
Home » Posts tagged 'fda' (Page 29)
Tag Archives: fda
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Celgene phase 3 – Oral Apremilast Achieves Statistical Significance for the Primary Endpoint of PASI-75 in the First Phase III Study in Patients with Psoriasis

APREMILAST, N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
mar02,2013
Celgene International Sàrl, a subsidiary of Celgene Corporation (NASDAQ: CELG) today presented the results from ESTEEM 1, the Company’s first phase III study in psoriasis, at the American Academy of Dermatology annual meeting in Miami, Florida.
“I see this as a prime candidate for future management of psoriasis that allows us to treat a range of patients, including more moderate cases earlier on”
The company previously announced statistical significance for the primary and major secondary endpoint of PASI-75 at Week 16 and the Static Physician Global Assessment for patients receiving apremilast in the ESTEEM 1&2 phase III studies. ESTEEM 1&2 are the phase III registrational randomized, placebo-controlled studies evaluating the Company’s oral small-molecule inhibitor of phosphodiesterase-4 (PDE4) in patients with moderate-to-severe chronic plaque psoriasis.
ESTEEM 1, presented today, evaluated efficacy and safety in a range of patients. Approximately one-third of the study population was systemic and/or phototherapy treatment-naïve. Nearly 30 percent of the overall study population had prior biologic therapy, which included biologic-failures.
In the ESTEEM 1 study, a significantly higher percentage of apremilast-treated patients demonstrated PASI-75 at week 16 than did placebo patients (33.1% vs. 5.3%; P<0.0001). Significantly higher PASI-75 scores at week 16 were demonstrated across all patient segments enrolled in this study, including systemic-naïve and biologic-naïve patients receiving apremilast 30 mg BID compared with placebo (38.7% vs. 7.6%; P<0.0001 and 35.8% vs. 5.9%; P<0.0001 respectively). Apremilast demonstrated maintenance of effect over time, as measured by the Mean Percent Change from Baseline in PASI score over 32 weeks, with apremilast demonstrating a 54.9% reduction at week 16 and a 61.9% reduction at week 32.
Statistical significance at week 16 was also demonstrated in the major secondary endpoint, Static Physician Global Assessment (sPGA) of clear or almost clear (P<0.0001), and other key secondary endpoints (change in BSA, Pruritus VAS, DLQI), as well as in assessments of difficult to treat areas (nail and scalp psoriasis).
“I see this as a prime candidate for future management of psoriasis that allows us to treat a range of patients, including more moderate cases earlier on,” said Kristian Reich, M.D., SCIderm Research Institute and Dermatologikum Hamburg, Germany.
The overall safety and tolerability profile was consistent with results from previously reported phase III psoriatic arthritis trials. No cases of tuberculosis or lymphoma were observed through week 16, and there was no increased risk of cardiovascular events or serious opportunistic infection. Apremilast was generally well tolerated. The most common adverse events (AEs) greater than placebo were diarrhea, nausea and headache. Greater than 96% of patients in the study reported no AEs or mild to moderate AEs. A similar percentage of patients reported both serious AEs and severe AEs in the apremilast 30 mg BID treatment group compared to placebo (2.1% vs. 2.8% and 3.6% vs. 3.2%, respectively).
An NDA submission to the U.S. Food and Drug Administration, based on the combined ESTEEM 1&2 studies for psoriasis, is expected in the second half of 2013. The Company previously announced it expects to file a separate NDA for psoriatic arthritis in the first quarter of 2013. A combined PsA/psoriasis MAA submission in Europe is also planned for the second half of 2013.
Top-line positive results from the two pivotal, randomized, placebo-controlled phase III studies of apremilast in psoriasis (ESTEEM 1&2) were released in January 2013. The studies included more than 1,200 patients with moderate-to-severe psoriasis and are ongoing. Results from PSOR-005, a phase IIb dose-range study, were recently published in The Lancet (http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(12)60642-4/fulltext).
About ESTEEM 1 & 2
ESTEEM 1 & 2 are two pivotal phase III randomized, placebo-controlled studies evaluating apremilast in subjects with a diagnosis of moderate-to-severe chronic plaque psoriasis for at least 12 months prior to the screening, and at baseline, and who were also candidates for phototherapy and/or systemic therapy. Approximately 1,250 patients were randomized 2:1 to receive either apremilast 30 mg BID or placebo for the first 16 weeks, followed by a maintenance phase from weeks 16-32 in which placebo subjects were switched to apremilast 30 mg BID through week 32, and a randomized withdrawal phase for responders from Week 32-Week 52 based on their initial apremilast randomization and PASI response.
Apremilast, an oral small-molecule inhibitor of phosphodiesterase 4 (PDE4), works intracellularly to modulate a network of pro-inflammatory and anti-inflammatory mediators. PDE4 is a cyclic adenosine monophosphate (cAMP)-specific PDE and the dominant PDE in inflammatory cells (see http://discoverpde4.com/). PDE4 inhibition elevates intracellular cAMP levels, which in turn down-regulates the inflammatory response by modulating the expression of TNF-α, IL-23, and other inflammatory cytokines. Elevation of cAMP also increases anti-inflammatory cytokines such as IL-10. To learn more go to www.discoverpde4.com/.
Top-line positive results from three pivotal randomized, placebo-controlled phase III studies of apremilast in PsA (PALACE 1, 2 & 3) were released in September 2012. PALACE 1 was also presented as an oral presentation at the ACR annual meeting in November 2012. Taken together, the PALACE program comprises the most comprehensive psoriatic arthritis studies to date intended for regulatory submission.
