
Home » Posts tagged 'Inc.' (Page 2)
Tag Archives: Inc.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BEXAGLIFLOZIN


Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
FDA APPROVED
| 1/20/2023 |
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise
Drug Trials Snapshot
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)

Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
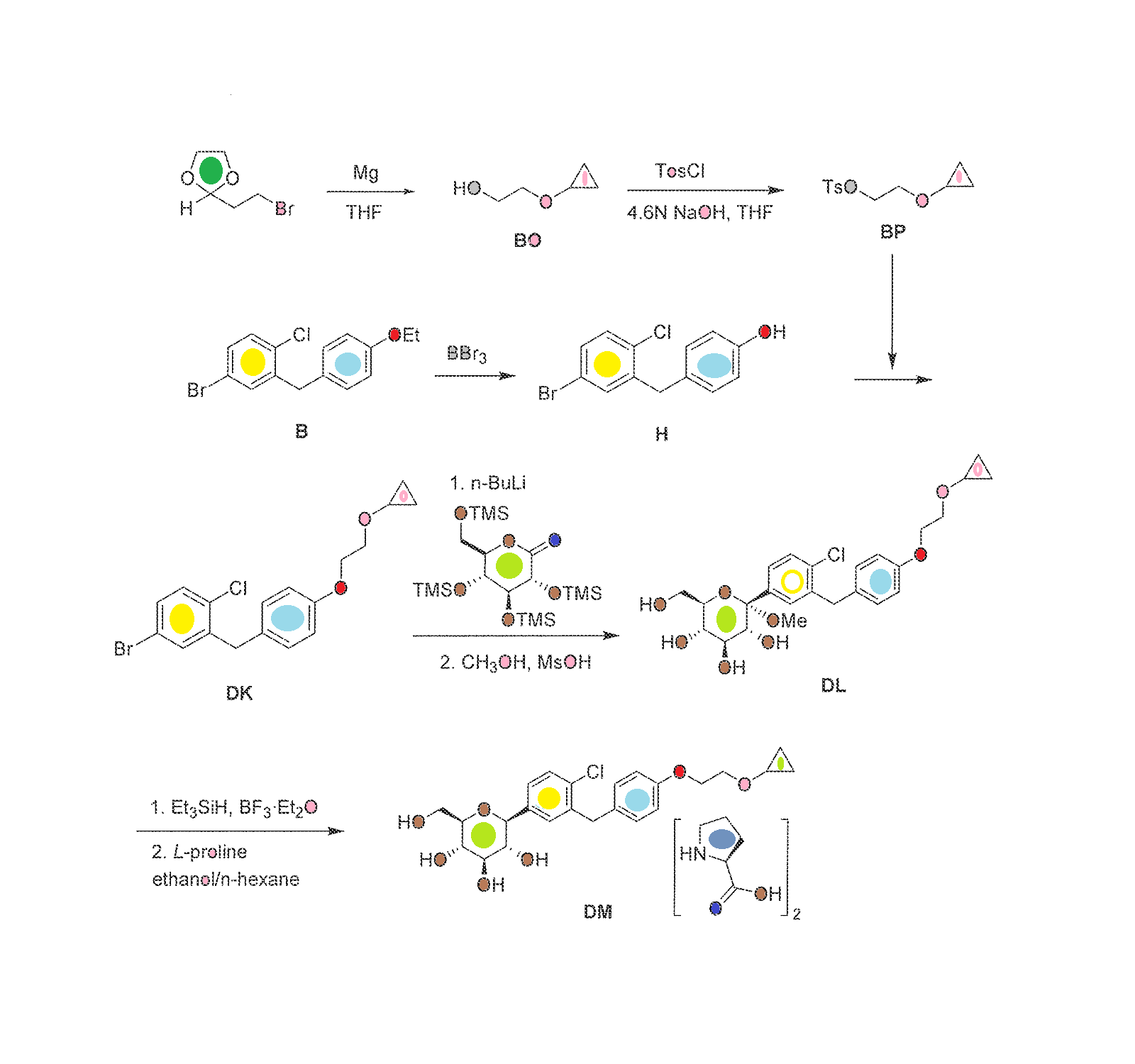
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.

Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694

Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.

Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.

(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Example 1A
Preparation of 2-cyclopropoxyethanol (1)

To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
////////BEXAGLIFLOZIN, APPROVALS 2023, FDA 2023
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079J.Med.Chem.2025,68,2147−2182
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof
type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting
SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe
reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates,
whichmaybeacommonchallengetootherSGLT2inhibitors.31
Anotherpatent disclosedbyPiramal Enterprises suggesteda
similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34
Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2
whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith
activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal
functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane,
whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was
reported in 92% yield. Details on stereoselectivity of this
approachwerenotdisclosed.
Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol
3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether
3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.

(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447−
453.
(29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium
glucose co-transporter-2 inhibitors for the treatment of diabetes
mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79.
(30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive
review of small-molecule drugs for the treatment of type 2 diabetes
mellitus: Synthetic approaches and clinical applications. Eur. J. Med.
Chem. 2024, 267, No. 116185.
(31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F.
Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A.
Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev.
2018, 22, 467−488.
(32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.;
Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′
position as potent and selective renal sodium-dependent glucose co
transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes.
Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470.
(33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the
preparation of benzyl-benzene C-glycosides via coupling reaction as
potential SGLT2 inhibitors. US 20130267694, 2013.
(34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal,
D. Aprocess for the preparation of SGLT2 inhibitor and intermediates
thereof. WO 2018207113, 2018.
(35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu,
B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside
derivatives as antidiabetic agents. WO 2009026537, 2009.
.
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
Bexagliflozin (Brenzavvy)
On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin
can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which
lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108].
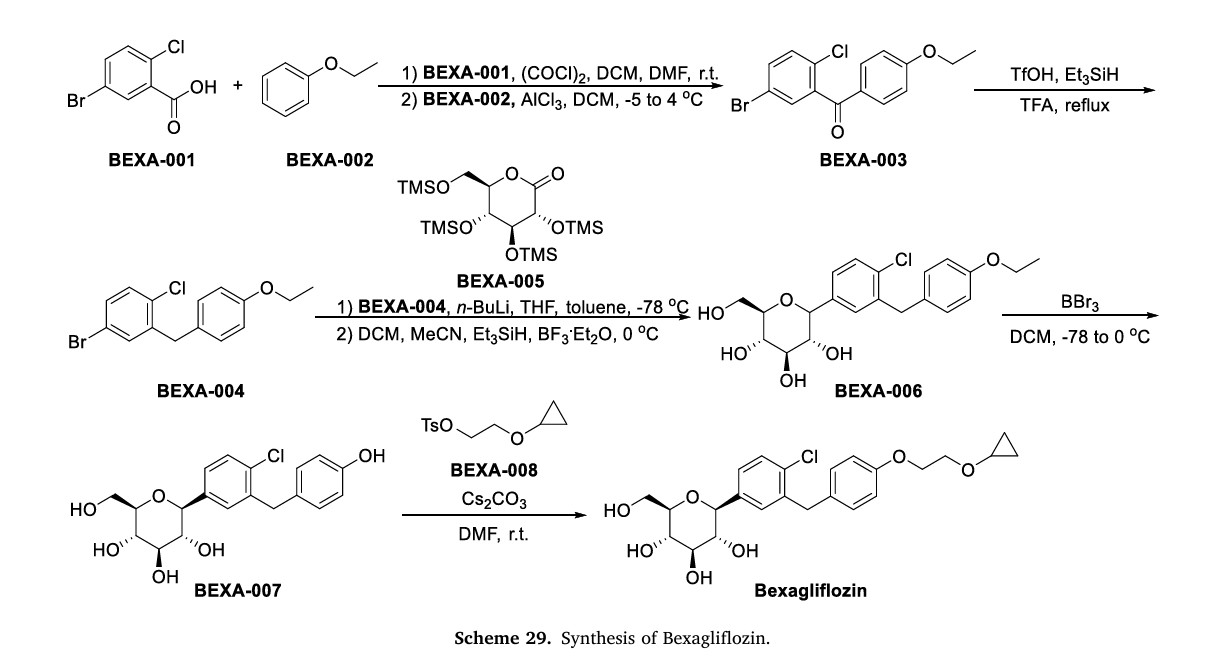
The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring.
BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol.
Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453.
[105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng,
B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and
prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293.
[106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M.
P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin.
Pharmacother. 22 (2021) 2095–2103.
[107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am.
Soc. Nephrol. 18 (2023) 279–289.
[108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr.
Drug Saf. 13 (2018) 84–91.
[109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd,
J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives
as Antidiabetic Agents, 2009. WO2009026537A1.
.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Bremelanotide, Female Libido Enhancer


Female Libido Enhancer – Bremelanotide
Bremelanotide is a compound that is currently under investigation for its potential uses in managing reperfusion injury, female sexual dysfunction or hemorrhagic shock. The chemical may also see success in managing modulate inflammation or limiting the effects of ischemia.
N-Acetyl-L-norleucyl-L-alpha-aspartyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-lysine (2-7)-lactam
Bremelanotide, PT 141, CAS NO.: 189691-06-3
Bremelanotide ![]() i/ˌbrɛmɨˈlænətaɪd/ (formerly PT-141) is a compound under drug development by Palatin Technologies as a treatment for female sexual dysfunction, hemorrhagic shock and reperfusion injury. It functions by activating the melanocortin receptors MC1R and MC4R, to modulate inflammation and limiting ischemia.[2] It was originally tested for intranasal administration in treating female sexual dysfunction but this application was temporarily discontinued in 2008 after concerns were raised over adverse side effects of increased blood pressure. As of December 2014, Palatin is conducting a human Phase 3 study[3] using a subcutaneous drug delivery system that appears to have little effect on blood pressure.
i/ˌbrɛmɨˈlænətaɪd/ (formerly PT-141) is a compound under drug development by Palatin Technologies as a treatment for female sexual dysfunction, hemorrhagic shock and reperfusion injury. It functions by activating the melanocortin receptors MC1R and MC4R, to modulate inflammation and limiting ischemia.[2] It was originally tested for intranasal administration in treating female sexual dysfunction but this application was temporarily discontinued in 2008 after concerns were raised over adverse side effects of increased blood pressure. As of December 2014, Palatin is conducting a human Phase 3 study[3] using a subcutaneous drug delivery system that appears to have little effect on blood pressure.
Palatin, in collaboration with European licensee Gedeon Richter, is developing an sc formulation of the synthetic peptide bremelanotide (PT-141; BMT), a melanocortin MCR-4 agonist and a synthetically modified analog of PT-14, also analogous to alpha-melanocyte-stimulating hormone (alpha-MSH), for the potential treatment of female sexual dysfunction (FSD) including hypoactive sexual desire disorder (HSDD)

The Bremelanotide or PT-141 is a mean that explains the revolution caused by the medical world in a silent but attractive manner in the human health related study. Bremelanotide is the latest arrival from the company called Palatin Technologies which forms the basic treatment for the hemorrhagic shock and reperfusion injury.( In short about the company, the Palatin Technologies is the owner of this research and is located in New Jersey. Hence this medicine is a Jersey based Product. And regarding the product under research, is waiting for the approval from the Food and Drug Association. Once this is done, the company has targeted to reach those customers, whom the Viagra has approached. This has the effect of helping the male patients suffering with an erectile dysfunction syndrome. Also if it gets the approval as a treatment measure for the female sexual dysfunction, then this medicine is expected to bring a relief to the post-menopausal and also supports or provides their sexual happiness and also they are checking regarding thehyposexual desire disorder. This is expected to be a blockbuster, if released. So this medicine is waiting for a confirmation as well as an approval.
In February 2015, a randomized, double-blind, placebo-controlled, open-label extension, phase III trial (NCT02338960; BMT-302, Reconnect Study) was initiated in the US in premenopausal women (expected n = 550) with hypoactive sexual desire disorder to evaluate the efficacy and safety of bremelanotide. At that time, the trial was expected to complete in July 2017
Study – Potential Use Erectile Dysfunction
One study has explored the potential use of bremelanotide as a replacement for natural peptide melanocyte stimulating hormones for the sake of treating erectile dysfunction.
- The goal of this study was to determine if the effects of bremelanotide stimulating sexual desire that was shown in male rats could be replicated in the brains of female rats. To do this, hormone primed female rats in a control group and a test group that were treated with bremelanotide and known to have consummatory sexual disorders was introduced to a group of male rats and the reactions were measured.
- Heart racing, hops and darts, pacing and customary sexual behaviors were assessed while the brain was stimulated. The stimulation of specific molecular markers within the brain was examined to determine arousal in the female subjects.
- Results indicated that the females saw an increase in sexual behavior when bremelanotide was applied to the limbic and hypothalamic regions of their brains. It is suggested that this was because the chemical that stimulated the mPOA terminals, leading to activated dopamine in the brain.
Additional study is necessary to determine the extent of the effects bremelanotide has on the brain and natural stimulating chemicals.

