
Home » Posts tagged 'patent expiry'
Tag Archives: patent expiry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
GADODIAMIDE, OMNISCAN Drug Patent Expiration, 1 st oct 2013

GADODIAMIDE
GE HEALTHCARE, OMNISCAN
Drug Patent Expiration
1 st oct 2013, US5560903, CAS 122795-43-1
| GADODIAMIDE | INJECTABLE; INJECTION | 287MG/ML | RX | NDA 020123 |
Gadodiamide is a gadolinium-based MRI contrast agent, used in MR imaging procedures to assist in the visualization of blood vessels. It is commonly marketed under the trade name Omniscan.
For intravenous use in MRI to visualize lesions with abnormal vascularity (or those thought to cause abnormalities in the blood-brain barrier) in the brain (intracranial lesions), spine, and associated tissues.

Gadodiamide is a contrast medium for cranial and spinal magnetic resonance imaging (MRI) and for general MRI of the body after intravenous administration. The product provides contrast enhancement and facilitates visualisation of abnormal structures or lesions in various parts of the body including the central nervous system (CNS). It does not cross an intactblood brain barrier but might give enhancement in pathological conditions.
Based on the behavior of protons when placed in a strong magnetic field, which is interpreted and transformed into images by magnetic resonance (MR) instruments. Paramagnetic agents have unpaired electrons that generate a magnetic field about 700 times larger than the proton’s field, thus disturbing the proton’s local magnetic field. When the local magnetic field around a proton is disturbed, its relaxation process is altered. MR images are based on proton density and proton relaxation dynamics. MR instruments can record 2 different relaxation processes, the T1 (spin-lattice or longitudinal relaxation time) and the T2 (spin-spin or transverse relaxation time). In magnetic resonance imaging (MRI), visualization of normal and pathological brain tissue depends in part on variations in the radiofrequency signal intensity that occur with changes in proton density, alteration of the T1, and variation in the T2. When placed in a magnetic field, gadodiamide shortens both the T1 and the T2 relaxation times in tissues where it accumulates. At clinical doses, gadodiamide primarily affects the T1 relaxation time, thus producing an increase in signal intensity. Gadodiamide does not cross the intact blood-brain barrier; therefore, it does not accumulate in normal brain tissue or in central nervous system (CNS) lesions that have not caused an abnormal blood-brain barrier (e.g., cysts, mature post-operative scars). Abnormal vascularity or disruption of the blood-brain barrier allows accumulation of gadodiamide in lesions such as neoplasms, abscesses, and subacute infarcts.

1.Schenker MP, Solomon JA, Roberts DA. (2001). Gadolinium Arteriography Complicated by Acute Pancreatitis and Acute Renal Failure, Journal of vascular and interventional radiology 12(3):393.[1]
2 Unal O, Arslan H. (1999). Cardiac arrest caused by IV gadopentetate dimeglumine. AJR Am J Roentgenol 172:1141.[2]
3 Cacheris WP, Quay SC, Rocklage SM. (1990). The relationship between thermodynamics and the toxicity of gadolinium complexes, Magn Reson Imaging 8(6):467-81. doi:10.1016/0730-725X(90)90055-7
4 Canavese, C; Mereu, MC; Aime, S; Lazzarich, E; Fenoglio, R; Quaglia, M; Stratta, P (2008). “Gadolinium-associated nephrogenic systemic fibrosis: the need for nephrologists’ awareness”. Journal of nephrology 21 (3): 324–36. PMID 18587720.
COUNTRY PATENT APPROVED, EXPIRY
| United States | 5560903 | 1993-10-01 | 2013-10-01 |
| Canada | 1335819 | 1995-06-06 | 2012-06-06 |
| United States | 5362475 | 1994-11-08 | 2011-11-08 |
| Canada | 1335819 | 1995-06-06 | 2012-06-06 |
| United States | 5560903 | 1993-10-01 | 2013-10-01 |
Gadolinium contrast agents are used as contrast media to enhance magnetic resonance imaging as they are paramagnetic. This compound has a low incidence of adverse side effects, although there is a rare association with nephrogenic systemic fibrosis (NSF) when given to people with severe renal impairment (ie, GFRglomerular filtration rate <30mL/min/1·73m2).It seems to be related to the liberation of free gadolinium ions, and UK CHM advice is against using the least stable of the agents – Omniscan (gadodiamide) – in patients with severe renal impairment, and carefully considering whether to use others where renal function is impaired.
OMNISCAN (gadodiamide) Injection is the formulation of the gadolinium complex of diethylenetriamine pentaacetic acid bismethylamide, and is an injectable, nonionic extracellular enhancing agent for magnetic resonance imaging. OMNISCAN is administered by intravenous injection. OMNISCAN is provided as a sterile, clear, colorless to slightly yellow, aqueous solution. Each 1 mL contains 287 mg gadodiamide and 12 mg caldiamide sodium in Water for Injection.
The pH is adjusted between 5.5 and 7.0 with hydrochloric acid and/or sodium hydroxide. OMNISCAN contains no antimicrobial preservative. OMNISCAN is a 0.5 mol/L solution of aqua[5,8-bis(carboxymethyl)11-[2-(methylamino)-2-oxoethyl]-3-oxo-2,5,8,11-tetraazatridecan-13-oato (3-)-N5, N8, N11, O3, O5, O8, O11, O13] gadolinium hydrate, with a molecular weight of 573.66 (anhydrous), an empirical formula of C16H28GdN5O9•xH2O, and the following structural formula:
 |
Pertinent physicochemical data for OMNISCAN are noted below:
PARAMETER
| Osmolality (mOsmol/kg water) | @ 37°C | 789 |
| Viscosity (cP) | @ 20°C | 2 |
| @ 37°C | 1.4 | |
| Density (g/mL) | @ 25°C | 1.14 |
| Specific gravity | @ 25°C | 1.15 |
OMNISCAN has an osmolality approximately 2.8 times that of plasma at 37°C and is hypertonic under conditions of use.
gadodiamide, chemical name: [5,8 _ bis (carboxymethyl) -11 – [2_ (methylamino)-2_ ethyl] -3 – O 2 ,5,8, 11 – tetraazacyclododecane-decane -13 – oxo-(3 -)] gadolinium trihydrate. Its structure is shown in formula one.
[0003] Structural Formula:
[0004]

