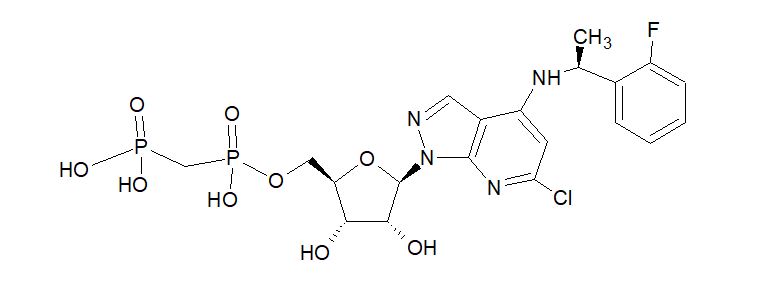
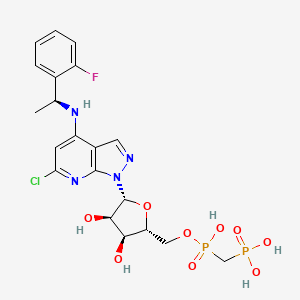
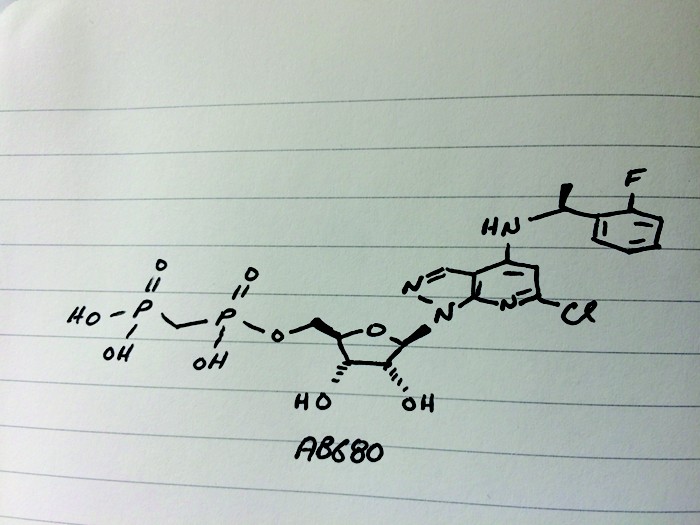
AB 680
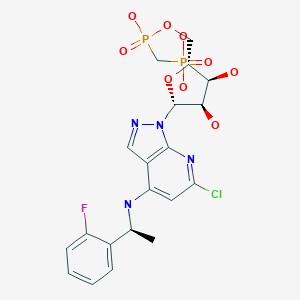

AB 680
C20H24ClFN4O9P2, 580.827 g/mol
Cas 2105904-82-1
1H-Pyrazolo[3,4-b]pyridin-4-amine, 6-chloro-N-[(1S)-1-(2-fluorophenyl)ethyl]-1-[5-O-[hydroxy(phosphonomethyl)phosphinyl]-β-D-ribofuranosyl]-
[[(2R,3S,4R,5R)-5-[6-chloro-4-[[(1S)-1-(2-fluorophenyl)ethyl]amino]pyrazolo[3,4-b]pyridin-1-yl]-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]methylphosphonic acid
[({[(2R,3S,4R,5R)-5-(6-chloro-4-{[(1S)-1-(2-fluorophenyl)ethyl]amino}-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid
- Originator C
- Class Antineoplastics; Small molecules
- Mechanism of Action 5-nucleotidase inhibitors; Adenosine A2 receptor antagonists
- Phase I Cancer
- 19 Nov 2018 Arcus Biosciences plans to initiate a clinical trial in Cancer in first half of 2019
- 16 Oct 2018 Phase-I clinical trials in Cancer (In volunteers) in Australia (IV) (NCT03677973)
- 30 Sep 2018 Preclinical pharmacodynamics data in Cancer presented at 4th CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference (CRI-CIMT-EATI-AACR – 2018)
Clip
Credit: Tien Nguyen/C&EN
Presenter: Kenneth V. Lawson, senior scientist at Arcus Biosciences
Target: Ecto-5’-nucleotidase (CD73)
Disease: Cancer
Reporter’s notes: In the first talk of the day, Lawson introduced the idea of cancer drugs that target the host’s immune system. “Checkpoint inhibitors changed the way we think of treating cancer,” he said. These drugs successfully disrupt the binding interaction between a protein and a checkpoint protein that stops immune T cells from killing cancer cells. As a result, these drugs turn immune cells loose to attack tumor cells. But the drugs work only in about 30-40% of patients—an issue pharmaceutical companies like Arcus hope to address with new immunotherapies that can be taken in combination with checkpoint inhibitors.
Lawson’s team set out to inhibit an enzyme commonly found in tumors called CD73, the second of two enzymes which break down extracellular adenosine trisphosphate (ATP) to adenosine. Adenosine then binds to immunosuppressive receptors on immune cells and shuts them down. Yet developing a small molecule inhibitor of CD73 proved challenging, Lawson said. After striking out with high-throughput screening, the team turned to CD73’s natural substrate for inspiration. However, the molecule possessed more than one phosphate group, which is notoriously a liability for drug molecules because small molecules with such negative changes struggle to cross cell membranes. The team’s goal was to remove the phosphate groups, Lawson says, but things didn’t exactly go according to plan. After showing the audience a series of compounds from structure-activity relationship (SAR) studies—slides no medicinal chemistry talk would be complete without—Lawson revealed the structure of their final clinical compound AB680 as the sound of people flipping notebook sheets rippled across the room. Synthesized in 34% overall yield, the candidate ultimately included two phosphate groups—a feature that surprised audience members.
Tests revealed that AB680 can be given intravenously but the compound also showed moderate oral bioavailability. Lawson suggested a possible route for how the molecule might pass from the digestive tract to the bloodstream, a paracellular mechanism by which molecules cross the epithelium by passing through the space between cells. AB680 showed “extraordinary potency,” inhibiting CD73 in human T-cells at a concentration of 0.008 nM. The compound has a 4 day half-life, which means it could be dosed every two weeks, coinciding with the dosing schedule for patients who receive a checkpoint inhibitor. AB680 is currently in Phase 1 clinical trials with healthy patients.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US204141996&tab=PCTDESCRIPTION&maxRec=1000
|
Purinergic signaling, a type of extracellular signaling mediated by purine nucleotides and nucleosides such as ATP and adenosine, involves the activation of purinergic receptors in the cell and/or in nearby cells, resulting in the regulation of cellular functions. Most cells have the ability to release nucleotides, which generally occurs via regulated exocytosis (see Praetorius, H. A.; Leipziger, J. (1 Mar. 2010) Ann Rev Physiology 72(1): 377-393). The released nucleotides can then be hydrolyzed extracellularly by a variety of cellular membrane-bound enzymes referred to as ectonucleotidases.
|
Example 92
Synthesis of [({[(2R,3S,4R,5R)-5-(6-chloro-4-{[(1S)-1-(2-fluorophenyl)ethyl]amino}-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy}(hydroxy)phosphoryl)methyl]phosphonic Acid
PATENT

////////////////ARCUS, AB 680, AB680, AB-680, PHASE 1
https://www.arcusbio.com/wp-content/uploads/2018/04/AACR_AB680_1756_final_90x42-abstract-4886.pdf
Fc1ccccc1[C@H](C)Nc4cc(Cl)nc3c4cnn3[C@@H]2O[C@H](COP(=O)(O)CP(=O)(O)O)[C@@H](O)[C@H]2O
CC(C1=CC=CC=C1F)NC2=CC(=NC3=C2C=NN3C4C(C(C(O4)COP(=O)(CP(=O)(O)O)O)O)O)Cl
CMX-8521, CMX-521
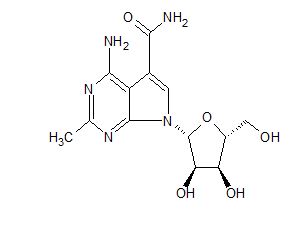
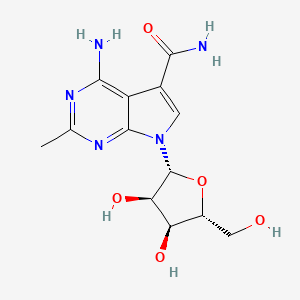
CMX-8521, CMX-521
MF C13 H17 N5 O5, MW 323.30
CAS Number 2077178-99-3
7H-Pyrrolo[2,3-d]pyrimidine-5-carboxamide, 4-amino-2-methyl-7-β-D-ribofuranosyl-
Nucleoside analogs (oral, norovirus infection), Chimerix
4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide
4-amino-7-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-methylpyrrolo[2,3-d]pyrimidine-5-carboxamide
CMX8521 is a nucleoside analog that inhibits the norovirus RNA polymerase. CMX8521 has in vitro activity against mouse and human norovirus.Where possible, Chimerix uses its lipid conjugate technology to build nucleoside-analog antivirals that are orally absorbed and have favorable tissue penetration.
CMX-8521 (presumed to be CMX-521) being developed by Chimerix for treating norovirus infection. In June 2018, a phase II efficacy trial was planned in 2019.
In January 2016, preclinical data were presented at the 34th Annual JP Morgan Healthcare Conference in San Francisco, CA. CMX-8521 had in vitro activity against mouse and human norovirus (EC50 = 2.1; CC50 = 114 microM). A 7-day non GLP toxicology/toxicokinetic study was completed in-life with no clinical or gross post mortem signs of toxicity. No off-target pharmacology was observed in vitro when screened against a panel of 87 receptors, transporters and enzymes associated with adverse pharmacology
PATENT
WO2017024310
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017024310
Scheme 1: General Synthesis of Compounds of the Invention

Scheme 2: General Synthesis of Compounds of the Invention

Example 7– Synthesis of Compound 1

[00315] Step 1 (Protocol #1): To a 100-L jacketed reactor were charged 4-amino-6- bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate (6.60 kg) and DCE (18.89 kg). Stirring was started and DBU (3.61) kg was added. Over a period of 03 h and 14 min, TMSOTf (8.01 kg) was added between 30.6 °C and 37.3 °C. IPC after 01 h and 30 min at approx.32 °C showed 4% of 4-amino-6-bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg),
(3R,4R,5R)-2-acetoxy-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate remaining. IPC after 03h and 16 min at approx.32 °C showed 2% 4-amino-6-bromo-2-methyl-7H- pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate remaining (spec:≤3%). The reaction mixture was diluted with DCM (39.81 kg) and quenched with potable water (15.02 kg) over an 11 min period between 9.5 °C and 15.6 °C. The extractive work-up (at approx.22 °C) was completed by a back extraction of the aqueous phase with DCM (19.90 kg), a wash with sat NaHCO3 (1.3 kg NaHCO3 in 14.9 kg potable water), a back extraction of the bicarbonate phase with DCM (19.71 kg) and a wash with brine (4.5 kg NaCl in 14.9 kg potable water). Note: the reactor was cleaned with potable water, acetone and DCM after each wash/back extraction.
[00316] The drummed organic phase containing the product was charged to the 100-L jacketed reactor through an in-line filter followed by a DCM rinse of the drum and filter with DCM (2.48 kg). The contents of the reactor were distilled to 31 L with the aid of vacuum over a period of 06 h and 04 min with a maximum temperature of 50.1 °C. At this point a thick suspension had formed. Next, over a period of 39 min, IPAc (41.88 kg) was added between 44.5 °C and 49.5 °C and the contents of the reactor were heated to 76.9 °C over a period of 01 h and 25 min. Next, the contents of the reactor were cooled to 9.9 °C over a period of 04 h and 21 min and stirred for 12 h and 26 min with a minimum temperature of 1.6 °C.
[00317] Step 1 (Protocol # 2): To a 100-L jacketed reactor were charged 4-amino-6- bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (3.00 kg), (3R,4R,5R)-2-acetoxy-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate (6.60 kg) and DCE (18.80 kg). Stirring was started and DBU (3.59) kg was added. Over a period of 01 h and 46 min, TMSOTf (7.90 kg) was added between 30.4 °C and 34.2 °C. IPC after 02 h and 49 min at approx.34 °C showed 1% of 4-amino-6-bromo-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile remaining (spec: ≤3%). The reaction mixture was diluted with DCM (40/70 kg) and quenched with potable water (14.97 kg) over an 04 min period between 9.9 °C and 18.0 °C. The extractive work-up (at approx.22 °C) was completed by a back extraction of the aqueous phase with DCM (20.34 kg), a wash with sat NaHCO3 (1.30 kg NaHCO3 in 14.90 kg potable water), a back extraction of the bicarbonate phase with DCM (20.65 kg) and a wash with brine (4.50 kg NaCl in 14.96 kg potable water). Note: the reactor was cleaned with potable water, acetone and DCM after each wash/back extraction.
[00318] The drummed organic phase containing the product was charged to the 100-L jacketed reactor through an in-line filter followed by a DCM rinse of the drum and filter with DCM (1.49 kg). The contents of the reactor were distilled to with the aid of vacuum over a period of 04 h and 49 min with a maximum temperature of 45.6 °C. At this point a thick suspension had formed. Next, over a period of 27 min, IPAc (41.70 kg) was added between 45.6 °C and 48.2 °C and the contents of the reactor were heated to 75.7 °C over a period of 01 h and 20 min. Next, the contents of the reactor were cooled to 9.4 °C over a period of 04 h and 15 min and stirred overnight with a minimum temperature of 2.3 °C.
[00319] Step 2: To the reactor were charged (2R,3R,4R,5R)-2-(4-amino-6-bromo-5- cyano-2-methyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4- diyl dibenzoate (10.0 kg), 10% Pd on C (Degussa, Type E101NE/W), trimethylamine (7.3 kg) and THF (44.5 kg). Hydrogen was submitted to the reactor and the mixture was stirred for 03 h and 54 min between 24.7 °C and 19.6 °C at approx.30.8 psig. IPC (HPLC) showed that
(2R,3R,4R,5R)-2-(4-amino-6-bromo-5-cyano-2-methyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5- ((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate could no longer be detected.
[00320] The reaction mixture was filtered over Celite (7.2 kg) and a polish filter and the filter residue was washed with THF (5.2 kg). The combined filtrate and wash was transferred to a 100-L jacketed reactor with the aid of a THF wash (2.12 kg). The contents of the reactor were vacuum distilled with a maximum batch temperature of 30.0 °C over a period of 05 h and 38 min to a final volume of 27 L. IPA (31.48 kg) was charged over a 40 min period to the reactor between 39.7 °C and 53.2 °C. The contents of the reactor were vacuum distilled with a maximum batch temperature of 53.2 °C over a period of 03 h and 02 min to a final volume of 33 L. IPA (48.99 kg) was charged over a 43 min period to the reactor between 53.1 °C and 57.1 °C. The contents of the reactor were heated to 60.2 °C, agitated for 12 min and cooled over a period of 04 and 28 min to 5.4 °C. Cold stirring was continued for a period of 08 h and 55 min with a minimum temperature of 1.1 °C. The slurry was filtered and washed with IPA (9.41 kg, at approx.4.5 °C). The residue was dried under vacuum with a nitrogen bleed for a period of 11 h and 44 min at a maximum temperature of 44.0 °C to provide an LOD of 0.36%. Yield: 6.58 kg (73.9 %).1H NMR confirms structure. Purity: 97.78 % (HPLC, AUC).
[00321] Step 3:

