
Home » Posts tagged 'Orphan Drug' (Page 2)
Tag Archives: Orphan Drug
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Omaveloxolone



Omaveloxolone
CAS
1474034-05-3
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-octadecahydropicen-4a-yl]-2,2-difluoropropanamide
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,3,4,5,6,7,8,8a,14a,14b-decahydropicen-4a-yl]-2,2-difluoropropanamide
FDA 2023, 2/28/2023, To treat Friedrich’s ataxia
Drug Trials Snapshot
WeightAverage: 554.723
Monoisotopic: 554.331999611
Chemical FormulaC33H44F2N2O3
- RTA 408
- RTA-408
- OriginatorDartmouth College; University of Texas M. D. Anderson Cancer Center
- DeveloperBiogen
- ClassAnalgesics; Anti-inflammatories; Antineoplastics; Eye disorder therapies; Neuroprotectants; Small molecules; Triterpenes
- Mechanism of ActionNF-E2-related factor 2 stimulants
- Orphan Drug StatusYes – Friedreich’s ataxia; Malignant melanoma
- MarketedFriedreich’s ataxia
- Phase IIMitochondrial disorders; Ocular inflammation; Ocular pain
- Phase I/IIMalignant melanoma
- PreclinicalBrain disorders
- DiscontinuedDuchenne muscular dystrophy; Non-small cell lung cancer; Radiation-induced skin damage
- 08 Apr 2025Biogen completes a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
- 17 Mar 2025Registered for Friedreich’s ataxia (In adolescents, In adults) in Canada (PO)
- 18 Oct 2024Biogen initiates enrolment in a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
Omaveloxolone, sold under the brand name Skyclarys, is a medication used for the treatment of Friedreich’s ataxia.[2][5] It is taken by mouth.[2]
The most common side effects include an increase in alanine transaminase and an increase of aspartate aminotransferase, which can be signs of liver damage, headache, nausea, abdominal pain, fatigue, diarrhea and musculoskeletal pain.[5]
Omaveloxolone was approved for medical use in the United States in February 2023,[2][5][6][7][8] and in the European Union in February 2024.[3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[9]
SYNTHESIS

PATENT
Sheikh, AY et al. (2018). Bardoxolonmethyl-2,2-difluoropropionamide derivatives, polymorphe forms and procedures for use thereof. DK/EP 2989114 T3. Danish Patent and Trademark Office. Available at https://patentimages.storage.googleapis.com/51/87/43/97d0fb3e69ee73/DK2989114T3.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP159939262&_cid=P21-MAKI10-93498-1

[0164] Reagents and conditions: (a) (PhO) 2PON 3 (DPPA), triethylamine, toluene, 0 °C for 5 minutes, then ambient temperature overnight, ∼94%; (b) benzene, 80 °C for 2 hours; (c) HCl, CH 3CN, ambient temperature for 1 hour; (d) CH 3CF 2CO 2H, dicyclohexylcarbodiimide, 4-(dimethylamino)pyridine, CH 2Cl 2, ambient temperature overnight, 73% from RTA 401 (4 steps).
[0165]Compound 1: RTA 401 (20.0 g, 40.6 mmol), triethylamine (17.0 mL, 122.0 mmol), and toluene (400 mL) were added into a reactor and cooled to 0 °C with stirring. Diphenyl phosphoryl azide (DPPA) (13.2 mL, 61.0 mmol) was added with stirring at 0 °C over 5 minutes, and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no RTA 401 left). The reaction mixture was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 5% ethyl acetate in CH 2Cl 2) to give compound 1 (19.7 g, ∼94%, partially converted into compound 2) as a white foam.
[0166]Compound 2: Compound 1 (19.7 g, ∼38.1 mmol) and benzene (250 mL) were added into a reactor and heated to 80 °C with stirring for 2 hours (HPLC-MS check shows no compound 1 left). The reaction mixture was concentrated at reduced pressure to afford crude compound 2 as a solid residue, which was used for the next step without purification.
[0167]Compound 3: Crude compound 2 (≤38.1 mmol) and CH 3CN (200 mL) were added into a reactor and cooled to 0 °C with stirring. HCl (12 N, 90 mL) was added at 0 °C over 1 minute, and the mixture was continually stirred at room temperature for 1 hour (HPLC-MS check shows no compound 2 left). The reaction mixture was cooled to 0 °C and 10% NaOH (∼500 mL) was added with stirring. Then, saturated NaHCO 3 (1 L) was added with stirring. The aqueous phase was extracted by ethyl acetate (2×500 mL). The combined organic phase was washed by H 2O (200 mL), saturated NaCl (200 mL), dried over Na 2SO 4, and concentrated to afford crude compound 3 (16.62 g) as a light yellow foam, which was used for the next step without purification.
[0168]RTA 408: Crude amine 3 (16.62 g, 35.9 mmol), CH 3CF 2CO 2H (4.7388 g, 43.1 mmol), and CH 2Cl 2 (360 mL) were added into a reactor with stirring at room temperature. Then, dicyclohexylcarbodiimide (DCC) (11.129 g, 53.9 mmol) and 4-(dimethylamino)pyridine (DMAP) (1.65 g, 13.64 mmol) were added and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no compound 3 left). The reaction mixture was filtered to remove solid by-products, and the filtrate was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 20% ethyl acetate in hexanes) twice to give compound RTA 408 (16.347 g, 73% from RTA 401 over 4 steps) as a white foam: 1H NMR (400 MHz, CD 3Cl) δ ppm 8.04 (s, 1H), 6.00 (s, 1H), 5.94 (s, br, 1H), 3.01 (d, 1H, J = 4.8 Hz), 2.75-2.82 (m, 1H), 1.92-2.18 (m, 4H), 1.69-1.85 (m, 7H), 1.53-1.64 (m, 1H), 1.60 (s, 3H), 1.50 (s, 3H), 1.42 (s, 3H), 1.11-1.38 (m, 3H), 1.27 (s, 3H), 1.18 (s, 3H), 1.06 (s, 3H), 1.04 (s, 3H), 0.92 (s, 3H); m/z 555 (M+1).
SYNTHESIS
J. Med. Chem. 2025, 68, 2147−2182
Omaveloxolone (Skyclarys). Omaveloxolone (6) was approved in February 2023 for the treatment of Friedreich’s Ataxia (FRDA), a genetic, neurodegenerative disease. Patients with FRDA have lowered activity of the frataxin gene (FXN), attributed to an expansion of a guanine-adenine-adenine (GAA)
triplet. The resulting decrease in frataxin limits the production of iron−sulfur clusters, leading to accumulation of iron in the mitochondria and oxidative stress which in turn leads to cell damageanddeath.49
Omaveloxoloneactivates the nuclear factor erythroid 2-related factor 2 (Nrf2), an important pathway in
oxidative stress. It acts by preventing ubiquitination and subsequent degradation of Nrf2, keeping levels high enough to counteract the oxidative stress associated with FRDA. 50
Omaveloxolone was developed by Reata Pharmaceuticals (which was acquired by Biogen in September 2023) and was granted orphan drug, fast track, priority review, and rare pediatric disease designations. 51Omaveloxolone (6) is a semisynthetic triterpenoid based on the oleanolic acid scaffold.52
advanced intermediate 6.1,The synthesis started from the53also known as CDDO orbardoxolone, which has individually been investigated fortherapeutic benefits from Nrf2 activation (Scheme 10).
Treatment of acid 6.1 with DPPA produced the azide, and subsequent heating in benzene generated isocyanate 6.2 via aCurtius rearrangement. Hydrolysis with aqueous acid generated amine 6.3, and an amidation with 2,2-difluoropropanoic acid produced omaveloxolone (6). A yield of 73% over the sequence was reported, and intermediates were used crude with no purification between steps.
(49) Ghanekar, S. D.; Miller, W. W.; Meyer, C. J.; Fenelon, K. J.;
Lacdao, A.; Zesiewicz, T. A. Orphan drugs in development for the
treatment of Friedreich’s ataxia: focus on omaveloxolone. Degener.
Neurol. Neuromuscular Dis. 2019, 9, 103−107.
(50) Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-inducer
prevents mitochondrial defects and oxidative stress in Friedreich’s
ataxia models. Front. Cell. Neurosci. 2018, 12, 188.
(51) Lee,A.Omaveloxolone:first approval. Drugs 2023, 83, 725−729.
(52) Anderson, E.; Decker, A.; Liu, X. Synthesis, pharmaceutical use,
and characterization of crystalline forms of 2,2-difluoropropionamide
derivatives of bardoxolone methyl. WO 2013163344, 2013.
(53) Honda, T.; Rounds, B. V.; Gribble, G. W.; Suh, N.; Wang, Y.;
Sporn, M. B. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9
dien-28-oic acid, a novel and highly active inhibitor of nitric oxide
production in mouse macrophages. Bioorg. Med. Chem. Lett. 1998, 8,
2711−2714.

SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Omaveloxolone (Skyclarys)
Omaveloxolone was granted FDA approval on February 28, 2023, to treat Friedrich’s ataxia in individuals aged 16 and older [2]. Omaveloxolone possesses antioxidant and anti-inflammatory properties, making it a semi-synthetic triterpenoid compound. It has the ability to function as a stimulator of nuclear factor-erythroid 2 related factor 2(Nrf2), a transcription factor that reduces oxidative stress. In individuals
suffering from FA, a genetic disorder characterized by mitochondrial dysfunction, the Nrf2 pathway is compromised, leading to a decrease in Nrf2 activity. Hence, Omaveloxolone, an Nrf2 activator, can be
employed as a therapeutic option for the management of these in dividuals [23].The process route of Omaveloxolone is described below in Scheme 724]. The substitution reaction of carboxylic acid OMAV-001 with diphenylphosphoryl azide (DPPA) gave the acyl azide OMAV-002,which underwent Curtius-rearrangement under heating conditions to produce isocyanate OMAV-003. The amine OMAV-004 was obtained under acidic conditions. OMAV-004 was condensed with 2,2-difluoro propionic acid to obtain the final product Omaveloxolone.
[23] B.L. Probst, I. Trevino, L. McCauley, R. Bumeister, I. Dulubova, W.C. Wigley, D.
A. Ferguson, RTA 408, A novel synthetic triterpenoid with broad anticancer and
anti-inflammatory activity, PLoS One 10 (2015) e0122942.
[24] E. Anderson, A. Decker, X. Liu Synthesis, Pharmaceutical Use, and
Characterization of Crystalline Forms of 2,2-difluoropropionamide Derivatives of
Bardoxolone Methyl, 2013. WO2013163344.

.
Medical uses
Omaveloxolone is indicated for the treatment of Friedreich’s ataxia.[2][5]
Friedreich’s ataxia causes progressive damage to the spinal cord, peripheral nerves, and the brain, resulting in uncoordinated muscle movement, poor balance, difficulty walking, changes in speech and swallowing, and a shortened lifespan.[5] The condition can also cause heart disease.[5] This disease tends to develop in children and teenagers and gradually worsens over time.[5]
Although rare, Friedreich’s ataxia is the most common form of hereditary ataxia in the United States, affecting about one in every 50,000 people.[5]
Mechanism of action
The mechanism of action of omaveloxolone and its related compounds has been demonstrated to be through a combination of activation of the antioxidative transcription factor Nrf2 and inhibition of the pro-inflammatory transcription factor NF-κB.[10]
Nrf2 transcriptionally regulates multiple genes that play both direct and indirect roles in producing antioxidative potential and the production of cellular energy (i.e., adenosine triphosphate or ATP) within the mitochondria. Consequently, unlike exogenously administered antioxidants (e.g., vitamin E or Coenzyme Q10), which provide a specific and finite antioxidative potential, omaveloxolone, through Nrf2, broadly activates intracellular and mitochondrial antioxidative pathways, in addition to pathways that may directly increase mitochondrial biogenesis (such as PGC1α) and bioenergetics.[11]
History
Omaveloxolone is a second generation member of the synthetic oleanane triterpenoid compounds and in clinical development by Reata Pharmaceuticals. Preclinical studies have demonstrated that omaveloxolone possesses antioxidative and anti-inflammatory activities[10][12] and the ability to improve mitochondrial bioenergetics.[11] Omaveloxolone is under clinical investigation for a variety of indications, including Friedreich’s ataxia, mitochondrial myopathies, immunooncology, and prevention of corneal endothelial cell loss following cataract surgery.
The efficacy and safety of omaveloxolone was evaluated in a 48-week randomized, placebo-controlled, and double-blind study [Study 1 (NCT02255435)] and an open-label extension.[5] Study 1 enrolled 103 individuals with Friedreich’s ataxia who received placebo (52 individuals) or omaveloxolone 150 mg (51 individuals) for 48 weeks.[5] Of the research participants, 53% were male, 97% were white, and the mean age was 24 years at study entry.[5] Nine (18%) patients were younger than age 18.[5] The primary objective was to evaluate the change in the modified Friedreich’s Ataxia Rating Scale (mFARS) score compared to placebo at week 48.[5] The mFARS is a clinical assessment that measures disease progression, namely swallowing and speech (bulbar), upper limb coordination, lower limb coordination, and upright stability.[5] Individuals receiving omaveloxolone performed better on the mFARS than people receiving placebo.[5]
The US Food and Drug Administration (FDA) granted the application for omaveloxolone orphan drug, fast track, priority review, and rare pediatric disease designations.[5][9]
Society and culture
Legal status
Omaveloxolone was approved for medical use in the United States in February 2023.[2][5]
In December 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Skyclarys, intended for the treatment of Friedreich’s ataxia.[3] The applicant for this medicinal product is Reata Ireland Limited.[3] Omaveloxolone was approved for medical use in the European Union in February 2024.[3][4]
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ Jump up to:a b c d e f “Skyclarys- omaveloxolone capsule”. DailyMed. 12 May 2023. Archived from the original on 1 July 2023. Retrieved 16 December 2023.
- ^ Jump up to:a b c d e “Skyclarys EPAR”. European Medicines Agency (EMA). 14 December 2023. Archived from the original on 15 December 2023. Retrieved 16 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Skyclarys product information”. Union Register of medicinal products. 12 February 2024. Retrieved 19 February 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA approves first treatment for Friedreich’s ataxia”. U.S. Food and Drug Administration (FDA). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Reata Pharmaceuticals Announces FDA Approval of Skyclarys (Omavaloxolone), the First and Only Drug Indicated for Patients with Friedreich’s Ataxia”. Reata Pharmaceuticals Inc. (Press release). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
- ^ Lee A (June 2023). “Omaveloxolone: First Approval”. Drugs. 83 (8): 725–729. doi:10.1007/s40265-023-01874-9. PMID 37155124. S2CID 258567442. Archived from the original on 9 December 2023. Retrieved 16 December 2023.
- ^ Subramony SH, Lynch DL (May 2023). “A Milestone in the Treatment of Ataxias: Approval of Omaveloxolone for Friedreich Ataxia”. Cerebellum. 23 (2): 775–777. doi:10.1007/s12311-023-01568-8. PMID 37219716. S2CID 258843532.
- ^ Jump up to:a b New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Jump up to:a b Reisman SA, Lee CY, Meyer CJ, Proksch JW, Ward KW (July 2014). “Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin”. Archives of Dermatological Research. 306 (5): 447–454. doi:10.1007/s00403-013-1433-7. PMID 24362512. S2CID 25733020.
- ^ Jump up to:a b Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, et al. (July 2011). “Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis”. Free Radical Biology & Medicine. 51 (1): 88–96. doi:10.1016/j.freeradbiomed.2011.03.027. PMC 3109235. PMID 21457778.
- ^ Reisman SA, Lee CY, Meyer CJ, Proksch JW, Sonis ST, Ward KW (May 2014). “Topical application of the synthetic triterpenoid RTA 408 protects mice from radiation-induced dermatitis”. Radiation Research. 181 (5): 512–520. Bibcode:2014RadR..181..512R. doi:10.1667/RR13578.1. PMID 24720753. S2CID 23906747.
External links
Clinical trial number NCT02255435 for “RTA 408 Capsules in Patients With Friedreich’s Ataxia – MOXIe” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Skyclarys |
| Other names | RTA 408 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Omaveloxolone |
| Routes of administration | By mouth |
| ATC code | N07XX25 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3][4] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1474034-05-3 |
| PubChem CID | 71811910 |
| IUPHAR/BPS | 7573 |
| DrugBank | DB12513 |
| ChemSpider | 34980948 |
| UNII | G69Z98951Q |
| KEGG | D10964 |
| ChEBI | CHEBI:229661 |
| CompTox Dashboard (EPA) | DTXSID101138251 |
| Chemical and physical data | |
| Formula | C33H44F2N2O3 |
| Molar mass | 554.723 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Zesiewicz TA, Hancock J, Ghanekar SD, Kuo SH, Dohse CA, Vega J: Emerging therapies in Friedreich’s Ataxia. Expert Rev Neurother. 2020 Dec;20(12):1215-1228. doi: 10.1080/14737175.2020.1821654. Epub 2020 Sep 21. [Article]
- Jiang Z, Qi G, Lu W, Wang H, Li D, Chen W, Ding L, Yang X, Yuan H, Zeng Q: Omaveloxolone inhibits IL-1beta-induced chondrocyte apoptosis through the Nrf2/ARE and NF-kappaB signalling pathways in vitro and attenuates osteoarthritis in vivo. Front Pharmacol. 2022 Sep 27;13:952950. doi: 10.3389/fphar.2022.952950. eCollection 2022. [Article]
- Shekh-Ahmad T, Eckel R, Dayalan Naidu S, Higgins M, Yamamoto M, Dinkova-Kostova AT, Kovac S, Abramov AY, Walker MC: KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain. 2018 May 1;141(5):1390-1403. doi: 10.1093/brain/awy071. [Article]
- Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, Ferguson DA: RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PLoS One. 2015 Apr 21;10(4):e0122942. doi: 10.1371/journal.pone.0122942. eCollection 2015. [Article]
- Lynch DR, Farmer J, Hauser L, Blair IA, Wang QQ, Mesaros C, Snyder N, Boesch S, Chin M, Delatycki MB, Giunti P, Goldsberry A, Hoyle C, McBride MG, Nachbauer W, O’Grady M, Perlman S, Subramony SH, Wilmot GR, Zesiewicz T, Meyer C: Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann Clin Transl Neurol. 2018 Nov 10;6(1):15-26. doi: 10.1002/acn3.660. eCollection 2019 Jan. [Article]
- Zighan M, Arkadir D, Douiev L, Keller G, Miller C, Saada A: Variable effects of omaveloxolone (RTA408) on primary fibroblasts with mitochondrial defects. Front Mol Biosci. 2022 Aug 12;9:890653. doi: 10.3389/fmolb.2022.890653. eCollection 2022. [Article]
- FDA Approved Drug Products: SKYCLARYS (omaveloxolone) capsules for oral use (February 2023) [Link]
- EMA Approved Drug Products: Skyclarys (omaveloxolone) Oral Capsules [Link]
- Health Canada Approved Drug Products: SKYCLARYS (Omaveloxolone) Capsules For Oral Use [Link]
///////////Omaveloxolone, Skyclarys, Friedrich’s ataxia, FDA 2023, APPROVALS 2023, RTA 408, RTA-408, omaveloxolona, RTA 408, 63415, PP415, orphan drug, fast track, priority review, rare pediatric disease



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Clofutriben



Clofutriben
Cas 1204178-50-6
HCL 1203941-88-1
- ASP 3662
- 4-(5-(2-(4-Chloro-2,6-difluorophenoxy)propan-2-yl)-4-methyl-4H-1,2,4-triazol-3-yl)-3-fluorobenzamide
- 4-{5-[2-(4-Chloro-2,6-difluorophenoxy)propan-2-yl]-4-methyl-4H-1,2,4-triazol-3-yl}-3-fluorobenzamide
- 4-[5-[2-(4-chloro-2,6-difluorophenoxy)propan-2-yl]-4-methyl-1,2,4-triazol-3-yl]-3-fluorobenzamide
- 4L1TY1U5VC
| Molecular Weight | 424.80 |
|---|---|
| Formula | C19H16ClF3N4O2 |
Clofutriben (ASP3662) is a 11β-hydroxysteroid dehydrogenase type 1 inhibitor.
Clofutriben is an orally bioavailable selective inhibitor of the enzyme 11beta-hydroxysteroid dehydrogenase type 1 (11b-HSD1; 11bHSD1; HSD11B1; HSD1; HSD-1), with potential protective activity for disorders of corticosteroid excess. Upon oral administration, clofutriben selectively binds to and inhibits the activity of HSD-1. This prevents the conversion of cortisone to the active hormone cortisol and thereby preventing the activation of the glucocorticoid receptors (GRs). By blocking cortisol production in metabolic tissues, clofutriben may inhibit the adverse metabolic effects that are caused by exogenous administration of glucocorticoids or in disorders in which cortisol is secreted in excess. HSD-1 is highly expressed in metabolic tissues, such as liver, skeletal muscle, and adipose tissue. It plays a crucial role in regulating the production of cortisol to activate the GRs.
SCHEME

