
Home » Posts tagged 'Orphan Drug'
Tag Archives: Orphan Drug
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Repinatrabit
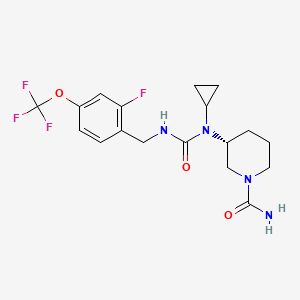
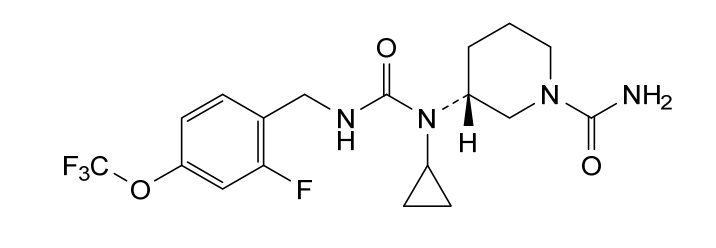
Repinatrabit
CAS 2837993-05-0
MF C18H22F4N4O3 MW 418.4 g/mol
(3R)-3-[cyclopropyl-[[2-fluoro-4-(trifluoromethoxy)phenyl]methylcarbamoyl]amino]piperidine-1-carboxamide
(3R)-3-[cyclopropyl({[2-fluoro-4-(trifluoromethoxy)phenyl]methyl}carbamoyl)amino]piperidine-1-
carboxamide
solute carrier family 6 member 19 (SLC6A19) inhibitor(phenylketonuria), JNT-517, JNT 517, orphan drug, rare pediatric disease designations, Jnana Therapeutics, 5P44NDU6AC, JN 11804, JN-11804
Repinatrabit (JNT-517) is an investigational, oral, small-molecule drug developed by Jnana Therapeutics (now part of Otsuka Pharmaceutical) to treat Phenylketonuria (PKU). It acts as a selective inhibitor of the SLC6A19 transporter, reducing blood phenylalanine (Phe) levels by increasing its urinary excretion.
Key Details About Repinatrabit:
- Mechanism: It targets a novel, cryptic allosteric site to block kidney reabsorption of phenylalanine, aiming to be a first-in-class oral therapy for all PKU patients, regardless of age or genotype.
- Clinical Trials: Otsuka initiated a global Phase 3 study (NCT06971731) in December 2025 to evaluate its safety and efficacy, following positive results from earlier studies.
- Status: The FDA has granted it orphan drug and rare pediatric disease designations.
- A Study to Evaluate the Safety and Efficacy of JNT-517 in Participants With Phenylketonuria (PKU)CTID: NCT06971731Phase: Phase 3Status: RecruitingDate: 2026-02-04
- A Phase 2 Study of JNT-517 in Adolescent Participants With PhenylketonuriaCTID: NCT06637514Phase: Phase 2Status: RecruitingDate: 2025-08-19
- First-in-Human, Multiple Part Clinical Study of JNT-517 in Healthy Participants and in Participants With PhenylketonuriaCTID: NCT05781399Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-07-31
- A Study to Evaluate the Long-Term Safety and Efficacy of JNT-517 in Participants With PhenylketonuriaCTID: NCT06628128Phase: Phase 3Status: Not yet recruitingDate: 2025-06-03
EMA Drug Information,
Type, Orphan designations
(R)-3-(1-Cyclopropyl-3-(2-fluoro-4-(trifluoromethoxy)benzyl)ureido)piperidine-1-carboxamide
Intended Use, Treatment of hyperphenylalaninaemia, Status of Orphan Designation, Positive
First Published Date, 2024-08-22
PAT
SYN
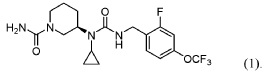
PAT
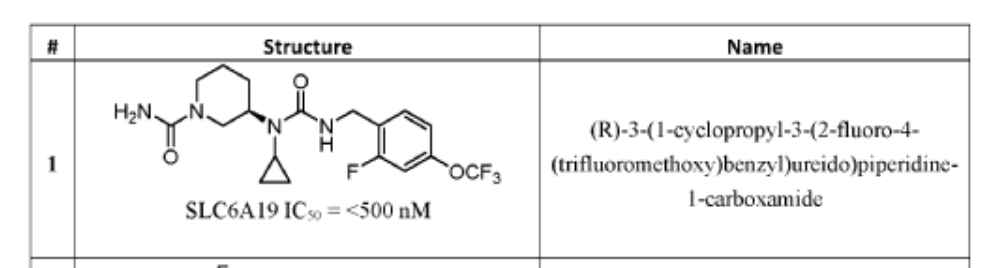
PAT
- Crystalline forms of a piperidine inhibitor of slc6a19 functionPublication Number: WO-2024118721-A1Priority Date: 2022-11-30
- Crystalline forms of a piperidine inhibitor of slc6a19 functionPublication Number: EP-4626420-A1Priority Date: 2022-11-30
- Dosing regimen for treatment of PKU with SLC6A19 functional piperidine inhibitorsPublication Number: CN-120091814-APriority Date: 2022-09-14
- Small molecule inhibitors of mammalian slc6a19 functionPublication Number: US-2024208923-A1Priority Date: 2021-03-10
- Small molecule inhibitors of mammalian slc6a19 functionPublication Number: US-2025289797-A1Priority Date: 2021-03-10
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

///////////repinatrabit, ANAX, JNT-517, JNT 517, orphan drug, rare pediatric disease designations, Jnana Therapeutics, 5P44NDU6AC, JN 11804, JN-11804
Cambritaxestat


Cambritaxestat
CAS 1979939-16-6
MFC25H22ClF3N4O2 MW502.9 g/mol
N-[(1S)-1-(4-chlorophenyl)ethyl]-3-[3-[[4-(trifluoromethoxy)phenyl]methyl]imidazo[4,5-b]pyridin-2-yl]propanamide
N-[(1S)-1-(4-chlorophenyl)ethyl]-3-(3-{[4-(trifluoromethoxy)phenyl]methyl}-3H-imidazo[4,5-b]pyridin-2-yl)propanamide
autotaxin inhibitor, antineoplastic, Orphan Drug, IOA 289, IOA-289, IOA289, LYY3P2KA27, CRT 0273750
- OriginatorCancer Research Technology; Merck & Co
- DeveloperiOnctura
- ClassAntifibrotics; Antineoplastics; Small molecules
- Mechanism of ActionAngiogenesis inhibitors; Cell proliferation inhibitors; ENPP2 protein inhibitors
- Orphan Drug StatusYes – Pancreatic cancer
- Phase I/IIPancreatic cancer
- Phase ISolid tumours
- PreclinicalNon-alcoholic steatohepatitis
- 14 Oct 2025Efficacy and adverse event data from a phase I/II trial in Pancreatic cancer released by iOnctura
- 04 Oct 2024Cambritaxestat is still in phase-I development in Solid-tumours (In volunteers) in Italy (PO, Capsule) (NCT05027568)
- 31 May 2024Efficacy and adverse event data from a phase I/II trial in Pancreatic cancer presented at the 60th Annual Meeting of the American Society of Clinical Oncology (ASCO-2024)
Cambritaxestat is an autotaxin inhibitor.
Cambritaxestat is an orally bioavailable small molecule inhibitor of autotaxin (ATX; ectonucleotide pyrophosphatase/phosphodiesterase family member 2; ENPP2), with potential antifibrotic and antineoplastic activities. Upon oral administration, cambritaxestat targets and binds to both the substrate pocket and the lysophosphatidic acid (LPA) carrier channel of ATX, thereby inhibiting the activity of ATX. This both directly inhibits the proliferation of tumor cells and reduces fibrosis in the tumor microenvironment (TME), allowing lymphocytes to infiltrate into the tumor and enhancing immune responses against tumor cells. ATX, a secreted glycoprotein with lysophospholipase D activity, hydrolyzes lysophosphatidylcholine (LPC) to LPA. LPA-mediated signaling plays an important role in cellular migration, proliferation and survival in fibrotic response. ATX and LPA are overexpressed in many tumors.
- A Study to Assess an ATX Inhibitor (IOA-289) in Healthy VolunteersCTID: NCT05027568Phase: Phase 1Status: CompletedDate: 2025-03-20
- A Study to Assess an ATX Inhibitor (IOA-289) in Patients with Metastatic Pancreatic CancerCTID: NCT05586516Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-03-20
SYN
WO2016/124939
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016124939&_cid=P22-MKBYYZ-98558-1


SYN

WO2016/124939 describes various ATX inhibitor compounds and their use in the treatment of proliferative disorders in which ATX activity is implicated, including Compound 1.
Compound 1 is example 40 in WO2016/124939, which document is incorporated herein by reference in its entirety. WO2016/124939 describes over 200 examples. Compound 1’s structure is according to Formula I.

PAT
- Autotaxin inhibitory compoundsPublication Number: US-10654846-B2Priority Date: 2015-02-06Grant Date: 2020-05-19
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-B3Priority Date: 2015-02-06Grant Date: 2024-05-29
- Autotaxin inhibitor compoundsPublication Number: ES-2778898-T7Priority Date: 2015-02-06Grant Date: 2024-11-15
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-A1Priority Date: 2015-02-06
- Home chemokine inhibiting compoundsPublication Number: CN-107428752-BPriority Date: 2015-02-06Grant Date: 2021-06-29
- Autotaxin inhibitory compoundsPublication Number: US-11453666-B2Priority Date: 2015-02-06Grant Date: 2022-09-27
- Autotaxin Inhibitory CompundsPublication Number: US-2020283435-A1Priority Date: 2015-02-06
- Autotaxin inhibitory compoundsPublication Number: WO-2016124939-A1Priority Date: 2015-02-06
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: WO-2022207648-A1Priority Date: 2021-03-29
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: EP-4313059-A1Priority Date: 2021-03-29
- A pi3k-delta inhibitor for the treatment of pancreatic cancerPublication Number: US-2024216385-A1Priority Date: 2021-03-29
- Autotaxin inhibitory compoundsPublication Number: EP-3253737-B1Priority Date: 2015-02-06Grant Date: 2020-01-08
- Autotaxin inhibitory compoundsPublication Number: US-2018016274-A1Priority Date: 2015-02-06
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: WO-2022258693-A1Priority Date: 2021-06-09
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: US-2025057820-A1Priority Date: 2021-06-09
- Autotaxin (atx) inhibitor for the treatment of pancreatic cancerPublication Number: EP-4351563-A1Priority Date: 2021-06-09
- Autotaxin (ATX) inhibitors for the treatment of pancreatic cancerPublication Number: CN-117295496-APriority Date: 2021-06-09
- PI3K-δ inhibitors for the treatment of pancreatic cancerPublication Number: CN-116997340-APriority Date: 2021-03-29