1100 g NaOH dissolved in potable water to a total volume of 1 L; 2 Diluted 500 mL conc. HCl in 2 L total with potable water [00322] A solution of (2R,3R,4R,5R)-2-(4-amino-5-cyano-2-methyl-7H-pyrrolo[2,3- d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate and THF was heated to 54 °C and the addition of 2.5 M NaOH was started. The initial addition gave a biphasic mixture and endothermic response (the temperature dropped to 50 °C) but as the addition continued a single phased, clear solution formed which was accompanied by a fast exotherm to 61 °C; the reaction temperature was maintained at 60 °C to 61 °C during the rest of the addition and for an additional 2 ½ h. IPC showed that no (2R,3R,4R,5R)-2-(4-amino-5-cyano-2-methyl- 7H-pyrrolo[2,3-d]pyrimidin-7-yl)-5-((benzoyloxy)methyl)tetrahydrofuran-3,4-diyl dibenzoate was left.
[00323] The reaction mixture was cooled to 21 °C and neutralized with 3 N HCl with external cooling to pH = 7.06 (Denver Instrument UB-10 pH meter equipped with a Sartorius P- P11 pH electrode, the electrode was checked with buffer solutions of pH = 4.00 and pH = 7.00); the mixture continued to cool to 8°C. The resulting neutralized mixture was distilled under vacuum with a pot temperature of 45 °C to 50 °C until the emergence of solids were observed in the pot. The suspension was cooled and stirred for 2 h at 2 °C. The beige suspension was filtered to afford a dark filtrate; the off-white residue was washed once with cold water (500 mL, 5 °C). A first LOD after 16 h gave a value of 18.73 %. HPLC) of the drying material showed the presence of 1.6% benzoate.
[00324] A brief rework study for compound 1, (containing 1.6% benzoic acid per AUC, HPLC) was executed in 10 vol of water (1 g in 10 mL):
● 3 h slurry at ambient
● 3h slurry at 50 °C
● 24 h slurry at ambient
[00325] All three experiments gave compound 1 with less than 0.1 % benzoic acid (UAC, HPLC). The slurries were fluid, were easily stirred and filtration was fast. Short term drying on the filter gave a powder-like solid indicating that a displacement wash with an organic solvent is not needed. Without wishing to be bound by theory, a loss of NMT than 1% is expected
(solubility 1 mg/mL).HPLC data for compound 1 were obtained with a method suitable for polar compounds using a Zorbax Eclipse Plus C18 column (water / ACN / TFA, 97.5 / 2.5 / 0.05). This is the same column used for steps 1 and 2.
[00326] The cold product suspension was filtered and the reactor and residue were washed with cold IPAc (approx.7.5 °C, 13.16 kg and 13.62 kg) until a colorless filtrate had been obtained. The residue was dried under vacuum and a nitrogen bleed≤ 45 °C for a period of 65 h and 19 min to an LOD of 0 %. Yield: 5.87 kg (70.7 %), 1H NMR confirmed identity; HPLC purity 98.84% (AUC). EQUIVALENTS
[0001] The disclosure can be embodied in other specific forms without departing from the spirit or essential characteristics thereof. The foregoing embodiments are therefore to be considered in all respects illustrative rather than limiting on the disclosure described herein. Scope of the disclosure is thus indicated by the appended claims rather than by the foregoing description, and all changes that come within the meaning and range of equivalency of the claims are intended to be embraced therein.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019060692&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline forms of 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide and their stable hemihydrate crystalline forms (designated as Form A-G), processes for their preparation and compositions comprising them are claimed. Also claimed is their use for treating viral infection.
Viral infections can have serious adverse effects on individuals and society as a whole. In addition to fatal viral infections such as Ebola, even non-fatal infections can have serious societal and economic consequences. For example, human noroviruses (NV) are the most common cause of epidemic acute gastroenteritis worldwide with an estimated 19-21 million cases each year in the United States including 56,000-71,000 hospitalizations and 570-800 deaths (Hall et al., Emerg.Infect.Dis. 2013 Aug; 19(8): 1198-205).
[0004] 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo [2,3-d]pyrimidine-5-carboxamide (Compound 1) is an antiviral drug.
Formula 1
[0065] As used herein, “Formula I” is understood to encompass all diastereomers of 4-amino-7-(3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide, and pharmaceutically acceptable salts and solvates thereof. The structure of Formula I is shown below:
(Formula I).
[0066] In some embodiments, a compound of Formula I can be 4-amino-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-2-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (“Compound 1”), or a pharmaceutically acceptable salt solvate, or isomers (e.g., enantiomers and diastereomers) thereof. The structure of Compound 1 is shown below:
| atent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9701706 | Pyrrolopyrimidine nucleosides and analogs thereof | 2016-11-22 | 2017-07-11 |
| US9708359 | PYRROLOPYRIMIDINE NUCLEOSIDES AND ANALOGS THEREOF | 2016-08-08 | |
| US2017253628 | PYRROLOPYRIMIDINE NUCLEOSIDES AND ANALOGS THEREOF | 2017-05-18 |
///////////CMX-8521, CMX 8521, CMX-521, PHASE 1
NC(=O)c2cn([C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O)c3nc(C)nc(N)c23
Epitinib
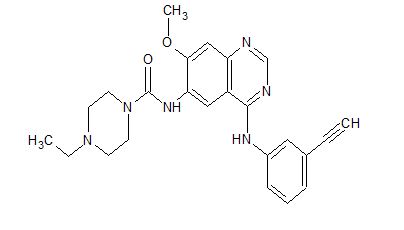
Epitinib succinate; HMPL-813; Huposuan yipitini
1203902-67-3, 430.50, C24 H26 N6 O2
1-Piperazinecarboxamide, 4-ethyl-N-[4-[(3-ethynylphenyl)amino]-7-methoxy-6-quinazolinyl]-
4-Ethyl-N-[4-[(3-ethynylphenyl)amino]-7-methoxy-6-quinazolinyl]-1-piperazinecarboxamide
Cancer; Glioblastoma; Non-small-cell lung cancer
Epitinib is in phase I clinical trials by Hutchison MediPharma for the treatment of solid tumours.
Epitinib succinate is an oral EGFR tyrosine kinase inhibitor in early clinical development at Hutchison China MediTech (Chi-Med) for the treatment of solid tumors and the treatment of glioblastoma patients with EGFR gene amplification.
- Originator Hutchison MediPharma
- Class Antineoplastics; Small molecules
- Mechanism of Action Epidermal growth factor receptor antagonists
- Phase I/II Glioblastoma; Non-small cell lung cancer
- No development reported Oesophageal cancer; Solid tumours
- 28 May 2018 No recent reports of development identified for preclinical development in Oesophageal-cancer in China (PO)
- 06 Mar 2018 Hutchison Medipharma plans a phase III pivotal study for Non-small cell lung cancer (NSCLC) patients with brain metastasis in China in 2018
- 06 Mar 2018 Phase-I/II clinical trials in Glioblastoma (Second-line therapy or greater) in China (PO)
PATENT
Binding of epidermal growth factor (EGF) to epidermal growth factor receptor (EGFR) activates tyrosine kinase activity and thereby triggers reactions that lead to cellular proliferation. Overexpression and/or overactivity of EGFR could result in uncontrolled cell division which may be a predisposition for cancer. Compounds that inhibit the overexpression and/or overactivity of EGFR are therefore candidates for treating cancer.
The relevant compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide of the present invention has the effect of effectively inhibiting the overexpression and/or overactivity of EGFR. Thus, it is useful in treating diseases associated with overexpression and/or overactivity of EGFR, such as the treatment of cancer.
The phenomenon that a compound could exist in two or more crystal structures is known as polymorphism. Many compounds may exist as various polymorph crystals and also in a solid amorphous form. Until polymorphism of a compound is discovered, it is highly unpredictable (1) whether a particular compound will exhibit polymorphism, (2) how to prepare any such unknown polymorphs, and (3) how are the properties, such as stability, of any such unknown polymorphs. See, e.g., J. Bernstein “Polymorphism in Molecular Crystals” , Oxford University Press, (2002)
Since the properties of a solid material depend on the structure as well as on the nature of the compound itself, different solid forms of a compound can and often do exhibit different physical and chemical properties as well as different biopharmaceutical properties. Differences in chemical properties can be determined, analyzed and compared through a variety of analytical techniques. Those differences may ultimately be used to differentiate among different solid forms. Furthermore, differences in physical properties, such as solubility, and biopharmaceutical properties, such as bioavailability, are also of importance when describing the solid state of a pharmaceutical compound. Similarly, in the development of a pharmaceutical compound, e.g., 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide, the new crystalline and amorphous forms of the pharmaceutical compound are also of importance.
The compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide as well as the preparation thereof was described in patent CN101619043A.
pon extensive explorations and researchs, we have found that compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide can be prepared into succinate salts, the chemical structure of its semisuccinate and monosuccinate being shown by Formula A. Studies have shown that, compared with its free base, the solubility of compound of Formula A is significantly increased, which is beneficial for improving the pharmacokinetic characteristics and in vivo bioavailability of the compound. We have also found that compound of Formula A can exist in different crystalline forms, and can form solvates with certain solvents. We have made extensive studies on the polymorphic forms of compound of Formula A and have finally prepared and determined the polymorphic forms which meet the requirement of pharmaceutical use. Based on these studies, the present invention provides the compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin -6-yl) piperazine-1-carboxamide succinate and the various crystalline forms thereof, solvates and the crystalline forms thereof, which are designated as Form I, Form IV and Form V respectively.
The compound 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide raw material used in the examples were prepared according to CN101619043A.
Example 1 Preparation of Form I of compound of Formula A
The 4-ethyl-N- (4- ( (3-ethynylphenyl) amino) -7-methoxyquinazolin-6-yl) piperazine-1-carboxamide (60g, 0.139mol) was dissolved in 150 times (volume/weight ratio) of tetrahydrofuran (9L) under refluxing. Then the obtained solution was cooled to 50℃, and succinic acid (65.8g, 0.557mol, 4 equivalents) was added in one portion. Then the obtained mixed solution was cooled naturally under stirring. The white precipitate was appeared at about 28℃. After further stirring for 18 hours, the white solid was collected by filtration, and dried at 40℃ under vacuum. A powder sample of 56.7g was obtained (yield 83%) .
1H NMR (400 MHz, cd3od) δ 8.52 (s, 1H) , 8.45 (s, 1H) , 7.93 –7.89 (m, 1H) , 7.77 –7.73 (m, 1H) , 7.35 (t, J = 7.9 Hz, 1H) , 7.24 (dd, J = 5.2, 3.8 Hz, 1H) , 7.19 (s, 1H) , 4.05 (s, 3H) , 3.69 –3.61 (m, 4H) , 3.49 (s, 1H) , 2.71 –2.64 (m, 4H) , 2.60 (q, J = 7.2 Hz, 2H) , 2.53 (s, 2H) , 1.18 (t, J = 7.2 Hz, 3H) .
The obtained powder sample is Form I of compound of Formula A, the X-ray powder diffractogram of which is shown in Figure 1. Peaks (2θ) chosen from the figure has the following values: 6.1, 7.9, 10.2, 11.6, 12.2, 13.6, 15.3, 15.9, 16.6, 17.8, 19.6, 20.4, 21.4, 21.7, 22.3, 23.5, 24.3, and 25.1 degrees, the measured 2θ values each having an error of about ± 0.2 degrees (2θ) , wherein characteristic peaks (2θ) are at 6.1, 7.9, 12.2, 15.3, 15.9, 16.6, and 20.4 degrees. DSC result is given in Figure 2, showing that the melting point range of Form I is about 193.4-197.3℃.
PATENT

PATENT
CN 108863951
PATENT
US 20100009958
PATENT
WO 2010002845
////////////Epitinib , PHASE 1, PHASE 2, Epitinib succinate, HMPL-813, Huposuan yipitini, 1203902-67-3,
CIFORADENANT

CIFORADENANT
1202402-40-1
Chemical Formula: C20H21N7O3
Molecular Weight: 407.434
CPI-444, CPI 444, CPI444, V81444, V-81444, V 81444,
UNII 8KFO2187CP
Corvus Pharmaceuticals, Inc. PHASE 1
(S)-7-(5-methylfuran-2-yl)-3-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)-3H-[1,2,3]triazolo[4,5-d]pyrimidin-5-amine
| 3H-1,2,3-TRIAZOLO(4,5-D)PYRIMIDIN-5-AMINE, 7-(5-METHYL-2-FURANYL)-3-((6-((((3S)-TETRAHYDRO-3-FURANYL)OXY)METHYL)-2-PYRIDINYL)METHYL)- |
(73 S)-15 -methyl-6-oxa-2(7,3)-[1,2,3]triazolo[4,5- d]pyrimidina-4(2,6)-pyridina-1(2)-furana-7(3)- oxolanaheptaphan-25 -amine adenosine receptor antagonist
Ciforadenant, also known as CPI-444 and V81444, is an orally administered antagonist of the adenosine A2A receptor. Upon oral administration, CPI-444 binds to adenosine A2A receptors expressed on the surface of immune cells, including T-lymphocytes, natural killer (NK) cells, macrophages and dendritic cells (DCs). This prevents tumor-released adenosine from interacting with the A2A receptors on these key immune surveillance cells, thereby abrogating adenosine-induced immunosuppression in the tumor microenvironment.
Ciforadenant is an antagonist of adenosine A2A being developed by Corvus , under license from Vernalis , for the oral treatment of advanced solid tumor; the company is also developing the drug in combination with atezolizumab , for non-small-cell lung cancer.
In 2015, Vernalis licensed the exclusive rights of the product for use of all therapeutic application to Corvus.
Synthesis
WO 2009156737

