








| FDA ORANGE BOOK PATENTS: 1 OF 2 | |
|---|---|
| Patent | 7459561 |
| Expiration | Oct 31, 2020 |
| Applicant | ASTELLAS |
| Drug Application | N207500 (Prescription Drug: CRESEMBA. Ingredients: ISAVUCONAZONIUM SULFATE) |
from FDA Orange Book
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Posts tagged 'Orphan Designation'
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

Bulevirtide acetate
(N-Myristoyl-glycyl-L-threonyl-L-asparaginyl-L-leucyl-L-seryl-L-valyl-Lprolyl-L-asparaginyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-aspartyl-L-histidyl-Lglutaminyl-L-leucyl-L-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparaginyl-L-seryl-Lasparaginyl-L-asparaginyl-Lprolyl-L-aspartyl-L-tryptophanyl-L-aspartyl-L-phenylalanyl-L-asparaginyl-L-prolylL-asparaginyl-L-lysyl-L-aspartyl-L-histidyl-L-tryptophanyl-L-prolyl-L-glutamyl-L-alanyl-L-asparaginyl-L-lysylL-valylglycinamide, acetate salt.
molecular formula C248H355N65O72,
molecular mass is 5398.9 g/mol
ブレビルチド酢酸塩;
APROVED 2020/7/31, EU, Hepcludex
MYR GmbH
|
Antiviral, Entry inhibitor
|
|
| Disease |
Hepatitis delta virus infection
|
|---|
Bulevirtide is a 47-amino acid peptide with a fatty acid, a myristoyl residue, at the N-terminus and an amidated C-terminus. The active substance is available as acetate salt. The counter ion acetate is bound in ionic form to basic groups of the peptide molecule and is present in a non-stoichiometric ratio. The chemical name of bulevirtide is (N-Myristoyl-glycyl-L-threonyl-L-asparaginyl-L-leucyl-L-seryl-L-valyl-Lprolyl-L-asparaginyl-L-prolyl-L-leucyl-glycyl-L-phenylalanyl-L-phenylalanyl-L-prolyl-L-aspartyl-L-histidyl-Lglutaminyl-L-leucyl-L-aspartyl-L-prolyl-L-alanyl-L-phenylalanyl-glycyl-L-alanyl-L-asparaginyl-L-seryl-Lasparaginyl-L-asparaginyl-Lprolyl-L-aspartyl-L-tryptophanyl-L-aspartyl-L-phenylalanyl-L-asparaginyl-L-prolylL-asparaginyl-L-lysyl-L-aspartyl-L-histidyl-L-tryptophanyl-L-prolyl-L-glutamyl-L-alanyl-L-asparaginyl-L-lysylL-valylglycinamide, acetate salt. It corresponds to the molecular formula C248H355N65O72, its relative molecular mass is 5398.9 g/mol
Bulevirtide appears as a white or off-white hygroscopic powder. It is practically insoluble in water and soluble at concentrations of 1 mg/ml in 50% acetic acid and about 7 mg/ml in carbonate buffer solution at pH 8.8, respectively. The structure of the active substance (AS) was elucidated by a combination of infrared spectroscopy (IR), mass spectrometry (MS), amino acid analysis and sequence analysis Other characteristics studied included ultraviolet (UV) spectrum, higher order structure (1D- and 2D- nuclear magnetic resonance spectroscopy (NMR)) and aggregation (Dynamic Light Scattering). Neither tertiary structure nor aggregation states of bulevirtide have been identified. With regard to enantiomeric purity, all amino acids are used in L-configuration except glycine, which is achiral by nature. Two batches of bulevirtide acetate were evaluated for enanatiomeric purity and no relevant change in configuration during synthesis was detected.
Bulevirtide is manufactured by a single manufacturer. It is a chemically synthesised linear peptide containing only naturally occurring amino acids. The manufacturing of this peptide is achieved using standard solidphase peptide synthesis (SPPS) on a 4-methylbenzhydrylamine resin (MBHA resin) derivatised with Rink amide linker in order to obtain a crude peptide mixture. This crude mixture is purified through a series of washing and preparative chromatography steps. Finally, the purified peptide is freeze-dried prior to final packaging and storage. The process involves further four main steps: synthesis of the protected peptide on the resin while side-chain functional groups are protected as applicable; cleavage of the peptide from the resin, together with the removal of the side chain protecting groups to obtain the crude peptide; purification; and lyophilisation. Two chromatographic systems are used for purification. No design space is claimed. Resin, Linker Fmoc protected amino acids and myristic acid are starting materials in line with ICH Q11. Sufficient information is provided on the source and the synthetic route of the starting materials. The active substance is obtained as a nonsterile, lyophilised powder. All critical steps and parameters were presented and clearly indicated in the description of the manufacturing process. The process description includes also sufficient information on the type of equipment for the SPPS, in-process controls (IPCs). The circumstances under which reprocessing might be performed were clearly presented. No holding times are proposed. Overall the process is sufficiently described.
The finished product is a white to off white lyophilised powder for solution for injection supplied in single-use vials. Each vial contains bulevirtide acetate equivalent to 2 mg bulevirtide. The composition of the finished product was presented. The powder is intended to be dissolved in 1 ml of water for injection per vial. After reconstitution the concentration of bulevirtide net peptide solution in the vial is 2 mg/ml. The components of the formulation were selected by literature review and knowledge of compositions of similar products available on the market at that time, containing HCl, water, mannitol, sodium carbonate, sodium hydrogen carbonate and sodium hydroxide. All excipients are normally used in the manufacture of lyophilisates. The quality of the excipients complies with their respective Ph. Eur monographs. The intrinsic properties of the active substance and the compounding formulation do not support microbiological growth as demonstrated by the stability data. No additional preservatives are therefore needed.
Hepcludex is an antiviral medicine used to treat chronic (long-term) hepatitis delta virus (HDV) infection in adults with compensated liver disease (when the liver is damaged but is still able to work), when the presence of viral RNA (genetic material) has been confirmed by blood tests.
HDV is an ‘incomplete’ virus, because it cannot replicate in cells without the help of another virus, the hepatitis B virus. Because of this, patients infected with the virus always also have hepatitis B.
HDV infection is rare, and Hepcludex was designated an ‘orphan medicine’ (a medicine used in rare diseases) on 19 June 2015. For further information on the orphan designation, see EU/3/15/1500.
Hepcludex contains the active substance bulevirtide.
Bulevirtide, sold under the brand name Hepcludex, is an antiviral medication for the treatment of chronic hepatitis D (in the presence of hepatitis B).[2]
The most common side effects include raised levels of bile salts in the blood and reactions at the site of injection.[2]
Bulevirtide works by attaching to and blocking a receptor (target) through which the hepatitis delta and hepatitis B viruses enter liver cells.[2] By blocking the entry of the virus into the cells, it limits the ability of HDV to replicate and its effects in the body, reducing symptoms of the disease.[2]
Bulevirtide was approved for medical use in the European Union in July 2020.[2]
Bulevirtide is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in plasma (or serum) HDV-RNA positive adult patients with compensated liver disease.[2][3]
Bulevirtide binds and inactivates the sodium/bile acid cotransporter, blocking both viruses from entering hepatocytes.[4]
The hepatitis B virus uses its surface lipopeptide pre-S1 for docking to mature liver cells via their sodium/bile acid cotransporter (NTCP) and subsequently entering the cells. Myrcludex B is a synthetic N-acylated pre-S1[5][6] that can also dock to NTCP, blocking the virus’s entry mechanism.[7]
The drug is also effective against hepatitis D because the hepatitis D virus is only infective in the presence of a hepatitis B virus infection.[7]
| Clinical data | |
|---|---|
| Trade names | Hepcludex |
| Other names | MyrB, Myrcludex-B[1] |
| License data | |
| Routes of administration |
Subcutaneous injection |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEMBL | |
/////////Bulevirtide acetate, ブレビルチド酢酸塩 , orphan designation, MYR GmbH, PEPTIDE, EU 2020, 2020 APPROVALS

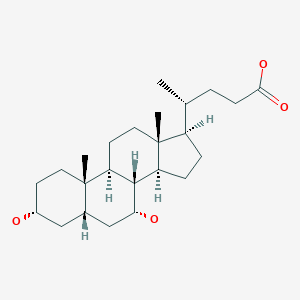
Chenodeoxycholic acid
Chenodiol
|
Chenodeoxycholate;
Chenodeoxycholic acid; 3alpha,7alpha-Dihydroxy-5beta-cholanic acid; Chenodiol |
Synthesis ReferenceHenry Francis Frost, Fritz Fabian, Christopher James Sharpe, William Arthur Jones, “Process for preparing chenodeoxycholic acid.” U.S. Patent US4022806, issued October, 1974. US4022806
First ref

Chenodeoxycholic acid (also known as chenodesoxycholic acid, chenocholic acid and 3α,7α-dihydroxy-5β-cholan-24-oic acid) is a bile acid. It occurs as a white crystalline substance insoluble in water but soluble in alcohol and acetic acid, with melting point at 165–167 °C. Salts of this carboxylic acid are called chenodeoxycholates. Chenodeoxycholic acid is one of the main bile acids produced by the liver.[1]
It was first isolated from the bile of the domestic goose, which gives it the “cheno” portion of its name (Greek: χήν = goose).[2]
Chenodeoxycholic acid and cholic acid are the two primary bile acids in humans. Some other mammals have muricholic acid or deoxycholic acid rather than chenodeoxycholic acid.[1]
Chenodeoxycholic acid is synthesized in the liver from cholesterol by a process which involves several enzymatic steps.[1] Like other bile acids, it can be conjugated in the liver with taurine or glycine, forming taurochenodeoxycholate or glycochenodeoxycholate. Conjugation results in a lower pKa. This means the conjugated bile acids are ionized at the usual pH in the intestine and will stay in the gastrointestinal tract until reaching the ileum where most will be reabsorbed. Bile acids form micelles which facilitate lipid digestion. After absorption, they are taken up by the liver and resecreted, so undergoing an enterohepatic circulation. Unabsorbed chenodeoxycholic acid can be metabolised by bacteria in the colon to form the secondary bile acid known as lithocholic acid.
Chenodeoxycholic acid is the most potent natural bile acid at stimulating the nuclear bile acid receptor, farnesoid X receptor (FXR).[3]The transcription of many genes is activated by FXR.
Indication
Chenodiol is indicated for patients with radiolucent stones in well-opacifying gallbladders, in whom selective surgery would be undertaken except for the presence of increased surgical risk due to systemic disease or age. Chenodiol will not dissolve calcified (radiopaque) or radiolucent bile pigment stones.
Associated Conditions
Pharmacodynamics
It acts by reducing levels of cholesterol in the bile, helping gallstones that are made predominantly of cholesterol to dissolve. Chenodeoxycholic acid is ineffective with stones of a high calcium or bile acid content.
Mechanism of action
Chenodiol suppresses hepatic synthesis of both cholesterol and cholic acid, gradually replacing the latter and its metabolite, deoxycholic acid in an expanded bile acid pool. These actions contribute to biliary cholesterol desaturation and gradual dissolution of radiolucent cholesterol gallstones in the presence of a gall-bladder visualized by oral cholecystography. Bile acids may also bind the the bile acid receptor (FXR) which regulates the synthesis and transport of bile acids.
EMA
On 16 December 2014, orphan designation (EU/3/14/1406) was granted by the European Commission to Sigma-Tau Pharma Ltd, United Kingdom, for chenodeoxycholic acid for the treatment of inborn errors in primary bile acid synthesis.
The sponsorship was transferred to sigma-tau Arzneimittel GmbH, Germany, in May 2015.
Chenodeoxycholic acid has been authorised in the EU as Chenodeoxycholic acid sigma-tau since 10 April 2017.
The name of the product changed to Chenodeoxycholic acid Leadiant in May 2017.
The sponsorship was transferred to Leadiant GmbH, Germany, in June 2017.
On 16 February 2017, the Committee for Orphan Medicinal Products (COMP) concluded its review of the designation EU/3/14/1406 for Chenodeoxycholic acid sigma-tau (chenodeoxycholic acid) as an orphan medicinal product for the treatment of inborn errors in primary bile acid synthesis. The COMP assessed whether, at the time of marketing authorisation, the medicinal product still met the criteria for orphan designation. The Committee looked at the seriousness and prevalence of the condition, and the existence of other methods of treatment. As other methods of treatment are authorised in the European Union (EU), the COMP also considered whether the medicine is of significant benefit to patients with inborn errors in primary bile acid synthesis. The COMP recommended that the orphan designation of the medicine be maintained1.
1 The maintenance of the orphan designation at time of marketing authorisation would, except in specific situations, give an orphan medicinal product 10 years of market exclusivity in the EU. This means that in the 10 years after its authorisation similar products with the same therapeutic indication cannot be placed on the market.
http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2015/02/WC500183233.pdf
Chenodeoxycholic acid has been used as medical therapy to dissolve gallstones.[4]
Chenodeoxycholic acid can be used in the treatment of cerebrotendineous xanthomatosis.[5]
The Australian biotechnology company Giaconda has tested a treatment for Hepatitis C infection that combines chenodeoxycholic acid with bezafibrate.[6]
As diarrhea is a complication of chenodeoxycholic acid therapy, it has also been used to treat constipation.[7][8]
In supramolecular chemistry, molecular tweezers based on a chenodeoxycholic acid scaffold is a urea receptor that can contain anionsin its binding pocket in order of affinity: H2PO4− (dihydrogen phosphate) > Cl− > Br− > I− reflecting their basicities (tetrabutylammonium counter ion).[9]