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- Characterization and translational development of IOA-289, a novel autotaxin inhibitor for the treatment of solid tumorsPublication Name: Immuno-Oncology and TechnologyPublication Date: 2023-06PMCID: PMC10205783PMID: 37234285DOI: 10.1016/j.iotech.2023.100384
- The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacologyPublication Name: ImmunologyPublication Date: 2020-03-02PMCID: PMC7160657PMID: 32020584DOI: 10.1111/imm.13175
- Discovery of potent inhibitors of the lysophospholipase autotaxinPublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2016-11-15PMID: 27780639DOI: 10.1016/j.bmcl.2016.10.036
///////Cambritaxestat, autotaxin inhibitor, antineoplastic, Orphan Drug, IOA 289, IOA-289, IOA289, LYY3P2KA27, CRT 0273750
Tambiciclib
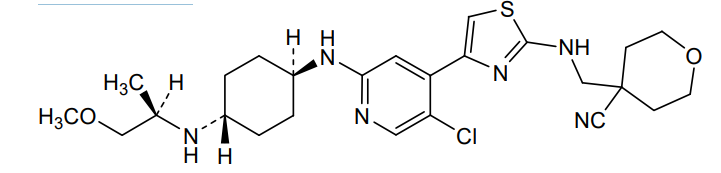
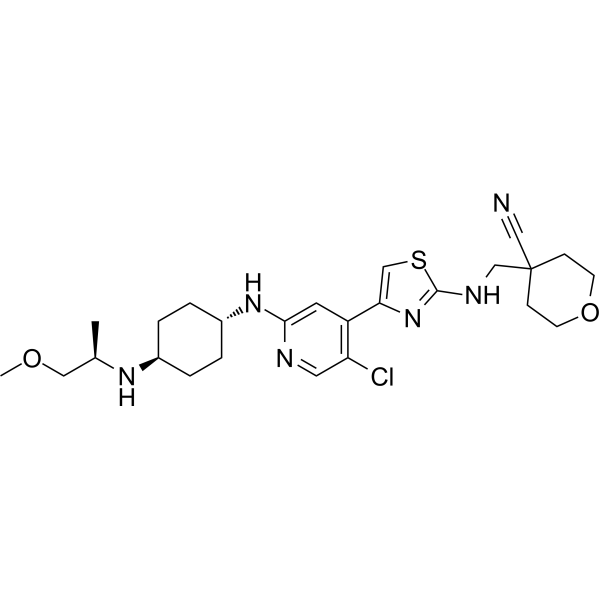
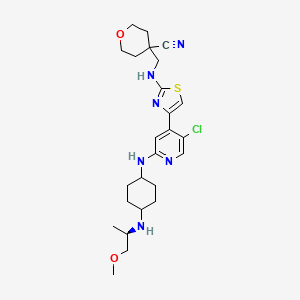
Tambiciclib
CAS 2247481-08-7
MF C25H35ClN6O2S, 519.10
4-[[[4-[5-chloro-2-[[4-[[(2R)-1-methoxypropan-2-yl]amino]cyclohexyl]amino]-4-pyridinyl]-1,3-thiazol-2-yl]amino]methyl]oxane-4-carbonitrile
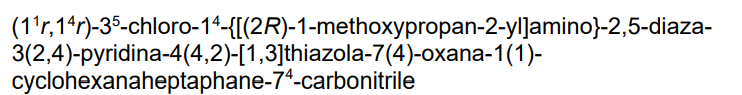
cyclin-dependent kinase inhibitor, antineoplastic, GFH 009, JSH 009, XDZ7VK8CXC, Orphan Drug , Acute myeloid leukaemia, Peripheral T-cell lymphoma
Tambiciclib (GFH009, JSH-009) is an orally active, highly potent and selective CDK9 inhibitor (IC50 = 1 nM), demonstrating >200-fold selectivity over other CDKs, >100-fold selectivity over DYRK1A/B, and excellent selectivity over 468 kinases/mutants. Tambiciclib demonstrates potent in vitro and in vivo antileukemic efficacy in acute myeloid leukemia (AML) mouse models by inhibiting RNA Pol II phosphorylation, downregulating MCL1 and MYC, and inducing apoptosis. Tambiciclib can be used for AML research.
Tambiciclib is a selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon administration, tambiciclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins. This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII). It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
- OriginatorGenFleet Therapeutics
- DeveloperGenFleet Therapeutics; Sellas Life Sciences Group
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase 9 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia; Peripheral T-cell lymphoma
- Phase IIAcute myeloid leukaemia
- Phase I/IIDiffuse large B cell lymphoma; Haematological malignancies; Peripheral T-cell lymphoma
- Phase ISolid tumours
- PreclinicalColorectal cancer; T-cell prolymphocytic leukaemia
- 13 Oct 2025Preclinical trials in T-cell prolymphocytic leukaemia (Combination therapy) in USA (Parenteral)
- 13 Oct 2025Preclinical trials in T-cell prolymphocytic leukaemia (Monotherapy) in USA (Parenteral)
- 13 Oct 2025Pharmacodynamics data from preclinical studies in T-cell prolymphocytic leukaemia released by SELLAS Life Sciences
CLINICAL
- A Study of GFH009 in Combination With Zanubrutinib in Subjects With Relapsed or Refractory DLBCLCTID: NCT06375733Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-08-12
- A Study of GFH009 Monotherapy in Patients with Relapsed or Refractory Peripheral T-cell Lymphoma (PTCL)CTID: NCT05934513Phase: Phase 1/Phase 2Status: RecruitingDate: 2024-12-13
Publication Name: European Journal of Medicinal Chemistry
Publication Date: 2018-10-05
PMID: 30253346
DOI: 10.1016/j.ejmech.2018.09.025
SYN
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018192273&_cid=P12-MJ18VV-17351-1
Example 1: Synthesis of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino) cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4- carboxynitrile
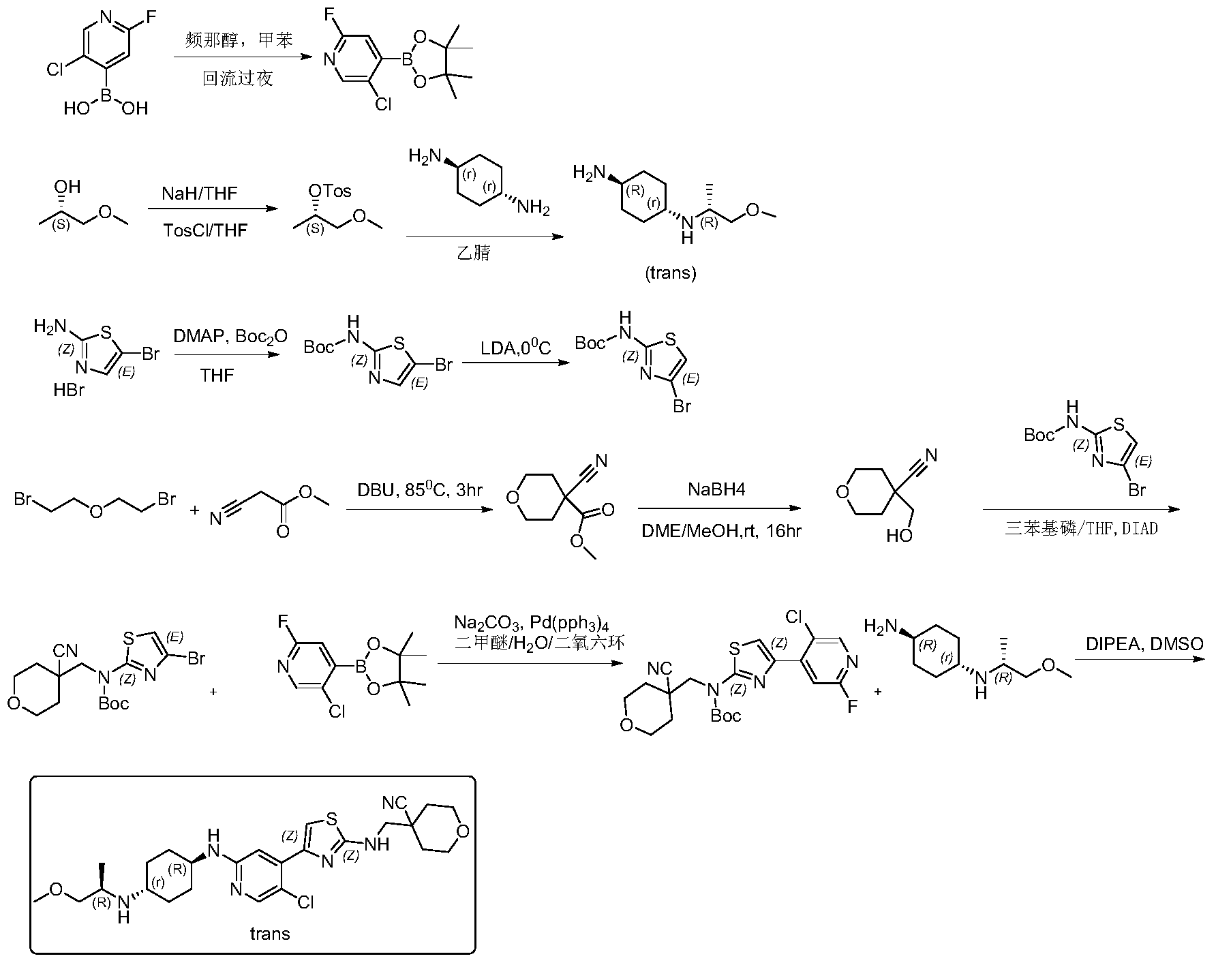
Step 1: Synthesis of 5-chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhecyclopentan-2-yl)pyridine
[0102]5-Chloro-2-fluoropyridine-4-boronic acid (0.7 g, 4.46 mmol) and pinacol (0.63 g, 5.35 mmol) were added to 50 mL of toluene, and the mixture was refluxed at 120 °C overnight. TLC showed a small amount of starting material remaining. The reaction mixture was cooled to room temperature and concentrated, then dried by an oil pump to give 0.92 g of a white solid compound, 5-chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhexacyclopentan-2-yl)pyridine, yield 80%, MS (ESI): m/z 258.1 (M+H) + .
[0103]Step 2: Synthesis of (S)-1-methoxypropyl-2-yl-4-toluenesulfonyl ester
[0104]60% sodium hydride (NaH) (6.52 g, 283 mmol) was added to anhydrous tetrahydrofuran (THF) (200 mL). The mixture was cooled to 0 °C in an ice bath under nitrogen protection, and (S)-(+)-1-methoxy-2-propanol (21 g, 233 mmol) was added dropwise. After the addition was complete, the mixture was brought to room temperature and stirred for 1.5 hours. The reaction mixture was then cooled back to 0 °C, and a tetrahydrofuran (THF) solution of p-toluenesulfonyl chloride (45.3 g, 283 mmol) (200 mL) was added dropwise. After the addition was complete, the mixture was stirred overnight at room temperature. TLC showed that the starting material had reacted completely. The reaction mixture was diluted with ethyl acetate (500 mL), and the reaction was quenched by adding water (500 mL) dropwise while cooling in an ice bath. The mixture was separated, and the aqueous phase was extracted once more with ethyl acetate (200 mL). The combined organic phases were washed with water (200 mL) and then with saturated brine (200 mL). The crude product was dried with anhydrous sodium sulfate, filtered, and concentrated to obtain 43 g of a pale yellow oily substance. Column separation (petroleum ether/ethyl acetate = 5/1) yielded 37 g of (S)-1-methoxypropyl-2-yl-4-toluenesulfonyl ester, a pale yellow oily substance, with a yield of 65.1%. MS (ESI): m/z 245.1 (M+H) + .
[0105]Step 3: Synthesis of (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine
[0106](S)-1-methoxypropyl-2-yl 4-toluenesulfonyl ester (5 g, 20.5 mmol) and trans-1,4-cyclohexanediamine (5.84 g, 51.2 mmol) were added to 50 mL of acetonitrile and heated to 90 °C overnight. The reaction was monitored by TLC until complete. After cooling, the reaction solution was filtered, the filtrate was concentrated, and the residue was dissolved in dichloromethane and separated by silica gel stirring column (dichloromethane/methanol = 10/1) to give 2.5 g of the pale yellow liquid compound (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine, yield 65%, MS (ESI): m/z 187.3 (M+H) + .
[0107]Step 4: Synthesis of tert-butyl 5-bromothiazol-2-ylcarbamate
[0108]105 g (403 mmol) of 5-bromothiazol-2-amine hydrobromide was suspended in 500 mL of tetrahydrofuran. Dimethylaminopyridine (2.41 g, 20 mmol) was added, resulting in a white turbidity. A tetrahydrofuran solution of di-tert-butyl dicarbonate (105.6 g, 484.6 mmol) was slowly added dropwise, and the reaction was allowed to proceed at room temperature for two days. The reaction solution was concentrated and dissolved in 300 mL of dichloromethane. The solution was mixed with silica gel and separated by column chromatography (petroleum ether/ethyl acetate = 10/1-6/1 gradient elution) to give 45 g of off-white solid, yield 40%. MS (ESI): m/z 278.98 (M+H) + .
[0109]Step 5: Synthesis of tert-butyl 4-bromothiazol-2-ylcarbamate
[0110]A 200 mL solution of diisopropylamine (64 mL, 446 mmol) in tetrahydrofuran was added to a dry three-necked flask. Under nitrogen protection, the mixture was cooled to 0 °C, and n-butyllithium (2.5 M, 173 mL, 431.7 mmol) was added dropwise. The reaction was allowed to proceed for 1 hour after the addition was complete. Then, a 400 mL solution of 5-bromothiazol-2-ylcarbamate in tetrahydrofuran was added dropwise at 0 °C. The reaction was allowed to proceed for 2 hours after the addition was complete. TLC showed that the reaction was complete. At 0℃, ice water (5 mL) was slowly added dropwise to quench the reaction. After stirring for 30 minutes, saturated ammonium chloride (500 mL) aqueous solution was added. The mixture was separated, and the aqueous layer was extracted with dichloromethane (2 × 300 mL). The organic layers were combined, washed with saturated brine, dried with anhydrous sodium sulfate, filtered, concentrated, and recrystallized from petroleum ether:ethyl acetate = 30:1. 31 g of tert-butyl 4-bromothiazol-2-ylcarbamate was obtained as a white solid, yield 77.5%. MS (ESI): m/z 278.98 (M+H) + .
[0111]Step Six: Synthesis of Methyl 4-cyano-tetrahydro-2H-pyran-4-carbonate
[0112]Methyl cyanoacetate (39.1 g, 395.3 mmol) and 2,2-dibromoethyl ether (100 g, 434.8 mmol) were added to 600 mL of dimethylformamide, followed by DBU (90 g, 593 mmol). The mixture was heated to 85 °C and reacted for 3 hours. TLC showed that the starting material reacted completely. The solid was filtered off, washed with ethyl acetate (2 × 300 mL), and the mother liquor was concentrated to obtain a brown oily substance. The oil was distilled under reduced pressure at an internal temperature of 65-70 °C, and the fraction collected was a colorless liquid. Crystallization was observed to give 42 g of a white solid, 4-cyano-tetrahydro-2H-pyran-4-carbonate. Yield: 62.8%, MS (ESI): m/z 178.2 (M+H) + .
[0113]Step 7: Synthesis of 4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile
[0114]4-Cyano-tetrahydro-2H-pyran-4-carbonate methyl ester (42 g, 248.4 mmol) was dissolved in 400 mL of ethylene glycol dimethyl ether and 40 mL of methanol. The mixture was cooled to 0 °C in an ice bath, and sodium borohydride (11.1 g, 149 mmol) was added in portions. After the addition was complete, the mixture was allowed to rise to room temperature and stirred for 16 hours. The reaction was completed by TLC. The reaction solution was concentrated, and methanol was added to quench excess sodium borohydride. The solution was then concentrated again. Column chromatography (petroleum ether/ethyl acetate = 5/1) yielded 28 g of 4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile, a pale yellow oil, yield: 79.5%, MS (ESI): m/z 142.1 (M+H) + .
[0115]Step 8: Synthesis of tert-butyl (4-bromothiazolyl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate
[0116]4-(hydroxymethyl)-tetrahydro-2H-pyran-4-carboxynitrile, 4-bromothiazol-2-ylcarbamate tert-butyl ester, and triphenylphosphine were added to anhydrous tetrahydrofuran (THF) and cooled to 0°C. Diisopropyl azodicarbonate (DIAD) was added dropwise. The mixture was stirred at room temperature for 10 minutes, then heated to 40°C overnight. The reaction solution was concentrated, and the residue was dissolved in dichloromethane. The solution was mixed with silica gel and separated by column chromatography (petroleum ether/ethyl acetate = 50/1, 30/1, 20/1) to obtain (4-bromothiazol-2-yl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate tert-butyl ester, a white solid of 365 mg, yield 50%. MS (ESI): m/z 402.1 (M+H) + .
[0117]Step Nine: Synthesis of tert-butyl (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate
[0118]5-Chloro-2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborhexacyclopentan-2-yl)pyridine and sodium carbonate were added to a mixture of dimethyl ether/H₂O
/ dioxane. The system was purged with nitrogen twice. Then, tert-butyl (4-bromothiazolyl)((4-cyanotetrahydro-2H-pyran-4-yl)methyl)carbamate and tetraphenylphosphine palladium Pd(pph 3 )
4 were added . The system was purged with nitrogen three times. The temperature was then raised to 70°C and the reaction was carried out for 6 hours. TLC showed that only half of the starting material remained. Heating was then stopped and the reaction was terminated. The reaction solution was cooled to room temperature, ethyl acetate and methanol were added, and the mixture was filtered. The filter cake was washed with ethyl acetate, the filtrate was concentrated, and the residue was dissolved in dichloromethane. The residue was washed with saturated brine, separated, and the organic phase was dried over anhydrous sodium sulfate. The mixture was filtered, and silica gel was added for mixing. The sample was separated by column chromatography (petroleum ether/ethyl acetate = 30/1) to give 3.2 g of (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate, a white foamy solid, with a yield of 55%. MS (ESI): m/z 453.1 (M+H) + .
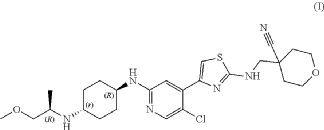
[0119]Step 10: Synthesis of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino)cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4-carboxynitrile
[0120]The tert-butyl carbamate (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate (3.2 g, 7.1 mmol) and (1r,4R)-N
1 -((R)-1-methoxypropyl-2-yl)cyclohexane-1,4-diamine (3.9 g, 21.2 mmol) and diisopropylethylamine (DIPEA) were added to 30 mL of dimethyl sulfoxide. Under nitrogen protection, the mixture was heated to 100-110 °C and reacted for two days. The reaction was monitored by TLC and LCMS. The starting material (4-(5-chloro-2-fluoropyridin-4-yl)thiazolyl)((4-cyano-tetrahydro-2H-pyran-4-yl)methyl)carbamate tert-butyl ester had completely disappeared, with some BOC-free intermediate remaining. The reaction was stopped, and the reaction solution was cooled and diluted with ethyl acetate (60 mL). Water (150 mL) was added under ice bath. The mixture was separated, and the aqueous layer was extracted again with ethyl acetate (2 × 50 mL). The organic layers were combined, washed with saturated brine (100 mL), dried with anhydrous sodium sulfate, filtered, and concentrated to obtain a crude product of yellowish-brown oil. Column separation (acetonitrile/water/trifluoroacetic acid = 80/20/0.001) yielded 700 mg of 4-(((4-(5-chloro-2-(((1R,4r)-4-(((R)-1-methoxypropyl-2-yl)amino)cyclohexyl)amino)pyridin-4-yl)thiazolyl)amino)methyl)tetrahydro-2H-pyran-4-carboxynitrile, a pale yellow solid. Yield: 19.1%. ¹H NMR (400 MHz, CDCl₃
) )δ8.06(s,1H),7.38(s,1H),6.97(s,1H),5.92(brs,1H),4.45(d,J=8.0Hz,1H),4.02(dd,J 1=2.8Hz, J2=12Hz,2H),3.71-3.74(m,4H),3.54-3.56(m,1H),3.35(s,3H),3.21-3.25(m,2 H),3.00-3.05(m,1H),2.50-2.60(m,1H),2.15(d,J=9.6Hz,2H),2.04-2.07(m,1H),1.95(d ,J=12.8Hz,3H),1.74-1.82(m,3H),1.10-1.30(m,4H),1.00(d,J=.4Hz,3H),MS(ESI):m/z 519.3(M+H) + .
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US376039987&_cid=P12-MJ18R0-12787-1
PAT
- A novel cyclin-dependent kinase CDK9 inhibitorPublication Number: CN-108727363-BPriority Date: 2017-04-19Grant Date: 2020-06-19
- Inhibitor of cyclin-dependent kinase CDK9Publication Number: US-10952999-B2Priority Date: 2017-04-19Grant Date: 2021-03-23
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: EP-3613737-B1Priority Date: 2017-04-19Grant Date: 2021-12-29
- Pharmaceutical combination and use thereof in treatment of cancerPublication Number: WO-2024239512-A1Priority Date: 2023-05-22
- Polymorph of cdk9 inhibitor and preparation method for polymorph and use thereofPublication Number: WO-2020244612-A1Priority Date: 2019-06-06
- Polymorphic substance of CDK9 inhibitor and preparation method and application thereofPublication Number: CN-113966332-APriority Date: 2019-06-06
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: EP-3613737-A1Priority Date: 2017-04-19
- Novel inhibitor of cyclin-dependent kinase cdk9Publication Number: US-2020078343-A1Priority Date: 2017-04-19
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//Tambiciclib, cyclin-dependent kinase inhibitor, antineoplastic, GFH 009, JSH 009, XDZ7VK8CXC, Orphan Drug , Acute myeloid leukaemia, Peripheral T-cell lymphoma
Privosegtor
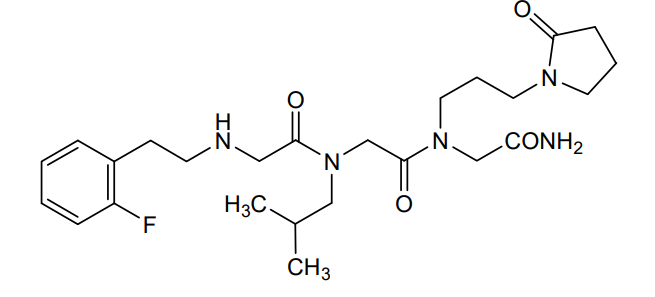
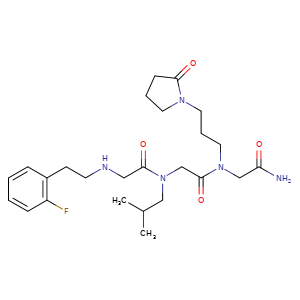
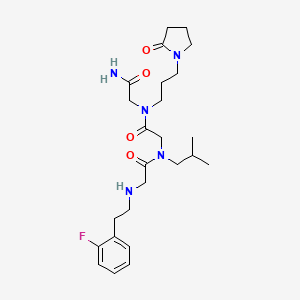
Privosegtor
CAS 1361200-34-1
MF C25H38FN5O4, MW 491.6 g/mol
GLYCINAMIDE, N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXO-1-PYRROLIDINYL)PROPYL)-
N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXO-1-PYRROLIDINYL)PROPYL)GLYCINAMIDE
N-(2-(2-FLUOROPHENYL)ETHYL)GLYCYL-N-(2-METHYLPROPYL)GLYCYL-N2-(3-(2-OXOPYRROLIDIN-1-YL)PROPYL)GLYCINAMIDE
N-[2-(2-fluorophenyl)ethyl]glycyl-N-(2-methylpropyl)glycyl-N2[3-(2-oxopyrrolidin-1-yl)propyl]glycinamide
serum/ glucocorticoid-regulated kinase 2 (Sgk2) activator, Phase 2, Optic neuritis, orphan drug, BN-201, BN 201, G-79, G 79, KCN37L7EIH
- OriginatorBionure
- DeveloperBionure; Oculis Pharma
- ClassAnti-inflammatories; Antiglaucomas; Eye disorder therapies; Neuroprotectants; Peptides; Small molecules
- Mechanism of ActionBrain derived neurotrophic factor agonists; Insulin-like growth factor I stimulants; Neuron modulators; Serum-glucocorticoid regulated kinase stimulants
- Orphan Drug StatusYes – Optic neuritis
- Phase IIOptic neuritis
- PreclinicalMultiple sclerosis; Neurotrophic keratopathy
- No development reportedGlaucoma; Neuromyelitis optica
- 06 Oct 2025Oculis Holding plans the PIONEER-2 trial in Optic neuritis in first half of 2026
- 06 Oct 2025Oculis Holding plans the PIONEER-3 trial in Optic nerve disorders in mid-2026
- 06 Oct 2025Oculis Holding completes End-of-phase II meeting with US FDA and receives positive feedback for registrational PIONEER program in Optic neuritis and Optic nerve disorders
OCS-05 in Patients With Optic Neuritis
CTID: NCT04762017
Phase: Phase 2
Status: Completed
Date: 2025-09-22
N-[2-[(2-amino-2-oxoethyl)-[3-(2-oxopyrrolidin-1-yl)propyl]amino]-2-oxoethyl]-2-[2-(2-fluorophenyl)ethylamino]-N-(2-methylpropyl)acetamide (BN201) is a small peptide molecule, a first-in-class neuroprotective compound. BN201 promotes the survival of cultured neural cells when subjected to oxidative stress or when deprived of trophic factors. BN201 promotes neuronal differentiation, the differentiation of precursor cells to mature oligodendrocytes in vitro, and the myelination of new axons. BN201 modulates several kinases participating in the insulin growth factor 1 pathway including serum-glucocorticoid kinase and midkine, inducing the phosphorylation of NDRG1 and the translocation of the transcription factor Foxo3 to the cytoplasm. In vivo, BN201 prevents axonal and neuronal loss, and it promotes remyelination in models of multiple sclerosis, chemically induced demyelination, and glaucoma. Bionure, a spin-off from Hospital Clínic de Barcelona that is based in California, is developing BN201 for multiple sclerosis, acute optic neuritis (AON) and glaucoma. BN201 was granted with orphan designation status for optic neuritis by the FDA. Optic neuritis is often an early sign of multiple sclerosis. The efficacy, safety, and capacity of the drug to cross the blood-brain barrier have been demonstrated in animal models, but the drug has not yet entered clinical testing.
PAT
Agonists of neurotrophin receptors and their use as medicaments
Publication Number: WO-2012028959-A1
Priority Date: 2010-08-31
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012028959&_cid=P10-MIDYQ0-58943-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021084013&_cid=P10-MIDYSN-60542-1
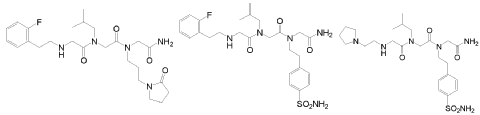
In another embodiment, optionally in combination with one or more features of the various embodiments described above or below throughout all the description, the compound of formula (I) is selected from the group consisting of G79 ([N-(2-(2′-fluorophenyl)ethyl)- glycyl]-[N-(2-methylpropyl)-glycyl]-N-[3-(2′-oxopyrrolidinyl)-propyl]glycinamide, BN201 , Chemical Formula: C25H38FN5O4; MW 491.5987), G-80 ([N-(2-(2′-fluorophenyl)ethyl)- glycyl]-[N-(2-methyl-propyl)glycyl]-N-[2-(4′-sulfamoyl-phenyl)ethyl]glycinamide, BN 119, Chemical Formula: C26H36FN5O5S; MW 549.658) and G81 ([N-(2-(1 -pyrrolidinyl)ethyl)- glycyl]-[N-(2-methyl-propyl)glycyl]-N-[2-(4′-sulfamoyl-phenyl)ethyl]glycinamide, BN 120, Chemical Formula: C24H4oN6OS; MW 524.6766):
G79 (BN201) G80 (BN119) G81 (BN120)
Compounds of formula (I) can be prepared as disclosed in WO2012028959.
PAT
- Agonists of Neurotrophin Receptors and Their Use as MedicamentsPublication Number: US-2012052094-A1Priority Date: 2010-08-31
- Agonists of Neurotrophin Receptors and Their Use as MedicamentsPublication Number: US-2015005239-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-2017121367-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-8791076-B2Priority Date: 2010-08-31Grant Date: 2014-07-29
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-9453047-B2Priority Date: 2010-08-31Grant Date: 2016-09-27
- Combination Therapy Methods, Compositions and KitsPublication Number: KR-20220109378-APriority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: US-2022378866-A1Priority Date: 2019-07-03
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: EP-2611775-A1Priority Date: 2010-08-31
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: EP-2611775-B1Priority Date: 2010-08-31Grant Date: 2016-03-16
- Agonists of neurotrophin receptors and their use as medicamentsPublication Number: US-10106577-B2Priority Date: 2010-08-31Grant Date: 2018-10-23
- Combination therapy methods, compositions and kitsPublication Number: WO-2021001464-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: AU-2020298782-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: CN-114206329-APriority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: EP-3993784-A1Priority Date: 2019-07-03
- Combination therapy methods, compositions and kitsPublication Number: JP-2022539999-APriority Date: 2019-07-03
- Boron-nitrogen compound, organic electroluminescence composition, and organic electroluminescence device containing samePublication Number: WO-2022121951-A1Priority Date: 2020-12-10
- New treatment regimen for the treatment of neurological diseases or conditionsPublication Number: WO-2021084013-A1Priority Date: 2019-10-30
- Novel Therapeutic Approaches for the Treatment of Neurological Diseases or ConditionsPublication Number: CN-115052595-APriority Date: 2019-10-30
- New treatment regimen for the treatment of neurological diseases or conditionsPublication Number: EP-4051263-A1Priority Date: 2019-10-30
- New treatment regiment for the treatment of neurological diseases or conditionsPublication Number: US-2022387385-A1Priority Date: 2019-10-30
- A plant zinc-increasing compound inoculant and its preparation method and applicationPublication Number: CN-117286034-APriority Date: 2023-09-11
- A plant zinc-enhancing composite bacterial agent and its preparation method and applicationPublication Number: CN-117286034-BPriority Date: 2023-09-11Grant Date: 2024-11-15
- Compound, pharmaceutical composition comprising the same, and process for synthesizing the samePublication Number: TW-202432095-APriority Date: 2022-12-22
- Synthesis of small molecule agonists of neuroptrophinPublication Number: WO-2024133860-A1Priority Date: 2022-12-22
- Boron-nitrogen compound, organic electroluminescent composition and organic electroluminescent device containing samePublication Number: WO-2022121920-A1Priority Date: 2020-12-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- Development and validation of PAMPA-BBB QSAR model to predict brain penetration potential of novel drug candidatesPublication Name: Frontiers in PharmacologyPublication Date: 2023-12-01PMCID: PMC10722238PMID: 38108064DOI: 10.3389/fphar.2023.1291246
- A Phase 1 randomized study on the safety and pharmacokinetics of OCS-05, a neuroprotective disease modifying treatment for Acute Optic Neuritis and Multiple SclerosisPublication Name: Scientific ReportsPublication Date: 2023-03-29PMCID: PMC10060579PMID: 36991169DOI: 10.1038/s41598-023-32278-0
- Retrospective assessment of rat liver microsomal stability at NCATS: data and QSAR modelsPublication Name: Scientific ReportsPublication Date: 2020-11-26PMCID: PMC7693334PMID: 33244000DOI: 10.1038/s41598-020-77327-0
- A High-Throughput Screen of a Library of Therapeutics Identifies Cytotoxic Substrates of P-glycoproteinPublication Name: Molecular PharmacologyPublication Date: 2019-11PMCID: PMC6790066PMID: 31515284DOI: 10.1124/mol.119.115964
- Predictive models of aqueous solubility of organic compounds built on A large dataset of high integrityPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2019-07-15PMCID: PMC8274818PMID: 31176566DOI: 10.1016/j.bmc.2019.05.037
/////////Privosegtor, Phase 2, Optic neuritis, orphan drug, BN-201, BN 201, G-79, G 79, KCN37L7EIH
Nuvisertib
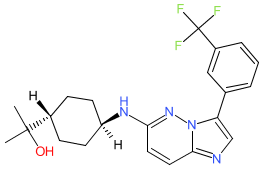
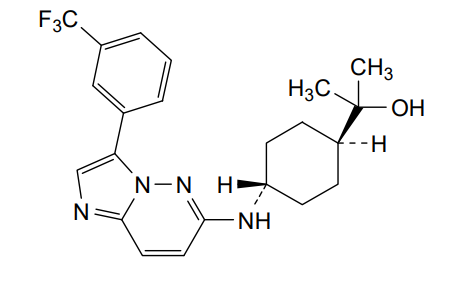