Bremelanotide and Ongoing Research

This is an advanced research involved even now. This functions by activating the Melanocortin, which is a group of peptide hormones which includes the adrenocorticotropic hormone and also the different forms of the melanocyte stimulating hormones. These melanocortins are produced or prepared from the proopiomelanocortin in the pituitary glands. The melanocortin releases or exert their effects by making a bind with the melanocortin and thereby activating it).The Bremelanotide functions by activating the melanocortin receptors and thereby makes a modulation in the inflammation. This is actually produced for making use in treating the sexual dysfunction. Due to certain reasons; the process of researching was kept under hold in recently, since it created some adverse side effects of increased blood pressure. In the chemistry of the preparation of the bremelanotide, the Peptide Melanaton II forms the basic compound. This compound is tested using a sunless tanning agent.
The actual information about the peptide melanaton has the effect of making sexual arousal and speed as well as sudden erections and some other side effects. However, there are several other measures taken to test the property of the same under several other health situations to make a detailed study about the chemical compound structure to make a change in the combination of the chemical structure. This medicine has made a revolution in the field of science of the human structure. When made a deep verification of the compound structure of the chemical study showed the following information. The structural design has an appearance of white colored powder like material, which has an accurate purity of nearly 98%. The actual molecular weight of the compound formed is around 1025.2. This compound has the collective share of Amino acids in the composition, peptide and acetate contents also.
The study of the compound structure PT-141 has an enhanced support of making a recombination that produces a different profile of the same medicine but in a different standard with different properties that may support the human requirement.
Bremelanotide PT-141 is known for its aphrodisiac properties
Development
![]()
Bremelanotide was developed from the peptide hormone Melanotan II which underwent testing as a sunless tanningagent. In initial testing, Melanotan II did induce tanning but additionally caused sexual arousal and spontaneous erections as unexpected side effects in nine out of the ten original male volunteer test subjects.[4]
In studies, bremelanotide was shown to induce lordosis in an animal model[5] and was also effective in treating sexual dysfunction in both men (erectile dysfunction or impotence) and women (sexual arousal disorder). Unlike Viagra and other related medications, it does not act upon the vascular system, but directly increases sexual desire via the nervous system.[6]
A Phase III clinical trial was scheduled to begin in the first half of 2007, but was delayed until August 2007. On August 30, Palatin announced that the U.S. Food and Drug Administration had expressed serious concerns regarding therisk/benefit ratio of bremelanotide with regards to the side effect of increased blood pressure. The FDA stated that it would consider alternate uses for bremelanotide, including as a treatment for individuals who do not respond to more established ED treatments. However, On May 13, 2008, Palatin Technologies announced it had “discontinued development of Bremelanotide for the treatment of male and female sexual dysfunction” while concurrently announcing plans to develop it as a treatment for hemorrhagic shock instead.[7] The company additionally announced intentions to focus its attention on another compound, PL-6983, that causes lower blood pressure in animal models.[8]Palatin has since re-initiated Bremelanotide studies for ED and FSD using a subcutaneous delivery method. On August 12, 2009, the company announced that in a double-blind study of 54 volunteers bremelanotide failed to evoke the hypertensive side effects seen with the nasal delivery system used in prior studies, concluding that “variability of uptake” inherent in intranasal administration of the drug resulted in “increases in blood pressure and gastrointestinal events…primarily related to high plasma levels in [only] a subset of patients” and that subcutaneous administration of the drug circumvented the potential for this side effect.[8] Palatin has completed a human Phase 2B study utilizing subcutaneous administration and reported positive results.[9]
Structure

Bremelanotide is a cyclic hepta-peptide lactam analog of alpha-melanocyte-stimulating hormone (alpha-MSH) that activates the melanocortin receptors MC3-R and MC4-R in thecentral nervous system. It has the amino acid sequence Ac-Nle-cyclo[Asp-His-D-Phe-Arg-Trp-Lys]-OH or cyclo-[Nle4, Asp5, D-Phe7, Lys10]alpha-MSH-(4-10). It is a metabolite of Melanotan II that lacks the C-terminal amide function.
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3S,6S,9R,12S,15S,23S)-15-[(N-acetyl-L-norleucyl)amino]-9-benzyl-6-{3-[(diaminomethylidene)amino]propyl}-12-(1H-imidazol-5-ylmethyl)-3-(1H-indol-3-ylmethyl)-2,5,8,11,14,17-hexaoxo-1,4,7,10,13,18-hexaa zacyclotricosane-23-carboxylic acid | |
| Clinical data | |
| Legal status |
|
| Pharmacokinetic data | |
| Half-life | 120 minutes[1] |
| Identifiers | |
| CAS number | 189691-06-3 |
| ATC code | None |
| PubChem | CID 9941379 |
| ChemSpider | 8116997 |
| UNII | 6Y24O4F92S |
| KEGG | D06569 |
| ChEMBL | CHEMBL2070241 |
| Chemical data | |
| Formula | C50H68N14O10 |
| Molecular mass | 1025.2 g/mol |
Sexual dysfunction, including both penile erectile dysfunction or impotence and female sexual dysfunction, are common medical problems. Significant effort has been devoted over the last twenty or more years to develop methods, devices and compounds for treatment of sexual dysfunction. While more effort has been undertaken for treatment of penile erectile dysfunction, female sexual dysfunction is also an area to which significant research and effort has been devoted.
At present, one commonly used orally administered drug for treatment of sexual dysfunction in the male is Viagra®, a brand of sildenafil, which is a phosphodiesterase 5 inhibitor, increasing the persistence of cyclic guanosine monophosphate and thereby enhancing erectile response. There are several other medical treatment alternatives currently available depending on the nature and cause of the impotence problem. Some men have abnormally low levels of the male hormone testosterone, and treatment with testosterone injections or pills may be beneficial. However, comparatively few impotent men have low testosterone levels. For many forms of erectile dysfunction, treatment may be undertaken with drugs injected directly into the penis, including drugs such as papaverin, prostaglandin E1, phenoxybenzamine or phentolamine. These all work primarily by dilating the arterial blood vessels and decreasing the venous drainage. Urethral inserts, such as with suppositories containing prostaglandin, may also be employed. In addition, a variety of mechanical aids are employed, including constriction devices and penile implants.
A variety of treatments have also been explored for female sexual dysfunction, including use of sildenafil, although the Food and Drug Administration has not specifically approved such use. Testosterone propionate has also been employed to increase or augment female libido.
Melanocortin receptor-specific compounds have been explored for use of treatment of sexual dysfunction. In one report, a cyclic α-melanocyte-stimulating hormone (“α-MSH”) analog, called Melanotan-II, was evaluated for erectogenic properties for treatment of men with psychogenic erectile dysfunction. Wessells H. et al., J Urology 160:389-393 (1998); see also U.S. Pat. No. 5,576,290, issued Nov. 19, 1996 to M. E. Hadley, entitled Compositions and Methods for the Diagnosis and Treatment of Psychogenic Erectile Dysfunction and U.S. Pat. No. 6,051,555, issued Apr. 18, 2000, also to M. E. Hadley, entitled Stimulating Sexual Response in Females. The peptides used in U.S. Pat. Nos. 5,576,290 and 6,051,555 are also described in U.S. Pat. No. 5,674,839, issued Oct. 7, 1997, to V. J. Hruby, M. E. Hadley and F. Al-Obeidi, entitled Cyclic Analogs of Alpha–MSH Fragments, and in U.S. Pat. No. 5,714,576, issued Feb. 3, 1998, to V. J. Hruby, M. E. Hadley and F. Al-Obeidi, entitled Linear Analogs of Alpha–MSH Fragments. Melanotan-II is a peptide of the following formula:

Additional related peptides are disclosed in U.S. Pat. Nos. 5,576,290, 5,674,839, 5,714,576 and 6,051,555. These peptides are described as being useful for both the diagnosis and treatment of psychogenic sexual dysfunction in males and females. These peptides are related to the structure of melanocortins.
In use of Melanotan-II, significant erectile responses were observed, with 8 of 10 treated men developing clinically apparent erections, and with a mean duration of tip rigidity greater than 80% for 38 minutes with Melanotan-II compared to 3.0 minutes with a placebo (p=0.0045). The drug was administered by subcutaneous abdominal wall injection, at doses ranging from 0.025 to 0.157 mg/kg body weight. Transient side effects were observed, including nausea, stretching and yawning, and decreased appetite.
The minimum peptide fragment of native α-MSH needed for erectile response is the central tetrapeptide sequence, His6-Phe7-Arg8-Trp9 (SEQ ID NO:1). In general, all melanocortin peptides share the same active core sequence, His-Phe-Arg-Trp (SEQ ID NO:1), including melanotropin neuropeptides and adrenocorticotropin. Five distinct melanocortin receptor subtypes have been identified, called MC1-R through MC5-R, and of these MC3-R and MC4-R are believed to be expressed in the human brain. MC3-R has the highest expression in the arcuate nucleus of the hypothalamus, while MC4-R is more widely expressed in the thalamus, hypothalamus and hippocampus. A central nervous system mechanism for melanocortins in the induction of penile erection has been suggested by experiments demonstrating penile erection resulting from central intracerebroventricular administration of melanocortins in rats. While the mechanism of His-Phe-Arg-Trp (SEQ ID NO:1) induction of erectile response has not been fully elucidated, it has been hypothesized that it involves the central nervous system, and probably binding to MC3-R and/or MC4-R.
Other peptides and constructs have been proposed which are ligands that alter or regulate the activity of one or more melanocortin receptors. For example, International Patent Application No. PCT/US99/09216, entitled Isoquinoline Compound Melanocortin Receptor Ligands and Methods of Using Same, discloses two compounds that induce penile erections in rats. However, these compounds were administered by injection at doses of 1.8 mg/kg and 3.6 mg/kg, respectively, and at least one compound resulted in observable side effects, including yawning and stretching. Other melanocortin receptor-specific compounds with claimed application for treatment of sexual dysfunction are disclosed in International Patent Application No. PCT/US99/13252, entitled Spiropiperidine Derivatives as Melanocortin Receptor Agonists.
Both cyclic and linear α-MSH peptides have been studied; however, the peptides heretofore evaluated have had an amide or —NH2 group at the carboxyl terminus. See, for example, Wessells H. et al., J Urology, cited above; Haskell-Luevano C. et al., J Med Chem 40:2133-39 (1997); Schiöth H. B. et al., Brit J Pharmacol 124:75-82 (1998); Schiöth H. B. et al., Eur J Pharmacol 349:359-66 (1998); Hadley M. E. et al., Pigment Cell Res 9:213-34 (1996); Bednarek M. A. et al., Peptides20:401-09 (1999); U.S. Pat. Nos. 6,054,556, 6,051,555 and 5,576,290; and, International Patent Applications PCT/US99/04111 and PCT/US98/03298. While significant research has been conducted in an effort to determine the optimal structure of α-MSH peptides, including a variety of structure-function, agonist-antagonist, molecular modeling and pharmacophore studies, such studies have relied upon peptides with an art conventional —NH2 group at the carboxyl terminus. Further, it has long been believed that biologically active neuropeptides, including α-MSH peptides, are amidated, with an —NH2 group at the carboxyl terminus, and that such amidation is required both for biological activity and stability. See, for example, Metabolism of Brain Peptides, Ed. G. O’Cuinn, CRC Press, New York, 1995, pp. 1-9 and 99-101.
…………………………………………….
Bioorganic and Medicinal Chemistry Letters, 2005 , vol. 15, 4 pg. 1065 – 1068
http://www.sciencedirect.com/science/article/pii/S0960894X04014842

Figure 2.
NMR structural analysis on compound 3.

