
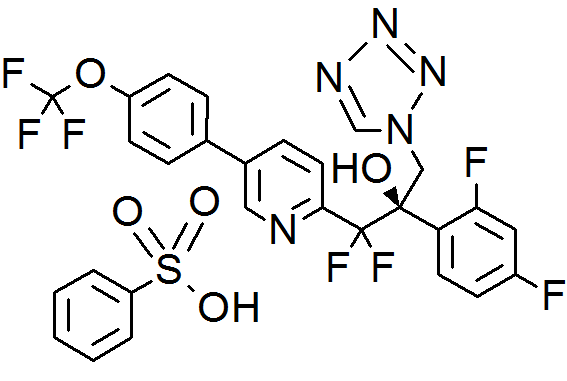
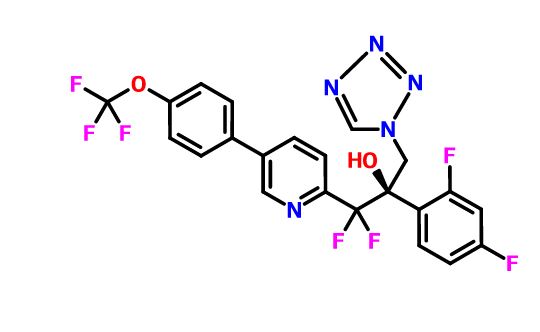
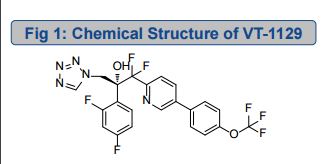
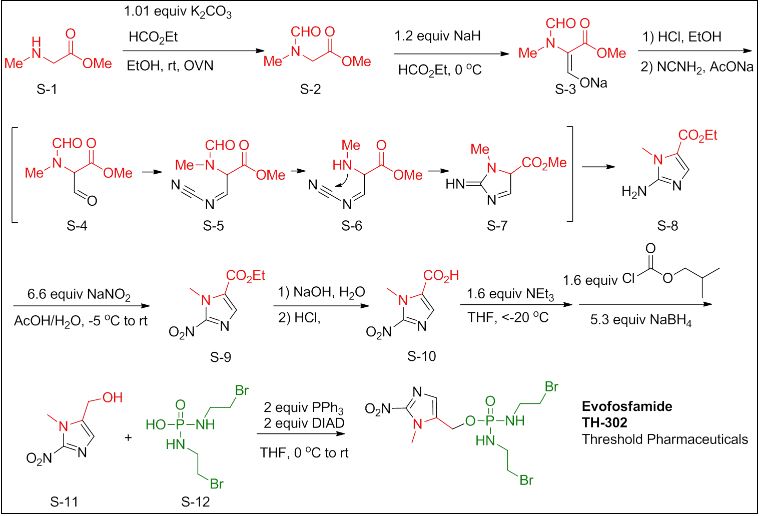
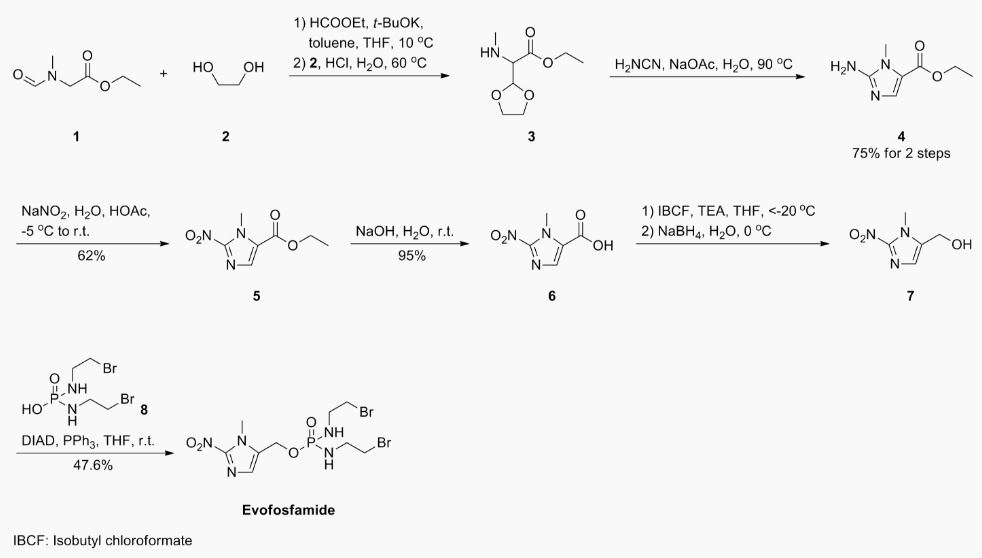
VT 1129 BENZENE SULFONATE
CAS 1809323-18-9


VT 1129
1340593-70-5 CAS
- Originator Viamet Pharmaceuticals
- Class Antifungals; Small molecules
- Mechanism of Action 14-alpha demethylase inhibitors
- Orphan Drug Status Yes – Cryptococcosis
- On Fast track Cryptococcosis
- Phase I Cryptococcosis
-
Most Recent Events
- 01 Jun 2016 VT 1129 receives Fast Track designation for Cryptococcosis [PO] (In volunteers) in USA
- 30 May 2016 Viamet Pharmaceuticals plans a phase II trial for Cryptococcal meningitis in USA (Viamet Pharmaceuticals pipeline; May 2016)
- 27 May 2016 Phase-I clinical trials in Cryptococcosis (In volunteers) in USA (PO) before May 2016 (Viamet Pharmaceuticals pipeline; May 2016)


Viamet, in collaboration with Therapeutics for Rare and Neglected diseases, is investigating VT-1129, a small-molecule lanosterol demethylase inhibitor, developed using the company’s Metallophile technology, for treating fungal infections, including Cryptococcus neoformans meningitis.
VT-1129 is a novel oral agent that we are developing for the treatment of cryptococcal meningitis, a life-threatening fungal infection of the brain and the spinal cord that occurs most frequently in patients with HIV infection, transplant recipients and oncology patients. Without treatment, the disease is almost always fatal.
 VT-1129 has shown high potency and selectivity in in vitro studies and is an orally administered inhibitor of fungal CYP51, ametalloenzyme important in fungal cell wall synthesis. In preclinical studies, VT-1129 has demonstrated substantial potency against Cryptococcus species, the fungal pathogens that cause cryptoccocal meningitis, and has also been shown to accumulate to high concentrations within the central nervous system, the primary site of infection.
VT-1129 has shown high potency and selectivity in in vitro studies and is an orally administered inhibitor of fungal CYP51, ametalloenzyme important in fungal cell wall synthesis. In preclinical studies, VT-1129 has demonstrated substantial potency against Cryptococcus species, the fungal pathogens that cause cryptoccocal meningitis, and has also been shown to accumulate to high concentrations within the central nervous system, the primary site of infection.
In in vitro studies, VT-1129 was significantly more potent against Cryptococcus isolates than fluconazole, which is commonly used for maintenance therapy of cryptococcal meningitis in the United States and as a primary therapy in the developing world. Oral VT-1129 has also been studied in a preclinical model of cryptococcal meningitis, where it was compared to fluconazole. At the conclusion of the study, there was no detectable evidence of Cryptococcus in the brain tissue of the high dose VT-1129 treated groups, in contrast to those groups treated with fluconazole. To our knowledge, this ability to reduce the Cryptococcus pathogen in the central nervous system to undetectable levels in this preclinical model is unique to VT-1129.
Opportunity
An estimated 3,400 hospitalizations related to cryptococcal meningitis occur annually in the United States and the FDA has granted orphan drug designation to VT-1129 for the treatment of this life-threatening disease. In addition, the FDA has granted Qualified Infectious Disease Product designation to VT-1129 for the treatment of Cryptococcus infections, which further underscores the unmet medical need. In developing regions such as Africa, cryptococcal meningitis is a major public health problem, with approximately one million cases and mortality rates estimated to be as high as 55-70%.
Current Status
VT-1129 has received orphan drug and Fast Track designations for the treatment of cryptococcal meningitis and has been designated a Qualified Infectious Disease Product (QIDP) by the U.S. Fod and Drug Administration. We are currently conducting a Phase 1 single-ascending dose study of VT-1129 in healthy volunteers.


Conclusions
• VT-1129 has robust activity against Cryptococcus isolates with elevated fluconazole MICs and may be a viable option in persons infected with such strains.
• A Phase 1 study of VT-1129 in healthy volunteers is scheduled to begin by the end of 2015. Phase 2 trials in persons with cryptococcal meningitis are targeted to begin by the end of 2016.

Living organisms have developed tightly regulated processes that specifically import metals, transport them to intracellular storage sites and ultimately transport them to sites of use. One of the most important functions of metals such as zinc and iron in biological systems is to enable the activity of metalloenzymes. Metalloenzymes are enzymes that incorporate metal ions into the enzyme active site and utilize the metal as a part of the catalytic process. More than one-third of all characterized enzymes are metalloenzymes.
The function of metalloenzymes is highly dependent on the presence of the metal ion in the active site of the enzyme. It is well recognized that agents which bind to and inactivate the active site metal ion dramatically decrease the activity of the enzyme. Nature employs this same strategy to decrease the activity of certain metalloenzymes during periods in which the enzymatic activity is undesirable. For example, the protein TIMP (tissue inhibitor of metalloproteases) binds to the zinc ion in the active site of various matrix metalloprotease enzymes and thereby arrests the enzymatic activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain a l-(l,2,4-triazole) group that binds to the heme iron present in the active site of the target enzyme lanosterol demethylase and thereby inactivates the enzyme.
In the design of clinically safe and effective metalloenzyme inhibitors, use of the most appropriate metal-binding group for the particular target and clinical indication is critical. If a weakly binding metal-binding group is utilized, potency may be suboptimal. On the other
hand, if a very tightly binding metal-binding group is utilized, selectivity for the target enzyme versus related metalloenzymes may be suboptimal. The lack of optimal selectivity can be a cause for clinical toxicity due to unintended inhibition of these off-target metalloenzymes. One example of such clinical toxicity is the unintended inhibition of human drug metabolizing enzymes such as CYP2C9, CYP2C19 and CYP3A4 by the currently- available azole antifungal agents such as fluconazole and voriconazole. It is believed that this off-target inhibition is caused primarily by the indiscriminate binding of the currently utilized l-(l,2,4-triazole) to iron in the active site of CYP2C9, CYP2C19 and CYP3A4. Another example of this is the joint pain that has been observed in many clinical trials of matrix metalloproteinase inhibitors. This toxicity is considered to be related to inhibition of off-target metalloenzymes due to indiscriminate binding of the hydroxamic acid group to zinc in the off-target active sites.
Therefore, the search for metal-binding groups that can achieve a better balance of potency and selectivity remains an important goal and would be significant in the realization of therapeutic agents and methods to address currently unmet needs in treating and preventing diseases, disorders and symptoms thereof. Similarly, methods of synthesizing such therapeutic agents on the laboratory and, ultimately, commercial scale is needed. Addition of metal-based nucleophiles (Zn, Zr, Ce, Ti, Mg, Mn, Li) to azole-methyl substituted ketones have been effected in the synthesis of voriconazole (M. Butters, Org. Process Res. Dev.2001, 5, 28-36). The nucleophile in these examples was an ethyl-pyrimidine substrate. Similarly, optically active azole-methyl epoxide has been prepared as precursor electrophile toward the synthesis of ravuconazole (A. Tsuruoka, Chem. Pharm. Bull.1998, 46, 623-630). Despite this, the development of methodology with improved efficiency and selectivity is desirable.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011133875
Scheme 1

