






- USUS5856529 A
- USUS8785492 B2
- US5856529
- US8785492
- US9060995
- US9549913
- US9539234
- US9730910
- USRE46604
- US9855241
Home » Posts tagged 'EU 2015'
Tag Archives: EU 2015
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tasimelteon, タシメルテオン

Tasimelteon
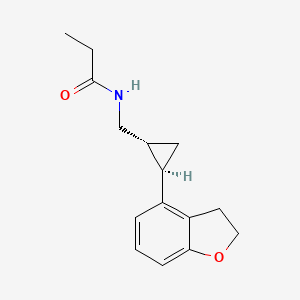
N-([(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl)propanamide,
609799-22-6 [RN]
8985
Hetlioz [Trade name]
N-{[(1R,2R)-2-(2,3-Dihydro-1-benzofuran-4-yl)cyclopropyl]methyl}propanamide [ACD/IUPAC Name]
Propanamide, N-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]methyl]- [ACD/Index Name]
SHS4PU80D9
609799-22-6 cas, BMS-214778; VEC-162, ATC:N05CH03
- Use:Treatment of sleep disorder; Melatonin receptor agonist
- (1R,2R)-N-[2-(2,3-dihydrobenzofuran-4-yl)cyclopropylmethyl]propanamide
- Formula:C15H19NO2, MW:245.3 g/mol
-
Hetlioz Vanda Pharmaceuticals, 2014
Approved fda 2014
EMA
Tasimelteon is a white to off-white crystalline powder, it is non hygroscopic, soluble in water across relevant pH values and freely soluble in alcohols, cyclohexane, and acetonitrile. Conducted in vivo studies demonstrate that tasimelteon is highly permeable substance. Photostability testing and testing on stress conditions demonstrated that the active substance degrades in light.
Tasimelteon exhibits stereoisomerism due to the presence of two chiral centres. Active substance is manufactured as a single, trans-1R,2R isomer. Enantiomeric purity is controlled routinely during manufacture of active substance intermediates by chiral HPLC/specific optical rotation and additionally controlled in the active substance. Stability data indicates tasimelteon is isomerically stable.
Polymorphism has been observed in polymorphic screening studies for tasimelteon and two forms have been identified. The thermodynamically more stable form has been chosen for development and the manufacturing process consistently yields active substance of single, desired polymorphic form. It was demonstrated that milling of the active substance does not affect polymorphic form. Polymorphism is additionally controlled in active substance release and shelf-life specifications using X-ray powder diffraction analysis.
Tasimelteon is synthesized in nine main steps using linear synthesis and using commercially available well-defined starting materials with acceptable specifications. Three intermediates are isolated for control of active substance quality including stereochemical control. The active substance is isolated by slow recrystallisation or precipitation of tasimelteon from an ethanol/water mixture which ensures the formation of desired polymorphic form. Up to two additional, optional recrystallisations may be performed for unmilled tasimelteon to ensure that milled tasimelteon active substance is of high purity. Seed crystals complying with active substance specifications can be used optionally. Active substance is jet milled (micronised) to reduce and control particle size, which is critical in finished product performance with regards to content uniformity and dissolution…….http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003870/WC500190309.pdf
launched in 2014 in the U.S. by Vanda Pharmaceuticals for the treatment of non-24-hour sleep-wake disorder in totally blind subjects. In 2015, the European Committee for Medicinal Products of the European Medicines Agency granted approval for the same indication. In 2010 and 2011, orphan drug designations were assigned for the treatment of non-24 hour sleep/wake disorder in blind individuals without light perception in the U.S. and the E.U., respectively.
Tasimelteon (trade name Hetlioz) is a drug approved by the U.S. Food and Drug Administration (FDA)[2] in January 2014 for the treatment of non-24-hour sleep–wake disorder (also called Non-24, N24 and N24HSWD).[3] In June 2014, the European Medicines Agency accepted an EU filing application for tasimelteon[4] and in July 2015, the drug was approved in Europe for the treatment of non-24-hour sleep-wake rhythm disorder in totally blind adults,[5] but not in the rarer case of non-24 in sighted people.
Tasimelteon is a selective agonist for the melatonin receptors MT1 and MT2, similar to other members of the melatonin receptor agonistclass of which ramelteon (2005) and agomelatine (2009) were the first approved.[6] As a treatment for N24HSWD, as with melatonin or other melatonin derivatives, the patient may experience improved sleep timing while taking the drug. Reversion to baseline sleep performance occurs within a month of discontinuation.[7]
![]()
Development
Tasimelteon (previously known as BMS-214,778) was developed for the treatment of insomnia and other sleep disorders. A phase II trial on circadian rhythm sleep disorders was concluded in March 2005.[8] A phase III insomnia trial was conducted in 2006.[9] A second phase III trial on insomnia, this time concerning primary insomnia, was completed in June 2008.[10] In 2010, the FDA granted orphan drug status to tasimelteon, then regarded as an investigational medication, for use in totally blind adults with N24HSWD.[11] (Through mechanisms such as easing the approval process and extending exclusivity periods, orphan drug status encourages development of drugs for rare conditions that otherwise might lack sufficient commercial incentive.)
On completion of Phase III trials, interpretations of the clinical trials by the research team concluded that the drug may have therapeutic potential for transient insomnia in circadian rhythm sleep disorders.[12] A year-long (2011–2012) study at Harvard tested the use of tasimelteon in blind subjects with non-24-hour sleep-wake disorder. The drug has not been tested in children nor in any non-blind people.
FDA approval
In May 2013 Vanda Pharmaceuticals submitted a New Drug Application to the Food and Drug Administration for tasimelteon for the treatment of non-24-hour sleep–wake disorder in totally blind people. It was approved by the FDA on January 31, 2014 under the brand name Hetlioz.[3] In the opinion of Public Citizen, an advocacy group, the FDA erroneously allowed it to be labelled without stating that it is only approved for use by totally blind people.[13] However, FDA updated its press release on Oct. 2, 2014 to clarify the approved use of Hetlioz, which includes both sighted and blind individuals. The update did not change the drug labeling (prescribing information).[14]
Toxicity
Experiments with rodents revealed fertility impairments, an increase in certain cancers, and serious adverse events during pregnancy at dosages in excess of what is considered the “human dose”.[15][16]
As expected, advisors to the US Food and Drug Administration have recommended approval of Vanda Pharmaceuticals’ tasimelteon, to be sold as Hetlioz, for the treatment of non-24-hour disorder in the totally blind.http://www.pharmatimes.com/Article/13-11-14/FDA_panel_backs_Vanda_body_clock_drug_for_blind.aspx
The master body clock controls the timing of many aspects of physiology, behavior and metabolism that show daily rhythms, including the sleep-wake cycles, body temperature, alertness and performance, metabolic rhythms and certain hormones which exhibit circadian variation. Outputs from the
suprachiasmatic nucleus (SCN) control many endocrine rhythms including those of melatonin secretion by the pineal gland as well as the control of Cortisol secretion via effects on the hypothalamus, the pituitary and the adrenal glands. This master body clock, located in the SCN, spontaneously generates rhythms of approximately 24.5 hours. These non-24-hour rhythms are synchronized each day to the 24-hour day-night cycle by light, the primary environmental time cue which is detected by specialized cells in the retina and transmitted to the SCN via the retino-hypothalamic tract. Inability to detect this light signal, as occurs in most totally blind individuals, leads to the inability of the master body clock to be reset daily and maintain entrainment to a 24-hour day.
Non-24-Hour Disorder, Non-24, also referred to as Non-24-Hour Sleep-Wake Disorder, (N24HSWD) or Non-24-Hour Disorder, is an orphan indication affecting approximately 65,000 to 95,000 people in the U.S. and 140,000 in Europe. Non- 24 occurs when individuals, primarily blind with no light perception, are unable to synchronize their endogenous circadian pacemaker to the 24-hour light/dark cycle. Without light as a synchronizer, and because the period of the internal clock is typically a little longer than 24 hours, individuals with Non-24 experience their circadian drive to initiate sleep drifting later and later each day. Individuals with Non-24 have abnormal night sleep patterns, accompanied by difficulty staying awake during the day. Non-24 leads to significant impairment, with chronic effects impacting the social and occupational functioning of these individuals.
In addition to problems sleeping at the desired time, individuals with Non-24 experience excessive daytime sleepiness that often results in daytime napping.
The severity of nighttime sleep complaints and/or daytime sleepiness complaints varies depending on where in the cycle the individual’s body clock is with respect to their social, work, or sleep schedule. The “free running” of the clock results in approximately a 1-4 month repeating cycle, the circadian cycle, where the circadian drive to initiate sleep continually shifts a little each day (about 15 minutes on average) until the cycle repeats itself. Initially, when the circadian cycle becomes desynchronous with the 24h day-night cycle, individuals with Non-24 have difficulty initiating sleep. As time progresses, the internal circadian rhythms of these individuals becomes 180 degrees out of synchrony with the 24h day-night cycle, which gradually makes sleeping at night virtually impossible, and leads to extreme sleepiness during daytime hours.
Eventually, the individual’s sleep-wake cycle becomes aligned with the night, and “free-running” individuals are able to sleep well during a conventional or socially acceptable time. However, the alignment between the internal circadian rhythm and the 24-hour day-night cycle is only temporary.
In addition to cyclical nighttime sleep and daytime sleepiness problems, this condition can cause deleterious daily shifts in body temperature and hormone secretion, may cause metabolic disruption and is sometimes associated with depressive symptoms and mood disorders.
It is estimated that 50-75% of totally blind people in the United States (approximately 65,000 to 95,000) have Non-24. This condition can also affect sighted people. However, cases are rarely reported in this population, and the true rate of Non-24 in the general population is not known.
The ultimate treatment goal for individuals with Non-24 is to entrain or synchronize their circadian rhythms into an appropriate phase relationship with the 24-hour day so that they will have increased sleepiness during the night and increased wakefulness during the daytime. Tasimelteon
Tasimelteon is a circadian regulator which binds specifically to two high affinity melatonin receptors, Mella (MT1R) and Mellb (MT2R). These receptors are found in high density in the suprachiasmatic nucleus of the brain (SCN), which is responsible for synchronizing our sleep/wake cycle. Tasimelteon has been shown to improve sleep parameters in prior clinical studies, which simulated a desynchronization of the circadian clock. Tasimelteon has so far been studied in hundreds of individuals and has shown a good tolerability profile.
Tasimelteon has the chemical name: tr ns-N-[[2-(2,3-dihydrobenzofuran- 4-yl)cycloprop-lyl] methyl] propanamide, has the structure of Formula I:

Formula I
and is disclosed in US 5856529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78°C (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG- 400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MTIR. It’s affinity (¾) for MTIR is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
Metabolites of tasimelteon include, for example, those described in “Preclinical Pharmacokinetics and Metabolism of BMS-214778, a Novel
Melatonin Receptor Agonist” by Vachharajani et al., J. Pharmaceutical Sci., 92(4):760-772, which is hereby incorporated herein by reference. The active metabolites of tasimelteon can also be used in the method of this invention, as can pharmaceutically acceptable salts of tasimelteon or of its active metabolites. For example, in addition to metabolites of Formula II and III, above, metabolites of tasimelteon also include the monohydroxylated analogs M13 of Formula IV, M12 of Formula V, and M14 of Formula VI.
Formula IV

Formula V
MO

Formula VI
Thus, it is apparent that this invention contemplates entrainment of patients suffering free running circadian rhythm to a 24 hour circadian rhythm by administration of a circadian rhythm regulator (i.e., circadian rhythm modifier) capable of phase advancing and/or entraining circadian rhythms, such as a melatonin agonist like tasimelteon or an active metabolite oftasimelteon or a pharmaceutically acceptable salt thereof. Other MT1R and MT2R agonists, i.e., melatonin agonists, can have similar effects on the master body clock. So, for example, this invention further contemplates the use of melatonin agonists such as but not limited to melatonin, N-[l-(2,3-dihydrobenzofuran-4- yl)pyrrolidin-3-yl]-N-ethylurea and structurally related compounds as disclosed in US 6,211,225, LY-156735 ((R)-N-(2-(6-chloro-5-methoxy-lH-indol- 3yl) propyl) acetamide) (disclosed in U.S. Patent No. 4,997,845), agomelatine (N- [2-(7-methoxy-l-naphthyl)ethyl]acetamide) (disclosed in U.S. Patent No.
5,225,442), ramelteon ((S)-N-[2-(l,6,7,8-tetrahydro-2H-indeno- [5,4-b] furan-8- yl)ethyl]propionamide), 2-phenylmelatonin, 8-M-PDOT, 2-iodomelatonin, and 6- chloromelatonin.
Additional melatonin agonists include, without limitation, those listed in U.S. Patent Application Publication No. 20050164987, which is incorporated herein by reference, specifically: TAK-375 (see Kato, K. et al. Int. J.
Neuropsychopharmacol. 2000, 3 (Suppl. 1): Abst P.03.130; see also abstracts P.03.125 and P.03.127), CGP 52608 (l-(3-allyl-4-oxothiazolidine-2-ylidene)-4- met- hylthiosemicarbazone) (See Missbach et al., J. Biol. Chem. 1996, 271, 13515-22), GR196429 (N-[2-[2,3,7,8-tetrahydro-lH-fur-o(2,3-g)indol-l- yl] ethyl] acetamide) (see Beresford et al., J. Pharmacol. Exp. Ther. 1998, 285, 1239-1245), S20242 (N-[2-(7-methoxy napth-l-yl) ethyl] propionamide) (see Depres-Brummer et al., Eur. J. Pharmacol. 1998, 347, 57-66), S-23478 (see Neuropharmacology July 2000), S24268 (see Naunyn Schmiedebergs Arch. June 2003), S25150 (see Naunyn Schmiedebergs Arch. June 2003), GW-290569, luzindole (2-benzyl-N-acetyltryptamine) (see U.S. Patent No. 5,093,352), GR135531 (5-methoxycarbonylamino-N-acetyltrypt- amine) (see U.S. Patent Application Publication No. 20010047016), Melatonin Research Compound A, Melatonin Agonist A (see IMSWorld R&D Focus August 2002), Melatonin
Analogue B (see Pharmaprojects August 1998), Melatonin Agonist C (see Chem. Pharm. Bull. (Tokyo) January 2002), Melatonin Agonist D (see J. Pineal Research November 2000), Melatonin Agonist E (see Chem. Pharm. Bull. (Tokyo) Febrary 2002), Melatonin Agonist F (see Reprod. Nutr. Dev. May 1999), Melatonin Agonist G (see J. Med. Chem. October 1993), Melatonin Agonist H (see Famaco March 2000), Melatonin Agonist I (see J. Med. Chem. March 2000), Melatonin Analog J (see Bioorg. Med. Chem. Lett. March 2003), Melatonin Analog K (see MedAd News September 2001), Melatonin Analog L, AH-001 (2-acetamido-8- methoxytetralin) (see U.S. Patent No. 5,151,446), GG-012 (4-methoxy-2- (methylene propylamide)indan) (see Drijfhout et al., Eur. J. Pharmacol. 1999, 382, 157-66), Enol-3-IPA, ML-23 (N-2,4-dinitrophenyl-5-methoxy-tryptamine ) (see U.S. Patent No. 4,880,826), SL-18.1616, IP-100-9 (US 5580878), Sleep Inducing Peptide A, AH-017 (see U.S. Patent No. 5,151,446), AH-002 (8-methoxy- 2-propionamido-tetralin) (see U.S. Patent No. 5,151,446), and IP-101.
Metabolites, prodrugs, stereoisomers, polymorphs, hydrates, solvates, and salts of the above compounds that are directly or indirectly active can, of course, also be used in the practice of this invention.
Melatonin agonists with a MT1R and MT2R binding profile similar to that of tasimelteon, which has 2 to 4 time greater specificity for MT2R, are preferred.
Tasimelteon can be synthesized by procedures known in the art. The preparation of a 4-vinyl-2,3-dihydrobenzofuran cyclopropyl intermediate can be carried out as described in US7754902, which is incorporated herein by reference as though fully set forth.
Pro-drugs, e.g., esters, and pharmaceutically acceptable salts can be prepared by exercise of routine skill in the art.
In patients suffering a Non-24, the melatonin and Cortisol circadian rhythms and the natural day/night cycle become desynchronized. For example, in patients suffering from a free-running circadian rhythm, melatonin and Cortisol acrophases occur more than 24 hours, e.g., >24.1 hours, prior to each previous day’s melatonin and Cortisol acrophase, respectively, resulting in desynchronization for days, weeks, or even months, depending upon the length of a patient’s circadian rhythm, before the melatonin, Cortisol, and day /night cycles are again temporarily synchronized.
Chronic misalignment of Cortisol has been associated with metabolic, cardiac, cognitive, neurologic, neoplastic, and hormonal disorders. Such disorders include, e.g., obesity, depression, neurological impairments.

