



A series of novel, potent orthopoxvirus egress inhibitors was identified during high-throughput screening of the ViroPharma small molecule collection. Using structure−activity relationship information inferred from early hits, several compounds were synthesized, and compound 14was identified as a potent, orally bioavailable first-in-class inhibitor of orthopoxvirus egress from infected cells. Compound 14 has shown comparable efficaciousness in three murine orthopoxvirus models and has entered Phase I clinical trials.
Home » Posts tagged 'FDA 2018' (Page 4)
Tag Archives: FDA 2018
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TAFENOQUINE タフェノキン


Tafenoquine
タフェノキン
N-[2,6-dimethoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinolin-8-yl]pentane-1,4-diamine
1,4-Pentanediamine, N4-[2,6-dimethoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]-8-quinolinyl]-
106635-80-7 [RN]
262P8GS9L9
7835
N4-{2,6-Dimethoxy-4-methyl-5-[3-(trifluormethyl)phenoxy]-8-chinolinyl}-1,4-pentandiamin
WR-238605, WR 238605, cas no 106635-80-7, Tafenoquine succinate, Etaquine, SB-252263, WR-238605
N(4)-(2,6-Dimethoxy-4-methyl-5-((3-trifluoromethyl)phenoxy)-8-quinolinyl)-1,4-pentanediamine
Molecular Formula: C24H28F3N3O3
Molecular Weight: 463.49263
Medicines for Malaria Venture
Walter Reed Army Institute (Originator)
PATENT US 4617394
Synonyms
New Drug Application (NDA): 210795
Company: GLAXOSMITHKLINE
FDA approved on July 20, 2018
FDA
Orphan
This new drug application provides for the use of KRINTAFEL (tafenoquine) tablets for the radical cure (prevention of relapse) of Plasmodium vivax malaria in patients aged 16 years and older who are receiving appropriate antimalarial therapy for acute P. vivax infection….https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/210795Orig1s000Ltr.pdf
Tafenoquine under the commercial name of Krintafel is an 8-aminoquinoline drug manufactured by GlaxoSmithKline that is being investigated as a potential treatment for malaria, as well as for malaria prevention.[2][3]
The proposed indication for tafenoquine is for treatment of the hypnozoite stages of Plasmodium vivax and Plasmodium ovale that are responsible for relapse of these malaria species even when the blood stages are successfully cleared. This is only now achieved by administration of daily primaquine for 14 days. The main advantage of tafenoquine is that it has a long half-life (2–3 weeks) and therefore a single treatment may be sufficient to clear hypnozoites. The shorter regimen has been described as an advantage.[4]
Like primaquine, tafenoquine causes hemolysis in people with G6PD deficiency.[2] Indeed, the long half-life of tafenoquine suggests that particular care should be taken to ensure that individuals with severe G6PD deficiency do not receive the drug.
The dose of tafenoquine has not been firmly established, but for the treatment of Plasmodium vivax malaria, a dose of 800 mg over three days has been used.[5]

In 2018 United States Food and Drug Administration (FDA) approved single dose tafenoquine for the radical cure (prevention of relapse) of Plasmodium vivax malaria[6].
Tafenoquine is used for the treatment and prevention of relapse of Vivax malaria in patients 16 years and older. Tafenoquine is not indicated to treat acute vivax malaria.[1]
Malaria is a disease that remains to occur in many tropical countries. Vivax malaria, caused by Plasmodium vivax, is known to be less virulent and seldom causes death. However, it causes a substantive illness-related burden in endemic areas and it is known to present dormant forms in the hepatocytes named hypnozoites which can remain dormant for weeks or even months. This dormant form produces ongoing relapses
FDA Approves Tafenoquine, First New P VivaxMalaria Treatment in 60 Years
JUL 23, 2018
The US Food and Drug Administration (FDA) has approved, under Priority Review, GlaxoSmithKline (GSK)’s tafenoquine (Krintafel), which is the first single-dose medicine for the prevention of Plasmodium vivax (P vivax) malaria relapse in patients over the age of 16 years who are receiving antimalarial therapy. This is the first drug to be approved for the treatment of P vivax in over 60 years.
“[The] approval of Krintafel, the first new treatment for Plasmodium vivax malaria in over 60 years, is a significant milestone for people living with this type of relapsing malaria.” Hal Barron, MD, chief scientific officer and president of research and development of GSK, said in the announcement, “Together with our partner, Medicines for Malaria Venture (MMV), we believe Krintafel will be an important medicine for patients with malaria and contribute to the ongoing effort to eradicate this disease.”
Tafenoquine is an 8-aminoquinoline derivative with activity against all stages of the P vivax lifecycle, including hypnozoites. It was first synthesized by scientists at the Walter Reed Army Institute of Research in 1978, and in 2008, GSK entered into a collaboration with MMV, to develop tafenoquine as an anti-relapse medicine.
After an infected mosquito bite, the P vivax parasite infects the blood and causes an acute malaria episode and can also lie dormant in the liver (in a form known as hypnozoite) from where it periodically reactivates to cause relapses, which can occur weeks, months, or years after the onset of the initial infection. The dormant liver forms cannot be readily treated with most anti-malarial treatments. Primaquine, an 8-aminoquinolone, has been the only FDA-approved medicine that targeted the dormant liver stage to prevent relapse; however, effectiveness only occurs after 14 days and the treatment has shown to have poor compliance.
“The US FDA’s approval of Krintafel is a major milestone and a significant contribution towards global efforts to eradicate malaria,” commented David Reddy, PhD, chief executive officer of MMV in a recent statement, “The world has waited decades for a new medicine to counter P vivax malaria relapse. Today, we can say the wait is over. Moreover, as the first ever single-dose for this indication, Krintafel will help improve patient compliance.”
Approval for tafenoquine was granted based on the efficacy and safety data gleaned from a comprehensive global clinical development program for P vivaxprevention of relapse which has been designed by GSK and MMV in agreement with the FDA. The program consisted of 13 studies assessing the safety of a 300 mg single-dose of tafenoquine, including 3 double-blind studies referred to as DETECTIVE Parts 1 and 2 and GATHER.
With the approval of tafenoquine, GSK has also been awarded a tropical disease priority review voucher by the FDA. Additionally, GSK is waiting for a decision from Australian Therapeutics Good Administration regarding the regulatory submission for the drug.
P vivax malaria has caused around 8.5 million clinical infections each year, primarily in South Asia, South-East Asia, Latin America, and the Horn of Africa, a peninsula in East Africa. Symptoms include fever, chills, vomiting, malaise, headache and muscle pain, and can lead to death in severe cases.
Tafenoquine should not be administered to: patients who have glucose-6-phosphate dehydrogenase (G6PD) deficiency or have not been tested for G6PD deficiency, patients who are breastfeeding a child known to have G6PD deficiency or one that has not been tested for G6PD deficiency, or patients who are allergic to tafenoquine or any of the ingredients in tafenoquine or who have had an allergic reaction to similar medicines containing 8-aminoquinolines
Stereochemistry
Tafenoquine contains a stereocenter and consists of two enantiomers. This is a mixture of (R) – and the (S) – Form:
| Enantiomers of tafenoquine | |
|---|---|
-Tafenoquin_Structural_Formula_V1.svg) (R)-Form |
-Tafenoquin_Structural_Formula_V1.svg) (S)-Form |
 CLIP
CLIP
US 4431807


Nitration of 1,2-dimethoxybenzene (XXIX) with HNO3/AcOH gives 4,5-dimethoxy-1,2-dinitrobenzene (XXX), which is treated with ammonia in hot methanol to yield 4,5-dimethoxy-2-nitroaniline (XXXI). Cyclization of compound (XXXI) with buten-2-one (XXXII) by means of H3PO4 and H3AsO4 affords 5,6-dimethoxy-4-methyl-8-nitroquinoline (XXXIII), which is selectively mono-demethylated by means of HCl in ethanol to provide 5-hydroxy-6-methoxy-4-methyl-8-nitroquinoline (XXXIV). Reaction of quinoline (XXXIV) with POCl3 gives the corresponding 5-chloro derivative (XXXV), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH to yield the diaryl ether (XXXVI). Finally, the nitro group of (XXXVI) is reduced by means of H2 over PtO2 in THF or H2 over Raney nickel.

Nitration of 2-fluoroanisole (XXXVII) with HNO3/Ac2O gives 3-fluoro-4-methoxynitrobenzene (XXXVIII), which is reduced to the corresponding aniline (XXXIX) with SnCl2/HCl. Reaction of compound (XXXIX) with Ac2O yields the acetanilide (XL), which is nitrated with HNO3 to afford 5-fluoro-4-methoxy-2-nitroacetanilide (XLI). Hydrolysis of (XLI) with NaOH provides 5-fluoro-4-methoxy-2-nitroaniline (XLII), which is cyclized with buten-2-one (XXXII) by means of As2O5 and H3PO4 to furnish 5-fluoro-6-methoxy-4-methyl-8-nitroquinoline (XLIII). Condensation of quinoline (XLIII) with 3-(trifluoromethyl)phenol (IV) by means of K2CO3 gives the diaryl ether (XXXIV), which is finally reduced by means of H2 over PtO2 in THF.
CLIP
US 4617394

Reaction of 8-amino-6-methoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinoline (XIV) with phthalic anhydride (XV) affords the phthalimido derivative (XVI), which is oxidized with MCPBA to yield the quinoline N-oxide (XVII). Treatment of compound (XVII) with neutral alumina gives the quinolone derivative (XVIII), which by reaction with POCl3 in refluxing CHCl3 provides the 2-chloroquinoline derivative (XIX). Alternatively, reaction of the quinoline N-oxide (XVII) with POCl3 as before also gives the 2-chloroquinoline derivative (XIX) The removal of the phthalimido group of compound (XIX) by means of hydrazine in refluxing ethanol gives the chlorinated aminoquinoline (XX), which is finally treated with MeONa in hot DMF.
CLIP
US 6479660; WO 9713753

Chlorination of 6-methoxy-4-methylquinolin-2(1H)-one (I) with SO2Cl2 in hot acetic acid gives the 5-chloro derivative (II), which is nitrated with HNO3 in H2SO4 to yield the 8-nitroquinolinone (III). Condensation of compound (III) with 3-(trifluoromethyl)phenol (IV) by means of KOH in NMP provides the diaryl ether (V), which is treated with refluxing POCl3 to afford the 2-chloroquinoline (VI). Reaction of compound (VI) with MeONa in refluxing methanol results in the 2,6-dimethoxyquinoline derivative (VII), which is reduced with hydrazine over Pd/C to give the 8-aminoquinoline derivative (VIII). Condensation of aminoquinoline (VIII) with N-(4-iodopentyl)phthalimide (IX) by means of diisopropylamine in hot NMP yields the phthalimido precursor (X), which is finally cleaved with hydrazine in refluxing ethanol.


Reaction of 1,4-dibromopentane (XI) with potassium phthalimide (XII) gives N-(4-bromopentyl)phthalimide (XIII), which is then treated with NaI in refluxing acetone.

Reaction of 4-methoxyaniline (XXI) with ethyl acetoacetate (XXII) by means of triethanolamine in refluxing xylene gives the acetoacetanilide (XXIII), which is cyclized by means of hot triethanolamine and H2SO4 to yield 6-methoxy-4-methylquinolin-2(1H)-one (I), which is treated with refluxing POCl3 to provide 2-chloro-6-methoxy-4-methylquinoline (XXIV). Reaction of compound (XXIV) with SO2Cl2 in hot AcOH affords 2,5-dichloro-6-methoxy-4-methylquinoline (XXV), which is treated with MeONa in refluxing methanol to furnish 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Alternatively, the reaction of compound (XXIV) with MeONa as before gives 2,6-dimethoxy-4-methylquinoline (XXVII), which is treated with SO2Cl2 in hot AcOH to give the already described 5-chloro-2,6-dimethoxy-4-methylquinoline (XXVI). Nitration of compound (XXVI) with KNO3 and P2O5 gives the 8-nitroquinoline derivative (XXVIII), which is condensed with 3-(trifluoromethyl)phenol (IV) by means of KOH in hot NMP to yield the diaryl ether (VII). Finally, the nitro group of compound (VII) is reduced with hydrazine over Pd/C.
PAPER
http://pubs.rsc.org/en/Content/ArticleLanding/2017/RA/C7RA04867J#!divAbstract
An antimalarial drug, tafenoquine, as a fluorescent receptor for ratiometric detection of hypochlorite
Abstract
Tafenoquine (TQ), a fluorescent antimalarial drug, was used as a receptor for the fluorometric detection of hypochlorite (OCl−). TQ itself exhibits a strong fluorescence at 476 nm, but OCl−-selective cyclization of its pentan-1,4-diamine moiety creates a blue-shifted fluorescence at 361 nm. This ratiometric response facilitates rapid, selective, and sensitive detection of OCl− in aqueous media with physiological pH. This response is also applicable to a simple test kit analysis and allows fluorometric OCl− imaging in living cells.




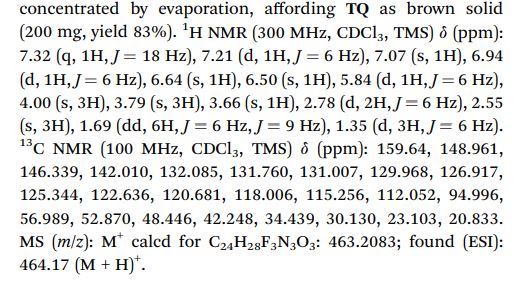
1 H NMR (300 MHz, CDCl3, TMS) d (ppm): 7.32 (q, 1H, J ¼ 18 Hz), 7.21 (d, 1H, J ¼ 6 Hz), 7.07 (s, 1H), 6.94 (d, 1H, J ¼ 6 Hz), 6.64 (s, 1H), 6.50 (s, 1H), 5.84 (d, 1H, J ¼ 6 Hz), 4.00 (s, 3H), 3.79 (s, 3H), 3.66 (s, 1H), 2.78 (d, 2H, J ¼ 6 Hz), 2.55 (s, 3H), 1.69 (dd, 6H, J ¼ 6 Hz, J ¼ 9 Hz), 1.35 (d, 3H, J ¼ 6 Hz).
13C NMR (100 MHz, CDCl3, TMS) d (ppm): 159.64, 148.961, 146.339, 142.010, 132.085, 131.760, 131.007, 129.968, 126.917, 125.344, 122.636, 120.681, 118.006, 115.256, 112.052, 94.996, 56.989, 52.870, 48.446, 42.248, 34.439, 30.130, 23.103, 20.833.
MS (m/z): M+ calcd for C24H28F3N3O3: 463.2083; found (ESI): 464.17 (M + H)+ .
PAPER
J Med Chem 1989,32(8),1728-32
https://pubs.acs.org/doi/pdf/10.1021/jm00128a010

Synthesis of the intermediate diazepinone (IV) is accomplished by a one-pot synthesis. Condensation of 2-chloro-3-aminopyridine (I) with the anthranilic ester (II) is effected in the presence of potassium tert-butoxide as a catalyst. The resulting anthranilic amide (III) is cyclized under the influence of catalytic amounts of sulfuric acid. Treatment of (IV) with chloroacetylchloride in toluene yields the corresponding choroacetamide (V). The side chain of AQ-RA 741 is prepared starting from 4-picoline, which is alkylated by reaction with 3-(diethylamino)propylchloride in the presence of n-butyllithium. Hydrogenation of (VIII) using platinum dioxide as a catalyst furnishes the diamine (IX), which is coupled with (V) in the presence of catalytic amounts of sodium iodide in acetone leading to AQ-RA 741 as its free base.