Figure 4.
NMR structural analysis of compound 1.
……………………………………………….
In a preferred embodiment, the invention provides the peptide
Ac-Nle-cyclo(-Asp-His-D-Phe-Arg-Trp-Lys)-OH Compound 1
The peptide of Compound 1 has a formula of C50H68N14O10, and a net molecular weight of 1025.18. This peptide may be synthesized by solid-phase means and purified to greater than 96% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of Compound 1 is:

In general, the peptide compounds of this invention may be synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of well-known procedures utilizing a variety of resins and reagents may be used to prepare the compounds of this invention.
The peptides of this invention may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, succinic or methanesulfonic. The acetate salt form is especially useful. Where the compounds of this invention include an acidic moiety, suitable pharmaceutically acceptable salts may include alkali metal salts, such as sodium or potassium salts, or alkaline earth metal salts, such as calcium or magnesium salts.
The invention provides a pharmaceutical composition that includes a peptide of this invention and a pharmaceutically acceptable carrier. The carrier may be a liquid formulation, and is preferably a buffered, isotonic, aqueous solution. Pharmaceutically acceptable carriers also include excipients, such as diluents, carriers and the like, and additives, such as stabilizing agents, preservatives, solubilizing agents, buffers and the like, as hereafter described.
EXAMPLE 1
Peptide Synthesis
The peptide Ac-Nle-cyclo(-Asp-His-D-Phe-Arg-Trp-Lys)-OH was synthesized by standard solid phase peptide synthesis methods, and is a cyclic heptapeptide melanocortin peptide analog with a free acid at the carboxyl terminus and an acetylated amino group at the amino terminus, with the structure:

The peptide has a net molecular weight of 1025.18, and is supplied in an acetate salt form. The peptide is a white, odorless amorphous hygroscopic powder, soluble in 0.9% saline, composed of C50H68N14O10. For synthesis, an Fmoc-Lys(R3)-p-alkoxybenzyl alcohol resin was transferred to a solid phase peptide synthesizer reactor with a mechanical stirrer. The R3group, such as 1-(1′-adamantyl)-1-methyl-ethoxycarbonyl (Adpoc), allyloxycarbonyl (Aloc) or 4-methyltrityl (Mtt), was removed and the next Fmoc-protected amino acid (Fmoc-Trp(Boc)-OH) was added to the resin through standard coupling procedures. The Fmoc protective group was removed and the remaining amino acids added individually in the correct sequence, by repeating coupling and deprotection procedures until the amino acid sequence was completed. After completion of coupling with the last Fmoc-amino acid derivative, Fmoc-Nle-OH, and cleavage of the Fmoc protective group, the exposed terminal amino group was acetylated with acetic anhydride and pyridine in N,N-dimethylformamide (DMF). The peptide-resin was dried and the Lys and Asp protective groups cleaved. The Lys and Asp deprotected peptide resin was suspended in a suitable solvent, such as DMF, dichloromethane (DCM) or 1-methyl-2-pyrrolidone (NMP), a suitable cyclic coupling reagent, such as 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU), 2-(7-aza-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TATU), 2-(2-oxo-1(2H)-pyridyl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) or N,N′-dicyclohexylcarbodiimide/1-hydroxybenzotriazole (DCCl/HOBt) was added, and coupling initiated by use of a suitable base, such as N,N-diispropylethylamine (DIPEA), sym-collidine or N-methylmorpholine (NMM). After cyclization, the peptide-resin was washed and the peptide cleaved from the resin and any remaining protective groups using trifluoroacetic acid (TFA) in the presence of water and 1,2-ethanedithiol (EDT). The final product was precipitated by adding cold ether and collected by filtration. Final purification was by reversed phase HPLC using a C18 column. The purified peptide was converted to acetate salt by passage through an ion-exchange column.
…………………………………………..
WO2014071339

Compounds of the Invention.
in a preferred embodiment of the present invention, fie rneianocortin receptor agonist is;
Ac-Nie”Cyc/o{-Asp-His–D–Phe-Arg–Trp»Lys)–OH (bremeianotide)
The peptide of bremeianotide has a formula of CsaHesN< C½, and a net mofecufar weight of 1025.18, This peptide may be synthesized by conventional means, including either solid-phase or Squid-phase techniques, and purified to greater than 99% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of bremeianotide is:

in one embodiment of the invention, bremeianotide is synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of ‘well-known procedures utilizing a variety of resins and reagents may be used to prepare bremeianotide.
Bremeianotide may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacefie, maieic, citric, tartaric, oxalic, succinic or methanesu!fonic acid. The acetate salt form is especially useful.
in a preferred embodiment, bremelanotide is an acetate salt form, and is formulated in a buffered aqueous solution including giycerin, and prepackaged in a syringe and auto-injector device. In alternative embodiments, bremelanotide is any pharmaceutically acceptable salt form, and is formulated in any pharmaceutically acceptable aqueous solution, the aqueous solution optionally including one or more salts, such as sodium chloride, one or more acids, such as citric acid, and one or more additional ingredients, including cellulose or derivatives thereof, saccharides o
polysaccharides such as dextrose, and any of a wide variety of surfactants, chelating agents and preservatives.
………………………………………….
In yet another embodiment of the present invention, the melanocortin receptor agonist is:
Ac–Nle-cyclo(-Asp–His–D–Phe–Arg–Trp–Lys)-OH PT-141
The peptide of PT-141 has a formula of C50H68N14O10, and a net molecular weight of 1025.18. This peptide may be synthesized by conventional means, including either solid-phase or liquid-phase techniques, and purified to greater than 99% purity by HPLC, yielding a white powder that is a clear, colorless solution in water. The structure of PT-141 is:

In one embodiment of the invention, PT-141 is synthesized by solid-phase synthesis and purified according to methods known in the art. Any of a number of well-known procedures utilizing a variety of resins and reagents may be used to prepare PT-141.
PT-141 may be in the form of any pharmaceutically acceptable salt. Acid addition salts of the compounds of this invention are prepared in a suitable solvent from the peptide and an excess of an acid, such as hydrochloric, hydrobromic, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, citric, tartaric, oxalic, succinic or methanesulfonic acid. The acetate salt form is especially useful. Where the compounds of this invention include an acidic moiety, suitable pharmaceutically acceptable salts may include alkali metal salts, such as sodium or potassium salts, or alkaline earth metal salts, such as calcium or magnesium salts.
In a preferred embodiment, PT-141 is an acetate salt form, and is formulated in a buffered aqueous solution including glycerin, prepackaged in a metered unit dose intranasal delivery device. In alternative embodiments, PT-141 is any pharmaceutically acceptable salt form, and is formulated in any pharmaceutically acceptable aqueous solution, the aqueous solution optionally including one or more salts, such as sodium chloride, one or more acids, such as citric acid, and one or more additional ingredients, including cellulose or derivatives thereof, saccharides or polysaccharides such as dextrose, and any of a wide variety of surfactants, chelating agents and preservatives. In one preferred embodiment, PT-141 is administered to patients in volumes of 100 μL, with the quantity of PT-141 delivered determined by the concentration thereof. As described hereafter, in one preferred embodiment a metered unit dose contains 7.5 mg of PT-141.
While certain embodiments of the present invention are described primarily in the context of PT-141, it is to be understood that other melanocortin receptor agonists may be employed. For example, the metallopeptide melanocortin receptor agonists disclosed in WO 02/064091, filed on Feb. 13, 2001, and U.S. Ser. No. 10/640,755, filed on Aug. 13, 2003, both entitled Melanocortin Metallopeptides for Treatment of Sexual Dysfunction; and WO 01/13112, filed on Jun. 14, 2000, entitled Melanocortin Metallopeptide Constructs, Combinatorial Libraries and Applications, may be employed. In addition, the peptidomimetic melanocortin receptor agonists disclosed in U.S. Ser. No. 10/776,419, filed on Feb. 10, 2004, entitled Peptidomimetics of Biologically Active Metallopeptides; the pyrrolidine melanocortin receptor agonists disclosed in U.S. Ser. No. 10/766,657, filed on Feb. 10, 2004, entitled Pyrrolidine Melanocortin-Specific Compounds; and the bicyclic melanocortin receptor agonists disclosed in PCT/US04/01505, filed on Jan. 20, 2004, entitled Bicyclic Melanocortin-Specific Compounds, may also be employed. Also particular preferred are the piperazine melanocortin agonists disclosed in PCT/US04/01462, filed on Jan. 20, 2004 and U.S. Ser. No. 10/762,079, filed on Jan. 20, 2004, both entitled piperazine Melanocortin-Specific Compounds; the melanocortin agonists disclosed in WO 03/006620, filed on Jul. 11, 2002, entitled Linear and Cyclic Melanocortin Receptor-Specific Peptides; WO 04/005324, filed on Jul. 9, 2003, entitled Peptide Compositions for Treatment of Sexual Dysfunction; PCT/US00/18217, filed on Jun. 29, 2000 and U.S. Ser. No. 10/040,547, filed on Jan. 4, 2002, entitled Compositions and Methods for Treatment of Sexual Dysfunction; and U.S. Ser. No. 10/638,071, filed on Aug. 8, 2003, entitled Cyclic Peptide Compositions and Methods for Treatment of Sexual Dysfunction. The entire disclosure of each of the foregoing are incorporated here by reference. It is to be understood that the foregoing listing of patent applications disclosing melanocortin receptor agonists is intended to only be exemplary, and that other melanocortin receptor agonists, whether heretofore known or hereafter developed, may similarly be used in the practice of this invention.
…………………….
NMR prediction


- King SH, Mayorov AV, Balse-Srinivasan P, Hruby VJ, Vanderah TW, Wessells H (2007).“Melanocortin receptors, melanotropic peptides and penile erection”. Current Topics in Medicinal Chemistry 7 (11): 1098–1106. doi:10.2174/1568026610707011111.PMC 2694735. PMID 17584130.
- Bremelanotide for Organ Protection and Related Indications, Palatin Technologies fact sheet. Retrieved on 2009-01-18.
- “Palatin Announces Start of Bremelanotide Phase 3 Program For Female Sexual Dysfunction”. PR Newswire. Retrieved 2015-02-17.
- “Tanning drug may find new life as Viagra alternative”. CNN. 1999. Retrieved2007-09-16.
- Pfaus JG, Shadiack A, Van Soest T, Tse M, Molinoff P (July 2004). “Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist”. Proc. Natl. Acad. Sci. U.S.A. 101 (27): 10201–4. doi:10.1073/pnas.0400491101. PMC 454387.PMID 15226502.
- Vicki Mabrey (2006). “ABC News “The Business of Desire – Love Potion””. ABC News. Retrieved 2009-01-24.
- “Palatin Technologies announces new strategic objectives and reports third quarter 2008 financial results”. Palatin Technologies press release. 2008. Retrieved 2008-08-21.
- “Palatin Technologies Announces New Strategic Objectives”. Retrieved 2008-05-13.
- http://www.palatin.com/news/news.asp?ud=306
External links
- Palatin Technologies The company that has developed bremelanotide.
- US 6,794,489 bremelanotide (PT-141) patent (Appl. No.:040547)
- US 6,579,968 bremelanotide (PT-141) patent (Appl. No.:066501)
- Bremelanotide Edguider Forum
| PALATIN TECHNOLOGIES, INC.: ‘Bremelanotide in Premenopausal Women With Female Sexual Arousal Disorder and/or Hypoactive Sexual Desire Disorder‘ CLINICALTRIALS.GOV (NCT01382719, [Online] 20 March 2012, page 1 Retrieved from the Internet: <URL:http://clinicaltrials.gov/archive/NCT0 1382719/ 2012-03 20> [retrieved on 2014-02-10] | ||
| 2 | * | PALATIN TECHNOLOGIES, INC.: ‘Reports Positive Bremelanotide Study; Improved Safety Profile with Subcutaneous Administration‘ PR NEWSWIRE., [Online] 12 August 2009, Retrieved from the Internet: <URL:http://www.thefreelibrary.com/Palatin +Technolo9ies,+Inc.+Reports+Positive+Bremel anotide+Study%38…-a020561 3302> [retrieved on 2014-02-10] |
| 3 | * | SAFARINEJAD, MR.: ‘Evaluation of the Safety and Efficacy of Bremelanotide, a Melanocortin Receptor Agonist, in Female Subjects with Arousal Disorder: A Double-Blind Placebo-Controlled, Fixed Dose, Randomized Study”.‘ INTERNATIONAL SOCIETY FOR SEXUAL MEDICINE. vol. 5, 2008, pages 887 – 897 |
| US8455617 | Jun 7, 2010 | Jun 4, 2013 | Astrazeneca Ab | Melanocortin receptor-specific peptides |
| US8455618 | Oct 26, 2011 | Jun 4, 2013 | Astrazeneca Ab | Melanocortin receptor-specific peptides |
| US8487073 | Nov 23, 2010 | Jul 16, 2013 | Palatin Technologies, Inc. | Melanocortin receptor-specific peptides for treatment of sexual dysfunction |
| US8729224 | Jun 5, 2013 | May 20, 2014 | Palatin Technologies, Inc. | Melanocortin receptor-specific peptides for treatment of female sexual dysfunction |
| EP2266567A1 | May 26, 2009 | Dec 29, 2010 | Æterna Zentaris GmbH | Use of cetrorelix in combination with PDE V inhibitors for the treatment of sex hormone dependent disorders |
| EP2266568A1 | May 26, 2009 | Dec 29, 2010 | Æterna Zentaris GmbH | Use of LHRH antagonists in combination with PDE V inhibitors for the treatment of sex hormone dependent disorders |
| WO2013067309A1 | Nov 2, 2012 | May 10, 2013 | Xion Pharmaceutical Corporation | Methods and compositions for oral administration of melanocortin receptor agonist compounds |
| WO2014071339A2 * | Nov 5, 2013 | May 8, 2014 | Palatin Technologies, Inc. | Uses of bremelanotide in therapy for female sexual dysfunction |
| WO2009151714A2 * | Mar 24, 2009 | Dec 17, 2009 | Palatin Technologies, Inc. | Therapeutic for treatment of circulatory shock, ischemia, inflammatory disease and related conditions |
| US6794489 * | Jan 4, 2002 | Sep 21, 2004 | Palatin Technologies, Inc. | Peptide sequence ac-nle-cyclo(-asp-his-d-phe-arg-trp-lys)-oh derived from a melanocyte-stimulating hormone (? alpha -msh?) analog, called melanotan-ii |
| US20050222014 * | May 26, 2005 | Oct 6, 2005 | Palatin Technologies, Inc. | Administering phosphodiestarase inhibitors and melanocortin receptor antagonist: synergistic mixture |
| US20110065652 * | Nov 23, 2010 | Mar 17, 2011 | Palatin Technologies, Inc. | Melanocortin Receptor-Specific Peptides for Treatment of Sexual Dysfunction |
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com