Nuvisertib
CAS 1361951-15-6
MF C22H26ClF3N4O MW418.5 g/mol
2-[(1r,4r)-4-({3-[3-(trifluoromethyl)phenyl]imidazo[1,2-b]pyridazin-6-yl}amino)cyclohexyl]propan-2-ol
serine/ threonine kinase inhibitor, antineoplastic, Orphan Drug, myelofibrosis, SGI-9481, SGI 9481, TP-3654, TP 3654, EOB0N7BOY4
The chemical structure for nuvisertib was obtained from proposed INN list 130 (Feb. 2024), in which the compound is described as a serine/ threonine kinase inhibitor with antineoplastic action. A structure match to clinical lead TP-3654 was made via PubChem. TP-3654 is declared as an orally available, second-generation pan-PIM kinase inhibitor [1-2].
| References |
| 1. Foulks JM, Carpenter KJ, Luo B, Xu Y, Senina A, Nix R, Chan A, Clifford A, Wilkes M, Vollmer D et al.. (2014) A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia, 16 (5): 403-12. [PMID:24953177] |
| 2. Wu CP, Li YQ, Chi YC, Huang YH, Hung TH, Wu YS. (2021) The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer Drugs. Int J Mol Sci, 22 (17). [PMID:34502348] |
Nuvisertib is an orally available, second-generation and selective ATP-competitive inhibitor of proviral integration site for Moloney murine leukemia virus (PIM) kinases, with potential antineoplastic activity. Upon oral administration, nuvisertib selectively binds to and prevents the activation of the PIM kinases. This prevents the activation of PIM-mediated signaling pathways and inhibits proliferation in cells that overexpress PIM. PIMs, constitutively active proto-oncogenic serine/threonine kinases, are upregulated in various types of cancers and play key roles in tumor cell proliferation and survival.
Nuvisertib, also known as TP-3654, is an oral, investigational, and highly selective PIM1 kinase inhibitor being studied in a Phase 1/2 clinical trial for intermediate- or high-risk myelofibrosis (MF). It is not currently an approved medication.
Key Information
- Mechanism of Action: Nuvisertib targets the PIM1 kinase pathway, which is often overactive in myelofibrosis and can promote cancer cell growth. By inhibiting this pathway, nuvisertib is being investigated for its potential to manage symptoms, reduce spleen size, improve blood counts, and slow the progression of bone marrow fibrosis.
- Current Status: Nuvisertib is in ongoing Phase 1/2 clinical trials (NCT04176198) as a monotherapy and in combination with JAK inhibitors like ruxolitinib and momelotinib.
- Designations: Nuvisertib has received Orphan Drug Designation for myelofibrosis
Study of TP-3654 in Patients With Advanced Solid Tumors
CTID: NCT03715504
Phase: Phase 1
Status: Completed
Date: 2023-11-14
SYN
WO2013013188
Example 31
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US427659372&_cid=P10-MHWTVL-76212-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US130491286&_cid=P10-MHWU33-81462-1
31. 4-((3-(3-(Trifluoromethyl)phenyl)imidazo[1,2-b]pyridazin-6-yl)amino)-trans-cyclohexyl)propan-2-ol (EX. 8-31)
| EX. 8-31 was prepared by similar procedures as in EX. 8-1 using 2-(trans-4-aminocyclohexyl)propan-2-ol. |

