
Home » Posts tagged 'Orphan Drug' (Page 4)
Tag Archives: Orphan Drug
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lumasiran
 |
The molecular formula of lumasiran sodium is C530H669F10N173O320P43S6Na43 and the molecular weight is 17,286 Da.
lumasiran
CAS 1834610-13-7
FDA APPROVED, 11/23/2020, Oxlumo
To treat hyperoxaluria type 1
Press Release
Drug Trials Snapshot
RNA, (Gm-sp-Am-sp-Cm-Um-Um-Um-(2′-deoxy-2′-fluoro)C-Am-(2′-deoxy-2′-fluoro)U-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)C-Um-Gm-Gm-Am-Am-Am-Um-Am-Um-Am), 3′-[[(2S,4R)-1-[29-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[3-[[5-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-1-oxopentyl]amino]propyl]amino]-3-oxopropoxy]methyl]-1,12,19,25-tetraoxo-16-oxa-13,20,24-triazanonacos-1-yl]-4-hydroxy-2-pyrrolidinyl]methyl hydrogen phosphate], complex with RNA (Um-sp-(2′-deoxy-2′-fluoro)A-sp-Um-Am-Um-(2′-deoxy-2′-fluoro)U-Um-(2′-deoxy-2′-fluoro)C-(2′-deoxy-2′-fluoro)C-Am-Gm-Gm-Am-(2′-deoxy-2′-fluoro)U-Gm-(2′-deoxy-2′-fluoro)A-Am-Am-Gm-Um-Cm-sp-Cm-sp-Am) (1:1)
Nucleic Acid Sequence
Sequence Length: 44, 23, 2115 a 8 c 7 g 14 umultistranded (2); modified
OXLUMO is supplied as a sterile, preservative-free, clear, colorless-to-yellow solution for subcutaneous administration containing the equivalent of 94.5 mg of lumasiran (provided as lumasiran sodium) in 0.5 Ml of water for injection and sodium hydroxide and/or phosphoric acid to adjust the pH to ~7.0.
Lumasiran An investigational RNAi Therapeutic for Primary Hyperoxaluria Type 1 (PH1)
Overview • Lumasiran (ALN-GO1) is an investigational, subcutaneously administered (under the skin) RNA interference (RNAi) therapeutic targeting glycolate oxidase (GO) in development for the treatment of primary hyperoxaluria type 1 (PH1).
• PH1 is a rare, life-threatening disease that can cause serious damage to kidneys and progressively to other organs.1
• PH1 is characterized by the pathologic overproduction of oxalate by the liver. Oxalate is an end product of metabolism that, when in excess, is toxic and accumulates in the kidneys forming calcium oxalate crystals.1,2
• Symptoms of PH1 are often associated with recurrent kidney stones and include flank pain, urinary tract infections, painful urination, and blood in the urine.2,3
• Currently, the only curative treatment is a liver transplant, to correct the metabolic defect, combined with a kidney transplant, to replace the terminally damaged kidneys.1,3 Clinical Development
• The safety and efficacy of lumasiran are being evaluated in a randomized, double-blind, placebo-controlled, global, multicenter Phase 3 study of approximately 30 PH1 patients, called ILLUMINATE-A (NCT03681184).
• The primary endpoint is percent change in 24-hour urinary oxalate excretion from baseline to Month 6.
• Key secondary and exploratory endpoints in ILLUMINATE-A will evaluate additional measures of urinary oxalate, estimated glomerular filtration rate (eGFR), safety, and tolerability.
Regulatory Designations • Breakthrough Therapy Designation by the U.S. Food and Drug Administration (FDA) • Priority Medicines (PRIME) Designation from the European Medicines Agency (EMA) • Orphan Drug Designations in both the U.S. and the European Union
/////////lumasiran, fda 2020, 2020 approvals, Oxlumo, Breakthrough Therapy Designation, Orphan Drug, Priority Medicines (PRIME) Designation
FDA approves first treatment Givlaari (givosiran) for inherited rare disease
///////////Givlaari, givosiran, fda 2019, Breakthrough Therapy designation, Priority Review, Orphan Drug
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
Today, the U.S. Food and Drug Administration approved Inrebic (fedratinib) capsules to treat adult patients with certain types of myelofibrosis.
“Prior to today, there was one FDA-approved drug to treat patients with myelofibrosis, a rare bone marrow disorder. Our approval today provides another option for patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The FDA is committed to encouraging the development of treatments for patients with rare diseases and providing alternative options, as not all patients respond in the same way.”
Myelofibrosis is a chronic disorder where scar tissue forms in the bone marrow and the production of the blood cells moves from the bone marrow to the spleen and liver, causing organ enlargement. It can cause extreme fatigue, shortness of breath, pain below the ribs, fever, night sweats, itching and bone pain. When myelofibrosis occurs on its own, it is called primary myelofibrosis. Secondary myelofibrosis occurs when there is excessive red blood cell production (polycythemia vera) or excessive platelet production (essential thrombocythemia) that evolves into myelofibrosis.
Jakafi (ruxolitinib) was approved by the FDA in 2011. The approval of Inrebic for intermediate-2 or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis was based on the results of a clinical trial where 289 patients with myelofibrosis were randomized to receive two different doses (400 mg or 500 mg daily by mouth) of fedratinib or placebo. The clinical trial showed that 35 of 96 patients treated with the fedratinib 400 mg daily dose (the dose recommended in the approved label) experienced a significant therapeutic effect (measured by greater than or equal to a 35% reduction from baseline in spleen volume at the end of cycle 6 (week 24) as measured by an MRI or CT scan with a follow-up scan four weeks later). As a result of treatment with Inrebic, 36 patients experienced greater than or equal to a 50% reduction in myelofibrosis-related symptoms, such as night sweats, itching, abdominal discomfort, feeling full sooner than normal, pain under ribs on left side, and bone or muscle pain.
The prescribing information for Inrebic includes a Boxed Warning to advise health care professionals and patients about the risk of serious and fatal encephalopathy (brain damage or malfunction), including Wernicke’s, which is a neurologic emergency related to a deficiency in thiamine. Health care professionals are advised to assess thiamine levels in all patients prior to starting Inrebic, during treatment and as clinically indicated. If encephalopathy is suspected, Inrebic should be immediately discontinued.
Common side effects for patients taking Inrebic are diarrhea, nausea, vomiting, fatigue and muscle spasms. Health care professionals are cautioned that patients may experience severe anemia (low iron levels) and thrombocytopenia (low level of platelets in the blood). Patients should be monitored for gastrointestinal toxicity and for hepatic toxicity (liver damage). The dose should be reduced or stopped if a patient develops severe diarrhea, nausea or vomiting. Treatment with anti-diarrhea medications may be recommended. Patients may develop high levels of amylase and lipase in their blood and should be managed by dose reduction or stopping the mediation. Inrebic must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
The FDA granted this application Priority Review designation. Inrebic also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Inrebic to Impact Biomedicines, Inc., a wholly-owned subsidiary of Celgene Corporation.
LINK
///////Inrebic , fedratinib, FDA 2019, Priority Review , Orphan Drug, Biomedicines, Celgene , bone marrow disorder
Tanzisertib
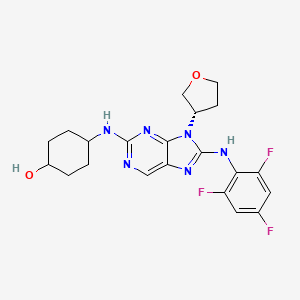
Tanzisertib
CAS 899805-25-5
trans-4-((9-((3S)-Tetrahydrofuran-3-yl)-8-((2,4,6-trifluorophenyl)amino)-9H-purin-2-yl)amino)cyclohexanol
4-[[9-[(3S)-oxolan-3-yl]-8-(2,4,6-trifluoroanilino)purin-2-yl]amino]cyclohexan-1-ol
C21-H23-F3-N6-O2, 448.4467
- CC 930
- CC-930
- Tanzisertib
- UNII-M5O06306UO
- A c-Jun amino-terminal kinase inhibitor.UNII, M5O06306UO
Treatment of Idiopathic Pulmonary Fibrosis (IPF)
- Originator Celgene Corporation
- Class Antifibrotics; Small molecules
- Mechanism of ActionJ NK mitogen-activated protein kinase inhibitors
- Orphan Drug Status Yes – Idiopathic pulmonary fibrosis
- Discontinued Discoid lupus erythematosus; Idiopathic pulmonary fibrosis
- 16 Jul 2012 Celgene Corporation terminates a phase II trial in Discoid lupus erythematosus in USA (NCT01466725)
- 23 Feb 2012 Celgene initiates enrolment in a phase II trial for Discoid lupus erythematosus in the USA (NCT01466725)
- 08 Nov 2011The Committee for Orphan Medicinal Products (COMP) recommends orphan drug designation for tanzisertib in European Union for Idiopathic pulmonary fibrosis
Tanzisertib has been granted orphan drug status by the FDA for the treatment of idiopathic pulmonary fibrosis. A positive opinion has been received from the EU Committee for Orphan Medicinal Products (COMP
Tanzisertib has been used in trials studying the treatment of Fibrosis, Discoid Lupus, Pulmonary Fibrosis, Interstitial Lung Disease, and Lung Diseases, Interstitial, among others.
PATENT
https://patents.google.com/patent/US20090048275A1/de

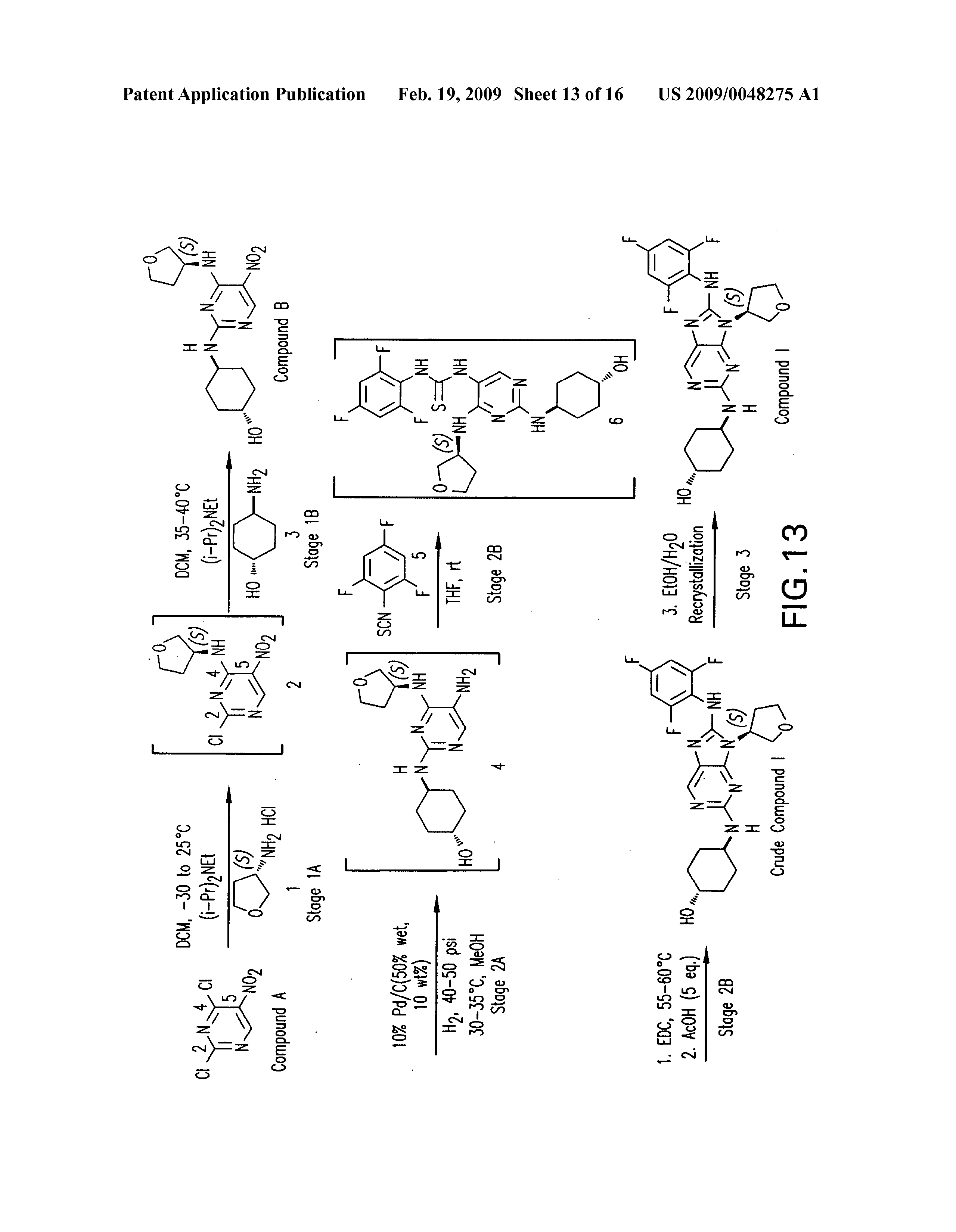

PATENT
WO 2006076595
US 20070060598
WO 2008057252
US 20080021048
US 20140094456
WO 2014055548
PATENT
WO 2015153683
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015153683
/////////Tanzisertib, CC 930, Idiopathic Pulmonary Fibrosis, Orphan Drug, phase II, CELGENE
c1c(c(c(cc1F)F)Nc2n(c3nc(ncc3n2)N[C@H]4CC[C@@H](CC4)O)[C@@H]5COCC5)F
FDA approves first treatment Soliris (eculizumab) for neuromyelitis optica spectrum disorder, a rare autoimmune disease of the central nervous system
The U.S. Food and Drug Administration today approved Soliris (eculizumab) injection for intravenous use for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adult patients who are anti-aquaporin-4 (AQP4) antibody positive. NMOSD is an autoimmune disease of the central nervous system that mainly affects the optic nerves and spinal cord.
“Soliris provides the first FDA-approved treatment for neuromyelitis optica spectrum disorder, a debilitating disease that profoundly impacts patients’ lives,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This approval changes the landscape of therapy for patients with NMOSD. Having an approved therapy for this condition is the culmination of extensive work we have engaged in with drug companies to …
- June 27, 2019
The U.S. Food and Drug Administration today approved Soliris (eculizumab) injection for intravenous use for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adult patients who are anti-aquaporin-4 (AQP4) antibody positive. NMOSD is an autoimmune disease of the central nervous system that mainly affects the optic nerves and spinal cord.
“Soliris provides the first FDA-approved treatment for neuromyelitis optica spectrum disorder, a debilitating disease that profoundly impacts patients’ lives,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This approval changes the landscape of therapy for patients with NMOSD. Having an approved therapy for this condition is the culmination of extensive work we have engaged in with drug companies to expedite the development and approval of safe and effective treatments for patients with NMOSD, and we remain committed to these efforts for other rare diseases.”
In patients with NMOSD, the body’s immune system mistakenly attacks healthy cells and proteins in the body, most often in the optic nerves and spinal cord. Individuals with NMOSD typically have attacks of optic neuritis, which causes eye pain and vision loss. Individuals also can have attacks resulting in transverse myelitis, which often causes numbness, weakness, or paralysis of the arms and legs, along with loss of bladder and bowel control. Most attacks occur in clusters, days to months to years apart, followed by partial recovery during periods of remission. Approximately 50% of patients with NMOSD have permanent visual impairment and paralysis caused by NMOSD attacks. According to the National Institutes of Health, women are more often affected by NMOSD than men and African Americans are at greater risk of the disease than Caucasians. Estimates vary, but NMOSD is thought to impact approximately 4,000 to 8,000 patients in the United States.
NMOSD can be associated with antibodies that bind to a protein called aquaporin-4 (AQP4). Binding of the anti-AQP4 antibody appears to activate other components of the immune system, causing inflammation and damage to the central nervous system.
