
Home » Posts tagged 'IND 2021'
Tag Archives: IND 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BIAPENEM

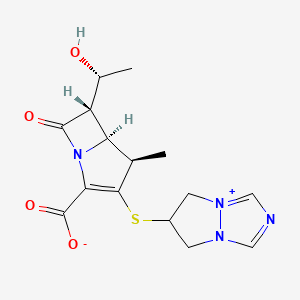
Biapenem
RPX7009
- Molecular FormulaC15H18N4O4S
- Average mass350.393 Da
Biapenern
CL 186-815LJ
C10,627LJ
C10627LJC 10627
omegacin
YR5U3L9ZH1
(4R,5S,6S)-3-((6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-yl)thio)-6-((R)-1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
[4R-[4a,5b,6b(R*)]]-6-[[2-Carboxy-6-(1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5 H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
120410-24-4[RN]
5H-Pyrazolo[1,2-a][1,2,4]triazol-4-ium, 6-[[(4R,5S,6S)-2-carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-, inner salt [ACD/Index Name]
6-[[(4R,5S,6S)-2-Carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
7074
(4R,5S,6S)-3-(6,7-Dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-ylsulfanyl)-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
TL8000539UNII:YR5U3L9ZH1UNII-YR5U3L9ZH1биапенем
بيابينام比
阿培南
INDIA CDSCO APPROVED 25 SEPT 2021, BDR PHARMA, 
https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/BDR.pdfhttps://medicaldialogues.in/news/industry/pharma/bdr-pharma-gets-dcgi-nod-for-generic-antibiotic-drug-biapenem-82384

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
Biapenem (INN) is a carbapenem antibiotic. It has in vitro activity against anaerobes.[1] 1-β-methyl-carbapenem antibiotic. Approved in Japan in 2001.
PATENT
EP 168707
EP 289801
JP 02088578
ZA 9100014
EP 533149
CN 1995040
IN 2006DE01555
CN 101121716
IN 2008CH00177
CN 101805359
CN 101851206
CN 101935321
CN 111875622
WO 2018074916
WO 2016059622
US 20150328323
WO 2015151081
WO 2015155753
WO 2015151078
US 20150284416
WO 2015151080
US 20150038726
WO 2014104488
IN 2013MU00181
WO 2014111957
CN 103570750
WO 2014097221
IN 2012CH01371
WO 2013150550
PAPERS
Journal of Organic Chemistry (1992), 57(15), 4243-9.
Heterocycles (1993), 36(8), 1729-34.
Journal of Antibiotics (1993), 46(12), 1866-82.
e-EROS Encyclopedia of Reagents for Organic Synthesis (2008), 1-3.
Bioorganic & medicinal chemistry letters (2009), 19(17), 5162-5.
IP.com Journal (2014), 14(12A), 1-3
IP.com Journal (2014), 14(10A), 1-2.
Bioorganic & medicinal chemistry (2013), 21(18), 5841-50.

NEW DRUG APPROVALS
one time
$10.00
PATENT
https://patents.google.com/patent/WO2014097221A1/esBiapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)- hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3 -yljthio] 6,7-dihydro-5H- pyrazolo[l,2-a][l,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.

Formula 1U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.EXAMPLESExample 1 : Purification of BiapenemBiapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.Yield: 84%HPLC Purity: 99.87% Example 2: Purification of BiapenemBiapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.Yield: 81.77%HPLC Purity: 99.80%
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013150550
The present invention relates to an improved process for the preparation of carbapenem antibiotic; more particularly relates to the preparation of Ertapenem monosodium salt of formula (I) having purity greater than 98.5% and having pharmaceutically acceptable level of residual solvent and palladium content.
The US patents namely US 5,478,820 and US 5,856,321 disclose various processes for preparing Ertapenem and its sodium salt. Example 12 of US 5,478,820 discloses a process in which the Ertapenem was isolated using column purification followed by freeze-drying technique. According to Example-4 of this patent disodium salt of Ertapenem was prepared by dissolving crude product in water using NaHCO3, followed by purification using column chromatography and subsequent lyophilization.
US 6,504,027 provides a process for preparing Ertapenem in crystalline form which comprises deprotecting and extracting a polar organic solution containing a crude mono-protected Ertapenem of formula
wherein P represents protecting group and X represents charge balancing group like sodium
with C4.10 alcohol in the presence of ion-pairing reagent followed by adjusting the pH of the aqueous layer to 5.5 and crystallizing using methanol and 1-propanol to produce a crystalline compound; this patent process involves operations like
multiple extractions which is cumbersome in plant and said operation affects the overall yield.
US 7,145,002 provides a process for producing Ertapenem or its sodium salt and/or its solvate in crystalline form. This patent states (refer para 3, lines 31-41) that contact of Ertapenem sodium with water and alcoholic solvents results in the formation of crystalline solvates. The processes reported in examples- 1 & 2 provide crystalline Ertapenem monosodium which is isolated from a mixture of methanol, 1-propanol and water followed by washing with aqueous isopropyl alcohol which results in the formation of crystalline solvate of Ertapenem sodium. Applicant found the Ertapenem monosodium obtained according to this process contain higher amount of residual solvent and palladium content.
US 7,022,841 provide a process for reducing the levels of organic solvents in Ertapenem to pharmaceutically acceptable levels. This patent discloses (Refer para 1, lines 52-60) that Ertapenem sodium obtained from water/alcohol mixture according to US 7, 145,002 becomes amorphous when water content of the solid is reduced and further the organic solvent present in the solid is not readily removed. In view of this drawback, this patent provides a process wherein the water content of Ertapenem sodium is maintained between 13-25% during the washing and drying process. This patent further discloses that (Refer para 9, lines 6-14) the washing of Ertapenem sodium can be carried out using anhydrous solvents which results in the formation of amorphous solid, which is then dried using hydrated nitrogen by increasing the water content of the solid. Due to the hygroscopic and unstable nature of Ertapenem sodium when in contact with water, the above processes result in more degradation of Ertapenem. The patent further discloses in example 5 that the degradation of Ertapenem sodium is more when it takes more time for drying.
Further this patent requires repetitive washing and control of moisture content to get the desired results.
For isolation of Ertapenem sodium from the reaction mass, all the above discussed prior art patents utilize methanol and 1-propanol as crystallization solvent. The filtration of Ertapenem sodium formed by using these solvents or their mixture takes longer time duration and subsequent drying for the removal of residual solvent also takes several hours due to occlusion of solvent into Ertapenem sodium. During these operations the Ertapenem sodium degrades an results in the formation of many impurities such as several dimers, methanolysis impurity etc., and hence the reported processes is not suitable to manufacture Ertapenem sodium on commercial scale with purity greater than 98.5% and with pharmaceutically acceptable level of residual solvent content.
Methanolysis impurity Dimer-I
Dimer-II
Further the applicant found that Ertapenem monosodium isolated by following the process reported in prior art was having palladium content above the pharmaceutically acceptable level. Hence the process reported in prior art is not suitable on manufacturing scale where maintaining stringent technological condition is cumbersome and involves higher operating cost.
Thus all the reported processes suffer in terms of one or more of the following facts:
■ Filtration time of Ertapenem sodium takes several hours.
■ Drying time takes several hours due to occlusion of solvent and nature of the solid.
■ Stringent technological condition is required for maintenance of moisture content during washing & drying operation.
■ Palladium content is found to be higher (greater than 25 ppm) which is not acceptable for pharmaceutical products.
■ The isolated Ertapenem sodium is having higher amount of residual solvents.
■ The purity is reduced over to several hours of filtration & drying.
With our continued research for developing a process for the preparation of Ertapenem monosodium of formula (I) to overcome the above mentioned drawbacks, we surprisingly found that when esters of organic acid were used as solvents in place of 1-propanol, the solid obtained was easily filterable with less cycle time. Further the washing with hydrocarbon solvents containing 0-75% alcoholic solvent followed by drying results in Ertapenem having residual solvent content well below the pharmaceutically acceptable levels. The use of thiourea, thiosemicarbazide or their N-substituted derivatives in the presence of organic solvents during isolation brings down the palladium content to pharmaceutically acceptable level.
The Ertapenem or its sodium salt can be prepared according the processes provi
(I)
P’ and P” represent carboxylic protecting groups and X is H or Na
Scheme-1
The present invention is illustrated with the following examples, which should not be construed to limit the scope of the invention.
Example- I
Preparation of Ertapenem monosodium of formula (I)
Step-I:
To a stirred solution of p-nitrobenzyl (4R,5S,6S)-3-(diphenyloxy)phosphoryloxy-6-[(lR)-l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound II) (100 g) and (2S,4S)-2-[[(3-carboxyphenyl) amino]carbonyl]-4-mercapto-l-(4-nitrobenzyl)pyrrolidinecarboxylate (compound III) (75 g) in N,N-dimethylformamide was added Ν,Ν-diisopropylethylamine at -30 to -40° C and stirred. The reaction mass, after completion of the reaction, was quenched with a mixture of phosphate buffer solution-ethyl acetate and the pH was adjusted to 5 – 6 with phosphoric acid. The organic layer was separated, washed with water and subjected to carbon treatment. To the organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl), a solution of sodium 2-ethylhexanoate (42 g in 500 mL methanol) was added and taken to next step as such. (If required the compound of formula (IV) is isolated either as sodium salt or as free acid by following the process reported in prior art and taken further)
Step-II:
To the Step-I organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl & X is Na), 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8- 10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered to remove palladium on carbon. To the filtrate, thiourea (5 g) and tetrahydrofuran were added and stirred. The aqueous layer was separated and treated with carbon and neutral alumina at 10-15° C while degassing and filtered. The filtrate was added to methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 46 g; Purity by HPLC: 98.93%; Palladium content: 1.8 ppm by ICP MS
The HPLC purity of Ertapenem monosodium was checked using the following parameters
Column : Zorbax Eclipse plus C8, (50 mm x 4.6 mm), 1.8μ).
Mobile phase : Ammoniam acetate buffer: Acetonitile: water
Detector : UV at 250 nm
Flow rate : 0.5 mL/min
Run time : 45 min.
Example- II
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon & neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with cyclohexane (200 ml) and
dried under vacuum. Yield: 44 g; Purity by HPLC: 98.84%; Palladium content: 0.93 ppm by ICP MS
The term ICP MS method refers to the inductively coupled plasma mass spectrometry. The following parameter was used to determine the content of palladium.
The carbapenem was digested in a closed vessel system in presence of reagents Nitric acid, Hydrogen peroxide and Hydrochloric acid by using Microwave reaction system with microwave radiation power 1200 Watts. The digested sample was introduced into inductively coupled plasma mass spectrometer by help of Peltier cooled spray chamber. The sample aerosol is getting atomized then ionized in the argon plasma. The ionized Palladium was estimated by using Quadrupole mass detector. The sample was quantified against NIST traceable reference standards at mass number ! 05.
Example- III
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was separated and treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of toluene: ethanol (200 ml) and dried under vacuum. Yield: 42 g; Purity by HPLC: 99.03%
Example- IV
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the filtrate was treated with thiosemicarbazide and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C followed by the addition of ethyl acetate and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 41 g; Purity by HPLC: 99.13%; Palladium content: 1.71 ppm by ICP MS
Example- V
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, a mixture of ethyl acetate containing 10% methyl acetate was added and stirred. The solid obtained was
filtered, washed with cyclohexane:ethanol and dried under vacuum. Yield: 40.5 g; Purity by HPLC: 98.77%; Palladium content: 1.43 ppm by ICP MS
Example-VI
(V ) (V I )
The diprotected Meropenem of formula (V) (where P and P’ were p-nitrobenzyl) was dissolved in tetrahydrofuran and 3-(N-morpholino)propanesulfonic acid buffer and hydrogenated using palladium on carbon at 9-10 kg hydrogen pressure. The mass was filtered and the filtrate was washed with ethyl acetate. The aqueous layer was treated with thiourea and 2-methyltetrahydrofuran. The aqueous layer was separated, treated with carbon and degassed. The carbon was filtered off and acetone was added to the filtrate to crystallize Meropenem trihydrate of formula (VI). The product was filtered and washed with aq. acetone and dried under vacuum to get Meropenem trihydrate. Purity: 99.8%; Pd content: 0.08 ppm
Reference example-I:
Preparation of Ertapenem monosodium of formula (I)
To Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered. The filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass ethyl acetate was added and stirred. The solid obtained was filtered, washed with ethanol (5 * 100 ml) and dried under vacuum. Yield: 31 g; Purity by HPLC: 96.76%
Reference example-II:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass , as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol and dried under vacuum. Yield: 43 g; Purity by HPLC: 98.6%; Palladium content: 35.8 ppm by ICP MS.
Reference example-HI:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with 1-propanol at -5° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass methanol and 1-propanol were added and stirred. The solid obtained was filtered, washed with ethanol and dried under nitrogen atmosphere in vacuum. Yield: 25 g; Purity by HPLC: 97 %.: palladium content: 38.2 ppm
The following tables illustrate the advantages of the present invention over prior art process:
Table-I: Comparison of present process with prior art process
The crystallization and washing method disclosed in US 7,022,841 was followed.
The above table indicates that the use of ethyl acetate as crystallization solvent results with improved yield and high purity with less filtration and drying time thereby increasing the productivity significantly on manufacturing scale. Further the use of thiourea or thiosemicarbazide as reagents in the present process results in the pharmaceutically acceptable level of palladium content.
Table-II: Comparison of solvents for washing Ertapenem monosodium
The above table indicates that the use of hydrocarbon solvents containing 0-75% of alcoholic solvent helps in washing to remove the residual solvent content in shorter duration and with single run wash. On the other hands the use of ethanol alone results in Ertapenem monosodium having less yield and purity requiring repetitive washing.
Table-IH: Effect of different reagent in reduction of palladium content
Reagent : thiourea, thiosemicarbazide or its N-substituted derivatives
Advantages of the process of the present invention:
> The use of ester of an organic acid for the crystallization of Ertapenem sodium results in fast filtration and reduced cycle time, thereby increasing the productivity.
> Washing of Ertapenem sodium with hydrocarbon solvent optionally containing alcohol results in improved physical nature of Ertapenem sodium resulting in reduced washing and drying time thereby avoid the degradation of Ertapenem and providing Ertapenem sodium with purity greater than 98.5% by HPLC.
Use of thiourea, thiosemicarbazide or their N-substituted derivatives in the process results in Ertapenem sodium having pharmaceutically acceptable level of palladium content.
PATENT
https://patents.google.com/patent/WO2002057266A1/enEXAMPLE