[0005] Magnetic resonance contrast agent gadodiamide resonance than ionic contrast agents safer generation of products, it is non-ionic structure significantly reduces the number of particles in solution, osmotic balance of body fluids is very small.Meanwhile, gadodiamide relatively low viscosity to bring the convenience of nursing staff, making it easier to bolus. In addition, gadodiamide pioneered the use of amide-substituted carboxyl part, not only reduces the toxicity of carboxyl groups and ensure the non-ionic nature of the product solution.
[0006] reported in the literature and their intermediates gadodiamide synthetic route is as follows:
[0007] 1. Compound III synthetic routes for its preparation in U.S. Patent No. US5508388 described as: In the synthesis process, the inventors using acetonitrile as solvent, acetic anhydride as dehydrating agent, pyridine as acid-binding agent, at 55 ~ 60 ° C, the reaction 18h. Anti-
See the reaction should be a process. The disadvantage of this synthesis are acetonitrile toxicity, not widely used.
[0008]

[0009] Reaction a
[0010] (2) Synthesis of Compound III in many articles are reported in the patent and its implementation method similar to the patent US5508388.
[0011] In US3660388, the diethylenetriamine pentaacetic acid (Compound II), pyridine, acetic anhydride, the mixture was reacted at 65 ° C or 20h at 125 ° C the reaction 5min, to give compound III.
[0012] In US4822594, the compounds II, pyridine, acetic anhydride mixture was reacted at 65 ° C 20h, to give compound III.
[0013] In US4698263, the compounds II, pyridine, acetic anhydride heated in a nitrogen or argon atmosphere under reflux for 18h, to give compound III. [0014] In the EPO183760B1, the compounds II, pyridine, acetic anhydride mixture was reacted at 55 ° C 24h, to give compound III.
[0015] In CN1894223A, the compounds II, pyridine, acetic anhydride, the mixture above 65 ° C the reaction mixture, and the pyridine of DTPA feed ratio is: 1: (0.5 to 3).
[0016] The above patents do not provide for the compound III is post-processing method.
[0017] 3 Synthesis of Compound IV.
[0018] In U.S. Patent US4859451, the diethylenetriamine pentaacetic acid dianhydride (compound III) and ammonia, methanol and the reaction of compounds IV, see Reaction Scheme II.
[0019]

[0020] Reaction two
[0021] In the patent US5087439, the compound III with methylamine in aqueous solution for several hours, or overnight reactions, see reaction formula III.
[0022]

[0023] Reactive three
[0024] These two patents using ammonia and methylamine, which can form explosive mixtures with air, in case of fire or high pressure can cause an explosion in the production process of great insecurity. Although raw material prices are lower, but higher production conditions (such as requiring sealed, low temperature, etc.). Compared to this synthesis process,
[0025] 4, gadodiamide (Compound I) synthesis.
[0026] In the patent US4859451, the use of gadolinium chloride with the compound IV is carried out under acidic conditions, complexing. Finally, tune
Section PH neutral, see reaction IV.
[0027]

[0028] Reaction formula tetrakis [0029] in the patent US5087439, the chlorides are used as reactants, and details of the post-processing method of Compound I.
[0030] In the patent US5508388, the use of gadolinium oxide with compound IV in acetonitrile, water with stirring, the resulting compound I.
[0032] The synthetic route is as follows:
[0033]

[0034] 1) Compound II (diethylenetriamine pentaacetic acid) in pyridine, acetic anhydride in the presence of a dehydration reaction into the acid anhydride, and the product was stirred with cold DMF, leaving the solid filtered, washed with ether reagents, drying , to obtain a white powdery solid compound III (diethylenetriamine pentaacetic acid anhydride);
[0035] 2) Compound III in DMF with methylamine hydrochloride, the reaction of the compound IV (5,8 _ bis carboxymethyl methyl-11 – [2 – (dimethylamino) -2 – oxoethyl] – 3 – oxo -2,5,8,11 – tetraazacyclododecane _13_ tridecyl acid); and the control compound III: MeNH2 · HCl molar ratio = 1: (1 to 4), control the temperature between 20 ~ 80 ° C, the reaction time is 4 ~ 6h, after the treatment, the method of distillation under reduced pressure to remove DMF, the product is dissolved in a polar solvent, methanol, and then adding a solvent polarity modulation, so that the target Compound IV from system completely precipitated;
[0036] 3) Compound IV with gadolinium oxide formed in the presence of hydrochloric acid of the complex, after the reaction, filtration and drying, to obtain a white powdery compound I, i.e. gadodiamide.
[0037] Existing gadodiamide Synthesis basically from the synthesis of Compound IV as a starting material, the present invention is first introduced to the compound II as a starting material to synthesize gadodiamide. Synthesis of the conventional method of gadodiamide, the present invention has the advantage of inexpensive starting materials, convenient and easy to get. In addition, the synthetic pathway intermediates are involved in the post-processing is simple, enabling continuous reaction, saving time and cost savings, the reaction becomes controlled step by step, and try to avoid the use of toxic reagents, reducing the possibility of operator injury , while also greatly reducing damage to the environment.
ATACAND, CANDESARTAN CILEXETIL, ASTRAZENECA.Drug Patent Expiration on 9 th Jan 2014

