
Home » Posts tagged 'organic reactions' (Page 4)
Tag Archives: organic reactions
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Biosimilar drugs step up complexity, by Phillip Broadwith

The first ever generic monoclonal antibody therapies have been recommended for approval in Europe. The two biosimilar versions of infliximab (Johnson & Johnson’s Remicade) have passed assessment by the European Medicines Agency’s committee for medicinal products for human use, but will need to be fully approved by the European commission before they can be marketed.
Monoclonal antibodies are significantly larger and more complex than previously approved biosimilars, which include growth hormones and erythropoietin. Proving that they are functionally similar to the original drug is therefore complex. Both manufacturers, Celltrion and Hospira, had to complete human trials to prove that their generic infliximab products were as safe and effective as Remicade in treating autoimmune diseases.
read all at
http://www.rsc.org/chemistryworld/2013/07/biosimilar-approval-steps-complexity
Array Starts First Phase 3 Trial, Shifts to Late-Stage Development

HY-15202
MEK162
(Synonyms ARRY-162; ARRY-438162; MEK 162; ARRY 162; ARRY 438162)
MEK162 M.Wt: 441.23
MEK162 Formula: C17H15BrF2N4O3
MEK162 Storage: at -20℃ 2 years
MEK162 CAS No.: 606143-89-9
http://clinicaltrials.gov/ct2/show/NCT00959127
|
Array Starts First Phase 3 Trial, Shifts to Late-Stage Development read all at |
FDA awards Activartis brain cancer drug Orphan Drug Designation

Austrian biotech company Activartis has received Orphan Drug Designation for its cancer immunotherapy AV0113 from the US Food and Drug Administration.
The experimental vaccine will be fast-tracked for evaluation after it was shown to improve survival rates in people with malignant glioma, an advanced form of brain cancer in a phase II clinical trial earlier this year.

Roche’s Perjeta Gets FDA Priority Review

The structure of HER2 and pertuzumab
Application follows proposed new FDA pathway designed to help bring promising medicines to people with earlier stages of breast cancer faster
Perjeta is one of the first medicines the FDA will evaluate as an option given before surgery (neoadjuvant treatment)
Perjeta, in combination with Herceptin and chemotherapy, was approved by the FDA in 2012 for HER2-positive metastatic breast cancer
Pertuzumab (also called 2C4, trade name Perjeta) is a monoclonal antibody. The first of its class in a line of agents called “HER dimerization inhibitors”. By binding to HER2, it inhibits the dimerization of HER2 with other HER receptors, which is hypothesized to result in slowed tumor growth. Pertuzumab received US FDA approval for the treatment of HER2-positive metastatic breast cancer on June 8, 2012. Pertuzumab was developed at Genentech and is now owned by Roche which acquired Genentech in 2009.
Early clinical trials of pertuzumab in prostate, breast, and ovarian cancers met with limited success.
The dosage of pertuzumab used in the pivotal phase III CLEOPATRA (Clinical Evaluation of Pertuzumab and Trastuzumab) trial was as follows: IV 840 mg loading dose followed by IV 420 mg every three weeks.
The pharmacokinetics of intravenous pertuzumab appear to be unaffected by age and no drug-drug interaction has been reported with docetaxel. The pharmacokinetics and pharmacodynamics of pertuzumab were summarized in a Feb 2012 review by Gillian Keating.
The combination of pertuzumab plus trastuzumab plus docetaxel, as compared with placebo plus trastuzumab plus docetaxel, when used as first-line treatment for HER2-positive metastatic breast cancer, significantly prolonged progression-free survival, with no increase in cardiac toxic effects in the randomized, double-blind, multinational, phase III CLEOPATRA trial.
Intravenous pertuzumab is currently being evaluated in patients with breast cancer in the following trials: MARIANNE (advanced breast cancer), NEOSPHERE (early breast cancer), TRYPHAENA (HER2-positive stage II/III breast cancer) and APHINITY (HER2-positive nonmetastatic breast cancer)
EU OKs Blood Cancer Drug Iclusig