PAPER
Improved Chemical Synthesis, X-Ray Crystallographic Analysis, and NMR Characterization of (22R)-/(22S)-Hydroxy Epimers of Bile Acids
Lipids (2014), 49, (11), 1169-1180.
A Practical and Eco-friendly Synthesis of Oxo-bile Acids
By Han, Young Taek and Yun, HwayoungFrom Organic Preparations and Procedures International, 48(1), 55-61; 2016
DOI:10.1080/00304948.2016.1127101
General Procedure
An aqueous solution of 0.2 M NaBrO3 (1.5 equiv. per hydroxy group) was added dropwise to a slurry of bile acid (1 equiv.) and ceric ammonium nitrate (0.05 equiv.) in 20% aqueous acetonitrile (0.2 M) at 80°C over 20 min. The bile acid slowly dissolved in a few minutes, and then the color of the reaction mixture changed to orange. The reaction mixture was stirred at the same temperature and the progress of the reaction was monitored by TLC on silica gel (1:20 MeOH-CH2Cl2) until disappearance of the starting material and partially oxidized intermediates. It was then cooled in an ice bath and quenched with aqueous Na2S2O3 solution. Water was added slowly to the resulting white suspension until no more oxo-bile acid precipitated. The white solid was collected, washed with water until the filtrate was colorless, and then dried in vacuo at 50°C. Methyl 3,7α-Diacetoxy-12-oxo-5β-cholanoate(3),21 was obtained in 92% yield (275 mg) as a white solid from 300 mg (0.590 mmol) of 2 via the general procedure. mp. 176-178°C, lit.22 mp. 178-179°C, IR (thin film, neat): 2947 (m), 2873 (s), 1736 (w), 1706 (w), 1436 (s), 1365 (m) cm-1; 1H-NMR (400 MHz, CDCl3): δ 4.96 (m, 1H, 7-CH), 4.55 (m, 1H, 3-CH), 3.64 (s, 3H), 2.49 (t, 1H, J = 12.6 Hz), 2.41-0.80 (m, 23H), 2.01 (s, 3H), 2.00 (s, 3H), 1.01 (s, 3H, 18-CH3), 1.00 (s, 3H, 19-CH3), 0.83 (d, 3H, J = 6.6 Hz, 21-CH3); 13C-NMR (CDCl3, 100 MHz): δ 214.0 (12-C), 174.6 (24-C), 170.7 (C = O), 170.2 (C = O), 73.5 (3-C), 70.5 (7-C), 57.1 (13-C), 53.1 (14-C), 51.5 (CH3O), 46.3 (17-C), 40.5 (5-C), 37.9 (11-C), 37.8 (4-C), 37.6 (8-C), 35.54 (9-C), 35.52 (20-C), 34.9 (1-C), 34.5 (10-C), 31.3 (6-C), 31.2 (22-C), 30.4 (23-C), 27.4 (16-C), 26.5 (2-C), 23.8 (15-C), 22.1 (19-C), 21.51 (CH3CO2), 21.46 (CH3CO2), 18.6 (21-C), 11.5 (18-C); LR-MS (FABC) m/z 505 (M+H +). HR-MS (FABC): Calcd for C29H45O7 (M+H +): 505.3165. Found 505.3161.
next step
R:KOH, R:N2H4

NOTE STARTING IS BILE ACID AS BELOW
Cholan-24-oic acid, 3,7-bis(acetyloxy)-12-oxo-, methyl ester, (3α,5β,7α)-

PAPER
https://pubs.acs.org/doi/pdf/10.1021/jo01091a623
Journal of Organic Chemistry
Volume24
Pages1367-8
Journal
1959
DOI:10.1021/jo01091a623
Chenodeoxycholic acid (V). Five hundred mg. of the above ester IV was hydrolyzed with 80 ml. of ethanolic 5% potassium hydroxide for 4 hr. After partial concentration of the volume and addition of water, the reaction product was acidified with hydrochloric acid. The resulting precipitate was collected, dried, and crystallized from ethyl acetate. A quantitative crop (400 mg.) of prisms melting at 143- 145° were obtained. Recrystallization from the same solvent yielded a product of m.p. 145-146°, [ ]2 +10.7° (dioxane). Anal. Caled, for C24H40O4: C, 73.43; H, 10.27. Found: C, 73.49; H, 10.31.

NOTE I IS BILE ACID
Cholan-24-oic acid, 3,7-bis(acetyloxy)-12-oxo-, methyl ester, (3α,5β,7α)-
PATENT
https://patents.google.com/patent/CN102060902A/en
chenodeoxycholic acid (3 α, 7 α – dihydroxy _5 β – cholestane-24-oic acid) Chenodeoxycholic Ac id (referred to as CDCA), clinically used to correct dissolving cholesterol calculi and bile saturation drugs, the main function is to reduce the cholesterol in the bile saturation, large doses can inhibit the synthesis of cholesterol CDCA and increasing bile gallstone patients cholesterol level in a non-saturated, thereby preventing the formation of cholesterol gallstones of cholesterol and promote stone dissolve and fall off. It also has significant anti-asthmatic, anti-inflammatory, antitussive and expectorant effects.
[0003] Synthesis of chenodeoxycholic acid or ursodeoxycholic acid (3 α, 7β_ -5β_ dihydroxy-cholestane-24-oic acid, ursodeoxycholic Acid, referred UDCA), a key intermediate. Ursodeoxycholic acid is the main active ingredient of precious Chinese medicine bear bile, used in a variety of clinical hepatobiliary disease and dyspepsia. Currently we bear bile resources are scarce, mainly used synthetic chemical ursodeoxycholic acid as a clinical treatment. Therefore, the preparation of chenodeoxycholic acid is also important for the preparation of ursodeoxycholic acid.
[0004] CDCA mainly come from poultry or livestock bile extraction. Traditional extraction process complicated operation, low yield, (pharmaceutical industry, 1987,18 (9), 416; Chinese Journal of Biochemical Pharmaceutics, 1996,17 (1), 17; Applied Technology, 1998, (4), 9; CN1850846A ) can not meet the needs of modern industry. Chemical synthesis of chenodeoxycholic acid have also been reported (Japanese Journal of Chemistry 1955,76 (3), 297 -J Org Chem 1982,47 (2): 2331; Journal of Biochemical Pharmaceutics 1987,1,6 -, Tap Chi Duoc ^ oc2004 , 44 (1), 11; CN1869043A), but lower yield widespread pollution major problem, especially in the oxidation reaction is often used to expensive, and polluting agents.Therefore, to reduce pollution, reduce environmental hazards, streamline operations, improve yield, reduce costs, important for the synthesis of chenodeoxycholic acid.

n particular by the following steps:
(1) Preparation of cholate: bile acid in alcohol, concentrated hydrochloric acid as catalyst, at reflux, cooling and crystallization, filtration, and washed with methanol.
[0008] (2) Preparation of 3α, 7α- diacetyl hydroxy -12α- cholate: bile acid ester was dissolved in dichloromethane and triethylamine was added with stirring acetic anhydride and the catalyst N, N- dimethyl pyridine, methylene chloride was distilled off, poured into water, filtered to give 3α, 7α- diacetyl -12 α – hydroxy cholate.
[0009] (3) 3α, 7α- diacetyl -12– Preparation oxo chenodeoxycholic acid ester: Take 3 α, 7 α – diacetyl -12 α – hydroxy cholate dissolved in ethyl acetate and methanol, bromide and tetrabutylammonium bromide as catalyst, and acetic acid was added dropwise under stirring hypochlorite, the organic solvent was distilled off and filtered, to give 12-oxo-3,7-diacetyl Chenodeoxy cholate.
[0010] (4) i2 – Preparation oxo chenodeoxycholic acid: 3,7-diacetyl-12-oxo-chenodeoxycholic acid ester added ethanol – sodium hydroxide solution, at reflux.PH adjusted with hydrochloric acid value of the reaction system acidic, ethanol was distilled off, and filtered to give 12- oxo crude chenodeoxycholic acid, fine recrystallization.
[0011] Preparation of chenodeoxycholic acid (5): 12- oxo take chenodeoxycholic acid, ethylene glycol and solid sodium hydroxide, hydrated corpus, refluxed for 2 hours, gradually warming evaporated partially hydrated corpus, continue to heat up to 150 ° C, continued to reflux, cooled to room temperature, poured into water, adjusting the PH with hydrochloric acid, the white precipitate was filtered, washed with water to give crude chenodeoxycholic acid, recrystallization
Azusa mouth
M ο not mesh
[0012] Step (1): cholic acid to alcohol weight to volume ratio of 1: 2 ~ 5, the volume ratio of concentrated hydrochloric acid to alcohol is 10 wide: 100, 5-5 hours reflux time was 0.5.
[0013] Step (2): cholate: acetic anhydride molar ratio = 1: 2 ~ 5, the reaction temperature, time; Tl2O hours; cholate was added per mole of N, N- dimethylpyridine wide 5g.
[0014] Step (; 3): The hypochlorite is sodium hypochlorite or calcium hypochlorite; bromide is sodium bromide, potassium bromide and the like.
[0015] Step (4): recrystallization from a solvent with an alcohol such: as methanol or ethanol.
[0016] Step (5): recrystallization solvent is a water-miscible organic solvents, such as: methanol, ethanol, acetonitrile, acetone and the like.
[0017] Step (cholate was used ¾ of methyl cholate, ethyl cholate, cholic acid or cholic acid propyl ester; Step (3) used as 3 [alpha], 7 α – diacetyl -12 α – hydroxy cholate as 3 α, 7 α – diacetyl -12 α – hydroxy methyl cholate, 3 α, 7α- diacetyl -12 α – hydroxy bile acid ethyl ester, 3 α, 7α- diacetyl yl -12 α – hydroxy acid or ester 3α, 7α- diacetyl -12 α – hydroxy acid ester.
[0018] The invention has the advantages: in cholic acid as raw materials, and the choice of bromide tetrabutylammonium bromide as catalyst, in a non-polluting oxidizing agent is hypochlorite, Intermediate 3 α, 7 α – Diacetyl _12_ oxo chenodeoxycholic acid ester yield of 90% or more, thereby improving the yield of the final product of chenodeoxycholic acid, 99% yield, low cost and no pollution, very convenient for industrial production. detailed description
[0019] The present invention will be better described, for example is as follows:
(1) Preparation of methyl cholate: bile acid 5. lg, 15ml of anhydrous methanol, heating the whole solution. Refluxed for 3 hours, was added 0. 4ml concentrated hydrochloric acid, the reaction was stopped after 30min, after slow cooling, and filtered to give methyl cholate 5. 05g, 95% yield. 1HNMR (CDCl3):. Δ 0. 70 (s, 3H, 18- CH3), 0.90 (s, 3H, 19- CH3), 0.98 (d, 3H, 21-CH3), 3 50 (m, 1H, 3 β -H), 3. 67 (s, 3H, OCH3), 3. 87 (s, 1H, 7 β -H), 3. 99 (s, 1H, 12 β -H).
[0020] (2) Preparation of 3α, 7α- methyl cholate diacetyl-hydroxy -12α-: bile acid methyl ester 4. 71g (Ilmmol) IOOml was placed in a flask, was added methylene chloride 30ml, triethylamine 3 . Chiu 1, stirred at room temperature, was added dropwise acetic anhydride 2. 7ml (28. 6mmo 1), followed by addition of 20mg N, N- dimethylpyridine catalyst, the reaction time of 7 hours, methylene chloride was distilled off, into the water, filtered to give a white solid. The crude product was recrystallized from methanol to give white crystals 4. 05g, yield 67.2%. 1H NMR (CDCl3) δ: 4.90 (m, 1H, 7 β -H), 4. 59 (s, 1H, 3 β -H), 4 01 (s, 1H, 12 β -H), 3 67.. (s, 3Η, OCH3), 2. 08 (s, 3Η, CH3CO), 2. 02 (s, 3Η, CH3CO), 0. 98 (s, 3Η, 21-CH3), 0. 93 (s, 3Η , 19-CH3), 0.69 (s, 3Η, 18_CH3).
[0021] (3) 3α, 7α – 12-oxo-diacetyl chenodeoxycholic acid methyl ester prepared: Take 3 α, 7 α – diacetyl -12 α- hydroxy methyl cholate 1.917 g ( 3. 79mmol) was placed in a 50ml round bottom flask, 12ml of ethyl acetate was added, 5ml methanol, stirring at room temperature, was added 0. 25g 0. Ig of potassium bromide and tetrabutylammonium bromide. Was added dropwise a solution of acetic acid and 6g of sodium hypochlorite (7%) (5.62mmol), for 10 hours. Methanol was distilled off under reduced pressure and ethyl acetate, filtered, washed with water, and dried to give crude 1.915g, 1.75g as a white solid after recrystallization from methanol, yield 91.2%. 1H bandit R (CDCl3) δ:.. 4. 99 (d, 1H, 7 β-H), 4 60 (m, 1H, 3 β-H), 3 67 (s, 3H, OCH3), 2. 07 (s, 6H, CH3CO), 1. 03 (s, 6H, I8-CH3 and 19-CH3), 0. 82 (d, 3H, 21-CH3) ο
[0022] (4) 12- oxo chenodeoxycholic acid Preparation: Take 3 α, 7 α – diacetyl _12_ oxo chenodeoxycholic acid methyl ester 1. 56g, was dissolved in 30ml 95% ethanol was added 3. 2g of sodium hydroxide, heated at reflux for 5 hours. PH adjusted with hydrochloric acid value of the reaction system, most of the ethanol was distilled off, filtered, washed with water, and dried to give a white solid 12- oxo-1 crude chenodeoxycholic acid, recrystallized from methanol ^ g 1. 25g, yield rate of 96%. Tun bandit R (CDCl3) δ:. 3.96 (d, 1H, 7 β-H), 3 47 (m, 1H, 3 β-H), 1.03 (s, 3H, 19_CH3), 0.89 (s, 3H, 18_CH3 ), 0 · 70 (d, 3 H, 21_CH3).
[0023] Preparation of chenodeoxycholic acid (5): 12- oxo take chenodeoxycholic acid 0. 9g, 15ml ethylene glycol was added solid sodium hydroxide and 1. 5g, 15ml hydrated corpus (80%) , 120 ° C reflux for 2 hours, change return device is a distillation apparatus, was gradually warmed evaporated amount hydrated corpus, continue to heat up to 150 ° C, continuing reflux for 4h, cooled to room temperature, poured into water, adjusted with HCl of PH3, white precipitated, was filtered cake was washed with water, and dried to give crude chenodeoxycholic acid 0. 92g, recrystallized from methanol to give 0. 86g, 99 (s, 1H, C00H).
Paper
https://pubs.acs.org/doi/abs/10.1021/ja01168a045
The Preparation of Chenodeoxycholic Acid and Its Glycine and Taurine Conjugates.Hofmann, Alan F.