PATENT
WO 2009156737
US 8450328
WO2017112917
WO 2018175473
WO 2018009972
WO 2018049271
WO 2018022992
PATENT
PATENT
WO-2018183965
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018183965&redirectedID=true
EXAMPLES
Reaction Scheme 1
[0314] Referring to Reaction Scheme 1 , the process to manufacture triazolo[4,5]pyramidine derivatives and intermediates thereof in accordance with the present disclosure, such as the compound known as CPI-444, consists of three chemical steps and uses starting materials known as CP-55, CP-56 and CP-60. The intermediate known as CP-57 is formed at step la without isolation (telescoped) and taken to the next step to form the compound known as CP-58 at step lb. Suzuki coupling using CP-60 during step 2 generates crude CPI-444 which undergoes crystallization during step 3 to form CPI-444.
[0315] Previously described processes for making triazolo[4,5]pyramidine derivatives and intermediates thereof utilized a compound known as CP-59:
[0316] Moreover, such previously described process utilize triethylamine which takes a longer time for the layers to separate where excessive rag layer is observed during phase separation. [0317] The present inventors unexpectedly and surpisingly found that the replacement of CP-59 with CP-60 improved ease of handling and improved process efficiency. In addition, the present inventors unexpectedly and surpisingly found that the use of potassium carbonate (K2CO3) during step 2 improves the phase separation and minimizes rag layer formation upon reaction completion. Finally, Step 3 employs the use of thermocycler in order to facilitate the removal of residual solvents such as isopropyl alcohol.
[0318] Accordingly, the processes in accordance with the teachings of the present disclosure are an improvement over, and are more suitable for commercial scale-up, than processes previously described.
[0319] Starting material (C-55) is commercially available through Astatech, Inc., Keystone Business Park, 2525 Pearl Buck Road, Bristol, PA, 19007, USA; or Suven, SDE Serene Chambers, Road No.5, Avenue 7 Banjara Hills, Hyderabad, 500034, India.
[0320] CP-60 is commercially available through ARK Pharma, Inc., 3860 North Ventura Drive, Arlington Heights, IL, 60004, USA; or Boron Technology Institute, Road No. 2, Building No. 10, room No. 259, Haidian District, Beijing, China.
EXAMPLE 1. Preparation of CP-56
Reaction Scheme 1
Boc20, CbzCI
[0321] Preparation of Dimethyl pyridine-2,6-dicarboxylate:
Pyridine-2,6-dicarboxylic acid (900g, leq) is suspended in methanol(5 volume) and added H2SO4. (19g). The mixture is heated to reflux for approximately 4hr. After reaction completion, the mixture is cooled to 5- 10°C to allow the solids to precipitate. The solids are stirred for an additional hour. The solids are collected by filtration. The wet-cake is re-dissolved in DCM (3 volume) and extract in sequence with an aqueous saturated solution of NaHC03 (2 Volume) followed by with a 5% brine solution (2 Volume). The organic layer is concentrated to dryness to obtain dimethyl pyridine-2,6-dicarboxylate; 914.85g, purity 100%, yield 87.%.
[0322] Preparation of pyridine-2,6-diyldimethanol:
Dimethyl pyridine-2,6-dicarboxylate (885g, leq) is dissolved in EtOH (4425g, 5 Volume) at room temperature. The NaBH4 (341 g, 2eq) is added slowly to the reaction while keeping the internal temperature below 30°C using an ice bath. The reaction is heated to 35°C for approximately 2hrs. After reaction completion, the mixture is cooled to room temperature and adjusted with 32% HCl solution to pH value of approximately 2.5. The mixture is stirred for
2hrs to allow the solids to precipitate. The mixture is then adjusted pH value of approximately 9 using 30% NaOH solution while maintaining an internal temperature below 30°C and stirred at room temperature for about 30 min. The solids are removed by filtration. The filtrate is concentrated at 50°C. The concentrated residual is suspended with isopropanol (4160g, 8 vol)
/water (416g, 0.8 vol) and heated to 70°C for about lhr. The solution is then cooled to room
temperature and stirred for 2hr before cooling to 5-10°C for 30min. The un-dissolved solids are
removed by filtration. The filtrate is concentrated at 50°C. The concentrated residue is charged
with dichloromefhane (2700g, 5vol) and heated to 40 °C for 30min. The suspension is cooled to 5-
10°C and stirred for 30mins. The solid is collected by filtration and dried under vacuum at 40°C to obtain pyridine-2,6-diyldimethanol; 540.77g, purity 100%, yield 85.86%.
[0323] Preparation of 2,6-6 s(chloromethyl)pyridine:
2,6-bis(chloromethyl)pyridine (400g, leq) is suspended in DCM (2000g) and then cooled to 10- 15°C. Thionyl chloride (SOCb; 775g, 3eq) is charged with CH2CI2 (775g) and then added drop- wised into the reaction vessel while maintaining the internal temperature below 20 °C. The reaction is then warmed to room temperature and held for approximately 2hrs. After reaction completion, the 15% aqueous solution of a2C03 (9038g) is pre-cooled to 10-15°C before charging the reaction mixture into the carbonate solution while maintaining internal temperature below 20 °C. The mixture is stirred until gas-evolution is no longer observed. The organic layer is extracted with water (2 x 3200g) and then concentrated at 50°C to a crude product. The concentrated crude is purified by recrystallization using heptane (946g). The mixture is cooled to 5-10°C for 30min. The solid is collected by filtration and wet-cake is washed with heptane and dried at 40°C under vacuum to obtain 2,6-6zs(chloromethyl)pyridine; 442.6g, purity 100%, yield 87.0%.
[0324] Preparation of (3r,5r,7r)-l-((6-(chloromethyl)pyridin-2-yl)methyl)-l,3,5,7-tetraazaadamantan-l-ium:
2,6-to(chloromethyl)pyridine (420g, leq) is dissolved in CH2CI2 (8400g), HMTA (336g, leq) is added into the reaction vessel. The reaction is heated to approximately 40 °C for about 3hrs. Additional HMTA (168g, 0.5eq) is added into the reaction mixture and stirred overnight at room
temperature. The product is collected by filtration. The wet-cake is washed with CthCkand dried under vacuumat 50°C to obtain (3r,5r,7r)-l -((6-(chloromethyl)pyridin-2-yl)methyl)- 1 ,3, 5,7-tetraazaadamantan- 1 -ium; 730g, purity 97.01%, yield 96.58%.
[0325] Preparation of (6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride:
(3r,5r,7r)- 1 -((6-(chloromethyl)pyridin-2-yl)methyl)- 1 ,3 ,5 ,7-tetraazaadamantan- 1 -ium (730g, leq) is suspended in EtOH (4380g) before charging 37% HC1 (159g). The mixture is heated to approximately 60 °C for about lhr. After reaction completion, it is cooled to 25°C. MTBE
(1200g) is charged into the suspension. The suspension is then stirred for about 30 min and cooled to 5-10°C for about lhr. The solids are collected by filtration and washed with MTBE and dried at 50°C under vacuum to obtain (6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride; 449.56g (after assay correction), purity 98.15%, yield85.23%.
[0326] Preparation of tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamate:
(6-(chloromethyl)pyridin-2-yl)methanamine dihydrochloride [422.56g (after assay correction), leq] is dissolved in CH2CI2 (5600g) and pre-cooled to 10-15°C. K2CO3 (1632g) pre-dissolved in water (4000g) is charged into the reaction solution solution. The mixture is stirred for about lOmin and then cooled to 10-15°C. Boc-anhydride (603g) is pre-dissolved in CH2CI2 (1808g) before charging into the reactor. The mixture is warmed to room temperature and held for about an hour. After reaction completion, the organic layer is extracted with water (4000g), The organic layer is concentrated to dryness at 50 °C to obtain tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamate; 382.93g [after assay correction); purity 99.01%; yield 81%].
[0327] Preparation of tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate:
tert-butyl ((6-(chloromethyl)pyridin-2-yl)methyl)carbamat [ 382.93g (after assay correction) , leq] is dissolved in THF (1 150) and Nal (720g) is added, the reaction is at room temperature for approximately 4hr. After reaction completion, excess Nal and NaCl are filtered off and the filtrate is concentrated at 40°C. The concentrated residue is re-dissolved in ethyl acetate (2300g) and extracted with water (2900g), the organic layer is washed with 10% aqueous solution of Na2S203 (2600g) followed by 5% brine solution (2900g). The organic layer is concentrated to a residue. The residue is re-dissolved in ethyl acetate (4200g), and then filtered. The filtrate is oncentrated and taken up in ethyl acetate (765g) and stirred at room temperature for about 2hr before slowly adding heptane (380g). The solids are filtered and dried at 50°C under vacuum to
obtain tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate; 440g; purity 100%, Yield 85%.
[0328] Preparation of tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate:
A solution of t-BuOK (113g in THF (1.1 kg) is pre-cooled to 5- 10°C, before charging asolutionof (S)-tetrahydrofuran-3-ol (166g) in THF (220g). The mixture is stirred at room temperature for about lhr. A solution of tert-butyl ((6-(iodomethyl)pyridin-2-yl)methyl)carbamate (440g, leq) in THF (880g) is pre-cooled to 10-15°C before. The tetrahydrofuranyl solution is slowly charged into reaction solution while maintaining an internal temperature below 1 °C. After about 1 hour another solution of pre-cooled solution of t-BuOK (50g) and (S)-tetrahydrofuran-3-ol (66g) in THF (405g) kg) is slowly added into reaction mixture while maintaining internal temperature below 10 °C. The mixture is stirred at about 10 °C for approximately 1 hour. After reaction completion, the mixture is quenched with water (2200g) and extracted with toluene (4400g). The organic layer is washed with 5% brine (2x 2200g). The organic layer is concentrated to dryness at 50°C under vacuum to obtain tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate; 389g, purity 89.63%, yield 105%.
[0329] Preparation of CP-56 free base:
tert-butyl (S)-((6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methyl)carbamate (389g, leq) is dissolved in CH2CI2 (1556g) and pre-cooled to 0-5°C before charging drop-wise methanesulfonic acid ( MSA; 600g) into the reaction solution while maintaining internal temperature below 20°C. The mixture is warmed to room temperature and hold for about lhr. After reaction completion, water (389g) is added and cooled to 5-10°C. 30% NaOH is charged to adjust the reactor pH to approximately 12.5. The mixture is stirred for about 30 min before extracting with CH2CI2 (1556g). The organic layer is collected and extracted with an aqueous saturated solution of brine (584g). The organic layer is concentrated under vacuum. The residue is re-dissolved in toluene (1560g andthenconcentrated. The concentrated residue is re-dissolved in toluene (1560g) and then filtered. The filtrate is concentrated to dryness at 50°C under vacuum to obtain CP-56 free base; 221g (after assay correction), purity 91%, yield 84.23%.
[0330] Preparation of CP-56:
CP-56 free base (22 lg (after assay correction), leq) is dissolved in MeOH (260g) and EtOH (1300g) and then cooled about 15°C. Oxalic acid (47), pre-dissolved in MeOH (1 lOg is charged into reaction mixture. The reaction is at 15-20°C for 3hr. The mixture is cooled to 0-5°C and
stirred for about an Ihr. The solid is collected by filtration and the wet-cake is washed with EtOH (390g). The solid is dried under vacuum at 50°C to obtain CP-56 crude. Crude CP-56 is re-crystallized from isopropanol (865g) and H20 (lOOg). The mixture is heated to about 70°C to obtain a solution. The solution is slowly cooled to 50°C for Ihr. The mixture is cooled to 0-5°C for about another Ihr. The solid is filtered and washed with isopropanol. The wet-cake is dried at 50°C under vacuum to obtain CP-56; 164g, purity 99%, yield 95%.
[0331] Alternatively, CP-56 can be formed using the following process:
Reaction Scheme 2
7 8 9
[0332] Preparation of Dimethyl pyridine-2,6-dicarboxylate (compound 2):
Charge diacid (1; 628g) into reactor containing methanol (2Kg) and heat to reflux. After reaction completion the reaction is cooled to 30 C and stirred. The wet-cake is filtered and washed with methanol (500g). The wet-cake is dried under vacuum at about 55 °C to obtain diester (680 g, purity >99%; yield 85%).
[0333] Preparation of 6-(hydroxymethyl)picolinamide (compound 4):
Charge diester (2; 600 g) into reactor containing methanol (1.8 kg) and tetrahydrofuran (1.2 kg). Charge slowly sodium borohydride ( aBH4; about 130 g) into the reaction solution while maintaining an internal temperature below 30 °C. After reaction completion aqueous hydrochloric acid (about 350 g of 32% HC1) is charged into the reaction solution. The mixture is concentrated and then charged with dichloromethane (1.8 kg). The organic solution is extracted with water (600 g) and then concentrated to obtain the crude product (3). Crude 3 was dissolved in methanol (1.3 kg) and then charge ammonium hydroxide (20%; 1.3 kg). The solution was stirred until reaction completion before concentrating solution. The residue was taken up in water (600g) and heated to about 60 °C before cooling to 0 °C. The wet-cake was filtered, washed with water and dried in vacuum oven to obtain 6-(hydroxymethyl)picolinamide (about 220 g, >99% purity).
[0334] Preparation of 6-(chloromethyl)picolinonitrile (compound 5):
Charge 6-(hydroxymethyl)picolinamide (about 220 g) into a rector containing acetonitrile (450 g). Charge POCb (519 g and agitate at about 70 °C. After reaction completion the solution is
cooled to about 30 °C before slowly charging into a pre-cool (about 10 °C) reactor with water
(305 g). Charge toluene (1.4 kg) to extract the solution mixture. The toluene phase is washed in sequence with 20 % NaOH (600 g), saturated NaHC03 (300 g) and water (300 g). Toluene is concentrated to obtain crude Cl-nitrile, 5. Isopropyl alcohol (400 g) is charged to dissolve the wet-cake at about 45 °C before cooling to about 0 °C. The wet-cake was filtrated and washed with heptane (150 g) and dried in vacuum oven to obtain 6-(chloromethyl)picolinonitrile (180 g; > 99%.
[0335] Preparation of (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (compound 7):
Charge Cl-nitrile (180 g) into a rector containing THF (540 g). Charge Nal (185.7 g) to the reactor and stirred at 50 °C. After reaction completion, the reactor is cooled to 0 °C. In another
reactor, charge t-BuOK (145.6 g) and THF (320 g). Add (S)-tetrahydrofuran-3-ol (31 1.9 g) into the reactor while maintaining internal temperature below 50 °Cto deprotonate the alcohol. Stir
until t-BuOK dissolves. Add THF-OK / THF solution into 6-(iodomethyl)picolinonitrile solution (compound 6) while maintaining internal temperature below 10 °C. Stir at room
temperature until reaction completion. Concentrate the solution to remove THF solvent. Add
ethyl acetate (630 g) and wash by water (420 g). Extract water phase by ethyl acetate (630 g). Combine organic layer and concentrate to obtain oil crude 374 g. The residue was distilled under vaccum (P=3~4 torr, internal temperature 174 °C to 188 °C) to obtain (S)-6-
(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (compound 7) as an oily product (204g, >96% purity; 74% yield).
[0336] Preparation of (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (compound 9):
Charge (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinonitrile (180 g) into a rector containing MeOH (1620 g). Charge NaOMe (95.3 g) to the reactor and stirred for 30 min at 30 °C until
reaction completion. The methyl (S)-6-(((tetrahydrofuran-3-yl)oxy)methyl)picolinimidate solution (compound 8) was transferred to hydrogenation apparatus containing 50% Ni (60 g). Purge with N2 and then increase the H2 pressure. Under H2 pressure of 5 kg / cm2 and temperature of 30 °C until reaction completion. The reaction is filtered through celite. The filtrate is concentrated. Toluene is charged (1kg) and then concentrated. Then add toluene (1000 g) and filter to remove salt by-products. The filtrate was concentrated to obtain the oil residue of (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (136 g; 85% yield, assay 80%, >91% purity).
[0337] Preparation of CP-56:
Charge (S)-(6-(((tetrahydrofuran-3-yl)oxy)methyl)pyridin-2-yl)methanamine (170 g) into a rector containing isopropyl alcohol (600 g). Set internal temperature of 75 °C. In another reactor,
charge oxalic acid (41.1 g) and water (60 g) and heat solution. Add oxalic acid solution into
CP-56 free-base solution. Cool to 30 °C for about 4 hours and agitate. The wet-cake was filtered
and washed with isopropyl alcohol (175 g) and dried under vacuum drying with heat to obtain crude CP-56 (136.2 g). Charge CP-56 crude (123 g) into a rector containing methanol (1295 g). Stir until CP-56 was dissolved completely. Filter through celite to remove insoluble salt. The filtrate is concentrated. Charge isopropyl alcohol (500 g) and water (50 g) to dissolve CP-56 using heat. Cool to about 30 °C for about 3 hours and stir. The wet-cake was filtrated and
washed by isopropyl alcohol (165 g) and dried under vacuum drying with heat to obtain CP-56 (1 13.4 g. purity = >99 %, > 99% ee).
EXAMPLE 4. Preparation of CPI-444
CP-58 CP-60
C15H16CIN702 CPI-444
1H-17BO3
W: 361 .79 MW: 208.06 C20H21N O3
MW: 407.43
[0349] It is to be noted that other Pd coupling reagents can also be used such as Pd(PPh3)4 or Pd(PPh3)2Cl2.
[0350] A solution of CP-58 (30.0 g, 1 equiv.), CP-60 (approximately 20.8 g, 1.2 equiv.), in THF (approximately 180 mL), K2C03 (approximately 17.5 g), Pd(dtbpf)Cl2(approximately 337 mg), and water (approximately 100 mL) were stirred and heated to about 60 °C until reaction completion. The reaction was cooled to about 50 °C and the layers were allowed to separate. The aqueous layer was removed and back extracted with THF (approximately 30 mL). The THF layers were combined and water (approximately 450 ml) was added to precipitate out crude CPI-444. The slurry was cooled to about 20 °C and stirred for approximately 60 min and the slurry was filtered. The cake was washed in sequence with water (approximately 120 ml) and 2-propanol (approximately 30 ml). The wet-cake was dried in the vacuum oven to provide an off- white solid (29.74 g, 88% yield) with a purity of 98.5 %. Crude CPI-444 conforms to reference.
-444 can be prepared by the following process:
EDA and DAP are used to remove Palladium during CPI-444 formation.
[0352] The solution of CP-58 (10 g), CP-60 (6.9 g) , Pd(dtbpf)C12 (approx. 0.0015 mol eq) and K2C03 (5.8 g) in THF (6V) and H20 (3V) is heated to approximately 60 °C. The reaction is complete after approximately 30 minutes. The solution is cooled to 50 °C and aqueous layer is separated. The aqueous layer is extracted with THF (9 mL); the THF layer is added to organic solution. The organics are cooled to 40 °C, 1 ,3-diaminopropane (DAP; approximately 50 g) or ethylene diamine (EDA; approximately 45 g) is added and the mixture stirred for 1 hour. H20 (15V) is added to the organic layer over 10 min. The slurry is cooled to 20 °C for 2 hours, and stirred for an additional 1 hour. The slurry is filtered and washed with H20 (2V x 2) and z‘PrOH (IV). CPI-444 wet-cake is dried at 50 °C under full vacuum. (Yield = 90 %; purity > 99.0%).
[0353] Alternatively, CPI-444 can be prepared by the following process:
using cysteine in TNF to remove Palladium during CPI-444 formation
[0354] CP-58 (1 kg), K2C03 (0.58 kg), water (3 kg), CP-60 (0.69 kg), and THF (5.3 kg),
Pd(dtbpf)Cb (3 g). The solution is heated to 60 °C. The reaction is complete after approximately 30 minutes. Charge THF (4.5 kg) and cool to 50 °C. The aqueous layer is separated. The organic layer is charged with cysteine (0.32 kg) and water (5 kg). The mixture is agitated. NH4OH (1.1 kg) is charged to the reaction mixture and agitate for approximately 15 minutes. The layers are allowed to separate and the lower aqueous layer is separated. The organic layer is charged with cysteine (0.32 kg) and water (5 kg). The mixture is agitated. NH4OH (1.1 kg) is charged to the reaction mixture and agitate for approximately 15 minutes. The layers are allowed to separate and the lower aqueous layer is separated. THF is distilled to approximately 7 volumes under atmospheric pressure. The solution is cooled to 50 °C before charging NH4OH (0.5 kg) and agitate for 30 min. Water (14.5 kg) is charged while maintaining the internal temperature >40 °C. The reactor is cooled to 20 °C for 2 hours and hold for an additional 1 hour. CPI-444 is filtered and washed with water followed by isopropanol. CPI-444 wet-cake is dried under vacuum at 50 °C. Purity > 99%, yield 85%.
EXAMPLE 5. Removal of Residual Palladium With Biocap Filter Cartridge
[0355] A mixture of CPI-444 crude (16.00 g), THF (approximately 190 ml), L-cysteine
(approximately 8 g), and H20 (approximately 90 ml) were mixed and heated to a solution at about 60 °C for 1 hour. A solution of 28% NH OH (approximately 20 ml) was added and heated for an additional 15 minutes. The agitation was turned off to allow the layers allowed to settle. The aqueous layer was removed; the THF layer was washed with brine solution (approximately 15 ml). The combined aqueous solutions were back extracted with THF (approximately 15 ml). A 3M Biocap filter (BC0025LR55SP; available from 3M) was pretreated with THF (approximately 150 ml) at about 50 °C. The combined organic layers were recirculated through the Biocap at about 10 ml/min for approximately 3 hours and then filtered forward. The Biocap filter was rinsed with THF (approximately 130 ml) at about 50 °C. The combined filtrates were concentrated. Water
(approximately 80 ml) was added, and distilled to remove residual THF. 2-Propanol (approximately 1 10 ml) was added to the slurry, and the mixture was heated to a solution. The solution was cooled to 20 °C and water (approximately 240 ml) was added. The slurry was performed in series by heating to about 55 °C and held that that temperature for approximately 30 minutes, cooled to 20 °C over 30 minutes, and held at 20 °C for 30 minutes. This heating cycle was repeated two more. The slurry was then held at 20 °C for approximately 12 hours. The slurry was filtered, and the product was washed with water (approximately 300 ml). The wet cake (about 23 g) was dried in the vacuum oven to obtain an off white solid (13.6 g; 85% yield;99.9% purity; Pd = 25 ppm).
[0356] Reprocess of step 4. AFC-825-106
[0357] CPI-444 (16.02 g, AFC-825-48) and THF (approximately 280 ml) were charged to a flask and heated to about 50 °C for about 30 minutes to obtain a solution. A 3M Biocap filter
(BC0025LR55SP) was pretreated with THF (approximately 150 ml) at about 50 °C . The CPI-444 solution was passed through the Biocap at aboutl O ml/min. The Biocap filter was rinsed with THF (approximately 130 ml) at about 50 °C. The combined filtrates were transferred to a reactor and concentrated. Water (approximately 80 ml) was added, and distilled to remove residual THF solvent. 2-Propanol (approximately 1 10 ml) was added to the slurry and heated to about 65 °C to obtain a solution. The solution was cooled to about 20 °C before adding water (approximately 240 ml). The slurry was heated to 55 °C over 30 minutes, held at 55 °C for 30 minutes, cooled to 20 °C over 30 minutes, and held at 20 °C for 30 minutes. This heating cycle was two more times. The slurry was then held at 20 °C for 12 hours. The slurry was filtered, and the product was washed with water (approximately 300 ml). The wet cake (26.6 g) was dried in the vacuum oven overnight to obtain 15 as a white solid (95% yield; 99% purity; Pd = 5 ppm).
EXAMPLE 6. Removal of Residual Palladium With Darco KB-G
Crude CPI-444
CPI-444 Drug Substance
[0358] Crude CPI-444 (475 g, 1.17 mol, 1.00 eq), 2-MeTHF (1 1.9 L, 25.0 vol) and WFI water (2.6 L, 5.5 vol) were charged to a 19 L jacketed reactor. The mixture was mechanically agitated under a nitrogen blanket. Nitrogen was bubbled through the solution for 20 minutes. L-Cysteine (242 g, 1.99 mol, 1.71 eq) was then charged. The solution in the reactor was heated to 55±5 °C. Upon reaching 50 °C, the reaction mixture was stirred for 1 hour. 28-30% NH4OH (594 mL, 1.25 vol) was charged via addition funnel, and then the reaction mixture was stirred for 15 min. Agitation was stopped and the reaction was allowed to separate for 1 hour. The aqueous layer was removed. The organic layer was allowed to cool to ambient. The organic layer was filtered and the frit was washed with 2-MeTHF (618 mL, 1.3 vol). The organics were concentrated off by rotary evaporation. WFI water (2.42 L, 5.1 vol) and IPA (2.38 L, 5.0 vol) were used to charge the concentrated slurry to a clean 19 L jacketed reactor under N2. The mixture was heated to 65±5 °C, and then was stirred for 1 hour to obtain solution. Darco KB-G activated carbon (71.3 g, 15 wt%) was charged. The reactor was heated to 75±5 °C and stirred for 15 hours. A I L pocket filter was prepared with filter cloth and a heating jacket and heated to 70±5 °C. Reactor contents were filtered through the pocket filter using N2 pressure. The pocket filter was rinsed with a mixture of IPA/WFI water (1 : 1, 950 mL, 2 vol) followed by a mixture of IPA/WFI water (1 : 1, 1.90 L, 4 vol) and IPA/WFI water ( 1 : 1 , 1.90 L, 4 vol). Inside a 22 L three neck round bottom flask the filtrates were mechanically agitated under a N2 blanket. WFI water (7.13 L, 15 vol) was slowly added via addition funnel over 1 h at ambient temperature, and aged for 1 h. The slurry was heated to 55±5 °C and maintained the temperature for 30 min. This heating and subsequent cooling were repeated twice more. After reaching ambient
temperature the final time, the mixture was stirred for at least 2 hours. The reaction mixture was filtered and the reactor rinsed with WFI water (2.38 L, 5.0 vol, 3x). The cake was dried under N2 for 30 minutes and then transferred to a glass dish. The material was dried under full vacuum at 55±5 °C. The desired product was obtained 368.1 g (77%) as light yellow solids. This material was 99.6% pure by HPLC and had a Pd content of 3.6 ppm.
EXAMPLE 7. Removal of Residual Palladium With Polymer-Bound Thiol (SiST)
[0359] Crude CPI-444 (24.48 g, pd = 1267 ppm) and THF (244.8 mL, 10 vol) were charged to a 500 mL 4-necked flask fitted with mechanical agitation, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 60 °C for 20 minutes and then slowly cooled to 45 °C. SiST (36.72 g) was added to the solution and the mixture was stirred at 42 °C for 14 h. The mixture was filtered and washed by THF (24 mL, 1 vol, twice; Pd= 13.12 ppm). H20 (120 mL, 5 vol) and IPA (120 mL, 5vol) were charged to the flask. The slurry was heated to 70 °C and maintained for 1 h (the slurry became solution). The solution was slowly cooled to room temperature and the slurry was added H20 (360 mL, 15 vol) and heated to 55 °C for 1 h. The slurry was cooled to room temperature and then heated to 55 °C for 1 h. The slurry was cooled to rt. and stirred at rt. for 2 h. The slurry was filtered and washed by H20 (100 mL, 4 vol, three times). The wet cake (28.36 g) was dried by 10 mmHg and 50 °C for overnight (14h) and the weight of CPI-444 was 19.31 g (79% recovery).
EXAMPLE 8. Removal of Residual Palladium By Recrystallization
[0360] CUNO Filter Cartridge 55 S
[0361] CPI-444 (5.0 g, Pd 14.06 ppm) and THF (50 mL, 10 vol) were charged to a 100 mL 3-necked flask fitted with stirring bar, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 60 °C for 20 minutes and added CUNO 55S filter (0.75 g, 15w%). The mixture was stirred at 60 °C for 1 h. The mixture was filtered and washed by THF (5 mL, 1 vol, twice). The filtrate was concentrated. The solid, H20 (25 mL, 5 vol) and IPA (25 mL, 5vol) were charged to 250 mL 3 -necked flask fitted with stirring bar, a condenser with nitrogen balloon and a thermometer. The slurry was heated to 70 °C and maintained for 1 h (the slurry became solution). The solution was slowly cooled to rt.(40 minutes) The slurry was added H20 (75 mL, 15 vol) and then heated to 55 °C for 1 h. The slurry was cooled to rt. (30 minutes) and stirred at rt. for 2 h. The slurry was filtered and washed by H20 (20 mL, 4 vol, three times). The cake (6.355 g) was dried by 10 mmHg and 50 °C
for overnight (16 h) and the weight of CPI-444 was 4.281 g (85% recovery). Pd content(ppm) = 2.02 ppm.
[0362] Polymer-bound Thiol: SiST
[0363] CPI-444(5 g; Pd 14.06ppm) was dissolved in THF (50 mL) at 60 °C. The solution was cooled to 55 °C and SiST (7.5 g) was added to the solution. The solution was stirred at 50-55 °C for 16 h. The solution was filtered through celite and a 0.2 micron filter. The filtrate was tested for Pd content. Result: 2.43 ppm.
Catalyst
Molecular Weight: 291.6990
Molecular Weight: 337.3430
[0364] 1. A solution of S.M., CP-60, Pd(PPh3)2Cl2 and K2C03 in THF – H20 (7.9 mL, 1 : 1) was put in oil-bath at 70-75 °C.
[0365] 2. After 2 h, 0.047 g CP-60 was added to the reaction at 70-75 °C.
[0366] 3. After 1 hr, the reaction was cooled to rt. and 10 mL H20 was added to the reaction.
[0367] 4. The reaction was filtered to provide wet cake (0.812 g).
[0368] 5. The solid wet cake was dried at 45 °C and 20 mmHg for 2h to provide weight 0.499 g. (86%).
[0369] 6. The solid wet cake was stirred in 2 mL DMF for 30 mins (slurry) and then filtered. The solid was dried by 45 °C and 10 mmHg for 12h to provide weight 0.40 g; 69% yield; 98.1% purity.
//////////CIFORADENANT, CPI-444, CPI 444, CPI444, V81444, V-81444, V 81444, UNII 8KFO2187CP, Corvus Pharmaceuticals, Inc., PHASE 1,
NC1=NC2=C(N=NN2CC3=NC(CO[C@H]4CCOC4)=CC=C3)C(C5=CC=C(O5)C)=N1
Linrodostat BMS 986205, ONO 7701
cas 2221034-29-1
- Linrodostat
- (2R)-N-(4-chlorophenyl)-2-(cis-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
- Linrodostat mesylate
- Linrodostat [USAN]
- UNII-OS7OBU191R
- OS7OBU191R
- Linrodostat mesylate [USAN]
- BMS-986205-04
- 2221034-29-1
- Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-alpha- methyl-, (alphaR,1alpha,4alpha)-, methanesulfonate (1:1)
Linrodostat; (2R)-N-(4-chlorophenyl)-2-(cis-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide; Linrodostat mesylate; Linrodostat [USAN]; UNII-OS7OBU191R; OS7OBU191R