Results from PSA-001, the phase II study of apremilast in psoriatic arthritis, were recently published online in the journal Arthritis & Rheumatism (http://onlinelibrary.wiley.com/doi/10.1002/art.34627/abstract).
A randomized, placebo-controlled phase III study (POSTURE) of apremilast in ankylosing spondylitis (AS) began enrolling patients in April 2012. AS, a debilitating disease, which may cause fusion of the spine, arthritis, inflammation of the eye and damage to the heart, affects approximately 1.5 million people in the U.S. and Europe. The trial will randomize approximately 450 patients to receive 20 mg or 30 mg apremilast BID, or placebo BID.
Psoriasis is an immune-mediated, non-contagious chronic inflammatory skin disorder of unknown cause. The disorder is a chronic recurring condition that varies in severity from minor localized patches to complete body coverage. Plaque psoriasis is the most common type of psoriasis. About 80 percent of people who develop psoriasis have plaque psoriasis, which appears as patches of raised, reddish skin covered by silvery-white scales. These patches, or plaques, frequently form on the elbows, knees, lower back, and scalp. Psoriasis occurs nearly equally in males and females. Recent studies show that there may be an ethnic link. Psoriasis is believed to be most common in Caucasians and slightly less common in other ethnic groups. Worldwide, psoriasis is most common in Scandinavia and other parts of northern Europe. About 10 percent to 30 percent of patients with psoriasis also develop a condition called psoriatic arthritis, which causes pain, stiffness and swelling in and around the joints.
Celgene International Sàrl, located in Boudry, in the Canton of Neuchâtel, Switzerland, is a wholly owned subsidiary and international headquarters of Celgene Corporation. Celgene Corporation, headquartered in Summit, New Jersey, is an integrated global pharmaceutical company engaged primarily in the discovery, development and commercialization of innovative therapies for the treatment of cancer and inflammatory diseases through gene and protein regulation. For more information, please visit the Company’s website at www.celgene.com.
Apremilast is an orally available small molecule inhibitor of PDE4 being developed by Celgene for ankylosing spondylitis, psoriasis, and psoriatic arthritis.[1][2] The drug is currently in phase III trials for the three indications. Apremilast, an anti-inflammatory drug, specifically inhibits phosphodiesterase 4. In general the drug works on an intra-cellular basis to moderate proinflammatory and anti-inflammatory mediator production.
Apremilast is being tested for its efficacy in treating “psoriasis, psoriatic arthritis and other chronic inflammatory diseases such as ankylosing spondylitis, Behcet’s disease, and rheutmatoid arthritis.”
- “Apremilast Palace Program Demonstrates Robust and Consistent Statistically Significant Clinical Benefit Across Three Pivotal Phase III Studies (PALACE-1, 2 & 3) in Psoriatic Arthritis” (Press release). Celgene Corporation. 6 September 2012. Retrieved 2012-09-10.
- “US HOT STOCKS: OCZ, VeriFone, Men’s Wearhouse, AK Steel, Celgene”. The Wall Street Journal. 6 September 2012. Retrieved 2012-09-06
Otsuka receives FDA approval for ABILIFY MAINTENA to treat schizophrenia
 |
|
|---|---|
| 7-{4-[4-(2,3-Dichlorophenyl)piperazin-1-yl]butoxy}-3,4-dihydroquinolin-2(1H)-one |
aripiprazole
mar 1, 2013
Otsuka Pharmaceutical Co., Ltd. (Otsuka) and H. Lundbeck A/S (Lundbeck) announced the U.S. Food and Drug Administration (FDA) has approved ABILIFY MAINTENA™ (aripiprazole) for extended- release injectable suspension, an intramuscular (IM) depot formulation indicated for the treatment of schizophrenia.
ABILIFY MAINTENA is the first dopamine D2 partial agonist approved as a once- monthly injection. It contributes a new treatment option to address the ongoing need for relapse prevention in patients with schizophrenia – a chronic, debilitating disease.
Efficacy was demonstrated in a 52-week, placebo-controlled, double-blind, randomized-withdrawal, Phase 3 maintenance trial of ABILIFY MAINTENA in patients with schizophrenia. The time to relapse was the primary endpoint. In the trial, ABILIFY MAINTENA>1 In a key secondary endpoint, the percentage of subjects experiencing relapse (i.e., meeting clinical trial criteria for exacerbation of psychotic symptoms/relapse) was also significantly lower with ABILIFY MAINTENA compared to placebo at the end of the study (10% vs. 40%, respectively; p<0.0001). Additional support for efficacy was derived from oral aripiprazole trials.
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. ABILIFY MAINTENA is not approved for the treatment of patients with dementia-related psychosis. ABILIFY MAINTENA is contraindicated in patients with a known hypersensitivity reaction to aripiprazole. Reactions have ranged from pruritus/urticaria to anaphylaxis (see Important Safety Information below).
ABILIFY MAINTENA will be the first commercialized product from the long-term global alliance between Otsuka and Lundbeck to develop CNS medicines worldwide. The companies expect the product will start becoming available in the U.S. on March 18.
Aripiprazolebrand names: Abilify, Aripiprex) is a partial dopamine agonist of the second generation class of atypical antipsychoticswith additional antidepressant properties that is used in the treatment of schizophrenia,bipolar disorder, and clinical depression. It was approved by the U.S. Food and Drug Administration (FDA) for schizophrenia on November 15, 2002 and the European Medicines Agency on 4 June 2004; for acute manic and mixed episodes associated with bipolar disorder on October 1, 2004; as an adjunct for major depressive disorder on November 20, 2007; and to treat irritability in children with autism on 20 November 2009.[1][2] Aripiprazole was developed by Otsuka in Japan, and in the United States,Otsuka America markets it jointly with Bristol-Myers Squibb.
Phase 3 Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease
 CAS Number:75172-81-5
CAS Number:75172-81-5-
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride (1:1), (2R,3S,4R,5S)-
- Molecular Structure:

- Formula:C6H14ClNO4
- Molecular Weight:199.63
- Synonyms:3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, (2R,3S,4R,5S)- (9CI);3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, [2R-(2a,3a,4a,5b)]-;Migalastat hydrochloride;Galactostatin hydrochloride;(2S,3R,4S,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol hydrochloride;
- Melting Point:260 °C
- Boiling Point:382.7 °C at 760 mmHg
- Flash Point:185.2 °C
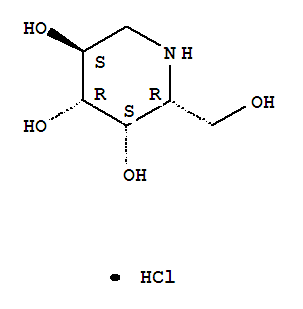
end feb 2013
About Amicus Therapeutics
Amicus Therapeutics is a biopharmaceutical company at the forefront of therapies for rare and orphan diseases. The Company is developing orally-administered, small molecule drugs called pharmacological chaperones, a novel, first-in-class approach to treating a broad range of human genetic diseases. Amicus’ late-stage programs for lysosomal storage disorders include migalastat HCl monotherapy in Phase 3 for Fabry disease; migalastat HCl co-administered with enzyme replacement therapy (ERT) in Phase 2 for Fabry disease; and AT2220 co-administered with ERT in Phase 2 for Pompe disease.
About Migalastat HCl
Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease. Amicus has commercial rights to all Fabry products in the United States and GSK has commercial rights to all of these products in the rest of world.
As a monotherapy, migalastat HCl is designed to bind to and stabilize, or “chaperone” a patient’s own alpha-galactosidase A (alpha-Gal A) enzyme in patients with genetic mutations that are amenable to this chaperone in a cell-based assay. Migalastat HCl monotherapy is in Phase 3 development (Study 011 and Study 012) for Fabry patients with genetic mutations that are amenable to this chaperone monotherapy in a cell-based assay. Study 011 is a placebo-controlled study intended primarily to support U.S. registration, and Study 012 compares migalastat HCl to ERT to primarily support global registration.
For patients currently receiving ERT for Fabry disease, migalastat HCl in combination with ERT may improve ERT outcomes by keeping the infused alpha-Gal A enzyme in its properly folded and active form thereby allowing more active enzyme to reach tissues.2 Migalastat HCl co-administered with ERT is in Phase 2 (Study 013) and migalastat HCl co-formulated with JCR Pharmaceutical Co. Ltd’s proprietary investigational ERT (JR-051, recombinant human alpha-Gal A enzyme) is in preclinical development.
About Fabry Disease
Fabry disease is an inherited lysosomal storage disorder caused by deficiency of an enzyme called alpha-galactosidase A (alpha-Gal A). The role of alpha-Gal A within the body is to break down specific lipids in lysosomes, including globotriaosylceramide (GL-3, also known as Gb3). Lipids that can be degraded by the action of α-Gal are called “substrates” of the enzyme. Reduced or absent levels of alpha-Gal A activity leads to the accumulation of GL-3 in the affected tissues, including the kidneys, heart, central nervous system, and skin. This accumulation of GL-3 is believed to cause the various symptoms of Fabry disease, including pain, kidney failure, and increased risk of heart attack and stroke.
It is currently estimated that Fabry disease affects approximately 5,000 to 10,000 people worldwide. However, several literature reports suggest that Fabry disease may be significantly under diagnosed, and the prevalence of the disease may be much higher.
2. Benjamin, et al., Molecular Therapy: April 2012, Vol. 20, No. 4, pp. 717–726.
http://clinicaltrials.gov/show/NCT01458119
http://www.docstoc.com/docs/129812511/migalastat-hcl
| Chemical Name: | DEOXYGALACTONOJIRIMYCIN, HYDROCHLORIDE |
| Synonyms: | DGJ;Amigal;Unii-cly7m0xd20;GALACTOSTATIN HCL;DGJ, HYDROCHLORIDE;Migalastat hydrochloride;Galactostatin hydrochloride;DEOXYGALACTONOJIRIMYCIN HCL;1-DEOXYGALACTONOJIRIMYCIN HCL;1,5-dideoxy-1,5-imino-d-galactitol |

Lumacaftor, VX-809 an experimental drug for the treatment of Late-Stage cystic fibrosis, being developed by Vertex Pharmaceuticals