PNB = p-nitrobenzyl

Ia’A hydrogenator is charged with 63 g of 10% Pd on carbon catalyst (dry weight) in 1.8 L of water. The vessel is placed under hydrogen then vented and placed under nitrogen. Sodium hydroxide (68 g, 50%) is charged adjusting the pH to about 7.5 with carbon dioxide.The enol phosphate (170 g) and the thiol (86 g) are dissolved in 1.3‘L of N- ethylpyrrolidinone (NEP). The mixture is cooled to below -40°C and 1,1,3,3- tetramethylguanidine (109 g) is added. After 3 hours, the reaction mixture is quenched into the hydrogenator at below 15°C adjusting the pH to about 8 with carbon dioxide. The vessel is placed under hydrogen. When the reaction is complete, the hydrogen is vented and the reaction mixture is treated with activated carbon and filtered. The filtrate is extracted with iso-amyl alcohol containing diphenylphosphoric acid (240 g) and 50% NaOH (44 g). The resulting aqueous solution is further extracted with iso-amyl alcohol to give an aqueous solution containing at least 90 mg/mL of the product. Both extractions are performed using two CINC centrifugal separators set in series for countercurrent extraction. The pH is adjusted to 5.5 with acetic acid. The product is crystallized by adding equal volumes of methanol and 1- propanol at below -5°C and isolated by filtration. The solid is washed with a mixture of 2-propanol and water (85: 15 v/v) then dried to yield a compound of formula la’.While certain preferred embodiments of the invention have been described herein in detail, numerous alternative embodiments are contemplated as falling within the scope of the appended claims. Consequently the invention is not to be limited thereby.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleUS4866171A1987-04-111989-09-12Lederle (Japan), Ltd.(1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazolium-6-yl)]thio-6-[R-1-hydroxyethyl]-1-methyl-carbapenum-3-carboxylateUS5241073A1990-10-121993-08-31Lederle (Japan)Process for preparing (1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo [1,2-a][1,2,4]triazolium-6-yl)]thio-6-[(R)-1-hydroxyethyl]-1-methyl-carbapenem-3-carboxylate and starting materials thereofUS5286856A1991-09-201994-02-15Takeda Chemical Industries, Ltd.Production of crystalline penemWO2002057266A1 *2001-01-162002-07-25Merck & Co., Inc.Improved process for carbapenem synthesisWO2009047604A1 *2007-10-082009-04-16Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of carbapenem antibioticCN102268025A *2011-07-152011-12-07海南美兰史克制药有限公司一种比阿培南化合物及其制法
References
- ^ Aldridge KE, Morice N, Schiro DD (April 1994). “In vitro activity of biapenem (L-627), a new carbapenem, against anaerobes”. Antimicrob. Agents Chemother. 38 (4): 889–93. doi:10.1128/aac.38.4.889. PMC 284564. PMID 8031067.
External links
- (in Japanese) Omegacin
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | IV |
| ATC code | J01DH05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120410-24-4 |
| PubChem CID | 71339 |
| ChemSpider | 64442 |
| UNII | YR5U3L9ZH1 |
| ChEBI | CHEBI:3089 |
| ChEMBL | ChEMBL285347 |
| CompTox Dashboard (EPA) | DTXSID5046435 |
| Chemical and physical data | |
| Formula | C15H18N4O4S |
| Molar mass | 350.39 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04552444 | Clinical Efficacy of Combination Therapy Based on High-dose Biapenem in CRKP Infections | Recruiting | 2020-09-17 | |
| NCT01772836 | Safety Study of Intravenous Biapenem (RPX2003) and RPX7009 Given Alone and in Combination | Phase 1 | Completed | 2013-07-11 |
| NCT01702649 | Safety, Tolerability, Pharmacokinetics of Intravenous RPX2003 (Biapenem) in Healthy Adult Subjects | Phase 1 | Completed | 2012-12-03 |
NIPH Clinical Trials Search of Japan
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| UMIN000017219 | Feasibility and efficacy of the de-escalation therapy by Biapenem for postoperative bacterial pneumonia. | None | Recruiting | 2015-04-22 |
| UMIN000003964 | Clinical evaluation of Biapenem 0.3g, three times daily dosing in eldery patients with pneumonia (moderate and severe infection) | Not applicable | Complete: follow-up complete | 2010-07-29 |
/////////BIAPENEM, TL8000539, UNII:YR5U3L9ZH1, UNII-YR5U3L9ZH1, биапенем, بيابينام ,比阿培南 , Biapenern, CL 186-815, CL 186815, L 627, LJC 10627, Omegacin, Antibacterial, Antibiotics, Lactams, Carbapenems, ind 2021, india 2021, approvals 2021
CC1C2C(C(=O)N2C(=C1SC3CN4C=NC=[N+]4C3)C(=O)[O-])C(C)O
https://clinicaltrials.gov/search/intervention=Biapenem

updated
Biapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)-hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3-yljthio] 6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.
Formula 1
U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.
U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.
U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.
The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.
The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.
EXAMPLES
Example 1 : Purification of Biapenem
Biapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.
Yield: 84%
HPLC Purity: 99.87%
Example 2: Purification of Biapenem
Biapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.
Yield: 81.77%
HPLC Purity: 99.80%
PATENT
Background of the Invention Biapenem is a synthetic broad-spectrum carbapenem antibiotic which suppresses bacterial growth by inhibiting the enzymes responsible for bacterial cell wall synthesis, and shows broad-spectrum antibacterial activity both against gram-positive bacteria and gram-negative bacteria. Biapenem is chemically known as (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][ 1,2,4] triazol-8-ium-6-ylsulfanyl)-6-( 1 -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo [3.2.0]hept-2-ene-2-carboxylate and marketed in Japan as OMEGACIN®.Various methods are reported in the prior art for the preparation of Biapenem of formula (I) which includes the condensation of compound of formula (II) with compound of formula (III) and subsequent deprotection of the protecting group as shown in scheme-1. wherein R1 is hydrogen or hydroxy protecting group such as tert-butyl dimethyl silyl and the like, R2 is hydrogen or carboxyl protecting group such as p-nitrobenzyl, p-methoxy benzyl, allyl and the like, A is an activating group such as P(0)(OR)2, SO2R and the like wherein R is selected from substituted or unsubstituted C1-6 alkyl, aralkyl or aryl to form the compound of formula (II). The X” in compound of formula (III) is halogen selected from Br or CI.Biapenem was first disclosed in US 4,866,171 and the said patent also discloses a process for the preparation of the same. US 5,241,073 disclosed the method for the preparation of compound of formula (III) followed by condensation with compound of general formula (II) using base such as N-ethyldiisopropylamine and subsequent deprotection yields Biapenem which was isolated by column chromatography followed by crystallization from ethanol.EP 0289801 discloses a process for the preparation of crystalline Biapenem wherein Biapenem was dissolved in water and lyophilized to get amorphous compound. The amorphous compound was dissolved in water at 40° C followed by cooling to get crystalline product. This patent further provides the PXRD values of the crystalline Biapenem. The Biapenem obtained according to the process provided in this patent takes longer time for reconstitution and hence not suitable.US 5,286,856 and US 5,424,069 provide a process for the crystallization of Biapenem which utilizes freeze-drying technique and vial lyophillisation method respectively. These patents disclose (refer para 1, lines 10-33 of US’ 856) that the process provided in EP 0289801 results with Biapenem crystals which take relatively longer time for dissolution during use. To overcome the above issues, these patents utilize the freeze-drying and vial lyophillisation methods. The said methods involve freezing of the solution containing Biapenem followed by raising the temperature and repeating the cooling and heating process followed by lyophillisation to get the crystalline product. Lyophillisation and related process are capital intensive techniques and uneconomical in commercial scale operations.All the above said prior arts utilize either the lyophillisation technique or preparing the amorphous material and crystallizing it from water to get crystalline Biapenem.Biapenem is available as powder for injection which needs to be reconstituted with water or saline solution before injection. The process of preparing a solution having an appropriate concentration of an active ingredient for the administration is called “reconstitution”. The reconstitution time (RCT) plays a critical role in injectable powders. Short reconstitution time is preferable for both a member of medical center and patients. If the reconstitution time is too long, it will increase the preparation time thus making it difficult to administrate it to many patients at the same, which will eventually lower the competitiveness of the drug. The problem before the applicants is to find economic and robust process for the preparation of Biapenem with high purity and yield which should dissolve in water in less than 25 seconds (reconstitution time). With our continued intensive and diligent research for developing a process for the preparation of Biapenem having high purity and yield with reconstitution time of less than 25 seconds, we have identified an improved process which is commercially viable and eliminates the issues associated with reconstitution time. The process of this invention is simple and obviates the use of freeze crystallization. Further the present invention fulfils the need for a process for the manufacture of Biapenem which is convenient to operate in commercial scale
Objectives of the inventionThe main objective of the present invention is to provide a simple and commercially viable, industrially scalable process for the crystallization of Biapenem of formula (I) with high purity and good yield.Yet another objective of the present invention is to provide a simple and commercially suitable process for the preparation of Biapenem of formula (I) with reconstitution time less than 25 seconds. The reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Summary of the inventionAccordingly the primary aspect of the present invention is to provide an improved process for the preparation of Biapenem of formula (I) the said process comprises;(i) obtaining a solution of Biapenem in water containing co-solvent; and(ii) adding anti-solvent in to the solution of step (i) or vice-versa to crystallize Biapenem followed by filtration. Detailed Description In an embodiment of the present invention, the co-solvent used in step (i) is selected from alcoholic solvents consisting of methanol, ethanol, isopropyl alcohol, n-propanol, n-butanol and iso-butanol or mixtures thereof; preferably methanol, ethanol and isopropyl alcohol; more preferably methanol.In another embodiment of the present invention the anti-solvent used in step (ii) is selected from acetone, methyl ethyl ketone, methyl isobutyl ketone, ethyl acetate, methyl acetate, butyl acetate, tetrahydrofuran or mixtures thereof; preferably acetone. In yet another embodiment of the present invention, the solution of Biapenem in step (i) can be obtained by (a) dissolving Biapenem in water followed by addition of co-solvent (b) dissolving Biapenem in water containing the co-solvent (c) the aqueous solution containing Biapenem can be obtained directly from the reaction mass followed by addition of co-solvent (d) the aqueous solution of Biapenem containing co-solvent can be obtained directly from the reaction mass. The said solutions, if necessary can be subjected to sterile filtration before the addition of anti-solvent. Thus the present invention provided a process for the preparation of sterile Biapenem having reconstitution time less than 25 seconds, more preferably less than 15 seconds.The prior art lyophillisation process for the preparation of Biapenem requires capital investment and high operating cost due to the involvement of repetitive heating and cooling process which is tedious technology in commercial scale operations. The reported prior art process for the crystallization of Biapenem of formula (I) from water results in the formation of crystalline powder which takes longer time for dissolution in water or saline solution (reconstitution time). Surprisingly, applicant found that the use of co-solvents during the crystallization of Biapenem results with Biapenem having reconstitution time of less than 25 seconds. This constitutes the novelty of the present invention.In this present invention the Biapenem of formula (I) is obtained as crystalline solid with purity above 99.0 % by HPLC with good stability and further can be easily filled in vials.
The following examples are provided by way of illustration only and should not be construed to limit the scope of the invention.
Crystallization of (4R,5S,6S)-3-(6,7-dihvdro-5H-pyrazolo[l,2-al 11,2,41 triazol-8-ium-6-vlsulfanvl)-6-(l-hydroxvethvl)-4-methvl-7-oxo-l-azabicyclo [3.2.01hept-2-ene-2-carboxvlate [Biapenem of formula (1)1:Example -1:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. Activated carbon and EDTA were added to the clear solution and filtered through hi-flow bed, washed with water followed by filtration through micron filters in sterile area. To the filtrate, methanol (600 mL) was added followed by acetone under stirring. To the reaction mass, Biapenem seed material was added and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 85 g Purity by HPLC: 99.5% Reconstitution time (RCT): < 15 seconds
Example -2:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. To the filtrate, isopropyl alcohol (500 ml) was added followed by acetone under stirring. The mass was cooled and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 83 g Purity by HPLC: 99.6% Reconstitution time: < 15 seconds
Example -3;To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through micron filters. To the filtrate, ethanol (600 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 84 g Purity by HPLC: 99.5% Reconstitution time : < 15 seconds
Example -4:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through hi-flow bed, washed with water followed by filtration through micron filters. To the filtrate, methanol (450 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem. Yield: 87 g Purity by HPLC: 99.4% Reconstitution time (RCT): < 15 seconds
Reference example -1:Preparation of Biapenem (Non-Sterile)Step-I: Preparation of p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pvrazolofl,2-al[l,2,41triazol-8-ium-6-vlsulfanvn-6-(l-hvdroxvethyl)-4-methvI-7-oxo-l-azabicvclo[3.2.01hept-2-ene-2-carboxylate [Compound of formula (IV)1To a mixture of acetonitrile and DMF, P-Nitrobenzyl (4R,5S,6S)-3-(dipheny loxy)phosphory loxy-6- [(1R)-1 -hydroxyethy 1] -4-methy 1-7-oxo-1 -azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound of formula II) and 6,7-dihydro-6-mercapto-5H-pyrazolo[l,2-a] [1,2,4] triazole chloride (compound of formula III) were added and cooled to 0-5° C. To this mixture, N-ethyldiisopropyl amine was added and stirred till the completion of the reaction, followed by the addition of dichloromethane to crystallize the p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo[3.2.0] hept-2-ene-2-carboxylate which was filtered and dried under nitrogen.
Step-II: Preparation of BiapenemTo a solution of MOPS buffer and THF, p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l-hydroxy ethyl)-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (Compound of formula-IV) was added at pH 7-8 and cooled to 5-10° C. The mixture was hydrogenated using palladium on carbon as catalyst. The catalyst was filtered and the filtrate was treated with activated carbon and filtered. The filtrate was extracted with dichloromethane and the layers separated. The aqueous layer was degassed. To the aqueous layer, acetone was added to crystallize Biapenem at 20-25° C. The product was filtered, washed with aqueous acetone and dried under vacuum to get Biapenem (Non-Sterile).
Reference example -2: Crystallization of Biapenem
Example -1 was repeated without the addition of methanol.Yield: 84 g Purity by HPLC: 99.5%Reconstitution time : > 90 secondsThe reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Table-1: Comparative Data:The comparative data provided in the table-1 clearly indicates that the addition of co-solvent during crystallization provides Biapenem with reconstitution time less than 25 seconds.
Benzonatate
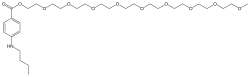