BMS 986205
(2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoro-4-quinolinyl)cyclohexyl]propanamide
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-(αR)-
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
CAS: 1923833-60-6
Phase III Head and neck cancer; Malignant melanoma
BMS-986205, ONO-7701, F- 001287
- Molecular Formula C24H24ClFN2O
- Average mass 410.912 Da
BMS986205, BMS 986205, ONO-7701
Cyclohexaneacetamide, N-(4-chlorophenyl)-4-(6-fluoro-4-quinolinyl)-α-methyl-, cis-(αR)-
A potent and selective IDO1 (indoleamine 2,3-dioxygenase 1) inhibitor.
| Alternate Name | (R)-N-(4-chlorophenyl)-2-((1s,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propenamide |
|---|---|
| Appearance | Crystalline solid |
| CAS # | 1923833-60-6 |
| Molecular Formula | C₂₄H₂₄ClFN₂O |
| Molecular Weight | 410.92
|
- Originator Bristol-Myers Squibb
- Developer Bristol-Myers Squibb; Ono Pharmaceutical
- Class Antineoplastics; Cyclohexanes; Quinolines; Small molecules
- Mechanism of Action Indoleamine-pyrrole 2,3-dioxygenase inhibitors
Highest Development Phases
- Phase II IHead and neck cancer; Malignant melanoma
- Phase I/II Cancer
- Phase I Solid tumours
Most Recent Events
- 01 Jun 2018Efficacy and adverse events data from a phase I/IIa trial in Bladder cancer (Combination therapy, Late-stage disease) presented at the 54th Annual Meeting of the American Society of Clinical Oncology (ASCO- 2018)
- 08 May 2018Bristol-Myers Squibb plans the CheckMate 9UT phase II trial for Bladder Cancer in USA, Canada, Italy, Mexico, Netherlands, Spain and United Kingdom , (NCT03519256)
- 30 Apr 2018Bristol-Myers Squibb withdraws a phase III trial for Non-small cell lung cancer (First-line therapy, Combination therapy, Late-stage disease) in USA, Austria, Australia, Brazil, Canada, Czech Republic, France, Germany, Greece, Italy, Japan, South Korea, Mexico, Spain, Switzerland, Taiwan and Turkey prior to enrolment (NCT03417037)
BMS , following its acquisition of Flexus Biosciences , and licensee Ono Pharmaceutical are developing linrodostat, a once-daily, indoleamine 2,3-dioxygenase 1 inhibitor for the potential oral treatment of cancer including renal cell carcinoma, muscle-invasive bladder cancer and melanoma. In October 2018, the trial was initiated in the US, Europe, Israel and Brazil.
WO2015031295 product pat
WO2016073770 first disclosed
WO2018209049
- WO 2016073770


Bristol-Myers Squibb, following its acquisition of Flexus Biosciences, is developing BMS-986205 (previously F- 001287), the lead from an immunotherapy program of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors for the potential treatment of cancer. In February 2016, a phase I/IIa trial was initiated .
BMS-986205 (ONO-7701) is being evaluated at Bristol-Myers Squibb in phase I/II clinical trials for the oral treatment of adult patients with advanced cancers in combination with nivolumab. Early clinical development is also ongoing at Ono in Japan for the treatment of hematologic cancer and for the treatment of solid tumors.
In April 2017, data from the trial were presented at the 108th AACR Annual Meeting in Washington DC. As of February 2017, the MTD had not been reached, but BMS-986205 plus nivolumab treatment was well tolerated, with only two patients discontinuing treatment due to DLTs. The most commonly reported treatment-related adverse events (TRAEs) were decreased appetite, fatigue, nausea, diarrhea, and vomiting. Grade 3 TRAEs were reported in three patients during the combination therapy; however, no grade 3 events were reported during BMS-986205 monotherapy lead-in. No grade 4 or 5 TRAEs were reported with BMS-986205 alone or in combination with nivolumab
Indoleamine 2,3-dioxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO is an oxidoreductase and one of two enzymes that catalyze the first and rate-limiting step in the conversion of tryptophan to N-formyl-kynurenine. It exists as a 41kD monomer that is found in several cell populations, including immune cells, endothelial cells, and fibroblasts. IDO is relatively well-conserved between species, with mouse and human sharing 63% sequence identity at the amino acid level. Data derived from its crystal structure and site-directed mutagenesis show that both substrate binding and the relationship between the substrate and iron-bound dioxygenase are necessary for activity. A homolog to IDO (ID02) has been identified that shares 44% amino acid sequence homology with IDO, but its function is largely distinct from that of IDO. (See, e.g., Serafini P, et al, Semin. Cancer Biol, 16(l):53-65 (Feb. 2006) and Ball, H.J. et al, Gene, 396(1):203-213 (Jul. 2007)).
IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. Importantly, IDO regulates immunity at the T cell level, and a nexus exists between IDO and cytokine production. In addition, tumors frequently manipulate immune function by upregulation of IDO. Thus, modulation of IDO can have a therapeutic impact on a number of diseases, disorders and conditions.
A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res., 12(4): 1144-1151 (Feb. 15, 2006)).
Treatment of cancer commonly entails surgical resection followed by chemotherapy and radiotherapy. The standard treatment regimens show highly variable degrees of long-term success because of the ability of tumor cells to essentially escape by regenerating primary tumor growth and, often more importantly, seeding distant metastasis. Recent advances in the treatment of cancer and cancer-related diseases, disorders and conditions comprise the use of combination therapy incorporating immunotherapy with more traditional chemotherapy and radiotherapy. Under most scenarios, immunotherapy is associated with less toxicity than traditional chemotherapy because it utilizes the patient’s own immune system to identify and eliminate tumor cells.
In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g. , rheumatoid arthritis). Thus, suppression of tryptophan degradation by inhibition of IDO activity has tremendous therapeutic value. Moreover, inhibitors of IDO can be used to enhance T cell activation when the T cells are suppressed by pregnancy, malignancy, or a virus (e.g., HIV). Although their roles are not as well defined, IDO inhibitors may also find use in the treatment of patients with neurological or neuropsychiatric diseases or disorders (e.g., depression).
Small molecule inhibitors of IDO have been developed to treat or prevent IDO-related diseases. For example, the IDO inhibitors 1-methyl-DL-tryptophan; p-(3-benzofuranyl)-DL-alanine; p-[3-benzo(b)thienyl]-DL-alanine; and 6-nitro-L-tryptophan have been used to modulate T cell-mediated immunity by altering local extracellular concentrations of tryptophan and tryptophan metabolites (WO 99/29310). Compounds having IDO inhibitory activity are further reported in WO 2004/094409.
In view of the role played by indoleamine 2,3-dioxygenase in a diverse array of diseases, disorders and conditions, and the limitations (e.g., efficacy) of current IDO inhibitors, new IDO modulators, and compositions and methods associated therewith, are needed.
In April 2017, preclinical data were presented at the 108th AACR Annual Meeting in Washington DC. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively
In April 2017, preclinical data were presented at the 253rd ACS National Meeting and Exhibition in San Francisco, CA. BMS-986205 showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. A good pharmacokinetic profile was seen at oral and iv doses in rats, dogs and monkeys. The compound showed good oral exposure and efficacy in in vivo assays
Preclinical studies were performed to evaluate the activity of BMS-986205, a potent and selective optimized indoleamine 2, 3-dioxygenase (IDO)- 1inhibitor, for the treatment of cancer. BMS-986205 inhibited kynurenine production with IC50 values of 1.7, 1.1 and > 2000 and 4.6, 6.3 and > 2000 nM in human (HeLa, HEK293 expressing human IDO-1 and tryptophan-2, 3-dioxygenase cell-based assays) and rat (M109, HEK293 expressing mouse ID0-1 and -2 cell-based assays) respectively. BMS-986205 was also found to be potent when compared with IDO-1from other species (human < dog equivalent monkey equivalent mouse > rat). In cell-free systems, incubation of inhibitor lead to loss of heme absorbance of IDO-1 which was observed in the presence of BMS-986205 (10 microM), while did not observed with epacadostat (10 microM). The check inhibitory activity and check reversibility (24 h after compound removal) of BMS-986205 was found to be < 1 and 18% in M109 (mouse) and < 1 and 12% SKOV3 (human) cells, respectively. In human whole blood IDO-1, human DC mixed lymphocyte reaction and human T cells cocultured with SKOV3 cells- cell based assays, BMS-986205 showed potent cellular effects (inhibition of kynurenine and T-cell proliferation 3H-thymidine) with IC50 values of 2 to 42 (median 9.4 months), 1 to 7 and 15 nM, respectively. In human SKOV-3 xenografts (serum and tumor) AUC (0 to 24h; pharmacokinetic and pharmacodynamic [PK and PD])) was 0.8, 4.2 and 23 and 3.5, 11 and 40 microM h, respectively; area under the effect curve (PK and PD) was 39, 32 and 41 and 60, 63 and 76% kyn, at BMS-986205 (5, 25 and 125 mg/kg, qd×5), respectively. In vivo human-SKOV3 and hWB-xenografts, IC50 values of BMS-986205 were 3.4 and 9.4 NM, respectively. The ADME of BMS-986205 at parameters iv/po dose was 0.5/2, 0.5/1.5 and 0.5/1.2 mg/kg, respectively; iv/clearance was 27, 25 and 19 ml, min/kg, respectively; iv Vss was 3.8, 5.7 and 4.1 l/kg, respectively; t1/2 (iv) was 3.9, 4.7 and 6.6 h, respectively; fraction (po) was 64, 39 and 10%, respectively. At the time of presentation, BMS-986205 was being evaluated in combination with nivolumab.
The chemical structure and preclinical profile was presented for BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.
BMS-986158: a BET inhibitor for cancerAshvinikumar Gavai of Bristol Myers Squibb (BMS) gave an overview of his company’s research into Bromodomian and extra-terminal domain (BET) as oncology target for transcriptional suppression of key oncogenes, such as MYC and BCL2. BET inhibition has been defined as strong rational strategy for the treatment of hematologic malignancies and solid tumors. From crystal-structure guided SAR studies, BMS-986158, 2-{3-(1,4-Dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol, was chosen as a potent BET inhibitor, showing IC50 values for BRD2, BRD3 and BRD4 activity of 1 nM; it also inhibited Myc oncogene (IC50 = 0.5 nM) and induced chlorogenic cancer cell death. In vitro the compound also displayed significant cytotoxicity against cancer cells. When administered at 0.25, 0.5 and 1 mg/kg po, qd to mice bearing human lung H187 SCLC cancer xenograft, BMS-986158 was robust and showed efficacy as a anticancer agent at low doses. In metabolic studies, it showed t1/2 of 36, 40 and 24 min in human, rat and mice, respectively, and it gave an efflux ratio of 3 in Caco-2 permeability assay. In phase 1/II studies, BMS-986158 was well tolerated at efficacious doses and regimens, and drug tolerable toxicity at efficacy doses and regimens. Selective Itk inhibitors for inflammatory disordersThe development of highly selective Itk inhibitors for the treatment of diseases related to T-cell function, such as inflammatory disorders, was described by Shigeyuki Takai (Ono Pharmaceutical). Inhibitory properties of a hit compound, ONO-8810443, were modified via X-ray structure and Molecular Dynamics stimulation to get ONO-212049 with significant kinase selectivity (140-fold) against Lck, a tyrosine kinase operating upstream of Itk in the TCR cascade. Further modifications identified final lead compound ONO-7790500 (N-[6-[3-amino-6-[2-(3-methoxyazetidin-1-yl)pyridin-4-yl]pyrazin-2-yl]pyridin-3-yl]-1-(3-methoxyphenyl)-2,3-dimethyl-5-oxopyrazole-4-carboxamide), which selectively inhibited Itk (IC50 = < 0.004 microM) over Lck (IC50 = 9.1 microM; SI 2000-fold) and suppressed Jurkat T-cell proliferation (IC50 = 0.014 microM). This compound suppressed alphaCD3/CDP28 CD4+T-cell stimulation (IC50 = 0.074 microM) with selectivity over PMA/Ionomycin (IC50 = > 10 microM). ONO-7790500 also exhibited in vivo IL-2 inhibitory properties (62% inhibition at 30 mg/kg po) in mice. In pharmacokinetic studies in balb/c mice, the compound administered orally (10 mg/kg) showed a Cmax of 1420 ng/ml, AUClast of 11,700 ng*h/ml, t1/2 of 5.3 h and oral bioavailability of 68%. Administration iv at 0.3 mg/kg gave an AUC last of 610 ng*h/ml, t1/2 of 3.8 h, Vss of 1260 ml/kg and Cl of 5.1 ml/min/kg. ADMET data showed ONO-7790500 did not have relevant activity in cytochromes and hERG channels (IC50 > 10 microM) in toxicological studies, and gave a PAMPA value of 5.0 x 10(-6) cm/s. Fused imidazole and pyrazole derivatives as TGF-beta inhibitorsDual growth and differentiation factor-8 (GDF-8; also known as myostatin) and TGF-beta inhibitors were described. Both targets belong to TGF-beta superfamily consisting of a large group of structurally related cell regulatory proteins involved in fundamental biological and pathological processes, such as cell proliferation or immunomodulation. Myostatin (GDF8) is a negative regulator negative regulator of skeletal muscle growth and has also been related to bone metabolism. Investigators at Rigel Pharmaceuticals found that compounds designed to be GDF-8 inhibitors were able to inhibit TGF-beta as well, this could be an advantage for the treatment of diseases associated with muscle and adipose tissue disorders, as well as potentially immunosuppressive disorders. Jiaxin Yu from the company described new fused imidazole derivatives, of which the best compound was 6-[2-(2,4,5-Trifluorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl]quinoxaline. This compound was very potent at TGF-beta Receptor Type-1 (ALK5) inhibition with an IC50 value of 1nM. In an in vivo mouse assay this compound showed good activity at 59.7 mg/kg, po, and good plasma exposure; inhibition of GDF-8 and TGFbeta growth factors was 90 and 81.6 %, respectively.Rigel’s Ihab Darwish described a series of fused pyrazole derivatives, with the best compound being 6-[2-(2,4-Difluorophenyl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl][1,2,4]triazolo[1,5-a]pyridine. This compound showed an IC50 of 0.06 and 0.23 microM for GDF-8 and TGFbeta, respectively, in the pSMAD (MPC-11) signaling inhibition test. The compound had a good pharmacokinetic profile, with 40% of bioavailability in mice after a 5-mg/kg po dose. An iv dose of 1 mg/kg showed t1/2 of 0.7 h and Vss of 1.0 l/h/kgDiscovery of selective inhibitor of IDO BMS-986205 for cancerIndoleamine-2,3-dioxygenase (IDO)-1 enzyme initiates and regulates the first step of the kynurenine pathway (KP) of tryptophan metabolism, and evidence has shown that overexpression of IDO-1 in cancer tumors is a crucial mechanism facilitating tumor immune evasion and persistence. The chemical structure and preclinical profile of BMS-986205 was presented by Aaron Balog from BMS. BMS-986205 ((2R)-N-(4-Chlorophenyl)-2-[cis-4-(6-fluoroquinolin-4-yl)cyclohexyl]propanamide), is a potent IDO-1 inhibitor in phase I for the treatment of cancer. This compound showed potent and selective inhibition of IDO-1 enzyme (IC50 = 1.7nM) and potent growth inhibition in cellular assays (IC50 = 3.4 nM) in SKOV3 cells. The pharmacokinetic profile in rats dosed at 0.5 mg/kg iv and 2 mg/kg po, with clearance, Vss, half-life and bioavailability of 27 ml/min/kg, 3.8 l/kg, 3.9 h and 4%, respectively; in dogs at 0.5 iv and 1.5 po mg/kg dosing results were 25 ml/min/kg, 5.7 l/kg, 4.7 h and 39%; and, in cynomolgus monkeys with the same doses as dogs results were 19 ml/min/kg, 4.1 l/kg, 6.6 h and 10%, respectively. The compound showed good oral exposure and efficacy in in vivo assays.Three further reports have been published from this meeting .The website for this meeting can be found at https://www.acs.org/content/acs/en/meetings/spring-2017.html.
SYNTHESIS
1 Wittig NaH
2 REDUCTION H2, Pd, AcOEt, 4 h, rt, 50 psi
3 Hydrolysis HCl, H2O, Me2CO, 2 h, reflux
4 4-Me-2,6-(t-Bu)2-Py, CH2Cl2, overnight, rt
5 SUZUKI AcOK, 72287-26-4, Dioxane, 16 h, 80°C
6 Heck Reaction, Suzuki Coupling, Hydrogenolysis of Carboxylic Esters, Reduction of Bonds, HYDROGEN
7 Et3N, THF, rt – -78°C , Pivaloyl chloride, 15 min, -78°C; 1 h, 0°C ,THF, 0°C – -78°C, BuLi, Me(CH2)4Me, 15 min, -78°C, R:(Me3Si)2NH •Na, THF, 10 min, -50°C , HYDROLYSIS, (PrP(=O)O)3, C5H5N, AcOEt, 5 min, rt