EXAMPLE 7

2-(2, 4-Difluorophenyl)-l, l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4- (trifluoromethoxy) phenyl) pyridin-2-yl) propan-2-ol (7)
To a stirred solution of bromo epoxide C (0.5 g, 1.38 mmol) in THF (30 mL) and water (14 mL) were added 4-(trifluoromethoxy) phenylboronic acid (0.22 g, 1.1 mmol), Na2C03 (0.32 g, 3.1 mmol) and Pd(dppf)2Cl2 (0.28 g, 0.34 mmol) at RT under inert atmosphere. After purged with argon for a period of 30 min, the reaction mixture was heated to 75°C and stirring was continued for 4 h. Progress of the reaction was monitored by TLC. The reaction mixture was cooled to RT and filtered through a pad of celite. The filtrate was concentrated under reduced pressure; obtained residue was dissolved in ethyl acetate (30 mL). The organic layer was washed with water, brine and dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude compound was purified by column chromatography to afford the coupled product (0.45 g, 1.0 mmol, 73%) as solid. 1H NMR (200 MHz, CDC13): δ 8.87 (s, 1 H), 7.90 (dd, / = 8.2, 2.2 Hz, 1 H), 7.66-7.54 (m, 3 H), 7.49-7.34 (m, 3 H), 6.90-6.70 (m, 2 H), 3.49 (d, / = 5.0 Hz, 1 H), 3.02-2.95 (m, 1 H). Mass: m/z 444 [M++l].
To a stirred solution of the coupled product (0.45 g, 1.0 mmol) in DMF (10 mL) was added K2C03 (70 mg, 0.5 mmol) followed by IH-tetrazole (70 mg, 1.0 mmol) at RT under inert atmosphere. The reaction mixture was stirred for 4 h at 80 °C. The volatiles were removed under reduced pressure and obtained residue was dissolved in water (15 mL) and extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with water, brine and dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude compound was purified by column chromatography to afford 7 (0.19 g, 0.37 mmol, 36 %) as white solid. 1H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.97 (dd, / = 8.0, 2.0 Hz, 1 H), 7.68 (d, / = 8.5 Hz, 1 H), 7.60-7.56 (m, 3 H), 7.43-7.36 (m, 3 H), 6.80-6.76 (m, 1 H), 6.70-6.67 (m, 1 H), 5.57 (d, / = 14.5 Hz, 1 H), 5.17 (d, / = 14.5 Hz, 1 H). HPLC: 98.3%. Mass: m/z 513.9 [M++l].
Chiral preparative HPLC of enantiomers:
The enantiomers of 7 (17.8 g, 34.6 mmol) were separated by normal-phase preparative high performance liquid chromatography (Chiralpak AD-H, 250 x 21.2 mm, 5μ; using (A) n-hexane – (B) IPA (A:B : 70:30) as a mobile phase; Flow rate: 15 mL/min) to obtain 7(+) (6.0 g) and 7(-) (5.8 g).
Analytical data for 7 (+):
HPLC: 99.8%.
Chiral HPLC: Rt = 9.88 min (Chiralpak AD-H, 250 x 4.6mm, 5μ; mobile phase (A) n-Hexane (B) IPA (7/3): A: B (70:30); flow Rate: 1.00 mL/min)
Optical rotation [a]D25: + 19° (C = 0.1 % in MeOH).
Patent
WO2015143137,
https://patentscope.wipo.int/search/ko/detail.jsf;jsessionid=61AAA66F887FDBB9CFC3F752AFF04016.wapp2nC?docId=WO2015143137&recNum=303&office=&queryString=&prevFilter=%26fq%3DICF_M%3A%22C07D%22&sortOption=%EA%B3%B5%EA%B0%9C%EC%9D%BC(%EB%82%B4%EB%A6%BC%EC%B0%A8%EC%88%9C)&maxRec=58609
Examples
The present invention will now be demonstrated using specific examples that are not to be construed as limiting.
General Experimental Procedures
Definitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.
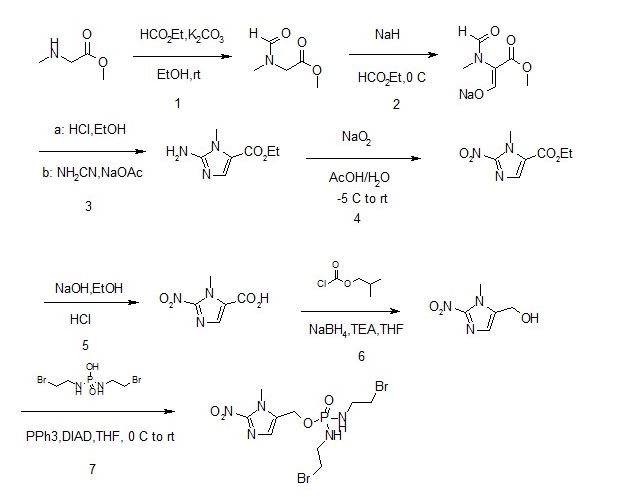
Synthesis of 1 or la

A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of 1 or la may be accomplished using the example syntheses that are shown below (Schemes 1-9). The preparation of precursor ketone 8 is performed starting with reaction of dibromo-pyridine 2-Br with ethyl 2-bromo-difluoroacetate to produce ester 3-Br. This ester is reacted with tetrazole reagent 4 via Claisen reaction to furnish 5-Br. Decarboxylation of 5-Br via a two-step process produces compound 6-Br. Suzukin coupling of 6-Br with boronate 7 furnishes 8.
Scheme 1. Synthesis of ketone 8

Ketone 8 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared using according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).
Scheme 2. Synthesis of ketone 8


= halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.
Compounds 6 or 8 may be reacted with a series of metallated derivatives of 2,4-difluoro-bromobenzene and chiral catalysts/reagents (e.g. BINOL) to effect enantiofacial-selective addition to the carbonyl group of 6 or 8 (Scheme 3). These additions can be performed on 6 or 8 to furnish 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof), respectively.
Scheme 3. Synthesis of 1 or la

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.
Alternatively, ketone 8 can be synthesized from aldehyde 10 (Scheme 4). Aldehyde 10 is coupled with 7 to produce 11. Compound 11 is then converted to 12 via treatment with diethylaminosulfurtrifluoride (DAST).
Scheme 4. Alternate synthesis of ketone 8


Scheme 5 outlines the synthesis of precursor ketone 15-Br. The ketone is prepared by conversion of 2-Br to 3-Br as described above. Next, ester 3-Br is converted to 15-Br by treatment via lithiation of 2,4-difluoro-bromobenzene.
Scheme 5. Synthesis of ketone 15-Br

Ketone 15 may be prepared in an analogous fashion as described for 15-Br in Scheme 5 starting from corresponding substituted 2-bromo-pyridines, which can be prepared using according to synthetic transformations known in the art and contained in the references cited herein (Scheme 6).
Scheme 6. Synthesis of ketone 15

F = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.
Ketone 15 may be used to prepare 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) by the following three-step process (Scheme 7). In the presence of a chiral catalyst/reagent (e.g. proline derivatives), base-treated nitromethane is added to 15 or 16 to furnish 17 (or 17a, the enantiomer of 17, or mixtures thereof) or 18 (or 18a, the enantiomer of 18, or mixtures thereof), respectively. Reduction of 17 (or 17a, the enantiomer of 17, or mixtures thereof) or 18 (or 18a, the enantiomer of 18, or mixtures thereof) (e.g. lithium aluminum hydride) produces 19 (or 19a, the enantiomer of 19, or mixtures thereof) or 20 (or 20a, the enantiomer of 20, or mixtures thereof). Annulation of 19 (or 19a, the enantiomer of 19, or mixtures thereof) or 20 (or 20a, the enantiomer of 20, or mixtures thereof) by treatment with sodium azide/triethylorthoformate furnishes tetrazoles 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof). Suzuki coupling of 9 (or 9a, the enantiomer of 9, or mixtures thereof) with 4-trifluoromethoxyphenyl-boronic acid produces 1 (or la, the enantiomer of 1, or mixtures thereof).
Scheme 7. Asymmetric Henry reaction

R-ι = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted a 0(S02)-aryl, or -0(S02)-substituted aryl.
Ketone 21 may be employed to prepare optically-active epoxides via Horner-Emmons reaction of a difluoromethyl substrate to produce 22 or 22a. Ketones related to 21 have been prepared (M. Butters, Org. Process Res. Dev. 2001, 5, 28-36). Nucleophilic addition of metalated 5-(4-trifluoromethoxy)phenyl-2-pyridine (M = metal) to epoxide 22 or 22a may furnish compound
1 or la.
Scheme 8. Enantioselective epoxidation strategy

Ketone 15 or 16 may be used to prepare 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) by an alternative three-step process to Scheme 7 (Scheme 9). In the presence of a chiral catalyst/reagent, trimethylsilyl-cyanide is added to 15 or 16 to furnish 23 (or 23a, the enantiomer of 23, or mixtures thereof) or 24 (or 24a, the enantiomer of 24, or mixtures thereof), respectively (S.M. Dankwardt, Tetrahedron Lett. 1998, 39, 4971-4974). Reduction of 23 (or 23a, the enantiomer of 23, or mixtures thereof) or 24 (or 24a, the enantiomer of 24, or mixtures thereof) (e.g. lithium aluminum hydride) produces 19 (or 19a, the enantiomer of 19, or mixtures thereof) or 20 (or 20a, the enantiomer of 20, or mixtures thereof). Annulation of 19 (or 19a, the enantiomer of 19, or mixtures thereof) or 20 (or 20a, the enantiomer of 20, or mixtures thereof) by treatment with sodium azide/triethylorthoformate furnishes tetrazoles 9 (or 9a, the enantiomer of 9, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof). Suzuki coupling of 9 (or 9a, the enantiomer of 9, or mixtures thereof) with 4-trifluoromethoxyphenyl-boronic acid produces 1 (or la, the enantiomer of 1, or mixtures thereof).
Scheme 9. Asymmetric cyanohydrin strategy

R’ = H or trimethylsilyl
Suzuki

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.