SAR
Figure : Melatonin receptor agonists. The applied colors indicate the mutual properties with the general melatonin receptor agonists pharmacophore.

INTRODUCTION
Tasimelteon has the chemical name: trans-N-[[2-(2,3-dihydrobenzofuran-4-yl)cycloprop-1yl]methyl]propanamide, has the structure of Formula I:

and is disclosed in U.S. Pat. No. 5,856,529 and in US 20090105333, both of which are incorporated herein by reference as though fully set forth.
Tasimelteon is a white to off-white powder with a melting point of about 78° C. (DSC) and is very soluble or freely soluble in 95% ethanol, methanol, acetonitrile, ethyl acetate, isopropanol, polyethylene glycols (PEG-300 and PEG-400), and only slightly soluble in water. The native pH of a saturated solution of tasimelteon in water is 8.5 and its aqueous solubility is practically unaffected by pH. Tasimelteon has 2-4 times greater affinity for MT2R relative to MT1R. It’s affinity (Ki) for MT1R is 0.3 to 0.4 and for MT2R, 0.1 to 0.2. Tasimelteon is useful in the practice of this invention because it is a melatonin agonist that has been demonstrated, among other activities, to entrain patients suffering from Non-24.
SYNTHESIS
(1R-trans)-N-[[2 – (2,3-dihydro-4 benzofuranyl) cyclopropyl] methyl] propanamide PATENT: BRISTOL-MYERS SQUIBB PRIORITY DATE: 1996 HYPNOTIC


PREPARATION OF XV
XXIV D-camphorsulfonic acid IS REACTED WITH THIONYL CHLORIDE TO GIVE
…………XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
TREATED WITH
XXVI ammonium hydroxide
TO GIVE
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
TREATED WITH AMBERLYST15
….XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
TREATED WITH LAH, ie double bond is reduced to get
…..XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide

Intermediate
I 3-hydroxybenzoic acid methyl ester
II 3-bromo-1-propene
III 3 – (2-propenyloxy) benzoic acid methyl ester
IV 3-hydroxy-2-(2-propenyl) benzoic acid methyl ester
V 2,3-dihydro-4-hydroxy-2-benzofurancarboxylic acid methyl ester
VI benzofuran-4-carboxylic acid methyl ester
VII benzofuran-4-carboxylic acid
VIII 2,3-dihydro-4-benzofurancarboxylic acid
IX 2,3-dihydro-4-benzofuranmethanol
X 2,3-dihydro-4-benzofurancarboxaldehyde
XI Propanedioic acid
XII (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoic acid
XIII thionyl chloride
XIV (E) -3 – (2,3-dihydro-4-benzofuranyl) propenoyl chloride
XV (3aS, 6R, 7aR)-hexahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
XVI (3aS,6R,7aR)-1-[(E)-3-(2,3-dihydro-4-benzofuranyl)-1-oxo-2-propenyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVII (3aS,6R,7aR)-1-[[(1R,2R)-2-(2,3-dihydro-4-benzofuranyl)cyclopropyl]carbonyl]hexahydro-8,8-dimethyl-3H-3a,6-methano-2,1-benzisothiazole-2,2-dioxide
XVIII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanol
XIX [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarboxaldehyde
XX hydroxylamine hydrochloride
XXI [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanecarbaldehyde oxime
XXII [R-(R *, R *)] -2 – (2,3-dihydro-4-benzofuranyl) cyclopropanemethanamine
XXIII propanoyl chloride
XXIV D-camphorsulfonic acid
XXV (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonyl chloride
XXVI ammonium hydroxide
XXVII (1S, 4R) -7,7-dimethyl-2-oxo-bicyclo [2.2.1] heptane-1-methanesulfonamide
XXVIII (3aS, 6R) -4,5,6,7-tetrahydro-8 ,8-dimethyl-3H-3a ,6-methano-2 ,1-benzisothiazole-2 ,2-dioxide
Bibliography
– Patents: Benzofuran and dihydrobenzofuran melatonergic agents: US5856529 (1999)
Priority: US19960032689P, 10 Dec. 1996 (Bristol-Myers Squibb Company, U.S.)
– Preparation III (quinazolines): US2004044015 (2004) Priority: EP20000402845, 13 Oct. 2000
– Preparation of VII (aminoalkylindols): Structure-Activity Relationships of Novel Cannabinoid Mimetics Eissenstat et al, J.. Med. Chem. 1995, 38, 3094-3105
– Preparation XXVIII: Towson et al. Organic Syntheses, Coll. Vol. 8, p.104 (1993) Vol. 69, p.158 (1990)
– Preparation XV: Weismiller et al. Organic Syntheses, Coll. Vol. 8, p.110 (1993) Vol. 69, p.154 (1990).
– G. Birznieks et al. Melatonin agonist VEC-162 Improves sleep onset and maintenance in a model of transient insomnia. Sleep 2007, 30, 0773 Abstract.
-. Rajaratnam SM et al, The melatonin agonist VEC-162 Phase time immediately advances the human circadian system, Sleep 2006, 29, 0159 Abstract.
-. AK Singh et al, Evolution of a manufacturing route for a highly potent drug candidate, 229th ACS Natl Meet, March 13-17, 2005, San Diego, Abstract MEDI 576.
– Vachharajani NN et al, Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist, J Pharm Sci. 2003 Apr; 92 (4) :760-72.
. – JW Scott et al, Catalytic Asymmetric Synthesis of a melotonin antagonist; synthesis and process optimization. 223rd ACS Natl Meet, April 7-11, Orlando, 2002, Abstract ORGN 186.
SYNTHESIS CONSTRUCTION AS IN PATENT
GENERAL SCHEMES
Reaction Scheme 1

The syntheses of the 4-aryl-propenoic acid derivatives, 2 and 3, are shown in Reaction Scheme 1. The starting aldehydes, 1 , can be prepared by methods well known to those skilled in the art. Condensation of malonic acid with the aldehydes, 1, in solvents such as pyridine with catalysts such as piperidine or pyrrolidine, gives the 4-aryl- propenoic acid, 2. Subsequent conversion of the acid to the acid chloride using reagents such as thionyl chloride, phosphoryl chloride, or the like, followed by reaction with N,0-dimethyl hydroxylamine gives the amide intermediate 3 in good yields. Alternatively, aldehyde 1 can be converted directly to amide 3 using reagents such as diethyl (N-methoxy- N-methyl-carbamoylmethyl)phosphonate with a strong base such as sodium hydride.
Reaction Scheme 2

The conversion of the amide intermediate 3 to the racemic, trans- cyclopropane carboxaldehyde intermediate, 4, is shown in Reaction Scheme 2. Intermediate 3 was allowed to react with cyclopropanating reagents such as trimethylsulfoxonium iodide and sodium hydride in solvents such as DMF, THF, or the like. Subsequent reduction using reagents such as LAH in solvents such as THF, ethyl ether, or the like, gives the racemic, trans-cyclopropane carboxaldehyde intermediates, 4.
Reaction Scheme 3

Racemic cyclopropane intermediate 5 (R = halogen) can be prepared from intermediate 2 as shown in Reaction Scheme 3. Intermediate 2 was converted to the corresponding allylic alcohol by treatment with reducing agents such as sodium borohydride plus iodine in solvents such as THF. Subsequent acylation using reagents such as acetic anhydride in pyridine or acetyl chloride gave the allylic acetate which was allowed to react with cyclopropanating reagents such as sodium chloro-difluoroacetate in diglyme to provide the racemic, trans- cyclopropane acetate intermediates, 5. Reaction Scheme 4

The conversion of the acid 2 to the chiral cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, is shown in Reaction Scheme 4. Intermediate 2 is condensed with (-)-2,10-camphorsultam under standard conditions, and then cyclopropanated in the presence of catalysts such as palladium acetate using diazomethane generated from reagents such as 1-methyl-3-nitro-1-nitrosoguanidine. Subsequent reduction using reagents such as LAH in solvents such as THF, followed by oxidation of the alcohol intermediates using reagents such as DMSO/oxalyl chloride, or PCC, gives the cyclopropane carboxaldehyde intermediate, (-)-(trans)-4, in good yields. The enantiomer, (+)-(trans)-4, can also be obtained employing a similar procedure using (+)-2,10- camphorsultam in place of (-)-2,10-camphorsultam.
When it is desired to prepare compounds of Formula I wherein m = 2, the alcohol intermediate may be activated in the conventional manner such as with mesyl chloride and treated with sodium cyanide followed by reduction of the nitrile group with a reducing agent such as LAH to produce the amine intermediate 6.
Reaction Scheme 5


Reaction Scheme 5 shows the conversion of intermediates 4 and 5 to the amine intermediate, 7, and the subsequent conversion of 6. or 7 to compounds of Formula I. The carboxaldehyde intermediate, 4, is condensed with hydroxylamine and then reduced with reagents such as LAH to give the amine intermediate, 7. The acetate intermediate 5 is hydrolyzed with potassium hydroxide to the alcohol, converted to the mesylate with methane sulfonyl chloride and triethyl amine in CH2CI2and then converted to the azide by treatment with sodium azide in solvents such as DMF. Subsequent reduction of the azide group with a reducing agent such as LAH produced the amine intermediate 7. Further reaction of 6 or 7 with acylating reagents gives compounds of Formula I. Suitable acylating agents include carboxylic acid halides, anhydrides, acyl imidazoles, alkyl isocyanates, alkyl isothiocyanates, and carboxylic acids in the presence of condensing agents, such as carbonyl imidazole, carbodiimides, and the like. Reaction Scheme 6

Reaction Scheme 6 shows the alkylation of secondary amides of Formula I (R2 = H) to give tertiary amides of Formula I (R2 = alkyl). The secondary amide is reacted with a base such as sodium hydride, potassium tert-butoxide, or the like, and then reacted with an alkylating reagent such as alkyl halides, alkyl sulfonate esters, or the like to produce tertiary amides of Formula I.
Reaction Scheme 7