3-{6-{[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino}-3-methylpyridin-2-yl}benzoic acid
26,FEB 2013
syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
Vertex Pharmaceuticals announced Tuesday night the design of two phase III studies for its combination therapy to treat the most common form of cystic fibrosis. The studies will each run for six months, so results could be ready as early as the end of 2013 or during first half of 2014.
The studies announced Tuesday will evaluate the two different doses of an experimental medicine VX-809 in combination with Kalydeco. Each study will enroll 500 cystic fibrosis patients randomized to either the VX-809/Kalydeco arms or a placebo for six months of treatment. The studies’ primary endpoint will be the relative improvement in lung function of VX-809/Kalydeco compared to placebo.
Last fall, Vertex presented data from a phase II study demonstrating that a 600 mg dose of VX-809 and Kalydeco worked synergistically to improve lung function in cystic fibrosis patients with the F508del mutation compared to placebo. This same dose combination will be tested in the phase III study along with a higher 800 mg (actually, 400 mg given twice a day) dose of VX-809 plus Kalydeco.
Vertex also announced new data from this phase II study on Tuesday night showing similar lung function improvements between the 800 mg and 600 mg doses of VX-809. For this reason, the higher dose was included in the phase III studies.
Along with the two phase III studies in adult patients, Vertex will also conduct a six-month study of the combination therapy in pediatric patients ages 6 to 11. This study, along with the data from the adult studies, may be used to expand the combination therapy’s approval into younger patients.
In January, FDA anointed Kalydeco and VX-809 with Breakthrough Therapy Designation as part of the agency’s efforts to accelerate the development and approval of drugs for serious and life-threatening disease. Vertex did not say whether Breakthrough Designation played a specific role in the VX-809/Kalydeco phase III program but the relatively short six-month duration of the studies plus the ability to test the combination in children at the same time does accelerate the development of the combination therapy. If the data from the studies are positive, the drugs could be approved sooner than expected and for more patients.
Lumacaftor (USAN, codenamed VX-809) is an experimental drug for the treatment of cystic fibrosis, being developed by Vertex Pharmaceuticals. The drug is designed to be effective in patients that have the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), the defective protein that causes the disease. F508del, meaning that the amino acid phenylalanine in position 508 is missing, is found in about 60% of cystic fibrosis patients.[1]
Interim results from a Phase II clinical trial indicate that patients with the most common form of genetic mutation causing cystic fibrosis homozygous F508del had an 8.5% increase in lung function (FEV1) after 56 days on a combination of lumacaftor and ivacaftor (Kalydeco).[2]
- Merk; Schubert-Zsilavecz. (in German)Pharmazeutische Zeitung 156 (37): 24–27.
- Vertex Pharmaceuticals. May 29,2012.
- syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
- syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
FDA Approves Stivarga, Regorafenib for Advanced Gastrointestinal Stromal Tumors

Regorafenib
cas 755037-03-7
4-[4-({[4-Chloro-3-(trifluoromethyl)phenyl]carbamoyl}amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide hydrate
February 25, 2013 — The U.S. Food and Drug Administration today expanded the approved use of Stivarga (regorafenib) to treat patients with advanced gastrointestinal stromal tumors (GIST) that cannot be surgically removed and no longer respond to other FDA-approved treatments for this disease.
GIST is a tumor in which cancerous cells form in the tissues of the gastrointestinal tract, part of the body’s digestive system. According to the National Cancer Institute, an estimated 3,300 to 6,000 new cases of GIST occur yearly in the United States, most often in older adults.
Stivarga, a multi-kinase inhibitor, blocks several enzymes that promote cancer growth. With this new approval, Stivarga is intended to be used in patients whose GIST cancer cannot be removed by surgery or has spread to other parts of the body (metastatic) and is no longer responding to Gleevec (imatinib) and Sutent (sunitinib), two other FDA-approved drugs to treat GIST.
“Stivarga is the third drug approved by the FDA to treat gastrointestinal stromal tumors,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “It provides an important new treatment option for patients with GIST in which other approved drugs are no longer effective.”
Stivarga was approved in September 2012 to treat colorectal cancer. It is marketed by Bayer HealthCare Pharmaceuticals, based in Wayne, N.J. Gleevec is marketed by East Hanover, N.J.-based Novartis, and Sutent is marketed by New York City-based Pfizer.
Regorafenib (BAY 73-4506, commercial name Stivarga) is an oral multi-kinase inhibitor developed by Bayer which targets angiogenic, stromal and oncogenic receptor tyrosine kinase (RTK). Regorafenib shows anti-angiogenic activity due to its dual targetedVEGFR2-TIE2 tyrosine kinase inhibition. It is currently being studied as a potential treatment option in multiple tumor types.[1]
Metastatic colorectal cancer
Regorafenib demonstrated to increase the overall survival of patients with metastaticcolorectal cancer[2] and has been approved by the US FDA on September 27, 2012.[3]Stivarga is being approved with a Boxed Warning alerting patients and health care professionals that severe and fatal liver toxicity occurred in patients treated with Stivarga during clinical studies. The most common side effects reported in patients treated with Stivarga include weakness or fatigue, loss of appetite, hand-foot syndrome (also called palmar-plantar erythrodysesthesia), diarrhea, mouth sores (mucositis), weight loss, infection, high blood pressure, and changes in voice volume or quality (dysphonia).[4]
- “Bayer Announces New Data on Oncology Portfolio To Be Presented at the ECCO-ESMO Congress 2009”. Retrieved 2009-09-19.
- “Phase III Trial of Regorafenib in Metastatic Colorectal Cancer Meets Primary Endpoint of Improving Overall Survival”. Retrieved 2011-10-26.
- “FDA approves new treatment for advanced colorectal cancer”. 27 Sep 2012.
- “FDA Prescribing Information”. 27 Sept 2012.
Medical Imaging Drugs Advisory Committee Recommends Approval of Guerbet NDA for Dotarem (gadoterate meglumine)

| Cas No. | 98059-18-8 |
| Name | 2-[4,7-bis(carboxylatomethyl)-10-(carboxymethyl)-1,4,7, 10-tetrazacyclododec-1-yl]acetate; gadolinium(3+); (2R,3R,4R,5S)-6-(methylamino)hexane-1,2,3,4,5-pentol |
Dotarem (gadoterate meglumine)
Company: Guerbet
Treatment for: Diagnostic
Dotarem (gadoterate meglumine) is a gadolinium-based contrast agent under review for use in magnetic resonance imaging (MRI).

VILLEPINTE, France, Feb. 14, 2013 Guerbet, the contrast agent specialist for medical imaging, today announced that the Medical Imaging Drugs Advisory Committee to US Food and Drug Administration (FDA) has voted unanimously by votes of 17 to 0 to recommend that FDA approve the New Drug Application (NDA) for Dotarem (gadoterate meglumine) for adults, and for pediatric use for children two years of age and older. The Committee voted 10 to 6 (with one member abstaining) not to recommend at this time approval of the indication for children under two years of age.
Dotarem is the only macrocyclic and ionic gadolinium-based contrast agent (GBCA) for the intravenous use with magnetic resonance imaging (MRI) in the brain (intracranial), spine and associated tissues in adults and pediatric patients to detect and visualize areas with disruption of the blood-brain barrier (BBB) and/or abnormal vascularity. The Guerbet NDA recommended dose is 0.1 mmol Gd/kg.