1
2-(2, 4-Difluorophenyl)-l, l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(trifluoromethoxy) phenyl) pyridin-2-yl) propan-2-ol (1 or la)
White powder: *H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.97 (dd, J = 8.0, 2.0 Hz, 1 H), 7.68 (d, / = 8.5 Hz, 1 H), 7.60-7.56 (m, 3 H), 7.43-7.36 (m, 3 H), 6.80-6.76 (m, 1 H), 6.70-6.67 (m, 1 H), 5.57 (d, J = 14.5 Hz, 1 H), 5.17 (d, J = 14.5 Hz, 1 H). HPLC: 98.3%. Mass: m/z 513.9 [M++l]. HPLC: 99.8%. Optical rotation [a]D25: + 19° (C = 0.1 % in MeOH).
INTERMEDIATE 3-Br Ri = Br)
To a clean and dry 100 L jacketed reactor was added copper powder (1375 g, 2.05 equiv, 10 micron, sphereoidal, SAFC Cat # 326453) and DMSO (17.5 L, 7 vol). Next, ethyl bromodifluoroacetate (2.25 kg, 1.05 equiv, Apollo lot # 102956) was added and the resulting slurry stirred at 20-25 °C for 1-2 hours. Then 2,5-dibromopyridine (2-Br, 2.5 kg, 1.0 equiv, Alfa Aesar lot # F14P38) was added to the batch and the mixture was immediately heated (using the glycol jacket) to 35 °C. After 70 hours at 35 °C, the mixture was sampled for CG/MS analysis. A sample of the reaction slurry was diluted with 1/1 CH3CN/water, filtered (0.45 micron), and the filtrate analyzed directly. Ideally, the reaction is deemed complete if <5% (AUC) of 2,5-dibromopyridine remains. In this particular batch, 10% (AUC) of 2,5-dibromopyridine remained. However due to the already lengthy reaction time, we felt that prolonging the batch would not help the conversion any further. The reaction was then deemed complete and diluted with EtOAc (35 L). The reaction mixture was stirred at 20-35 °C for 1 hour and then the solids (copper salts) were removed by filtration through a pad of Celite. The residual solids inside the reactor were rinsed forward using EtOAc (2 x 10 L) and then this was filtered through the Celite. The filter cake was washed with additional EtOAc (3 x 10 L) and the EtOAc filtrates were combined. A buffer solution was prepared by dissolving NH4CI (10 kg) in DI water (100 L), followed by the addition of aqueous 28% NH4OH (2.0 L) to reach pH = 9. Then the combined EtOAc filtrates were added slowly to a pre-cooled (0 to 15 °C) solution of NH4C1 and NH4OH (35 L, pH = 9) buffer while maintaining T<30 °C. The mixture was then stirred for 15-30 minutes and the phases were allowed to separate. The aqueous layer (blue in color) was removed and the organic layer was washed with the buffer solution until no blue color was discernable in the aqueous layer. This experiment required 3 x 17.5 L washes. The organic layer was then washed with a 1/1 mixture of Brine (12.5 L) and the pH = 9 NH4C1 buffer solution (12.5 L), dried over MgS04, filtered, and concentrated to dryness. This provided crude compound 3-Br [2.29 kg, 77% yield, 88% (AUC) by GC/MS] as a yellow oil. The major impurity present in crude 3-Br was unreacted 2,5-dibromopyridine [10% (AUC) by GC/MS]. ‘ll NMR (CDC13) was consistent with previous lots of crude compound 3-Br. Crude compound 3-Br was then combined with similar purity lots and purified by column chromatography (5/95 EtO Ac/heptane on S1O2 gel).
INTERMEDIATE 15-Br (R, = Br)
To a clean and dry 72 L round bottom flask was added l-bromo-2,4-difluorobenzene (1586 g,
1.15 equiv, Oakwood lot # H4460) and MTBE (20 L, 12.6 vol). This solution was cooled to -70 to -75 °C and treated with n-BuLi (3286 mL, 1.15 equiv, 2.5 M in hexanes, SAFC lot # 32799MJ), added as rapidly as possible while maintaining -75 to -55 °C. This addition typically required 35-45 minutes to complete. (NOTE: If the n-BuLi is added slowly, an white slurry will form and this typically gives poor results). After stirring at -70 to -65 °C for 45 minutes, a solution of compound 3-Br (2000 g, 1.0 equiv, AMRI lot # 15CL049A) in MTBE (3 vol) was added rapidly (20-30 min) by addition funnel to the aryl lithium solution while maintaining -75 to -55 °C. After stirring for 30-60 minutes at -75 to -55 °C, the reaction was analyzed by GC/MS and showed only trace (0.5% AUC) l-bromo-2,4-difluorobenzene present. The reaction was slowly quenched with aqueous 2 M HC1 (3.6 L) and allowed to warm to room temperature. The mixture was adjusted to pH = 6.5 to 8.5 using NaHCC>3 (4 L), and the organic layer was separated. The MTBE layer was washed with brine (5% NaCl in water, 4 L), dried over MgS04, filtered, and concentrated. In order to convert the intermediate hemi-acetal to 4-Br, the crude mixture was heated inside the 20 L rotovap flask at 60-65 °C for 3 hours (under vacuum), at this point all the hemi-acetal was converted to the desired ketone 4 by !Η NMR (CDC13). This provided crude compound 4-Br [2.36 kg, 75% (AUC) by HPLC] as a brown oil that solidified upon standing. This material can then be used “as-is” in the next step without further purification.
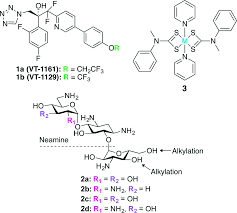
PATENT FOR VT1161 SIMILAR TO VT 1129
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016149486&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
Synthesis of 1 or la

EXAMPLE 1
Preparation of Compound 1 X-Hydrate
Compound 1 and its preparation are described in the art, including in US Patent 8,236,962 (incorporated by reference herein). Compound 1 can then be partitioned between ethanol and water to afford Compound 1 X-hydrate.
EXAMPLE 2
Compound 1 Anhydrous Form Recrystallization
Compound 1 X-hydrate (29.1 g, 28.0 g contained 1) was suspended in 2-propanol (150 ml) and heated to 56 °C. The solution was filtered through a 0.45 μιη Nylon membrane with 2-propanol rinses. The combined filtrate was concentrated to 96.5 g of a light amber solution. The solution was transferred to a 1-L flask equipped with overhead stirring, thermocouple and addition funnel, using 2-propanol (30 ml total) to complete the transfer. The combined solution contained about 116 ml 2-propanol.
The solution was heated to 50 °C and n-heptane (234 ml) was added over 22 minutes. The resulting hazy mixture was seeded with 1 anhydrous form. After about 1 hour a good
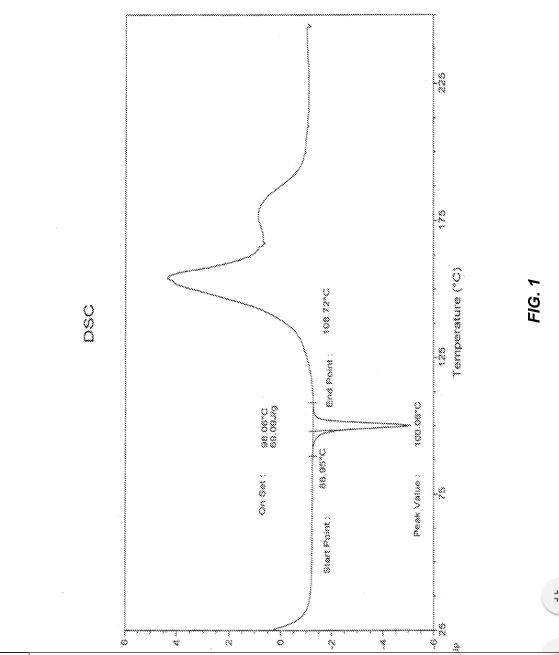
suspension had formed. Additional n-heptane (230 ml) was added over 48 minutes. Some granular material separated but most of the suspension was a finely divided pale beige solid. After about ½ hour at 50 °C the suspension was cooled at 10 °C/h to room temperature and stirred overnight. The product was collected at 22 °C on a vacuum filter and washed with 1:4 (v/v) 2-PrOH/ n-heptane (2 x 50 ml). After drying on the filter for 1-2 hours the weight of product was 25.5 g. The material was homogenized in a stainless steel blender to pulverize and blend the more granular solid component. The resulting pale beige powder (25.37 g) was dried in a vacuum oven at 50 °C. The dry weight was 25.34 g. The residual 2-propanol and n- heptane were estimated at <0.05 wt% each by 1H NMR analysis. The yield was 90.5% after correcting the X-hydrate for solvent and water content. Residual Pd was 21 ppm. The water content was 209 ppm by KF titration. The melting point was 100.7 °C by DSC analysis.
Table 1: Data for the isolated and dried Compound 1 – X-hydrate and anhydrous forms

M.P. by DSC; Pd by ICP; Purity by the API HPLC method; Chiral purity by HPLC; water content by KF titration; residual solvent estimated from :H NMR.
Table 2: Characterisation Data for Compounds 1 (X-hydrate) and 1 (anhydrous)

Needle like crystals Needle like crystals and agglomerates
PLM
particle size >100μιη particle size range from 5μπι-100μιη
0.59%w/w water uptake at 90%RH. 0.14%w/w water uptake at 90%RH.
GVS
No sample hysteresis No sample hysteresis
XRPD
No form change after GVS experiment No form change after GVS experiment post GVS
KF 2.4%w/w H20 Not obtained
<0.001mg/ml <0.001mg/ml
Solubility
pH of saturated solution = 8.6 pH of saturated solution = 8.7
Spectral Pattern 1 Spectral Pattern 2
Charcteristic bands/ cm“1: Charcteristic bands/ cm 1:
FT-IR 3499, 3378, 3213, 3172 3162
1612, 1598, 1588, 1522, 1502 1610, 1518, 1501 931, 903, 875, 855, 828, 816 927, 858, 841, 829, 812

The structure solution of Compound 1 anhydrous form was obtained by direct methods, full-matrix least-squares refinement on F 2 with weighting w‘1 = <52{F02) + (0.0474P)2 + (0.3258P), where P = (F02+2F 2)/3, anisotropic displacement parameters, empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. Final wR2
= {∑[w(F02-Fc2)2]/∑[w(F02)2]m} = 0.0877 for all data, conventional Ri = 0.0343 on F values of 8390 reflections with F0 > 4a( F0), S = 1.051 for all data and 675 parameters. Final Δ/a (max) 0.001, A/a(mean), 0.000. Final difference map between +0.311 and -0.344 e A“3.
Below shows a view of two molecules of Compound 1 in the asymmetric unit of the anhydrous form showing the numbering scheme employed. Anisotropic atomic displacement ellipsoids for the non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are displayed with an arbitrarily small radius. The absolute configuration of the molecules has been determined to be R.

EXAMPLE 3
Compound 1 Ethanol Solvate Recrystallization
Compound 1 X-hydrate (50 mg) was suspended in -40 volumes of 15% H20/EtOH. The suspension was then placed in an incubation chamber for maturation. The maturation protocol involved treating the suspension to a two-temperature cycle of 50 °C/ ambient temperature at 8 hours per cycle for 3 days with constant agitation. After maturation, the suspension was cooled in a fridge at 4°C for up to 2 days to encourage the formation of crystals. Then, the solvent was removed at RT and the sample was vacuum dried at 30°C -35°C for up to 1 day. Suitable crystals formed on cooling were harvested and characterized.
Table 4: Single Crystal Structure of 1 Ethanol solvate
Molecular formula C25H22F7N5O3

The structure solution of Compound 1 ethanol solvate was obtained by direct methods, full-matrix least-squares refinement on F 2 with weighting w‘1 = σ2^2) + (0.0450P)2 + (0.5000P), where P = (F02+2F 2)/3, anisotropic displacement parameters, empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. Final wR2 = {∑[w(F02-F 2)2]/∑[w(F02)2]m} = 0.0777 for all data, conventional Ri = 0.0272 on F values of 4591 reflections with F0 > 4σ( F0), S = 1.006 for all data and 370 parameters. Final Δ/σ (max) 0.000, A/a(mean), 0.000. Final difference map between +0.217 and -0.199 e A“3.
Below shows a view of the asymmetric unit of the ethanol solvate from the crystal structure showing the numbering scheme employed. Anisotropic atomic displacement ellipsoids for the non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are displayed with an arbitrarily small radius. The asymmetric unit shows stoichiometry of 1 : 1 for solvent of crystallisation to Compound 1.