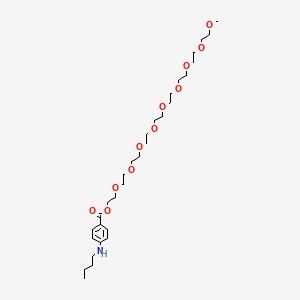

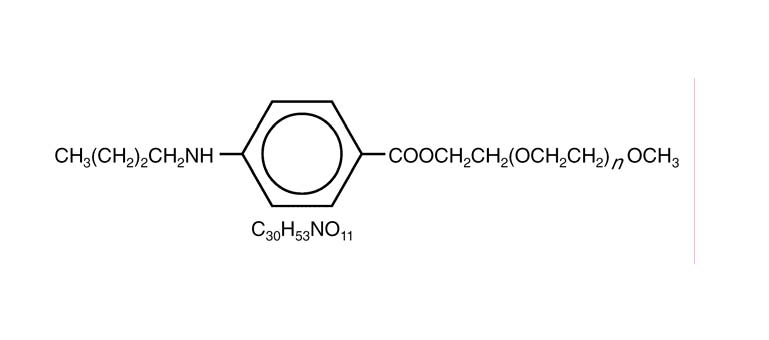
Benzonatate
- Molecular FormulaC30H53NO11
- Average mass603.742 Da
104-31-4[RN]2,5,8,11,14,17,20,23,26-Nonaoxaoctacosan-28-yl 4-(butylamino)benzoateбензонататبنزوناتات苯佐那酯ベンゾナテート;KM 652,5,8,11,14,17,20,23,26-nonaoxaoctacosan-28-yl 4-(butylamino)benzoate2-[2-[2-[2-[2-[2-[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethyl 4-(butylamino)benzoate
Benzonatate bulk and Benzonatate capsules 100mg, cdsco india 2021, 15.07.2021
For the treatment of refractory coughCAS Registry Number: 104-31-4CAS Name: 4-(Butylamino)benzoic acid 3,6,9,12,15,18,21,24,27-nonaoxaoctacos-1-yl esterAdditional Names: nonaethyleneglycol monomethyl ether p-n-butylaminobenzoate; p-butylaminobenzoic acid w-O-methylnonaethyleneglycol ester; benzononatineTrademarks: Exangit; Tessalon (Forest)Molecular Formula: C30H53NO11Molecular Weight: 603.74Percent Composition: C 59.68%, H 8.85%, N 2.32%, O 29.15%Literature References: Prepn: Matter, US2714608 (1955 to Ciba).Properties: Colorless to faintly yellow oil. Soluble in most organic solvents except aliphatic hydrocarbons.Therap-Cat: Antitussive.Keywords: Antitussive.
Synthesis Reference
Matter, M.; U.S. Patent 2,714,608; August 2, 1955; assigned to Ciba Pharmaceutical Products, Inc.
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 94-32-6 | C13H19NO2 | ethyl 4-butylaminobenzoate | Benzoic acid, 4-(butylamino)-, ethyl ester |
| 6048-68-6 | C19H40O10 | nonaethylene glycol monomethyl ether | 2,5,8,11,14,17,20,23,26-Nonaoxaoctacosan-28-ol |
Benzonatate, sold under the brand name Tessalon among others, is a medication used to try to help with the symptoms of cough and hiccups.[1][2] It is taken by mouth.[1] Use is not recommended in those under the age of ten.[3] Effects generally begin within 20 minutes and last up to eight hours.[1][4]
Side effects include sleepiness, dizziness, headache, upset stomach, skin rash, hallucinations, and allergic reactions.[1] Excessive doses may cause seizures, irregular heartbeat, and death.[3] Chewing or sucking on the capsule can lead to laryngospasm, bronchospasm, and circulatory collapse.[1] It is unclear if use in pregnancy or breastfeeding is safe.[5] It works by numbing stretch receptors in the lungs and suppressing the cough reflex in the brain.[1]
Benzonatate was approved for medical use in the United States in 1958.[1] It is available as a generic medication.[3] It is not available in many countries.[6] In 2018, it was the 113th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[7][8]
Medical uses

100mg generic benzonatate capsules
Cough
Benzonatate is a prescription non-opioid alternative for the symptomatic relief of cough.[1][3] It has been shown to improve cough associated with a variety of respiratory conditions including asthma, bronchitis, pneumonia, tuberculosis, pneumothorax, opiate-resistant cough in lung cancer, and emphysema.[1][9][10]
Benzonatate also reduces the consistency and volume of sputum production associated with cough in those with chronic obstructive pulmonary disorder (COPD).[9]
Compared to codeine, benzonatate has been shown to be more effective in reducing the frequency of induced cough in experiments.[1]
Benzonatate does not treat the underlying cause of the cough.[11]
Hiccups
Benzonatate has been shown to have use in the suppression of hiccups.[2]
Intubation
Benzonatate acts as a local anesthetic and the liquid inside the capsule can be applied in the mouth to numb the oropharynx for awake intubation.[1] However, there can be life-threatening adverse effects when the medication is absorbed by the oral mucosa, including choking, hypersensitivity reactions, and circulatory collapse.[1]
Contraindications
Hypersensitivity to benzonatate or any related compounds is a contraindication to its administration.[4]
Side effects
Benzonatate is generally well-tolerated[vague][specify] if the liquid-capsule is swallowed intact.[1] Potential adverse effects to benzonatate include:
- Constipation, dizziness, fatigue, stuffy nose, nausea, headache are frequently reported.[12]
- Sedation, a feeling of numbness in the chest, sensation of burning in the eyes, a vague “chilly” sensation, itchiness, and rashes are also possible.[1][4]
- Ingestion of a small handful of capsules has caused seizures, cardiac arrhythmia, and death in adults.[13]
Hypersensitivity reactions
Benzonatate is structurally related to anesthetic medications of the para-aminobenzoic acid (PABA) class which includes procaine and tetracaine.[4][13] Procaine and tetracaine, previously used heavily in the fields of dentistry and anesthesiology, have fallen out of favor due to allergies associated with their metabolites.[13] Similarly, severe hypersensitivity reactions to benzonatate have been reported and include symptoms of laryngospasm, bronchospasm, and cardiovascular collapse.[4][14] These reactions are possibly associated with chewing, sucking, or crushing the capsule in the mouth.[4][13]
Improper use
Benzonatate should be swallowed whole.[4] Crushing or sucking on the liquid-filled capsule, or “softgel,” will cause release of benzonatate from the capsule and can produce a temporary local anesthesia of the oral mucosa.[4] Rapid development of numbness of the tongue and choking can occur.[4][13] In severe cases, excessive absorption can lead to laryngospasm, bronchospasm, seizures, and circulatory collapse.[4][13] This may be due to a hypersensitivity reaction to benzonatate or a systemic local anesthetic toxicity, both of which have similar symptoms.[13] There is a potential for these adverse effects to occur at a therapeutic dose, that is, a single capsule, if chewed or sucked on in the mouth.[13]
Psychiatric effects
Isolated cases of bizarre behavior, mental confusion, and visual hallucinations have been reported during concurrent use with other prescribed medications.[4] Central nervous system effects associated with other para-aminobenozic acid (PABA) derivative local anesthetics, for example procaine or tetracaine, could occur with benzonatate and should be considered.[1]
Children
Safety and efficacy in children below the age of 10 have not been established.[4] Accidental ingestion resulting in death has been reported in children below the age of 10.[4] Benzonatate may be attractive to children due to its appearance, a round-shaped liquid-filled gelatin capsule, which looks like candy.[14][15] Chewing or sucking of a single capsule can cause death of a small child.[4][15] Signs and symptoms can occur rapidly after ingestion (within 15–20 minutes) and include restlessness, tremors, convulsions, coma, and cardiac arrest.[15] Death has been reported within one hour of ingestion.[12][15]
Pregnancy and breast feeding
In the U.S., benzonatate is classified by the U.S. Food and Drug Administration (FDA) as pregnancy category C.[5] It is not known if benzonatate can cause fetal harm to a pregnant woman or if it can affect reproduction capacity.[4][5] Animal reproductive studies have not yet been conducted with benzonatate to evaluate its teratogenicity.[4] Benzonatate should only be given to a pregnant woman if it is clearly needed.[4][5]
It is not known whether benzonatate is excreted in human milk.[4][5] It is recommended to exercise caution when benzonatate is given to a nursing woman.[4][5]
Overdose
Benzonatate is chemically similar to other local anesthetics such as tetracaine and procaine, and shares their pharmacology and toxicology.[13]
Benzonatate overdose is characterized by symptoms of restlessness, tremors, seizures, abnormal heart rhythms (cardiac arrhythmia), cerebral edema, absent breathing (apnea), fast heart beat (tachycardia), and in severe cases, coma and death.[1][4][16][11] Symptoms develop rapidly, typically within 1 hour of ingestion.[4][11] Treatment focuses on removal of gastric contents and on managing symptoms of sedation, convulsions, apnea, and cardiac arrhythmia.[4]
Despite a long history of safe and appropriate usage, the safety margin of benzonatate is reportedly narrow.[13] Toxicity above the therapeutic dose is relatively low and ingestion of a small handful of pills can cause symptoms of overdose.[13][11] Children are at an increased risk for toxicity, which have occurred with administration of only one or two capsules.[15][16][11]
Due to increasing usage of benzonatate and rapid onset of symptoms, there are accumulating cases of benzonatate overdose deaths, especially in children.[11]
Pharmacology
Benzonatate is chemically similar to other local anesthetics such as tetracaine and procaine, and shares their pharmacology.[13]
Mechanism of action
Similar to other local anesthetics, benzonatate is a potent voltage-gated sodium channel inhibitor.[13] After absorption and circulation to the respiratory tract, benzonatate acts as a local anesthetic, decreasing the sensitivity of vagal afferent fibers and stretch receptors in the bronchi, alveoli, and pleura in the lower airway and lung.[1][2] This dampens their activity and reduces the cough reflex.[1][4] Benzonatate also has central antitussive activity on the cough center in central nervous system at the level of the medulla.[1][9] However, there is minimal inhibition of the respiratory center at a therapeutic dosage.[4]
Pharmacokinetics
The antitussive effect of benzonatate begins within 15 to 20 minutes after oral administration and typically lasts between 3 and 8 hours.[4][9]
Benzonatate is hydrolyzed by plasma butyrylcholinesterase (BChE) to the metabolite 4-(butylamino)benzoic acid (BABA) as well as polyethylene glycol monomethyl esters.[13] Like many other local anesthetic esters, the hydrolysis of the parent compound is rapid.[13] There are concerns that those with pseudocholinesterase deficiencies may have an increased sensitivity to benzonatate as this hydrolysis is impaired, leading to increased levels of circulating medication.[13]
Chemical structure
Benzonatate is a butylamine, structurally related to other polyglycol ester local anesthetics such as procaine and tetracaine.[13] The molecular weight of benzonatate is 603.7 g/mol.[4] However, the reference standard for benzonatate is a mixture of n-ethoxy compounds, differing in the abundance of 7-9 repeating units, with an average molecular weight of 612.23 g/mol.[13] There is also evidence that the compound is not uniform between manufacturers.[13]
Society and culture
Benzonatate was first made available in the U.S. in 1958 as a prescription medication for the treatment of cough in individuals over the age of 10.[15][16] There are a variety of prescription opioid-based cough relievers, such as hydrocodone and codeine, but have unwanted side effects and potential of abuse and diversion.[13] However, benzonatate is currently the only prescription non-opioid antitussive and its usage has been rapidly increasing.[13][11] The exact reasons of this increase are unclear.[11]
Economics
In the United States between 2004 and 2009, prescriptions increased 50% from 3.1 million to 4.7 million, the market share of benzonatate among antitussives increased from 6.3% to 13%, and the estimated number of children under the age of 10 years receiving benzonatate increased from 10,000 to 19,000.[13][11] Throughout this same period, greater than 90% of prescriptions were given to those 18 or older.[11] The majority of prescriptions were given by general, family, internal, and osteopathic physicians with pediatricians account for about 3% of prescribed benzonatate.[11]
In 2018, it was the 113th most commonly prescribed medication in the United States, with more than 6 million prescriptions.[7][8]
Brand names
Tessalon is a brand name version of benzonatate manufactured by Pfizer, Inc.[13][11] It is available as perles or capsules.[17] Zonatuss was a brand name manufactured by Atley Pharmaceuticals, Inc. and Vertical Pharmaceuticals, Inc.[18][19]
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s “Benzonatate Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 23 March 2019.
- ^ Jump up to:a b c Becker, DE (2010). “Nausea, vomiting, and hiccups: a review of mechanisms and treatment”. Anesthesia Progress. 57 (4): 150–6, quiz 157. doi:10.2344/0003-3006-57.4.150. PMC 3006663. PMID 21174569.
- ^ Jump up to:a b c d “Drugs for cough”. The Medical Letter on Drugs and Therapeutics. 60 (1562): 206–208. 17 December 2018. PMID 30625123.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z “Tessalon – benzonatate capsule”. DailyMed. 20 November 2019. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f “Benzonatate Use During Pregnancy”. Drugs.com. 10 October 2019. Retrieved 20 February 2020.
- ^ Walsh, T. Declan; Caraceni, Augusto T.; Fainsinger, Robin; Foley, Kathleen M.; Glare, Paul; Goh, Cynthia; Lloyd-Williams, Mari; Olarte, Juan Nunez; Radbruch, Lukas (2008). Palliative Medicine E-Book. Elsevier Health Sciences. p. 751. ISBN 9781437721942.
- ^ Jump up to:a b “The Top 300 of 2021”. ClinCalc. Retrieved 18 February2021.
- ^ Jump up to:a b “Benzonatate – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b c d Homsi, J.; Walsh, D.; Nelson, K. A. (November 2001). “Important drugs for cough in advanced cancer”. Supportive Care in Cancer. 9 (8): 565–574. doi:10.1007/s005200100252. ISSN 0941-4355. PMID 11762966. S2CID 25881426.
- ^ Estfan, Bassam; LeGrand, Susan (November 2004). “Management of cough in advanced cancer”. The Journal of Supportive Oncology. 2 (6): 523–527. ISSN 1544-6794. PMID 16302303.
- ^ Jump up to:a b c d e f g h i j k l McLawhorn, Melinda W.; Goulding, Margie R.; Gill, Rajdeep K.; Michele, Theresa M. (January 2013). “Analysis of benzonatate overdoses among adults and children from 1969-2010 by the United States Food and Drug Administration”. Pharmacotherapy. 33 (1): 38–43. doi:10.1002/phar.1153. ISSN 1875-9114. PMID 23307543. S2CID 35165660.
- ^ Jump up to:a b “Benzonatate (Professional Patient Advice)”. Drugs.com. 4 March 2020. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w Bishop-Freeman SC, Shonsey EM, Friederich LW, Beuhler MC, Winecker RE (June 2017). “Benzonatate Toxicity: Nothing to Cough At”. J Anal Toxicol. 41 (5): 461–463. doi:10.1093/jat/bkx021. PMID 28334901.
- ^ Jump up to:a b “Drugs for Cough”. The Medical Letter on Drugs and Therapeutics. 60 (1562): 206–208. 17 December 2018. PMID 30625123.
- ^ Jump up to:a b c d e f “FDA Drug Safety Communication: Death resulting from overdose after accidental ingestion of Tessalon (benzonatate) by children under 10 years of age”. U.S. Food and Drug Administration (FDA). 28 June 2019. Retrieved 22 April 2020.
- ^ Jump up to:a b c “In brief: benzonatate warning”. The Medical Letter on Drugs and Therapeutics. 53 (1357): 9. 7 February 2011. ISSN 1523-2859. PMID 21304443.
- ^ “Tessalon- benzonatate capsule”. DailyMed. 20 November 2019. Retrieved 25 April 2020.
- ^ “Zonatuss (Benzonatate Capsules USP, 150 mg)”. DailyMed. 2 June 2010. Retrieved 20 August 2020.
- ^ “Zonatuss (Benzonatate Capsules USP, 150 mg)”. DailyMed. 31 October 2016. Retrieved 20 August 2020.
External links
- “Benzonatate”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Tessalon, Zonatuss, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682640 |
| License data | US DailyMed: Benzonatate |
| Routes of administration | By mouth |
| ATC code | R05DB01 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Elimination half-life | 3-8 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 32760-16-0 |
| PubChem CID | 7699 |
| IUPHAR/BPS | 7611 |
| DrugBank | DB00868 |
| ChemSpider | 7413 |
| UNII | 5P4DHS6ENR |
| KEGG | D00242 |
| ChEBI | CHEBI:3032 |
| ChEMBL | ChEMBL1374379 |
| CompTox Dashboard (EPA) | DTXSID9022655 |
| ECHA InfoCard | 100.002.904 |
| Chemical and physical data | |
| Formula | C30H53NO11 |
| Molar mass | 603.750 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////Benzonatate, refractory cough , INDIA 2021, APPROVALS 2021, бензонатат , بنزوناتات , 苯佐那酯 , KM 65 , ベンゾナテート, ANTITUSSIVE, IND 2021
CCCCNC1=CC=C(C=C1)C(=O)OCCOCCOCCOCCOCCOCCOCCOCCOCCOC