PATENTS
Clinical and Translational Science (2019), 12(3), 291-301
British Journal of Pharmacology (2018), 175(19), 3784-3796
Sparrow Pharmaceuticals, Inc. WO2020106337
WO2019075394
WO2018117063
WO2010001946
PATENT
PDT PAT FOR HCL SALT, WO2012033070
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012033070
PATENT
PDT PAT FOR BASE, WO2018117063
PATENT
WO2010001946
[1]. Kiso T, et al. Analgesic effects of ASP3662, a novel 11尾-hydroxysteroid dehydrogenase 1 inhibitor, in rat models of neuropathic and dysfunctional pain. Br J Pharmacol. 2018 Oct;175(19):3784-3796. [Content Brief]
////////////Clofutriben, ASP 3662, orphan drug, 4L1TY1U5VC, Sparrow Pharmaceuticals,
Levacetylleucine
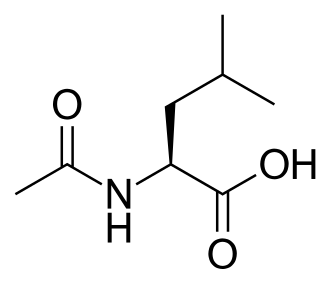

Levacetylleucine
WeightAverage: 173.212
Monoisotopic: 173.105193347
Chemical FormulaC8H15NO3
- N-Acetyl-L-leucine
- CAS 1188-21-2
- acetyl-L-leucine
- Ac-Leu-OH
- N-Acetylleucine
- NSC 206316
- UNII-E915HL7K2O
NSC-206316
(2S)-2-acetamido-4-methylpentanoic acid
FDA APPROVED 9/24/2024, To treat Niemann-Pick disease type C
Press Release
Drug Trials Snapshot
- Originator University of Munich; University of Oxford
- Developer IntraBio
- Class Acetamides; Amino acids; Esters; Neuroprotectants; Pentanoic acids; Small molecules; Vestibular disorder therapies
- Mechanism of Action Calcium channel modulators
- Orphan Drug StatusYes – Tay-Sachs disease; Niemann-Pick disease type C; Ataxia telangiectasia
Registered Niemann-Pick disease type C
- Phase IIIAtaxia telangiectasia
- Phase IISandhoff disease; Tay-Sachs disease
18 Mar 2025Phase-III clinical trials in Ataxia telangiectasia (In adolescents, In children, In the elderly, In adults) in Switzerland, Slovakia, Spain, Germany, USA, United Kingdom (PO) (NCT06673056)
- 04 Nov 2024IntraBio plans a phase III trial for Ataxia telangiectasia (In children, In adolescents, In adults, In elderly) in the US, Germany, Slovakia, Spain and Switzerland (PO, Suspension) in March 2025 (NCT06673056)
- 24 Sep 2024Registered for Niemann-Pick disease type C (In adolescents, In children, In adults) in USA (PO)
Levacetylleucine (N-acetyl-L-leucine), sold under the brand name Aqneursa, is a medication used for the treatment of neurological manifestations of Niemann-Pick disease type C.[1][2] Levacetylleucine is a modified version of the amino acid leucine.[1] It is the L-form of acetylleucine. It is taken by mouth.[1]
The most common side effects include abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting.[1][2]
Levacetylleucine was approved for medical use in the United States in September 2024.[1][2][3] Levacetylleucine is the second medication approved by the US Food and Drug Administration (FDA) for the treatment of Niemann-Pick disease type C.[2] The FDA considers it to be a first-in-class medication.[4]
DATA
N-acetyl-D, L-leucine is the active ingredient of Tanganil ® which helps treat vertigo attacks.

N-Acetyl-D, L-leucine
Unlike the majority of chemical syntheses of active principles where it is desirable to separate the enanti omers and / or to retain the selective stereo information during the synthesis steps, the synthesis of N-acetyl-D, L-leucine is carried out from L-leucine and therefore involves a racemization step. This racemization takes place before the acetylation step, via a Schiff base formed in situ with salicylic aldehyde (Yamada et al., J. Org. Chem., 1983 48, 843- 846).

Two competitive reactions are then involved: the acetylation of leucine, the main reaction, where acetic anhydride reacts with the amine function of leucinate of sodium to give N-acetyleucinate and the hydrolysis of acetic anhydride to acetic acid, a side reaction described below.

This synthesis has a molar yield of 70%. The limiting steps are essentially the secondary reaction of hydrolysis of acetic anhydride and the step of isolation of the racemized leucine before the acetylation reaction. Indeed, on an industrial scale, the quantities of products brought into play for isolations prove to be very restrictive.
There is therefore a real need to develop a new process for the preparation of N-actéyl-D, L-leucine which is faster and more economical.
The inventors thus discovered that the racemization step could be carried out after the L-leucine acetylation step making it possible to avoid a step of isolating the intermediate product and that this process could be carried out in continuous flow. Du Vigneaud & Meyer (J. Biol Chem, 1932, 98, 295-308) had already shown that it was possible to racemize different acetylated amino acids by bringing them into the presence of acetic anhydride for several hours. However, no examples had been made with acetyl leucine. By attempting to reproduce this process with acetyl-leucine, the inventors have thus found that this racemization reaction did not give satisfactory results with acetyl-leucine because of a competitive hydrolysis reaction of acetic anhydride. used. The inventors have also surprisingly discovered that the racemization reaction of N-acetyl-L-leucine could be improved by producing it in a continuous flow. It seems indeed that the realization of this continuous flow process allows better control of the mixing of the reagents and therefore to better control the reaction. The inventors have also shown that the racemization of N-acetyl-L-Leucine in continuous flow was obtained in a very short time of the order of a few minutes.
Furthermore, there is also a need to develop a new method of acetylation of leucine for the preparation of N-actyle-leucine which is faster and more economical. The inventors have discovered that the acetylation reaction of leucine can be improved by making it in a continuous flow. The process according to the invention gives good yields, in a very short time and using fewer reagents compared to the method known hitherto.
Indeed, DeWitt et al. (J Am Chem Soc (1951) 73 (7) 3359-60) described the preparation of N-acetyl-L-Leucine by reacting L-Leucine with 3 molar equivalents of acetic anhydride and sodium hydroxide for 2 hours 20 minutes. . N-acetyl-L-leucine is then obtained in a yield of only 70-80%. In addition, the authors of this publication clearly indicated that a molar ratio between L-Leucine and acetic anhydride below 2 resulted in much lower yields.
SYNTHESIS
H. D. DeWitt and A. W. Ingersoll. The Preparation of Pure N-Acetyl-L-leucine and L-Leucine. Journal of the American Chemical Society 1951 73 (7), 3359-3360. DOI: 10.1021/ja01151a108
PATENT
https://patents.google.com/patent/WO2012038515A1/en
EXAMPLES
A. Acetylation of L-Leucine in Continuous Flow

A. L. Study of the molar ratio of acetic anhydride to leucine
The objective of this study is to define the necessary molar ratio of acetic anhydride so that the acetylation reaction with acetic anhydride is complete and is not disadvantageous by competition with the acetic anhydride hydrolysis reaction. In this study, the residence time in the reactor / exchanger (1 process plate) was set at 9 seconds, for a temperature of the reaction medium of between 25 and 30 ° C.
The ratio range studied is between 0.9 and 2.0 molar equivalents. The optimum is obtained for a ratio between 1.20 and 2.00, more particularly between 1.30 and 1.60. Below this ratio, the acetylation reaction is disadvantageous compared to the acetic hydrolysis reaction. Beyond this, the drop in pH (acid instead of base) also disadvantages the acetylation reaction.
EXAMPLES 1-10:
A solution of sodium L-leucinate, for passage in continuous flow reactor, is prepared in the following manner: 700 g of L-leucine are dissolved in a solution of 576 g of sodium hydroxide and 3.5 liters of Demineralized Water. This solution is the main fluid process. The reaction between this solution and the acetic anhydride is carried out in a continuous flow in a Boostec® reactor, made of silicon carbide. The reactor / exchanger is configured with an injection-type process plate comprised between two utility plates. The volume of the process plate is 10 mL. The temperature in the reactor is maintained by the circulation of a coolant heated by a thermostatic bath. The transformation of L-leucine to N-acetyl-L-leucine is monitored online by quantitative Raman spectroscopy. This method of analysis is calibrated beforehand with solutions of known concentration prepared with pure L-leucine and N-acetyl-L-leucine.
Example 1
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 4.06 kg.h -1 and 0.42 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 0.91 equivalents. The total flow rate is therefore 4.48 kg.h -1 , which corresponds to a residence time (equivalent to the reaction time) of 8.7 s The yield of acetyl-L-leucinate determined by Raman spectroscopy online at the outlet of the reactor is 40% Example 2:
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.95 kg · h -1 and 0.45 kg · h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.01 equivalents. The total flow rate is therefore 4.40 kg.h -1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 52.degree. %.
Example 3
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.89 kg · h -1 and 0.52 kg · h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.18 equivalents. The total flow rate is therefore 4.41 kg.h -1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 57.degree. %. Example 4
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.82 kg. h -1 and 0.57 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.32 equivalents. The total flow is therefore 4.39 kg. h “1 , which corresponds to a residence time of 8.9 S. The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 83%.
Example 5
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective rates set at 3.64 kg. h -1 and 0.55 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.34 equivalents. The total flow is therefore 4, 19 kg. h “1 , which corresponds to a residence time of 9.4 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 98%.
Example 6
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective rates set at 3.66 kg. h 1 and 0.62 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.50 equivalents. The total flow is therefore 4.28 kg. h “1 , which corresponds to a residence time of 9.2 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 96%.
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates fixed at 3.67 kg. h -1 and 0.64 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.54 equivalents. The total flow is therefore 4.31 kg. h “1 , which corresponds to a residence time of 9.1 sec The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 100%. Example 8
The temperature of the thermostated bath is set at 25 ° C. The sodium leucinate solution and pure acetic anhydride are introduced into the reactor at respective flow rates set at 3.63 kg. h -1 and 0.73 kg h -1 . These flow rates correspond to a molar ratio of acetic anhydride to leucine of 1.78 equivalents. The total flow is therefore 4.36 kg. h “1 , which corresponds to a residence time of 9.0 s The yield of acetyl-L-leucinate determined by in-line Raman spectroscopy at the outlet of the reactor is 90%.
PATENT
https://patents.google.com/patent/CN104592052A/en
Example 1:
100gL-leucine adds 1000ML2NNaOH rising temperature for dissolving, adds 1ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add 160ML HCl and adjust PH 2.5, be cooled to 4 degree, suction filtration, the 118g. of oven dry
Example 2:
100gL-leucine adds 1200ML 2NNaOH rising temperature for dissolving, adds 3ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add the 3.0. that 180ML HCl adjusts PH, be cooled to 4 degree, suction filtration, the 110g. of oven dry
Example 3:
100gL-leucine adds 1000ML 2NNaOH rising temperature for dissolving, adds 2ML salicylic aldehyde, 95 degree of insulations of intensification 3 hours, recording optically-active is 0, be cooled to 5 degree and keep, dripping 80ML diacetyl oxide, dropwise maintenance 0.5 hour, be warmed up to 60 degree, add proper amount of active carbon decolouring, add 180ML HCl and adjust PH 3.0, be cooled to 4 degree, suction filtration, the 120g. of oven dry
Medical uses
Levacetylleucine is indicated for the treatment of neurological manifestations of Niemann-Pick disease type C in people weighing at least 15 kilograms (33 lb).[1][2]
Adverse effects
The most common side effects include abdominal pain, difficulty swallowing, upper respiratory tract infections, and vomiting.[2]
Levacetylleucine may cause embryo-fetal harm if used during pregnancy.[1][2]
History
The safety and efficacy of levacetylleucine for the treatment of Niemann-Pick disease type C were evaluated in a randomized, double-blind, placebo-controlled, two-period, 24-week crossover study.[2] The duration was twelve weeks for each treatment period.[2] The study enrolled 60 participants.[2] To be eligible for the study participants had to be four years of age or older with a confirmed diagnosis of Niemann-Pick disease type C and at least mild disease-related neurological symptoms.[2] Participants could receive miglustat, an enzyme inhibitor, as background treatment in the study.[2]
The US Food and Drug Administration (FDA) granted the application for levacetylleucine priority review, fast track, orphan drug, and rare pediatric disease designations.[2] The FDA granted approval of Aqneursa to IntraBio Inc.[2]
Society and culture
Legal status
Levacetylleucine was approved for medical use in the United States in September 2024.[1][2][5]
Names
Levacetylleucine is the international nonproprietary name.[6]
Research
Levacetylleucine is being studied for the treatment of GM2 gangliosidoses (Tay-Sachs and Sandhoff diseases),[7] ataxia-telangiectasia,[8] Lewy body dementia,[9] amyotrophic lateral sclerosis, restless legs syndrome, multiple sclerosis, and migraine.[10]
References
- ^ Jump up to:a b c d e f g h i “Aqneursa- levacetylleucine granule, for suspension”. DailyMed. 24 September 2024. Retrieved 5 October 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves New Drug to Treat Niemann-Pick Disease, Type C”. U.S. Food and Drug Administration (Press release). 24 September 2024. Retrieved 25 September 2024. This article incorporates text from this source, which is in the public domain.
- ^ “IntraBio Announces U.S. FDA Approval of Aqneursa for the Treatment of Niemann-Pick Disease Type C”. IntraBio (Press release). 25 September 2024. Retrieved 26 September 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 29 November 2024.
- ^ World Health Organization (2024). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 131”. WHO Drug Information. 38 (2). hdl:10665/378367. ISBN 9789240098558.
- ^ Martakis K, Claassen J, Gascon-Bayari J, Goldschagg N, Hahn A, Hassan A, et al. (March 2023). “Efficacy and Safety of N-Acetyl-l-Leucine in Children and Adults With GM2 Gangliosidoses”. Neurology. 100 (10): e1072 – e1083. doi:10.1212/WNL.0000000000201660. PMC 9990862. PMID 36456200.
- ^ Fields T, Patterson M, Bremova-Ertl T, Belcher G, Billington I, Churchill GC, et al. (January 2021). “A master protocol to investigate a novel therapy acetyl-L-leucine for three ultra-rare neurodegenerative diseases: Niemann-Pick type C, the GM2 gangliosidoses, and ataxia telangiectasia”. Trials. 22 (1): 84. doi:10.1186/s13063-020-05009-3. PMC 7821839. PMID 33482890.
- ^ Passmore P (15 April 2014). A clinical trial to test amlodipine as a new treatment for vascular dementia. ISRCTN registry (Report). doi:10.1186/isrctn31208535.
- ^ Strupp M, Bayer O, Feil K, Straube A (February 2019). “Prophylactic treatment of migraine with and without aura with acetyl-DL-leucine: a case series”. Journal of Neurology. 266 (2): 525–529. doi:10.1007/s00415-018-9155-6. PMID 30547273. S2CID 56148131.
Further reading
- Churchill GC, Strupp M, Factor C, Bremova-Ertl T, Factor M, Patterson MC, et al. (August 2021). “Acetylation turns leucine into a drug by membrane transporter switching”. Scientific Reports. 11 (1): 15812. Bibcode:2021NatSR..1115812C. doi:10.1038/s41598-021-95255-5. PMC 8338929. PMID 34349180.
- Bremova-Ertl T, Ramaswami U, Brands M, Foltan T, Gautschi M, Gissen P, et al. (February 2024). “Trial of N-Acetyl-l-Leucine in Niemann-Pick Disease Type C”. The New England Journal of Medicine. 390 (5): 421–431. doi:10.1056/NEJMoa2310151. PMID 38294974.
- Tifft CJ (February 2024). “N-Acetyl-l-Leucine and Neurodegenerative Disease”. The New England Journal of Medicine. 390 (5): 467–470. doi:10.1056/NEJMe2313791. PMID 38294981.
External links
- Clinical trial number NCT05163288 for “A Pivotal Study of N-Acetyl-L-Leucine on Niemann-Pick Disease Type C” at ClinicalTrials.gov
- Bremova-Ertl T, Ramaswami U, Brands M, Foltan T, Gautschi M, Gissen P, Gowing F, Hahn A, Jones S, Kay R, Kolnikova M, Arash-Kaps L, Marquardt T, Mengel E, Park JH, Reichmannova S, Schneider SA, Sivananthan S, Walterfang M, Wibawa P, Strupp M, Martakis K: Trial of N-Acetyl-l-Leucine in Niemann-Pick Disease Type C. N Engl J Med. 2024 Feb 1;390(5):421-431. doi: 10.1056/NEJMoa2310151. [Article]
- Fields T, M Bremova T, Billington I, Churchill GC, Evans W, Fields C, Galione A, Kay R, Mathieson T, Martakis K, Patterson M, Platt F, Factor M, Strupp M: N-acetyl-L-leucine for Niemann-Pick type C: a multinational double-blind randomized placebo-controlled crossover study. Trials. 2023 May 29;24(1):361. doi: 10.1186/s13063-023-07399-6. [Article]
- FDA Approved Drug Products: Aqneursa (levacetylleucine) for oral suspension (September 2024) [Link]
- FDA News Release: FDA Approves New Drug to Treat Niemann-Pick Disease, Type C [Link]
| Clinical data | |
|---|---|
| Trade names | Aqneursa |
| Other names | IB1001 |
| AHFS/Drugs.com | Aqneursa |
| License data | US DailyMed: Levacetylleucine |
| Pregnancy category | Not recommended |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1188-21-2 |
| PubChem CID | 70912 |
| DrugBank | DB16956 |
| ChemSpider | 1918 |
| UNII | E915HL7K2O |
| KEGG | D12967 |
| ChEBI | CHEBI:17786 |
| ChEMBL | ChEMBL56021 |
| PDB ligand | LAY (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID6045870 |
| ECHA InfoCard | 100.013.370 |
| Chemical and physical data | |
| Formula | C8H15NO3 |
| Molar mass | 173.212 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Levacetylleucine, Aqneursa, Niemann-Pick disease type C, FDA 2024, APPROVALS 2024, N-Acetyl-L-leucine, 1188-21-2, acetyl-L-leucine, Ac-Leu-OH, N-Acetylleucine, NSC 206316, UNII-E915HL7K2O, ORPHAN DRUG, NSC-206316, NSC 206316
Crinecerfont



Crinecerfont
CAS 752253-39-7
SSR125543
SSR 125543
SSR-125543, WHO 10958, UNII-MFT24BX55I, 06-RORI,NBI-74788
- (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methyl-N-(prop-2-yn-1-yl)thiazol-2-amine
- 2-Thiazolamine, 4-(2-chloro-4-methoxy-5-methylphenyl)-N-((1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methyl-N-2-propynyl-
- 2-Thiazolamine, 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-2-propyn-1-yl
FDA APPROVED 12/13/2024, Crenessity, To treat classic congenital adrenal hyperplasia
Press Release
4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine
WeightAverage: 483.04
Monoisotopic: 482.1594906
Chemical FormulaC27H28ClFN2OS

CAS No. : 321839-75-2
| Molecular Weight | 519.50 |
|---|---|
| Formula | C27H29Cl2FN2OS |
Crinecerfont, sold under the brand name Crenessity, is a medication used for the treatment of congenital adrenal hyperplasia.[1] It is a corticotropin-releasing factor type 1 receptor (CRF1R) antagonist developed to treat classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency (21OHD).[1] It is taken by mouth.[1]
The most common side effects of crinecerfont in adults include fatigue, dizziness, and arthralgia (joint pain).[2] For children, the most common side effects include headache, abdominal pain, and fatigue.[2]
Crinecerfont was approved for medical use in the United States in December 2024.[2][3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
A medication used to reduce the amount of steroid replacement required in patients with a genetic disease that causes, amongst other symptoms, a steroid deficiency.
- OriginatorSanofi
- DeveloperNeurocrine Biosciences; Sanofi
- ClassAmines; Antidepressants; Anxiolytics; Chlorobenzenes; Cyclopropanes; Fluorobenzenes; Halogenated hydrocarbons; Phenyl ethers; Small molecules; Thiazines; Thiazoles
- Mechanism of ActionCorticotropin releasing factor receptor 1 antagonists
- Orphan Drug StatusYes – Congenital adrenal hyperplasia
- MarketedCongenital adrenal hyperplasia
- DiscontinuedMajor depressive disorder; Post-traumatic stress disorders
20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In adolescents, In children) in USA (PO)
- 20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In adolescents, In children, In the elderly, In adults) in USA (PO)
- 20 Dec 2024Launched for Congenital adrenal hyperplasia (Adjunctive treatment, In the elderly, In adults) in USA (PO)



SYN

https://patents.google.com/patent/US12128033
Example processes and certain intermediates of the present invention are shown in Scheme I to Scheme VII below.
A representative Coupling-Step of 2-cyclopropylacetic acid (Compound
1A) with N,O-dimethylhydroxylamine or a salt thereof in the presence of a coupling-step reagent (e.g., 1,1′-carbonyldiimidazole), a coupling-step base (e.g., triethylamine), and a coupling-step solvent (e.g., dichloromethane) to prepare 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A) is provided below in Scheme I.

