
Home » Posts tagged 'eisai'
Tag Archives: eisai
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Avatrombopag

| C29H34Cl2N6O3S2 | |
| Molecular Weight: | 649.65466 g/mol |
|---|
Elemental Analysis: C, 53.61; H, 5.28; Cl, 10.91; N, 12.94; O, 7.39; S, 9.87
1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid,
1-(3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]pyridin-2-yl)piperidine-4-carboxylic acid,
1-[3-Chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl]-2-pyridyl]piperidine-4-carboxylic acid
4-Piperidinecarboxylic acid, 1-[3-chloro-5-[[[4-(4-chloro-2-thienyl)-5-(4-cyclohexyl-1-piperazinyl)-2-thiazolyl]amino]carbonyl]-2-pyridinyl]-
Phase III Clinical Trials
Drugs used in platelet disorders
Idiopathic thrombocytopenic purpura (ITP)
small-molecule thrombopoietin receptor (c-Mpl) agonist that stimulates platelet production
INNOVATOR: YAMANOUCHI PHARMACEUTICAL
DEVELOPER: Eisai

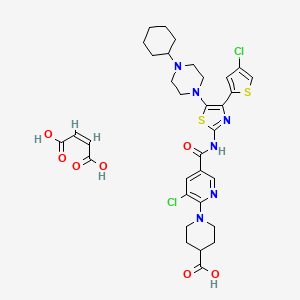
| C33H38Cl2N6O7S2 | |
| Molecular Weight: | 765.72682 g/mol |
|---|
UNIIGDW7M2P1IS
(Z)-but-2-enedioic acid;1-[3-chloro-5-[[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl]pyridin-2-yl]piperidine-4-carboxylic acid
INTRODUCTION
Avatrombopag, also known as AKR-501, YM477, AS 1670542 or E5501, is a novel orally-active thrombopoietin (TPO) receptor agonist. AKR-501 specifically targeted the TPO receptor and stimulated megakaryocytopoiesis throughout the development and maturation of megakaryocytes just as rhTPO did. Daily oral administration of AKR-501 dose-dependently increased the number of human platelets in these mice, with significance achieved at doses of 1 mg/kg and above. The peak unbound plasma concentrations of AKR-501 after administration at 1 mg/kg in NOD/SCID mice were similar to those observed following administration of an active oral dose in human subjects. AKR-501 may be useful in the treatment of patients with thrombocytopenia. (source: Eur J Haematol. 2009 Apr;82(4):247-54).

Avatrombopag is a thrombopoietin receptor (c-Mpl) agonist in phase III clinical evaluation at Eisai for the oral treatment of chronic immune thrombocytopenia (idiopathic thrombocytopenia purpura) and for the treatment of thrombocytopenia associated with liver diseases. Phase II studies are ongoing for the treatment of thrombocytopenia during antiviral therapy (inhibition and maintenance) with Interferon for hepatitis C.
The drug candidate may hold potential in treating thrombocytopenia of diverse etiologies, including idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia of myelodysplastic syndromes (MDS), in combination with or as a substitute for platelet transfusion.
AKR-501, a novel, small-molecule thrombopoietin mimetic being investigated for the treatment of thrombocytopenia. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture AKR-501. AKR-501 is an investigational thrombopoietin receptor agonist that, based on preclinical studies, increases platelet production by stimulating megakaryocytic proliferation and differentiation. Eisai is currently conducting Phase II clinical trials of AKR-501 in the United States as a potential treatment for idiopathic thrombocytopenic purpura (ITP) and thrombocytopenia associated with liver diseases (TLD), and has confirmed proof of concept in the clinical studies for ITP. In addition, Eisai will explore the compound’s potential as a treatment for chemotherapy-induced thrombocytopenia (CIT).

E-5501 stimulates the production of thrombopoietin (TPO), a glycoprotein hormone that stimulates the production and differentiation of megakaryocytes, the bone marrow cells that fragment into large numbers of platelets. The drug candidate was originally developed at Yamanouchi, and development responsibilities were passed to AkaRx when it was formed in 2005 as a spin-off following the creation of Astellas Pharma subsequent to the merger of Yamanouchi Pharmaceutical and Fujisawa Healthcare.
In 2007, MGI Pharma was granted a license to E-5501 for the treatment of thrombocytopenia. Eisai eventually gained the rights to the product as results of its acquisition of MGI Pharma. In 2010, Eisai acquired AkaRx. AkaRx is now a wholly-owned subsidiary of Eisai Inc. and Eisai has the exclusive worldwide rights to develop, market and manufacture E-5501. In 2011, orphan drug designation was assigned by the FDA for the treatment of idiopathic thrombocytopenic purpura.
E5501 (or AKR-501 or YM477) is a small molecule agonist c-Mpl, orally available. It is in clinical trials for the treatment of chronic idiopathic thrombocytopenic purpura (ITP). It acts as an agonist of the thrombopoietin receptor active orally, mimicking its biological effect. Thrombocytopenic purpura The is the idiopathic consequence of a low number of platelets (thrombocytopenia) of unknown cause. A very low platelets can even lead to purpura (bruises), or bleeding diathesis.
February 2012: A Phase III, multicenter, randomized, double-blind, controlled against placebo, parallel group, with an open-label extension phase to assess the efficacy and safety of combined oral E5501 to standard treatment for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia, is underway.
January 2010: Eisai Inc. announced its successful acquisition of the biopharmaceutical company, AkaRx Inc. Following this acquisition, AkaRx became a wholly owned subsidiary of Eisai Inc. Eisai now owns the worldwide exclusive rights to develop , marketing and manufacture AKR-501.
October 2009: Eisai Research Institute of Boston, Inc. (established in 1987) and Eisai Medical Research Inc. (established in 2002) were merged into Eisai Inc. 2005: AkaRx was founded as a spin-out of the merger of Yamanouchi Pharmaceutical Company Ltd. and Fujisawa Pharmaceutical Company Ltd. to form Astellas Pharma Inc. AKR-501 was discovered by Yamanouchi and was licensed to AkaRx as part of the foundation of the company in 2005.
In a Phase I trial in healthy volunteers, 10 mg of AKR-501 for 14 days, increased platelet count by 50%.AKR-501 was well tolerated in both studies, mono- and multi-dose. No adverse effects were reported, even at the highest doses.
……………………
Patent
Compound A is a compound of the present invention has the following chemical structure.

That is, compounds useful as a platelet 增多 agent according to the present invention A, as well as medicaments for the Compound A as an active ingredient, in particular increasing platelets agents and Z or thrombocytopenia treating agent.


………………
PATENT

……………………
JP 2014144916/WO 2013018362
https://www.google.co.in/patents/WO2013018362A1?cl=en
1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)thiazol-2-yl]carbamoyl}pyridin-2-yl)piperidine-4-carboxylic acid as expressed by the following chemical formula (hereinafter referred to as “Compound X”) and pharmaceutically acceptable salts are known to have excellent thrombocytosis effects (patent literature 1, patent literature 2).
[Formula 1]

Patent literature 1 discloses a hydrochloride of compound X as example 16 (hereinafter referred to as “compound X hydrochloride”).
Furthermore, patent literature 2 discloses a maleic acid salt of compound X that has endothermic peaks near 198 degree C and 271 degree C in thermo gravimetric analysis (hereinafter referred to as “maleic acid salt of compound X”). However, patent literature 2 neither discloses nor suggests that the maleic acid salt of compound X exhibits crystal polymorphism.
On the other hand, compounds exhibiting crystal polymorphism demonstrate entirely different effects regardless of being the same compound, because various physical properties including physicochemical properties differ depending on the crystalline form. In pharmaceutical products in particular, if compounds that have different functional effects are expected to have the same effect, a different functional effect than expected will occur, which is thought to induce unexpected circumstances, and therefore there is demand for supply of a drug substance with constant quality. Therefore, when a compound which has crystal polymorphism is used as a medicine, one type of crystal of that compound must always be constantly provided in order to ensure constant quality and constant effects that are required of the medicine.
Under the aforementioned conditions, from the perspective of supplying a drug substance for medicines, there is a need for compound X or crystals of pharmaceutically acceptable salts thereof, which can ensure constant quality and constant effects and which can be stably supplied in mass production such as industrial production or the like, as well as for establishment of a manufacturing method thereof.
International patent publication WO 03/062233 International patent publication WO 2004/029049
The crystals of compound X maleic acid salt disclosed in patent literature 2 (hereinafter referred to as “compound X maleic acid salt A type crystals”) cannot be isolated as compound X maleic acid salt A type crystals when scaled up for mass production using the method disclosed in example 1 of patent literature 2, and therefore must be isolated in a different crystal form. (This other crystal form is referred to as “compound X maleic acid salt B type crystals”). Therefore, the compound X maleic acid salt A type crystals have a possibility that the crystal form will morph depending on the scale of production, and is clearly inappropriate as a drug substance for medicines which require constant quality and constant effects.
Preparation Example 1: Manufacture of Compound X Maleic Acid Salt B Type Crystal
310 mL of a 1 M aqueous solution of sodium hydroxide at room temperature was added to a mixture of 70.0 g of the ethyl ester of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid and 1.2 L of ethanol, the insoluble matter was filtered out, and then washed with 200 mL of ethanol. The reaction solution was stirred for 90 minutes at 60 degree C. After cooling to room temperature, 1.4 L of an aqueous solution containing 24.11 g of maleic acid was added to the solution obtained, and then the precipitate was collected by filtering.
The same operation was repeated and when combined with the previously obtained precipitate, 136.05 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid was obtained.
18.9 g of maleic acid and 2.1 L of 80% ethanol water were added to 88.90 g of the carboxylic acid obtained, and the solution was stirred for one hour at room temperature and for another hour at 100 degree C. After cooling to room temperature and further cooling with ice, the precipitated solid was filtered out to obtain 87.79 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
6.84 g of maleic acid was added to 231 g of the crude product containing the crude product obtained above and those manufactured in a similar manner, dissolved in 5.5 L of 80% ethanol water, and then the precipitated solid was collected by filtering to obtain 203 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 1: Manufacture of Compound X Maleic Acid Salt C Type Crystals (1)
1.52 L of ethanol, 0.38 L of water, and 15.7 g of maleic acid were added to 78.59 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, and heated while stirring. After cooling to room temperature and further cooling with ice, the precipitated solid was collected by filtering to obtain 71.60 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt as a crude product.
296 mg of maleic acid was added to 10.0 g of the crude product obtained, dissolved in 60 mL of acetone, 60 mL of DMSO, and 30 mL of water, and then the precipitated solids were collected to obtain 8.41 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 2: Manufacture of Compound X Maleic Acid Salt C Type Crystals (2)
A mixture containing 80.1 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 580 mL of DMSO, 580 mL of acetone, 17.2 g of maleic acid, and 290 mL of water was stirred at 69 degree C. The insoluble matter was filtered out, washed with a mixture of 32 mL of DMSO, 32 mL of acetone, and 16 mL of water, and then the filtrate was cooled and the precipitate was collected by filtering. Washing was successively performed using 150 mL of water, 80 mL of acetone, 650 mL of water, and 80 mL of acetone, followed by drying, to obtain 70.66 g of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
Example 3: Manufacture of Compound X Maleic Acid Salt C Type Crystals (3)
A mixture containing 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid, 100 L of DMSO, 100 L of acetone, 4.29 kg of maleic acid, and 50 L of water is stirred at 65 degree C, and then the insoluble matter is filtered out and washed with a mixture of 8 L of DMSO, 8 L of acetone, and 4 L of water, and then the filtrate is cooled, the precipitate is collected by filtering, successively washed using 40 L of acetone, 100 L of water, and 40 L of acetone, and then dried to obtain approximately 20 kg of 1-(3-chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexyl piperazin-1-yl) thiazol-2-yl] carbamoyl} pyridin-2-yl) piperidine-4-carboxylic acid maleic acid salt.
…………………………….