| 1H-NMR (CD 3OD/400 MHz): δ 8.82 (s, 1H), 8.19 (m, 1H), 7.88 (s, 1H), 7.62 (m, 3H), 6.70 (d, J=9.6 Hz, 1H), 3.71 (m, 1H), 2.26 (m, 2H), 1.95 (m, 2H), 1.36 (m, 1H), 1.27 (m, 4H), 1.21 (s, 6H). MS (ES +, m/z): (M+H) +: 419.6. |
| To a solution of trans-4-((tert-butoxycarbonyl)amino)cyclohexanecarboxylic acid (823 g, 3.38 mol) in EtOAc (4000 mL) was added EA/HCl (2500 mL). The mixture was stirred at 0° C. overnight. The reaction mixture was filtered and dried in vacuo to give a product of hydrochloride salt of trans-4-aminocyclohexanecarboxylic acid as white solid (604 g, 99.42% yield). |
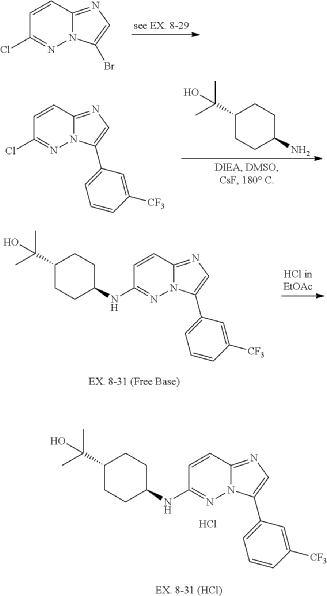
| 6-chloro-3-(3-(trifluoromethyl)phenyl)imidazo[1,2-b]pyridazine was prepared according to procedure in EX. 8-29. |
PAT
- Heterocyclic protein kinase inhibitorsPublication Number: ES-2834093-T3Priority Date: 2011-07-21Grant Date: 2021-06-16
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2021238183-A1Priority Date: 2011-07-21
- Imidazo[1,2-b]pyridazine and pyrazolo[1,5-a]pyrimidine derivatives and their use as protein kinase inhibitorsPublication Number: US-2012058997-A1Priority Date: 2006-11-06
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-9416132-B2Priority Date: 2011-07-21Grant Date: 2016-08-16
- Heterocyclic protein kinase inhibitorsPublication Number: WO-2013013188-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3409278-B1Priority Date: 2011-07-21Grant Date: 2020-09-16
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10875864-B2Priority Date: 2011-07-21Grant Date: 2020-12-29
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3812387-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10392392-B2Priority Date: 2011-07-21Grant Date: 2019-08-27
- Heterocyclic protein kinase inhibitorsPublication Number: US-2014329807-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2017002014-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2019071446-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2020102313-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-2734205-B1Priority Date: 2011-07-21Grant Date: 2018-03-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3409278-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: JP-2014520898-APriority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: JP-6105578-B2Priority Date: 2011-07-21Grant Date: 2017-03-29
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10047093-B2Priority Date: 2011-07-21Grant Date: 2018-08-14
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
REF
– Nuvisertib (TP-3654), an investigational highly selective oral PIM1 kinase inhibitor, is being evaluated in patients with relapsed or refractory myelofibrosis (MF) –
– Nuvisertib demonstrated symptom and spleen responses correlating with cytokine modulation in the preliminary Phase 1/2 data recently presented at the European Hematology Association (EHA) 2025 Congress –
MARLBOROUGH, Mass., June 12, 2025 /PRNewswire/ — Sumitomo Pharma America, Inc. (SMPA) today announced that the U.S. Food and Drug Administration (FDA) granted Fast Track Designation to nuvisertib (TP-3654) for the treatment of patients with intermediate or high-risk myelofibrosis (MF). The FDA Fast Track Designation is granted to investigational therapies being developed to treat serious or life-threatening conditions that demonstrate the potential to address unmet medical needs. Nuvisertib is an oral, investigational, highly selective inhibitor of PIM1 kinase, which demonstrated clinical activity including symptom and spleen responses correlating with cytokine modulation in the updated preliminary Phase 1/2 data presented at the European Hematology Association (EHA) 2025 Congress in Milan, Italy.
MF, a serious and rare type of blood cancer, is characterized by the buildup of fibrous tissues in the bone marrow which is caused by dysregulation in the Janus-associated kinase (JAK) signaling pathway. The clinical manifestations of MF include an enlarged spleen, debilitating symptoms and reduction in hemoglobin and/or platelets. MF affects 1 in 500,000 people worldwide.1
“This positive momentum for nuvisertib signals strong promise in our pipeline and reflects our dedication to addressing unmet medical needs on behalf of patients with myelofibrosis and their families,” said Tsutomu Nakagawa, Ph.D, President and Chief Executive Officer of SMPA. “Receiving FDA Fast Track Designation for nuvisertib in the treatment of myelofibrosis reinforces our confidence in its potential as a treatment option for patients facing a poor prognosis with limited treatment options. We are committed to working closely with the FDA to progress the clinical development of nuvisertib and bring an alternative treatment option to patients with myelofibrosis.”
Updated data from the ongoing Phase 1/2 study of nuvisertib in patients with relapsed/refractory MF were presented at the EHA Congress on June 12, 2025. Preliminary data showed that nuvisertib monotherapy appears to be well tolerated with no dose-limiting toxicities (DLTs). Evaluable patients showed clinical activity including a ≥25% spleen volume reduction (SVR25) in 22.2% of patients and a ≥50% reduction in total symptom score (TSS50) of 44.4% of patients, as well as improvement of bone marrow fibrosis (42.9% patients), hemoglobin (24% patients) and platelet count (26.7% patients). Data also showed that nuvisertib treatment led to significant cytokine modulation [reduction of pro-inflammatory cytokines (e.g. EN-RAGE, MIP-1β) and increase of anti-inflammatory cytokines (e.g. adiponectin)], which demonstrated significant (p<0.001) correlation with symptom and spleen responses. Preclinical2 and emerging clinical data support the development of nuvisertib in combination with JAK inhibitors for the treatment of patients with MF.
“The data observed to date demonstrate promising clinical activity for nuvisertib and the strong potential for selective PIM1 inhibition to slow the progression of myelofibrosis,” said Jatin Shah, MD, Chief Medical Officer, Oncology. “Patients with myelofibrosis are in need of new therapeutic approaches, including combination treatment options, that can provide increased and durable response rates with limited hematologic adverse events. The FDA Fast Track Designation reinforces the potential of nuvisertib to provide clinical benefits for patients with myelofibrosis, an unmet medical need.”
About Nuvisertib (TP-3654)
Nuvisertib (TP-3654) is an oral investigational selective inhibitor of PIM1 kinase, which has shown potential antitumor and antifibrotic activity through multiple pathways, including induction of apoptosis in preclinical models.2,3 Nuvisertib was observed to inhibit proliferation and increase apoptosis in murine and human hematopoietic cells expressing the clinically relevant JAK2 V617F mutation.3 Nuvisertib alone and in combination with ruxolitinib showed white blood cell and neutrophil count normalization, and also reduced spleen size and bone marrow fibrosis in JAK2 V617F and MPLW515L murine models of myelofibrosis.2 The safety and efficacy of nuvisertib is currently being clinically evaluated in a Phase 1/2 study in patients with intermediate and high-risk myelofibrosis (NCT04176198). The U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation to nuvisertib for the indication of myelofibrosis in May 2022. The Japan Ministry of Health, Labour and Welfare (MHLW) granted Orphan Drug Designation to nuvisertib for the treatment of myelofibrosis in November 2024.
About Sumitomo Pharma
Sumitomo Pharma Co., Ltd., is a global pharmaceutical company based in Japan with key operations in the U.S. (Sumitomo Pharma America, Inc.), Canada (Sumitomo Pharma Canada, Inc.), and Europe (Sumitomo Pharma Switzerland GmbH) focused on addressing patient needs in oncology, urology, women’s health, rare diseases, psychiatry & neurology, and cell & gene therapies. With several marketed products in the U.S., Canada, and Europe, a diverse pipeline of early- to late-stage assets, we aim to accelerate discovery, research, and development to bring novel therapies to patients sooner. For more information on SMPA, visit our website https://www.us.sumitomo-pharma.com or follow us on LinkedIn.
The Sumitomo corporate symbol mark is a trademark of Sumitomo Pharma Co., Ltd., used under license. SUMITOMO PHARMA is a trademark of Sumitomo Pharma Co., Ltd., used under license. SUMITOMO is a registered trademark of Sumitomo Chemical Co., Ltd., used under license. Sumitomo Pharma America, Inc. is a U.S. subsidiary of Sumitomo Pharma Co., Ltd.
©2025 Sumitomo Pharma America, Inc. All rights reserved.
References
- U.S. National Library of Medicine. (n.d.). Primary myelofibrosis: Medlineplus Genetics. MedlinePlus. https://medlineplus.gov/genetics/condition/primary-myelofibrosis/
- Dutta A., Nath D, Yang Y, et al. Genetic ablation of Pim1 or pharmacologic inhibition with TP-3654 ameliorates myelofibrosis in murine models. Leukemia. 2022; 36 (3): 746-759. doi: 10.1038/s41375-021-01464-2.
- Foulks JM, Carpenter KJ, Luo B, et al. A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia. 2014;16(5):403-412.
SOURCE Sumitomo Pharma America
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer DrugsPublication Name: International Journal of Molecular SciencesPublication Date: 2021-08-30PMCID: PMC8431370PMID: 34502348DOI: 10.3390/ijms22179440
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- PIM kinase inhibitors: Structural and pharmacological perspectivesPublication Name: European Journal of Medicinal ChemistryPublication Date: 2019-06-15PMID: 30954777DOI: 10.1016/j.ejmech.2019.03.050
- A Small-Molecule Inhibitor of PIM Kinases as a Potential Treatment for Urothelial CarcinomasPublication Name: Neoplasia (New York, N.Y.)Publication Date: 2014-05PMCID: PMC4198696PMID: 24953177DOI: 10.1016/j.neo.2014.05.004
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer DrugsPublication Name: International Journal of Molecular SciencesPublication Date: 2021-08-30PMCID: PMC8431370PMID: 34502348DOI: 10.3390/ijms22179440
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- PIM kinase inhibitors: Structural and pharmacological perspectivesPublication Name: European Journal of Medicinal ChemistryPublication Date: 2019-06-15PMID: 30954777DOI: 10.1016/j.ejmech.2019.03.050
- A Small-Molecule Inhibitor of PIM Kinases as a Potential Treatment for Urothelial CarcinomasPublication Name: Neoplasia (New York, N.Y.)Publication Date: 2014-05PMCID: PMC4198696PMID: 24953177DOI: 10.1016/j.neo.2014.05.004
///////Nuvisertib, serine/ threonine kinase inhibitor, antineoplastic, Orphan Drug, myelofibrosis, SGI-9481, SGI 9481, TP-3654, TP 3654, EOB0N7BOY4
Lirodegimod
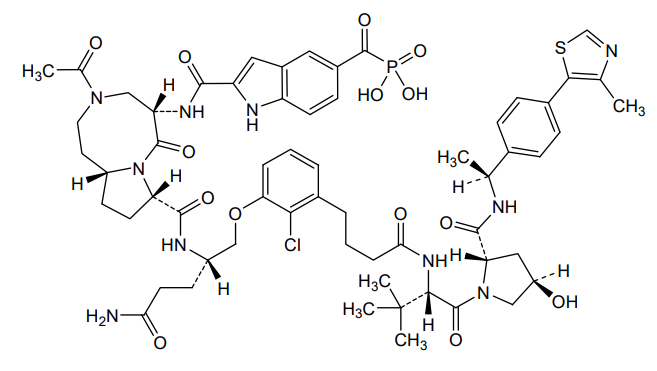
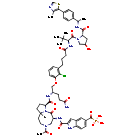
Lirodegimod
CAS 2502186-79-8
MF C60H74ClN10O14PS, MW 1257.79