Candesartan cilexetil Candesartan cilexetil, Candesartan hexetil, H212/91, TCV-116, Kenzen, Blopress 16 mg Plus, Parapres, Ratacand, Blopress, Amias, Atacand
ATACAND
ATACAND (candesartan cilexetil), a prodrug, is hydrolyzed to candesartan during absorption from the gastrointestinal tract. Candesartan is a selective AT1 subtype angiotensin II receptor antagonist. Candesartan cilexetil, a nonpeptide, is chemically described as (±)-1-Hydroxyethyl 2-ethoxy-1-[p-(o-1H-tetrazol-5ylphenyl)benzyl]-7-benzimidazolecarboxylate, cyclohexyl carbonate (ester). Its empirical formula is C33H34N6O6, and its structural formula is:
 |
Candesartan cilexetil is a white to off-white powder with a molecular weight of 610.67. It is practically insoluble in water and sparingly soluble in methanol. Candesartan cilexetil is a racemic mixture containing one chiral center at the cyclohexyloxycarbonyloxy ethyl ester group. Following oral administration, candesartan cilexetil undergoes hydrolysis at the ester link to form the active drug, candesartan, which is achiral. ATACAND is available for oral use as tablets containing either 4 mg, 8 mg, 16 mg, or 32 mg of candesartan cilexetil and the following inactive ingredients: hydroxypropyl cellulose, polyethylene glycol, lactose, corn starch, carboxymethylcellulose calcium, and magnesium stearate. Ferric oxide (reddish brown) is added to the 8-mg, 16-mg, and 32-mg tablets as a colorant.
Drug Patent Expiration and Exclusivity
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| CANDESARTAN CILEXETIL | TABLET; ORAL | 4MG | RX | 020838 | 001 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 8MG | RX | 020838 | 002 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 16MG | RX | 020838 | 003 | |
| CANDESARTAN CILEXETIL | TABLET; ORAL | 32MG | RX | 020838 | 004 |
Patents
There are 6 patent(s) protecting ASTRAZENECA’s ATACAND. The last patent 5534534*PED expires on 2014-01-09.View patent at USPTO
| Patent US | US | Expiration |
|---|---|---|
| 5534534*PED | 2014-1-9 | |
| 5534534 | Pharmaceutical compositions for oral use and method of preparing them
A pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) having antagonistic action to angiotensin II ##STR1## (wherein the ring W is an optionally substituted N-containing heterocyclic residue; R.sup.3 is a group capable of forming an anion or a group convertible thereinto; X is a direct bond or a spacer having an atomic length of two or less between the phenylene group and the phenyl group; and n is an integer of 1 or 2) and an oily substance having a lower melting point, and a method for preparing a pharmaceutical composition for oral use comprising an effective amount of a compound of the formula (I) and an oily substance having a lower melting point, which comprises admixing the compound of the formula (I) with an oily substance having a lower melting point and then subjecting the mixture to molding.
|
2013-7-9(expired) |
| 5196444*PED | 2012-12-4(expired) | |
| 5196444 | 1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-c arboxylate and compositions and methods of pharmaceutical use thereof
1-(Cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-car boxylate or a pharmaceutically acceptable salt thereof has potent angiotensin II antihypertensive activity, thus being useful as therapeutic agents for treating circulatory system diseases such as hypertensive diseases, heart diseases (e.g. hypercardia, heart failure, cardiac infarction, etc.), strokes, cerebral apoplexy, nephritis, etc.
|
2012-6-4(expired) |
| 7538133*PED | 2011-10-18(expired) | |
| 5705517*PED | 2011-10-18(expired) |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ASTRAZENECA.
| Date | Supplement No. | Action | Documents |
|---|---|---|---|
| 2013-04-26 | 038 | Labeling Revision | |
| 2012-04-27 | 035 | Labeling Revision | |
| 2012-04-13 | 032 | Labeling Revision | |
| 1998-06-04 | 000 | Approval | |
| 2011-06-24 | 033 | Labeling Revision | |
| 2009-10-22 | 031 | Patient Population Altered | |
| 2006-08-17 | 026 | Labeling Revision | |
| 2005-05-18 | 022 | New or Modified Indication | |
| 2005-02-22 | 024 | New or Modified Indication | |
| 2004-12-16 | 023 | Labeling Revision | |
| 2000-06-14 | 008 | Labeling Revision | |
| 2002-09-13 | 015 | Comparative Efficacy Claim | |
| 2003-01-22 | 017 | Labeling Revision | |
| 2003-04-23 | 019 | Labeling Revision | |
| 2013-02-21 | 037 | Manufacturing Change or Addition | |
| 1999-08-11 | 005 | Package Change | |
| 2000-12-27 | 009 | Manufacturing Change or Addition | |
| 2001-05-24 | 012 | Manufacturing Change or Addition | |
| 2001-11-28 | 016 | Labeling Revision | |
| 1999-07-28 | 004 | Control Supplement | |
| 2001-04-02 | 011 | Manufacturing Change or Addition | |
| 2001-10-04 | 014 | Control Supplement | |
| 1998-11-16 | 002 | Manufacturing Change or Addition | |
| 1999-12-08 | 006 | Package Change | |
| 2001-06-07 | 010 | Manufacturing Change or Addition | |
| 2001-03-29 | 013 | Package Change | |
| 1998-12-07 | 001 | Manufacturing Change or Addition |
 Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Candesartan is marketed as the cyclohexyl 1-hydroxyethyl carbonate (cilexetil) ester, known ascandesartan cilexetil. Candesartan cilexetil is metabolised completely by esterases in theintestinal wall during absorption to the active candesartan moieity. The use of a prodrug form increases the bioavailability of candesartan. Despite this, absolute bioavailability is relatively poor at 15% (candesartan cilexetil tablets) to 40% (candesartan cilexetil solution). Its IC50 is 15 µg/kg. U.S. Patent Nos. 5,196,444 and 5,578,733 describe the removal of a trityl protecting group of the N-protected tetrazolyl compounds using methanol in the presence of a mineral acid, such as hydrochloric acid, which requires complex extractions or chromatographic purification to produce pure candesartan cilexetil. U.S. Patent No. 7,345,072 describes the deprotection of tetrazolyl compounds, including candesartan cilexetil, in the presence of an anhydrous mineral acid or aqueous mineral acid at a concentration higher than 20% w/w. The strong acidic conditions produce more decomposition products and thereby reduces the overall purity of the final product. WO 05/021535 discloses the preparation of candesartan cilexetil by the deprotection of trityl moiety at a reflux temperature in the presence of anhydrous Ci to C5 alcohol under neutral or slightly basic conditions involving longer reaction time (for e.g. stirring for several hours, such as 18-24 hours); this is followed by removal of the triphenylmethylether moiety precipitated as a solid, and thereby increases the number of reaction steps. WO 05/037821 describes the deprotection of the trityl candesartan cilexetil by the use of methane sulphonic acid, p-toluene sulphonic acid, formic and trifluoroacetic acid in solvent mixture or by refluxing candesartan cilexetil in mixture of toluene, water, and methanol. The initial product obtained by these procedures is mostly a viscous oil or a semi solid, which is difficult to handle. WO 07/074399 and WO 07/042161 disclose the preparation of candesartancilexetil from trityl candesartan cilexetil involving Lewis acids such as boron trifluoride, zinc chloride, aluminium trihalide, or titanium tetrachloride which are costly and thus are not commercially viable.
Synthesis
Candesartan is synthesised as follows:  kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
kubo, K.; Kohara, Y.; Imamiya, E.; Sugiura, Y.; Inada, Y.; Furukawa, Y.; Nishikawa, K.; Naka, T. (1993). “Nonpeptide angiotensin II receptor antagonists. Synthesis and biological activity of benzimidazolecarboxylic acids”. Journal of Medicinal Chemistry 36 (15): 2182–2195. doi:10.1021/jm00067a016. PMID 8340921. Candesartan, a blocking agent against angiotensin II receptor, has been used for years for treating high blood pressure and heart failure. Candesartan cilexetil, a prodrug of candesartan is commercially available from AstraZeneca and Takeda Pharmaceuticals Ltd. European Patent No. 0459136B1 of Takeda Chemical Industries discloses that methods for preparing candesartan cilexetil schematically represented by the following Reaction Scheme 1: Reaction Scheme 1
The method has technical problems as follows: a) the starting material is obtained by a minor reaction, b) its yield is relatively low and its industrial applicability is poor (due to N2 gas formation) because the Curtius rearrangement reaction is involved, and c) materials industrially hard to handle such as SOCI2 or NaH are used. In addition, methods for preparing an intermediate of candesartan cilexetil are disclosed in Organic Process Research & Development 11:490-493(2007), as represented by the following Reaction Scheme 2: Reaction Scheme 2