A representative Reacting-Step between 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A) with an organomagnesium reagent of 4-bromo-2-fluoro-1-methylbenzene in the presence of a reacting-step solvent (e.g., tetrahydrofuran (THF)) to prepare 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A) is provided below in Scheme II.

A representative Condensing-Step of 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A) with a Compound of Formula (Ic) or a salt thereof, in the presence of a condensing-step acid (e.g., p-toluenesulfonic acid) and a condensing-step solvent (e.g., toluene) to prepare a Compound of Formula (Ie) is provided below in Scheme III.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Reducing-Step of a Compound of Formula (Ie) in the presence of a reducing-catalyst (e.g., sponge nickel and Pd/Cu—C), hydrogen, and a reducing-step solvent (e.g., ethanol) to prepare a Compound of Formula (Ig) is provided below in Scheme IV.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Deprotecting-Step of a Compound of Formula (Ig), or a salt thereof, in the presence of a deprotecting-catalyst (e.g., Pd), hydrogen, and a deprotecting-step solvent (e.g., ethanol) to prepare (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound
6A) or a salt thereof is provided below in Scheme V.

-
- wherein:
- R1c, R2c, and R3c are each independently selected from: H, C1-C6 alkoxy, C1-C6 alkyl, C1-C6 haloalkyl, and halogen.
A representative Cyclizing-Step of (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound
6A) or a salt thereof, with 1-(2-chloro-4-methoxy-5-methylphenyl)-2-thiocyanatopropan-1-one (Compound
8A) or a tautomeric form thereof, in the presence of a cyclizing-step solvent (e.g., n-heptane) to prepare (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound
9A) or a salt thereof is provided below in Scheme VI.

A representative Alkylating-Step of (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound
9A) or a salt thereof, with a Compound of Formula (Ii), wherein LG is suitable leaving group (e.g., Br), in the presence of an alkylating-step solvent (e.g., methyl tert-butyl ether (MTBE), toluene, and mixtures thereof), a phase-transfer catalyst (e.g., tetra-n-butylammonium bromide (TBAB)), an alkylating-step base (e.g., potassium hydroxide), and water to prepare 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1) or a pharmaceutically acceptable salt thereof is provided below in Scheme VII.

One aspect of the present invention includes every combination of one or more process steps and intermediates related thereto used in the preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1), and/or pharmaceutically acceptable salts, and crystalline forms thereof, such as those processes exemplified by Schemes I, II, III, IV, V, VI, VII, and VII (supra) and Compounds contained therein.
8A was previously described in International Publication Number WO2010/125414 by Sanofi-Aventis.Example 1: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1), See FIG. 5 for a general synthetic schemeStep1A: Preparation of 2-Cyclopropyl-N-methoxy-N-methylacetamide (Compound 2A)

A suspension of 1,1′-carbonyldiimidazole (CDI, 152.6 kg, 1.01 eq.) in DCM (682 kg, 513 L, 7.3 w/w relative to 2-cyclopropylacetic acid) was treated with a solution of 2-cyclopropylacetic acid (Compound
1A, 93.6 kg, 1 eq.) in DCM (248 kg, 186 L, 2.65 w/w) over at least 1 h, keeping the temperature ≤25° C. and compensating for significant effervescence. The resulting mixture was stirred for 15 min at 22° C. and then N,O-dimethylhydroxylamine-HCl (93.6 kg, 1.03 eq.) was added in portions, keeping the temperature ≤30° C. Subsequently, triethylamine (46.4 kg, 0.49 eq.) was added to the stirring mixture at 20-25° C. The resulting mixture was stirred at 22° C. at least 1 h. The mixture was washed once with KHSO4 solution (0.24 M, 357.1 kg, 0.09 eq.), once with KHSO4 solution (0.40 M, 365.4 kg, 0.15 eq.), once with KHSO4 solution (0.80 M, 384.5 kg, 0.30 eq.), and once with NaHCO3 solution (0.60 M, 393.1 kg, 0.24 eq.). Residual DCM was removed by two put-and-takes of THF (166.6 kg, 1.78 w/w) and vacuum distillation (50-60° C., to minimum volume/until distillation stops) to provide Compound
2A. THF (333.2 kg. 3.56 w/w) was added and the yield was determined by correcting for the LOD and GC-FID purity of the sample (131.5 kg, 98.2% corrected). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) −0.01-0.03 (m, 2H), 0.32-0.36 (m, 2H), 0.81-0.90 (br m, 1H), 2.18 (d, J=6.80 Hz, 2H), 2.97 (s, 3H), 3.53 (s, 3H). ESI-MS: 144.0 [M+H]+.Step 1B: Preparation of 2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound 3A)

Mg (turnings, 28.6 kg, 1.37 eq.) were suspended in THF (244.7 kg, 2.0 w/w) and DIBAL-H (1 M in n-heptane, 18.9 kg, 0.03 eq.) was added dropwise at 30° C. The resulting mixture was stirred at 30° C. for at least 10 min and then 4-bromo-2-fluoro-1-methylbenzene (neat, 21.1 kg, 0.13 eq.) was added over at least 30 min at 30-50° C. Subsequently, the mixture was treated with a solution of 4-bromo-2-fluoro-1-methylbenzene (191.6 kg, 1.18 eq.) in THF (414.5 kg, 3.37 w/w) at 30-50° C. over 3 h or less. The mixture was stirred at 30° C. for at least 1 h. The mixture was cooled to 12-18° C. and subsequently treated with 2-cyclopropyl-N-methoxy-N-methylacetamide (Compound
2A, 123.0 kg, 1 eq., 25.9% w/w solution in THF) over at least 1 h at 15-25° C. The resulting mixture was stirred at 20-25° C. for at least 1 h. The stirring mixture was then treated with aqueous HCl (3 M, 10.3% w/w, 668.9 kg, 2.24 eq.) at 10-25° C. and the resulting mixture was stirred at least 2 h until no Mg turnings were observed (check pH 3.0-3.5). The layers were separated, and the aqueous layer discarded. The organic layer was distilled at 55-65° C. and 400 mbar until distillation halts. Heptane (290.3 kg, 2.36 w/w) was added. The layers were separated, and the organic layer was washed once with NaHCO3 solution (0.63 M, 211.6 kg, 0.15 eq.) and once with NaCl solution (2.57 M, 213.0 kg, 0.55 eq.). The residual solvents were removed by vacuum distillation at 58-62° C. until distillation stops and then one put-and-take of toluene (275.5 kg, 2.24 w/w) at 107-117° C. until distillation stops. Toluene (275.5 kg, 2.24 w/w) was added and the yield was determined by correcting for the LOD and GC-FID purity of the sample (150.7 kg, 91.3% corrected). 1H NMR (400 MHz, DMSO-d6) β (ppm) 0.07-0.21 (m, 2H), 0.40-0.54 (m, 2H), 1.02 (ttt, J=8.16, 8.16, 6.68, 6.68, 4.86, 4.86 Hz, 1H), 2.30 (d, J=1.77 Hz, 3H), 2.91 (d, J=6.57 Hz, 2H), 7.44 (t, J=7.83 Hz, 1H), 7.57-7.78 (m, 2H). ESI-MS: 193.1 [M+H]+.Step 1C: Preparation of (S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)-N-(1-phenylethyl)ethan-1-imine (Compound 4A)

A mixture of 2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-one (Compound
3A, 150.7 kg, 1 eq., as a 27.6% w/w solution in toluene), (S)-(−)-1-phenylethylamine (112.9 kg, 1.19 eq.), and p-toluenesulfonic acid (7.4 kg, 0.05 eq.) was heated to reflux at 110-120° C. for 23-25 h in a reactor set up in a Dean-Stark configuration. The solvent was then removed at 125-135° C. under atmospheric pressure until distillation halts and a portion of toluene (275 kg, 2.24 w/w) was added to afford a suspension. The suspension was heated to reflux at 110-120° C. for 23-25 h. The mixture was cooled to 22° C. and washed twice with aqueous NH4Cl (10%, 301.2 kg, 0.72 eq.) and once with aqueous NaHCO3 (5%, 301.2 kg, 0.23 eq., check pH 8-9). The solvent was removed at 125-135° C. and atmospheric pressure to a target volume of 256 L, the mixture was filtered over CELITE®, and the cake was washed with toluene (25 kg). The resulting mixture containing Compound 4A was used directly in the next step without further isolation. The yield was determined by correcting for the LOD and GC-FID purity of the sample (208.4 kg, 90.0% corrected). EL-MS: 294.1 [M−H]*, 190.1 [M-C6H5CH(CH3)]+, 105.1 [C6H5CH(CH3)]+.Step 1D: Preparation of (S)-2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)-N—((S)-1-phenylethyl)ethan-1-amine (Compound 5A) as the Hydrochloride Salt

Sponge nickel catalyst (144 kg, 0.70 w/w, shipped as a 50% w/w suspension in water) was added to a hydrogenation reactor, equipped with a dip tube capable of removing material from the top of the mass inside, minimizing the amount of water introduced. The supernatant was discarded, ethanol (329.3 kg, 1.58 w/w, anhydrous) was added, the suspension was stirred and then allowed to settle. This process was repeated four more times and the supernatant is checked; ≤1% H2O w/w (Karl Fisher (KF)). Compound 4A (208.4 kg, 1 eq., as a 62.6% solution in toluene) was added to the mixture in the hydrogenation reactor. Ethanol (389.4 kg, 1.86 w/w) was used to rinse the addition flask into the hydrogenation reactor. The hydrogenation reactor was pressurized/depressurized twice with nitrogen (2 bar), twice with hydrogen (5 bar), and then pressurized with hydrogen (9.8-10.2 bar). The resulting mixture was heated to 33-37° C. and stirred for 17-19 h. The system was depressurized/pressurized three times with nitrogen (1 bar). The suspension was filtered and washed three times with ethanol (total amount, 493.8 kg, 2.37 w/w). The filtrate was combined with HCl (concentrated, 83.4 kg, 1.07 eq.) and the resulting mixture stirred 25-35 min at 20-24° C. The mixture was concentrated by distillation at 78-80° C. and atmospheric pressure to remove water with a distillate target volume of 1167 L (5.6 L/kg based on imine Compound 4A) and the KF of the solution checked (≤1.5% H2O w/w). The mixture was stirred at 48-52° C. for 55-65 min, then 68-72° C. for 55-65 min, then cooled to 20-24° C. at a rate of 12° C./h and stirred for 25-35 min, then cooled to 0-4° C. at a rate of 10° C./h and stirred for 55-65 min. The suspension was filtered, the cake was washed twice with precooled ethanol (total amount, 329.2 kg, 1.58 w/w, 0° C.), and the collected solid was dried at 40° C. to afford Compound
5A as the HCl salt (156.5 kg, 66.4% uncorrected). 1H NMR (400 MHz, DMSO-d6) δ (ppm) −0.33–0.06 (m, 2H), 0.11-0.31 (m, 3H), 1.57 (d, J=6.57 Hz, 3H), 1.95 (br t, J=7.07 Hz, 2H), 2.26 (d, J=1.26 Hz, 3H), 3.68 (br d, J=7.83 Hz, 1H), 3.92 (br t, J=6.44 Hz, 1H), 6.98 (dd, J=7.71, 1.14 Hz, 1H), 7.28-7.36 (m, 2H), 7.37-7.50 (m, 5H). EST-MS: 298.2 m/z [M+H]+.Step 1E: Preparation of (S)-2-Cyclopropyl-1-(3-fluoro-4-methylphenyl)ethan-1-amine (Compound 6A) as the Hydrochloride Salt

5A (HCl salt, 156.5 kg, 1.00 eq.) and Pd/C (7.8 kg, 10% Pd basis) were added to an inerted hydrogenation reactor. The reactor was then pressurized/depressurized twice with nitrogen (2 bar) and then methanol (494.5 kg, 3.16 w/w) was added. The reactor was depressurized/pressurized three times with nitrogen (2 bar) then three times with hydrogen (5 bar), pressurized with hydrogen (9.8-10.2 bar), heated to 58-62° C. and stirred for 7-9 h. The reaction mixture was cooled to 20-24° C. The reactor was depressurized/pressurized three times with nitrogen (1 bar) and the suspension was filtered and washed three times with methanol (total amount, 492.9 kg, 3.15 w/w). The solution was concentrated at 63-67° C. and atmospheric pressure to a distillate target volume of 1408 L (9.0 L/kg Compound
6A), n-Heptane (1173.8 kg, 7.5 w/w) was added and the resulting mixture was heated to reflux at 65-80° C. and atmospheric pressure in Dean-Stark configuration to remove methanol. The suspension was cooled to 31-35° C. and filtered, the cake washed with n-heptane (147.1 kg, 0.94 w/w), and the solid dried at 40° C. to provide Compound
6A as the HCl salt (101.0 kg, 93.8% uncorrected, 99.6% ee). 1H NMR (400 MHz, DMSO-d6) δ (ppm) −0.12-0.14 (m, 2H), 0.26-0.42 (m, 2H), 0.44-0.55 (m, 1H), 1.70-1.83 (m, 2H), 2.23 (d, J=1.52 Hz, 3H), 4.24 (t, J=7.33 Hz, 1H), 7.22-7.29 (m, 1H), 7.29-7.36 (m, 1H), 7.40 (dd, J=10.99, 1.39 Hz, 1H). ESI-MS: 194.2 [M+H]+, 177.0 [M-NH2]+.Step 1F: Preparation of (S)-4-(2-chloro-4-methoxy-5-methylphenyl)-N-(2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl)-5-methylthiazol-2-amine (Compound 9A)

A mixture of n-heptane (146 kg), water (142 kg), Compound
6A (HCl salt, 57.4 kg), and aqueous sodium hydroxide (30% w/w, 41.0 kg) was stirred together. The layers were partitioned, and the aqueous layer removed. The organic layer was washed with water (170 kg) and the layers partitioned. The organic layer was set aside, n-Heptane (145 kg) and 1-(2-chloro-4-methoxy-5-methylphenyl)-2-thiocyanatopropan-1-one (Compound
8A, 66.1 kg, the preparation of Compound
8A has been previously described in International Publication Number WO2010/125414) were added to the reactor and heated to 85° C. The previously set aside organic layer containing the free base of Compound
6A was added at 84-85° C. to the reactor and rinsed with n-heptane (20 kg). The resulting mixture was stirred for 2 h at 83° C. Subsequently, the solvent was switched to methanol by four put-and-take additions/vacuum distillations of methanol (180 kg) at 55° C. with the target volume being 287 L remaining in the reactor. The suspension was cooled to 5° C. and water (570 kg) was added over 4 h at 5-10° C., with the first 60 kg added very slowly. The suspension was aged 2 h at 5° C. and then isolated by filtration, washed with a mixture of methanol/water (91/115 kg) and then a mixture of methanol/water (134/57 kg). The yellow solid was dried at 25° C. and 1 mbar for 17 h then 40° C. and 1 mbar for 22 h to afford Compound
9A (97.4 kg, 87.5% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm −0.01-0.14 (m, 2H), 0.29-0.42 (m, 2H), 0.61-0.73 (m, 1H), 1.47 (dt, J=13.83, 6.85 Hz, 1H), 1.76 (dt, J=13.89, 7.20 Hz, 1H), 2.00 (s, 3H), 2.11 (s, 3H), 2.19 (d, J=1.01 Hz, 3H), 3.82 (s, 3H), 4.54 (q, J=7.58 Hz, 1H), 7.00 (s, 1H), 7.06 (d, J=0.76 Hz, 1H), 7.08-7.14 (m, 2H), 7.18-7.23 (m, 1H), 7.89 (d, 1=8.08 Hz, 1H). ESI-MS: 445.3 m/z [M+H]+.Step 1G: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)