Product Patent
WO2016073770
Example 19
(i?)-N-(4-chlorophenyl)-2- c 5-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
Example 19 : (i?)-N-(4-chlorophenyl)-2-(cz5-4-(6-fluoroquinolin-4- yl)cyclohexyl)propanamide
[0277] Prepared using General Procedures K, B, E, L, M, N, and O. General Procedure L employed 2-(4-(6-fluoroquinolin-4-yl)-cyclohexyl)acetic acid (mixture of
diastereomers), and ( ?)-2-phenyl-oxazolidinone. General Procedure M employed the cis product and iodomethane. The auxiliary was removed following General Procedure N and the desired product formed employing General Procedure O with 4-chloroaniline.
Purified using silica gel chromatography (0% to 100% ethyl acetate in hexanes) to afford Example 19. 1H NMR of czs-isomer (400 MHz; CDC13): δ 9.14 (s, 1H), 8.70 (d, J= 4.6 Hz, 1H), 8.06 (dd, J= 9.2 Hz, J= 5.6 Hz, 1H), 7.58-7.64 (m, 3H), 7.45 (ddd, J= 9.3 Hz, J= 7.8 Hz, J= 2.7 Hz, 1H), 7.19-7.24 (m, 2H), 7.15 (d, J= 4.6Hz, 1H), 3.16-3.26 (m, 1H), 2.59-2.69 (m, 1H), 2.08-2.16 (m, 1H), 1.66-1.86 (m, 7H), 1.31-1.42 (m, 1H), 1.21 (d, J= 6.8Hz, 3H) ppm. m/z 411.2 (M+H)+.
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(3), 319-329.
https://www.sciencedirect.com/science/article/pii/S0960894X17312180

PATENT
WO 2018022992
PATENT
WO 2018071500
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018071500&redirectedID=true
WO-2019006292
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006292&tab=PCTDESCRIPTION&maxRec=1000
Improved methods for the preparation of substituted quinolinycyclohexylpropanamide compounds, such as linrodostat claiming substituted pyridine compounds as IDO1 inhibitors, useful for treating cancers.
Indoleamine 2,3 -di oxygenase (IDO; also known as IDOl) is an IFN-γ target gene that plays a role in immunomodulation. IDO plays a major role in immune regulation, and its immunosuppressive function manifests in several manners. A pathophysiological link exists between IDO and cancer. Disruption of immune homeostasis is intimately involved with tumor growth and progression, and the production of IDO in the tumor microenvironment appears to aid in tumor growth and metastasis. Moreover, increased levels of IDO activity are associated with a variety of different tumors (Brandacher, G. et al, Clin. Cancer Res. , 12(4): 1144-1151 (Feb. 15, 2006)). In addition to cancer, IDO has been implicated in, among other conditions, immunosuppression, chronic infections, and autoimmune diseases or disorders (e.g., rheumatoid arthritis).
Substituted quinolinylcyclohexylpropanamide pharmaceutical compounds that inhibit IDO and are useful for the treatment of cancer have been previously described. See, e.g., WO2016/073770. Improved methods of making such compounds, which reduce production costs and improve production safety, are, therefore, needed.
Scheme 4
[0076] The disclosure is also directed to methods of preparing intermediate compounds of formula IV. Methods to produce compounds of formula IV are depicted in Schemes 5 and 6.
Scheme 5
IX-A
Scheme 6
IX-B IV
Compounds of the disclosure that include one or more radioisotopes can be used in imaging. See, e.g., WO2018017529. For example, radiolabeled compounds of the disclosure can be used in Positron Emission Tomography (PET). Such methods are useful in the imaging of cancer in a subject. A preferred radiolabeled compound is
1
Pharmaceutically acceptable salts of [18F]-Compound 1 are also within the scope of the disclosure. An exemplary method for the preparation of [18F]-Compound 1 is depicted in Scheme below.
1 . reaction
[18F]-Compound 1
Example 9
(R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
[00258] To a 10 L glass-lined reactor under a blanket of nitrogen was charged 349 g Ν,Ν,Ν’,Ν’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and 2 L acetonitrile. 245 g N-methylimidazole was added followed by 0.3 L acetonitrile. 300 g (R)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanoic acid was added followed by 0.3 L acetonitrile. The mixture was held for 0.5 h then 139 g 4-chloroaniline charged followed by 0.4 L acetonitrile. The mixture was maintained at 20 °C until the reaction was deemed complete by HPLC analysis. The solution was then heated to 60°C, and 1.2 L water was charged. The solution was then cooled to 40 °C, seeds (3 g) were charged, and the resulting slurry was maintained for 1 h. The slurry was then cooled to 20 °C and 2.7 L water was charged. The slurry was filtered and the cake was washed three times with 3 L of 2: 1 water: acetonitrile. The cake was dissolved with 5.1 L ethyl acetate and the solution was distilled to a volume of 4.2 L at 41 °C under vacuum. The slurry was cooled to 20 °C, 4.14 g seeds were charged, and a solution of 95.7 g methanesulfonic acid in 2.9 L ethyl acetate was added. The slurry was then filtered and washed two times with 1.65 L ethyl acetate and dried under vacuum at 50°C to yield 445 g of (R)-N-(4-chlorophenyl)-2-((l s,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide methanesulfonate as a white solid in 88% yield.
[00259] ¾ NMR (600 MHz, DMSO-de) δ 10.19 (s, IH), 9.24 (d, J=5.7 Hz, IH), 8.40 (dd, J=10.3, 2.6 Hz, IH), 8.33 (dd, J=9A, 5.3 Hz, IH), 8.09 (d, J=5.7 Hz, IH), 8.04 (t, J=8.6 Hz, IH), 7.71 – 7.64 (m, 2H), 7.37 – 7.30 (m, 2H), 3.64 (ddt, J=10.8, 7.3, 3.8 Hz, IH), 2.98 – 2.89 (m, IH), 2.43 (s, 3H), 2.05 – 1.60 (m, 9H), 1.14 (d, J=6.7 Hz, 3H); 13C NMR (126 MHz, DMSO-de) δ 175.0, 162.7, 161.1 , 145.4, 138.2, 136.8, 128.6, 128.1 , 126.7, 126.4, 123.3, 120.8, 119.8, 109.0, 39.8, 39.7, 38.6, 35.5, 28.3, 27.6, 27.2, 26.1 , 16.2 MS (ESI): calcd for C24H24CIFN2O
([M + H]+), 410.16; found, 410.15.
[00260] HPLC analysis: Column: Sigma-Aldrich Supelco Ascentis Express CI 8 2.7um, 150 x 4.6 mm ID; Solvent A: 0.05% TFA with MeCN:water (5/95 v/v); Solvent B: 0.05% TFA with MeCN: water (95/5 v/v); Gradient: %B: 0 Min. 15%; 1 Min. 15%; 13 Min. 55%; 19 Min. 65%; 24 Min. 100%; 24.1 15%; 28 Min. 15%; Stop Time: 24 Min; Flow Rate: 1.0 ml/min;
Column temperature: 30 °C; wavelength: 218 nm. The retention time (R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide peak was 12.6 min.
Example 7
(R)-N-(4-chlorophenyl)-2-((ls, -4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide
[00252] To a 50 L glass-lined reactor under a blanket of nitrogen was charged 13.75 kg acetonitrile, then 2.68 Kg Ν,Ν,Ν’,Ν’-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and rinsed with 2.0 Kg acetonitrile. 2.03 Kg N-methylimidazole was added followed by 1.95 Kg acetonitrile. 2.48 Kg (R)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanoic acid was added followed by 1.05 Kg acetonitrile. The mixture was held for 0.5 h then 1.21 Kg 4-chloroaniline charged followed by 1.0 Kg acetonitrile. The mixture was maintained at 20 °C until the reaction was deemed complete by HPLC analysis. The solution was then heated to 60°C, and 9.25 Kg water was charged. The solution was then cooled to 40 °C, the mixture was aged
for 1 h, seeds (32 g) were charged and rinsed with 1.15 Kg 2: 1 water: acetonitrile, and the resulting slurry was maintained for 1 h. The slurry was then cooled to 20 °C and 25.75 Kg water was charged. The slurry was filtered and the cake was washed three times with 6.9 Kg of 2: 1 water: acetonitrile. The cake was dried under vacuum at 50°C to yield 3.33 Kg of (R)-N-(4-chlorophenyl)-2-((ls,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide hydrate as a white solid in 94.1% yield.
[00253] ¾ NMR (600 MHz, DMSO-de) δ 10.09 (s, 1H), 8.86 (d, J=4.5 Hz, 1H), 8.08 (dd, J=9.0, 5.6 Hz, 1H), 7.95 (dd, J=10.9, 2.6 Hz, 1H), 7.70 – 7.60 (m, 3H), 7.54 (d, J=4.5 Hz, 1H), 7.33 (d, J=9.0 Hz, 2H), 3.43 – 3.31 (m, 3H), 2.90 – 2.80 (m, 1H), 1.99 – 1.55 (m, 9H), 1.13 (d, J=6.8 Hz, 3H); 13C NMR (151 MHz, DMSO-de) δ 175.0, 159.9, 152.4, 149.7, 145.2, 138.1, 132.7, 128.5, 127.2, 126.7, 120.8, 119.0, 118.6, 107.2, 40.2, 37.4, 35.6, 28.5, 27.6, 27.4, 26.3, 16.1 ; HRMS (ESI); calcd for C24H24CIFN2O ([M + H]+), 411.1619; found 411.1649.
WO-2019006283
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006283&redirectedID=true
Novel crystalline forms of linrodostat , its salts and hydrates, designated as Forms 1, 2 and 4 (first disclosed in WO2016073770 ), processes for their preparation and compositions comprising them are claimed. Also claims are their use for treating prostate cancer, liver cancer, brain cancer, bladder cancer, ovary cancer and breast cancer.
(R)-N-(4-chlorophenyl)-2-((l S,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanami the below structure:
[0003] Compound 1 is a potent inhibitor of indoleamine 2,3-dioxygenase (IDO; also known as IDOl), which is an IFN-γ target gene that plays a role in immunomodulation.
Compound 1 is being investigated as a treatment for cancer and other diseases. Compound 1 has been previously described in WO2016/073770.
[0004] A compound, as a free base, hydrate, solvate, or salt, can exist in amorphous form and/or one or more crystalline forms, each having different physical properties, for example, different X-ray diffraction patterns (XRPD or PXRD) and different thermal behavior. The free base, hydrate, solvate, and salt forms of a compound can also differ with respect to their individual stabilities, processing, formulation, dissolution profile, bioavailability, and the like. [0005] New forms of (R)-N-(4-chlorophenyl)-2-((l S,4S)-4-(6-fluoroquinolin-4-yl)cyclohexyl)propanamide, having desirable and beneficial chemical and physical properties are needed. There is also a need for reliable and reproducible methods for the manufacture, purification, and formulation of Compound 1 (and its hydrates, solvates, salt,, and hydrated salt forms) to facilitate commercialization. The present disclosure is directed to these, as well as other important aspects.
REFERENCES
23-Feb-2015
Bristol-Myers Squibb To Expand Its Immuno-Oncology Pipeline with Agreement to Acquire Flexus Biosciences, Inc
Bristol-Myers Squibb Co; Flexus Biosciences Inc
17-Dec-2014
Flexus Biosciences, a Cancer Immunotherapy Company Focused on Agents for the Reversal of Tumor Immunosuppression (ARTIS), Announces $38M Financing
Flexus Biosciences Inc
2015106thApril 21Abs 4290
Potent and selective next generation inhibitors of indoleamine-2,3-dioxygenase (IDO1) for the treatment of cancer
American Association for Cancer Research Annual Meeting
Jay P. Powers, Matthew J. Walters, Rajkumar Noubade, Stephen W. Young, Lisa Marshall, Jan Melom, Adam Park, Nick Shah, Pia Bjork, Jordan S. Fridman, Hilary P. Beck, David Chian, Jenny V. McKinnell, Maksim Osipov, Maureen K. Reilly, Hunter P. Shunatona, James R. Walker, Mikhail Zibinsky, Juan C. Jaen
2017108thApril 04Abs 4964
Structure, in vitro biology and in vivo pharmacodynamic characterization of a novel clinical IDO1 inhibitor
American Association for Cancer Research Annual Meeting
John T Hunt, Aaron Balog, Christine Huang, Tai-An Lin, Tai-An Lin, Derrick Maley, Johnni Gullo-Brown, Jesse Swanson, Jennifer Brown
2017253rdApril 05Abs MEDI 368
Discovery of a selective inhibitor of indoleamine-2,3-dioxygenase for use in the therapy of cancer
American Chemical Society National Meeting and Exposition
Aaron Balog
April 2-62017
American Chemical Society – 253rd National Meeting and Exhibition (Part IV) – OVERNIGHT REPORT, San Francisco, CA, USA
Casellas J, Carceller V
////////////////PHASE 1, BMS 986205, 1923833-60-6, BMS-986205, ONO-7701,Bristol-Myers Squibb, Antineoplastics, F- 001287
C[C@H]([C@H]1CC[C@@H](C2=CC=NC3=CC=C(F)C=C23)CC1)C(NC4=CC=C(Cl)C=C4)=O
Wrapping up #MEDI‘s 1st time disclosures is Aaron Balog of @bmsnews talking about an IOD-1 inhibitor to treat cancer #ACSSanFran
////////////////BMS986205, BMS 986205, BM-986205, ONO-7701, Phase III, Head and neck cancer, Malignant melanoma, 1923833-60-6, Linrodostat
CC(C1CCC(CC1)C2=C3C=C(C=CC3=NC=C2)F)C(=O)NC4=CC=C(C=C4)Cl
YINLITINIB