3:1 s

However, the preparation process has also shortcomings of a) undesired byproducts formed by nitrogenation at ortho- or para-position, b) safety problems from strong acids (sulfuric acid and nitric acid) used twice when introducing and rearranging nitrogen groups, and c) utilization of high-flammable Raney Ni.
cut paste
Novel and Practical Synthesis of Candesartan Cilexetil
Yongjun Mao, Ruisheng Xiong, Zheng Liu, Haihong Li, Jingkang Shen, and Jingshan Shen* *Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Rd., Zhangjiang Hi-Tech Park, Shanghai, 201203, China
Abstract
A novel and convergent synthetic route of candesartan cilexetil (API of Atacand), an effective angiotensin II receptor blocker, is described. Cleavage of the N-Boc and N-trityl protective group are implemented simultaneously and formation of the benzimidazole ring is conducted at the last step of this route, which gives candesartan cilexetil in 55% yield over six steps with 99.1% purity (HPLC). Full Text HTMLPDF (567KB)PDF with Links (932KB)
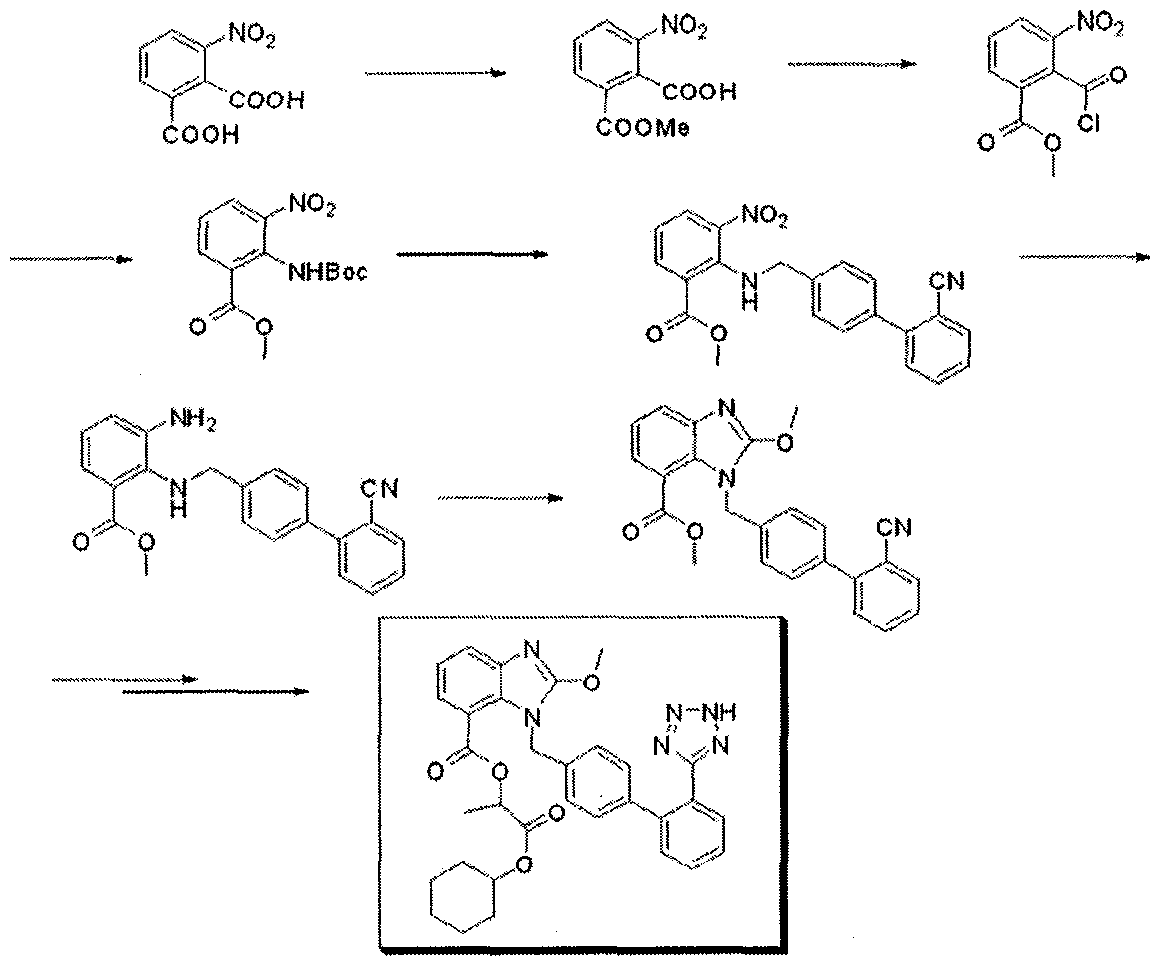
 This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
This compound can be obtained by two related ways: 1) The partial esterification of 3-nitrophthalic acid (I) with ethanol and H2SO4 gives 3-nitrophthalic acid 1-monoethyl ester (II), which is treated with SOCl2 in refluxing benzene to yield the corresponding acyl chloride (III). The reaction of (III) first with sodium azide in DMF and then with refluxing tert-butanol affords 2-(tert-butoxycarbonylamino)-3-nitrobenzoic acid ethyl ester (IV), which is condensed with 4-(2-cyanophenyl)benzyl bromide (V) by means of NaH in THF giving 2-(2′-cyanobiphenyl-4-ylmethylamino)-3-nitrobenzoic acid ethyl ester (VI). The reduction of (VI) with SnCl2.2H2O in ethanol yields the corresponding 3-amino derivative (VII), which is cyclocondensed with ethyl orthocarbonate and acetic acid affording 1-(2′-cyanobiphenyl-4-ylmethyl)-2-ethoxybenzimidazole-7-carboxylic acid ethyl ester (VIII). The reaction of (VIII) with trimethyltin azide in refluxing toluene gives the 2′-(1H-tetrazol-5-yl) derivative (IX), which is saponified with NaOH in ethanol to the corresponding free acid (X). Protection of (X) with trityl chloride and triethylamine in dichloromethane gives the protected compound (XI), which is finally esterified with cyclohexyl 1-iodoethyl carbonate (XII) by means of K2CO3 in DMF. 2) Compound (VIII) can also be obtained by reaction of 2-chloro-1-(2′-cyanobiphenyl-4-ylmethyl)benzimidazole-7-carboxylic acid ethyl ester (XIII) with sodium ethoxide in refluxing ethanol.
| Benzimidazole derivs., their production and use | |
| Naka, T.; Nishikawa, K.; Kato, T. (Takeda Chemical Industries, Ltd.) | |
| EP 0459136; EP 0720982; JP 1992364171; JP 1996099960; US 5196444; US 5328919; US 5401764; US 5703110; US 5705517; US 5962491; US 6004989 |
more info Candesartan cilexetil of Formula I, disclosed in U.S. Patent No. 5,196,444 as crystalline form, i.e., Form-I (C-type crystals), is chemically described as 1- cyclohexyloxycarbonyloxyethyl 2-ethoxy-3-[[4-[2-(2H-tetrazol-5- yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate.