Reaction Scheme 7 shows the halogenation of compounds of Formula I. The carboxamides, i (Q1 = Q2 = H), are reacted with excess amounts of halogenating agents such as iodine, N-bromosuccinimide, or the like to give the dihalo-compounds of Formula I (Q1 = Q2 = halogen). Alternatively, a stoichiometric amount of these halogenating agents can be used to give the monohalo-compounds of Formula I (Q1 = H, Q2 = halogen; or Q1 = halogen, Q2 = H). In both cases, additives such as lead IV tetraacetate can be used to facilitate the reaction. Biological Activity of the Compounds
The compounds of the invention are melatonergic agents. They have been found to bind human melatonergic receptors expressed in a stable cell line with good affinity. Further, the compounds are agonists as determined by their ability, like melatonin, to block the forskolin- stimulated accumulation of cAMP in certain cells. Due to these properties, the compounds and compositions of the invention should be useful as sedatives, chronobiotic agents, anxiolytics, antipsychotics, analgesics, and the like. Specifically, these agents should find use in the treatment of stress, sleep disorders, seasonal depression, appetite regulation, shifts in circadian cycles, melancholia, benign prostatic hyperplasia and related conditions
EXPERIMENTAL PROCEDURES
SEE ORIGINAL PATENT FOR CORECTIONS
Preparation 1
Benzofuran-4-carboxaldehyde
Step 1 : N-Methoxy-N-methyl-benzofuran-4-carboxamide
A mixture of benzofuran-4-carboxylic acid [Eissenstat, et al.. J. Medicinal Chemistry, 38 (16) 3094-3105 (1995)] (2.8 g, 17.4 mmol) and thionyl chloride (25 mL) was heated to reflux for 2 h and then concentrated in vacuo. The solid residue was dissolved in ethyl acetate (50 mL) and a solution of N,O-dimethylhydroxylamine hydrochloride (2.8 g) in saturated NaHC03(60 mL) was added with stirring. After stirring for 1.5 h, the ethyl acetate layer was separated. The aqueous layer was extracted with ethyl acetate. The ethyl acetate extracts were combined, washed with saturated NaHCO3 and concentrated in vacuo to give an oil (3.2 g, 95.4%).
Step 2: Benzofuran-4-carboxaldehyde
A solution of N-methoxy-N-methyl-benzofuran-4-carboxamide (3.2 g, 16.6 mmol) in THF (100 mL) was cooled to -45°C and then LAH (0.7 g, 18.7 mmol) was added. The mixture was stirred for 15 min, allowed to warm to -5°C, and then recooled to -45°C. Saturated KHS04 (25 mL) was added with vigorous stirring, and the mixture was allowed to warm to room temperature. The precipitate was filtered and washed with acetone. The filtrate was concentrated in vacuo to give an oil (2.3 g, 94%). Preparation 2
2,3-Dihydrobenzofuran-4-carboxaldehyde
Step 1 : 2,3-Dihydrobenzofuran-4-carboxylic acid
Benzofuran-4-carboxylic acid (10.0 g, 61 .7 mmol) was hydrogenated (60 psi) in acetic acid (100 mL) over 10% Pd/C (2 g) for 12 hr. The mixture was filtered and the filtrate was diluted with water (500 mL) to give 2,3- dihydrobenzofuran-4-carboxylic acid as a white powder (8.4 g, 83%). A sample was recrystallized from isopropanol to give fine white needles (mp: 185.5-187.5°C).
Step 2: (2,3-Dihydrobenzofuran-4-yl)methanol
A solution of 2,3-dihydrobenzofuran-4-carboxylic acid (10 g, 61 mmol) in THF (100 mL) was stirred as LAH (4.64 g, 122 mmol) was slowly added. The mixture was heated to reflux for 30 min. The mixture was cooled and quenched cautiously with ethyl acetate and then with 1 N HCI (150 mL). The mixture was then made acidic with 12 N HCI until all the inorganic precipitate dissolved. The organic layer was separated, and the inorganic layer was extracted twice with ethyl acetate. The organic layers were combined, washed twice with brine, and then concentrated in vacuo. This oil was Kϋgelrohr distilled to a clear oil that crystallized upon cooling (8.53 g, 87.6%).
Step 3: 2.3-Dihydrobenzofuran-4-carboxaldehyde
DMSO (8.10 mL, 1 14 mmol) was added at -78°C to a stirred solution of oxalyl chloride in CH2CI2 (40 mL of a 2M solution). A solution of (2,3- dihydrobenzofuran-4-yl)methanol (8.53 g, 56.9 mmol) in CH2CI2 (35 mL) was added dropwise, and the solution stirred at -78°C for 30 min. Triethyl amine (33 mL, 228 mmol) was added cautiously to quench the reaction. The resulting suspension was stirred at room temperature for 30 min and diluted with CH2CI2 (100 mL). The organic layer was washed three times with water, and twice with brine, and then concentrated in vacuo to an oil (8.42 g, 100%) that was used without purification.
Preparation 16
(±)-(trans)-2-(2,3-Dihyd robenzofuran-4-yl)cyclopropane- carboxaldehyde
Step 1 : (±Htrans)-N-Methoxy-N-methyl-2-(2.3-dihydrobenzofuran-4- yhcyclopropanecarboxamide
Trimethylsulfoxonium iodide (9.9 g, 45 mmol) was added in small portions to a suspension of sodium hydride (1 .8 g, 45 mmol) in DMF (120 mL). After the foaming had subsided (10 min), a solution of (trans)- N-methoxy-N-methyl-3-(2,3-dihydrobenzofuran-4-yl)propenamide (3.5 g, 15 mmol) in DMF (60 mL) was added dropwise, with the temperature maintained between 35-40°C. The mixture was stirred for 3 h at room temperature. Saturated NH4CI (50 mL) was added dropwise and the mixture was extracted three times with ethyl acetate. The organic extracts were combined, washed with H2O and brine, dried over K2CO3, and concentrated in vacuo to give a white wax (3.7 g, 100%).
Step 2: (±)-(trans)- 2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde
A solution of (±)-(trans)-N-methoxy-N-methyl-2-(2,3-dihydrobenzofuran- 4-yl)cyclopropanecarboxamide (3.7 g, 15 mmol) in THF (10 mL) was added dropwise to a rapidly stirred suspension of LAH (683 mg, 18 mmol) in THF (50 mL) at -45°C, maintaining the temperature below -40°C throughout. The cooling bath was removed, the reaction was allowed to warm to 5°C, and then the reaction was immediately recooled to -45°C. Potassium hydrogen sulfate (3.4 g, 25.5 mmol) in H20 (50 mL) was cautiously added dropwise, the temperature maintained below – 30°C throughout. The cooling bath was removed and the suspension was stirred at room temperature for 30 min. The mixture was filtered through Celite and the filter cake was washed with ether. The combined filtrates were then washed with cold 1 N HCI, 1 N NaOH, and brine. The filtrates were dried over MgSO4, and concentrated in vacuo to give a clear oil (2.6 g, 99%).
Preparation 18
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde
Step 1 : (-Htrans)-N-[3-(2.3-Dihvdrobenzofuran-4-yl)-propenoyll-2.10- camphorsultam
To a solution of (-)-2,10-camphorsultam (8.15 g, 37.9 mmol) in 50 mL toluene at 0°C was added sodium hydride (1.67 g, 41.7 mmol). After stirring for 0.33 h at 0°C and 0.5 h at 20°C and recooling to 0°C, a solution of 3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl chloride
(37.9 mmol), prepared in situ from the corresponding acid and thionyl chloride (75 mL), in toluene (50 mL), was added dropwise. After stirring for 18 h at 20°C, the mixture was diluted with ethyl acetate and washed with water, 1 N HCI, and 1 N NaOH. The organic solution was dried and concentrated in vacuo to give 15.8 g of crude product. Recrystallization form ethanol-methanol (600 mL, 1 :1) gave the product (13.5 g, 92%, mp 199.5-200°C).
Step 2: (-)-N-[[(trans)-2-(2,3-Dihydrobenzofuran-4-yl)-cyclopropylj- carbonylj-2, 10-camphorsultam
1 -Methyl-3-nitro-1 -nitrosoguanidine (23.88g 163 mmol) was added in portions to a mixture of 10 N sodium hydroxide (60 mL) and ether (200 mL) at 0°C. The mixture was shaken vigorously for 0.25 h and the ether layer carefully decanted into a solution of (-)-N-[3-(2,3-dihydrobenzofuran-4-yl)-2-propenoyl]-2,10-camphorsultam (9.67 g, 25 mmol) and palladium acetate (35 mg) in methylene chloride (200 mL). After stirring for 18 h, acetic acid (5 mL) was added to the reaction and the mixture stirred for 0.5 h. The mixture was washed with 1 N HCI, 1 N NaOH and brine. The solution was dried, concentrated in vacuo and the residue crystallized twice from ethanol to give the product (6.67 g, 66.5%, mp 157-159°C).
Step 3: (-)-(trans)-2-(2,3-Dihydrobenzofuran-4-yl)cyclopropane- methanol
A solution of (-)-N-[(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclo-propanecarbonylj-2,10-camphorsultam (4.3 g, 10.7 mmol) in THF (50 mL) was added dropwise to a mixture of LAH (0.81 g, 21.4 mmol) in THF (50 mL) at -45°C. The mixture was stirred for 2 hr while it warmed to 10°C. The mixture was recooled to -40°C and hydrolyzed by the addition of saturated KHS0 (20 mL). The mixture was stirred at room temperature for 30 minutes and filtered. The precipitate was washed twice with acetone. The combined filtrate and acetone washes were concentrated in vacuo. The gummy residue was dissolved in ether, washed with 1 N NaOH and 1 N HCI, and then dried in vacuo to give the product (2.0 g, 98.4%).
Step 4: (-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane- carboxaldehyde DMSO (1.6 g, 21 mmol) was added to oxalyl chloride in CH2CI2(7.4 mL of 2 M solution, 14.8 mmole) at -78°C. The (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)-cyclopropylmethanol (2.0 g, 10.5 mmol) in CH2CI2(15 mL) was added. The mixture was stirred for 20 min and then triethylamine (4.24 g, 42 mmol) was added. The mixture was warmed to room temperature and stirred for 30 min. The mixture was diluted with CH2CI2 and washed with water, 1 N HCI, and then 1 N NaOH. The organic layer was dried and concentrated iι> vacuo to give the aldehyde product (1.98 g, 100%).
Preparation 24
(-)-(trans)-2-(2.3-Dihydrobenzofuran-4-yl)cyclopropane-methanamine A mixture of (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)cyclopropane-carboxaldehyde (1.98 g, 10.5 mmol), hydroxylamine hydrochloride (2.29 g, 33 mmol), and 30% NaOH (3.5 mL, 35 mmol), in 5:1
ethanol/water (50 mL) was heated on a steam bath for 2 h. The solution was concentrated in vacuo. and the residue mixed with water. The mixture was extracted with CH2CI2. The organic extracts were dried and concentrated in vacuo to give a solid which NMR analysis showed to be a mixture of the cis and trans oximes. This material was dissolved in THF (20 mL) and added to solution of alane in THF [prepared from LAH (1.14 g, 30 mmol) and H2S04 (1.47 g, 15 mmol) at 0°Cj. The reaction was stirred for 18 h, and quenched successively with water (1.15 mL), 15% NaOH (1.15 mL), and then water (3.45 mL). The mixture was filtered and the filtrate was concentrated in vacuo. The residue was mixed with ether and washed with water and then 1 N HCI. The acid washes were made basic and extracted with CH2CI . The extracts were dried and concentrated in vacuo to give the amine product (1.4 g, 70.5%). The amine was converted to the fumarate salt in ethanol (mp: 197-198°C).
Anal. Calc’d for C12H15NO • C4H404: C, 62.94; H, 6.27; N, 4.59.
Found: C, 62.87; H, 6.31 ; N, 4.52.
FINAL PRODUCT TASIMELTEON
Example 2
(-)-(trans)-N-[[2-(2,3-Dihydrobenzofuran-4-yl)cycloprop-1-yl]methyl]propanamide
This compound was prepared similar to the above procedure using propionyl chloride and (-)-(trans)-2-(2,3-dihydrobenzofuran-4-yl)- cyclopropanemethanamine to give an oil that solidified upon standing to an off-white solid (61 %, mp: 71-72°C). IR (NaCI Film): 3298, 1645, 1548, 1459, 1235 cm“1.
Mo5 : -17.3°
Anal. Calc’d for C15H19N02: C, 73.44; H, 7.87; N, 5.71 . Found: C, 73.28; H, 7.68; N, 5.58
SYNTHESIS
Synthesis Path
SYN