 Location in Madhya Pradesh
Location in Madhya Pradesh
-
Khajuraho Group of Monuments – Wikipedia, the free …
en.wikipedia.org/wiki/Khajuraho_Group_of_Monuments
The Khajuraho Group of Monuments are a group of Hindu and Jain temples in Madhya Pradesh, India. About 620 kilometres (385 mi) southeast of New Delhi, …








Hotel Chandela – A Taj Leisure Hotel


LATUR, MAHARASHTRA, INDIA
http://en.wikipedia.org/wiki/Latur
| Latur लातूर Lattalur, Ratnapur |
|
|---|---|
| City | |
|
Latur
Location in Maharashtra, India |
|
| Coordinates: 18.40°N 76.56°ECoordinates: 18.40°N 76.56°E | |
| Country | |
| State | Maharashtra |
| Region | Aurangabad Division |
| District | Latur |
| Settled | Possibly 7th century AD |
| Government | |
| • Body | Latur Municipal Corporation |
| • Mayor | Akhtar Shaikh |
| Area[1] | |
| • Total | 117.78 km2(45.48 sq mi) |
| Area rank | 89 |
| Elevation | 515 m (1,690 ft) |
| Population (2011) | |
| • Total | 382,754 |
| • Rank | 89th |
| • Density | 3,200/km2(8,400/sq mi) |
| Demonym | Laturkar |
| Languages | |
| • Official | Marathi |
| Time zone | IST (UTC+5:30) |
| PIN |
|
| Telephone code | 91-2382 |
| Vehicle registration | MH-24 |
| Sex ratio | 923.54 ♀/1000 ♂ |
| Literacy | 89.67 |
| Distance from Mumbai | 497 kilometres (309 mi) E (land) |
| Distance fromHyderabad | 337 kilometres (209 mi) NW (land) |
| Distance fromAurangabad, Maharashtra | 294 kilometres (183 mi) SE (land) |
| Climate | BSh (Köppen) |
| Precipitation | 666 millimetres (26.2 in) |
| Avg. summer temperature | 41 °C (106 °F) |
| Avg. winter temperature | 13 °C (55 °F) |
| http://www.citypopulation.de/world/Agglomerations.html | |





his Is The Famous ‘Ganj-Golai’ As The Central Place Of The Latur City. There Are 16 Roads Connecting To This Place And Seperate Markets i.e. Jewellers …

लातूर जिल्हयातील चित्र संग्रह
LATUR AIRPORT
 LATUR AIRPORT
LATUR AIRPORT


2012 Navratri Mahotsav in Latur

SOS Children’s Village Latur


Latur, India: Carnival Resort


Ausa Near Latur

Chakur near Latur

Vilasrao Deshmukh’s ancestral home at Babhalgaon village in Latur. Machindra Amle



UDGIR: Udgir is one of the most important towns of Latur district. Udgir has a great historical significance. It has witnessed the war between the Marathas …
 The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
The city of Latur is located in India’s welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
Radius Announces Positive Phase 3 Top-Line Results for Its Investigational Drug Abaloparatide-SC in Postmenopausal Women With Severe Osteoporosis

Abaloparatide
WALTHAM, Mass., Dec. 21, 2014 (GLOBE NEWSWIRE) — Radius Health, Inc. today announced positive top-line 18-month fracture results from the Company’s Phase 3 clinical trial (ACTIVE) evaluating the investigational drug abaloparatide-SC for potential use in the reduction of fractures in postmenopausal osteoporosis.

https://in.finance.yahoo.com/news/radius-announces-positive-phase-3-042531179.html
Abaloparatide
BA058
BIM-44058
UNII-AVK0I6HY2U
BA058; BIM-44058; CAS 247062-33-5
MW 3960.5896, MF C174 H300 N56 O49
NAME………C2.29-methyl(22-L-glutamic acid(F>E),23-L-leucine(F>L),25-L-glutamic acid(H>E),26-L-lysine(H>K),28-L-leucine(I>L),30-L-lysine(E>K),31-L-leucine(I>L))human parathyroid hormone-related protein-(1-34)-proteinamide
L-Alaninamide, L-alanyl-L-valyl-L-seryl-L-alpha-glutamyl-L-histidyl-L-glutaminyl-L-leucyl-L-leucyl-L-histidyl-L-alpha-aspartyl-L-lysylglycyl-L-lysyl-L-seryl-L-isoleucyl-L-glutaminyl-L-alpha-aspartyl-L-leucyl-L-arginyl-L-arginyl-L-arginyl-L-alpha-glutamyl-L-leucyl-L-leucyl-L-alpha-glutamyl-L-lysyl-L-leucyl-L-leucyl-2-methylalanyl-L-lysyl-L-leucyl-L-histidyl-L-threonyl-
L-Alaninamide, L-alanyl-L-valyl-L-seryl-L-α-glutamyl-L-histidyl-L-glutaminyl-L-leucyl-L-leucyl-L-histidyl-L-α-aspartyl-L-lysylglycyl-L-lysyl-L-seryl-L-isoleucyl-L-glutaminyl-L-α-aspartyl-L-leucyl-L-arginyl-L-arginyl-L-arginyl-L-α-glutamyl-L-leucyl-L-leucyl-L-α-glutamyl-L-lysyl-L-leucyl-L-leucyl-2-methylalanyl-L-lysyl-L-leucyl-L-histidyl-L-threonyl-
CLINICAL……….https://clinicaltrials.gov/search/intervention=Abaloparatide%20OR%20BA058%20OR%20BIM-44058
BIM-44058 is a 34 amino acid analog of native human PTHrP currently in phase III clinical trials at Radius Health for the treatment of postmenopausal osteoporosis. Radius is also developing a microneedle transdermal patch using a 3M drug delivery system in phase II clinical trials. The drug candidate was originally developed at Biomeasure (a subsidiary of Ipsen), and was subsequently licensed to Radius and Teijin Pharma.
………………………….
PATENT
http://www.google.com/patents/EP2206725A1?cl=en
-
A peptide of the formula:[Glu22, 25, Leu23, 28, 31, Lys26, Aib29, Nle30]hPTHrP(1-34)NH2;[Glu22, 25, Leu23, 28, 30, 31, Lys26, Aib29]hPTHrP(1-34)NH2; [Glu22, 25,29, Leu23, 28, 30, 31, Lys26]hpTHrP(1-34)NH2; [Glu22, 25, 29, Leu23, 28, 31, Lys26, Nle30]hPTHrP(1-34)NH2; [Ser1, Ile5, Met8, Asn10, Leu11, 23, 28, 31, His14, Cha15, Glu22, 25, Lys26, 30, Aib29]hPTHrP (1-34)NH2; [Cha22, Leu23, 28, 31, Glu25, 29, Lys26, Nle30]hPTHrP(1-34)NH2; [Cha7, 11, 15]hPTHrP(1-34)NH2; [Cha7, 8, 15]hPTHrP(1-34)NH2; [Glu22, Leu23, 28, Aib25, 29, Lys26]hpTHrP(1-34)NH2; [Aib29]hPTHrP(1-34)NH2; [Glu22, 25, Leu23, 28, 31, Lys26, Aib29, 30]hPTHrP(1-34)NH2; [Glu22, 25, Leu23, 28, 31, Lys26, Aib29]hPTHrP(1-34)NH2; [Glu22, 25, Leu23, 28, 31, Aib26, 29, Lys30] hPTHrP(1-34)NH2; or [Leu27, Aib29]hPTH(1-34)NH2; or a pharmaceutically acceptable salt thereof.
…………………
SEE……http://www.google.com.ar/patents/US8148333?cl=en
………………..
SEE…………http://www.google.im/patents/US20090227498?cl=pt
| EP5026436A | Title not available | |||
| US3773919 | Oct 8, 1970 | Nov 20, 1973 | Du Pont | Polylactide-drug mixtures |
| US4767628 | Jun 29, 1987 | Aug 30, 1988 | Imperial Chemical Industries Plc | Polylactone and acid stable polypeptide |
| WO1994001460A1 * | Jul 13, 1993 | Jan 20, 1994 | Syntex Inc | Analogs of pth and pthrp, their synthesis and use for the treatment of osteoporosis |
| WO1994015587A2 | Jan 5, 1994 | Jul 21, 1994 | Steven A Jackson | Ionic molecular conjugates of biodegradable polyesters and bioactive polypeptides |
| WO1997002834A1 * | Jul 3, 1996 | Jan 30, 1997 | Biomeasure Inc | Analogs of parathyroid hormone |
| WO1997002834A1 * | 3 Jul 1996 | 30 Jan 1997 | Biomeasure Inc | Analogs of parathyroid hormone |
| WO2008063279A2 * | 3 Oct 2007 | 29 May 2008 | Radius Health Inc | A stable composition comprising a bone anabolic protein, namely a pthrp analogue, and uses thereof |
| US5695955 * | 23 May 1995 | 9 Dec 1997 | Syntex (U.S.A.) Inc. | Gene expressing a nucleotide sequence encoding a polypeptide for treating bone disorder |
| US20030166836 * | 6 Nov 2002 | 4 Sep 2003 | Societe De Conseils De Recherches Et D’application Scientefiques, S.A.S., A France Corporation | Analogs of parathyroid hormone |
| US20050282749 * | 14 Jan 2005 | 22 Dec 2005 | Henriksen Dennis B | Glucagon-like peptide-1 (GLP-1); immunotherapy; for treatment of obesity |
FDA Approves Spiriva Respimat (tiotropium) for the Maintenance Treatment of COPD

Ridgefield, Conn., September 25, 2014 – Boehringer Ingelheim Pharmaceuticals, Inc. announced today that the U.S. Food and Drug Administration (FDA) approved Spiriva Respimat (tiotropium bromide) inhalation spray for the long-term, once-daily maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema and to reduce exacerbations in COPD patients. Boehringer Ingelheim anticipates Spiriva Respimat to be available in January 2015.

Spiriva Respimat provides a pre-measured amount of medicine in a slow-moving mist that helps patients inhale the medicine. Spiriva Respimat was developed to actively deliver medication in a way that does not depend of how fast air is breathed in from the inhaler.
READ AT




MAKE IN INDIA
Tecadenoson…………Atrial Fibrillation





CVT-510 (tecadenoson) has chemical structure (8 :


EXAMPLE 1
The compounds of this invention may be prepared by conventional methods of organic chemistry. The reaction sequence outlined below, is a general method, useful for the preparation of compounds of this invention.