H Formula I It is useful in the treatment of cardiovascular complaints such as hypertension and heart failure. Candesartan cilexetil is poorly soluble in water, which is attributed to its hydrophobic nature. Solubility plays an important role in achieving the desired concentration of a drug in systemic circulation for accomplishing the pharmacological response. Various techniques are known in literature to increase the solubility of poorly-soluble drugs, including decreasing the particle size, complexation, changing the surface characteristics of the particles, and incorporation of drug particles into colloidal systems like nanoparticles and liposomes. Among these, the most commonly used technique to increase the solubility is particle size reduction. Sometimes the rate of dissolution of a poorly-soluble drug is the rate limiting factor in its rate of absorption by the body. These drugs may be more readily bioavailable if administered in a finely divided state. Particle size reduction increases the surface area causing an increase in the dissolution rate of the compound, and hence, its bioavailability. There are certain techniques reported in literature to reduce the particle size of such poorly-soluble drugs. PCT Publication No. WO 2006/122254 discloses stable candesartan cilexetil of fine particle size, wherein the stable micronized candesartan cilexetil is prepared by slurrying a sample of candesartan cilexetil of fine particle size in a suitable solvent for a suitable amount of time. In this application, candesartan cilexetil of fine particle size is obtained directly from the synthesis of candesartan cilexetil or by comminuting candesartan cilexetil using milling. PCT Publication No. WO 2005/123720 describes fine particles of candesartan cilexetil having improved pharmacokinetic profile and a process for their production, wherein fine particle size is obtained by a) dissolving candesartan cilexetil in an organic solvent; b) cooling the solution obtained in step a) under stirring to crystallize candesartan cilexetil from the solution; and c) isolating candesartan cilexetil having a particle size of with d90 not more than about 25 μ. U.S. Patent Application No. 2006/0165806 describes compositions comprising a candesartan, such as candesartan cilexitil. The candesartan particles of the composition have an effective average particle size of less than about 2000 nm. U.S. Patent Application No. 2008/0038359 describes a nanoparticle pharmaceutical formulation comprising a poorly soluble drug substance having an average particle size of less than about 1000 nm, a solid or semisolid dispersion vehicle, and optionally a non-surface modifying excipient. U.S. Patent No. 7,828,996 discloses the methods for forming nanoparticles of a material of narrow polydispersity with ultrasonic waves using a partially submersed sonicator that does not touch any part of the apparatus and the point of addition of organic solvent is in the wave funnel produced by sonication and within the selected distance from the wave-source depending on the desired particle size. U.S. Patent No. 7,780,989 discloses the preparation of a dispersion of nanocrystalline particles in an aqueous medium using ultrasound. U.S. Patent No. 5,314,506 describes a crystallization process in which a jet of a solution containing a substance is impinged with a second jet containing an anti-solvent for the substance. The rapid mixing produced by the impinging jets results in a reduction of the crystals so formed compared to conventional slow crystallization processes. The smallest crystals disclosed are about 3 μ and the majority are in the range of from about 3 μ to about 20 μ. PCT Publication No. WO 00/44468 describes a modification to the apparatus described in U.S. Patent No. 5,314,506, wherein ultrasound energy is applied at the point of impingement of the two jets to further enhance localized mixing and is stated to give direct formation of small crystals with a diameter of less than 1 μ. Generally, the crystalline particles described have an average size of 0.5 μ. Conventional particle size reduction methods such as high energy milling may result in loss of yield, noise and dusting, as well as unwanted exposure to highly potent pharmaceutical compounds. Also, in the case of crystalline compounds, stress generated on crystal surfaces during milling can adversely affect labile compounds. Therefore, there is a need for a process for particle size reduction of candesartan cilexetil, which is industrially advantageous, easy to handle and is cost effective.
-
Candesartan is a potent, selective AT1 subtype angiotensin II receptor antagonist and used for treatment of hypertension. Due to poor absorption of Candesartan in body, the prodrug candesartan cilexetilwas developed. The candesartan cilexetil is rapidly and completely hydrolyzed to candesartan in gastrointestinal tract.
-
[0004]U.S. Pat. No. 5,196,444 discloses Candesartan cilexetil and a process for its preparation by the reaction of 2-ethoxy-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylic acid with trityl chloride in presence of triethyl amine in methylene chloride and purification by column chromatography gives 2-ethoxy-1-[[2′-(N-triphenylmethyltetrazol-5-yl)-biphenyl-4-yl]methyl] benzimidazole -7-carboxylic acid, which upon condensation with cyclohexyl 1-iodoethyl carbonate in presence of potassium carbonate in DMF followed by purification with column chromatography gives a colorless powder which is recrystallized in ethanol yields ‘C’ type crystals of Candesartancilexitil.
-
[0005]U.S. Pat. Application No. 2005/131027 discloses a process for preparation of candesartan cilexetil by reaction of trityl candesartanwith cilexetil halide and at least one base in a low boiling solvent in presence of phase transfer catalyst to give Trityl candesartan cilexetil, which upon deprotection with at least one organic acid in at least one organic solvent. U.S. Pat. Application 2005/131027 further discloses the deprotection of Trityl candesartan cilexetil in methanol without an acid.
-
[0006]The PCT publication WO 2005/021535 discloses the deprotection of Trityl candesartan cilexetil with neutral or slightly basic medium in alcohol.
-
[0007]Chem.Pharm.Bull. 47(2), 182-186 (1999) discloses two novel crystalline forms of Candesartan cilexetil, form-I and form-II.
-
[0008]PCT publication WO 04/085426 discloses Candesartan cilexetil 1,4-Dioxane solvate and two more crystalline forms, designated as form-III and form-IV. The disclosed process for preparation of form-III involves crystallization of Candesartan cilexetil in toluene and for form-IV involves crystallization in a mixture of methyl tert-butyl ether and methanol.
-
[0009]PCT publication WO 2005/077941 discloses several crystalline forms, solvates of Candesartan cilexetil along with a process for preparation of form-I (type-C).
-
[0010]The prior art disclosed methods for preparation of Candesartan cilexetilinvolves purification of Trityl candesartan and Candesartan cilexetil by column chromatography or involves the use of strong acids like IN HCl or the use of organic acids or without an acid in methanol for detrytilation of Trityl candesartan cilexetil.
-
[0011]There is a requirement of a process for preparation of Candesartancilexetil which yields a pure Candesartan cilexetil without involving the purification by column chromatography and the usage of strong acids for deprotection.
Candesartan cilexetil of formula (I) shown beiow is chemicaily described as (+/-)-1- [[(cyclohexyloxy)carbonyl]oxy]ethyl-2-ethoxy-1 -[[2′-(1 H-tetrazol-5-yl)-1 , 1 ‘-biphenyl- 4-yl]methyl]-1 H-benzimidazoie-7-carboxylate. An alternative designation is (+-)-1- hydroxyethyf 2~Ethoxy-1 -{p-(o-1 H-tetrazo!-5-yIphenyi)benzyJ)-7-benzϊmidazoie~ carboxyiic acid cyclohexyl carbonate (ester), with candesartan being the underlying carboxylic acid, i.e. 2-Ethoxy-1 -(p-(o-1 H-tetrazol-5-ylphenyl)benzy!)-7-benz- imidazolecarboxylic acid.