Gadoteric acid
Gadoteric acid (trade names Artirem, Dotarem) is a macrocycle-structured gadolinium-based MRI contrast agent. It consists of the organic acid DOTA as a chelating agent, and gadolinium (Gd3+), and is used in form of the meglumine salt.[1] The drug is approved and used in a number of countries worldwide.[2]
References
- Herborn, C. U.; Honold, E.; Wolf, M.; Kemper, J.; Kinner, S.; Adam, G.; Barkhausen, J. (2007). “Clinical Safety and Diagnostic Value of the Gadolinium Chelate Gadoterate Meglumine (Gd-DOTA)”. Investigative Radiology 42 (1): 58–62. doi:10.1097/01.rli.0000248893.01067.e5. PMID 17213750. edit
- Drugs.com: Gadoteric Acid

A gadolinium chelate paramagnetic contrast agent. When placed in a magnetic field, gadoterate meglumine produces a large magnetic moment and so a large local magnetic field, which can enhance the relaxation rate of nearby protons; as a result, the signal intensity of tissue images observed with magnetic resonance imaging (MRI) may be enhanced. Because this agent is preferentially taken up by normal functioning hepatocytes, normal hepatic tissue is enhanced with MRI while tumor tissue is unenhanced. In addition, because gadobenate dimeglumine is excreted in the bile, it may be used to visualize the biliary system using MRI.
FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC).
Bevacizumab, CAS NO 216974-75-3
A MONOCLONAL ANTIBODY
January 23, 2013
Avastin (bevacizumab) is a recombinant humanized monoclonal IgG1 antibody that binds to and inhibits the biologic activity of human vascular endothelial growth factor (VEGF) in in vitro and in vivo assay systems. Bevacizumab contains human framework regions and the complementarity-determining regions of a murine antibody that binds to VEGF. Avastin has an approximate molecular weight of 149 kD. Bevacizumab is produced in a mammalian cell (Chinese Hamster Ovary) expression system in a nutrient medium containing the antibiotic gentamicin. Gentamicin is not detectable in the final product.
FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC).
On January 23, 2013, the FDA has approved a new use of Avastin® (bevacizumab) in combination with fluoropyrimidine-based irinotecan or oxaliplatin chemotherapy for people with metastatic colorectal cancer (mCRC). The new indication will allow people who received Avastin plus an irinotecan or oxaliplatin containing chemotherapy as an initial treatment (first-line) for mCRC to continue to receive Avastin plus a different irinotecan or oxaliplatin containing chemotherapy after their cancer worsens (second-line treatment).
People who start on Avastin for mCRC can now stay on Avastin after their cancer worsens.
Bevacizumab (trade name Avastin, Genentech/Roche) is an angiogenesis inhibitor, a drug that slows the growth of new blood vessels. It is licensed to treat various cancers, including colorectal, lung, breast (outside the USA), glioblastoma (USA only), kidney and ovarian.
Bevacizumab is a humanized monoclonal antibody that inhibits vascular endothelial growth factor A (VEGF-A).[1] VEGF-A is a chemical signal that stimulates angiogenesis in a variety of diseases, especially in cancer. Bevacizumab was the first clinically available angiogenesis inhibitor in the United States.[citation needed]
Bevacizumab was approved by the U.S. Food and Drug Administration (FDA) for certain metastatic cancers. It received its first approval in 2004, for combination use with standard chemotherapy for metastatic colon cancer.[2] It has since been approved for use in certain lung cancers, renal cancers, and glioblastoma multiforme of the brain.
At one point bevacizumab was approved for breast cancer by the FDA, but the approval was revoked on 18 November 2011.[3][4]
- Los, M.; Roodhart, J. M. L.; Voest, E. E. (2007). “Target Practice: Lessons from Phase III Trials with Bevacizumab and Vatalanib in the Treatment of Advanced Colorectal Cancer”. The Oncologist 12 (4): 443–50. doi:10.1634/theoncologist.12-4-443. PMID 17470687.
- http://www.gene.com/gene/products/information/pdf/avastin-prescribing.pdf
- Pollack, Andrew (18 November 2011). “F.D.A. Revokes Approval of Avastin for Breast Cancer”. New York Times.
- “Cancer drug Avastin loses US approval”. BBC. November 18, 2011.
SEQUENCE
>1bj1_H|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||VH-CH1 (VH(1-123)+CH1(124-215))|||||||231||||MW 24867.8|MW 24867.8| EVQLVESGGGLVQPGGSLRLSCAASGYTFTNYGMNWVRQAPGKGLEWVGWINTYTGEPTY AADFKRRFTFSLDTSKSTAYLQMNSLRAEDTAVYYCAKYPHYYGSSHWYFDVWGQGTLVT VSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVL QSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHT >1bj1_L|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||L-KAPPA (V-KAPPA(1-107)+C-KAPPA(108-213))|||||||214||||MW 23451.0|MW 23451.0| DIQMTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQKPGKAPKVLIYFTSSLHSGVPS RFSGSGSGTDFTLTISSLQPEDFATYYCQQYSTVPWTFGQGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC >1bj1_J|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||L-KAPPA (V-KAPPA(1-107)+C-KAPPA(108-213))|||||||214||||MW 23451.0|MW 23451.0| DIQMTQSPSSLSASVGDRVTITCSASQDISNYLNWYQQKPGKAPKVLIYFTSSLHSGVPS RFSGSGSGTDFTLTISSLQPEDFATYYCQQYSTVPWTFGQGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC >1bj1_K|Fab-12, F(ab)-12, 12-IgG1, rhuMAb-VEGF|||VH-CH1 (VH(1-123)+CH1(124-215))|||||||231||||MW 24867.8|MW 24867.8| EVQLVESGGGLVQPGGSLRLSCAASGYTFTNYGMNWVRQAPGKGLEWVGWINTYTGEPTY AADFKRRFTFSLDTSKSTAYLQMNSLRAEDTAVYYCAKYPHYYGSSHWYFDVWGQGTLVT VSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVL QSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHT
FDA approves Kadcyla (ado-trastuzumab emtansine), a new therapy for patients with HER2-positive, late-stage (metastatic) breast cancer.