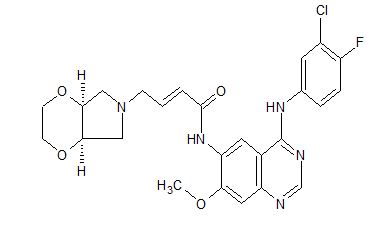
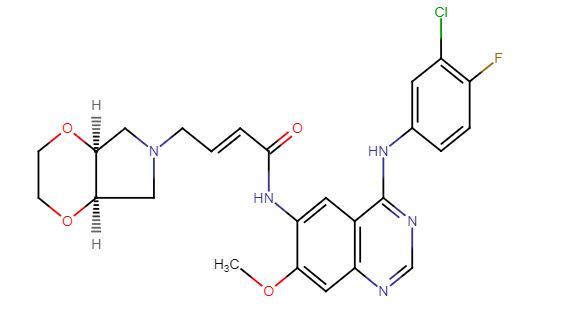
YINLITINIB
error EMAIL ME amcrasto@gmail.com
(E)-4-[(4aR,7aS)-2,3,4a,5,7,7a-hexahydro-[1,4]dioxino[2,3-c]pyrrol-6-yl]-N-[4-(3-chloro-4-fluoroanilino)-7-methoxyquinazolin-6-yl]but-2-enamide
(E)-N-(4-((3-Chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
CAS 1637253-79-2
2-Butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-methoxy-6-quinazolinyl]-4-[(4aR,7aS)-hexahydro-6H-1,4-dioxino[2,3-c]pyrrol-6-yl]-, (2E)-rel–
- C25 H25 Cl F N5 O4, 513.95
DNT-04110 ; yinlitinib maleate , Guangdong Hec Pharmaceutical
Use for treating proliferative diseases, atherosclerosis and pulmonary fibrosis
Phase I CHINA
NOTE AND USE YOUR JUDGMENT ON DRUG SUBSTANCE, EMAIL ME amcrasto@gmail.com
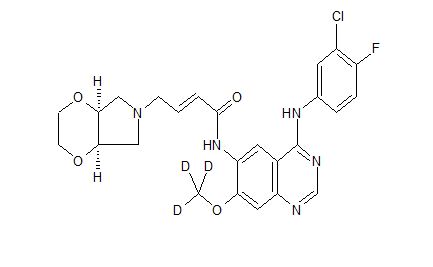
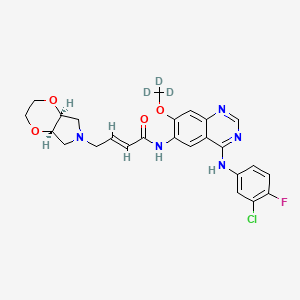
| Molecular Formula: | C25H25ClFN5O4 |
|---|---|
| Molecular Weight: | 516.973 g/mol |
Yinlitinib methoxy-d3
CAS 1637254-71-7
C25 H22 Cl D3 F N5 O4
2-Butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-(methoxy-d3)-6-quinazolinyl]-4-[(4aR,7aS)-hexahydro-6H-1,4-dioxino[2,3-c]pyrrol-6-yl]-, (2E)-rel–
CN 104119350
YINLITINIB MALEATE methoxy-d3
CAS ?
EMAIL ME amcrasto@gmail.com
MAY BE DRUG COMD
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9556191 | AMINOQUINAZOLINE DERIVATIVES AND THEIR SALTS AND METHODS OF USE THEREOF |
2014-04-28
|
2016-02-11
|
In March 2015, an IND was filed in China ; in February 2016, approval to conduct a clinical trial was obtained
Guangdong Hec Pharmaceutical is investigating an oral capsule formulation of yinlitinib maleate (DNT-04110), an irreversible pan-ErbB inhibitor, for the potential treatment of solid tumors . In March 2015, an IND was filed in China ; in February 2016, approval to conduct a clinical trial was obtained . In December 2016, a phase I trial was planned in China
Protein kinases (PKs) represent a large family of proteins, which play an important role in the regulation of a wide variety of cellular processes and maintaining control over cellular functions. There are two classes of protein kinases (PKs): the protein tyrosine kinases (PTKs) and the serine-threonine kinases (STKs). The protein tyrosine kinase is an enzyme that catalytically transfers the phosphate group from ATP to the tyrosine residue located at the protein substrate, and has a play in the normal cell growth. Many growth factor receptor proteins operate via the tyrosine kinase, and influence the conduction of signal passage and further regulate the cell growth by this process. However, in some circumstances, these receptors become abnormal due to either mutation or overexpression, which cause the uncontrolled cell multiplication, cause the tumor growth, and finally initiate the well-known disease, i.e., cancer. The growth factor receptor protein tyrosine kinase inhibitor, via the inhibition of the above phosphorylation process, may treat cancers and other diseases characterized by the uncontrolled or abnormal cell growth.
Epidermal growth factor receptor (EGFR), a kind of receptor tyrosine kinases, is a multifunction glycoprotein that is widely distributed on the cell membranes of the tissues of the human body, and is an oncogene analog of avian erythroblastic leukemia viral (v-erb-b). Human EGFR/HER1/ErbB-1 and HER2 (human epidermal growth factor receptor-2)/ErbB-2/Teu/p185, HER3/ErbB-3, HER4/ErbB-4 and the like are grouped into the HER/ErbB family, and belong to protein tyrosine kinases (PTKs). They are single polypeptide chains, and each is encoded respectively by genes located on different chromosomes. EGFR and the like are expressed in the epithelia-derived tumors such as squamous cell carcinoma of head and neck, mammary cancer, rectal cancer, ovarian cancer, prostate carcinoma, non-small cell lung cancer, and the like, which are associated with cell proliferation, metastasis, and the like. Pan-HER tyrosine kinase inhibitor, via the competitive binding to the kinase catalytic sites in the intracellular region against ATP, blocks the autophosphorylation of intramolecular tyrosine, blocks the tyrosine kinase activation, inhibits HER-2 family activation, and therefore inhibits cell cycle progression, accelerates cell apoptosis, and exerts the therapeutic action.
EGFR, after binding to the ligand, forms a dimer with a subgroup of HER family, and then combines with ATP to activate the tyrosine kinase activity of the EGFR itself. Therefore, the autophosphorylation occurs in several tyrosine sites of the intracellular kinase region. Pan-HER tyrosine kinase inhibitor, via simultaneity acting on EGFR and HER2/4, inhibits the activation of HER family, and plays a good role in the tumor growth inhibition.
It is indicated in the study that Pan-HER tyrosine kinase irreversible inhibitor has an inhibition effect on HER2/4, besides it effectively inhibits EGFR. The pharmaceutical drugs of this kind, having an irreversible inhibition to both of HER/ErbB families, not only increase the drug activity, but also reduce the drug resistance, and have a substantial inhibition effect on H1975 cell lines which are resistant to erlotinib.
The pharmaceutical drugs that are now commercially available include selective EGFR tyrosine kinase inhibitor gefitinb (IRESSA®, ZD1839), erlotinib (TARCEVA®, OSI-774), double EGFR/HER2 inhibitor Lapatinib (TYKERB®, GW572016), and the like. These three drugs are all reversible EGF receptor tyrosine phosphorylation kinase inhibitor. It has been found in the study that they have good therapeutic response to some tumors initially. However, several months after the treatment, the disease progression appears again and therefore a natural or secondary drug resistance forms. For example, about half of the patients administered with gefitinib or erlotinib develop resistance to gefitinib or erlotinib, which can not lead to the desired therapeutic effect. And it has been indicated by study that the development of drug resistance to selective EGFR tyrosine kinase inhibitor relates to mutations in EGFR.
The mutations of EGFR gene mostly located in the tyrosing kinase coding domain (TK, exons 18-21) are mainly deletion mutation in exon 19 and point mutation in exon 21, both of which are drug-sensitive, and few are point mutation in exon 18 and insertion mutation in exon 20. T790M mutation recognized as one of the mechanism of drug resistance is a point mutation in exon 20 of EGFR. The presence of a second-site EGFR mutation leads to the substitution of methionine for threonine at position 790 (T790M) and changes in the structure of EGFR, which hinder the binding of EGFR inhibitors to EGFR or greatly increase the affinity between EGFR and ATP, so that ATP affinity back to the level of wild-type EGFR, thus resulting in drug resistance. Further studies shows that the pre-treatment tumor samples with mutations of EGFR contain T790M mutation, which indicates that T790M mutation is not just associated with drug resistance and it may have the carcinogenic potential itself.
Irreversible inhibitor can bind to EGFR tyrosine kinase by covalent bond. Thus, the drugs can act on the entire link of epidermal growth factor signal transduction pathway, and improve efficiency of drug blocking. Many clinical studies show that some irreversible inhibitors in current development can against T790M mutation, and overcome the drug resistance caused by T790M. Meanwhile, listed drug Afatinib (BIBW 2992) and some irreversible inhibitors in clinical development (e.g., Dacomitinib, PF00299804, etc.), can inhibit multiple members of EGFR receptor family, especially to the role of EGFR and HER-2, possibly by blocking collaborative signal pathway activated by homodimer and heterodimer to enhance inhibitory effect (Oncologist, 2009, 14 (11): 1116-1130).
Upon developing the drug having an excellent antineoplastic effect, being able to reduce the drug resistance and having a good tolerance, the present inventors discover a quinazoline derivatives as tyrosine kinase inhibitors having a Pan-HER irreversible inhibition function.
PATENT
https://patents.google.com/patent/US9556191
EXAMPLES Example 1 (E)-N-(4-((3-Chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
Step 1) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine
A solution of N-(3-chloro-4-fluorophenyl)-7-fluoro-6-nitroquinazolin-4-amine (10.00 g, 29.8 mmol) and sodium methanolate (2.80 g, 51.8 mmol) in methanol (150 mL) was heated to 70° C. and stirred for 4.0 hours. The reaction mixture was then cooled to 25° C. The resulting mixture was poured into ice water (500 mL), and a yellow solid precipitated out. The mixture was filtered and the filter cake was dried under vacuum to give the title compound as a yellow solid (9.00 g, 86.9%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 349.1 [M+1]+; and 1H NMR (400 MHz, DMSO-d6) δ: 11.60 (s, 1H), 9.55 (s, 1H), 8.08 (dd, J1=6.6 Hz, J2=2.4 Hz, 1H), 7.90 (s, 1H), 7.76-7.71 (m, 1H), 7.58 (s, 1H), 7.55 (t, J=9.4 Hz, 1H), 4.10 (s, 3H).
Step 2) N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine
To a solution of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine (9.00 g, 25.9 mmol) in ethanol (100 mL) were added iron powder (14.50 g, 259.0 mmol) and concentrated hydrochloric acid (3.0 mL) at 25° C. The reaction mixture was heated to 90° C. and stirred for 3.0 hours. Then heating was stopped, and the resulting mixture was adjusted to pH 11 with aqueous sodium hydroxide solution (1 M) while the mixture was still at a temperature of about 60±10° C. The pH-adjusted resulting mixture was then immediately filtered hot to remove iron mud. The filtrate was concentrated in vacuo. The residue was triturated with ethanol (50 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a yellow solid (6.00 g, 73.0%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 319.1 [M+1]+.
Step 3) (E)-4-bromobut-2-enoyl chloride
To a solution of 4-bromocrotonic acid (2.47 g, 15.0 mmol) and DMF (0.05 mL) in DCM (60 mL) was added oxalyl chloride (4.19 g, 33.0 mmol) dropwise at 0° C. The reaction mixture was stirred at 0° C. for 3.0 hours, and then concentrated in vacuo. The residue was stored in a refrigerator for the next step.
Step 4) (E)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide
To a solution of N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine (4.00 g, 12.6 mmol) and TEA (6.0 mL, 37.8 mmol) in anhydrous tetrahydrofuran (80 mL) was added (E)-4-bromobut-2-enoyl chloride (2.74 g, 15.1 mmol) slowly at 0° C. The reaction mixture was then heated to 25° C. and stirred for 2.0 hours. The resulting mixture was poured into water (100 mL) and extracted with DCM (50 mL×3). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was triturated with DCM (30 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a brownish yellow solid (2.00 g, 34.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 465.1 [M+1]+.
Step 5) (E)-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4-((4aR,7aS)-tetrahydro-2H-[1,4]dioxin[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
To a solution of (E)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide (0.50 g, 1.1 mmol) and diisopropylethylamine (0.6 mL, 3.2 mmol) in N,N-dimethylacetamide (10 mL) was added (4aR,7aS)-hexahydro-2H-[1,4]dioxino[2,3-c]pyrrole (0.42 g, 3.2 mmol) at 25° C., and the reaction mixture was then stirred at 25° C. for 5.0 hours. The resulting mixture was poured into water (70 mL) and extracted with DCM (40 mL×3). The combined organic phases were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH (v/v)=20/1) to give the title compound as a brownish yellow solid (0.30 g, 54.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z: 514.1 [M+1]+; and 1H NMR (400 MHz, DMSO-d6) δ: 10.60 (s, 1H), 9.35 (s, 1H), 8.90 (s, 1H), 8.08 (dd, J1=6.6 Hz, J2=2.4 Hz, 1H), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J=8.4 Hz, 1H), 6.75-6.65 (m, 1H), 6.63 (d, J=16.2 Hz, 1H), 4.10 (s, 3H), 3.78 (t, J=6.2 Hz, 4H), 3.26 (t, J=4.4 Hz, 2H), 3.20 (dd, J1=7.8 Hz, J2=2.6 Hz, 2H), 2.20 (d, J=4.6 Hz, 4H).
PATENT
WO2017067447
DIFFERENT COMPD
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017067447
claiming novel crystalline polymorphic forms of a similar EGFR, useful for treating cancer. One of these two compounds is probably yinlitinib maleate , an irreversible pan-ErbB inhibitor, being developed by Guangdong Hec Pharmaceutical , another subsidiary of HEC Pharm , for treating solid tumors; in April 2017, yinlitinib maleate was reported to be in preclinical development
Chinese patent CN 103102344 A (publication number) have disclosed the structure of 4- [ (3-chloro-4-fluorophenyl) amino] -7-methoxy-6- [3- [ (1R, 6S) -2, 5-dioxa-8-azabicyclo [4.3.0] nonan-8-yl] propoxy] quinazoline in example 6 of specification, page 57, and the structure is shown as Formula (II) . The compound of Formula (II) has a high inhibition activity against EGFR, and can be used for treating proliferative disorders.
PATENT
https://patents.google.com/patent/WO2014177038A1/en
InventorYingjun ZhangBing LiuJinlei LiuJiancun ZhangChangchun Zheng
Original AssigneeSunshine Lake Pharma Co., Ltd.
PATENT
Inventor张英俊刘兵刘金雷张健存郑常春 Original Assignee广东东阳光药业有限公司
https://patents.google.com/patent/CN104119350B/en