REFERENCES
Garabet, L.; Ghanima, W.; Lee, S.; Mowinckel, M.C.; Liebman, H.; Jonassen, C.M.; Bussel, J.; Sandset, P.M.
Thrombopoietin receptor agonists do no not cause coagulation activation: In patients with immune thrombocytopenia
25th Congr Int Soc Thromb Haemost (ISTH) (June 20-25, Toronto) 2015, Abst PO311-MON
Terrault, N.; Hassanein, T.; Joshi, S.; Lake, J.R.; Sher, L.S.; Vargas, H.E.; McIntosh, J.W.; Tang, S.; Jenkins, T.
Once-daily oral avatrombopag (E5501) prior to elective surgical or diagnostic procedures in patients with chronic liver disease and thrombocytopenia: Results from a phase 2, randomized, double-blind, placebo-controlled study (study 202)
63rd Annu Meet Am Assoc Study Liver Dis (November 9-13, Boston) 2012, Abst
Thiophenyl Triazol-3-one Derivatives As Smooth Muscle relaxers: US6613786 (2003) Priority: US20010336865P, Nov. 2, 2001 (Bristol-Myers Squibb CO, US)
Preparation Of Avatrombopag: 2-Acylaminothiazole derivative or salt thereof: EP1466912 (2004) Priority: JP20020010413, 18 Jan. 2002 (Yamanouchi Pharma Co Ltd, Japan)
Synthesis And Use Of MSE Framework-Type Molecular Sieves: US2009318696 (2009) Priority: US20080214631 20 Jun. 2008 (Exxon Mobil, US).
5,6-Dichloro-Nicotinic Acid Production By Reacting 6-Hydroxy-Nicotinic Acid With Acid Chloride Reacting With Chlorine Products, Then With Acid Chloride And Hydrolysing Products: CH664754 (1988) Priority: CH19850002692, 25 Jun. 1985 (Lonza AG, Switzerland).
David J. Kuter, New Thrombopoietic Growth Factors, Lymphoma and Myeloma Clinical Journal Volume 9, Supplement 3, S347-S356
| WO2003062233A1 | 15 Jan 2003 | 31 Jul 2003 | Yamanouchi Pharma Co Ltd | 2-acylaminothiazole derivative or salt thereof |
| WO2004029049A1 | 29 Sep 2003 | 8 Apr 2004 | Yuuji Awamura | Novel salt of 2-acylaminothiazole derivative |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP2764866A1 | 4 Feb 2014 | 13 Aug 2014 | IP Gesellschaft für Management mbH | Inhibitors of nedd8-activating enzyme |
| Patent | Submitted | Granted |
|---|---|---|
| CANCER TREATMENT METHOD [US2011160130] | 2011-06-30 | |
| METHOD FOR STIMULATING PLATELET PRODUCTION [US2011166112] | 2011-07-07 | |
| COMPOSITIONS AND METHODS FOR INCREASING BLOOD PLATELET LEVELS IN HUMANS [US2011224226] | 2011-09-15 | |
| Method of treating viral diseases with combinations of TPO receptor agonist and anti-viral agents [US2012020923] | 2012-01-26 |
| Patent | Submitted | Granted |
|---|---|---|
| 2-Acylaminothiazole derivative or salt thereof [US7638536] | 2005-07-14 | 2009-12-29 |
| Compositions and methods for treating thrombocytopenia [US2007203153] | 2007-08-30 | |
| Novel Combinations [US2009304634] | 2009-12-10 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222329] | 2010-09-02 | |
| 2-ACYLAMINOTHIAZOLE DERIVATIVE OR SALT THEREOF [US2010222361] | 2010-09-02 | |
| Compositions and methods for increasing blood platelet levels in humans [US2008039475] | 2008-02-14 | |
| CANCER TREATMENT METHOD [US2009022814] | 2009-01-22 | |
| Compositions and methods for treating thrombocytopenia [US2010041668] | 2010-02-18 | |
| CANCER TREATMENT METHOD [US2010075928] | 2010-03-25 |
///////E 5501, AKR 501, Phase III, eisai, Avatrombopag, y 477, orphan drug, ym 477, AS 1670542, Yamanouchi Pharma Co Ltd, Japan
UPDATE MAY 2018
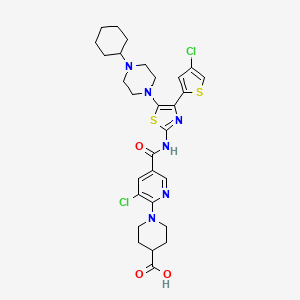
Avatrombopag
https://newdrugapprovals.org/2015/08/24/avatrombopag/
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.Continue reading.
May 21, 2018
Release
The U.S. Food and Drug Administration today approved Doptelet (avatrombopag) tablets to treat low blood platelet count (thrombocytopenia) in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure. This is the first drug approved by the FDA for this use.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
Platelets (thrombocytes) are colorless cells produced in the bone marrow that help form blood clots in the vascular system and prevent bleeding. Thrombocytopenia is a condition in which there is a lower-than-normal number of circulating platelets in the blood. When patients have moderately to severely reduced platelet counts, serious or life-threatening bleeding can occur, especially during invasive procedures. Patients with significant thrombocytopenia typically receive platelet transfusions immediately prior to a procedure to increase the platelet count.
The safety and efficacy of Doptelet was studied in two trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. The trials investigated two dose levels of Doptelet administered orally over five days as compared to placebo (no treatment). The trial results showed that for both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to seven days following the procedure as compared to those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and swelling in the hands or feet (edema). People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet.
This product was granted Priority Review, under which the FDA’s goal is to take action on an application within six months where the agency determines that the drug, if approved, would significantly improve the safety or effectiveness of treating, diagnosing or preventing a serious condition.
The FDA granted this approval to AkaRx Inc.
//////////////Doptelet, avatrombopag, fda 2018, akarx, priority review,
Tazemetostat

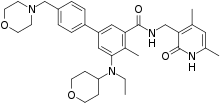
Tazemetostat
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
HBR 1467052-75-0
タゼメトスタット臭化水素酸塩
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
HBR
Chemical Formula: C34H44N4O4
Exact Mass: 572.33626
USFDA APPROVED 23/1/2020 AS HBR SALT, TAZVERIK, EPIZYME
N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[1,1′-biphenyl]-3-carboxamide
SIMLES: O=C(C1=CC(C2=CC=C(CN3CCOCC3)C=C2)=CC(N(CC)C4CCOCC4)=C1C)NCC5=C(C)C=C(C)NC5=O
(1,1′-Biphenyl)-3-carboxamide, N-((1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(4-morpholinylmethyl)-
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(oxan-4-yl)amino)-4-methyl-4′-((morpholin-4-yl)methyl)(1,1′-biphenyl)-3-carboxamide
UNII-Q40W93WPE1
Tazemetostat, sold under the brand name Tazverik, is a medication used for the treatment of adults and adolescents aged 16 years and older with metastatic (when cancer cells spread to other parts of the body) or locally advanced (when cancer has grown outside the organ it started in, but has not yet spread to distant parts of the body) epithelioid sarcoma not eligible for complete resection (surgically removing all of a tissue, structure, or organ).[1]
Tazemetostat is a cancer drug that acts as a potent selective EZH2 inhibitor.[2]
Tazemetostat blocks activity of the EZH2 methyltransferase, which may help keep the cancer cells from growing.[1] Most cases of epithelioid sarcoma begin in the soft tissue under the skin of an extremity, though it can start in other areas of the body.[1] Surgical removal is considered the main treatment when the cancer is localized to one area of the body.[1] Chemotherapy or radiation may also be given.[1] However, there is a high likelihood for local and regional spread of the disease even with treatment and approximately 50% of patients have metastatic disease at the time of diagnosis.[1] Metastatic disease is considered life-threatening to the patient.[1]
The most common side effects are pain, fatigue, nausea, decreased appetite, vomiting and constipation.[1] People taking tazemetostat are at increased risk of developing secondary malignancies including: T-cell lymphoblastic lymphoma (a type of blood cancer that affects the lymphatic system usually found in the lymph nodes), myelodysplastic syndrome (a disorder resulting from poorly formed or dysfunctional blood cells) and acute myeloid leukemia (a cancer of the blood and bone marrow).[1]
According to the NCI Drug Dictionary, “tazemetostat is an orally available, small molecule selective and S-adenosyl methionine (SAM) competitive inhibitor of histone methyl transferase EZH2, with potential antineoplastic activity. Upon oral administration, tazemetostat selectively inhibits the activity of both wild-type and mutated forms of EZH2. Inhibition of EZH2 specifically prevents the methylation of histone H3 lysine 27 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways and results in decreased tumor cell proliferation in EZH2 mutated cancer cells. EZH2, which belongs to the class of histone methyltransferases (HMTs), is overexpressed or mutated in a variety of cancer cells and plays a key role in tumor cell proliferation.”[3]
History
The U.S. Food and Drug Administration (FDA) approved tazemetostat in January 2020,[1] based on the results of a clinical trial (NCT02601950) enrolling 62 subjects with metastatic or locally advanced epithelioid sarcoma.[1][4] During the clinical trial, subjects received 800 milligrams (mg) of tazemetostat twice a day until the disease progressed or the subject reached an unacceptable level of toxicity.[1][4] Tumor response assessments were performed every eight weeks during the clinical trial.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] The overall response rate was 15%, with 1.6% of subjects having a complete response and 13% having a partial response.[1] Of the nine subjects that had a response, six (67%) subjects had a response lasting six months or longer.[1]
The trial was conducted at 22 sites in France, United Kingdom, Taiwan, Italy, Canada, Belgium, and the United States.[4]
The FDA granted the application for tazemetostat accelerated approval and orphan drug designation.[1] The FDA granted the approval of Tazverik to Epizyme Inc.[1]
PATENT
PRODUCT PAT
US 8410088 EXP 21/1/2034
WO 2012142504
US 9090562 EXP 13/4/32
SEE Proceedings of the National Academy of Sciences of the United States of America (2013), 110(19), 7922-7927, S7922/1-S7922/5….http://www.pnas.org/content/110/19/7922.abstract
http://www.epizyme.com/wp-content/uploads/2014/11/Ribrag-ENA-FINAL.pdf
Tazemetostat, also known as EPZ-6438, is a potent, selective, and orally bioavailable small-molecule inhibitor of EZH2 enzymatic activity. EPZ-6438 induces apoptosis and differentiation specifically in SMARCB1-deleted MRT cells.
Treatment of xenograft-bearing mice with EPZ-6438 leads to dose-dependent regression of MRTs with correlative diminution of intratumoral trimethylation levels of lysine 27 on histone H3, and prevention of tumor regrowth after dosing cessation.
These data demonstrate the dependency of SMARCB1 mutant MRTs on EZH2 enzymatic activity and portend the utility of EZH2-targeted drugs for the treatment of these genetically defined cancers. EPZ-6438 is currently in clinical trials.
Epizyme, Inc., Eisai R&D Management Co.Ltd.
Epizyme is developing tazemetostat, a lead from several small molecule EZH2 inhibitors, for treating cancer (phase 1 clinical, as of April 2015). Japanese licensee Eisai was developing the program for the potential oral treatment of cancers, including non-Hodgkin’s lymphoma; however, in March 2015, Epizyme regained worldwide, ex-Japan, rights to the program.
It appeared that Eisai was planning to investigate the program in Japan .
WO-2015057859 From, Eisai Research Institute; Epizyme Inc, indicates Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
see WO2013155317, claiming novel hydrobromide salt of tazemetostat.
PREDICT

………………………………….
PATENT
WO 2012142504
http://www.google.com/patents/WO2012142504A1?cl=en

Example 44: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(moφholinomethyl)-[l , – biphenyl]-3-carboxamide

Compound 44
[Step 1 : Synthesis of 5-brom -2-methyl-3-nitrobenzoic acid

To stirred solution of 2-methyl-3-nitrobenzoic acid ( 100 g, 552 mmol) in cone. H2S04 (400 mL), 1 ,3-dibromo-5,5-dimethyl-2,4-imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid ( 140 g, 98%). The isolated compound was taken directly into the next step. Ή NMR (DMSO-4$, 400 MHz) δ 8.31 (s, 1 H), 8.17 (s, 1 H), 2.43 (s, 3H).
Step 2: Synthesis of methyl -bromo-2-methyl-3-nitrobenzoate

To a stirred solution of 5-bromo-2-methyl-3-nitrobenzoic acid (285 g, 1 105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate ( 1 L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).
Step 3: Synthesis of methyl 3-amino-5-bromo-2-methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-nitrobenzoate (290 g,
1058 mmol) in ethanol (1 .5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in-vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 7.37 (s, 1 H), 6.92 (s, 1 H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).
Step 4: Synthesis of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate

To a stirred solution of methyl 3-amino-5-bromo-2-methylbenzoate (15 g, 61 .5 mmol) and dihydro-2H-pyran-4(3)-one (9.2 g, 92 mmol) in dichloroethane (300 mL) was added acetic acid (22 g, 369 mmol) and the reaction mixture stirred at room temperature for 15 minutes, then the reaction mixture was cooled to 0°C and sodium triacetoxyborohydnde (39 g, 184 mmol) was added. The reaction mixture was stirred overnight at room temperature. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH of 7-8 was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a solid ( 14 g, 69%). ‘H NMR (DMSO-<fc, 400 MHz) δ 7.01 (s, 1 H), 6.98 (s, 1 H), 5.00 (d, 1 H, J=7.6 Hz), 3.84-3.87 (m, 2H), 3.79 (s, 31 1), 3.54-3.56 (mf 1 H), 3.43 (L 21 1, J 12 Hz), 2.14 (s. 31 1). 1 . 1 – 1 .84 (m: 211). 1 .47- 1 .55 (m, 2H).
Step 5: Synthesis of methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid ( 15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium
triacetoxyborohydnde (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). Ή NMR (DMSO-cfo 400 MHz) δ 7.62 (s, 1 H), 7.52 (s, 1 H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1 H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1 .37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).
Step 6: Synthesis of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3 -(ethyl (tetrahydro-2H-pyra -4-yl) amino)-2-methylbenzamide