The effectiveness of Soliris for the treatment of NMOSD was demonstrated in a clinical study of 143 patients with NMOSD who had antibodies against AQP4 (anti-AQP4 positive) who were randomized to receive either Soliris treatment or placebo. Compared to treatment with placebo, the study showed that treatment with Soliris reduced the number of NMOSD relapses by 94 percent over the 48-week course of the trial. Soliris also reduced the need for hospitalizations and the need for treatment of acute attacks with corticosteroids and plasma exchange.
Soliris has a boxed warning to alert health care professionals and patients that life-threatening and fatal meningococcal infections have occurred in patients treated with Soliris, and that such infections may become rapidly life-threatening or fatal if not recognized and treated early. Patients should be monitored for early signs of meningococcal infections and evaluated immediately if infection is suspected. Use should be discontinued in patients who are being treated for serious meningococcal infections. Health care professionals should use caution when administering Soliris to patients with any other infection. In the NMOSD clinical trial, no cases of meningococcal infection were observed.
Soliris is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS). Prescribers must enroll in the REMS program. Prescribers must counsel patients about the risk of meningococcal infection, provide the patients with the REMS educational materials and ensure patients are vaccinated with meningococcal vaccine(s). The drug must be dispensed with the FDA-approved patient Medication Guide that provides important information about the drug’s uses and risks.
The most frequently reported adverse reactions reported by patients in the NMOSD clinical trial were: upper respiratory infection, common cold (nasopharyngitis), diarrhea, back pain, dizziness, influenza, joint pain (arthralgia), sore throat (pharyngitis) and contusion.
The FDA granted the approval of Soliris to Alexion Pharmaceuticals.
Soliris was first approved by the FDA in 2007. The drug is approved to reduce destruction of red blood cells in adults with a rare blood disease called paroxysmal nocturnal hemoglobinuria, for the treatment of adults and children with a rare disease that causes abnormal blood clots to form in small blood vessels in the kidneys (atypical hemolytic uremic syndrome to inhibit complement-mediated thrombotic microangiopathy), and for the treatment of adults with Myasthenia Gravis who are anti-acetylcholine receptor antibody positive.
The FDA granted this application Priority Review. The use for NMOSD received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
///////////////fda 2019, Soliris, eculizumab, neuromyelitis optica spectrum disorder, Orphan Drug, Priority Review
Solriamfetol hydrochloride, ソルリアムフェトル塩酸塩 , солриамфетол , سولريامفيتول , 索安非托 ,
Solriamfetol hydrochloride
FDA APPROVED 2019/3/20, Sunosi
ソルリアムフェトル塩酸塩; R228060, R 228060
| Formula |
C10H14N2O2. HCl
|
|---|---|
| CAS |
178429-65-7 HCL
|
| Mol weight |
230.6913
|
- Developer Jazz Pharmaceuticals plc; SK biopharmaceuticals
- Class Carbamates; Sleep disorder therapies; Small molecules
- Mechanism of Action Adrenergic uptake inhibitors; Dopamine uptake inhibitors
- Orphan Drug Status Yes – Narcolepsy
- Registered Hypersomnia
- Discontinued Depressive disorders
- 26 Mar 2019 Discontinued – Phase-I for Depressive disorders (Adjunctive treatment) in USA (PO) (Jazz Pharmaceuticals pipeline, March 2019)
- 20 Mar 2019 Registered for Hypersomnia (excessive daytime sleepiness) in patients with obstructive sleep apnoea and narcolepsy in USA (PO) – First global approval
- 20 Mar 2019 US FDA approves solriamfetol to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnoea(OSA)
- New Drug Application (NDA): 211230
Company: JAZZ PHARMA IRELAND LTD
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Solriamfetol hydrochloride | K7RO88SP7A | 178429-65-7 | KAOVAAHCFNYXNJ-SBSPUUFOSA-N |
Solriamfetol, sold under the brand name Sunosi, is a medication used for the treatment of excessive sleepiness associated with narcolepsy and sleep apnea.[1]
Common side effects include headache, nausea, anxiety, and trouble sleeping.[1] It is a norepinephrine–dopamine reuptake inhibitor(NDRI). It is derived from phenylalanine and its chemical name is (R)-2-amino-3-phenylpropylcarbamate hydrochloride.[2]
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of 11 countries in Asia to Aerial Pharma in 2011.[3]
History
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of 11 countries in Asia to Aerial Pharma in 2011.[3]Aerial ran two Phase II trials of the drug in narcolepsy[4] before selling the license to solriamfetol to Jazz in 2014; Jazz Pharmaceuticalspaid Aerial $125 million up front and will pay Aerial and SK up to $272 million in milestone payments, and will pay double digit royalties to SK.[3][5]
In March 2019 the FDA accepted SK’s and Jazz’ NDA for use of solriamfetol to treat excessive sleepiness in people with narcolepsy or obstructuve sleep apnea; the drug has an orphan designation for narcolepsy.[3][6]
Names
During development it has been called SKL-N05, ADX-N05, ARL-N05, and JZP-110.[6]
Research
Solriamfetol had also been tested in animal models of depression, but as of 2017 that work had not been advanced to clinical trials.[7]
PATENT
WO 9607637
https://patents.google.com/patent/WO1996007637A1/e
Organic alkyl carbamates have been effectively used for controlling various central nervous system (CNS) disorders. For example, U.S. Pat. Nos . 2,884,444, 2,937,119 and 3,313,697 disclose function of carbamate in CNS disorders, especially as antiepileptic and centrally acting muscle relaxant.
Phenylethylamine derivatives, one important class of therapeutical medicines useful for managing CNS diseases, have been used mainly to treat obesity, narcolepsy, minimal brain dysfunction and mild depression.
Recent design of pharmacologically useful compounds has been based on amino acids or the derivatives thereof, which is mainly attributable to the fact that many of the compounds found in biological systems come from amino acids or the derivatives thereof. In addition, in most cases, the function of a pharmaceutically useful compound is effected after it binds to an enzyme or receptor, which may trigger the regulatory mechanisms of the enzyme or receptor.
REACTION SCHEME I
REACTION SCHEME II
REACTION SCHEME III
EXAMPLE I
Preparation of N-Benzyloxycarbonyl-D-phenylalaninol
In a 500 mL RB flask equipped with a mechanical stirrer and a dropping funnel, D-phenylalaninol (45.4 g, 300 mmol) was dissolved in 220 mL of distilled water, and cooled in an ice-bath. The pH of the solution was adjusted with 50 % sodium hydroxide to 14. Benzyl chloroformate (49.3 mL, 345 mmol) was charged into the dropping funnel and added slowly to the well stirred solution over 0.5 hr. After the completion of the addition, the reaction mixture was stirred for 1 hr. at 0 *C. The product precipitated from the reaction mixture as a white solid. It was collected by filtration and washed completely with distilled water. After being dried in vacuo, the solid thus obtained weighed 104 grams without any further purification: 99.8% Yield.
Melting point = 90 – 92 *C
[α]D20 = + 43.4 (c = 1.0, EtOH)
Analysis calc: C, 71.56; H, 6.71; N,4.91
Found: C, 71.35; H, 6.71; N,4.91
EXAMPLE II
Preparation of N-Benzyloxycarbonyl-D-phenylalaninol
carbamate
In a 500 mL RB flask, N-benzyloxycarbonyl-D- phenylalaninol (13.56 g, 50 mmol) was charged with antipyrine (11.29 g, 60 mmol) in 250 mL of dry THF under a nitrogen atmosphere. The reaction mixture was cooled in an ice-bath and phosgene (30.3 mL of 1.93 M solution in toluene, 58.5 mmol) was added quickly while vigorously stirring. After stirring for 1 hr. , the formation of a corresponding chloroformate from the starting material was monitored by TLC. The chloroformate solution thus prepared, was slowly added to a well stirred and ice-chilled aqueous ammonium hydroxide solution (75 mL, 28-30 %, 1,190 mmol) via cannula over 0.5 hr. The resulting reaction mixture was stirred for an extra 0.5 hr. The organic phase separated was collected. The aqueous phase was extracted twice with methylene chloride (100 mL). The combined organic phase was washed with brine (50 mL), dried over sodium sulfate, and concentrated to yield 17.8 g (113%) of foamy solid. It was purified a flash column chromatography to give 14.8 g of the title compound, white solid: 94% Yield.
Melting point = 121 – 125 *C
[α]D20 = + 28.6 (c = 2.0, EtOH)
Analysis calc. : C, 65.84; H, 6.14; N, 8.53
Found: C, 66.68; H, 6.21; N, 7.80
EXAMPLE III
Preparation of D-Phenylalaninol carbamate hydrochloric
acid salt In a 160 mL Parr reactor, N-benzyloxycarbonyl-D-phenylalaninol carbamate (9.43 g) was added with 75 mL of anhydrous methanol and 10 % palladium on charcoal (0.32 g). Then, the reactor was closed and purged with hydrogen for 1 in. The reaction was completed in 2 hrs . under 40 psi pressure of hydrogen at 45 #C. The catalyst was filtered off. Thereafter, the organic layer was concentrated into 5.97 g (102 %) of pale yellow thick liquid. The liquid was poured in 50 mL of anhydrous THF and cooled to 0 “C. Anhydrous hydrogen chloride gas was then purged through the solution with slowly stirring for
0.5 hr. 50 mL of anhydrous ether was added, to give a precipitate. Filtration with THF-ether (1:1) mixture provided 6.1 g of the title compound as a white solid: 88 % Yield.
Melting point = 172 – 174 “C
[α]D20 = – 12.9 (c = 2.0, H20)
Analysis calc. : C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.90; H, 6.60; N, 12.15; Cl ,
15.52
EXAMPLE IV
Preparation of N-benzyloxγcarbonyl-L-Phenγlalaninol
The title compound was prepared in the same manner as that of Example I, except that (L)-phenylalaninol was used as the starting material.
Melting point = 90 – 92 *C
[α]D20 = – 42.0 (c = 1.0, EtOH)
Analysis calc. : C, 71.56; H, 6.71; N,4.91
Found: C, 70.98; H, 6.67; N,4.95
EXAMPLE V
Preparation of -N-benzyloxycarbonyl-L-Phenylalaninol
carbamate
The title compound was prepared in the same manner as that of Example II, except that N-benzyloxycarbonyl-L-phenylalaninol was used as the starting material.
Melting point = 121 – 128 ‘C
[α]D20 = – 28.9 (c = 2.0, EtOH)
Analysis calc: C, 65.84; H, 6.14; N, 8.53
Found: C, 65.45; H, 6.15; N, 8.32
EXAMPLE VI
Preparation of L-Phenylalaninol carbamate hydrochloric
acid salt
The title compound was prepared in the same manner as that of Example III, except that N-benzyloxycarbonyl-L-phenylalaninol carbamate was used as the starting material.
Melting point = 175 – 177 *C [α]D20 = + 13.1 (c = 1.0, H20)
Analysis calc : C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.95; H, 6.58; N, 12.09; Cl , 15.37
EXAMPLE VII
Preparation of N-benzyloxycarbonyl-D,L-Phenylalaninol
The title compound was prepared in the same manner as that of Example I, except that (D,L)-phenylalaninol was used as the starting material.
Melting point = 72 – 75 #C
Analysis calc: C, 71.56; H, 6.71; N,4.91
Found: C, 71.37; H, 6.74; N,4.84
EXAMPLE VIII
Preparation of N-benzyloxycarbonyl-D,L-Phenylalaninol
carbamate
The title compound was prepared in the same manner as that of Example II, except that N-benzyloxycarbonyl-D,L-phenylalaninol was used as the starting material.
Melting point = 130 – 133 *C
Analysis calc: C, 65.84; H, 6.14; N, 8.53
Found: C, 65.85; H, 6.14; N, 8.49 EXAMPLE IX
Preparation of D,L-Phenylalaninol carbamate hydrochloric
acid salt
The title compound was prepared in the same manner as that of Example III, except that N-benzyloxycarbonyl-D,L-phenylalaninol carbamate was used as the starting material.
Melting point = 163 – 165 *C
Analysis calc: C, 52.60; H, 6.55; N, 12.14; Cl, 15.37
Found: C, 51.92; H, 6.56; N, 11.95; Cl , 15.82
PATENT
US 20050080268
PATENT
WO 2018133703
https://patents.google.com/patent/WO2018133703A1/en
Excessive daytime sleepiness (Excessive Daytime Sleepiness, EDS) or pathological somnolence refers to excessive daytime sleep and wakefulness associated with various sleep disorders. These disorders can be the basis for a sleep disorder or sleep have side effects caused by some other medical conditions. Excessive daytime sleep, also known as narcolepsy, sleep clinics is seen mainly in patients with disease that affects 12% of the general population. EDS patients may be manifested as mental distress, poor work or school performance, increasing the risk of accidents, the impact of EDS can debilitating, even life-threatening.
R228060, also known JZP-110, is a selective dopamine and norepinephrine reuptake inhibitor, originally developed by R & D, SK biopharmaceutical, 2014 Sir ownership of the pharmaceutical compound. R228060 has the potential to treat narcolepsy and sleep apnea syndrome, in three multi-center study in two global reached the primary endpoint, and achieved positive results, significantly improved adult obstructive sleep apnea patients excessive sleepiness in patients with narcolepsy and excessive sleep problems.
R228060 chemical name is O- carbamoyl – (D) – phenylalaninol, as shown in the structural formula of formula (I):

Solid Form different chemicals, can cause varying their solubility and stability, and thus affects the absorption and bioavailability of the drug, and can lead to differences in clinical efficacy. Improve the candidate compound has a solubility by salt way become an important means of drug development. Compared to the free form of the drug, suitable pharmaceutically acceptable salts can improve the solubility of the drug type, increased physical and chemical stability, and also to improve the drug-salt having a melting point, hygroscopicity, crystal type and other physical properties, further development of the pharmaceutical dosage form It plays an important role. Patent Document WO1996007637A1 discloses R228060 hydrochloride and its preparation method, and other characteristics of the obtained having a melting point of 172-174 deg.] C as a white solid, the solid was not given in the text data. Further, the present inventors found no other relevant R228060 hydrochloride polymorph or patent literature. Accordingly, the present need in the art to develop a comprehensive system R228060 hydrochloride polymorph, found to be suitable to the development of crystalline form. The present inventors after many experiments, found that polymorph CS1 R228060 hydrochloride CS2 and a melting point polymorph, Form CS1 and CS2 is Form 183 ℃, much higher than the melting point disclosed in prior art solid. It provides a better alternative preparation of pharmaceutical preparations containing R228060 is, has very important implications for drug development.
PATENT
WO 2019027941
(i?)-2-amino-3-phenylpropyl carbamate (APC) is a phenylalanine analog that has been demonstrated to be useful in the treatment of a variety of disorders, including excessive daytime sleepiness, cataplexy, narcolepsy, fatigue, depression, bipolar disorder, fibromyalgia, and others. See, for example, US Patent Nos. 8,232,315; 8,440,715; 8,552,060; 8,623,913; 8,729,120; 8,741,950; 8,895,609; 8,927,602; 9,226,910; and 9,359,290; and U.S. Publication Nos. 2012/0004300 and 2015/0018414. Methods for producing APC (which also has other names) and related compounds can be found in US Patent Nos. 5,955,499; 5,705,640; 6,140,532 and 5,756,817. All of the above patents and applications are hereby incorporated by reference in their entireties for all purposes.
EXAMPLE 1
Synthesis of Compounds
Compound 8 (110CR002)
1 B 110CR002
[0083] tert- utyl (if)-(l-(Carbamothioyloxy)-3-phenylpropan-2-yl)carbamate (IB): A
60% dispersion of sodium hydride (0.36 g, 4.78 mmol, 1.2 equiv) in mineral oil was added in portions to compound 1A (1.0 g. 3.98 mmol, 1 equiv) in THF (20 mL) at 0 °C. After stirring for 1 hour, carbon disulfide (0.191 g, 4.78 mmol, 1.2 equiv) was added at 0 °C. After an additional hour of stirring, methyl iodide (0.3 mL, 4.78 mmol, 1.2 equiv) was added and the reaction was warmed to room temperature. After stirring two additional hours, concentrated ammonium hydroxide (1.6 mL, 7.98 mmol, 2 equiv) was added and the reaction was stirred overnight at room temperature. The reaction was diluted with water (50 mL) and extracted with dichloromethane (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to give crude compound IB. The solid was triturated in diethyl ether (20 mL) to give compound IB (0.17 g, 14% yield) as a light yellow solid.