According to this method, oxacycloalkyl carboxylic acid is heated in a mixture of dioxane, diphenylphosphoryazide and triethylamine for 1 hour. To this mixture is added benzyl alcohol and the reaction is further heated over night to give intermediate compound 1. Compound 1 is dissolved in methanol. Next, concentrated HC1, Pd/C is added and the mixture is placed under hydrogen at 1 atm. The mixture is stirred overnight at room temperature and filtered. The residue is recrystallized to give intermediate compound 2. 6-chloropurine riboside is combined and the mixture is compound 2 dissolved in methanol and treated with triethylamine. The reaction is heated to 80° C for 30 hours. Isolation and purification leads to Compound 3.
EXAMPLE 2
Compounds of this invention prepared according to the method of Example 1 were tested in two functional models specific for adenosine A, receptor agonist function. The first was the A , receptor mediated inhibition of isoproterenol stimulated cAMP accumulation in DDT cells. The EC50 of each derivative is shown in Table I. Also shown in Table I is the ability of each derivative to stimulate cAMP production in PC 12 cells, a function of agonist stimulation of adenosine A2 receptors. The ratio of the relative potency of each compound in stimulating either an A, receptor or an A2 receptor effect is termed the selectivity of each compound for the A, receptor. As can be seen in Table I, each derivative is relatively selective as an A, receptor agonist. The use of measuring cAMP metabolism as an assay for adenosine A , receptor function has been previously described (Scammells, P., Baker, S., Belardinelli, L., and Olsson, R. , 1994, Substituted 1 ,3-dipropylxanthines as irreversible antagonists of A, adenosine receptors. J. Med. Chem 37: 2794-2712, 1994).
Table I
Compound R EC50 (nM) ECS, (nM) A,/A2 A-/A, DDT cells PC 12 cells
I 4-arninopyran 12 970 0.012 80.0
II (±)-3-aminotetrahydrofuran 13 1400 0.0093 107.6
III (R)-3-aminotetrahydrofuran 1.08 448 0.0024 414
IV ( 1 )-caprolactam 161 181 0.889 1.12
V (S)-3-aminotetrahydrofuran 3.40 7680 0.00044 2258
Compounds were also tested in a whole organ model of A, receptor activation with respect to atrial and AV nodal function. In this model, guinea pig hearts are isolated and perfused with saline containing compound while atrial rate and AV nodal conduction time are assessed by electrographic measurement of atrial cycle length and AV intervals, as detailed in Belardinelli, L, Lu, J. Dennis, D. Martens, J, and Shryock J. (1994); The cardiac effects of a novel A,-adenosine receptor agonist in guinea pig isolated heart. J. Pharm. Exp. Therap. 271:1371-1382 (1994). As shown in Figure 1, each derivative was effective in slowing the atrial rate and prolonging the AV nodal conduction time of spontaneously beating hearts in a concentration-dependent manner, demonstrating efficacy as adenosine A, receptor agonists in the intact heart.
EXAMPLE 3
Preparation ofN-benzyloxycarbonyl-4-aminopyran.
A mixture of 4-pyranylcarboxylic acid (2.28 gm, 20 mmol), diphenylphosphorylazide (4.31 ml, 20 mmol), triethylamine (2.78 ml, 20 mmol) in dioxane (40 ml) was heated in a 100° C oil bath under dry nitrogen for 1 hour. Benzyl alcohol (2.7 ml, 26 mmol) was added, and heating was continued at 100° C for 22 hours. The mixture was cooled, filtered from a white precipitate and concentrated. The residue was dissolved in 2N HC1 and extracted twice with EtOAc. The extracts were washed with water, sodium bicarbonate, brine and then dried over MgSO4, and concentrated to an oil which solidified upon standing. The oil was chromatographed (30% to 60% EtO Ac/Hex) to give 1.85 g of a white solid (40%).
Preparation of 4-aminopyran.
N-benzyloxycarbonyl-4-aminopyran (1.85 gm, 7.87 mmol) was dissolved in MeOH (50 ml) along with cone. HC1 and Pd-C ( 10%, 300 mg). The vessel was charged with hydrogen at 1 atm and the mixture was allowed to stir for 18 hours at room temperature. The mixture was filtered through a pad of eelite and concentrated. The residue was co-evaporated twice with MeOH/EtOAc and recrystallized from MeOH/EtOAc to afford 980 mg (91 %) of white needles (mp 228-230° C).
Preparation of 6-(4-aminopyran)-purine riboside. A mixture of 6-chloropurine riboside (0.318 gm, 1. 1 mmol), 4-aminopyran-HCl
(0.220 mg,
1.6 mmol) and triethylamine (0.385 ml, 2.5 mmol) in methanol (10 ml) was heated to 80° C for 30 hours. The mixture was cooled, concentrated and the residue chromatographed (90: 10: 1, CH2 Cl2/MeOH/PrNH2). The appropriate fractions were collected and recliromatographed using a chromatotron
(2 mm plate, 90: 10: 1, CH2 Cl2/MeOH/PrNH2) to give an off white foam (0.37 gm, 95%).
EXAMPLE 4
Preparation of N-benzyloxycarbonyl-3-aminotetrahydrofuran. A mixture of 3-tetrahydrofuroic acid (3.5 gm, 30 mmol), diphenylphosphorylazide (6.82 ml, 32 mmol), triethylamine (5 ml, 36 mmol) in dioxane (35 ml) was stirred at RT for 20 min then heated in a 100° C oil bath under dry nitrogen for 2 hours. Benzyl alcohol (4.7 ml, 45 mmol) was added, and continued heating at 100° C for 22 hours. The mixture was cooled, filtered from a white precipitate and concentrated. The residue was dissolved in 2N HC1 and extracted twice using EtOAc. The extracts were washed with water, sodium bicarbonate, brine dried over MgSO4, and then concentrated to an oil which solidifies upon standing. The oil was chromatographed (30% to 60% EtO Ac/Hex) to give 3.4 g of an oil (51
%).
Preparation of 3-aminotetrahydrofuran.
N-benzyloxycarbonyl-3-aminotetrahydrofuran (3.4 gm, 15 mmol) was dissolved in MeOH (50 ml) along with cone. HC1 and Pd-C (10%, 300 mg). The vessel was charged with hydrogen at 1 atm and the mixture was allowed to stir for 18 hours at room temperature. The mixture was filtered through a pad of celite and concentrated. The residue was co-evaporated two times with MeOH/EtOAc and recrystallized from MeOH/EtOAc to give 1.9 g of a yellow solid.
Preparation of 6-(3-aminotetrahydrofuranyl)purine riboside. A mixture of 6-chloropurine riboside (0.5 gm, 1.74 mmol), 3-aminotetrahydrofuran
(0.325 gm, 2.6 mmol) and triethylamine (0.73 ml, 5.22 mmol) in methanol (10 ml) was heated to 80° C for 40 hours. The mixture was cooled, and concentrated. The residue was filtered through a short column of silica gel eluting with 90/10/1 (CH2Cl2/MeOH/PrNH2), the fractions containing the product were combined and concentrated. The residue was chromatorgraphed on the chromatotron (2 mm plate, 92.5/7.5/1 , CH2CL2/MeOH/P.NH2). The resulting white solid was recrystallized from MeOH/EtOAc to give 0.27 gm of white crystals (mp 128-130° C).
EXAMPLE 5
Resolution of 3-arninotetrahydrofuran hydrochloride
A mixture of 3-aminotetrahydrofuran hydrochloride (0.5 gm, 4 mmol) and
(S)-(+)-10-camphorsulfonyl chloride (1.1 gm, 4.4 mmol) in pyridine (10 ml) was stirred for 4 hours at room temperature and then concentrated. The residue was dissolved in EtOAc and washed with 0.5N HC1, sodium bicarbonate and brine. The organic layer was dried over MgSO4, filtered and concentrated to give 1. 17 g of a brown oil (97%) which was chromatographed on silica gel (25% to 70% EtOAc/Hex). The white solid obtained was repeatedly recrystallized from acetone and the crystals and supernatant pooled until an enhancement of greater than 90% by 1H NMR was acheived.
Preparation of 3-(S)-aminotetrahydrofuran hydrochloride.
The sulfonamide (170 mg, 0.56 mmol) was dissolved in cone. HCl/AcOH (2 mL each), stirred for 20 hours at room temperature, washed three times with CH2C12 (10 ml) and concentrated to dryness to give 75 mg (qaunt ) of a white solid
Preparation of 6-(3-(S)-aminotetrahydrofuranyl)puπne riboside.
A mixture of 6-chloropurιne riboside (30 mg, 0.10 mmol),
3-(S)-amιnotetrahydrofuran hydrochloride (19 mg, 0.15 mmol) and triethylamine (45 ml, 0.32 mmol) in methanol
(0.5 ml) was heated to 80° C for 18 hours. The mixture was cooled, concentrated and chromatographed with 95/5 (CH2Cl /MeOH) to give 8 mg (24%) of a white solid.
| US7144871 * | 19 Feb 2003 | 5 Dec 2006 | Cv Therapeutics, Inc. | Partial and full agonists of A1 adenosine receptors |
| US7696181 * | 24 Aug 2006 | 13 Apr 2010 | Cv Therapeutics, Inc. | Partial and full agonists of A1 adenosine receptors |
EMA grants orphan drug designations to Alnylam’s ALN-AT3 for haemophilia treatment

EMA grants orphan drug designations to Alnylam’s ALN-AT3 for haemophilia treatment
Biopharmaceutical company Alnylam Pharmaceuticals has received orphan drug designations for ALN-AT3 from the European Medicines Agency (EMA) Committee to treat haemophilia A and B
SEE