Tasimelteon (Hetlioz)Tasimelteon, which is marketed by Vanda Pharmaceuticals as Hetlioz and developed in partnership with Bristol-Myers Squibb,is a drug that was approved by the US FDA in January 2014 for the treatment of non-24-hour sleep–wake disorder (also called Non-24, N24 and N24HSWD).234 Tasimelteon is a melatonin MT1
and MT2 receptor agonist; because it exhibits a greater affinity to the MT2 receptor than MT1, is also known as Dual Melatonin
Receptor Agonist.234 Two randomized controlled trials (phases II
and III) demonstrated that tasimelteon improved sleep latency
and maintenance of sleep with a shift in circadian rhythms, and
therefore has the potential to treat patients with transient insomnia
associated with circadian rhythm sleep disorders.235 Preclinical
studies showed that the drug has similar phase-shifting properties
to melatonin, but with less vasoconstrictive effects.236 The most
likely scale preparation of the drug, much of which has been published
in the chemical literature, is described below in Scheme 44.
Activation of commercial bis-ethanol 250 with 2.5 equivalents
of the Vilsmeier salt 251 followed by treatment with base resulted
an intramolecular cyclization reaction with the proximal phenol
and concomitant elimination of the remaining imidate to deliver
the vinylated dihydrobenzofuran 252 in 76% yield.237 Interestingly,
this reaction could be performed on multi-kilogram scale, required
no chromatographic purification, and generated environmentallyfriendly
DMF and HCl as byproducts.237 Sharpless asymmetric
dihydroxylation of olefin 252 delivered diol 253 in 86% yield and
impressive enantioselectivity (>99% ee). This diol was then activated
with trimethylsilyl chloride and then treated with base to generate epoxide 254.238 Next, a modified Horner–Wadsworth–
Emmons reaction involving triethylphosphonoacetate (TEPA, 255)
was employed to convert epoxide 254 to cyclopropane 256.239
The reaction presumably proceeds through removal of the acidic
TEPA proton followed by nucleophilic attack at the terminal epoxide
carbon. The resulting alkoxide undergoes an intramolecular
phosphoryl transfer reaction resulting in an enolate, which then attacked the newly formed phosphonate ester in an SN2 fashion
resulting in the trans-cyclopropane ester, which was ultimately
saponified and re-acidified to furnish cyclopropane acid 256.239
Conversion of this acid to the corresponding primary amide preceded
carbonyl reduction with sodium borohydride. The resulting
amine was acylated with propionyl chloride to furnish tasimelteon
(XXXI) as the final product in 86% yield across the four-step
sequence.
PATENTS
| US2010261786 | 10-15-2010 | PREDICTION OF SLEEP PARAMETER AND RESPONSE TO SLEEP-INDUCING COMPOUND BASED ON PER3 VNTR GENOTYPE |
| US2009209638 | 8-21-2009 | TREATMENT FOR DEPRESSIVE DISORDERS |
| US6060506 | 5-10-2000 | Benzopyran derivatives as melatonergic agents |
| US5981571 | 11-10-1999 | Benzodioxa alkylene ethers as melatonergic agents |
| WO9825606 | 6-19-1998 | BENZODIOXOLE, BENZOFURAN, DIHYDROBENZOFURAN, AND BENZODIOXANE MELATONERGIC AGENTS |
| WO2007137244A1 * | May 22, 2007 | Nov 29, 2007 | Gunther Birznieks | Melatonin agonist treatment |
| US4880826 | Jun 25, 1987 | Nov 14, 1989 | Nava Zisapel | Melatonin antagonist |
| US4997845 | May 10, 1990 | Mar 5, 1991 | Eli Lilly And Company | β-alkylmelatonins as ovulation inhibitors |
| US5093352 | May 16, 1990 | Mar 3, 1992 | Whitby Research, Inc. | Antidepressant agents |
| US5151446 | Mar 28, 1991 | Sep 29, 1992 | Northwestern University | Substituted 2-amidotetralins as melatonin agonists and antagonists |
| US5225442 | Jan 3, 1992 | Jul 6, 1993 | Adir Et Compagnie | Compounds having a naphthalene structure |
| US5580878 | Jun 7, 1995 | Dec 3, 1996 | Interneuron Pharmaceuticals, Inc. | Substituted tryptamines phenalkylamines and related compounds |
| US5856529 | Dec 9, 1997 | Jan 5, 1999 | Bristol-Myers Squibb Company | Benzofuran and dihydrobenzofuran melatonergic agents |
| US6211225 | Jun 6, 2000 | Apr 3, 2001 | Bristol-Meyers Squibb | Heterocyclic aminopyrrolidine derivatives as melatonergic agents |
| US7754902 | May 18, 2006 | Jul 13, 2010 | Vanda Pharmaceuticals, Inc. | Ruthenium(II) catalysts for use in stereoselective cyclopropanations |
| US20010047016 | Apr 12, 2001 | Nov 29, 2001 | Gregory Oxenkrug | Method for treating depression |
| US20050164987 | Dec 22, 2004 | Jul 28, 2005 | Barberich Timothy J. | Melatonin combination therapy for improving sleep quality |
| US20090105333 | May 22, 2007 | Apr 23, 2009 | Gunther Birznieks | Melatonin agonist treatment |
extra info
Org. Synth.1990, 69, 154
(−)-D-2,10-CAMPHORSULTAM
[3H-3a,6-Methano-2,1-benzisothiazole, 4,5,6,7-tetrahydro-8,8-dimethyl-2,2-dioxide, (3aS)-]
|
Submitted by Michael C. Weismiller, James C. Towson, and Franklin A. Davis1.
Checked by David I. Magee and Robert K. Boeckman, Jr..
1. Procedure
(−)-2,10-Camphorsultam. A dry, 2-L, three-necked, round-bottomed flask is equipped with a 1.5-in egg-shaped Teflon stirring bar, a 250-mL addition funnel, and a 300-mL Soxhlet extraction apparatus equipped with a mineral oil bubbler connected to an inert-gas source. The flask is charged with 600 mL of dry tetrahydrofuran (THF) (Note 1) and6.2 g (0.16 mol) of lithium aluminum hydride (Note 2). Into the 50-mL Soxhlet extraction thimble is placed 35.0 g (0.16 mol) of (−)-(camphorsulfonyl)imine (Note 3) and the reaction mixture is stirred and heated at reflux. After all of the(camphorsulfonyl)imine has been siphoned into the reaction flask (3–4 hr), the mixture is allowed to cool to room temperature. The unreacted lithium aluminum hydride is cautiously hydrolyzed by dropwise addition of 200 mL of 1 Nhydrochloric acid via the addition funnel (Note 4). After the hydrolysis is complete the contents of the flask are transferred to a 1-L separatory funnel, the lower, silver-colored aqueous layer is separated, and the upper layer placed in a 1-L Erlenmeyer flask. The aqueous phase is returned to the separatory funnel and washed with methylene chloride (3 × 100 mL). After the reaction flask is rinsed with methylene chloride (50 mL), the organic washings are combined with the THF phase and dried over anhydrous magnesium sulfate for 10–15 min. Filtration through a 300-mL sintered-glass funnel of coarse porosity into a 1-L round-bottomed flask followed by removal of the solvent on arotary evaporator gives 33.5 g (95%) of the crude (−)-2,10-camphorsultam. The crude sultam is placed in a 250-mL Erlenmeyer flask and crystallized from approximately 60 mL of absolute ethanol. The product is collected on a 150-mL sintered-glass funnel of coarse porosity and dried in a vacuum desiccator to give 31.1 g (88%) of the pure sultam. A second crop of crystals can be gained by evaporating approximately half the filtrate; the residue is crystallized as above to give 1.4 g (4%). The combined yield of white crystalline solid, mp 183–184°C, [α]D −30.7° (CHCl3, c 2.3) is92% (Note 5) and (Note 6).
2. Notes
1. Tetrahydrofuran (Aldrich Chemical Company, Inc.) was distilled from sodium benzophenone.
2. Lithium aluminum hydride was purchased from Aldrich Chemical Company, Inc.
3. (−)-(Camphorsulfonyl)imine, [(7S)-(−)-10,10-dimethyl-5-thia-4-azatricyclo[5.2.1.03,7]dec-3-ene 5,5-dioxide] was prepared by the procedure of Towson, Weismiller, Lal, Sheppard, and Davis, Org. Synth., Coll. Vol. VIII, 1993, 104.
4. The addition must be very slow at first (1 drop/5 sec) until the vigorous reaction has subsided.
5. The NMR spectrum of (−)-2,10-camphorsultam is as follows: 1H NMR (CDCl3) δ: 0.94 (s, 3 H, CH3), 1.14 (s, 3 H, CH3), 1.33 (m, 1 H), 1.47 (m,, 1 H), 1.80–2.05 (5 H), 3.09 (d, 1 H, J = 14), 3.14 (d, 1 H, J = 14), 3.43 (m, 1 H), 4.05 (br s, 1 H, NH); 13C NMR (CDCl3) δ: 20.17 (q, CH3), 26.51 (t), 31.55 (t), 35.72 (t), 44.44 (d), 47.15 (s), 50.08 (t), 54.46 (s), 62.48 (d).
6. Checkers obtained material having the same mp (183–184°C) and [α]D − 31.8° (CHCl3, c 2.3).
3. Discussion
(−)-2,10-Camphorsultam was first prepared by the catalytic hydrogenation of (−)-(camphorsulfonyl)imine overRaney nickel.2 Lithium aluminum hydride reduction was used by Oppolzer and co-workers in their synthesis of the sultam.3,4 However, because of the low solubility of the sultam in tetrahydrofuran, a large amount of solvent was required.4 In the procedure described here the amount of solvent is significantly reduced by using a Soxhlet extractor to convey the imine slowly into the reducing medium.5
Oppolzer’s chiral auxiliary,6 (−)-2,10-camphorsultam, is useful in the asymmetric Diels–Alder reaction,3,4 and for the preparation of enantiomerically pure β-substituted carboxylic acids7 and diols,8 in the stereoselective synthesis of Δ2-isoxazolines,9 and in the preparation of N-fluoro-(−)-2,10-camphorsultam, an enantioselective fluorinating reagent.10
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Shriner, R. L.; Shotton, J. A.; Sutherland, H. J. Am. Chem. Soc.1938, 60, 2794.
- Oppolzer, W.; Chapuis, C.; Bernardinelli, G. Helv. Chim. Acta1984, 67, 1397.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, G.; Carroll,, P. J. J. Am. Chem. Soc.1988, 110, 8477.
- Oppolzer, W. Tetrahedron1987, 43, 1969.
- Oppolzer, W.; Mills, R. J.; Pachinger, W.; Stevenson, T. Helv. Chim. Acta1986, 69, 1542; Oppolzer, W.; Schneider, P. Helv. Chim. Acta1986, 69, 1817; Oppolzer, W.; Mills, R. J.; Réglier, M. Tetrahedron Lett.1986, 27, 183; Oppolzer, W.; Poli. G.Tetrahedron Lett.1986, 27, 4717; Oppolzer, W.; Poli, G.; Starkemann, C.; Bernardinelli, G. Tetrahedron Lett.1988, 29, 3559.
- Oppolzer, W.; Barras, J-P. Helv. Chim. Acta1987, 70, 1666.
- Curran, D. P.; Kim, B. H.; Daugherty, J.; Heffner, T. A. Tetrahedron Lett.1988, 29, 3555.
- Differding, E.; Lang, R. W. Tetrahedron Lett.1988, 29, 6087.
Org. Synth.1990, 69, 158
(+)-(2R,8aS)-10-(CAMPHORYLSULFONYL)OXAZIRIDINE
[4H-4A,7-Methanooxazirino[3,2-i][2,1]benzisothiazole, tetrahydro-9,9-dimethyl-, 3,3-dioxide, [4aS-(4aα,7α,8aR*)]]
 |
Submitted by James C. Towson, Michael C. Weismiller, G. Sankar Lal, Aurelia C. Sheppard, Anil Kumar, and Franklin A. Davis1.
Checked by David I. Magee and Robert K. Boeckman, Jr..
1. Procedure
A. (+)-(1S)-10-Camphorsulfonamide. Into a 2-L, two-necked, round-bottomed flask, equipped with a 250-mL dropping funnel, a magnetic stirring bar, and a reflux condenser fitted with an outlet connected to a disposable pipettedipped in 2 mL of chloroform in a test tube for monitoring gas evolution, were placed 116 g (0.5 mol) ofcamphorsulfonic acid (Note 1) and 750 mL of reagent-grade chloroform. The suspension of camphorsulfonic acid was heated to reflux and 71.4 g (43.77 mL, 0.6 mol, 1.2 equiv) of freshly distilled thionyl chloride was added dropwise over a 1-hr period. Heating was continued until gas evolution (sulfur dioxide and hydrogen chloride) had ceased (approximately 9–10 hr). The resultant solution of camphorsulfonyl chloride in chloroform was converted tocamphorsulfonamide without further purification.
In a 5-L, two-necked, round-bottomed flask fitted with a 250-mL dropping funnel and a mechanical stirrer was placed a solution of 1.6 L of reagent-grade ammonium hydroxide solution and the flask was cooled to 0°C in an ice bath. The solution of the crude camphorsulfonyl chloride, prepared in the preceding section, was added dropwise to the ammonium hydroxide solution at 0–10°C over a period of 1 hr. The reaction mixture was warmed to room temperature, stirred for 4 hr, the organic layer separated, and the aqueous layer was extracted with methylene chloride (3 × 250 mL). The combined organic layers were washed with brine (250 mL) and dried over anhydrousmagnesium sulfate. Removal of the solvent on the rotary evaporator gave 104.0 g (90%) of the crudecamphorsulfonamide (Note 2) and (Note 3).
B. (−)-(Camphorsulfonyl)imine. A 1-L, round-bottomed flask is equipped with a 2-in. egg-shaped magnetic stirring bar, a Dean–Stark water separator, and a double-walled condenser containing a mineral oil bubbler connected to an inert gas source. Into the flask are placed 5 g of Amberlyst 15 ion-exchange resin (Note 4) and 41.5 g of the crude(+)-(1S)-camphorsulfonamide in 500 mL of toluene. The reaction mixture is heated at reflux for 4 hr. After the reaction flask is cooled, but while it is still warm (40–50°C), 200 mL of methylene chloride is slowly added to dissolve any(camphorsulfonyl)imine that crystallizes. The solution is filtered through a 150-mL sintered glass funnel of coarse porosity an the reaction flask and filter funnel are washed with an additional 75 mL of methylene chloride.
Isolation of the (−)-(camphorsulfonyl)imine is accomplished by removal of the toluene on the rotary evaporator. The resulting solid is recrystallized from absolute ethanol (750 mL) to give white crystals, 34.5–36.4 g (90–95%), mp225–228°C; [α]D −32.7° (CHCl3, c 1.9) (Note 5).
C. (+)-(2R, 8aS)-10-Camphorylsulfonyloxaziridine. A 5-L, three-necked, round-bottomed Morton flask is equipped with an efficient mechanical stirrer, a 125-mm Teflon stirring blade, a Safe Lab stirring bearing (Note 6), and a 500-mL addition funnel. Into the flask are placed the toluene solution of (−)-(camphorsulfonyl)imine (39.9 g, 0.187 mol)prepared in Step B and a room-temperature solution of 543 g (3.93 mol, 7 equiv based on oxone) of anhydrouspotassium carbonate dissolved in 750 mL of water. The reaction mixture is stirred vigorously and a solution of 345 g (0.56 mol, 6 equiv of KHSO5) of oxone dissolved in 1250 mL of water is added dropwise in three portions over 45 min(Note 7) and (Note 8). Completion of the oxidation is determined by TLC (Note 9) and the reaction mixture is filtered through a 150-mL sintered-glass funnel of coarse porosity to remove solids. The filtrate is transferred to a 3-L separatory funnel, the toluene phase is separated and the aqueous phase is washed with methylene chloride (3 × 100 mL). The filtered solids and any solids remaining in the Morton flask are washed with an additional 200 mL of methylene chloride. The organic extracts are combined and washed with 100 mL of saturated sodium sulfite, dried over anhydrousmagnesium sulfate for 15–20 min, filtered, and concentrated on the rotary evaporator. The resulting white solid is crystallized from approximately 500 mL of hot 2-propanol to afford, after drying under vacuum in a desiccator, 35.9 g(84%) of white needles, mp 165–167°C, [α]D +44.6° (CHCl3, c 2.2) (Note 10) and (Note 11).
