
Home » Posts tagged 'LOSARTAN'
Tag Archives: LOSARTAN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
LOSARTAN

E-3340
L-158086
MK-0954
MK-954
Ex-89 (free acid)
COZAAR (losartan potassium, cas 124750-99-8) is an angiotensin II receptor (type AT1)antagonist. Losartan potassium, a nonpeptide molecule, is chemically described as 2-butyl-4-chloro-1-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]imidazole-5-methanol monopotassium salt. Its empirical formula is C22H22ClKN6O, and its structural formula is:
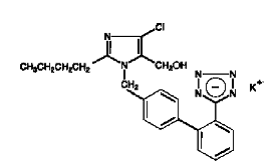 |
Losartan potassium is a white to off-white free-flowing crystalline powder with a molecular weight of 461.01. It is freely soluble in water, soluble in alcohols, and slightly soluble in common organic solvents, such as acetonitrile and methyl ethyl ketone. Oxidation of the 5-hydroxymethyl group on the imidazole ring results in the active metabolite of losartan.
COZAAR is available as tablets for oral administration containing either 25 mg, 50 mg or 100 mg of losartan potassium and the following inactive ingredients: microcrystalline cellulose, lactose hydrous, pregelatinized starch, magnesium stearate, hydroxypropyl cellulose, hypromellose, and titanium dioxide.
COZAAR 25 mg, 50 mg and 100 mg tablets contain potassium in the following amounts: 2.12 mg (0.054 mEq), 4.24 mg (0.108 mEq) and 8.48 mg (0.216 mEq), respectively. COZAAR 25 mg, COZAAR 50 mg, and COZAAR 100 mg may also contain carnauba wax.
|
Losartan (rINN) /loʊˈsɑrtən/ is an angiotensin II receptor antagonist drug used mainly to treat high blood pressure (hypertension). Losartan was the first angiotensin II antagonist to be marketed. Losartan potassium is marketed by Merck & Co. Inc. under the trade nameCozaar. Losartan is available in generic form.
As with all angiotensin II type 1 receptor (AT1) antagonists, losartan is indicated for the treatment of hypertension. It may also delay progression of diabetic nephropathy, and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes, hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).
Although clinical evidence shows calcium channel blockers and thiazide-type diuretics are preferred first-line treatments for most patients (from both efficacy and cost points of view), an angiotensin II receptor antagonist such as losartan is recommended as first-line treatment in patients under the age of 55 who cannot tolerate an ACE inhibitor.The LIFE study demonstrated losartan was significantly superior to atenolol in the primary prevention of adverse cardiovascular events (myocardial infarction or stroke), with a significant reduction in cardiovascular morbidity and mortality for a comparable reduction in blood pressure. A study hints that losartan has a beneficial effect on mitochondria by reversing age related dysfunction in maintaining normal blood pressure and cellular energy usage. The maximal effects on blood pressure usually occur within 3–6 weeks upon starting losartan.
Losartan is also available as hydrochlorothiazide/losartan, a combination drug with a low dose thiazide diuretic to achieve an additive antihypertensive effect.

-
Activation of AT1 receptors in the outer membrane of vascular smooth muscle cells of the heart and arteries causes those tissues to constrict. Blocking of vasoconstriction mediated by AT1 receptors has been found to be beneficial to patients with hypertension.
-
[0003]AT1 receptors are activated by an octa-peptide, angiotensin II. Angiotensin II helps to maintain constant blood pressure despite fluctuations in a person’s state of hydration, sodium intake and other physiological variables. Angiotensin II also performs the regulatory tasks of inhibiting excretion of sodium by the kidneys, inhibiting norephedrin reuptake and stimulating aldosterone biosynthesis.
-
[0004]Inhibiting angiotensin II binding to AT1 receptors with an AT1 receptor antagonist disrupts the vasoconstriction mediated by AT1 receptors that contributes to hypertension.
-
[0005]In the early 1970s, it was discovered that certain oligopeptides competitively inhibited angiotensin receptors (at that time the existence of two receptor subtypes, AT1 and AT2, was unknown). This discovery spurred interest in development of therapeutic oligopeptides with increased potency, but interest in peptide analogs waned due in part to their poor oral bioavailability.
-
[0006]In 1982, Furukawa. Kishimoto and Nishikawa of Taketa Chemical Indus. discovered a class of non-peptide-containing imidazoles that also inhibited the vasoconstriction effect of angiotensin II. See U.S. Patents Nos. 4,340,598 and 4,355,040. Later, U.S. Patent No. 5,138,069 was obtained by Carini, Denucia and Pancras of E.I. DuPont de Nemours on another class of imidazoles, which encompasses the compound losartan. In 1995, losartan (CA Index: 2-butyl-4-chloro-1-[[2′-(1H-tetrazol-5-yl) [1,1′-biphenyl] -4-yl]methyl]-1H-imidazole-5-methanol) (formula I):