Because of its ability to inhibit the angiotensin-converting enzyme it is widely used for the treatment of hypertension and related diseases and conditions. As an angiotensin Ii receptor antagonist, candesartan ciiexetil avoids the side-effects of calcium antagonists, and shows high stability and obvious curative effects. Currently candesartan ciiexetil is soid as racemic mixture, it is produced according to published patents, e.g. EP 0 720 982 B1 and EP 0 459 136. in Chem. Pharm. Bull. 47(2), 182-186 (1999) two crystalline forms (Form I and II), together with an amorphous form, are disclosed and characterized by their DSC thermograms, X-ray diffraction patterns and IR spectra. US 5,196,444 disclosed the C-type crystal (Form I) of candesartan cilexetif, and processes for producing it under acidic conditions. WO 04/085426 discloses the dioxane solvate of candesartan ciiexetil, together with two additional crystalline forms. WO 2005/077941 discloses hydrates and solvates of candesartan ciiexetil, together with processes for their preparation. WO 2006/048237 also describes the preparation of new polymorphic forms ofcandesartan ciiexetil, together with processes for their preparation, including the preparation of amorphous candesartan ciiexetil by precipitating it with a liquid cyclic hydrocarbon from a solution of candesartan ciiexetil in a chlorinated solvent. in WO 2005/123721 processes for the preparation of amorphous candesartanciiexetil are provided, comprised of spray-drying and precipitation. HPLC CUT PASTE , READER TO PICK ONLY REQUIRED INFO Candesartan cilexetil (60 g) is dissolved in isopropanol (900 m!_) at 60-65 0C. Solution is hot filtered into reactor and quickly cooled to 35 0C. At this temperature nucleation is provoked with 300 mg of candesartan cilexetil form I and stirring is enforced. Suspension is cooled to 3O0C in 1 hour and rigorous stirring is continued at this temperature for additional 5 hours. Then stirring power is reduced and the suspension is cooled to 2O0C in 8 hours. The product is filtered, washed with isopropanoi and dried for 2 hours at 38°C. Yield: 48.7 g of candesartan cilexetil form I. Area % HPLC: candesartan cilexetil: 99.73%, alky ester of candesartan cilexetil 0.08%, candesartan cilexetii pyran below 0.05%, tritylcandesartan cϋexetil 0.09% Average particle size: 19 /vm, no agglomerates present (see Figure 2) B) Detection of impurities in candesartan cilexetil Example 6 Detection of candesartan cϊlexetil pyran in candesartan cilexetii by HPLC HPLC (external standard method) was performed using the following specifications : Column: Zorbax Eclipse XDB-C18, 50 mm x 4.6 mm i.d.τ 1.8 μm particles Eluent A: 0.01 M NaH2PO4, pH 2.5 Eluent B: acetonitriie Gradient of Eluent:

Flow rate: about 1.2 ml/min Diluent: acetonitriie : water = 70 : 30 (V/V). Detection: UV, wavelength 225 nm injection volume: 5 μl Column temperature : 500C Autosampler temperature: 7°C Example 7 Detection of cilexetil pyran in 1 -chloroethyl cyclohexylcarbonate by GC GC/FID (area percent method) was performed using the following specifications: Column: capillary (fused-silica) AT-WAX or adequate Length: 30 m ID: 0.32 mm Film thickness: 0.25 μm Carrier gas: helium Carrier gas flow rate: 2.0 ml/mi n Split ratio: 10 : 1 Air flow rate: 400 ml/min Hydrogen flow rate: 40 ml/min Make up gas flow ISb rate: 25 ml/min Column temperature 100°C (0 min) → 10°C/min → 2000C (10 min or prolonged if necessary) Injector temperature: 21 O0C Detector temperature: 250OC Injection volume : 1 μl Diluent: Acetonithle: chromatography grade. Chromatographic system suitability Signal/noise of 1 -chloroethyl cyclohexyl carbonate: not less than 10
………………
Seki M * Mitsubishi Tanabe Pharma Corporation, Osaka, Japan An Efficient C–H Arylation of a 5-Phenyl-1H-tetrazole Derivative: A Practical Synthesis of an Angiotensin II Receptor Blocker. Synthesis 2012; 44: 3231-3237 
Significance
Candesartan cilexetil (Atacand®) is an angiotensin II receptor antagonist that is prescribed for the treatment of hypertension. It is a prodrug that is hydrolyzed to candesartan in the gut. The synthesis depicted, features an efficient protocol for ruthenium-catalyzed C–H arylation of the tetrazole A. Comment A significant challenge in this small-scale synthesis was the final removal of the benzyl protecting group from the tetrazole unit using transfer hydrogenation. Best results were obtained using a ‘thickshell’ Pd/C catalyst from Evonik
EMEDASTINE DIFUMARATE, EMADINE, 8 TH DEC 2013 PATENT EXPIRY

EMEDASTINE DIFUMARATE
Emedastine difumarate (Emadine) is a second generation antihistamine used in eye drops to treat allergic conjunctivitis. Its mechanism of action is a H1 receptor antagonist.