(−)-(2S,8aR)-10-(camphorylsulfonyl)oxaziridine is prepared in a similar manner starting from (−)-10-camphorsulfonic acid; mp 166–167°C, [α]D +43.6° (CHCl3, c 2.2).
2. Notes
1. (1S)-(+)-10-Camphorsulfonic acid was purchased from Aldrich Chemical Company, Inc.
2. The crude sulfonamide is contaminated with 5–10% of the (camphorsulfonyl)imine, the yield of which increases on standing.
3. The 1H NMR spectrum of (+)-(1S)-10-camphorsulfonamide is as follows: (CDCl3) δ: 0.93 (s, 3 H, CH3), 1.07 (s, 3 H, CH3), 1.40–2.50 (m, 7 H), 3.14 and 3.53 (AB quartet, 2 H, CH2-SO2, J = 15.1), 5.54 (br s, 2 H, NH2).
4. Amberlyst 15 ion-exchange resin is a strongly acidic, macroreticular resin purchased from Aldrich Chemical Company, Inc.
5. The spectral properties of (−)-(camphorsulfonyl)imine are as follows: 1H NMR (CDCl3) δ: 1.03 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 1.45–2.18 (m, 6 H), 2.65 (m, 1 H), 3.10 and 3.28 (AB quartet, 2 H, CH2-SO2, J = 14.0); 13C NMR (CDCl3) δ: 19.01 (q, CH3), 19.45 (q, CH3), 26.64 (t), 28.44 (t), 35.92 (t), 44.64 (d), 48.00 (s), 49.46 (t), 64.52 (s), 195.52 (s); IR (CHCl3) cm−1: 3030, 2967, 1366. Checkers obtained material having identical melting point and [α]D−32.3° (CHCl3, c 1.8).
6. The SafeLab Teflon bearing can be purchased from Aldrich Chemical Company, Inc. A glass stirring bearing lubricated with silicone grease is unsatisfactory because the dissolved salts solidify in the shaft, causing freezing.
7. Efficient stirring is important and indicated by a milky white appearance of the solution.
8. Occasionally batches of oxone purchased from Aldrich Chemical Company, Inc., have exhibited reduced reactivity in this oxidation. Oxone exposed to moisture prior to use also gives reduced reactivity in this oxidation. If this occurs, oxone is added until oxidation is complete as determined by TLC (Note 9). Potassium carbonate is added as needed to maintain the pH at approximately 9.0. Oxone stored in the refrigerator under an inert atmosphere has shown no loss in reactivity for up to 6 months.
9. Oxidation is generally complete after addition of the oxone solution. The oxidation is monitored by TLC as follows. Remove approximately 0.5 mL of the toluene solution from the nonstirring solution, spot a 250-μm TLC silica gel plate, elute with methylene chloride, and develop with 10% molybdophosphoric acid in ethanol and heating(camphorsulfonyl)imine Rf = 0.28 and (camphorylsulfonyl)oxaziridine Rf = 0.62. If (camphorsulfonyl)imine is detected, stirring is continued at room temperature until the reaction is complete (see (Note 8)). If the reaction mixture takes on a brownish color after addition of oxone and has not gone to completion after 30 min, the reaction mixture is filtered through a 150-mL sintered-glass funnel of coarse porosity, and the solids are washed with 50 mL of methylene chloride. The aqueous/organic extracts are returned to the 5-L Morton flask and stirred vigorously and 52 g (0.08 mol, 1 equiv KHSO5) of oxone is added over 5 min and stirring continued until oxidation is complete (approximately 10–15 min).
10. The submitters employed a toluene solution of crude imine prepared in Part B and obtained somewhat higher yields (90–95%). However, the checkers obtained yields in this range on one half the scale using isolatedsulfonylimine.
11. The spectral properties of (+)-(camphorsulfonyl)oxaziridine are as follows: 1H NMR (CDCl3) δ: 1.03 (s, 3 H, CH3), 1.18 (s, 3 H, CH3), 1.45–2.18 (m, 6 H), 2.65 (d, 1 H), 3.10 and 3.28 (AB quartet, 2 H, CH2-SO2, J = 14.0); 13C NMR (CDCl3) δ: 19.45 (q, CH3), 20.42 (q, CH3), 26.55 (t), 28.39 (t), 33.64 (t), 45.78 (d), 48.16 (s), 48.32 (t), 54.07 (s), 98.76 (s). The checkers obtained material (mp 165–167°C) having [α]D +44.7° (CHCl3, c 2.2).
3. Discussion
Camphorsulfonamide, required for the preparation of the (camphorsulfonyl)imine, was previously prepared in two steps. The first step involved conversion of camphorsulfonic acid to the sulfonyl chloride with PCl5 or SOCl2. The isolated sulfonyl chloride was converted in a second step to the sulfonamide by reaction with ammonium hydroxide. This modified procedure is more efficient because it transforms camphorsulfonic acid directly to camphorsulfonamide, avoiding isolation of the camphorsulfonyl chloride.
(Camphorsulfonyl)imine has been reported as a by-product of reactions involving the camphorsulfonamide.2,3,4,5Reychler in 1898 isolated two isomeric camphorsulfonamides,2 one of which was shown to be the(camphorsulfonyl)imine by Armstrong and Lowry in 1902.3 Vandewalle, Van der Eycken, Oppolzer, and Vullioud described the preparation of (camphorsulfonyl)imine in 74% overall yield from 0.42 mol of the camphorsulfonyl chloride.6 The advantage of the procedure described here is that, by using ammonium hydroxide, the camphorsulfonyl chloride is converted to the sulfonamide in >95% yield.7 The sulfonamide is of sufficient purity that it can be used directly in the cyclization step, which, under acidic conditions, is quantitative in less than 4 hr. These modifications result in production of the (camphorsulfonyl)imine in 86% overall yield from the sulfonyl chloride.
In addition to the synthesis of enantiomerically pure (camphorylsulfonyl)oxaziridine7 and its derivatives,8 the(camphorsulfonyl)imine has been used in the preparation of (−)-2,10-camphorsultam (Oppolzers’ auxiliary),6,9 (+)-(3-oxocamphorysulfonyl) oxaziridine,10 and the N-fluoro-2,10-camphorsultam, an enantioselective fluorinating reagent.11
The N-sulfonyloxaziridines are an important class of selective, aprotic oxidizing reagents.121314 Enantiomerically pure N-sulfonyloxaziridines have been used in the asymmetric oxidation of sulfides to sulfoxides (30–91% ee),15selenides to selenoxides (8–9% ee).16 disulfides to thiosulfinates (2–13% ee),5 and in the asymmetric epoxidation of alkenes (19–65% ee).17,18 Oxidation of optically active sulfonimines (R*SO2N=CHAr) affords mixtures of N-sulfonyloxaziridine diastereoisomers requiring separation by crystallization and/or chromatography.3
(+)-(Camphorylsulfonyl)oxaziridine described here is prepared in four steps from inexpensive (1S)-(+)- or (1R)-(+)-10-camphorsulfonic acid in 77% overall yield.7 Separation of the oxaziridine diastereoisomers is not required because oxidation is sterically blocked from the exo face of the C-N double bond in the (camphorsulfonyl)imine. In general, (camphorsulfonyl)oxaziridine exhibits reduced reactivity compared to other N-sulfonyloxaziridines. For example, while sulfides are asymmetrically oxidized to sulfoxides (3–77% ee), this oxaziridine does not react with amines or alkenes.7 However, this oxaziridine is the reagent of choice for the hydroxylation of lithium and Grignard reagents to give alcohols and phenols because yields are good to excellent and side reactions are minimized.19 This reagent has also been used for the stereoselective oxidation of vinyllithiums to enolates.20
The most important synthetic application of the (camphorylsulfonyl)oxaziridines is the asymmetric oxidation of enolates to optically active α-hydroxy carbonyl compounds.14,21,22,23,24 Chiral, nonracemic α-hydroxy carbonylcompounds have been used extensively in asymmetric synthesis, for example, as chiral synthons, chiral auxiliaries, and chiral ligands. This structural array is also featured in many biologically active natural products. This oxidizing reagent gives uniformly high chemical yields regardless of the counterion, and stereoselectivities are good to excellent (50–95% ee).9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24 Since the configuration of the oxaziridine three-membered ring controls the stereochemistry, both α-hydroxy carbonyl optical isomers are readily available. Representative examples of the asymmetric oxidation of prochiral enolates by (+)-(2R,8aS)-camphorylsulfonyl)oxaziridine are given in Tables I and II.
This preparation is referenced from:
- Org. Syn. Coll. Vol. 8, 110
- Org. Syn. Coll. Vol. 9, 212
-
References and Notes
- Department of Chemistry, Drexel University, Philadelphia, PA 19104.
- Reychler, M. A. Bull. Soc. Chim. III1889, 19, 120.
- Armstrong, H. E.; Lowry, T. M. J. Chem. Soc., Trans.1902, 81, 1441.
- Dauphin, G.; Kergomard, A.; Scarset, A. Bull. Soc. Chim. Fr.1976, 862.
- Davis, F. A.; Jenkins, Jr., R. H.; Awad, S. B.; Stringer, O. D.; Watson, W. H.; Galloy, J. J. Am. Chem. Soc.1982, 104, 5412.
- Vandewalle, M.; Van der Eycken, J.; Oppolzer, W.; Vullioud, C. Tetrahedron, 1986, 42, 4035.
- Davis, F. A.; Towson, J. C.; Weismiller, M. C.; Lal, S.; Carroll, P. J. J. Am. Chem. Soc.1988, 110, 8477.
- Davis, F. A.; Weismiller, M. C.; Lal, G. S.; Chen, B. C.; Przeslawski, R. M. Tetrahedron Lett., 1989, 30, 1613.
- Oppolzer, W. Tetrahedron1987, 43, 1969.
- Glahsl, G.; Herrmann, R. J. Chem. Soc., Perkin Trans. I1988, 1753.
- Differding, E.; Lang, R. W. Tetrahedron Lett.1988, 29, 6087.
- For recent reviews on the chemistry of N-sulfonyloxaziridines, see: (a) Davis, F. A.; Jenkins, Jr., R. H. in “Asymmetric Synthesis,” Morrison, J. D., Ed.; Academic Press: Orlando, FL, 1984, Vol. 4, Chapter 4;
- Davis, F. A.; Haque, S. M. in “Advances in Oxygenated Processes,” Baumstark, A. L., Ed.; JAI Press: London, Vol. 2;
- Davis, F. A.; Sheppard, A. C. Tetrahedron1989, 45, 5703.
- Davis, F. A.; McCauley, Jr., J. P.; Chattopadhyay, S.; Harakal, M. E.; Towson, J. C.; Watson, W. H.; Tavanaiepour, I. J. Am. Chem. Soc.1987, 109, 3370.
- Davis, F. A.; Stringer, O. D.; McCauley, Jr., J. M. Tetrahedron1985, 41, 4747.
- Davis, F. A.; Chattopadhyay, S. Tetrahedron Lett.1986, 27, 5079.
- Davis, F. A.; Harakal, M. E.; Awad, S. B. J. Am. Chem. Soc.1983, 105, 3123.
- Davis, F. A.; Wei, J.; Sheppard, A. C.; Gubernick S. Tetrahedron Lett.1987, 28, 5115.
- Davis, F. A.; Lal, G. S.; Wei, J. Tetrahedron Lett.1988, 29, 4269.
- Davis, F. A.; Haque, M. S.; Ulatowski, T. G.; Towson, J. C. J. Org. Chem.1986, 51, 2402.
- Davis, F. A.; Haque, M. S. J. Org. Chem.1986, 51, 4083; Davis, F. A.; Haque, M. S.; Przeslawski, R. M. J. Org. Chem.1989, 54, 2021.
- Davis, F. A.; Ulatowski, T. G.; Haque, M. S. J. Org. Chem.1987, 52, 5288.
- Davis, F. A.; Sheppard, A. C., Lal, G. S. Tetrahedron Lett.1989, 30, 779.
- Davis, F. A.; Sheppard, A. C.; Chen, B. C.; Haque, M. S. J. Am. Chem. Soc.1990, 112, 6679.
a US 5 856 529 (Bristol-Myers Squibb; 5.1.1999; appl. 9.12.1997; USA-prior. 10.12.1996).
-
- b US 7 754 902 (Vanda Pharms.; 13.7.2010; appl. 18.5.2006).
-
treatment of circadian rhythm disorders:
- US 8 785 492 (Vanda Pharms.; 22.7.2014; appl. 25.1.2013; USA-prior. 26.1.2012).
-
synthesis cis-isomer:
- US 6 214 869 (Bristol-Myers Squibb; 10.4.2001; appl. 25.5.1999; USA-prior. 5.6.1998).
Patents
References
- Jump up^ “Tasimelteon Advisory Committee Meeting Briefing Materials”(PDF). Vanda Pharmaceuticals Inc. November 2013.
- Jump up^ “FDA transcript approval minutes” (PDF). FDA. November 14, 2013.
- ^ Jump up to:a b Food and Drug Administration (January 31, 2014). “FDA approves Hetlioz: first treatment for non-24 hour sleep-wake disorder”. FDA.
- Jump up^ “tasimelteon (Hetlioz) UKMi New Drugs Online Database”. Retrieved August 6, 2014.
- Jump up^ “HETLIOZ® Receives European Commission Approval for the Treatment of Non-24-Hour Sleep-Wake Disorder in the Totally Blind”. MarketWatch. PR Newswire. 7 July 2015. Retrieved 8 July 2015.
- Jump up^ Vachharajani, Nimish N.; Yeleswaram, Krishnaswamy; Boulton, David W. (April 2003). “Preclinical pharmacokinetics and metabolism of BMS-214778, a novel melatonin receptor agonist”. Journal of Pharmaceutical Sciences. 92 (4): 760–72. doi:10.1002/jps.10348. PMID 12661062.
- Jump up^ Sack, R. L.; Brandes, R. W.; Kendall, A. R.; Lewy, A. J. (2000). “Entrainment of Free-Running Circadian Rhythms by Melatonin in Blind People”. New England Journal of Medicine. 343 (15): 1070–7. doi:10.1056/NEJM200010123431503. PMID 11027741.
- Jump up^ “Safety and Efficacy of VEC-162 on Circadian Rhythm in Healthy Adult Volunteers”. ClinicalTrials.gov. |accessdate=May 15, 2014
- Jump up^ “VEC-162 Study in Healthy Adult Volunteers in a Model of Insomnia”. ClinicalTrials.gov. Retrieved May 15, 2014.
- Jump up^ “VEC-162 Study in Adult Patients With Primary Insomnia”. ClinicalTrials.gov. Retrieved May 15, 2014.
- Jump up^ Lynne Lamberg. “Improving Sleep and Alertness in the Blind (Part 5)”. Matilda Ziegler Magazine for the Blind. Retrieved May 15, 2014.
- Jump up^ Shantha MW Rajaratnam; Mihael H Polymeropoulos; Dennis M Fisher; Thomas Roth; Christin Scott; Gunther Birznieks; Elizabeth B Klerman (2009-02-07). “Melatonin agonist tasimelteon (VEC-162) for transient insomnia after sleep-time shift: two randomised controlled multicentre trials”. The Lancet. 373 (9662): 482–491. doi:10.1016/S0140-6736(08)61812-7. PMID 19054552. Retrieved 2010-02-23.
- Jump up^ Carome, Michael (1 July 2015). “Outrage of the Month: FDA Makes Major Blunder After Approving Drug for Rare Sleep Disorder”. Huffington Post. Retrieved 8 July 2015.
- Jump up^ Food and Drug Administration (January 31, 2014). “FDA NEWS RELEASE: FDA approves Hetlioz: first treatment for non-24 hour sleep–wake disorder in blind individuals”. FDA.
- Jump up^ “Side Effects Drug Center: Hetlioz Clinical Pharmacology”. RxList. February 10, 2014.
- Jump up^ “Side Effects Drug Center: Hetlioz Warnings and Precautions”. RxList. February 10, 2014.
In animal studies, administration of tasimelteon during pregnancy resulted in developmental toxicity (embryofetal mortality, neurobehavioral impairment, and decreased growth and development in offspring) at doses greater than those used clinically.
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Hetlioz |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | not determined in humans[1] |
| Protein binding | 89–90% |
| Metabolism | extensive hepatic, primarily CYP1A2 and CYP3A4-mediated |
| Elimination half-life | 0.9–1.7 h / 0.8–5.9 h (terminal) |
| Excretion | 80% in urine, 4% in feces |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ECHA InfoCard | 100.114.889 |
| Chemical and physical data | |
| Formula | C15H19NO2 |
| Molar mass | 245.32 g/mol |
| 3D model (JSmol) | |

DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
//////////////BMS-214778, VEC-162, Tasimelteon, Hetlioz, FDA 2014, 609799-22-6 , BMS-214778, VEC-162, ATC N05CH03, タシメルテオン , EU 2015, VANDA, BMS, orphan drug designations
CCC(=O)NCC1CC1C2=C3CCOC3=CC=C2
Chemical and physical properties
Tasimelteon has two stereogenic centers. Besides the medically used trans-1 R , 2 R isomer (in the picture above left), there are thus three further stereoisomers that do not arise in the synthesis.

Tasimelteon is a white to off-white crystalline non-hygroscopic substance, soluble in water at physiologically relevant pH levels and readily soluble in alcohols, cyclohexane and acetonitrile. The compound occurs in two crystal forms. It is an anhydrate melting at 74 ° C and a hemihydrate . [4] The hemihydrate is from about 35 ° C the water of hydration and converts thereby in the anhydrate form to. [4] The anhydrate crystallizes in a monoclinic lattice with the space group P 2 1 , and the hemihydrate crystallizes in a tetragonal lattice with the space group P 4 3 21 2. [4]
4 Kaihang Liu, Zhou Xinbo, Zhejing Xu, Bai Hongzhen, Jianrong Zhu Jianming Gu, Guping Tang, Liu Xingang, Hu Xiurong: anhydrate and hemihydrate of Tasimelteon: Synthesis, structure, and pharmacokinetic study in J. Pharm. Biomed. Anal. 151 (2018) 235-243, doi : 10.1016 / j.jpba.2017.12.035 .
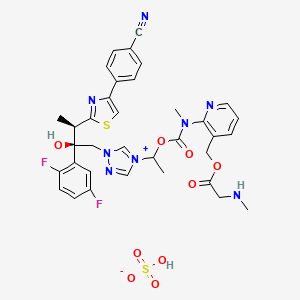
Isavuconazonium sulfate, Изавуконазониев сулфат


Isavuconazonium sulfate
Изавуконазониев сулфат
CAS 946075-13-4
| MOLECULAR FORMULA: | C35H36F2N8O9S2 |
|---|---|
| MOLECULAR WEIGHT: | 814.837 g/mol |
BAL-8557-002, BAL 8557
[2-[1-[1-[(2R,3R)-3-[4-(4-cyanophenyl)-1,3-thiazol-2-yl]-2-(2,5-difluorophenyl)-2-hydroxybutyl]-1,2,4-triazol-4-ium-4-yl]ethoxycarbonyl-methylamino]pyridin-3-yl]methyl 2-(methylamino)acetate;hydrogen sulfate
UNII:31Q44514JV
(2-{[(1-{1-[(2R,3R)-3-[4-(4-cyanophenyl)-1,3-thiazol-2-yl]-2-(2,5-difluorophenyl)-2-hydroxybutyl]-1H-1,2,4-triazol-4-ium-4-yl}ethoxy)carbonyl](methyl)amino}pyridin-3-yl)methyl N-methylglycinate hydrogen sulfate
(2-{[(1-{1-[(2R,3R)-3-[4-(4-Cyanophenyl)-1,3-thiazol-2-yl]-2-(2,5-difluorophenyl)-2-hydroxybutyl]-1H-1,2,4-triazol-4-ium-4-yl}ethoxy)carbonyl](methyl)amino}-3-pyridinyl)methyl N-methylglycinate hydrog en sulfate
FDA 2015, EU 2015, BAL8557-002, BCS CLASS I, RO-0098557 , AK-1820
fast track designation
QIDP
ORPHAN DRUG EU

1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47.
Isavuconazonium is a second-generation triazole antifungal approved on March 6, 2015 by the FDA for the treatment of invasive aspergillosis and invasive mucormycosis, marketed by Astellas under the brand Cresemba. It is the prodrug form of isavuconazole, the active moiety, and it is available in oral and parenteral formulations. Due to low solubility in waterof isavuconazole on its own, the isovuconazonium formulation is favorable as it has high solubility in water and allows for intravenous administration. This formulation also avoids the use of a cyclodextrin vehicle for solubilization required for intravenous administration of other antifungals such as voriconazole and posaconazole, eliminating concerns of nephrotoxicity associated with cyclodextrin. Isovuconazonium has excellent oral bioavailability, predictable pharmacokinetics, and a good safety profile, making it a reasonable alternative to its few other competitors on the market.
Originally developed at Roche, the drug candidate was subsequently acquired by Basilea. In 2010, the product was licensed to Astellas Pharma by Basilea Pharmaceutica for codevelopment and copromotion worldwide, including an option for Japan, for the treatment of fungal infection.

03/06/2015 02:10 PM EST
The U.S. Food and Drug Administration today approved Cresemba (isavuconazonium sulfate), a new antifungal drug product used to treat adults with invasive aspergillosis and invasive mucormycosis, rare but serious infections.
Syn……https://newdrugapprovals.org/2013/10/02/isavuconazole-basilea-reports-positive-results-from-study/
PRODUCT PATENT
https://patents.google.com/patent/US6300353
InventorTadakatsu HayaseShigeyasu IchiharaYoshiaki IsshikiPingli LiuJun OhwadaToshiya SakaiNobuo ShimmaMasao TsukazakiIsao UmedaToshikazu Yamazaki
Current Assignee Basilea Pharmaceutica International Ltd Original
AssigneeBasilea Pharmaceutica AG Priority date 1998-03-06
https://patents.google.com/patent/WO1999045008A1/en
POLYMORPHS OF BASE
WO 2016055918
https://patents.google.com/patent/WO2016055918A1/en
PATENT
IN 2014MU03189
WOCKHARDT
Isavuconazole, isavuconazonium, Voriconazole, and Ravuconazole are azole derivatives and known as antifungal drugs for treatment of systemic mycoses as reported in US 5,648,372, US 5,792,781, US 6,300,353 and US 6,812,238. The US patent No. 6,300,353 discloses Isavuconazole and its process. It has chemical name [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5- difluorophenyl)-butan-2-ol;
The Isavuconazonium iodide hydrochloride and Isavuconazonium sulfate can be prepared according to known methods, e.g. pending Indian Patent Applications IN 2424/MUM/2014 and IN 2588/MUM/2014.
Example-1: Preparation of Amorphous Isavuconazole

4-cyano Phenacyl bromide F F N N N OH N S CN Formula-I Formula-III In a round bottomed flask charged ethanol (250 ml), thioamide compound of formula-II (25.0 gm) and 4-cyano phenacyl bromide (18.4 gm) under stirring. The reaction mixture were heated to 70 0C. After completion of reaction the solvent was removed under vacuum distillation and water (250 ml) and Ethyl acetate (350 ml) were added to reaction mass. The reaction mixture was stirred and its pH was adjusted between 7 to 7.5 by 10 % solution of sodium bicarbonate. The layer aqueous layer was discarded and organic layer was washed with saturated sodium chloride solution (100 ml) and concentrated under vacuum to get residue. The residue was suspended in methyl tert-butyl ether (250 ml) and the reaction mixture was heated to at 40°C to make crystals uniform and finally reaction mass is cooled to room temperature filtered and washed with the methyl tert-butyl ether. The product was isolated dried to get pale yellowish solid product. Yield: 26.5 gm HPLC purity: 92.7%
CLIP
March 6, 2015
Release
The U.S. Food and Drug Administration today approved Cresemba (isavuconazonium sulfate), a new antifungal drug product used to treat adults with invasive aspergillosis and invasive mucormycosis, rare but serious infections.
Aspergillosis is a fungal infection caused by Aspergillus species, and mucormycosis is caused by the Mucorales fungi. These infections occur most often in people with weakened immune systems.
Cresemba belongs to a class of drugs called azole antifungal agents, which target the cell wall of a fungus. Cresemba is available in oral and intravenous formulations.
“Today’s approval provides a new treatment option for patients with serious fungal infections and underscores the importance of having available safe and effective antifungal drugs,” said Edward Cox, M.D., M.P.H, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research.
Cresemba is the sixth approved antibacterial or antifungal drug product designated as a Qualified Infectious Disease Product (QIDP). This designation is given to antibacterial or antifungal drug products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act.
As part of its QIDP designation, Cresemba was given priority review, which provides an expedited review of the drug’s application. The QIDP designation also qualifies Cresemba for an additional five years of marketing exclusivity to be added to certain exclusivity periods already provided by the Food, Drug, and Cosmetic Act. As these types of fungal infections are rare, the FDA also granted Cresemba orphan drug designations for invasive aspergillosis and invasive mucormycosis.
The approval of Cresemba to treat invasive aspergillosis was based on a clinical trial involving 516 participants randomly assigned to receive either Cresemba or voriconazole, another drug approved to treat invasive aspergillosis. Cresemba’s approval to treat invasive mucormycosis was based on a single-arm clinical trial involving 37 participants treated with Cresemba and compared with the natural disease progression associated with untreated mucormycosis. Both studies showed Cresemba was safe and effective in treating these serious fungal infections.
The most common side effects associated with Cresemba include nausea, vomiting, diarrhea, headache, abnormal liver blood tests, low potassium levels in the blood (hypokalemia), constipation, shortness of breath (dyspnea), coughing and tissue swelling (peripheral edema). Cresemba may also cause serious side effects including liver problems, infusion reactions and severe allergic and skin reactions.
Cresemba is marketed by Astellas Pharma US, Inc., based in Northbrook, Illinois.

The active substance is isavuconazonium sulfate, a highly water soluble pro-drug of the active triazole isavuconazole. The chemical name of the active substance isavuconazonium sulfate is 1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47. The active substance has the following structure:
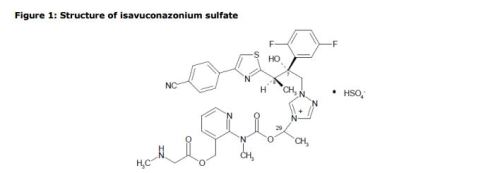
The structure of the active substance has been confirmed by elemental analysis, mass spectrometry, UV, IR, 1H-, 13C- and 19F-NMR spectrometry, and single crystal X-ray analysis, all of which support the chemical structure. It appears as a white, amorphous, hygroscopic powder. It is very soluble in water and over the pH range 1-7. It is also very soluble in methanol and sparingly soluble in ethanol. Two pKa values have been found and calculated to be 2.0 and 7.3. Its logPoct/wat calculated by software is 1.31.
Isavuconazonium sulfate has three chiral centres. The stereochemistry of the active substance is introduced by one of the starting materials which is controlled by appropriate specification. The two centres, C7 and C8 in the isavuconazole moiety and in an intermediate of the active substance, have R configuration. The third chiral centre, C29, is not located on isavuconazole moiety and has both the R and S configurations. The nondefined stereo centre at C29 has been found in all batches produced so far to be racemic. Erosion of stereochemical purity has not been observed in the current process. The active substance is a mixture of two epimers of C29.
An enantiomer of drug substance was identified as C7 (S), C8 (S) and C29 (R/S) structure. The control of the stereochemistry of isavuconazonium sulfate is performed by chiral HPLC on the active substance and its two precursors. Subsequent intermediates are also controlled by relevant specification in the corresponding steps. Two crystal forms have been observed by recrystallisation studies. However the manufacturing process as described yields amorphous form only.
Two different salt forms of isavuconazonuium (chloride and sulfate) were identified during development. The sulfate salt was selected for further development. A polymorph screening study was also performed. None of the investigated salts could be obtained in crystalline Form………http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002734/WC500196130.pdf

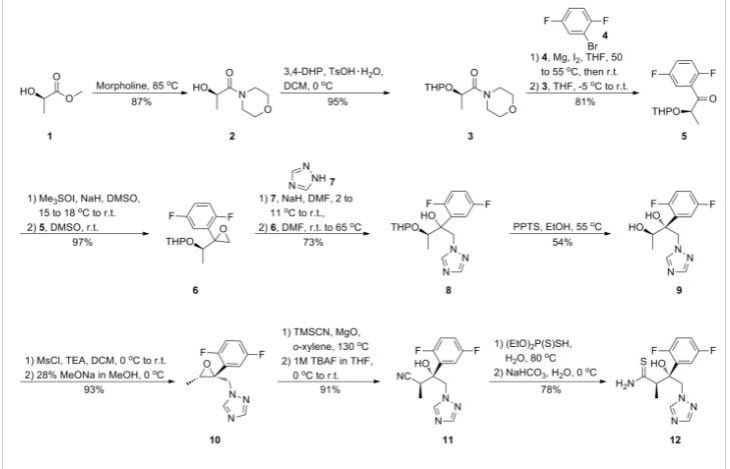
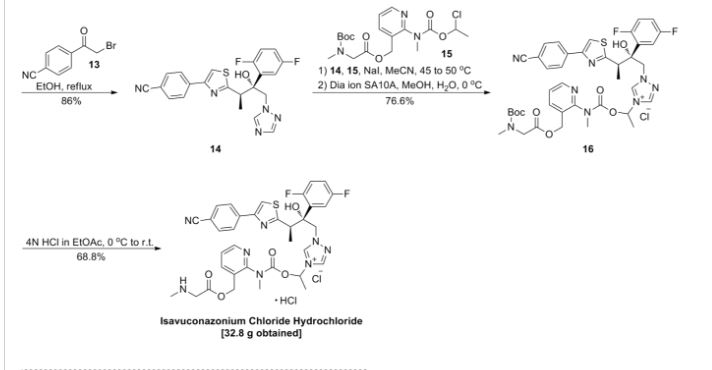