Chenodeoxycholic acid (3α, 7α- -5β- dihydroxy-cholestane acid) Chenodeoxycholic Acid (referred to as CDCA), a medicine for treating gallstones. 1848 first discovered in goose bile, 1924, known as the CDCA. By reducing cholesterol absorption, synthesis, the bile cholesterol decreased, thereby suppressing cholesterol gallstone formation and promote dissolution, and can reduce cholesterol saturation.
Chenodeoxycholic acid addition pharmaceutically itself, but also as the preparation of ursodeoxycholic acid (3α, 7β- -5β- dihydroxy bile acid, abbreviated UDCA) starting material. Ursodeoxycholic acid is the main active ingredient contained bile valuable medicine, in clinical treatment of various gastrointestinal diseases and bladder diseases. But the limited sources of bear bile medicine, and contrary to the principles of animal protection. So, dwindling source of natural bear bile, can not meet the medical requirements. Therefore, the preparation of chenodeoxycholic acid is also of great significance for further preparation of ursodeoxycholic acid.
CDCA bile extracted mainly from poultry or animal bile extraction methods in the past as it involves toxic chemicals (animal biological pharmacy, 1981, People’s Medical Publishing House, P259; pharmaceutical industry, 1987,18 (2): 75-76; ) or unsafe to use a large amount of organic solvent (Chinese Journal of biochemical Pharmaceutics, 1996,17 (1): 17; application technology, 1998,4: 9-10; US Patent, 3,965,131; US Patent, 4,331,607; USPatent, 4,163,017), can not be meet the requirements of modern industry, CDCA and low purity prepared costly.
PATENT
https://patents.google.com/patent/WO2007069814A1/en
Chenodeoxycholic acid is generally contained in bile of cow, swine, bear, or poultry such as chicken or goose, as well as in bile of human. Chenodeoxycholic acid is used as starting material for the preparation of ursodeoxycholic acid which is effective to alleviate biliary system diseases, hyperlipidemia, cholelithiasis, and chronic liver diseases, and a typical process for preparing ursodeoxycholic acid known in the art is as follows.
A typical process for preparing chenodeoxycholic acid comprises the steps of: esterifying cholic acid (3α,7α,12θ!-trihydroxy cholic acid) with methyl; protecting the hydroxyl group of 3α and Ia position by acetylating them with anhydrous acetic acid; oxidizing the hydroxyl group of 12α position to carbonyl group by using chromic acid, and then removing the carbonyl group by Wolff-kichner reduction reaction; hydrolyzing and deprotecting the obtained product to yield chenodeoxycholic acid. The above process requires the reaction to be maintained at a high temperature of more than 200 °C , and the supply of raw material may be interrupted by bovine spongiform encephalopathy, etc. Bile ,of poultry contains chenodeoxycholic acid, lithocholic acid, and a small amount of cholic acid. Thus, the process for separating chenodeoxycholic acid from poultry is well known in the art, but is not economically reasonable due to the supply decrease of raw material and low yield [see, Windhaus et al, I Physiol. Chem., 140, 177-185 (1924)].
US Patent No. 4,186,143 disclosed a process for purely separating and purifying chenodeoxycholic acid from chenodeoxycholic acid mixture derived from natural swine bile. This process comprises the major steps of: pre-treatment to remove 3ohydroxy-6- oxo-5/3-cholic acid by saponification of bile; esterification of bile acid; acetylation of bile acid ester; removal of intermediate product by using non-polar organic solvent; crystallization of acetylated ester of formula I; deprotection; and production of the compound of formula I by using crystallization in organic solvent. However, this patent does not describe HPLC content for acetylated ester of formula I, and the purity of the final product is very low since the specific rotatory power is [ofo25 +13.8° (c=l, CHCl3), and the melting point is 119-121 °C [STD: [α]D 25 +15.2°(c=l, CHCl3), melting point 127- 129 “C]. Also, the crystallization for purifying the final product requires a very long time (i.e., 16-48 hours), and the entire process is complex as eight (8) steps. Thus, when purifying the compound of formula I by using the above process, the yield of the final product becomes low, and the reaction time is as long as 12 days. Therefore, the process is not economically reasonable.
Step 6: Deprotection and crystallization of chenodeoxycholic acid
To 220ml of water were added 24.5g of chenodeoxycholic acid-diacetate-ester and 29.5g of sodium hydroxide, and then the solution was stirred with reflux for 4 hours. To the solution was added 370ml of water. The solution’s pH is adjusted to 2.0-3.0 by using 59ml of hydrochloric acid. Then, the solution was stirred at 35-45 °C for 1 hour, and then filtered. The filtered material was washed with 24.5ml of water and dried in vacuum at 70 °C to obtain 19.5g of pure chenodeoxycholic acid, m.p.: 160-161 °C, [α]o25 +13.0°(c=l, CHCl3).
Step 8: Production of the compound of formula I
The reaction solution was extracted by using ethyl acetate, and aqueous layer was discarded therefrom. Ethyl acetate layer in the solution was washed with 6% saline, and the solution was distilled to about 90ml. This solution was cooled, kept cool for one day after adding 90ml of hexane, and filtered. Thus filtered material was washed with 20ml of hexane, and dried in vacuum at 60 °C to produce 12.7g of chenodeoxycholic acid. m.p. 142-1450C; [α]D 25 +13.0°(c=l, CHCl3). INDUSTRIAL APPLICABILITY The present invention can purify chenodeoxycholic acid of formula I from swine bile solid in high yield and purity. Also, the present invention is suitable for industrial purification by reducing the purification time.
PATENT
https://patents.google.com/patent/CN102060902A/en
chenodeoxycholic acid (3 α, 7 α – dihydroxy _5 β – cholestane-24-oic acid) Chenodeoxycholic Ac id (referred to as CDCA), clinically used to correct dissolving cholesterol calculi and bile saturation drugs, the main function is to reduce the cholesterol in the bile saturation, large doses can inhibit the synthesis of cholesterol CDCA and increasing bile gallstone patients cholesterol level in a non-saturated, thereby preventing the formation of cholesterol gallstones of cholesterol and promote stone dissolve and fall off. It also has significant anti-asthmatic, anti-inflammatory, antitussive and expectorant effects.
[0003] Synthesis of chenodeoxycholic acid or ursodeoxycholic acid (3 α, 7β_ -5β_ dihydroxy-cholestane-24-oic acid, ursodeoxycholic Acid, referred UDCA), a key intermediate. Ursodeoxycholic acid is the main active ingredient of precious Chinese medicine bear bile, used in a variety of clinical hepatobiliary disease and dyspepsia. Currently we bear bile resources are scarce, mainly used synthetic chemical ursodeoxycholic acid as a clinical treatment. Therefore, the preparation of chenodeoxycholic acid is also important for the preparation of ursodeoxycholic acid.
[0004] CDCA mainly come from poultry or livestock bile extraction. Traditional extraction process complicated operation, low yield, (pharmaceutical industry, 1987,18 (9), 416; Chinese Journal of Biochemical Pharmaceutics, 1996,17 (1), 17; Applied Technology, 1998, (4), 9; CN1850846A ) can not meet the needs of modern industry. Chemical synthesis of chenodeoxycholic acid have also been reported (Japanese Journal of Chemistry 1955,76 (3), 297 -J Org Chem 1982,47 (2): 2331; Journal of Biochemical Pharmaceutics 1987,1,6 -, Tap Chi Duoc ^ oc2004 , 44 (1), 11; CN1869043A), but lower yield widespread pollution major problem, especially in the oxidation reaction is often used to expensive, and polluting agents.Therefore, to reduce pollution, reduce environmental hazards, streamline operations, improve yield, reduce costs, important for the synthesis of chenodeoxycholic acid.
Preparation of chenodeoxycholic acid.
[0007]
Cholic acid esters prepared by (1) Weigh 50 g of cholic acid, dissolved in 150 ml of anhydrous methanol was added 5 ml of concentrated hydrochloric acid was refluxed for 30 minutes, cooled slowly into the freezer, the available capacity methyl cholate It was 95%.
(2) hydroxy -12α- diacetyl – Preparation of methyl cholate methyl cholate weighed 50 g, was dissolved in 100 ml of pyridine was purified, dissolved completely, 100 ml of acetic anhydride was stirred at room temperature for 3 to 4 hours, poured into 500 ml of water, a white precipitate in the refrigerator, filtered the next day, diacetyl -12α- available hydroxy – methyl cholate, yield 40%.
(3) 3α, 7α–diacetoxy-12-oxo – Preparation of methyl cholanic acid prepared above was weighed 25 g of crude product, dissolved in 250 ml of acetone, filtered to remove insolubles, the stirring conditions , the Jones reagent was slowly added, at room temperature for 30 minutes, filtered, water was added to the filtrate precipitated white precipitate was filtered available 3α, 7α–diacetoxy-12-oxo – methyl-cholanic acid. The yield was 100%.
(4) 12- oxo – Preparation of chenodeoxycholic acid in ethanol 10% – sodium hydroxide solution and saponified for 1 hour at room temperature, the solution was acidified, poured into water to give 12- oxo – chenodeoxycholic acid , 100% yield.Recrystallized in absolute ethanol.
Preparation of chenodeoxycholic acid (5) was weighed 12- oxo – chenodeoxycholic acid, 20 grams, was added 300 ml of ethylene glycol and 30 g of solid sodium hydroxide and 300 ml of hydrazine hydrate (85%), 100 ℃ refluxed for 2 hours, warming gradually raised to 130. ℃, generated by hydrazine hydrate was distilled off, continue to heat up to 185 ~ 190 ℃, continued reflux for 4 hours, cooled to a lower temperature, poured into water and heat, PH adjusted with hydrochloric acid (20%) 3, a white precipitate was filtered cake was washed with water to give chenodeoxycholic acid.
(6) Purification of chenodeoxycholic acid obtained weighed amount of chenodeoxycholic acid, dissolved with a small amount of ethanol, was impregnated on a silica gel column petroleum ether, liquid flow linear velocity by column chromatography 1 ~ 5cm / control points, with petroleum ether: acetone = 2, begins to elute, detected by TLC chromatography therebetween, Junichi appearance of spots to be chenodeoxycholic acid appears to start collecting the eluate until no Chenodeoxy acid spots, distillation under reduced pressure and dried to give pure higher chenodeoxycholic acid.
PATENTS
 |
|
 |
|
| Names | |
|---|---|
| IUPAC names
chenodiol
OR 3α,7α-dihydroxy-5β-cholanic acid OR 5β-cholanic acid-3α,7α-diol OR (R)-((3R,5S,7R,8R,9S,10S,13R,14S,17R)-3,7-dihydroxy-10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid |
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| DrugBank | |
| ECHA InfoCard | 100.006.803 |
| EC Number | 207-481-8 |
| KEGG | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H40O4 | |
| Molar mass | 392.57 g/mol |
| Melting point | 165 to 167 °C (329 to 333 °F; 438 to 440 K) |
| Pharmacology | |
| A05AA01 (WHO) | |
| License data | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
////////////////////Chenodeoxycholic acid, ケノデオキシコール酸 , orphan designation
[H][C@@]1(CC[C@@]2([H])[C@]3([H])[C@H](O)C[C@]4([H])C[C@H](O)CC[C@]4(C)[C@@]3([H])CC[C@]12C)[C@H](C)CCC(O)=O

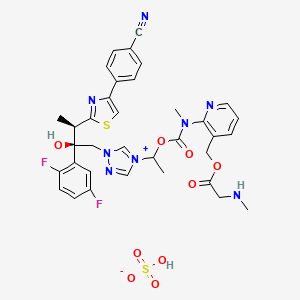

| MOLECULAR FORMULA: | C35H36F2N8O9S2 |
|---|---|
| MOLECULAR WEIGHT: | 814.837 g/mol |


Syn……https://newdrugapprovals.org/2013/10/02/isavuconazole-basilea-reports-positive-results-from-study/
PRODUCT PATENT
https://patents.google.com/patent/US6300353
InventorTadakatsu HayaseShigeyasu IchiharaYoshiaki IsshikiPingli LiuJun OhwadaToshiya SakaiNobuo ShimmaMasao TsukazakiIsao UmedaToshikazu Yamazaki
Current Assignee Basilea Pharmaceutica International Ltd Original
AssigneeBasilea Pharmaceutica AG Priority date 1998-03-06
https://patents.google.com/patent/WO1999045008A1/en
POLYMORPHS OF BASE
WO 2016055918
https://patents.google.com/patent/WO2016055918A1/en
PATENT
IN 2014MU03189
WOCKHARDT
Isavuconazole, isavuconazonium, Voriconazole, and Ravuconazole are azole derivatives and known as antifungal drugs for treatment of systemic mycoses as reported in US 5,648,372, US 5,792,781, US 6,300,353 and US 6,812,238. The US patent No. 6,300,353 discloses Isavuconazole and its process. It has chemical name [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5- difluorophenyl)-butan-2-ol;
The Isavuconazonium iodide hydrochloride and Isavuconazonium sulfate can be prepared according to known methods, e.g. pending Indian Patent Applications IN 2424/MUM/2014 and IN 2588/MUM/2014.
Example-1: Preparation of Amorphous Isavuconazole

4-cyano Phenacyl bromide F F N N N OH N S CN Formula-I Formula-III In a round bottomed flask charged ethanol (250 ml), thioamide compound of formula-II (25.0 gm) and 4-cyano phenacyl bromide (18.4 gm) under stirring. The reaction mixture were heated to 70 0C. After completion of reaction the solvent was removed under vacuum distillation and water (250 ml) and Ethyl acetate (350 ml) were added to reaction mass. The reaction mixture was stirred and its pH was adjusted between 7 to 7.5 by 10 % solution of sodium bicarbonate. The layer aqueous layer was discarded and organic layer was washed with saturated sodium chloride solution (100 ml) and concentrated under vacuum to get residue. The residue was suspended in methyl tert-butyl ether (250 ml) and the reaction mixture was heated to at 40°C to make crystals uniform and finally reaction mass is cooled to room temperature filtered and washed with the methyl tert-butyl ether. The product was isolated dried to get pale yellowish solid product. Yield: 26.5 gm HPLC purity: 92.7%
CLIP
March 6, 2015
The U.S. Food and Drug Administration today approved Cresemba (isavuconazonium sulfate), a new antifungal drug product used to treat adults with invasive aspergillosis and invasive mucormycosis, rare but serious infections.
Aspergillosis is a fungal infection caused by Aspergillus species, and mucormycosis is caused by the Mucorales fungi. These infections occur most often in people with weakened immune systems.
Cresemba belongs to a class of drugs called azole antifungal agents, which target the cell wall of a fungus. Cresemba is available in oral and intravenous formulations.
“Today’s approval provides a new treatment option for patients with serious fungal infections and underscores the importance of having available safe and effective antifungal drugs,” said Edward Cox, M.D., M.P.H, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Cresemba is the sixth approved antibacterial or antifungal drug product designated as a Qualified Infectious Disease Product (QIDP). This designation is given to antibacterial or antifungal drug products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act.
As part of its QIDP designation, Cresemba was given priority review, which provides an expedited review of the drug’s application. The QIDP designation also qualifies Cresemba for an additional five years of marketing exclusivity to be added to certain exclusivity periods already provided by the Food, Drug, and Cosmetic Act. As these types of fungal infections are rare, the FDA also granted Cresemba orphan drug designations for invasive aspergillosis and invasive mucormycosis.
The approval of Cresemba to treat invasive aspergillosis was based on a clinical trial involving 516 participants randomly assigned to receive either Cresemba or voriconazole, another drug approved to treat invasive aspergillosis. Cresemba’s approval to treat invasive mucormycosis was based on a single-arm clinical trial involving 37 participants treated with Cresemba and compared with the natural disease progression associated with untreated mucormycosis. Both studies showed Cresemba was safe and effective in treating these serious fungal infections.
The most common side effects associated with Cresemba include nausea, vomiting, diarrhea, headache, abnormal liver blood tests, low potassium levels in the blood (hypokalemia), constipation, shortness of breath (dyspnea), coughing and tissue swelling (peripheral edema). Cresemba may also cause serious side effects including liver problems, infusion reactions and severe allergic and skin reactions.
Cresemba is marketed by Astellas Pharma US, Inc., based in Northbrook, Illinois.