EMADINE
Drug Patent Expiration and Exclusivity
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| EMEDASTINE DIFUMARATE | SOLUTION/DROPS; OPHTHALMIC | 0.05% | RX | 020706 | 001 |
Patents
There are 1 patent(s) protecting ALCON’s EMADINE.
The last patent expires on 2013-12-08.
| Patent | Expiration | |
|---|---|---|
| US5441958 | Ophthalmic compositions comprising emedastine and methods for their use
Topical ophthalmic compositions comprising 1-(2-ethoxyethyl)-2-(4-methyl-1-homopiperazinyl)-benzimidazole and its ophthalmically acceptable acid addition salts have been found to be useful in treating allergic conjunctivitis and related ailments.
|
2013-12-8 |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ALCON.
EMADINE ® (emedastine difumarate ophthalmic solution) 0.05% is a sterile ophthalmic solution containing emedastine, a relatively selective, H1-receptorantagonist for topical administration to the eyes. Emedastine difumarate is a white, crystalline, water-soluble fine powder with a molecular weight of 534.57. The chemical structure is presented below:
Structural Formula:
 |
Chemical Name:
lH-Benzimidazole, 1-(2-ethoxyethyl)-2-(hexahydro-4-methyl-1H-1,4-diazepin-1-yl), (E)-2-butenedioate (1:2)
Each mL of EMADINE contains: Active: 0.884 mg emedastine difumarate equivalent to 0.5 mg emedastine. Preservative: benzalkonium chloride0.01%. Inactives: tromethamine; sodium chloride; hydroxypropyl methylcellulose; hydrochloric acid/sodium hydroxide (adjust pH); and purified water. It has a pH of approximately 7.4 and an osmolality of approximately 300 mOsm/kg.
l-(2- ethoxyethyl)-2-(4-methyl-l-homopiperazinyl)-benzimidazole, otherwise known asemedastine, and its ophthalmically acceptable acid addition salts and methods for their use.
Allergic conjunctivitis is frequently characterized by ocular pruritus
(itching), erythema (inflammatory redness), edema and tearing. This condition is one of the most frequently treated by ophthalmologists, optometrists and allergists. To date, treatment has been primarily through the use of topically applied histamine t antagonists in combination with α-agonists. See, for example, the following articles:
1. Miller, J. and E.H. Wolf, “Antazoline phosphate and naphazoline hydrochloride, singly and in combination for the treatment of allergic conjunctivitis – a controlled, double-blind clinical trial.” Ann. Allergy, 35:81-86 (1975). 2. Vandewalker, M.L. et al., “Efficacy of Vasocon-A and its components with conjunctival provocation testing (CPT).” j± Allergy Clin. Immunol., 83:302 (1989). 3. Abelson, M.B. et al., “Effects of topically applied ocular decongestant and antihistamine.” Am. I. Ophthalmol., 90:254- 257 (1980).
Recent studies indicate that the antihistamine levocabastine exhibits clinical activity in patients with allergic conjunctivitis without the addition of a vasoconstrictor. See, Dechant, K.L. and K.L. Goa, “Levocabastine. A review of its pharmacological properties and therapeutic potential as a topical antihistamine in allergic rhinitis and conjunctivitis/’ Drugs, 41:202-224 (1991). In addition, it has recently been demonstrated that Hα antagonists are effective in relieving conjunctival injection (hyperemia) and erythema, as well as pruritus. See, Berdy, G.J. et al., “Allergic conjunctivitis: A survey of new antihistamines.” T. Ocular Pharmacol.. 7:313-324 (1991).
Although there are many different antihistamines available for systemic treatment of allergies and related ailments, many such antihistamines are not suitable for topical ophthalmic use because of limited ocular bioavailability. For example, terfenadine (Seldane®, made by Marion Merrell Dow), astemizole (Hismanal®, made by Janssen Pharmaceutica) and loratadine (Claritin®, made by Schering) all have good systemic activity; however, terfenadine has little or no local ocular activity, and astemizole and loratadine each have greatly reduced local ocular activity (as compared to its systemic activity).
LASTACAFT, ALCAFTADINE.. Drug Patent Expiration, 21st Nov 2013