[0084] (R)-0-(2-Amino-3-phenylpropyl) carbamothioate dihydrochloride (110CR002):
4M HCI in dioxane (0.68 mL, 2.74 mmol, 5 equiv) was added to neat compound IB (0.17 g, 0.548 mmol, 1 equiv) and the reaction was stirred overnight. The solution was diluted with diethyl ether (20 mL) and the resulting suspension was filtered. The solid was triturated in diethyl ether (20 mL) and the filtered solid was dried under vacuum at room temperature for two hours to give compound 110CR003 (140 mg, 93% yield, 96.9% purity) as a white solid.
Compound 9 (110CR003)
Scheme 2
2A 2B 110CR003
[0085] (R)-2-((ter^Butoxycarbonyl)amino)-3-phenylpropyl sulfamate (2B): A solution of sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added dropwise to a solution of compound 2 A (1.0 g, 3.98 mmol, 1 equiv) and triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) in N,N-dimethylacetamide (20 mL) at 0 °C. After stirring at room temperature for 4 hours, additional triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) and sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added at 0 °C. The reaction was stirred at room temperature overnight, at which point LCMS indicated a 3 :2 mixture of product to starting material. Additional triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) and sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added at 0 °C and the reaction was stirred at room temperature for an additional 6 hours. LCMS indicated a 4: 1 mixture of product to starting material. The reaction was quenched with saturated sodium bicarbonate (5 mL) and stirred for an additional hour at room temperature. The reaction was diluted with saturated sodium bicarbonate (25 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The product still contained unreacted starting material which could not be easily separated. Sulfamoyl chloride (1.15 g, 9.95 mmol, 2.5 equiv) in acetonitrile (2 mL) was added dropwise to a solution of crude compound 2B (0.9 g) and triethylamine (2.1 mL, 14.95 mmol, 3.75 equiv) in N,N-dimethylacetamide (20 mL) at 0 °C. After stirring at room temperature for two hours, the reaction was quenched with saturated sodium bicarbonate (5 mL) and the reaction was stirred for an additional hour at room temperature. The reaction was diluted with saturated sodium bicarbonate (25 mL) and extracted with ethyl acetate (3 x 50 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (Redisep 24 g silica gel column), eluting with a gradient of 25 to 50% ethyl acetate in heptanes, to give compound 2B (0.37 g, 28% yield) as a white solid.
[0086] (R)-2-Amino-3-phenylpropyl sulfamate hydrochloride (110CR003): 4M HC1 in dioxane (1.4 mL, 5.6 mmol, 5 equiv) was added to neat compound 2B (0.37 g, 1.12 mmol, 1 equiv) and the reaction was stirred overnight. The solution was diluted with diethyl ether (20 mL) and the resulting suspension was filtered. The solid was triturated in diethyl ether (20 mL) and the filtered solid was dried under a vacuum at room temperature for two hours to give compound 110CR003 (250 mg, 84% yield, 97.8% purity) as a white solid.
Com ound 3 (110CR007)
[0087] (Benzyl (R)-(l-phenyl-3-ureidopropan-2-yl)carbamate) (3B): Concentrated hydrochloric acid (0.06 mL, 0.68 mmol, 0.12 equiv) was added to a solution of benzyl (ft)-(l -amino-3-phenylpropan-2-yl)carbamate ( 1.5 g, 5.28 mmol, 1 equiv) and urea (1.26 g, 21.21 mmol, 4 equiv) in toluene (150 mL) under nitrogen. After refluxing overnight, LCMS indicated the reaction was complete. The reaction was concentrated under reduced pressure, diluted with water (150 mL) and stirred for 30 minutes. The resulting solid was filtered and washed with water (25 mL) to give crude compound 3B (1.4 g, 4.27 mmol, 80% yield) as a white solid, which was used sequentially.
[0088] ((R)-l-(2-mino-3-phenylpropyl)urea) (3C): Compound 3B (0.5 g, 1.5 mmol, 1 equiv) and 10% palladium on carbon (0.09 g) in methanol (60 mL) was hydrogenated at 30 psi for 1 hour at which time LC-MS determined that the reaction was incomplete. The solution was filtered and fresh catalyst (0.09 g) was added. The solution was hydrogenated at 30 psi for an additional 45 minutes resulting in complete conversion. Two identical scale reactions were run for 105 minutes each, both resulting in complete conversion. The three runs were combined and filtered through celite, which was washed with methanol (50 mL). The filtrate was concentrated under reduced pressure to give crude compound 3C (0.9 g), which was used sequentially.
[0089] (R)-l-(2-Amino-3-phenylpropyI)urea hydrochloride (110CR007): Compound 3C (0.88 g, 4.58 mmol, 1 equiv) was dissolved diethyl ether (10 mL) and 4 N HCl in dioxane (2.31 mL, 9.27 mmol, 2 equiv) was added. The reaction was stirred overnight and then concentrated under reduced pressure to give crude 110CR007 as a white solid. The material was twice recrystallized from 10% methanol in ethanol (30 mL) to give 110CR007 (0.163 g, 16 % yield, 93.7 % purity) as a white solid.
Compound 4 (110CR009)
Scheme 4
[0090] Ethyl (R^)-4-((tert-butoxycarbonyI)amino)-5-phenylpent-2-enoate (4B): A solution of compound 4A (4.0 g, 16.1 mmol, 1 equiv) and ethyl (triphenylphos-phoranylidene)acetate (5.6 g, 16.1 mmol, 1 equiv) in dichloromethane (40 mL) was stirred at room temperature overnight. The reaction was concentrated under reduce pressure to remove the organic solvent and the resulting residue was purified on an AnaLogix automated system (40 g Sorbtech silica gel column), eluting with gradient of 50 to 100% ethyl acetate in heptanes, to give compound 4B (4.8 g, 94% yield) as a white solid.
[0091] (R^E)-4-((te *i-ButoxycarbonyI)amino)-5-phenylpent-2-enoic acid (4C): Lithium hydroxide (1.4 g, 60 mmol, 4 equiv) in water (15 mL) was added to compound 4B (4.8 g, 15 mmol, 1 equiv) in THF (60 mL) at room temperature and the reaction was stirred overnight. After 16 hours, the reaction was adjusted to pH 4 with IN hydrochloric acid. The organic layer was removed and the aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers was washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure to give compound 4C (4.2 g, 97% yield) as a light cream solid, which was used subsequently.
[0092] Methyl (R E)-4-((½ -i-butoxycarbonyl)amino)-5-phenylpent-2-enoate (4D1):
Isobutyl chloro formate (1.3 mL, 10 mmol, 1 equiv) in THF (4 mL) was added dropwise to a solution of compound 4C (3.0 g, 10 mmol, 1 equiv) and N-methyl-morpholine (1.1 mL, 10 mmol, 1 equiv) in THF (12 mL) at -15 °C. After 30 minutes of stirring, LCMS indicated complete conversion to the anhydride intermediate. 2M Ammonia in methanol (5 mL, 10 mmol, 1 equiv) was added dropwise over 20 minutes, keeping the internal temperature between -25 to -15 °C. After 30 minutes of stirring, the reaction was warmed to room
temperature and stirred overnight. The reaction mixture was concentrated at reduced pressure to remove the organic solvent. The resulting residue was dissolved in ethyl acetate (50 mL) and washed with water (100 mL). The aqueous layer was extracted with ethyl acetate (2 x 50 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (80 g Sorbtech silica gel column), eluting with a gradient of 25 to 50% ethyl acetate in heptanes, to give compound 4D1 (1.1 g, 35 % yield) as a white solid.
[0093] Methyl (S)-4-((te^-butoxycarbonyl)amino)-5-phenylpentanoate (4D2): A mixture of compound 4D1 (1.1 g, 3.6 mmol, 1 equiv) and 10% palladium on carbon (0.33 g, 50% wet) in methanol (40 mL) was hydrogenated at 40 psi at room temperature for 4 hours. The mixture was filtered through celite, which was washed with methanol (100 mL). The filtrate was concentrated under reduced pressure to give compound 4D2 (1.1 g, 99% yield) as a white solid.
[0094] (S)-4-((ii? i-Butoxycarbonyl)amino)-5-phenylpentanoic acid (4D3): Lithium hydroxide (73 mg, 3 mmol, 1.5 equiv) in water (1 mL) was added to compound 4B (0.6 g, 2 mmol, 1 equiv) in THF (9 mL) at room temperature. After stirring overnight, the reaction was adjusted to pH 4 with IN hydrochloric acid. The organic layer was removed and the aqueous layer was extracted with ethyl acetate (3 x 25 mL). The combined organic layers was washed with saturated brine (25 mL), dried over sodium sulfate and concentrated under reduced pressure to give compound 4D3 (0.56 g, 98% yield) as a white solid, which was used subsequently.
[0095] tert-Butyl (S)-(5-amino-5-oxo-l-phenylpentan-2-yl)carbamate (4E): Isobutyl chloroformate (0.23 mL, 1.8 mmol, 1 equiv) in THF (0.5 mL) was added drop-wise to a solution of compound 4C (0.54 g, 1.8 mmol, 1 equiv) and N-methylmorpholine (0.2 mL, 1.8 mmol, 1 equiv) in THF (1 mL) at -15 °C. After 20 minutes of stirring, LCMS indicated complete conversion to the anhydride intermediate. 0.4M Ammonia in THF (9 mL, 3.6 mmol, 2 equiv) was added drop-wise over 20 minutes, keeping the internal temperature between -25 to -15 °C. After 30 minutes of stirring the reaction was warmed to room temperature and stirred overnight. The reaction mixture was concentrated under reduced pressure to remove the organic solvent. The resulting residue was dissolved in ethyl acetate (25 mL) and washed with water (25 mL). The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2 x 25 mL). The combined organic layers were washed with saturated brine (50 mL), dried over sodium sulfate and concentrated under
reduced pressure to give compound 4E (0.5 g, 93% yield) as a white solid, which was used subsequently.
[0096] (S)-4-Amino-5-phenylpentanamide hydrochloride (110CR009): 4M HC1 in dioxane (6 mL, 25 mmol, 10 equiv) was added to compound 4E (0.73 g, 1.12 mmol, 1 equiv) After stirring overnight at room temperature, the reaction was diluted with diethyl ether (20 mL) and stirred for 6 hours. The resulting suspension was filtered and the solid was washed with diethyl ether (20 mL). The filtered solid was dried under vacuum at room temperature for two hours to give compound 110CR009 (340 mg, 60% yield, 97.9 % purity) as a white solid.
Compound 10 (110CR012)
[0097] tert-Butyl (R)-(l-(carbamoylthio)-3-phenyIpropan-2-yI)carbamate (5B):
Compound 5 A (0.15 g, 0.56 mmol, 1 equiv) was dissolved in THF (8 mL) and sparged with nitrogen for 15 minutes. Trichloroacetyl isocyanate (0.1 mL, 0.84 mmol, 1.5 equiv) was added and the solution stirred for 3 hours, at which point TLC (30% ethyl acetate in heptane) indicated absence of starting material. The reaction was cooled to 0°C and concentrated ammonium hydroxide (0.15 mL) was added. After stirring overnight at room temperature, TLC indicated that the reaction was complete. The reaction was washed with a 10% ammonium hydroxide (10 mL). The organic layer was concentrated under reduced pressure. The residue was purified on an AnaLogix automated system (12 g silica gel column), eluting with a gradient of 0 to 30% ethyl acetate in heptane, to give compound 5B. This reaction was repeated an additional two times 0.15 g and 0.18 g). The products were to give compound 5B (0.35 g, 1.12 mmol, 62.2% yield) as a white solid.
[0098] (R)-S-(2-Amino-3-phenylpropyl) carbamothioate hydrochloride (110CR012):
Compound 5B (0.35 g, 1.12 mmol, 1 equiv) was dissolved in 4N HCI in dioxane (2 mL). The reaction was stirred for two hours and then concentrated under reduced pressure to give crude 110CR012 as a white solid. The material was triturated in diethyl ether (15 mL) to give 110CR012 (0.215 g, 78 % yield, 98.0 % purity) as a white solid.
References
- ^ Jump up to:a b “SUNOSI™ (solriamfetol) Tablets, for Oral Use. Full Prescribing Information” (PDF). Jazz Pharmaceuticals. 2019. Retrieved 21 March2019.
- ^ Abad, VC; Guilleminault, C (2017). “New developments in the management of narcolepsy”. Nature and Science of Sleep. 9: 39–57. doi:10.2147/NSS.S103467. PMC 5344488. PMID 28424564.
- ^ Jump up to:a b c d Ji-young, Sohn (5 March 2018). “SK Biopharmaceuticals’ narcolepsy drug on track to hitting US market”. The Korea Herald.
- ^ Sullivan, SS; Guilleminault, C (2015). “Emerging drugs for common conditions of sleepiness: obstructive sleep apnea and narcolepsy”. Expert Opinion on Emerging Drugs. 20 (4): 571–82. doi:10.1517/14728214.2015.1115480. PMID 26558298.
- ^ Garde, Damian (January 14, 2014). “Jazz bets up to $397M on Aerial’s narcolepsy drug”. FierceBiotech.
- ^ Jump up to:a b “Solriamfetol – Jazz Pharmaceuticals/SK Biopharmaceuticals”. AdisInsight. Retrieved 15 April 2018.
- ^ de Biase, S; Nilo, A; Gigli, GL; Valente, M (August 2017). “Investigational therapies for the treatment of narcolepsy”. Expert Opinion on Investigational Drugs. 26 (8): 953–963. doi:10.1080/13543784.2017.1356819. PMID 28726523.
 |
|
| Clinical data | |
|---|---|
| Trade names | Sunosi |
| Synonyms | SKL-N05, ADX-N05, ARL-N05, and JZP-110; (R)-2-amino-3-phenylpropylcarbamate hydrochloride |
| Routes of administration |
By mouth |
| ATC code | |
| Pharmacokinetic data | |
| Bioavailability | ~95% |
| Protein binding | 13.3–19.4% |
| Metabolism | negligible |
| Elimination half-life | ~7.1 h |
| Excretion | urine (95% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C10H14N2O2 |
| Molar mass | 194.234 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////Solriamfetol hydrochloride, Solriamfetol, ソルリアムフェトル塩酸塩; солриамфетол , سولريامفيتول , 索安非托 , JZP-110, Orphan Drug, fda 2019, R228060, R 228060
UPDATE MAR 2022
Solriamfetol, sold under the brand name Sunosi, is a medication used for the treatment of excessive sleepiness associated with narcolepsy and sleep apnea.[1] It is derived from d-phenylalanine and its chemical name is (R)-2-amino-3-phenylpropylcarbamate hydrochloride.[3] It is a norepinephrine–dopamine reuptake inhibitor (NDRI). Common side effects include headache, nausea, anxiety, and trouble sleeping.[1]
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of eleven countries in Asia to Aerial Pharma in 2011.[4]
Synthetic Description
Reference: Choi, Yong-Moon; Kim, Min Woo. Process for preparing O-carbamoyl amino alcohols by treatment of amino alcohols with cyanates in the presence of acid. US 20050080268. (2005).