A mixture of MTBE (279 kg), tetra-n-butylammonium bromide (10.5 kg), and Compound
9A (95.4 kg) were heated at 60° C. external temperature for 30 min and then cooled to 0° C. Aqueous potassium hydroxide (52.4% w/w, 364 kg) and propargyl bromide (39.4 kg as an 80% w/w solution in toluene, 1.19 eq.) were added at 0-5° C. The propargyl bromide additional funnel was washed with MTBE (25 kg) and the biphasic mixture was aged 14.5 h at 4-6° C. with vigorous stirring. Subsequently, water (191 kg) was added and the aqueous layer was discharged at 20° C. The organic layer was washed twice with water (382 kg) and once with aqueous acetic acid (5.26% w/w, 190 kg) at 20° C. The mixture is polish filtered, rinsed with ethanol (11 kg) and then the solvent switched to ethanol by 3 put-and-take additions/vacuum distillations of ethanol (300 kg) at 25-30° C. for the first cycle and then 35-40° C. with the target volume of each cycle being 250 L remaining in the reactor. Ethanol (164 kg) was added and the mixture heated at 60° C. external for 0.5 h before it was cooled to 25° C. in 1 h and seeded with authentic Form I (free base) of Compound 1 (0.340 kg) which can be prepared as described below in Example 2 and Example 3. The suspension was aged for 5 h, cooled to 0° C. in 2 h, aged 12 h, filtered, and washed twice with ethanol (24 kg each) pre-cooled to 0° C. The white solid was dried at 40° C. and 1 mbar for 19 h to yield 80.15 kg of Compound 1 (77.2% yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 0.14 (qt, J=8.59, 4.42 Hz, 2H), 0.29-0.48 (m, 2H), 0.61-0.82 (m, 1H), 1.89 (dt, J=14.08, 6.98 Hz, 1H), 2.07 (br d, J=7.83 Hz, 1H), 2.10 (s, 3H), 2.14 (s, 3H), 2.20 (d, J=1.01 Hz, 3H), 3.11 (t, J=2.27 Hz, 1H), 3.83 (s, 3H), 3.94-4.22 (m, 2H), 5.26 (t, J=7.58 Hz, 1H), 7.05 (s, 1H), 7.10-7.36 (m, 4H). ESI-MS: 483.2 m/z [M+H]+.Example 2: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)
A mixture of MTBE (2 mL), tetra-n-butylammonium bromide (110 mg), and Compound
9A (1.003 g) at 0° C. was treated with aqueous potassium hydroxide (52.4% w/w, 1.80 mL, 2.73 g) and propargyl bromide (405 mg as an 80% w/w solution in toluene) maintaining the temperature at 0-5° C. The resulting biphasic mixture was aged 23 h at 4-6° C. Subsequently, water (2 mL) and MTBE (2 mL) were added and the aqueous layer was discharged. The organic layer was washed twice with water (4 mL) and once with aqueous acetic acid (5% w/w, 2 mL) at 20° C. Ethanol (4 mL) was added and then the solvent was switched to ethanol by 3 put-and-take additions/vacuum distillations of ethanol (6 mL) at 35-40° C. with the target volume of each cycle being 2 mL remaining in the vessel, except for the third cycle where the mixture was concentrated to dryness. Ethanol (4 mL) was added to the vessel and the mixture heated at 60° C. (external) for 0.5 h before it was cooled to 20° C. in 1 h and aged 18 h. The resulting suspension was cooled to 0° C., aged 6 h, filtered, and washed twice with ethanol (2 mL each) pre-cooled to 0° C. to afford a solid. The solid was dried at 40° C. under vacuum to afford Compound 1 (506 mg, 46% yield) as Form I. The 1H NMR and ESI-MS data matches that as described above in Example 1, Step 1G.Example 3: Preparation of 4-(2-chloro-4-methoxy-5-methylphenyl)-N-1(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]-5-methyl-N-prop-2-ynyl-1,3-thiazol-2-amine (Compound 1)
A mixture of MTBE (40 mL), tetra-n-butylammonium bromide (1.1 g), and Compound
9A (10.0 g) was heated to 45° C., aged for 10 min, then cooled to 0° C. The solution was treated with aqueous potassium hydroxide (52.4% w/w, 38.2 g) and propargyl bromide (3.36 g as an 80% w/w solution in toluene) maintaining the temperature at 0-5° C. The resulting biphasic mixture stirred vigorously for 16 h at 4-6° C. Subsequently, water (20 mL) was added and the aqueous layer was discharged. The organic layer was washed twice with water (40 mL) and once with aqueous acetic acid (5.2% w/w, 20 mL) at 20° C. The solvent was switched to ethanol by 4 put-and-take additions/vacuum distillations of ethanol (15 mL) at 35-40° C. with the target volume of each cycle being 15 mL remaining in the vessel. The solution was weighed to approximate the amount of ethanol remaining, and ethanol (26 mL) was added to the vessel to bring the total amount of ethanol to 40 mL. The solution was cooled to 4° C. and stirred for 45 min to afford a suspension. The suspension was heated to 38° C. in 15 min, aged 10 min, then cooled to 20° C. over 14 h. The suspension was cooled to 0° C., aged 1.5 h, filtered, and the solids washed twice with ethanol (7.5 mL each) pre-cooled to 0° C. The solid was dried at 40° C. under vacuum to afford Compound 1 (8.27 g, 76% yield) as Form I. The 1H NMR and ESL-MS data matches that as described above in Example 1, Step 1G.
The crystalline free base Compound
1, Form I was characterized by X-ray powder diffraction (XRPD) (FIG. 1
, Table 2) and DSC (FIG. 2
). The DSC indicated the crystalline Compound
1, Form I has an onset of melt (temperature) at about 83.7° C. (76.6 J/g). The Thermogravimetric Analysis (TGA) (FIG. 2
) of the crystalline free base exhibited substantially no weight loss (about 0.2%) from room temperature to ˜125° C. indicating Form I for the free base of Compound
1 is anhydrous.
Medical uses
Crinecerfont is indicated as adjunctive treatment to glucocorticoid replacement to control androgens in people four years of age and older with classic congenital adrenal hyperplasia.[1][2]
Adverse effects
The US Food and Drug Administration prescription label for crinecerfont has a warning for acute adrenal insufficiency or adrenal crisis.[2]
History
Crinecerfont’s approval is based on two randomized, double-blind, placebo-controlled trials in 182 adults and 103 children with classic congenital adrenal hyperplasia.[2] In the first trial, 122 adults received crinecerfont twice daily and 60 received placebo twice daily for 24 weeks.[2] After the first four weeks of the trial, the glucocorticoid dose was reduced to replacement levels, then adjusted based on levels of androstenedione, an androgen hormone.[2] The primary measure of efficacy was the change from baseline in the total glucocorticoid daily dose while maintaining androstenedione control at the end of the trial.[2] The group that received crinecerfont reduced their daily glucocorticoid dose by 27% while maintaining control of androstenedione levels, compared to a 10% daily glucocorticoid dose reduction in the group that received placebo.[2]
In the second trial, 69 children received crinecerfont twice daily and 34 received placebo twice daily for 28 weeks.[2] The primary measure of efficacy was the change from baseline in serum androstenedione at week four.[2] The group that received crinecerfont experienced a statistically significant reduction from baseline in serum androstenedione, compared to an average increase from baseline in the placebo group.[2] At the end of the trial, children assigned to crinecerfont were able to reduce their daily glucocorticoid dose by 18% while maintaining control of androstenedione levels compared to an almost 6% daily glucocorticoid dose increase in children assigned to placebo.[2]
The US Food and Drug Administration (FDA) granted the application for crinecerfont fast track, breakthrough therapy, orphan drug, and priority review designations.[2] The FDA granted the approval of Crenessity to Neurocrine Biosciences, Inc.[2]
Society and culture
Legal status
Crinecerfont was approved for medical use in the United States in December 2024.[1][2][5]
Names
Crinecerfont is the international nonproprietary name.[6]
Crinecerfont is sold under the brand name Crenessity.[1]
References
- ^ Jump up to:a b c d e f g “Crenessity- crinecerfont; capsule Crenessity- crinecerfont solution”. DailyMed. 1 December 2024. Retrieved 25 January 2025.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA Approves New Treatment for Congenital Adrenal Hyperplasia”. U.S. Food and Drug Administration (FDA) (Press release). 1 October 2024. Retrieved 16 December 2024. This article incorporates text from this source, which is in the public domain.
- ^ “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ “Neurocrine Biosciences Announces FDA Approval of Crenessity (crinecerfont), a First-in-Class Treatment for Children and Adults With Classic Congenital Adrenal Hyperplasia” (Press release). Neurocrine Biosciences. 13 December 2024. Retrieved 16 December 2024 – via PR Newswire.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
Further reading
- Auchus, Richard; Chan, Jean; Farber, Robert; Fechner, Patricia; Giri, Nagdeep; Nokoff, Natalie; et al. (1 November 2022). “OR18-4 Crinecerfont (NBI-74788), a Novel CRF1 Receptor Antagonist, Lowers Adrenal Androgens and Precursors in Adolescents with Classic Congenital Adrenal Hyperplasia”. Journal of the Endocrine Society. 6 (Supplement_1): A618. doi:10.1210/jendso/bvac150.1281. PMC 9625506.
- Auchus, Richard J; Sarafoglou, Kyriakie; Fechner, Patricia Y; Vogiatzi, Maria; Giri, Nagdeep; Roberts, Eiry; et al. (8 May 2020). “OR25-03 The Effects of Crinecerfont (NBI-74788), a Novel CRF1 Receptor Antagonist, on Adrenal Androgens and Precursors in Patients with Classic Congenital Adrenal Hyperplasia: Results from A Multiple-Dose Phase 2 Study”. Journal of the Endocrine Society. 4 (Supplement_1): OR25-03. doi:10.1210/jendso/bvaa046.221. PMC 7209526.
- Auchus, Richard J; Sarafoglou, Kyriakie; Fechner, Patricia Y; Vogiatzi, Maria G; Imel, Erik A; Davis, Shanlee M; et al. (17 February 2022). “Crinecerfont Lowers Elevated Hormone Markers in Adults With 21-Hydroxylase Deficiency Congenital Adrenal Hyperplasia”. The Journal of Clinical Endocrinology & Metabolism. 107 (3): 801–812. doi:10.1210/clinem/dgab749. PMC 8851935. PMID 34653252.
- Newfield, Ron S; Sarafoglou, Kyriakie; Fechner, Patricia Y; Nokoff, Natalie J; Auchus, Richard J; Vogiatzi, Maria G; et al. (18 October 2023). “Crinecerfont, a CRF1 Receptor Antagonist, Lowers Adrenal Androgens in Adolescents With Congenital Adrenal Hyperplasia”. The Journal of Clinical Endocrinology & Metabolism. 108 (11): 2871–2878. doi:10.1210/clinem/dgad270. PMC 10583973. PMID 37216921.
External links
- “Crinecerfont (Code C174708)”. NCI Thesaurus.
- Clinical trial number NCT03525886 for “Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of NBI-74788 in Adults With Congenital Adrenal Hyperplasia” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Crenessity |
| Other names | SSR-125543, NBI-74788 |
| AHFS/Drugs.com | Crenessity |
| License data | US DailyMed: Crinecerfont |
| Routes of administration | By mouth |
| Drug class | Corticotropin-releasing factor type 1 receptor antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 752253-39-7 |
| PubChem CID | 5282340 |
| DrugBank | DB18518 |
| ChemSpider | 4445507 |
| UNII | MFT24BX55I |
| KEGG | D12366 |
| ChEBI | CHEBI:34969 |
| ChEMBL | ChEMBL291657 |
| CompTox Dashboard (EPA) | DTXSID10996687 |
| Chemical and physical data | |
| Formula | C27H28ClFN2OS |
| Molar mass | 483.04 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Prete A, Auchus RJ, Ross RJ: Clinical advances in the pharmacotherapy of congenital adrenal hyperplasia. Eur J Endocrinol. 2021 Nov 30;186(1):R1-R14. doi: 10.1530/EJE-21-0794. [Article]
- Yogi A, Kashimada K: Current and future perspectives on clinical management of classic 21-hydroxylase deficiency. Endocr J. 2023 Oct 30;70(10):945-957. doi: 10.1507/endocrj.EJ23-0075. Epub 2023 Jun 29. [Article]
- FDA Approved Drug Products: Crenessity (crinecerfont) capsules/solution for oral administration (December 2024) [Link]
- FDA News Release: FDA Approves New Treatment for Congenital Adrenal Hyperplasia [Link]
///////Crinecerfont, Crenessity, FDA 2024, APPROVALS 2024, 752253-39-7, SSR125543, SSR 125543, SSR-125543, WHO 10958, 06-RORI, NBI-74788, ORPHAN DRUG
RELACORILANT
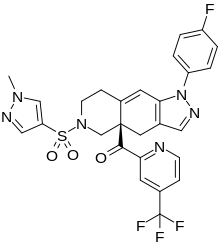
Relacorilant
- Molecular FormulaC27H22F4N6O3S
- Average mass586.561 Da
CAS 1496510-51-0
Phase III
[(4aR)-1-(4-fluorophenyl)-6-(1-methylpyrazol-4-yl)sulfonyl-4,5,7,8-tetrahydropyrazolo[3,4-g]isoquinolin-4a-yl]-[4-(trifluoromethyl)pyridin-2-yl]methanone
релакорилант[Russian][INN]
ريلاكوريلانت[Arabic][INN]
瑞拉可兰[Chinese][INN]
- OriginatorCorcept Therapeutics
- ClassAntineoplastics; Fluorine compounds; Isoquinolines; Ketones; Organic sulfur compounds; Pyrazoles; Pyridines; Small molecules
- Mechanism of ActionGlucocorticoid receptor antagonists
- Orphan Drug StatusYes – Pancreatic cancer; Cushing syndrome
- Phase IIICushing syndrome; Ovarian cancer; Pancreatic cancer
- Phase IIFallopian tube cancer; Peritoneal cancer; Prostate cancer
- Phase I/IISolid tumours
- Phase IAdrenocortical carcinoma
Most Recent Events
- 09 Sep 2022Subgroup analysis efficacy data from a phase-II trial in Ovarian cancer presented at the 47th European Society for Medical Oncology Congress (ESMO-2022)
- 29 Jun 2022Phase-III clinical trials in Ovarian cancer (Combination therapy, Recurrent, Second-line therapy or greater) in USA (PO)
- 06 Jun 2022Corcept Therapeutics announces intentions to submit a NDA for Ovarian cancer
Relacorilant (developmental code name CORT-125134) is an antiglucocorticoid which is under development by Corcept Therapeutics for the treatment of Cushing’s syndrome.[1] It is also under development for the treatment of solid tumors and alcoholism.[1][2] The drug is a nonsteroidal compound and acts as an antagonist of the glucocorticoid receptor.[1] As of December 2017, it is in phase II clinical trials for Cushing’s syndrome and phase I/II clinical studies for solid tumors, while the clinical phase for alcoholism is unknown.[1]
Relacorilant is an orally available antagonist of the glucocorticoid receptor (GR), with potential antineoplastic activity. Upon administration, relacorilant competitively binds to and blocks GRs. This inhibits the activity of GRs, and prevents both the translocation of the ligand-GR complexes to the nucleus and gene expression of GR-associated genes. This decreases the negative effects that result from excess levels of endogenous glucocorticoids, like those seen when tumors overproduce glucocorticoids. In addition, by binding to GRs and preventing their activity, inhibition with CORT125134 also inhibits the proliferation of GR-overexpressing cancer cells. GRs are overexpressed in certain tumor cell types and promote tumor cell proliferation.
SCHEME

CLIP
https://europepmc.org/article/pmc/pmc8175224
Relacorilant (CORT125134)118) is being developed by Corcept Therapeutics, Inc. It is an orally active, high-affinity, selective antagonist of the glucocorticoid receptor that may benefit from the modulation of cortisol activity. In structural optimization, the introduction of a trifluoromethyl group to the 4-position on the pyridyl moiety was found to increase HepG2 tyrosine amino transferase assay potency by a factor of four. Relacorilant is currently being evaluated in a phase II clinical study in patients with Cushing’s syndrome.119)
2-Bromo-4-(trifluoromethyl)pyridine (17) prepared from (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one is employed as a key intermediate for the preparation of relacorilant as shown in Scheme 31.120)

Scheme31. Synthesis of relacorilant.118)
118) H. Hunt, T. Johnson, N. Ray and I. Walters (Corcept Therapeutics, Inc.): PCT Int. Appl. WO2013/177559 (2013).
119) H. J. Hunt, J. K. Belanoff, I. Walters, B. Gourdet, J. Thomas, N. Barton, J. Unitt, T. Phillips, D. Swift and E. Eaton: Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexahydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methanone (CORT125134): A Selective Glucocorticoid Receptor (GR) Antagonist. J. Med. Chem. 60, 3405–3421 (2017). [Abstract] [Google Scholar]
120) B. Lehnemann, J. Jung and A. Meudt (Archimica GmbH): PCT Int. Appl. WO 2007/000249 (2007).
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.7b00162
The nonselective glucocorticoid receptor (GR) antagonist mifepristone has been approved in the U.S. for the treatment of selected patients with Cushing’s syndrome. While this drug is highly effective, lack of selectivity for GR leads to unwanted side effects in some patients. Optimization of the previously described fused azadecalin series of selective GR antagonists led to the identification of CORT125134, which is currently being evaluated in a phase 2 clinical study in patients with Cushing’s syndrome.

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013177559


SYN

////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Cushing’s syndrome (CS) is a metabolic disorder caused by chronic hypercortisolism. CS is associated with cardiovascular, metabolic, skeletal and psychological dysfunctions and can be fatal if left untreated. The first-line treatment for all forms of CS is a surgery. However, medical therapy has to be chosen if surgical resection is not an option or is deemed ineffective. Currently available therapeutics are either not selective and have side effects or are only available as an injection (pasireotide).
References
- ^ Jump up to:a b c d “Relacorilant – Corcept Therapeutics – AdisInsight”.
- ^ Veneris JT, Darcy KM, Mhawech-Fauceglia P, Tian C, Lengyel E, Lastra RR, Pejovic T, Conzen SD, Fleming GF (2017). “High glucocorticoid receptor expression predicts short progression-free survival in ovarian cancer”. Gynecol. Oncol. 146 (1): 153–160. doi:10.1016/j.ygyno.2017.04.012. PMC 5955699. PMID 28456378.
External links
| Clinical data | |
|---|---|
| Other names | CORT-125134 |
| Routes of administration | By mouth |
| Drug class | Antiglucocorticoid |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1496510-51-0 |
| PubChem CID | 73051463 |
| ChemSpider | 57617720 |
| UNII | 2158753C7E |
| KEGG | D11336 |
| Chemical and physical data | |
| Formula | C27H22F4N6O3S |
| Molar mass | 586.57 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
//////////////Relacorilant, Phase III , Orphan Drug, Cushing syndrome, Ovarian cancer, Pancreatic cancer, релакорилант , ريلاكوريلانت , 瑞拉可兰 ,
CN1C=C(C=N1)S(=O)(=O)N2CCC3=CC4=C(CC3(C2)C(=O)C5=NC=CC(=C5)C(F)(F)F)C=NN4C6=CC=C(C=C6)F

NEW DRUG APPROVALS
ONE TIME
$10.00
ELRAGLUSIB
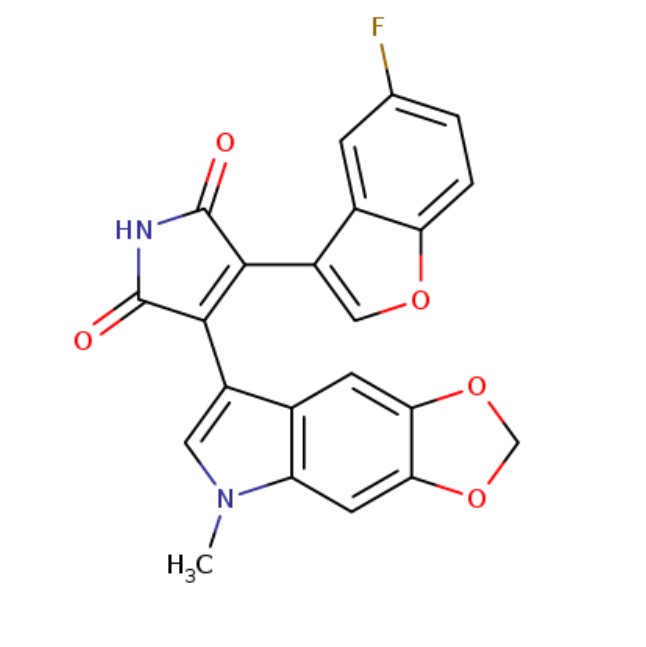
![3-(5-fluorobenzofuran-3-yl)-4-(5-methyl-5H-[1,3]dioxolo[4,5-f]indol-7-yl)-1H-pyrrole-2,5-dione.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=44582816&t=l)
ELRAGLUSIB
RN: 1034895-42-5
UNII: ND1SOF0DLU, WHO 11553, 9-ING-41
- 1H-Pyrrole-2,5-dione, 3-(5-fluoro-3-benzofuranyl)-4-(5-methyl-5H-1,3-dioxolo(4,5-F)indol-7-yl)-
- 3-(5-Fluoro-benzofuran-3-yl)-4-(5-methyl-5H-(1,3)dioxolo(4,5-F)indol-7-yl)-pyrrole-2,5-dioneAntineoplastic
Molecular Formula
- C22-H13-F-N2-O5
Molecular Weight
- 404.3517
- OriginatorNorthwestern University; University of Illinois at Chicago
- DeveloperActuate Therapeutics; Incyte Corporation; Levine Cancer Institute; University of Kansas Medical Center
- ClassAntineoplastics; Benzofurans; Dioxolanes; Indoles; Pyrroles; Small molecules
- Mechanism of ActionGlycogen synthase kinase 3 beta inhibitors
- Orphan Drug StatusYes – Glioblastoma; Neuroblastoma
- Phase IIAdenoid cystic carcinoma; Myelofibrosis; Neuroblastoma; Pancreatic cancer; Salivary gland cancer
- Phase I/IICancer
- PreclinicalBrain cancer; Chronic lymphocytic leukaemia; Colorectal cancer
- 20 Sep 2022Elraglusib – Actuate Therapeutics receives Fast Track designation for Pancreatic cancer [IV] (Combination therapy, First-line therapy, Late-stage disease, Metastatic disease, Recurrent) in USA
- 03 Jun 2022Efficacy and safety data from a phase I trial in cancer presented at the 58th Annual Meeting of the American Society of Clinical Oncology (ASCO-2022)
- 08 Apr 2022Preclinical trials in Brain cancer in USA (unspecified route)
9-ING-41 is under investigation in clinical trial NCT04218071 (Actuate 1901: 9-ING-41 in Myelofibrosis).
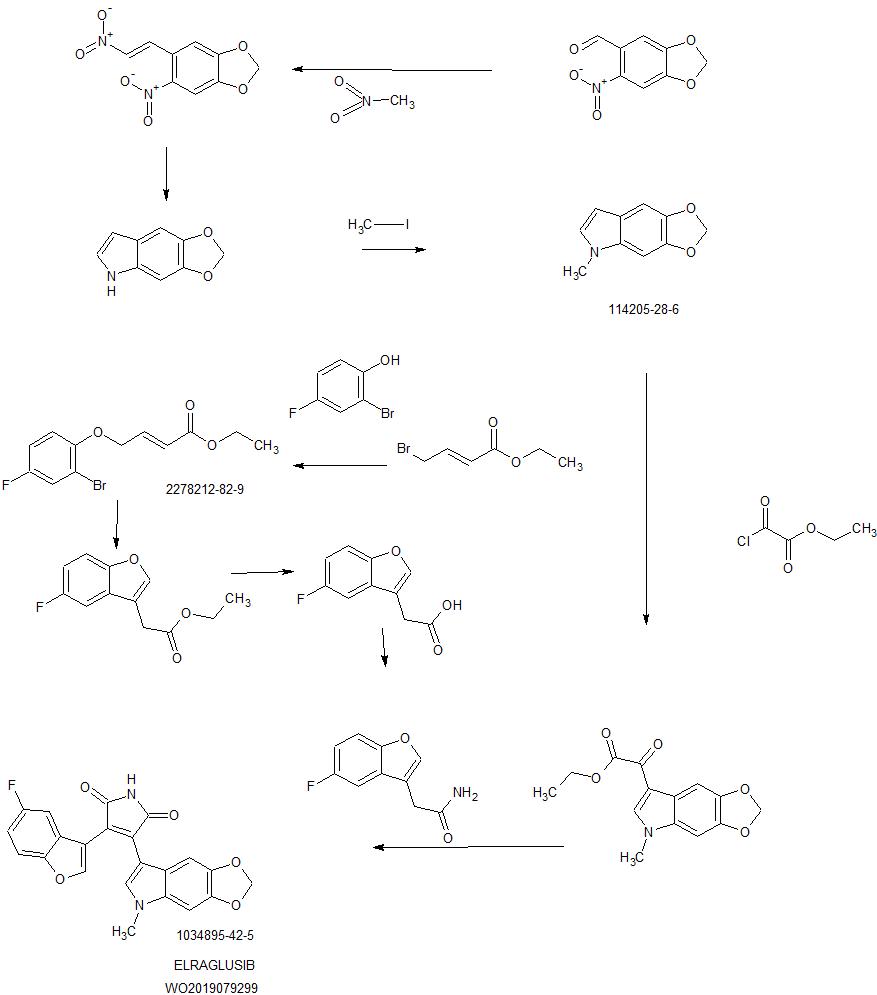