ALCAFTADINE
Alcaftadine is used to prevent eye irritation brought on by allergic conjunctivitis. It is a H1histamine receptor antagonist.
It was approved by the U.S. Food and Drug Administration in 2010 under the trade name Lastacaft.
LASTACAFT, ALLERGAN
Drug Patent Expiration and Exclusivity
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| ALCAFTADINE | SOLUTION/DROPS; OPHTHALMIC | 0.25% | RX | 022134 | 001 |
Patents
There are 1 patent(s) protecting ALLERGAN’s LASTACAFT.
The last patent expires on 2013-11-21.
| Patent | Expiration | |
|---|---|---|
| US5468743 | Imidazo[2,1-b]benzazepine derivatives, compositions and method of use
The present invention is concerned with novel imidazo[2, 1-b][3]benzazepines of formula ##STR1## the pharmaceutically acceptable addition salts and stereochemically isomeric forms thereof, wherein each of the dotted lines independently represents an optional bond; R.sup.1 represents hydrogen, halo, C.sub.1-4 alkyl or C.sub.1-4 alkyloxy; R.sup.2 represents hydrogen, halo, C.sub.1-4 alkyl or C.sub.1-4 alkyloxy; R.sup.3 represents hydrogen, C.sub.1-4 alkyl, ethenyl substituted with hydroxycarbonyl or C.sub.1-4 alkyloxycarbonyl, C.sub.1-4 alkyl substituted with hydroxycarbonyl or C.sub.1-4 alkyloxycarbonyl, hydroxyC.sub.1-4 alkyl, formyl or hydroxycarbonyl; R.sup.4 represents hydrogen, C.sub.1-4 alkyl, hydroxyC.sub.1-4 alkyl, phenyl or halo; R.sup.5 represents hydrogen, C.sub.1-4 alkyl or halo; L represents hydrogen; C.sub.1-6 alkyl; C.sub.1-6 alkyl substituted with one substituent selected from the group consisting of hydroxy, halo, C.sub.1-4 alkyloxy, hydroxycarbonyl, C.sub.1-4 alkyloxycarbonyl, C.sub.1-4 alkyloxycarbonyl-C.sub.1-4 alkyloxy, hydroxycarbonylC.sub.1-4 alkyloxy, C.sub.1-4 alkyloxycarbonylamino, C.sub.1-4 alkylaminocarbonyl, C.sub.1-4 alkylaminocarbonylamino, C.sub.1-4 alkylaminothiocarbonylamino, aryl, aryloxy and arylcarbonyl; C.sub.1-6 alkyl substituted with both hydroxy and aryloxy; C.sub.3-6 alkenyl; C.sub.3-6 alkenyl substituted with aryl; or, L represents a radical of formula –Alk–Y–Het.sup.1 (a-1),–Alk–NH–CO–Het.sup.2 (a-2)or –Alk–Het.sup.3 (a-3); provided that 6,11-dihydro-11-(4-piperidinylidene)-5H-imidazo[2,1-b][3]benzazepine is ecxluded, which are useful antiallergic compounds.Compositions comprising said compounds, methods of using and processes for preparing the same.
|
2013-11-21 |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the ALLERGAN.
Exclusivity ends on 2015-07-28.
| Date | Supplement No. | Action | Documents |
|---|---|---|---|
| 2010-07-28 | 000 | Approval |
KAPVAY, CLONIDINE HYDROCHLORIDE, Patent expiry…13 th oct 2013


CLONIDINE

C9H9Cl2N3•HCl Mol. Wt. 266.56
Clonidine hydrochloride is an imidazoline derivative and exists as a mesomeric compound. The chemical name is 2-(2,6-dichlorophenylamino)-2-imidazoline hydrochloride. The following is the structural formula:
Clonidine hydrochloride is an odorless, bitter, white, crystalline substance soluble in water and alcohol.
KAPVAY
SHIONOGI INC
Drug Patent Expiration and Exclusivity
Drug Information
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| CLONIDINE HYDROCHLORIDE | TABLET, EXTENDED RELEASE; ORAL | 0.1MG | RX | 022331 | 003 | |
| CLONIDINE HYDROCHLORIDE | TABLET, EXTENDED RELEASE; ORAL | 0.2MG | RX | 022331 | 004 |
Patents
There are 1 patent(s) protecting SHIONOGI INC’s KAPVAY.
The last patent expires on 2013-10-13.
| Patent | Expiration | |
|---|---|---|
| US5869100 | Extended release clonidine formulation (tablet)
A method of providing a patient needing clonidine with an extended dosage of clonidine over a prolonged period of time. Such method involves administering to the patient an oral dosage unit comprising a homogenous mixture of a therapeutically effective amount of clonidine, about 30 to about 70 percent by weight of one or more cellulose ethers such as hydroxypropyl methylcellulose, and about 30 to about 70 percent by weight of an inert substance such as cornstarch. The oral dosage unit may be contained in a gelatin capsule or in the form of a tablet.
|
2013-10-13(expired) |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the SHIONOGI INC.
Exclusivity ends on 2013-09-28.

loteprednol etabonate…Patent expiry this week……of October 20, 2013
loteprednol etabonate
82034-46-6 cas
Drug Patent Expiry for the week of October 20, 2013
Tradename….LOTEMAX
Applicant………Pharmos
SUSPENSION/DROPS; OPHTHALMIC, o.5%
Generic Name………loteprednol etabonate
Patent No.US 5,540,930
http://www.google.co.in/patents/US5540930
The invention provides novel compositions of matter containing water-insoluble steroid drugs suitable for therapeutic use. The invention provides stable aqueous suspensions of water-insoluble steroid drugs of particle sizes of ≦15 μm which remain in such a state so as to allow for immediate suspension, when desired, even after extended periods of settling.
| Date | Supplement No. | Action | Documents |
|---|---|---|---|
| 1998-03-09 | 000 | Approval |
| Publication number | US5540930 A |
| Publication type | Grant |
| Application number | US 08/142,743 |
| Publication date | 30 Jul 1996 |
| Filing date | 25 Oct 1993 |
| Priority date | 25 Oct 1993 |
| Fee status | Paid |
| Also published as | CA2174550A1, CA2174550C, DE69430635D1, DE69430635T2, EP0730443A1, EP0730443A4, EP0730443B1, US5747061, WO1995011669A1, Less «8 More » |
| Publication number | 08142743, 142743, US 5540930 A, US 5540930A, US-A-5540930, US5540930 A, US5540930A |
| Inventors | Doron I. Friedman, Yaacov J. Guy |
| Original Assignee | Pharmos Corporation |
| Company | |||
|---|---|---|---|
| LOTEMAX NDA (020583) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE | |
| ALREX NDA (020803) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE | |
| LOTEMAX NDA (020841) | PHARMOS | LOTEPREDNOL ETABONATE…expired | |
| ZYLET NDA (050804) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE; TOBRAMYCIN | |
| LOTEMAX NDA (200738) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE | |
| LOTEMAX NDA (202872) | BAUSCH AND LOMB | LOTEPREDNOL ETABONATE |
Loteprednol (as the ester loteprednol etabonate) is a corticosteroid used in optometry and ophthalmology. Marketed by Bausch and Lomb as Lotemax in the U.S., ocular applications for this drug include the treatment of inflammation of the eye due to allergies (according to the prescription information sheet), as well as chronic forms of keratitis (e.g.: adenoviral and Thygeson’s keratitis), vernal keratoconjunctivitis, pingueculitis, and episcleritis. The drug has little or no effect on intraocular pressure.

Druzgala, P.; Hochhaus, G.; Bodor, N.; J. Steroid Biochem. Mol. Biol. 1991, 38, 149.
http://dx.doi.org/10.1016/0960-0760(91)90120-T
- Steward, R; et al. (November 1998). “Double-masked, placebo-controlled evaluation of loteprednol etabonate 0.5% for postoperative inflammation”. J Cataract Surg 24: 1480–1489.
- Pavesio, CE; Decory, HH (2008). “Treatment of ocular inflammatory conditions with loteprednol etabonate”. Br J Ophthalmol 92 (4): 455–459. doi:10.1136/bjo.2007.132621. PMID 18245274.