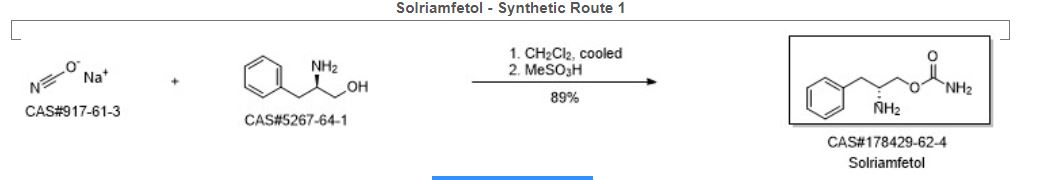
SYN
https://www.researchgate.net/figure/Synthesis-of-solriamfetol-173_fig37_344079894

SYN
Cite this article
Yin, Z., Hu, W., Zhang, W. et al. Tailor-made amino acid-derived pharmaceuticals approved by the FDA in 2019. Amino Acids 52, 1227–1261 (2020). https://doi.org/10.1007/s00726-020-02887-4
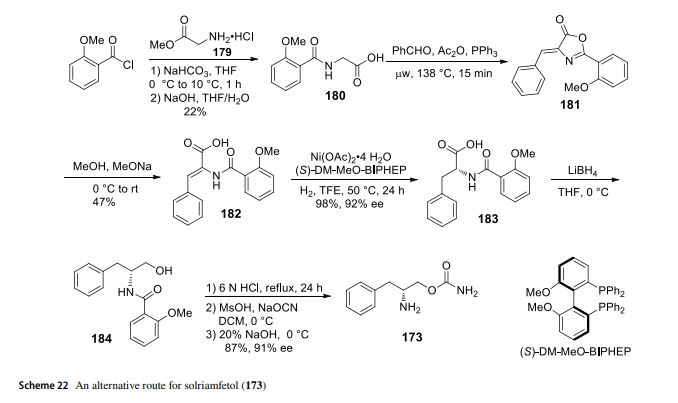
Solriamfetol (Sunosi™) Solriamfetol (Sunosi™) (173), formerly known as JZP-110, is a selective dopamine and norepinephrine reuptake inhibitor (DNRI) (Fig. 23). It was discovered by SK Biopharmaceuticals and developed by Jazz Pharmaceuticals (Markham 2019c). The afnity of solriamfetol for these monoamine transporters dopamine transporter (DAT, Ki=14.2 μM), norepinephrine transporter (NET, Ki=3.7 μM), and serotonin transporter (SERT, Ki=81.5 μM) was lower than that of cocaine in transfected cells and inhibits dopamine and norepinephrine reuptake with low potency (IC50=2.9 and 4.4 μM, respectively) (Baladi et al. 2018). In 2019, US FDA approved solriamfetol for using as an oral drug to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnoea (OSA). It was granted as an orphan drug (Schweitzer et al. 2019). The systematic name of solriamfetol is (R)-2-amino3-phenylpropylcarbamate hydrochloride, which contains a phenylalanine (171)-derived (R)-2-amino-3-phenylpropan1-ol (172) moiety (Fig. 23). Some alkyl carbamates have been introduced for controlling various central nervous system (CNS) disorders. Phenylethylamine derivatives are one of the important class of therapeutical medicines, useful for managing CNS diseases. After an intensive research, these two skeletons were combined to produce solriamfetol (173) as a drug for the treatment of CNS disorder, especially for depression. The compound 174 with a (S) carbon center showed almost no activity at all, which the racemic compound 175 displayed a half potency of the activity (Fig. 24) (Yang and Gao 2019; Choi and Byun 1996). Solriamfetol (173) was discovered and patented by SK Biopharmaceuticals in 1996 (Choi and Byun 1996). The synthesis of solriamfetol using (D)-phenylalaninol (176) as a starting material is highlighted in Scheme 21. (D)-Phenylalaninol (176) was frst converted to Cbz-protected D-phenylalaninol 177 by reacting with benzyl chloroformate. Carbamoylation of 177 with phosgene followed by ammonolysis with excess of concentrated ammonium hydroxide aqueous solation aforded (D)-O-carbamoyl-N-benzyloxycarbonylphenylalaninol 178. Hydrogenolysis removal of the Cbz protection group gave solriamfetol 173 which was treated

with HCl (gas) to provide (D)-O-carbamoylphenylalaninol hydrochloride salt. In 2020, the Zhang lab reported a method of Ni-catalyzehd asymmetric hydrogenation of 2-amidoacrylates for making solriamfetol (173) (Hu et al. 2020). In this method, o-methoxybenzoyl chloride reacted with glycine methyl ester hydrochloride 179 under a base condition and then hydrolysised in the presence of NaOH to aford desired o-methoxyhippuric acid 180. The one-step construction of oxazolone 181 was accomplished by cyclization and condensation of 180 with benzaldehyde in acetic anhydride and PPh3. Oxazolone 181 was then treated with MeOH and NaOMe to aford 2-amidoacrylate 182. Hydrogenation of 182 using Ni salt and ligand (S)-DM-MeO-BIPHEP gave product 183 in 92% ee. The reduction of 183 with LiBH4 followed by hydrolysised in the presence of NaOH provided intermediate (D)-phenylalaninol 184. Then, (D)-phenylalaninol 184 was reacted with NaOCN yielded solriamfetol (173) in 91% ee (Scheme 22). As a general comment related to this and other chiral compounds discussed here, we would like to emphasize the growing awareness about the Self-Disproportionation of Enantiomers (SDE) phenomenon and the problems related to accurate determination of the stereochemical outcome of enantioselective catalytic reactions (Han et al. 2018, 2019b, 2011a; Soloshonok et al. 2017; Sorochinsky et al. 2013c,
2013d). It was demonstrated that the SDE phenomenon is ubiquitous, being manifested virtually by all types of chiral compounds subjected to physicochemical phase transfer under totally achiral conditions (Han et al. 2019b; Sorochinsky et al. 2013c, d). One of the most frequent cases is a separation of more and less enantiomerically enriched fractions as compared with the original enantiomeric purity of a chiral compound. Consequently, to ensure the accuracy in the %ee determination, it was suggested to perform SDE tests, in particular, under the conditions of achiral column chromatography (Soroshinsky et al. 2013c) and sublimation (Han et al. 2011a).
(Soroshinsky et al. 2013c) Sorochinsky AE, Katagiri T, Ono T, Wzorek A, Aceña JL, Soloshonok VA (2013c) Optical purifcations via self-disproportionation of enantiomers by achiral chromatography; case study of a series of α-CF3-containing secondary alcohols. Chirality 25:365–368
SYN
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 63-91-2 | C9H11NO2 | D-Phenylalanine | |
| 5267-64-1 | C9H13NO | D-Phenylalaninol | |
| 75-44-5 | CCl2O | phosgene |
SYN
European Journal of Medicinal Chemistry

Pharmacology
Pharmacodynamics
Solriamfetol is a norepinephrine–dopamine reuptake inhibitor (NDRI).[1] It binds to the dopamine transporter and the norepinephrine transporter with affinities (Ki) of 14.2 μM and 3.7 μM, respectively).[1] It inhibits the reuptake of dopamine and norepinephrine with IC50 values of 2.9 μM and 4.4 μM, respectively.[1] It has weak affinity for the serotonin transporter (Ki = 81.5 μM) and does not appreciably inhibit serotonin reuptake (IC50 > 100 μM).[1] Solriamfetol has no appreciable affinity for a variety of other targets, including the dopamine, serotonin, adrenergic, GABA, adenosine, histamine, orexin, benzodiazepine, and acetylcholine receptors.[1]
Pharmacokinetics
The elimination half-life of solriamfetol is about 7.1 hours.[1]
History
The drug was discovered by a subsidiary of SK Group, which licensed rights outside of eleven countries in Asia to Aerial Pharma in 2011.[4] Aerial ran two Phase II trials of the drug in narcolepsy[5] before selling the license to solriamfetol to Jazz in 2014; Jazz Pharmaceuticals paid Aerial $125 million up front and will pay Aerial and SK up to $272 million in milestone payments, and will pay double-digit royalties to SK.[4][6]
In 2019, solriamfetol was approved in the United States to improve wakefulness in adults with narcolepsy or obstructive sleep apnea (OSA).[7][8] It was granted orphan drug designation.[9]
The U.S. Food and Drug Administration (FDA) approved solriamfetol based primarily on evidence from five clinical trials (Trial 1/NCT02348593, Trial 2/NCT02348606, Trial 3/NCT02348619, Trial 4/NCT02348632, Trial 5 NCT01681121) of 622 patients with narcolepsy or obstructive sleep apnea (OSA).[7] The trials were conducted in Canada, Europe, and the United States.[7]
Solriamfetol was approved for medical use in the European Union in January 2020.[2]
Society and culture
Names
During development it has been called SKL-N05, ADX-N05, ARL-N05, and JZP-110.[10]
Legal status
In the United States, solriamfetol is a Schedule IV controlled substance,[1] meaning that it has an accepted medical use and a low potential for abuse, but that abuse may lead to physical or psychological dependence.[11] A prescription is required, and can only be refilled up to five times in a six-month period.[12] In countries of the European Union, a prescription is required.[2]
Research
A case report of solriamfetol for the treatment of attention deficit hyperactivity disorder (ADHD) exists.[13]
References
- ^ Jump up to:abcdefghijklmn “Sunosi – solriamfetol tablet, film coated”. DailyMed. 16 October 2019. Retrieved 24 November 2019.
- ^ Jump up to:abc “Sunosi EPAR”. European Medicines Agency (EMA). 12 November 2019. Retrieved 26 September 2020.
- ^ Abad VC, Guilleminault C (2017). “New developments in the management of narcolepsy”. Nature and Science of Sleep. 9: 39–57. doi:10.2147/NSS.S103467. PMC5344488. PMID28424564.
- ^ Jump up to:abc Ji-young S (5 March 2018). “SK Biopharmaceuticals’ narcolepsy drug on track to hitting US market”. The Korea Herald.
- ^ Sullivan SS, Guilleminault C (2015). “Emerging drugs for common conditions of sleepiness: obstructive sleep apnea and narcolepsy”. Expert Opinion on Emerging Drugs. 20 (4): 571–82. doi:10.1517/14728214.2015.1115480. PMID26558298. S2CID7951307.
- ^ Garde D (14 January 2014). “Jazz bets up to $397M on Aerial’s narcolepsy drug”. FierceBiotech.
- ^ Jump up to:abc “Drug Trials Snapshots: Sunosi”. U.S. Food and Drug Administration (FDA). 16 April 2019. Archived from the original on 28 September 2019. Retrieved 24 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Drug Approval Package: Sunosi”. U.S. Food and Drug Administration (FDA). 29 April 2019. Retrieved 24 November 2019.This article incorporates text from this source, which is in the public domain.
- ^ “Solriamfetol Orphan Drug Approval”. U.S. Food and Drug Administration (FDA). Retrieved 24 November 2019.This article incorporates text from this source, which is in the public domain.
- ^ “Solriamfetol – Jazz Pharmaceuticals/SK Biopharmaceuticals”. AdisInsight. Retrieved 15 April 2018.
- ^ 21 U.S.C.§ 812 – Schedules of controlled substances
- ^ “Manuals – Practitioner’s Manual – Section V”. Retrieved 2014-01-07
- ^ Naguy A, El-Sheshaie A, Elsori DH, Alamiri B (April 2021). “Solriamfetol for attention deficit hyperactivity disorder”. CNS Spectr: 1–2. doi:10.1017/S1092852921000328. PMID33870884.
External links
- “Solriamfetol”. Drug Information Portal. U.S. National Library of Medicine.
- * “Solriamfetol hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
//////////////////
N[C@@H](COC(N)=O)CC1=CC=CC=C1
RISDIPLAM , リスジプラム
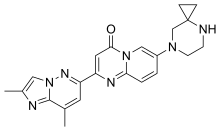
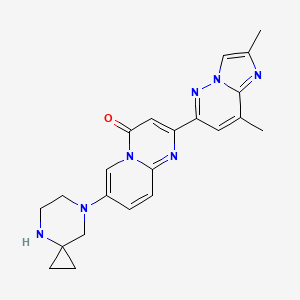
RISDIPLAM
RG-7916, RO-7034067, リスジプラム
| Formula |
C22H23N7O
|
|---|---|
| Cas |
1825352-65-5
|
| Mol weight |
401.4643
|
| US9969754 |
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido[1,2-a]pyrimidin-4-one
The compound was originally claimed in WO2015173181 , for treating spinal muscular atrophy (SMA). Roche , under license from PTC Therapeutics , and Chugai , are developing risdiplam (RO-7034067; RG-7916), a small-molecule survival motor neuron (SMN)2 gene splicing modulator and a lead from an SMN2 gene modulator program initiated by PTC Therapeutics in collaboration with the SMA Foundation , for the oral treatment of spinal muscular atrophy
The product was granted orphan drug designation in the U.S., E.U. and in Japan for the treatment of spinal muscular atrophy. In 2018, it also received PRIME designation in the E.U. for the same indication.
Risdiplam (RG7916, RO7034067) is a highly potent, selective and orally active small molecule experimental drug being developed by F. Hoffmann-La Roche, PTC Therapeutics and SMA Foundation to treat spinal muscular atrophy (SMA). It is a pyridazine derivative that works by increasing the amount of functional survival of motor neuron protein produced by the SMN2 gene through modifying its splicing pattern.[1][2]
As of September 2018, risdiplam is undergoing late-stage clinical trials across the spectrum of spinal muscular atrophy[3][4][5] where it has shown promising preliminary results.[6][7]
PATENT
WO2015173181
Example 20
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6- yl)pyrido[l,2-a]pyrimidin-4-one
In a sealed tube, 2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-7-fluoro-pyrido[l,2-a]pyrimidin-4-one (Intermediate 2; 50 mg, 0.162 mmol), DIPEA (0.22 mL, 1.29 mmol, 4 eq.) and 4,7-diazaspiro[2.5]octane dihydrochloride (32 mg, 0.320 mmol, 3.0 eq.) were stirred in
DMSO (2 mL) at 130°C for 48 hours. The solvent was removed under high vacuum. The residue was taken up in CH2CI2 and washed with an aqueous saturated solution of NaHC03. The organic layer was separated and dried over Na2S04 and concentrated in vacuo. The crude was purified by column chromatography (Si02, CH2Cl2/MeOH=98/2 to 95/5) to afford the title product (12 mg, 18%) as a light yellow solid. MS m/z 402.3 [M+H+].

PATENT
WO-2019057740
Process for the preparation of risdiplam and its derivatives.
Scheme 1:
Scheme 3:
Scheme 4:
xample 1: tert-Butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
5-Bromo-2-chloropyridine (85.0 g, 442 mmol), tert-butyl 4,7-diazaspiro[2.5]octane-4-carboxylate (102 g, 442 mmol) and Me-THF (722 g) were charged into a reaction vessel. After 10 minutes stirring, most of the solids were dissolved and [Pd(Xantphos)Cl2] (3.34 g) was added followed after 5 minutes by a solution of sodium tert-butanolate (56.3 g, 574 mmol) in Me-THF (173 g). The reaction mixture was stirred at 70 °C for 1.25 hours, cooled to room temperature and water (595 g) and 1-propylacetate (378 g) were added. After vigorous stirring, the phases were separated, the organic phase was washed with a second portion of water (425 g) and with a mixture of water (425 g) and brine (25 mL). The organic phase was treated with active charcoal (6.8 g), filtered and concentrated under reduced pressure to afford a brown oil, which was dissolved in tert-amyl-methyl-ether (347 g) at reflux. The solution was cooled slowly to room temperature. After stirring 18 hours at room temperature, n-heptane (205 g) was added and the suspension was further cooled to -10 °C. The precipitate was filtered off and dried under high vacuum to afford tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (110.9 g, 77.5%) as a beige solid.