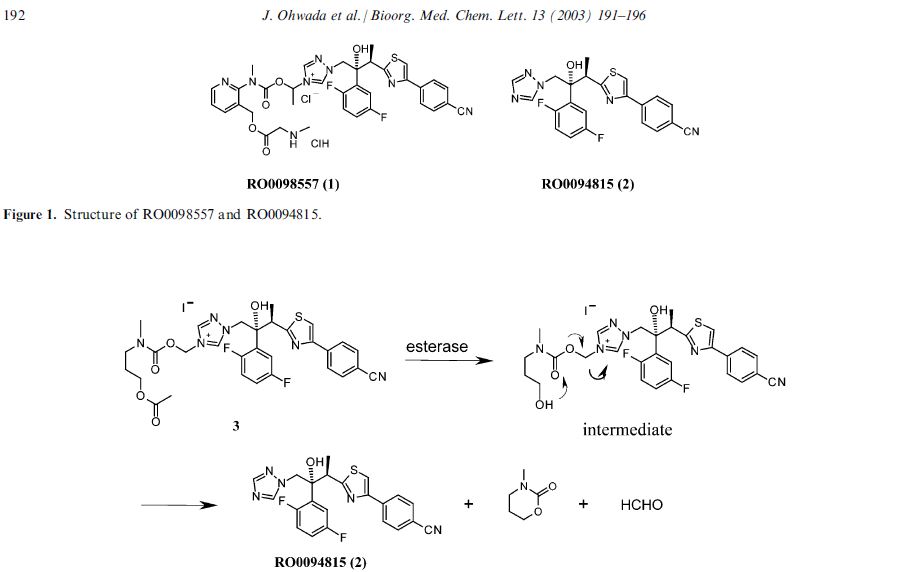
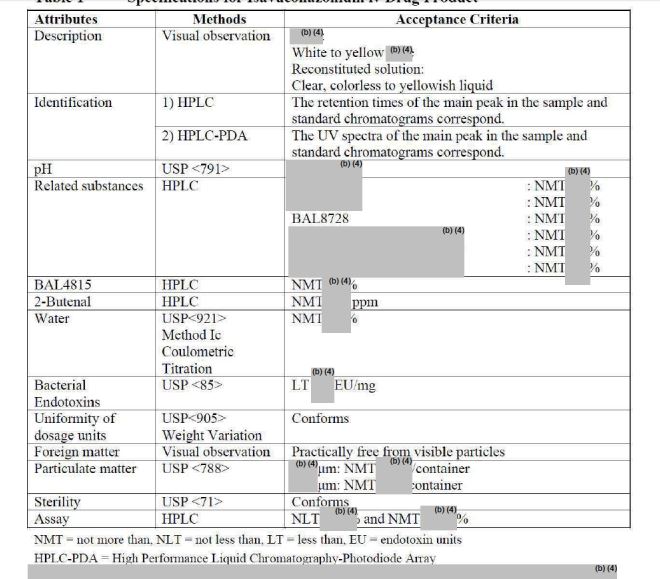
The active substance is isavuconazonium sulfate, a highly water soluble pro-drug of the active triazole isavuconazole. The chemical name of the active substance isavuconazonium sulfate is 1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47. The active substance has the following structure:
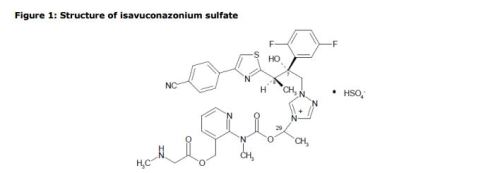
The structure of the active substance has been confirmed by elemental analysis, mass spectrometry, UV, IR, 1H-, 13C- and 19F-NMR spectrometry, and single crystal X-ray analysis, all of which support the chemical structure. It appears as a white, amorphous, hygroscopic powder. It is very soluble in water and over the pH range 1-7. It is also very soluble in methanol and sparingly soluble in ethanol. Two pKa values have been found and calculated to be 2.0 and 7.3. Its logPoct/wat calculated by software is 1.31.
Isavuconazonium sulfate has three chiral centres. The stereochemistry of the active substance is introduced by one of the starting materials which is controlled by appropriate specification. The two centres, C7 and C8 in the isavuconazole moiety and in an intermediate of the active substance, have R configuration. The third chiral centre, C29, is not located on isavuconazole moiety and has both the R and S configurations. The nondefined stereo centre at C29 has been found in all batches produced so far to be racemic. Erosion of stereochemical purity has not been observed in the current process. The active substance is a mixture of two epimers of C29.
An enantiomer of drug substance was identified as C7 (S), C8 (S) and C29 (R/S) structure. The control of the stereochemistry of isavuconazonium sulfate is performed by chiral HPLC on the active substance and its two precursors. Subsequent intermediates are also controlled by relevant specification in the corresponding steps. Two crystal forms have been observed by recrystallisation studies. However the manufacturing process as described yields amorphous form only.
Two different salt forms of isavuconazonuium (chloride and sulfate) were identified during development. The sulfate salt was selected for further development. A polymorph screening study was also performed. None of the investigated salts could be obtained in crystalline Form………http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002734/WC500196130.pdf

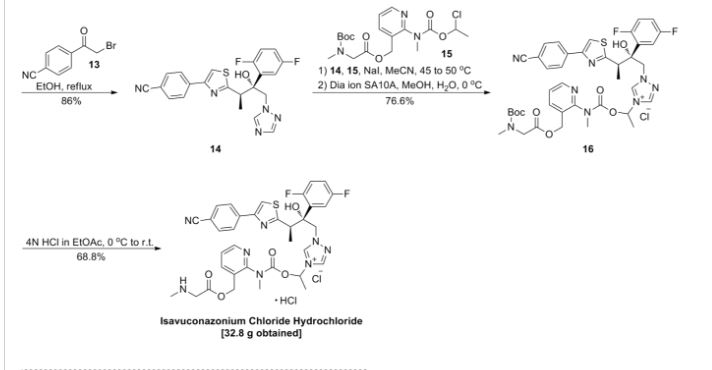

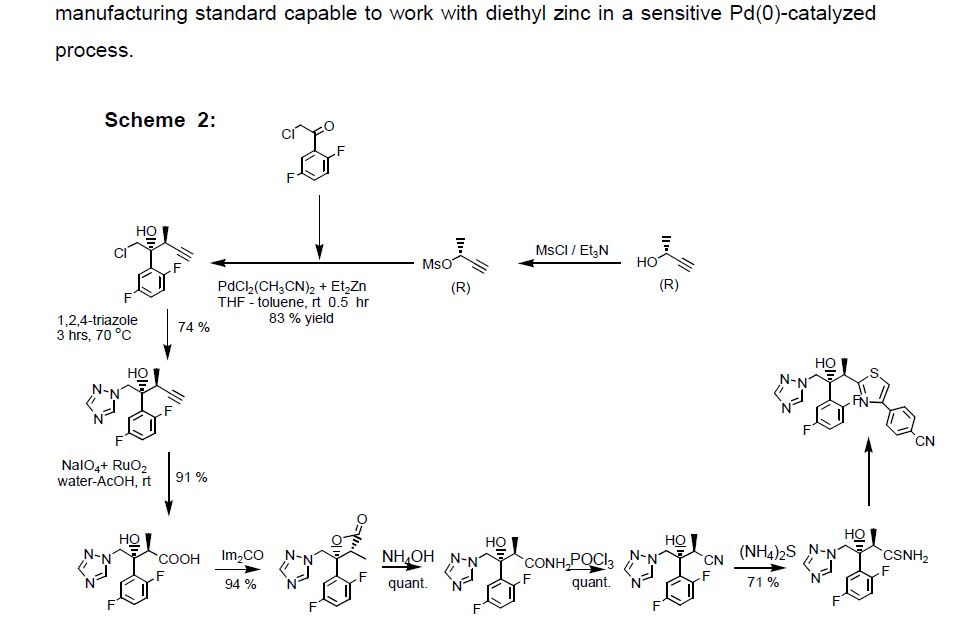
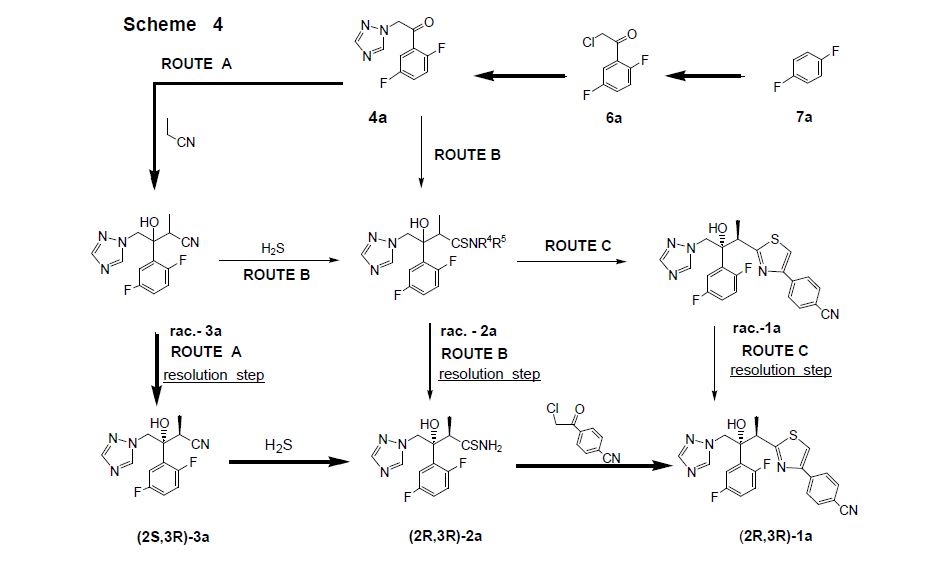
Clip








Isavuconazonium (Cresemba ) is a water-soluble prodrug of the triazole antifungal isavuconazole (BAL4815), a 14-a-demethylase inhibitor, under development byBasilea Pharmaceutica International Ltd and Astellas Pharma Inc. Isavuconazonium, in both its intravenous and oral formulations, was approved for the treatment of invasive aspergillosis and invasive mucormycosis (formerly termed zygomycosis) in the US in March 2015. Isavuconazonium is under regulatory review in the EU for invasive aspergillosis and mucormycosis. It is also under phase III development worldwide for the treatment of invasive candidiasis and candidaemia. This article summarizes the milestones in the development of isavuconazonium leading to the first approval for invasive spergillosis and mucormycosis.
Introduction
The availability of both an intravenous (IV) and an oral formulation of isavuconazonium (Cresemba ), as a result of its water solubility, rapid hydrolysis to the active entity isavuconazole and very high oral bioavailability, provides maximum flexibility to clinicians for treating seriously ill patients with invasive fungal infections [1]. Both the IV and oral formulations have been approved by the US Food and Drug Administration (FDA) to treat adults with invasive aspergillosis and invasive mucormycosis [2]. The recommended dosages of each formulation are identical, consisting of loading doses of 372 mg (equivalent to 200 mg of isavuconazole) every eight hours for six doses, followed by maintenance therapy with 372 mg administered once daily [3]. The Qualified Infectious Disease Product (QIDP) designation of the drug with priority review status by the FDA isavuconazonium in the US provided and a five year extension of market exclusivity from launch. Owing to the rarity of the approved infections,
isavuconazonium was also granted orphan drug designation by the FDA for these indications [2]. It has also been granted orphan drug and QIDP designation in the US for the treatment of invasive candidiasis [4]. In July 2014, Basilea Pharmaceutica International Ltd submitted a Marketing Authorization Application to the European Medicines Agency (EMA) for isavuconazonium in the treatment of invasive aspergillosis and invasive mucormycosis, indications for which the EMA has granted isavuconazonium orphan designation [5, 6]. Isavuconazonium is under phase III development in many countries worldwide for the treatment of invasive candidiasis and candidaemia.
1.1 Company agreements
In 2010, Basilea Pharmaceutica International Ltd (a spinoff from Roche, founded in 2000) entered into a licence agreement with Astellas Pharma Inc in which the latter would co-develop and co-promote isavuconazonium worldwide, including an option for Japan. In return for milestone payments, Astellas Pharma was granted an exclusive right to commercialize isavuconazonium, while Basilea Pharmaceutica retained an option to co-promote the drug in the US, Canada, major European countries and China [7]. The companies amended their agreement in 2014, making Astellas Pharma responsible for all regulatory filings, commercialization and manufacturing of isavuconazonium in the US and Canada. Basilea Pharmaceutica waived its right to co-promote the product in the US and Canada, in order to assume all rights in the rest of the world [8]. However, Astellas Pharma remains as sponsor of the multinational, phase III ACTIVE trial in patients with invasive candidiasis.
2 Scientific Summary
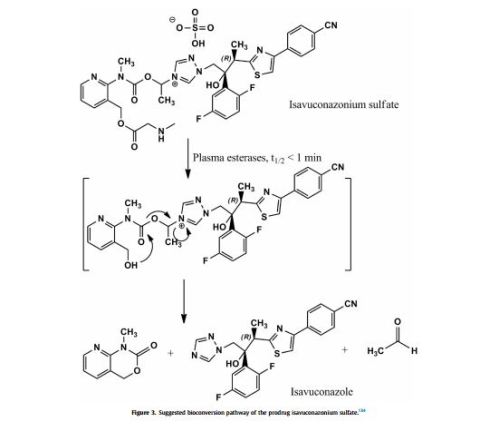
Isavuconazonium (as the sulphate; BAL 8557) is a prodrug that is rapidly hydrolyzed by esterases (mainly butylcholinesterase) in plasma into the active moiety isavuconazole
(BAL 4815) and an inactive cleavage product (BAL 8728).
References
1. Falci DR, Pasqualotto AC. Profile of isavuconazole and its potential in the treatment of severe invasive fungal infections. Infect Drug Resist. 2013;6:163–74.
2. US Food and Drug Administration. FDA approves new antifungal drug Cresemba. 2015. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm437106.htm. Accessed 12 Mar 2015.
3. US Food and Drug Administration. Cresemba (isavuconazonium sulfate): US prescribing information. 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207500Orig1s000lbl.pdf. Accessed 18 Mar 2015.
4. Astellas Pharma US Inc. FDA grants Astellas Qualified Infectious Disease Product designation for isavuconazole for the treatment of invasive candidiasis (media release). 2014. http://newsroom astellas.us/2014-07-16-FDA-Grants-Astellas-Qualified-Infectious-Disease-Product-Designation-for-Isavuconazole-for-the-Treatmentof-Invasive-Candidiasis.
5. European Medicines Agency. Public summary of opinion on orphan designation: isavuconazonium sulfate for the treatment of invasive aspergillosis. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2014/07/WC500169890.pdf. Accessed 18 Mar 2015.
European Medicines Agency. Public summary of opinion on orphan designation: isavuconazonium sulfate for the treatment of mucormycosis. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2014/07/WC500169714.pdf. Accessed 18 Mar 2015.
7. Basilea Pharmaceutica. Basilea announces global partnership with Astellas for its antifungal isavuconazole (media release).2010. http://www.basilea.com/News-and-Media/Basilea-announcesglobal-partnership-with-Astellas-for-its-antifungal-isavuconazole/343.
8. Basilea Pharmaceutica. Basilea swaps its isavuconazole North American co-promote rights for full isavuconazole rights outside of North America (media release). 2014. http://www.basilea.com/News-and-Media/Basilea-swaps-its-isavuconazole-North-Americanco-promote-rights-for-full-isavuconazole-rights-outside-