SYN
WO2019079299
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019079299
3-(5-Fluorobenzofuran-3-yl)-4-(5-methyl-5H-[l,3]dioxolo[4,5-f]indol-7-yl)pyrrole-2,5-dione (“9-ING-41”) has the following chemical structure:
[0004] 9-ING-41 has been reported as being useful for the treatment of cancers, including brain, lung, breast, ovarian, bladder, neuroblastoma, renal, and pancreatic cancers, as well as for treatment of traumatic brain injury.
[0005] The structure, properties, and/or biological activity of 9-ING-41 are set forth in U.S. Patent Number 8,207,216; Gaisina et al., From a Natural Product Lead to the Identification of Potent and Selective Benzofuran-3-yl-(indol-3-yl)maleimides as Glycogen Synthase Kinase 3β Inhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells, J. Med. Chem. 2009, 52, 1853-1863; and Hilliard, et al., Glycogen synthase kinase 3β inhibitors induce apoptosis in ovarian cancer cells and inhibit in-vivo tumor growth, Anti-Cancer Drugs 2011, 22:978-985.
Example 1: Preparation of 9-ING-41
[0056] Crude 9-ING-41 can be obtained by the general methods described in U.S. Patent Number 8,207,216, and in Gaisina et al., From a Natural Product Lead to the
Identification of Potent and Selective Benzofuran-3-yl-(indol-3-yl)maleimides as Glycogen Synthase Kinase 3β Inhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells, J. Med. Chem. 2009, 52, 1853-1863.
Example 2: Preparation of 9-ING-41 Crystalline Form I
[0057] Crystalline Form I of 9-ING-41 may also be prepared as follows.
Synthesis of Intermediate 1
[0058] Into a 3-L 4-necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed 6-nitro-2H-l,3-benzodioxole-5-carbaldehyde (200 g, 1.02 mol, 1.00 equiv), ammonium acetate (200 g, 2.59 mol, 2.53 equiv), acetic acid (2 L), and nitromethane (313 g, 5.13 mol, 5.00 equiv). The solution was stirred for 12 h at lOOoC. The reaction repeated three times. The solutions were combined and diluted with 20 L of water. The resulting solution was extracted with 3×10 L of ethyl acetate and the organic layers were combined. The mixture was washed with 3×10 L of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in 450 g (crude) of 5-nitro-6-[(E)-2-nitroethenyl]-2H-l,3-benzodioxole (1) as a dark green solid.
Synthesis of Intermediate 2
[0059] Fe (120 g, 2.14 mol, 17.01 equiv) was slowly added in portions into a suspension of 5-nitro-6-[(Z)-2-nitroethenyl]-2H-l,3-benzodioxole (30 g, 125.97 mmol, 1.00 equiv), silica gel (120 g) in acetic acid (300 mL), toluene (200 mL), and cyclohexane (400 mL) at 80oC under nitrogen. The resulting black mixture was stirred for 8h at 80oC.The reaction repeated ten times. The reaction mixtures were combined. The solids were filtrated out. The filtrate was concentrated under vacuum and the residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1/5). The collected fractions were combined and concentrated under vacuum to give 67.3 g (33%) of 2H, 5H-[1, 3] dioxolo [4, 5-f] indole (2) as an off-white solid.
Synthesis of Intermediate 3
[0060] Sodium hydride (19.9 g, 497.50 mmol, 1.18 equiv, 60%) was added in portions into a solution of 2H,3H,5H-furo[2,3-f]indole (67.3 g, 422.78 mmol, 1.00 equiv) in N,N-
dimethylformamide (1.3 L) at 0°C under nitrogen. The mixture was stirred for lh at 0°C and CH3I (70.9 g, 499.51 mmol, 1.18 equiv) was added dropwise. The resulting solution was stirred for 3 h at room temperature. The solution was quenched by added 1 L of ice water. The resulting solution was extracted with 3×1 L of ethyl acetate and the organic layers were combined. The mixture was washed with 3×1 L of brine, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1/10). The collected fractions were combined and concentrated under vacuum to give 71 g (97%) of 5-methyl-2H,3H,5H-furo[2,3-f]indole (3) as a light yellow solid.
Synthesis of Int rmediate 4
[0061] Ethyl 2-chloro-2-oxoacetate (220 g, 1.61 mol, 3.96 equiv) was added dropwise into a solution of 5-methyl-2H,3H,5H-furo[2,3-f]indole (70.4 g, 406.44 mmol, 1.00 equiv) in ethyl ether (1.6 L) at OoC under nitrogen. The resulting solution was warmed to room temperature and stirred for 4 h. The reaction was quenched slowly by the addition of 2 L of ice water and the pH value of the resulting solution was adjusted to 9 by Na2C03. The resulted mixture was extracted with 3×1.5 L of ethyl acetate. The organic layers were combined and dried over anhydrous sodium sulfate and concentrated under vacuum to give 92.8 g (84%) of ethyl 2-[5-methyl-2H,3H,5H-furo[2,3-f]indol-7-yl]-2-oxoacetate (4) as a light yellow solid.
[0062] 1H MR (300 MHz, DMSO-d6): δ 8.28 (s, 4H), 7.56 (s, 4H), 7.27 (s, 4H), 6.17 (s, 1H), 6.08 (s, 8H), 4.35 (q, J = 7.1 Hz, 7H), 3.85 (s, 11H), 3.35 (s, 2H), 1.35 (t, J = 7.1 Hz, 11H), 1.25 (s, 2H).
Synthesis of Intermediate 5
5
[0063] Into a 10-L 4-necked round-bottom flask was placed 2-bromo-4-fluorophenol (500 g, 2.62 mol, 1.00 equiv), N,N-dimethylformamide (5 L), potassium carbonate (1253 g, 9.07 mol, 3.46 equiv), and ethyl (2E)-4-bromobut-2-enoate (1010 g, 5.23 mol, 2.00 equiv). The resulting solution was stirred for 12 h at room temperature. The solids were collected by filtration. The reaction was then quenched by the addition of 15 L of water and extracted with 3×10 L of ethyl acetate. The organic layers were combined and washed with 4×20 L of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1/20). The collected fractions were combined and concentrated under vacuum to give 500 g (63%) of ethyl (2E)-4-(2-bromo-4-fluorophenoxy)but-2-enoate (5) as a white solid.
Synthesis of Intermediate 6
[0064] Into a 2-L 3 -necked round-bottom flask, purged and maintained with an inert atmosphere of nitrogen, was placed ethyl (2E)-4-(2-bromo-4-fluorophenoxy)but-2-enoate (125 g, 412.37 mmol, 1.00 equiv), benzyltri ethyl azanium chloride (99 g, 434.64 mmol, 1.05 equiv), sodium formate dihydrate (45.1 g), Pd(OAc)2 (2.9 g, 12.92 mmol, 0.03 equiv), sodium carbonate (92 g, 868.01 mmol, 2.10 equiv), and N,N-dimethylformamide (1.25 L). The resulting solution was stirred for 12 h at 80°C. The reaction repeated four times. The reaction mixtures were combined and the solids were filtrated out. The filtrate was diluted with 10 L of brine and extracted with 3×5 L of ethyl acetate. The organic layers were combined and washed with 4×6 L of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1/20). The collected fractions were combined and concentrated under vacuum. This resulted in 258 g (crude) of ethyl 2-(5-fluoro-l-benzofuran-3-yl)acetate (6) as light yellow oil.
Synthesis of Intermediate 7
7
[0065] Into a 5-L round-bottom flask was placed ethyl 2-(5-fluoro-l-benzofuran-3-yl)acetate (147 g, 661.53 mmol, 1.00 equiv), methanol (1 L), tetrahydrofuran (1 L), water (1 L), and Li OH (47.7 g, 1.99 mol, 3.01 equiv). The resulting solution was stirred for 3 h at room temperature. The reaction repeated twice. The mixture was concentrated under vacuum and then extracted with 1 L of dichloromethane. The aqueous layer was collected and the pH of the layer was adjust to 1-3 by hydrogen chloride (1 mol/L). The resulting solution was extracted with 3×1 L of ethyl acetate and the combined organic layers were dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in 160 g (62%) of 2-(5-fluoro-l-benzofuran-3-yl)acetic acid (7) as a white solid.
Synthesis of Intermediate 8
[0066] Into a 10-L round-bottom flask was placed 2-(5-fluoro-l-benzofuran-3-yl) acetic acid (160 g, 824.1 mmol, 1.00 equiv), H4C1 (436 g, 8.16 mol, 9.89 equiv), N,N-dimethylformamide (6L), DIEA (1064 g, 8.24 mol, 9.99 equiv), and HATU (376 g, 988.88 mmol, 1.20 equiv). The resulting solution was stirred for 12 h at room temperature. The resulting solution was diluted with 10 L of water. The solids were collected by filtration to give in 126 g (78%) of 2-(5-fluoro-l-benzofuran-3-yl) acetamide (8) as a white solid.
Synthesis of 9-ING-41 in cr stalline Form I
8 9-ING-41
[0067] t-BuOK (1200 mL, 1 mol/L in THF) was added dropwise into a solution of ethyl 2-[5-methyl-2H,3H,5H-furo[2,3-f]indol-7-yl]-2-oxoacetate (100 g, 365.9 mmol, 1.00 equiv), 2-(5-fluoro-l-benzofuran-3-yl)acetamide (72 g, 372.7 mmol, 1.02 equiv) in tetrahydrofuran (3 L) at 0°C under nitrogen. The reaction was stirred for 2h at room temperature. The reaction was cooled to 0°C and poured into of 2 L of H4C1 (saturated solution in water) and extracted with 4×2 L of dichloromethane. The organic layers were combined, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/dichloromethane/petroleum ether (1/1/5). The collected fractions were combined and concentrated under vacuum to give 107.9 g (74%) of 3-(5-fluoro-l-benzofuran-3-yl)-4-[5-methyl-2H,5H-[l,3]dioxolo[4,5-f]indol-7-yl]-2,5-dihydro-lH-pyrrole-2,5-dione as a red solid. This red solid is 9-ING-41 crystalline Form I. MS-ESI: [M+H]+ = 405.
PATENT
WO2019032958
PATENT
US20100004308
REF
Journal of Medicinal Chemistry (2009), 52(7), 1853-1863
PATENT
WO2008077138
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Elraglusib is a maleimide-based, small molecule inhibitor of glycogen synthase kinase-3 (GSK-3; serine/threonine-protein kinase GSK3) with potential antineoplastic activity. Upon intravenous administration, elraglusib binds to and competitively inhibits GSK-3, which may lead to downregulation of nuclear factor kappa B (NF-kappaB) and decreased expression of NF-kappaB target genes including cyclin D1, B-cell lymphoma 2 (Bcl-2), anti-apoptotic protein XIAP, and B-cell lymphoma extra-large (Bcl-XL). This may inhibit NF-kappaB-mediated survival and chemoresistance in certain tumor types. GSK-3, a constitutively active serine/threonine kinase that plays a role in numerous pathways involved in protein synthesis, cellular proliferation, differentiation, and metabolism, is aberrantly overexpressed in certain tumor types and may promote tumor cell survival and resistance to chemotherapy and radiotherapy.
Actuate Therapeutics Announces Initiation of a Multicenter Randomized Trial of Elraglusib Plus FOLFIRINOX As First Line Therapy for Advanced Pancreatic Cancer
Published: Feb 07, 2022
CHICAGO and FORT WORTH, Texas, Feb. 07, 2022 (GLOBE NEWSWIRE) — Actuate Therapeutics (Actuate), a clinical stage biopharmaceutical company, today announced the opening of a randomized study of elraglusib (9-ING-41) plus FOLFIRINOX alone or with Losartan for patients with advanced pancreatic cancer in the first-line setting (NCT05077800). Elraglusib is Actuate’s proprietary small molecule glycogen synthase kinase-3 beta (GSK-3β) inhibitor which is being developed for adults and children with advanced refractory cancers. This multicenter investigator-initiated study, which is receiving substantial support from the Lustgarten Foundation for Pancreatic Cancer Research, is being led by Colin D. Weekes MD at the Massachusetts General Hospital and will also enroll patients at the University of Washington, University of Colorado Denver, and Johns Hopkins University.
“Novel approaches for patients with advanced pancreatic cancer are urgently required,” said Dr Weekes. “The pre-clinical and clinical data being generated with elraglusib in a spectrum of cancers, including pancreatic cancer, is extremely encouraging and we are delighted to have initiated this study with elraglusib. Elraglusib is the first clinically relevant specific GSK-3β inhibitor that we can thoroughly investigate. In preclinical models, elraglusib has multiple biologic effects relevant to targeting pancreatic cancer including direct cytotoxicity, reversal of chemoresistance, reversal of pathologic fibrosis, and there is increasing evidence of its immune-modulatory activity. In our study, we are particularly focused on elraglusib’s potential to synergize with TGF-β suppression mediated by Losartan. This study builds on the work of our investigative teams demonstrating the roles of TGF-β and GSK-3β in acquired chemotherapy resistance. This study uniquely attempts to harness the mechanisms that pancreatic cancer utilizes to combat the effects of chemotherapy as an Achilles heel for therapeutic intent. We believe that a multi-pronged attack as represented by elraglusib plus Losartan is a potentially sophisticated approach to a complex, often lethal, situation. It is an honour to lead this multicenter collaboration with my clinical and pre-clinical colleagues across the US and Europe. We are very grateful for the critical support of this program by the Lustgarten Foundation.”
“At the Lustgarten Foundation, we understand time is everything for patients and their families,” said Andrew Rakeman, PhD, VP of Research. “Dr. Weekes’ study will help us understand and address a critical issue in pancreatic cancer treatment—acquired chemotherapy resistance. This trial builds on exciting observations from previous preclinical and clinical research. The Foundation established the Clinical Accelerator Initiative for projects like this; bringing more trials based on the best science to the clinic and expanding our understanding of pancreatic cancer biology and treatment. We believe Dr. Weekes’ trial and others like it have the potential to change the way we think about treating pancreatic cancer, ultimately transforming it into a curable disease.”
“We are honored and excited to collaborate with Dr. Weekes, his colleagues at world-leading cancer research centers, and the Lustgarten Foundation on this important trial, which will advance the development of elraglusib for treating patients with one of the most challenging types of cancer,” said Daniel Schmitt, Actuate’s President & CEO. “The results we have seen to date with elraglusib combined with chemotherapy in pancreatic cancer are very promising, and this Phase 2 trial in combination with FOLFIRINOX leverages significant positive preclinical and clinical experience for potentially better outcomes for patients.”
Based on positive data from a prior Phase 2 open-label single arm study of elraglusib plus gemcitabine/nab-paclitaxel, Actuate has also recently initiated an international randomized controlled study of elraglusib in combination with gemcitabine/nab-paclitaxel, in patients with advanced pancreatic cancer in the first-line setting (NCT03678883, EudraCT#:2018-003739-32). Actuate is also conducting studies in pediatric patients with refractory tumors in preparation for a neuroblastoma-specific clinical program (NCT04239092). Actuate is also collaborating with investigators at the Dana-Farber Cancer Institute and Brigham and Women’s Hospital on a Phase 2 study focused on elraglusib combined with cytotoxic therapy for patients with advanced salivary gland carcinomas (NCT05010629).
About Actuate Therapeutics, Inc.
Actuate is a clinical stage pharmaceutical company focused on the development and commercialization of novel therapeutics for cancers and inflammatory diseases. For additional information, please visit the Company’s website at http://www.actuatetherapeutics.com.
///////////ELRAGLUSIB, WHO 11553, 9-ING-41, Orphan Drug
Cn1cc(C2=C(C(=O)NC2=O)c3coc4ccc(F)cc34)c5cc6OCOc6cc15

NEW DRUG APPROVALS
one time
$10.00
Valemetostat tosilate
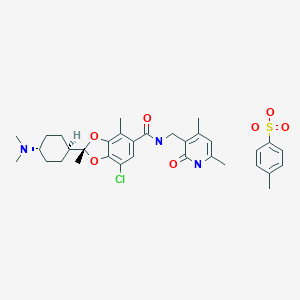
Valemetostat tosilate
バレメトスタットトシル酸塩
| Formula | C26H34ClN3O4. C7H8O3S |
|---|---|
| CAS | 1809336-93-3 |
| Mol weight | 660.2205 |
PMDA JAPAN approved 2022/9/26, Ezharmia
- 1,3-Benzodioxole-5-carboxamide, 7-chloro-N-((1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl)-2-(trans-4-(dimethylamino)cyclohexyl)-2,4-dimethyl-, (2R)-, compd. with 4-methylbenzenesulfonate (1:1)
Antineoplastic, histone methyltransferase inhibitor
1809336-39-7 (free base). 1809336-93-3 (tosylate) 1809336-92-2 (mesylate) 1809336-94-4 (fumarate) 1809336-95-5 (tarate)
Synonym: Valemetostat; DS-3201; DS 3201; DS3201; DS-3201b
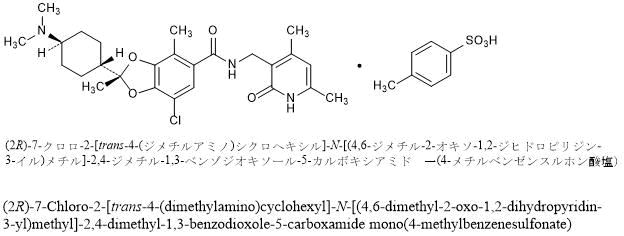
(2R)-7-Chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide mono(4-methylbenzenesulfonate)
C26H34ClN3O4▪C7H8O3S : 660.22
[1809336-93-3]
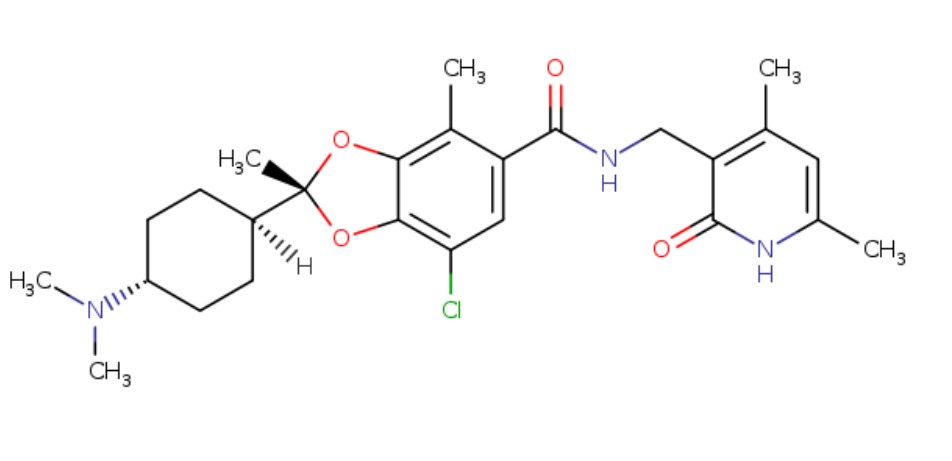
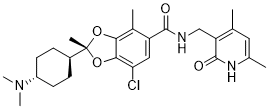
1809336-39-7 (free base)
Chemical Formula: C26H34ClN3O4
Exact Mass: 487.2238
Molecular Weight: 488.02
(2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide
Valemetostat, also known as DS-3201 is a potent, selective and orally active EZH1/2 inhibitor. DS-3201 selectively inhibits the activity of both wild-type and mutated forms of EZH1 and EZH2. Inhibition of EZH1/2 specifically prevents the methylation of lysine 27 on histone H3 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways, enhances transcription of certain target genes, and results in decreased proliferation of EZH1/2-expressing cancer cells.
- OriginatorDaiichi Sankyo Inc
- DeveloperCALYM Carnot Institute; Daiichi Sankyo Inc; Lymphoma Academic Research Organisation; Lymphoma Study Association; University of Texas M. D. Anderson Cancer Center
- ClassAmides; Amines; Antineoplastics; Benzodioxoles; Chlorinated hydrocarbons; Cyclohexanes; Pyridones; Small molecules
- Mechanism of ActionEnhancer of zeste homolog 1 protein inhibitors; Enhancer of zeste homolog 2 protein inhibitors
- Orphan Drug StatusYes – Adult T-cell leukaemia-lymphoma; Peripheral T-cell lymphoma
- New Molecular EntityYes
- RegisteredAdult T-cell leukaemia-lymphoma
- Phase IIB-cell lymphoma; Peripheral T-cell lymphoma
- Phase I/IISmall cell lung cancer
- Phase INon-Hodgkin’s lymphoma; Prostate cancer; Renal cell carcinoma; Urogenital cancer
- PreclinicalDiffuse large B cell lymphoma
- No development reportedAcute myeloid leukaemia; Precursor cell lymphoblastic leukaemia-lymphoma
- 26 Sep 2022First global approval – Registered for Adult T-cell leukaemia-lymphoma (Monotherapy, Second-line therapy or greater) in Japan (PO)
- 26 Sep 2022Updated efficacy and adverse events data from a phase II trial in Adult T-cell leukaemia-lymphoma released by Daiichi Sankyo
- 28 Dec 2021Preregistration for Adult T-cell leukaemia-lymphoma (Monotherapy, Second-line therapy or greater) in Japan (PO