NEW DRUG APPROVALS
ONE TIME
$10.00
TRIENTINE HYDROCHLORIDE, 塩酸トリエンチン , 曲恩汀
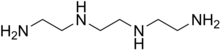
TRIENTINE
- Molecular Formula C6H18N4
- Average mass 146.234 Da
112-24-3 CAS
曲恩汀, KD-034, MK-0681, MK-681, TECZA, TETA, TJA-250

TRIENTINE HYDROCHLORIDE
- Molecular Formula C6H19ClN4
- Average mass 182.695 Da
38260-01-4 CAS
Launched – 1986 VALEANT, WILSONS DISEASE

塩酸トリエンチン
Trientine Hydrochloride

C6H18N4 2HCl : 219.16
2HCl : 219.16
[38260-01-4]
UPDATE CDSCO INDIA Trientine 08.06.2021 APPROVED
Trientine Tetrahydrochloride bulk and
Trientine Tetrahydrochloride capsules 333 mg
(Each capsule contains Trientine
tetrahydrochloride 333mg equivalent to
Trientine 167mg base)
For the treatment of Wilson’s disease
(hepatolenticular degeneration) in patients
intolerant to Penicillamine. It should be
used when continued treatment with
Penicillamine is no longer possible because
of intolerable or life endangering side
effects.
Aton Pharma, a subsidiary of Valeant Pharmaceuticals, has developed and launched Syprine, a capsule formulation of trientine hydrochloride, for treating Wilson disease.

Triethylenetetramine, abbreviated TETA and trien and also called trientine (INN), is an organic compound with the formula [CH2NHCH2CH2NH2]2. This oily liquid is colorless but, like many amines, assumes a yellowish color due to impurities resulting from air-oxidation. It is soluble in polar solvents. The branched isomer tris(2-aminoethyl)amine and piperazine derivatives may also be present in commercial samples of TETA.[1]
Trientine hydrochloride is a metal antagonist that was first launched by Merck, Sharp & Dohme in the U.S. in 1986 under the brand name Syprine for the oral treatment of Wilson’s disease.
Orphan drug designation has also been assigned in the U.S. for the treatment of patients with Wilson’s disease who are intolerant or inadequately responsive to penicillamine and in the E.U. by Univar for the treatment of Wilson’s disease

By condensation of ethylenediamine (I) with 1,2-dichloroethane (II)
Trientine hydrochloride is N,N’-bis (2-aminoethyl)-1,2-ethanediamine dihydrochloride. It is a white to pale yellow crystalline hygroscopic powder. It is freely soluble in water, soluble in methanol, slightly soluble in ethanol, and insoluble in chloroform and ether.
The empirical formula is C6H18N4·2HCI with a molecular weight of 219.2. The structural formula is:
NH2(CH2)2NH(CH2)2NH(CH2)2NH2•2HCI
Trientine hydrochloride is a chelating compound for removal of excess copper from the body. SYPRINE (Trientine Hydrochloride) is available as 250 mg capsules for oral administration. Capsules SYPRINE contain gelatin, iron oxides, stearic acid, and titanium dioxide as inactive ingredients.