Structure of trastuzumab emtansine. An ADC is a three-block “engine” — antibody-linker-drug — and each part of the composite molecule has to be carefully selected and assembled. Considered as an armed-antibody, an ADC is a bi-dentate construction where both parts (antibody and drug) of the molecule combine their effect to ensure selectivity and potency. The role of the linker arm is of paramount importance demanding a fine tuning to execute the controlled release and delivery of the two active components in the tumor environment.
Feb. 22, 2013
FDA approves new treatment for late-stage breast cancer
The U.S. Food and Drug Administration today approved Kadcyla (ado-trastuzumab emtansine), a new therapy for patients with HER2-positive, late-stage (metastatic) breast cancer.
HER2 is a protein involved in normal cell growth. It is found in increased amounts on some types of cancer cells (HER2-positive), including some breast cancers. In these HER2-positive breast cancers, the increased amount of the HER2 protein contributes to cancer cell growth and survival.
Kadcyla is intended for patients who were previously treated with trastuzumab, another anti-HER2 therapy, and taxanes, a class of chemotherapy drugs commonly used for the treatment of breast cancer.
“Kadcyla is trastuzumab connected to a drug called DM1 that interferes with cancer cell growth,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Kadcyla delivers the drug to the cancer site to shrink the tumor, slow disease progression and prolong survival. It is the fourth approved drug that targets the HER2 protein.”

Referred to as T-DM1 during clinical research, Kadcyla was reviewed under the FDA’s priority review program, which provides for an expedited six-month review of drugs that may provide safe and effective therapy when no satisfactory alternative therapy exists, or offer significant improvement compared to marketed products. Other FDA-approved drugs used to treat HER2-positive breast cancer include trastuzumab (1998), lapatinib (2007) and pertuzumab (2012).
Kadcyla, trastuzumab and pertuzumab are marketed by South San Francisco, Calif.-based Genentech, a member of the Roche Group. Lapatinib is marketed by GlaxoSmithKline, based in Research Triangle Park, N.C
ImmunoGen, Inc. a biotechnology company that develops anticancer therapeutics using its TAP technology, today announced that Roche has reported that the U.S. Food and Drug Administration (FDA) has granted marketing approval to Kadcyla for the treatment of people with HER2-positive metastatic breast cancer who have received prior treatment with Herceptin® (trastuzumab) and a taxane chemotherapy.
“This is a big day for the patients with this cancer and for ImmunoGen,” commented Daniel Junius, President and CEO. “In clinical testing, the findings with Kadcyla in this patient population have been impressive, and we’re delighted the product can now be used by practicing oncologists across the US. In addition to its importance from a medical perspective, commercialization of Kadcyla also marks the start of ImmunoGen earning royalty income.”

Mr. Junius continued, “The efficacy and tolerability seen with Kadcyla underscores the transformative potential of our technology. Kadcyla is the most advanced of ten compounds with our TAP technology already in the clinic, with more in earlier stages of development. We are hopeful that in the future many different types of cancers will be routinely treated with TAP compounds.”
Genentech licensed from ImmunoGen exclusive rights to use the Company’s maytansinoid TAP technology to develop anticancer products targeting HER2.
 as a new therapy for patients with HER2-positive, late-stage (metastatic) breast cancer")
According to Genentech, Kadcyla will cost $9,800 per month, compared to $4,500 per month for regular Herceptin. The company estimates a full course of Kadcyla, about nine months of medicine, will cost $94,000. Thus, the cost of the drug is beyond the reach of many women unless they have an insurance plan.
”””””’
Afatinib
-
- Synonyms:BIBW 2992
- ATC:L01XE13
- Use:anticancer; tyrosine kinase inhibitor
- Chemical name:N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-2-butenamide; N-[(3-chloro-4-fluorophenyl)amino]-6-{[4-(N,N-dimethylamino)-1-oxo-2-buten-1-yl]amino}-7-((S)-tetrahydrofuran-3-yloxy)-quinazoline
- Formula:C24H25ClFN5O3
- MW:485.9 g/mol
- CAS-RN:439081-18-2; 850140-72-6
Derivatives
dimaleate
- Formula:C32H33ClFN5O11
- MW:718.1 g/mol
- CAS-RN:850140-73-7
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 446-32-2 | C7H6FNO2 | 4-fluoro-anthranilic acid | |
| 162012-70-6 | C8H3ClFN3O2 | 4-chloro-7-fluoro-6-nitroquinazoline | |
| 367-21-5 | C6H5ClFN | 3-chloro-4-fluoroaniline | |
| 86087-23-2 | C4H8O2 | (S)-(+)-3-hydroxytetrahydrofuran | |
| 314771-76-1 | C18H16ClFN4O2 | N-(3-chloro-4-fluorophenyl)-7-((tetrahydrofuran-3-yl)oxy)quinazoline-4,6-diamine | |
| 13991-36-1 | C4H5BrO2 | bromocrotonic acid | |
| 3095-95-2 | C6H13O5P | diethylphophonoacetic acid | |
| 618061-76-0 | C24H27ClFN4O6P | Diethyl-{[4-((3-chloro-4-fluorophenyl)amino)-7-(((S)-tetrahydro- furan-3-yloxy)quinazolin-6-yl)carbamoyl]-methyl}phosphonate |
|
| 3616-56-6 | C8H19NO2 | (dimethylamino)-acetaldehyde diethylacetate |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| USA | Gilotrif | Boehringer Ingelheim, 2013 | |
| EU | Giotrif | Boehringer Ingelheim, 2013 |
Formulations
- tabs.; 20, 30 and 40 mg
References
-
- a US 6 251 912 (American Cyanamid; 26.6.2001; appl. 29.7.1998; USA-prior. 1.8.1997).
- WO 0 250 043 (Boehringer Ingelheim; 27.6.2002; appl. 12.12.2001; DE-prior. 20.12.2000).
- US RE 43431 (Boehringer Ingelheim; 29.5.2012; appl. 18.8.2009; DE-prior. 20.12.2000).
- b US 8 426 586 (Boehringer Ingelheim; 1.2.2007; appl. 14.7.2006; DE-prior. 17.10.2003).
-
crystalline forms of Afatinib di-maleate:
- Solca, F. et al., J. Pharmacol. Exp. Ther., (2012) 343(2), 342-350.
- WO 2013 052157 (Ratiopharm/Teva; 11.4.2013; appl. 25.4.2012; USA-prior. 6.10.2011).
Chelsea encouraged by FDA talks on Northera(droxidopa)