Ή-ΝΜΡν (CDC13, 600 MHz): 7.95 (d, 1H); 7.18 – 7.14 (m, 1H); 7.13 – 7.09 (m, 1H); 3.79 – 3.63 (m, 2H); 3.24 – 3.12 (m, 2H); 2.96 (s, 2H); 1.47 (s, 9H); 1.11 – 1.04 (m, 2H); 0.90 -0.79 (m, 2H); LCMS: 324.15, 326.15 (M+H+)
Example 2: tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
An autoclave equipped with an ascending pipe was filled with ammonia (78.7 g, 15 eq; 10 eq are sufficient) at -70 °C. Another autoclave was charged with tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (100 g, 309 mmol), sodium tert-butanolate (32.6 g, 340 mmol) and dioxane (800 mL). After 10 minutes stirring at room temperature under Ar, a solution of Pd2(dba)3 (1.41 g, 1.54 mmol) and tBuBrettPhos (1.50 g, 3.09 mmol) in dioxane (180 mL) was added. Thereafter, the connected ammonia vessel was warmed with a warm water bath and the connecting valve was opened. The autoclave was warmed to 30 °C and the reaction mixture stirred 5 hours at this temperature. The ammonia vessel was closed and disconnected. The excess ammonia was washed out of the autoclave with Argon. The reaction solution was poured into a separating funnel, the autoclave washed with ethyl acetate (300 mL) and water (100 mL) and these two solvent portions were added to the separating funnel. The biphasic mixture was further diluted with ethyl acetate (900 mL) and water (1000 mL). After vigorous stirring, the phases were separated. The organic phase was washed with a mixture of water (500 mL) and brine (10 mL). The combined aqueous phases were extracted twice with ethyl acetate (500 mL). The combined organic phases were treated with active charcoal (3.70 g, 309 mmol), filtered and the filtrate was concentrated under reduced pressure to afford a thick brown oil. This oil was dissolved in 1 -propyl acetate (160 mL) at 45-50°C and n-heptane (940 mL) was added drop wise within 1.5 hours. The suspension was cooled slowly to -5°C, stirred 4 hours at -5 °C and filtered. The precipitate was washed with cold n-heptane and dried under high vacuum at 50°C to afford tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (81.4 g, 86.5%) as a beige solid.
Ή-ΝΜΡν (CDCb, 600 MHz): 7.71 (d, 1H); 7.12 (dd, 1H); 6.47 (d, 1H); 4.18 (br s, 2H); 3.74 – 3.58 (m, 2H); 3.09 – 2.94 (m, 2H); 2.81 (s, 2H); 1.52 – 1.39 (m, 9H); 1.17 – 0.98 (m, 2H); 0.92 – 0.75 (m, 2H); LCMS: 305.20 (M+H+)
Example 3: tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
An autoclave was charged with tert-butyl 7-(6-chloro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (339 mg, 1 mmol), sodium tert-butanolate (109 mg, 1.1 mmol) and dioxane (5 mL). After 5 minutes stirring at room temperature under Argon [Pd(allyl)(tBuBrettPhos)]OTf (4 mg, 5 μιηοΐ) was added. Thereafter, the autoclave was closed and connected to an ammonia tank, the valve was open and ammonia (230 mg, 13.5 mmol) was introduced into the autoclave. The valve was closed and the autoclave disconnected. The autoclave was warmed to 30 °C and the reaction mixture stirred 4 hours at this temperature. Then the autoclave was opened and the excess ammonia was washed out of the autoclave with Argon. The reaction solution was poured into a flask and taken to dryness under reduced pressure. The residue was purified by chromatography over silica gel (eluent: dichloromethane/ethyl acetate to dichloromethane/methanol). After evaporation of the solvents tert-butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (283 mg, 93%) was isolated as a brown oil containing 4% dichloromethane and 3% ethyl acetate.
Example 4: tert-butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 4,7-diazaspiro[2.5]octane-4-carboxylate oxalate salt (2.46 kg, 8.13 mol), 5-bromo-2-nitro-pyridine (1.50 kg, 7.39 mol) and dimethyl sulfoxide (7.80 L) were char; into a reaction vessel pre-heated to 35 °C. With stirring, and keeping the temperature below 40°C, lithium chloride (1.25 kg, 25.6 mol) was added portion- wise followed by tetramethylguanidine (2.98 kg, 25.9 mol). Dimethyl sulfoxide (450 mL) was used to rinse the feed line. The reaction mixture was stirred at 79 °C for 8 hours, cooled to 70°C and water (2.48 L) was added within 2 hours. After stirring at 70 °C for an additional 1 hour, the precipitate was filtered off and washed with water (4.5 L) three times. The precipitate was dissolved in ethyl acetate (15 L) and water (7.5 L) at reflux temperature. The phases were separated at 60°C and n-heptane (7.5 L) was added to the organic layer at 60°C within 30 minutes. The solution was cooled to 0°C in 2 hours and further stirred at 0°C for 1 hour. The precipitate was filtered off, washed with a mixture of ethyl acetate (750 mL)/n-heptane (375 mL) twice and dried under reduced pressure to afford 1.89 kg (76.4%) of tert-butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate as a yellow to light brown solid.
!H-NMR (CDCls, 600 MHz): 8.16 (d, 1H); 8.07 (d, 1H); 7.15 (dd, 1H); 3.80 – 3.72 (m, 2H); 3.49 – 3.41 (m, 2H); 3.23 (s, 2H); 1.48 (s, 9H); 1.16 – 1.08 (m, 2H); 0.92 – 0.85 (m, 2H); LCMS: 335.17 (M+H+)
Example 5: tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-(6-amino-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (80.0 g, 263 mmol) was dissolved in anisole (800 mL) and di-tert-butyl malonate (71.1 g, 315 mmol) was added. The solution was stirred 3.5 hours at 145 °C then cooled to room temperature. The precipitate was filtered off, washed with toluene (in portions, 320 mL in total) and dried under high vacuum at 50°C to afford tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (65.6 g, 67%) as a light pink powder.
Ή-ΝΜΡν (CDCI3, 600 MHz): 8.46 (d, 1H); 7.74 (dd, 1H); 7.52 (d, 1H); 5.37 (s, 2H); 3.83 – 3.69 (m, 2H); 3.23 (t, 2H); 3.01 (s, 2H); 1.48 (s, 9H); 1.17 – 1.03 (m, 2H); 0.95 – 0.75 (m, 2H); LCMS: 373.19 (M+H+)
Example 6: tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-(6-nitro-3-pyridyl)-4,7-diazaspiro[2.5]octane-4-carboxylate (950 g, 2.84 mol), Pt 1%, V 2% on active charcoal (95.1 g, 2 mmol) and ethyl acetate (9.5 L) were charged into an autoclave that was pressurized with hydrogen gas to 3 bar. The reaction mixture was stirred at room temperature for 6 hours. The excess hydrogen was vented. The reaction mixture was filtered, the catalyst was washed with ethyl acetate (0.95 L) three times. The filtrate was concentrated under reduced pressure and the solvent exchanged to anisole (add two portions of 2.85 L and 5.18 L) by distillation. Di tert-butyl malonate (921.7 g, 4.26 mol) was added and the charging line was rinsed with anisole (618 mL) and the reaction mixture was stirred at 125-135 °C for 8 hours. It may be necessary to distill off the by-product tert-butanol to reach this temperature. The progress of the reaction was followed eg.by HPLC. If the reaction stalls, the temperature is increased to 135-145°C and checked for progress after 1 hour. When the reaction was complete, the batch was cooled to room temperature and stirred at room temperature for 4 hours. The precipitate was filtered off, washed with toluene (3.55 L) and dried under vacuum at 60°C to afford tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (861.0 g, 81.4%) as a yellow to light brown solid.
Example 7: tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
A reactor was charged with tert-butyl 7-(2-hydroxy-4-oxo-pyrido[l,2-a]pyrimidin-7-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate (920 g, 2.47 mol) and then triethylamine (325 g, 3.21 mol), followed by tosyl chloride (527.1 g, 2.77 mol) and dichloromethane (4.6 L). The reaction mixture was stirred at 20-25 °C for at least three hours. Upon complete reaction, the organic solution was washed with a prepared solution of HC1 (32%, 247.8 mL) and water (4.6 L), followed by a prepared solution of sodium hydroxide (432.3 mL of a 30% stock solution) and water (3.9 L) in that order. The organic phase was finally washed with water (4.8 L) and then dichloromethane was nearly completely distilled off under reduced pressure at 50-55°C. Ethyl acetate (920 mL) was added and distilled twice at this temperature under reduced pressure, and then ethyl acetate (4.8 L) was added and the suspension cooled to 20-25 °C over two hours. n-Heptane (944.4 mL) was added and the mixture was cooled to 0-5 °C and then stirred for an additional 3 hours. The precipitate was filtered off, washed with a prepared solution of ethyl acetate (772.8 mL) and n-heptane (147.2 mL), and then twice with n-heptane (2.6 L). The solid was dried under vacuum at 45-50°C to afford 1122.6 g (86.3%) tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate as yellow crystals.
!H-NMR (CDCls, 600 MHz): 8.32 (d, 1H); 8.00 – 7.89 (m, 2H); 7.66 (dd, 1H); 7.50 (d, 1H); 7.36 (d, 2H); 6.04 (s, 1H); 3.80 – 3.68 (m, 2H); 3.23 (t, 2H); 3.01 (s, 2H); 1.48 (s, 9H); 1.15 – 1.04 (m, 2H); 0.92 – 0.82 (m, 2H); LCMS: 527.20 (M+H+)
Example 8: 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine
6-Chloro-2,8-dimethylimidazo[l,2-b]pyridazine (40.0 g, 220 mmol), bis pinacol diborane (69.9 g, 275 mmol) and potassium acetate (43.2 g, 440 mmol) were suspended in acetonitrile (440 mL). The suspension was heated to reflux and stirred 30 minutes at reflux, then a suspension of PdCl2(dppf) (4.03 g, 5.51 mmol) and dppf (610 mg, 1.1 mmol) in acetonitrile (40 mL) was added. The vessel was rinsed with acetonitrile (20 mL), which were also poured into the reaction mixture. The orange suspension was further stirred at reflux, whereby acetonitrile (50 mL) were distilled off. After 4 hours, the reaction mixture was filtered off, the filter was washed with several portions of acetonitrile (in total 150 mL). The filtrate was diluted to obtain a volume of 700 mL. The 314 mmolar solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in acetonitrile was used as such in the next step.
Example 9: 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine
6-chloro-2,8-dimethylimidazo[l,2-b]pyridazine (29.0 g, 22.8 mmol), bis pinacol diborane (44.6, 25.1 mmol) and potassium acetate (31.3 g, 45.6 mmol) were suspended in 1-propyl acetate (365 mL). The suspension was heated to 80°C and a solution of
tricyclohexylphosphine (448 mg, 0.23 mmol) and Pd(OAc)2 (179 mg, 0.11 mmol) in 1-propyl acetate (37 mL) was added within 20 minutes. After 2.5 hours further stirring at 80°C, the suspension was cooled to 40°C and filtered at this temperature. The precipitate was washed with 1-propyl acetate (200 mL). The filtrate corresponds to 516.4 g of a 8.5% solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1 -propyl acetate.
Example 10: Isolation of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[ 1 ,2-b]pyridazine
In another experiment, the above solution obtained was cooled to 0-5 °C within 3 hours. The precipitate was filtered off, washed with cold 1 -propyl acetate and dried under high vacuum at 60°C to afford 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine (24. Og, 55%) as a colourless solid.
lH NMR (CDCls, 600 MHz, ) δ ppm 7.86 (d, J=0.7 Hz, 1 H), 7.20 (d, J=1.0 Hz, 1 H), 2.63 (d, J=1.0 Hz, 3 H), 2.51 (d, J=0.7 Hz, 3 H), 1.33 – 1.49 (m, 12 H)
Example 11: (step 6) tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
tert-Butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5] octane-4-carboxylate (25 g, 47.5 mmol), 2,8-dimethyl-6-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine (314 mM in acetonitrile, 191 mL, 59.8 mmol), PdCi2(dppf) (868 mg, 1.19 mmol) and aqueous potassium carbonate 4.07 M (17.1 mL, 69.8 mmol) were charged into a reaction vessel. The reaction mixture was stirred at reflux for 3 hours, cooled overnight to room temperature and filtered. The precipitate was washed with several portions of acetonitrile (146 mL in total), then suspended in methyl-THF (750 mL) and methanol (75 mL). Aqueous sodium hydrogen carbonate 5% (250 mL) was added, the mixture was vigorously stirred at 35°C. The phases were separated, the organic phase was washed again with aqueous sodium hydrogen carbonate 5% (250 mL). The organic phase was treated with active charcoal for 1 hour at room temperature, filtered and the filtrate was concentrated under reduced pressure at 60 °C to a volume of 225 mL, heated to reflux then cooled to room temperature, stirred at room temperature for 16 hours, then cooled to 0°C and stirred at 0°C for 3 hours. The precipitate was filtered off, washed with n-heptane (60 mL) and dried under high vacuum at 55°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (20.13 g, 84.5%) as a yellow solid.
This solid could be recrystallized in the following manner: 15 g of the above solid was dissolved at reflux in toluene (135 mL) and ethanol (15 mL). The solution was slowly cooled to room temperature, stirred 16 hours at room temperature, then cooled to 0°C and stirred at 0°C for 4 hours. The precipitate was filtered off, washed with cold toluene and dried under high vacuum at 55°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (11.92 g, 79.5%) as a yellow-green solid.
!H-NMR (CDCls, 600 MHz): 8.44 (d, 1H); 7.93 (d, 1H); 7.96 – 7.89 (m, 1H); 7.80 (d, 1H); 7.76 – 7.72 (m, 1H); 7.70 – 7.63 (m, 1H); 7.38 (s, 1H); 3.85 – 3.69 (m, 2H); 3.28 (t, 2H); 3.07 (s, 2H); 2.74 (d, 3H); 2.55 (s, 3H); 1.49 (s, 9H); 1.16 – 1.09 (m, 2H); 0.93 – 0.86 (m, 2H); LCMS: 502.26 (M+H+)
Example 12: tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate
6-chloro-2,8-dimethylimidazo[l,2-b]pyridazine (4.14 g, 22.8 mmol), bis pinacol diborane (6.37g, 25.1 mmol) and potassium acetate (4.47 g, 45.6 mmol) were suspended in 1-propyl acetate (59 mL). The suspension was heated to 80°C and a solution of
tricyclohexylphosphine (63.9 mg, 0.23 mmol) and Pd(OAc)2 (25.6 mg, 0.11 mmol) in 1-propyl acetate (6 mL) was added within 20 minutes. After 2.5 hours further stirring at 80°C, the suspension was cooled to 40°C and filtered at this temperature. The precipitate was washed with 1-propyl acetate (32 mL). The filtrate corresponds to 74.6 g of a 8.5% solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1-propyl acetate.
A reaction vessel was charged with tert-butyl 7-[4-oxo-2-(p-tolylsulfonyloxy)pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (10.0 g, 19.0 mmol), tricyclohexylphosphine (58.6 mg, 0.21 mmol) and Pd(OAc)2 (21.3 mg, 0.10 mmol) and 1-propyl acetate (42 mL) and a solution of potassium carbonate (5.25 g, 38.0 mmol) in water (19.0 mL) was added. The suspension was heated to 70°C and the solution of 2,8-dimethyl-6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)imidazo[l,2-b]pyridazine in 1-propyl acetate was added within 30 minutes. The mixture was stirred for 2 hours at 70-75°C. The suspension was cooled to 40°C, water (10 mL) was added. The suspension was aged for 30 minutes. The crude product was filtered off and rinsed with 1-propyl acetate (41 mL). The crude product was taken up in toluene (100 mL), 5% aqueous NaHC03-solution (30 mL) and 1-propanol (20.0 mL). The mixture was heated to 60-65 °C, the phases were separated and the organic phase was washed with 2 more portions of water (30.0 mL). The organic phase was filtered on active charcoal, the filter washed with toluene (60.0 mL). The filtrate was concentrated under reduced pressure to a volume of ca. 120 mL, heated to reflux and 1-propanol (0.8 mL) was added to obtain a solution. The solution was cooled to 0-5°C within 4-6 hours, stirred at 0-5°C for 1 hour. The precipitate was filtered off, washed with toluene (30 mL) and dried under reduced pressure at 70-80°C to afford tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (7.7 g, 80.8%) as a yellowish solid.