May 13,2014
Alnylam Pharmaceuticals, Inc., a leading RNAi therapeutics company, announced today positive top-line results from its ongoing Phase 1 trial of ALN-AT3, a subcutaneously administered RNAi therapeutic targeting antithrombin (AT) in development for the treatment of hemophilia and rare bleeding disorders (RBD). These top-line results are being presented at the World Federation of Hemophilia (WFH) 2014 World Congress being held May 11 – 15, 2014 in Melbourne, Australia. In Part A of the Phase 1 study, human volunteer subjects received a single subcutaneous dose of ALN-AT3 and, per protocol, the maximum allowable level of AT knockdown was set at 40%. Initial results show that a single, low subcutaneous dose of ALN-AT3 at 0.03 mg/kg resulted in an up to 28-32% knockdown of AT at nadir that was statistically significant relative to placebo (p < 0.01 by ANOVA). This led to a statistically significant (p < 0.01) increase in peak thrombin generation, that was temporally associated and consistent with the degree of AT knockdown. ALN-AT3 was found to be well tolerated with no significant adverse events reported. With these data, the company has transitioned to the Multiple Ascending Dose (MAD) Part B of the study in moderate-to-severe hemophilia subjects. Consistent with previous guidance, the company plans to present initial clinical results from the Phase 1 study, including results in hemophilia subjects, by the end of the year. These human study results are the first to be reported for Alnylam’s Enhanced Stabilization Chemistry (ESC)-GalNAc conjugate technology, which enables subcutaneous dosing with increased potency, durability, and a wide therapeutic index. Further, these initial clinical results demonstrate a greater than 50-fold potency improvement with ESC-GalNAc conjugates relative to standard template chemistry conjugates.
“We are excited by these initial positive results for ALN-AT3 in the human volunteer ‘Part A’ of our Phase 1 study. Indeed, within the protocol-defined boundaries of single doses that provide no more than a 40% knockdown of AT in normal subjects, we were able to demonstrate a statistically-significant knockdown of AT of up to 28-32% and an associated increase in thrombin generation. Remarkably, this result was achieved at the lowest dose tested of 0.03 mg/kg, demonstrating a high and better than expected level of potency for ALN-AT3, our first ESC-GalNAc conjugate to enter clinical development,” said Akshay Vaishnaw, M.D., Ph.D., Executive Vice President and Chief Medical Officer of Alnylam. “With these results in hand, we are now proceeding to ‘Part B’ of the study, where we will administer multiple ascending doses to up to 18 patients with moderate-to-severe hemophilia A or B. Patients will receive three weekly doses, and we fully expect to achieve robust levels of AT knockdown as we dose escalate. In addition, we will aim to evaluate a once-monthly dosing regimen in future clinical studies, as we believe this could provide a highly attractive prophylactic regimen for patients. We look forward to sharing our detailed Phase 1 results, including data in hemophilia subjects, later this year, consistent with our original guidance.”
“There are several notable implications of these exciting initial results with ALN-AT3. First, ALN-AT3 now becomes the fourth program in our ‘Alnylam 5×15’ pipeline to demonstrate clinical activity. As such, these results increase our confidence level yet further across the entirety of our pipeline efforts, where we remain focused on genetically defined, liver-expressed disease targets with a modular and reproducible delivery platform. Moreover, these results with ALN-AT3 establish human proof of concept for our ESC-GalNAc conjugate technology, extending and broadening the human results we have previously shown with ALN-TTRsc which employs our standard template chemistry. Our ESC-GalNAc conjugate technology enables subcutaneous dosing with increased potency and durability and a wide therapeutic index, and has now become our primary approach for the delivery of RNAi therapeutics,” said John Maraganore, Ph.D., Chief Executive Officer of Alnylam. “Finally, the achievement of target knockdown at such a low dose of 0.03 mg/kg is unprecedented. Based on our evaluation of datasets from non-human primate (NHP) and human studies, these results demonstrate a 10-fold improved potency for ALN-AT3 as compared with NHP and a 50-fold improved potency in humans as compared with ALN-TTRsc. Based on data we announced earlier this week at TIDES, we believe that this increased potency is the combined result of enhanced stability for ESC-GalNAc conjugates and an attenuated nuclease environment in human tissue compared with other species. If these results extend to other ESC-GalNAc-siRNA conjugates, such as those in our complement C5 and PCSK9 programs, we believe we can expect highly potent clinical activities with very durable target knockdown effects.”
The ongoing Phase 1 trial of ALN-AT3 is being conducted in the U.K. as a single- and multi-dose, dose-escalation study comprised of two parts. Part A – which has now been completed – was a randomized, single-blind, placebo-controlled, single-dose, dose-escalation study, intended to enroll up to 24 healthy volunteer subjects. The primary objective of this part of the study was to evaluate the safety and tolerability of a single dose of ALN-AT3, with the potential secondarily to show changes in AT plasma levels at sub-pharmacologic doses. This part of the study evaluated only low doses of ALN-AT3, with a dose-escalation stopping rule at no more than a 40% level of AT knockdown. Based on the pharmacologic response achieved in this part of the study, only the lowest dose cohort (n=4; 3:1 randomization of ALN-AT3:placebo) was enrolled. Part B of the study is an open-label, multi-dose, dose-escalation study enrolling up to 18 people with moderate-to-severe hemophilia A or B. The primary objective of this part of the study is to evaluate the safety and tolerability of multiple doses, specifically three doses, of subcutaneously administered ALN-AT3 in hemophilia subjects. Secondary objectives include assessment of clinical activity as determined by knockdown of circulating AT levels and increase in thrombin generation at pharmacologic doses of ALN-AT3; thrombin generation is known to be a biomarker for bleeding frequency and severity in people with hemophilia (Dargaud, et al., Thromb Haemost; 93, 475-480 (2005)). In this part of the study, dose-escalation will be allowed to proceed beyond the 40% AT knockdown level.
In addition to reporting positive top-line results from the Phase 1 trial with ALN-AT3, Alnylam presented new pre-clinical data with ALN-AT3. First, in a saphenous vein bleeding model performed in hemophilia A (HA) mice, a single subcutaneous dose of ALN-AT3 that resulted in an approximately 70% AT knockdown led to a statistically significant (p < 0.0001) improvement in hemostasis compared to saline-treated HA mice. The improved hemostasis was comparable to that observed in HA mice receiving recombinant factor VIII. These are the first results in what can be considered a genuine bleeding model showing that AT knockdown with ALN-AT3 can control bleeding. Second, a number of in vitro studies were performed in plasma from hemophilia donors. Stepwise AT depletion in these plasma samples was shown to achieve stepwise increases in thrombin generation. Furthermore, it was shown that a 40-60% reduction of AT resulted in peak thrombin levels equivalent to those achieved with 10-15% levels of factor VIII in HA plasma and factor IX in hemophilia B (HB) plasma. These levels of factor VIII or IX are known to significantly reduce bleeding in hemophilia subjects. As such, these results support the hypothesis that a 40-60% knockdown of AT with ALN-AT3 could be fully prophylactic. Finally, a modified Activated Partial Thromboplastin Time (APTT) assay – an ex vivomeasure of blood coagulation that is significantly prolonged in hemophilia – was developed, demonstrating sensitivity to AT levels. Specifically, depletion of AT in HA plasma led to a shortening of modified APTT. This modified APTT assay can be used to routinely and simply monitor functional activity of AT knockdown in further ALN-AT3 clinical studies.
“The unmet need for new therapeutic options to treat hemophilia patients remains very high, particularly in those patients who experience multiple annual bleeds such as patients receiving replacement factor ‘on demand’ or patients who have developed inhibitory antibodies. Indeed, I believe the availability of a safe and effective subcutaneously administered therapeutic with a long duration of action would represent a marked improvement over currently available approaches for prophylaxis,” said Claude Negrier, M.D., head of the Hematology Department and director of the Haemophilia Comprehensive Care Centre at Edouard Herriot University Hospital in Lyon. “I continue to be encouraged by Alnylam’s progress to date with ALN-AT3, including these initial data reported from the Phase 1 trial showing statistically significant knockdown of antithrombin and increased thrombin generation, which has been shown to correlate with bleeding frequency and severity in hemophilia. I look forward to the advancement of this innovative therapeutic candidate in hemophilia subjects.”
About Hemophilia and Rare Bleeding Disorders
Hemophilias are hereditary disorders caused by genetic deficiencies of various blood clotting factors, resulting in recurrent bleeds into joints, muscles, and other major internal organs. Hemophilia A is defined by loss-of-function mutations in Factor VIII, and there are greater than 40,000 registered patients in the U.S. and E.U. Hemophilia B, defined by loss-of-function mutations in Factor IX, affects greater than 9,500 registered patients in the U.S. and E.U. Other Rare Bleeding Disorders (RBD) are defined by congenital deficiencies of other blood coagulation factors, including Factors II, V, VII, X, and XI, and there are about 1,000 patients worldwide with a severe bleeding phenotype. Standard treatment for hemophilia patients involves replacement of the missing clotting factor either as prophylaxis or on-demand therapy. However, as many as one third of people with severe hemophilia A will develop an antibody to their replacement factor – a very serious complication; these ‘inhibitor’ patients become refractory to standard replacement therapy. There exists a small subset of hemophilia patients who have co-inherited a prothrombotic mutation, such as Factor V Leiden, antithrombin deficiency, protein C deficiency, and prothrombin G20210A. Hemophilia patients that have co-inherited these prothrombotic mutations are characterized as having a later onset of disease, lower risk of bleeding, and reduced requirements for Factor VIII or Factor IX treatment as part of their disease management. There exists a significant need for novel therapeutics to treat hemophilia patients.
About Antithrombin (AT)
Antithrombin (AT, also known as “antithrombin III” and “SERPINC1″) is a liver expressed plasma protein and member of the “serpin” family of proteins that acts as an important endogenous anticoagulant by inactivating Factor Xa and thrombin. AT plays a key role in normal hemostasis, which has evolved to balance the need to control blood loss through clotting with the need to prevent pathologic thrombosis through anticoagulation. In hemophilia, the loss of certain procoagulant factors (Factor VIII and Factor IX, in the case of hemophilia A and B, respectively) results in an imbalance of the hemostatic system toward a bleeding phenotype. In contrast, in thrombophilia (e.g., Factor V Leiden, protein C deficiency, antithrombin deficiency, amongst others), certain mutations result in an imbalance in the hemostatic system toward a thrombotic phenotype. Since co-inheritance of prothrombotic mutations may ameliorate the clinical phenotype in hemophilia, inhibition of AT defines a novel strategy for improving hemostasis.
About GalNAc Conjugates and Enhanced Stabilization Chemistry (ESC)-GalNAc Conjugates
GalNAc-siRNA conjugates are a proprietary Alnylam delivery platform and are designed to achieve targeted delivery of RNAi therapeutics to hepatocytes through uptake by the asialoglycoprotein receptor. Alnylam’s Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate technology enables subcutaneous dosing with increased potency and durability, and a wide therapeutic index. This delivery platform is being employed in several of Alnylam’s genetic medicine programs, including programs in clinical development.
About RNAi
RNAi (RNA interference) is a revolution in biology, representing a breakthrough in understanding how genes are turned on and off in cells, and a completely new approach to drug discovery and development. Its discovery has been heralded as “a major scientific breakthrough that happens once every decade or so,” and represents one of the most promising and rapidly advancing frontiers in biology and drug discovery today which was awarded the 2006 Nobel Prize for Physiology or Medicine. RNAi is a natural process of gene silencing that occurs in organisms ranging from plants to mammals. By harnessing the natural biological process of RNAi occurring in our cells, the creation of a major new class of medicines, known as RNAi therapeutics, is on the horizon. Small interfering RNA (siRNA), the molecules that mediate RNAi and comprise Alnylam’s RNAi therapeutic platform, target the cause of diseases by potently silencing specific mRNAs, thereby preventing disease-causing proteins from being made. RNAi therapeutics have the potential to treat disease and help patients in a fundamentally new way.
About Alnylam Pharmaceuticals
Alnylam is a biopharmaceutical company developing novel therapeutics based on RNA interference, or RNAi. The company is leading the translation of RNAi as a new class of innovative medicines with a core focus on RNAi therapeutics as genetic medicines, including programs as part of the company’s “Alnylam 5x15TM” product strategy. Alnylam’s genetic medicine programs are RNAi therapeutics directed toward genetically defined targets for the treatment of serious, life-threatening diseases with limited treatment options for patients and their caregivers. These include: patisiran (ALN-TTR02), an intravenously delivered RNAi therapeutic targeting transthyretin (TTR) for the treatment of TTR-mediated amyloidosis (ATTR) in patients with familial amyloidotic polyneuropathy (FAP); ALN-TTRsc, a subcutaneously delivered RNAi therapeutic targeting TTR for the treatment of ATTR in patients with TTR cardiac amyloidosis, including familial amyloidotic cardiomyopathy (FAC) and senile systemic amyloidosis (SSA); ALN-AT3, an RNAi therapeutic targeting antithrombin (AT) for the treatment of hemophilia and rare bleeding disorders (RBD); ALN-CC5, an RNAi therapeutic targeting complement component C5 for the treatment of complement-mediated diseases; ALN-AS1, an RNAi therapeutic targeting aminolevulinate synthase-1 (ALAS-1) for the treatment of hepatic porphyrias including acute intermittent porphyria (AIP); ALN-PCS, an RNAi therapeutic targeting PCSK9 for the treatment of hypercholesterolemia; ALN-AAT, an RNAi therapeutic targeting alpha-1 antitrypsin (AAT) for the treatment of AAT deficiency-associated liver disease; ALN-TMP, an RNAi therapeutic targeting TMPRSS6 for the treatment of beta-thalassemia and iron-overload disorders; ALN-ANG, an RNAi therapeutic targeting angiopoietin-like 3 (ANGPTL3) for the treatment of genetic forms of mixed hyperlipidemia and severe hypertriglyceridemia; ALN-AC3, an RNAi therapeutic targeting apolipoprotein C-III (apoCIII) for the treatment of hypertriglyceridemia; and other programs yet to be disclosed. As part of its “Alnylam 5×15” strategy, as updated in early 2014, the company expects to have six to seven genetic medicine product candidates in clinical development – including at least two programs in Phase 3 and five to six programs with human proof of concept – by the end of 2015. Alnylam is also developing ALN-HBV, an RNAi therapeutic targeting the hepatitis B virus (HBV) genome for the treatment of HBV infection. The company’s demonstrated commitment to RNAi therapeutics has enabled it to form major alliances with leading companies including Merck, Medtronic, Novartis, Biogen Idec, Roche, Takeda, Kyowa Hakko Kirin, Cubist, GlaxoSmithKline, Ascletis, Monsanto, The Medicines Company, and Genzyme, a Sanofi company. In March 2014, Alnylam acquired Sirna Therapeutics, a wholly owned subsidiary of Merck. In addition, Alnylam holds an equity position in Regulus Therapeutics Inc., a company focused on discovery, development, and commercialization of microRNA therapeutics. Alnylam scientists and collaborators have published their research on RNAi therapeutics in over 200 peer-reviewed papers, including many in the world’s top scientific journals such as Nature, Nature Medicine, Nature Biotechnology, Cell, the New England Journal of Medicine, and The Lancet. Founded in 2002, Alnylam maintains headquarters in Cambridge, Massachusetts. For more information, please visit www.alnylam.com.
Pharmacyclics Announces Third Breakthrough Therapy Designation for Ibrutinib from the U.S. Food and Drug Administration