PATENT
WO 2015141616
Watson, W. D. J. Org. Chem. 1985, 50, 2145.
Lengyel, I. ; Cesare, V. ; Stephani, R. Synth. Common. 1998, 28, 1891.
PATENT
WO2022009911
The equipment and measurement conditions for the powder X-ray diffraction measurement in the examples are as follows.
Model: Rigaku Rint TTR-III
Specimen: Appropriate
X-ray generation conditions: 50 kV, 300 mA
Wavelength: 1.54 Å (Copper Kα ray)
Measurement temperature: Room temperature
Scanning speed: 20°/min
Scanning range: 2 to 40°
Sampling width: 0.02°
[0043]
(Reference Example 1) Production of ethyl trans-4-[(tert-butoxycarbonyl)amino]cyclohexanecarboxylate
[0044]
[hua 6]
[0045]
Under a nitrogen atmosphere, ethanol (624 L) and ethyl trans-4-aminocyclohexanecarboxylate monohydrochloride (138.7 kg, 667.8 mol) were added to a reaction vessel and cooled. Triethylamine (151.2 kg, 1495 .5 mol) and di-tert-butyl dicarbonate (160.9 kg, 737.2 mol) were added dropwise while maintaining the temperature below 20°C. After stirring at 20-25°C for 4 hours, water (1526 kg) was added dropwise at 25°C or lower, and the mixture was further stirred for 2 hours. The precipitated solid was collected by filtration, washed with a mixture of ethanol:water 1:4 (500 L), and dried under reduced pressure at 40°C to obtain 169.2 kg of the title compound (yield 93.4%). .
1 H NMR (500 MHz, CDCl 3 ): δ 4.37 (br, 1H), 4.11 (q, J = 2.8 Hz, 2H), 3.41 (br, 1H), 2.20 (tt, J = 4.8, 1.4 Hz, 1H),2.07(m,2H),2.00(m,2H),1.52(dq,J=4.6,1.4Hz,2H),1.44(s,9H),1.24(t,J=2.8Hz,3H), 1.11(dq,J=4.6,1.4Hz,2H)
[0046]
(Reference Example 2) Production of tert-butyl = [trans-4-(hydroxymethyl)cyclohexyl]carbamate
[0047]
[hua 7]
[0048]
Under a nitrogen atmosphere, tetrahydrofuran (968 kg), ethyl = trans-4-[(tert-butoxycarbonyl)amino]cyclohexanecarboxylate (110 kg, 405.4 mol), lithium chloride (27.5 kg, 648 kg) were placed in a reaction vessel. .6 mol), potassium borohydride (32.8 kg, 608.1 mol), and water (2.9 L, 162.2 mol) were added, the temperature was slowly raised to 50°C, and the mixture was further stirred for 6 hours. Cooled to 0-5°C. Acetone (66 L) and 9 wt % ammonium chloride aqueous solution (1210 kg) were added dropwise while maintaining the temperature at 20° C. or lower, and the mixture was stirred at 20-25° C. for 1 hour. Additional ethyl acetate (550 L) was added, the aqueous layer was discarded and the organic layer was concentrated to 550 L. Ethyl acetate (1650 L) and 9 wt% aqueous ammonium chloride solution (605 kg) were added to the residue, and the aqueous layer was discarded after stirring. Washed sequentially with water (550 L). The organic layer was concentrated to 880 L, ethyl acetate (660 L) was added to the residue, and the mixture was concentrated to 880 L while maintaining the internal temperature at 40-50°C. The residue was cooled to 0-5° C. and stirred for 1 hour, petroleum ether (1760 L) was added dropwise over 30 minutes, and the mixture was stirred at the same temperature for 2 hours. The precipitated solid was collected by filtration, washed with a petroleum ether:ethyl acetate 3:1 mixture (220 L) cooled to 0-5°C, and dried at 40°C under reduced pressure to give 86.0 kg of the title compound (yield: obtained at a rate of 92.3%).
1 H NMR (500 MHz, CDCl 3 ): δ 4.37 (br, 1H), 3.45 (d, J = 2.2 Hz, 2H), 3.38 (br, 1H), 2.04 (m, 2H),
1.84(m,2H),1.44(m,10H),1.28-1.31(m,1H),1.00-1.13(m,4H)
[0049]
(Reference Example 3) Production of tert-butyl = [trans-4-(2,2-dibromoethenyl)cyclohexyl]carbamate
[0050]
[hua 8]
[0051]
(Step 1)
Under a nitrogen atmosphere, ethyl acetate (50 L), tert-butyl = [trans-4-(hydroxymethyl)cyclohexyl]carbamate (2.5 kg, 10.90 mol), potassium bromide ( 39.3 g, 0.33 mol), 2,2,6,6-tetramethylpiperidine 1-oxyl (51.1 g, 0.33 mol), 4.8% aqueous sodium hydrogen carbonate solution (26.25 kg ) was added and cooled to 0-5°C, 9.9% sodium hypochlorite (8.62 kg, 11.45 mol) was added at 5°C or lower, and the mixture was further stirred at 0°C for 4 hours. Sodium sulfite (250 g) was added to the mixture and stirred at 0-5°C for 30 minutes before warming to 20-25°C. Thereafter, the aqueous layer was discarded and washed with a 20% aqueous sodium chloride solution (12.5 kg), then the organic layer was dried over sodium sulfate and concentrated to 7.5 L. Ethyl acetate (12.5 L) was added to the residue, the mixture was concentrated again to 7.5 L, and used in the next reaction as a tert-butyl=(trans-4-formylcyclohexyl)carbamate solution.
[0052]
(Step 2)
Under a nitrogen atmosphere, tetrahydrofuran (30 L) and triphenylphosphine (5.72 kg, 21.8 mol) were added to a reaction vessel, heated to 40°C, and stirred for 5 minutes. Carbon tetrabromide (3.61 kg, 10.9 mol) was added over 30 minutes and stirred at 40-45° C. for another 30 minutes. A mixture of tert-butyl (trans-4-formylcyclohexyl)carbamate solution and triethylamine (2.54 kg, 25.1 mol) was added below 45°C over 20 minutes and stirred at 40°C for an additional 15 hours. After cooling the reaction solution to 0° C., water (0.2 L) was added at 10° C. or lower, and water (25 L) was added. After heating to 20-25° C., the aqueous layer was discarded, ethyl acetate (4.5 kg) and 10% aqueous sodium chloride solution (25 kg) were added, and after stirring, the aqueous layer was discarded again. After the obtained organic layer was concentrated to 15 L, 2-propanol (19.65 kg) was added and concentrated to 17.5 L. 2-Propanol (11.78 kg) and 5 mol/L hydrochloric acid (151.6 g) were added to the residue, and the mixture was stirred at 25-35°C for 2.5 hours. Water (16.8 L) was added dropwise to the resulting solution, and the mixture was stirred at 20-25°C for 30 minutes and then stirred at 0°C for 2 hours. The precipitated solid was collected by filtration, washed with a mixture (11 kg) of acetonitrile:water 60:40 cooled to 0-5°C, and dried at 40°C under reduced pressure to give 3.05 kg of the title compound (yield 73%). .0%).
1 H NMR (500 MHz, CDCl3):δ6.20(d,J=3.6Hz,1H),4.37(br,1H),3.38(br,1H),2.21(dtt,J=3.6,4.6,1.4Hz,1H),2.05-2.00(m,2H),1.80-1.83(m,2H),1.44(s,9H),1.23(ddd,J=9.9,5.3,1.2 Hz,2H), 1.13(ddt,J=4.6,1.4,5.2 Hz,2H)
[0053]
(Reference Example 4) Production of tert-butyl = (trans-4-ethynylcyclohexyl) carbamate
[0054]
[Chemical 9]
[0055]
Under a nitrogen atmosphere, toluene (1436 kg), tert-butyl = [trans-4-(2,2-dibromoethenyl)cyclohexyl]carbamate (110 kg, 287.1 mol), and N,N,N ‘,N’-Tetramethylethane-1,2-diamine (106.7 kg, 918.8 mol) was added and cooled to -10°C. An isopropylmagnesium chloride-tetrahydrofuran solution (2.0 mol/L, 418 kg, 863 mol) was added dropwise at -5°C or lower, and stirred at -10°C for 30 minutes. After the reaction, 5 mol/L hydrochloric acid (465 kg) was added at 5°C or lower, heated to 20-25°C, and further 5 mol/L hydrochloric acid (41.8 kg) was used to adjust the pH to 5.0-. adjusted to 6.0. After discarding the aqueous layer, the organic layer was washed twice with water (550 L) and concentrated to 550 L. 2-Propanol (1296 kg) was added to the concentrate and concentrated to 550 L again. Further, 2-propanol (1296 kg) was added to the residue, and after concentrating to 550 L, water (770 L) was added dropwise in 4 portions. At that time, it was stirred for 30 minutes after each addition. After the addition, the mixture was stirred for 1 hour and further stirred at 0° C. for 1 hour. The precipitated solid was collected by filtration, washed with a 5:7 mixture of 2-propanol:water (550 L) cooled to 0-5°C, and dried at 40°C under reduced pressure to yield 57.8 kg of the title compound. obtained at a rate of 90.2%).
1 H NMR (500 MHz, CDCl 3 ): δ 4.36 (br, 1H), 3.43 (br, 1H), 2.18-2.23 (m, 1H), 1.97-2.04 (m, 5H), 1.44-1.56 (m, 11H ),1.06-1.14(m,2H)
[0056]
(Reference Example 5) Production of 4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile
[0057]
[Chemical 10]
[0058]
Under a nitrogen atmosphere, water (300 L), 2-cyanoacetamide (20 kg, 238 mol), 1-pentane-2-4-dione (26.2 kg, 262 mol), potassium carbonate (3.29 mol) were added to a reaction vessel. kg, 23.8 mol) was added and stirred at room temperature for 6 hours or longer. After the reaction, the precipitated solid was collected by filtration, washed with water (60 L), further washed with a mixture of methanol (40 L) and water (40 L), and dried under reduced pressure at 40°C to give the title compound as 34 Obtained in .3 kg (97.3% yield).
1 H NMR (500 MHz, DMSO-d 6 ): δ 2.22 (s, 3H), 2.30 (s, 3H), 6.16 (s, 1H), 12.3 (brs, 1H)
[0059]
(Reference Example 6) Production of 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one monohydrochloride
[0060]
[Chemical 11]
[0061]
Under a nitrogen atmosphere, water (171 L), methanol (171 L), 4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (17.1 kg, 116 mol), concentrated After adding hydrochloric acid (15.8 kg, 152 mol) and 5% palladium carbon (55% wet) (3.82 kg), the inside of the reaction vessel was replaced with hydrogen. Then, the mixture was pressurized with hydrogen and stirred overnight at 30°C. After the reaction, the reaction vessel was purged with nitrogen, the palladium on carbon was removed by filtration, and the palladium on carbon was washed with a 70% aqueous solution of 2-propanol (51 L). Activated carbon (0.86 kg) was added to the filtrate and stirred for 30 minutes. Activated carbon was removed by filtration and washed with 70% aqueous 2-propanol solution (51 L). The filtrate was concentrated under reduced pressure until the liquid volume became 103 L, and 2-propanol (171 L) was added. The mixture was again concentrated under reduced pressure until the liquid volume reached 103 L, then 2-propanol (171 L) was added, and the mixture was stirred for 1 hour or longer. Precipitation of a solid was confirmed, and the solution was concentrated to a volume of 103 L. Further, 2-propanol (51 L) was added, and after concentration under reduced pressure until the liquid volume reached 103 L, the mixture was stirred at 50° C. for 30 minutes. After adding acetone (171 L) over 1 hour while keeping the internal temperature at 40° C. or higher, the mixture was stirred at 40 to 45° C. for 30 minutes. The solution was cooled to 25°C and stirred for 2 hours or longer, and the precipitated solid was collected by filtration, washed with acetone (86 L) and dried under reduced pressure at 40°C to give 19.7 kg of the title compound (yield 90.4%). ).
1 H NMR (500 MHz, methanol-d 4 ): δ 2.27 (s, 3H), 2.30 (s, 3H), 4.02 (s, 2H), 6.16 (s, 1H)
[0062]
(Example 1-1) Production of methyl 5-chloro-3,4-dihydroxy-2-methylbenzoate
[0063]
[Chemical 12]
[0064]
Under a nitrogen atmosphere, water (420 L), toluene (420 L), acetonitrile (420 L), and methyl 3,4-dihydroxy-2-methylbenzoate (1) (60 kg, 329 mol) were added to the reactor and cooled. After that, sulfuryl chloride (133.4 kg, 988 mol) was added dropwise while maintaining the temperature at 20°C or lower. After the reaction, the mixture was separated into an organic layer 1 and an aqueous layer, acetonitrile (60 L) and toluene (120 L) were added to the aqueous layer, and the mixture was stirred. Water (420 L) and acetonitrile (210 L) were added to the organic layer 1, and after cooling, sulfuryl chloride (88.9 kg, 659 mol) was added dropwise at 20°C or lower, and sulfuryl chloride (53.2 kg, 394 mol) was added. ) was added in portions. After the reaction, the mixture was separated into an organic layer 3 and an aqueous layer, and the organic layer 2 was added to the aqueous layer and stirred. Water (420 L), acetonitrile (210 L) were added to the combined organic layer, sulfuryl chloride (44.5 kg, 329 mol) was added dropwise below 20°C, and sulfuryl chloride (106.4 kg, 788 mol) was added. ) was added in portions. After the reaction, the organic layer 4 and the aqueous layer were separated, acetonitrile (60 L) and toluene (120 L) were added to the aqueous layer, and the mixture was stirred. The combined organic layers were washed three times with 20 wt % aqueous sodium chloride solution (300 L) and then concentrated under reduced pressure to 600 L. After repeating the operation of adding toluene (300 L) and concentrating under reduced pressure to 600 L again twice, the mixture was heated and stirred at 60° C. for 1 hour. After cooling to room temperature, the precipitated solid was collected by filtration, washed with toluene (120 L), and dried under reduced pressure at 40°C to give 52.1 kg of the crude title compound (2) (yield: 73.0%). ).
[0065]
Under a nitrogen atmosphere, toluene (782 L) and crude title compound (52.1 kg, 241 mol) were added to a reactor and heated to 80°C. After confirming that the crystals were completely dissolved, they were filtered and washed with heated toluene (261 L). The mixture was cooled to 60° C. and stirred for 0.5 hours after crystallization. After cooling to 10°C, the precipitated solid was collected by filtration, washed with toluene (156 L), and dried under reduced pressure at 40°C to give 47.9 kg of the title compound (2) (yield 91.9%). Acquired.
1 H NMR (500 MHz, methanol-d 4 ): δ 2.41 (s, 3H), 3.82 (s, 3H), 7.41 (s, 1H)
[0066]
(Example 1-2) Examination of chlorination conditions 1 Since
it is difficult to remove compound (1), which is the starting material, and compound (4), which is a by-product of the reaction, even in subsequent steps, need to control. Therefore, chlorination was investigated in the same manner as in Example 1-1 using compound (1) as a starting material. Table 1 shows the results.
[0067]
[Chemical 13]
[0068]
[Table 1]
[0069]
HPLC condition
detection: 220 nm
column: ACQUITY UPLC BEH C18 (2.1 mm ID x 50 mm, 1.7 μm, Waters)
column temperature: 40 ° C
mobile phase: A: 0.1 vol% trifluoroacetic acid aqueous solution, B: acetonitrile
Gradient conditions:
[0070]
[Table 2]
[0071]
Flow rate: 1.0 mL/min
Injection volume: 1 μL
Sample solution: acetonitrile/water (1:1)
wash solution: acetonitrile/water (1:1)
purge solution: acetonitrile/water (1:1)
seal wash solution : Acetonitrile/water (1:1)
Sample cooler temperature: None
Measurement time: 5 minutes
Area measurement time: about 0.5 minutes – 4.0 minutes
Comp. 1: 1.11 min, Comp. 2: 1.55 min,
Comp. 3: 1.44 min, Comp. 4: 1.70 min
[0072]
(Example 1-3) Examination of chlorination conditions 2
Compound (1) was used as a starting material, sulfuryl chloride was used as a chlorination reagent, and chlorination in various solvents was examined. Table 3 shows the results.
[0073]
[table 3]
[0074]
(Example 2) Methyl (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5- Manufacture of carboxylates
[0075]
[Chemical 14]
[0076]
Toluene (9.0 L), tert-butyl = (trans-4-ethynylcyclohexyl) carbamate (2.23 kg, 9.99 mol), methyl = 5-chloro-3,4- were added to a reaction vessel under a nitrogen atmosphere. Dihydroxy-2-methylbenzoate (1.80 kg, 8.31 mol), tri(o-tolyl)phosphine (76.0 g, 250 mmol), triruthenium dodecacarbonyl (53.0 g, 82.9 mmol) ) was added, and the mixture was heated and stirred at 80 to 90° C. for 7 hours under an oxygen-containing nitrogen stream. The reaction solution was cooled to room temperature to obtain a toluene solution of the title compound.
[0077]
(Example 3) (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carvone acid production
[0078]
[Chemical 15]
[0079]
Methyl = (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole obtained in Example 2 -5-carboxylate toluene solution (13 L, equivalent to 7.83 mol), methanol (9.0 L), 1,2-dimethoxyethane (3.6 L), 5 mol / L sodium hydroxide aqueous solution ( 2.50 L, 12.5 mol) was added and stirred at 55-65° C. for 3 hours. After adding water (5.4 L), the mixture was allowed to stand and separated into an organic layer and an aqueous layer. After cooling to room temperature, 1,2-dimethoxyethane (16.2 L) was added to the aqueous layer, and after adjusting the pH to 4.0 to 4.5 with 3 mol/L hydrochloric acid, toluene (5.4 L) was added. added. After heating to 50-60° C., the organic layer and aqueous layer were separated, and the organic layer was washed with a 20 wt % sodium chloride aqueous solution (7.2 L). Then, 1,2-dimethoxyethane (21.6 L) was added to the organic layer, and after concentration under reduced pressure to 9 L, 1,2-dimethoxyethane (21.6 L) was added and heated to 50-60°C. After that, filtration was performed to remove inorganic substances. Then, after washing with 1,2-dimethoxyethane (1.8 L), the 1,2-dimethoxyethane solution of the title compound (quantitative value 89.6% (Example 2 total yield from ), corresponding to 7.45 mol).
[0080]
(Example 4) (1S)-1-phenylethanaminium (2R)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1, Preparation of 3-benzodioxole-5-carboxylate
[0081]
[Chemical 16]
[0082]
(2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5 obtained in Example 3 – A solution of carboxylic acid in dimethoxyethane (21.6 L, corresponding to 7.45 mol) was heated to 75-80°C, and then (1S)-1-phenylethanamine (1.02 kg, 8.42 mmol). was added and stirred for 4 hours. A mixture of 1,2-dimethoxyethane (9.2 L) and water (3.4 L) heated to 50-60° C. was added, stirred, and then cooled to room temperature. The precipitated solid was collected by filtration and washed with 1,2-dimethoxyethane (9 L) to give a crude title compound (1.75 kg (converted to dry matter), yield 38.5% (Example 2 total yield from ) and an optical purity of 93.8% ee).
[0083]
Under a nitrogen atmosphere, a 1,2-dimethoxyethane aqueous solution (13.6 L) was placed in a reaction vessel, and (1S)-1-phenylethanaminium obtained in step 1 (2R)-2-{trans-4-[(tert -Butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate crude (1.70 kg equivalent, 3.11 mol) was added. After that, 5 mol/L hydrochloric acid (0.56 L, 2.8 mol) was added dropwise. After stirring at room temperature for 10 minutes or longer, the mixture was heated to 75° C. or higher, and (1S)-1-phenylethanamine (360 g, 2.97 mmol) was dissolved in 1,2-dimethoxyethane (2.6 L). The solution was added dropwise over 1 hour. It was then washed with 1,2-dimethoxyethane (0.9 L), stirred for 2 hours and cooled to 0-5°C. The slurry was collected by filtration and washed with 1,2-dimethoxyethane (5.1 L) cooled to 0-5° C. to give the title compound (1.56 kg, yield 91.9%, obtained with an optical purity of 99.5% ee).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.15-1.23(m,2H), 1.28-1.35(m,2H), 1.42(s,9H),
1.59(s,3H), 1.60-1.61(d ,3H,J=7.0Hz,3H),1.80-1.86(dt,J=12.0,3.0Hz,1H),1.95-1.96(m,4H),2.27(s,3H),3.24-3.28(m,1H ),4.39-4.43(q,J=7.0Hz,1H),7.07(s,1H),7.37-7.45(m,5H)
[0084]
(Example 5) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride Manufacturing A
[0085]
[Chemical 17]
[0086]
(Step 1)
Under a nitrogen atmosphere, 1,2-dimethoxyethane (200 L) and (1S)-1-phenylethanaminium (2R)-2-{trans-4-[(tert-butoxycarbonyl) were placed in a reaction vessel. Amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate (equivalent to 87.64 kg, 160 mol), 35% hydrochloric acid (16.7 kg, 160 mol) was added and heated to 45-55° C., 35% hydrochloric acid (36.7 kg, 352 mol) was added dropwise in 7 portions and stirred for 3 hours after dropping. After cooling to room temperature, the reaction solution was added to a mixture of water (982 L) and 5 mol/L sodium hydroxide (166.34 kg, 702 mol). 3 mol/L hydrochloric acid (22.4 kg) was added dropwise to the resulting solution at 30°C, crystal precipitation was confirmed, and the mixture was stirred for 30 minutes or more, cooled to 10°C, and further stirred for 2 hours. After stirring, 3 mol/L hydrochloric acid (95.1 kg) was added dropwise at 10°C to adjust the pH to 7.0. The slurry liquid was collected by filtration, washed with water (293 L) cooled to 10° C., and (2R)-2-(trans-4-aminocyclohexyl)-7-chloro-2,4-dimethyl-1,3- Benzodioxol-5-carboxylic acid trihydrate was obtained (57.63 kg (converted to dry matter), yield 94.7%).
1 H NMR (500 MHz, methanol- d4 + D2O): 1.32-1.44 ( m, 4H), 1.61 (s, 3H), 1.89-1.94 (m, 1H), 2.01-2.13 (m, 4H) ,2.27(s,3H),2.99-3.07(m,1H),7.06(s,3H)
[0087]
(Step 2)
Under nitrogen atmosphere, 1,2-dimethoxyethane (115 L), (2R)-2-(trans-4-aminocyclohexyl)-7-chloro-2,4-dimethyl-1,3 -benzodioxole-5-carboxylic acid trihydrate (57.63 kg equivalent, 152 mmol), formic acid (34.92 kg, 759 mol), 37% formaldehyde aqueous solution (93.59 kg, 1153 mol) was added and stirred at 55-65°C for 2 hours. Cool to room temperature, add 2-propanol (864 L) and concentrate to 576 L under reduced pressure. 2-Propanol (231 L) was added thereto and concentrated under reduced pressure to 576 L. Further, 2-propanol (231 L) was added and concentrated under reduced pressure to 576 L. After concentration, 35% hydrochloric acid (20.40 kg, 196 mol) was added dropwise over 2 hours and stirred at room temperature for 30 minutes. Ethyl acetate (576 L) was added to the resulting slurry over 30 minutes and concentrated to 692 L. Ethyl acetate (461 L) was added followed by further concentration to 519 L. Ethyl acetate (634 L) was added to the residue and the mixture was stirred at room temperature for 2 hours. The precipitated solid was collected by filtration, washed with ethyl acetate (491 L) and dried under reduced pressure at 40°C to give the title compound (51. 56 kg, 87.1% yield).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.38-1.47 (m, 2H), 1.53-1.61 (m, 2H), 1.67 (s, 3H), 1.99-2.05 (m, 1H), 2.13 -2.18(m,4H),2.38(s,3H),2.84(s,6H),3.19-3.25(dt,J=12.5,3.5Hz,1H),
7.53(s,1H)
[0088]
(Example 6) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride Manufacturing B
[0089]
[Chemical 18]
[0090]
Under a nitrogen atmosphere, formic acid (20 mL), 37% formaldehyde aqueous solution (15 mL), dimethoxyethane (10 mL), (1S)-1-phenylethanaminium (2R)-2-{trans-4- [(tert-Butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate (10 g, 18.3 mmol) was added and Stirred for 10 hours. After cooling to room temperature and filtering the insolubles, 2-propanol (100 mL) was added and the mixture was concentrated under reduced pressure until the liquid volume became 30 mL. While stirring at room temperature, ethyl acetate (120 mL) and concentrated hydrochloric acid (6.1 mL) were added to form a slurry. This was concentrated under reduced pressure to 30 mL, ethyl acetate (120 mL) was added, and then concentrated under reduced pressure to 30 mL again. After adding ethyl acetate (120 mL), the precipitated solid was collected by filtration, washed with ethyl acetate (50 mL) and dried under reduced pressure at 40°C to give 6.56 g of the title compound (yield 92.0%). Acquired.
[0091]
(Example 7) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl ) Preparation of methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide p-toluenesulfonate
[0092]
[Chemical 19]
[0093]
Under nitrogen atmosphere, acetone (6.5 L), purified water (1.3 L), (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4- Dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride (650.4 g, 1.67 mol), 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one Monohydrochloride (330.1 g, 1.75 mol) and triethylamine (337 g, 3.33 mol) were added and stirred at room temperature for 30 minutes. After that, 1-hydroxybenzotriazole monohydrate (255 g, 1.67 mol), 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (383 g, 2.00 mmol) were added, and the mixture was stirred overnight at room temperature. Stirred. After adjusting the pH to 11 with 5 mol/L sodium hydroxide, toluene (9.8 L) was added, and after stirring, the mixture was separated into an organic layer 1 and an aqueous layer. Toluene (3.3 L) was added to the aqueous layer, and after stirring, the aqueous layer was discarded, and the obtained organic layer was combined with the previous organic layer 1. The combined organic layers were concentrated under reduced pressure to 9.75 L, toluene (6.5 L) was added and washed twice with purified water (3.25 L). The resulting organic layer was concentrated under reduced pressure to 4.875 L and 2-propanol (1.625 L) was added. A solution of p-toluenesulfonic acid monohydrate (0.12 kg, 0.631 mol) dissolved in 4-methyl-2-pentanone (1.14 L) was added to the organic layer heated to 68°C. The mixture was added dropwise over 5 hours and stirred at 68°C for 30 minutes. Furthermore, a solution of p-toluenesulfonic acid monohydrate (0.215 kg, 1.13 mol) dissolved in 4-methyl-2-pentanone (2.11 L) was added dropwise over 3.5 hours, Stirred at 68° C. for 30 minutes. After that, 4-methyl-2-pentanone (6.5 L) was added dropwise over 1 hour. After cooling to room temperature, the precipitated solid was collected by filtration, washed with 4-methyl-2-pentanone (3.25 L) and dried under reduced pressure at 40°C to give 1.035 kg of the crude title compound (yield 94%). .2%).
[0094]
Under a nitrogen atmosphere, 2-propanol (6.65 L) and the obtained crude title compound (950 g) were added to the reactor and stirred. Purified water (0.23 L) was added to completely dissolve the solid at 68° C., filtered, and washed with warm 2-propanol (0.95 L). After confirming that the solid was completely dissolved at an internal temperature of 68°C, the solution was cooled to 50°C. After cooling, seed crystals* (9.5 g, 0.01 wt) were added and stirred at 50° C. overnight. tert-Butyl methyl ether (11.4 L) was added dropwise thereto in 4 portions over 30 minutes each. At that time, it was stirred for 30 minutes after each addition. After cooling to room temperature, the precipitated solid was collected by filtration, washed with a mixture of 2-propanol (0.38 L) and tert-butyl methyl ether (3.42 L), and further treated with tert-butyl methyl ether (4.75 L). ) and dried under reduced pressure at 40° C. to obtain the title compound (915.6 g, yield 96.4%).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.35-1.43 (m, 2H), 1.49-1.57 (m, 2H), 1.62 (s, 3H),
1.94-2.00 (dt, J = 12.5, 3.0Hz ,1H),2.09-2.13(m,4H),2.17(s,3H),2.24(s,3H),2.35(s,3H),2.36(s,3H),2.82(s,6H),3.16- 3.22(dt,J=12.0,3.5Hz,1H),4.42(s,2H),
6.10(s,1H),6.89(s,1H),7.22-7.24(d,J=8.0Hz,2H),7.69 -7.71(dt,J=8.0,1.5 Hz,2H)
*Seed crystal preparation method
Under a nitrogen atmosphere, 2-propanol (79.0 L) and the obtained crude title compound (7.90 kg) were added to a reactor and stirred. Purified water (7.9 L) was added to completely dissolve the solid, and activated carbon (0.40 kg) was added and stirred. After filtering the activated carbon, it was washed with 2-propanol (79.0 L) and concentrated to 58 L. 2-Propanol (5 L) was added to the residue, and after heating to 64° C., tert-butyl methyl ether (19.8 L) was added, and after crystal precipitation was confirmed, tert-butyl methyl ether (75. 1 L) was added in three portions. At that time, it was stirred for 30 minutes after each addition. After cooling to room temperature, the precipitated solid was collected by filtration, washed with a mixture of 2-propanol (7.9 L) and tert-butyl methyl ether (15.8 L), and dried under reduced pressure at 40°C to obtain seed crystals. The title compound was obtained (7.08 kg, 89.6% yield).
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
///////Valemetostat tosilate, japan 2022, approvals 2022, Ezharmia, バレメトスタットトシル酸塩 , DS-3201, DS 3201, DS3201, DS-3201b, Orphan Drug
CN(C)[C@@H]1CC[C@H](CC1)[C@]2(C)Oc3c(C)c(cc(Cl)c3O2)C(=O)NCC4=C(C)C=C(C)NC4=O