To a stirred solution of 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate (14 g, 39.4 mmol) in ethanol ( 100 mL) was added aqueous NaOH (2.36 g, 59.2 mmol in 25mL water) and the resulting mixture was stirred at 60 °C for 1 h. Upon completion of the reaction as determined by TLC, the solvent was removed under reduced pressure and the residue obtained was acidified with IN HC1 until a pH 7 was obtained and then aqueous citric acid solution was added until a pH 5-6 was obtained. The aqueous layer was extracted with 10% MeOH in DCM (200mL X 3), the combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the respective acid (14 g, 100%).
The above acid (14 g, 40.9 mmol) was then dissolved in DMSO (70 mL) and 3- (amino methyl)-4, 6-dimethylpyridin-2( l H)-one ( 12.4 g, 81 .9 mmol) was added to it. The reaction mixture was stirred at room temperature for 15 minutes, then PYBOP (31.9 g, 61.4 mmol) was added and stirring was continued for overnight at room temperature. Upon completion of the reaction as determined by TLC, the reaction mixture was poured onto ice- cold water (700 mL), stirred for 30 minutes and the precipitated solid was collected by filtration, washed with water (500 mL) and air dried. The solid obtained was stirred with acetonitrile (75mL X 2), filtered and air dried. The solid obtained was again stirred with 5% MeOH in DCM ( l OOmL), filtered and dried completely under vacuum to afford the title compound as a solid ( 14 g, 74 %). Ή NMR (DMSO- 6, 400 MHz) δ 1 1.47 (s, 1 H), 8.23 (t, 1 H), 7.30 (s, 1 H), 7.08 (s, 1 H), 5.85 (s, 1 H), 4.23 (d, 2H, J=4.4 Hz), 3.81 (d, 2H, J=l 0.4 Hz), 3.20-3.26 (m, 2H), 3.00-3.07 (m, I H), 2.91 -2.96 (m, 2H), 2.18 (s, 3H), 2.14 (s, 3H), 2.10 (s, 3H), 1.58-1.60 (m, 2H), 1.45-1.50 (m, 2H), 0.78 (t, 3H, J=6.8 Hz).
Step 7: Synthesis of N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl) amino)-4-methyl-4′-(morpholinomethyl)-[l , l ‘-biphenyl]-3- carboxamide
 TITLE COMPD
TITLE COMPDTo a stirred solution of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzamide (14 g, 29.5 mmol) in dioxane/ water mixture (70 mL/ 14 mL) was added 4-(4-(4, 4, 5, 5-tetramethyl- l , 3, 2- dioxaborolan-2-yl) benzyl) morpholine (13.4 g, 44.2 mmol) followed by addition of Na2C03 (1 1 .2 g, 106.1 mmol). The solution was purged with argon for 15 minutes and then Pd (PPh3)4 (3.40 g, 2.94 mmol) was added and the solution was again purged with argon for a further 10 min. The reaction mixture was heated at 100°C for 4 h. After completion (monitored by TLC), the reaction mixture was diluted with water and extracted with 10% MeOH/DCM.
The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with methanol: DCM to the title compound as a solid (12 g, 71 %).
Analytical Data: LCMS: 573.35 (M + 1 )+; HPLC: 99.5% (@ 254 nm) (R,;3.999; Method: Column: YMC ODS-A 1 50 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol : 10 μΐ, Col. Temp.: 30 °C; Flow rate: 1 .4 mL/min.;
Gradient: 5% B to 95% B in 8 min, Hold for 1 .5 min, 9.51 -12 min 5% B);
Ή NMR (DMSO-i 6, 400 MHz) 5 1 1 .46 (s, I H), 8. 19 (t, 1 H), 7.57 (d, 2H, J=7.2 Hz), 7.36-7.39 (m, 3H), 7.21 (s, I H), 5.85 (s, I H), 4.28 (d, 2H, J=2.8 Hz), 3.82 (d, 2H, J=9.6 Hz), 3.57 (bs, 4H), 3.48 (s, 2H), 3.24 (t, 2H, J=10.8Hz), 3.07-3.09 (m, 2H), 3.01 (m, I H), 2.36 (m, 4H), 2.24 (s, 3H), 2.20 (s, 3H), 2.10 (s, 3H), 1 .64-1 .67 (m, 2H), 1 .51 – 1 .53 (m, 2H), 0.83 (t, 3H, J=6.4 Hz).
TRIHYDROCHLORIDE
Step 8: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3-yl)methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[ 1 , 1 ‘-biphenyl]-3- carboxamide trihydrochloride

N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5-(ethyl (tetrahydro- 21 l-pyran-4-yl) amino)-4-methyI-4′-(niorpholinornethyl)-[ 1 , 1 ‘-biphenyl]-3-carboxamide ( 12 g, 21.0 mmol) was dissolved in methanolic HC1 (200 mL) and stirred at room temperature for 3 h. After three hours of stirring, the reaction mixture was concentrated under reduced pressure. The solid obtained was stirred with ether ( l OOmL X 2) to afford the desired salt as a solid ( 1 1 g, 77 %).
Analytical Data of the tri-HCl salt: LCMS: 573.40 (M + 1 )+; HPLC: 99.1 % (@ 254 nm) (R,;3.961 ; Method: Column: YMC ODS-A 150 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol: 10 pL, Col. Temp.: 30 °C; Flow rate: 1.4 mL/min.; Gradient: 5% B to 95% B in 8 min, Hold for 1.5 min, 9.51 -12 min 5% B);
Ή NMR (D20 400 MHz) δ 7.92 (bs, I H,) 7.80 (s, I H), 7.77 (d, 2H, J=8 Hz), 7.63 (s, I H), 7.61 (s, I H), 6.30 (s, I H), 4.48 (s, 2H), 4.42 (s, 2H), 4.09-4.1 1 (m, 4H), 3.95-3.97 (m, 2H), 3.77 (t, 3H, J=10.4 Hz), 3.44-3.47 (m, 3H), 3.24-3.32 (m, 3H), 2.42 (s, 3H), 2.35 (s, 3H), 2.26 (s, 3H), 2.01 (m, 2H), 1 .76 (m, 2H), 1 .04 (t, 3H, J=6.8 Hz).
…………………………………………
PATENT
WO2013155317
http://www.google.com/patents/WO2013155317A1?cl=en
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl] -3-carboxamide hydrobromide:

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl
(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3- carboxamide hydrobromide:

As used herein, “Compound I” refers to N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxamide. The hydrobromide of Compound I can be used to inhibit the histone methyltransferase activity of EZH2, either in a subject or in vitro. The hydrobromide of Compound I can also be used to treat cancer in a subject in need thereof.
Scheme 1

……………………………………..Compound I Compound I – HBr
HPLC
HPLC was conducted on an Agilent 1200 HPLC quaternary pump, low pressure mixing, with an in-line degasser. Analytical method conditions: 8 μΐ^ sample (20 mg of ER-581982-06 diluted with 50 mL of a methanol to provide approximately 0.4 mg/mL solution) was injected onto a Agilent Zorbax Eclipse XDB-C18 (4.6 x 150 mm, 3.5 um), Chromatography conditions: mobile phase A, water with 5mM ammonium formate; mobile phase B, 5 mM ammonium formate in 50/45/5 acetonitrile/methanol/water; flow rate, 1.5 ml/min.; gradient: isocratic at 10% B from 0 to 3 min; linear increase to 70% B from 3 to 7 min; isocratic at 70% B from 7 to 12 min; linear increase to 100% B from 12 to 15 min isocratic at 100% B from 15 to 20 min;
column temperature, 35 °C; detection, UV 230 nm. Approximate retention time of Compound I = 10.7 min.
Synthesis of Polymorph A

5-bromo-2-methyl-3-nitrobenzoic acid stirred solution of 2-methyl-3-nitrobenzoic acid (100 g, 552 mmol) in cone. H2S04 (400 mL), l,3-dibromo-5,5-dimethyl-2,4- imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid (140 g, 98%). The isolated compound was taken directly into the next step. 1H NMR (DMSO-J6, 400 MHz) δ 8.31 (s, 1H), 8.17 (s, 1H), 2.43 (s, 3H).

Methyl 5-bromo-2-methyl-3-nitrobenzoate To a stirred solution of 5-bromo-2- methyl-3-nitrobenzoic acid (285 g, 1105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate (1L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. 1H NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).

Methyl 3-amino-5-bromo-2-methylbenzoate (1) To a stirred solution of methyl 5- bromo-2-methyl-3-nitrobenzoate (290 g, 1058 mmol) in ethanol (1.5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in- vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. 1H
NMR (CDCI3, 400 MHz) δ 7.37 (s, 1H), 6.92 (s, 1H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).

Methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (2) A reactor was charged with methyl 3-amino-5-bromo-2-methylbenzoate (455.8 g, 1.87 mol), 1,2- Dichloroethane (4.56 L), and acetic acid (535 ml, 9.34 mol). To the mixture were added dihydro-2H-pyran-4(3H)-one (280 g, 2.80 mol) and sodium triacetoxyborohydride (594 g, 2.80 mol) maintaining the internal temperature below 40 °C. The mixture was stirred at 25 °C for 2.5 h and then the reaction was quenched with a solution of sodium hydroxide (448 g, 11.20 mol) in water (5.61 L). After stirring for 20 minutes at ambient temperature, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (3.65 L). The organic layers were combined, washed with brine (1.5 L), and concentrated under vacuum.
The residue was treated with ethyl acetate (1.8 L) and heated to 65-70 °C. The mixture was stirred at 65-70 °C for 15 minutes to give a clear solution and then treated with n-heptane (7.3 L) maintaining the temperature between 60-70 °C. Once the heptane was completely added to the solution, the mixture was held at 65-70 °C for 15 minutes and then allowed to cool to 18- 22 °C over 3 h. The resulting suspension was stirred at 18-22 °C for 4 h, cooled to 0-5 °C over 1 h, and held at 0-5 °C for 2 h. The precipitate was filtered, washed twice with n-heptane (1.4 L), and dried under vacuum to give the title compound (540 g, 88%). The XRPD pattern of this compound is shown in Figure 17.

Methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzoate (3)
To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid (15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium triacetoxyborohydride (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). 1H NMR DMSO-d6, 400 MHz) δ 7.62 (s, 1H), 7.52 (s, 1H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1.37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).

Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-
[l,l’-biphenyl]-3-carboxylate (4): A mixture of methyl 5-bromo-3-(ethyl(tetrahydro-2H-pyran- 4-yl)amino)-2-methylbenzoate (580 g, 1.63 mol), 4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)benzyl)morpholine (592 g, 1.95 mol), 1,4-dioxane (3.86 L), sodium carbonate (618 g, 5.83 mol), and water (771 ml) was degassed by bubbling nitrogen through the mixture at 20 °C for 20 minutes and treated with tetrakis(triphenylphosphine)palladium(0) (14.11 g, 12.21 mmol). The resulting mixture was degassed for an additional 20 minutes and then heated to 87-89 °C for 17 h. After cooling to 20 °C, the mixture was diluted with ethyl acetate (5.80 L) and a solution of (R)-2-Amino-3-mercaptopropionic acid (232 g) in water (2.320 L). After stirring for 1 h at 20 °C, the organic layer was separated and washed again with a solution of (R)-2-Amino-3- mercaptopropionic acid (232 g) in water (2.320 L). The aqueous layers were combined and extracted with ethyl acetate (5.80 L). The organic layers were combined, washed with a solution of sodium hydroxide (93 g) in water (2.32 L), and concentrated under vacuum at 35 °C to give the title compound as an orange oil (1.21 kg, 164% yield).

5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxylic acid (5): Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylate (69.0 g, 152.5 mmol) (based on the theoretical yield from the previous step) was suspended in ethanol (380 mL) and treated with a solution of sodium hydroxide (24.84 g, 621.0 mmol) in water (207 mL). The mixture was stirred at 40°C for 18 h. After cooling to 0-5 °C, the mixture was neutralized to pH 6.5 with 1 N hydrochloric acid (580 mL) maintaining the temperature below 25 °C. Then, the mixture was extracted twice with a mixture of dichloromethane (690 mL) and methanol (69.0 mL). The organic layers were combined and concentrated under vacuum to give a crude product as a yellow solid (127g).
The crude product was dissolved in 2-methyltetrahydrofuran (656 mL) at 70 °C and then treated with IPA (828 mL). The mixture was allowed to cool to rt over 3-4 h and then stirred overnight at rt. The precipitate was filtered, washed twice with IPA (207 mL), and dried under vacuum to give the title compound as an off white solid (53.54 g, 80%). The XRPD pattern of this compound is shown in Figure 9.

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H- pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3-carboxamide
(Compound I): A mixture of 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylic acid (540 g, 1.23 mol) and 3-(aminomethyl)- 4,6-dimethyl-dihydro-pyridin-2(lH)-one hydrochloride (279 g, 1.48 mol) was suspended in DMSO (2.70 L) and treated with triethylamine (223 ml, 1.60 mol). The mixture was stirred at 25 °C for 30 min and treated with EDC-HC1 (354 g, 1.85 mol) and HOBT hydrate (283 g, 1.85 mol). The reaction mixture was stirred at rt for 16 h. After addition of triethylamine (292 ml, 2.09 mol), the mixture was cooled to 15 °C, diluted with water (10.1 L) maintaining the temperature below 30 °C, and stirred at 19-25 °C for 4 h. The resulting precipitate was filtered, washed twice with water (2.70 L), and dried under vacuum to give a crude product (695 g, wt-wt analysis = 78%).
For the further purification of the product, recrystallization was conducted. A crude product (20.00 g, 34.92 mmol) was suspended in a mixture of ethanol (190 ml) and water (10.00 ml) and heated to 75°C until a clear solution was obtained. The solution was allowed to cool to rt overnight. The precipitate was filtered, washed twice with a mixture of ethanol (30.0 ml) and water (30.0 ml), and dried under vacuum at 35 °C to give the title compound as an off white solid (14.0 g, 70% recovery from the crude and 90% yield based on wt-wt assay).