Production
TETA is prepared by heating ethylenediamine or ethanolamine/ammonia mixtures over an oxide catalyst. This process gives a variety of amines, which are separated by distillation and sublimation.[2]
Uses
The reactivity and uses of TETA are similar to those for the related polyamines ethylenediamine and diethylenetriamine. It was primarily used as a crosslinker (“hardener”) in epoxy curing.[2]
The hydrochloride salt of TETA, referred to as trientine hydrochloride, is a chelating agent that is used to bind and remove copper in the body to treat Wilson’s disease, particularly in those who are intolerant to penicillamine. Some recommend trientine as first-line treatment, but experience with penicillamine is more extensive.[3]
Coordination chemistry
TETA is a tetradentate ligand in coordination chemistry, where it is referred to as trien.[4] Octahedral complexes of the type M(trien)Cl3 can adopt several diastereomeric structures, most of which are chiral.[5]
Trientine, chemically known as triethylenetetramine or N,N’-bis(2-aminoethyl)-l,2-ethanediamine belongs to the class of polyethylene polyamines. Trientine dihydrochloride is a chelating agent which is used to bind and remove copper in the body in the treatment of Wilson’s disease.
Trientine dihydrochloride (1)
Trientine dihydrochloride formulation, developed by Aton with the proprietary name SYPRINE, was approved by USFDA on November 8, 1985 for the treatment of patients with Wilson’s disease, who are intolerant to penicillamine. Trientine dihydrochloride, due to its activity on copper homeostasis, is being studied for various potential applications in the treatment of internal organs damage in diabetics, Alzheimer’s disease and cancer.
Various synthetic methods for preparation of triethylenetetramine (TETA) and the corresponding dihydrochloride salt have been disclosed in the prior art.
U.S. 4,806,517 discloses the synthesis of triethylenetetramine from ethylenediamine and monoethanolamine using Titania supported phosphorous catalyst while U.S. 4,550,209 and U.S. 5,225,599 disclose catalytic condensation of ethylenediamine and ethylene glycol for the synthesis of linear triethylenetetramine using catalysts like zirconium trimethylene diphosphonate, or metatungstate composites of titanium dioxide and zirconium dioxide.
U.S. 4,503,253 discloses the preparation of triethylenetetramine by reaction of an alkanolamine compound with ammonia and an alkyleneamine having two primary amino groups in the presence of a catalyst, such as supported phosphoric acid wherein the support is comprised of silica, alumina or carbon.
The methods described above for preparation of triethylenetetramine require high temperatures and pressure. Further, due to the various possible side reactions and consequent associated impurities, it is difficult to control the purity of the desired amine.
CN 102924289 discloses a process for trientine dihydrochloride comprising reduction of Ν,Ν’-dibenzyl-,N,N’-bis[2-(l,3-dioxo-2H-isoindolyl)ethyl]ethanediamine using hydrazine hydrate to give N,N’-dibenzyl-,N,N’-bis(2-aminoethyl)ethanediamine, which, upon condensation with benzyl chloroformate gave N,N’-dibenzyl-,N,N’-bis[2-(Cbz-amino)ethyl]ethanediamine, and further reductive deprotection to give the desired compound.
CS 197,093 discloses a process comprising reaction of triethylenetetramine with concentrated hydrochloric acid to obtain the crystalline tetrahydrochlonde salt. Further reaction of the salt with sodium ethoxide in solvent ethanol, filtration of the solid sodium chloride which is generated in the process, followed by slow cooling and crystallization of the filtrate provided the dihydrochloride salt. Optionally, aqueous solution of the tetrahydrochloride salt was passed through a column of an anion exchanger and the eluate containing free base was treated with a calculated amount of the tetrahydrochloride, evaporated, and the residue was crystallized from aqueous ethanol to yield the dihydrochloride salt.
The process is quite circuitous and cumbersome, requiring use of strong bases, filtration of sodium chloride and results in yields as low as 60%.
US 8,394,992 discloses a method for preparation of triethylenetetramine dihydrochloride wherein tertiary butoxycarbonyl (boc) protected triethylenetetramine is first converted to its tetrahydrochloride salt using large excess of hydrochloric acid in solvent isopropanol, followed by treatment of the resulting tetrahydrochloride salt with a strong base like sodium alkoxide to produce the amine free base (TETA) and sodium chloride salt in anhydrous conditions. The free amine is extracted with tertiary butyl methyl ether (TBME), followed by removal of sodium chloride salt and finally the amine free base TETA is treated with hydrochloric acid in solvent ethanol to give trientine hydrochloride salt.
PATENT
WO-2017046695
EXAMPLES
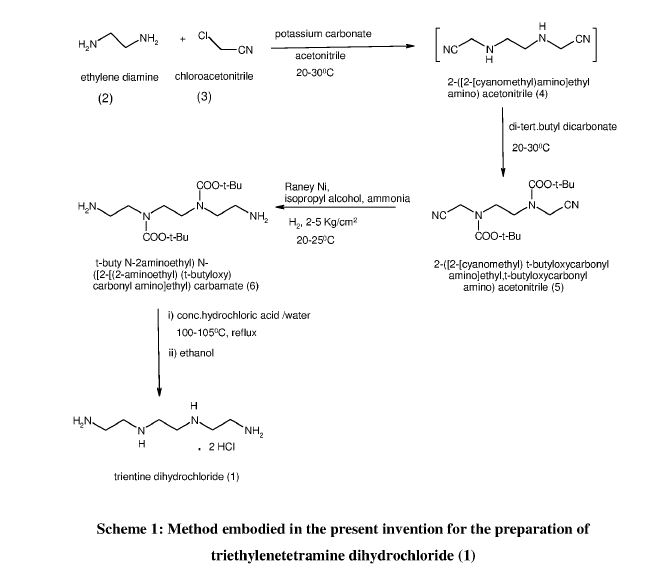
Example 1: Preparation of 2-([2-[cyanomethyl]-t-butyloxycarbonylamino]ethyl- 1-butyloxy carbonylamino)acetonitrile (5)
Potassium carbonate (481.9 g) was added to a stirred mixture of ethylenediamine (100.0 g) in acetonitrile (800 ml) and cooled to around 10°C. Chloroacetonitrile (263.8 g) was gradually added at same temperature and stirred at 25-30°C, till completion of the reaction, as monitored by HPLC. The mixture was cooled to 5-15°C and Boc-anhydride (762. lg) was added to it, followed by stirring at the same temperature. The temperature was raised to 25-30°C and the mass was stirred till completion of the reaction, as monitored by HPLC.
The reaction mass was filtered and the filtrate was concentrated. Toluene was added to the residue, and the mixture was heated to around 70°C followed by cooling and filtration to give 2-([2-[cyanomethyl)-t-butyloxycarbonylamino]ethyl-t-butyloxycarbonylamino) acetonitrile (5).
Yield: 506.8 g
% Yield: 89.9 %
Example 2: Preparation of t-butyl( N-2-aminoethyl)N-([2-[(2-aminoethyl)t-butyloxy)carbonylamino] ethyl) carbamate (6)
Raney nickel (120.0 g) in isopropanol (100 ml) was charged into an autoclave, followed by a mixture of Compound 5 (200 g) in isopropanol (400 ml). Cooled ammonia solution prepared by purging ammonia gas in 1400 ml isopropanol, equivalent to 125 g ammonia was gradually charged to the autoclave and the reaction was carried out around 15-25°C under hydrogen pressure of 2-5 Kg/cm2.
After completion of the reaction, as monitored by HPLC, the mass was filtered, concentrated, and methyl tertiary butyl ether was added to the residue. The mixture was heated to around 50°C, followed by cooling of the mass, stirring, optional seeding with compound 6 and filtration to give tertiary butyl-(N-2-aminoethyl)N-([2-[(2-aminoethyl)-(tert-butyloxy) carbonylamino] ethyl) carbamate.
Yield: 174 g
%Yield: 85 %
Example 3: Preparation of triethylenetetramine dihydrochloride (1)
Concentrated hydrochloric acid (121.5 g) was gradually added to a stirred mixture of tertiary-butyl-N-(2-aminoethyl)-N-2-[(2-aminoethyl)-(tert-butoxy) carbonyl] amino] ethyl} carbamate (Compound 6, 200.0 g) and water (1400 ml) at 20-30°C. The reaction mixture was heated in the temperature range of 100-105°C till completion of the reaction, as monitored by HPLC, with optionally distilling out water, if so required.
The reaction mass was concentrated and ethanol (600 ml) was added to the residue, followed by heating till a clear solution was obtained. The reaction mixture was gradually cooled with stirring, filtered and dried to provide triethylenetetramine dihydrochloride (1).
Yield: 88.9 g, (70 %)
Purity : > 99%
Patent
https://www.google.com/patents/US8394992
Trientine was said to be used in the synthesis of benzylidene-(2-{3-[2-(benzylidene-amino)-ethyl]-2-phenyl-imidazolidin-1-yl}-ethyl)-amine in French Patent No. FR2810035 to Guilard et al. Cetinkaya, E., et al., “Synthesis and characterization of unusual tetraminoalkenes,” J. Chem. Soc. 5:561-7 (1992), is said to be directed to synthesis of benzylidene-(2-{3-[2-(benzylidene-amino)-ethyl]-2-phenyl-imidazolidin-1-yl}-ethyl)-amine from trientine, as is Araki T., et al., “Site-selective derivatization of oligoethyleneimines using five-membered-ring protection method,” Macromol., 21:1995-2001 (1988). Triethylenetetramine may reportedly also be used in the synthesis of N-methylated triethylenetetramine, as reported in U.S. Pat. No. 2,390,766, to Zellhoefer et al.
Synthesis of polyethylenepolyamines, including triethylenetetramines, from ethylenediamine and monoethanolamine using pelleted group IVb metal oxide-phosphate type catalysts was reported by Vanderpool et al. in U.S. Pat. No. 4,806,517. Synthesis of triethylenetetramine from ethylenediamine and ethanolamine was also proposed in U.S. Pat. No. 4,550,209, to Unvert et al. U.S. Pat. No. 5,225,599, to King et al. is said to be directed to the synthesis of linear triethylene tetramine by condensation of ethylenediamine and ethylene glycol in the presence of a catalyst. Joint production of triethylenetetramine and 1-(2-aminoethyl)-aminoethyl-piperazine was proposed by Borisenko et al. in U.S.S.R. Patent No. SU1541204. U.S. Pat. No. 4,766,247 and European Patent No. EP262562, both to Ford et al., reported the preparation of triethylenetetramine by reaction of an alkanolamine compound, an alkaline amine and optionally either a primary or secondary amine in the presence of a phosphorous containing catalyst, for example phosphoric acid on silica-alumina or Group IIIB metal acid phosphate, at a temperature from about 175° C. to 400° C. under pressure. These patents indicate that the synthetic method used therein was as set forth in U.S. Pat. No. 4,463,193, to Johnson. The Ford et al. ‘247 patent is also said to be directed to color reduction of polyamines by reaction at elevated temperature and pressure in the presence of a hydrogenation catalyst and a hydrogen atmosphere. European Patent No. EP450709 to King et al. is said to be directed to a process for the preparation of triethylenetetramine and N-(2-aminoethyl)ethanolamine by condensation of an alkylenamine and an alkylene glycol in the presence of a condensation catalyst and a catalyst promoter at a temperature in excess of 260° C.
Russian Patent No. RU2186761, to Zagidullin, proposed synthesis of diethylenetriamine by reaction of dichloroethane with ethylenediamine. Ethylenediamine has previously been said to have been used in the synthesis of N-carboxylic acid esters as reported in U.S. Pat. No. 1,527,868, to Hartmann et al.
Japanese Patent No. 06065161 to Hara et al. is said to be directed to the synthesis of polyethylenepolyamines by reacting ethylenediamine with ethanolamine in the presence of silica-treated Nb205 supported on a carrier. Japanese Patent No. JP03047154 to Watanabe et al., is said to be directed to production of noncyclic polyethylenepolyamines by reaction of ammonia with monoethanolamine and ethylenediamine. Production of non-cyclic polyethylenepolyamines by reaction of ethylenediamine and monoethanolamine in the presence of hydrogen or a phosphorous-containing substance was said to be reported in Japanese Patent No. JP03048644. Regenerative preparation of linear polyethylenepolyamines using a phosphorous-bonded catalyst was proposed in European Patent No. EP115,138, to Larkin et al.
A process for preparation of alkyleneamines in the presence of a niobium catalyst was said to be provided in European Patent No. 256,516, to Tsutsumi et al. U.S. Pat. No. 4,584,405, to Vanderpool, reported the continuous synthesis of essentially noncyclic polyethylenepolyamines by reaction of monoethanolamine with ethylenediamine in the presence of an activated carbon catalyst under a pressure between about 500 to about 3000 psig., and at a temperature of between about 200° C. to about 400° C. Templeton, et al., reported on the preparation of linear polyethylenepolyamides asserted to result from reactions employing silica-alumina catalysts in European Patent No. EP150,558.
Production of triethylenetetramine dihydrochloride was said to have been reported in Kuhr et al., Czech Patent No. 197,093, via conversion of triethylenetetramine to crystalline tetrahydrochloride and subsequently to triethylenetetramine dihydrochloride. “A study of efficient preparation of triethylenetetramine dihydrochloride for the treatment of Wilson’s disease and hygroscopicity of its capsule,” Fujito, et al., Yakuzaigaku, 50:402-8 (1990), is also said to be directed to production of triethylenetetramine.
Preparation of triethylenetetramine salts used for the treatment of Wilson’s disease was said to be reported in “Treatment of Wilson’s Disease with Triethylene Tetramine Hydrochloride (Trientine),” Dubois, et al., J. Pediatric Gastro. & Nutrition, 10:77-81 (1990); “Preparation of Triethylenetetramine Dihydrochloride for the Treatment of Wilson’s Disease,” Dixon, et al., Lancet, 1(1775):853 (1972); “Determination of Triethylenetetramine in Plasma of Patients by High-Performance Liquid Chromatography,” Miyazaki, et al., Chem. Pharm. Bull., 38(4):1035-1038 (1990); “Preparation of and Clinical Experiences with Trien for the Treatment of Wilson’s Disease in Absolute Intolerance of D-penicillamine,” Harders, et al., Proc. Roy. Soc. Med., 70:10-12 (1977); “Tetramine cupruretic agents: A comparison in dogs,” Allen, et al., Am. J. Vet. Res., 48(1):28-30 (1987); and “Potentiometric and Spectroscopic Study of the Equilibria in the Aqueous Copper(II)-3,6-Diazaoctane-1,8-diamine System,” Laurie, et al., J.C.S. Dalton, 1882 (1976).
Preparation of Triethylenetetramine Salts by Reaction of Alcohol Solutions of Amines and acids was said to be reported in Polish Patent No. 105793, to Witek. Preparation of triethylenetetramine salts was also asserted in “Polycondensation of polyethylene polyamines with aliphatic dicarboxylic acids,” Witek, et al., Polimery, 20(3):118-119 (1975).
Baganz, H., and Peissker, H., Chem. Ber., 1957; 90:2944-2949; Haydock, D. B., and Mulholland, T. P. C., J. Chem. Soc., 1971; 2389-2395; and Rehse, K., et al., Arch. Pharm., 1994; 393-398, report on Strecker syntheses. Use of Boc and other protecting groups has been described. See, for example, Spicer, J. A. et al., Bioorganic & Medicinal Chemistry, 2002; 10: 19-29; Klenke, B. and Gilbert, I. H., J. Org. Chem., 2001; 66: 2480-2483.
FIG. 6 shows an 1H-NMR spectrum of a triethylenetetramine hydrochloride salt in D2O, as synthesized in Example 3. NMR values include a frequency of 400.13 Mhz, a 1H nucleus, number of transients is 16, points count of 32768, pulse sequence of zg30, and sweep width of 8278.15 H
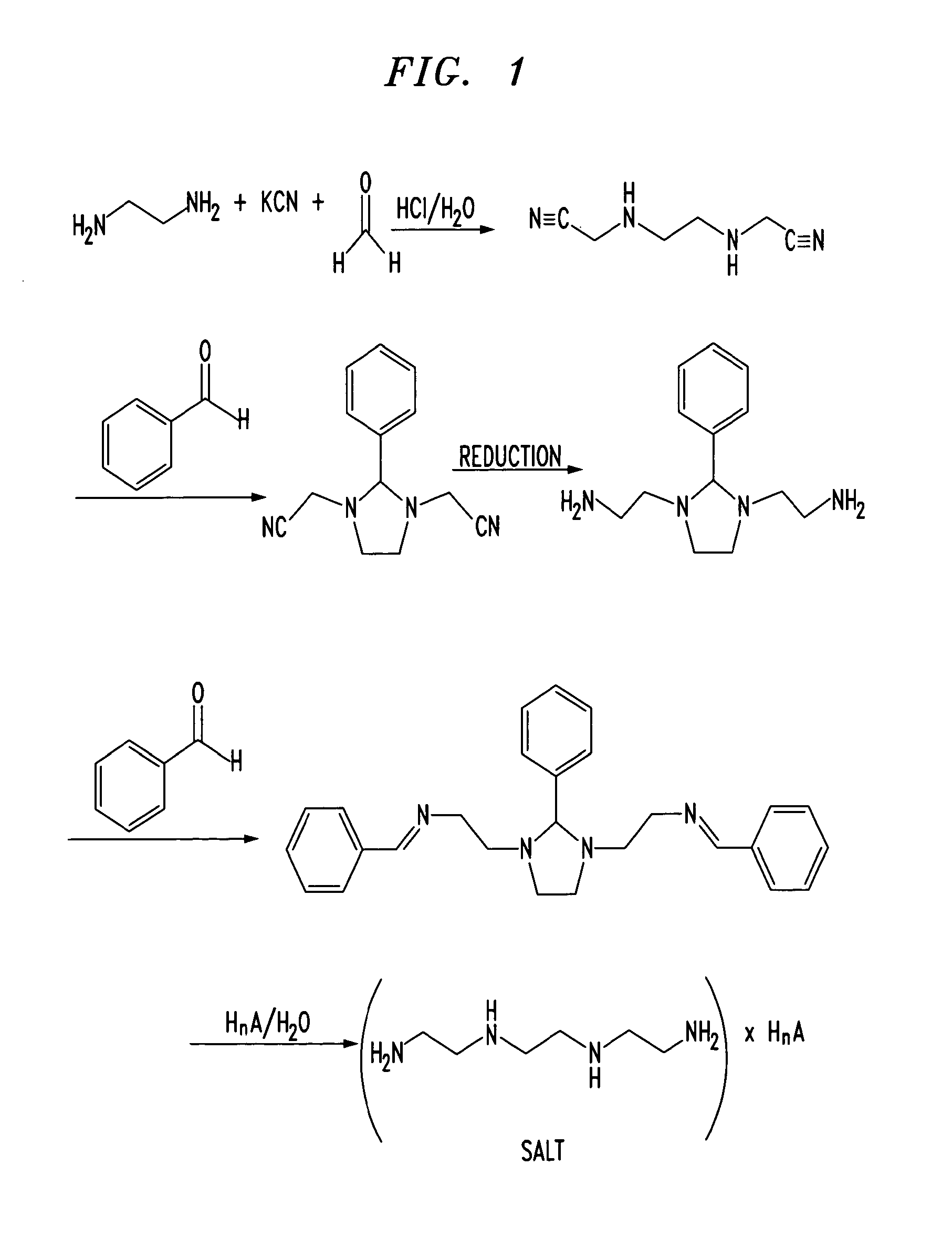
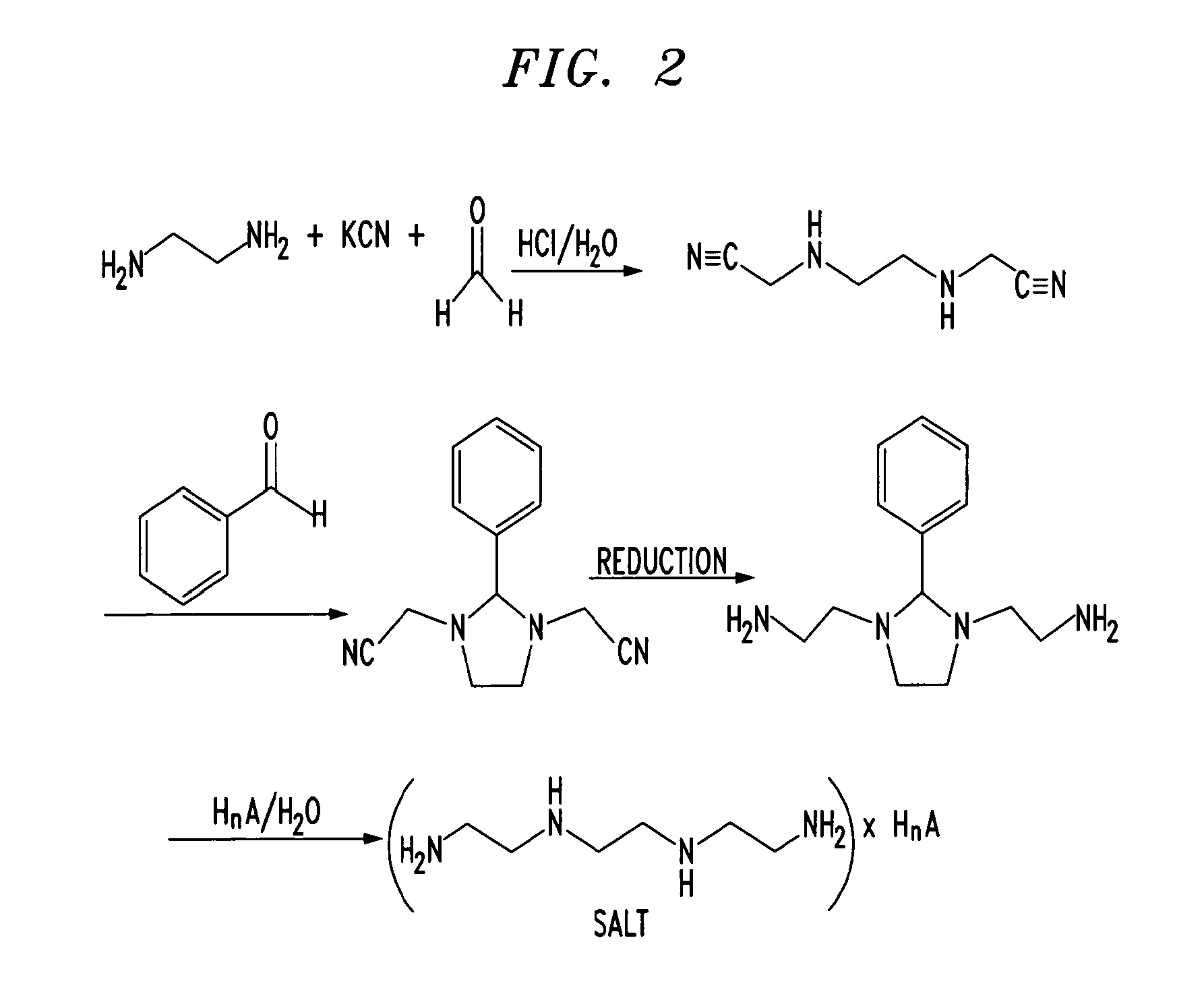

CLIP
Method of purification: Dissolve Trientine Hydrochloride in water while warming, and recrystallize by addition of ethanol (99.5). Or dissolve Trientine Hydrochloride in water while warming, allow to stand after addition of activated charcoal in a cool and dark place for one night, and filter. To the filtrate add ethanol (99.5), allow to stand in a cool and dark place, and recrystallize. Dry the crystals under reduced pressure not exceeding 0.67 kPa at 409C until ethanol odor disappears.
References
- “Ethyleneamines” (PDF). Huntsman. 2007.
- ^ Jump up to:a b Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. (2005). “Amines, Aliphatic”. Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a02_001.
- Jump up^ Roberts, E. A.; Schilsky, M. L. (2003). “A practice guideline on Wilson disease” (pdf). Hepatology. 37 (6): 1475–1492. doi:10.1053/jhep.2003.50252. PMID 12774027.
- Jump up^ von Zelewsky, A. (1995). Stereochemistry of Coordination Compounds. Chichester: John Wiley. ISBN 047195599X.
- Utsuno, S.; Sakai, Y.; Yoshikawa, Y.; Yamatera, H. (1985). “Three Isomers of the Trans-Diammine-[N,N′-bis(2-Aminoethyl)-1,2-Ethanediamine]-Cobalt(III) Complex Cation”. Inorganic Syntheses. 23: 79–82. doi:10.1002/9780470132548.ch16.
 |
|
 |
|
 |
|
| Names | |
|---|---|
| Other names
N,N’-Bis(2-aminoethyl)ethane-1,2-diamine; TETA; Trien; Trientine (INN); Syprine (brand name)
|
|
| Identifiers | |
|
3D model (Jmol)
|
|
| 605448 | |
| ChEBI | |
| ChemSpider | |
| ECHA InfoCard | 100.003.591 |
| EC Number | 203-950-6 |
| 27008 | |
| KEGG | |
| MeSH | Trientine |
|
PubChem CID
|
|
| RTECS number | YE6650000 |
| UNII | |
| UN number | 2259 |
| Properties | |
| C6H18N4 | |
| Molar mass | 146.24 g·mol−1 |
| Appearance | Colorless liquid |
| Odor | Fishy, ammoniacal |
| Density | 982 mg mL−1 |
| Melting point | −34.6 °C; −30.4 °F; 238.5 K |
| Boiling point | 266.6 °C; 511.8 °F; 539.7 K |
| Miscible | |
| log P | 1.985 |
| Vapor pressure | <1 Pa (at 20 °C) |
|
Refractive index (nD)
|
1.496 |
| Thermochemistry | |
| 376 J K−1 mol−1 (at 60 °C) | |
| Pharmacology | |
| A16AX12 (WHO) | |
| Hazards | |
| GHS pictograms |   |
| GHS signal word | DANGER |
| H312, H314, H317, H412 | |
| P273, P280, P305+351+338, P310 | |
|
EU classification (DSD)
|
|
| R-phrases | R21, R34, R43, R52/53 |
| S-phrases | (S1/2), S26, S36/37/39, S45 |
| Flash point | 129 °C (264 °F; 402 K) |
| Lethal dose or concentration (LD, LC): | |
|
LD50 (median dose)
|
|
| Related compounds | |
|
Related amines
|
|
|
Related compounds
|
|
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////////////TRIENTINE, 112-24-3, 曲恩汀 , KD-034 , MK-0681, MK-681, TECZA, TETA, TJA-250, Orphan drug
NCCNCCNCCN
Cetilistat, セチリスタット
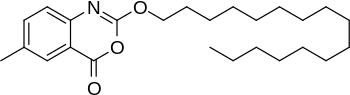
Cetilistat, セチリスタット
- Molecular FormulaC25H39NO3
- Average mass401.582 Da