EXAMPLE 4
Compound 1 1.5 Hydrate Recrystallization
Compound 1 X-hydrate (50 mg) was suspended in -40 volumes of 15% Η20/ΙΡΑ. The suspension was then placed in an incubation chamber for maturation. The maturation protocol involved treating the suspension to a two-temperature cycle of 50 °C/ ambient temperature at 8 hours per cycle for 3 days with constant agitation. After maturation, the suspension was cooled in a fridge at 4°C for up to 2 days to encourage the formation of crystals. Then, the solvent was removed at RT and the sample was vacuum dried at 30°C -35°C for up to 1 day. Suitable crystals formed on cooling were harvested and characterized.
Table 5: Single Crystal Structure of 1 1.5 Hydrate


The structure solution of Compound 1 1.5 hydrate was obtained by direct methods, full-matrix least-squares refinement on F 2 with weighting w‘1 = ^(F 2) + (0.1269P)2 + (0.0000P), where P = (F02+2F 2)/3, anisotropic displacement parameters, empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. Final wR2 = {∑[w(F 2-F 2)2]/∑[w(F 2)2] m} = 0.1574 for all data, conventional Ri = 0.0668 on F values of 2106 reflections with F0 > 4σ( F0), S = 1.106 for all data and 361 parameters. Final Δ/σ (max) 0.000, A/a(mean), 0.000. Final difference map between +0.439 and -0.598 e A“3.
Below shows a view of the asymmetric unit of the 1.5 hydrate from the crystal structure showing the numbering scheme employed. Anisotropic atomic displacement ellipsoids for the non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are displayed with an arbitrarily small radius. The asymmetric unit shows stoichiometry of 1.5: 1 for water to Compound 1.

EXAMPLE 5
Human Pharmacokinetic Comparison of Compound 1 X-Hydrate and Compound 1 Anhydrous Form
Table 6 compares human multiple-dose pharmacokinetic (PK) parameters between dosing with Compound 1 X-hydrate and Compound 1 Anhydrous form. Compound 1 X-hydrate was dosed at 600 mg twice daily (bid) for three days followed by dosing at 300 mg once daily (qd) for 10 days. Compound 1 Anhydrous form was dosed at 300 mg qd for 14 days. Despite the higher initial dosing amount and frequency (i.e., 600 mg bid) of Compound 1 X-hydrate, Compound 1 Anhydrous form surprisingly displayed higher maximal concentration (Cmax) and higher area-under-the-curve (AUC) than Compound 1 X-hydrate.
Table 6. Comparison of Multiple Dose PK between Compound 1 X-Hydrate and Compound 1
Anhydrous Polymorph

Further characterization of the various polymorph forms of compound 1 are detailed in the accompanying figures.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015143154
Examples
General Experimental Procedures
Definitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.
Synthesis of 1 or la

la
A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of lor la may be accomplished using the example syntheses that are shown below (Schemes 1-4). The preparation of precursor ketone 3-Br is performed starting with reaction of 2,5-dibromo-pyridine with ethyl 2-bromo-difluoroacetate to produce ester 2-Br. This ester is reacted with morpholine to furnish morpholine amide 2b-Br, followed by arylation to provide ketone 3-Br.
Scheme 1. Synthesis of ketone 3-Br

Ketone 3 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared using according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).
Scheme 2. Synthesis of ketone 3

R1 = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, 0(S02)-aryl, or -0(S02)-substituted aryl.
Alternatively, compound 1 (or la, the enantiomer of 1, or mixtures thereof) can be prepared according to Scheme 3 utilizing amino-alcohols ±4b or ±1-6. Epoxides 4 and 5 can be prepared by reacting ketones 3 and 1-4 with trimethylsulfoxonium iodide (TMSI) in the presence of a base (e.g., potassium i-butoxide) in a suitable solvent or a mixture of solvents (e.g., DMSO or THF). Also, as indicated in Scheme 3, any of pyridine compounds, 3, 4, ±4b, 4b, or 6, can be converted to the corresponding 4-CF3O-PI1 analogs (e.g., 1-4, 5, ±1-6, 1-6*, or 1 or the corresponding enantiomers, or mixtures thereof) by cross-coupling with (4-trifluoromethoxyphenyl)boronic acid (or the corresponding alkyl boronates or pinnacol boronates or the like), in a suitable solvent system (e.g., an organic-aqueous solvent mixture), in the presence of a transition metal catalyst (e.g., (dppf)PdCl2; dppf = 1,1′-(diphenylphosphino)ferrocene), and in the presence of a base (e.g., KHCO3, K2CO3, CS2CO3, or Na2CC>3, or the like). Epoxides 4 and 5 can then be converted into amino-alcohols ±4b and ±1-6 through ammonia-mediated epoxide opening using ammonia in a suitable solvent (e.g., MeOH, EtOH, or water). Racemic amino-alcohols ±4b and ±1-6 can then be enantio-enriched by exposure to a chiral acid (e.g., tartaric acid, di-benzoyltartaric acid, or di-p-toluoyltartaric acid or the like) in a suitable solvent (e.g., acetonitrile, isopropanol, EtOH, or mixtures thereof, or a mixture of any of these with water or MeOH; preferably acetonitrile or a mixture of acetonitrile and MeOH, such as 90:10, 85: 15, or 80:20 mixture) to afford compounds 4b (or 4c, the enantiomer of 4b, or mixtures thereof) or 1-6* (or 1-7*, the enantiomer of 1-6*, or mixtures thereof). Subsequent treatment with TMS-azide in the presence of trimethylorthoformate and sodium acetate in acetic acid would yield compounds 20 (or 20a, the enantiomer of 20, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) (US 4,426,531).
Scheme 3. Synthesis of 1 or la via TMSI Epoxidation Method


R-ι = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)- substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0- aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.
Compound 1 (or la, the enantiomer of 1, or mixtures thereof) prepared by any of the methods presented herein can be converted to a sulfonic salt of formula IX (or IXa, the enantiomer of
IX, or mixtures thereof), as shown in Scheme 4. This can be accomplished by a) combining compound 1 (or la, the enantiomer of 1, or mixtures thereof), a crystallization solvent or crystallization solvent mixture (e.g., EtOAc, i‘PrOAc, EtOH, MeOH, or acetonitrile, or o
Z-S-OH
combinations thereof), and a sulfonic acid o (e.g., Z = Ph, p-tolyl, Me, or Et), b) diluting the mixture with an appropriate crystallization co-solvent or crystallization co-solvent mixture (e.g., pentane, methyl i-butylether, hexane, heptane, or toluene, or combinations thereof), and c) filtering the mixture to obtain a sulfonic acid salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof).
Scheme 4. Synthesis of a Sulfonic Acid Salt of Compound 1 or la

EXAMPLE 1: Preparation of l-(2,4-difluorophenyl)-2,2-difluoro-2-(5-(4- (trifluoromethoxy)phenyl)pyridin-2-yl)ethanone (1-4).
la. ethyl 2-(5-bromopyridin-2-yl)-2,2-difluoroacetate (2)

2-Br
Typical Procedure for Preparing 2-Br
Copper ( 45μιη, 149g, 0.198moles, 2.5 equiv) was placed into a 3L, 3-neck round bottom flask equipped with a condenser, thermocouple, and an overhead stirrer. DMSO (890 mL, 4.7 vol. based on ethyl 2-bromo-2,2-difluoroacetate) and 14mL of concentrated sulfuric acid was added and the mixture stirred for 30 minutes. The mixture self-heated to about 31°C during the stir time. After cooling the contents to 23°C, 2,5-dibromopyridine 1 (277g, 1.17 moles, 1.5 eq) was added to the reaction mixture. The temperature of the contents decreased to 16°C during a 10 minute stir time. 2-bromo-2,2-difluoroacetate (190 g, 0.936 moles, 1.0 eq) was added in one portion and the mixture stirred for 10 min. The flask contents were warmed to 35°C and the internal temperature was maintained between 35-38° for 18 h. In-process HPLC showed 72% desired 2-Br. The warm reaction mixture was filtered through filter paper and the collected solids washed with 300mL of 35°C DMSO. The solids were then washed with 450mL of n-heptane and 450mL of MTBE. The collected filtrate was cooled to about 10°C and was slowly added 900mL of a cold 20% aqueous NH4C1 solution, maintaining an internal temperature of <16°C during the addition. After stirring for 15 minutes, the layers were settled and separated. The aqueous layer was extracted 2 X 450mL of a 1: 1 MTBE: n-heptane mixture. The combined organic layers were washed 2 X 450mL of aqueous 20% NH4CI and with 200mL of aqueous 20% NaCl. The organic layer was dried with 50g MgS04 and the solvent removed to yield 2-Br as a dark oil. Weight of oil = 183g ( 70% yield by weight) HPLC purity ( by area %) = 85%. *H NMR (400 MHz, d6-DMSO) : 58.86 (m, 1H), 8.35 ( dd, J= 8.4, 2.3Hz, 1H), 7.84 (dd, J= 8.3, 0.6Hz, 1H), 4.34 ( q, J= 7.1Hz, 2H), 1.23 ( t, J= 7.1Hz, 3H). MS m/z 280 ( M+H+), 282 (M+2+H+).
lb. 2-(5-bromopyridin-2-yl)-2,2-difluoro-l-morpholinoethanone (2b-Br)

Table 2 illustrates the effects of the relative proportions of each of the reagents and reactants, and the effect of varying the solvent had on the overall performance of the transformation as measured by the overall yield and purity of the reaction.
Table 2. Process Development for the Preparation of compound 2b-Br

Note: All reactions were conducted at 22- 25°C
Typical Procedure for Converting 2-Br to 2b-Br
Crude ester 2-Br (183g, 0.65moles) was dissolved in 1.5L of n-heptane and transferred to a 5L 3-neck round bottom flask equipped with a condenser, an overhead stirrer and a thermocouple. Morpholine ( 248g, 2.85 moles, 4.4 equiv.) was charged to the flask and the mixture warmed to 60°C and stirred for 16 hours. In-process HPLC showed <1 % of ester 2-Br. The reaction mixture was cooled to 22-25 °C and 1.5L of MTBE was added with continued cooling of the mixture to 4°C and slowly added 700mL of a 30%, by weight, aqueous citric acid solution. The temperature of the reaction mixture was kept < 15°C during the addition. The reaction was stirred at about 14°C for one hour and then the layers were separated. The organic layer was washed with 400mL of 30%, by weight, aqueous citric acid solution and then with 400mL of aqueous 9% NaHC03. The solvent was slowly removed until 565g of the reaction mixture
remained. This mixture was stirred with overhead stirring for about 16 hours. The slurry was filtered and the solids washed with 250mL of n-heptane. Weight of 2b-Br = 133g. HPLC purity (by area %) 98%.
This is a 44% overall yield from 2,5-dibromopyridine.
*H NMR (400 MHz, d6-DMSO): 58.86 (d, J= 2.3Hz, 1H), 8.34 (dd, J= 8.5, 2.3Hz, 1H), 7.81 (dd, J = 8.5, 0.5Hz, 1H), 3.63-3.54 ( m, 4H), 3.44-3.39 (m, 2H), 3.34-3.30 ( m, 2H). MS m/z 321 (M+H+), 323 (M+2+H+).
lc. 2-(5-bromopyridin-2-yl)-l-(2,4-difluorophenyl)-2,2-difluoroethanone (3-Br)
Process Development