CLIP
Journal of Fluorine Chemistry
Volume 167, November 2014, Pages 37-54


References
- ^ Jump up to:a b Peters W (1999). “The evolution of tafenoquine–antimalarial for a new millennium?”. J R Soc Med. 92 (7): 345–352. PMC 1297286
 . PMID 10615272.
. PMID 10615272. - ^ Jump up to:a b Shanks GD, Oloo AJ, Aleman GM, et al. (2001). “A New Primaquine Analogue, Tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria”. Clin Infect Dis. 33 (12): 1968–74. doi:10.1086/324081. JSTOR 4482936. PMID 11700577.
- Jump up^ Lell B, Faucher JF, Missinou MA, et al. (2000). “Malaria chemoprophylaxis with tafenoquine: a randomised study”. Lancet. 355 (9220): 2041–5. doi:10.1016/S0140-6736(00)02352-7. PMID 10885356.
- Jump up^ Elmes NJ, Nasveld PE, Kitchener SJ, Kocisko DA, Edstein MD (November 2008). “The efficacy and tolerability of three different regimens of tafenoquine versus primaquine for post-exposure prophylaxis of Plasmodium vivax malaria in the Southwest Pacific”. Transactions of the Royal Society of Tropical Medicine and Hygiene. 102 (11): 1095–101. doi:10.1016/j.trstmh.2008.04.024. PMID 18541280.
- Jump up^ Nasveld P, Kitchener S (2005). “Treatment of acute vivax malaria with tafenoquine”. Trans R Soc Trop Med Hyg. 99 (1): 2–5. doi:10.1016/j.trstmh.2004.01.013. PMID 15550254.
- Jump up^ “Drugs@FDA: FDA Approved Drug Products”. http://www.accessdata.fda.gov. Retrieved 2018-07-23.
- Shanks GD, Oloo AJ, Aleman GM et al. (2001). “A New Primaquine Analogue, Tafenoquine (WR 238605), for prophylaxis against Plasmodium falciparum malaria”. Clin Infect Dis 33 (12): 1968–74. doi:10.1086/324081. JSTOR 4482936.PMID 11700577.
- Lell B, Faucher JF, Missinou MA et al. (2000). “Malaria chemoprophylaxis with tafenoquine: a randomised study”.Lancet 355 (9220): 2041–5. doi:10.1016/S0140-6736(00)02352-7. PMID 10885356.
- Elmes NJ, Nasveld PE, Kitchener SJ, Kocisko DA, Edstein MD (November 2008). “The efficacy and tolerability of three different regimens of tafenoquine versus primaquine for post-exposure prophylaxis of Plasmodium vivax malaria in the Southwest Pacific”. Transactions of the Royal Society of Tropical Medicine and Hygiene 102 (11): 1095–101.doi:10.1016/j.trstmh.2008.04.024. PMID 18541280.
- Nasvelda P, Kitchener S. (2005). “Treatment of acute vivax malaria with tafenoquine”. Trans R Soc Trop Med Hyg 99 (1): 2–5. doi:10.1016/j.trstmh.2004.01.013. PMID 15550254.
- Peters W (1999). “The evolution of tafenoquine–antimalarial for a new millennium?”. J R Soc Med 92 (7): 345–352.PMID 10615272.
- J Med Chem 1982,25(9),1094
|
8-3-2007
|
Methods and compositions for treating diseases associated with pathogenic proteins
|
|
|
12-6-2006
|
Process for the preparation of quinoline derivatives
|
|
|
3-14-2002
|
PROCESS FOR THE PREPARATION OF ANTI-MALARIAL DRUGS
|
|
|
4-2-1998
|
MULTIDENTATE METAL COMPLEXES AND METHODS OF MAKING AND USING THEREOF
|
|
|
4-18-1997
|
PROCESS FOR THE PREPARATION OF ANTI-MALARIAL DRUGS
|
|
|
12-20-1996
|
MULTIDENTATE METAL COMPLEXES AND METHODS OF MAKING AND USING THEREOF
|
|
|
12-15-1993
|
Use of interferon and a substance with an antimalarial activity for the treatment of malaria infections
|
|
|
10-15-1986
|
4-methyl-5-(unsubstituted and substituted phenoxy)-2,6-dimethoxy-8-(aminoalkylamino) quinolines
|
Title: Tafenoquine
CAS Registry Number: 106635-80-7
CAS Name: N4–[2,6-Dimethoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]-8-quinolinyl]-1,4-pentanediamine
Additional Names: 8-[(4-amino-1-methylbutyl)amino]-2,6-dimethoxy-4-methyl-5-[3-(trifluoromethyl)phenoxy]quinoline
Manufacturers’ Codes: WR-238605
Molecular Formula: C24H28F3N3O3
Molecular Weight: 463.49
Percent Composition: C 62.19%, H 6.09%, F 12.30%, N 9.07%, O 10.36%
Literature References: Analog of primaquine, q.v. Prepn: P. Blumbergs, M. P. LaMontagne, US 4617394 (1986 to U.S. Sec. Army); M. P. LaMontagne et al., J. Med. Chem. 32, 1728 (1989). HPLC determn in blood and plasma: D. A. Kocisko et al., Ther. Drug Monit. 22, 184 (2000). Metabolism: O. R. Idowu et al., Drug Metab. Dispos. 23, 1 (1995). Clinical pharmacokinetics: M. D. Edstein et al., Br. J. Pharmacol. 52, 663 (2001). Clinical evaluation in prevention of malaria relapse: D. S. Walsh et al., J. Infect. Dis. 180, 1282 (1999); in malaria prophylaxis: B. Lell et al., Lancet 355, 2041 (2000); B. R. Hale et al., Clin. Infect. Dis. 36, 541 (2003).
Derivative Type: Succinate
CAS Registry Number: 106635-81-8
Trademarks: Etaquine (GSK)
Molecular Formula: C24H28F3N3O3.C4H6O4
Molecular Weight: 581.58
Percent Composition: C 57.83%, H 5.89%, F 9.80%, N 7.23%, O 19.26%
Properties: Crystals from acetonitrile, mp 146-149°. LD50 in male, female rats (mg/kg): 102, 71 i.p.; 429, 416 orally (LaMontagne).
Melting point: mp 146-149°
Toxicity data: LD50 in male, female rats (mg/kg): 102, 71 i.p.; 429, 416 orally (LaMontagne)
Therap-Cat: Antimalarial.
Keywords: Antimalarial.
-Tafenoquin_Structural_Formula_V1.svg) |
|
| Clinical data | |
|---|---|
| Synonyms | Etaquine,[1] WR 238605,[1] SB-252263 |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| NIAID ChemDB | |
| Chemical and physical data | |
| Formula | C24H28F3N3O3 |
| Molar mass | 463.493 g/mol |
| 3D model (JSmol) | |
OLD CLIP
April 28, 2014
GlaxoSmithKline (GSK) and Medicines for Malaria Venture (MMV) announced the start of a Phase 3 global program to evaluate the efficacy and safety of tafenoquine, an investigational medicine which is being developed for the treatment and relapse prevention (radical cure) of Plasmodium vivax (P. vivax) malaria.
P. vivax malaria, a form of the disease caused by one of several species of Plasmodium parasites known to infect humans, occurs primarily in South and South East Asia, Latin America and the horn of Africa. Severe anemia, malnutrition and respiratory distress are among the most serious consequences described to be caused by the infection.
The Phase 3 program includes two randomized, double-blind treatment studies to investigate tafenoquine in adult patients with P. vivax malaria. The DETECTIVE study (TAF112582) aims to evaluate the efficacy, safety and tolerability of tafenoquine as a radical cure for P. vivax malaria, co-administered with chloroquine, a blood stage anti-malarial treatment. The GATHER study (TAF116564) aims to assess the incidence of hemolysis and safety and efficacy of tafenoquine compared to primaquine, the only approved treatment currently available for the radical cure of P. vivax malaria.
Tafenoquine is not yet approved or licensed for use anywhere in the world.
“P. vivax malaria can affect people of all ages and is particularly insidious because it has the potential to remain dormant within the body in excess of a year, and causes some patients to experience repeated episodes of illness after the first mosquito bite,” said Nicholas Cammack, head, Tres Cantos Medicines Development Center for Diseases of the Developing World. “Our investigation of tafenoquine for the treatment of P. vivax malaria is part of GSK’s efforts to tackle the global burden of malaria. Working with our partners, including MMV, we are determined to stop malaria in all its forms.”
“One of the big challenges we face in tackling malaria is to have new medicines to prevent relapse, caused by dormant forms of P. vivax,” said Dr. Timothy Wells, MMV’s chief scientific officer. “The Phase 3 program is designed to build upon the promising results of the Phase 2b study which showed that treatment with tafenoquine prevented relapses. If successful, tafenoquine has the potential to become a major contributor to malaria elimination. It’s a great privilege to be working with GSK on this project; they have a clear commitment to changing the face of public health in the countries in which we are working.”
/////////////Tafenoquine, タフェノキン , Orphan, FDA 2018, KRINTAFEL, Priority Review, GlaxoSmithKline
COC1=CC(C)=C2C(OC3=CC=CC(=C3)C(F)(F)F)=C(OC)C=C(NC(C)CCCN)C2=N1
Ivosidenib, ивосидениб , إيفوزيدينيب , 艾伏尼布 ,

Ivosidenib
AG-120; TIBSOVO
FDA approves first targeted treatment Tibsovo (ivosidenib) for patients with relapsed or refractory acute myeloid leukemia who have a certain genetic mutation
The U.S. Food and Drug Administration today approved Tibsovo (ivosidenib) tablets for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. This is the first drug in its class (IDH1 inhibitors) and is approved for use with an FDA-approved companion diagnostic used to detect specific mutations in the IDH1 gene in patients with AML.
“Tibsovo is a targeted therapy that fills an unmet need for patients with relapsed or refractory AML who have an IDH1 mutation,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The use of Tibsovo is associated with a complete remission in some patients and a reduction in the need for both red cell and platelet transfusions.”
July 20, 2018
Release
The U.S. Food and Drug Administration today approved Tibsovo (ivosidenib) tablets for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) who have a specific genetic mutation. This is the first drug in its class (IDH1 inhibitors) and is approved for use with an FDA-approved companion diagnostic used to detect specific mutations in the IDH1 gene in patients with AML.
“Tibsovo is a targeted therapy that fills an unmet need for patients with relapsed or refractory AML who have an IDH1 mutation,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The use of Tibsovo is associated with a complete remission in some patients and a reduction in the need for both red cell and platelet transfusions.”
AML is a rapidly progressing cancer that forms in the bone marrow and results in an increased number of abnormal white blood cells in the bloodstream and bone marrow. The National Cancer Institute at the National Institutes of Health estimates that approximately 19,520 people will be diagnosed with AML this year; approximately 10,670 patients with AML will die of the disease in 2018.
Tibsovo is an isocitrate dehydrogenase-1 inhibitor that works by decreasing abnormal production of the oncometabolite 2-hydroxyglutarate (2-HG), leading to differentiation of malignant cells. If the IDH1 mutation is detected in blood or bone marrow samples using an FDA-approved test, the patient may be eligible for treatment with Tibsovo. Today the agency also approved the RealTime IDH1 Assay, a companion diagnostic that can be used to detect this mutation.
The efficacy of Tibsovo was studied in a single-arm trial of 174 adult patients with relapsed or refractory AML with an IDH1 mutation. The trial measured the percentage of patients with no evidence of disease and full recovery of blood counts after treatment (complete remission or CR), as well as patients with no evidence of disease and partial recovery of blood counts after treatment (complete remission with partial hematologic recovery or CRh). With a median follow-up of 8.3 months, 32.8 percent of patients experienced a CR orCRh that lasted a median 8.2 months. Of the 110 patients who required transfusions of blood or platelets due to AML at the start of the study, 37 percent went at least 56 days without requiring a transfusion after treatment with Tibsovo.
Common side effects of Tibsovo include fatigue, increase in white blood cells, joint pain, diarrhea, shortness of breath, swelling in the arms or legs, nausea, pain or sores in the mouth or throat, irregular heartbeat (QT prolongation), rash, fever, cough and constipation. Women who are breastfeeding should not take Tibsovo because it may cause harm to a newborn baby.
Tibsovo must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. The prescribing information for Tibsovo includes a boxed warning that an adverse reaction known as differentiation syndrome can occur and can be fatal if not treated. Signs and symptoms of differentiation syndrome may include fever, difficulty breathing (dyspnea), acute respiratory distress, inflammation in the lungs (radiographic pulmonary infiltrates), fluid around the lungs or heart (pleural or pericardial effusions), rapid weight gain, swelling (peripheral edema) or liver (hepatic), kidney (renal) or multi-organ dysfunction. At first suspicion of symptoms, doctors should treat patients with corticosteroids and monitor patients closely until symptoms go away.
Other serious warnings include a QT prolongation, which can be life-threatening. Electrical activity of the heart should be tested with an electrocardiogram during treatment. Guillain-Barré syndrome, a rare neurological disorder in which the body’s immune system mistakenly attacks part of its peripheral nervous system, has happened in people treated with Tibsovo, so patients should be monitored for nervous system problems.
The FDA granted this application Fast Track and Priority Review designations. Tibsovo also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Tibsovo to Agios Pharmaceuticals, Inc. The FDA granted the approval of the RealTime IDH1 Assay to Abbott Laboratories.
ivosidenib
- Molecular FormulaC28H22ClF3N6O3
- Average mass582.961 Da
1448347-49-6 [RN]
2-Pyrrolidinecarboxamide, N-[(1S)-1-(2-chlorophenyl)-2-[(3,3-difluorocyclobutyl)amino]-2-oxoethyl]-1-(4-cyano-2-pyridinyl)-N-(5-fluoro-3-pyridinyl)-5-oxo-, (2S)-
AG-120
Ivosidenib is an experimental drug for treatment of cancer. It is a small molecule inhibitor of IDH1, which is mutated in several forms of cancer. The drug is being developed by Agios Pharmaceuticals and is in phase III clinical trials. The FDA awarded orphan drug statusfor acute myeloid leukemia and cholangiocarcinoma.[1][better source needed]
It is in a phase III clinical trial for acute myeloid leukemia (AML) with an IDH1 mutation and a phase III clinical trial for cholangiocarcinoma with an IDH1 mutation.[2]
- OriginatorAgios Pharmaceuticals
- DeveloperAbbVie; Agios Pharmaceuticals; University of Texas M. D. Anderson Cancer Center
- ClassAntineoplastics; Cyclobutanes; Nitriles; Pyridines; Pyrrolidines; Small molecules
- Mechanism of ActionIsocitrate dehydrogenase 1 inhibitors
- Orphan Drug StatusYes – Acute myeloid leukaemia; Cholangiocarcinoma
- New Molecular EntityYes
Highest Development Phases
- PreregistrationAcute myeloid leukaemia
- Phase IIICholangiocarcinoma
- Phase IGlioma; Myelodysplastic syndromes; Solid tumours
Most Recent Events
- 28 Jun 2018Massachusetts General Hospital and Agios Pharmaceuticals plan a phase I trial for Acute myeloid leukaemia; Myelodysplastic syndromes and Chronic myelomonocytic leukaemia (Maintenance therapy) in USA (NCT03564821)
- 26 Jun 2018Ivosidenib licensed to CStone Pharmaceuticals in China, Hong Kong, Macau and Taiwan
- 14 Jun 2018Efficacy and adverse events data from a phase I trial in Acute myeloid leukaemia presented at the 23rd Congress of the European Haematology Association (EHA-2018)
References
External links
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C28H22ClF3N6O3 |
| Molar mass | 582.97 g·mol−1 |
| 3D model (JSmol) | |
///////////////Tibsovo, ivosidenib, fda 2018, Fast Track, Priority Review , Orphan Drug designation, UNII:Q2PCN8MAM6, ивосидениб , إيفوزيدينيب , 艾伏尼布 ,
FDA approves first cancer drug Kisqali (ribociclib) through new oncology review pilot that enables greater development efficiency FDA expands the use of breast cancer drug
FDA approves first cancer drug through new oncology review pilot that enables greater development efficiency FDA expands the use of breast cancer drug
The U.S. Food and Drug Administration today approved Kisqali (ribociclib) in combination with an aromatase inhibitor for the treatment of pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer, as initial endocrine-based therapy. The FDA also approved Kisqali in combination with fulvestrant for the treatment of postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer, as initial endocrine based therapy or following disease progression on endocrine therapy.
July 18, 2018
Release
The U.S. Food and Drug Administration today approved Kisqali (ribociclib) in combination with an aromatase inhibitor for the treatment of pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer, as initial endocrine-based therapy. The FDA also approved Kisqali in combination with fulvestrant for the treatment of postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer, as initial endocrine based therapy or following disease progression on endocrine therapy.
This is the first approval that FDA has granted as a part of two new pilot programs announced earlier this year that collectively aim to make the development and review of cancer drugs more efficient, while improving FDA’s rigorous standard for evaluating efficacy and safety. With this real-time review, the FDA was able to start evaluating the clinical data as soon as the trial results become available, enabling FDA to be ready to approve the new indication upon filing of a formal application with the Agency.
The first new program, called Real-Time Oncology Review, allows for the FDA to review much of the data earlier, after the clinical trial results become available and the database is locked, before the information is formally submitted to the FDA. This allows the FDA to begin its analysis of the data earlier, and provide feedback to the sponsor on how they can most effectively analyze the data to answer key regulatory questions. The pilot focuses on early submission of data that are the most relevant to assessing safety and effectiveness of the product. Then, when the sponsor submits the application with the FDA, the review team will already be familiar with the data and in a better position to conduct a more efficient, timely, and thorough review.
The second program is a new templated Assessment Aid that the applicant uses to organize its submission into a structured format to facilitate FDA’s review of the application. By using a structured template, the FDA is able to layer its assessment into the same file submitted by the sponsor, allowing this annotated application to serve as the document that contains the FDA review. This voluntary submission form provides for a more streamlined approach to reviewing data and illustrating FDA’s analysis. It allows for drug reviewers to focus on the key benefit-risk and labeling issues rather than administrative issues.
“With this approval, we’ve demonstrated some of the benefits of the new programs that we’re piloting for our review of cancer drugs, to improve regulatory efficiency while enhancing the process for evaluating the data submitted to us. This shows that, with smart policy approaches, we can gain efficiency while also improving the rigor of our process. These new programs were designed to reduce some of the administrative issues that can add to the time and cost of the review process, including the staffing burdens on the FDA. For example, by analyzing data earlier in the process, before formal submission to the FDA, and evaluating submissions in a structured template, we can make it easier to identify earlier when applications are missing key analysis or information that can delay reviews,” said FDA Commissioner Scott Gottlieb, M.D. “With today’s approval, the FDA used these new approaches to allow the review team to start analyzing data before the actual submission of the application and help guide the sponsor’s analysis of the top-line data to tease out the most relevant information. This enabled our approval less than one month after the June 28 submission date and several months ahead of the goal date.”
These new processes are good for patients, good for health care providers, good for product developers, and good for the FDA, by allowing our staff to have more time to engage with product developers and focus on the key aspects of drug reviews. We can improve efficiency and solidify our gold standard for review.”
Currently the two pilot programs are being used for supplemental applications for already-approved cancer drugs and could later be expanded to original drugs and biologics.
Kisqali was first approved in March 2017 for use with an AI to treat HR-positive, HER2-negative breast cancer in post-menopausal women whose cancer is advanced or has spread to other parts of the body.
“The approval adds a new treatment choice for patients with breast cancer,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to continuing to bring more treatment options to patients.”
Breast cancer is the most common form of cancer in the United States. The National Cancer Institute at the National Institutes of Health estimates approximately 266,120 women will be diagnosed with breast cancer this year and 40,920 will die of the disease. Approximately 72 percent of patients with breast cancer have tumors that are HR-positive and HER2-negative.
The efficacy of Kisqali in combination with an AI for pre/perimenopausal women was demonstrated in a clinical trial of 495 participants who received either Kisqali and an AI or placebo and an AI. All pre- or peri-menopausal patients on this study received ovarian suppression with goserelin. The trial measured progression-free survival (PFS), which is generally the amount of time after the start of this treatment during which the cancer does not substantially grow and the patient is alive. PFS was longer for patients taking Kisqali plus an AI (median PFS of 27.5 months) compared to patients who received placebo plus an AI (median PFS of 13.8 months).
The efficacy of Kisqali in combination with fulvestrant in treating advanced or metastatic breast cancer was demonstrated in a clinical trial that included 726 participants who received either Kisqali and fulvestrant or placebo and fulvestrant. The trial measured PFS, which was longer for patients taking Kisqali plus fulvestrant (median PFS of 20.5 months) compared to patients who received placebo plus fulvestrant (median PFS of 12.8 months).
The common side effects of Kisqali are infections, abnormally low count of a type of white blood cell (neutropenia), a reduction in the number of white cells in the blood (leukopenia), headache, cough, nausea, fatigue, diarrhea, vomiting, constipation, hair loss and rash.
Warnings include the risk of a heart problem known as QT prolongation that can cause an abnormal heartbeat and may lead to death, serious liver problems, low white blood cell counts that may result in infections that may be severe, and fetal harm.
The FDA granted Priority Review and Breakthrough Therapy designation for this indication.
The FDA granted this approval to Novartis Pharmaceuticals Corporation.
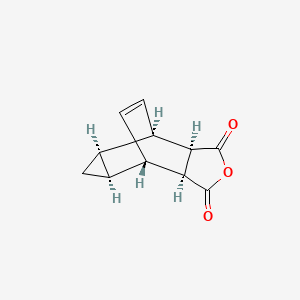
Tecovirimat

Tecovirimat
- Molecular FormulaC19H15F3N2O3
- Average mass376.329 Da
816458-31-8 [RN]
869572-92-9 [RN]
UNII-F925RR824R
тековиримат [Russian]
تيكوفيريمات [Arabic]
替韦立马 [Chinese]
Benzamide, N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-
N-[(1R,2R,6S,7S,8S,10R)-3,5-Dioxo-4-azatetracyclo[5.3.2.02,6.08,10]dodec-11-en-4-yl]-4-(trifluoromethyl)benzamide
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Tecovirimat monohydrate | SB96YO2BR8 | 1162664-19-8 | QRHXYGPOQKLBJP-NPIFKJBVSA-N |
Tecovirimat, sold under the brand name Tpoxx among others,[6] is an antiviral medication with activity against orthopoxviruses such as smallpox and monkeypox.[4][7][8] It is the first antipoxviral drug approved in the United States.[9][10] It is an inhibitor of the orthopoxvirus VP37 envelope wrapping protein.[4]
The drug works by blocking cellular transmission of the virus, thus preventing the disease.[11] Tecovirimat has been effective in laboratory testing; it has been shown to protect animals from monkeypox and rabbitpox and causes no serious side effects in humans.[6] Tecovirimat was first used for treatment in December 2018, after a laboratory-acquired vaccinia virus infection.[12]
Two million doses of tecovirimat are stockpiled in the US Strategic National Stockpile should an orthopoxvirus-based bioterror attack occur.[13][14] The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[15]
The World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980. However, there have been longstanding concerns that smallpox may be used as a bioweapon.2,5 Tecovirimat is an antiviral drug that was identified via a high-throughput screen in 2002.2 It is effective against all orthopoxviruses, including vaccinia, cowpox, ectromelia, rabbitpox, monkeypox, and Variola (smallpox) virus.1,4
Tecovirimat was approved by the FDA in July 2018 as the first drug ever approved to treat smallpox.6,5 Tecovirimat was later approved by Health Canada in December 2021,7 followed by the approval from the European Commission in January 2022.9 Other than smallpox, tecovirimat is also indicated to treat complications due to replication of the vaccinia virus following vaccination against smallpox, and to treat monkeypox and cowpox in adults and children.8 Tecovirimat is available as both oral and intravenous formulations.10
Medical uses
In the United States, tecovirimat is indicated for the treatment of human smallpox disease.[4] In the European Union it is indicated for the treatment of smallpox, monkeypox, and cowpox.[5]
Mechanism of action
Tecovirimat inhibits the function of a major envelope protein required for the production of extracellular virus. The drug prevents the virus from leaving an infected cell, hindering the spread of the virus within the body.[16]
Chemistry
The first synthesis of tecovirimat was published in a patent filed by scientists at Siga Technologies in 2004. It is made in two steps from cycloheptatriene.[17]
A Diels–Alder reaction with maleic anhydride forms the main ring system[18] and subsequent reaction with 4-trifluormethylbenzhydrazide gives the cyclic imide of the drug.[17][19]
Synthesis
US 9,546,137 [2017, to SIGA TECH INC]