patients with both type 2 diabetes mellitus
and dyslipidaemia, and with a BMI ≥ 25
kg/m2 inspite of dietary treatment and /or
excersise therapy)…………Cetilistat bulk and Cetilistat 120 mg tablets
Cetilistat was approved by Pharmaceuticals Medical Devices Agency of Japan (PMDA) on September 20, 2013. It was developed by Norgine and Takeda, then marketed as Oblean® by Takeda in Japan.
Cetilistat is a pancreatic lipase inhibitor, and it acts in the same way as the older drug orlistat (Xenical) by inhibiting pancreatic lipase, an enzyme that breaks down triglycerides in the intestine. Without this enzyme, triglycerides from the diet are prevented from being hydrolyzed into absorbable free fatty acids and are excreted undigested. It is usually used for the treatment of obesity (limited to patients with both type 2 diabetes mellitus and dyslipidemia, and with a BMI≥25 kg/m2 in spite of dietary treatment and/or exercise therapy).
Oblean® is available as tablet for oral use, containing 120 mg of free Cetilistat. The recommended dose is 120 mg three times a day immediately after each meal.
Cetilistat is a drug designed to treat obesity. It acts in the same way as the older drug orlistat (Xenical) by inhibitingpancreatic lipase, an enzyme that breaks down triglycerides in the intestine. Without this enzyme, triglycerides from the diet are prevented from being hydrolyzed into absorbable free fatty acids and are excreted undigested.[1]
In human trials, cetilistat was shown to produce similar weight loss to orlistat, but also produced similar side effects such as oily, loose stools, fecal incontinence, frequent bowel movements, and flatulence.[2][3] It is likely that the same precautions would apply in that absorption of fat-soluble vitamins and other fat-soluble nutrients may be inhibited, requiring vitamin supplements to be used to avoid deficiencies.
Central obesity have an important impact on the development of risk factors for coronary heart disease, including dislipidemia, glucose intolerance, insulin resistance and hypertension. These factors contribute to building cardiovascular (CV) disease as a major cause of death. The approach to obesity therapy should be designed to reduce CV risk and mortality. Diet and lifestyle changes remain the cornerstones of therapy for obesity, but the resultant weight loss is often small and long-term success is uncommon and disappointing. Drug therapy is considered for individuals with a body mass index greater than 30 kg/m2 or ranging from 25 to 30 kg/m2 if they have comorbid conditions. Antiobesity agents can be helpful to some patients in achieving and maintaining meaningful weight loss, but yet our pharmaceutical tools are of limited effectiveness considering the magnitude of the problem. At the present, only two drugs, orlistat and sibutramine, are approved for long-term treatment of obesity and promote no more than 5 to 10% of weight loss.
Rimonabant, a cannabinoid-1 receptor antagonist, was withdrawn from the market because of concerns about its safety, including risk of suicidal and seizures, although very effective in promoting clinically meaningful weight loss, reduction in waist circumference, and improvements in several metabolic risk factors, rimonabant, a cannabinoid-1 receptor antagonist was withdrawn from the market because it concerns about its safety, including risk of suicidal and seizures. Fortunately, recent fundamental insights into the neuroendocrine mechanisms regulating body weight provide an expanding list of molecular targets for novel, rationally designed antiobesity drugs. In this review, the therapeutic potential of some antiobesity molecules in the development will be analyzed based on an understanding of energy homeostasis.


Cetilistat has completed Phase 1 and 2 trials in the West and is currently in Phase 3 trials in Japan where it is partnered with Takeda.[4] Norgina BV has now acquired the full global rights to cetilistat from Alizyme after the latter went into administration.[5]
A published phase 2 trial found cetilistat significantly reduced weight with and was better tolerated than orlistat.[6
CLIP
Cetilistat (Oblean®)
Cetilistat is a selective pancreatic lipase inhibitor which was approved in Japan in September 2013
for the treatment of obesity. The drug was discovered by Alizyme PLC and later co-developed with
Takeda. Cetilistat demonstrated a lower incidence of adverse gastrointestinal events during a 12 week clinical trial, and the degree of weight loss associated with cetilistat is comparable to that of other approved antiobesity therapies.30 The most likely process-scale preparation of cetilistat is described below in Scheme. 4.31
Commercially available hexadecanol (21) was treated with phosgene in THF/toluene to give the
corresponding chloroformate (22), which was immediately subjected to commercial 2-amino-5-
methylbenzoic acid (23) in pyridine. Subsequent slow addition of methyl chloroformate at room
temperature resulted in the formation of cetilistat (IV), which was produced in 31% overall yield from
hexadecanol.31
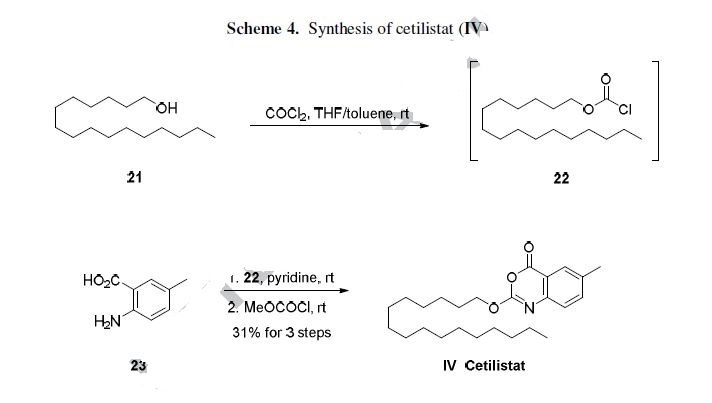
REF FOR ABOVE ONLY
30 Kopelman, P.; Groot, G. d. H.; Rissanen, A.; Rossner, S.; Toubro, S.; Palmer, R.; Hallam, R.;
Bryson, A.; Hickling, R. I. Obesity 2010, 18, 108.
31. Hodson, H. F.; Downham, R.; Mitchell, T. J.; Carr, B. J.; Dunk, C. R.; Palmer, R. M. J. US
Patent 20030027821A1, 2003.
SYNTHESIS
Route 1
WO2000040569
AND
http://www.google.co.in/patents/US6624161

WO0040569A1 / US6656934B2.2. WO0040247A1 / US6624161B2.
Route 2
http://www.google.co.in/patents/US7396952
Carbamic ester derivatives of the general formula (1) and especially (2-carboxy-4-methylphenyl)carbamic esters of the general formula (1′)

are suitable intermediates for active pharmaceutical ingredients.
Thus, for example, hexadecyl (2-carboxy-4-methylphenyl)carbamate as compound of the formula (1′) with R═C16H33 is disclosed as an intermediate in the preparation of 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one of the formula (3)

from the originally published version of WO-A 00/40569.
2-Hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one of the formula (3) is described therein as potential active ingredient for the treatment of obesity and type II diabetes.
In this originally published version of WO-A 00/40569, two synthetic routes 1 and 2 are described for preparing 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one (3), each of which starts from the 5-methyl-substituted anthranilic acid (4).
In the two-stage synthetic route 1, the 5-methyl-substituted anthranilic acid (4) is reacted with hexadecyl chloroformate (5) and subsequently with methyl chloroformate to give 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one (3), although the overall yield obtained is only 31%.
The one-stage synthetic route 2 with an excess of pyridine affords 2-hexadecyl-oxy-6-methyl-4H-3,1-benzoxazin-4-one (3) in an even lower yield of 15%.

The starting compound which is required for both the synthetic routes 1 and 2, the 5-methyl-substituted anthranilic acid (4), is not easily obtainable, however.
It is prepared by the method described in J. Org. Chem. 1952, 17, 141. This starts from p-toluidine, which is reacted with chloral hydrate and hydroxylamine hydrochloride. The resulting oxime is cyclized with acid catalysis, and subsequently the ring is cleaved again by oxidation under basic conditions.

The disadvantages of this synthesis are the low yields and the fact that only very low concentrations can be used. For this reason, this synthetic route is unattractive for an industrial reaction.
Further alternative routes known in principle for obtaining anthranilic acids are as follows:
J. Org. Chem. 1978, 43, 220 and Chem. Ber. 1909, 42, 430 disclose initial nitration of 3-cyanotoluene, then reduction of the nitro group and subsequent hydrolysis of the nitrile to the carboxylic acid.



Because of the diverse difficulties, described above, associated with the known processes for preparing optionally substituted anthranilic acids and the yields, which are only unsatisfactory and thus limiting for the overall process, of the subsequent synthetic routes 1 and 2, the object of the present invention was to provide an improved process for preparing carbamic ester derivatives of the general formula (1).

EXAMPLES Example 1 Synthesis of hexadecyl 4-methylphenylcarbamate

91 g (375 mmol) of 1-hexadecanol were added to a solution of 50 g (375 mmol) of p-tolyl isocyanate in 50 ml of toluene, and the resulting solution was heated under reflux for 8 h. After cooling to room temperature and stirring at this temperature for 12 h, the precipitated solid was filtered off. The colourless solid was washed twice with 10 ml of toluene each time and then dried in vacuo. 80 g (213 mmol, 57%) of the desired carbamate were obtained in the form of a colourless solid with a melting point of 75° C. The melting point agreed with literature data (75-76° C., Microchem J. 1962, 6, 179).
1H-NMR (CDCl3, 400 MHz): δ=0.88 ppm (t, J=7.3 Hz, 3H), 1.25-1.40 (m, 26 H), 1.66 (sext, J=6.9 Hz, 2H), 2.30 (s, 3H), 4.14 (t, J=6.9 Hz, 2H), 6.53 (br, 1 H), 7.10 (d, J=7.8 Hz, 2H), 7.25 (d, J=8.3 Hz, 2H). Elemental Analysis Showed: Calculated: C 76.8%, H 11.0%, N 3.7% Found: C 76.9%, H 11.2%, N 3.7%.
Example 2 Synthesis of hexadecyl (2-bromo-4-methylphenyl)carbamate

19 g (119 mmol) of bromine were added dropwise to a solution of 45 g (119 mmol) of the carbamate in 225 ml (235 g) of glacial acetic acid at room temperature over the course of 1 h, and then the resulting solution was stirred at room temperature for 1 h. After addition of a further 25 ml (26 g, 437 mmol) of glacial acetic acid, the reaction mixture was stirred at 40° C. for 5 h and then cooled to room temperature. The precipitated solid was filtered off and washed with 20 ml of glacial acetic acid. Drying in vacuo resulted in 40 g (88 mmol, 74%) of the desired bromo compound in the form of a colourless solid with a melting point of 57° C.
1H-NMR (CDCl3, 400 MHz): δ=0.93 ppm (t, J=6.6 Hz, 3H), 1.25-1.43 (m, 26 H), 1.73 (sext, J=6.8 Hz, 2H), 2.33 (s, 3H), 4.21 (t, J=6.7 Hz, 2H), 7.04 (br, 1H), 7.14 (d, J=8.4 Hz, 1H), 7.37 (s, 1H), 8.02 (d, J=8.3 Hz, 1H). 13C-NMR (CDCl3, 100 MHz): δ=14.2 ppm, 20.4, 22.7. 25.9, 29.0, 29.3, 29.4, 29.6 (2C), 29.7 (2C), 29.8 (4C), 32.0, 65.7, 112.5, 120.3, 129.0, 132.5, 133.5, 134.1, 153.5. Elemental Analysis Showed: Calculated: C 63.4%, H 8.9%, N 3.1% Found: C 63.6%, H 8.9%, N 3.1%.
Example 3 Synthesis of 2-hexadecyloxycarbonylamino-5-methylbenzoic acid

217.5 g (478.5 mmol) of hexadecyl (2-bromo-4-methylphenyl)carbamate, 0.5 g (0.7 mmol) of bis(triphenylphosphine)palladium dichloride and 2.5 g (9.3 mmol) of triphenylphosphine were introduced into an autoclave. The autoclave was closed, flushed with nitrogen and an oxygen-free solution of 78.1 g (565.3 mmol) of potassium carbonate in 400 ml of water is added. The autoclave is evacuated and then 2 bar of carbon monoxide are injected and heated to 115° C. The pressure is subsequently adjusted to 8 bar. After CO uptake ceases, the mixture is cooled to RT and 200 ml of toluene are added. The pH is adjusted to 2 with 2M aqueous HCl solution, and the organic phase is separated off. The aqueous phase is extracted anew with 100 ml of toluene, the organic phase is separated off, and the two toluene extracts are combined. Removal of the solvent in vacuo results in 154.9 g (369.2 mmol, 77%) of 2-hexadecyloxycarbonylamino-5-methylbenzoic acid in the form of a pale yellow-coloured solid.
1H-NMR (CDCl3, 400 MHz): δ=0.88 ppm (t, J=6.7 Hz, 3H), 1.24-1.40 (m, 26 H), 1.70 (sext, J=6.8 Hz, 2H), 2.33 (s, 3H), 4.17 (t, J=6.8 Hz, 2H), 7.38 (d, J=8.7 Hz, 1H), 7.90 (s, 1H), 8.35 (d, J=8.6 Hz, 1H). Signal of the NH proton not identifiable.13C-NMR (CDCl3, 100 MHz): δ=14.1 ppm, 20.5, 22.7. 25.9, 29.0, 29.3, 29.4, 29.6 (2 C), 29.7 (6 C), 32.0, 65.5, 113.6, 119.0, 131.1, 131.8, 136.3, 140.1, 153.9, 172.5.
Example 4 Synthesis of 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one

4.0 g (10.0 mmol) of 2-hexadecyloxycarbonylamino-5-methylbenzoic acid are introduced into 20 ml of pyridine at 0° C. under a nitrogen atmosphere, and 4.93 g (45.4 mmol) of ethyl chloroformate are added dropwise to the resulting solution at 0° C. over the course of 20 min. After the reaction mixture has been stirred at 0° C. for 1 h and at room temperature for 2 h it is added to 30 ml of ice-water. The solid is filtered off and dried in vacuo. 3.3 g (8.2 mmol, 82%) of 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one are obtained in the form of a pale yellow coloured solid with a melting point of 67° C. (literature: 72-73° C., WO 00/40569).
1H-NMR (CDCl3, 400 MHz): δ=0.86 ppm (t, J=6.6 Hz, 3H), 1.24-1.42 (m, 26 H), 1.75-1.82 (m, 2H), 2.40 (s, 3H), 4.41 (t, J=6.8 Hz, 2H), 7.30 (d, J=8.3 Hz, 1H), 7.51 (dd, J=8.2, 1.9 Hz, 1H), 7.90 (d, J=0.9 Hz, 1H).
The 1H-NMR data agree with the literature data from WO-A 00/40569.