4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′- (ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4-yl)methyl)morpholin- 4-ium bromide (Polymorph A): A crude N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)am
biphenyl]-3-carboxamide (595 g, 464 g based on wt-wt assay, 810.3 mmol) was suspended in ethanol (3.33 L). After heating to 70 °C, the mixture was treated with 48% aqueous HBr (97 ml, 850.8 mmol) and stirred at 70 °C for 30 min. The resulting orange-red solution was treated with ethyl acetate (3.33 L) maintaining the temperature above 60 °C. The mixture was slowly cooled to rt over 18 h. The mixture was cooled to 0 °C over 1 h and stirred at that temperature for 5.5 h. The resulting precipitate was filtered, washed twice with ethyl acetate (1.39 L), and dried under vacuum to give the title compound as an off white solid (515 g, 97% yield).
Recrystallization of Polymorph A: 4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4- yl)methyl)morpholin-4-ium bromide (0.50 g, 0.77 mmol; 95.6% pure by HPLC) was suspended in ethanol (3.0 mL) and heated to 80 °C until a clear solution was obtained. To the solution was added MTBE (5.0 mL) slowly. The resulting solution was allowed to cool to 18-22 °C over 3 h and stirred at 18-22 °C for 15 h. The precipitate was filtered, washed twice with MTBE (2 mL) and dried under vacuum to give 0.45 g of the title compound (89% recovery, 96.6% pure by HPLC).
Compound I is protonated at the nitrogen of the morpholino substituent, providing a monohydrobromide of Compound I having the following structure:

This particular monohydrobromide can be referred to as “4-((3′-(((4,6-dimethyl-2-oxo- l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′- methyl-[l, -biphenyl]-4-yl)methyl)morpholin-4-ium bromide.” Figure 11 depicts the X-ray crystal structure of this particular salt form.
…………………………………………………………..
see
WO-2015057859
Eisai Research Institute; Epizyme Inc
Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
…………………
Synthesis
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| USA | Tazverik | Epizyme, 2020 |
Formulations
- oral tabs. and suspension
References
-
- Knutson, S. K. et al., Proc. Natl. Acad. Sci USA, (2013) 110(19), 7922-7927.
- WO 2012 142504 (Epizyme/Eisai Co; 18.10.2012; appl. 13.4.2012; USA-prior. 13.4.2011).
- WO 2013 155317 (Epizyme/Eisai Co; 17.10.2013; appl. 11.4.2013).
- WO 2015 057859 (Epizyme/Eisai Co; 23.4.2015; appl. 15.10.2014; USA-prior. 16.10.2013).
-
EZH2 inhibitors for treating lymphona:
- WO 2015 195848 (Epizyme; 23.12.2015; appl. 17.6.2015; USA-prior. 17.6.2014).
////////
PAPER
RSC Advances (2015), 5(33), 25967-25978
http://pubs.rsc.org/en/content/articlelanding/2015/ra/c5ra02365c#!divAbstract
DOI: 10.1039/C5RA02365C
The histone lysine methyltransferase EZH2 has been implicated as a key component in cancer aggressiveness, metastasis and poor prognosis. This study discovered a new class of hexahydroisoquinolin derivatives as EZH2 inhibitors. A structure–activity relationship study showed that the steric hindrance was important to the activity for EZH2. A preliminary optimization study led to the discovery of several potent compounds with low nanomolar to sub-nanomolar potency for EZH2. Biological evaluation indicated that SKLB1049 was a highly potent with improved solubility compared to EPZ6438, SAM-competitive, and cell-active EZH2 inhibitor that decreased global H3K27me3 in SU-DHL-6 and Pfeiffer lymphoma cells in a concentration- and time-dependent manner. Further study indicated that SKLB1049 caused cell arrest in G0/G1 phase. These compounds would be useful as chemical tools to further explore the biology of EZH2 and provided us with a start point to develop new EZH2 inhibitors.

|
In vitro protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
In vivo protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
References |
1: Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, Copeland RA, Pollock RM, Kuntz KW, Raimondi A, Keilhack H. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS One. 2014 Dec 10;9(12):e111840. doi: 10.1371/journal.pone.0111840. eCollection 2014. PubMed PMID: 25493630; PubMed Central PMCID: PMC4262195.
2: Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014 Apr;13(4):842-54. doi: 10.1158/1535-7163.MCT-13-0773. Epub 2014 Feb 21. PubMed PMID: 24563539
3. Inhibitors of human histone methyltransferase EZH2, and methods of use thereof for treating cancer. By Kuntz, Kevin W.; Knutson, Sarah K.; Wigle, Timothy James Nelson . From U.S. Pat. Appl. Publ. (2013), US 20130040906 A1 20130214.
4. Aryl-or heteroaryl-substituted benzamide compounds as anticancer agents and their preparation By Kuntz, Kevin Wayne; Chesworth, Richard; Duncan, Kenneth William; Keilhack, Heike; Warholic, Natalie; Klaus, Christine; Zheng, Wanjun; Seki, Masashi; Shirotori, Syuji; Kawano, Satoshi From PCT Int. Appl. (2012), WO 2012142504 A1 20121018.
5: Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. doi: 10.1073/pnas.1303800110. Epub 2013 Apr 25. PubMed PMID: 23620515; PubMed Central PMCID: PMC3651445.
| WO2013155317A1 * | Apr 11, 2013 | Oct 17, 2013 | Epizyme, Inc. | Salt form of a human hi stone methyltransf erase ezh2 inhibitor |
| WO2013155464A1 * | Apr 12, 2013 | Oct 17, 2013 | Epizyme, Inc. | Combination therapy for treating cancer |
| WO2014049488A1 * | Sep 16, 2013 | Apr 3, 2014 | Pfizer Inc. | Benzamide and heterobenzamide compounds |
| WO2014062732A1 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014062733A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014172044A1 * | Mar 14, 2014 | Oct 23, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015004618A1 * | Jul 9, 2014 | Jan 15, 2015 | Glaxosmithkline Intellectual Property (No.2) Limited | Enhancer of zeste homolog 2 inhibitors |
| WO2015010049A1 * | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015010078A2 | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted 6,5-fused bicyclic heteroaryl compounds |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012142504A1 * | Apr 13, 2012 | Oct 18, 2012 | Eisai Co., Ltd. | Aryl-or heteroaryl-substituted benzene compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014062720A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Methods of treating cancer |
| WO2011140324A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indoles |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012005805A1 * | May 5, 2011 | Jan 12, 2012 | Glaxosmithkline Llc | Azaindazoles |
| US4522811 | Jul 8, 1982 | Jun 11, 1985 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US5763263 | Jul 24, 1996 | Jun 9, 1998 | Dehlinger; Peter J. | Method and apparatus for producing position addressable combinatorial libraries |
| US7563589 | May 27, 2005 | Jul 21, 2009 | The University Of North Carolina At Chapel Hill | Including EED, EZH2 and SUZ12 wherein the reconstituted complex has histone methyltransferase (HMTase) activity for lysine 27 of histone H3 (H3-K27); cancer |
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment option specifically for patients with epithelioid sarcoma, a rare soft tissue cancer”. U.S. Food and Drug Administration (FDA) (Press release). 23 January 2020. Retrieved 23 January 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Lue JK, Amengual JE (October 2018). “Emerging EZH2 Inhibitors and Their Application in Lymphoma”. Curr Hematol Malig Rep. 13 (5): 369–382. doi:10.1007/s11899-018-0466-6. PMID 30112706. S2CID 52010283.
- ^ “Tazemetostat”. NCI Drug Dictionary. National Cancer Institute.
- ^ Jump up to:a b c “Drug Trials Snapshots: Tazverik”. U.S. Food and Drug Administration (FDA). 23 January 2020. Retrieved 22 February 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Tazemetostat”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Tazverik |
| Other names | EPZ-6438 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620018 |
| License data |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C34H44N4O4 |
| Molar mass | 572.750 g·mol−1 |
| 3D model (JSmol) | |
Epizyme®, Inc.
400 Technology Square, 4th Floor
Cambridge, MA 02139
Phone: (617) 229-5872
Fax: (617) 349-0707
contact@Epizyme.com
![]()



 Jason Rhodes (left) has been appointed to president of Epizyme Inc.,
Jason Rhodes (left) has been appointed to president of Epizyme Inc.,

100 Technology Square

PRI-724, ICG 001, What is correct structure?

PRI 724 AND ICG001 do confuse us, my efforts to unlock this confusion
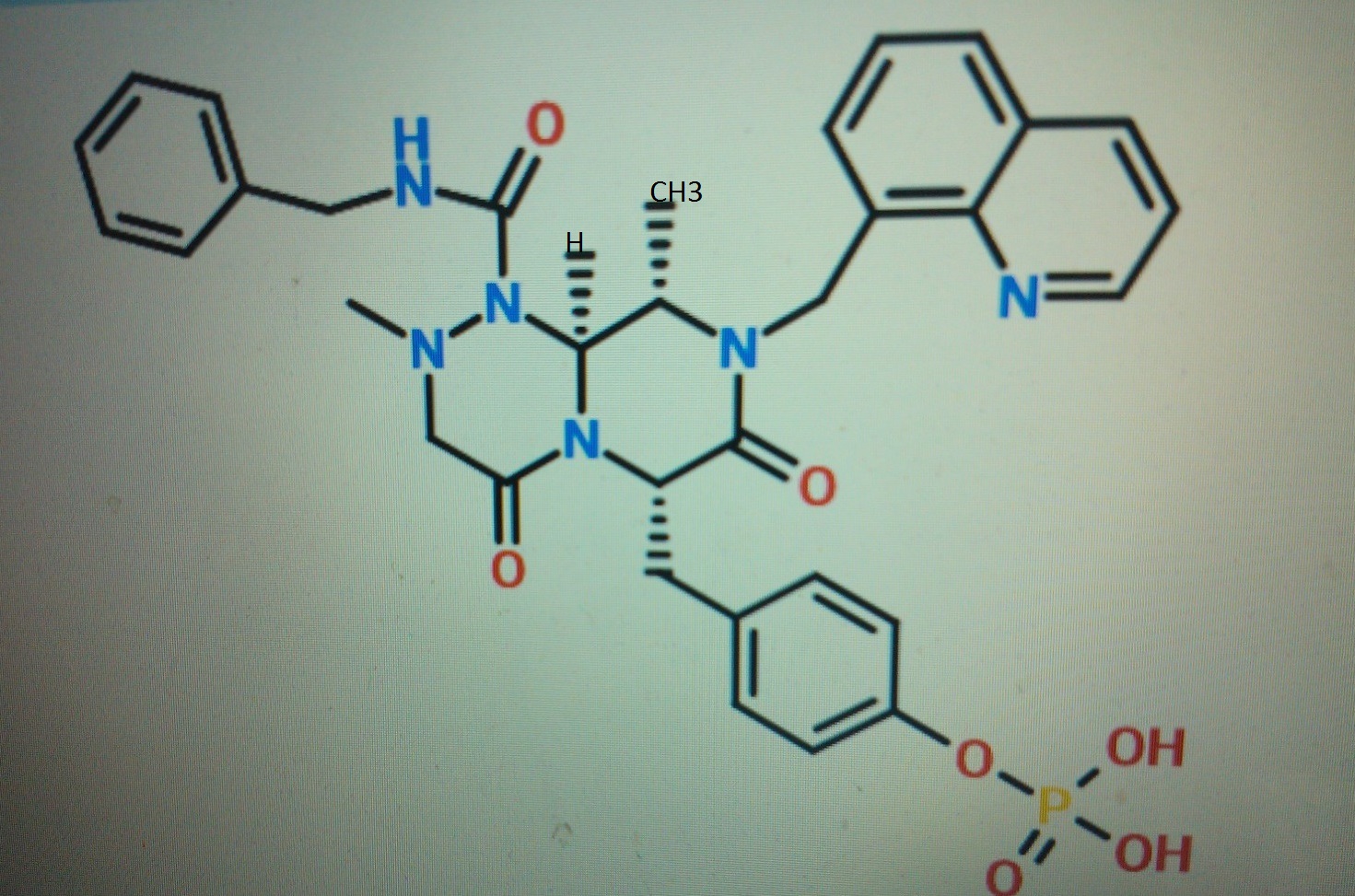
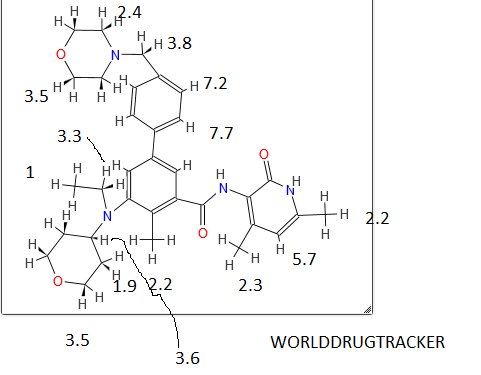
STRUCTURE 4
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate……………seems most likely PRI 724