Clip




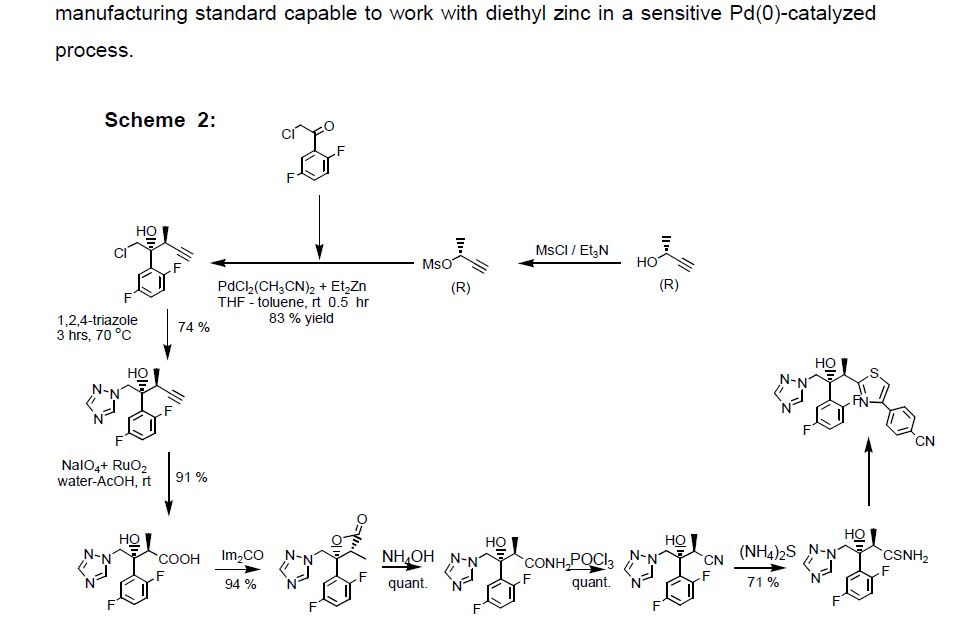


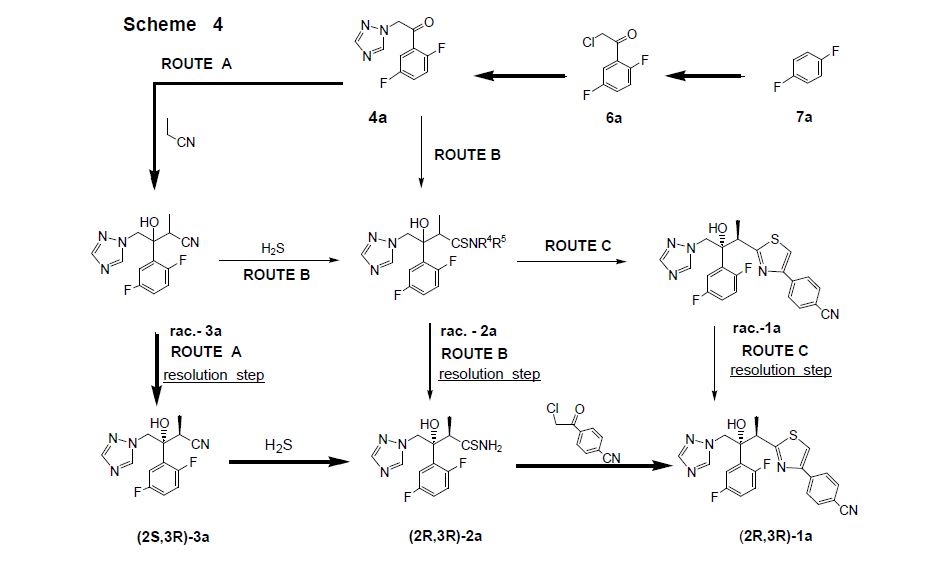
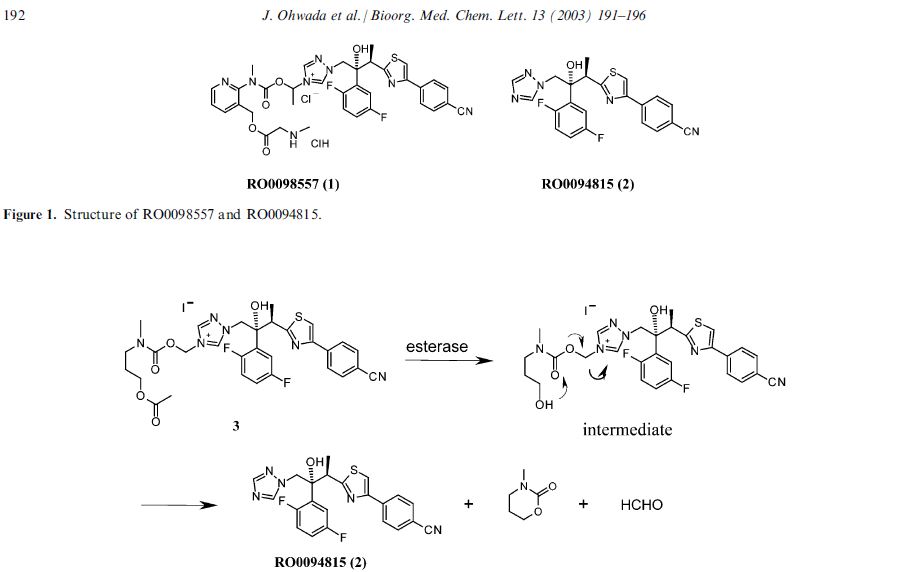
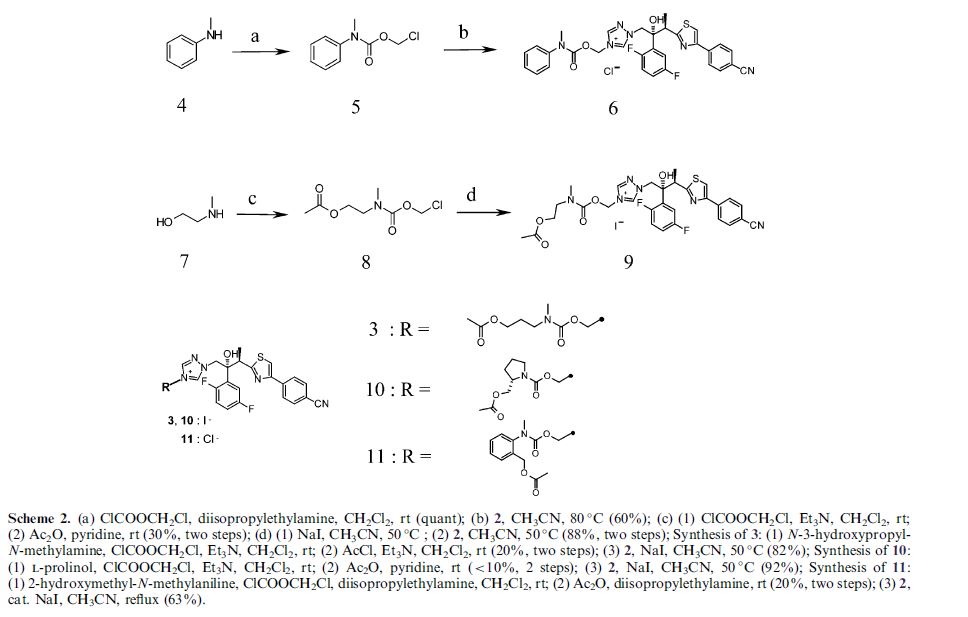

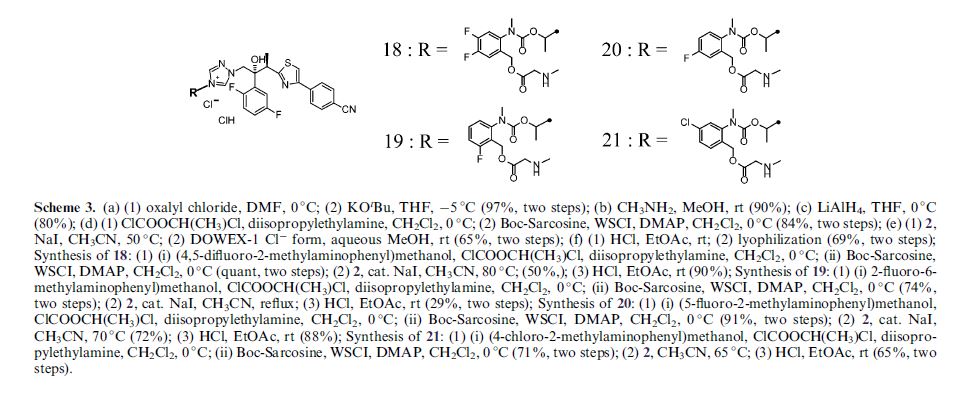

Isavuconazonium (Cresemba ) is a water-soluble prodrug of the triazole antifungal isavuconazole (BAL4815), a 14-a-demethylase inhibitor, under development byBasilea Pharmaceutica International Ltd and Astellas Pharma Inc. Isavuconazonium, in both its intravenous and oral formulations, was approved for the treatment of invasive aspergillosis and invasive mucormycosis (formerly termed zygomycosis) in the US in March 2015. Isavuconazonium is under regulatory review in the EU for invasive aspergillosis and mucormycosis. It is also under phase III development worldwide for the treatment of invasive candidiasis and candidaemia. This article summarizes the milestones in the development of isavuconazonium leading to the first approval for invasive spergillosis and mucormycosis.
Introduction
The availability of both an intravenous (IV) and an oral formulation of isavuconazonium (Cresemba ), as a result of its water solubility, rapid hydrolysis to the active entity isavuconazole and very high oral bioavailability, provides maximum flexibility to clinicians for treating seriously ill patients with invasive fungal infections [1]. Both the IV and oral formulations have been approved by the US Food and Drug Administration (FDA) to treat adults with invasive aspergillosis and invasive mucormycosis [2]. The recommended dosages of each formulation are identical, consisting of loading doses of 372 mg (equivalent to 200 mg of isavuconazole) every eight hours for six doses, followed by maintenance therapy with 372 mg administered once daily [3]. The Qualified Infectious Disease Product (QIDP) designation of the drug with priority review status by the FDA isavuconazonium in the US provided and a five year extension of market exclusivity from launch. Owing to the rarity of the approved infections,
isavuconazonium was also granted orphan drug designation by the FDA for these indications [2]. It has also been granted orphan drug and QIDP designation in the US for the treatment of invasive candidiasis [4]. In July 2014, Basilea Pharmaceutica International Ltd submitted a Marketing Authorization Application to the European Medicines Agency (EMA) for isavuconazonium in the treatment of invasive aspergillosis and invasive mucormycosis, indications for which the EMA has granted isavuconazonium orphan designation [5, 6]. Isavuconazonium is under phase III development in many countries worldwide for the treatment of invasive candidiasis and candidaemia.
1.1 Company agreements
In 2010, Basilea Pharmaceutica International Ltd (a spinoff from Roche, founded in 2000) entered into a licence agreement with Astellas Pharma Inc in which the latter would co-develop and co-promote isavuconazonium worldwide, including an option for Japan. In return for milestone payments, Astellas Pharma was granted an exclusive right to commercialize isavuconazonium, while Basilea Pharmaceutica retained an option to co-promote the drug in the US, Canada, major European countries and China [7]. The companies amended their agreement in 2014, making Astellas Pharma responsible for all regulatory filings, commercialization and manufacturing of isavuconazonium in the US and Canada. Basilea Pharmaceutica waived its right to co-promote the product in the US and Canada, in order to assume all rights in the rest of the world [8]. However, Astellas Pharma remains as sponsor of the multinational, phase III ACTIVE trial in patients with invasive candidiasis.
2 Scientific Summary
Isavuconazonium (as the sulphate; BAL 8557) is a prodrug that is rapidly hydrolyzed by esterases (mainly butylcholinesterase) in plasma into the active moiety isavuconazole
(BAL 4815) and an inactive cleavage product (BAL 8728).
References
1. Falci DR, Pasqualotto AC. Profile of isavuconazole and its potential in the treatment of severe invasive fungal infections. Infect Drug Resist. 2013;6:163–74.
2. US Food and Drug Administration. FDA approves new antifungal drug Cresemba. 2015. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm437106.htm. Accessed 12 Mar 2015.
3. US Food and Drug Administration. Cresemba (isavuconazonium sulfate): US prescribing information. 2015. http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207500Orig1s000lbl.pdf. Accessed 18 Mar 2015.
4. Astellas Pharma US Inc. FDA grants Astellas Qualified Infectious Disease Product designation for isavuconazole for the treatment of invasive candidiasis (media release). 2014. http://newsroom astellas.us/2014-07-16-FDA-Grants-Astellas-Qualified-Infectious-Disease-Product-Designation-for-Isavuconazole-for-the-Treatmentof-Invasive-Candidiasis.
5. European Medicines Agency. Public summary of opinion on orphan designation: isavuconazonium sulfate for the treatment of invasive aspergillosis. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2014/07/WC500169890.pdf. Accessed 18 Mar 2015.
European Medicines Agency. Public summary of opinion on orphan designation: isavuconazonium sulfate for the treatment of mucormycosis. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2014/07/WC500169714.pdf. Accessed 18 Mar 2015.
7. Basilea Pharmaceutica. Basilea announces global partnership with Astellas for its antifungal isavuconazole (media release).2010. http://www.basilea.com/News-and-Media/Basilea-announcesglobal-partnership-with-Astellas-for-its-antifungal-isavuconazole/343.
8. Basilea Pharmaceutica. Basilea swaps its isavuconazole North American co-promote rights for full isavuconazole rights outside of North America (media release). 2014. http://www.basilea.com/News-and-Media/Basilea-swaps-its-isavuconazole-North-Americanco-promote-rights-for-full-isavuconazole-rights-outside-


CLIP
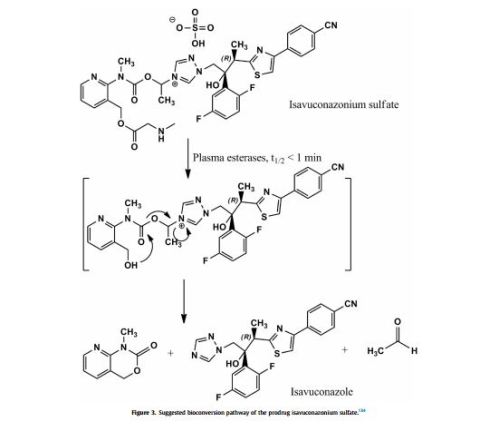
http://www.jpharmsci.org/article/S0022-3549(15)00035-0/pdf
A CLIP
http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207500Orig1207501Orig1s000ChemR.pdf