Patent
https://www.google.com/patents/CN104341370A?cl=en
cetirizine orlistat (2-methyl-6-firing sixteen -4H-3, 1- benzo ah winded -4- Korea, cetilistat) is a long-acting Alizyme developed and potent specific gastrointestinal lipase inhibitor, with the active serine site of the gastric and intestinal lumen gastric lipase and lipase membrane forms a covalent bond to inactivate the enzyme, and to reduce calorie intake, weight control therapeutic effect. The biggest advantage of the drug is not acting on the nervous system, does not affect other activity in the gastrointestinal tract, it is more secure than existing similar drugs orlistat. Its structural formula is as follows:

West Division for the benefit of his synthesis and intermediates have been described in U.S. Patent US2007232825 and US2003027821, domestic literature orlistat no cetirizine synthesis of relevant reports.
U.S. Patent US2007232825 2-amino-5-methyl-benzoic acid starting material, direct and vilify chloroformate cetyl alcohol vinegar into the ring, get cetirizine orlistat. The reaction byproducts and more difficult W purification needs over baby gel column, resulting in a low yield, suitable for mass industrialization. Directions are as follows:

Patent US2003027821 W toluene different acid vinegar as raw material to produce amino acid vinegar intermediate chloroformate, cetyl alcohol and vinegar reaction, after the desert generation essays glycosylation chloroformate caprolactone ring closure to give cetirizine orlistat. This method requires a great deal of glacial acetic acid, the presence of H waste discharge more harsh reaction conditions, equipment requirements, is not conducive to industrial production and other defects.

The present invention is a W under the technical program realization:






The following combination of embodiments of the present invention will be further described below.
(Sixteen essays firing oxygen-amino) -5- Preparation of 2-methyl benzoate desert vinegar; [0041] Example 1

4. 9g H phosgene will be added to 50 blood dichloromethane firing, the temperature was lowered to OC, a solution of 2-amino-5 Desert benzoic acid methyl ester (5g) and H hexylamine (13.8 blood) dichloro A firing (20 blood) solution, the addition was complete OC to maintain 15min, warmed to room temperature the reaction mix of football.

[0042] The 5. 26g cetyl alcohol was added to the reaction solution at room temperature the reaction of. After completion of the reaction, filtered and the filtrate was concentrated in vacuo spin dry, dry methanol residue fight starched coating, filtration, the filter cake is dried to constant weight. To give a white solid powder 9. Ig, namely 2- (sixteen essays firing oxygen-ylamino) -5-benzoic acid methyl ester desert; Yield; 85%.
2- (grilled oxygen sixteen essays) -5-methyl-benzoic acid methyl ester prepared; [0043] Example 2

Under nitrogen blanket IOg 2- (sixteen grilled oxygen essays) -5- desert benzoic acid methyl ester was dissolved in 1,4-dioxane (50mL) and water Qiao blood), and Ilg anhydrous carbonate Bell, 1.44g methacrylic acid test, 0. 731g Pd (dppf) 2Cl2, the mixture at 105C for 3 hours. Completion of the reaction, cool down, filtered and the filtrate spin dry, the residue of anhydrous methanol wash coating, the filter cake dried to give a gray solid 6. 5g, is 2- (xvi grilled oxygen essays) -5-methyl benzoic acid methyl ester in 75% yield.
2- (grilled oxygen sixteen essays) -5-methyl-benzoic acid; [0044] Example 3

The 7g 2- (sixteen grilled oxygen essays) -5-methyl-benzoic acid methyl ester was added to 35mL tetraammine clever furans and 7mL water mixture, adding ammonia oxidation in 20. Ig, 6 (TC reaction of the reaction is completed, the reaction mixture was concentrated, the residue was added 70mL of ice water, 6M hydrochloric suppression of 7, the filter cake was dried to constant weight to give a gray solid 6. 2g, namely 2- (sixteen firing oxygen-ylamino essays ) -5-methyl-benzoic acid, yield 92%.
Preparation of 2-methyl-6-firing sixteen -4H-3, 1- benzo Lai ah winded -4- (cetirizine Division him); 4 [0045] Example

The 66g 2- (XVI essays firing oxo-ylamino) -5-methylbenzoic acid in 330mL of information coincidence floating in an ice bath, was slowly added dropwise 45mL chloroformate caprolactone, after the addition was complete, naturally rise to room temperature The reaction of. After completion of the reaction, the reaction solution was poured into 700mL ice water, filtered, and the filter cake was dried to constant weight to give a gray solid 56g, that is, sixteen firing-6-methyl-2- -4H-3, 1- benzo Lai ah winded -4- (cetirizine orlistat), a yield of 85%. Mass spectrum shown in Figure 2, ESI-MS〇b / z): 402 [M + Tin +; X- ray diffraction as shown in (3 consistent with the data reported in FIG patent US2012101090), analyzed as shown in Table 1, Figure 1 FIG. 2 W and W Table 1 confirm that the product was obtained as cetirizine orlistat.
[0046] Table 1







| Synthesis of Cetilistat |
| 1. Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050; 2. Beijing Union Pharmaceutical Factory, Beijing 102600 |
CLIP
http://amogsobgy.com/downloads/AkumentisChechwt/CurrOpinInvestigDrugs.pdf

 CLICK ON IMAGE
CLICK ON IMAGE
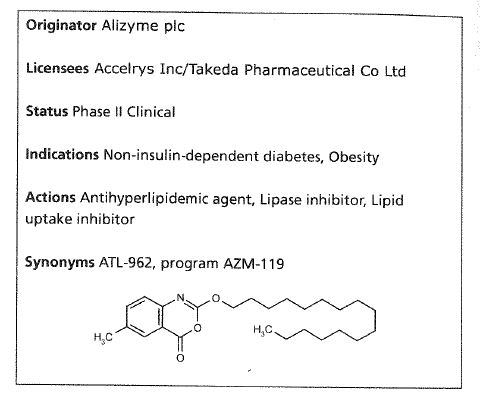
Cetilistat, a new lipase inhibitor for the treatment of obesity – AMOGS
amogsobgy.com/downloads/AkumentisChechwt/CurrOpinInvestigDrugs.pdf
clinical trials, and the above-mentioned lipase inhibitor cetilistat, which is the focus of this review.Synthesis and SAR. Cetilistat (2-hexadecyloxy-6-methyl-4H-3 …
PATENT
SEE
http://www.google.co.in/patents/WO1999016758A1?cl=en
CLIP
Taken from Ayurajan

https://ayurajan.blogspot.in/2016_01_01_archive.html
Cetilistat | Inhibitor of Gastrointestinal Lipases | Inhibitor of Pancreatic Lipases | Anti-Obesity Drug
Cetilistat [2-(Hexadecyloxy)-6-methyl-4H-3,1-benzoxazin-4-one] is a novel highly lipophilic benzoxazinone that inhibits gastrointestinal (GI) and pancreatic lipases, and is chemically distinct from Orlistat [1].
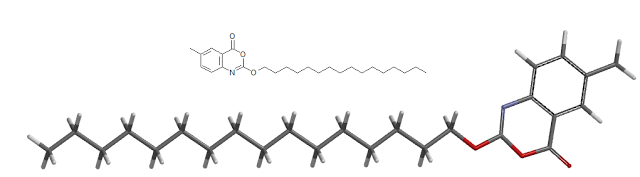 |
| Cetilistat: 2D and 3D Structure |
Pancreatic lipase is the enzyme that breaks down triglycerides in the intestine. Inhibition of this enzyme ensures that triglycerides from the diet are prevented from being hydrolyzed into absorbable free fatty acids and are excreted undigested.
In Phase I clinical trials in healthy volunteers, Cetilistat increased faecal fat excretion and was well tolerated. Cetilistat produced a clinically and statistically significant weight loss in obese patients in this short-term 12-week study. This was accompanied by significant improvements in other obesity-related parameters. Cetilistat treatment was well tolerated. The risk-benefit demonstrated in this study in terms of weight loss vs intolerable GI adverse effects shows that Cetilistat merits further evaluation for the pharmacotherapy of obesity and related disorders.
The NDA submission is based on the results of three Phase 3 clinical trials in obese patients with type 2 diabetes and dyslipidemia: a 52-week placebo-controlled, double-blind study to evaluate the efficacy and safety, and two open-label studies to evaluate safety, 24-week and 52-week respectively. The results of the 52-week placebo-controlled, double-blind study demonstrate that Cetilistat 120mg three times daily is superior to placebo in the primary endpoint, with a mean reduction in body weight from baseline of -2.776% with Cetilistat versus -1.103% with placebo (p=0.0020). Greater reduction in HbA1c and low-density lipoprotein cholesterol were also observed in patients treated with Cetilistat, compared to placebo. In all these three studies, Cetilistat showed a good safety profile and was well tolerated.
Cetilistat was approved in Japan in September 2013 for the treatment of obesity. Cetilistat (Tradename: Oblean) is approved for a dosage of 120 mg three times a day for the treatment of obesity with complications.
The drug was discovered by UK based Alizyme PLC and in 2003 Takeda acquired the rights for development and commercialisation for Japan. Norgine acquired all rights to the product from Alizyme in October 2009 [3].
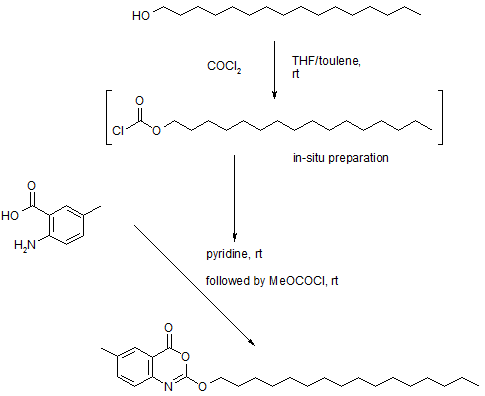
Cetilistat Synthesis
US20030027821A1: It appears to be the industrial process. The yields are in the range of 30-35%.
Identification:
 |
| 1H NMR (Estimated) for Cetilistat |
Experimental: 1H-NMR δH (400 MHz, CDCl3) 0.87 (3H, t, J 6.8, CH2CH3), 1.24-1.45 (26H, m, 13×CH2), 1.75-1.83 (2H, m, OCH2CH2), 2.41 (3H, s, ArCH3), 4.41 (2H, t, J 6.7, OCH2), 7.3 (1H, d, J 8.3, ArH), 7.51 (1H, dd, J 8.5, 2.0, ArH), 7.90 (1H, d, J 1.1, ArH); m/z (ES+) 402 (MH+); M Pt. 72-73° C.
Sideeffects: The most frequently experienced adverse events were those involving the gastrointestinal (GI) tract. The proportion of patients and the total number of GI adverse events reported in each of the active treatment groups were higher compared to the placebo group. However, GI adverse events were predominantly mild to moderate in intensity, with no evidence of a dose relationship.
The most frequently reported GI-related adverse events included increased defecation, soft stools, abdominal pain, flatulence and fatty/oily stool, which were all reported more frequently in the treatment arms compared to the placebo arm.
Faecal incontinence, flatus with discharge, oily evacuation and oily spotting occurred in only 1.8-2.8% of subjects in the active treatment arms and was not dose-related. Adverse events generally occurred on only one occasion and resolved rapidly.
Serum vitamin D, vitamin E and β-carotene levels were decreased significantly in the Cetilistat treatment arms. Generally, these reductions in vitamin levels did not take the levels outside the normal range and none required the use of vitamin supplements.
References FOR ABOVE ONLY
- Kopelman, P.; et. al. Cetilistat (ATL-962), a novel lipase inhibitor: a 12-week randomized, placebo-controlled study of weight reduction in obese patients. Int J Obes (Lond) 2007, 31(3), 494-499.
- Hodson, H.; et. al. 2-Oxy-benzoxazinone derivatives for the treatment of obesity.US20030027821A1
- Cetilistat Approval (here).

| CN1359378A * | Jan 6, 2000 | Jul 17, 2002 | 阿利茨默治疗学有限公司 | 2-oxy-benzoxazine derivatives for the treatment of obesity |
| CN1785967A * | Dec 12, 2005 | Jun 14, 2006 | 兰爱克谢斯德国有限责任公司 | Process for the preparation of carbamic acid derivatives |
| CN103936687A * | Mar 24, 2014 | Jul 23, 2014 | 重庆东得医药科技有限公司 | Method for preparing cetilistat |
| WO2013166037A1 * | Apr 30, 2013 | Nov 7, 2013 | The Trustees Of Columbia University In The City Of New York | Non-retinoid antagonists for treatment of eye disorders |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20030013707 | 6 Jul 2001 | 16 Jan 2003 | Hodson Harold Francis | 2-amino-benzoxazinone derivatives for the treatment of obesity |
| EP0034292A2 | 31 Jan 1981 | 26 Aug 1981 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Process for the preparation of anthranilic acid derivatives |
| WO1997028118A1 | 30 Jan 1997 | 7 Aug 1997 | Hoechst Celanese Corporation | Process for preparing anthranilic acids |
| Reference | ||
|---|---|---|
| 1 | Chem. Ber. 1909, 42, 430. | |
| 2 | J. Chem. Soc. Perkin I, 1973, 2940; Peter H. Gore et al. Friedel-Crafts Reactions, Part XXV.<SUP>1 </SUP>Acetylation and Benzoylation of Iodobenzene and of o-, m-, and p- Iodotoluenes. | |
| 3 | J. Org. Chem. 1952, 17, 141 B. R. Baker et al.; “An Antimalarial Alkaloid From Hydrangea, XIV, Synthesis of 5- ,6-,7-, and 8-Monosubstituted Derivatives“. | |
| 4 | J. Org. Chem. 1978, vol. 43, No. 2, 220 T.H. Fisher et al.; “Kinetic Study of the N-Bromosuccin-imide Bromination of Some 4-Substituted 3-Cyanotoluenes“. | |
| 5 | J. Org. Chem. 1981, 46, 4614-4617 Donald Valentine, Jr. et al; “Practical, Catalytic Synthesis of Anthranilic Acids“. | |
| 6 | Monatsch. Chem. 1920, 41, 155. | |
| 7 | Thomas G. Back et al.: “Conjugate Additions of o-Iodoanilines and Methyl Anthranilates to Acetylenic Sulfones. A New Route to Quinolones Including First Syntheses of Two Alkaloids from the Medical Herb Ruta chalepensis” Journal of Organic Chemistry., Bd. 68, 2003, Seiten 2223-2233, XP002371555 USAmerican Chemical Society, Easton. Seite 2227, Spalte 1, Reaktionsschema 4 und Spalte 2, Zeile 8-Zeile 9; Seite 2231, Spalte 2, Zeile 43-Zeile 54. | |
| 8 | * | Yadav et al., New Journal of Chemistry (2000), 24(8), 571-573. |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8883780 | 22 Apr 2010 | 11 Nov 2014 | Norgine B.V. | Crystal of a benzoxazinone compound |
References
- Yamada Y, Kato T, Ogino H, Ashina S, Kato K (2008). “Cetilistat (ATL-962), a novel pancreatic lipase inhibitor, ameliorates body weight gain and improves lipid profiles in rats”. Hormone and Metabolic Research. 40 (8): 539–43. doi:10.1055/s-2008-1076699. PMID 18500680.
- Kopelman, P; Bryson, A; Hickling, R; Rissanen, A; Rossner, S; Toubro, S; Valensi, P (2007). “Cetilistat (ATL-962), a novel lipase inhibitor: A 12-week randomized, placebo-controlled study of weight reduction in obese patients”. International journal of obesity (2005). 31 (3): 494–9. doi:10.1038/sj.ijo.0803446. PMID 16953261.
- Padwal, R (2008). “Cetilistat, a new lipase inhibitor for the treatment of obesity”. Current opinion in investigational drugs (London, England : 2000). 9 (4): 414–21. PMID 18393108.
- http://www.alizyme.com/alizyme/products/cetilistat/ Archived January 7, 2009, at the Wayback Machine.
- Norgine acquires cetilistat
- “Weight loss, HbA1c reduction, and tolerability of cetilistat in a randomized, placebo-controlled phase 2 trial in obese diabetics: comparison with orlistat (Xenical).”. Obesity. 18: 108–15. Jan 2010. doi:10.1038/oby.2009.155. PMID 19461584.
- Japan PMDA.
セチリスタット
Cetilistat