STRUCTURE 5
Cas 1422253-37-9
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.

compd 2 and 1
OR
COMPD 3
 http://www.medkoo.com/Anticancer-trials/PRI-724.htm and similar/Same
http://www.medkoo.com/Anticancer-trials/PRI-724.htm and similar/Same
http://www.nature.com/nrc/journal/v14/n4/fig_tab/nrc3690_T1.html
compd 3.both above str are same
One of compd 1,2, 3, 4, 5 see at the end as an update , CAN BE ICG001, PRI-724,
Beta-catenin (CTNNB1) inhibitor
ICG001, also known as PRI-724, is a potent, specific inhibitor of the canonical Wnt signaling pathway in cancer stem cells with potential antineoplastic activity. Wnt signaling pathway inhibitor PRI-724 specifically inhibits the recruiting of beta-catenin with its coactivator CBP (the binding protein of the cAMP response element-binding protein CREB); together with other transcription factors beta-catenin/CBP binds to WRE (Wnt-responsive element) and activates transcription of a wide range of target genes of Wnt/beta-catenin signaling. Blocking the interaction of CBP and beta-catenin by this agent prevents gene expression of many proteins necessary for growth, thereby potentially suppressing cancer cell growth. The Wnt/beta-catenin signaling pathway regulates cell morphology, motility, and proliferation; aberrant regulation of this pathway leads to neoplastic proliferation.
JAPAN
4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate
(6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide,
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.
Compound A as in wo 2014061827……..4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyI dihydrogen phosphate in WO2014061827
4-(((6S,9S)-1-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl)octahydro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl)phenyl dihydrogen phosphate (presumed to be PRI-724; first disclosed in WO2009148192), useful for treating cancer, neurodegenerative diseases, glaucoma and idiopathic pulmonary fibrosis.
Eisai, under license from PRISM Pharma, is developing PRI-724, an inhibitor of CREB binding protein or beta-catenin complex formation, for treating cancer (phase 1, as of March 2015) and HCV-induced cirrhosis (preclinical trial).
Follows on from WO2014061827, claiming the use of PRI-724 for treating pulmonary fibrosis.
IS IT

cas 847591-62-2…………http://www.medkoo.com/Anticancer-trials/PRI-724.htm
(6S,9aS)-N-Benzyl-6-(4-hydroxybenzyl)-8-(naphthalen-1-ylmethyl)-4,7-dioxoperhydropyrazino[1,2-a]pyrimidine-1-carboxamide
COMPD 3
OR

COMPD 2
PRI724
1198780-43-6, 578.66, C33 H34 N6 O4
(6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide,

COMPD1
PRI 724
4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate
COMPD 1
SEE
http://www.google.co.in/patents/WO2009148192A1?cl=en

About PRI-724
PRI-724 is an antiproliferative small molecule that selectively inhibits the CBP/beta-catenin complex, which modulates the beta-catenin dependent pathway of Wnt signaling. Activation of the Wnt/beta-catenin signaling pathway is observed in various tumor cells and results in proliferation and metastasis. PRI-724 exhibits a selective antiproliferative effect, inhibiting various cancer cell lines in vitroand substantially inhibiting tumor growth in animal studies. PRI-724 is currently in clinical trials in oncology indications, partnered with Eisai Co., Ltd. PRI-724 also has potential to provide therapeutic benefit in non-oncology areas such as fibrosis and clinical trials in that indication are targeted to start in the second half of 2013.
About PRISM Pharma Co., Ltd.
PRISM Pharma Co., Ltd. has developed its platform technology to modulate inter-cellular protein-protein interactions using peptide mimetic small molecules and found various hit compounds including PRI-724.
SEE
Eisai Research Institute; PRISM Pharma Co Ltd
出願人:エ_ ザイ■ ア_ ル■ アンド■ ディ_ ■
マネジメン卜株式会社(EISAI R&D MANAGEMENT
CO., LTD.) [JP /JP ];亍1128088 東京都文京区
小石川四丁目6 番1 O 号Tokyo (JP).株式会社P
R I S M P h a r m a (PRISM PHARMA CO.,
LTD.) [JP /JP ];亍2268510神奈川県横浜市緑区長津
田町 4 2 5 9 — 3 Kanagawa (JP)
(IO) 国際公開番号
2 0 1 5 ^ ® S 3 .2 0 1 5 )
WO 2015/037587 Al
This method of producing 4-(((6S,9S)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahy- dro-1H-pyrazino[2,1-c][1,2,4]triazine-6-yl)methyl) phenyl dihydrogen phosphate involves a step for adding a reaction solution (I) comprising (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinoline-8-ylmethyl) octahydro-1H-pyrazino[2,1-c] [I,z,4]triazine-1-carboxamide, triethylamine and a solvent to a reaction solution (2) comprising a phosphorylating agent and a solvent.
1
1H-NMR (600MHz, METHAN0L-d4) δ (ppm):1.15 (d, J=6 Hz, 3H), 2.65 (s, 3H), 3.12 (d, J=18 Hz, 1H), 3.35 (d, J=7 Hz, 2H), 3.48 (d, J=18 Hz,1H), 4.15 (m,1H), 4.32 (d, J=15 Hz, 1H), 4.40 (d, J=15 Hz, 1H), 5.33(d, J=16 Hz, 1H), 5.41(d, J=16 Hz, 1H), 5.44 (d, J=7 Hz, 1H), 5.64 (d, J=10 Hz, 1H), 7.07 (dd, J=9,1 Hz, 2H), 7.15 (d, J=9 Hz, 2H), 7.24 (t, J=7 Hz, 1H), 7.27 (d, J=7 Hz, 2H), 7.34 (t, J=8 Hz, 2H), 7.55 (d d, J=8, 4 Hz, 1H), 7.60 (brd, J=6 Hz, 1H), 7.62 (dd, J=8, 7 Hz, 1H), 7.88 (dd, J=8,1 Hz, 1H), 8.38 (dd, J=8, 2 Hz, 1H), 8.90 (dd, J =4, 2 Hz, 1H).
…………………………………………………………………….
SEE
http://www.google.co.in/patents/WO2009148192A1?cl=en
SYNTHESIS OF COMPD 2
PART A
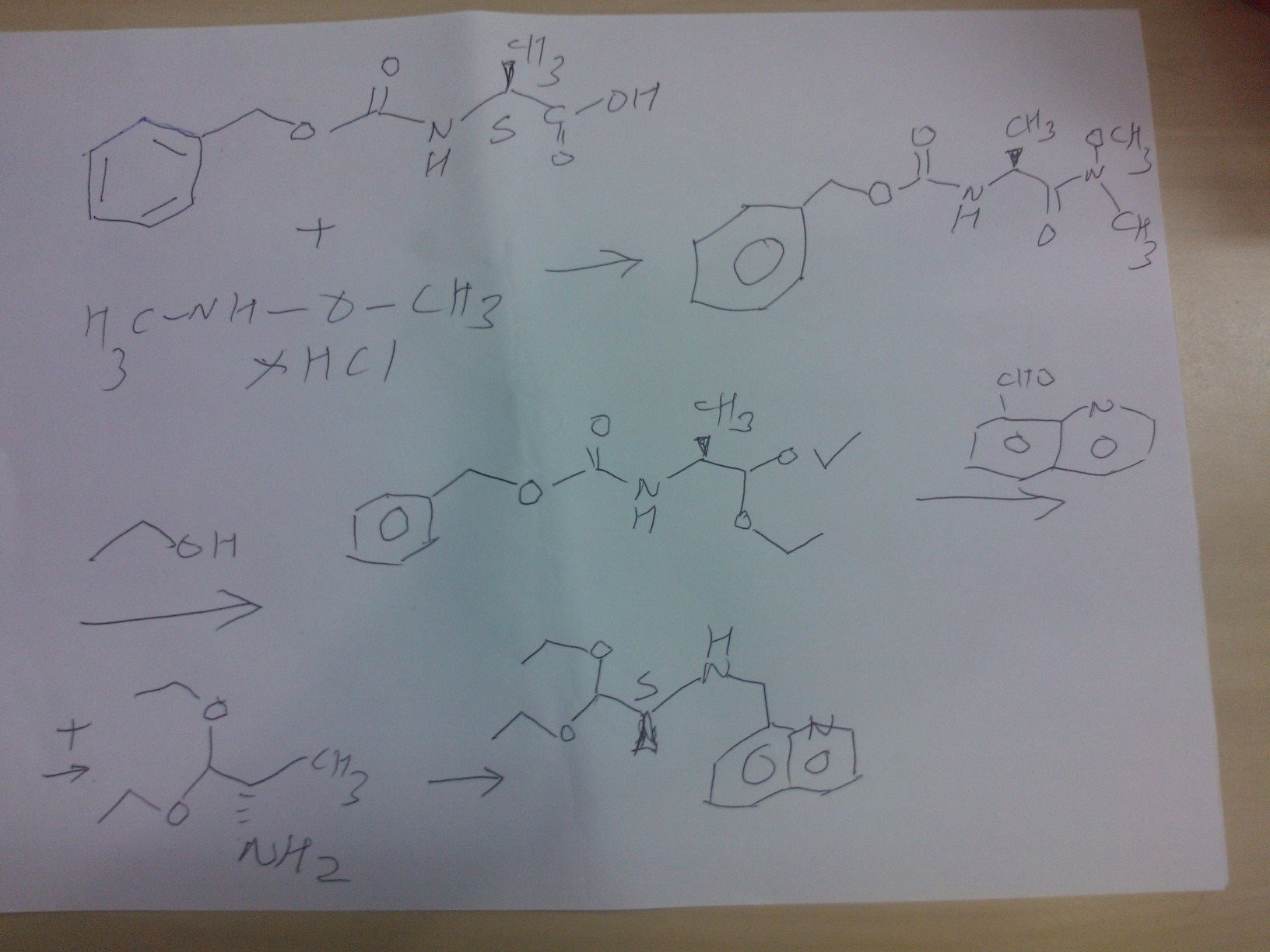
Synthesis Part A
step A
(S)-benzyl 1-(methoxy(methyl)amino)-1-oxopropan-2-ylcarbamate
STEP B
(S)-benzyl 1,1-diethoxypropan-2-ylcarbamate
STEP C
(S)-1,1-diethoxypropan-2-amine
Reaction of the foll……………….(S)-benzyl 1,1-diethoxypropan-2-ylcarbamate, 5% palladium on carbon title compound . (S)-1,1-diethoxypropan-2-amine,
STEP D
PART B
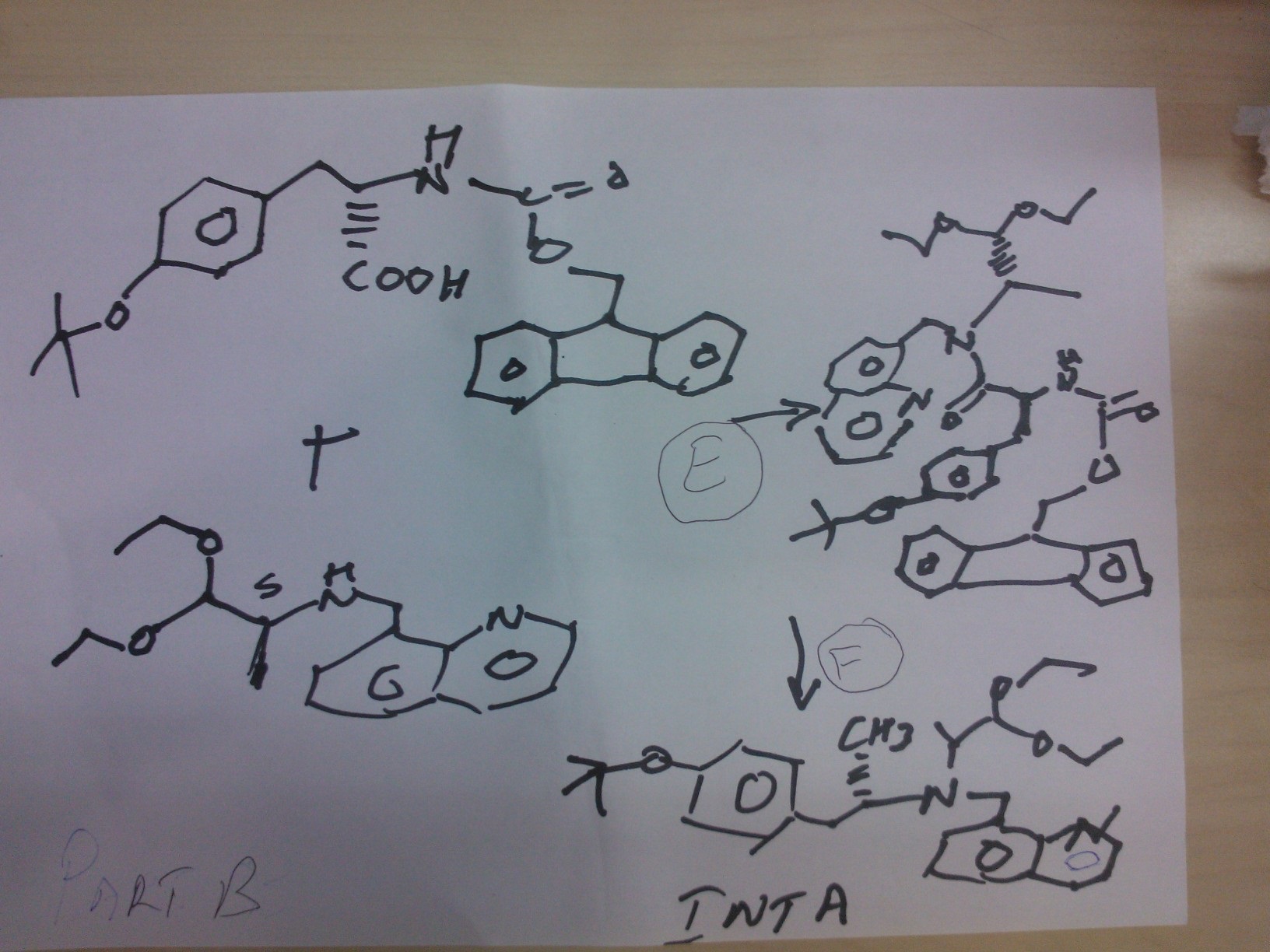
STEP E
(9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate
Reaction of the foll………………. (S)-1,1-diethoxy-N-(quinolin-8-ylmethyl)propan-2-amine, (S)-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-3-(4-tertbutoxyphenyl)propanoic acid to obtain the title compound (9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate
STEP f
(S)-2-amino-3-(4-tertbutoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide INT A
Reaction of the foll………………. (9H-fluoren-9-yl)methyl (S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylcarbamate and piperidine to
obtain the title compound (S)-2-amino-3-(4-tertbutoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide INT A
PART C