ponatinib
ARIAD Announces Marketing Authorization for Iclusig® (ponatinib) in the European Union
CAMBRIDGE, Mass. & LAUSANNE, Switzerland–(BUSINESS WIRE)–Jul. 2, 2013– ARIAD Pharmaceuticals, Inc. (NASDAQ: ARIA) today announced that the European Commission (EC) has granted a marketing authorization for Iclusig® (ponatinib) as an orphan medicinal product for two indications:read all at
http://www.pharmalive.com/eu-oks-blood-cancer-drug-iclusig
Ponatinib (Iclusig, previously AP24534) is an FDA approved oral drug candidate developed by ARIAD Pharmaceuticals for the treatment of chronic myeloid leukemia (CML) and Philadelphia chromosome positive (Ph+) acute lymphoblastic leukemia (ALL). It is a multi-targeted tyrosine-kinase inhibitor. Some forms of CML, those that have the T315I mutation, are resistant to current therapies such as imatinib. Ponatinib has been designed to be effective against these types of tumors.
Oncologists have complained, however, that many patients can’t afford the “astronomical” cost of $138,000 a year, which makes it one of the most expensive drugs in medicine, and far more expensive than what is needed to pay the development costs.
Ponatinib was approved by the US FDA on December 14, 2012, for patients with resistant or intolerant CML and Ph+ ALL, based on results of the PACE phase II trial reported days earlier at the annual ASH meeting.Because the approval was under the FDA’s accelerated approval program the applicant will be required to carry out additional studies.
The PACE (Ponatinb Ph+ ALL and CML Evaluation) pivotal phase II trial started enrolling patients in September 2010 and is designed to provide definitive clinical data for regulatory approval in this setting. Good results were reported in Dec 2012.
At the 2010 annual meeting of the American Society of Hematology, ARIAD announced from a Phase I study of ponatinib in patients with resistant and refractory chronic myeloid leukemia (CML) and Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). The study demonstrated that in chronic-phase CML patients treated with ponatinib, 66 percent of patients in the trial achieved a major cytogenetic response, including 100 percent of patients who also had a T315I mutation
BMS, Pfizer: Eliquis Meets Phase III Goal


Eliquis® (apixaban) Demonstrated Comparable Efficacy and Significantly Lower Rates of Major Bleeding in Patients Compared to Current Standard of Care for the Treatment of Acute Venous Thromboembolism
Phase 3 AMPLIFY Results Published in New England Journal of Medicine and Presented as a Late-Breaker at the Congress of the International Society on Thrombosis and Haemostasis Show:
•Eliquis Was Noninferior to Current Standard of Care for Treatment of Both Symptomatic Deep Vein Thrombosis and Pulmonary Embolism Conditions
•69 Percent Relative Risk Reduction for Major Bleeding in Patients on Eliquis Compared to Current Standard of Care “The study results showed that apixaban, as a single-agent, has comparable efficacy with significantly fewer major bleeding events with respect to the standard of care.
These results complement the previously published results for the AMPLIFY-EXT study” PRINCETON, N.J. and NEW YORK, June 30, 2013
READ ALL AT
http://www.pharmalive.com/bms-pfizer-eliquis-meets-phase-iii-goal

Flamel Technologies Announces FDA Approval of Bloxiverz

LYON, FRANCE — (Marketwire) — 06/03/2013 — Flamel Technologies today announced that the U.S. Food and Drug Administration (FDA) has approved the company’s New Drug Application (NDA) for Bloxiverz (neostigmine methylsulfate), a drug used intravenously in the operating room for the reversal of the effects of non-depolarizing neuromuscular blocking agents after surgery. Flamel expects to launch Bloxiverz in July 2013 in 0.5 and 1.0 mg/mL strengths
read all at
.http://www.drugs.com/newdrugs/flamel-technologies-announces-fda-approval-bloxiverz-3802.html
Sunovion Pharmaceuticals Inc. Announces FDA Approval of Latuda® (lurasidone HCl) as Monotherapy and Adjunctive Therapy in Adult Patients with Bipolar Depression