Example 1
[0442] (E) -N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) -4- ((4aR, 7aS) – tetrahydro _2H_ [1,4] dioxin burning and [2,3_c] R ratio slightly -6 (3H) – yl) butyric acid amide dilute _2_
[0443]
[0444] Synthesis Step Shu: N- (3- chloro-4-fluorophenyl) -7-methoxy-6-nitro quinazolin-4-amine
[0445] The N- (3- chloro-4-fluorophenyl) -7-fluoro-6-nitro-quinazolin-4-amine (10 • 0g, 29 • 8mmol) and sodium methoxide (2.80g, 51.8 mmol) was dissolved in methanol (150 mL), the reaction was warmed to 70 ° C 4. Oh. Was cooled to 25 ° C, the reaction mixture was poured into ice-water (500 mL), the precipitated yellow solid was filtered, the filter cake was dried in vacuo to give a yellow solid 9.00g, yield 86.9%.
[0446] MS (. ESI, pos ion) m / z: 349.1 [M + l] +;
[0447] bandit R (400MHz, DMS〇-d6) S: 11 • 60 (s, 1H), 9 • 55 (s, 1H), 8 • 08 (dd, Ji = 6 • 6Hz, J2 = 2.4Hz, lH), 7.90 (s, lH), 7.76-7.71 (m, lH), 7.58 (s, lH), 7.55 (t, J = 9.4Hz, 1H), 4.10 (s, 3H) square
[0448] Synthesis Step 2: n4- (3- chloro-4-fluorophenyl) -7-methoxy-quinazolin-4,6-diamine
[0449] The N- (3- chloro-4-fluorophenyl) -7-methoxy-6-nitro quinazolin-4-amine (9.00g, 25.9mmol) was dissolved in ethanol (100 mL), the was added reduced iron powder (14.5g, 259. Ommol) and concentrated hydrochloric acid (3mL) at 25 ° C, the reaction was warmed to 90 ° C 3.Oh. With 1M aqueous sodium hydroxide solution adjusted to pH 11, filtered hot to remove iron sludge, the mother liquor was concentrated and the residue was purified slurried with ethanol (50 mL), filtered, and the filter cake was dried in vacuo to a yellow solid 6.00g, yield 73.0%.
[0450] MS (ESI, pos ion.) M / z: 319.1 [M + l] + square
[0451] Synthesis Step 3: (E) -4- bromo-but-2-enoyl chloride
The [0452] square ° C Oxalyl chloride (4.19g, 33. Ommol) was slowly added dropwise to a solution containing 4-bromo crotonic acid (2.47g, 15. Ommol) and DMF (0.05mL) in dichloromethane (60 mL) solution of in 3. Oh reaction was stirred at 0 ° C. The reaction solution was concentrated, the residue was stored in a refrigerator until use.
[0453] Synthesis Step 4: (E) -4- bromo–N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) butan – 2_ dilute amide
[0454] The N4- (3- chloro-4-fluorophenyl) -7-methoxy-quinazolin-4,6-diamine (4.00g, 12.6mmol) and triethylamine (6.0mL, 37.8mmol ) was dissolved in anhydrous tetrahydro-furan in Misaki (80 mL), cooled to 0 ° C, was slowly added (E) -4- bromo-2-dilute acid chloride (2.748,15.12 dirty 〇1), warmed to 25 ° ( : 2.011 reaction the reaction mixture was poured into water (1001 ^) and extracted with methylene chloride (50mL X 3), the organic phases were combined, dried over anhydrous sodium sulfate filtered, concentrated and the residue with dichloromethane (30 mL). beating purified filtered, the filter cake was dried in vacuo 2.00g tan solid, yield 34.5%.
[0455] MS (ESI, pos ion.) M / z: 465.1 [M + l] + square
[0456] Synthesis Step 5: (E) -N- (4 _ ((3- chloro-4-fluorophenyl) amino) -7_ methoxy-quinazolin-6-yl) _4_ ((4aR, 7aS) – tetrahydro -2H- [1,4] dioxin burning and [2,3_c] P ratio slightly -6 (3H) – yl) butyric acid amide dilute _2_
[0457] The (E) -4- bromo–N- (4- ((3- chloro-4-fluorophenyl) amino) -7-methoxy-quinazolin-6-yl) but-2-ene amide (0.50g, 1.08mmol) and diisopropylethylamine (0.6mL, 3.24mmol) was dissolved in dimethylacetamide (10 mL) was added at 25 ° C (4aR, 7aS) – hexahydro–2H- [1,4] dioxane, and [2,3-c] pyrrole (0 • 42g, 3 • 24mmol) 5. Oh reaction was continued under stirring, 25 ° C. The reaction mixture was poured into water (70 mL) and extracted with methylene chloride (40mL X 3), the organic phases were combined, dried over anhydrous sodium sulfate. Filtered, concentrated and the residue purified by column chromatography (CH2Cl2 / MeOH (V / v) = 20/1), to give 0.30g tan solid, yield 54.5%.
[0458] MS (. ESI, pos ion) m / z: 514.1 [M + l] +;
[0459] XH NMR (400MHz, DMS0-d6) 8: 10.60 (s, lH), 9.35 (s, lH), 8.90 (s, lH), 8.08 (dd, Ji = 6.6Hz, J2 = 2.4Hz, 1H ), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 8.4Hz, 1H), 6.75-6.65 (m, lH), 6.63 (d, J = 16.2Hz, lH) , 4.10 (s, 3H), 3.78 (t, J = 6.2Hz, 4H), 3.26 (t, J = 4.4Hz, 2H), 3.20 (dd, Ji = 7.8Hz, J2 = 2.6Hz, 2H), 2.20 (d, J = 4.6Hz, 4H)
PATENT
Patent applications WO 2014/177038 and CN 104119350 discloses aminoquinazoline tyrosine kinase inhibitors with irreversible inhibition effect on Pan-HER, wherein the compound (E) -N- (4- (3-chloro-4-fluorophenyl) amino) -7- (methyloxy-D3) -quinazolin-6-yl) -4- ( (4aR, 7aS) -tetra hydro-2H- [l, 4] dioxino [2, 3-c] pyrrole-6 (3H) -yl) butyl-2-enamide (i.e. compound (I) ) has an excellent antitumor effect. It can reduce the generation of drug resistance and also have good tolerance.
[0011]
EXPERIMENTAL PART
[0184]
The specific synthetic method for compound (I) (E) -N- (4- (3-chloro-4-fluorophenyl) amino) -7- (methyloxy-D3) -quinazolin-6-yl) -4- ( (4aR, 7aS) -tetra hydro-2H- [l, 4] dioxino [2, 3-c] pyrrole-6 (3H) -yl) butyl-2-enamide refers to Example 20 of Patent CN 104119350 A (Application Publication No. ) .
[0185]
EXAMPLES
[0186]
Example 1
[0187]
(E) -N- (4- (3-chloro-4-fluorophenyl) amino) -7- (methyloxy-D3) -quinazolin-6-yl) -4- ( (4aR, 7aS) -t etrahydro-2H- [l, 4] dioxino [2, 3-c] pyrrole-6 (3H) -yl) butyl-2-enamide dimesylate having crystalline form A
[0188]
1. Preparation of dimesylatesulfonate having crystalline form A
[0189]
(E) -N- (4- (3-Chloro-4-fluorophenyl) amino) -7- (methyloxy-D3) -quinazolin-6-yl) -4- ( (4a R, 7aS) -tetrahydro-2H- [l, 4] dioxino [2, 3-c] pyrrole-6 (3H) -yl) butyl-2-enamide (1.032 g, 2.0 mmol) was added to acetone (80 mL) , the mixture was heated to reflux for 30 minutes and filtered. The filtrate was refluxed, and mesylate (0.481 g, 5.0 mmol) was added. The resulting mixture was refluxed overnight. A part of solvent was evaporated under reduced pressure, then the temperature of the residue was gradually cooled to room temperature and maintained at this temperature overnight. The resulting mixture was filtered with suction. The filter cake was washed with acetone and dried at 50 ℃ for 8 hours in vacuo to give a white solid (1.15 g, 81.3%) .
PATENT
Example 6
[00221] N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-(tetrahvdro-2H-n,41dioxinor2,3-clpyrrol-6(3H -vn propoxy quinazolin-4-amine

[00222] Step Ubenzyl 3,4-dihvdroxypyrrolidine-l -carboxylate

To a solution of N- carbobenzoxy-3-pyrroline ( 1.00 g, 4.92 mmol, 1.0 eq) in acetone (20 mL) was added NMO ( 1.0 g, 7.38 mmol, 1.5 eq) followed by Os04 (cat. 10 mg in 1 mL ‘PrOH). The mixture was stirred for 3 h. To this, saturated NaHS03aqueous solution (5 mL) was added, and the mixture was stirred for another 0.5 h. The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mL x 3). The combined organic phases were dried over anhydrous Na2S04 and filtered. The filtrate was concentrated in vacuo and the residue was purified by a silica gel column chromatography (EtOAc) to give the compound as colorless oil (1.16 g, 100 %).
[00223] Step 2) benzyl tetrahvdro-2H-n.41dioxino[2.3-c1pyrrole-6(3H)- carboxylate

A mixture of NaOH aqueous solution (35 w/w %, 21 mL, aq.), C1CH2CH2C1 (21 mL), benzyl 3,4-dihydroxypyrrolidine-l -carboxylate (1.16 g, 4.9 mmol, 1.0 eq) and TBAB (0.31 g, 0.98 mmol, 0.2 eq) was heated at 55 °C for 48h in a round-bottom flask. The reaction mixture was cooled to room temperature and poured into water (50 mL), extracted with EtOAc (50 mL). The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mLx3). The combined organic phases were dried over anhydrous Na2S04 and filtered. The filtrate was concentrated in vacuo and the residue was purified with a silica gel column chromatography ( 1 : 1 (v/v) PE/EtOAc) to give the product as colorless oil (0.50 g, 39 %).
[00224] Step 3) hexahvdro-2H-n.41dioxinor2.3-clpyrrole

To a solution of benzyl tetrahydro-2H-[l ,4]dioxino[2,3-c] pyrrole-6(3H)-carboxylate (0.46 g, 1 .94 mmol) in MeOH (20 mL) was added two drops of HC02H followed by 20 % Pd(OH)2 (50mg). The reaction mixture was stirred under H2 for 4h at rt and was filtered. The filtrate was concentrated in vacuo to give the crude product, which was used for the next step without further purification.
[00225] Step 4) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-(tetrahvdro-2H-n,41 dioxinor2,3-clpyrrol-6(3H) -yl)propoxy)quinazolin-4-amine

A mixture of hexahydro-2H-[ l ,4]dioxino[2,3-c]pyrrole (1.0 eq), N-(3-chloro-4-fluorophenyI)-6- (3-chloropropoxy)-7-methoxyquinazolin-4-amine (710 mg, 1.8 mmol, 0.95 eq), 2C03 (524 mg, 3.8 mmol, 2.0 eq) and KI (16 mg, 0.095 mmol, 0.05 eq) in DMF (12 mL) was heated at 60 °C for 3 h and cooled to room temperature. The reaction mixture was quenched with water (10 mL) and diluted with EtOAc (20 mL). The organic phase was separated from the mixture, and the water phase was extracted with EtOAc (20 mLx3). The combined organic phases were dried over anhydrous Na2S04 and concentrated in vacuo. The residue was purified by a silica gel column chromatography (20: 1 (v/v) CH2Cl2/CH3OH) to give the crude product, which was recrystallized from CH2C12/PE to afford the title compound as a grayish-white solid (230 mg, 25.00 %), HPLC:99.1 1 % . The compound was characterized by the following spectroscopic data: MS (ESI, pos. ion) m/z: 489.9 (M+1 );’H NMR (400 MHz, CDC13) δ: 2.09 (2H, m), 2.74 (4H, m), 2.99 (2H, dd, = 3.3, 10.4 Hz), 3.56 (2H, m), 3.80 (2H, m), 3.99 (3H, s), 4.12 (2H, t, J = 3.5 Hz), 4.22 (2H, t, J = 6.8 Hz), 7.14 (1 H, t, J = 8.8 Hz), 7.23 (1 H, s), 7.29 ( 1 H, d, J = 15.8 Hz), 7.60 (1 H, m), 7.89 (1 H, dd, J = 2.5, 6.5 Hz), 8.63 (1 H, s) ppm.
PATENT
Example 1
[00192] (^-N 4 (3-Chloro -fluorophenyl)amino)-7-methoxyquinazolin-6-yl)-4 (4aR,7a5)-tetrahydro-2H-[ l,4]dioxino[2,3-c]pyrrol-6(3H)

[00193] Step 1) N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine
A solution of N-(3-chloro-4-fluorophenyl)-7-fluoro-6-nitroquinazolin-4-amine (10.00 g, 29.8 mmol) and sodium methanolate (2.80 g, 51.8 mmol) in methanol (150 mL) was heated to 70 °C and stirred for 4.0 hours. The reaction mixture was then cooled to 25 °C. The resulting mixture was poured into ice water (500 mL), and a yellow solid precipitated out. The mixture was filtered and the filter cake was dried under vacuum to give the title compound as a yellow solid (9.00 g, 86.9%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 349.1 [M+l]+; and ‘H NMR (400 MHz, DMSO-<&) δ: 11.60 (s, 1H), 9.55 (s, 1H), 8.08 (dd, Jx = 6.6 Hz, J2 = 2.4 Hz, 1H), 7.90 (s, 1H), 7.76-7.71 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 9.4 Hz, lH ), 4.10 (s, 3H).
[00194] Step 2) N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine
To a solution of N-(3-chloro-4-fluorophenyl)-7-methoxy-6-nitroquinazolin-4-amine (9.00 g, 25.9 mmol) in ethanol (100 mL) were added iron powder (14.50 g, 259.0 mmol) and concentrated hydrochloric acid (3.0 mL) at 25 °C. The reaction mixture was heated to 90 °C and stirred for 3.0 hours. Then heating was stopped, and the resulting mixture was adjusted to pH 11 with aqueous sodium hydroxide solution (1 M) while the mixture was still at a temperature of about 60 ± 10 °C. The pH-adjusted resulting mixture was then immediately filtered hot to remove iron mud. The filtrate was concentrated in vacuo. The residue was triturated with ethanol (50 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a yellow solid (6.00 g, 73.0%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 319.1 [M+l]+.
[00195] Step 3) (£)-4-bromobut-2-enoyl chloride
To a solution of 4-bromocrotonic acid (2.47 g, 15.0 mmol) and DMF (0.05 mL) in DCM (60 mL) was added oxalyl chloride (4.19 g, 33.0 mmol) dropwise at 0 °C. The reaction mixture was stirred at 0 °C for 3.0 hours, and then concentrated in vacuo. The residue was stored in a refrigerator for the next step.
[00196] Step 4) (ii)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide
To a solution of N4-(3-chloro-4-fluorophenyl)-7-methoxyquinazoline-4,6-diamine (4.00 g, 12.6 mmol) and TEA (6.0 mL, 37.8 mmol) in anhydrous tetrahydrofuran (80 mL) was added (E)-4-bromobut-2-enoyl chloride (2.74 g, 15.1 mmol) slowly at 0 °C. The reaction mixture was then heated to 25 °C and stirred for 2.0 hours. The resulting mixture was poured into water (100 mL) and extracted with DCM (50 mL x 3). The combined organic phases were dried over anhydrous NaaSOzi, filtered and concentrated in vacuo. The residue was triturated with DCM (30 mL) and filtered. The filter cake was dried under vacuum to give the title compound as a brownish yellow solid (2.00 g, 34.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 465.1 [M+l]+.
[00197] Step 5) (^-N 4 (3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl) (4aR,7aS)-tetrahydro-2H-[l,4]dioxino[2,3-c]pyrrol-6(3H)-yl)but-2-enamide
To a solution of (iT)-4-bromo-N-(4-((3-chloro-4-fluorophenyl)amino)-7-methoxyquinazolin-6-yl)but-2-enamide (0.50 g, 1.1 mmol) and diisopropylethylamine (0.6 mL, 3.2 mmol) in N^V-dimethylacetamide (10 mL) was added (4aR,7aS)-hexahydro-2H-[l,4]dioxino[2,3-c]pyrrole (0.42 g, 3.2 mmol) at 25 °C, and the reaction mixture was then stirred at 25 °C for 5.0 hours. The resulting mixture was poured into water (70 mL) and extracted with DCM (40 mL x 3). The combined organic phases were dried over anhydrous Na2S04, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2 MeOH (v/v) = 20/1) to give the title compound as a brownish yellow solid (0.30 g, 54.5%). The compound was characterized by the following spectroscopic data: MS (ESI, pos.ion) m/z : 514.1 [M+l]+; and lH NMR (400 MHz, DMSO-t/tf) δ: 10.60 (s, 1H), 9.35 (s, 1H) , 8.90 (s, 1H), 8.08 (dd, Jx = 6.6 Hz, J2 = 2.4 Hz, 1H), 7.76-7.70 (m, 1H), 7.58 (s, 1H), 7.55 (t, J = 8.4 Hz, 1H ), 6.75-6.65 (m, 1H), 6.63(d, J = 16.2 Hz, 1H), 4.10 (s, 3H), 3.78 (t, J= 6.2 Hz, 4H), 3.26 (t, J = 4.4 Hz, 2H), 3.20 (dd, Jx = 7.8 Hz, J2 = 2.6 Hz, 2H), 2.20 (d, J= 4.6 Hz, 4H).
////////////DNT-04110, yinlitinib maleate , Guangdong Hec Pharmaceutical, PHASE 1, CHINA, yinlitinib
Fc1ccc(cc1Cl)Nc2ncnc3cc(OC)c(cc23)NC(=O)/C=C/CN4C[C@H]5OCCO[C@H]5C4
Fc1ccc(cc1Cl)Nc2ncnc3cc(OC([2H])([2H])[2H])c(cc23)NC(=O)/C=C/CN4C[C@H]5OCCO[C@H]5C4
SIMILAR COMPDS
|
1
|
MW: 485.9445 – |
|
|
2
|
MW: 558.8663 |
|
|
3
|
MW: 469.9455 |
|
|
4
|
MW: 485.9445 |
|
|
5
|
MW: 485.9445 |
PF-06409577