SYNTHESIS FROM SMARTCHEM
The scheme has taken from SmartChem a knowledgebase by ROW2 Technologies, Inc. (www.row2technologies.com)
A perfect amalgamation of information on chemicals and global suppliers. A database where you can search for information on more than 150,000 chemicals and around 15,000 Global chemicals suppliers, including routes of synthesis, Applications, end uses, and validated contact details of global suppliers. For more information, please visit www.row2technologies.com or contact,
Anand Ramakrishnan (VP- Sales)
Tel : +1 973 795 1141
Mobile: +91 9821384045
Harshada Shivalkar (Manager-Business Development)
Mobile: +91 9323945301
hshivalkar@row2technologies.com
SYN 1

Synthetic Description
Reference: Dong, Ming-xin; Li, Hai-tao; Wang, Xiao-hua; Mao, Wen-xiang; Zhou, Shang-min; Dai, Qiu-yun. Preparation and structural determination of tecovirimat monohydrate crystal. Zhongguo Xinyao Zazhi. Volume 21. Issue 23. Pages 2736-2739. (2012).
SYN 2

Synthetic Description
Reference: Dai, Dongcheng. Process for the preparation of tecovirimat. Assignee Siga Technologies, Inc., USA. WO 2014028545. (2014).
SYN 3

Synthetic Description
Reference: Medical composition containing ST-246, its preparation and anti-poxvirus application. Assignee Institute of Microbiology and Epidemiology, Academy of Military Medical Sciences, PLA, Peop. Rep. China. CN 101912389. (2010).
EMA
Click to access tecovirimat-siga-epar-public-assessment-report_en.pdf
PATENT
https://patents.google.com/patent/US9546137B2/en
The present invention provides a process for making ST-246 outlined in Scheme 1

The present invention also provides a process for making ST-246 outlined in Scheme 2
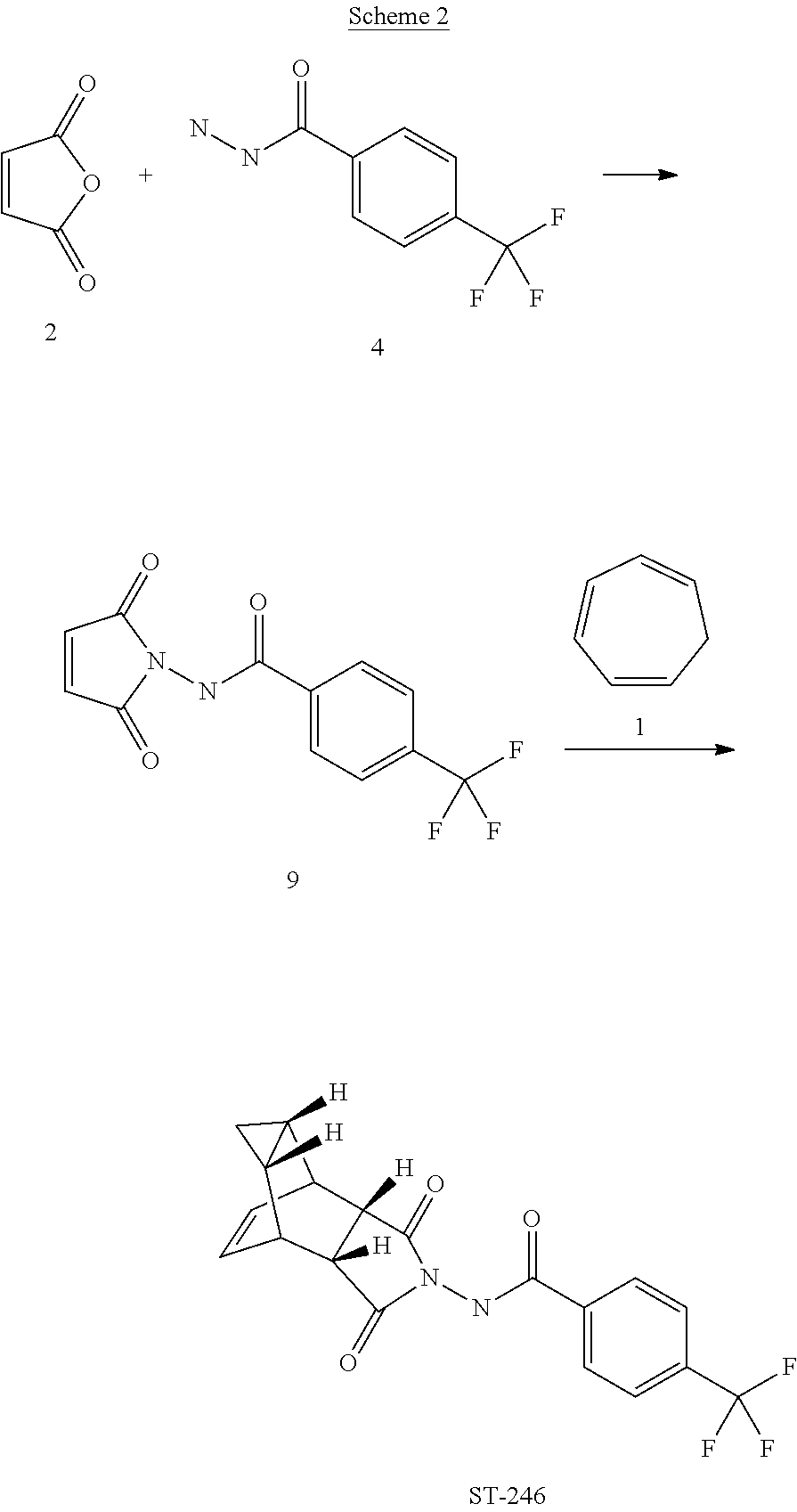
The present invention further provides a process for making ST-246 outlined in Scheme 3
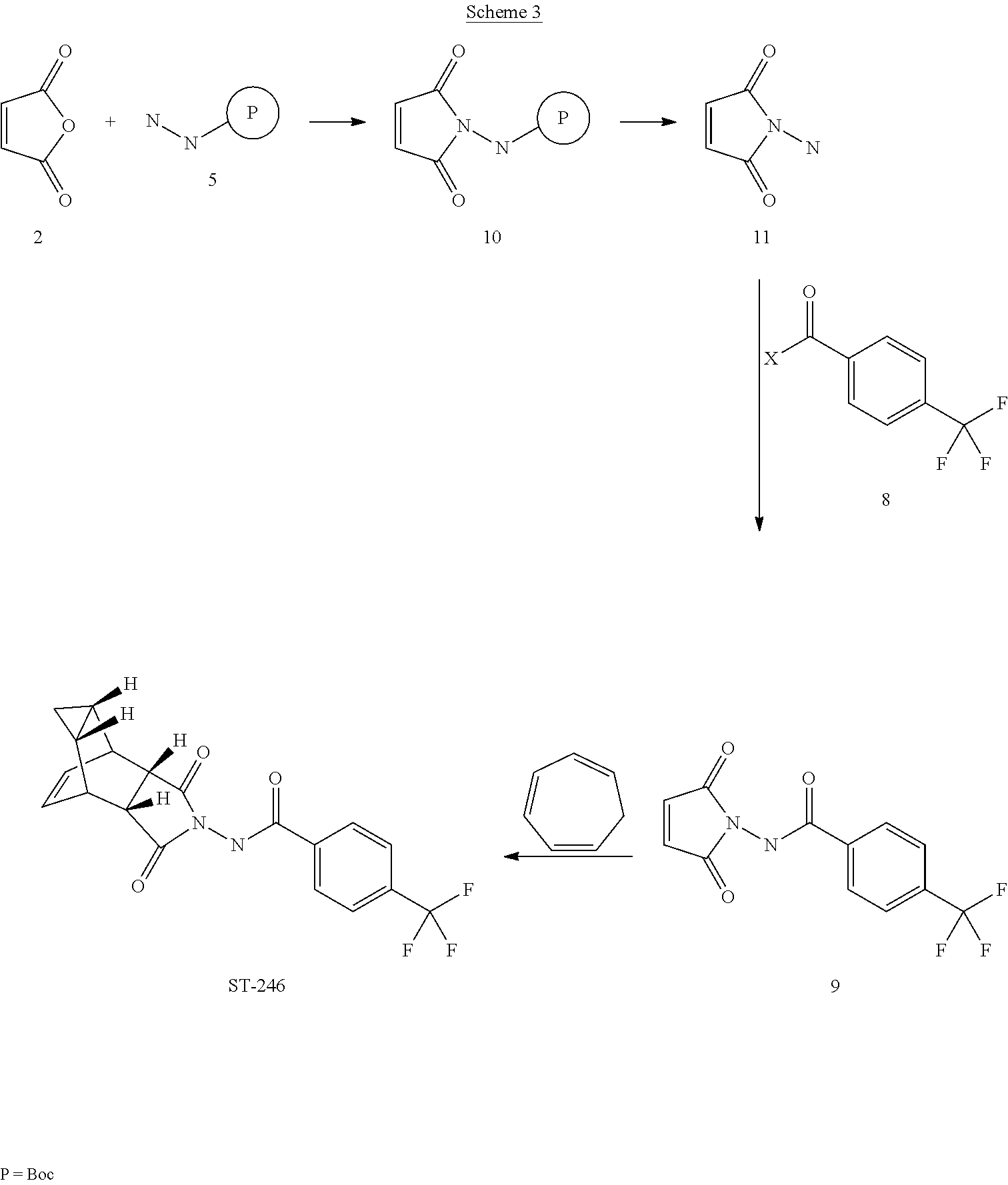
The present invention also provides a process for making ST-246 outlined in Scheme 4

The present invention further provides a process for making ST-246 outlined in Scheme 5

The present invention also provides the following compounds useful in the synthesis of ST-246:
EXAMPLE 1Synthetic Route I

Step A. Synthesis of Compound 6 (P=Boc)

Step A. Synthesis of Compound 9


Step A. Synthesis of Compound 10

Step B. Synthesis of Compound 11 (HCl salt)

Step A. Synthesis of Compound 10

Step B. Synthesis of Compound 6

Step A. Synthesis of Compound 13
Step A. Synthesis of Compound 6 (P=Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO04112718) in EtOH (80 mL, EMD, AX0441-3) was added tert-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc-hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCl3: δ 6.30 (br s, 1H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1.46 (s, 9H), 1.06-1.16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCl Salt)
Compound 6 (3.6 g, 11.83 mmol) was dissolved in i-PrOAc (65 mL, Aldrich, 99.6%). 4M HCl in dioxane (10.4 mL, 41.4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (15 mL) and dried under vacuum to yield HCl salt of compound 7 (1.9 g, 67% yield) as a white solid. The filtrate was concentrated to ⅓ its volume and stirred at 10-15° C. for 30 min. The solid was filtered, washed with minimal volume of i-PrOAc and dried to afford additional 0.6 g (21% yield) of compound 7. Total yield: 2.5 g (88% yield). 1H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1.07-1.17 (m, 2H), 0.18-0.29 (m, 1H), −0.01-0.07 (m, 1H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1.17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20° C. The resulting solution was stirred for 5 minutes at 15-20° C., to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15-20° C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH4Cl (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO04112718 and were consistent.
EXAMPLE 2Synthetic Route II
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (11.6%).
The reaction mixture was cooled to 45° C. and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1.5 g, 54% yield) as an off-white solid. 1H NMR in CDCl3: δ 8.44 (s, 1H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95° C. under nitrogen atmosphere. After 1.5 h at 95° C., LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo=94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95° C., LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 110° C. and the reaction was monitored. After heating at 110° C. for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo=94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO04112718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo=97:3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+
EXAMPLE 3Synthetic Route III
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and tert-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine by-product (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1H NMR in DMSO-d6: δ 9.61 (s, 1H), 7.16 (s, 2H), 1.42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
Step B. Synthesis of Compound 11 (HCl salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in i-PrOAc (57 mL, Aldrich, 99.6%). 4M HCl in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (10 mL) and dried at 45° C. under vacuum for 1 h to afford HCl salt of compound 11 (2.39 g, 89% yield) as a white solid. 1H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 113.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1.19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylamine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20° C. The resulting solution was stirred for 5 minute at 15-20° C. and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1.31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15-20° C. and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4Cl (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1.76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 110-115° C. under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo=94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo=93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25-35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo=99:1) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO04112718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo=91:9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
EXAMPLE 4Synthetic Route IV
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and tert-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 mL, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1.0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31.1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95° C. under nitrogen atmosphere. After 15 h at 95° C., LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105° C. overnight. After total 40 h at 95-105° C., LC-MS analysis at 254 nm showed ˜99% conversion to the desired product (endo:exo=93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25-50% EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo=91:9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCl salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in i-PrOAc (26 mL, Aldrich, 99.6%). 4M HCl in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20° C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with i-PrOAc (5 mL) and dried under vacuum to yield HCl salt of compound 7 (1.57 g, 97% yield) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1.34 mL, 7.7 mmol) keeping the temperature below 20° C. and the resulting solution was stirred for 5-10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20° C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4Cl (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30-35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO04112718.
EXAMPLE 5Synthetic Route V
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1.6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 mL,) was added triethylamine (2.04 mL, 14.63 mmol) keeping the temperature below 20° C. and the resulting solution was stirred for 5-10 minute. 4-Iodobenzoyl chloride 12 (1.95 g, 7.31 mmol, 1.1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20° C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20° C. After 24 h, additional 0.18 g (0.1 equiv, used total 1.6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and ˜5% of compound 7. The reaction was diluted with dichloromethane (100 mL). The organic phase was washed with saturated aqueous NH4Cl (100 mL), water (100 mL) and saturated aqueous NaHCO3 (100 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25-50% EtOAc in hexanes to afford compound 13 (1.63 g, 57% yield, HPLC area 93% pure) as a white solid. 1H NMR in DMSO-d6: δ 11.19 and 10.93 (two singlets with integration ratio of 1.73:1, total of 1H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1.18 (s, 2H), 0.27 (q, 1H), 0.06 (s, 1H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 mL) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (0.44 mL, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at −90° C. for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45° C. and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25-35% EtOAc in hexanes to afford ST-246 (55 mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO04112718.
PATENT
Example 1 : Synthetic Route I:

P = Boc
Scheme 1
Step A. Synthesis of Compound 6 (P = Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt) Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. Example 2: Synthetic Route II

Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).

Uncyclized product (MS = 303) Dimer by-product (MS = 489)
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co- injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III

ST-246 9
P = Boc Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+. duct

C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+ Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:

P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III. Im ine by-product

Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV) To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:

Scheme 5 Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55 mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
History
Originally researched by the National Institute of Allergy and Infectious Diseases, the drug was owned by Viropharma and discovered in collaboration with scientists at the United States Army Medical Research Institute of Infectious Diseases.[] It is owned and manufactured by Siga Technologies. Siga and Viropharma were issued a patent for tecovirimat in 2012.[20]
Clinical trials
As of 2009, the results of clinical trials support its use against smallpox and other related orthopoxviruses. It shows potential for a variety of uses including preventive healthcare, as a post-exposure therapeutic, as a therapeutic, and an adjunct to vaccination.[21][
Tecovirimat can be taken by mouth and as of 2008, was permitted for phase II trials by the U.S. Food and Drug Administration (FDA). In phase I trials, tecovirimat was generally well tolerated with no serious adverse events.[22] Due to its importance for biodefense, the FDA designated tecovirimat for fast-track status, creating a path for expedited FDA review and eventual regulatory approval. On 13 July 2018, the FDA announced approval of tecovirimat.[23]
Society and culture
Legal status
In November 2021, the Committee for Medicinal Products for Human Use of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization under exceptional circumstances for the medicinal product tecovirimat siga, intended for the treatment of orthopoxvirus disease (smallpox, monkeypox, cowpox, and vaccinia complications) in adults and in children who weigh at least 13 kilograms (29 lb)[24] The applicant for this medicinal product is Siga Technologies Netherlands B.V.[24] Tecovirimat was approved for medical use in the European Union in January 2022.[5][25]
In December 2021, Health Canada approved oral tecovirimat for the treatment of smallpox in people weighing at least 13 kilograms (29 lb).[26][27]
As of August 2022, Tpoxx is available in the US only through the Strategic National Stockpile as a Centers for Disease Control and Prevention investigational new drug.[28][29] Intravenous Tpoxx has no lower weight cap and can be used in infants under the investigational new drug protocol.[30]
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ “Notice: Multiple Additions to the Prescription Drug List (PDL) [2022-01-24]”. Health Canada. 24 January 2022. Archived from the original on 29 May 2022. Retrieved 28 May 2022.
- ^ “New Medicines Approved in 2018”. Health Canada. 15 January 2020. Archived from the original on 29 May 2022. Retrieved 28 May 2022.
- ^ “Summary Basis of Decision (SBD) for Tpoxx”. Health Canada. 23 October 2014. Archived from the original on 29 May 2022. Retrieved 29 May 2022.
- ^ Jump up to:a b c d “Tpoxx- tecovirimat monohydrate capsule”. DailyMed. 2 December 2021. Archived from the original on 23 April 2022. Retrieved 23 April 2022.
- ^ Jump up to:a b c “Tecovirimat Siga EPAR”. European Medicines Agency. 10 November 2021. Archived from the original on 16 May 2022. Retrieved 23 April 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b McNeil Jr DG (13 July 2018). “Drug to Treat Smallpox Approved by F.D.A., a Move Against Bioterrorism”. The New York Times. Archived from the original on 28 March 2019. Retrieved 16 July 2018.
- ^ Nakoune E, Olliaro P (May 2022). “Waking up to monkeypox”. BMJ. 377: o1321. doi:10.1136/bmj.o1321. PMID 35613732. S2CID 249047112.
- ^ Adler H, Gould S, Hine P, Snell LB, Wong W, Houlihan CF, et al. (May 2022). “Clinical features and management of human monkeypox: a retrospective observational study in the UK”. The Lancet. Infectious Diseases. 22 (8): 1153–1162. doi:10.1016/S1473-3099(22)00228-6. PMC 9300470. PMID 35623380. S2CID 249057804.
- ^ “FDA approves the first drug with an indication for treatment of smallpox”. U.S. Food and Drug Administration (FDA) (Press release). 13 July 2018. Archived from the original on 23 April 2019. Retrieved 1 August 2018.
- ^ “U.S. Food and Drug Administration Approves Siga Technologies’ Tpoxx (tecovirimat) for the Treatment of Smallpox”. Siga (Press release). Archived from the original on 21 September 2018. Retrieved 14 July 2018.
- ^ Grosenbach DW, Honeychurch K, Rose EA, Chinsangaram J, Frimm A, Maiti B, et al. (July 2018). “Oral Tecovirimat for the Treatment of Smallpox”. The New England Journal of Medicine. 379 (1): 44–53. doi:10.1056/NEJMoa1705688. PMC 6086581. PMID 29972742.
- ^ Whitehouse ER, Rao AK, Yu YC, Yu PA, Griffin M, Gorman S, et al. (October 2019). “Novel Treatment of a Vaccinia Virus Infection from an Occupational Needlestick – San Diego, California, 2019” (PDF). MMWR. Morbidity and Mortality Weekly Report. 68 (42): 943–946. doi:10.15585/mmwr.mm6842a2. PMC 6812835. PMID 31647789. Archived (PDF) from the original on 2 August 2022. Retrieved 2 August 2022.
- ^ Damon IK, Damaso CR, McFadden G (May 2014). “Are we there yet? The smallpox research agenda using variola virus”. PLOS Pathogens. 10 (5): e1004108. doi:10.1371/journal.ppat.1004108. PMC 4006926. PMID 24789223.
- ^ Cunningham A (13 July 2018). “FDA approves the first smallpox treatment”. Archived from the original on 12 July 2018. Retrieved 4 May 2018.
- ^ New Drug Therapy Approvals 2018 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2019. Archived from the original on 17 September 2020. Retrieved 16 September 2020.
- ^ Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, et al. (October 2005). “An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus Challenge”. Journal of Virology. 79 (20): 13139–13149. doi:10.1128/JVI.79.20.13139-13149.2005. PMC 1235851. PMID 16189015.
- ^ Jump up to:a b AU patent 2004249250, Bailey, Thomas R.; Jordan, Robert & Rippin, Susan R., “Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases”, published 2004-12-29, assigned to Siga Pharmaceuticals Inc
- ^ Ishitobi, Hiroyuki; Tanida, Hiroshi; Tori, Kazuo; Tsuji, Teruji (1971). “Re-examination of the Cycloaddition of Cycloheptatriene with Maleic Anhydride”. Bulletin of the Chemical Society of Japan. 44 (11): 2993–3000. doi:10.1246/bcsj.44.2993.
- ^ Hughes, David L. (2019). “Review of the Patent Literature: Synthesis and Final Forms of Antiviral Drugs Tecovirimat and Baloxavir Marboxil”. Organic Process Research & Development. 23 (7): 1298–1307. doi:10.1021/acs.oprd.9b00144. S2CID 197172102.
- ^ U.S. Patent 8,124,643
- ^ “Siga Technologies”. Archived from the original on 20 February 2012. Retrieved 18 September 2009.
- ^ Jordan R, Tien D, Bolken TC, Jones KF, Tyavanagimatt SR, Strasser J, et al. (May 2008). “Single-dose safety and pharmacokinetics of ST-246, a novel orthopoxvirus egress inhibitor”. Antimicrobial Agents and Chemotherapy. 52 (5): 1721–1727. doi:10.1128/AAC.01303-07. PMC 2346641. PMID 18316519.
- ^ Commissioner, Office of the (24 March 2020). “Press Announcements – FDA approves the first drug with an indication for treatment of smallpox”. U.S. Food and Drug Administration (FDA). Archived from the original on 23 April 2019. Retrieved 14 July 2018.
- ^ Jump up to:a b “Tecovirimat Siga: Pending EC decision”. European Medicines Agency. 11 November 2021. Archived from the original on 13 November 2021. Retrieved 13 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Summary of Product Characteristics” (PDF). European Medicines Agency. Archived (PDF) from the original on 21 May 2022. Retrieved 24 May 2022.
- ^ “Notice: Multiple Additions to the Prescription Drug List (PDL) [2022-01-24]”. Health Canada. 24 January 2022. Archived from the original on 29 May 2022. Retrieved 28 May 2022.
- ^ “Siga Announces Health Canada Regulatory Approval of Oral Tpoxx” (Press release). Siga Technologies. 1 December 2021. Archived from the original on 24 May 2022. Retrieved 24 May 2022.
- ^ “Information for Healthcare Providers on Obtaining and Using TPOXX (Tecovirimat) for Treatment of Monkeypox”. U.S. Centers for Disease Control and Prevention (CDC). 22 July 2022. Archived from the original on 31 July 2022. Retrieved 1 August 2022.
- ^ “Steps for Clinicians to Order Medication to Treat Monkeypox”. Coca Now. 19 July 2022. Archived from the original on 2 August 2022. Retrieved 24 July 2022.
- ^ “Monkeypox Outbreak: Updates on the Epidemiology, Testing, Treatment, and Vaccination” (PDF). U.S. Centers for Disease Control and Prevention. Archived (PDF) from the original on 2 August 2022. Retrieved 27 July 2022.
External links
- “Tecovirimat”. Drug Information Portal. U.S. National Library of Medicine.
- “Tecovirimat monohydrate”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Tpoxx |
| Other names | ST-246 |
| AHFS/Drugs.com | Monograph |
| License data |
|
| Routes of administration |
By mouth, intravenous |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C19H15F3N2O3 |
| Molar mass | 376.335 g·mol−1 |
| 3D model (JSmol) | |
FDA approves the first drug with an indication for treatment of smallpox
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
July 13, 2018
Release
The U.S. Food and Drug Administration today approved TPOXX (tecovirimat), the first drug with an indication for treatment of smallpox. Though the World Health Organization declared smallpox, a contagious and sometimes fatal infectious disease, eradicated in 1980, there have been longstanding concerns that smallpox could be used as a bioweapon.
“To address the risk of bioterrorism, Congress has taken steps to enable the development and approval of countermeasures to thwart pathogens that could be employed as weapons. Today’s approval provides an important milestone in these efforts. This new treatment affords us an additional option should smallpox ever be used as a bioweapon,” said FDA Commissioner Scott Gottlieb, M.D. “This is the first product to be awarded a Material Threat Medical Countermeasure priority review voucher. Today’s action reflects the FDA’s commitment to ensuring that the U.S. is prepared for any public health emergency with timely, safe and effective medical products.”
Prior to its eradication in 1980, variola virus, the virus that causes smallpox, was mainly spread by direct contact between people. Symptoms typically began 10 to 14 days after infection and included fever, exhaustion, headache and backache. A rash initially consisting of small, pink bumps progressed to pus-filled sores before finally crusting over and scarring. Complications of smallpox could include encephalitis (inflammation of the brain), corneal ulcerations (an open sore on the clear, front surface of the eye) and blindness.
TPOXX’s effectiveness against smallpox was established by studies conducted in animals infected with viruses that are closely related to the virus that causes smallpox, and was based on measuring survival at the end of the studies. More animals treated with TPOXX lived compared to the animals treated with placebo. TPOXX was approved under the FDA’s Animal Rule, which allows efficacy findings from adequate and well-controlled animal studies to support an FDA approval when it is not feasible or ethical to conduct efficacy trials in humans.
The safety of TPOXX was evaluated in 359 healthy human volunteers without a smallpox infection. The most frequently reported side effects were headache, nausea and abdominal pain.
The FDA granted this application Fast Track and Priority Review designations. TPOXX also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases and a Material Threat Medical Countermeasure Priority Review Voucher, which provides additional incentives for certain medical products intended to treat or prevent harm from specific chemical, biological, radiological and nuclear threats.
The FDA granted approval of TPOXX to SIGA Technologies Inc.
TPOXX was developed in conjunction with the U.S. Department of Health and Human Services’ Biomedical Advanced Research and Development Authority (BARDA).
Tecovirimat