STEP g
ethyl 2-(1-methylhydrazinyl)acetate
Reaction of the foll……………….methylhydrazine 7 was reacted with ethyl 2-bromoacetate 1to obtain the title compound
STEP h
ethyl 2-(1-Methyl-2-(benzylcarbamoyl)hydrazinyl)acetate
Reaction of the foll………………. ethyl 2-(1-methylhydrazinyl)acetateand benzyl isocyanate to obtain the title
compound ethyl 2-(1-Methyl-2-(benzylcarbamoyl)hydrazinyl)acetate
STEP i
2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid
Reaction of the foll………………. ethyl 2-(1-allyl-2-
(benzylcarbamoyl)hydrazinyl)acetate and lithium hydroxide monohydrate to obtain the title compound 2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid
STEP j
N-benzyl-2-(2-((S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-
diethoxypropan-2-yl)(quinolin-8-ylmethyl)amino)-1-oxopropan-2-ylamino)-2-oxoethyl)-2-
methylhydrazinecarboxamide……… precursor
Reaction of the foll………………. 2-(2-(benzylcarbamoyl)-1-methylhydrazinyl)acetic acid and (S)-2-amino-3-(4-tert-butoxyphenyl)-N-((S)-1,1-diethoxypropan-2-yl)-N-(quinolin-8-ylmethyl)propanamide ( INT A )yielded the title compound ie the precursor
PART D
THIS PRECURSOR GIVES FINAL PRODUCT
Synthesis of (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-
dimethyl-8-(naphthalen-1-ylmethyl)-4,7-dioxooctahydro-1H-pyrazino[2,1-c][1,2,4]triazine-1-
carboxamide ……….final
fOLL reactants……….. N-benzyl-2-(2-((S)-3-(4-tert-butoxyphenyl)-1-(((S)-1,1-diethoxypropan-2-yl)(naphthalen-1-ylmethyl)amino)-1-oxopropan-2-ylamino)-2-oxoethyl)-2-methylhydrazinecarboxamide, ie the precursor and 10%-water/HCOOH gave (6S,9S)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro-1Hpyrazino[2,1-c][1,2,4]triazine-1-carboxamide
RT 4.22; Mass 578.9
COMPD 3

(6S,9aS)-N-Benzyl-6-(4-hydroxybenzyl)-8-(naphthalen-1-ylmethyl)-4,7-dioxoperhydropyrazino[1,2-a]pyrimidine-1-carboxamide
SEE
US 6762185
……………………………..
SEE
http://www.google.com/patents/WO2012141038A1?cl=en

novel compounds, agent for inducing differentiation into hepatocytes of mesenchymal stem cells, Wnt / β- catenin signaling pathway inhibitor, method for producing hepatocytes with them on hepatocytes such as by their production.
Liver disease is said to be Japan’s national disease, a large number of patients suffering from liver disease. In addition, the annual number of deaths from hepatocellular carcinoma amounts to about 30 004 thousand people. Recently, hepatocellular cancer outcome is improved by advances in treatment, but the increase of advanced cancer, with hepatic dysfunction cirrhosis to merge, so-called hepatic failure death has increased. Liver failure therapy, although liver transplantation is ideal, it is difficult in Japan to obtain sufficient donors, it is necessary to develop a liver regeneration therapy with stem cells.
As stem cells that have the potential to differentiate into liver cells, bone marrow cells, tissue stem cells, such as umbilical cord blood cells can be expected.Therefore, a number of research institutions, for the realization of by regenerative medicine liver cell transplantation treatment of chronic liver failure patient, to differentiate human tissue stem cells into functional hepatocytes, truly clinically applicable efficient differentiation induction technology you are conducting research and development with the goal of developing a.
For example, in the laboratory of Shioda Professor of Tottori University Graduate School of Medicine, reported that the Wnt / β- catenin signaling pathway were differentiated into hepatocytes showed that suppressed by RNA interference at the time of induction of differentiation from human mesenchymal stem cells into hepatocytes you are (Non-Patent Document 1 and Non-Patent Documents 3-5).Furthermore, studies to induce differentiation of hepatocytes in other institutions have been conducted (Non-Patent Document 2, Patent Documents 1 and 2).
On the other hand, recently, from 4,000 or more screening of large compound libraries, Wnt / β- catenin signaling pathway inhibitory low molecular compound 5 types have been identified (Non-Patent Documents 6-9).
Kohyo 2009-535035 JP Patent Publication No. 2010-75631
Atsushi Yanagitani et al., ” retinoic Acid Receptor Dominant Level Negative Form Causes steatohepatitis and Liver Tumors in Transgenic Mice “, Hepatology, Vol. 40, No. 2, 2004, P. 366-375 Seoyoung Park et al.,”Hexachlorophene Inhibits Wnt / beta-catenin Pathway by Promoting Siah-Mediated beta-catenin Degradation “, Mol Pharmacol Vol. 70, No. 3, 960-966, 2006 Yoko Yoshida et al.,” A role of Wnt / beta-catenin Signals in hepatic fate Specification of human umbilical cord blood-derived mesenchymal stem cells “, Am J Physiol Gastrointest Liver Physiol 293:. G1089-G1098, 2007 Shimomura T et al,” Hepatic differentiation of human bone marrow-derived UE7T-13 cells: Effects of cytokines and CCN family Gene expression “, Hepatol Res., 37, 1068-79, 2007 Ishii K et al.,” Hepatic differentiation of human bone marrow-derived mesenchymal stem cells by tetracycline-regulated Hepatocyte Nuclear factor 3Beta “Hepatology, 48, 597- 606, 2008 Maina Lepourcelet et al., ” Small-molecule Antagonists of the oncogenic Tcf / beta-catenin protein complex “, CANCER CELL, JANUARY 2004, VOL. 5, 91-102 Emami KH et al.,” A Small molecule inhibitor of beta-catenin / CREB-binding protein Transcription “, Proc Natl Acad Sci US A. 2004 Aug 24; 101 (34):.. 12682-7 Jufang Shan et al,”Identification of a Specific Inhibitor of the Dishevelled PDZ Domain ” , Biochemistry 2005 Nov 29; 44 (47):.. 15495-503 Trosset JY et al, ” Inhibition of protein-protein Interactions: the discovery of beta-catenin Druglike Inhibitors by combining virtual and Biophysical Screening . “, Proteins 2006 Jul 1 ; 64 (1): 60-7
However, the conventional techniques described above literature, had a room for improvement in the following points.
Patent Documents 1 and 2, it has been described for proteins to induce stem cells from Hikimomiki cells, due to the use of the protein formulation as a differentiation inducing agent, a room for further improvement in terms of stability and safety and there was.
Non-Patent Document 1 and Non-Patent Document 3 to 5, and have reported that induced differentiated hepatocytes from human mesenchymal stem cells, the use of siRNA as a differentiation inducing agent, such as stability and safety there is room for further improvement in the surface. Non-Patent Document 2, 6 to 9, is not described with respect to method of inducing differentiation into hepatocytes.
The present invention has been made in view of the above circumstances, and an object thereof is to provide an effective low-molecular compounds that induce differentiation into hepatocytes from mesenchymal stem cells. Or, it is intended that the low-molecular compound was used to provide a secure differentiation inducing method is excellent from the mesenchymal stem cell differentiation efficiency of liver cells.
According to the present invention, there is provided formula (1) and one or more compounds selected from the group of compounds represented by the formula (2), a salt thereof or a solvate thereof.



<Example 1> synthetic ICG-001 of synthesis (1) ICG-001 of the IC-2 is an oligopeptide having two rings of β- turn mimic structure in central skeleton, and transcription by β-catenin / Tcf complex can function as a potent antagonist for activation has been reported (Drug Discov. Today 2005, 10, 1467-1474). Synthesis of ICG-001 in accordance with the literature (Tetrahedron 2007, 63, 12912-12916), was subjected to examination.

(1-1) of Compound 1 Synthesis 1-naphtaldehyde (Wako Pure Chemical) (1.56 g, 10 mmol) and 2,2-diethoxyethanamine (Tokyo Kasei Kogyo) (1.33 g, 10 mmol) were mixed 100 I was stirred 20 min at o C. After cooling to room temperature, diluted with EtOH (20 mL), was added portionwise NaBH 4 (0.38 g, 10 mmol), at room temperature, and stirred for 16 h. After completion of the reaction, was distilled off by concentration under reduced pressure EtOH, the product was extracted with AcOEt. The resulting product was purified by silica gel column chromatography (hexane / AcOEt = 5/1) to give the to give compound 1 (2.29 g, 8.5 mmol, 85%).

(1-2) Synthesis of Compound 3 Fmoc-L-Tyr (t-Bu) -OH (0.87 g, 1.9 mmol) in DMF (7 mL) solution of a condensing agent HATU (0.76 g, 2.0 mmol) and diisopropylethylamine (DIEA) (0.35 mL, 2.0 mmol) was added and after stirring for 20 min, compound 1 (0.54 g, a 2.0 mmol) was added, at room temperature, 16 h the mixture was stirred. After the reaction, DMF was distilled off by concentration under reduced pressure, and the resulting product was purified by column chromatography (hexane / AcOEt = 10/1), compound 2 was obtained (1.33 g, 1.9 mmol, 93%). The resulting compound 2 (1.33 g, 1.9 mmol) was dissolved in CH 2 Cl 2 (20 mL), was added diethylamine (DEA) (10 ml, excess), at room temperature, was 2 h stirring.After confirming the completion of the reaction by TLC, vacuum was distilled off CH 2 Cl 2 by concentration, the resulting product was purified by silica gel column chromatography (AcOEt), to give compound 3 (0.92 g, 1. 8 mmol, 92%).

(1-3) Synthesis Fmoc-β-Ala-OH (0.53 g, 1.7 mmol) of compound 5 in DMF (8 mL) solution of a condensing agent HATU (0.70 g, 1.8 mmol) and diisopropylethylamine (DIEA) (0.32 mL, 1.8 mmol) was added and after stirring for 20 min, compound 3 (0.92 g, 1.8 mmol) was added, at room temperature, and stirred for 14 h. After the reaction, DMF was distilled off by concentration under reduced pressure, the resulting product was purified by column chromatography (hexane / AcOEt = 1/1), compound 4 was obtained (1.2 g, 1.5 mmol, 82%). Obtained compound 4 (1.2 g, 1.5 mmol) was dissolved in CH 2 Cl 2 (20 mL), was added diethylamine (DEA) (9 mL, excess), at room temperature, and stirred for 1 h. After confirming the completion of the reaction by TLC, was distilled off CH 2 Cl 2 by concentration under reduced pressure, and the resulting product was purified by silica gel column chromatography (AcOEt / EtOH = 1/1), to give compound 5 (0 .66 g, 1.2 mmol, 80%).

(1-4) synthetic compounds 5 (0.66 g, 1.2 mmol) of compound 7 CH 2 Cl 2 of solution (8 mL) to benzylisocyanate (0.16 g, 1.2 mmol) of CH 2 Cl 2 solution (8 mL) was added, at room temperature, and stirred for 12 h. After confirming the completion of the reaction by TLC, was distilled off CH 2 Cl 2 by concentration under reduced pressure, and the resulting product was purified by column chromatography (AcOEt / EtOH = 1/1), to give compound 6 (0. 59 g, 0.85 mmol, 73%). The obtained compound 6 (0.59 g, 0.85 mmol) at room temperature in the formic acid (9 ml), I was stirred 20 h. Was evaporated formic acid by concentration under reduced pressure, the resulting product was purified by column chromatography (AcOEt), Compound 7a to (ICG-001) was obtained as a white solid (0.26 g, 0.48 mmol, 57 %).
The resulting product, MS spectra and were identified from the 1 H NMR spectrum (with the literature value) (Fig. 1).