(2R,3S)-2-amino-3-(3,4-dihydroxyphenyl)-3-hydroxypropanoic acid
L-DOPS (L-threo-dihydroxyphenylserine; Droxidopa; SM-5688) is a psychoactive drugand synthetic amino acid precursor which acts as a prodrug to the neurotransmittersnorepinephrine (noradrenaline) and epinephrine (adrenaline).[1] Unlike norepinephrine and epinephrine themselves, L-DOPS is capable of crossing the protective blood–brain barrier(BBB).[1]
L-DOPS was developed by Sumitomo Pharmaceuticals under the trade name Droxidopafor the treatment of hypotension, including NOH,[2] and NOH associated with variousdisorders such as MSA, FAP, and PD, as well as IDH. The drug has been used in Japanand some surrounding Asian areas for these indications since 1989. Following a merge with Dainippon Pharmaceuticals in 2006, Dainippon Sumitomo Pharma licensed L-DOPS to Chelsea Therapeutics to develop and market it worldwide except in Japan, Korea, China, and Taiwan.
Clinical trials
Though L-DOPS has been used in Japan and Southeast Asia already for some time, it is also currently in clinical trials at the phase IIIpoint in the United States (U.S.), Canada, Australia, and throughout Europe. Provided L-DOPS successfully completes clinical trials, it could be approved for the treatment of NOH as early as 2011.[4] Additionally, phase II clinical trials for IDH are also underway. Chelsea Therapeutics obtained orphan drug status (ODS) for L-DOPS in the U.S. for NOH, and that of which associated with PD, PAF, and MSA, and is the pharmaceutical company developing it in that country.
FEBRUARY 21, 2013
Shares in Chelsea Therapeutics International have leapt after the company said it will resubmit its previously-rejected treatment of neurogenic orthostatic hypotension, Northera, after helpful discussions with US regulators.
A year ago, the Food and Drug Administration issued Chelsea with a complete response letter asking for more data regarding its filing for Northera (droxidopa)for NOH. That came as something of a surprise given that the agency’s Cardiovascular and Renal Drugs Advisory Committee had earlier voted 7-4 in favour of the therapy.
Now Chelsea says that following a meeting, it has received written guidance from the Director of the Office of New Drugs at the FDA stating that an ongoing study has the potential to serve as the basis for a resubmission.
The guidance suggests that “data strongly demonstrating a short-term clinical benefit of droxidopa in patients with NOH would be adequate for approval, with a possible requirement to verify durable clinical benefit post-approval”.
Encouraged by this, Chelsea plans to refile Northera in the late second quarter of 2013. Chief executive Joseph Oliveto said the firm looks forward to submitting the totality of our clinical experience to date to the agency for review…we now have a regulatory path forward”.
Chelsea also intends to initiate a new clinical trial in the fourth quarter of 2013.
- Goldstein, DS (2006). “L-Dihydroxyphenylserine (L-DOPS): a norepinephrine prodrug”. Cardiovasc Drug Rev 24 (3-4): 189–203. doi:10.1111/j.1527-3466.2006.00189.x.PMID 17214596.
- Mathias, Christopher J (2008). “L-dihydroxyphenylserine (Droxidopa) in the treatment of orthostatic hypotension”. Clin Auton Res 18 (Supplement 1): 25–29. doi:10.1007/s10286-007-1005-z.
- Crofford, LJ (2008). “Pain management in fibromyalgia”. Curr Opin Rheumatol 20 (3): 246–250.doi:10.1097/BOR.0b013e3282fb0268. PMID 18388513.
- Search of: “Droxidopa” – List Results – ClinicalTrials.gov
- Robertson, David (2008). “The pathophysiology and diagnosis of orthostatic hypotension”. Clin Auton Res 18(Supplement 1): 2–7. doi:10.1007/s10286-007-1004-0.