Tecovirimat
4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop(f)isoindol-2(1H)-yl)-benzamide
N- [(3aR,4R,4aR,5aS,6S, 6aS)- 3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6- ethenocycloprop[f]iso- indol-2(1H)-yl]-4- (trifluoromethyl)- benzamide
4 -trifluoromethyl -N- (3, 3a, 4, 4a, 5, 5a, 6, 6a- octahydro-1, 3 -dioxo-4, 6 -ethenocycloprop [f] isoindol -2 ( 1H) -yl ) – benzamide
Details
NDA FILED IN US
2006 ORPHAN DRUG DESIGNATION IN US FOR SMALL POX
2010 ORPHAN DRUG DESIGNATION IN US FOR ORTHOPOX VIRUS
A core protein cysteine protease inhibitor potentially for treatment of smallpox infection.
SIGA TECHNOLOGIES INNOVATOR
SIGA-246; ST-246
SYN
Tecovirimat (Tpoxx)
Tecovirimat is a drug used for the
treatment or prophylaxis of viral infections, particularly those caused by the
orthopoxvirus (Figure 12).
In 2015, Dai described a procedure
for the preparation of tecovirimat in a
US patent (Scheme 33).[57 ] The developed method started with a cycloaddition reaction of cycloheptatriene
with maleic anhydride in xylene to
yield adduct 192, which after reaction
with tert-butyl carbazate provided compound 193. Deprotection in acidic media gave rise to hydrazine derivative 194 and
subsequent reaction with p-trifluoromethylbenzoyl chloride afforded tecovirimat (191).
57 [57] D. Dai, US Patent 0322010, 2015.
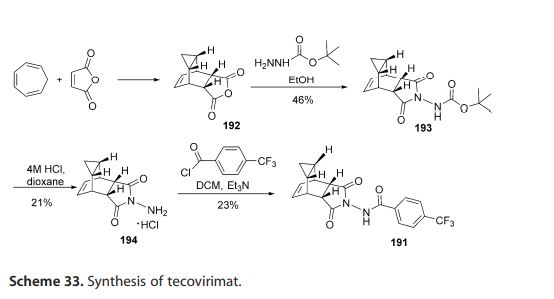
Synthesis
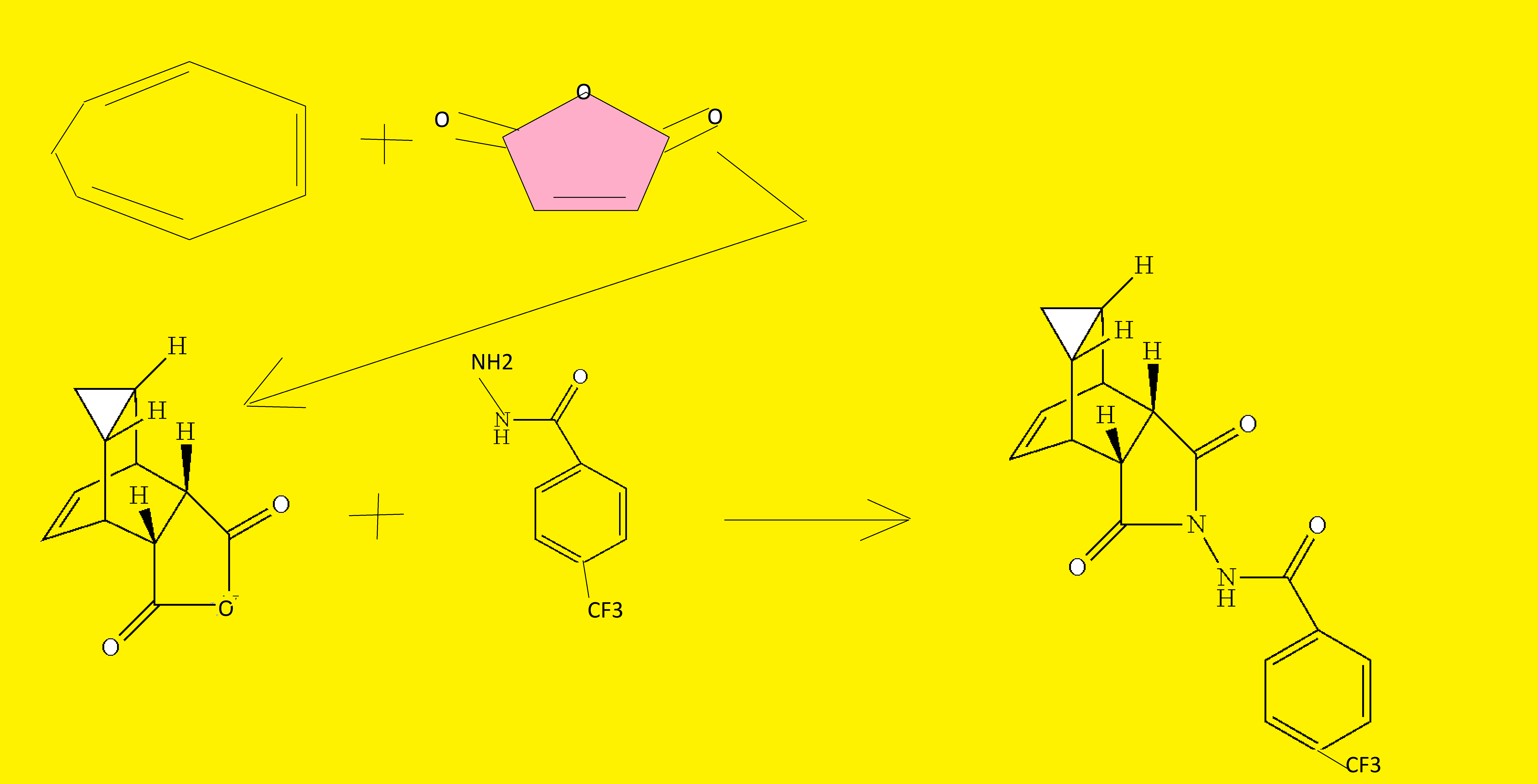
RAW MATERIAL
Key RM is, 4,6-Etheno-1H-cycloprop[f]isobenzofuran-1,3(3aH)-dione, 3a,4,4a,5,5a,6-hexahydro-, (3aR,4R,4aR,5aS,6S,6aS)-rel–
cas 944-41-2, [US7655688]
| Molecular Formula: | C11H10O3 |
|---|---|
| Molecular Weight: | 190.1953 g/mol |
- 4,6-Etheno-1H-cycloprop[f]isobenzofuran-1,3(3aH)-dione, 4,4a,5,5a,6,6a-hexahydro-, (3aα,4β,4aα,5aα,6β,6aα)-
- Tricyclo[3.2.2.02,4]non-8-ene-6,7-dicarboxylic anhydride, stereoisomer (8CI)
- 3,6-Cyclopropylene-Δ4-tetrahydrophthalic anhydride
MP 94-96 °C
Ref, Dong, Ming-xin; European Journal of Medicinal Chemistry 2010, V45(9), Pg 4096-4103
SMILES……….
O=C1OC(=O)[C@H]4[C@@H]1[C@H]3C=C[C@@H]4[C@@H]2C[C@@H]23
SYNTHESIS CONTINUED…….
ST-246
Patent
WO2014028545
The present invention provides a process for making ST-246 outlined in Scheme 1
P = Boc
Scheme 1
The present invention also provides a process for making ST-246 outlined in, Scheme 2
Scheme 2
The present invention further provides a process for making ST-246 outlined in Scheme 3
ST-246
P = Boc
Scheme 3
P = Boc
Scheme 4
The present invention further provides a process for making ST-246 outlined in
Scheme 5
Scheme 5
Example 1 : Synthetic Route I:
P = Boc
Scheme 1
Step A. Synthesis of Compound 6 (P = Boc)
To a mixture of compound 3 (5.0 g, 26.3 mmol, synthesized according to WO041 12718) in EtOH (80 mL, EMD, AX0441 -3) was added terf-butyl carbazate 5 (3.65 g, 27.6 mmol, Aldrich, 98%). The reaction mixture was heated to reflux for 4 h under nitrogen atmosphere. LC-MS analysis of the reaction mixture showed less than 5% of compound 3 remained. The reaction mixture was evaporated under reduced pressure. The residue was recrystallized from EtOAc – hexanes, the solid was filtered, washed with hexanes (50 mL) and dried under vacuum to afford compound 6 (3.1 g, 39% yield) as a white solid. The filtrate was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to give an additional 3.64 g (46% yield) of compound 6 as a white solid. Total yield: 6.74 g (84% yield). 1H NMR in CDCI3: δ 6.30 (br s, 1 H), 5.79 (t, 2H), 3.43 (s, 2H), 3.04 (s, 2H), 1 .46 (s, 9H), 1 .06-1 .16 (m, 2H), 0.18-0.36 (m, 2H); Mass Spec: 327.2 (M+Na)+
Step B. Synthesis of Compound 7 (HCI salt)
Compound 6 (3.6 g, 1 1 .83 mmol) was dissolved in /‘-PrOAc (65 mL, Aldrich, 99.6%). 4M HCI in dioxane (10.4 mL, 41 .4 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight (18 h) under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (15 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .9 g, 67% yield) as a white solid. The filtrate was concentrated to 1/3 its volume and stirred at 10 – 15 °C for 30 min. The solid was filtered, washed with minimal volume of /‘-PrOAc and dried to afford additional 0.6 g (21 % yield) of compound 7. Total yield: 2.5 g (88% yield). 1 H NMR in DMSO-d6: δ 6.72 (br s, 3H), 5.68 (m, 2H), 3.20 (s, 2H), 3.01 (s, 2H), 1 .07-1 .17 (m, 2H), 0.18-0.29 (m, 1 H), -0.01 -0.07 (m, 1 H); Mass Spec: 205.1 (M+H)+
Step C. Synthesis of ST-246
To a mixture of compound 7 (0.96 g, 4 mmol) in dry dichloromethane (19 mL) was added triethylamine (1 .17 mL, 8.4 mmol, Aldrich) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minutes at 15 – 20 °C, to it was added drop-wise 4-(trifluoromethyl)benzoyl chloride 8 (0.63 mL, 4.2 mmol, Aldrich, 97%) and the reaction mixture was stirred at room temperature overnight (18 h). LC-MS and TLC analysis showed the correct molecular weight and Rf value of ST-246 but the reaction was not complete. Additional 0.3 mL (2 mmol, 0.5 eq) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C. The reaction was then stirred at room temperature overnight (19 h). LC-MS analysis indicated ca. 5% of starting material 7 still remained. The reaction was stopped and dichloromethane (30 mL) was added. The organic phase was washed with water (30 mL), saturated aqueous NH CI (30 mL), water (15 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 -50% EtOAc in hexanes to afford ST-246 (0.34 g, 23% yield) as an off-white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent.
Example 2: Synthetic Route II
Scheme 2
Step A. Synthesis of Compound 9
A mixture of compound 4 (2.0 g, 9.8 mmol) and maleic anhydride 2 (0.96 g, 9.8 mmol, Aldrich powder, 95%) in o-xylene (100 mL, Aldrich anhydrous, 97%) was heated to reflux using a Dean-Stark trap apparatus overnight. After 18 h, LC-MS analysis at 215 nm showed the desired product 9 (86%), an uncyclized product (2.6%) and a dimer by-product (1 1 .6%).
Uncyclized product (MS = 303) Dimer by-product (MS = 489)
The reaction mixture was cooled to 45 °C and evaporated under reduced pressure. The residue was dissolved in EtOAc (50 mL) and the insoluble solid (mostly uncyclized product) was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 50% EtOAc in hexanes to yield compound 9 (1 .5 g, 54% yield) as an off-white solid. 1 H NMR in CDCI3: δ 8.44 (s, 1 H), 7.91 (d, 2H), 7.68 (d, 2H), 6.88 (s, 2H); Mass Spec: 285.1 (M+H)+
Step B. Synthesis of ST-246 (Route II)
A mixture of compound 9 (0.97 g, 3.4 mmol) and cycloheptatriene 1 (0.51 mL, 4.42 mmol, distilled before use, Aldrich tech 90%) in toluene (50 mL, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 1 .5 h at 95 °C, LC-MS analysis at 254 nm showed 29% conversion to the desired product (endo:exo = 94:6). The resulting solution was continued to be heated at same temperature overnight. After 18 h at 95 °C, LC-MS analysis indicated 75% conversion with an endo:exo ratio of 94:6. The reaction temperature was increased to 1 10 °C and the reaction was monitored. After heating at 1 10 °C for 7 h, LC-MS analysis at 254 nm showed 96.4% conversion to the desired product (endo:exo = 94:6). The volatiles were removed by evaporation under reduced pressure and the reside was purified by column chromatography eluting with 30% EtOAc in hexanes to afford ST-246 (0.29 g, 22.6% yield, HPLC area 99.7% pure and 100% endo isomer) as a white solid. Analytical data (1H NMR, LC-MS and HPLC by co-injection) were matched with those of ST-246 synthesized according to WO041 12718 and were consistent. An additional 0.5 g of ST-246 (38.9% yield, endo:exo = 97: 3) was recovered from column chromatography. Total Yield: 0.84 g (65.4% yield). 1H NMR of ST-246 exo isomer in CDCI3: δ 8.62 (s, 1 H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1 .17 (s, 2H), 0.24 (q, 1 H), 0.13 (m, 1 H); Mass Spec: 377.1 (M+H)+
Example 3: Synthetic Route III
ST-246 9
P = Boc
Scheme 3
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (15.2 g, 155 mmol, Aldrich powder 95%) and terf-butyl carbazate 5 (20.5 g, 155 mmol, Aldrich, 98%) in anhydrous toluene (150 mL, Aldrich anhydrous) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (20% by HPLC area), imine byproduct (18%) and disubstituted by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 25% EtOAc in hexanes to afford compound 10 (5.98 g, 18% yield, HPLC area >99.5% pure) as a white solid. 1 H NMR in DMSO-d6: δ 9.61 (s, 1 H), 7.16 (s, 2H), 1 .42 (s, 9H); Mass Spec: 235.1 (M+Na)+.
duct
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
C9H12N204 C14H22N405
Mol. Wt.: 212.2 Mol. Wt.: 326.35
Step B. Synthesis of Compound 11 (HCI salt)
Compound 10 (3.82 g, 18 mmol) was dissolved in /‘-PrOAc (57 mL, Aldrich, 99.6%). 4M HCI in dioxane (15.8 mL, 63 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (24 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (10 mL) and dried at 45 °C under vacuum for 1 h to afford HCI salt of compound 11 (2.39 g, 89% yield) as a white solid. 1 H NMR in CD3OD: δ 6.98 (s, 2H); Mass Spec: 1 13.0 (M+H)+
Step C. Synthesis of Compound 9 (Route III)
To a mixture of compound 11 (1 .19 g, 8 mmol) in dry dichloromethane (24 mL) was added diisopropylethylannine (2.93 mL, 16.8 mmol, Aldrich redistilled grade) keeping the temperature below 20 °C. The resulting solution was stirred for 5 minute at 15 – 20 °C and to it was added 4-(trifluoromethyl)benzoyl chloride 8 (1 .31 mL, 8.8 mmol, Aldrich, 97%) drop-wise. The reaction was stirred at room temperature for 5 h. LC-MS analysis showed the correct MW but the reaction was not complete. Additional 0.48 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction mixture at 15 – 20 °C and the reaction mixture was stirred at room temperature overnight (21 h). The reaction was stopped and dichloromethane (50 mL) was added. The organic phase was washed with water (50 mL), saturated aqueous NH4CI (50 mL), water (30 mL) and saturated aqueous NaHCO3 (30 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford compound 9 (0.8 g, 35% yield) as a light pink solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 9 obtained in Synthetic Route II.
Step D. Synthesis of ST-246 (Route III)
A mixture of compound 9 (0.5 g, 1 .76 mmol) and cycloheptatriene 1 (0.33 mL, 3.17 mmol, distilled before to use, Aldrich tech 90%) in toluene (10 mL, Aldrich anhydrous) was heated at 1 10 – 1 15 °C under nitrogen atmosphere. After 6 h, LC-MS analysis at 254 nm showed 95% conversion to the desired product (endo:exo = 94:6). The resulting solution was heated at same temperature overnight (22 h). LC-MS analysis at 254 nm showed no starting material 9 remained and the desired product (endo:exo = 93:7). The reaction mixture was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (0.39 g, HPLC area >99.5% pure with a ratio of endo:exo = 99:1 ) as a white solid. Analytical data (1 H NMR, LC-MS and HPLC by co-injection) were compared with those of ST-246 synthesized according to WO041 12718 and were found to be consistent. An additional 0.18 g of ST-246 (HPLC area >99.5% pure, endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 0.57 g (86% yield).
Example 4 ; Synthetic Route IV:
P = Boc
Scheme 4
Step A. Synthesis of Compound 10
A mixture of maleic anhydride 2 (3.4 g, 34.67 mmol, Aldrich powder, 95%) and terf-butyl carbazate 5 (4.6 g, 34.67 mmol, Aldrich, 98%) in anhydrous toluene (51 ml_, Aldrich) was heated to reflux using a Dean-Stark trap apparatus under nitrogen atmosphere. After refluxing for 2.5 h, no starting material 2 remained and LC-MS analysis at 254 nm showed the desired product 10 (19% HPLC area), imine by-product (18%) and another by-product (56%). The reaction mixture was concentrated and purified by column chromatography eluting with 30% EtOAc in hexanes to afford compound 10 (1 .0 g, 13.6% yield, HPLC area >99% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of compound 10 obtained in Synthetic Route III.
Im ine by-product
Mol. Wt.: 212.2
Step B. Synthesis of Compound 6
A mixture of compound 10 (4.4 g, 20.74 mmol) and cycloheptatriene 1 (3.22 mL, 31 .1 mmol, distilled before to use, Aldrich tech 90%) in toluene (88 mL, 20 volume, Aldrich anhydrous) was heated at 95 °C under nitrogen atmosphere. After 15 h at 95 °C, LC-MS analysis showed 83% conversion to the desired product. The reaction mixture was heated at 105 °C overnight. After total 40 h at 95 – 105 °C, LC-MS analysis at 254 nm showed -99% conversion to the desired product (endo:exo = 93:7). The reaction mixture was concentrated and the crude was purified by column chromatography eluting with 25 – 50 % EtOAc in hexanes to afford compound 6 (2.06 g, 32.6% yield, HPLC area 99.9% pure and 100% endo isomer) as a white solid. 1 H NMR and LC-MS were consistent with those of compound 6 obtained in Synthetic Route I. An additional 4.0 g of 6 (63.4% yield, HPLC area 93% pure with a ratio of endo:exo = 91 : 9) was recovered from column chromatography. Total Yield: 6.06 g (96% yield).
Step C. Synthesis of Compound 7 (HCI salt)
Compound 6 (2.05 g, 6.74 mmol) was dissolved in /‘-PrOAc (26 mL, Aldrich, 99.6%). 4M HCI in dioxane (5.9 mL, 23.58 mmol, Aldrich) was added drop-wise to the above solution keeping the temperature below 20 °C. The solution was stirred overnight (18 h) at room temperature under nitrogen atmosphere. The resulting solid was filtered, washed with /‘-PrOAc (5 mL) and dried under vacuum to yield HCI salt of compound 7 (1 .57 g, 97% yield) as a white solid. Analytical data (1 H NMR and LC-MS) were consistent with those of compound 7 in Synthetic Route I.
Step D. Synthesis of ST-246 (Route IV)
To a mixture of compound 7 (0.84 g, 3.5 mmol) in dichloromethane (13 mL) was added diisopropylethylamine (1 .34 mL, 7.7 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minutes. 4-(Trifluoromethyl)benzoyl chloride 8 (0.57 mL, 3.85 mmol, Aldrich, 97%) was added to above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature for 2 h. Additional 0.2 mL (0.4 equiv) of 4-(trifluoromethyl)benzoyl chloride 8 was added to the reaction keeping the temperature below 20 °C. The reaction was stirred at room temperature overnight (24 h). The reaction mixture was diluted with dichloromethane (20 mL). The organic phase was washed with water (20 mL), saturated aqueous NH4CI (20 mL), water (20 mL) and saturated aqueous NaHCO3 (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 30 – 35% EtOAc in hexanes to afford ST-246 (0.25 g, 19% yield, HPLC area >99.5% pure) as a white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
Example 5: Synthetic Route V:
Scheme 5
Step A. Synthesis of Compound 13
To a mixture of compound 7 (1 .6 g, 6.65 mmol, synthesized according to Synthetic Route I) in dichloromethane (80 ml_,) was added triethylamine (2.04 ml_, 14.63 mmol) keeping the temperature below 20 °C and the resulting solution was stirred for 5 – 10 minute. 4-lodobenzoyl chloride 12 (1 .95 g, 7.31 mmol, 1 .1 equiv, Aldrich) was added portion-wise under nitrogen atmosphere to the above solution keeping the temperature below 20 °C. The reaction mixture was stirred at room temperature overnight. After 17 h and 19 h, additional 0.35 g (0.2 equiv) of acid chloride 12 was added to the reaction keeping the temperature below 20 °C. After 24 h, additional 0.18 g (0.1 equiv, used total 1 .6 equiv) of acid chloride 12 was added and the reaction was continued to stir at room temperature overnight (total 43 h). LC-MS analysis at 215 nm showed 43% of the desired product (13) and -5% of compound 7. The reaction was diluted with dichloromethane (100 ml_). The organic phase was washed with saturated aqueous NH4CI (100 ml_), water (100 ml_) and saturated aqueous NaHCO3 (100 ml_). The organic phase was separated, dried over Na2SO4, filtered and concentrated to give crude product. The crude product was purified by column chromatography eluting with 25 – 50% EtOAc in hexanes to afford compound 13 (1 .63 g, 57% yield, HPLC area 93% pure) as a white solid. 1 H NMR in DMSO-d6: δ 1 1 .19 and 10.93 (two singlets with integration ratio of 1 .73:1 , total of 1 H, same proton of two rotamers), 7.93 (d, 2H), 7.66 (d, 2H), 5.80 (s, 2H), 3.36 (s, 2H), 3.27 (s, 2H), 1 .18 (s, 2H), 0.27 (q, 1 H), 0.06 (s,1 H); Mass Spec: 435.0 (M+H)+
Step B. Synthesis of ST-246 (Route V)
Anhydrous DMF (6 ml_) was added to a mixture of compound 13 (0.2 g, 0.46 mmol), methyl 2, 2-difluoro-2-(fluorosulfonyl)acetate (0.44 ml_, 3.45 mmol, Aldrich) and copper (I) iodide (90 mg, 0.47 mmol). The reaction mixture was stirred at -90 °C for 4 h. LC-MS analysis at 254 nm indicated no starting material 13 remained and showed 48% HPLC area of ST-246. The reaction mixture was cooled to 45 °C and DMF was removed under reduced pressure. The residue was slurried in EtOAc (30 mL) and insoluble solid was removed by filtration. The filtrate was concentrated and purified by column chromatography eluting with 25 – 35% EtOAc in hexanes to afford ST-246 (55
mg, 32% yield, 95% pure by HPLC at 254 nm) as off-white solid. Analytical data (1H NMR and LC-MS) were consistent with those of ST-246 synthesized according to WO041 12718.
PAPER
N-(3,3a,4,4a,5,5a,6,6a-Octahydro-1,3-dioxo-4,6- ethenocycloprop[f]isoindol-2-(1H)-yl)carboxamides: Identification of Novel Orthopoxvirus Egress Inhibitors
ViroPharma Incorporated, 397 Eagleview Boulevard, Exton, Pennsylvania 19341, United States Army Medical Research Institute of Infectious Diseases, 1425 Porter Street, Frederick, Maryland 21702, University of Alabama, Birmingham, Alabama 35294, and SIGA Technologies, Inc., 4575 SW Research Way, Corvallis, Oregon 97333
J. Med. Chem., 2007, 50 (7), pp 1442–1444
DOI: 10.1021/jm061484y
http://pubs.acs.org/doi/suppl/10.1021/jm061484y/suppl_file/jm061484ysi20070204_060607.pdf
General Procedure for synthesis of compounds 2-14, 16-18.
N-(3,3a,4,4a,5,5a,6,6aoctahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-4- (trifluoromethyl)benzamide (14).
A mixture of 2.00 g (9.8 mmol) of 4-(trifluoromethyl) benzoic acid hydrazide, 1.86 g (9.8 mmol) of 4,4a,5,5a,6,6a-hexahydro-4,6-etheno-1Hcycloprop[f]isobenzofuran-1,3(3aH)-dione, and one drop of diisopropylethylamine in 40 mL of absolute ethanol was refluxed for 4.5 h. Upon cooling to rt, 4 mL of water was added, and the product began to crystallize. The suspension was cooled in an ice bath, and the precipitate collected by filtration. The crystalline solid was air-dried affording 3.20 g (87%) of the product as a white solid;
Mp 194-195 ºC. 1 H NMR, (300 MHz, d6 -DMSO) δ 11.20, 11.09 (2 brs from rotamers, 1H), 8.06 (d, J= 7.8 Hz, 2H), 7.90 (d, J= 7.8 Hz, 2H), 5.78 (m, 2H), 3.26 (m, 4H), 1.15 (m, 2H), 0.24 (dd, J= 7.2, 12.9 Hz, 1H), 0.04 (m, 1H).
Anal. calcd. for C19H15F3N2O3● 0.25H2O: %C, 59.92; %H, 4.10; %F, 14.97; %N, 7.36; %O, 13.65. Found: %C, 59.97; %H, 4.02; %F, 14.94; %N, 7.36; %O, 13.71.
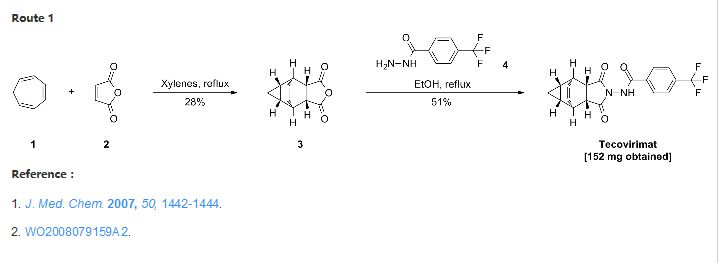
CLICK ON IMAGE
PATENT
US20140316145

CLICK ON IMAGE
http://www.google.com/patents/US8802714
Example 1
Preparation of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide
a. Preparation of Compounds 1(a) and 1(b).

A mixture of cycloheptatriene (5 g, 54.26 mmol) and maleic anhydride (6.13 g, 62.40 mmol) in xylenes (35 mL) was heated at reflux under argon overnight. The reaction was cooled to room temperature and a tan precipitate was collected by filtration and dried to give 2.94 grams (28%) of the desired product, which is a mixture of compounds 1(a) and 1(b). Compound 1(a) is normally predominant in this mixture and is at least 80% by weight. The purity of Compound 1(a) may be further enhanced by recrystallization if necessary. Compound 1(b), an isomer of compound 1(a) is normally less than 20% by weight and varies depending on the conditions of the reaction. Pure Compound 1(b) was obtained by concentrating the mother liquid to dryness and then subjecting the residue to column chromatography. Further purification can be carried out by recrystallization if necessary. 1H NMR (500 MHz) in CDCl3: δ 5.95 (m, 2H), 3.42 (m, 2H), 3.09 (m, 2H), 1.12 (m, 2H), 0.22 (m, 1H), 0.14 (m, 1H).
b. Preparation of N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. desired
A mixture of compound 1(a) (150 mg, 0.788 mmol) and 4-trifluoromethylbenzhydrazide (169 mg, 0.827 mmol) in ethanol (10 mL) was heated under argon overnight. The solvent was removed by rotary evaporation. Purification by column chromatography on silica gel using 1/1 hexane/ethyl acetate provided 152 mg (51%) of the product as a white solid.
c. Preparation of N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide. UNWANTED
N-[(3aR,4S,4aS,5aR,6R,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]4-(trifluoromethyl)-benzamide was prepared and purified in the same fashion as for N-[(3aR,4R,4aR,5aS,6S,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide by replacing 1(a) with 1(b) and was obtained as a white solid. 1H NMR (300 MHz) in CDCl3: δ 8.62 (s, 1H), 7.92 (d, 2H), 7.68 (d, 2H), 5.96 (m, 2H), 3.43 (s, 2H), 2.88 (s, 2H), 1.17 (s, 2H), 0.24 (q, 1H), 0.13 (m, 1H); Mass Spec: 377.1 (M+H)+.
FINAL COMPD SYNTHESIS
| TABLE 1 | ||||
| Example | **Mass | |||
| Number | R6 | *NMR | Spec | Name |
| 1 |
|
1H NMR in DMSO-d6: δ 11.35 (d, 1H); 11.09 (d, 1H); 8.08 (d, 2H); 7.92 (d, 2H); 5.799 (s, 2H); 3.29 (brs, 4H); 1.17 (m, 2H); 0.26 (m, 1H); 0.078 (s, 1H) | 375 (M − H)− | N-[(3aR,4R,4aR,5aS,6S, 6aS)-3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6-ethenocycloprop[f] isoindol-2(1H)-yl]-4- (trifluoromethyl)- benzamide |
TABLE 1 EXAMPLE 1
N- [(3aR,4R,4aR,5aS,6S, 6aS)- 3,3a,4,4a,5,5a,6,6a- octahydro-1,3-dioxo- 4,6- ethenocycloprop[f]iso- indol-2(1H)-yl]-4- (trifluoromethyl)- benzamide
1H NMR in DMSO-d6: δ 11.35 (d, 1H); 11.09 (d, 1H); 8.08 (d, 2H); 7.92 (d, 2H); 5.799 (s, 2H); 3.29 (brs, 4H); 1.17 (m, 2H); 0.26 (m, 1H); 0.078 (s, 1H), 375 (M − H)−
EXAMPLE 42 Characterization of 4-trifluoromethyl-N-(3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl)-benzamide (“ ”)
In the present application, ST-246 refers to: N-[(3aR,4R,4aR,5aS,65,6aS)-3,3a,4,4a,5,5a,6,6a-octahydro-1,3-dioxo-4,6-ethenocycloprop[f]isoindol-2(1H)-yl]-4-(trifluoromethyl)-benzamide.
Physico-Chemical Properties
Appearance: ST-246 is a white to off-white powder.
Melting Point: Approximately 196° C. by DSC.
Permeability: The calculated log P is 2.94. Based on the partition coefficient, ST-246 is expected to have good permeability.
Particle Size: The drug substance is micronized to improve its dissolution in the gastrointestinal fluids. The typical particle size of the micronized material is 50% less than 5 microns.
Solubility: The solubility of ST-246 is low in water (0.026 mg/mL) and buffers of the gastric pH range. Surfactant increases its solubility slightly. ST-246 is very soluble in organic solvents. The solubility data are given in Table 5.