CLIP
http://www.jpharmsci.org/article/S0022-3549(15)00035-0/pdf
http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207500Orig1207501Orig1s000ChemR.pdf








On 4 July 2014 orphan designation (EU/3/14/1284) was granted by the European Commission to Basilea Medical Ltd, United Kingdom, for isavuconazonium sulfate for the treatment of invasive aspergillosis.
Update: isavuconazonium sulfate (Cresemba) has been authorised in the EU since 15 October 2015. Cresemba is indicated in adults for the treatment of invasive aspergillosis.
Consideration should be given to official guidance on the appropriate use of antifungal agents.
The active substance is isavuconazonium sulfate, a highly water soluble pro-drug of the active triazole isavuconazole. The chemical name of the active substance isavuconazonium sulfate is 1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47. The active substance has the following structure
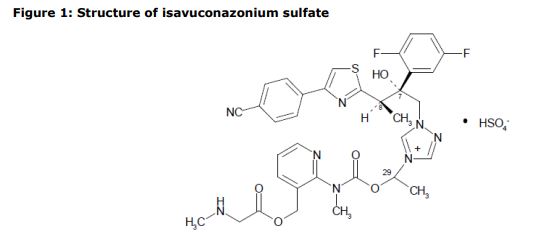
It appears as a white, amorphous, hygroscopic powder. It is very soluble in water and over the pH range 1-7. It is also very soluble in methanol and sparingly soluble in ethanol. Two pKa values have been found and calculated to be 2.0 and 7.3. Its logPoct/wat calculated by software is 1.31.
Isavuconazonium sulfate has three chiral centres. The stereochemistry of the active substance is introduced by one of the starting materials which is controlled by appropriate specification. The two centres, C7 and C8 in the isavuconazole moiety and in an intermediate of the active substance, have R configuration. The third chiral centre, C29, is not located on isavuconazole moiety and has both the R and S configurations. The nondefined stereo centre at C29 has been found in all batches produced so far to be racemic. Erosion of stereochemical purity has not been observed in the current process. The active substance is a mixture of two epimers of C29. An enantiomer of drug substance was identified as C7 (S), C8 (S) and C29 (R/S) structure. The control of the stereochemistry of isavuconazonium sulfate is performed by chiral HPLC on the active substance and its two precursors.
US 6812238
US 7459561
| FDA ORANGE BOOK PATENTS: 1 OF 2 | |
|---|---|
| Patent | 7459561 |
| Expiration | Oct 31, 2020 |
| Applicant | ASTELLAS |
| Drug Application | N207500 (Prescription Drug: CRESEMBA. Ingredients: ISAVUCONAZONIUM SULFATE) |
FREE FORM
Isavuconazonium; Isavuconazonium ion; Cresemba; BAL-8557; 742049-41-8;
[2-[1-[1-[(2R,3R)-3-[4-(4-cyanophenyl)-1,3-thiazol-2-yl]-2-(2,5-difluorophenyl)-2-hydroxybutyl]-1,2,4-triazol-4-ium-4-yl]ethoxycarbonyl-methylamino]pyridin-3-yl]methyl 2-(methylamino)acetate
| MOLECULAR FORMULA: | C35H35F2N8O5S+ |
|---|---|
| MOLECULAR WEIGHT: | 717.773 g/mol |
Patent IDDatePatent Title
US20102494262010-09-30STABILIZED PHARMACEUTICAL COMPOSITION
US74595612008-12-02N-substituted carbamoyloxyalkyl-azolium derivativesUS71898582007-03-13N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
US71511822006-12-19Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives
US68122382004-11-02N-substituted carbamoyloxyalkyl-azolium derivatives
REF
http://www.drugbank.ca/drugs/DB06636
////////// BAL 8557, BAL-8557-002, CRESEMBA, ISAVUCONAZONIUM SULFATE, QIDP designation, Cresemba , priority review, FDA 2015, EU 2015, BAL8557-002, BCS CLASS I, orphan designation, invasive aspergillosis, invasive mucormycosis, RO-0098557 , AK-1820, fast track designation, QIDP, 946075-13-4
CC(C1=NC(=CS1)C2=CC=C(C=C2)C#N)C(CN3C=[N+](C=N3)C(C)OC(=O)N(C)C4=C(C=CC=N4)COC(=O)CNC)(C5=C(C=CC(=C5)F)F)O
CC(C1=NC(=CS1)C2=CC=C(C=C2)C#N)C(CN3C=[N+](C=N3)C(C)OC(=O)N(C)C4=C(C=CC=N4)COC(=O)CNC)(C5=C(C=CC(=C5)F)F)O.OS(=O)(=O)[O-]
UPDATE NEW PATENT

![]()
(WO2016016766) A PROCESS FOR THE PREPARATION OF ISAVUCONAZONIUM OR ITS SALT THEREOF
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
KHUNT, Rupesh Chhaganbhai; (IN).
RAFEEQ, Mohammad; (IN).
MERWADE, Arvind Yekanathsa; (IN).
DEO, Keshav; (IN)
The present invention relates to a process for the preparation of stable Isavuconazonium or its salt thereof. In particular of the present invention relates to process for the preparing of isavuconazonium sulfate, Isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide has purity more than 90%. The process is directed to preparation of solid amorphous form of isavuconazonium sulfate, isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide. The present invention process of Isavuconazonium or its salt thereof is industrially feasible, simple and cost effective to manufacture of isavuconazonium sulfate with the higher purity and better yield.
Isavuconazonium sulfate is chemically known l-[[N-methyl-N-3-[(methylamino) acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl)thiazol-2-yl]butyl]-lH-[l,2,4]-triazo-4-ium Sulfate and is structurally represented by formula (I):


Formula I
Isavuconazonium sulfate (BAL8557) is indicated for the treatment of antifungal infection. Isavuconazonium sulfate is a prodrug of Isavuconazole (BAL4815), which is chemically known 4-{2-[(lR,2R)-(2,5-Difluorophenyl)-2-hydroxy-l-methyl-3-(lH-l ,2,4-triazol-l-yl)propyl]-l ,3-thiazol-4-yl}benzonitrile compound of Formula II


Formula II
US Ppatent No. 6,812,238 (referred to herein as ‘238); 7,189,858 (referred to herein as ‘858); 7,459,561 (referred to herein as ‘561) describe Isavuconazonium and its process for the preparation thereof.
The US Pat. ‘238 patent describes the process of preparation of Isavuconazonium chloride hydrochloride.
The US Pat. ‘238 described the process for the Isavuconazonium chloride hydrochloride, involves the condensation of Isavuconazole and [N-methyl-N-3((tert-butoxycarbonyl methylamino) acetoxymethyl) pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester. The prior art reported process require almost 15-16 hours, whereas the present invention process requires only 8-10 hours. Inter alia prior art reported process requires too many step to prepare isavuconazonium sulfate, whereas the present invention process requires fewer steps.
Moreover, the US Pat. ‘238 describes the process for the preparation Isavuconazonium hydrochloride, which may be used as the key intermediate for the synthesis of isavuconazonium sulfate, compound of formula I. There are several drawbacks in the said process, which includes the use of anionic resin to prepare Isavuconazonium chloride hydrochloride, consequently it requires multiple time lyophilization, which makes the said prior art process industrially, not feasible.
The inventors of the present invention surprisingly found that Isavuconazonium or a pharmaceutically acceptable salt thereof in yield and purity could be prepared by using substantially pure intermediates in suitable solvent.
Thus, an object of the present invention is to provide simple, cost effective and industrially feasible processes for manufacture of isavuconazonium sulfate. Inventors of the present invention surprisingly found that isavuconazonium sulfate prepared from isavuconazonium iodide hydrochloride, provides enhanced yield as well as purity.
The process of the present invention is depicted in the following scheme:


Formula I
Formula-IA
The present invention is further illustrated by the following example, which does not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present application.
Examples
Example-1: Synthesis of l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino) acetoxymethyl]pyridin-2-yl]carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3 – [4-(4-cyanophenyl)thiazol-2-yl]butyl] – 1 H-[ 1 ,2,4] -triazo-4-ium iodide
Isavuconazole (20 g) and [N-methyl-N-3((tert-butoxycarbonylmethylamino)acetoxy methyl)pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester (24.7 g) were dissolved in acetonitrile (200ml). The reaction mixture was stirred to add potassium iodide (9.9 g). The reaction mixture was stirred at 47-50°C for 10-13 hour. The reaction mixture was cooled to room temperature. The reaction mass was filtered through celite bed and washed acetonitrile. Residue was concentrated under reduced pressure to give the crude solid product (47.7 g). The crude product was purified by column chromatography to get its pure iodide form (36.5 g).
Yield: 84.5 %
HPLC Purity: 87%
Mass: m/z 817.4 (M- 1)+
Example-2: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride
l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide (36.5 g) was dissolved in ethyl acetate (600 ml). The reaction mixture was cooled to -5 to 0 °C. The ethyl acetate hydrochloride (150 ml) solution was added to reaction mixture. The reaction mixture was stirred for 4-5 hours at room temperature. The reaction mixture was filtered and obtained solid residue washed with ethyl acetate. The solid dried under vacuum at room temperature for 20-24 hrs to give 32.0 gm solid.
Yield: 93 %
HPLC Purity: 86%
Mass: m/z 717.3 (M-HC1- 1)
Example-3: Preparation of Strong anion exchange resin (Sulfate).
Indion GS-300 was treated with aqueous sulfate anion solution and then washed with DM water. It is directly used for sulfate salt.
Example-4: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium Sulfate
Dissolved 10.0 g l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride in 200 ml deminerahzed water and 30 ml methanol. The solution was cooled to about 0 to 5°C. The strong anion exchange resin (sulfate) was added to the cooled solution. The reaction mixture was stirred to about 60-80 minutes. The reaction was filtered and washed with 50ml of demineralized water and methylene chloride. The aqueous layer was lyophilized to obtain
(8.0 g) white solid.
Yield: 93 %
HPLC Purity: > 90%
Mass: m/z 717.4 (M- HS04) +
PATENT
CN 105288648
PATENT
CN 106883226
https://patents.google.com/patent/CN106883226A/en
PATENT
CN 107982221
PAPER
| Title: Introduction of New Drugs Approved by the U.S. FDA in 2015 |
| Author: Ma Shuai; Wenying Ling; Zhou Weicheng; |
| Source: China Pharmaceutical Industry |
| Publisher: Tongfangzhiwang Beijing Technology Co., Ltd. |
| Year of publication: |
| DOI code: 10.16522/j.cnki.cjph.2016.01.022 |
| Registration Time: 2016-02-19 02:04:15 |
///////////////

(R)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine

ASLAN001 , Varlitinib
C22H19ClN6O2S
Molecular Weight: 466.94
Elemental Analysis: C, 56.59; H, 4.10; Cl, 7.59; N, 18.00; O, 6.85; S, 6.87
CAS: 845272-21-1 (Varlitinib); 1146629-86-8 (Varlitinib tosylate).
ASLAN001; ASLAN-001; ASLAN 001; AR 00334543; ARRY-334543; ARRY334543; ARRY-543; ARRY543; ARRY 543.
(R)-N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)-N6-(4-methyl-4,5-dihydrooxazol-2-yl)quinazoline-4,6-diamine.
(R)-4-[[3-Chloro-4-[(thiazol-2-yl)methoxy]phenyl]amino]-6-[(4-methyl-4,5-dihydrooxazol-2-yl)amino]quinazoline
4,6-Quinazolinediamine, N4-[3-chloro-4-(2-thiazolylmethoxy)phenyl]-N6-[(4R)-4,5-dihydro-4-methyl-2-oxazolyl]-
ASLAN Pharmaceuticals, a Singapore-based drugmaker, announced The Food and Drug Administration (FDA) gave an orphan drug designation on August 13 to its pan-HER inhibitor ASLAN001 (varlitinib), a drug candidate created to treat a destructive form of bile duct cancer called cholangiocarcinoma that has no known cure. ………http://www.dddmag.com/news/2015/08/aslan-pharmaceuticals-gains-orphan-designation-rare-cancer-drug
Current developer: Array Biopharma Inc,
![]()
Varlitinib, also known as ARRY-543 and ASLAN001, is an orally bioavailable inhibitor of the epidermal growth factor receptor family with potential antineoplastic activity.
Varlitinib (ASLAN-001) is an oncolytic drug in phase II clinical trials at ASLAN Pharmaceuticals for the treatment of gastric cancer and for the treatment of metastatic breast cancer in combination with capecitabine. Clinical development is also ongoing for the treatment of solid tumors in combination with cisplatin/FU and cisplatin/capecitabine. The product had been in phase I/II clinical trials at Array BioPharma for the treatment of patients with advanced pancreatic cancer. Phase II clinical trials had also been ongoing for the treatment of solid tumors. No recent development has been reported for this research
Varlitinib selectively and reversibly binds to both EGFR (ErbB-1) and Her-2/neu (ErbB-2) and prevents their phosphorylation and activation, which may result in inhibition of the associated signal transduction pathways, inhibition of cellular proliferation and cell death. EGFR and Her-2 play important roles in cell proliferation and differentiation and are upregulated in various human tumor cell types. Due to the dual inhibition of both EGFR and Her-2, this agent may be therapeutically more effective than agents that inhibit EGFR or Her-2 alone.
The drug is a dual inhibitor of the ErB-2 and EGFR receptor kinases, both of which have been shown to stimulate aberrant growth, prolong survival and promote differentiation of many tumor types. The compound behaves as a reversible ATP-competitive inhibitor with nanomolar potency both in vitro and in cell-based proliferation assays.
In 2011, the compound was licensed to Aslan Pharmaceuticals by Array BioPharma worldwide for the treatment of solid tumors, initially targeting patients with gastric cancer through a development program conducted in Asia.
In 2015, orphan drug designation was assigned to the compound in the U.S. for the treatment of cholangiocarcinoma.
SEE NMR ………….http://www.medkoo.com/Product-Data/Varlitinib/Varlitinib-QC-KB20121128web.pdf
……………..
https://www.google.co.in/patents/US20050043334
Example 52
(R)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine
Prepared using (R)-2-aminopropan-1-o1. MS APCI (+) m/z 467, 469 (M+1, Cl pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, 1H), 8.47 (s, 1H), 8.09 (s, 1H), 7.86 (d, 1H), 7.81 (d, 1H), 7.77 (d, 1H), 7.69 (m, 3H), 7.32 (d, 1H), 7.02 (s, 1H), 5.54 (s, 2H), 4.47 (m, 1H), 3.99 (m, 1H), 3.90 (m, 1H), 1.18 (d, 3H).
Example 53