[2-[[(5S,8S,10aR)-3-acetyl-8-[[(2S)-5-amino-1-[2-chloro-3-[4-[[(2S)-1-[(2S,4R)-4-hydroxy-2-[[(1S)-1-[4-(4-methyl-1,3-thiazol-5-yl)phenyl]ethyl]carbamoyl]pyrrolidin-1-yl]-3,3-dimethyl-1-oxobutan-2-yl]amino]-4-oxobutyl]phenoxy]-5-oxopentan-2-yl]carbamoyl]-6-oxo-1,2,4,5,8,9,10,10a-octahydropyrrolo[1,2-a][1,5]diazocin-5-yl]carbamoyl]-1H-indole-5-carbonyl]phosphonic acid
KT 333, KT333, ANTINEOPLASTIC, Fast Track (United States), Orphan Drug (United States), 4Q6ZHJ2MNA
Lirodegimod is a small molecule drug. The usage of the INN stem ‘-imod’ in the name indicates that Lirodegimod is a immunomodulator, both stimulant/suppressive and stimulant. Lirodegimod has a monoisotopic molecular weight of 1256.45 Da.
Safety, PK, PD, Clinical Activity of KT-333 in Adult Patients With Refractory Lymphoma, Large Granular Lymphocytic Leukemia, Solid Tumors
CTID: NCT05225584
Phase: Phase 1
Status: Completed
Date: 2025-03-19
PAT
- Stat3 degraders and uses thereofPublication Number: US-2023212201-A1Priority Date: 2021-12-11
- Stat3 degraders and uses thereofPublication Number: US-2025019388-A1Priority Date: 2021-12-11
- Stat degraders and uses thereofPublication Number: US-2024016942-A1Priority Date: 2020-03-17
- Stat degraders and uses thereofPublication Number: WO-2020206424-A1Priority Date: 2019-04-05
- Stat degraders and uses thereofPublication Number: US-11746120-B2Priority Date: 2019-04-05Grant Date: 2023-09-05
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Lirodegimod, KT 333, KT333, ANTINEOPLASTIC, Fast Track, Orphan Drug, 4Q6ZHJ2MNA
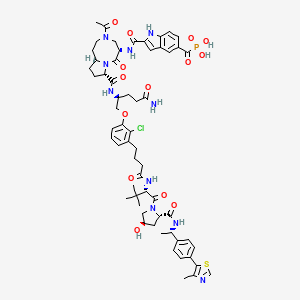
Gildeuretinol
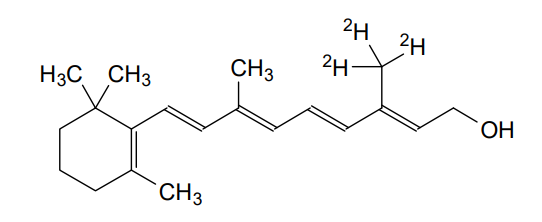
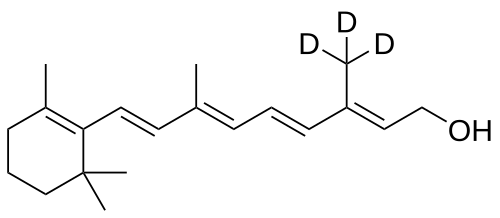
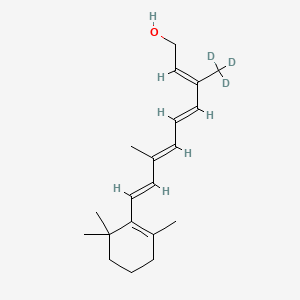
Gildeuretinol
CAS118139-35-8
MF C20H272H3O, MW 289.5 g/mol
(2E,4E,6E,8E)-3-(2H3)methyl-7-methyl-9-(2,6,6-trimethylcyclohex-1-en-1-yl)nona-2,4,6,8-tetraen-1-ol; (20,20,20-2H3)retinol
(2E,4E,6E,8E)-7-methyl-3-(trideuteriomethyl)-9-(2,6,6-trimethylcyclohexen-1-yl)nona-2,4,6,8-tetraen-1-ol
vitamin A analogue, Orphan Drug, Stargardt disease, breakthrough therapy, Pediatric Rare Disease designations, ALK-001, KL-49, ALK 001, KL 49
- OriginatorColumbia University
- DeveloperAlkeus Pharmaceuticals
- ClassEye disorder therapies; Retinoids; Vitamins
- Mechanism of ActionDimerisation inhibitors; Vitamin A replacements
- Orphan Drug StatusYes – Stargardt disease
- Phase II/IIIDry age-related macular degeneration
- Phase IIStargardt disease
- No development reportedRetinal dystrophies
- 08 Sep 2025Gildeuretinol – Alkeus Pharmaceuticals receives Orphan Drug status for Stargardt disease in European Union
- 09 Jan 2025Alkeus Pharmaceuticals announces intention to submit an NDA to US FDA for Stargardt disease in 2025
- 09 Jan 2025Efficacy and adverse event data from phase II trial for Stargardt disease released by Alkeus Pharmaceuticals
Gildeuretinol is an investigational new drug being developed by Alkeus Pharmaceuticals, Inc. for the treatment of retinal diseases, particularly Stargardt disease and geographic atrophy secondary to age-related macular degeneration (AMD). Stargardt disease is caused by a defect in the ABCA4 gene that clears toxic byproducts resulting from the dimerization of vitamin A. Gildeuretinol is new molecular entity designed to reduce the dimerization of vitamin A in the eye without affecting the visual cycle.[1]
Gildeuretinol has received breakthrough therapy, orphan drug and Pediatric Rare Disease designations from the U.S. Food and Drug Administration.[2]

AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Zaydon YA, Tsang SH (July 2024). “The ABCs of Stargardt disease: the latest advances in precision medicine”. Cell & Bioscience. 14 (1) 98. doi:10.1186/s13578-024-01272-y. PMC 11282698. PMID 39060921.
- Fitch J (22 November 2024). “Gildeuretinol for Stargardt disease receives Rare Pediatric Disease, Fast Track Designations”. Contemporary Pediatrics.
| Clinical data | |
|---|---|
| Other names | ALK-001, KL-49 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 118139-35-8 |
| PubChem CID | 169490774 |
| UNII | PSZ7W5NR24 |
| KEGG | D12713 |
| ChEMBL | ChEMBL5314606 |
| Chemical and physical data | |
| Formula | C20H30D3O |
| Molar mass | 292.500 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Gildeuretinol, vitamin A analogue, Orphan Drug, Stargardt disease, breakthrough therapy, Pediatric Rare Disease designations, ALK-001, KL-49, ALK 001, KL 49, PSZ7W5NR24
Tebapivat
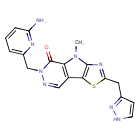
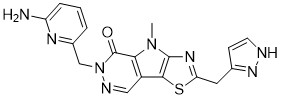
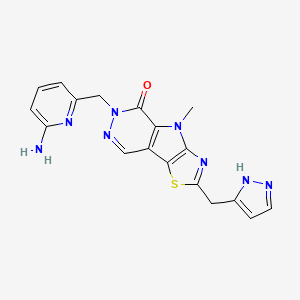
Tebapivat
CAS 2283422-04-6
WeightAverage: 392.44
Monoisotopic: 392.116778341
Chemical FormulaC18H16N8OS
10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
6-[(6-aminopyridin-2-yl)methyl]-4-methyl-2-[(1H-pyrazol-3-yl)methyl]-4,6-dihydro-5H-[1,3]thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5-one
- AG946
- CS-0115951
- HY-135884
- ORG4KGP5ZS
- OriginatorAgios Pharmaceuticals
- ClassAntianaemics; Small molecules
- Mechanism of ActionPyruvate kinase stimulants
- Orphan Drug StatusYes – Myelodysplastic syndromes
- Phase IIAnaemia; Sickle cell anaemia
- 01 May 2025Phase-II clinical trials in Sickle cell anaemia in USA (PO) (NCT06924970)
- 01 May 2025Agios plans to initiate a phase II clinical trial for Sickle cell disease(PO) in mid-2025.
- 21 Feb 2025Agios Pharmaceuticals completes a phase I bioavailability trial (In volunteers) in USA (PO, capsule) (NCT06745271)
Tebapivat is under investigation in clinical trial NCT05490446 (A Study of Tebapivat (AG-946) in Participants With Anemia Due to Lower-risk Myelodysplastic Syndromes (LR-MDS)).
Tebapivat is an orally available activator of the red cell isoform of pyruvate kinase (PK-R; PKR), with potential to improve hemolytic anemia and related-symptoms in patients with pyruvate kinase deficiency (PKD). Upon oral administration, tebapivat binds to and activates PKR, thereby enhancing glycolytic pathway activity in red blood cells (RBCs), improving adenosine triphosphate (ATP) levels and reducing 2,3-diphosphoglycerate (2,3-DPG) levels. This may result in increased oxygen affinity, improved RBC deformability, decreased sickle RBC hemolysis, increased hemoglobin (Hb) levels and improved RBC membrane function. Mutations in PKR cause deficiency in PKR which prevents adequate RBC glycolysis, leading to a build-up of the upstream glycolytic intermediate 2,3-DPG and deficiency in the PKR product ATP.
SCHEME
COUPLER
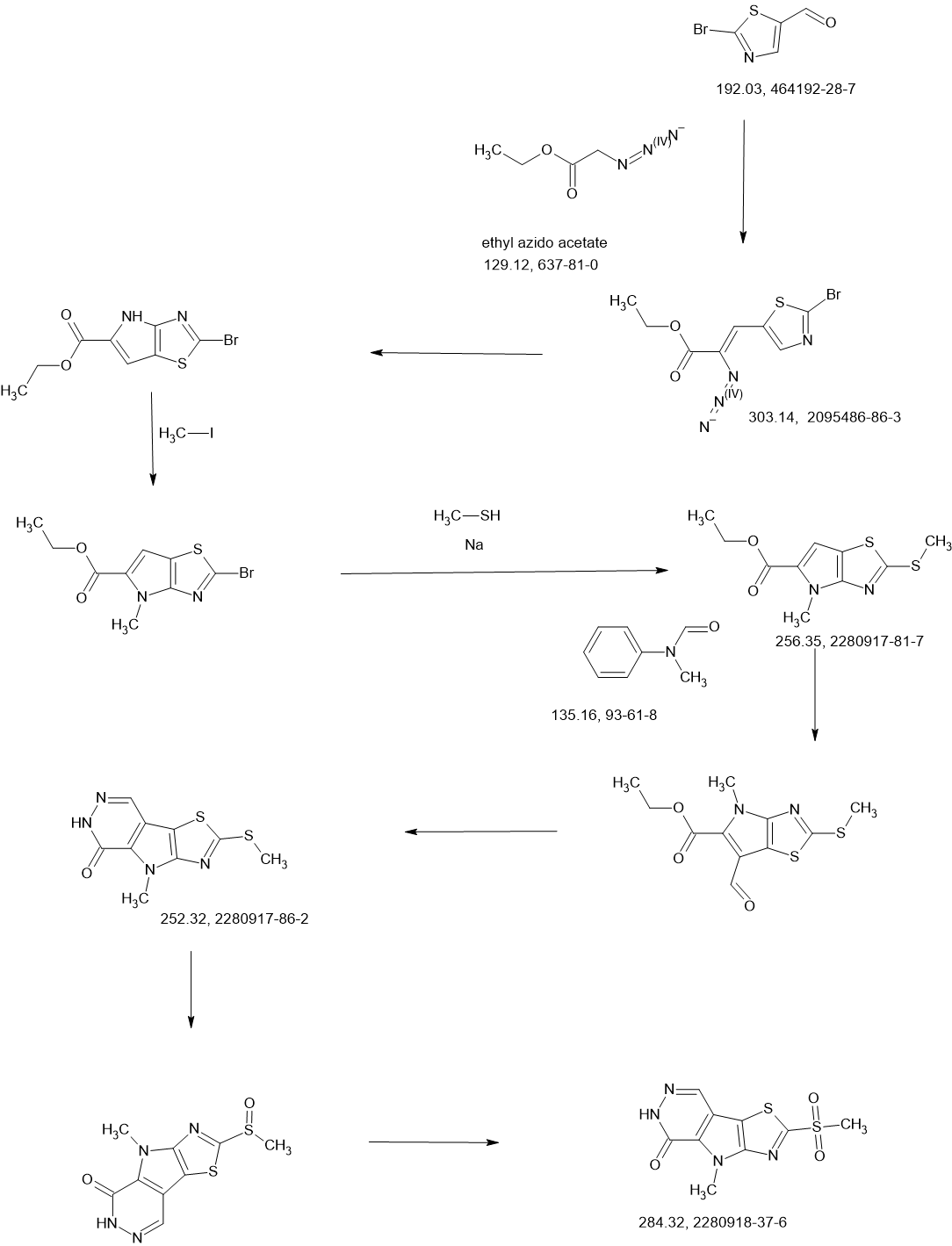
COUPLER
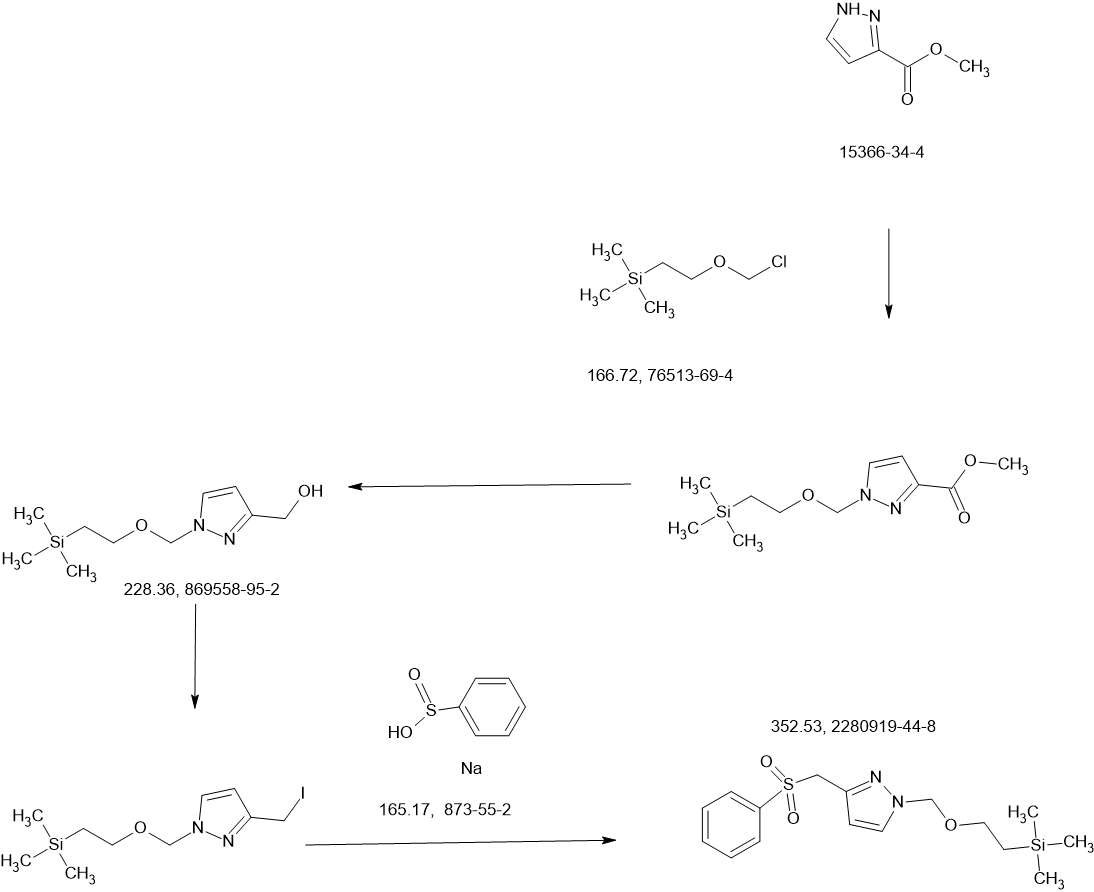
MAIN
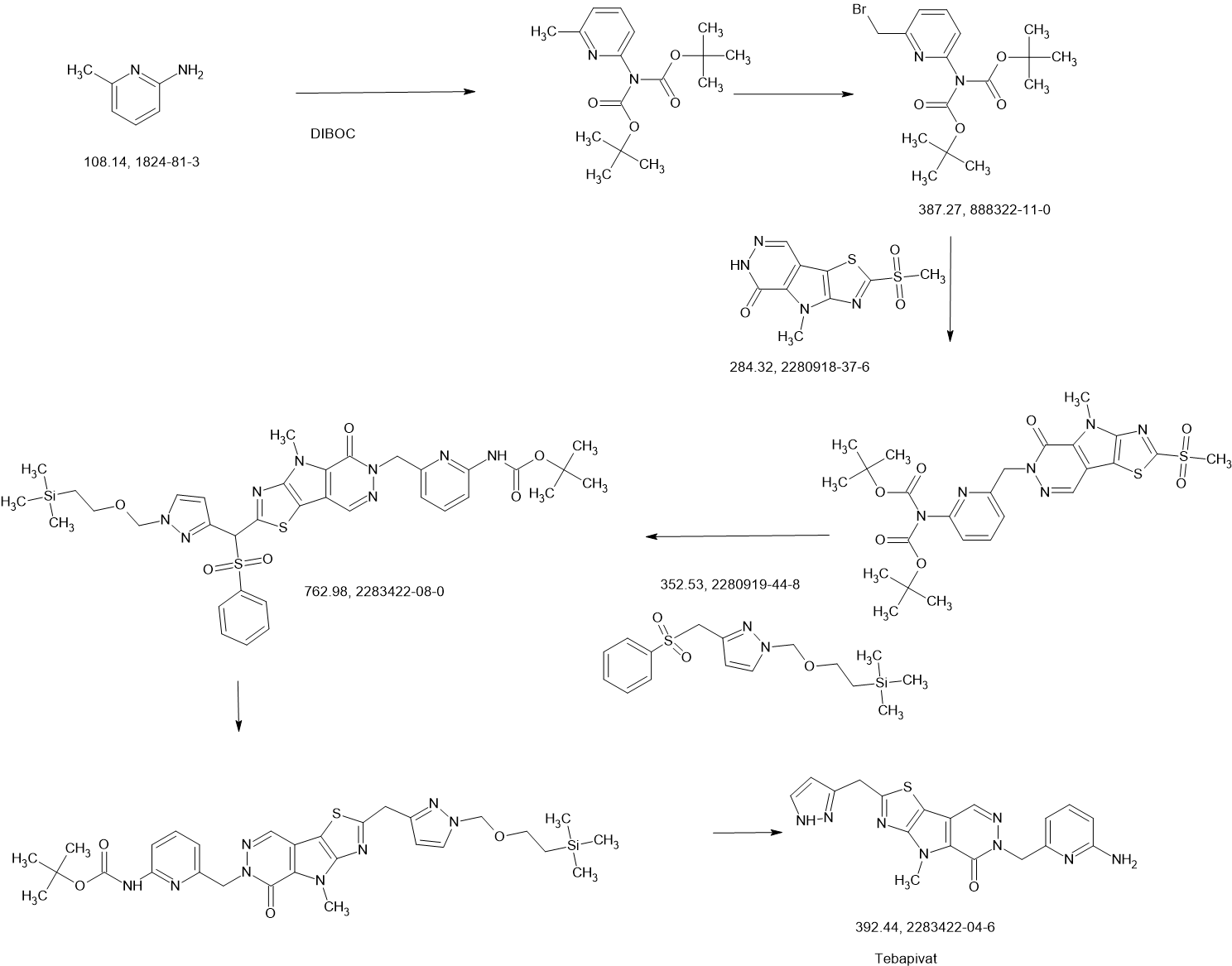
PATENT
Agios Pharmaceuticals, Inc.
WO2019035864
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019035864&_cid=P22-MDGSEF-03229-1
Example 8A. Synthesis of 2-((1H-pyrazol-3-yl)methyl)-6-((6-aminopyridin-2-yl)methyl)- 4-methyl-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one and 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrroIo[2,3-d]pyridazin-5(6H)-one
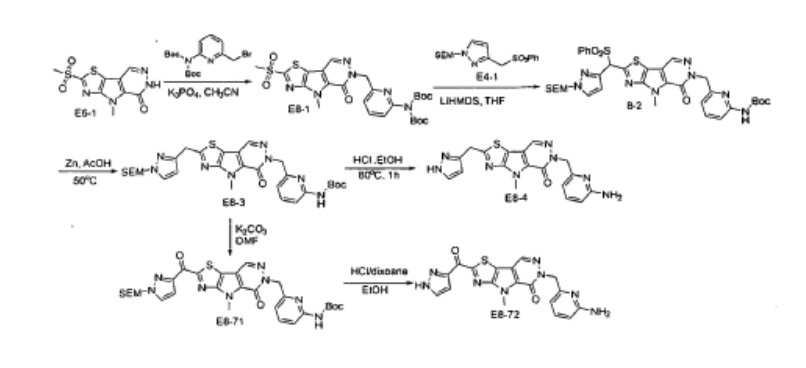