Example 13: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one di-hydrochloride salt
To prepare a solution of HC1 in in 1-propyl acetate/ 1-propanol, acetyl chloride (15.8 g, 199 mmol) was slowly added to a mixture of 1-propyl acetate (60 mL) and 1-propanol (30 mL) at 0°C, and stirring was pursued for an additional 2 hours at room temperature.
tert-Butyl 7-[2-(2,8-dimethylimidazo[ 1 ,2-b]pyridazin-6-yl)-4-oxo-pyrido[ 1 ,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (20 g, 39.9 mmol) was suspended in 1-propyl acetate (60 mL) and 1-propanol (30 mL) at room temperature and the HC1 solution in 1-propyl acetate and 1-propanol was added. The reaction mixture was heated within 3 hours to 70°C and stirred 16 hours at this temperature, then cooled to 20°C. The precipitate was filtered off, washed with 1-propyl acetate (50 mL) in several portions and dried under vacuum at 55 °C to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one hydrochloride salt (18.8 g, 99%) as yellow crystals.
^-NMR (CDCls, 600 MHz): 8.34 (s, 1H); 8.22(s, 1H); 8.05 (s, 1H); 8.01 (dd, 1H); 7.80 (d, 1H); 7.16 (s, 1H); 3.71 – 3.67 (m, 2H); 3.64 – 3.59 (m, 2H); 3.52 (s, 2H); 2.69 (s, 3H); 2.54 (s, 3H); 1.23- 1.20 (m, 2H); 1.14 – 1.08 (m, 2H); LCMS: 402.20 (M+H+)
Example 14: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[ 1 ,2-a]pyrimidin-4-one
To a suspension of tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (25 g, 50 mmol) in 1-propyl acetate (375 mL) was added a solution of HC1 in 1-propanol (prepared by adding slowly at 5°C acetyl chloride (18.0 mL) to 1-propanol (37.6 mL) and stirring 1 hour at room temperature). The stirred suspension was heated to 75°C within 10 hours and stirred a further 5 hours at 75 °C. Water (160.0 mL) was added and the phases were separated at 75°C. Aqueous sodium hydroxide 32% (27.8 mL) was added to the aqueous phase. The suspension obtained was cooled to room temperature within 5 hours and stirred one hour at room temperature. The precipitate was filtered off, washed with water (100.0 mL) and dried under reduced pressure at 50°C for 18 hours to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one (19.7 g, 98.3%) as yellow crystals.
!H-NMR (CDCb, 600 MHz): 8. 45 (d, 1H); 7.92 (d, 1H); 7.80 (s, 1H); 7.75 – 7.71 (m, 1H); 7.71 – 7.67 (m, 1H); 7.37 (s, 1H); 3.31 – 3.24 (m, 2H); 3.22 – 3.16 (m, 2H); 3.09 (s, 2H); 2.73 (s, 3H); 2.55 (s, 3H); 0.82- 0.76 (m, 2H); 0.71 – 0.63 (m, 2H); LCMS: 402.20
(M+H+)
Example 15: 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[ 1 ,2-a]pyrimidin-4-one
A suspension of tert-butyl 7-[2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)-4-oxo-pyrido[l,2-a]pyrimidin-7-yl]-4,7-diazaspiro[2.5]octane-4-carboxylate (13.5 g, 26.9
in toluene (237.0 g) was stirred at 75°C and a 21.9% solution of HCl in 1-propanol (21.4 g, 134.5 mmol) was added within 2.5 hours. The reaction mixture was stirred further at 75 °C until complete conversion. The reaction mixture was cooled to 20-25°C. Water (70 g) was added. The biphasic mixture was stirred another 10 minutes at 20-25 °C and the phases were separated. The organic phase was extracted with water (17 g) twice and the combined aqueous phases were added into mixture of aqueous sodium hydroxide 28% (15.0 g) and water (45.0 g). The suspension obtained was cooled to 20°C. The precipitate was filtered off , washed with water (25 g) three times and dried under reduced pressure at 60°C to afford 7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[l,2-b]pyridazin-6-yl)pyrido[l,2-a]pyrimidin-4-one (9.5 g, 95.1%) as yellow crystals.
Example 16: 4-bromo-6-chloro-pyridazin-3-amine
3-amino-6-chloropyridazine (20 g, 154 mmol), sodium bicarbonate (25.9 g, 309 mmol) and methanol (158 g) were charged in a reaction vessel and cooled to 0-10°C. Bromine (34.5 g, 216 mmol) was added drop wise and the reaction mixture was stirred 3 days at room temperature. 10% Aqueous sodium sulfate was added. The suspension was filtered off. The filtrate was washed with ethyl acetate (300 mL) twice. The combined organic layers were dried and evaporated. A suspension of the residue in methanol (50 mL) was heated to reflux, water (120 mL) was added and the suspension was stirred 16 hours at room temperature. The precipitate was filtered off and dried. The residue was suspended in n-heptane (50 mL), stirred 2 hours at room temperature, filtered off and dried to afford 4-bromo-6-chloro-pyridazin-3-amine (14.5 g, 46.2%) as a light brown solid.
!H-NMR (CDCls, 600 MHz): 7.55 (s, 1H); 5.83-4.89 (m, 2H); LCMS: 209.93 (M+H+)
Example 17: 4-bromo-6-chloro-pyridazin-3-amine
3-amino-6-chloropyridazine (50 g, 360 mmol), acetic acid (5.8 g, 96.5 mmol), sodium acetate (28.7 g, 289.5 mmol) and methanol (395 g) were charged in a reaction vessel and heated to 25-35°C. Dibromodimethylhydatoin (66.0 g, 231.6 mmol) was added in several portions and the reaction mixture was stirred 3 hours at 30°C. Completion is checked by IPC and if the conversion is incomplete, dibromodimethylhydantoin is added (5.5g). At reaction completion, 38% aqueous sodium sulfate (77.2 mmol NaHS03) was added slowly. The suspension was concentrated under reduced pressure and water (500 g) was added slowly at 45°C, then 30% aqueous sodium hydroxide (31.5 g, 231.6 mmol NaOH) was added at 20°C to adjust pH to 7-8. The precipitate was filtered off, washed with water and dried under reduced pressure to afford 4-bromo-6-chloro-pyridazin-3-amine (50.2 g, 62.5%) as a grey solid.
Example 18: 6-chloro-4-methyl-pyridazin-3-amine
4-bromo-6-chloro-pyridazin-3-amine (3.0 g, 14.4 mmol) and
tetrakis(triphenylphosphine)palladium (1666 mg, 144 μιηοΐ) were suspended in THF (13.2 g) and a solution of zinc chloride in Me-THF (2.0 M, 9 mL, 18 mmol) was added. The reaction mixture was cooled to -5°C and methyllithium in diethoxymethane (3.1 M, 11.6 mL, 36 mmol) was added. The reaction mixture was stirred at 45°C for 4 hours. Sodium sulfate decahydrate (11.7 g, 36 mmol) was added at room temperature, the mixture was stirred 1.5 hours at 60°C, diluted with water (100 mL) and after 30 minutes the precipitate was filtered off. The precipitate was dissolved in aqueous HC1 2M (100 mL) and ethyl acetate (140 mL). The biphasic system was filtered, the phases were separated and the pH of the water layer adjusted to 7 with aqueous NaOH 32% (18 mL). The precipitate was filtered and dried. The solid obtained was digested twice in methanol (20 mL) at room temperature. The two filtrates were combined, evaporated and dried under high vacuum to afford 6-chloro-4-methyl-pyridazin-3-amine (1.2 g, 58.1%) as a red solid.
Ή-ΝΜΡν (CDCb, 600 MHz): 7.09 (d, 1H); 4.90 (br s, 2H), 2.17 (d, 3H)
Example 19: 6-chloro-4-methyl-pyridazin-3-amine
4-bromo-6-chloro-pyridazin-3-amine (30.02 g, 143 mmol) and THF (180 mL) were charged into a reaction vessel. Methylmagnesium chloride (22% in THF, 50.0 mL, 1.03 eq.) was added at 20°C over 60 minutes, followed by zinc chloride in Me-THF (25%, 37 mL, 0.50 eq.) and palladium tetrakis(triphenyphosphine) (1.66 g, lmol%). The reaction mixture was heated to 50°C and methylmagnesium chloride (22% in THF, 81 mL, 1.7 eq.) was added slowly. The reaction mixture was stirred at 50°C until complete conversion, then at 10°C for 14.5 hours and poured into a mixture of water (90 g), aqueous HCl 33% (52.5 g) and toluene (150 mL) maintained at 20-30°C. The aqueous phase was separated and the organic phase was extracted with a solution of aqueous HCl 33% (2.0 g) and water (45 g). The aqueous layers were combined and washed with toluene (30 mL) twice and the pH was adjusted by addition of 25% aqueous ammonia solution. When a pH of 2.4 was reached, seeding crystals were added, the mixture was stirred further for 15 minutes and thereafter the pH was brought to 4.0. The suspension was stirred at 20°C for 2 hours, the precipitate was filtered off, washed with water (20 mL) three times to afford crude 6-chloro-4-methyl-pyridazin-3-amine (29 g) as a brown solid.
29 g crude product was transferred to a reaction vessel and methanol (20 mL) was added. The mixture was refluxed for 30 minutes and 12 g water was added. The solution was cooled to 0°C and stirred for 2 hours at this temperature. The precipitate was filtered off, washed with water three times and dried under reduced pressure at 40°C to afford purified 6-chloro-4-methyl-pyridazin-3-amine (13.8 g, 66%) as a light brown solid.
Alternative purification:
50 g crude 6-chloro-4-methyl-pyridazin-3-amine were dissolved in methanol (250 mL) and active charcoal (4.0 g) and diatomaceous earth (2.5 g) were added. The suspension was stirred at 45°C for 1 hour, cooled to 30°C and potassium hydrogenophosphate (2.1 g) was added. The suspension was stirred at 30°C for another 90 minutes, filtered and the precipitate washed with methanol (100 mL). The filtrate was concentrated to a residual volume of 175 mL and water (120 mL) was added. The resulting suspension was heated
to reflux affording a solution which was cooled to 20°C resulting in a suspension. The precipitate was filtered off, washed with water (90 mL) and dried under reduced pressure to afford pure 6-chloro-4-methyl-pyridazin-3-amine (38 g, 76%) as a light yellow solid.
Example 20: 6-chloro-2,8-dimethyl-imidazo[l,2-b]pyridazine
6-chloro-4-methyl-pyridazin-3-amine (70.95 kg, 494.2 mol), sodium bromide (35 kg, 345.9 mol), isopropyl acetate (611 kg), isopropanol (28 kg and water (35 kg) were charged into a reaction vessel. The reaction mixture was stirred at 80-85 °C for 8 hours. Isopropyl acetate (310 kg) and water (420 kg) were added. 30% Aqueous NaOH was added at 45-55 °C and the system was stirred for 2 hours. The phases were separated at 25-35 °C. The organic layer was washed with water (370 kg), filtered on diatomite (7 kg) and the filter washed with isopropyl acetate (35 kg). The organic phase was extracted with two portions of 5.4% aqueous sulfuric acid (910 kg followed by 579 kg). The combined aqueous phases were basified with 30% aqueous NaOH (158 kg). The suspension was stirred 2 hours at 15-25 °C. The precipitate was isolated by centrifugation in three portions, each washed with water (31 kg). The wet solid was dissolved in isopropyl acetate (980 kg) at 25-35 °C, the solution washed with water (210 kg), three times. The organic phase was treated with active charcoal for 12 hours at 45-50 °C, concentrated to ca. 300 kg and heated to 70-80 °C to obtain a clear solution. This solution was cooled to 50-60 °C, stirred at this temperature for 1 hour, n-heptane (378 kg) was added and stirring was pursued for 1 hour. The mixture was cooled to -10- -5°C and stirred for another 3 hours. The precipitate was isolated by centrifuging, washed with n-heptane (33 kg) and dried under reduced pressure at 30-50 °C for 15 hours to afford 67.4 kg (76%) 6-chloro-2,8-dimethyl-imidazo[l,2-b]pyridazine as an off-white solid.
XH-NMR (CDCls, 600 MHz): 7.67 (s, 1H); 6.86 (s, 1H); 2.65 (s, 3H), 2.50 (s, 3H)
Paper
https://pubs.acs.org/doi/pdf/10.1021/acs.jmedchem.8b00741

SMA is an inherited disease that leads to loss of motor function and ambulation and a reduced life expectancy. We have been working to develop orally administrated, systemically distributed small molecules to increase levels of functional SMN protein. Compound 2 was the first SMN2 splicing modifier tested in clinical trials in healthy volunteers and SMA patients. It was safe and well tolerated and increased SMN protein levels up to 2-fold in patients. Nevertheless, its development was stopped as a precautionary measure because retinal toxicity was observed in cynomolgus monkeys after chronic daily oral dosing (39 weeks) at exposures in excess of those investigated in patients. Herein, we describe the discovery of 1 (risdiplam, RG7916, RO7034067) that focused on thorough pharmacology, DMPK and safety characterization and optimization. This compound is undergoing pivotal clinical trials and is a promising medicine for the treatment of patients in all ages and stages with SMA.
7-(4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)pyrido[1,2-a]pyrimidin-4-one 1 (12 mg, 18%) as a pale yellow solid. 1H NMR (600 MHz,CDCl3) δ ppm 8.45 (d, J = 2.4 Hz, 1H), 7.92 (d, J = 1.0 Hz, 1H), 7.73 (d, J = 9.6 Hz, 1H) 7.80 (s, 1H), 7.70 (dd, J = 9.7, 2.5 Hz, 1H), 7.38 (s, 1H), 3.31–3.22 (m, 2H), 3.20–3.16 (m, 2H), 3.08 (s, 2H), 2.74 (d, J = 0.9 Hz, 3H) 2.55 (s, 3H), 1.68 (br s, 1H), 0.77–0.75 (m, 2H), 0.67–0.64 (m, 2 H);
13C NMR (151 MHz,CDCl3) δ ppm 158.2, 156.3, 148.5, 147.2, 144.1, 142.2, 140.0, 135.6, 131.2, 126.7, 114.9, 114.7, 110.1, 99.3, 56.7, 49.9, 44.5, 36.5, 16.9, 15.0, 13.0. LC–HRMS: m/z = 402.2051 [(M + H)+ calcd for C22H24N7O, 402.2042; Diff 0.9 mDa].


References
- ^ Maria Joao Almeida (2016-09-08). “RG7916”. BioNews Services. Retrieved 2017-10-08.
- ^ Zhao, Xin; Feng, Zhihua; Ling, Karen K. Y; Mollin, Anna; Sheedy, Josephine; Yeh, Shirley; Petruska, Janet; Narasimhan, Jana; Dakka, Amal; Welch, Ellen M; Karp, Gary; Chen, Karen S; Metzger, Friedrich; Ratni, Hasane; Lotti, Francesco; Tisdale, Sarah; Naryshkin, Nikolai A; Pellizzoni, Livio; Paushkin, Sergey; Ko, Chien-Ping; Weetall, Marla (2016). “Pharmacokinetics, pharmacodynamics, and efficacy of a small-molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy”. Human Molecular Genetics. 25 (10): 1885. doi:10.1093/hmg/ddw062. PMC 5062580. PMID 26931466.
- ^ “Genentech/Roche Releases Clinical Trial Update for RG7916”. CureSMA. 2017-09-15. Retrieved 2017-10-08.
- ^ “A Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7034067 in Infants With Type1 Spinal Muscular Atrophy (Firefish)”.
- ^ “A Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7034067 in Type 2 and 3 Spinal Muscular Atrophy Participants (Sunfish)”.