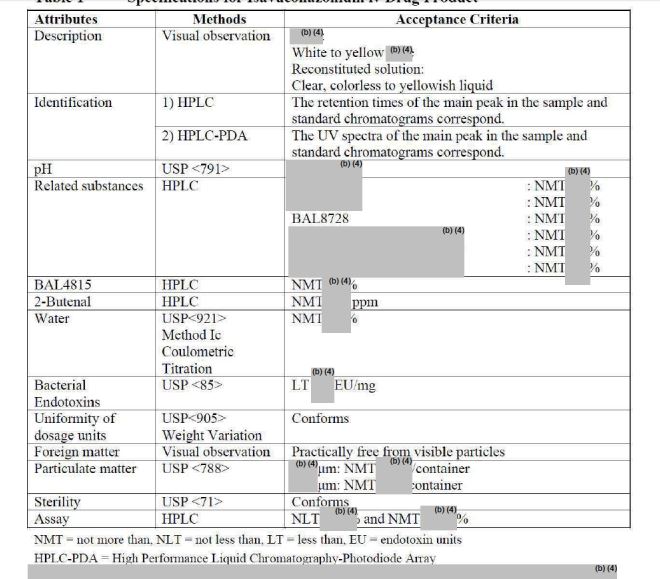
EMA
On 4 July 2014 orphan designation (EU/3/14/1284) was granted by the European Commission to Basilea Medical Ltd, United Kingdom, for isavuconazonium sulfate for the treatment of invasive aspergillosis.
Update: isavuconazonium sulfate (Cresemba) has been authorised in the EU since 15 October 2015. Cresemba is indicated in adults for the treatment of invasive aspergillosis.
Consideration should be given to official guidance on the appropriate use of antifungal agents.
The active substance is isavuconazonium sulfate, a highly water soluble pro-drug of the active triazole isavuconazole. The chemical name of the active substance isavuconazonium sulfate is 1-{(2R,3R)-3-[4-(4-cyanophenyl)-1,3- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxybutyl}-4-[(1RS)-1-({methyl[3-({[(methylamino)acetyl] oxy}methyl) pyridin-2-yl]carbamoyl}oxy)ethyl]-1H-1,2,4-triazol-4-ium monosulfate (IUPAC), corresponding to the molecular formula C35H35F2N8O5S·HSO4 and has a relative molecular mass of 814.84 g/mol. The relative molecular mass of isavuconazole is 437.47. The active substance has the following structure
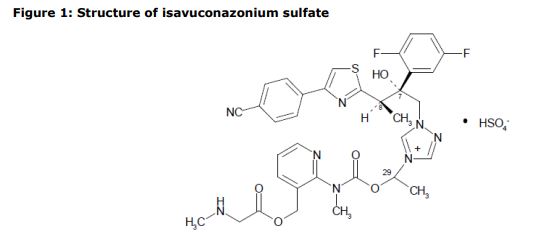
It appears as a white, amorphous, hygroscopic powder. It is very soluble in water and over the pH range 1-7. It is also very soluble in methanol and sparingly soluble in ethanol. Two pKa values have been found and calculated to be 2.0 and 7.3. Its logPoct/wat calculated by software is 1.31.
Isavuconazonium sulfate has three chiral centres. The stereochemistry of the active substance is introduced by one of the starting materials which is controlled by appropriate specification. The two centres, C7 and C8 in the isavuconazole moiety and in an intermediate of the active substance, have R configuration. The third chiral centre, C29, is not located on isavuconazole moiety and has both the R and S configurations. The nondefined stereo centre at C29 has been found in all batches produced so far to be racemic. Erosion of stereochemical purity has not been observed in the current process. The active substance is a mixture of two epimers of C29. An enantiomer of drug substance was identified as C7 (S), C8 (S) and C29 (R/S) structure. The control of the stereochemistry of isavuconazonium sulfate is performed by chiral HPLC on the active substance and its two precursors.
FDA Orange Book Patents
US 6812238
US 7459561
| FDA ORANGE BOOK PATENTS: 1 OF 2 | |
|---|---|
| Patent | 7459561 |
| Expiration | Oct 31, 2020 |
| Applicant | ASTELLAS |
| Drug Application | N207500 (Prescription Drug: CRESEMBA. Ingredients: ISAVUCONAZONIUM SULFATE) |
from FDA Orange Book
FREE FORM
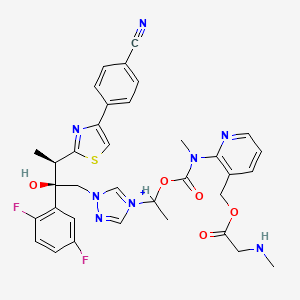
Isavuconazonium; Isavuconazonium ion; Cresemba; BAL-8557; 742049-41-8;
[2-[1-[1-[(2R,3R)-3-[4-(4-cyanophenyl)-1,3-thiazol-2-yl]-2-(2,5-difluorophenyl)-2-hydroxybutyl]-1,2,4-triazol-4-ium-4-yl]ethoxycarbonyl-methylamino]pyridin-3-yl]methyl 2-(methylamino)acetate
| MOLECULAR FORMULA: | C35H35F2N8O5S+ |
|---|---|
| MOLECULAR WEIGHT: | 717.773 g/mol |
Patent IDDatePatent Title
US20102494262010-09-30STABILIZED PHARMACEUTICAL COMPOSITION
US74595612008-12-02N-substituted carbamoyloxyalkyl-azolium derivativesUS71898582007-03-13N-phenyl substituted carbamoyloxyalkyl-azolium derivatives
US71511822006-12-19Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives
US68122382004-11-02N-substituted carbamoyloxyalkyl-azolium derivatives
REF
http://www.drugbank.ca/drugs/DB06636
////////// BAL 8557, BAL-8557-002, CRESEMBA, ISAVUCONAZONIUM SULFATE, QIDP designation, Cresemba , priority review, FDA 2015, EU 2015, BAL8557-002, BCS CLASS I, orphan designation, invasive aspergillosis, invasive mucormycosis, RO-0098557 , AK-1820, fast track designation, QIDP, 946075-13-4
CC(C1=NC(=CS1)C2=CC=C(C=C2)C#N)C(CN3C=[N+](C=N3)C(C)OC(=O)N(C)C4=C(C=CC=N4)COC(=O)CNC)(C5=C(C=CC(=C5)F)F)O
CC(C1=NC(=CS1)C2=CC=C(C=C2)C#N)C(CN3C=[N+](C=N3)C(C)OC(=O)N(C)C4=C(C=CC=N4)COC(=O)CNC)(C5=C(C=CC(=C5)F)F)O.OS(=O)(=O)[O-]
UPDATE NEW PATENT
WOCKHARDT, WO 2016016766, ISAVUCONAZONIUM SULPHATE, NEW PATENT

![]()
(WO2016016766) A PROCESS FOR THE PREPARATION OF ISAVUCONAZONIUM OR ITS SALT THEREOF
WOCKHARDT LIMITED [IN/IN]; D-4, MIDC Area, Chikalthana, Aurangabad 431006 (IN)
KHUNT, Rupesh Chhaganbhai; (IN).
RAFEEQ, Mohammad; (IN).
MERWADE, Arvind Yekanathsa; (IN).
DEO, Keshav; (IN)
The present invention relates to a process for the preparation of stable Isavuconazonium or its salt thereof. In particular of the present invention relates to process for the preparing of isavuconazonium sulfate, Isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide has purity more than 90%. The process is directed to preparation of solid amorphous form of isavuconazonium sulfate, isavuconazonium iodide hydrochloride and Boc-protected isavuconazonium iodide. The present invention process of Isavuconazonium or its salt thereof is industrially feasible, simple and cost effective to manufacture of isavuconazonium sulfate with the higher purity and better yield.
Isavuconazonium sulfate is chemically known l-[[N-methyl-N-3-[(methylamino) acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl)thiazol-2-yl]butyl]-lH-[l,2,4]-triazo-4-ium Sulfate and is structurally represented by formula (I):


Formula I
Isavuconazonium sulfate (BAL8557) is indicated for the treatment of antifungal infection. Isavuconazonium sulfate is a prodrug of Isavuconazole (BAL4815), which is chemically known 4-{2-[(lR,2R)-(2,5-Difluorophenyl)-2-hydroxy-l-methyl-3-(lH-l ,2,4-triazol-l-yl)propyl]-l ,3-thiazol-4-yl}benzonitrile compound of Formula II


Formula II
US Ppatent No. 6,812,238 (referred to herein as ‘238); 7,189,858 (referred to herein as ‘858); 7,459,561 (referred to herein as ‘561) describe Isavuconazonium and its process for the preparation thereof.
The US Pat. ‘238 patent describes the process of preparation of Isavuconazonium chloride hydrochloride.
The US Pat. ‘238 described the process for the Isavuconazonium chloride hydrochloride, involves the condensation of Isavuconazole and [N-methyl-N-3((tert-butoxycarbonyl methylamino) acetoxymethyl) pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester. The prior art reported process require almost 15-16 hours, whereas the present invention process requires only 8-10 hours. Inter alia prior art reported process requires too many step to prepare isavuconazonium sulfate, whereas the present invention process requires fewer steps.
Moreover, the US Pat. ‘238 describes the process for the preparation Isavuconazonium hydrochloride, which may be used as the key intermediate for the synthesis of isavuconazonium sulfate, compound of formula I. There are several drawbacks in the said process, which includes the use of anionic resin to prepare Isavuconazonium chloride hydrochloride, consequently it requires multiple time lyophilization, which makes the said prior art process industrially, not feasible.
The inventors of the present invention surprisingly found that Isavuconazonium or a pharmaceutically acceptable salt thereof in yield and purity could be prepared by using substantially pure intermediates in suitable solvent.
Thus, an object of the present invention is to provide simple, cost effective and industrially feasible processes for manufacture of isavuconazonium sulfate. Inventors of the present invention surprisingly found that isavuconazonium sulfate prepared from isavuconazonium iodide hydrochloride, provides enhanced yield as well as purity.
The process of the present invention is depicted in the following scheme:


Formula I
Formula-IA
The present invention is further illustrated by the following example, which does not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present application.
Examples
Example-1: Synthesis of l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino) acetoxymethyl]pyridin-2-yl]carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3 – [4-(4-cyanophenyl)thiazol-2-yl]butyl] – 1 H-[ 1 ,2,4] -triazo-4-ium iodide
Isavuconazole (20 g) and [N-methyl-N-3((tert-butoxycarbonylmethylamino)acetoxy methyl)pyridine-2-yl]carbamic acid 1 -chloro-ethyl ester (24.7 g) were dissolved in acetonitrile (200ml). The reaction mixture was stirred to add potassium iodide (9.9 g). The reaction mixture was stirred at 47-50°C for 10-13 hour. The reaction mixture was cooled to room temperature. The reaction mass was filtered through celite bed and washed acetonitrile. Residue was concentrated under reduced pressure to give the crude solid product (47.7 g). The crude product was purified by column chromatography to get its pure iodide form (36.5 g).
Yield: 84.5 %
HPLC Purity: 87%
Mass: m/z 817.4 (M- 1)+
Example-2: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride
l-[[N-methyl-N-3-[(t-butoxycarbonylmethylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide (36.5 g) was dissolved in ethyl acetate (600 ml). The reaction mixture was cooled to -5 to 0 °C. The ethyl acetate hydrochloride (150 ml) solution was added to reaction mixture. The reaction mixture was stirred for 4-5 hours at room temperature. The reaction mixture was filtered and obtained solid residue washed with ethyl acetate. The solid dried under vacuum at room temperature for 20-24 hrs to give 32.0 gm solid.
Yield: 93 %
HPLC Purity: 86%
Mass: m/z 717.3 (M-HC1- 1)
Example-3: Preparation of Strong anion exchange resin (Sulfate).
Indion GS-300 was treated with aqueous sulfate anion solution and then washed with DM water. It is directly used for sulfate salt.
Example-4: Synthesis of l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium Sulfate
Dissolved 10.0 g l-[[N-methyl-N-3-[(methylamino)acetoxymethyl]pyridin-2-yl] carbamoyloxy]ethyl-l-[(2R,3R)-2-(2,5-difluorophenyl)-2-hydroxy-3-[4-(4-cyanophenyl) thiazol-2-yl]butyl]-lH-[l ,2,4]-triazo-4-ium iodide hydrochloride in 200 ml deminerahzed water and 30 ml methanol. The solution was cooled to about 0 to 5°C. The strong anion exchange resin (sulfate) was added to the cooled solution. The reaction mixture was stirred to about 60-80 minutes. The reaction was filtered and washed with 50ml of demineralized water and methylene chloride. The aqueous layer was lyophilized to obtain
(8.0 g) white solid.
Yield: 93 %
HPLC Purity: > 90%
Mass: m/z 717.4 (M- HS04) +
PATENT
CN 105288648
PATENT
CN 106883226
https://patents.google.com/patent/CN106883226A/en
PATENT
CN 107982221
PAPER
| Title: Introduction of New Drugs Approved by the U.S. FDA in 2015 |
| Author: Ma Shuai; Wenying Ling; Zhou Weicheng; |
| Source: China Pharmaceutical Industry |
| Publisher: Tongfangzhiwang Beijing Technology Co., Ltd. |
| Year of publication: |
| DOI code: 10.16522/j.cnki.cjph.2016.01.022 |
| Registration Time: 2016-02-19 02:04:15 |
///////////////
EMA approves AstraZeneca’s lesinurad to treat gout patients

EMA approves AstraZeneca’s lesinurad to treat gout patients
British-Swedish drugmaker AstraZeneca has received approval from European Medicines Agency (EMA) for its lesinurad 200mg tablets to treat gout patients. READ AT…..[LINK]

“The company submitted a MAA based on data from the Clear1, Clear2 and Crystal pivotal Phase III combination therapy studies.”
AstraZeneca’s subsidiary Ardea Biosciences carried out Clear1, Clear2 and Crystal trials.


Novartis obtains European approval for Cosentyx to treat psoriasis

Novartis obtains European approval for Cosentyx to treat psoriasis
Swiss drug-maker Novartis has received approval from the European Commission (EC) for its Cosentyx (secukinumab, formerly known as AIN457) to treat moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy.SEE
 PSORIAIS
PSORIAIS
secukinumab

Secukinumab is a human monoclonal antibody designed for the treatments of uveitis, rheumatoid arthritis, ankylosing spondylitis, and psoriasis. It targets member A from the cytokine family of interleukin 17.[1][2] At present, Novartis Pharma AG, the drug’s developer, plans to market it under the trade name “Cosentyx.” [3] It is highly specific to the human immunoglobulin G1k (IgG1k) subclass.[2]
In July 2014 secukinumab established superiority to placebo and to etanercept for the treatment of chronic plaque psoriasis in Phase III clinical trials.[4] In October 2014, the FDA Dermatologic and Ophthalmic Drugs Advisory Committee unanimously voted to recommend the drug for FDA approval, although this vote in and of itself does not constitute an approval. However, the FDA typically follows recommendations from these committees.[5] In October 2014, Novartis announced that the drug had achieved a primary clinical endpoint in two phase III clinical trials for ankylosing spondylitis.[6] As of 28 October, the relevant FDA committee had not yet responded to these results. In early November 2014, Novartis also released the results of a Phase 3 study on Psoriatic Arthritis that yielded very promising results.[7]

Although the drug was originally intended to treat rheumatoid arthritis, phase II clinical trials for this condition yielded disappointing results.[8] Similarly, while patients in a phase II clinical trial for [psoriatic arthritis] did show improvement over placebo, the improvement did not meet adequate endpoints and Novartis is considering whether to do more research for this condition.[9] Novartis has said that it is targeting approval and release in early 2015 for plaque psoriasis and ankyloding spondylitis indications.
It is also in a phase II clinical trial for Multiple Sclerosis [10] as it has exhibited efficacy in treating experimental autoimmune encephalomyelitis (EAE), an animal model of MS.
CAS registry numbers
- 875356-43-7 (heavy chain)
- 875356-44-8 (light chain)
References
- “Statement On A Nonproprietary Name Adopted By The USAN Council: Secukinumab”. American Medical Association.
- Hueber, W.; Patel, D. D.; Dryja, T.; Wright, A. M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M. H.; Psoriasis Study, P.; Durez, P. P.; Tak, J. J.; Gomez-Reino, C. S.; Rheumatoid Arthritis Study, R. Y.; Foster, C. M.; Kim, N. S.; Samson, D. S.; Falk, D.; Chu, Q. D.; Callanan, K.; Nguyen, A.; Uveitis Study, F.; Rose, K.; Haider, A.; Di Padova, F. (2010). “Effects of AIN457, a Fully Human Antibody to Interleukin-17A, on Psoriasis, Rheumatoid Arthritis, and Uveitis”. Science Translational Medicine 2 (52): 52ra72.doi:10.1126/scitranslmed.3001107. PMID 20926833.
- http://www.medscape.com/viewarticle/835331
- Langley RG, Elewski BE, Mark Lebwohl M, et al., for the ERASURE and FIXTURE Study Groups (July 24, 2014). “Secukinumab in Plaque Psoriasis — Results of Two Phase 3 Trials”. N Engl J Med 371: 326–338. doi:10.1056/NEJMoa1314258.
- committees.http://www.familypracticenews.com/index.php?id=2934&type=98&tx_ttnews=306073[dead link]
- http://inpublic.globenewswire.com/2014/10/23/Novartis+AIN457+secukinumab+meets+primary+endpoint+in+two+Phase+III+studies+in+ankylosing+spondylitis+a+debilitating+joint+condition+of+the+spine+HUG1864939.html
- http://www.medpagetoday.com/MeetingCoverage/ACR/48743
- http://www.medscape.com/viewarticle/806510_6
- http://www.ncbi.nlm.nih.gov/pubmed/23361084
- http://clinicaltrials.gov/show/NCT01874340
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | IL17A |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | |
| ATC code | L04AC10 |
| DrugBank | DB09029 |
| Synonyms | AIN457 |
| Chemical data | |
| Formula | C6584H10134N1754O2042S44 |
| Molecular mass | 147.94 kDa |

{kind=link}