became the first nonpeptide AT1 antagonist approved by the U.S. Food and Drug Administration for clinical use. Losartan can be administered orally as its monopotassium salt. Losartan potassium is available by prescription in tablet form as a sole active ingredient (Cozaar®: Merck) and as a co-active ingredient with hydrochlorothiazide (Hyzaar®: Merck).
-
[0007]Losartan has been prepared by a variety of synthetic pathways. In several of these synthetic pathways, the penultimate product is 2-butyl-4-chloro-1-[[2′-(2-triphenylmethyl-2H-tetrazol-5-yl) [1,1′-biphenyl] -4-yl]methyl]-1H-imidazole-5-methanol (“trityl losartan”). Trityl losartan is an intermediate in processes described in U.S. Patents Nos. 5,138,069; 5,962,500 and 5,206,374.
-
[0008]In a process described in Example 316 of U.S. Patent No. 5,138,069, the tetrazole ring of losartan is formed by reacting 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-butyl-4-chloro-5-hydroxymethylimidazole with trimethyltin azide. The reaction gives a trimethylstannyl substituted tetrazole compound directly. The trimethylstannyl group is cleaved from the product by reacting with trityl chloride. This reaction results in attachment of the trityl group to the tetrazole ring. In the last step, the trityl group is cleaved with acid to give losartan (Scheme 1).

-
[0009]In the last step, trityl losartan was suspended in methanol and cooled to ~10°C. 3.4 N Hydrochloric acid was added to the slurry. After a period of time, the pH of the reaction mixture was raised to 13 with 10 N NaOH. Methanol was then distilled off while makeup water was added. After distillation, additional water and toluene were added. The toluene phase was separated and the aqueous phase was extracted once more with toluene. Ethyl acetate and acetic acid were then added to the aqueous phase. Losartan was recovered from the aqueous phase as a solid and further purified by slurrying in ethyl acetate. Losartan was obtained in 88.5% yield and 98.8% purity as determined by HPLC. This process is also described in U.S. Patents Nos. 5,128,355 and 5,155,188.
-
[0010]U.S. Patent No. 5,962,500, Examples 3-5, describe a process for preparing losartan in which the tetrazole ring of losartan is present in the starting material, 5-phenyltetrazole. The ‘500 patent process, depicted in Scheme 2, is convergent and uses a Suzuki coupling reaction (Miyaura, N.; Suzuki, A. Chem. Rev., 1995, 95, 2457) in the convergent step. On one branch of the synthesis, 5-phenyltetrazole is converted into the boronic acid coupling partner for the Suzuki reaction by ortho metalation with n-butyl lithium, followed by reaction with trisopropylborate. The tetrazole ring is protected from reacting with the strong allcyl lithium base with a trityl group. The trityl group is conventionally attached by reacting the tetrazole with trityl chloride in the presence of a non-nucleophilic base. On the other branch of the convergent synthesis, 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde is alkylated with 4-bromobenzylbromide, followed by reduction of the aldehyde with sodium borohydride to yield the other Suzuki coupling partner.