IBRUTINIB
1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]piperidin-1-yl]prop-2-en-1-one
SUNNYVALE, Calif., April 8, 2013
Pharmacyclics, Inc. announced today that the U.S. Food and Drug Administration (FDA) has granted an additional Breakthrough Therapy Designation for the investigational oral agent ibrutinib as monotherapy for the treatment of chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) patients with deletion of the short arm of chromosome 17 (deletion 17p). Patients harboring a deletion within chromosome 17 generally have poor response to chemoimmunotherapy and have limited treatment options. The presence of deletion 17p is one of the worst prognostic factors in patients with CLL.
In February 2013, FDA granted Breakthrough Therapy Designations for ibrutinib as a monotherapy for the treatment of patients with relapsed or refractory mantle cell lymphoma (MCL) and as a monotherapy for the treatment of patients with Waldenstrom’s macroglobulinemia (WM), both of which are also B-cell malignancies. Ibrutinib is jointly being developed by Pharmacyclics and Janssen for treatment of B-cell malignancies.
The Breakthrough Therapy Designation is intended to expedite the development and review of a potential new drug for serious or life-threatening diseases where “preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.” The designation of a drug as a Breakthrough Therapy was enacted as part of the 2012 Food and Drug Administration Safety and Innovation Act. Pharmacyclics, together with Janssen, is working with the FDA to determine the implications of this Breakthrough Therapy Designation to the ongoing and planned development and the FDA filing requirements for the use of ibrutinib in CLL patients with deletion 17p.
The FDA Breakthrough Therapy Designation for ibrutinib in CLL patients with deletion 17p was based on data from pre-clinical and clinical studies where ibrutinib as a monotherapy was used to treat patients with this disease. Ibrutinib has the potential to improve the outcome in this serious and life-threatening disease which has a poor prognosis. In addition, Pharmacyclics and Janssen have recently initiated a Phase II study of ibrutinib in patients with CLL deletion 17p, RESONATE™ -17, which is a single-arm, open-label, multi-center trial using ibrutinib as a monotherapy in patients who have deletion 17p and who did not respond to or relapsed after at least one prior CLL treatment (a high unmet need population). The primary endpoint of the study will be overall response rate. This global study opened this year and Pharmacyclics plans to enroll 111 patients worldwide.
About Chronic Lymphocytic Leukemia
Chronic Lymphocytic Leukemia (CLL) is a slow-growing blood cancer that starts in the white blood cells (lymphocytes), most commonly from B-cells. CLL is the second most common adult leukemia. Approximately 16,000 patients in the US are diagnosed each year with CLL. The prevalence of CLL is approximately 113,000 in the US. The disease is a chronic disease of the elderly with an average survival of about 5 years. Patients commonly receive multiple lines of treatment over the course of their disease.
In CLL the genetic mutation 17p deletion occurs when the short arm of chromosome 17 is missing. Del 17p is associated with abnormalities of a key tumor suppressor gene, TP53, which results in poor response to chemoimmunotherapy and worse treatment outcomes. It occurs in about 7% of treatment naive CLL patients and is estimated to be approximately 20% to 40% of relapsed or refractory patients harboring the mutation.
About Ibrutinib
Ibrutinib , previously publicly known as PCI-32765, is an experimental drug candidate for the treatment of various types of cancer. It was first synthesized at Celera Genomics as a selective inhibitor of Bruton’s tyrosine kinase (Btk).It was later discovered to have anti-lymphoma properties in vivo by scientists at Pharmacyclics, Inc.Ibrutinib is currently under development by Pharmacyclics, Inc and Johnson & Johnson‘sJanssen Pharmaceutical division for chronic lymphocytic leukemia, mantle cell lymphoma,diffuse large B-cell lymphoma, and multiple myeloma. It also has potential effects against autoimmune arthritis.
Janssen Biotech, Inc. and Pharmacyclics entered a collaboration and license agreement in December 2011 to co-develop and co-commercialize ibrutinib. Ibrutinib was designed to specifically target and selectively inhibit an enzyme called Bruton’s tyrosine kinase (BTK). BTK is a key mediator of at least three critical B-cell pro-survival mechanisms occurring in parallel – regulation of apoptosis, adhesion, and cell migration and homing. Through these multiple signals, BTK regulation helps to direct malignant B-cells to lymphoid tissues, thus allowing access to a micro environment necessary for survival.
The effectiveness of ibrutinib alone or in combination with other treatments is being studied in several B-cell malignancies, including chronic lymphocytic leukemia/small lymphocytic lymphoma, mantle cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, Waldenstrom’s macroglobulinemia and multiple myeloma. To date five Phase III trials have been initiated with ibrutinib and a total of 26 trials are currently registered on www.clinicaltrials.gov.
About Pharmacyclics
Pharmacyclics® is a clinical-stage biopharmaceutical company focused on developing and commercializing innovative small-molecule drugs for the treatment of cancer and immune mediated diseases. Our mission and goal is to build a viable biopharmaceutical company that designs, develops and commercializes novel therapies intended to improve quality of life, increase duration of life and resolve serious unmet medical healthcare needs; and to identify promising product candidates based on scientific development and administrational expertise, develop our products in a rapid, cost-efficient manner and pursue commercialization and/or development partners when and where appropriate.
Presently, Pharmacyclics has three product candidates in clinical development and several preclinical molecules in lead optimization. The Company is committed to high standards of ethics, scientific rigor, and operational efficiency as it moves each of these programs to viable commercialization.
The Company is headquartered in Sunnyvale, California and is listed on NASDAQ under the symbol PCYC. To learn more about how Pharmacyclics advances science to improve human healthcare visit at http://www.pharmacyclics.com.
Lesinurad
Lesinurad
Acetic acid, 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]-,
sodium salt (1:1)
Sodium 2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-
yl]sulfanyl}acetate
2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid
MOLECULAR FORMULA C17H13BrN3NaO2S
MOLECULAR WEIGHT 426.3
http://clinicaltrials.gov/show/NCT01508702
http://www.ama-assn.org/resources/doc/usan/lesinurad.pdf
Ardea Biosciences, Inc.
- Lesinurad
- RDEA 594
- RDEA594
- UNII-09ERP08I3W
Gout phase 3
Gout is associated with elevated levels of uric acid that crystallize and deposit in joints, tendons, and surrounding tissues. Gout is marked by recurrent attacks of red, tender, hot, and/or swollen joints.
This study will assess the serum uric acid lowering effects and safety of lesinurad compared to placebo in patients who are intolerant or have a contraindication to allopurinol or febuxostat.
http://euroscan.org.uk/technologies/technology/view/2386
Lesinurad (RDEA-594, lesinurad sodium) is a selective urate transporter-1 (URAT-1) inhibitor, which blocks the reabsorption of urate within the renal proximal tubule. It is intended for the treatment of gout after failure of first line therapy and is administered orally at 400mg once daily
A Phase 3 Randomized, Double-Blind, Multicenter, Placebo- Controlled Study to Assess the Efficacy and Safety of Lesinurad Monotherapy Compared to Placebo in Subjects With Gout and an Intolerance or Contraindication to a Xanthine Oxidase Inhibitor
AstraZeneca’s lesinurad (formerly known as RDEA-594) is a selective oral Uric Acid Transporter URAT1 inhibitor currently in Phase III development for the treatment of of gout. The regulatory filings for lesinurad in the US and Europe are expected for the first half of 2014.

Gout (also known as podagra when it involves the big toe), while not life-threatening, is an excruciatingly painful condition caused by a buildup of a waste product in the blood called uric acid, which is normally eliminated from the body through urine. Excess Uric acid crystallizes and get deposited in the joints (usually the big toes), creating symptoms similar to an acute arthritis flare. Gout has seen a recent gradual resurgence as a result of rising obesity rates and poor diet according to a study in the journal Annals of the Rheumatic Diseases.
The current Standard treatment for gout works by inhibiting a protein called xanthine oxidase that helps in the formation of the uric acid. These therapies, some of which have been used for more than 50 years, are not effective in all patients. One is a generic drug called allopurinol that was approved in the U.S. in 1966. The other is febuxostat, marketed by Takeda Pharmaceutical Co. in the U.S. asUloric and by Ipsen SA and others in Europe as Adenuric and approved in the U.S. in 2009.
AstraZeneca’s new product Lesinurad, a selective uric acid re-absorption inhibitor (SURI), tackles gout by blocking a protein called Uric acid trasporter 1 (URAT1) that otherwise would cause the body to reabsorb the uric acid. AstraZeneca acquired lesinurad (aka RDEA-594) as part of its $1.26 billion takeouver of San Diego-based Ardea Biosciences in 2012. RDEA594 is a metabolite of RDEA806, a non-nucleoside reverse transcriptase inhibitor originally developed for HIV.

In top-line results from a Phase III LIGHT study released by AstraZeneca in December 2013 on gout patients who get no benefit from Zyloprim (allopurinol) and febuxostat, lesinurad alone significantly reduced serum levels of uric acid. The company has three other phase III studies ongoing that are testing the use of the drug alongside allopurinol and febuxostat, and these should generate results in the middle of 2014. Analysts at JPMorgan Chase forecast lesinurad alone may have peak sales of $1 billion a year. AstraZeneca also has a second, more potent drug called RDEA3179 to treat elevated levels of uric acid or hyperuricemia. Pfizer’s KUX-1151, licensed from Japan’s Kissei Phmarceuticals, is in early stage development.
Gout is not an automatic success indication of drugmakers. Savient Pharmaceuticals filed for Chapter 11 bankruptcy in October 2013 in the face of a severe cash crisis, having spent hundreds of millions of dollars on its would-be flagship — the gout-fighting drug Krystexxa (pegloticase) — with limited results. Krystexxa (pegloticase), a twice-monthly infusion designed to treat severe chronic gout that doesn’t respond to conventional therapy, was approved by the U.S. Food and Drug Administration in September 2010. Crealta Pharmaceuticals acquired Savient for $120.4 million in December 2013.
Lesinurad
RDEA-594
2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]sulfanyl}acetic acid
CAS number: 878672-00-5 (Lesinurad), 1151516-14-1 (Lesinurad sodium)
Mechanism of Action:once-daily inhibitor of URAT1, a transporter in the kidney that regulates uric acid excretion from the body
US patents:US8242154 , US8173690, US808448
Indication: Gout
Developmental Status: Phase III (US, UK, EU)
Originator: Ardea Biosciences (Acquired by AstraZeneca for $1.26 billion in 2012)
Developer: AstraZeneca
…………………………
http://www.google.co.in/patents/US8242154
Example 8 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)-N-(2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume, to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 mins and the organic layer removed. The aqueous layer was cooled to 0° C. and acidified by treatment with HCl (1N) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3×) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
Example 102 Methyl 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate


Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0° C., and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%).

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).

A solution of 1-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropylnaphthalene (3.1 g, 73%).

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of 1-amino-4-cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) at 0° C. The reaction mixture was stirred for 5 min at 0° C. and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanatonaphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50° C. for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.0 g, 49%).

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 mins to a suspension of 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
Example 104 Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate

Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 mins to a solution of 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10° C. The mixture was stirred at 10° C. for a further 10 mins. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 103 2-(5-Bromo-4-(1-cyclopropylnapthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 mins to a solution of methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (prepared as described in example 1 above; 1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0° C. The mixture was stirred at 0° C. for a further 45 mins and then neutralized to pH 7 by the addition of 0.5N HCl solution at 0° C. The resulting mixture was concentrated in vacuo to ⅕th of its original volume, then diluted with water (˜20 mL) and acidified to pH 2-3 by the addition of 0.5N HCl to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with DCM is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (1.02 g, 93%).
 |
1H NMR (400 MHz, DMSO-d6) δ ppm 0.84-0.91 (m, 2 H) 1.12-1.19 (m, 2 H) 2.54-2.61 (m, 1 H) 3.99 (d, J = 1.45 Hz, 2 H) 7.16 (d, J = 7.88 Hz, 1 H) 7.44 (d, J = 7.46 Hz, 1 H) 7.59-7.70 (m, 2 H) 7.75 (td, J = 7.62, 1.14 Hz, 1 H) 8.59 (d, J = 8.50 Hz, 1 H) 12.94 (br. s., 1 H) | Mass found: 404.5 (M + 1) | B |
……
POLYMORPHS AND SYNTHESIS
Described herein are various polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate which decreases uric acid levels, (see for example US patent publication 2009/0197825, US patent publication 2010/0056464 and US patent publication 2010/0056465). Details of clinical studies involving sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate have been described in International patent application
PCT/US2010/052958.
Polymorph Form A
In one embodiment, sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H- l,2,4-triazol-3-ylthio)acetate polymorph Form A exhibits an x-ray powder diffraction pattern characterized by the diffraction pattern summarized in Table 1 A or Table IB. In some embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4- cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 3 peaks of (±0.1°2Θ) of Table 1A or IB. In certain embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 4 peaks of (±0.1°2Θ) of Table 1A or IB, at least 5 peaks of (±0.1°2Θ) of Table 1A or IB, at least 6 peaks of (±0.1°2Θ) of Table 1A or IB, at least 8 peaks of
(±0. Γ2Θ) of Table 1A or IB, at least 10 peaks of (±0. Γ2Θ) of Table 1A, at least 15 peaks of (±0. Γ2Θ) of Table 1A, at least 20 peaks of (±0. Γ2Θ) of Table 1A, at least 25 peaks of (±0.1 °2Θ) of Table 1A, or at least 30 peaks of (±0.1 °2Θ) of Table 1A.


Examples
I Preparation of compounds
Example 1: Preparation of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate
Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetate was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.