FDA Approves Pomalyst for Advanced Multiple Myeloma – February 8, 2013

Pomalyst (pomalidomide) Capsules
Company: Celgene Corporation
Date of Approval: February 8, 2013
Treatment for: Multiple Myeloma
Pomalyst (pomalidomide) is a thalidomide analogue indicated for the treatment of patients with multiple myeloma.
The U.S. Food and Drug Administration today approved Pomalyst (pomalidomide) to treat patients with multiple myeloma whose disease progressed after being treated with other cancer drugs.
Multiple myeloma is a form of blood cancer that primarily affects older adults and arises from plasma cells in the bone marrow. According to the National Cancer Institute, approximately 21,700 Americans are diagnosed with multiple myeloma and 10,710 die yearly from the disease.![]()
Pomalyst is a pill that modulates the body’s immune system to destroy cancerous cells and inhibit their growth. It is intended for patients who have received at least two prior therapies, including lenalidomide and bortezomib, and whose disease did not respond to treatment and progressed within 60 days of the last treatment (relapsed and refractory).
“Pomalyst is the third drug in a class of immunomodulatory agents that includes lenalidomide and thalidomide, and is the second drug approved in the past year to treat multiple myeloma,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in FDA’s Center for Drug Evaluation and Research. “Treatment for multiple myeloma is tailored to meet individual patient’s needs, and today’s approval provides an additional treatment option for patients who have not responded to other drugs.”
- FDA Approves Pomalyst for Advanced Multiple Myeloma – February 8, 2013
- Celgene Corporation Provides Update on FDA Advisory Committee for Pomalidomide – October 3, 2012
- The International Myeloma Foundation Says Pomalidomide, an Important New Drug for Patients, Has Been Submitted for FDA Approval – April 27, 2012
pomalidomide. 4-Amino-2-(2,6-dioxopiperidin-3-yl)isoindole-1,3-dione
Pomalidomide (INN, originally CC-4047 or 3-amino-thalidomide, marketed as Pomalyst by Celgene), is a derivative of thalidomide that is anti-angiogenic and also acts as an immunomodulator. Pomalidomide was approved on February 8, 2013 by the U.S. Food and Drug Administration (FDA) as a treatment for relapsed and refractory multiple myeloma.[1] An application for approval to treat multiple myeloma also has been submitted by Celgene to the European Medicines Agency, and a decision on that application is expected by the second half of 2013.[1]
Origin and development
The parent compound of pomalidomide, thalidomide, was originally discovered to inhibit angiogenesis in 1994.[2] Based upon this discovery, thalidomide was taken into clinical trials for cancer, leading to its ultimate FDA approval for multiple myeloma. Further structure activity studies done in Dr. Robert D’Amato’s lab at Boston Children’s Hospital led to the first report in 2001[3] that 3-amino-thalidomide was able to directly inhibit both the tumor cell and vascular compartments of myeloma cancers. This dual activity of pomalidomide makes it more efficacious than thalidomide in vitro and in vivo.[4]
Clinical trials
Phase I trial results showed tolerable side effects.[5]
Phase II clinical trials for multiple myeloma and myelofibrosis reported ‘promising results’.[6][7]
Phase III results were reported at ASH in 2012 and showed significant extension of progression-free survival (median 3.6 months vs. 1.8 months; P < 0.001), and overall survival in patients taking pomalidomide and dexamethasone.[8]
- “Pomalyst (Pomalidomide) Approved By FDA For Relapsed And Refractory Multiple Myeloma”. The Myeloma Beacon. Retrieved 2013-02-08.
- D’Amato, Robert J.; Loughnan, Michael S.; Flynn, Evelyn; Folkman, Judah (1994). “Thalidomide is an inhibitor of angiogenesis”. Proceedings of the National Academy of Sciences of the United States of America 91 (9): 4082–5. Bibcode 1994PNAS…91.4082D. doi:10.1073/pnas.91.9.4082. JSTOR 2364596. PMC 43727. PMID 7513432.
- D’Amato, R; Lentzsch, S; Anderson, KC; Rogers, MS (2001). “Mechanism of action of thalidomide and 3-aminothalidomide in multiple myeloma”. Seminars in Oncology 28 (6): 597–601. doi:10.1016/S0093-7754(01)90031-4. PMID 11740816.
- Lentzsch, S; Rogers, MS; Leblanc, R; Birsner, AE; Shah, JH; Treston, AM; Anderson, KC; D’Amato, RJ (2002). “S-3-Amino-phthalimido-glutarimide inhibits angiogenesis and growth of B-cell neoplasias in mice”. Cancer research 62 (8): 2300–5. PMID 11956087.
- Streetly, Matthew J.; Gyertson, Kylie; Daniel, Yvonne; Zeldis, Jerome B.; Kazmi, Majid; Schey, Stephen A. (2008). “Alternate day pomalidomide retains anti-myeloma effect with reduced adverse events and evidence of in vivo immunomodulation”. British Journal of Haematology 141 (1): 41–51. doi:10.1111/j.1365-2141.2008.07013.x. PMID 18324965.
- “Promising Results From 2 Trials Highlighting Pomalidomide Presented At ASH” (Press release). Celgene. December 11, 2008. Retrieved October 28, 2012.
- Tefferi, Ayalew (December 8, 2008). “Pomalidomide Therapy in Anemic Patients with Myelofibrosis: Results from a Phase-2 Randomized Multicenter Study”. 50th ASH Annual Meeting and Exposition. San Francisco. Retrieved October 28, 2012.
- “Phase III Study (MM-003) of Pomalidomide Plus Low-Dose Dexamethasone Demonstrates Significant Progression-Free and Overall Survival Improvement for Patients with Relapsed or Refractory Multiple Myeloma.”. 11 Dec 2012.
- This new drug is specifically indicated for patients who have received at least 2 prior therapies, including lenalidomide (Revlimid, Celgene) and bortezomib (Velcade, Millennium Pharmaceuticals), and whose disease did not respond to treatment and progressed within 60 days of the last treatment.