Table 3 illustrates the effects of the relative proportions of each of the reagents and reactants, and the effect of varying the temperature had on the overall performance of the transformation as measured by the overall yield and purity of the reaction.
Table 3. Process Development for the Preparation of bromo-pyridine 3-Br

Typical Procedure for Converting 2b-Br to 3-Br
Grignard formation:
Magnesium turnings (13.63 g, 0.56 moles) were charged to a 3-neck round bottom flask equipped with a condenser, thermocouple, addition funnel, and a stir bar. 540 mL of anhydrous tetrahydrofuran was added followed by l-Bromo-2,4-difluorobenzene (16.3 mL, 0.144 moles). The contents were stirred at 22-25°C and allowed to self -heat to 44°C. 1- Bromo-2,4-difluorobenzene ( 47mL, 0.416 moles) was added to the reaction mixture at a rate that maintained the internal temperature between 40-44°C during the addition. Once the addition was complete, the mixture was stirred for 2 hours and allowed to cool to about 25° during the stir time.
This mixture was held at 22-25°C and used within 3-4 hours after the addition of l-bromo-2,4-difluorobenzene was completed.
Coupling Reaction
Compound 2b-Br (120 g, 0.0374 moles) was charged to a 3-neck round bottom flask equipped with a condenser, thermocouple, and an overhead stirrer. 600 mL of anhydrous
tetrahydrofuran was added. The flask contents were stirred at 22°C until a clear solution was obtained. The solution was cooled to 0-5°C. The previously prepared solution of the Grignard reagent was then added slowly while maintaining the reaction temperature at 0-2°C. Reaction progress was monitored by HPLC. In-process check after 45 minutes showed <1% amide 2b-Br remaining. 2 N aqueous HC1 (600 mL, 3 vol) was added slowly maintaining the temperature below 18°C during the addition. The reaction was stirred for 30 minutes and the layers were separated. The aqueous layer was extracted with 240mL MTBE. The combined organic layers were washed with 240mL of aqueous 9% NaHCC>3 and 240mL of aqueous 20% NaCl. The organic layer was dried over 28g of MgS04 and removed the solvent to yield 3-Br (137g) as an amber oil.
HPLC purity ( by area %) = -90%; *H NMR (400 MHz, d6-DMSO) : 58.80 (d, J= 2.2Hz, 1H), 8.41 ( dd, J= 8.3, 2.3Hz, 1H), 8.00 (m, 2H), 7.45 ( m, 1H), 7.30 ( m, 1H). MS m/z 348 (M+H+), 350 (M+2+H+).
Id. l-(2,4-difluorophenyl)-2,2-difluoro-2-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)ethanone (1-4)

Typical Procedure for Converting 3-Br to 1-4
Into a 250 mL reactor were charged THF (45 mL), water (9.8 mL), bromo-pyridine 3-Br (6.0 g, 17.2 mmoles), 4-(trifluoromethoxy)phenylboronic acid (3.57 g, 17.3 mmoles), and Na2CC>3 (4.55 g, 42.9 mmoles). The stirred mixture was purged with nitrogen for 15 min. The catalyst (Pd(dppf)Cl2 as a CH2C12 adduct, 0.72 g, 0.88 mmoles) was added, and the reaction mixture was heated to 65 °C and held for 2.5 h. The heat was shut off and the reaction mixture was allowed to cool to 20-25 °C and stir overnight. HPLC analysis showed -90% ketone 1-4/hydrate and no unreacted bromo-pyridine 3-Br. MTBE (45 mL) and DI H20 (20 mL) were added, and the quenched reaction was stirred for 45 min. The mixture was passed through a plug of Celite (3 g) to remove solids and was rinsed with MTBE (25 mL). The filtrate was transferred to a separatory funnel, and the aqueous layer drained. The organic layer was washed with 20% brine (25 mL). and split into two portions. Both were concentrated by rotovap to give oils (7.05 g and 1.84 g, 8.89 g total, >100% yield, HPLC purity -90%). The larger aliquot was used to generate hetone 1-4 as is. The smaller aliquot was dissolved in DCM (3.7 g, 2 parts) and placed on a pad of Si02 (5.5 g, 3 parts). The flask was rinsed with DCM (1.8 g), and the rinse added to the pad. The pad was eluted with DCM (90 mL), and the collected filtrate concentrated to give an oil (1.52 g). To this was added heptanes (6 g, 4 parts) and the mixture stirred. The oil crystallized, resulting in a slurry. The slurry was stirred at 20-25 °C overnight. The solid was isolated by vacuum filtration, and the cake washed with heptanes (-1.5 mL). The cake was dried in the vacuum oven (40-45 °C) with a N2 sweep. 0.92 g of ketone 1-4 was obtained, 60.1% yield (corrected for aliquot size), HPLC purity = 99.9%.
TMSI Epoxidation Method
3d. 2-((2-(2,4-difluorophenyl)oxiran-2-yl)difluoromethyl)-5-(4-(trifluoromethoxy)phenyl)pyridine (5)

Typical Procedure for Converting 1-4 to 5
i-BuOK (2.22 g, 19.9 mmoles), TMSI (4.41 g, 20.0 mmoles), and THF (58.5 mL) were charged to a reaction flask, and the cloudy mixture was stirred. DMSO (35.2 mL) was added, and the clearing mixture was stirred at 20-25°C for 30 min before being cooled to 1-2°C.
Ketone 1-4 (crude, 5.85 g, 13.6 mmoles) was dissolved in THF (7.8 mL), and the 1-4 solution was added to the TMSI mixture over 12.75 min, maintaining the temperature between 1.5 and 2.0°C. The reaction was held at 0-2°C. After 1 h a sample was taken for HPLC analysis, which showed 77.6% epoxide 5, and no unreacted ketone 1-4. The reaction was quenched by the slow addition of 1 N HC1 (17.6 mL), keeping the temperature below 5°C. After 5 min 8% NaHCC>3 (11.8 mL) was added slowly below 5°C to afford a pH of 8. The reaction mixture was transferred to a separatory funnel, and the layers were separated. The aqueous layer was extracted with MTBE (78 mL), and the combined organic layers were washed with 20% NaCl (2 x 20 mL). After concentration, 7.36 g of a dark oil was obtained. HPLC of the crude oil shows it contained 75% epoxide 5. The oil was dissolved in DCM (14.7 g, 2 parts) and the solution placed on a pad of Si02 (22 g, 3 parts). The flask was rinsed with DCM (7.4 g, 1 part) and the rinse placed on the pad. The pad was eluted with DCM (350 mL) to give an amber filtrate. The filtrate was concentrated by rotovap, and when space in the flask allowed, heptane (100 mL) was added. The mixture was concentrated until 39.4 g remained in the flask, causing solid to form. The suspension was stirred for 70 min at 20-25°C. Solid was isolated by vacuum filtration, and the cake washed with heptane (10 mL) and pulled dry on the funnel. After drying in a vacuum oven (40-45 °C) with a N2 sweep, 3.33 g solid was obtained, 55.1% yield from bromo-pyridine 3, HPLC purity = 99.8%.
3e. 3-amino-2-(2,4-difluorophenyl)-l,l-difluoro-l-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)propan-2-ol (±1-6)

Process Development
Table 8 illustrates the effects of the relative proportions of each of the reagents and reactants, the effect of varying the solvent, and the effect of varying the temperature had on the overall performance of the transformation as measured by the overall yield and purity of the reaction. Table 8. Process Development for the Preparation of ±1-6

Typical Procedure for Converting 5 to +1-6
Epoxide 5 (2.17 g, 4.89 mmoles) was combined in a glass pressure tube with methanol (48 mL) and aqueous ammonia (19.5 mL). The tube was sealed and placed in an oil bath held at 54°C, with stirring. After 15 h the tube was removed from the bath, cooled, and the reaction sampled for HPLC, which showed 93.6% amino-alcohol ±1-6 and 6.0% di-adducts. To the reaction were added MTBE (48 mL) and 20% NaCl (20 mL). The layers were separated and the aqueous layer extracted with MTBE (20 mL). The combined organic layers were washed with H20 (20 mL) and transferred to a rotovap flask. Heptane (20 mL) was added, and the solution was concentrated until 16.9 g remained in the flask. An H20 layer appeared in the flask, and was pipetted out, leaving 12.8 g. Compound 1-6 seed was added, and the crystallizing mixture was stirred at 20-25 °C overnight. The flask was cooled in an ice bath for 2 h prior to filtration, and the isolated solid was washed with cold heptane (5 mL), and pulled dry on the funnel. After drying in a vacuum oven (40-45°C) for several hours 1.37 g of amino-alcohol ±1-6 was obtained, 60.8% yield, HPLC purity = 98.0%.
3f . 3-amino-2-(2,4-difluorophenyl)- 1 , 1-difluoro- 1 -(5-(4-(trifluoromethoxy)phenyl)pyridin-2- yl)propan-2-ol (1-6* or 1-7*)

Process Development
Table 9 illustrates the initial screen performed surveying various chiral acid/solvent combinations. All entries in Table 9 were generated using 0.1 mmoles of amino-alcohol ±1-6, 1 equivalent of the chiral acid, and 1ml of solvent.
Table 9. Resolution of ±1-6 (Initial Screen)

Since the best results from Table 9 were generated using tartaric acid and di-p-toluoyltartaric acid, Table 10 captures the results from a focused screen using these two chiral acids and various solvent combinations. All entries in Table 10 were performed with 0.2 mmoles of amino-alcohol ±1-6, 87 volumes of solvent, and each entry was exposed to heating at 51 °C for lh, cooled to RT, and stirred at RT for 24h.
Table 10. Resolution of ±1-6 (Focused Screen)

Each of the three entries using di-p-toluoyltartaric acid in Table 10 resulted in higher levels of enantio-enrichment when compared to tartaric acid. As such, efforts to further optimize the enantio-enrichment were focusing on conditions using di-p-toluoyltartaric acid (Table 11).

Ό.6 equivalents used
ee sense was opposite from the other entries in the table (i.e., enantiomer of 1-6*)
Typical Procedure for Converting +1-6 to 1-6* or 1-7*
(This experimental procedure describes resolution of ±1-6, but conditions used for DPPTA resolution of 1-6 or 1-7 are essentially the same.)
Amino-alcohol ±1-6 (7.0 g, 15 mmoles) was dissolved in a mixture of acetonitrile (84 mL) and methanol (21 mL). (D)-DPTTA (5.89 g, 15 mmoles) was added, and the reaction was warmed to 50°C and held for 2.5 h. The heat was then removed and the suspension was allowed to cool and stir at 20-25 °C for 65 h. The suspension was cooled in an ice bath and stirred for an additional 2 h. Solid was isolated by vacuum filtration, and the cake was washed with cold 8:2 ACN/MeOH (35 mL). After drying at 50°C, 5.18 g of 1-6* or l-7*/DPPTA salt was isolated, HPLC purity = 99.0, ee = 74.
The 1-6* or l-7*/DPPTA salt (5.18 g) was combined with 8:2 ACN/MeOH (68 mL) and the suspension was heated to 50°C and held for 20 min. After cooling to 20-25 °C the mixture was stirred for 16 h. Solids were isolated by vacuum filtration, and the cake washed with cold 8:2 ACN/MeOH (30 mL), and pulled dry on the funnel. 2.82 g of 1-6* or l-7*/DPPTA salt was obtained, 44.4% yield (from crude ±1-6), ee = 97.5. The resulting solids were freebased to provide 1-6* or 1-7* with the same achiral and chiral purity as the DPPTA salt.
EXAMPLE 4: Preparation of 2-(2.4-difluorophenyl -l.l-difluoro-3-(lH-tetrazol-l-yl -l-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la).