(S)-N4-[3-Chloro-4-(thiazol-2-ylmethoxy)-phenyl]-N6-(4-methyl-4,5-dihydro-oxazol-2-yl)-quinazoline-4,6-diamine
Prepared using (S)-2-amino-propan-1-o1. MS APCI (+) m/z 467, 469 (M+1, Cl pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, 1H), 8.47 (s, 1H), 8.09 (s, 1H), 7.86 (d, 1H), 7.81 (d, 1H), 7.77 (d, 1H), 7.69 (m, 3H), 7.32 (d, 1H), 7.02 (s, 1H), 5.54 (s, 2H), 4.47 (m, 1H), 3.99 (m, 1H), 3.90 (m, 1H), 1.18 (d, 3H).
………………
PATENT
http://www.google.co.in/patents/WO2005016346A1?cl=en
Example 52

R VN4-r3-Chloro-4-(‘thiazol-2-v-metho-xy)-phenyll-N6-(4-methyl-4,5-dihvdro-oxazol- 2-yl)-quinazoUne-4,6-diamine
[00194] Prepared using (R)-2-aminopropan- 1 -ol. MS APCI (+) m/z 467, 469
(M+l, CI pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, IH), 8.47 (s, IH), 8.09 (s, IH), 7.86 (d, IH), 7.81 (d, IH), 7.77 (d, IH), 7.69 (m, 3H), 7.32 (d, IH), 7.02 (s, IH), 5.54 (s, 2H), 4.47 (m, IH), 3.99 (m, IH), 3.90 (m, IH), 1.18 (d, 3H). Example 53