-
[0011]The direct product of Suzuki coupling is trityl losartan. In the next and last step, the tetrazole ring of trityl losartan is deprotected with 4N H2SO4 in THF. In that step, the acidic solution was aged overnight at 20 to 25°C. The solution was then extracted with isopropyl acetate and residual organic solvent was removed from the aqueous phase under vacuum. The solution was then carried forward to from the potassium salt without intermediate isolation of losartan. This process is also described in U.S.Patents Nos, 5,206,374, Example 21, and 5,310,928, Example 21.
-
[0012]Larsen, R.D et al. [J. Org. Chem. (1994), 59, 6391-6394] discloses a similar convergent synthesis of lasartan, whereby the trityl lasartan, generated by Suzuki coupling, is deprotected using 0.7 M H2SO4 in a 50 : 50 mixture of acetonitrile /water.
-
[0013]U.S. Patent No. 5,206,374 Examples 1 and 4-8 describe another process for making Iosartan that also involves a Suzuki coupling reaction. However, unlike the ‘500 patent process, the ‘374 patent process is not convergent. The ‘374 patent process is depicted in Scheme 3.

-
[0014]In the ‘374 patent process, as in the `500 patent process, the tetrazole ring of 5-phenyltetrazole is protected with a trityl group before orthometallation of the phenyl moiety with n-butyl lithium in preparation for making the boronic acid Suzuki coupling partner. In the Suzuki coupling step, the boronic acid is reacted with 4-bromotoluene. The methyl group attached to one of the phenyl rings of the Suzuki product is then halogenated with N-bromosuccinamide and the benzylic bromine atom of that product is displaced with 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde. Reduction of the aldehyde group with sodium borohydride yields trityl losartan. The tetrazole group of trityl losartan was deprotected with 12% aqueous HCl in THF. After 12 hours, the pH of the reaction mixture was raised to 12.5 with 30% NaOH. The THF was then distilled off while make-up water was added to the mixture. After distillation, the mixture was cooled and the triphenyl methanol byproduct of deprotection, which had precipitated, was removed by filtration. The filtrate and rinsate, with which it was combined, were extracted with toluene. Then, ethyl acetate was added and 36% HCI was added until the pH of the reaction mixture was lowered to 3.8. The mixture was cooled, causing losartan to precipitate from the solution. Losartan was obtained in 83% theoretical yield starting from trityl losartan.




EP 253310 discloses a process, wherein 2-n-butyl-4-chloro-1H-imidazolyl-5-methanol (III) is coupled with 5-(4′-bromomethyl-1,1′-biphenyl-2-yl)-2-triphenylmethyl-2H-tetrazole (IV) in N,N-dimethylformamide as solvent in presence of sodium methoxide as the base to furnish trityl losartan. The other bases that have been claimed are sodium hydride, alkali metal carbonates such as sodium carbonate and potassium carbonate and amine bases such as triethyl amine and pyridine.

The coupling reaction results in a mixture of trityl losartan and its regio isomer (V). These are separated by column chromatography.
U.S. Pat. Nos. 5,130,439 and 5,310,928 disclose a method for coupling (IV) and (VI) in N,N-dimethylacetamide solvent in the presence of anhydrous potassium carbonate as base. The imidazole aldehyde (VI) gives predominantly the desired regio isomer (VII). The intermediate VII is then reduced with sodium borohydride to furnish the trityl losartan. The product is isolated by extraction into toluene from aqueous N,N-dimethylacetamide, concentration of the toluene solution and crystallization using ethyl acetate or ethanol as solvent. The synthesis steps are depicted as follows.

In a process published in J. Med. Chem. (1991), 34, 2525-2547, Losartan is prepared by coupling (III) and (IV) in N,N-dimethylformamide in the presence of sodium methoxide. The desired compound is isolated after vacuum distillation of solvent followed by extractive work-up. The resultant product mixture is purified by chronmatography.
The U.S. Pat. Nos. 5,138,069, 5,128,355 and 5,155,118 describe a process for the preparation of losartan, wherein the tetrazole ring of losartan is formed by reacting 1-((2′-cyanobiphenyl-4-yl)methyl)-2-butyl-4-chloro-5-hydroxymethylimidazole with trimethyltin azide. The reaction results in trimethylstannyl substituted tetrazole compound, which is then reacted with trityl chloride and sodium hydroxide.