Step F. Synthesis of 6-((6-aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3- carbonyl)-4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one To a solution of tert- butyl (6-((4-methyl-5-oxo-2-(1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrazole-3-carbonyl)- 4H-thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-6(5H)-yl)methyl)pyridin-2-yl)carbamate (20 mg, 0.03 mmol) in EtOH (1 mL) was added HCl (1 mL, 4 mol/L in dioxane). The mixture was stirred at 80 °C for lhr and cooled down. The precipitate was collected by filtration and neutralized with sat. NaHCO3, washed with water and dried to afford 5 mg of 6-((6- aminopyridin-2-yl)methyl)-4-methyl-2-(1H-pyrazole-3-carbonyl)-4H- thiazolo[5′,4′:4,5]pyrrolo[2,3-d]pyridazin-5(6H)-one. LC-MS (ESI): m/z 407 (M+H)+. 1H NMR (400 MHz, DMSO-d6) δ: 8.75 (s, 1H), 7.96 (s, 1H), 7.50 (s, 1H), 7.31-7.22 (m, 1H), 6.31 (d, 1H), 6.14 (d, 1H), 5.91 (s, 2H), 5.23 (s, 2H), 4.38 (s, 3H).
PATENT
WO2023091414
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023091414&_cid=P22-MDGSRV-15431-1
PATENT
WO2019035863
WO2019035865
WO2019035864
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Tebapivat, 2283422-04-6, AG946, CS-0115951, HY-135884, AG 946, CS 0115951, HY 135884, ORG4KGP5ZS, AGIOS, Orphan Drug, PHASE 2,
PALTUSOTINE



PALTUSOTINE
CAS 2172870-89-0
- CRN00808
- F2IBD1GMD3
WeightAverage: 456.497
Monoisotopic: 456.17616767
Chemical FormulaC27H22F2N4O
3-[4-(4-Amino-1-piperidinyl)-3-(3,5-difluorophenyl)-6-quinolinyl]-2-hydroxybenzonitrile
fda 2025, approvals 2025, To treat acromegaly in adults who had an inadequate response to surgery and/or for whom surgery is not an option
- OriginatorCrinetics Pharmaceuticals
- ClassAmines; Antineoplastics; Antisecretories; Fluorobenzenes; Nitriles; Piperidines; Quinolines; Small molecules
- Mechanism of ActionSomatostatin receptor 2 agonists
- Orphan Drug Status – Acromegaly
- PreregistrationAcromegaly
- Phase IIMalignant carcinoid syndrome
- 08 May 2025Crinetics Pharmaceuticals expects potential EMA decision for paltusotine in Acromegaly, in the first half of 2026
- 08 May 2025FDA assigns PDUFA action date of 25/09/2025 for paltusotine for acromegaly
- 08 May 2025Crinetics Pharamceuticals plans the phase III CAREFNDR trial for Malignant carcinoid syndrome (PO), in the second quarter of 2025
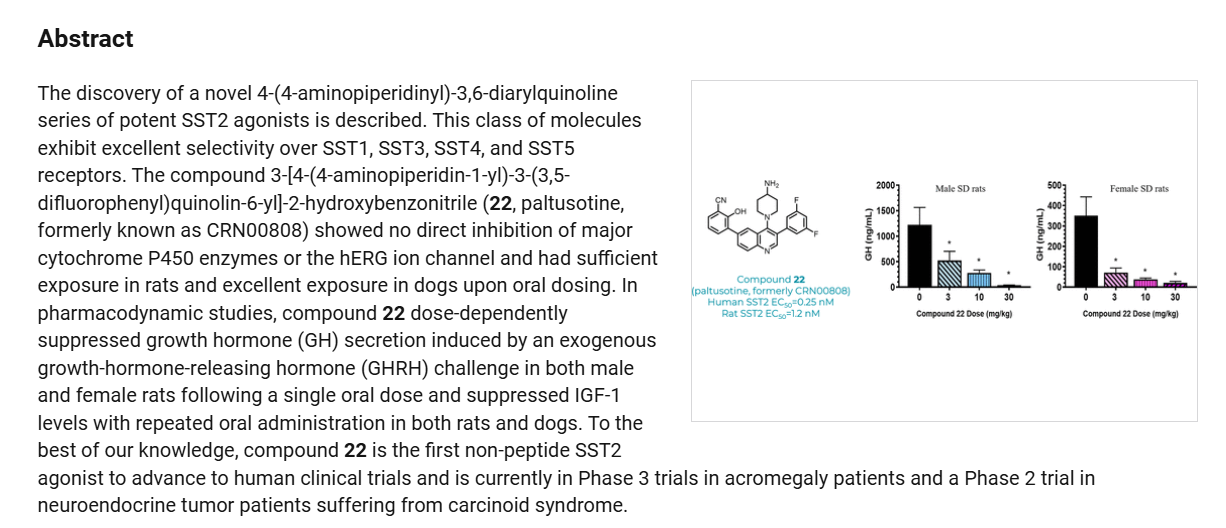
Paltusotine is a selective somatostatin receptor type 2 (SST2) agonist in development by Crinetics Pharmaceuticals for the treatment of acromegaly and certain neuroendocrine tumors. It is a small molecule delivered orally.[1][2][3][4]
SCHEME
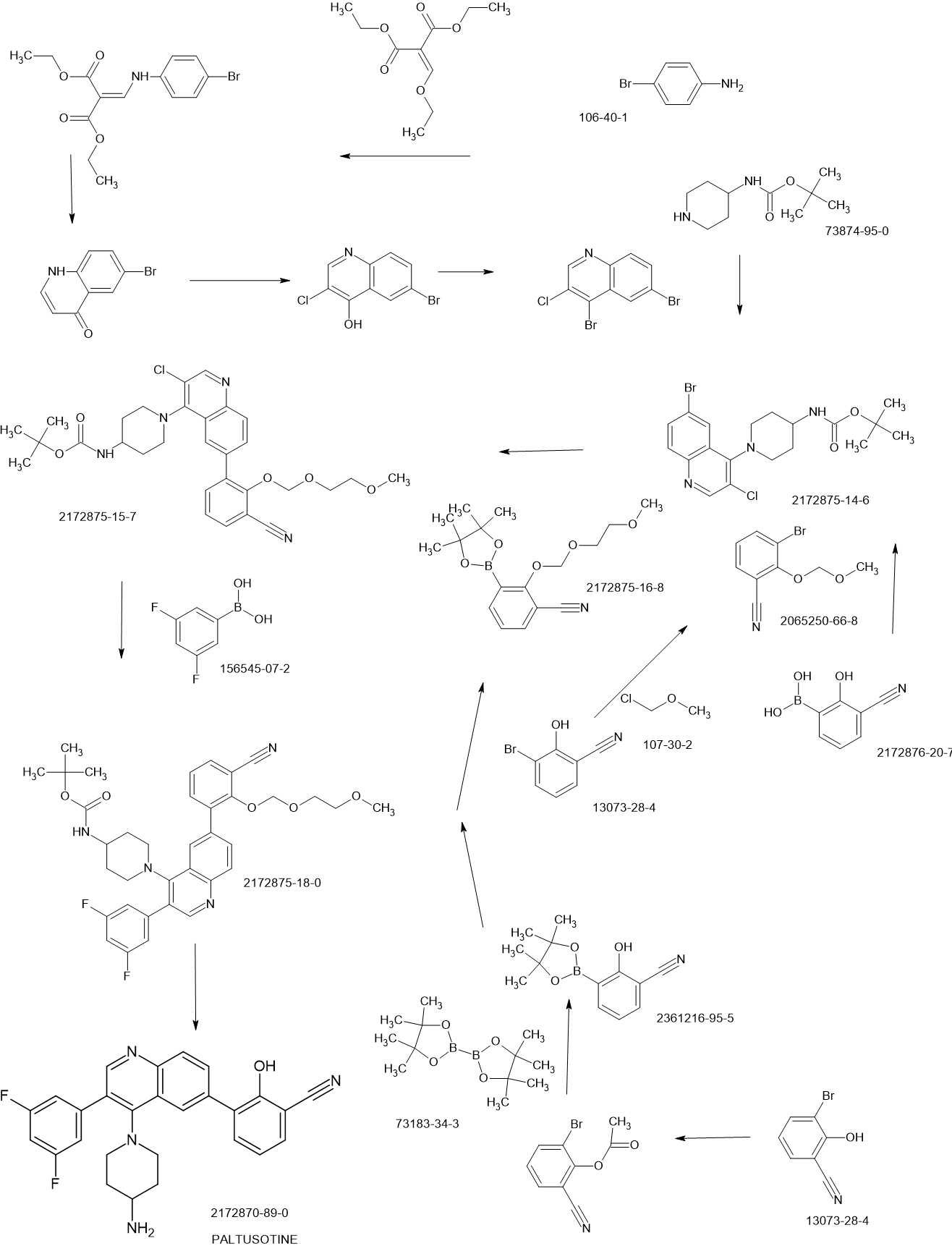

PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00431
Discovery of Paltusotine (CRN00808), a Potent, Selective, and Orally Bioavailable Non-peptide SST2 Agonist