NEW DRUG APPROVALS
ONE TIME
$10.00
Danavorexton, TAK 925


Danavorexton, TAK 925
2114324-48-8
- Molecular FormulaC21H32N2O5S
- Average mass424.554 Da
1-Piperidinecarboxylic acid, 3-[(methylsulfonyl)amino]-2-[[(cis-4-phenylcyclohexyl)oxy]methyl]-, methyl ester, (2R,3S)-
Methyl (2R,3S)-3-[(methylsulfonyl)amino]-2-[[(cis-4-phenylcyclohexyl)oxy]methyl]-1-piperidinecarboxylate
- OriginatorTakeda
- ClassCyclohexanes; Esters; Ethers; Piperidines; Sleep disorder therapies; Small molecules; Sulfonamides
- Mechanism of ActionOrexin receptor type 2 agonists
- Orphan Drug StatusYes – Narcolepsy
- Phase IHypersomnia; Narcolepsy; Respiration disorders; Sleep apnoea syndrome
- 01 Jun 2022Takeda Pharmaceuticals completes a phase I clinical trials in Respiratory disorder (In adults) in Netherlands (IV) (ISRCTN63027076)
- 02 Apr 2022Efficacy and safety data from phase a Ib trial in Hypersomnia presented at the 74th Annual Meeting of the American Academy of Neurology 2022 (AAN-2022)
- 10 Mar 2022Phase-I clinical trials in Sleep apnoea syndrome in Australia (IV) (NCT05180890)
Danavorexton (developmental code name TAK-925) is a selective orexin 2 receptor agonist.[1] It is a small-molecule compound and is administered intravenously.[1][2] The compound was found to dose-dependently produce wakefulness to a similar degree as modafinil in a phase 1 clinical trial.[1][3] As of March 2021, danavorexton is under development for the treatment of narcolepsy, idiopathic hypersomnia, and sleep apnea.[2][1][4] It is related to another orexin receptor agonist known as TAK-994, the development of which was discontinued for safety reasons in October 2021.[1][5]
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00626
TAK-925, a potent, selective, and brain-penetrant orexin 2 receptor (OX2R) agonist, [methyl (2R,3S)-3-((methylsulfonyl)amino)-2-(((cis-4-phenylcyclohexyl)oxy)methyl)piperidine-1-carboxylate, 16], was identified through the optimization of compound 2, which was discovered by a high throughput screening (HTS) campaign. Subcutaneous administration of compound 16 produced wake-promoting effects in mice during the sleep phase. Compound 16 (TAK-925) is being developed for the treatment of narcolepsy and other related disorders.


aReagents and conditions: (a) chiral column separation; (b) RCOCl, Et3N, THF, rt (for 15 and 16); (c) ethyl chlorocarbonate, DIEA, THF, rt (for 17); (d) isocyanatoethane, Et3N, THF, 0 °C−rt (for 18).
Methyl (2R,3S)-3-((methylsulfonyl)amino)-2-(((cis-4- phenylcyclohexyl)oxy)methyl)piperidine-1-carboxylate (16) To a mixture of 14 (58 mg, 0.16 mmol) and Et3N (0.044 mL, 0.32 mmol) in THF (3 mL) was added methyl chlorocarbonate (0.024 mL, 0.32 mmol) at rt. The mixture was stirred at rt overnight. The mixture was quenched with water and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane/EtOAc, 1:1 to 0:100) to give 16 (64 mg, 0.15 mmol, 95%) as a colorless oil. Crystallization of 16 (1.8 g, 4.1 mmol) from EtOH-H2O gave 16 (1.7 g, 3.9 mmol, 95%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 1.40−1.55 (5H, m), 1.56−1.73 (5H, m), 1.87 (1H, brd, J = 13.2 Hz), 1.96 (1H, brd, J = 13.6 Hz), 2.44−2.57 (1H, m), 2.83 (1H, brs), 2.95 (3H, s), 3.40 (1H, brs), 3.53−3.62 (5H, m), 3.73 (1H, brt, J = 9.7 Hz), 3.84 (1H, brs), 4.47 (1H, brs), 7.15 (1H, brt, J = 7.2 Hz), 7.18 (1H, brs), 7.19 (2H, brd, J = 8.1 Hz), 7.27 (2H, brt, J = 7.4 Hz). 13C NMR (151 MHz, DMSO-d6, the minor rotamer’s signals are marked with an asterisk) δ24.05, 24.39*, 26.00, 26.17*, 27.60*, 27.79, 28.68, 30.15*, 37.54, 38.13*, 39.91, 42.99, 51.01, 52.07, 53.90*, 54.49, 61.48, 61.89*, 71.68, 125.68, 126.51, 128.14, 147.34, 155.27*, 156.08. MS (ESI/APCI) mass calculated for [M + H]+ (C21H33N2O5S) requires m/z 424.6, found m/z 425.2. mp 113 °C. Anal. Calcd for C21H32N2O5S: C, 59.41; H, 7.60; N, 6.60. Found: C, 59.45; H, 7.59; N, 6.55. [α] 20 D +16.3 (c 0.1, CHCl3
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Other names | TAK-925 |
| Routes of administration | Intravenous[1][2] |
| Drug class | Orexin receptor agonist |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2114324-48-8 |
| PubChem CID | 130310079 |
| ChemSpider | 68011464 |
| UNII | 1QMD83K4YN |
| ChEMBL | ChEMBL4650341 |
| Chemical and physical data | |
| Formula | C21H32N2O5S |
| Molar mass | 424.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
References
- ^ Jump up to:a b c d e f Jacobson LH, Hoyer D, de Lecea L (January 2022). “Hypocretins (orexins): The ultimate translational neuropeptides”. J Intern Med. doi:10.1111/joim.13406. PMID 35043499.
- ^ Jump up to:a b c “Danavorexton – Takeda”. Adis Insight. Springer Nature Switzerland AG. Retrieved 7 March 2021.
- ^ Evans, R., Hazel, J., Faessel, H., Wu, J., Hang, Y., Alexander, R., … & Hartman, D. (2019). Results of a phase 1, 4-period crossover, placebo-controlled, randomized, single dose study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of TAK-925, a novel orexin 2 receptor agonist, in sleep-deprived healthy adults, utilizing modafinil as an active comparator. Sleep Medicine, 64, S106. https://scholar.google.com/scholar?cluster=10933819770107034612
- ^ Evans R, Tanaka S, Tanaka S, Touno S, Shimizu K, Sakui S, et al. (December 2019). “A Phase 1 single ascending dose study of a novel orexin 2 receptor agonist, TAK-925, in healthy volunteers (HV) and subjects with narcolepsy type 1 (NT1) to assess safety, tolerability, pharmacokinetics, and pharmacodynamic outcomes”. Sleep Medicine. 64: S105–S106. doi:10.1016/j.sleep.2019.11.290.
- ^ Tong A (6 October 2021). “Takeda flashes red light on ‘breakthrough’ narcolepsy drug after PhII trials turned up mysterious safety signal”. Endpoints News.
External links
///////////////Danavorexton, TAK 925, ORPHAN DRUG, PHASE 1

NEW DRUG APPROVALS
ONE TIME
$10.00
Darinaparsin
Darinaparsin
ダリナパルシン , Darvias
JAPAN 2022 APPROVED, PMDA 2022/6/20
(2S)-2-amino-5-[[(2R)-1-(carboxymethylamino)-3-dimethylarsanylsulfanyl-1-oxopropan-2-yl]amino]-5-oxopentanoic acid
Glycine, L-gamma-glutaMyl-S-(diMethylarsino)-L-cysteinyl-
| Formula | C12H22AsN3O6S |
|---|---|
| CAS | 69819-86-9 |
| Mol weight | 411.3062 |
| Efficacy | Antineoplastic |
|---|---|
| Comment | organic arsenical |
Zinapar, ZIO-101, DMAs(III)G, clarinaparsin, UNII-9XX54M675G, SP-02L
- OriginatorTexas A&M University; University of Texas M. D. Anderson Cancer Center
- DeveloperSolasia Pharma; ZIOPHARM Oncology
- ClassAmines; Antineoplastics; Arsenicals; Oligopeptides; Pentanoic acids; Small molecules; Sulfides
- Mechanism of ActionApoptosis stimulants; Cell cycle inhibitors; Reactive oxygen species stimulants
- Orphan Drug StatusYes – Peripheral T-cell lymphoma
- PreregistrationPeripheral T-cell lymphoma
- DiscontinuedLiver cancer; Lymphoma; Multiple myeloma; Non-Hodgkin’s lymphoma; Solid tumours
- 28 Mar 2022No recent reports of development identified for phase-I development in Peripheral-T-cell-lymphoma in China (IV, Injection)
- 26 Jan 2022ZIOPHARM Oncology is now called Alaunos Therapeutics
- 11 Dec 2021Safety and efficacy data from a phase II trial in Peripheral T-cell lymphoma presented at the 63rd American Society of Hematology Annual Meeting and Exposition (ASH-2021)
Darinaparsin is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects. Compared to inorganic arsenic compounds such as arsenic trioxide (As2O3), darinaparsin appears to exhibit a wide therapeutic window.
Darinaparsin, also know as ZIO-101 and SP-02, is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects.
Darinaparsin is an organic arsenical composed of dimethylated arsenic linked to glutathione, and is being investigated for antitumor properties in vitro and in vivo. While other arsenicals, including arsenic trioxide, have been used clinically, none have shown significant activity in malignancies outside of acute promyelocytic leukemia. Darinaparsin has significant activity in a broad spectrum of hematologic and solid tumors in preclinical models. Here, we review the literature describing the signaling pathways and mechanisms of action of darinaparsin and compare them to mechanisms of cell death induced by arsenic trioxide. Darinaparsin has overlapping, but distinct, signaling mechanisms. We also review the current results of clinical trials with darinaparsin (both intravenous and oral formulations) that demonstrate significant antitumor activity.
PAPER
Biochemical Pharmacology (Amsterdam, Netherlands), 126, 79-86; 2017