[00103] Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 min to a solution of 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H- l,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10 °C. The mixture was stirred at 10 °C for a further 10 min. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 2: Preparation of 2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol- 3-ylthio)acetic acid
2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetic acid was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.
[00104] Route i:

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)-N- (2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures, see US 2009/0197825; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 min and the organic layer removed. The aqueous layer was cooled to 0 °C and acidified by treatment with HCl (IN) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3x) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5- bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
[00105] Route ii:

STEP A: 1-Cyclopropylnaphthalene

Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [l,3-bis(diphenylphosphino)propane] dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0 °C, and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%). ] STEP B: l-Cyclopropyl-4-nitronaphthalene

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0 °C. The reaction mixture was stirred at 0 °C for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).
[00108] STEP C: l-Amino-4-cyclopropylnaphthalene

A solution of l-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-amino-4-cyclopropylnaphthalene (3.1 g, 73%).
STEP D: l-Cyclopropyl-4-isothiocvanatonaphthalene

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of l-amino-4- cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in
dichloromethane (50 mL) at 0 °C. The reaction mixture was stirred for 5 min at 0 °C and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield l-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%>).
[00110] STEP E: 5-Amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol

A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), l-cyclopropyl-4- isothiocyanato naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50 °C for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50 °C for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol (2.0 g, 49%). [00111] STEP F: Methyl 2-(5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- -3 -ylthio)acetate

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 min to a suspension of 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room
temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the presence of P2O5 to yield methyl 2-(5- amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).
[00112] STEP G: Methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3 -ylthio)acetate

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room
temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
[00113] STEP H: 2-(5-Bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- )acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 min to a solution of methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3-ylthio)acetate (1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0 °C. The mixture was stirred at 0 °C for a further 45 min and then neutralized to pH 7 by the addition of 0.5N HC1 solution at 0 °C. The resulting mixture was concentrated in vacuo to l/5th of its original volume, then diluted with water (~20 mL) and acidified to pH 2-3 by the addition of 0.5N HC1 to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with dichloromethane is recommended.) The tan solid was
collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the
presence of P2O5 to yield 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- ylthio)acetic acid (1.02 g, 93%).
………………………….
EXAMPLES
The following experiments are provided only by way of example, and should not be understood as limiting the scope of the invention.
COMPOUNDS OF THE INVENTION 2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method A)

1-Cyclopropyl-naphthalene
Cyclopropylmagnesium bromide (150 mL, 0.5 M in tetrahydrofuran) was slowly added to a solution of 1-bromo-naphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloronickel(II) in tetrahydrofuran (10 mL) stirred at 0° C. The reaction mixture was stirred at room temperature for 16 hours and the solvent was evaporated under reduced pressure. EtOAc and ammonium chloride in water were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-naphthalene (6.4 g, 76%).
1-Cyclopropyl-4-nitro-naphthalene
Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropyl-naphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then was slowly poured into ice. Water was added, followed by EtOAc. After extraction, the organic layer was washed with a 1% aqueous solution of NaOH, then washed with water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitro-naphthalene (5.2 g, 64%).
1-Amino-4-cyclopropyl-naphthalene
A solution of 1-cyclopropyl-4-nitro-naphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, then filtered over celite. The solvent was evaporated, and the residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropyl-naphthalene (3.1 g, 73%).
1-Cyclopropyl-4-isothiocyanato-naphthalene
Thiophosgene (1.1 g, 9.7 mmol) was added to a solution of 1-amino-4-cyclopropyl-naphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) stirred at 0° C. The reaction mixture was stirred for 5 min at this temperature, then a 1% solution of HCl in water was added and the organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent was evaporated under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanato-naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was evaporated, toluene was added, and the solvent was evaporated again. A 2.0 M aqueous solution of sodium hydroxide (30 mL) was added and the reaction mixture was heated at 50° C. for 60 hours. The reaction mixture was filtered, and the filtrate was neutralized with a 2.0 M aqueous solution of HCl. New filtration, then evaporation of solvent and purification of the residue by silica gel chromatography to yield 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (2.0 g, 49%).
2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)Acetamide
In a solution of 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (708 mg, 2.5 mmol), K2CO3 (380 mg, 2.5 mmol) in DMF (20 mL) was added 2-chloro-N-(2-chloro-4-sulfamoylphenyl)acetamide (710 mg, 2.5 mmol). The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction, the solvent was evaporated. The residue was purified by silica gel chromatography to yield 2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (1.26 g, 95%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide
Dichloroacetic acid (180 uL, 2.2 mmol) was added to a suspension of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (0.59 g, 1.1 mmol), sodium nitrite (1.5 g, 22 mmol) and BTEABr (0.91 g, 3.3 mmol) in dibromomethane (30 mL). The reaction mixture was stirred at room temperature for 4 hours, then extracted with dichloromethane and sodium bicarbonate in water. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (224 mg, 31%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method B)

2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole | 2.24 | g | 282.36 | 7.9 |
| methyl chloroacetate | 0.73 | ml | 108.52 | 8.3 (1.05 eq) |
| potassium carbonate | 1.21 | g | 138.21 | 8.7 (1.1 eq) |
| dimethylformamide | 40 | ml | (5 mL/mmol) | |
Procedure:
To a suspension of thiotriazole and potassium carbonate in DMF was added methyl chloroacetate dropwise at room temperature for 5 min. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2.24 g (80%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | ||
| thiotriazole L10183-58 | 709 | mg | 354.43 | 2.0 | |
| bromoform | 10 | ml | (5 ml/mmol) | ||
| sodium nitrite | 2.76 | g | 69.00 | 40 | (20 eq) |
| benzyltriethylammonium | 1.63 | g | 272.24 | 6.0 | (3 eq) |
| bromide | |||||
| dichloroacetic acid | 0.33 | ml | 128.94 | 4.0 | (2 eq) |
Procedure:
To a solution of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester and benzyltriethylammonium chloride in bromoform was added sodium nitrite. To the mixture was added dichloroacetic acid and the reaction mixture was stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel that was packed with CH2Cl2. The column was first eluted with CH2Cl2 until all CHBr3 eluted, and was then eluted with acetone/CH2Cl2 (5:95) to give 713 mg (85%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole methyl ester | 1.14 | g | 418.31 | 2.7 |
| tetrahydrofuran | 10 | ml | (~3 ml/mmol) | |
| ethanol | 10 | ml | (~3 ml/mmol) | |
| water | 10 | ml | (~3 ml/mmol) | |
| lithium hydroxide | 98 | mg | 23.95 | 4.1 (1.5 eq) |
Procedure:
To a solution of 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester, in a mixture of THF and EtOH at 0° C., was added a solution of LiOH in H2O dropwise over 5 min. The reaction was complete after stirring at 0° C. for an additional 45 min. The reaction was neutralized to pH 7 by the addition of 0.5 N HCl solution at 0° C., and the resulting mixture was concentrated in vacuo to ⅕th of its original volume. The mixture was diluted with H2O (˜20 mL) and acidified to pH 2-3 by the addition of 0.5 N HCl to produce sticky solid. (If the product comes out as an oil during acidification, extraction with CH2Cl2 is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 1.02 g (93%) of the title compound.
REF:
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8242154 B2 ;Also published as US20100056465, US20130040907;Original Assignee: Ardea Biosciences, Inc
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8173690 B2;Also published as US20100056464;Original Assignee: Ardea Biosciences, Inc
Barry D. Quart, Jean-Luc Girardet, Esmir Gunic, Li-Tain Yeh;Compounds and compositions and methods of use;US patent number US8084483 B2; Also published as CA2706858A1, CA2706858C, CN101918377A, CN102643241A, CN103058944A, EP2217577A2, EP2217577A4, US8283369, US8357713, US8546437, US20090197825, US20110268801, US20110293719, US20120164222, US20140005136, WO2009070740A2, WO2009070740A3;Original Assignee:Ardea Biosciences, Inc.
Gunic, Esmir; Galvin, Gabriel;Manufacture of 2-[5-bromo-4-(cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio]acetic acid and related compounds;PCT Int. Appl., WO2014008295 A1
Zamansky, Irina et al;Process for preparation of polymorphic, crystalline, and mesophase forms of 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]acetic acid sodium salt; PCT Int. Appl., WO2011085009
Gunic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels; U.S. Pat. Appl. Publ., 20100056465
unic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels;U.S. Pat. Appl. Publ., 20100056464
Quart, Barry D. et al;Preparation of azole carboxylates as modulators of blood uric acid levels;PCT Int. Appl., 2009070740, 04 Jun 2009
Girardet, Jean-Luc et al;Preparation of S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase;PCT Int. Appl., WO2006026356
US20100056465 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
US20100056542 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
WO2009070740A2 * Nov 26, 2008 Jun 4, 2009 Ardea Biosciences Inc Novel compounds and compositions and methods of use
WO2011085009A2 * Jan 5, 2011 Jul 14, 2011 Ardea Biosciences, Inc. Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof
| WO2011159732A1 * | Jun 14, 2011 | Dec 22, 2011 | Ardea Biosciences,Inc. | Treatment of gout and hyperuricemia |
| WO2012092395A2 * | Dec 28, 2011 | Jul 5, 2012 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio) acetic acid and uses thereof |
| EP2560642A2 * | Mar 29, 2011 | Feb 27, 2013 | Ardea Biosciences, Inc. | Treatment of gout |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US20100056465 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20100056542 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| WO2011085009A2 * | Jan 5, 2011 | Jul 14, 2011 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof |
| US8283369 | 30 Jun 2011 | 9 Oct 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | 30 Jun 2011 | 22 Jan 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8546437 | 30 Jun 2011 | 1 Oct 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8633232 * | 4 May 2012 | 21 Jan 2014 | Ardea Biosciences, Inc. | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20130040907 * | 4 May 2012 | 14 Feb 2013 | Ardea Biosciences Inc. | Compounds, Compositions and Methods of Using Same for Modulating Uric Acid Levels |
| WO2006026356A2 * | Aug 25, 2005 | Mar 9, 2006 | La Rosa Martha De | S-TRIAZOLYL α-MERCAPTOACETANILDES AS INHIBITORS OF HIV REVERSE TRANSCRIPTASE |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| US20090197825 * | Nov 26, 2008 | Aug 6, 2009 | Ardea Biosciences, Inc. | Novel compounds and compositions and methods of use |
| US7947721 | Aug 25, 2005 | May 24, 2011 | Ardes Biosciences Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8084483 | Nov 26, 2008 | Dec 27, 2011 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8106205 | Feb 2, 2010 | Jan 31, 2012 | Ardea Biosciences, Inc. | N[S(4-aryl-triazol-3-yl)α-mercaptoacetyl]p-amino benzoic acids as HIV reverse transcriptase inhibitors |
| US8252828 | Jun 30, 2011 | Aug 28, 2012 | Ardea Biosciences, Inc. | S-triazolyl α-mercapto acetanilides as inhibitors of HIV reverse transcriptase |
| US8283369 | Jun 30, 2011 | Oct 9, 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | Jun 30, 2011 | Jan 22, 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8372807 | May 20, 2010 | Feb 12, 2013 | Ardea Biosciences, Inc. | Methods of modulating uric acid levels |
| US8481581 | Jul 18, 2012 | Jul 9, 2013 | Ardea Biosciences, Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8524754 | Jan 5, 2011 | Sep 3, 2013 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio) acetate, and uses thereof |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US8546437 | Jun 30, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |

An unexpected phenomenon concerning an otherwise common impurity of lesinurad has been observed in the context of synthetic process development. A new industrial process was designed as a chlorine-free process, but the critical chlorinated impurity 10 was surprisingly detected in the isolated product. Because of the structural similarity of the impurity and the product, no efficient separation of 10 by conventional methods (e.g., crystallization) was discovered. The formation of the impurity was explained by a chlorine impurity in a commercial brominating agent. This communication also describes control of the critical impurity.
Identification of an Unexpected Impurity in a New Improved Synthesis of Lesinurad

2-[[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]thio]acetic Acid (1)
1H NMR (DMSO-d6), δ (ppm): 0.87 (m, 2H); 1.15 (m, 2H); 2.56 (m, 1H); 4.00 (m, 2H); 7.16 (d, J = 8.5 Hz, 1H); 7.44 (d, J = 7.3, 1H); 7.64 (d, J = 7.3 Hz, 1H); 7.66 (t, J= 7.6 Hz, 1H); 7.74 (t, J = 7.6 Hz, 1H); 8.58 (d, J = 8.5 Hz, 1H); 12.99 (s, COOH). 13C NMR (DMSO-d6), δ (ppm): 7.3; 7.4; 12.9; 34.1; 121.8; 122.7; 125.2; 126.6; 126.8; 127.3; 128.1; 128.6; 131.4; 133.5; 143.2; 153.5; 169.0.