(S)-N4-|“3-Chloro-4- thiazol-2-ylmethoxy)-phenyll-N6-(‘4-methyl-4,5-dihvdro-oxazol- 2-yl)-quinazoline-4,6-diamine [00195] Prepared using (S)-2-amino-propan- 1 -ol. MS APCI (+) m z 467, 469
(M+l, CI pattern) detected; 1H NMR (400 mHz, DMSO-D6) δ 9.53 (s, IH), 8.47 (s, IH), 8.09 (s, IH), 7.86 (d, IH), 7.81 (d, IH), 7.77 (d, IH), 7.69 (m, 3H), 7.32 (d, IH), 7.02 (s, IH), 5.54 (s, 2H), 4.47 (m, IH), 3.99 (m, IH), 3.90 (m, IH), 1.18 (d, 3H).
………
CAUTION a very similar molecule but not same
 NOTE……..METHYL NEXT TO OXYGEN ATOM
NOTE……..METHYL NEXT TO OXYGEN ATOM
DOI: 10.1002/cjoc.201400271,…………http://onlinelibrary.wiley.com/doi/10.1002/cjoc.201400271/abstract;jsessionid=04211D7526D97240F44E4355C6C57F50.f01t01
CAUTION …THIS IS NOT SAME
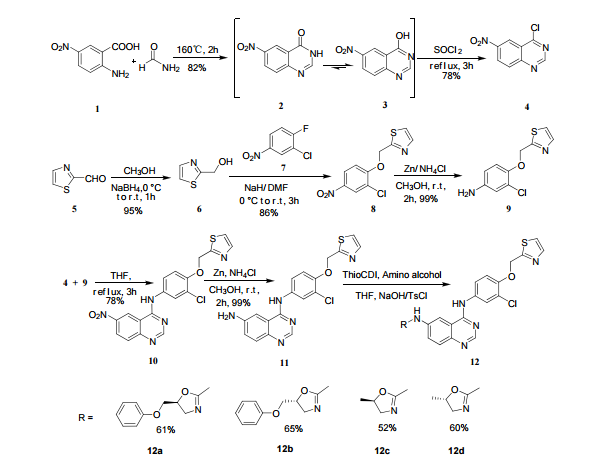
A novel type of quinazoline derivatives, which were designed by the combination of quinazoline as the backbone and oxazole scaffold as the substituent, have been synthesized and their biological activities were evaluated for anti-proliferative activities and EGFR inhibitory potency. Compound 12b demonstrated the most potent inhibitory activity (IC50=0.95 µmol/L for EGFR), which could be optimized as a potential EGFR inhibitor in the further study. The structures of the synthesized quinazoline analogs and all intermediates were comfirmed by 1H and 13C NMR, 2D NMR spectra, IR spectra and MS spectra.
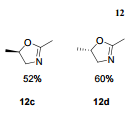
12c: Employing the same method as above, compound 12c was prepared and the amino alcohol was (S)-2-amino-propan-1-ol. Yellow solid, yield 52 %. m.p. 243-244 °C; [α] 20D =﹢22.5 ° (c 1.0, CH3CN); 1 H NMR (DMSO-D6): δ 9.54 (s, 1 H), 8.46 (s, 1 H), 8.06 (s, 2 H), 7.85 (d, 2 H, J=3.3 Hz), 7.79 (d, 2 H, J=3.3 Hz), 7.75 (d, 1 H, J=8.9 Hz), 7.64 (d, 1 H, J=8.3 Hz), 7.30 (d, 1 H, J=9.0 Hz), 5.54 (s, 2 H), 4.76 (m, 1 H), 3.72 (s, 1 H), 3.19 (s, 1 H), 1.34 (d, 3 H, J=6.15 Hz). 13C NMR (DMSO-D6) δ: 165.8, 156.9, 152.0, 148.8, 145.3, 142.6, 134.3, 128.7, 128.0, 123.5, 121.7, 121.3, 121.0, 115.6, 114.6, 72.5, 67.7, 63.0, 29.8, 29.0, 20.0, 13.9. IR (KBr) ν: 3439, 3278, 3101, 2925, 1660, 1631, 1601, 1557, 1500, 1428, 1404, 1384, 1329, 1291, 1257, 1225, 1052 cm-1. Anal. calcd for C22H19N6O2SCl: C 55.59, H 4.10, N 18.00, O 6.85; found C 55.55, H 4.13, N 18.02, O 6.78; MS (ESI) m/z: 467.2 (M+H).
12d: Employing the same method as above, compound 12d was prepared and the amino alcohol was (R)-2-amino-propan-1-ol. Yellow solid, yield 60%. m.p. 242-243 °C; [α] 20D = ﹣22.3 ° (c 1.0, CH3CN); 1 H NMR (DMSO-D6): δ 9.52 (s, 1 H), 8.80 (s, 1 H), 8.52 (dd, 1 H, J=2.7 Hz, J=8.9 Hz), 8.45 (s, 1 H), 8.30 (s, 1 H), 8.07 (s, 1 H), 7.85 (d, 1 H, J=3.2 Hz), 7.79 (d, 1 H, J=3.2 Hz), 7.75 (s, 1 H), 7.63 (d, 1 H, J=8.2 Hz), 7.31 (d, 1 H, J=9.0 Hz), 5.53 (s, 2 H), 4.76 (m, 1 H), 3.81 (s, 1 H), 3.19 (s, 1 H), 1.34 (d, 3 H, J=6.2 Hz). 13C NMR (DMSO-D6) δ: 165.8, 156.9, 152.0, 148.8, 145.3, 142.6, 134.3, 128.7, 128.0, 123.5, 121.7, 121.3, 121.0, 115.6, 114.6, 72.5, 67.7, 63.0, 29.8, 29.0, 20.0, 13.9. IR (KBr) ν: 3439, 3278, 3101, 2925, 1660, 1631, 1601, 1557, 1500, 1428, 1404, 1384, 1329, 1291, 1257, 1225, 1052 cm-1. Anal. calcd for C22H19N6O2SCl: C 55.59, H 4.10, N 18.00, O 6.85; found C 55.55, H 4.13, N 18.02, O 6.78; MS (ESI) m/z: 467.20 (M+H).
The above paper allows you to synthesize the key amino int 11 ………N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)quinazoline-4,6-diamine (11)
this can be applied to varlitinib till int 11
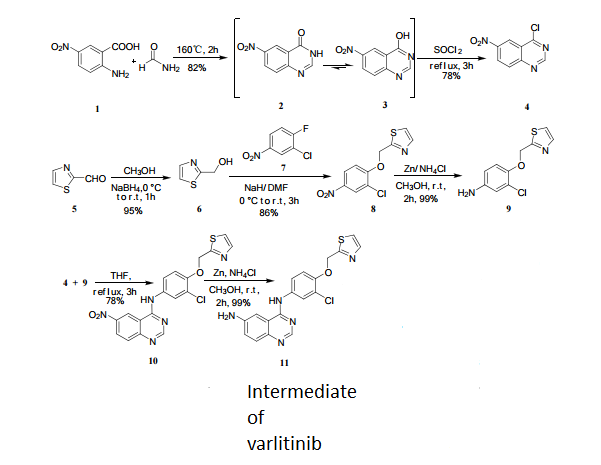
6-Nitro-4-hydroxyquinazoline (3)
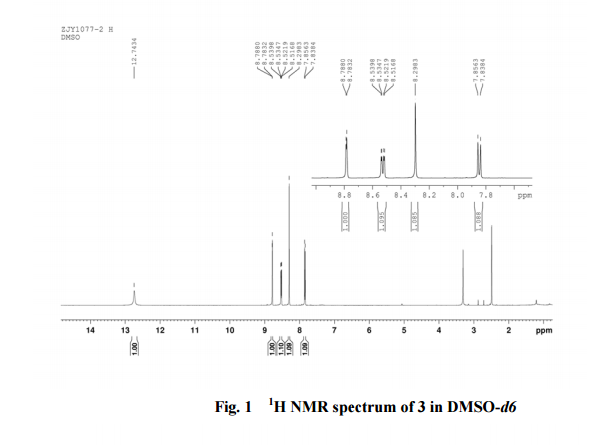
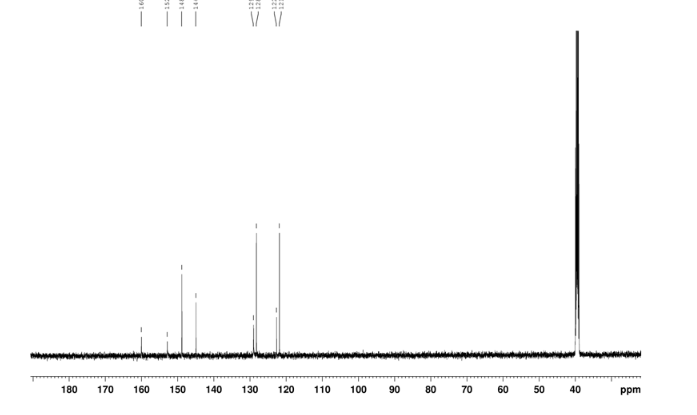
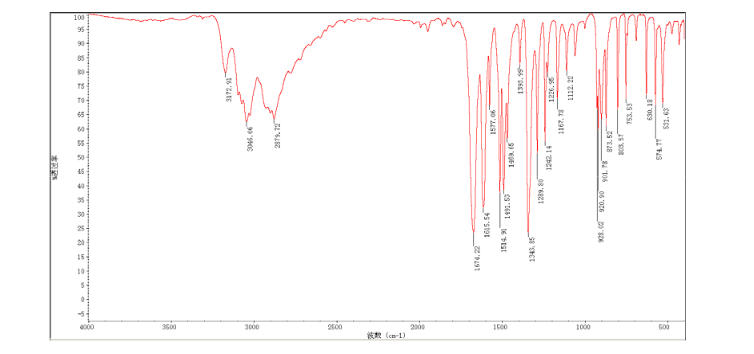
2-amino-5-nitrobenzoic acid (5.46 g, 30 mmol) was added to a 250 mL flask equipped with a reflux condenser. Then 50 mL formamide was added. The mixture was heated with vigorous stirring at 160 °C for 3 h. After cooling the solution was poured in ice-water to give 3 in almost pure form (Yellow solid 4.70 g, yield 82.0%). m.p. 317-318 °C; 1 H NMR (DMSO-d6): δ 12.74 (1 H, s, OH, exchangeable), 8.78 (1 H, d, J=2.4 Hz), 8.53 (1 H, dd, J=2.6 Hz, 9.0 Hz), 8.30 (s, 1 H), 7.84 (1 H, d, J=9.0 Hz); 13C NMR (DMSO-d6) δ: 160.1, 152.9, 148.9, 145.0, 129.1, 128.3, 122.7, 121.9. IR (KBr) ν: 3172, 3046, 2879, 1674, 1615, 1577, 1514, 1491, 1469, 1343, 1289, 1242, 1167, 1112, 928, 920, 901, 803, 753, 630, 574, 531 cm-1. Anal. calcd for C8H5N3O3: C 50.27, H 2.64, N 21.98; found C 50.30, H 2.65, N 21.96; MS (ESI) m/z: 189.97 (M-H).
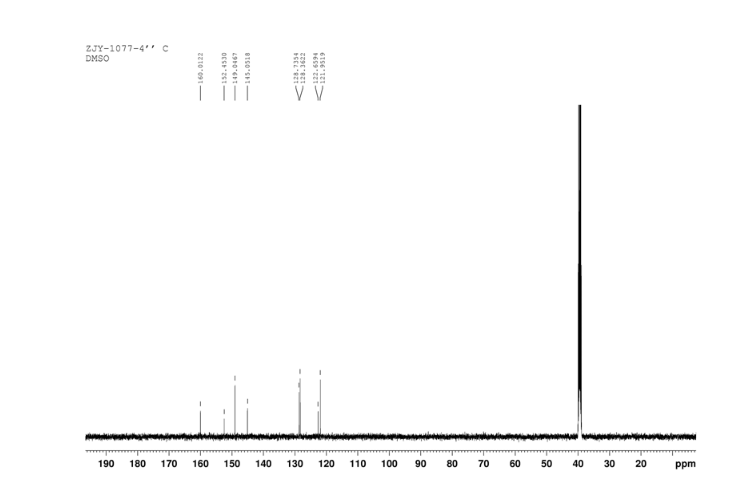
 13C NMR OF 3 IN DMSOD6
13C NMR OF 3 IN DMSOD6
IR
4-chloro-6-Nitroquinazoline (4)
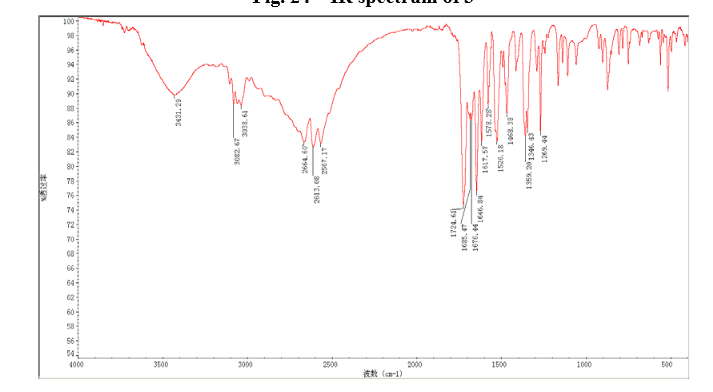
In a 100 mL flask equipped with a reflux condenser, 6-nitroquinazolin-4-one (2.86 g, 15 mmol) and thionyl chloride (SOCl2) 25 mL were added. The mixture was heated under reflux with vigorous stirring for 2 h. After the solution was clear, the reaction mixture was heated for another 2 h. Then, 150 mL of ice MeOH was dropped into it carefully, the mixture was extracted with CH2Cl2. The organic layer was S3 dried under MgSO4, filtered and the solvent removed to give 4-chloro-6-nitroquinazoline (4). Yellow solid 2.45 g, yield 78%. m.p. 134-135 °C; 1 H NMR (DMSO-d6): δ 8.80 (1 H, d, J=3.0 Hz), 8.54(1 H, dd, J=2.7 Hz, 9.0 Hz), 8.35(s, 1 H), 7.87 (1 H, d, J= 9.0 Hz); 13C NMR (DMSO-d6) δ: 160.0, 152.5, 149.1, 145.1, 128.7, 128.4, 122.7, 122.0. IR (KBr) ν: 3431, 3082, 3038, 2664, 2613, 2567, 1724, 1685, 1676, 1646, 1617, 1578, 1526, 1468, 1359, 1346, 1269 cm-1. Anal. calcd for C8H4N3O2Cl: C 45.84, H 1.92, N 20.05, O 15.27; found C 45.81, H 1.97, N 20.02, O 15.21; MS (ESI) m/z: 207.96 (M-H).
13C NMR OF4 IN DMSOD6
IR
Thiazol-2-yl-methano1 (6)
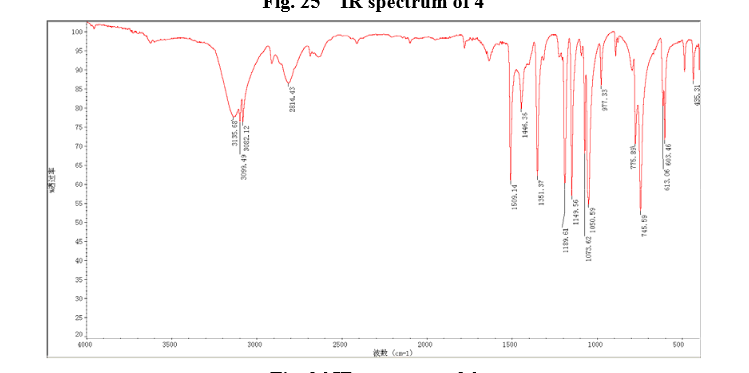
Sodium borohydride (16.0 g, 140 mmol) was added to a stirred solution of thiazole-2-carbaldehyde (24.2 g, 214 mmol) in MeOH (400 mL) at 0 °C . The reaction mixture was warmed to room temperature. After 1 hour, the reaction mixture was quenched by the addition of water and the organics were removed by concentration. The resulting aqueous mixture was extracted with EtOAc. The combined organic extracts were dried under Na2SO4 and concentrated to give thiazol-2-yl-methano1 (23.39 g, 95%). bp:75-76 °C (0.2 mmHg) [lit.[19] bp:70-80 °C (0.2 mmHg)]; m. p. 63-64 °C. 1 H NMR (CDCl3) δ 4.91 (s, 2 H), 5.1(br, l H), 7.28(d, 1 H, J=3.2 Hz), 7.68 (d, 1 H, J=2.9 Hz). IR (KBr) ν: 3135, 3099, 3082, 2814, 1509, 1446, 1351, 1189, 1149, 1073, 1050, 977, 775, 745, 613, 603 cm-1. Anal. calcd for C4H5NOS: C 41.72, H 4.38, N 12.16; found C 41.74, H 4.33, N 12.18; MS (ESI) m/z: 116.11 (M+H).
2-((2-Chloro-4-nitrophenoxy)methyl)thiazole (8)
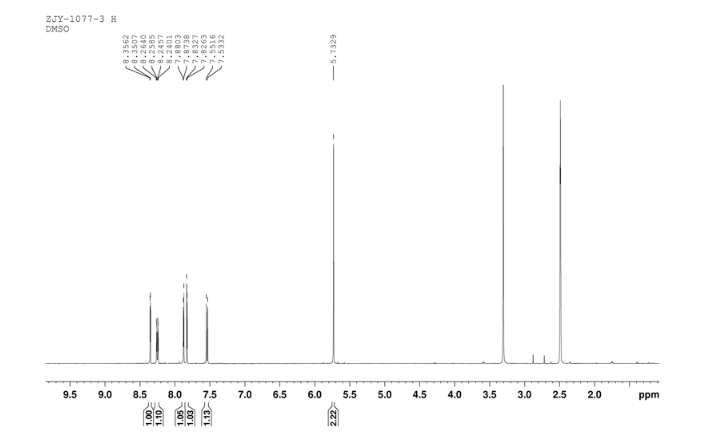
2-(2-chloro-4-nitro-phenoxymethy1)-thiazole was prepared by adding thiazol-2-yl-methanol (5.48 g, 47.65 mmol) to a slurry of sodium hydride (2.42 g of a 60% dispersion in oil, 60.5 mmol) in THF (50 ml) at 0 °C After several minutes, 2-chloro-1-fluoro- 4-nitro-benzene (7.58 g, 43.60 mmol) was added and the reaction mixture warmed to room temperature. The reaction mixture was stirred at room temperature for 3 h, and 60 °C for 16 h. After cooling to room temperature, the reaction mixture was poured into 300 mL water. The resulting precipitate was collected by filtration, washed with water, and dried in vacuo to give 2-(2- chloro-4-nitrophenoxymethy1)-thiazole (11.06 g, 86%) which was used in next step without further purification. m.p. 170-171 °C; 1 H NMR (DMSO-d6): δ 8.35 (1 H, d, J=2.8 Hz), 8.25 (1 H, dd, J=2.8 Hz, 9.15 Hz), 7.87 (1 H, d, J=3.3 Hz), 7.83(1 H, d, J=3.3 Hz), 7.54 (1 H, d, J=9.2 Hz), 5.73(s, 1 H); 13C NMR (DMSO-d6) δ: 164.2, 158.5, 143.2, 141.7, 125.9, 124.9, 122.4, 122.2, 114.6, 68.4; IR (KBr) ν: 3112, 3009, 1587, 1509, 1500, 1354, 1319, 1284, 1255, 1154, 1125, 1054, 1006, 894, 780, 746, 728 cm-1. Anal. calcd for C10H7N2O3SCl: C 44.37, H 2.61, N 10.35, O 17.73; found C 44.31, H 2.67, N 10.29; MS (ESI) m/z: 268.89 (M-H).
 1H NMR 8 DMSOD6
1H NMR 8 DMSOD6
13C NMR OF 8 IN DMSOD6
3-Chloro-4-(thiazol-2-ylmethoxy)aniline (9)
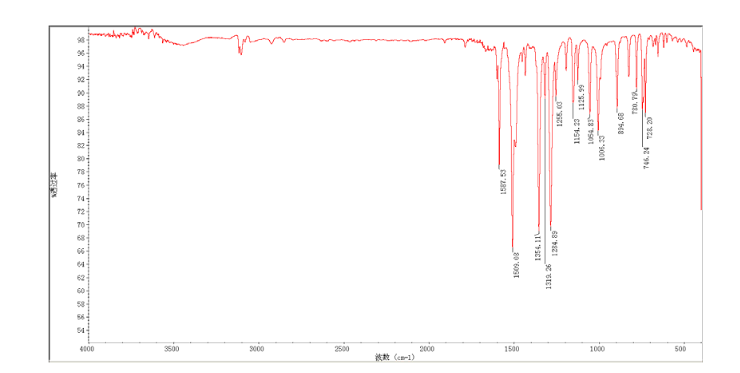
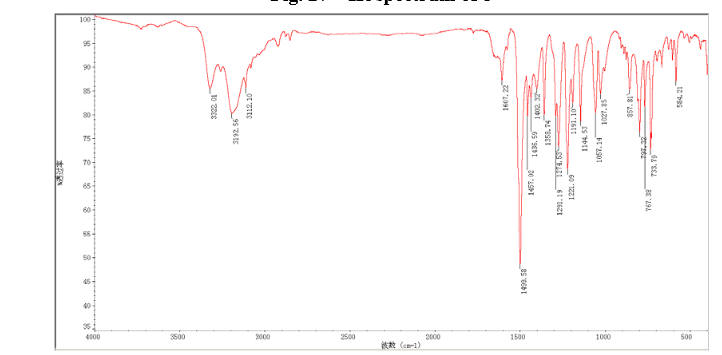
In a flask equipped with a reflux condenser, the compound 8 15.00 g (55.6 mmol), reduced zinc powder 14.44 g (222.0 mmo1, 4 eq), saturated ammonia chloride (5 mL) and methanol (100 mL) were mixed. The mixture was stirred at a temperature of 40 °C for 1.5 h. Then the zinc powder was filtered off, the filtrate was concentrated to obtain yellow solid 13.21 g, yield 99%. m.p. 60-61 °C; 1 H NMR (DMSO-d6): δ 7.80 (1 H, d, J=3.3 Hz), 7.75 (1 H, d, J=3.3 Hz), 6.96 (1 H, d, J=8.8 Hz), 6.64(1 H, d, J=2.7 Hz), 6.46 (1 H, dd, J=2.7 Hz, J=8.7 Hz), 5.30 (s, 2 H), 5.04 (s, 2 H, NH2, exchangeable); 13C NMR (DMSO-d6) δ: 166.8, 145.1, 144.1, 142.80, 123.1, 121.5, 117.7, 115.2, 113.6, 69.1. IR (KBr) ν: 3322, 3192, 3112, 1607, 1499, 1457, 1436, 1291, 1274, 1221, 1191, 1144, 1057, 1027, 857, 797, 767, 733, 584 cm-1. Anal. calcd for C10H9N2OSCl: C 49.90, H 3.77, N 11.64, O 6.65; found C 49.95, H 3.76, N 11.66, O 6.60; MS (ESI) m/z: 239.01 (M-H).
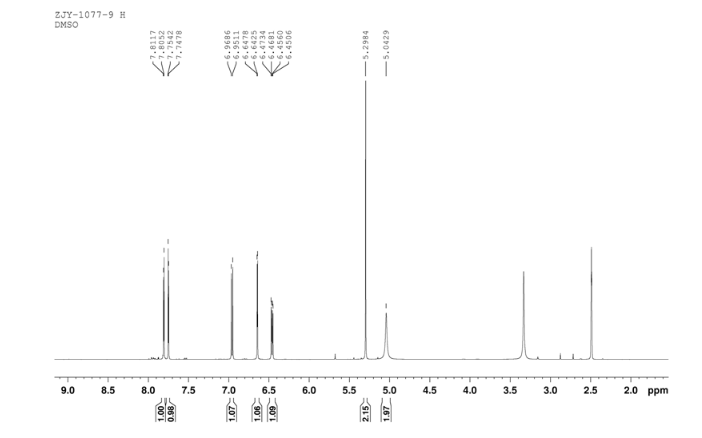 1H NMR DMSOD6 OF 9
1H NMR DMSOD6 OF 9
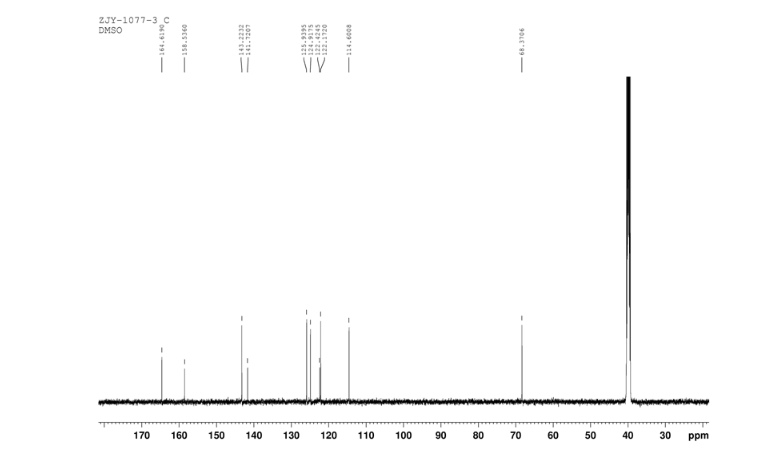
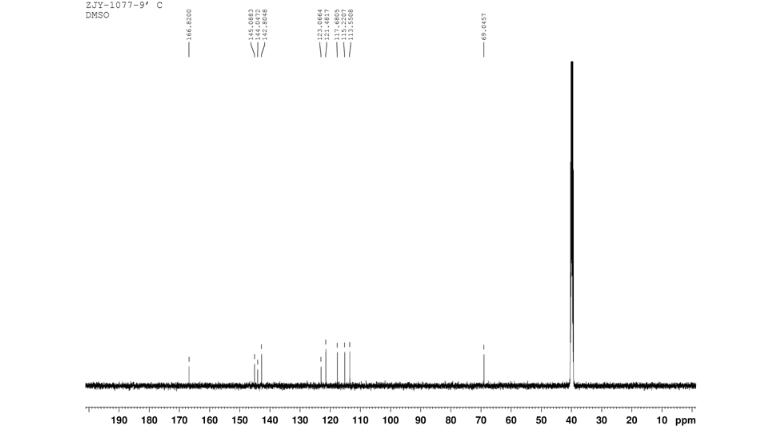 13C NMR OF 9 IN DMSOD6
13C NMR OF 9 IN DMSOD6
N-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)-6-nitro- quinazolin-4-amine(10)
In a flask equipped with a reflux condenser, 6-nitro-4-chloro- quinazoline 8.0 g (38.3 mmol) and 3-Chloro-4-(thiazol-2-ylmethoxy)aniline 8.9 g (37.0 mmol) were dissolved into 150 mL of THF, and the solution was refluxed for 3 h.Then a lot of yellow solid was deposited. Then it was filtered affording to yellow solid 12.8 g, yield 81%. m.p. 183-184 °C (decompose); 1 H NMR (DMSO-d6): δ 11.97(s, 1 H, exchangeable), 9.84 (s, 1 H), 9.00 (s, 1 H), 8.76 (1 H, d, J=9.1 Hz), 8.12-8.14 (m, 1 H), 7.94 (1 H, d, J=2.3 Hz), 7.87 (1 H, d, J=3.2 Hz), 7.81 (1 H, d, J=3.2 Hz), 7.44 (1 H, d, J=9.0 Hz), 7.69 (1 H, dd, J=2.5 Hz, J=8.9 Hz), 5.61 (s, 2 H); 13C NMR (DMSO-d6) δ: 166.8, 145.1, 144.1, 142.8, 123.1, 121.5, 117.7, 115.2, 113.7, 69.1. IR (KBr) ν: 3442, 3100, 1636, 1618, 1570, 1552, 1523, 1492, 1442, 1400, 1377, 1344, 1301, 1267, 1069, 805 cm-1. Anal. calcd for C18H12N5O3SCl: C 52.24, H 2.92, N 16.92, O 11.60; found C 52.26, H 2.93, N 16.96, O 11.58; MS (ESI) m/z: 412.84 (M-H).
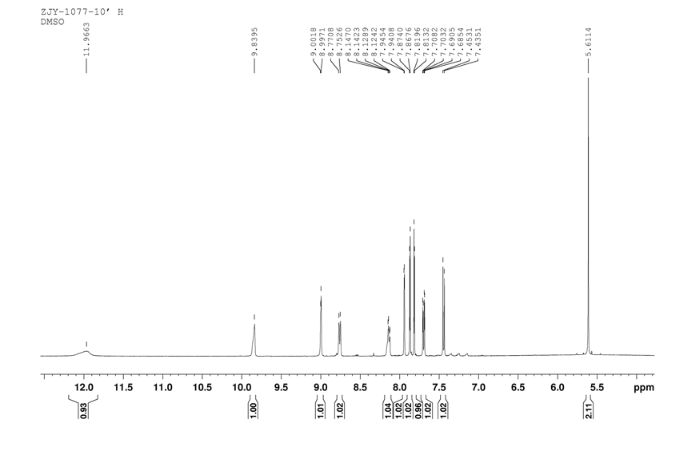 1H NMR DMSOD6 OF 10
1H NMR DMSOD6 OF 10
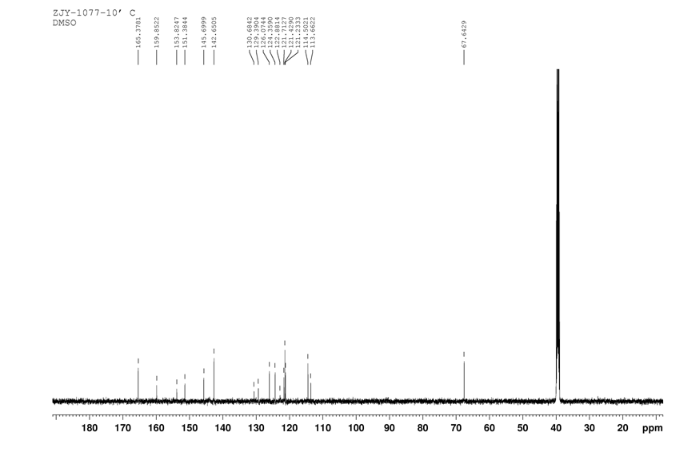 13C NMR OF 10 IN DMSOD6
13C NMR OF 10 IN DMSOD6
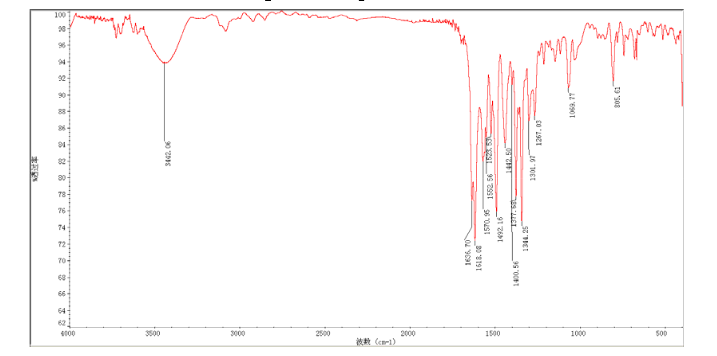
N4-(3-chloro-4-(thiazol-2-ylmethoxy)phenyl)quinazoline-4,6-diamine (11)
In a flask equipped with a reflux condenser, the compound 10 5.00 g (12.1 mmol), reduced zinc powder 3.2 g (48.5 mmo1, 4 eq), saturated ammonia chloride (3 mL) and methanol (60 mL) were mixed. The mixture was stirred at room temperature for 30 min. Then the zinc powder was filtered off, the filtrate was concentrated to obtain yellow solid 4.58 g, yield 98%. m.p. 197-198 °C (decompose); 1 H S4 NMR (DMSO-d6): δ 9.33(s, 1 H, exchangeable), 8.31 (s, 1 H), 8.05 (d, 1 H, J=2.6 Hz), 7.85 (d, 1 H, J=3.3 Hz), 7.79 (1 H, d, J=3.3 Hz), 7.73 (1 H, dd, J=2.5 Hz, J=9.0 Hz), 7.51 (1 H, d, J=8.9 Hz), 7.30 (1 H, d, J=2.4 Hz), 7.29 (1 H, d, J=4.7 Hz), 7.23 (1 H, dd, J=2.3 Hz, J=8.9 Hz), 5.57 (s, 2 H, exchangeable), 5.52 (s, 2 H); 13C NMR (DMSO-d6) δ: 165.9, 155.8, 149.7, 148.5, 147.3, 142.6, 142.5, 134.6, 128.7, 123.6, 123.2, 121.4, 121.3, 121.1, 116.5, 114. 7, 100.9, 67.8. IR (KBr) ν: 3443, 3358, 3211, 3100, 1631, 1596, 1577, 1560, 1530, 1494, 1431, 1383, 1217, 910 cm-1. Anal. calcd for C18H14N5OSCl: C 56.32, H 3.68, N 18.24, O 4.17; found C 56.34, H 3.70, N 18.22, O 4.14; MS (ESI) m/z: 382.66 (M-H).
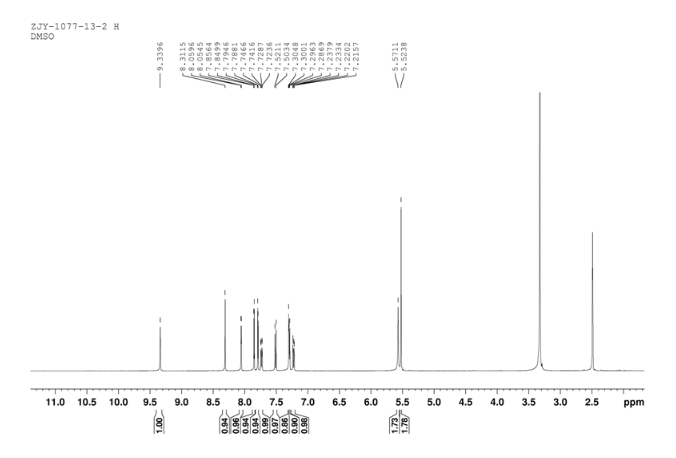 11 1HNMR DMSOD6
11 1HNMR DMSOD6
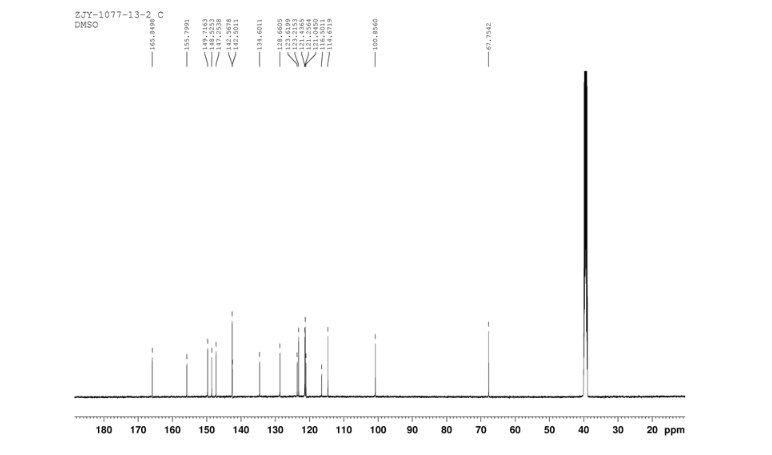 13C NMR OF 11 IN DMSOD6
13C NMR OF 11 IN DMSOD6
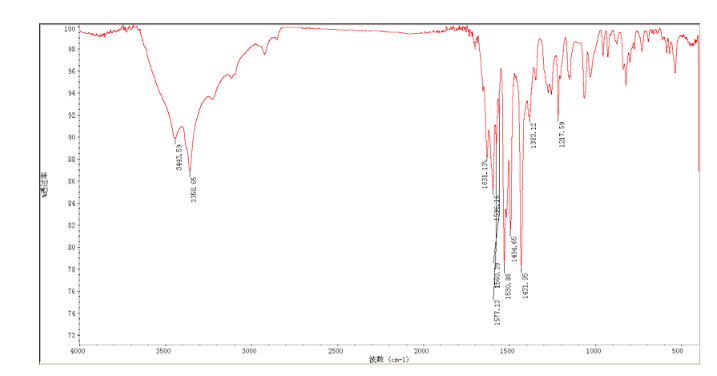
Construction finally as per patent ……….US20050043334
Treatment of N4-[3-chloro-4-(thiazol-2-ylmethoxy)phenyl]quinazoline-4,6-diamine (11) with 1,1′-thiocarbonyldiimidazole , followed by condensation with 2(R)-amino-1-propanol in THF/CH2Cl2 affords thiourea derivative , which finally undergoes cyclization in the presence of TsCl and NaOH in THF/H2O to furnish varlitinib .
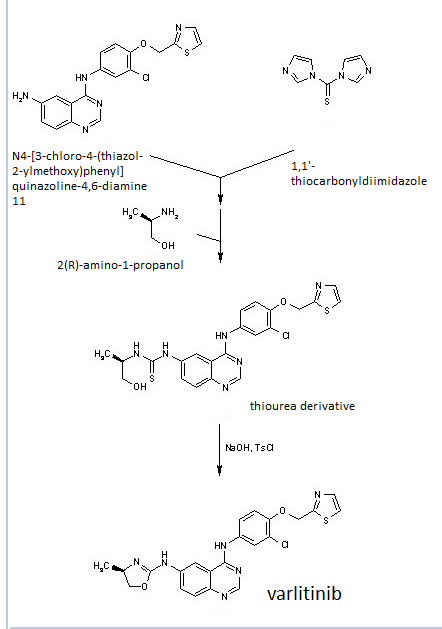