CLICK ON IMAGE
PATENT
http://www.google.com/patents/CN101445478A?cl=en


References
- Damon, Inger K.; Damaso, Clarissa R.; McFadden, Grant (2014). “Are We There Yet? The Smallpox Research Agenda Using Variola Virus”. PLoS Pathogens 10 (5): e1004108.doi:10.1371/journal.ppat.1004108. PMID 24789223.
- Siga Technologies
- Jordan, R; Tien, D; Bolken, T. C.; Jones, K. F.; Tyavanagimatt, S. R.; Strasser, J; Frimm, A; Corrado, M. L.; Strome, P. G.; Hruby, D. E. (2008). “Single-Dose Safety and Pharmacokinetics of ST-246, a Novel Orthopoxvirus Egress Inhibitor”. Antimicrobial Agents and Chemotherapy 52 (5): 1721–1727. doi:10.1128/AAC.01303-07. PMC 2346641. PMID 18316519.
- Yang, G; Pevear, D. C.; Davies, M. H.; Collett, M. S.; Bailey, T; Rippen, S; Barone, L; Burns, C; Rhodes, G; Tohan, S; Huggins, J. W.; Baker, R. O.; Buller, R. L.; Touchette, E; Waller, K; Schriewer, J; Neyts, J; Declercq, E; Jones, K; Hruby, D; Jordan, R (2005). “An Orally Bioavailable Antipoxvirus Compound (ST-246) Inhibits Extracellular Virus Formation and Protects Mice from Lethal Orthopoxvirus Challenge”. Journal of Virology 79 (20): 13139–13149. doi:10.1128/JVI.79.20.13139-13149.2005. PMC 1235851. PMID 16189015.
Referenced by
Citing Patent Filing date Publication date Applicant Title
CN101912389A * Aug 9, 2010 Dec 15, 2010 中国人民解放军军事医学科学院微生物流行病研究所 Pharmaceutical composition containing ST-246 and preparation method and application thereof
CN102406617A * Nov 30, 2011 Apr 11, 2012 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof
CN102406617B Nov 30, 2011 Aug 28, 2013 中国人民解放军军事医学科学院生物工程研究所 Tecovirimat dry suspension and preparation method thereof
CN103068232B * Mar 23, 2011 Aug 26, 2015 西佳科技股份有限公司 多晶型物形式st-246和制备方法
US8530509 Jul 29, 2011 Sep 10, 2013 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases
US8802714 Aug 14, 2013 Aug 12, 2014 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases
US9045418 Jul 3, 2014 Jun 2, 2015 Siga Technologies, Inc. Compounds, compositions and methods for treatment and prevention of Orthopoxvirus infections and associated diseases
Patent Citations
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20070287735 * | Apr 23, 2007 | Dec 13, 2007 | Siga Technologies, Inc. | Chemicals, compositions, and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US20090011037 * | Apr 23, 2008 | Jan 8, 2009 | Cydex Pharmaceuticals, Inc. | Sulfoalkyl Ether Cyclodextrin Compositions and Methods of Preparation Thereof |
| Citing Patent | Filing date | Publication date | Applicant | Title |
| US8530509 | Jul 29, 2011 | Sep 10, 2013 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US8802714 | Aug 14, 2013 | Aug 12, 2014 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases |
| US9045418 | Jul 3, 2014 | Jun 2, 2015 | Siga Technologies, Inc. | Compounds, compositions and methods for treatment and prevention of Orthopoxvirus infections and associated diseases |
//////////////////Tecovirimat, FDA 2018, ORPHAN DRUG DESIGNATION, TPOXX, SIGA Technologies Inc, Fast Track, Priority Review,
UNII-F925RR824R, тековиримат , تيكوفيريمات , 替韦立马 ,
FC(F)(F)c1ccc(cc1)C(=O)NN1C(=O)C2C(C3C=CC2C2CC32)C1=O

FDA approves novel device Zephyr Endobronchial Valve (Zephyr Valve) for treating breathing difficulty from severe emphysema

Depiction of the Zephyr ® endobronchial valve. Image courtesy of Pulmonx, Inc.
FDA approves novel device for treating breathing difficulty from severe emphysema
The U.S. Food and Drug Administration today approved a new device, the Zephyr Endobronchial Valve (Zephyr Valve), intended to treat breathing difficulty associated with severe emphysema.
The U.S. Food and Drug Administration today approved a new device, the Zephyr Endobronchial Valve (Zephyr Valve), intended to treat breathing difficulty associated with severe emphysema.
“Treatment options are limited for people with emphysema who have severe symptoms that have not improved from taking medicines. These have included lung surgery, such as lung volume reduction or lung transplants, which may not be suitable or appropriate for all patients,” said Tina Kiang, Ph.D., acting director, Division of Anesthesiology, General Hospital, Respiratory, Infection Control and Dental Devices, in the FDA’s Center for Devices and Radiological Health. “This novel device is a less invasive treatment that expands the options available to patients.”
June 29, 2018
Release
The U.S. Food and Drug Administration today approved a new device, the Zephyr Endobronchial Valve (Zephyr Valve), intended to treat breathing difficulty associated with severe emphysema.
“Treatment options are limited for people with emphysema who have severe symptoms that have not improved from taking medicines. These have included lung surgery, such as lung volume reduction or lung transplants, which may not be suitable or appropriate for all patients,” said Tina Kiang, Ph.D., acting director, Division of Anesthesiology, General Hospital, Respiratory, Infection Control and Dental Devices, in the FDA’s Center for Devices and Radiological Health. “This novel device is a less invasive treatment that expands the options available to patients.”
The Centers for Disease Control and Prevention estimates that 3.5 million American adults have been diagnosed with emphysema. Emphysema, including severe emphysema, is a type of chronic obstructive pulmonary disease (COPD) due to damage to the air sacs (alveoli) in the lungs. Lung damage from emphysema is irreversible. The damaged alveoli can cause used air to become trapped in the lungs during exhalation. This can cause the diseased parts of the lung to get larger and put pressure on the healthy part of the lung, which makes it difficult to breathe. As a result, the body may not get the oxygen it needs.
Using a flexible bronchoscope, a doctor places Zephyr Valves, similar in size to pencil erasers, into the diseased areas of the lung airways during a procedure in a hospital setting. Design of the device is intended to prevent air from entering the damaged parts of the lung and allow trapped air and fluids to escape. During inhalation, the valves close, preventing air from entering the damaged part of the lung and during exhalation, the valves open, letting out trapped air, which is intended to relieve pressure.
The FDA reviewed data from a multi-center study of 190 patients with severe emphysema. In this study, 128 patients were treated with Zephyr Valves and medical management according to current clinical guidelines, including medications (bronchodilators, corticosteroids, antibiotics or anti-inflammatory maintenance medications) and pulmonary rehabilitation, while 62 patients (the control group) received medical management only. Results of treatment were measured by how many patients in each arm of the study had at least a 15 percent improvement in pulmonary function scores (the volume of air that can forcibly be blown out in one second after full inhalation). At one year, 47.7 percent of patients treated with Zephyr Valves experienced at least a 15 percent improvement in their pulmonary function scores, compared with 16.8 percent of patients in the control group. Adverse events observed in the study include death, air leak (pneumothorax), pneumonia, worsening of emphysema, coughing up blood, shortness of breath and chest pain.
The Zephyr Valve device is contraindicated for patients with active lung infections; those who are allergic to nitinol, nickel, titanium or silicone; active smokers and those who are not able to tolerate the bronchoscopic procedure. Patients who have had major lung procedures, heart disease, large bubbles of air trapped in the lung or who have not responded to other treatments should talk with their providers to determine if the Zephyr Valve device is appropriate for them.
The Zephyr Valve was granted Breakthrough Device designation, meaning the FDA provided intensive interaction and guidance to the company on efficient device development, to expedite evidence generation and the agency’s review of the device. To qualify for such designation, a device must provide for more effective treatment or diagnosis of a life-threatening or irreversibly debilitating disease or condition, and meet one of the following criteria: the device must represent a breakthrough technology; there must be no approved or cleared alternatives; the device must offer significant advantages over existing approved or cleared alternatives; or the availability of the device is in the best interest of patients.
The FDA reviewed the Zephyr Valve device through the premarket approval review pathway, a regulatory pathway for the highest risk class of devices.
The FDA granted approval of the Zephyr Valve device to Pulmonx Inc.
////////////fda 2018, medical devices, Zephyr Valve device, Pulmonx Inc, Breakthrough Device designation, Zephyr Endobronchial Valve, Zephyr Valve,
FDA approves first drug Epidiolex (cannabidiol) comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
June 25, 2018
Release
The U.S. Food and Drug Administration today approved Epidiolex (cannabidiol) [CBD] oral solution for the treatment of seizures associated with two rare and severe forms of epilepsy, Lennox-Gastaut syndrome and Dravet syndrome, in patients two years of age and older. This is the first FDA-approved drug that contains a purified drug substance derived from marijuana. It is also the first FDA approval of a drug for the treatment of patients with Dravet syndrome.
CBD is a chemical component of the Cannabis sativa plant, more commonly known as marijuana. However, CBD does not cause intoxication or euphoria (the “high”) that comes from tetrahydrocannabinol (THC).
It is THC (and not CBD) that is the primary psychoactive component of marijuana.
“This approval serves as a reminder that advancing sound development programs that properly evaluate active ingredients contained in marijuana can lead to important medical therapies. And, the FDA is committed to this kind of careful scientific research and drug development,” said FDA Commissioner Scott Gottlieb, M.D. “Controlled clinical trials testing the safety and efficacy of a drug, along with careful review through the FDA’s drug approval process, is the most appropriate way to bring marijuana-derived treatments to patients. Because of the adequate and well-controlled clinical studies that supported this approval, prescribers can have confidence in the drug’s uniform strength and consistent delivery that support appropriate dosing needed for treating patients with these complex and serious epilepsy syndromes. We’ll continue to support rigorous scientific research on the potential medical uses of marijuana-derived products and work with product developers who are interested in bringing patients safe and effective, high quality products. But, at the same time, we are prepared to take action when we see the illegal marketing of CBD-containing products with serious, unproven medical claims. Marketing unapproved products, with uncertain dosages and formulations can keep patients from accessing appropriate, recognized therapies to treat serious and even fatal diseases.”
Dravet syndrome is a rare genetic condition that appears during the first year of life with frequent fever-related seizures (febrile seizures). Later, other types of seizures typically arise, including myoclonic seizures (involuntary muscle spasms). Additionally, status epilepticus, a potentially life-threatening state of continuous seizure activity requiring emergency medical care, may occur. Children with Dravet syndrome typically experience poor development of language and motor skills, hyperactivity and difficulty relating to others.
Lennox-Gastaut syndrome begins in childhood. It is characterized by multiple types of seizures. People with Lennox-Gastaut syndrome begin having frequent seizures in early childhood, usually between ages 3 and 5. More than three-quarters of affected individuals have tonic seizures, which cause the muscles to contract uncontrollably. Almost all children with Lennox-Gastaut syndrome develop learning problems and intellectual disability. Many also have delayed development of motor skills such as sitting and crawling. Most people with Lennox-Gastaut syndrome require help with usual activities of daily living.
“The difficult-to-control seizures that patients with Dravet syndrome and Lennox-Gastaut syndrome experience have a profound impact on these patients’ quality of life,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition.”
Epidiolex’s effectiveness was studied in three randomized, double-blind, placebo-controlled clinical trials involving 516 patients with either Lennox-Gastaut syndrome or Dravet syndrome. Epidiolex, taken along with other medications, was shown to be effective in reducing the frequency of seizures when compared with placebo.
The most common side effects that occurred in Epidiolex-treated patients in the clinical trials were: sleepiness, sedation and lethargy; elevated liver enzymes; decreased appetite; diarrhea; rash; fatigue, malaise and weakness; insomnia, sleep disorder and poor quality sleep; and infections.
Epidiolex must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. As is true for all drugs that treat epilepsy, the most serious risks include thoughts about suicide, attempts to commit suicide, feelings of agitation, new or worsening depression, aggression and panic attacks. Epidiolex also caused liver injury, generally mild, but raising the possibility of rare, but more severe injury. More severe liver injury can cause nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice and/or dark urine.
Under the Controlled Substances Act (CSA), CBD is currently a Schedule I substance because it is a chemical component of the cannabis plant. In support of this application, the company conducted nonclinical and clinical studies to assess the abuse potential of CBD.
The FDA prepares and transmits, through the U.S. Department of Health and Human Services, a medical and scientific analysis of substances subject to scheduling, like CBD, and provides recommendations to the Drug Enforcement Administration (DEA) regarding controls under the CSA. DEA is required to make a scheduling determination.
The FDA granted Priority Review designation for this application. Fast-Track designation was granted for Dravet syndrome. Orphan Drug designation was granted for both the Dravet syndrome and Lennox-Gastaut syndrome indications.
The FDA granted approval of Epidiolex to GW Research Ltd.

/////////// Epidiolex, cannabidiol, fda 2018, Dravet syndrome, epilepsy, Priority Review , Fast-Track designation, Orphan Drug designation
Tildrakizumab-asmn
| Heavy chain: | |
| QVQLVQSGAEVKKPGASVKVSCKASGYIFITYWMTWVRQAPGQGL | |
| EWMGQIFPASGSADYNEKFEGRVTMTTDTSTSTAYMELRSLRSDD | |
| TAVYYCARGGGGFAYWGQGTLVTVSSASTKGPSVFPLAPSSKSTS | |
| GGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS | |
| LSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTC | |
| PPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDP | |
| EVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNG | |
| KEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKN | |
| QVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFL | |
| YSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK | |
| Light chain: | |
| DIQMTQSPSSLSASVGDRVTITCRTSENIYSYLAWYQQKPGKAPK | |
| LLIYNAKTLAEGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQH | |
| HYGIPFTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCL | |
| LNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT | |
| LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC |
Tildrakizumab-asmn
Immunoglobulin G1, anti-(human interleukin 23) (human-Mus musculus monoclonal heavy chain), disulfide with human-Mus musculus monoclonal light chain, dimer
CAS 1326244-10-3, BLA 761067
Tildrakizumab (SCH 900222/MK-3222)
ILUMYA; MK-3222; SCH-900222; SUNPG 1622; SUNPG 1622 I; SUNPG 1623 I; SUNPG 1623 II; SUNPG 1623 III; SUNPG 1623 IV; SUNPG1623; Tildrakizumab-asmn
DRUG BANK https://www.drugbank.ca/drugs/DB14004
Company Sun Pharmaceuticals
Approval Status FDA Approved March 2018 FOR Psoriasis, plaque
Treatments plaque psoriasis
Protein chemical formulaC6426H9918N1698O2000S46
Protein average weight144400.0 DaSequences
>Tildrakizumab Sequence MLGSRAVMLLLLLPWTAQGRAVPGGSSPAWTQCQQLSQKLCTLAWSAHPLVGHMDLREEG DEETTNDVPHIQCGDGCDPQGLRDNSQFCLQRIHQGLIFYEKLLGSDIFTGEPSLLPDSP VGQLHASLLGLSQLLQPEGHHWETQQIPSLSPSQPWQRLLLRFKILRSLQAFVAVAARVF AHGAATLSP
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | IL23 |
| Clinical data | |
| Trade names | Ilumya |
| Synonyms | Tildrakizumab-asmn |
| Routes of administration |
Subcutaneous injection |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
| KEGG | |
| Chemical and physical data | |
| Formula | C6426H9918N1698O2000S46 |
| Molar mass | 144.4 kg/mol |
- Originator Schering-Plough
- Developer Almirall S.A.; Merck & Co; Schering-Plough; Sun Pharmaceutical Industries
- Class Antipsoriatics; Monoclonal antibodies
- Mechanism of Action Interleukin 23 inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- Registered Plaque psoriasis
- Phase II Ankylosing spondylitis; Psoriatic arthritis
- Discontinued Autoimmune disorders
Most Recent Events
- 21 Mar 2018 Registered for Plaque psoriasis in USA (SC) – First global approval
- 16 Feb 2018 Adverse events data from two phase III trials (reSURFACE 1 and 2) in chronic Plaque psoriasis presented at the 76th Annual Meeting of the American Academy of Dermatology (AAD-2018)
- 16 Feb 2018 Pharmacokinetics data from population PK model in healthy volunteers and patients with psoriasis presented at the 76th Annual Meeting of the American Academy of Dermatology (AAD-2018)
Ilumya (tildrakizumab-asmn) is an interleukin-23 antagonist.
Humanized monoclonal IgG1-kappa antibody against IL-23p19; produced in CHO cells
Immunoglobulin G1, anti-(human interleukin 23) (human-Mus musculus monoclonal heavy chain), disulfide with human-Mus musculus monoclonal light chain, dimer
Ilumya is specifically indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
Ilumya is supplied as a solution for subcutaneous injection. The recommended dose is 100 mg at Weeks 0, 4, and every twelve weeks thereafter.

Tildrakizumab (Ilumya) is a monoclonal antibody designed for the treatment of immunologically mediated inflammatory disorders.[1] In the United States, it is approved for the treatment of moderate-to-severe plaque psoriasis.[2]
Tildrakizumab was designed to block interleukin-23, a cytokine that plays an important role in managing the immune system and autoimmune disease. Originally developed by Schering-Plough, this drug is now part of Merck‘s clinical program, following that company’s acquisition of Schering-Plough.
Sun Pharmaceutical acquired worldwide rights to tildrakizumab for use in all human indications from Merck in exchange for an upfront payment of U.S. $80 million. Upon product approval, Sun Pharmaceutical will be responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. [3]

As of March 2014, the drug was in phase III clinical trials for plaque psoriasis. The two trials enrolled nearly 2000 patients. [4][5]
In 2016, tildrakizumab became the first IL-23p19 inhibitor to demonstrate positive results in Phase-3 clinical trials for the treatment of moderate-to-severe plaque psoriasis, further validating the importance of the role of IL-23 in psoriasis. Sun Pharma signed a licensing pact with Spain’s Almirall for marketing tildrakizumab in Europe [6]
In March 2018, it was approved by the Food and Drug Administration for the treatment of moderate-to-severe plaque psoriasis as an injection for subcutaneous use in the United States.[2]
In 2014, Sun Pharma acquired worldwide rights to tildrakizumab from Merck; upon product approval, Sun Pharma is responsible for regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the product. In 2016, Almirall sublicensed the product for the development and marketing in Europe for the treatment of psoriasis.
See also
- Ustekinumab, a monoclonal antibody targeting both IL-12 and IL-23 and used to treat plaque psoriasis, launched in the United States under the brand name Stelara
- Guselkumab, another experimental, IL-23-specific monoclonal antibody. (FDA approved in 2017)
- Risankizumab, another experimental, IL-23-specific monoclonal antibody. (In Phase 3 clinical trials for plaque psoriasis as of 2017)
References
- Jump up^ Statement On A Nonproprietary Name Adopted By The USAN Council – Tildrakizumab, American Medical Association.
- ^ Jump up to:a b “FDA approves Ilumya for plaque psoriasis”. National Psoriasis Foundation. March 22, 2018.
- Jump up^ http://www.merck.com/licensing/our-partnership/sunpharma_partnership.html
- Jump up^ http://clinicaltrials.gov/ct2/show/NCT01729754?term=SCH-900222&phase=2&fund=2&rank=1
- Jump up^ http://clinicaltrials.gov/ct2/show/NCT01722331?term=SCH-900222&phase=2&fund=2&rank=2
- Jump up^ http://www.business-standard.com/content/b2b-pharma/sun-pharma-signs-licensing-pact-with-spain-s-almirall-for-tildrakizumab-in-europe-116072800225_1.html
Mechanism of Action
Tildrakizumab is a humanized IgG1/k monoclonal antibody that selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor. IL-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses. Tildrakizumab inhibits the release of proinflammatory cytokines and chemokines.
FDA APPROVAL DATA
BLA 761067
https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/761067Orig1s000REPLACEMENT_ltr.pdf
Please refer to your Biologics License Application (BLA) dated and received March 23, 2017 and your amendments, submitted under section 351(a) of the Public Health Service Act for ILUMYA (tildrakizumab-asmn) injection. We also refer to our approval letter dated March 20, 2018 which contained the following error: the Final Report Submission date was incorrectly listed for postmarketing requirement 3357-3. This replacement approval letter incorporates the correction of the error. The effective approval date will remain March 20, 2018, the date of the original approval letter.
LICENSING We have approved your BLA for ILUMYA (tildrakizumab-asmn) effective this date. You are hereby authorized to introduce or deliver for introduction into interstate commerce, ILUMYA under your existing Department of Health and Human Services U.S. License No. 0002. ILUMYA is indicated for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy.
MANUFACTURING LOCATIONS Under this license, you are approved to manufacture ILUMYA drug substance at . The final formulated drug product will be manufactured, filled, labeled, and packaged at MSD Ireland, Carlow, Ireland. You may label your product with the proprietary name, ILUMYA, and market it in 100 mg/1 mL single-dose prefilled syringe
DATING PERIOD The dating period for ILUMYA drug product shall be 36 months from the date of manufacture when stored at 2-8°C. The date of manufacture shall be defined as the date of final sterile filtration of the formulated drug product. The dating period for your drug substance shall be months from the date of manufacture when stored at We have approved the stability protocols in your license application for the purpose of extending the expiration dating period of your drug substance and drug product under 21 CFR 601.12.
PATENTS
WO 2014109927
PAPER
https://www.tandfonline.com/doi/full/10.4161/19420862.2015.988944
Tildrakizumab (SCH 900222/MK-3222) targets the p19 subunit of IL-23. The mAb was developed by Schering-Plough, which was acquired by Merck & Co. in 2009, and it was then licensed by Merck to Sun Pharmaceutical Industries Ltd in September 2014. Clinical development and regulatory activities will be conducted by Merck, but funded by Sun Pharma. As of October 2014, the safety and efficacy of tildrakizumab are being evaluated in 2 Phase 3 studies that are ongoing but not recruiting patients. Both studies include patients with moderate-to-severe chronic plaque psoriasis and subcutaneously administered drug. The 52-week Phase 3 NCT01729754 study has 4 arms (200 mg tildrakizumab; 100 mg tildrakizumab; 50 mg etanercept; and placebo only), and includes an optional long-term safety extension study. The estimated enrollment is 1050, and the estimated primary completion date is October 2019. The 64-week Phase 3 NCT01722331 study is evaluating the effects of either 200 mg or 100 mg tildrakizumab to placebo; it includes an optional long-term safety extension study. The estimated enrollment is 885, and the estimated primary completion date is June 2015.