The trityl losartan thus formed is treated with 3.4N hydrochloric acid in methanol at about 10° C. to give losartan.
The U.S. Pat. Nos. 5,138,069, 5,128,355 and 5,155,118 also disclose another process for making trityl losartan, where in the coupling between IV and VI is carried out in a biphasic solvent system comprising of chlorinated solvent and water. The reaction is carried out at room temperature in presence of sodium hydroxide as the base and aliquat 336 as the phase transfer catalyst. The resulting intermediate VII is then reduced in situ with sodium borohydride to furnish trityl losartan.

U.S. Pat. No. 5,206,374, 5,310,928 and 5,962,500 disclose another process for preparing losartan in which 5-phenyltetrazole (X) is converted into the boronic acid coupling partner (XII) for the Suzuki reaction by tritylation of phenyltetrazole with trityl chloride in presence of a non-nucleophilic base, ortho metalation with n-butyl lithium, followed by reaction with triisopropylborate. 2-n-butyl-4-chloro-1H-imidazole-5-carboxaldehyde (VI) is alkylated with 4-bromobenzylbromide, followed by reduction of the aldehyde with sodium borohydride to yield the other Suzuki coupling partner (XIII). The product of Suzuki coupling is trityl losartan. This process is published in J. Org. Chem. (1994), 59, 6391-6394.


European patents EP 470,794 and EP 470,795 describe a method for the manufacture of biphenyl carbonitriles (XVI). These patents also describe a method of preparation of trityl losartan by coupling of intermediates (III) and (IV) employing the procedure described in EP 253,310.