 carl fith
carl fith
Mr Carl Firth, CEO, Aslan Pharmaceuticals, Singapore (left) and Mr Dan Devine, CEO, Patrys, Australia (right)
///////ASLAN001, varlitinib, ASLAN Pharmaceuticals, Orphan Designation, ARRY-534, ARRY-334543 , PHASE 2, ORPHAN DRUG DESIGNATION, array
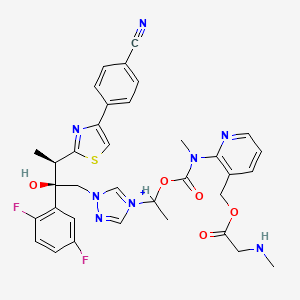
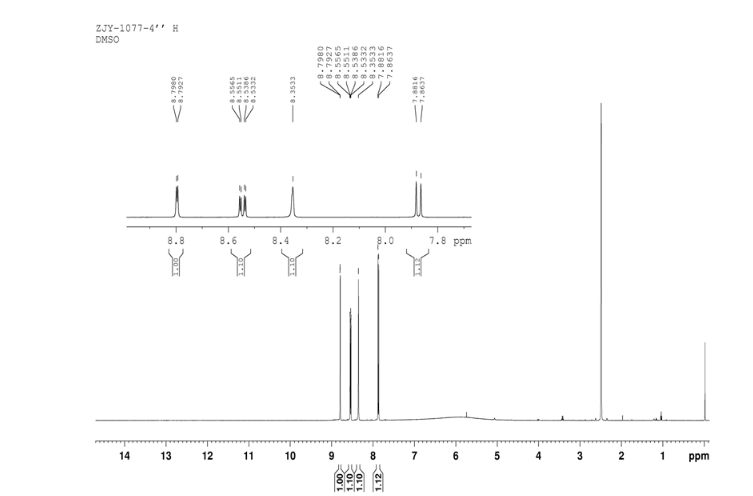 4 nmr dmsod6
4 nmr dmsod6 6 in dmsod6 1H NMR
6 in dmsod6 1H NMR