- ^ “Updated Preliminary Data from SMA FIREFISH Program in Type 1 Babies Presented at the CureSMA Conference”. http://www.prnewswire.com. Retrieved 2018-09-11.
 |
|
| Clinical data | |
|---|---|
| Synonyms | RG7916; RO7034067 |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C22H23N7O |
| Molar mass | 401.474 g/mol g·mol−1 |
| 3D model (JSmol) | |
///////////RISDIPLAM, RG-7916, RO-7034067, リスジプラム , PHASE 3, PRIME designation, ORPHAN DRUG
76RS4S2ET1 (UNII code)
CC1=CC(=NN2C1=NC(=C2)C)C3=CC(=O)N4C=C(C=CC4=N3)N5CCNC6(C5)CC6
Voretigene neparvovec , ボレチジーンネパルボベック;
Voretigene neparvovec
Voretigene neparvovec-rzyl;
Luxturna (TN)
ボレチジーンネパルボベック;
DNA (synthetic adeno-associated virus 2 vector AAV2-hRPE65v2)
CAS: 1646819-03-5
2017/12/19, FDA Luxturna, SPARK THERAPEUTICS
Vision loss treatment, Retinal dystrophy
AAV2-hRPE65v2
AAV2.RPE65
LTW-888
SPK-RPE65
rAAV.hRPE65v2
rAAV2-CBSB-hRPE65
2SPI046IKD (UNII code)
| melting point (°C) | 72-90ºC | Rayaprolu V. et al. J. Virol. vol. 87. no. 24. (2013) |
FDA
LUXTURNA
STN: 125610
Proper Name: voretigene neparvovec-rzyl
Trade Name: LUXTURNA
Manufacturer: Spark Therapeutics, Inc.
Indication:
- Is an adeno-associated virus vector-based gene therapy indicated for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Patients must have viable retinal cells as determined by the treating physician(s).
Product Information
- Package Insert – LUXTURNA (PDF – 602KB)
- Demographic Subgroup Information – voretigene neparvovec [LUXTURNA] (PDF – 2.7MB)
Refer to Section 1.1 of the clinical reviewer memo for information about participation in the clinical trials and any analysis of demographic subgroup outcomes that is notable.
Supporting Documents
Related Information
Voretigene neparvovec (Luxturna) is a novel gene therapy for the treatment of Leber’s congenital amaurosis.[1] It was developed by Spark Therapeutics and Children’s Hospital of Philadelphia.[2][3] It is the first in vivo gene therapy approved by the FDA.[4]
Leber’s congenital amaurosis, or biallelic RPE65-mediated inherited retinal disease, is an inherited disorder causing progressive blindness. Voretigene is the first treatment available for this condition.[5] The gene therapy is not a cure for the condition, but substantially improves vision in those treated.[6] It is given as an subretinal injection.
It was developed by collaboration between the University of Pennsylvania, Yale University, the University of Florida and Cornell University. In 2018, the product was launched in the U.S. by Spark Therapeutics for the treatment of children and adult patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. The same year, Spark Therapeutics received approval for the product in the E.U. for the same indication.
Chemistry and production
Voretigene neparvovec is an AAV2 vector containing human RPE65 cDNA with a modified Kozak sequence. The virus is grown in HEK 293 cells and purified for administration.[7]
History
Married researchers Jean Bennett and Albert Maguire, among others, worked for decades on studies of congenital blindness, culminating in approval of a novel therapy, Luxturna.[8]
It was granted orphan drug status for Leber congenital amaurosis and retinitis pigmentosa.[9][10] A biologics license application was submitted to the FDA in July 2017 with Priority Review.[5] Phase III clinical trial results were published in August 2017.[11] On 12 October 2017, a key advisory panel to the Food and Drug Administration (FDA), composed of 16 experts, unanimously recommended approval of the treatment.[12] The US FDA approved the drug on December 19, 2017. With the approval, Spark Therapeutics received a pediatric disease priority review voucher.[13]
The first commercial sale of voretigene neparvovec — the first for any gene therapy product in the US — occurred in March 2018.[14][14][4] The price of the treatment has been announced at $425,000 per eye.[15]
INDICATION
LUXTURNA (voretigene neparvovec-rzyl) is an adeno-associated virus vector-based gene therapy indicated for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy.
Patients must have viable retinal cells as determined by the treating physicians.
IMPORTANT SAFETY INFORMATION FOR LUXTURNA
Warnings and Precautions
-
Endophthalmitis may occur following any intraocular surgical procedure or injection. Use proper aseptic injection technique when administering LUXTURNA, and monitor for and advise patients to report any signs or symptoms of infection or inflammation to permit early treatment of any infection.
-
Permanent decline in visual acuity may occur following subretinal injection of LUXTURNA. Monitor patients for visual disturbances.
-
Retinal abnormalities may occur during or following the subretinal injection of LUXTURNA, including macular holes, foveal thinning, loss of foveal function, foveal dehiscence, and retinal hemorrhage. Monitor and manage these retinal abnormalities appropriately. Do not administer LUXTURNA in the immediate vicinity of the fovea. Retinal abnormalities may occur during or following vitrectomy, including retinal tears, epiretinal membrane, or retinal detachment. Monitor patients during and following the injection to permit early treatment of these retinal abnormalities. Advise patients to report any signs or symptoms of retinal tears and/or detachment without delay.
-
Increased intraocular pressure may occur after subretinal injection of LUXTURNA. Monitor and manage intraocular pressure appropriately.
-
Expansion of intraocular air bubbles Instruct patients to avoid air travel, travel to high elevations or scuba diving until the air bubble formed following administration of LUXTURNA has completely dissipated from the eye. It may take one week or more following injection for the air bubble to dissipate. A change in altitude while the air bubble is still present can result in irreversible vision loss. Verify the dissipation of the air bubble through ophthalmic examination.
-
Cataract Subretinal injection of LUXTURNA, especially vitrectomy surgery, is associated with an increased incidence of cataract development and/or progression.
Adverse Reactions
-
In clinical studies, ocular adverse reactions occurred in 66% of study participants (57% of injected eyes), and may have been related to LUXTURNA, the subretinal injection procedure, the concomitant use of corticosteroids, or a combination of these procedures and products.
-
The most common adverse reactions (incidence ≥5% of study participants) were conjunctival hyperemia (22%), cataract (20%), increased intraocular pressure (15%), retinal tear (10%), dellen (thinning of the corneal stroma) (7%), macular hole (7%), subretinal deposits (7%), eye inflammation (5%), eye irritation (5%), eye pain (5%), and maculopathy (wrinkling on the surface of the macula) (5%).
Immunogenicity
Immune reactions and extra-ocular exposure to LUXTURNA in clinical studies were mild. No clinically significant cytotoxic T-cell response to either AAV2 or RPE65 has been observed.
In clinical studies, the interval between the subretinal injections into the two eyes ranged from 7 to 14 days and 1.7 to 4.6 years. Study participants received systemic corticosteroids before and after subretinal injection of LUXTURNA to each eye, which may have decreased the potential immune reaction to either AAV2 or RPE65.
Pediatric Use
Treatment with LUXTURNA is not recommended for patients younger than 12 months of age, because the retinal cells are still undergoing cell proliferation, and LUXTURNA would potentially be diluted or lost during the cell proliferation. The safety and efficacy of LUXTURNA have been established in pediatric patients. There were no significant differences in safety between the different age subgroups.
Please see US Full Prescribing Information for LUXTURNA.
References:
1. LUXTURNA [package insert]. Philadelphia, PA: Spark Therapeutics, Inc; 2017. 2. Gupta PR, Huckfeldt RM. Gene therapy for inherited retinal degenerations: initial successes and future challenges. J Neural Eng. 2017;14(5):051002. 3. Kay C. Gene therapy: the new frontier for inherited retinal disease. Retina Specialist. March 2017. http://www.retina-specialist.com/CMSDocuments/2017/03/RS/rs0317I.pdf. Accessed November 14, 2017 4. Polinski NK, Gombash SE, Manfredsson FP, et al. Recombinant adeno-associated virus 2/5-mediated gene transfer is reduced in the aged rat midbrain. Neurobiol Aging. 2015;36(2):1110-1120. 5. Moore T. Restoring retinal function in a mouse model of hereditary blindness. PLoS Med. 2005;2(11):e399. 6. McBee JK, Van Hooser JP, Jang GF, Palczewski K. Isomerization of 11-cis-retinoids to all-trans-retinoids in vitro and in vivo. J Biol Chem. 2001;276(51):48483-48493. 7. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346-358. 8. Trapani I, Puppo A, Auricchio A. Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res. 2014;43:108-128. 9. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849-860.


PAPERS
Progress in Retinal and Eye Research (2018), 63, 107-131
Lancet (2017), 390(10097), 849-860.
References
- ^ “Luxturna (voretigene neparvovec-rzyl) label” (PDF). FDA. December 2017. Retrieved 31 December 2017. (for label updates, see FDA index page)
- ^ “Spark’s gene therapy for blindness is racing to a historic date with the FDA”. Statnews.com. 9 October 2017. Retrieved 9 October 2017.
- ^ Clarke,Reuters, Toni. “Gene Therapy for Blindness Appears Initially Effective, Says U.S. FDA”. Scientific American. Retrieved 2017-10-12.
- ^ Jump up to:a b “First Gene Therapy For Inherited Disease Gets FDA Approval”. NPR.org. 19 Dec 2017.
- ^ Jump up to:a b “Press Release – Investors & Media – Spark Therapeutics”. Ir.sparktx.com. Retrieved 9 October 2017.
- ^ McGinley, Laurie (19 December 2017). “FDA approves first gene therapy for an inherited disease”. Washington Post.
- ^ Russell, Stephen; Bennett, Jean; Wellman, Jennifer A.; Chung, Daniel C.; Yu, Zi-Fan; Tillman, Amy; Wittes, Janet; Pappas, Julie; Elci, Okan; McCague, Sarah; Cross, Dominique; Marshall, Kathleen A.; Walshire, Jean; Kehoe, Taylor L.; Reichert, Hannah; Davis, Maria; Raffini, Leslie; George, Lindsey A.; Hudson, F Parker; Dingfield, Laura; Zhu, Xiaosong; Haller, Julia A.; Sohn, Elliott H.; Mahajan, Vinit B.; Pfeifer, Wanda; Weckmann, Michelle; Johnson, Chris; Gewaily, Dina; Drack, Arlene; et al. (2017). “Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 -mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial”. The Lancet. 390 (10097): 849–860. doi:10.1016/S0140-6736(17)31868-8. PMC 5726391. PMID 28712537.
- ^ “FDA approves Spark’s gene therapy for rare blindness pioneered at CHOP – Philly”. Philly.com. Retrieved 2018-03-24.
- ^ “Voretigene neparvovec – Spark Therapeutics – AdisInsight”. adisinsight.springer.com.
- ^ Ricki Lewis, PhD (October 13, 2017). “FDA Panel Backs Gene Therapy for Inherited Blindness”. Medscape.
- ^ Lee, Helena; Lotery, Andrew (2017). “Gene therapy for RPE65 -mediated inherited retinal dystrophy completes phase 3”. The Lancet. 390 (10097): 823–824. doi:10.1016/S0140-6736(17)31622-7. PMID 28712536.
- ^ “Landmark Therapy to Treat Blindness Gets One Step Closer to FDA Approval”. Bloomberg.com. 2017-10-12. Retrieved 2017-10-12.
- ^ “Spark grabs FDA nod for Luxturna, a breakthrough gene therapy likely bearing a pioneering price”. FiercePharma.
- ^ Jump up to:a b “The anxious launch of Luxturna, a gene therapy with a record sticker price”. STAT. 2018-03-21. Retrieved 2018-03-24.
- ^ Tirrell, Meg (3 January 2018). “A US drugmaker offers to cure rare blindness for $850,000”. CNBC. Retrieved 3 January 2018.
Further reading
- Ledford, Heidi (2017). “FDA advisers back gene therapy for rare form of blindness”. Nature. 550 (7676): 314. doi:10.1038/nature.2017.22819. PMID 29052639.
- Wilson, James M. (2018). “Interview with Jean Bennett, MD, PhD”. Human Gene Therapy Clinical Development. 29 (1): 7–9. doi:10.1089/humc.2018.29032.int. PMID 29641279.
- Ameri, Hossein (2018). “Prospect of retinal gene therapy following commercialization of voretigene neparvovec-rzyl for retinal dystrophy mediated by RPE65 mutation”. Journal of Current Ophthalmology. 30 (1): 1–2. doi:10.1016/j.joco.2018.01.006. PMC 5859497. PMID 29564403.
- Russell, Stephen; Bennett, Jean; Maguire, Albert M.; High, Katherine A. (2018). “Voretigene neparvovec-rzyl for the treatment of biallelic RPE65 mutation–associated retinal dystrophy”. Expert Opinion on Orphan Drugs. 6 (8): 457–464. doi:10.1080/21678707.2018.1508340.
- Bakall, Benjamin; Hariprasad, Seenu M.; Klein, Kendra A. (2018). “Emerging Gene Therapy Treatments for Inherited Retinal Diseases”. Ophthalmic Surgery, Lasers and Imaging Retina. 49 (7): 472–478. doi:10.3928/23258160-20180628-02. PMID 30021033.
- “Drug and Device News”. P & T. 43 (2): 74–104. 2018. PMC 5768294. PMID 29386862.
| Gene therapy | |
|---|---|
| Vector | Adeno-associated virusserotype 2 |
| Nucleic acid type | DNA |
| Editing method | RPE65 |
| Clinical data | |
| Trade names | Luxturna |
| Pregnancy category |
|
| Routes of administration |
subretinal injection |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| KEGG | |
//////////FDA 2017, Voretigene neparvovec , Voretigene neparvovec-rzyl, Luxturna, ボレチジーンネパルボベック, 1646819-03-5 , FDA Luxturna, SPARK THERAPEUTICS, Vision loss treatment, Retinal dystrophy., AAV2-hRPE65v2, LTW-888, SPK-RPE65, Orphan drug,
Elapegademase, エラペグアデマーゼ (遺伝子組換え)
AQTPAFNKPK VELHVHLDGA IKPETILYYG RKRGIALPAD TPEELQNIIG MDKPLSLPEF
LAKFDYYMPA IAGSREAVKR IAYEFVEMKA KDGVVYVEVR YSPHLLANSK VEPIPWNQAE
GDLTPDEVVS LVNQGLQEGE RDFGVKVRSI LCCMRHQPSW SSEVVELCKK YREQTVVAID
LAGDETIEGS SLFPGHVKAY AEAVKSGVHR TVHAGEVGSA NVVKEAVDTL KTERLGHGYH
TLEDTTLYNR LRQENMHFEV CPWSSYLTGA WKPDTEHPVV RFKNDQVNYS LNTDDPLIFK
STLDTDYQMT KNEMGFTEEE FKRLNINAAK SSFLPEDEKK ELLDLLYKAY GMPSPA

>>Elapegademase<<< AQTPAFNKPKVELHVHLDGAIKPETILYYGRKRGIALPADTPEELQNIIGMDKPLSLPEF LAKFDYYMPAIAGSREAVKRIAYEFVEMKAKDGVVYVEVRYSPHLLANSKVEPIPWNQAE GDLTPDEVVSLVNQGLQEGERDFGVKVRSILCCMRHQPSWSSEVVELCKKYREQTVVAID LAGDETIEGSSLFPGHVKAYAEAVKSGVHRTVHAGEVGSANVVKEAVDTLKTERLGHGYH TLEDTTLYNRLRQENMHFEVCPWSSYLTGAWKPDTEHPVVRFKNDQVNYSLNTDDPLIFK STLDTDYQMTKNEMGFTEEEFKRLNINAAKSSFLPEDEKKELLDLLYKAYGMPSPA
Elapegademase, エラペグアデマーゼ (遺伝子組換え)
EZN-2279
Protein chemical formula C1797H2795N477O544S12
Protein average weight 115000.0 Da
Peptide
APPROVED, FDA, Revcovi, 2018/10/5
CAS: 1709806-75-6
Elapegademase-lvlr, Poly(oxy-1,2-ethanediyl), alpha-carboxy-omega-methoxy-, amide with adenosine deaminase (synthetic)
EZN-2279; PEG-rADA; Pegademase recombinant – Leadiant Biosciences; Pegylated recombinant adenosine deaminase; Polyethylene glycol recombinant adenosine deaminase; STM-279, UNII: 9R3D3Y0UHS
- Originator Sigma-Tau Pharmaceuticals
- Developer Leadiant Biosciences; Teijin Pharma
- Class Antivirals; Polyethylene glycols
- Mechanism of Action Adenosine deaminase stimulants
- Orphan Drug Status Yes – Immunodeficiency disorders; Adenosine deaminase deficiency
- Registered Adenosine deaminase deficiency; Immunodeficiency disorders
- 05 Oct 2018 Registered for Adenosine deaminase deficiency (In adults, In children) in USA (IM)
- 05 Oct 2018 Registered for Immunodeficiency disorders (In adults, In children) in USA (IM)
- 04 Oct 2018 Elapegademase receives priority review status for Immunodeficiency disorders and Adenosine deaminase deficiency in USA
検索キーワード:Elapegademase (Genetical Recombination)
検索件数:1
| エラペグアデマーゼ(遺伝子組換え) Elapegademase (Genetical Recombination)  [1709806-75-6] |
Elapegademase is a PEGylated recombinant adenosine deaminase. It can be defined molecularly as a genetically modified bovine adenosine deaminase with a modification in cysteine 74 for serine and with about 13 methoxy polyethylene glycol chains bound via carbonyl group in alanine and lysine residues.[4] Elapegademase is generated in E. coli, developed by Leadiant Biosciences and FDA approved on October 5, 2018.[1, 5]
Indication
Elapegademase is approved for the treatment of adenosine deaminase severe combined immune deficiency (ADA-SCID) in pediatric and adult patients.[1] This condition was previously treated by the use of pegamedase bovine as part of an enzyme replacement therapy.[2]
ADA-SCID is a genetically inherited disorder that is very rare and characterized by a deficiency in the adenosine deaminase enzyme. The patients suffering from this disease often present a compromised immune system. This condition is characterized by very low levels of white blood cells and immunoglobulin levels which results in severe and recurring infections.[3]
Pharmacodynamics
In clinical trials, elapegademase was shown to increase adenosine deaminase activity while reducing the concentrations of toxic metabolites which are the hallmark of ADA-SCID. As well, it was shown to improve the total lymphocyte count.[6]
Mechanism of action
The ADA-SCID is caused by the presence of mutations in the ADA gene which is responsible for the synthesis of adenosine deaminase. This enzyme is found throughout the body but it is mainly active in lymphocytes. The normal function of adenosine deaminase is to eliminate deoxyadenosine, created when DNA is degraded, by converting it into deoxyinosine. This degradation process is very important as deoxyadenosine is cytotoxic, especially for lymphocytes. Immature lymphocytes are particularly vulnerable as deoxyadenosine kills them before maturation making them unable to produce their immune function.[3]
Therefore, based on the causes of ADA-SCID, elapegademase works by supplementing the levels of adenosine deaminase. Being a recombinant and an E. coli-produced molecule, the use of this drug eliminates the need to source the enzyme from animals, as it was used previously.[1]
Absorption
Elapegademase is administered intramuscularly and the reported Tmax, Cmax and AUC are approximately 60 hours, 240 mmol.h/L and 33000 hr.mmol/L as reported during a week.[Label]
Volume of distribution
This pharmacokinetic property has not been fully studied.