lurasidone
MARLBOROUGH, Mass. – Monday, July 1st 2013 [ME NewsWire]
First Atypical Antipsychotic Indicated for the Treatment of Major Depressive Episodes Associated with Bipolar I Disorder (Bipolar Depression) Both as Monotherapy and as Adjunctive Therapy with Either Lithium or Valproate
(BUSINESS WIRE)– Sunovion Pharmaceuticals Inc. today announced that the U.S. Food and Drug Administration (FDA) approved two new indications for the use of Latuda® (lurasidone HCl) as 1) monotherapy and 2) adjunctive therapy with either lithium or valproate, both to treat adult patients with major depressive episodes associated with bipolar I disorder (bipolar depression).
read all at
http://www.me-newswire.net/news/7829/en
Lurasidone (trade name Latuda) is an atypical antipsychotic developed by Dainippon Sumitomo Pharma and marketed by Sunovion in the USA. It was approved by the U.S. Food and Drug Administration (FDA) for treatment of schizophrenia on October 28, 2010 after a review that found that two of the four Phase III clinical trials supported efficacy, while one showed only marginal efficacy and one was not interpretable because of high drop-out rates. It is currently pending approval for the treatment of bipolar disorder in the United States. It was launched in Canada for the treatment of schizophrenia in September 2012, Health Canada giving their Summary Basis of Decision (SBD) as favourable on October 15, 2012
FDA Approves Brisdelle, paroxetine mesylate- First Non-Hormonal Treatment for Hot Flashes Associated with Menopause
June 28, 2013 –The U.S. Food and Drug Administration today approved Brisdelle (paroxetine) to treat moderate to severe hot flashes (vasomotor symptoms) associated with menopause. Brisdelle, which contains the selective serotonin reuptake inhibitor paroxetine mesylate, is currently the only non-hormonal treatment for hot flashes approved by the FDA.
There are a variety of FDA-approved treatments for hot flashes, but all contain either estrogen alone or estrogen plus a progestin.
read all at
more info
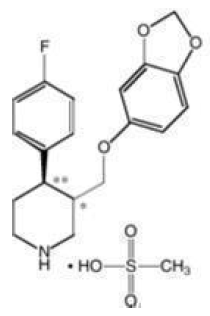
PEXEVA® (paroxetine mesylate) is an orally administered psychotropic drug with a chemical structure related to paroxetine hydrochloride (Paxil®). It is the mesylate salt of a phenylpiperidine compound identified chemically as (-)-trans-4R-(4′-fluorophenyl)-3S-[(3′,4′-methylenedioxyphenoxy) methyl] piperidine mesylate and has the empirical formula of C19H20FNO3•CH3SO3H. The molecular weight is 425.5 (329.4 as free base). The structural formula is: paroxetine mesylate
|
Paroxetine mesylate is an odorless, off-white powder, having a melting point range of 147° to 150°C and a solubility of more than 1 g/ml in water.
Tablets
Each oval, film-coated tablet contains paroxetine mesylate equivalent to paroxetine as follows: 10 mg (white); 20 mg (scored, dark orange); 30 mg (yellow); 40 mg (rose). Inactive ingredients consist of dibasic calcium phosphate, hydroxypropyl methylcellulose, hydroxypropylcellulose, magnesium stearate, sodium starch glycolate, titanium dioxide, ferric oxide red (C.I. 77491) (20 mg and 40 mg only) and ferric oxide yellow (C.I. 77492) (20 mg, 30 mg, and 40 mg only).
EP1286965B1



Amneal Pharma the 7th largest generic drug manufacturer in the US drug market has received ANDA approval for generic Skelaxin (metaxalone).

metaxalone
READ ALL AT
Metaxalone (marketed by King Pharmaceuticals under the brand name Skelaxin) is a muscle relaxant used to relax muscles and relieve pain caused by strains, sprains, and other musculoskeletal conditions. Its exact mechanism of action is not known, but it may be due to general central nervous system depression. It is considered to be a moderately strong muscle relaxant, with relatively low incidence of side effects. Skelaxin is available in an 800 mg scored tablet. Possible side effects include nausea, vomiting, drowsiness and CNS side effects, such as dizziness, headache, and irritability.
Metaxalone exhibits increased bioavailability when taken with food.[1] Specifically, in one study, compared to fasted conditions, the presence of food at the time of drug administration increased Cmax by 77.5%, AUC0-t by 23.5%, and AUC0-∞ by 15.4%.[2] Thus, based on the information in the labeling, patients receiving metaxalone therapy are directed to take metaxalone with food, and are informed that taking metaxalone with food results in an increase in the oral bioavailability of metaxalone compared to taking metaxalone without food.[3][4][5]
The metabolism of metaxalone involves the liver cytochrome P450 system. Based on the information in the labeling, patients receiving metaxalone therapy and physicians prescribing metaxalone are directed to take precaution when coadministering it with other medications involving the P450 system.[6][7]
Because of potential for side effects, this drug is on the list for high risk medications in the elderly.
A literature survey reveals very few methods are reported for the determination of metaxalone to date. Nirogi et al.[2] reported a liquid chromatographic method coupled to tandem mass spectrometry for the quantification of metaxalone in human plasma. A stability-indicating HPLC method was introduced by P.K. Sahu et al.[8] Metaxalone has been used as an internal standard for few analytical methods[9][10]

- Skelaxin Package Insert
- Nirogi RV, Kandikere VN, Shukla M, Mudigonda K, Shrivastava W, Datla PV (May 2006). “Quantification of Metaxalone in Human Plasma by Liquid Chromatography Coupled to Tandem Mass Spectrometry”. J Anal Toxicol 30 (4): 245–51. PMID 16803662.
- id.
- United States Patent No. 6,407,128
- United States Patent No. 6,683,102
- United States Patent No. 7,122,566, by Jie Du, et al
- United States Patent No. 7,378,434, by Jie Du, et al
- Prafulla Kumar Sahu, M. Mathrusri Annapurna and Dillip Kumar Sahoo, Development and Validation of Stability Indicating RP-HPLC Method for the Determination of Metaxalone in Bulk and its Pharmaceutical Formulations; E-Journal of Chemistry, 2011, 8(S1), S439-S447.[1]
- Mistri H N, Jangid A G, Pudage A, Gomes N, Sanyal M and Shrivastav P, J Chromatogr B, 2007, 853(1), 320-332.
- Mistri H N, Jangid A G, Pudage A and Shrivastav P, J Chromatogr B, 2008, 864(1-2), 137-148.