Step 2-1, preparation of [1-(6-bromo-3-chloro-quinolin-4-yl)-piperidin-4-yl]-carbamic acid tertbutyl ester: To a DMSO solution of 6-bromo-3,4-dichloroquinoline (950 mg, 1 Eq, 3.43 mmol)
was added tert-butyl piperidin-4-ylcarbamate (841 mg, 98% Wt, 1.2 Eq, 4.12 mmol) and DIPEA
(1.19 g, 1.60 mL, 3 Eq, 10.3 mmol). The resulting mixture was heated at 60 °C for overnight.
The reaction crude was quenched with water, extracted with EtOAc, washed with brine,
concentrated and purified by silica gel chromatography to afford tert-butyl (1-(6-bromo-3-
chloroquinolin-4-yl)piperidin-4-yl)carbamate (0.95 g, 2.2 mmol, 63 %) as an off-white solid. 1H
NMR (500 MHz, CDCl3) δ 8.66 (s, 1H), 8.25 (d, J=5 Hz, 1H), 7.94 (d, J=10 Hz, 1H), 7.74 (d,
J=10 Hz, 1H), 4.61 (s, 1H), 3.76 (s, 1H), 3.51 (m, 2H), 3.37 (m, 2H), 2.13-2.15 (m, 2H), 1.73-
1.65 (m, 2H), 1.48 (s, 9H). MS [M+H]
+= 442.0.
Step 4-2, preparation of 1-{3-chloro-6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-
quinolin-4-yl}-piperidin-4-yl)-carbamic acid tert-butyl ester: To a THF (5.0 mL) solution of [1-
(6-bromo-3-chloro-quinolin-4-yl)-piperidin-4-yl]-carbamic acid tert-butyl ester (1.0 mmol, 440
mg) and 2-(2-methoxy-ethoxymethoxy)-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-
benzonitrile (1.4 eq., 1.4 mmol, 460 mg) was added PdCl2dppf (0.1 eq., 0.1 mmol, 75 mg) and
KOAc (3.0 eq., 3.0 mmol, 300 mg). N2 was bubbled through the reaction solution for 5 min and
0.5 mL water was added. The resulting mixture was heated at 80 °C for 1 h. LCMS analysis
showed about 50% of the starting material has been converted to the desired product. Additional
2-(2-methoxy-ethoxymethoxy)-3-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzonitrile
(1.4 eq., 1.4 mmol, 460 mg), PdCl2dppf (0.1 eq., 0.1 mmol, 75 mg) and KOAc (3.0 eq., 3.0
mmol, 300 mg) were added and the resulting solution was heated at 80 °C for another 2 h. The
reaction solution was combined with silica gel and concentrated. The residue obtained was
purified by silica gel chromatography eluting with ethyl acetate/hexane (0~50%) to give 0.512 g
of the desired product as white solid. MS [M+H]
+= 567.6.
Step 4-3, preparation of {1-[6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-3-(3,5-difluorophenyl)-quinolin-4-yl]-piperidin-4-yl}-carbamic acid tert-butyl ester: To a dioxane (5 mL)
solution of (1-{3-chloro-6-[3-cyano-2-(2-methoxy-ethoxymethoxy)-phenyl]-quinolin-4-yl}-
piperidin-4-yl)-carbamic acid tert-butyl ester (0.5 mmol, 283 mg) was added Pd(amphos)Cl2 (0.1
eq., 0.05 mmol, 37 mg), 3, 5-difluorophenyl boronic acid (3.0 eq., 1.5 mmol, 250 mg) and
K2CO3 (4.0 eq., 2.0 mmol, 276 mg). N2 was bubbled through the reaction solution for 5 min and
0.5 mL water was added. The resulting mixture was heated at 95 °C for 0.5 h and LCMS analysis
showed that starting material was completely consumed. The reaction solution was concentrated
with silica gel and purified by silica gel chromatography eluting with ethyl acetate/hexane
(0~50%) to give 0.170 g of the desired product as white solid. MS (M+H)+= 645.6.
Step 4-4, preparation of 3-[4-(4-amino-piperidin-1-yl)-3-(3,5-difluoro-phenyl)-quinolin-6-yl]-2-hydroxybenzonitrile: to the dichloromethane (5.0 mL) solution of {1-[6-[3-cyano-2-(2-methoxyethoxymethoxy)-phenyl]-3-(3,5-difluoro-phenyl)-quinolin-4-yl]-piperidin-4-yl}-carbamic acid
tert-butyl ester (0.264 mmol, 170 mg) was added trifluroroacetic acid (2.0 mL) and the resulting
mixture was stirred at ambient temperature for 2 h. The reaction solution was concentrated and
purified by C18 reversed phase chromatography eluting with MeCN/water (0~40%). Pure
fractions were combined, neutralized with saturated NaHCO3, extracted with ethyl acetate and
dried with MgSO4. The organic solution was concentrated with HCl in ether (2.0 M) to give the
final compound as HCl salt (68 mg, 0.138 mmol, 52%).
1H NMR (500 MHz, DMSO-d6) δ 10.77
(br s, 1H), 8.78 (s, 1H), 8.29-8.15 (m, 5H), 7.79 (dd, J=20 Hz, 5 Hz, 2H), 7.41 (m, 1H), 7.26-
7.19 (m, 3H), 3.59 (t, J=12 Hz, 2H), 3.31 (m, 1H), 3.00 (t, J=12 Hz, 2H), 2.05-1.99 (m, 2H),
1.76-1.74 (m, 2H). MS [M+H]
+= 457.5. 13C NMR (DMSO-d6) δ 30.2, 47.4, 50.8, 102.4, 103.2,
113.4, 117.2, 121.4, 124.6, 130.7, 133.1, 134.6, 136.0, 141.7, 156.6, 161.2, 163.2. LCMS purity
98% (254&220 nM). HRMS m/z [M+H]+ Calcd for C27H23F2N4O 457.1834; found 457.1833.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US235548187&_cid=P20-MCSHXW-73235-1
PATENTS
WO2021011641
WO2018013676
References
- ^ Madan, Ajay; Markison, Stacy; Betz, Stephen F.; Krasner, Alan; Luo, Rosa; Jochelson, Theresa; Lickliter, Jason; Struthers, R. Scott (April 2022). “Paltusotine, a novel oral once-daily nonpeptide SST2 receptor agonist, suppresses GH and IGF-1 in healthy volunteers”. Pituitary. 25 (2): 328–339. doi:10.1007/s11102-021-01201-z. PMC 8894159. PMID 35000098.
- ^ Zhao, Jian; Wang, Shimiao; Markison, Stacy; Kim, Sun Hee; Han, Sangdon; Chen, Mi; Kusnetzow, Ana Karin; Rico-Bautista, Elizabeth; Johns, Michael; Luo, Rosa; Struthers, R. Scott; Madan, Ajay; Zhu, Yunfei; Betz, Stephen F. (12 January 2023). “Discovery of Paltusotine (CRN00808), a Potent, Selective, and Orally Bioavailable Non-peptide SST2 Agonist”. ACS Medicinal Chemistry Letters. 14 (1): 66–74. doi:10.1021/acsmedchemlett.2c00431. PMC 9841592. PMID 36655128.
- ^ Gadelha, Monica R; Gordon, Murray B; Doknic, Mirjana; Mezősi, Emese; Tóth, Miklós; Randeva, Harpal; Marmon, Tonya; Jochelson, Theresa; Luo, Rosa; Monahan, Michael; Madan, Ajay; Ferrara-Cook, Christine; Struthers, R Scott; Krasner, Alan (13 April 2023). “ACROBAT Edge: Safety and Efficacy of Switching Injected SRLs to Oral Paltusotine in Patients With Acromegaly”. The Journal of Clinical Endocrinology & Metabolism. 108 (5): e148 – e159. doi:10.1210/clinem/dgac643. PMC 10099171. PMID 36353760. S2CID 253445337.
- ^ Zhao, Jie; Fu, Hong; Yu, Jingjing; Hong, Weiqi; Tian, Xiaowen; Qi, Jieyu; Sun, Suyue; Zhao, Chang; Wu, Chao; Xu, Zheng; Cheng, Lin; Chai, Renjie; Yan, Wei; Wei, Xiawei; Shao, Zhenhua (21 February 2023). “Prospect of acromegaly therapy: molecular mechanism of clinical drugs octreotide and paltusotine”. Nature Communications. 14 (1): 962. Bibcode:2023NatCo..14..962Z. doi:10.1038/s41467-023-36673-z. ISSN 2041-1723. PMC 9944328. PMID 36810324.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2172870-89-0 |
| PubChem CID | 134168328 |
| ChemSpider | 81367268 |
| UNII | F2IBD1GMD3 |
| Chemical and physical data | |
| Formula | C27H22F2N4O |
| Molar mass | 456.497 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////PALTUSOTINE, ORPHAN DRUG, Acromegaly, CRN 00808, F2IBD1GMD3, fda 2025, approvals 2025
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
HEXASODIUM PHYTATE





HEXASODIUM PHYTATE
cas 34367-89-0
myo-Inositol, 1,2,3,4,5,6-hexakis(dihydrogen phosphate) sodium salt (1:6)
hexasodium;[2,3,4,5,6-pentakis[[hydroxy(oxido)phosphoryl]oxy]cyclohexyl] hydrogen phosphate
free form RN: 83-86-3
C6H12Na6O24P6, 791.93
- Inositol, hexakis(dihydrogen phosphate) hexasodium salt, myo– (8CI)
- myo-Inositol, hexakis(dihydrogen phosphate), hexasodium salt (9CI)
- Hexasodium fytate
- Hexasodium phytate
- SNF 472
- UNII-ZBX50UG81V
- CSL-525; Hexasodium phytate; Myo-inositol hexaphosphate; SNF-472
x Na salt
14306-25-3
C6H18O24P6.xNa
free form : 83-86-3
myo-Inositol, 1,2,3,4,5,6-hexakis(dihydrogen phosphate), sodium salt
| (1R,2R,3S,4S,5R,6S)-CYCLOHEXANE-1,2,3,4,5,6-HEXAYL-HEXAKIS(DIHYDROGEN PHOSPHATE) |
- Inositol, hexakis(dihydrogen phosphate) sodium salt, myo– (8CI)
- myo-Inositol, hexakis(dihydrogen phosphate), sodium salt (9CI)
- Inositol hexaphosphate sodium salt
- Phytic acid sodium salt
- Sodium inositol hexaphosphate
- Sodium phytate
- OriginatorLaboratoris Sanifit
- DeveloperCSL Vifor; Laboratoris Sanifit
- ClassAntineoplastics; Calcium regulators; Cardiovascular therapies; Phosphates; Small molecules; Sodium compounds; Sugar alcohols
- Mechanism of ActionUndefined mechanism
- Orphan Drug StatusYes – Peripheral arterial disorders; Calciphylaxis
- Phase IIICalciphylaxis; Peripheral arterial disorders
- 09 Nov 2022Phase-III clinical trials in Peripheral arterial disorders in USA (IV) (CSL Behring pipeline, November 2022)
- 24 Oct 2022Sanifit Therapeutics completes a phase III trial in Calciphylaxis in Belgium, Poland, United Kingdom, Germany, Spain, USA (IV) (NCT04195906)
- 28 Sep 2022Hexasodium fytate is still in phase III trials for Calciphylaxis in USA (IV) (NCT04195906)
- You need to be a logged in or subscribed to view this
Hexasodium phytate (also known as SNF472) is a compound being developed as a potential treatment for calciphylaxis, a condition causing skin damage and tissue death in patients with end-stage renal disease. It works by inhibiting the formation and growth of hydroxyapatite crystals, which are implicated in calciphylaxis.
What it is:Hexasodium phytate is the hexasodium salt of myo-inositol hexaphosphate (IP6), a naturally occurring substance found in foods like beans and grains.
- How it works:It binds to hydroxyapatite crystals, the main component of vascular calcification, and prevents their growth, potentially disrupting the calciphylaxis process.
- Why it’s used:Hexasodium phytate is being investigated as a treatment for calciphylaxis, a serious complication of end-stage renal disease characterized by skin and tissue damage due to calcification of small blood vessels.
- Mechanism:It is believed to work by inhibiting the formation and growth of calcium-phosphate crystals (hydroxyapatite) in the blood vessels, thus preventing the calcification that leads to calciphylaxis.
- Clinical trials:Clinical trials have demonstrated the safety and potential efficacy of hexasodium phytate in reducing hydroxyapatite crystallization in patients undergoing hemodialysis, providing a basis for its use in treating calciphylaxis.
- Intravenous administration:It is administered intravenously during dialysis sessions to achieve supra-physiological plasma concentrations, which are thought to be necessary for its therapeutic effect.
- Benefits:It has shown promise in preclinical studies and clinical trials, potentially improving wound healing, pain, and health-related quality of life in patients with calciphylaxis.
- Active development:It is currently in active development as a novel experimental drug for the treatment of calciphylaxis and other related conditions.
Phytic acid is a major phosphorus storage compound of most seeds and cereal grains. It has the strong ability to chelate multivalent metal ions, especially zinc, calcium, and iron. Phytic acid is also considered to be a natural antioxidant and is suggested to have potential functions of reducing lipid peroxidation and as a preservative in foods. Clathrin-associated adaprot complex AP-2 has it been suggested may act as one of the receptor sites for Phytic acid. Both in vivo and in vitro experiments have demonstrated striking anticancer (preventive as well as therapeutic) effects of Phytic acid.
SCHEME

contd………

References
WO2022129148
EP4015494
CN114874473
iScience (2022), 25(3), 103950
CN111718463
CN110483240
Uzbekskii Khimicheskii Zhurnal (1995), (5-6), 72-75 JP61056142
////////HEXASODIUM PHYTATE, Hexasodium fytate, Hexasodium phytate, SNF 472, calciphylaxis, UNII-ZBX50UG81V, CSL-525, Hexasodium phytate, Myo-inositol hexaphosphate, SNF-472, ORPHAN DRUG

{kind=link}
{kind=link}
{kind=link}