Protein binding
This pharmacokinetic property is not significant as the main effect is in the blood cells.
Metabolism
Metabolism studies have not been performed but it is thought to be degraded by proteases to small peptides and individual amino acids.
Route of elimination
This pharmacokinetic property has not been fully studied.
Half life
This pharmacokinetic property has not been fully studied.
Clearance
This pharmacokinetic property has not been fully studied.
Toxicity
As elapegademase is a therapeutic protein, there is a potential risk of immunogenicity.
There are no studies related to overdose but the highest weekly prescribed dose in clinical trials was 0.4 mg/kg. In nonclinical studies, a dosage of 1.8 fold of the clinical dose produced a slight increase in the activated partial thromboplastin time.[Label]
FDA label. Download (145 KB)
General References
- Rare DR [Link]
- Globe News Wire [Link]
- NIH [Link]
- NIHS reports [File]
- WHO Drug Information 2017 [File]
- Revcovi information [File]
/////////////Elapegademase, Peptide, エラペグアデマーゼ (遺伝子組換え) , EZN-2279, Elapegademase-lvlr, Orphan Drug, STM 279, FDA 2018
COCCOC(=O)NCCCC[C@H](N)C(=O)O
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent

READ
ANTHONY MELVIN CRASTO

DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
CALL +919323115463 INDIA
//////////////
Calaspargase pegol, カラスパルガーゼペゴル
|
LPNITILATG GTIAGGGDSA TKSNYTAGKV GVENLVNAVP QLKDIANVKG EQVVNIGSQD
MNDDVWLTLA KKINTDCDKT DGFVITHGTD TMEETAYFLD LTVKCDKPVV MVGAMRPSTS MSADGPFNLY NAVVTAADKA SANRGVLVVM NDTVLDGRDV TKTNTTDVAT FKSVNYGPLG YIHNGKIDYQ RTPARKHTSD TPFDVSKLNE LPKVGIVYNY ANASDLPAKA LVDAGYDGIV SAGVGNGNLY KTVFDTLATA AKNGTAVVRS SRVPTGATTQ DAEVDDAKYG FVASGTLNPQ KARVLLQLAL TQTKDPQQIQ QIFNQY (tetramer; disulfide bridge 77-105, 77′-105′, 77”-105”, 77”’-105”’) |

Calaspargase pegol
Molecular Formula, C1516-H2423-N415-O492-S8 (peptide monomer), Molecular Weight, 10261.2163
APPROVED, Asparlas, FDA 2018/12/20
CAS 941577-06-6
UNII T9FVH03HMZ
カラスパルガーゼペゴル;
(27-Alanine,64-aspartic acid,252-threonine,263-asparagine)-L-asparaginase 2 (EC 3.5.1.1, L-asparagineamidohydrolase II) Escherichia coli (strain K12) tetramer alpha4, carbamates with alpha-carboxy-omega-methoxypoly(oxyethylene)
Asparaginase (Escherichia coli isoenzyme II), conjugate with alpha-(((2,5-dioxo-1-pyrrolidinyl)oxy)carbonyl)-omega-methoxypoly(oxy-1,2-ethanediyl)
|
Peptide
|
- Calaspargase pegol
- calaspargase pegol-mknl
- EZN-2285
- Used to treat acute lymphoblastic leukemia., Antineoplastic
- BAX-2303
SC-PEG E. Coli L-asparaginase
SHP-663
Calaspargase pegol-mknl (trade name Asparlas) is a drug for the treatment of acute lymphoblastic leukemia (ALL). It is approved by the Food and Drug Administration for use in the United States as a component of a multi-agent chemotherapeutic regimen for ALL in pediatric and young adult patients aged 1 month to 21 years.[1]
Calaspargase pegol was first approved in 2018 in the U.S. as part of a multi-agent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia.
In 2008, orphan drug designation was assigned in the E.U.
Calaspargase pegol is an engineered protein consisting of the E. coli-derived enzyme L-asparaginase II conjugated with succinimidyl carbonate monomethoxypolyethylene glycol (pegol).[2] The L-asparaginase portion hydrolyzes L-asparagine to L-aspartic acid depriving the tumor cell of the L-asparagine it needs for survival.[2] The conjugation with the pegol group increases the half-life of the drug making it longer acting.
Asparaginase is an important agent used to treat acute lymphoblastic leukemia (ALL) [1]. Asparagine is incorporated into most proteins, and the synthesis of proteins is stopped when asparagine is absent, which inhibits RNA and DNA synthesis, resulting in a halt in cellular proliferation. This forms the basis of asparaginase treatment in ALL [1], [2], [6].
Calaspargase pegol, also known as asparlas, is an asparagine specific enzyme which is indicated as a part of a multi-agent chemotherapy regimen for the treatment of ALL [3]. The asparagine specific enzyme is derived from Escherichia coli, as a conjugate of L-asparaginase (L-asparagine amidohydrolase) and monomethoxypolyethylene glycol (mPEG) with a succinimidyl carbonate (SC) linker to create a stable molecule which increases the half-life and decreases the dosing frequency [Label], [1].
Calaspargase pegol, by Shire pharmaceuticals, was approved by the FDA on December 20, 2018 for acute lymphoblastic anemia (ALL) [3].
Indication
This drug is is an asparagine specific enzyme indicated as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia in pediatric and young adult patients age 1 month to 21 years [Label].
The pharmacokinetics of calaspargase pegol were examined when given in combination with multiagent chemotherapy in 124 patients with B-cell lineage ALL [3]. The FDA approval of this drug was based on the achievement and maintenance of nadir serum asparaginase activity above the level of 0.1 U/mL when administering calaspargase, 2500 U/m2 intravenously, at 3-week intervals.
Associated Conditions
Pharmacodynamics
The effect of this drug is believed to occur by selective killing of leukemic cells due to depletion of plasma L-asparagine. Leukemic cells with low expression of asparagine synthetase are less capable of producing L-asparagine, and therefore rely on exogenous L-asparagine for survival [Label]. When asparagine is depleted, tumor cells cannot proliferate [6].
During remission induction, one dose of SC-PEG (2500 IU/m2) results in a sustained therapeutic serum asparaginase activity (SAA) without excessive toxicity or marked differences in the proportion of patients with low end-induction minimum residual disease (MRD) [5].
Pharmacodynamic (PD) response was studied through measurement of plasma and cerebrospinal fluid (CSF) asparagine concentrations with an LC-MS/MS assay (liquid chromatography–mass spectrometry). Asparagine concentration in plasma was sustained below the assay limit of quantification for more than 18 days after one dose of calaspargase pegol, 2,500 U/m2, during the induction phase of treatment. Average cerebrospinal asparagine concentrations decreased from a pretreatment concentration of 0.8 μg/mL (N=10) to 0.2 μg/mL on Day 4 (N=37) and stayed decreased at 0.2 μg/mL (N=35) 25 days after the administration of one of 2,500 U/m2 in the induction phase [Label].
Mechanism of action
L-asparaginase (the main component of this drug) is an enzyme that catalyzes the conversion of the amino acid L-asparagine into both aspartic acid and ammonia [Label], [2]. This process depletes malignant cells of their required asparagine. The depletion of asparagine then blocks protein synthesis and tumor cell proliferation, especially in the G1 phase of the cell cycle. As a result, tumor cell death occurs. Asparagine is important in protein synthesis in acute lymphoblastic leukemia (ALL) cells which, unlike normal cells, cannot produce this amino acid due to lack of the enzyme asparagine synthase [2], [Label].
Pegylation decreases enzyme antigenicity and increases its half-life. Succinimidyl carbamate (SC) is used as a PEG linker to facilitate attachment to asparaginase and enhances the stability of the formulation [4], [1]. SC-PEG urethane linkages formed with lysine groups are more hydrolytically stable [2].
Toxicity
Pancreatitis, hepatotoxicity, hemorrhage, and thrombosis have been observed with calaspargase pegol use [Label].
Pancreatitis: Discontinue this drug in patients with pancreatitis, and monitor blood glucose.
Hepatotoxicity: Hepatic function should be tested regularly, and trough levels of this drug should be measured during the recovery phase of the drug cycle [Label].
Hemorrhage or Thrombosis: Discontinue this drug in serious or life-threatening hemorrhage or thrombosis. In cases of hemorrhage, identify the cause of hemorrhage and treat appropriately. Administer anticoagulant therapy as indicated in thrombotic events [Label].
A note on hypersensitivity:
Observe the patient for 1 hour after administration of calaspargase pegol for possible hypersensitivity [Label]. In cases of previous hypersensitivity to this drug, discontinue this drug immediately.
Lactation: Advise women not to breastfeed while taking this drug [Label].
Pregnancy: There are no available data on the use of calaspargase pegol in pregnant women to confirm a risk of drug-associated major birth defects and miscarriage. Published literature studies in pregnant animals suggest asparagine depletion can cause harm to the animal offspring. It is therefore advisable to inform women of childbearing age of this risk. The background risk of major birth defects and miscarriage for humans is unknown at this time [Label].
Pregnancy testing should occur before initiating treatment. Advise females of reproductive potential to avoid becoming pregnant while taking this drug. Females should use effective contraceptive methods, including a barrier methods, during treatment and for at least 3 months after the last dose. There is a risk for an interaction between calaspargase pegol and oral contraceptives. The concurrent use of this drug with oral contraceptives should be avoided. Other non-oral contraceptive methods should be used in women of childbearing potential [Label].
References
- Angiolillo AL, Schore RJ, Devidas M, Borowitz MJ, Carroll AJ, Gastier-Foster JM, Heerema NA, Keilani T, Lane AR, Loh ML, Reaman GH, Adamson PC, Wood B, Wood C, Zheng HW, Raetz EA, Winick NJ, Carroll WL, Hunger SP: Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children’s Oncology Group Study AALL07P4. J Clin Oncol. 2014 Dec 1;32(34):3874-82. doi: 10.1200/JCO.2014.55.5763. Epub 2014 Oct 27. [PubMed:25348002]
- Appel IM, Kazemier KM, Boos J, Lanvers C, Huijmans J, Veerman AJ, van Wering E, den Boer ML, Pieters R: Pharmacokinetic, pharmacodynamic and intracellular effects of PEG-asparaginase in newly diagnosed childhood acute lymphoblastic leukemia: results from a single agent window study. Leukemia. 2008 Sep;22(9):1665-79. doi: 10.1038/leu.2008.165. Epub 2008 Jun 26. [PubMed:18580955]
- Blood Journal: Randomized Study of Pegaspargase (SS-PEG) and Calaspargase Pegol (SPC-PEG) in Pediatric Patients with Newly Diagnosed Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results of DFCI ALL Consortium Protocol 11-001 [Link]
References
- ^ “FDA approves longer-acting calaspargase pegol-mknl for ALL” (Press release). Food and Drug Administration. December 20, 2018.
- ^ Jump up to:a b “Calaspargase pegol-mknl”. NCI Drug Dictionary. National Cancer Institute.
FDA label, Download(300 KB)
General References
- Angiolillo AL, Schore RJ, Devidas M, Borowitz MJ, Carroll AJ, Gastier-Foster JM, Heerema NA, Keilani T, Lane AR, Loh ML, Reaman GH, Adamson PC, Wood B, Wood C, Zheng HW, Raetz EA, Winick NJ, Carroll WL, Hunger SP: Pharmacokinetic and pharmacodynamic properties of calaspargase pegol Escherichia coli L-asparaginase in the treatment of patients with acute lymphoblastic leukemia: results from Children’s Oncology Group Study AALL07P4. J Clin Oncol. 2014 Dec 1;32(34):3874-82. doi: 10.1200/JCO.2014.55.5763. Epub 2014 Oct 27. [PubMed:25348002]
- Appel IM, Kazemier KM, Boos J, Lanvers C, Huijmans J, Veerman AJ, van Wering E, den Boer ML, Pieters R: Pharmacokinetic, pharmacodynamic and intracellular effects of PEG-asparaginase in newly diagnosed childhood acute lymphoblastic leukemia: results from a single agent window study. Leukemia. 2008 Sep;22(9):1665-79. doi: 10.1038/leu.2008.165. Epub 2008 Jun 26. [PubMed:18580955]
- Asparlas Approval History [Link]
- NCI: Calaspargase Pegol [Link]
- Blood Journal: Randomized Study of Pegaspargase (SS-PEG) and Calaspargase Pegol (SPC-PEG) in Pediatric Patients with Newly Diagnosed Acute Lymphoblastic Leukemia or Lymphoblastic Lymphoma: Results of DFCI ALL Consortium Protocol 11-001 [Link]
- Medsafe NZ: Erwinaze inj [File]
| Clinical data | |
|---|---|
| Trade names | Asparlas |
| Synonyms | EZN-2285 |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
/////////////Calaspargase pegol, Peptide, FDA 2018, EZN-2285, カラスパルガーゼペゴル , BAX-2303, SC-PEG E. Coli L-asparaginase , SHP-663, orphan drug
CC(C)C[C@@H](C(=O)O)NC(=O)OCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOC.COCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOC(=O)NCCCC[C@@H](C(=O)O)N