The procedure used to generate compound 1 or la is as described in US 4,426,531. Table 13 illustrates the efficient and quantitative nature of this procedure as performed on amino- alcohol 1-6* or 1-7* produced from both the TMS-cyanohydrin method and the TMSI- epoxidation method.
Table 13. Formation of Compound 1 or la

EXAMPLE 5: 2-(2.4-difluorophenyl -l.l-difluoro-3-(lH-tetrazol-l-yl -l-(5-(4- (trifluoromethoxy)phenyl)pyridin-2-yl)propan-2-ol benzenesulfonate (1 or la-BSA).

Typical Procedure for Converting 1 or la to 1 or la-BSA
46.6 g of compound 1 or la was dissolved in ethylacetate (360ml). The solution was filtered through a glass microfiber filter and placed in a 2 L reaction flask equipped with an overhead stirrer, condenser, and a J-Kem thermocouple. Pharma-grade benzenesulfonic acid (BSA, 14.39g, leq) was dissolved in ethyl acetate (100ml). The BSA solution was filtered through a glass microfiber filter and added to the stirred 1 or la solution in one portion. The mixture was warmed to 60-65 °C; precipitation of the 1 or la/BSA salt occurred during the warm up period. The slurry was held for 60 minutes at 60-65 °C. The suspension was allowed to slowly cool to 22 °C and was stirred at 20-25 °C for 16 hours. n-Heptane (920ml) was charged in one portion and the suspension was stirred at 22 °C for an additional 90 minutes. The slurry was filtered and the collected solids washed with n-heptane (250ml). The isolated solids were placed in a vacuum oven at 50 °C for 16 hours. 52.26g (86% yield) of 1 or la
benzenesulfonate was obtained.
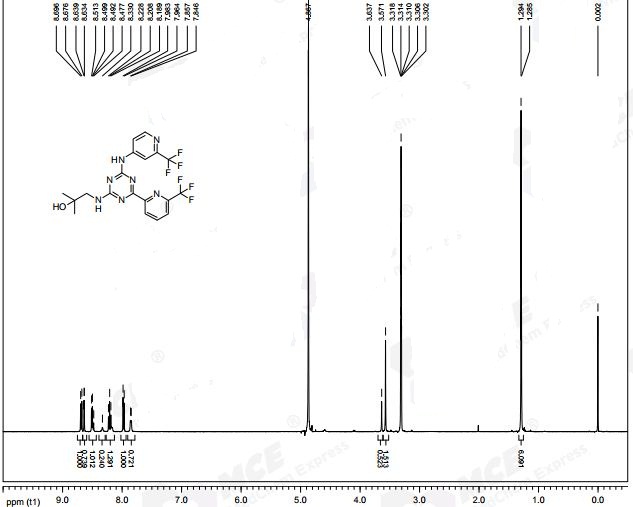
*H NMR (400 MHz, DMSO-d6 + D20): 89.16 (s, 1H), 8.95 (d, J = 2.1 Hz, 1H), 8.26 (dd, J = 8.2, 2.3 Hz, 1H), 7.96-7.89 (m, 2H), 7.66-7.61 (m, 2H), 7.59 (dd, J = 8.3, 0.4 Hz, 1H), 7.53 (br d, J = 8.0 Hz, 2H), 7.38-7.15 (m, 5H), 6.90 (dt, J = 8.3, 2.5 Hz, 1H), 5.69 (d, J = 14.8 Hz, 1H), 5.15 (d, J = 15.2 Hz, 1H).
Further results are in Table 14.
Table 14. Formation of 1 or la-BSA

( ) (%ee) Yield Purity (%) ee
97.9 95.9 84% 98.2 97.1
Figures 1-2 contain the analytical data for 1 or la-BSA prepared by the TMSI-epoxidation process.
EXAMPLE 6: 5-bromo-2-((2-(2,4-difluorophenyl)oxiran-2-yl)difluoromethyl)pyridine -Br).

Typical Procedure for Converting 3-Br to 4-Br
KOtBu ( 41.7g, 0.372moles, 1.05 equiv) and trimethylsulfoxonium iodide ( 85.7g,
0.389moles, 1.1 equiv) were charged to a 3L 3-neck round bottom flask equipped with an overhead stirrer, a thermocouple and an addition funnel. 1.2L of anhydrous THF and 740mL of DMSO were added to the flask and stirred at 22-25 °C for 70 minutes. The contents were cooled to 0°C. Crude ketone 3 was dissolved in 250mL of anhydrous THF and slowly added the ketone 3-Br solution to the reaction mixture over 20 minutes while maintaining a reaction temperature at < 3°C during the addition and stirred at 0°C for one hour. In-process HPLC showed <1% ketone 3-Br remaining. 200mL of IN HC1 was slowly added maintaining a reaction temperature of < 6°C during the addition. After stirring for 30 minutes the layers were separated and the aqueous layer was extracted with 375mL of MTBE. The combined organic layers were washed with 375mL of aqueous 9% NaHCC>3 and with 375mL of aqueous 20% NaCl. The solvent was removed to yield 4-Br as a brown waxy solid.
Weight of crude epoxide 4-Br = 124.6g; *H NMR (400 MHz, d6-DMSO) : 58.82 (d, J= 2.3Hz, 1H), 8.21 ( dd, J= 8.3, 2.3Hz, 1H), 7.50 (dd, J= 8.3, 0.5Hz, 1H), 7.41 ( m, 1H), 7.25 ( m, 1H), 7.10 (m,lH), 3.40 ( d, J= 4.5Hz, 1H), 3.14 ( m, 1H). MS m/z 362 (M+H+), 364 (M+2+H+).
EXAMPLE 7: 3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan-2-ol (4b-Br).

Typical Procedure for Converting 4-Br to 4b-Br
Crude epoxide 4-Br ( 54.4g, 0.15moles) was placed into a Schott autoclave bottle equipped with a stir bar. 550mL of MeOH was added to the bottle and stirred for 90 minutes at 22-25 °C. Concentrated NH4OH ( 550mL, 7.98 moles, 53 equiv) was added to the epoxide 4-Br
solution. The bottle was sealed and placed in an oil bath at 55 °C. The mixture was stirred at 55°C for 17 hours. The bottle was removed from the oil bath and cooled to 22-25°C. In-process HPLC showed <1% epoxide 4-Br remaining. The solvent was removed via rotary evaporation until 362g ( 37%) of the reaction mass remained. 500mL of MTBE was added and cooled the mixture to 8°C. 500mL of 6N HCl was slowly added maintaining the reaction temperature between 8 – 12°C during the addition. After stirring for 10 minutes, the layers were separated. The MTBE layer was extracted with 350mL of 6N HCl. The combined aqueous layers were washed with 250mL MTBE and 2 X 250mL heptane. MTBE, 250mL, was added to the aqueous layer and the mixture was cooled to 2°C. 344g of KOH was dissolved in 500mL of water. The KOH solution was slowly added to the reaction mixture over one hour while maintaining the temperature at <19°C. After stirring for 15 minutes, the layers were separated. The aqueous layer was extracted with 250mL MTBE. The combined organic layers were washed with 250mL of aqueous 20% NaCl and the solvent was removed to yield ±4b-Br as a dark oil. Weight of crude amino alcohol ±4b-Br = 46.0g. HPLC purity ( by area %) = 92%; *H NMR (400 MHz, d6-DMSO) : 58.67 (d, J= 2.2Hz, 1H), 8.15 ( dd, J= 8.6, 2.4Hz, 1H), 7.46 (m, 1H), 7.40 ( dd, J= 8.5, 0.7Hz, 1H), 7.10 ( m, 1H), 7.00 (m,lH), 3.37 (dd, J= 13.7, 2.1Hz, 1H), 3.23 ( dd, J= 13.7, 2.7, 1H). MS m/z 379 (M+H+), 381 (M+2+H+).
EXAMPLE 8: 3-amino-l-(5-bromopyridin-2-yl -2-(2.4-difluorophenyl -l.l-difluoropropan-2-ol (4b-Br or 4c-Br).

Typical Procedure for Converting 4-Br to 4b-Br or 4c-Br
Crude amino alcohol ±4b-Br ( 42.4, O. llmoles) was dissolved in 425mL of 8:2 IPA: CH3CN. The solution was charged to a 1L 3-neck round bottom flask equipped with a condenser, overhead stirrer and a thermocouple. Charged di-p-toluoyl-L-tartaric acid ( 21.6g, 0.056moles, 0.5 equiv) to the flask and warmed the contents to 52°C. The reaction mixture was stirred at 52°C for 5 hours, cooled to 22-25°C and stirred for 12 hours. The slurry was cooled to 5-10°C and stirred for 90 minutes. The mixture was filtered and collected solids washed with 80mL of cold CH3CN. The solids were dried in a vacuum oven 45-50°C. Weight of amino alcohol/ DPTTA salt = 17.4g
Chemical purity by HPLC ( area %) = 98.5%; Chiral HPLC= 98.0% ee.
13.60g of the amino alcohol/ DPTTA salt was placed into a 250mL flask with a stir bar and to this was added lOOmL of MTBE and lOOmL of 10% aqueous K2CO3solution. The reaction was stirred until complete dissolution was observed. The layers were separated and the aqueous layer was extracted with 50mL of MTBE. The combined MTBE layers were washed with 50mL of 20% aqueous NaCl and the solvent removed to yield 8.84 (98%) of 4b-Br or 4c-Br as a light yellow oil.
EXAMPLE 9: 3-amino-2-(2,4-difluorophenyl)-l J-difluoro-l-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)propan-2-ol (1-6* or 1-7*).