PATENT
WO 2015085208
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015085208
Preparation of Darinaparsin
[0071] Sterile water (15.5 L) and ethyl alcohol (200 proof, 15.5 L) were charged in a reaction flask prior to the addition of L-glutathione (3.10 kg). While being stirred, the reaction mixture was cooled to 0-5 °C prior to the addition of triethylamine (1.71 L). Stirring was continued until most of the solids were dissolved and the solution was filtered. After filtration, the reaction mixture was cooled to 0-5 °C prior to the addition of chlorodimethylarsine (1.89 kg) over 115 minutes while maintaining the temperature at 0-5 °C. Stirring continued at 0-5 °C for 4 hours before acetone (30.6 L) was added over 54 minutes while maintaining the temperature at 0-5 °C. The suspension was stored at 0-5°C overnight prior to filtration. The solid was collected in a filter funnel, washed successively with ethyl alcohol (200 proof, 13.5 L) and acetone (13.5 L) and dried in suction for 23 minutes. A second similar run was performed and the collected solids from both runs were combined. Ethyl alcohol (200 proof, 124 L) and the combined solids (11.08 kg) were charged in a vessel. The slurry was stirred at ambient temperature for 2 hours before filtration, washing successively with ethyl alcohol (200 proof, 27 L) and acetone (27 L) and dried in suction for 60 minutes. The resulting solid was transferred to drying trays and dried in a vacuum oven at ambient temperature for 66 hours to provide darinaparsin as a solid with the differential scanning calorimetry (DSC) thermogram of Figure 1, with an extrapolated onset temperature at about 191.36° C and a peak temperature at about 195.65° C.
PATENT
WO 2010021928
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109°C was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
SGLU-1: Glutathione (14.0 g, 45.6 mmol) was stirred rapidly in glyme while dimethylchoroarsine (6.5 g, 45.6 mmol) was added dropwise. Pyridine (6.9 g, 91.2 mmol) was then added to the slurry and the mixture was subsequently heated to reflux. The heat was removed immediately and the mixture stirred at room temperature for 4 h. Isolation of the resultant insoluble solid and recrystallization from ethanol afforded 4 as the pyridine hydrochloride complex (75% yield). mp 115-118°C; NMR (D20) δ1.35 (s, 6H), 1.9-4.1 (m’s, 10H), 7.8-9.0 (m, 5H); mass spectrum (m/e) 140, 125, 110, 105, 79, 52, 45, 36.
PATENT
WO 2009075870
Step 1
Example 1. Preparation of Dimethylchloroarsine (DMCA). A 3-neck round-bottom flask (500 mL) equipped with mechanical stirrer, inlet for nitrogen, thermometer, and an ice bath was charged with cacodylic acid (33 g, 0.23 mol) and cone. hydrochloric acid (67 mL). In a separate flask, a solution of SnCl2·2H2O (54 g, 0.239 mol) in cone. hydrochloric acid (10 mL) was prepared. The SnCl2·2 H2O solution was added to the cacodylic acid in HCl solution under nitrogen while maintaining the temperature between 5 °C and 10 °C. After the addition was complete, the ice bath was removed and the reaction mixture was stirred at ambient temperature for 1 h. The reaction mixture was transferred to a separatory funnel and the upper layer (organic) collected. The bottom layer was extracted with dichloromethane (DCM) (2 × 25 mL). The combined organic extract was washed with 1 N HCl (2 × 10 mL) and water (2 × 20 mL). The organic extract was dried over MgSO4 and DCM was removed by rotary evaporation (bath temperature 80 °C, under nitrogen, atmospheric pressure). The residue was further distilled under nitrogen. Two tractions of DMCA were collected. The first fraction contained some DCM and the second fraction was of suitable quality (8.5 g, 26% yield). The GC analysis confirmed the identity and purity of the product.
Step 2
Example 3. Preparation of S-Dimethylarsinoglutathione (SGLU-1). In a 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel and thermometer under an inert atmosphere was prepared a suspension of glutathione (114.5 g, 0.37 mol) in a 1:1 (v/v) mixture of water/ethanol (1140 mL) and cooled to below 5 °C. The mixture was treated slowly (over 15 min) with triethylamine (63.6 mL, 0.46 mol) while maintaining the temperature below 20 °C. The mixture was cooled to 4 °C and stirred for 15 min and then the traces of undissolved material removed by filtration. The filtrate was transferred in a clean 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel, nitrogen inlet, and thermometer and DMCA (70 g, 0.49 mol) (lot # 543-07-01-44) was added slowly while maintaining the temperature at 3-4°C. The reaction mixture was stirred at 1-4°C for 4 h, and acetone (1.2 L) was added over a period of 1 h. The mixture was stirred for 90 min between 2 and 3°C and the resulting solid was isolated by filtration. The product was washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and the wet solids were suspended in ethanol 200 Proof (2000 mL). The product was isolated by filtration, washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and dried in vacuum for 2 days at RT to give 115 g (75%) of SGLU-1, HPLC purity > 99.5% (in process testing).
PATENT
WO 2007027344
Example 2 Preparation of S-Dimethylarsinoglutathione A 5 L, three necked round bottom flask was equipped with a mechanical stirrer assembly, thermometer, addition funnel, nitrogen inlet, and a drying tube was placed in a cooling bath. A polyethylene crock was charged with glutathione-reduced (200 g) and deionized water (2 L) and stirred under a nitrogen atmosphere to dissolve all solids. The mixture was filtered to remove any insoluble material and the filtrate was transferred to the 5 L flask. While stirring, ethanol, 200 proof (2 L) was added and the clear solution was cooled to 0-5° C. using an ice/methanol bath. Pyridine (120 g) was added followed by a dropwise addition of Me2AsCl (120 g) over a minimum of 1 hour. The reaction mixture was stirred at 0-5° C. for a minimum of 2 hours prior to removal of the cooling bath and allowing the mixture to warm to room temperature under a nitrogen atmosphere with stirring. The reaction mixture was stirred overnight (>15 hrs) at room temperature under a nitrogen atmosphere at which time a white solid may precipitate. The reaction mixture was concentrated to a slurry (liquid and solid) at 35-45° C. using oil pump vacuum to provide a white solid residue. As much water as possible is removed, followed by two coevaporations with ethanol to azeotrope the last traces of water. The white solid residue was slurried in ethanol, 200 pf. (5 L) under a nitrogen atmosphere at room temperature overnight. The white solid was filtered and washed with ethanol, 200 pf. (2×500 mL) followed by acetone, ACS (2×500 mL). The resulting solid was transferred to drying trays and vacuum oven dried overnight at 25-35° C. using oil pump vacuum to provide pyridinium hydrochloride-free S-dimethylarsinoglutathione as a white solid. melting point of 189-190° C.
PATENT
WO 20060128682
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109° C. was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
Pyridine Hydrochloride Free Synthesis of S-Dimethylarsinoglutathione (GLU) Dimethylarsinoglutathione is made using an adapted of Chen (Chen, G. C., et al. Carbohydrate Res. (1976) 50: 53-62) the contents of which are hereby incorporated by reference in their entirety. Briefly, dithiobis(dimethylarsinoglutamine) is dissolved in dichloromethane under nitrogen. Tetramethyldiarsine is added dropwise to the solution and the reaction is stirred overnight at room temperature under nitrogen and then exposed to air for 1 h. The mixture is then evaporated to dryness and the residue is washed with water and dried to give a crude solid that is recrystallized from methanol to give S-dimethylarsinoglutathione.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

NEW DRUG APPROVALS
ONE TIME TO PAY BLOG SUBSCRIPTIONS
$10.00
Solasia Announces Submission of New Drug Application for Anti-cancer Drug DARINAPARSIN for Peripheral T-Cell Lymphoma in Japan
Solasia Pharma K.K. (TSE: 4597, Headquarters: Tokyo, Japan, President & CEO: Yoshihiro Arai, hereinafter “Solasia”) today announced submission of a New Drug Application (NDA) for its new anti-cancer drug darinaparsin (generic name, development code: SP-02) as a treatment for relapsed or refractory peripheral T-cell lymphoma to the Ministry of Health, Labour and Welfare (MHLW). Based on positive results of R&D on darinaparsin, centered primarily on the results of the Asian Multinational Phase 2 Study (study results released in June 2020), Solasia filed an NDA for the drug with the regulatory authority in Japan ahead of anywhere else in the world.
Solasia expects to obtain regulatory approval in 2022 and to also launch in the same year. If approved and launched, darinaparsin would be the third drug Solasia successfully developed and brought to market since its founding and is expected to contribute to the treatment of PTCL.
Mr. Yoshihiro Arai, President and CEO of Solasia, commented as follows:
“No standard treatment has been established for relapsed or refractory PTCL as of yet. I firmly believe that darinaparsin, with its novel mechanism of action that differs from those of already approved drugs, will contribute to patients and healthcare providers at clinical sites as a new treatment option for relapsed or refractory PTCL. Since founding, Solasia has conducted R&D on five pipeline drugs. Of the five, we have successfully developed and brought to market two drugs, i.e., began providing them to patients, and today, we submitted an NDA for our first anti-cancer drug. Under our mission to provide patients with ‘Better Medicine for a Brighter Tomorrow’, we will continue aiming to contribute to patients’ treatment and enhanced quality of life. ”
About darinaparsin (SP-02)
Darinaparsin, an organoarsenic compound with anticancer activity, is a novel mitochondrial-targeted agent being developed for the treatment of various hematologic and solid tumors. The proposed mechanism of action of the drug involves the disruption of mitochondrial function, increased production of reactive oxygen species, and modulation of intracellular signal transduction pathways. Darinaparsin is believed to exert anticancer effect by inducing cell cycle arrest and apoptosis. Darinaparsin has been granted orphan drug designation in the US and EU.
For more information, please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Asian Multinational Phase 2 Study
The Asian Multinational Phase 2 Study was a multinational, multicenter, single-arm, open-label, non-randomized study to evaluate the efficacy and safety of darinaparsin monotherapy in patients with relapsed or refractory PTCL conducted in Japan, Korea, Taiwan, and Hong Kong. (CT.gov Identifier: NCT02653976).
Solasia plans to present the results of the study at an international academic conference to be held in the near future.
About peripheral T-cell lymphoma (PTCL)
Please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Solasia
Please visit at https://solasia.co.jp/en/
/////////////Darinaparsin, Darvias, JAPAN 2022, APPROVALS 2022, PMDA, ダリナパルシン , Zinapar, ZIO-101, DMAs(III)G, clarinaparsin, UNII-9XX54M675G, SP-02L, Orphan Drug
C[As](C)SCC(C(=O)NCC(=O)O)NC(=O)CCC(C(=O)O)N
Olipudase alfa
| HPLSPQGHPA RLHRIVPRLR DVFGWGNLTC PICKGLFTAI NLGLKKEPNV ARVGSVAIKL CNLLKIAPPA VCQSIVHLFE DDMVEVWRRS VLSPSEACGL LLGSTCGHWD IFSSWNISLP TVPKPPPKPP SPPAPGAPVS RILFLTDLHW DHDYLEGTDP DCADPLCCRR GSGLPPASRP GAGYWGEYSK CDLPLRTLES LLSGLGPAGP FDMVYWTGDI PAHDVWHQTR QDQLRALTTV TALVRKFLGP VPVYPAVGNH ESTPVNSFPP PFIEGNHSSR WLYEAMAKAW EPWLPAEALR TLRIGGFYAL SPYPGLRLIS LNMNFCSREN FWLLINSTDP AGQLQWLVGE LQAAEDRGDK VHIIGHIPPG HCLKSWSWNY YRIVARYENT LAAQFFGHTH VDEFEVFYDE ETLSRPLAVA FLAPSATTYI GLNPGYRVYQ IDGNYSGSSH VVLDHETYIL NLTQANIPGA IPHWQLLYRA RETYGLPNTL PTAWHNLVYR MRGDMQLFQT FWFLYHKGHP PSEPCGTPCR LATLCAQLSA RADSPALCRH LMPDGSLPEA QSLWPRPLFC (Disulfide bridge: 43-119, 46-111, 74-85, 175-180, 181-204, 339-385, 538-542, 548-561) |
Olipudase alfa
Xenpozyme, Japan 2022, APPROVALS 2022, 2022/3/28
PEPTIDE, オリプダーゼアルファ (遺伝子組換え)
Alternative Names: Acid sphingomyelinase Niemann Pick disease type B – Sanofi; Acid-sphingomyelinase – Sanofi; GZ-402665; Recombinant human acid sphingomyelinase – Sanofi; rhASM – Sanofi; Sphingomyelinase-C (synthetic human) – Sanofi; Synthetic human sphingomyelinase-C – Sanofi; Xenpozyme
| Formula | C2900H4373N783O791S24 |
|---|---|
| CAS | 927883-84-9 |
| Mol weight | 63631.0831 |
| Efficacy | Lysosomal storage disease treatment, Enzyme replacement (acid sphingomyelinase) |
|---|---|
| Comment | Enzyme replacement therapy product Treatment of Niemann-Pick disease type A/B |
- OriginatorGenzyme Corporation
- DeveloperSanofi
- ClassRecombinant proteins; Sphingomyelin phosphodiesterases
- Mechanism of ActionSphingomyelin-phosphodiesterase replacements
- Orphan Drug StatusYes – Niemann-Pick diseases
- RegisteredNiemann-Pick diseases
- 28 Mar 2022Registered for Niemann-Pick diseases (In adolescents, In children, In adults) in Japan (IV) – First global approval
- 09 Feb 2022FDA assigns PDUFA action date of (03/07/2022) for Olipudase alfa (In children, In adults) for Niemann-Pick diseases
- 09 Feb 2022Adverse e
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
Olipudase Alfa Improves Lung Function, Spleen Volume in ASMD
Olipudase Alfa Improves Lung Function, Spleen Volume in ASMD
Olipudase alfa was associated with significant improvements in clinically relevant disease end points among patients with chronic visceral acid sphingomyelinase (ASM) deficiency (ASMD), according to results from the phase 2/3 ASCEND trial presented at the 17th Annual WORLDSymposium.
ASMD is a rare, debilitating lysosomal storage disease characterized by a deficiency of the enzyme acid sphingomyelinase, which results in the accumulation of sphingomyelin in various tissues of the body. Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM.
The multicenter, randomized, double-blind, placebo-controlled ASCEND trial evaluated the efficacy and safety of olipudase alfa in 36 adults with chronic visceral ASMD. Patients were randomly assigned 1:1 to receive olipudase alfa 3mg/kg intravenously every 2 weeks or placebo for 52 weeks. The coprimary end points were the percent change in spleen volume and percent-predicted diffusing capacity of the lung for carbon monoxide (DLCO).
At week 52, treatment with olipudase alfa resulted in a 39.45% reduction in spleen volume, compared with a 0.5% increase for placebo (P <.0001). A decrease in spleen volume of at least 30% was observed in 17 patients (94%) treated with olipudase afla compared with no patients treated with placebo. Additionally, olipudase alfa significantly improved lung function by 22% from baseline compared with 3% for patients receiving placebo (P =.0004), as measured by percent predicted DLCO.
Olipudase alfa also met key secondary end points including a 31.7% reduction in liver volume (vs a 1.4% reduction for placebo; P <.0001) and a 16.8% improvement in mean platelet counts (vs 2.5% with placebo; P =.019) at week 52. Significant improvements in HDL, LDL, AST, ALT, chitotriosidase (54% vs 12% with placebo; P =.0003), and lyso-sphingomyelin (78% vs 6% with placebo) were also observed in the olipudase alfa group at week 52.
With regard to Splenomegaly Related Score, a patient-reported outcome measurement that evaluates patient symptoms associated with an enlarged spleen, findings showed no meaningful difference between olipudase alfa and placebo (-8 point vs -9.3 points, respectively).
As for safety, olipudase alfa was well tolerated with most adverse events being mild to moderate in severity. There were no treatment-related serious adverse events and no adverse event-related discontinuations.
Disclosure: Some authors have declared affiliations with or received funding from the pharmaceutical industry. Please refer to the original study for a full list of disclosures.
Reference
Wasserstein M, Arash-Kaps L, Barbato A, et al. Adults with chronic acid sphingomyelinase deficiency show significant visceral, pulmonary, and hematologic improvements after enzyme replacement therapy with olipudase-alfa: 1-year results of the ASCEND placebo-controlled trial. Presented at: 17th Annual WORLDSymposium; February 8-12, 2021. Abstract 265.
CLIP
https://www.sanofi.com/en/media-room/press-releases/2021/2021-12-06-14-00-00-2346501
EMA accepts regulatory submission for olipudase alfa, the first potential therapy for ASMD
- Olipudase alfa has been granted PRIority MEdicines (PRIME) designation in Europe, Breakthrough Therapy designation in the United States, and SAKIGAKE designation in Japan
- European regulatory decision anticipated second half of 2022
DECEMBER 6, 2021
The European Medicines Agency (EMA) has accepted for review under an accelerated assessment procedure the Marketing Authorization Application (MAA) for olipudase alfa, Sanofi’s investigational enzyme replacement therapy which is being evaluated for the treatment of acid sphingomyelinase deficiency (ASMD). Historically referred to as Niemann-Pick disease (NPD) type A and type B, ASMD is a rare, progressive, and potentially life-threatening disease for which no treatments are currently approved. The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan. If approved, olipudase alfa will become the first and only therapy for the treatment of ASMD.
“Today’s milestone has been decades in the making and our gratitude goes to the ASMD community who has stood by us with endless patience while olipudase alfa advanced through clinical development,” said Alaa Hamed, MD, MPH, MBA, Global Head of Medical Affairs, Rare Diseases, Sanofi. “Olipudase alfa represents the kind of potentially life-changing innovation that is possible when industry, medical professionals and the patient community work together toward a common goal.”
The MAA is based on positive results from two separate clinical trials (ASCEND and ASCEND-Peds) evaluating olipudase alfa in adult and pediatric patients with non-central nervous system (CNS) manifestations of ASMD type A/B and ASMD type B.
Olipudase alfa has received special designations from regulatory agencies worldwide, recognizing the innovation potential of the investigational therapy.
“Scientific innovation is the greatest source of hope for people living with diseases like ASMD where there are no approved treatments and is a critical component for ensuring a viable healthcare ecosystem,” said Bill Sibold, Executive Vice President of Sanofi Genzyme. “At Sanofi, we have a long history of pioneering scientific innovation, and we remain committed to finding solutions to address unmet medical needs, including those of the rare disease community.”
The EMA awarded olipudase alfa the PRIority MEdicines designation, also known as PRIME, intended to aid and expedite the regulatory process for investigational medicines that may offer a major therapeutic advantage over existing treatments, or benefit patients without treatment options.
The U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy designation to olipudase alfa. This designation is intended to expedite the development and review of drugs intended to treat serious or life-threatening diseases and conditions. The criteria for granting Breakthrough Therapy designation include preliminary clinical evidence indicating that the molecule may demonstrate substantial improvement on a clinically significant endpoint over available therapies.
In Japan, olipudase alfa was awarded the SAKIGAKE designation, which is intended to promote research and development in Japan for innovative new medical products that satisfy certain criteria, such as the severity of the intended indication. In September, Sanofi filed the J-NDA submission for olipudase alfa.
About ASMD
ASMD results from a deficient activity of the enzyme acid sphingomyelinase (ASM), which is found in special compartments within cells called lysosomes and is required to breakdown lipids called sphingomyelin. If ASM is absent or not functioning as it should, sphingomyelin cannot be metabolized properly and accumulates within cells, eventually causing cell death and the malfunction of major organ systems. The deficiency of the lysosomal enzyme ASM is due to disease-causing variants in the sphingomyelin phosphodiesterase 1 gene (SMPD1). The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan.
ASMD represents a spectrum of disease caused by the same enzymatic deficiency, with two types that may represent opposite ends of a continuum sometimes referred to as ASMD type A and ASMD type B. ASMD type A is a rapidly progressive neurological form of the disease resulting in death in early childhood due to central nervous system complications. ASMD type B is a serious and potentially life-threatening disease that predominantly impacts the lungs, liver, and spleen, as well as other organs. ASMD type A/B represents an intermediate form that includes varying degrees of neurologic involvement. Patients with ASMD type A/B or ASMD type B were studied in the ASCEND trial program. Another type of NPD is NPD type C, which is unrelated to ASMD.
About olipudase alfa
Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM, allowing for the breakdown of sphingomyelin. Olipudase alfa is currently being investigated to treat non-CNS manifestations of ASMD. Olipudase alfa has not been studied in ASMD type A patients. Olipudase alfa is an investigational agent and the safety and efficacy have not been evaluated by the FDA, EMA, or any other regulatory authority worldwide.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions.
With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
///////Olipudase alfa, japan 2022, APPROVALS 2022, Xenpozyme, PEPTIDE, オリプダーゼアルファ (遺伝子組換え) , ORPHAN DRUG, GZ-402665 , GZ 402665

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}