Mar 21, 2018, 09:04 ET
MUMBAI, India and PRINCETON, N.J., March 21, 2018 /PRNewswire/ — Sun Pharmaceutical Industries Ltd. (Reuters: SUN.BO, Bloomberg: SUNP IN, NSE: SUNPHARMA, BSE: 524715, “Sun Pharma” and includes its subsidiaries and/or associate companies) today announced that the U.S. Food and Drug Administration (FDA) has approved ILUMYA™ (tildrakizumab-asmn) for the treatment of adults with moderate-to-severe plaque psoriasis who are candidates for systemic therapy or phototherapy. ILUMYA selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor leading to inhibition of the release of pro-inflammatory cytokines and chemokines. ILUMYA is administered at a dose of 100 mg by subcutaneous injection every 12 weeks, after the completion of initial doses at weeks 0 and 4. ILUMYA is contraindicated in patients with a previous serious hypersensitivity reaction to tildrakizumab or to any of the excipients.
“With the approval of ILUMYA and our long-standing commitment in dermatology, we are focused on making a difference for people living with moderate-to-severe plaque psoriasis,” said Abhay Gandhi, President and Chief Executive Officer, North America, Sun Pharma. “We are committed to working with all relevant stakeholders to make ILUMYA available to appropriate people with plaque psoriasis.”
The FDA approval of ILUMYA for the treatment of adults with moderate-to-severe plaque psoriasis was supported by data from the pivotal Phase-3 reSURFACE clinical development program. In the two multicenter, randomized, double-blind, placebo-controlled trials (reSURFACE 1 and reSURFACE 2), 926 adult patients were treated with ILUMYA (N=616) or placebo (N=310). Results from these studies were published in The Lancet in July 2017, with primary endpoints presented at the 25th European Academy of Dermatology and Venereology (EADV) Congress.
Both Phase-3 studies met the primary efficacy endpoints, demonstrating significant clinical improvement with ILUMYA 100 mg compared to placebo when measured by at least 75 percent of skin clearance (Psoriasis Area Sensitivity Index or PASI 75) and Physician’s Global Assessment (PGA) score of “clear” or “minimal” at week 12 after two doses.
|
Efficacy Primary Endpoint at Week 12 in Adults with Plaque Psoriasis (NRI*) |
||||
|
reSURFACE 1 Study (NCT01722331) |
reSURFACE 2 Study (NCT01729754) |
|||
|
ILUMYA 100 mg n=309 |
Placebo n=154 |
ILUMYA 100 mg n=307 |
Placebo n=156 |
|
|
PGA of “clear” (0) or “minimal” (1)† |
179 (58%) |
11 (7%) |
168 (55%) |
7 (4%) |
|
PASI 75† |
197 (64%) |
9 (6%) |
188 (61%) |
9 (6%) |
|
PASI 90 |
107 (35%) |
4 (3%) |
119 (39%) |
2 (1%) |
|
PASI 100 |
43 (14%) |
2 (1%) |
38 (12%) |
0 (0%) |
* NRI = Non-Responder Imputation † Co-Primary Endpoints
Of the patients in the reSURFACE 1 study 74 percent (229 patients) achieved 75 percent skin clearance at week 28 after three doses, and 84 percent of patients who continued receiving ILUMYA 100 mg maintained PASI 75 at week 64 compared to 22 percent of patients who were re-randomized to placebo. In addition, 69 percent of the patients receiving ILUMYA 100 mg who had a PGA score of “clear” or “minimal” at week 28 maintained this response at week 64 compared to 14 percent of patients who were re-randomized to placebo.
Full Prescribing Information and Medication Guide for ILUMYA are attached:
PDF: https://mma.prnewswire.com/media/656994/Sun_Pharma_ILUMYA_US_Prescribing_Information.pdf
PDF: https://mma.prnewswire.com/media/656995/Sun_Pharma_ILUMYA_US_Medication_Guide.pdf
IMPORTANT SAFETY INFORMATION (continued)
Cases of angioedema and urticaria occurred in ILUMYA treated subjects in clinical trial. If a serious hypersensitivity reaction occurs, discontinue ILUMYA immediately and initiate appropriate therapy.
ILUMYA may increase the risk of infection. Treatment with ILUMYA should not be initiated in patients with a clinically important active infection until the infection resolves or is adequately treated. Consider the risks and benefits of treatment prior to prescribing ILUMYA in patients with a chronic infection or a history of recurrent infection. Instruct patients receiving ILUMYA to seek medical help if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a clinically important or serious infection, or is not responding to standard therapy, closely monitor and discontinue ILUMYA until the infection resolves.
Evaluate patients for TB infection prior to initiating treatment with ILUMYA. Initiate treatment of latent TB prior to administering ILUMYA. Monitor patients for signs and symptoms of active TB during and after ILUMYA treatment. Do not administer ILUMYA to patients with active TB infection.
Prior to initiating ILUMYA, consider completion of all age-appropriate immunizations according to current immunization guidelines. Avoid use of live vaccines in patients treated with ILUMYA.
The most common (≥1%) adverse reactions associated with ILUMYA include upper respiratory infections, injection site reactions, and diarrhea. Adverse reactions that occurred at rates less than 1% but greater than 0.1% in the ILUMYA group and at a higher rate than in the placebo group included dizziness and pain in extremity.
About the Phase-3 reSURFACE Trials
The Phase-3 studies (reSURFACE 1 and reSURFACE 2) were randomized, placebo-controlled, multicenter, three-part studies designed to demonstrate efficacy of ILUMYA in moderate-to-severe plaque psoriasis compared to placebo and comparative drug and to assess safety and tolerability. Part one of the studies randomized patients into three or four treatment arms, including ILUMYA 100 mg, ILUMYA 200 mg, placebo and etanercept (reSURFACE 2 only). After Week 12, patients on placebo were then re-randomized into ILUMYA 100 mg and 200 mg treatment arms to proceed into part two of the studies. Finally, in part three of the reSURFACE 1 study, responders (PASI ≥75) and partial responders (PASI ≥50 and PASI <75) to ILUMYA were re-randomized after Week 28 to continue the same treatment, a different dose of ILUMYA or placebo. Partial and non-responders to etanercept were treated with ILUMYA 200 mg in part three of the reSURFACE 2 study. Patients with guttate, erythrodermic, or pustular psoriasis were excluded.
About Psoriasis
Psoriasis is a chronic immune disease that appears on the skin. It is a non-contagious disorder that speeds the growth cycle of skin cells1 and results in thick scaly areas of skin2. The most common form, affecting about 80 to 90 percent of people living with psoriasis, is called plaque psoriasis3. It appears as red, raised areas of skin covered with flaky white scales, which may be itchy and painful and can crack and bleed2. Many people with plaque psoriasis continue to struggle with the ongoing, persistent nature of this chronic disease.
About Sun Dermatology
Sun Dermatology (the branded dermatology division of a wholly owned subsidiary of Sun Pharma) is committed to expanding its dermatology portfolio to bring healthcare providers and patients around the world more treatment options and ongoing support for conditions like moderate-to-severe plaque psoriasis. Sun Pharma, along with its subsidiaries, is ranked fourth in dermatology prescription volume within the U.S. per IMS and is fifth largest specialty generic pharmaceutical company globally. In addition to ILUMYA, Sun Dermatology is comprised of several branded products indicated for the treatment of acne and actinic keratosis with a focus on other dermatologic conditions.
About Sun Pharma, Merck & Co., Inc., Kenilworth, NJ, USA, Agreement
Sun Pharmaceutical Industries Ltd.’s wholly owned subsidiary licensed worldwide rights to ILUMYA from a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, in 2014. Funded by a Sun Pharma subsidiary, Merck & Co., Inc., Kenilworth, NJ, USA was responsible for the completion of Phase-3 trials and submission of a Biologics License Application to the United States Food and Drug Administration (FDA), as well as manufacturing finished goods to support Sun Pharma’s initial product launch. Sun Pharma will be responsible for all post-approval regulatory activities, including subsequent submissions, pharmacovigilance, post approval studies, manufacturing and commercialization of the approved product. Sun Pharma will also be responsible for all regulatory, pharmacovigilance, post approval studies, manufacturing and commercialization of approved products for all non-U.S. markets. Merck & Co., Inc., Kenilworth, NJ, USA is eligible to receive milestone payments and royalties on sales of ILUMYA.
About Sun Pharma, Almirall S.A, Europe, Agreement
Sun Pharma and its wholly owned subsidiary and Almirall (Spanish Stock Exchange ticker: ALM) closed on July 2016 a licensing agreement on the development and commercialization of tildrakizumab-asmn for psoriasis in Europe. Under the terms of the licensing agreement, Almirall is able to lead European studies, and participate in larger Global clinical studies for plaque psoriasis indication subject to the terms of the Sun Pharma – Merck & Co., Inc., Kenilworth, NJ, USA agreements, as well as certain cost sharing agreements. Sun Pharma will be eligible to receive development and regulatory milestone payments and, additionally, sales milestone payments and royalties on net sales. Sun Pharma will continue to lead development of tildrakizumab-asmn for other indications, where Almirall will have right of first negotiation for certain indications in Europe. The agreement between Sun Pharma and Almirall remains subject to the exclusive licensing agreement between Sun Pharma and Merck & Co., Inc., Kenilworth, NJ, USA.
About Sun Pharmaceutical Industries Ltd. (CIN – L24230GJ1993PLC019050)
Sun Pharma is the world’s fifth largest specialty generic pharmaceutical company and India’s top pharmaceutical company. A vertically integrated business, economies of scale and an extremely skilled team enable us to deliver quality products in a timely manner at affordable prices. It provides high-quality, affordable medicines trusted by customers and patients in over 150 countries across the world. Sun Pharma’s global presence is supported by 41 manufacturing facilities spread across 6 continents, R&D centres across the globe and a multi-cultural workforce comprising over 50 nationalities. In India, the company enjoys leadership across 11 different classes of doctors with 30 brands featuring amongst top 300 pharmaceutical brands in India. Its footprint across emerging markets covers over 100 markets and 6 markets in Western Europe. Its Global Consumer Healthcare business is ranked amongst Top 10 across 3 global markets. Its API business footprint is strengthened through 14 world class API manufacturing facilities across the globe. Sun Pharma fosters excellence through innovation supported by strong R&D capabilities comprising about 2,000 scientists and R&D investments of approximately 8% of annual revenues. For further information, please visit www.sunpharma.com & follow us on Twitter @SunPharma_Live.
References
1. National Psoriasis Foundation. Facts about psoriasis. www.psoriasis.org/sites/default/files/for-media/MediaKit.pdf. Accessed on February 22, 2018.
2. National Psoriasis Foundation. About Psoriasis. www.psoriasis.org/about-psoriasis. Accessed on February 22, 2018.
3. Menter A, Gottlieb A, Feldman SR, Van Voorhees AS et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 2008 May; 58(5):826-50.
////////////////tildrakizumab-asmn, FDA 2018, MERCK, Schering-Plough, MONOCLONAL ANTIBODY, SCH 900222, MK-3222, Psoriasis, plaque, BLA 761067, SCH-900222, SUNPG 1622, SUNPG 1622 I, SUNPG 1623 I, SUNPG 1623 II, SUNPG 1623 III, SUNPG 1623 IV, SUNPG1623,
Lofexidine, лофексидин , لوفيكسيدين , 洛非西定 ,
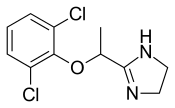
Lofexidine
- Molecular FormulaC11H12Cl2N2O
- Average mass259.132 Da
- (±)-2-[1-(2,6-Dichlorophenoxy)ethyl]-2-imidazoline
UNII:UI82K0T627
лофексидин [Russian] [INN]
لوفيكسيدين [Arabic] [INN]
洛非西定 [Chinese] [INN]
1H-Imidazole, 2-(1-(2,6-dichlorophenoxy)ethyl)-4,5-dihydro- (9CI)
2-{1-[(2,6-dichlorophenyl)oxy]ethyl}-4,5-dihydro-1H-imidazole
CAS 31036-80-3 [RN]
Lofetensin, Loxacor
Synthesis Reference ZA 6800850; eidem, US 3966757 (1968, 1976 both to Nordmark)
DE 1935479, Jan 21, 1971
U.S. Patent 3,966,757.
FDA Approved May 2018
Lofexidine was developed by US Woldmeds LLC and it got approved by the FDA on May 16, 2018

Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 221-223 | U.S. Patent 3,966,757. |
| boiling point (°C) | 421.5 ºC at 760 mm Hg | ‘MSDS’ |
| water solubility | Soluble | ‘MSDS’ |
| logP | 5.37 | FDA Advisory Committee Briefing Document. |
| pKa | 9.43 | FDA Advisory Committee Briefing Document. |
SYN
Organic Process Research & Development, 13(3), 415-419; 2009

Title: Lofexidine
CAS Registry Number: 31036-80-3
CAS Name: 2-[1-(2,6-Dichlorophenoxy)ethyl]-4,5-dihydro-1H-imidazole
Additional Names: 2-[1-(2,6-dichlorophenoxy)ethyl]-2-imidazoline
Molecular Formula: C11H12Cl2N2O
Molecular Weight: 259.13
Percent Composition: C 50.99%, H 4.67%, Cl 27.36%, N 10.81%, O 6.17%
Literature References: a2-Adrenoceptor agonist related structurally to clonidine, q.v. Prepn of the HCl salt: H. Baganz, H. J. May, ZA 6800850; eidem, US 3966757 (1968, 1976 both to Nordmark); of the free base: eidem, DE 1935479 (1971 to Nordmark), C.A.74, 87979 (1971). Pharmacological studies: J. Velly, J. Pharmacol. 8, 351 (1977); B. Jarrot et al., Biochem. Pharmacol. 28, 141 (1979). NMR data and cardiovascular effects: P. B. M. Timmermans, P. A. Van Zwieten, Eur. J. Med. Chem. 15, 323 (1980). Hypotensive and sedative properties: P. Birch et al., Br. J. Pharmacol. 68, 107 (1980). Effects in hypertension: N. D. Vlachakis et al., Fed. Proc. 39, 4844 (1980). Series of articles on pharmacology, toxicology, clinical studies: Arzneim.-Forsch. 32, 915-993 (1982). Toxicity studies: T. H. Tsai et al., ibid. 955. Review of clinical trials in treatment of opiate withdrawal: J. Strang et al., Am. J. Addict. 8, 337-348 (1999).
Properties: Crystals, mp 126-128°.
Melting point: mp 126-128°
Derivative Type: Hydrochloride
CAS Registry Number: 21498-08-8
Manufacturers’ Codes: MDL-14042A; Ba-168
Trademarks: BritLofex (Britannia); Lofetensin (Nattermann)
Molecular Formula: C11H12Cl2N2O.HCl
Molecular Weight: 295.59
Percent Composition: C 44.70%, H 4.43%, Cl 35.98%, N 9.48%, O 5.41%
Properties: Crystals from ethanol/ether or 2-propanol, mp 221-223° (U.S. patent); also reported as mp 230-232° (Ger. patent). Very sol in water, ethanol. Slightly sol in 2-propanol. Practically insol in ether. LD50 in mice, rats, dogs (mg/kg): between 74-147 orally (all species); between 8-18 i.v. (all species) (Tsai).
Melting point: mp 221-223° (U.S. patent); mp 230-232° (Ger. patent)
Toxicity data: LD50 in mice, rats, dogs (mg/kg): between 74-147 orally (all species); between 8-18 i.v. (all species) (Tsai)
Therap-Cat: In treatment of opioid withdrawal symptoms; antihypertensive.
Keywords: Antihypertensive; Imidazole Derivatives.