Losartan potassium exhibits polymorphism. Several polymorphic forms have been prepared and characterized. The following paragraphs briefly describe various polymorphs.
U.S. Pat. No. 5,608,075 discloses the polymorphic forms of losartan, wherein the trityl losartan is deprotected with H2SO4 in 50:50 acetonitrile:water and the free acid is treated with KOH solution. The aqueous solution containing losartan potassium is added slowly to a refluxing azeotropic mixture of cyclohexane/iso propanol and the ternary azeotrope cyclohexane/iso propanol/water is distilled till the water content of the pot is less than 0.05%. The white crystalline solid thus obtained is polymorphic form-I, which is characterized by DSC, XRD and IR. Polymorphic form-II is prepared by heating form-I in a DSC cell. This process is also described in U.S. Pat. No. 5,859,258.
U.S. Pat. No. 6,710,183 discloses the synthesis of losartan potassium starting from trityl losartan, wherein trityl losartan is reacted in an alcohol of formula R—OH (where R is C1 to C4 straight chain alkyl group) with 0.1 to 1 equivalent KOH. Losartan potassium thus formed is isolated after crystallizing out by changing the solvent to an aprotic or weakly protic solvent. The alcohol used is preferably methanol and the protic dipolar solvent used for the crystallization of the final product is preferably acetonitrile or straight or branched chain or cyclic aliphatic hydrocarbons.
EP 1294712 (WO 02/094816) discloses the process to manufacture losartan potassium form-I, wherein trityl losartan or losartan is suspended in a solvent and KOH is added to obtain a clear solution, which is then concentrated under reduced pressure to remove most of the solvent. An anti solvent is added to crystallize losartan potassium. The solvents to prepare losartan potassium include methanol, ethanol, and butanol but preferably the salt formation is carried out in methanol. Anti solvent is selected from common solvents such as ethyl acetate, acetonitrile, toluene and acetone, but the preferred anti solvent is acetone.
US application 2004/0006237 (WO 03/048135) relates to novel amorphous and novel crystalline forms III, IV, V of losartan potassium and the processes for their preparation. The patent also discloses novel processes for preparing losartan potassium forms I and II. The preparation of amorphous losartan includes the step of dissolving losartan potassium in a solvent to form a solution and distilling the solvent form the solution to dryness. Losartan form III (hydrated) is obtained by exposing losartan potassium amorphous or form I to an atmosphere having high relative humidity. Losartan potassium form IV is obtained by treating a saturated solution of losartan potassium in ethanol with methylene chloride. Losartan form V is obtained by treating a saturated solution of losartan potassium in ethanol with hexane. Losartan potassium form II is obtained by adding a saturated solution of losartan potassium in ethanol to xylene to form a mixture and evaporating ethanol from the mixture. Losartan form I is obtained by treating a saturated solution of losartan potassium in ethanol or iso propanol, with less soluble solvent like ethyl acetate, toluene, acetone, methyl ethyl ketone, methylene chloride, acetonitrile, dimethyl carbonate or hexane.
US application 2004/0034077 (WO 03/093262) discloses a process for preparing losartan and losartan potassium, wherein trityl losartan is treated with an acid in a diluent comprising a ketone. Especially preferred liquid ketones are acetone, methyl ethyl ketone and methyl isobutyl ketone, and acetone being the most preferred. Acids, which have been found suitable, include hydrochloric acid, sulphuric acid, acetic acid, trifluoroacetic acid, hydrobromic acid and formic acid. After the trityl losartan has been substantially converted to losartan, reaction mixture is basified. Preferred bases are alkali metal hydroxides and alkoxides. After addition of the base, the liquid ketone is evaporated under vacuum. After separation of triaryl methyl alcohol the residue is acidified to yield losartan. Free losartan is suspended in an alcohol and treated with a solution of potassium ions. Finally losartan potassium is precipitated from the alcohol. The alcohol is selected from the group consisting of isopropyl alcohol, butyl alcohol and isobutyl alcohol. The potassium ion solution is prepared by dissolving potassium iso propoxide, potassium butoxide and potassium iso butoxide or potassium hydroxide in the diluent.
US application 2004/0097568 discloses a process for preparing form III of losartan potassium, wherein trityl losartan is treated with aqueous solution of potassium hydroxide in methanol to obtain losartan potassium. The solvent is evaporated under vacuum and traces of water are removed as an azeotrope with toluene. Methanol and carbon are added to the resulting mixture. The carbon is filtered and the methanol is distilled. The resulting mixture is cooled to 20-25° C. to obtain crystalline form III losartan potassium.
US 5,138,069 and
WO 93/10106. The advantages provided by pharmaceutical products in the crystalline form in terms of easiness of processes for the preparation of related medicaments are well known. Crystalline compounds are in fact known to be more suited to the formulation of galenic forms, thanks both to their flowability in the form of powders or granulates, and to the surface properties of the crystals which promote adhesion, for example during the preparation of tablets. Furthermore, the solubility of crystalline compounds in aqueous solutions, in particular in the gastric juices, can also be significantly different than that of the corresponding amorphous compounds. There is therefore the need to discriminate between the crystalline and the amorphous forms of biologically active compounds, so as to fulfil the various pharmaceutical requirements.
A number of crystalline and amorphous forms of losartan potassium are known from
WO 95/17396 and
WO 03/048135. According to
WO 95/17396, crystalline losartan potassium is prepared by salification of acid losartan with an alkali hydroxide. The losartan potassium aqueous solution is then added to a isopropanol-cyclohexane azeotropic mixture under reflux. Water is then removed by azeotropic distillation of the resulting water-isopropanol-cyclohexane ternary mixture, which boils at 64°C. When the solution is anhydrous, the head temperature raises to 69°C and losartan potassium crystallizes.
US 5,859,258 discloses another crystallization process which comprises dissolution of losartan potassium in isopropanol-water, distillation of the binary azeotrope to an approx. 2.6% water content, precipitation by addition of a losartan potassium suspension in cyclohexane, subsequent distillation of the ternary azeotrope to a water content ranging from 0.02 to 0.11 %, and finally drying crystalline losartan potassium under vacuum at a temperature of approx. 45-50°C.

 , Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author email
, Ian R. Baxendale, Steven V. Ley and Nikzad Nikbin Corresponding author emailThe imidazole ring of losartan, an antihypertensive and angiotensin II blocker is formed in a condensation reaction between valeroamidine 160 and dihydroxyacetone [50]. It was found that direct chlorination of the imidazole 162also forms the dichlorination product 164 (as shown in Scheme 33) with formaldehyde as a by-product which proved difficult to suppress and made purification of the reaction mixture problematic. Hence, a sequence involving silyl protection, chlorination and deprotection was established which gave the desired product in 90% overall yield (Scheme 33).
![[1860-5397-7-57-i33]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i33.png)
Alternatively, glycine can be reacted with methyl pentanimidate 169 to form the corresponding amidine 171 in high yield. Cyclisation, followed by a Vilsmeier-type reaction then furnishes the key chloroimidazolyl building block 172in good yield (Scheme 34) [51].
![[1860-5397-7-57-i34]](https://i0.wp.com/beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i34.png)
- 50———Shi, Y.-J.; Frey, L. F.; Tschaen, D. M.; Verhoeven, T. R. Synth. Commun. 1993, 23, 2623–2630.doi:10.1080/00397919308012598
- 51—-Griffiths, G. J.; Hauck, M. B.; Imwinkelried, R.; Kohr, J.; Roten, C. A.; Stucky, G. C.; Gosteli, J. J. Org. Chem. 1999,64, 8084–8089. doi:10.1021/jo9824910
- 52–Zhong, Y.-L.; Lee, J.; Reamer, R. A.; Askin, D. Org. Lett. 2004, 6, 929–931. doi:10.1021/ol036423y
NMR: (1H, DMSO, 300 mHz): δ 0.80 (3H, t, J=10. CH3), 1.25 (2H, sext, J=10. CH3CH2), 1.45 (2H, quin, J=10. CH3CH2CH2), 2.45-2.55 (2H, m, CH3CH2CH2CH2), 4.25 (2H, d, J= 3, CH2OH), 5.15-5.25 (3H, m, CH2Ar and OH), 6.88 (d, 2H, J=12, ArH), 7.08 (d, 2H, J=12, ArH), 7.23-7.36 (3H, m, ArH), 7.50-7.55 (1H, ArH).
SEACOND SET
http://www.google.co.in/patents/US7915425
IR v max (KBR): 3201.01, 1580.73, 1460.18, 764.81, 540.09
1H NMR (MeOD) δ, 0.87 (t, 3H), 1.33 (sext, 2H), 1.53 (quint, 2H), 2.56 (t, 2H), 4.43 (s, 2H), 5.24 (s, 2H), 6.89-7.53 (m, 8H).
13C NMR (MeOD) δ, 14.07, 23.24, 27.40, 30.92, 126.71, 126.86, 127.35, 128.21, 130, 130.8, 131, 131.19, 131.81, 136.09, 142.21, 149.97, 162.72
MS (m/z)=423.3 (M+1).
……………………………..
Melting point: 179-180.2
IR, v max (KBR): 3376.27, 1579.77, 1468.86, 762.88, 556.4
1H NMR (CDCl3) δ, 0.87 (t, 3H), 1.31 (sext, 2H), 1.54 (quint, 2H), 2.57 (t, 2H), 4.45 (s, 2H), 5.30 (s, 2H), 7.01-7.68 (m, 8H).
13C NMR (CDCl3) δ, 14.07, 23.24, 27.40, 30.92, 126.71, 126.86, 127.35, 128.21, 130, 130.8, 131, 131.19, 131.81, 136.09, 142.21, 149.97, 162.72
MS (m/z)=423.5 (M+1).
……………………………..
ADDITIONAL WRITEUP FOR READERS, NUMBERINGS ARE ALL NEW
Losartan and its potassium salt, having the formulae (1) & (2) respectively are angiotensin-II receptor (Type AT1) antagonists.