Typical Procedure for Converting 4b-Br or 4c-Br to 1-6* or 1-7*
Amino alcohol 4b-Br or 4c-Br (8.84g, 0.023moles, 1 equiv) was dissolved in 73mL of n-propanol. The solution was transferred to a 250mL 3-neck round bottom flask equipped with a condenser, thermocouple, stir bar and septum. 17mL of water was added and stirred at 22-25°C for 5 minutes. To the reaction was added K2CO3 ( 9.67g, 0.07moles, 3 equiv), 4-(trifluoromethoxy)phenylboronic acid ( 5.76g, 0.028moles, 1.2 equiv.) and Pd(dppf)Cl2 as a CH2Cl2 adduct ( 0.38g, 0.47mmoles, 0.02 equiv) to the flask. After the mixture was purged with nitrogen for 10 minutes, the reaction was then warmed to 85-87°C and stirred at 85-87°C for 16 hours. HPLC analysis showed < 1% of the amino alcohol 4b-Br or 4c-Br remaining. The mixture was cooled to 22-25 °C, then 115mL of MTBE and 115mL of water were added and stirred for 30 minutes. The layers were separated and the organic layer was washed with 2 X 60mL of 20% aqueous NaCl. The solvent was removed to yield 12.96g ( 121% yield) of 1-6* or 1-7* as a crude dark oil. It should be noted that the oil contains residual solvent, Pd and boronic acid impurity.
‘ll NMR (400 MHz, d6-DMSO) : 58.90 (d, J= 2.2Hz, 1H), 8.22 ( dd, J= 8.3, 2.3Hz, 1H), 7.91 (m, 2H), 7.54 ( m, 4H), 7.14 ( m, 1H), 7.02 (m,lH), 3.41 (m, 1H), 3.27 ( dd, J= 14.0, 2.7, 1H). MS m/z 461 (M+H+)
CLIP
Med. Chem. Commun., 2016,7, 1285-1306
DOI: 10.1039/C6MD00222F
Fungal infections directly affect millions of people each year. In addition to the invasive fungal infections of humans, the plants and animals that comprise our primary food source are also susceptible to diseases caused by these eukaryotic microbes. The need for antifungals, not only for our medical needs, but also for use in agriculture and livestock causes a high demand for novel antimycotics. Herein, we provide an overview of the most commonly used antifungals in medicine and agriculture. We also present a summary of the recent progress (from 2010–2016) in the discovery/development of new agents against fungal strains of medical/agricultural relevance, as well as information related to their biological activity, their mode(s) of action, and their mechanism(s) of resistance.
CLIP
Design and optimization of highly-selective fungal CYP51 inhibitors
- Viamet Pharmaceuticals Inc., Durham, NC 27703, USA
- http://dx.doi.org/10.1016/j.bmcl.2014.05.068

able 3.Antifungal activity of difluoromethyl-pyridyl-benzenes
| Compound |
R |
C. albicans MICa |
T. rubrum MICa |
CYP3A4 IC50b |
Selectivity indexc |
| 7a |
Cl |
⩽0.001 |
0.004 |
36 |
9000 |
| 7b |
CF3 |
⩽0.001 |
0.002 |
54 |
27,000 |
| 7c
VT 1129 |
OCF3 |
⩽0.001 |
⩽0.001 |
79 |
>79,000 |
| 7d
VT 1161 |
OCH2CF3 |
⩽0.001 |
⩽0.001 |
65 |
>65,000 |
| Itraconazole |
— |
0.016 |
0.062 |
0.07 |
1.1 |
update………….


<p>Formula (I)</p> <p>Crizotinib is a potent small-molecule inhibitor of c-Met/HGFR (hepatocyte growth factor receptor) kinase and ALK (anaplastic lymphoma kinase) activity. Enantiomerically pure compound of formula I was first disclosed in US Patent No. 7,858,643. Additionally, the racemate of compound of formula I was disclosed in U.S. patent application 2006/0128724, both of these references discloses similar methods for the synthesis of Compound of Formula I.</p> <p>Conventionally, the compounds of formula I are prepared by reacting Bis(pinacolato)diboron with protected 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-pyridin-2-ylamine in the presence of Pd catalyst. The obtained product after deprotection is reacted with N- protected 4-(4-bromo-pyrazol-l-yl)-piperidine in the presence of Pd Catalyst. The obtained product is filtered through celite pad and purified by Column Chromatography. The final product of formula I was obtained by deprotection of the purified compound by using HCl/dioxane. US Patent No. 7,858,643 provides enantiomerically pure aminoheteroaryl compounds, particularly aminopyridines and aminopyrazines, having protein tyrosine kinase activity. More particularly, US 7,858,643 describes process for the preparation of 3-[(lR)-l-(2,6- dichloro-3-fluorophenyl)ethoxy]-5-(l-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine. The Scheme is summarized below in Scheme- 1 :</p>


<p>Scheme-1</p> <p>wherein, “Boc” means tert-butoxycarbonyl; and a) (Boc)<sub>2</sub>, DMF, Dimethylaminopyridine b) Pd(dppf)Cl<sub>2</sub>, KOAc, Dichloromethane; c) HC1, Dioxane, Dichloromethane; d) Pd(PPh<sub>3</sub>)<sub>2</sub>Cl<sub>2</sub>, Na<sub>2</sub>C0<sub>3</sub>, DME/H<sub>2</sub>0; e) 4M HCl/Dioxane, Dichloromethane</p> <p>A similar process has been disclosed in the U.S. patent application 2006/0128724 for the preparation of Crizotinib. J. Jean Cui et. al. in J. Med. Chem. 2011, 54, 6342-6363, also provides a similar process for the preparation of Crizotinib and its derivatives.</p> <p>However, above mentioned synthetic process requires stringent operational conditions such as filtration at several steps through celite pad. Also column chromatography is required at various steps which is not only tedious but also results in significant yield loss. Another disadvantage of above process involves extensive use of palladium catalysts, hence metal scavengers are required to remove palladium content from the desired product at various steps which makes this process inefficient for commercial scale.</p> <p>Yet another disadvantage of above process is the cost of Bis(pinacolato)diboron. This reagent is used in excess in the reaction mixture resulting in considerable cost, especially during large-scale syntheses.</p> <p>US Patent No. 7,825,137 also discloses a process for the preparation of Crizotinib where Boc protected 4-(4-iodo-pyrazol-l-yl)-piperidine is first reacted with Bis(pinacolato)diboron in the presence of Pd catalyst. The reaction mixture is filtered through a bed of celite and the obtained filtrate is concentrated and purified by silica gel chromatography to give to form tert-butyl-4-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]piperidine-l- carboxylate. To this compound, 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]- pyridin-2-ylamine is added in the presence of a Pd catalyst. The reaction mixture is stirred for 16h at 87°C. The reaction mixture is filtered through celite pad and the concentrated filtrate is purified on silica gel column to obtain (4-{6-amino-5-[(R)-l-(2,6-dichloro-3-fluoro- phenyl)-ethoxy]-pyri- din-3-yl}-pyrazol-l-yl)-piperidine-l-carboxylic acid tert-butyl ester of 95% purity. To the solution of resulting compound in dichloromethane 4N HCl/Dioxane is added and thereby getting the reaction suspension is filtered in Buchner funnel lined with filter paper. The obtained solid is dissolved in HPLC water and pH is adjusted to 10 with the addition of Na<sub>2</sub>C0<sub>3</sub> Compound is extracted using dichloroform and is purified on a silica gel column by eluting with CH<sub>2</sub>Cl<sub>2</sub> MeOH/NEt<sub>3</sub> system to obtain Crizotinib. The scheme is summarized below in scheme 2:</p>

<p>Formula (i) Formula (ii)</p>
<p>Formula (iii) Formula (ii) ula (iv)</p>

<p>Formula (v) Formula (I)</p> <p>Scheme-2</p> <p><span style=”color:#ff0000;”>Preparation of Crizotinib:</span></p> <p>To a stirred solution of Tert-butyl 4-(4-{ 6-amino-5-[(li?)-l-(2,6-dichloro-3- fluorophenyl)ethoxy]pyridin-3 -yl } – lH-pyrazol- 1 -yl)piperidine- 1 -carboxylate (material obtained in Example 3) (l.Og, 0.00181 moles) in dichloromethane (-13 ml) at 0°C was added 4.0 M dioxane HQ (6.7 ml, 0.0272 moles). Reaction mixture was stirred at room temperature for 4h. After the completion of reaction monitored by TLC, solid was filtered and washed with dichloromethane (10 ml). The obtained solid was dissolved in water (20 ml); aqueous layer was extracted with dichloromethane (10×2). The pH of aqueous layer was adjusted to 9-10 with Na<sub>2</sub>C03 and compound was extracted with dichloromethane (10 x 3), combined organic layers were washed with water (20 ml), evaporated under vacuum to get solid product. The solid was stirred with ether (10 ml), filtered off, washed well with ether, dried under vacuum to get <span style=”color:#ff0000;”>Crizotinib.</span></p> <p>Yield: 0.45g (55 %)</p> <p>HPLC Purity: 99.35 %</p> <p><span style=”color:#ff0000;”>1HNMR (400 MHz, CDC1<sub>3</sub>) δ: 7.76 (d, J = 1.6 Hz, 1H), 7.56 (s, 1H), 7.49 (s, 1H), 7.30 (dd, J = 9.2 Hz), 7.0 (m, 1H), 6.86 (d, J = 1.6 Hz, 1H), 6.09 ( q, J= 6.8 Hz, 1H), 4.75 (brs, 1H), 4.19 (m, 1H), 3.25 (m, 2H), 2.76 (m, 2H), 2.16 (m, 2H), 1.92 (m, 2H), 1.85 (d, J= 6.8 Hz, 3H), 1.67 (brs, 1H)</span></p> <p>…………………………</p> <p><a href=”http://www.sciencedirect.com/science/article/pii/S0040403914000872″>http://www.sciencedirect.com/science/article/pii/S0040403914000872</a></p>
Abstract
A novel approach for the synthesis of Crizotinib (1) is described. In addition, new efficient procedures have been developed for the preparation of (S)-1-(2,6-dichloro-3-fluorophenyl)ethanol (2) and tert-butyl 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)piperidine-1-carboxylate (4), the key intermediates required for the synthesis of Crizotinib.
<hr id=”absgraphicalab0051″ class=”artHeader” />
Graphical abstract
textboxdefaultfig
“>

- …………………
- http://www.sciencedirect.com/science/article/pii/S0040403911021745
-
Abstract
4-(4-Iodo-1H-pyrazol-1-yl)piperidine is a key intermediate in the synthesis of Crizotinib. We report a robust three-step synthesis that has successfully delivered multi-kilogram quantities of the key intermediate. The process includes nucleophilic aromatic substitution of 4-chloropyridine with pyrazole, followed by hydrogenation of the pyridine moiety and subsequent iodination of the pyrazole which all required optimization to ensure successful scale-up.
<hr id=”absgraphical1″ class=”artHeader” />
Graphical abstract
textboxdefaultfig
“>

</div> </div> </dt> </dl> </div> </div> </div> <p>……………………</p>
Org. Process Res. Dev., 2011, 15 (5), pp 1018–1026
DOI: 10.1021/op200131n