C25H39NO3 : 401.58
[282526-98-1]
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(Hexadecyloxy)-6-methyl-4H-3,1-benzoxazin-4-one
|
|
| Identifiers | |
| CAS Number | 282526-98-1 |
| ATC code | none |
| PubChem | CID 9952916 |
| ChemSpider | 8128526 |
| UNII | LC5G1JUA39 |
| KEGG | D09208 |
| ChEMBL | CHEMBL2103825 |
| Chemical data | |
| Formula | C25H39NO3 |
| Molar mass | 401.582 g/mol |
///////////////Cetilistat, ATL-962, ATL962, ATL 962, 2013-09-20, JAPAN, APPROVED, Japan PMDA, 282526-98-1, セチリスタット
 SEE
SEE
Annual Reports in Medicinal Chemistry
… versus vehicle-treated mice.34Noteworthy in the multistep synthesis of canagliflozin is …CETILISTAT (ANTIOBESITY)43–52 Class: Pancreatic lipase inhibitor …
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US
 GADOBUTROL
GADOBUTROL
gadolinium(III) 2,2′,2”-(10-((2R,3S)-1,3,4-trihydroxybutan-2-yl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate
Gadobutrol, SH-L-562, Gadovist,138071-82-6
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US.
The agency has approved the new indication for Gadavist injection for intravenous use with magnetic resonance imaging of the breast to assess the presence and extent of malignant breast disease.
Approval is based on priority review of two Phase III studies with identical design (GEMMA-1 and GEMMA-2).
Bayer HealthCare’s Gadavist (gadobutrol)
Bayer’s Gadavist injection cleared for breast cancer evaluation
UPDATE……. Gadoteridol 279.3 mg/ml for injection , CDSCO INDIA 29.07.2021
For intravenous use in magnetic
reasonance imaging (MRI) in adults and
pediatric patients over 2 years of age for
whole body MRI including the head, neck,
liver, breast, musculoskeletal system and
soft tissue pathologies
The US Food and Drug Administration (FDA) has approved Bayer HealthCare’s Gadavist (gadobutrol) injection as the first magnetic resonance contrast agent for evaluation of breast cancer in the US.
GADOBUTROL
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Licence data | US FDA:link |
| Pregnancy cat. | C (US) |
| Legal status | POM (UK) ℞-only (US) |
| Routes | IV |
| Identifiers | |
| CAS number | 138071-82-6 |
| ATC code | V08CA09 |
| PubChem | CID 72057 |
| DrugBank | DB06703 |
| UNII | 1BJ477IO2L |
| KEGG | D07420 |
| Chemical data | |
| Formula | C18H31GdN4O9 |
| Mol. mass | 604.710 g/mol |
………………………..
Gadobutrol (INN) (Gd-DO3A-butrol) is a gadolinium-based MRI contrast agent (GBCA).
It received marketing approval in Canada[1] and in the United States.[2][3][4]
As of 2007, it was the only GBCA approved at 1.0 molar concentrations.[5]
Gadobutrol is marketed by Bayer Schering Pharma as Gadovist, and by Bayer HealthCare Pharmaceuticals as Gadavist.[6]





 WORLDCUP FOOTBALL WEEK 2014 BRAZIL
WORLDCUP FOOTBALL WEEK 2014 BRAZIL
……………………………………………….
http://www.google.com/patents/EP0988294B1?cl=en
-
This type of complexes with metal ions, in particular with paramagnetic metal ions; is used for the preparation of non-ionic contrast agents for the diagnostic technique known as magnetic resonance (MRI, Magnetic Resonance Imaging), among which are ProHance(R) (Gadoteridol, gadolinium complex of 10-(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid), and Gadobutrol (gadolinium complex of [10-[2,3-dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid).

-
[0003]Two different synthetic approaches are described in literature for the preparation of this kind of complexes, said approaches differing in the strategy taken to discriminate one of the four nitrogen atoms: the first one (Dischino et al., Inorg. Chem., 1991, 30, 1265 or EP 448191, EP 292689, EP 255471) is based on the selective protection of one of the nitrogen atoms by formation of the compound of formula (III), 5H,9bH-2a,4a,7-tetraazacycloocta[cd]pentalene, and on the subsequent hydrolysis to compound of formula (IV), 1-formyl-1,4,7,10-tetraazacyclododecane, followed by the carboxymethylation of the still free nitrogen atoms and by the deprotection and alkylation of the fourth nitrogen atom, according to scheme 1.

-
[0004]The step from 1,4,7,10-tetraazacyclododecane disulfate (a commercially available product) to compound (III) is effected according to the conventional method disclosed in US 4,085,106, followed by formation of the compound of formula (IV) in water-alcohol medium.
-
[0005]This intermediate is subsequently tricarboxymethylated with tert-butyl bromoacetate (TBBA) in dimethylformamide at 2.5°C and then treated with a toluene-sodium hydroxide diphasic mixture to give the compound of formula (V), 10-formyl-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic, tris(1,1-dimethylethyl) ester, which is subsequently hydrolysed to compound of formula (II) in acidic solution.
-
[0006]In the process described in WO 93/24469 for the synthesis of Gadobutrol, at first one of the nitrogen atoms is alkylated in conditions such as to minimize the formation of polyalkylated derivatives, then the monoalkylderivative is purified and carboxymethylated, according to scheme 2.

-
[0007]The alkylation of 1,4,7,1,0-tetraazacyclododecane with the epoxide of formula (VI), 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]octane, is carried out in anhydrous n-BuOH under reflux and the reaction mixture is extracted with water, evaporated to dryness and the residue is subsequently diluted with water and extracted with methylene chloride.
-
[0008]The aqueous phase containing the mono-alkylated product (65% yield in Example 7 which reports the procedure for the preparation of 5 kg of Gadobutrol) is directly carboxymethylated at 70°C with chloroacetic acid, keeping pH 9.5 by addition of NaOH. The reaction mixture is adjusted to pH 1, concentrated to dryness and dissolved in methanol to remove the undissolved salts. The filtrate is then concentrated under vacuum, dissolved in water, and loaded onto a cation exchanger in the H+ form to fix the product. The subsequent elution with ammonia displaces the desired product, which is concentrated to small volume and subsequently complexed with gadolinium oxide according to conventional methods, and the resulting complex is purified by means of ion exchange resins. The overall yield is 42%.
-
[0009]Although the first of these two processes could theoretically provide a higher yield, in that all the single steps (protection, carboxymethylation and deprotection) are highly selective, the complexity of the operations required to remove salts and solvents and to purify the reaction intermediates makes such theoretical advantage ineffective: the overall yield is in fact, in the case of Gadoteridol, slightly higher than 37%.
-
[0010]The preparation of Gadobutrol according to the alternative process (WO 93/24469) provides a markedly better yield (72%) only on laboratory scale (example 2): example 7 (represented in the above Scheme 2) actually evidences that, when scaling-up, the yield of this process also remarkably decreases (42%).
-
[0011]In addition to the drawback of an about 40% yield, both processes of the prior art are characterized by troublesome operations, which often involve the handling of solids, the use of remarkable amounts of a number of different solvents, some of them having undesirable toxicological or anyway hazardous characteristics.
-
[0012]Moreover, the synthesis described by Dischino makes use of reagents which are extremely toxic, such as tert-butyl bromoacetate, or harmful and dangerous from the reactivity point of view, such as dimethylformamide dimethylacetal.
-
[0013]An alternative to the use of dimethyl formamide dimethylacetal is suggested by J. Am. Chem. Soc. 102(20), 6365-6369 (1980), which discloses the preparation of orthoamides by means of triethyl orthoformate.
-
[0014]EP 0596 586 discloses a process for the preparation of substituted tetraazacyclododecanes, among them compounds of formula (XII), comprising:
- formation of the tricyclo[5.5.1.0] ring;
- alkylation with an epoxide;
- hydrolysis of the 10-formyl substituent;
- reaction with an acetoxy derivative bearing a leaving group at the alpha-position.
-
[0015]Nevertheless, this method requires quite a laborious procedure in order to isolate the product of step b).
-
[0016]It is the object of the present invention a process for the preparation of the complexes of general formula (XII)

wherein
- R1 and R2
- are independently a hydrogen atom, a (C1-C20) alkyl containing 1 to 10 oxygen atoms, or a phenyl, phenyloxy group, which can be unsubstituted or substituted with a (C1-C5) alkyl or hydroxy, (C1-C5) alkoxy, carbamoyl or carboxylic groups,
- Me3+
- is the trivalent ion of a paramagnetic metal;
comprising the steps represented in the following Scheme 3:






-
The process of the present invention keeps the high selectivity typical of the protection/deprotection strategy described by Dischino in the above mentioned paper, while removing all its drawbacks, thus providing for the first time a reproducible industrial process for the preparation of the concerned compounds in high yields and without use of hazardous substances.
-
[0019]The preparation of the gadolinium complex of 10-(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane-1,4,7-tri-acetic) acid (Gadoteridol), according to scheme 4, is particularly preferred:

in which the synthetic steps a), b), c), d), e), and f) have the meanings defined above and the epoxide of formula (XI) in step d) is propylene oxide.
-
[0020]The preparation of the gadolinium complex of [10-[2,3-dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic) acid (Gadobutrol), according to the scheme 5, is also preferred.

in which the synthetic steps a), b), c), d), e), and f) have the meanings defined above and the epoxide of formula (XI) in step d) corresponds to the one of formula (VI), defined above.
-
[0021]On the other hand, step a) of the process of the present invention involves the use of triethyl orthoformate in the presence of an acid catalyst, instead of dialkylformamide-dialkylacetal.
-
[0022]Triethyl orthoformate can be added in amounts ranging from 105% to 200% on the stoichiometric value.
-
[0023]The reaction temperature can range from 110 to 150°C and the reaction time from 5 to 24 h.
-
[0024]The catalyst is a carboxylic acid having at least 3 carbon atoms, C3-C18, preferably selected from the group consisting of propionic, butyric and pivalic acids.
-
[0025]Triethyl orthoformate is a less toxic and less expensive product than N,N-dimethylformamide-dimethylacetal and does not involve the formation of harmful, not-condensable gaseous by-products. Moreover, triethyl orthoformate is less reactive than N,N-dimethylformamide-dimethylacetal, which makes it possible to carry out the loading procedures of the reactives as well as the reaction itself in utterly safe conditions even on a large scale, allows to better monitor the progress of the reaction on the basis of such operative parameters as time and temperature, without checking the progress by gas chromatography, and makes dosing the reactive less critical, in that it can be added from the very beginning without causing the formation of undesired by-products: all that rendering the process suitable for the production of compound (III) on the industrial scale in easily reproducible conditions.
-
[0026]The subsequent step b) involves the carboxymethylation of compound (III) in aqueous solution, using a haloacetic acid, to give compound (IX), i.e. the 10-formyl-1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid salt with an alkali or alkaline-earth metal, the salts of compound (IX) with sodium, potassium or calcium being most preferred.




Example 2
-
[0065]

-
[0066]The procedure of Example 1 is followed until step C included, to obtain a solution of DO3A trisodium salt.
-
[0067]pH is adjusted to 12.3 with conc. HCl and 57.7 kg (0.4 kmol) of 4,4-dimethyl-3,5,8-trioxabicyclo[5.1.0]-octane are added. After reaction for 4 h at 40°C and for 8 h at 80°C, the solution is cooled to 50°C, 120 kg of an aqueous solution containing 0.135 kmol of gadolinium trichloride are added. After 1 h the mixture is cooled at 17°C and acidified to pH 1.7 with conc. HCl, keeping this pH for 2 h. The solution is subsequently warmed to 50°C, pH is adjusted to 7 with sodium hydroxide, keeping these conditions for 1 h.
-
[0068]After that, the resulting crude Gadobutrol is purified repeating exactly the same process as in steps E and F of Example 1.

Recovery of the product (Gadobutrol)
-
[0069]The product-rich fraction is then thermally concentrated to a viscous residue and the residue is added with 350 kg of ethanol at 79°C.
-
[0070]The resulting suspension is refluxed for 1 h, then cooled, centrifuged and dried under reduced pressure to obtain 66.0 kg of Gadobutrol (0.109 kmol), HPLC assay 99.5% (A%).
Overall yield: 79.1% -
[0071]The IR and MS spectra are consistent with the indicated structure.

References
- Cheng, KT (2007). “Gadobutrol”. Molecular Imaging and Contrast Agent Database (MICAD) (Bethesda, MD: National Center for Biotechnology Information (NCBI)). PMID 20641787. NBK23589.
- http://bayerimaging.com/products/gadavist/index.php
- “FDA approves imaging agent for central nervous system scans” (Press release). U.S. Food and Drug Administration (FDA). March 15, 2011. Retrieved March 31, 2011.
- “U.S. FDA Approves Bayer’s Gadavist (Gadobutrol) Injection for MRI of the Central Nervous System” (Press release). Bayer HealthCare Pharmaceuticals. March 14, 2011. Retrieved March 31, 2011.
- “Gadobutrol 1.0-molar in Cardiac Magnetic Resonance Imaging (MRI) – Further Enhancing the Capabilities of Contrast-enhanced MRI in Ischaemic and Non-ischaemic Heart Disease?”
- “Gadavist full prescribing information”. Retrieved 2011-03-14.


{kind=link}