LOFEXIDINE HYDROCHLORIDE
Cas No. 21498-08-8
Lofexidine, sold under the brand name Lucemyra among others,[1] is a medication historically used to treat high blood pressure, but more commonly used to help with the physical symptoms of opioid withdrawal.[2] It is taken by mouth.[3] It is an α2A adrenergic receptoragonist.[3] It was approved for use by the Food and Drug Administration in the United States in 2018.[3]
Medical uses
In the United States, the brand name Lucemyra (lofexidine HCl) is approved for the “mitigation of withdrawal symptoms to facilitate abrupt discontinuation of opioids in adults,” for a treatment duration of 14 days.[1] In the United Kingdom, lofexidine is commonly used in conjunction with the opioid receptor antagonist naltrexone in rapid detoxification cases. When these two drugs are paired, naltrexone is administered to induce an opioid-receptor blockade sending the subject into immediate withdrawal and accelerating the detoxificationprocess, while lofexidine is given to relieve the symptoms associated with the withdrawal including chills, sweating, stomach cramps, muscle pain, and runny nose.[citation needed]
Opioid withdrawal
The United Kingdom’s National Institute for Health and Care Excellence (NICE) guidelines recommend the use of methadone or buprenorphine as first-line agents in the management of opioid use disorder. However, lofexidine is considered an acceptable alternative for people with mild or uncertain opioid dependence in need of short-term detoxification.[4]
Lofexidine is not an opioid.[3] It does not eliminate the symptoms of opioid withdrawal but reduces them.[3] Indeed, one suggested use for lofexidine is to ease withdrawal symptoms of methadone dependence. Its use is approved in the United States for up to 14 days.[3]
Other clinical uses
The possibility of using lofexidine to treat alcohol withdrawal symptoms has been investigated, and has not yet been shown to be an effective treatment.[5] It is also used in treatment of cases suffering from postmenopausal hot flashes.
Special populations
Lofexidine’s safety in pregnancy or in the setting of breastfeeding are unknown.[6] Caution is warranted if chronic kidney impairment is present.[6]
Adverse effects
Adverse effects that have occurred after taking lofexidine include the following:[6]
In addition, people may experience a sudden jump in blood pressure after stopping lofexidine.[1]
Overdose
The LD50 of lofexidine is above 77 mg/kg in animals. Studies of high-dose, single administrations of lofexidine proved tolerable for animals, but repeat administration induced symptoms consistent with toxicity. In studies on mice, rats, and dogs, these included ataxia, somnolence, and tremors. It is expected that an overdose of lofexidine would result in symptoms akin to its pharmacological side effects in humans, such as bradycardia and hypotension.[7]
Interactions
Many drug-drug interactions with lofexidine are possible.[8]
QT prolongation
Lofexidine prolongs the QT interval, which can result in a severe interaction (torsade de pointes) when combined with other drugs that also prolong the QT interval. Patient-specific characteristics that increase the risk for a clinically-significant drug-drug interaction include:[8]
- increasing age
- female sex
- cardiac disease
- electrolyte disturbances (low blood potassium)
As a result, there are many QT-prolonging drugs that may interact with lofexidine. These include medications such as amiodarone, citalopram, and fluconazole. Other medications may increase the risk for a low level of potassium in the blood, thereby indirectly increasing the risk for QT prolongation. For example, dexamethasone, hydrochlorothiazide, and theophylline can lower the level of potassium in the blood.[8]
CNS depression
Lofexidine can depress the central nervous system (CNS), which, in combination with other CNS depressants, may reduce a person’s ability to perform tasks that require skills and attention. For example, clobazam, gabapentin, and levetiracetam all can depress the CNS.[8]
Hypotension
The risk of hypotension (low blood pressure) is increased when lofexidine is combined with other drugs that lower blood pressure. These may include losartan, metoprolol, and pramipexole.[8]
Pharmacology
Lofexidine is an agonist at the α-2A, 2B, and 2C adrenergic receptor subtypes, with the highest activity at the alpha-2A receptor.[9]
-
-
Ki for lofexidine[9] Adrenergic receptor Ki (nM) α-2A 4 α-2B 67 α-2C 69
-
Ki represents the dissociation constant[10] for lofexidine’s binding to a specific subtype of alpha-2 receptor. The smaller the Ki value, the stronger the drug binds to the receptor to exert its activity.
Lofexidine inhibits the release of norepinephrine in the central and peripheral nervous system, thereby reducing some of the symptoms of opioid withdrawal, but it has no documented effect on drug craving and endogenous opioid levels.[2]
Pharmacokinetics
Lofexidine’s oral bioavailability is about 90%, with extensive oral absorption. Peak plasma concentrations occur at 3 hours after a single administration, with a half-life of 11 hours. Lofexidine is extensively metabolized by the liver, and primarily cleared by the kidney. It is 80-90% plasma protein bound.[7]
Chemistry
Lofexidine exists as a solid at room temperature, with a melting point of 127 degrees C.[7] The pair of ortho chlorine (Cl–) atoms on the phenyl ring are necessary for lofexidine’s agonism at the α2a adrenergic receptor subtype; removal of either chlorine atom results in antagonism at the receptor.[9]
Comparison to clonidine

Structure of clonidine and lofexidine
Lofexidine is structurally analogous to clonidine, another α2 adrenergic receptor agonist used for treatment of opioid withdrawal symptoms. A comparison of the two structures is shown at right. Both contain an imidazoline ring and a 2,6-dichlorinated phenyl ring. The differences in structure are shown in red, while the similarities are in black. In addition to the structural differences, administration of lofexidine to people who abuse opioids has been shown to be more effective for a longer duration, with fewer withdrawal symptoms than clonidine even after one day.[11] However, clonidine is often preferred as it is substantially cheaper than lofexidine when purchased with a private (non-NHS) prescription. This factor is exacerbated by the considerable number of and quantities of medications prescribed to alleviate the constellation of withdrawal signs and symptoms. Additionally, clonidine has been shown to significantly lower blood pressure. Therefore, although similar to lofexidine, clonidine is most frequently prescribed to treat high blood pressure.[citation needed]
Society and culture
Britannia Pharmaceuticals has licensed lofexidine to be sold by US WorldMeds for sale in North America.[12] In the United Kingdom, the hydrochloride form, lofexidine HCl, has been licensed and sold since 1992 for opioid withdrawal relief in tablet form as BritLofex by Britannia Pharmaceuticals.[2] BritLofex is only available by prescription. Lofexidine was first approved by the US FDA on May 16, 2018 under the brand name Lucemyra, produced by US WorldMeds.[13] It was noted as the first, non-opioid drug approved in the US for the treatment of opioid withdrawal.[1]
Heroin has been reported to be the most prominent illicit drug of abuse among admissions at public!} -funded substance abuse treatment facilities in the US. At some time in their lives, about 2.4 million people have used heroin; in 1997, there were 81 ,000 new heroin users of whom 87% were less than 26 years of age. In spite of efforts to decrease illicit drug abuse, the problem escalates and the abusing population is increasingly younger. Hospital emergency room episodes from 21 metropolitan areas show that 14% of drug-related emergency room episodes involved heroin, and such episodes increased more than 2-fold from 1991 to 1996. Additionally, prescription opioid abuse escalates; the number of people addicted to prescription pain relievers is 3 -fold higher than those addicted to heroin. For example, from 1999 to 2001, the non-medical use of OxyContin®increased 4-fold, and its use continues to escalate.
[0003] Generally, opioid addiction has been associated with high morbidity and mortality, with a 15-20 fold increase in risk of death for intravenous drug users compared with their same age peers. Clearly, the medical and social importance of the development of effective treatments for opioid addiction is well recognized. Surprisingly, few treatment options for opioid addiction are available.
[0004] Withdrawal, maintenance and relapse are considered the progressive stages for treatment of opioid addiction. There are two predominant management strategies for the treatment of opioid addiction, detoxification and substitution therapy, which are typically combined with medical, social and psychological support. A majority of individuals may benefit from remaining in the maintenance phase for an indefinite period of time, while others may be able to directly undergo medically-supervised detoxification and/or relapse therapy, without the need for maintenance therapy. Methadone and buprenorphine constitute the most commonly used pharmacotherapies. Although patients continue to be successfully treated with methadone, a mμ opioid receptor agonist, several disadvantages of methadone treatment include the length of time for withdrawal, the difficulty of obtaining complete abstinence, and liability for its abuse. Due to the abuse liability of methadone and its consequent Schedule II classification by the Drug Enforcement Administration (DEA), methadone has additional disadvantages with respect to its prescription requirements, the carefully controlled conditions under which it is dispensed, and the annoyance experienced by patients who must frequently visit the dispensing unit to obtain their methadone dosages.
[0005] BritLofex™ (Lofexidine hydrochloride 0.2 mg tablet), an α2-adrenergic agonist, is used as a non-opioid medication for opioid detoxification in the United Kingdom (UK). There is no non-opioid medication approved by the Food and Drug Administration (FDA) for this indication in the US. The only medications currently approved by the FDA for opioid detoxification are methadone and buprenorphine, both opioid receptor agonists and both associated with abuse liability. Clonidine, an 012-adrenergic agonist, is often used “off-label” for this indication in the U.S. However, clonidine has not been approved by the FDA for this indication. However, the use of clonidine is limited by its side-effect profile, i.e., significant hypotension at doses effective in alleviating opioid withdrawal symptoms.
[0006] In contrast, Lofexidine HCl is the only non-opiate, non-addictive treatment approved for use in the UK to manage withdrawal symptoms in patients undergoing opiate detoxification. Lofexidine has been found to be effective in reducing the symptoms associated with heroin withdrawal such as chills, vomiting, sweating, stomach cramps, diarrhea, muscle pain, and runny nose and eyes. In the UK, the treatment is responsible for approximately 20,000 detoxifications per year. The drug’s proven level of safety permits its use in an outpatient situation. This is of great importance to patients in the US who are located in parts of the country where treatment clinics are not readily available.
[0007] Although naltrexone, methadone and more recently buprenorphine are FDA approved in the treatment of opioid addiction, these opioid treatments are associated with high relapse rates. Furthermore, there is currently insufficient availability of methadone and buprenorphine treatment for patients who abuse opioids. A significant number of these patients are undergoing detoxification treatments. However, the great risk of abuse and several other existing restrictions, such as medical prescribing and pharmaceutical dispensing, limit the use of methadone and buprenorphine for outpatient detoxification. In addition, the unapproved status of clonidine, its side effects, such as the lowering of blood pressure, and moderate efficacy limit its use. A substantial amount of research is ongoing to understand the mechanisms that may underline the high rates of relapse associated with opioid addiction. There is growing evidence that chronic drug use results in neuroadaptive changes in brain stress and reward circuits that may be associated with increased drug craving and risk of relapse particularly in the face of environmental triggers such as stressful life events and drug cues.
PATENT
https://patents.google.com/patent/EP2334297A1/en
The lofexidine hydrochloride tablets available in the UK market (BritLofex™) contain the racemic mixture of the drug. However, since lofexidine enantiomers exhibit different affinities for central the nervous system neurotransmitter receptors involved in (±)-lofexidine’s action as a medication for opioid detoxification, each of these enantiomers may have therapeutic benefits in the treatment of opioid addiction.
Experimental
[0028] 1) Resolution of (-)-lofexidine and (+)-lofexidine enantiomers found in the racemic mixture using chiral stationary phases by HPLC method:
[0029] A chiral chromatographic matrix was used to separate a racemic mixture of lofexidine into its component enantiomers by a process of HPLC to obtain optically pure (-)- lofexidine and optically pure (+)-lofexidine. The separation was performed using a chiral stationary phase consisted of D-glucose cyclodextran complex (Cyclobond HP-RSP) from Astec
Company (Whippany, NJ, USA) using a mobile phase consisted of 1OmM ammonium acetate
(88%), acetonitrile (8%), and methanol (8%) at 0.85 ml/min flow rate. Analysis was performed using Agilent series 1100 HPLC system comprising a solvent degasser unit, quaternary pump, autosampler, and DAD detector. Using such chiral stationary phase in a preparative scale enables the yield of gram quantities of desired enantiomers.
[0030] Resolution of (-)-lofexidine and (+)-lofexidine enantiomers found in the racemic mixture using a chiral acid, not only diastereomeric salt formation but also preferential crystallization: [0031] Optical resolution of (±)-lofexidine hydrochloride by using the classical methods of salt formation with a chiral acid such as, [( Di-p-toluoyl-D-tartaric acid [D]D20 +142° (c=l, CH3OH)] as shown in Figure 1, yielded (-)-lofexidine hydrochloride and (+)-lofexidine hydrochloride enantiomers (yield = 87%). The method comprised the following steps: [0032] A racemic form of lofexidine (10 mmol) was placed in ethanol (100 mL), and the chiral acid (+)-Di-p-toluoyl-D-tartaric acid was added in order to form a mixture of the (+)(-) and (+)(+) diastereomeric lofexidine salts. The diastereomeric salts i.e.: (+)(-) lofexidine Di-p- toluoyl-D-tartarate salt was separated from the (+)(+) lofexidine Di-p-toluoyl-D-tartarate salt by a process of fractional crystallization. 10 mL methanol and 1 ml water was added and the mixture was heated for 1 hour at 55-65 0C. After the mixture became clear it was left to cool down at room temperature. The crystals were isolated after two days, dried under vacuum. Recrystallization was performed using ethanol (20 volumes). Final yield was 87%. [0033] Chiral purity of the resulting crystals was tested by the chiral HPLC method. The
(+)(-) lofexidine Di-p-toluoyl-D-tartarate salt or the(+)(+) lofexidine Di-p-toluoyl-D-tartarate salt obtained was treated with a base such as 0.1 N sodium carbonate to liberate (-)-lofexidine and (+)-lofexidine. The resulting enantiomerically pure free base of (-)-lofexidine and (+)-lofexidine was converted to lofexidine hydrochloride salt.
PAPER
A Scalable, Enantioselective Synthesis of the α2-Adrenergic Agonist, Lofexidine
Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 725 Rose Street, Lexington, Kentucky 40536, U.S.A.
Org. Process Res. Dev., 2009, 13 (3), pp 415–419
DOI: 10.1021/op8002689
* Author to whom correspondence may be sent. Tel: 859-257-1718. Fax: 859-257-7585. E-mail: pcrooks@email.uky.edu.

A scalable and high-yielding synthetic route toward pure enantiomers of the α2-adrenergic agonist, lofexidine hydrochloride, is presented. Salient features include a rapid one-pot amide alkylation-imidazoline formation sequence on the carboxamide function of α-(2,6-dichlorophenoxy)propionamide, while preserving the sensitive configuration about the α-carbon of the resulting product. A means to accelerate the sluggish O-alkylation of the carboxamide function of α-(2,6-dichlorophenoxy)propionamide by Me3O+BF4− is also described, which may be of general applicability.
PATENTS
US8101779B2 *2008-10-062012-01-24University Of Kentucky Research FoundationEnantioselective synthesis of (+) and (–)-2-[1-(2,6-dichlorophenoxy)-ethyl]-1,3-diazacyclopent-2-ene
DE3149009A1 *1981-12-101983-06-23Nattermann A & Cie(-) – 2- (1- (2,6-dichlorophenoxy) ethyl) -1,3-diazacyclopent-2-ene, its preparation and its use in pharmaceutical preparations
DE3149010A1 *1981-12-101983-07-07Nattermann A & Cie(+) – 2- (1- (2,6-dichlorophenoxy) ethyl) -1,3-diazacyclopent-2-ene, its preparation and its use in preparations pharamazeutischen
References
- ^ Jump up to:a b c d “Press Announcements – FDA approves the first non-opioid treatment for management of opioid withdrawal symptoms in adults”. http://www.fda.gov. U.S. Food and Drug Administration. Retrieved 16 May 2018.
- ^ Jump up to:a b c Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. p. 330. ISBN 978-0-85711-084-8.
- ^ Jump up to:a b c d e f “Press Announcements – FDA approves the first non-opioid treatment for management of opioid withdrawal symptoms in adults”. http://www.fda.gov. Retrieved 18 May2018.
- Jump up^ “Pharmacological interventions in opioid detoxification for drug misuse in people over 16”. pathways.nice.org.uk. NICE. Retrieved 16 May 2018.
- Jump up^ Keaney F, Strang J, Gossop M, Marshall EJ, Farrell M, Welch S, Hahn B, Gonzalez A. A double-blind randomized placebo-controlled trial of lofexidine in alcohol withdrawal: lofexidine is not a useful adjunct to chlordiazepoxide. Alcohol Alcohol (2001) 36:426–30.
- ^ Jump up to:a b c “LOFEXIDINE HYDROCHLORIDE”. bnf.nice.org.uk. NICE. Retrieved 16 May2018.
- ^ Jump up to:a b c “Lofexidine”. pubchem.ncbi.nlm.nih.gov. National Center for Biotechnology Information. Retrieved 16 May 2018.
- ^ Jump up to:a b c d e “Lofexidine | Interactions | BNF”. bnf.nice.org.uk. NICE. Retrieved 16 May 2018.
- ^ Jump up to:a b c Fulton, Brian (2014). Drug Discovery for the Treatment of Addiction: Medicinal Chemistry Strategies. John Wiley & Sons. p. 151. ISBN 0470614161.
- Jump up^ Neubig, R. R. (1 December 2003). “International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology”. Pharmacological Reviews. 55 (4): 597–606. doi:10.1124/pr.55.4.4.
- Jump up^ G. Gerra, et al., Lofexidine versus clonidine in rapid opioid detoxification, Journal of Substance Abuse TreatmentVolume 21, Issue 1, , July 2001, Pages 11-17.
- Jump up^ Britannia Pharmaceuticals Limited
- Jump up^ “Lucemyra (lofexidine hydrochloride) FDA Approval History – Drugs.com”. Drugs.com. Retrieved 16 May 2018.
 |
|
| Clinical data | |
|---|---|
| Trade names | BritLofex, Lucemyra, Kai Er Ding, others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
By mouth (tablets) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | >90% |
| Protein binding | 80–90% |
| Metabolism | Liver (glucuronidation) |
| Elimination half-life | 11 hours |
| Excretion | Kidney |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H12Cl2N2O |
| Molar mass | 259.131 g/mol |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
/////////////lofexidine, FDA 2018, лофексидин , لوفيكسيدين , 洛非西定 , Lofetensin, Loxacor
CC(C1=NCCN1)OC2=C(C=CC=C2Cl)Cl
FDA approves new drug Doptelet (avatrombopag) for patients with chronic liver disease who have low blood platelets and are undergoing a medical procedure
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
FDA approves new uses for two drugs Tafinlar (dabrafenib) and Mekinist (trametinib) administered together for the treatment of BRAF-positive anaplastic thyroid cancer


FDA approves new uses for two drugs Tafinlar (dabrafenib) and Mekinist (trametinib) administered together for the treatment of BRAF-positive anaplastic thyroid cancer
The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive). Continue reading.
May 4, 2018
Release
The U.S. Food and Drug Administration approved Tafinlar (dabrafenib) and Mekinist (trametinib), administered together, for the treatment of anaplastic thyroid cancer (ATC) that cannot be removed by surgery or has spread to other parts of the body (metastatic), and has a type of abnormal gene, BRAF V600E (BRAF V600E mutation-positive).
“This is the first FDA-approved treatment for patients with this aggressive form of thyroid cancer, and the third cancer with this specific gene mutation that this drug combination has been approved to treat,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “This approval demonstrates that targeting the same molecular pathway in diverse diseases is an effective way to expedite the development of treatments that may help more patients.”
Thyroid cancer is a disease in which cancer cells form in the tissues of the thyroid gland. Anaplastic thyroid cancer is a rare, aggressive type of thyroid cancer. The National Institutes of Health estimates there will be 53,990 new cases of thyroid cancer and an estimated 2,060 deaths from the disease in the United States in 2018. Anaplastic thyroid cancer accounts for about 1 to 2 percent of all thyroid cancers.
Both Tafinlar and Mekinist are also approved for use, alone or in combination, to treat BRAF V600 mutation-positive metastatic melanoma. Additionally, Tafinlar and Mekinist are approved for use, in combination, to treat BRAF V600E mutation-positive, metastatic non-small cell lung cancer.
The efficacy of Tafinlar and Mekinist in treating ATC was shown in an open-label clinical trial of patients with rare cancers with the BRAF V600E mutation. Data from trials in BRAF V600E mutation-positive, metastatic melanoma or lung cancer and results in other BRAF V600E mutation-positive rare cancers provided confidence in the results seen in patients with ATC. The trial measured the percent of patients with a complete or partial reduction in tumor size (overall response rate). Of 23 evaluable patients, 57 percent experienced a partial response and 4 percent experienced a complete response; in nine (64 percent) of the 14 patients with responses, there were no significant tumor growths for six months or longer.
The side effects of Tafinlar and Mekinist in patients with ATC are consistent with those seen in other cancers when the two drugs are used together. Common side effects include fever (pyrexia), rash, chills, headache, joint pain (arthralgia), cough, fatigue, nausea, vomiting, diarrhea, myalgia (muscle pain), dry skin, decreased appetite, edema, hemorrhage, high blood pressure (hypertension) and difficulty breathing (dyspnea).
Severe side effects of Tafinlar include the development of new cancers, growth of tumors in patients with BRAF wild-type tumors, serious bleeding problems, heart problems, severe eye problems, fever that may be severe, serious skin reactions, high blood sugar or worsening diabetes, and serious anemia.
Severe side effects of Mekinist include the development of new cancers; serious bleeding problems; inflammation of intestines and perforation of the intestines; blood clots in the arms, legs or lungs; heart problems; severe eye problems; lung or breathing problems; fever that may be severe; serious skin reactions; and high blood sugar or worsening diabetes.
Both Tafinlar and Mekinist can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
The FDA granted Priority Review and Breakthrough Therapy designation for this indication. Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases, was also granted for this indication.
The FDA granted this approval to Novartis Pharmaceuticals Corporation.
///////////////Tafinlar, dabrafenib, Mekinist, trametinib, fda 2018, Priority Review, Breakthrough Therapy designation, Orphan Drug designation, Novartis Pharmaceuticals Corporation,

{kind=link}