In adults Losartan is currently indicated for the treatment of hypertension (in hypertensive patients with left ventricular hypertrophy, it is also indicated to reduce the risk of stroke).
Losartan Potassium having the formula 2 and its principle active metabolite block the vasoconstrictor and aldosterone. Secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor found in many tissues (e.g., vasicular smooth muscle, adrenal gland) otherwise called as angiotensin receptor blockers (ARBs).
The present invention relates to a short, simple and practical process for the preparation of Losartan 1 which belongs to a novel class of tetrazole-imidazole compounds.
There are many processes recorded in literature. The latest prior art information for the preparation of Losartan is the disclosure made in the patent application of Novartis in their PCT WO 2005/014602 dated 17 Feb. 2005.
The process described in the application comprises the reaction of 4′-(Bromomethyl)-2-cyanobiphenyl (BromoOTBN) of the formula 3 with 2-n-butyl-4-chloro-5-formyl imidazole (BCFI) of the formula of 4 in the presence of Potassium carbonate and acetonitrile to give ‘cyano aldehyde’ of the formula 5. The Cyano aldehyde of the formula 5 is reduced with sodium borohydride to get ‘cyano alcohol’ of the formula 6. The Cyano alcohol is reacted with diethyl aluminium azide in the presence of triethyl aluminium to give Losartan of the formula 1.
The reaction scheme of the process is shown in the Scheme 1

Even though the process is simple, handling of triethyl aluminium used needs special attention like very anhydrous conditions, reactions are to be performed under nitrogen or argon and transferring of triethyl aluminium from the containers needs anhydrous systems. The neat liquid and dense solutions of triethyl aluminium are known to ignite very easily at room temperature in presence of air (Pyrophoric). So handling of both triethyl aluminium and diethyl aluminium needs special attention like anhydrous conditions, nitrogen atmosphere etc.,
In EP 0578125A1 of Takeda Chemical Industries dated 12 Jan. 1994, yet another method for the preparation of Losartan has been disclosed in which Trioctadecyl or Trioctyl tin azide has been used as a tetrazole-forming agent. This method also uses the Cyano alcohol of the formula (6). The process comprises reacting the cyano alcohol of the formula (6) with tri-n-octyl tin azide in presence of toluene to give tri-n-octyl tetrazole derivative, which was treated with nitrous acid to give Losartan of the formula (1) in 94.7% yield. The process is shown in the reaction scheme 2

Even though the yields are better (94.7%) in this process again handling of tri-n-octyl tin azide is involved.
Dupont/Merck in their patents and papers always described that trityl Losartan of the formula 7 is detritylated to get Losartan 1 For example they described in J. Med. Chem., 1991, 34, 2525-2547, the preparation of Losartan of the formula 1, from trityl Losartan of the formula 7 using mineral acids such as Hydrochloric acid and sulfuric acid in 93% yield. The reaction scheme of the process is shown in the scheme 3

In this paper ‘Aldehyde Tetrazole’ of the formula 8 is isolated from trityl tetrazole aldehyde of the formula 21 and were further used for preparing derivatives of aldehyde such as benzene sulfonyl hydrazones of the formula 9 but not for Losartan. This process is shown in the scheme 4

In J. Org. Chem 1994, 59, 6391-6394 again by Merck team reported Trityl Losartan and Losartan synthesis by coupling of boronic acid derivative 11 with 3-(4-bromobenzyl) derivative of BCBMI of the formula 10. The formed trityl Losartan of the formula 7 is converted to Losartan of the formula 1 with acid. The whole process is described in Scheme 5

The Compound of the formula 10 is prepared from the reaction of BCFI of the formula 4 with p-bromo benzyl bromide of the formula 12 in potassium carbonate and Dimethyl formamide followed by reduction with sodium borohydride (NaBH4). The details are given in the Scheme 6

The Compound of the formula 11 is prepared from 5-phenyl tetrazole of the formula 14 by reacting with trityl chloride to get N-trityl-5-phenyl tetrazole of the formula 13, which on reaction with butyl lithium and triisopropyl borate followed by hydrolysis to give compound of the formula 11. This process is shown in the Scheme 7