<p class=”articleBody_abstractText”>A robust six-step process for the synthesis of crizotinib, a novel c-Met/ALK inhibitor currently in phase III clinical trials, has been developed and used to deliver over 100 kg of API. The process includes a Mitsunobu reaction, a chemoselective reduction of an arylnitro group, and a Suzuki coupling, all of which required optimization to ensure successful scale-up. Conducting the Mitsunobu reaction in toluene and then crystallizing the product from ethanol efficiently purged the reaction byproduct. A chemoselective arylnitro reduction and subsequent bromination reaction afforded the key intermediate <b>6</b>. A highly selective Suzuki reaction between <b>6</b> and pinacol boronate <b>8</b>, followed by Boc deprotection, completed the synthesis of crizotinib <b>1</b>.</p> </div> <p><span id=”d43162769e1806″ class=”title2″>3-[(1<i>R</i>)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1<i>H</i>-pyrazol-4-yl]pyridin-2-amine <b>1</b></span></p> <p><span style=”color:#ff0000;”> <i>crizotinib</i><b>1</b> (20.7 kg, 80%) as a white solid. </span></p> <p><span style=”color:#ff0000;”>Mp 192 °C;</span></p> <p><span style=”color:#ff0000;”><sup>1</sup>H NMR (400 MHz, CDCl<sub>3</sub>) δ: 7.78 (d, <i>J</i> = 1.8 Hz, 1H), 7.58 (s, 1H), 7.52 (s, 1H), 7.31 (dd, <i>J</i> = 9.0, 4.9 Hz, 1H), 7.06 (m, 1H), 6.89 (d, <i>J</i> = 1.7 Hz, 1H), 6.09 (q, 1H), 4.79 (br s, 2H), 4.21 (m, 1H), 3.26 (m, 2H), 2.78 (m, 2H), 2.17 (m, 2H), 1.90 (m, 2H), 1.87 (d, <i>J</i> = 6.7 Hz, 3H), 1.63 (br s, 1H).</span></p> <p><span style=”color:#ff0000;”> <sup>13</sup>C NMR (100.6 MHz, CDCl<sub>3</sub>) δ: 157.5 (d, <i>J</i> = 250.7 Hz), 148.9, 139.8, 137.0, 135.7, 135.6, 129.9, 129.0 (d, <i>J</i> = 3.7 Hz), 122.4, 122.1 (d, <i>J</i> = 19.0 Hz), 119.9, 119.3, 116.7 (d, <i>J</i> = 23.3 Hz), 115.0, 72.4, 59.9, 45.7, 34.0, 18.9.</span></p> <p><span style=”color:#ff0000;”> LC-MS: found <i>m</i>/<i>z</i> 450.0, 451.0, 452.0, 453.0, 454.0, 455.0. </span></p> <p><span style=”color:#ff0000;”>Anal. Calcd for C<sub>21</sub>H<sub>22</sub>Cl<sub>2</sub>FN<sub>5</sub>O: C, 56.01; H, 4.92; N, 15.55. Found: C, 56.08; H, 4.94; N, 15.80.</span></p>
Cui, J. J.; Botrous, I.; Shen, H.; Tran-Dube, M. B.; Nambu, M. D.; Kung, P.-P.; Funk, L. A.; Jia, L.; Meng, J. J.; Pairish, M. A.; McTigue, M.; Grodsky, N.; Ryan, K.; Alton, G.; Yamazaki, S.; Zou, H.; Christensen, J. G.; Mroczkowski, B.Abstracts of Papers; 235th ACS National Meeting, New Orleans, LA, United States, April 6–10, 2008.
</div>
Cui, J. J.; Funk, L. A.; Jia, L.; Kung, P.-P.; Meng, J. J.; Nambu, M. D.; Pairish, M. A.; Shen, H.; Tran-Dube, M. B. U.S. Pat. Appl. U. S. 2006/0046991 A1, 2006.
Cosy predict above
1H NMR PREDICT
13C NMR PREDICT
| WO2006021881A2 * |
15 Aug 2005 |
2 Mar 2006 |
Pfizer |
Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
| WO2006021884A2 * |
15 Aug 2005 |
2 Mar 2006 |
Pfizer |
Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors |
| WO2013181251A1 * |
29 May 2013 |
5 Dec 2013 |
Ratiopharm Gmbh |
Crizotinib hydrochloride salt in crystalline |
| EP2620140A1 * |
26 Jan 2012 |
31 Jul 2013 |
ratiopharm GmbH |
Crizotinib containing compositions |
textboxdefaultfig
“>
-
textboxdefaultfig
“>
| WO2010048131A1 * |
Oct 20, 2009 |
Apr 29, 2010 |
Vertex Pharmaceuticals Incorporated |
C-met protein kinase inhibitors |
| WO2011042389A2 * |
Oct 4, 2010 |
Apr 14, 2011 |
Bayer Cropscience Ag |
Phenylpyri(mi)dinylazoles |
| US7825137 |
Nov 23, 2006 |
Nov 2, 2010 |
Pfizer Inc. |
Method of treating abnormal cell growth |
| US7858643 |
Aug 26, 2005 |
Dec 28, 2010 |
Agouron Pharmaceuticals, Inc. |
Crizotinib, a c-Met protein kinase inhibitor anticancer agent; 3-[(R)-1-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-5-(1-piperidin-4-yl-1H-pyrazol-4-yl)-pyridin-2-ylamine is crizotinib |
| US20060128724 |
Aug 26, 2005 |
Jun 15, 2006 |
Agouron Pharmaceuticals, Inc. |
Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
| 1 |
J. JEAN CUI J. MED. CHEM. vol. 54, 2011, pages 6342 – 6363 |
| 2 |
ORG. PROCESS RES. DEV. vol. 15, 2011, pages 1018 – 1026 |
| 3 |
* |
PIETER D. DE KONING ET AL: “Fit-for-Purpose Development of the Enabling Route to Crizotinib (PF-02341066)“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 15, no. 5, 16 September 2011 (2011-09-16), pages 1018-1026, XP055078841, ISSN: 1083-6160, DOI: 10.1021/op200131n |
VT 1129 BENZENE SULFONATE
CAS 1809323-18-9

VT 1129
1340593-70-5 CAS
QUILSECONAZOLE, VT-1129
Viamet, in collaboration with Therapeutics for Rare and Neglected diseases, is investigating quilseconazole benzenesulfonate (VT-1129), a small-molecule lanosterol demethylase (CYP51) inhibitor, developed using the company’s Metallophile technology, for treating fungal infections, including Cryptococcus neoformans meningitis.
WO-2017049080



////////VT 1129, VIAMET, WO 2016149486, Viamet Pharmaceuticals, Antifungals, Small molecules, 14-alpha demethylase inhibitors, Orphan Drug Status, Cryptococcosis, On Fast track, PHASE 1, VT-1129, QUILSECONAZOLE
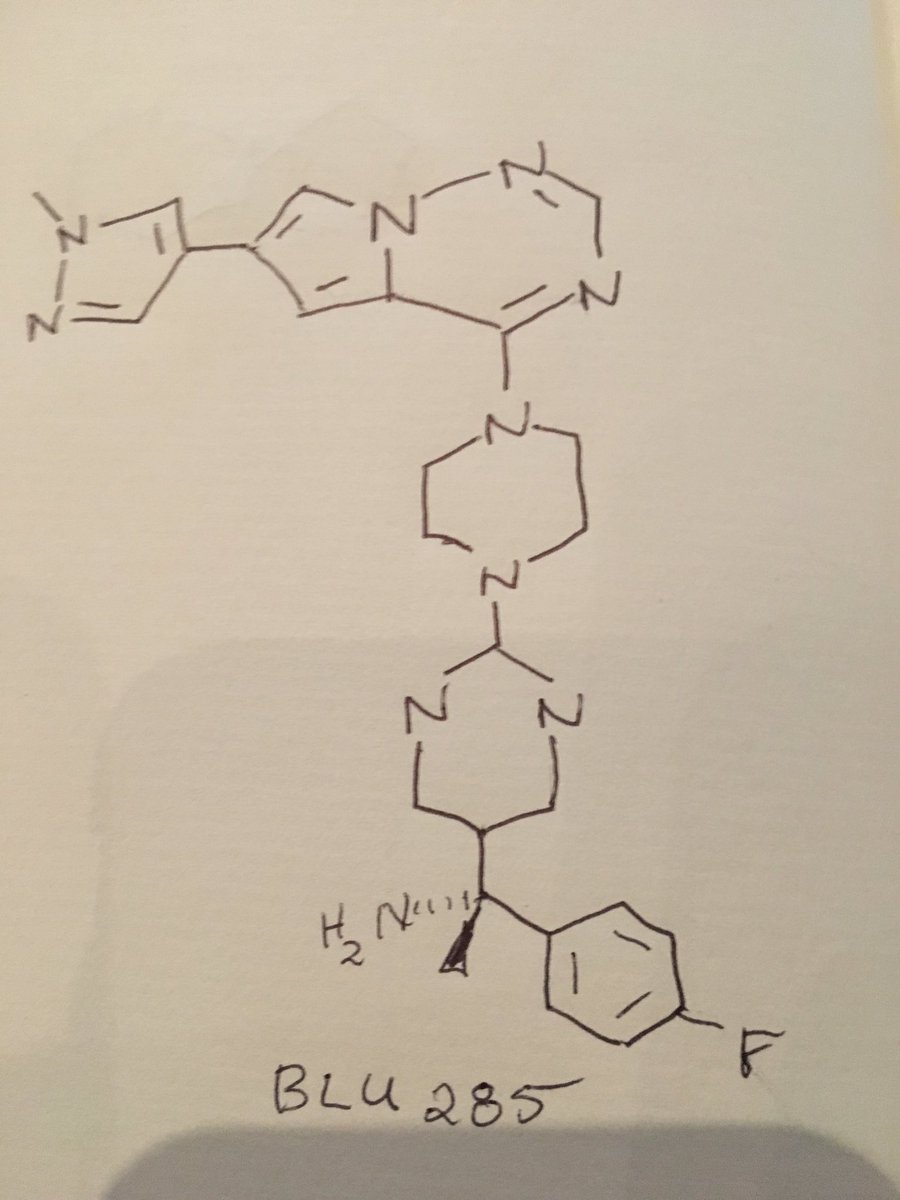
O[C@@](Cn1cnnn1)(c2ccc(F)cc2F)C(F)(F)c3ccc(cn3)c4ccc(OC(F)(F)F)cc4

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....















 Cavosonstat (N91115) was an experimental therapy being developed by Nivalis Therapeutics. Its primary mechanism of action was to inhibit the S-nitrosoglutathione reductase (GSNOR) enzyme and to stabilize cystic fibrosis transmembrane regulator (CFTR) protein activity. A press release published in February announced the end of research for this therapy in cystic fibrosis (CF) patients with F508del mutations. The drug, which did not meet primary endpoints in a Phase 2 trial, had been referred to as the first of a new class of compounds that stabilizes the CFTR activity.
Cavosonstat (N91115) was an experimental therapy being developed by Nivalis Therapeutics. Its primary mechanism of action was to inhibit the S-nitrosoglutathione reductase (GSNOR) enzyme and to stabilize cystic fibrosis transmembrane regulator (CFTR) protein activity. A press release published in February announced the end of research for this therapy in cystic fibrosis (CF) patients with F508del mutations. The drug, which did not meet primary endpoints in a Phase 2 trial, had been referred to as the first of a new class of compounds that stabilizes the CFTR activity.



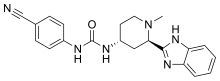
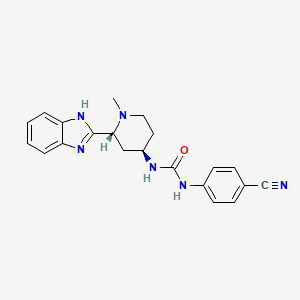










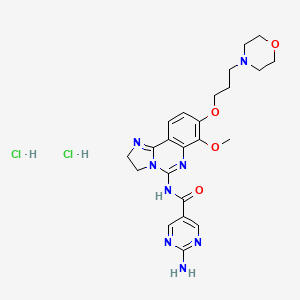 Copanlisib dihydrochloride; UNII-03ZI7RZ52O; 03ZI7RZ52O; 1402152-13-9; BAY 80-6946 dihydrochloride;
Copanlisib dihydrochloride; UNII-03ZI7RZ52O; 03ZI7RZ52O; 1402152-13-9; BAY 80-6946 dihydrochloride;






















































































![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-1h.png)
![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-13c.png)
































{kind=link}