PF-06409577
6-Chloro-5-[4-(1-hydroxycyclobutyl)phenyl]-1H-indole-3-carboxylic acid
CAS Number 1467057-23-3, C19H16ClNO3, 341.79
Biochem/physiol Actions
PF-06409577 is a potent and selective activator of 5′ adenosine monophosphate-activated protein kinase (AMPK).
PF-06409577 potently activates a1β1γ1 AMPK (5′ adenosine monophosphate-activated protein kinase) isoform, and prevents its dephosphorylation. It is similarly potent for β1 containing isoforms, but shows significantly lower potency for β2-containing isoforms of AMPK. Patch-clamp assays show that this compound does not inhibit hERG (human ether-a-go-go gene). It interacts with the allosteric drug and metabolite site (ADaM) of AMPK.
General description
PF-06409577 is a 6-chloro-indole derivative obtained from 5-bromo-6-chloro-indole.
PF-06409577 is a potent and selective activator of 5′ adenosine monophosphate-activated protein kinase (AMPK) for the Potential Treatment of diabetic nephropathy. PF-06409577 has AMPK α1β1γ1 Kd=9.0 nM. AMPK α1β1γ1 EC50 = 7.0 nM; AMPK α1β2γ1 EC50 > 40000 nM. PF-06409577 showed efficacy in a preclinical model of diabetic nephropathy. Upon the basis of its potent and selective AMPK activation, low metabolic turnover in human hepatocytes, clean off-target profile, and favorable preclinical in vivo efficacy results, PF-06409577 was profiled in regulatory toxicology studies and was subsequently advanced to clinical trials to assess human pharmacokinetics and safety/ tolerability.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by high levels of blood glucose resulting from defects in insulin production, insulin action, or both. Two major forms of diabetes are recognized, type I and type II. Type I diabetes develops when the body’s immune system destroys pancreatic beta cells, the only cells in the body that make the hormone insulin that regulates blood glucose. To survive, people with type 1 diabetes must have insulin delivered by injection or a pump. Type II diabetes accounts for about 90 to 95 percent of all diagnosed cases of diabetes. Type II diabetes usually begins as insulin resistance, a disorder in which the cells do not use insulin properly. Key target tissues, including liver, muscle, and adipose tissue, are resistant to the effects of insulin in stimulating glucose and lipid metabolism. As the need for insulin rises, the pancreas gradually loses its ability to produce insulin. Controlling type II diabetes with medication is essential; otherwise it can progress into pancreatic beta-cell failure requiring complete dependence on insulin.
Obesity increases the risk of type II diabetes as well as many other health conditions including coronary heart disease, stroke, and high blood pressure. More than one-third of U.S. adults (over 72 million people) and 17% of U.S. children are obese. During 1980-2008, obesity rates doubled for adults and tripled for children. During the past several decades, obesity rates for all population groups— regardless of age, sex, race, ethnicity, socioeconomic status, education level, or geographic region— have increased markedly.
Research has identified the enzyme 5′ adenosine monophosphate-activated protein kinase (AMPK) as a regulator of cellular and whole-body energy homeostasis. AMPK is activated by cellular stress resulting in downstream events that serve to conserve or generate ATP. AMPK is composed of three distinct subunits, each with multiple isoforms: the alpha subunit (alpha 1 or 2); the beta subunit (beta 1 or 2); and the gamma subunit (gamma 1, 2, or 3); for a total of twelve possible heterotrimeric isoforms.
In the liver, activated AMPK phosphorylates a variety of substrates including 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (Clarke, P.R. & Hardie, D.G., EMBO J 9, 2439-2446 (1990)) and acetyl-CoA carboxylase (Carling, D. et al. FEBS Letters 223, 217-222 (1987)) which inhibits cholesterol biosynthesis and decreases fatty acid synthesis, respectively. Therefore, activation of AMPK should lead to decreases in the levels of triglycerides and cholesterol. AMPK is also thought to regulate plasma glucose levels by decreasing hepatic gluconeogenesis through downregulation of key gene products following phosphorylation of CRTC2 (Koo S.H. et. AL, Nature 437, 1109-1111 (2005)). In muscle and myocardial tissues, AMPK activates the transport activity of glucose transporter 4 (GLUT4) increasing glucose uptake into cells thereby producing an additional avenue for decreasing plasma glucose (Kurth-Kraczek, E.J. et. al., Diabetes 48, 1667-1671 (1999)). AMPK activation has also been shown to enhance mitochondrial biogenesis improving fatty acid oxidation and decreasing circulating lipids (Merrill, G.M. et. al., Am. J. Physiol. 273, E1107-E1112 (1997)). Direct activation of AMPK using AICAR (5-aminoimidazole-4-carboxamide riboside) has been shown to lead to beneficial effects on several metabolic endpoints including improved glucose disposal, decreased hepatic glucose output and decreases in plasma triglycerides and free fatty acids (Song, X.M. et. al., Diabetologia 45, 56-65 (2002); Bergeron, R. et. al., Diabetes 50, 1076-1082 (2001); Buhl, E.S.et. al., Diabetes 50, 12-17 (2001); Iglesias, M.A. et. al., Diabetes 51, 2886-2894 (2002), Fogarty, S. & Hardie, D.G., Biochim et Biophys Acta 1804, 581-591 (2010)). Because of AMPK’s pluripotent effects on carbohydrate, lipid, and cholesterol metabolism and biosynthesis, agents that activate AMPK are attractive therapeutic targets for treating metabolic syndrome disorders such as diabetes, obesity, and dyslipidemia.
Decreases in renal AMPK activation have been implicated in the etiology of diseases of the kidney, including diabetic nephropathy, acute kidney injury (AKI), and polycystic kidney disease (PKD); activation of AMPK through hormonal (adiponectin) or pharmacological (AICAR) mechanisms has been shown to be protective in rodent models of these diseases. In diabetic nephropathy decreased AMPK activation in podocytes occurs early in the disease and is associated with increased expression of the NADPH-Oxidase protein Nox4 and increased proteinuria. These effects were reduced following administration of the AMPK activators AICAR, metformin, and Adiponectin (Lee, MJ. et.al. American Journal of Physiology – Renal Physiology. 292.
F617-F627 (2007); Sharma, K. et.al. Journal of Clinical Investigation.118. 1645-1656. (2008)). In ischemia/reperfusion models of AKI the AMPK activators metformin and AICAR were shown to dose-dependently reduce subsequent proteinuria, oxidative tissue damage, and kidney macrophage infiltration (Lempiainen, J. et.al. British Journal of Pharmacology 166. 1905-1915 (2012); Seo-Mayer, P.W. et.al. American Journal of Physiology – Renal Physiology, 301, F1346-F1357 (2011)). In two rodent models of PKD the AMPK activator metformin was shown to reduce renal cyst expansion (Takiar, V. et. al. PNAS 108, 2462-2467 (2011)). These studies suggest a broad benefit of AMPK activators in multiple renal diseases.
The compounds of the present invention activate AMPK and are, therefore, useful in treating metabolic disorders such as diabetes, obesity, and dyslipidemia as well as the renal diseases chronic kidney disease, diabetic nephropathy, acute kidney injury and polycystic kidney disease.
PATENT
US 20130267493
WO 2014140704
Example 5
6-Chloro-5-(4-(3-hydroxyoxetan-3-yl)phenyl)-1H-indole-3-carboxylic acid

Step 1
6-chloro-5-(4-(3-hydroxyoxetan-3-yl)phenyl)-1H-indole-3-carbaldehyde
A mixture of 5,5,5′,5′-tetramethyl-[2,2′]bi[[1,3,2]dioxaborinanyl] (149.0 mg, 0.44 mmol), oven dried potassium acetate (173.0 mg, 1.75 mmol) and 3-(4-bromo-phenyl)-oxetan-3-ol (100.0 mg, 0.44 mmol) in 1,4-dioxane (2 mL) was degassed with N2 for 5 minutes, treated with [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (33.0 mg, 0.044 mmol) and subjected to microwave irradiation at 110 °C for 1 hour. The cooled reaction mixture was filtered through celite and concentrated in vacuo to give a black oil. To the dark oil was added 5-bromo-6-chloro-1H-indole-3-carbaldehyde (112.0 mg, 0.43 mmol), 2 N aqueous potassium carbonate (0.4 mL, 0.80 mmol), toluene (1.5 mL) and EtOH (0.5 mL). The reaction mixture was degassed with N2 for 10 minutes, treated with [1, 1′-bis(diphenylphosphino)ferrocene] dichloropalladium(II) (25.0 mg, 0.034 mmol), and heated in a pressure tube to 110 °C for 2 hours. The cooled reaction mixture was purified by flash chromatography (33-100% EtOAc/ heptanes) to give a solid. The solid was triturated in MeOH and filtered to afford the title compound (50 mg, 35%) as a yellow solid. MS (ES+) 328.0 (M+H)+. 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1 H), 9.92 (s, 1 H), 8.35 (s, 1 H), 8.02 (s, 1 H), 7.66 (d, J = 9.4 Hz, 2 H), 7.44 (d, J = 8.2 Hz, 2 H), 6.36 (s, 1 H), 4.80 – 4.76 (m, 2 H), 4.75 – 4.71 (m, 2 H).
Step 2
6-Chloro-5-(4-(3-hydroxyoxetan-3-yl)phenyl)-1 H-indole-3-carboxylic acid To the mixture of 6-chloro-5-[4-(3-hydroxy-oxetan-3-yl)-phenyl]-1H-indole-3-carbaldehyde (50.0 mg, 0.15 mmol) in MeCN (2 mL) was added 2-methyl-2-butene (2.0 mL, 13.7 mmol), followed by sodium chlorite (138 mg, 1.53 mmol) and sodium phosphate monobasic hydrate (211.0 mg, 1.53 mmol) in water (1 mL). The reaction mixture was stirred at room temperature for 20 hours, and concentrated in vacuo. The residue was acidified with 1 N aqueous citric acid (1 mL) and extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated in vacuo. The crude material was purified by flash chromatography (34-80% EtOAc/heptanes, with 0.2% formic acid modifier) to afford the title compound (18 mg, 34%) as a brown solid. MS (ES-) 342.3 (M-H)-. 1H NMR (400 MHz, CD3OD) δ 8.02 (s, 1 H), 7.98 (s, 1 H), 7.66 (d, J = 8.20 Hz, 2 H), 7.56 (s, 1 H), 7.47 (d, J = 8.20 Hz, 2 H), 4.87 – 4.80 (m, 4 H).
Paper
Discovery and Preclinical Characterization of 6-Chloro-5-[4-(1-hydroxycyclobutyl)phenyl]-1H-indole-3-carboxylic Acid (PF-06409577), a Direct Activator of Adenosine Monophosphate-activated Protein Kinase (AMPK), for the Potential Treatment of Diabetic Nephropathy. Cameron KO et al Journal of Medicinal Chemistry 59(17), 8068-8081, (2016)

Adenosine monophosphate-activated protein kinase (AMPK) is a protein kinase involved in maintaining energy homeostasis within cells. On the basis of human genetic association data, AMPK activators were pursued for the treatment of diabetic nephropathy. Identification of an indazole amide high throughput screening (HTS) hit followed by truncation to its minimal pharmacophore provided an indazole acid lead compound. Optimization of the core and aryl appendage improved oral absorption and culminated in the identification of indole acid, PF-06409577 (7). Compound 7 was advanced to first-in-human trials for the treatment of diabetic nephropathy.
Discovery and Preclinical Characterization of 6-Chloro-5-[4-(1-hydroxycyclobutyl)phenyl]-1H-indole-3-carboxylic Acid (PF-06409577), a Direct Activator of Adenosine Monophosphate-activated Protein Kinase (AMPK), for the Potential Treatment of Diabetic Nephropathy
†Cardiovascular, Metabolic and Endocrine Diseases Medicinal Chemistry, ‡Cardiovascular, Metabolic and Endocrine Diseases Research Unit, and §Pharmacokinetics, Dynamics and Metabolism, Pfizer Worldwide Research & Development, 610 Main Street, Cambridge, Massachusetts 02139, United States
∥Cardiovascular, Metabolic and Endocrine Diseases Medicinal Chemistry, ⊥Cardiovascular, Metabolic and Endocrine Diseases Research Unit, #Worldwide Medicinal Chemistry, ∇Pharmacokinetics, Dynamics and Metabolism, ○Pharmaceutical Sciences, Pfizer Worldwide Research & Development, Groton, Connecticut 06340, United States
J. Med. Chem., 2016, 59 (17), pp 8068–8081
DOI: 10.1021/acs.jmedchem.6b00866
*For K.O.C.: phone, 617-551-3234; E-mail, Kimberly.O.Cameron@pfizer.com., *For D.W.K.: E-mail, Daniel.W.Kung@pfizer.com.
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.
6-Chloro-5-[4-(1-hydroxycyclobutyl)phenyl]-1H-indole-3-carboxylic Acid (7)
7 as a crystalline off-white solid (72.4 g, 58%). The mother liquor was concentrated to ∼30% of the initial volume, and a precipitate formed. The solids were collected by filtration and were dried under vacuum to obtain an additional batch of off-white solid (14.5 g, 12%). MS (ES−) 340.3 (M – H)−. 1H NMR (400 MHz, DMSO-d6) δ 12.12 (s, 1H), 11.95 (br s, 1H), 8.09 (d, J = 2.3 Hz, 1H), 7.96 (s, 1H), 7.65 (s, 1H), 7.58 (d, J = 7.8 Hz, 2H), 7.42 (d, J = 8.2 Hz, 2H), 5.53 (s, 1H), 2.42–2.48 (m, 2H), 2.28–2.35 (m, 2H), 1.91–2.01 (m, 1H), 1.62–1.79 (m, 1H). Analytical % Calcd: C, 66.77; H, 4.72; N, 4.10. Found: C, 66.59; H, 4.56; N, 3.96. mp 220–222 °C.
PAPER
Evolution of the Synthesis of AMPK Activators for the Treatment of Diabetic Nephropathy: From Three Preclinical Candidates to the Investigational New Drug PF-06409577
Aaron C. Smith*†  , Daniel W. Kung*† , Andre Shavnya†, Thomas A. Brandt†, Philip D. Dent†, Nathan E. Genung†, Shawn Cabral†, Jane Panteleev†, Michael Herr†, Ka Ning Yip‡, Gary E. Aspnes‡, Edward L. Conn†, Matthew S. Dowling† , David J. Edmonds‡ , Ian D. Edmonds§, Dilinie P. Fernando†, Paul M. Herrinton∥, Nandell F. Keene†, Sophie Y. Lavergne†, Qifang Li†, Jana Polivkova†, Colin R. Rose†, Benjamin A. Thuma†, Michael G. Vetelino†, Guoqiang Wang†, John D. Weaver, III†, Daniel W. Widlicka†, Kristin E. Price Wiglesworth†, Jun Xiao†, Todd Zahn∥, and Yingxin Zhang†
, Daniel W. Kung*† , Andre Shavnya†, Thomas A. Brandt†, Philip D. Dent†, Nathan E. Genung†, Shawn Cabral†, Jane Panteleev†, Michael Herr†, Ka Ning Yip‡, Gary E. Aspnes‡, Edward L. Conn†, Matthew S. Dowling† , David J. Edmonds‡ , Ian D. Edmonds§, Dilinie P. Fernando†, Paul M. Herrinton∥, Nandell F. Keene†, Sophie Y. Lavergne†, Qifang Li†, Jana Polivkova†, Colin R. Rose†, Benjamin A. Thuma†, Michael G. Vetelino†, Guoqiang Wang†, John D. Weaver, III†, Daniel W. Widlicka†, Kristin E. Price Wiglesworth†, Jun Xiao†, Todd Zahn∥, and Yingxin Zhang†
, Daniel W. Kung*† , Andre Shavnya†, Thomas A. Brandt†, Philip D. Dent†, Nathan E. Genung†, Shawn Cabral†, Jane Panteleev†, Michael Herr†, Ka Ning Yip‡, Gary E. Aspnes‡, Edward L. Conn†, Matthew S. Dowling† , David J. Edmonds‡ , Ian D. Edmonds§, Dilinie P. Fernando†, Paul M. Herrinton∥, Nandell F. Keene†, Sophie Y. Lavergne†, Qifang Li†, Jana Polivkova†, Colin R. Rose†, Benjamin A. Thuma†, Michael G. Vetelino†, Guoqiang Wang†, John D. Weaver, III†, Daniel W. Widlicka†, Kristin E. Price Wiglesworth†, Jun Xiao†, Todd Zahn∥, and Yingxin Zhang†† Pfizer Worldwide Research & Development, Eastern Point Road, Groton, Connecticut 06340, United States
‡ Pfizer Worldwide Research & Development, 610 Main Street, Cambridge, Massachusetts 02139, United States
§ Bridge Organics, 311 West Washington Street, Vicksburg, Michigan 49097, United States
∥ BoroPharm, Inc., 39555 Orchard Hill Place, Suite 600, Novi, Michigan 48375, United States
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.8b00059
*E-mail for Aaron C. Smith: Aaron.Smith2@pfizer.com., *E-mail for Daniel W. Kung: Daniel.W.Kung@pfizer.com.
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00059

Indole acids 1, 2, and 3 are potent 5′-adenosine monophosphate-activated protein kinase (AMPK) activators for the potential treatment of diabetic nephropathy. Compounds 1–3 were scaled to supply material for preclinical studies, and indole 3 was selected for advancement to first-in-human clinical trials and scaled to kilogram quantities. The progression of the synthesis strategy for these AMPK activators is described, as routes were selected for efficient structure–activity relationship generation and then improved for larger scales. The developed sequences employed practical isolations of intermediates and APIs, reproducible cross-coupling, hydrolysis, and other transformations, and enhanced safety and purity profiles and led to the production of 40–50 g of 1and 2 and 2.4 kg of 3. Multiple polymorphs of 3 were observed, and conditions for the reproducible formation of crystalline material suitable for clinical development were identified.

Mp: 192–194 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.12 (s, 1H), 11.94 (br d, J = 2.2 Hz, 1H), 8.08 (d, J = 2.9 Hz, 1H), 7.95 (s, 1H), 7.64 (s, 1H), 7.57 (d, J = 8.3 Hz, 2H), 7.40 (d, J = 8.1 Hz, 2H), 5.52 (s, 1H), 2.48–2.40 (m, 2H), 2.35–2.26 (m, 2H), 2.00–1.89 (m, 1H), 1.74–1.63 (m, 1H). 13C NMR (101 MHz, DMSO-d6): δ 165.6, 146.6, 138.1, 136.0, 133.8, 133.0, 129.2, 125.6, 125.3, 124.6, 122.8, 112.9, 107.6, 75.1, 37.3, 12.8. MS (ES): calcd for C19H17ClNO3 ([M – H]−) 340.1; found 340.3. Anal. Calcd (%): C, 66.77; H, 4.72; N, 4.10. Found: C, 66.59; H, 4.71; N, 3.96.
///////////////////PF-06409577, PHASE 1
O=C(C1=CNC2=C1C=C(C3=CC=C(C4(O)CCC4)C=C3)C(Cl)=C2)O