In one of the first patent filed by Dupont/Merck (date of filing 9 Jul. 1987, priority 11 January 1986 EP0253310) reported a procedure for the preparation of Losartan. Bromo OTBN of the formula 3 is reacted with BCHMI of the formula 15 in the presence of a base to give cyano alcohol of the formula 6, and its regioisomer of the formula 14. Separation of the isomer needs column chromatography. The cyano alcohol 6 is reacted with sodium/ammonium azide in DMF for 13 days to get Losartan 1 in 21% yield. The process is shown in the Scheme 8

The drawbacks of the above process are
- 1). Separation of the regioisomer using column chromatography which is industrially not feasible for the preparation of large scale (ton) material/product
- 2). The tetrazole formation takes 13 days with 21% yield, which is unproductive.
- 3). Dupont/Merck uses BCHMI 15 as the starting material for preparing cyano alcohol of the formula 6. BCHMI 15 is an expensive intermediate compared to BCFI 4, and also the formation of unwanted regio isomer 14 is higher. The process is schematically described in scheme 8. Even though the process looks simple it has two problems.
First: Cyano alcohol is produced as a mixture of regioisomers and needs column chromatography for purification.
Second: Tetrazole formation. This takes 13 days with 21% yield, which limits commercialization of the process.
In U.S. Pat. No. 4,820,843 and U.S. Pat. No. 4,879,186, Dupont prepares Losartan by reaction of BCFI of the formula 4 and N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16 in the presence of base, followed by reduction with sodium borohydride to give Trityl Losartan of the formula 7, which is treated with mineral acid to give Losartan 1.
The process is shown in scheme 9

In U.S. Pat. No. 4,874,867 of Dupont/Merck, a process for the preparation of N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16 is described by the reaction of OTBN of the formula 20 with trimethyl tin azide to give the compound 17, which is treated with Hydrochloric acid to give tetrazole derivative of OTBN of the formula 18. The tetrazole derivative of OTBN of the formula 18 is protected with trityl chloride to give compound of the formula 19, followed by bromination with N-bromosuccinimide to give N-Triphenylmethyl-5-[2-(4′-bromomethyl biphenyl)]tetrazole of the formula 16.
The process is shown in the scheme 10.

In all the above papers and patents by Dupont/Merck, the process yields in many steps are good 75-95% and in some steps are less to moderate 21-49%. The drawbacks, or the problems in all these processes is, the number of unit operations.
For example:
- 1). In J. Med. Chem 1991, 34, 2525-2547 the number of steps are six (6) to prepare Losartan of the formula 1 from the readily available intermediates.
- 2). In J. Org. Chem 1994, 59, 6391-6394 the number of steps are nine (9) to prepare Losartan of the formula 1 from the readily available intermediates.
- 3). In EP 0253310 patent the number of operations are two (2) but the problem is time & yields i.e., 13 days and poor yield (21%), also the uneconomical column chromatographic separation of regioisomer.
- 4). In U.S. Pat. Nos. 4,820,843 and 4,879,186 the number of steps are six (6).
- 5). In U.S. Pat. No. 4,874,867 the number of steps are seven (7).
………………………..
INTERMEDIATES
(1-(2′-Cyano biphenyl-4-methyl)-2-butyl-4-chloro-5-formyl imidazole) of the formula 5.

Melting point: 107-108° C.
HPLC Purity: >98%
IR. v max (KBR): 2218 (—CN), 1662.40 (—CHO)
1H NMR (CDCl3) δ, 0.91 (t, 3H), 1.38 (sext, 2H), 1.73 (quint, 2H), 2.67 (t, 2H), 5.61 (s, 2H), 7.16-7.77 (m, 8H), 9.77 (s, 1H).
13C NMR (CDCl3) δ, 13.51, 22.18, 26.33, 29.04, 47.74, 110.05, 118.36, 124.11, 126.59, 127.65, 129.16, 129.81, 132.76, 133.61, 136.01, 137.69, 142.96, 144.33, 154.46, 177.73