STRUCTURE 4
4-(((6S,9S,9aS)-l-(benzylcarbamoyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-ylmethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazin-6-yl)methyl)phenyl dihydrogen phosphate
STRUCTURE 5
(6S,9S,9aS)-N-benzyl-6-(4-hydroxybenzyl)-2,9-dimethyl-4,7-dioxo-8-(quinolin-8-yImethyl)octahydro- 1 H-pyrazino[2, 1 -c] [ 1 ,2,4]triazine- 1 -carboxamide.
Cas 1422253-37-9
2H-Pyrazino[2,1-c][1,2,4]triazine-1(6H)-carboxamide, hexahydro-6-[(4-hydroxyphenyl)methyl]-2,9-dimethyl-4,7-dioxo-N-(phenylmethyl)-8-(8-quinolinylmethyl)-, (6S,9S,9aS)-
Structure can represented as

P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

US priority review for Eisai cancer drug lenvatinib
![]()
US priority review for Eisai cancer drug lenvatinib
Eisai has been boosted by news that regulators in the USA have agreed to a quicker review of its anticancer agent lenvatinib.
The US Food and Drug Administration has granted a priority review to Eisai’s New Drug Application for lenvatinib as a treatment for progressive radioiodine-refractory differentiated thyroid cancer. This means that the agency has assigned a Prescription Drug User Fee Act action date of April 14 next year, eight months after the NDA was submitted.
Read more at: http://www.pharmatimes.com/Article/14-10-15/US_priority_review_for_Eisai_cancer_drug_lenvatinib.aspx#ixzz3GH3iXiDU
SEE SYNTHESIS




Eisai’s lenvatinib 兰伐替尼 レンバチニブ to get speedy review in Europe

Lenvatinib
For the treatment of patients with progressive radioiodine-refractory, differentiated thyroid cancer (RR-DTC).
European regulators have agreed to undertake an accelerated assessment of Eisai’s lenvatinib as a treatment for progressive radioiodine-refractory, differentiated thyroid cancer.
The drug, which carries Orphan Status in the EU, is to be filed “imminently” and could become the first in a new class of tyrosine kinase inhibitors, the drugmaker said.

Read more at: http://www.pharmatimes.com/Article/14-07-31/Eisai_s_lenvatinib_to_get_speedy_review_in_Europe.aspx#ixzz39OGhRHas
Lenvatinib was granted Orphan Drug Designation for thyroid cancer by the health authorities in Japan in 2012, and in Europe and the U.S in 2013. The first application for marketing authorization of lenvatinib in the world was submitted in Japan on June 2014. Eisai is planning to submit applications for marketing authorization in Europe and the U.S. in the second quarter of fiscal 2014.
Lenvatinib is an oral multiple receptor tyrosine kinase (RTK) inhibitor with a novel binding mode that selectively inhibits the kinase activities of vascular endothelial growth factor receptors (VEGFR), in addition to other proangiogenic and oncogenic pathway-related RTKs including fibroblast growth factor receptors (FGFR), the platelet-derived growth factor (PDGF) receptor PDGFRalpha, KIT and RET that are involved in tumor proliferation. This potentially makes lenvatinib a first-in-class treatment, especially given that it simultaneously inhibits the kinase activities of FGFR as well as VEGFR.
LENVATINIB BASE
COSY PREDICT
| Systematic (IUPAC) name | |
|---|---|
| 4-[3-chloro-4-(cyclopropylcarbamoylamino)phenoxy]-7-methoxy-quinoline-6-carboxamide | |
| Clinical data | |
| Legal status | ℞ Prescription only |
| Identifiers | |
| CAS number | |
| ATC code | None |
| PubChem | CID 9823820 |
| ChemSpider | 7999567 |
| UNII | EE083865G2 |
| Chemical data | |
| Formula | C21H19ClN4O4 |
| Mol. mass | 426.853 g/mol |
Lenvatinib (E7080) is a multi-kinase inhibitor that is being investigated for the treatment of various types of cancer by Eisai Co. It inhibits both VEGFR2 and VEGFR3 kinases.[1]
The substence was granted orphan drug status for the treatment of various types of thyroid cancer that do not respond toradioiodine; in the US and Japan in 2012 and in Europe in 2013[2] and is now approved for this use.

Clinical trials
Lenvatinib has had promising results from a phase I clinical trial in 2006[3] and is being tested in several phase II trials as of October 2011, for example against hepatocellular carcinoma.[4] After a phase II trial testing the treatment of thyroid cancer has been completed with modestly encouraging results,[5] the manufacturer launched a phase III trial in March 2011.[6]

Lenvatinib Mesilate
Molecular formula: C21H19ClN4O4,CH4O3S =523.0.
CAS: 857890-39-2.
UNII code: 3J78384F61.
About the Lenvatinib (E7080) Phase II Study
The open-label, global, single-arm Phase II study of multi-targeted kinase inhibitor lenvatinib (E7080) in advanced radioiodine (RAI)-refractory differentiated thyroid cancer involved 58 patients with advanced RAI refractory DTC (papillary, follicular or Hurthle Cell) whose disease had progressed during the prior 12 months. (Disease progression was measured using Response Evaluation Criteria in Solid Tumors (RECIST).) The starting dose of lenvatinib was 24 mg once daily in repeated 28 day cycles until disease progression or development of unmanageable toxicities.
2. About Thyroid Cancer
Thyroid cancer refers to cancer that forms in the tissues of the thyroid gland, located at the base of the throat or near the trachea. It affects more women than men and usually occurs between the ages of 25 and 65.
The most common types of thyroid cancer, papillary and follicular (including Hurthle Cell), are classified as differentiated thyroid cancer and account for 95 percent of all cases. While most of these are curable with surgery and radioactive iodine treatment, a small percentage of patients do not respond to therapy.
3. About Lenvatinib (E7080)
Lenvatinib is multi-targeted kinase inhibitor with a unique receptor tyrosine kinase inhibitory profile that was discovered and developed by the Discovery Research team of Eisai’s Oncology Unit using medicinal chemistry technology. As an anti-angiogenic agent, it inhibits tyrosine kinase of the VEGF (Vascular Endothelial Growth Factor) receptor, VEGFR2, and a number of other types of kinase involved in angiogenesis and tumor proliferation in balanced manner. It is a small molecular targeting drug that is currently being studied in a wide array of cancer types.
4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (additional name: 4-[3-chloro-4-(N′-cyclopropylureido)phenoxy]-7-methoxyquinoline-6-carboxamide) is known to exhibit an excellent angiogenesis inhibition as a free-form product, as described in Example 368 of Patent Document 1. 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide is also known to exhibit a strong inhibitory action for c-Kit kinase (Non-Patent Document 1, Patent Document 2).
However, there has been a long-felt need for the provision of a c-Kit kinase inhibitor or angiogenesis inhibitor that has high usability as a medicament and superior characteristics in terms of physical properties and pharmacokinetics in comparison with the free-form product of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide.
[Patent Document 1] WO 02/32872
[Patent Document 2] WO 2004/080462
[Non-Patent Document 1] 95th Annual Meeting Proceedings, AACR (American Association for Cancer Research), Volume 45, Page 1070-1071, 2004
………………………..
PATENT
http://www.google.co.in/patents/US8058474
EXAMPLES
Examples will now be described to facilitate understanding of the invention, but the invention is not limited to these examples.
Example 1Phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

After suspending 4-amino-3-chlorophenol (23.7 g) in N,N-dimethylformamide (100 mL) and adding pyridine (23.4 mL) while cooling on ice, phenyl chloroformate (23.2 ml) was added dropwise below 20° C. Stirring was performed at room temperature for 30 minutes, and then water (400 mL), ethyl acetate (300 mL) and 6N HCl (48 mL) were added, the mixture was stirred and the organic layer was separated. The organic layer was washed twice with 10% brine (200 mL), and dried over magnesium sulfate. The solvent was removed to give 46 g of the title compound as a solid.
1H-NMR (CDCl3): 5.12 (1h, br s), 6.75 (1H, dd, J=9.2, 2.8 Hz), 6.92 (1H, d, J=2.8 Hz), 7.18-7.28 (4H, m), 7.37-7.43 (2H, m), 7.94 (1H, br s)
Example 21-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea

After dissolving phenyl N-(2-chloro-4-hydroxyphenyl)carbamate in N,N-dimethylformamide (100 mL), cyclopropylamine (22.7 mL) was added while cooling on ice and the mixture was stirred overnight at room temperature. Water (400 mL), ethyl acetate (300 mL) and 6N HCl (55 mL) were then added, the mixture was stirred and the organic layer was separated. The organic layer was washed twice with 10% brine (200 mL), and dried over magnesium sulfate. Prism crystals obtained by concentrating the solvent were filtered and washed with heptane to give 22.8 g of the title compound (77% yield from 4-amino-3-chlorophenol).
1H-NMR (CDCl3): 0.72-0.77 (2H, m), 0.87-0.95 (2H, m), 2.60-2.65 (1H, m), 4.89 (1H, br s), 5.60 (1H, br s), 6.71 (1H, dd, J=8.8, 2.8 Hz), 6.88 (1H, d, J=2.8 Hz), 7.24-7.30 (1H, br s), 7.90 (1H, d, J=8.8H)
Example 34-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide

To dimethylsulfoxide (20 mL) were added 7-methoxy-4-chloro-quinoline-6-carboxamide (0.983 g), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (1.13 g) and cesium carbonate (2.71 g), followed by heating and stirring at 70° C. for 23 hours. After the reaction mixture was allowed to cool down to room temperature, water (50 mL) was added, and the produced crystals were collected by filtration to give 1.56 g of the title compound (88% yield).
1H-NMR (d6-DMSO): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz)
Example 44-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
In a reaction vessel were placed 7-methoxy-4-chloro-quinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethylsulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) in that order, under a nitrogen atmosphere. After stirring at 20° C. for 30 minutes, the temperature was raised to 65° C. over a period of 2.5 hours. After stirring at the same temperature for 19 hours, 33% (v/v) acetone water (5.0 L) and water (10.0 L) were added dropwise over a period of 3.5 hours. Upon completion of the dropwise addition, the mixture was stirred at 60° C. for 2 hours, and 33% (v/v) acetone water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or higher over a period of 1 hour. After then stirring at 40° C. for 16 hours, the precipitated crystals were collected by filtration using a nitrogen pressure filter, and the crystals were washed with 33% (v/v) acetone water (33.3 L), water (66.7 L) and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum drier to give 7.78 kg of the title compound (96.3% yield).
…………………………
SYNTHESIS
1H NMR PREDICT

13 C NMR PREDICT


…………………..
PATENT
http://www.google.co.in/patents/US7253286
EX 368

Example 368
4-(3-Chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
The title compound (22.4 mg, 0.052 mmol, 34.8%) was obtained as white crystals from phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (70 mg, 0.15 mmol) and cyclopropylamine, by the same procedure as in Example 11.
1H-NMR Spectrum (DMSO-d6) δ (ppm): 0.41 (2H, m), 0.66 (2H, m), 2.56 (1H, m), 4.01 (3H, s), 6.51 (1H, d, J=5.6 Hz), 7.18 (1H, d, J=2.8 Hz), 7.23 (1H, dd, J=2.8, 8.8 Hz), 7.48 (1H, d, J=2.8 Hz), 7.50 (1H, s), 7.72 (1H, s), 7.84 (1H, s), 7.97 (1H, s), 8.25 (1H, d, J=8.8 Hz), 8.64 (1H, s), 8.65 (1H, d, J=5.6 Hz).
The starting material was synthesized in the following manner.
Production Example 368-1Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate
The title compound (708 mg, 1.526 mmol, 87.4%) was obtained as light brown crystals from 4-(4-amino-3-chlorophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.745 mmol), by the same procedure as in Production Example 17.
1H-NMR Spectrum (CDCl3) δ (ppm): 4.14 (3H, s), 5.89 (1H, br), 6.50 (1H, d, J=5.6 Hz), 7.16 (2H, dd, J=2.4, 8.8 Hz), 7.22–7.30 (4H, m), 7.44 (2H, m), 7.55 (1H, s), 7.81 (1H, br), 8.31 (1H, d, J=8.8 Hz), 8.68 (1H, d, J=5.6 Hz), 9.27 (1H, s).
……………………
CRYSTALLINE FORM
http://www.google.co.in/patents/US7612208
Preparation Example 1
Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (1)
Phenyl N-(4-(6-carbamoyl-7-methoxy-4-quinolyl)oxy-2-chlorophenyl)carbamate (17.5 g, 37.7 mmol) disclosed in WO 02/32872 was dissolved in N,N-dimethylformamide (350 mL), and then cyclopropylamine (6.53 mL, 94.25 mmol) was added to the reaction mixture under a nitrogen atmosphere, followed by stirring overnight at room temperature. To the mixture was added water (1.75 L), and the mixture was stirred. Precipitated crude crystals were filtered off, washed with water, and dried at 70° C. for 50 min. To the obtained crude crystals was added ethanol (300 mL), and then the mixture was heated under reflux for 30 min to dissolve, followed by stirring overnight to cool slowly down to room temperature. Precipitated crystals was filtered off and dried under vacuum, and then further dried at 70° C. for 8 hours to give the titled crystals (12.91 g; 80.2%).
Preparation Example 2Preparation of 4-(3-cloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (2)
(1) Preparation of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate

To a suspension of 4-amino-3-chlorophenol (23.7 g) in N,N-dimethylformamide (100 mL) was added pyridine (23.4 mL) while cooling in an ice bath, and phenyl chloroformate (23.2 mL) was added dropwise below 20° C. After stirring at room temperature for 30 min, water (400 mL), ethyl acetate (300 mL), and 6N-HCl (48 mL) were added and stirred. The organic layer was separated off, washed twice with a 10% aqueous sodium chloride solution (200 mL), and dried over magnesium sulfate. The solvent was evaporated to give 46 g of the titled compound as a solid.
- 1H-NMR Spectrum (CDCl3) δ(ppm): 5.12 (1H, br s), 6.75 (1H, dd, J=9.2, 2.8 Hz), 6.92 (1H, d, J=2.8 Hz), 7.18-7.28 (4H, m), 7.37-7.43 (2H, m), 7.94 (1H, br s).
(2) Preparation of 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea

To a solution of phenyl N-(2-chloro-4-hydroxyphenyl)carbamate in N,N-dimethylformamide (100 mL) was added cyclopropylamine (22.7 mL) while cooling in an ice bath, and the stirring was continued at room temperature overnight. Water (400 mL), ethyl acetate (300 mL), and 6N-HCl (55 mL) were added thereto, and the mixture was stirred. The organic layer was then separated off, washed twice with a 10% aqueous sodium chloride solution (200 mL), and dried over magnesium sulfate. The solvent was evaporated to give prism crystals, which were filtered off and washed with heptane to give 22.8 g of the titled compound (yield from 4-amino-3-chlorophenol: 77%).
- 1H-NMR Spectrum (CDCl3) δ(ppm): 0.72-0.77 (2H, m), 0.87-0.95 (2H, m), 2.60-2.65 (1H, m), 4.89 (1H, br s), 5.60 (1H, br s), 6.71 (1H, dd, J=8.8, 2.8 Hz), 6.88 (1H, d, J=2.8 Hz), 7.24-7.30 (1H, br s), 7.90 (1H, d, J=8.8 Hz)
(3) Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide
To dimethyl sulfoxide (20 mL) were added 7-methoxy-4-chloroquinoline-6-carboxamide (0.983 g), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea (1.13 g) and cesium carbonate (2.71 g), and the mixture was heated and stirred at 70° C. for 23 hours. The reaction mixture was cooled to room temperature, and water (50 mL) was added, and the resultant crystals were then filtered off to give 1.56 g of the titled compound (yield: 88%).
Preparation Example 3Preparation of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (3)
7-Methoxy-4-chloroquinoline-6-carboxamide (5.00 kg, 21.13 mol), dimethyl sulfoxide (55.05 kg), 1-(2-chloro-4-hydroxyphenyl)-3-cyclopropylurea 5.75 kg, 25.35 mol) and potassium t-butoxide (2.85 kg, 25.35 mol) were introduced in this order into a reaction vessel under a nitrogen atmosphere. The mixture was stirred for 30 min at 20° C., and the temperature was raised to 65° C. over 2.5 hours. The mixture was stirred at the same temperature for 19 hours. 33% (v/v) acetone-water (5.0 L) and water (10.0 L) were added dropwise over 3.5 hours. After the addition was completed, the mixture was stirred at 60° C. for 2 hours. 33% (v/v) acetone-water (20.0 L) and water (40.0 L) were added dropwise at 55° C. or more over 1 hour. After stirring at 40° C. for 16 hours, precipitated crystals were filtered off using a nitrogen pressure filter, and was washed with 33% (v/v) acetone-water (33.3 L), water (66.7 L), and acetone (50.0 L) in that order. The obtained crystals were dried at 60° C. for 22 hours using a conical vacuum dryer to give 7.78 kg of the titled compound (yield: 96.3%).
1H-NMR chemical, shift values for 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamides obtained in Preparation Examples 1 to 3 corresponded to those for 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide disclosed in WO 02/32872.
Example 5
A Crystalline Form of the Methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form A)
(Preparation Method 1)
In a mixed solution of methanol (14 mL) and methanesulfonic acid (143 μL, 1.97 mmol) was dissolved 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (700 mg, 1.64 mmol) at 70° C. After confirming the dissolution of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide, the reaction mixture was cooled to room temperature over 5.5 hours, further stirred at room temperature for 18.5 hours, and crystals were filtered off. The resultant crystals were dried at 60° C. to give the titled crystals (647 mg).
(Preparation Method 2)
In a mixed solution of acetic acid (6 mL) and methanesulfonic acid (200 μL, 3.08 mmol) was dissolved 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (600 mg, 1.41 mmol) at 50° C. After confirming the dissolution of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide, ethanol (7.2 mL) and seed crystals of a crystalline form of the methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form A) (12 mg) were added in this order to the reaction mixture, and ethanol (4.8 mL) was further added dropwise over 2 hours. After the addition was completed, the reaction mixture was stirred at 40° C. for 1 hour then at room temperature for 9 hours, and crystals were filtered off. The resultant crystals were dried at 60° C. to give the titled crystals (545 mg).
Example 6A Crystalline Form of the Methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form B)
A crystalline form of the acetic acid solvate of the methanesulfonate of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinolinecarboxamide (Form I) (250 mg) obtained in Example 10 was dried under aeration at 30° C. for 3 hours and at 40° C. for 16 hours to give the titled crystals (240 mg)…………MORE IN PATENT
……………………………..
PATENT
https://www.google.com/patents/WO2014098176A1?cl=en
According to the present invention 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide amorphous is excellent in solubility in water.
Example 1 4- (3-chloro-4- (cyclopropylamino-carbonyl) aminophenoxy) -7-methoxy-6-quinolinecarboxamide manufacture of amorphous amide
4- (3-chloro-4- (cyclopropylamino-carbonyl) amino phenoxy) -7-methoxy-6-quinolinecarboxamide B-type crystals (Patent Document 2) were weighed to 300mg, is placed in a beaker of 200mL volume, it was added tert- butyl alcohol (tBA) 40mL. This was heated to boiling on a hot plate, an appropriate amount of tBA to Compound A is dissolved, water was added 10mL. Then, the weakened heated to the extent that the solution does not boil, to obtain a sample solution. It should be noted, finally the solvent amount I was 60mL. 200mL capacity eggplant type flask (egg-plant shaped flask), and rotated in a state of being immersed in ethanol which had been cooled with dry ice. It was added dropwise a sample solution into the interior of the flask and frozen. After freezing the sample solution total volume, to cover the opening of the flask in wiping cloth, and freeze-dried. We got an amorphous A of 290mg.
Patent Document 2: US Patent Application Publication No. 2007/0117842 Patent specification 
………………………..
Paper
ACS Medicinal Chemistry Letters (2015), 6(1), 89-94
http://pubs.acs.org/doi/full/10.1021/ml500394m
……………..
Paper
Journal of Pharmaceutical and Biomedical Analysis (2015), 114, 82-87
http://www.sciencedirect.com/science/article/pii/S0731708515002940
KEEP WATCHING WILL BE UPDATED………….most of my posts are updated regularly
References
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. (2008). “Multi-Kinase Inhibitor E7080 Suppresses Lymph Node and Lung Metastases of Human Mammary Breast Tumor MDA-MB-231 via Inhibition of Vascular Endothelial Growth Factor-Receptor (VEGF-R) 2 and VEGF-R3 Kinase”. Clinical Cancer Research 14 (17): 5459–65.doi:10.1158/1078-0432.CCR-07-5270. PMID 18765537.
- “Phae III trial shows lenvatinib meets primary endpoint of progression free surival benefit in treatment of radioiodine-refactory differentiated thyroid cancer”. Eisai. 3 February 2014.
- Glen, H; D. Boss; T. R. Evans; M. Roelvink; J. M. Saro; P. Bezodis; W. Copalu; A. Das; G. Crosswell; J. H. Schellens (2007). “A phase I dose finding study of E7080 in patients (pts) with advanced malignancies”. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings Part I 25 (18S): 14073.
- ClinicalTrials.gov NCT00946153 Study of E7080 in Patients With Advanced Hepatocellular Carcinoma (HCC)
- Gild, M. L.; Bullock, M.; Robinson, B. G.; Clifton-Bligh, R. (2011). “Multikinase inhibitors: A new option for the treatment of thyroid cancer”. Nature Reviews Endocrinology 7 (10): 617–624.doi:10.1038/nrendo.2011.141. PMID 21862995.
- ClinicalTrials.gov NCT01321554 A Trial of E7080 in 131I-Refractory Differentiated Thyroid Cancer
UPDATES
EXTRAS……………
Martin Schlumberger et al. A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib(E7080) in patients with 131I-refractory differentiated thyroid cancer (SELECT). 2014 ASCO Annual Meeting. Abstract Number:LBA6008. Presented June 2, 2014. Citation: J Clin Oncol 32:5s, 2014 (suppl; abstr LBA6008). Clinical trial information: NCT01321554.
Bando, Masashi. Quinoline derivative-containing pharmaceutical composition. PCT Int. Appl. (2011), WO 2011021597 A1
Tomohiro Matsushima, four Nakamura, Kazuhiro Murakami, Atsushi Hoteido, Yusuke Ayat, Naoko Suzuki, Itaru Arimoto, Pinche Hirose, Masaharu Gotoda.Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor. US patent number US7612208 Also published as: CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1 , US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date: Aug 7, 2007 Original Assignee: Eisai Co., Ltd
Funahashi, Yasuhiro et al.Preparation of urea derivatives containing nitrogenous aromatic ring compounds as inhibitors of angiogenesis. US patent number US7253286, Also published as:CA2426461A1, CA2426461C, CN1308310C, CN1478078A, CN101024627A, DE60126997D1, DE60126997T2, DE60134679D1, DE60137273D1, EP1415987A1, EP1415987A4, EP1415987B1, EP1506962A2, EP1506962A3, EP1506962B1, EP1777218A1, EP1777218B1, US7612092, US7973160, US8372981, US20040053908, US20060160832, US20060247259, US20100197911, US20110118470, WO2002032872A1, WO2002032872A8.Publication date:Aug 7, 2007. Original Assignee:Eisai Co., Ltd
Sakaguchi, Takahisa; Tsuruoka, Akihiko. Preparation of amorphous salts of 4-[3-chloro-4-[(cyclopropylaminocarbonyl)amino]phenoxy]-7-methoxy-6-quinolinecarboxamide as antitumor agents. PCT Int. Appl. (2006), WO2006137474 A1 20061228.
Naito, Toshihiko and Yoshizawa, Kazuhiro. Preparation of urea moiety-containing quinolinecarboxamide derivatives. PCT Int. Appl., WO2005044788, 19 May 2005
Itaru Arimoto et al. Crystal of salt of 4-[3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy]-7-methoxy-6-quinolinecarboxamide or solvate thereof and processes for producing these. PCT Int. Appl. (2005), WO2005063713 A1 20050714.
|
10-23-2009
|
ANTITUMOR AGENT FOR UNDIFFERENTIATED GASTRIC CANCER
|
|
|
10-2-2009
|
ANTI-TUMOR AGENT FOR MULTIPLE MYELOMA
|
|
|
8-21-2009
|
ANTITUMOR AGENT FOR THYROID CANCER
|
|
|
8-14-2009
|
THERAPEUTIC AGENT FOR LIVER FIBROSIS
|
|
|
2-27-2009
|
USE OF COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND c-kit KINASE INHIBITOR
|
|
|
9-5-2008
|
Medicinal Composition
|
|
|
8-8-2007
|
Nitrogen-containing aromatic derivatives
|
|
|
5-25-2007
|
Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same
|
|
|
7-21-2006
|
Nitrogen-containing aromatic derivatives
|
|
|
6-23-2006
|
Use of sulfonamide-including compounds in combination with angiogenesis inhibitors
|
|
11-16-2011
|
UREA DERIVATIVE AND PROCESS FOR PREPARING THE SAME
|
|
|
8-10-2011
|
c-Kit kinase inhibitor
|
|
|
7-6-2011
|
Nitrogen-Containing Aromatic Derivatives
|
|
|
12-24-2010
|
COMBINED USE OF ANGIOGENESIS INHIBITOR AND TAXANE
|
|
|
9-24-2010
|
COMBINATION OF ANTI-ANGIOGENIC SUBSTANCE AND ANTI-TUMOR PLATINUM COMPLEX
|
|
|
4-30-2010
|
METHOD FOR PREDICTION OF THE EFFICACY OF VASCULARIZATION INHIBITOR
|
|
|
4-16-2010
|
METHOD FOR ASSAY ON THE EFFECT OF VASCULARIZATION INHIBITOR
|
|
|
3-24-2010
|
Urea derivative and process for preparing the same
|
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF PANCREATIC CANCER
|
|
|
2-26-2010
|
COMPOSITION FOR TREATMENT OF UNDIFFERENTIATED GASTRIC CANCER
|
| US7253286 * | 18 Apr 2003 | 7 Aug 2007 | Eisai Co., Ltd | Nitrogen-containing aromatic derivatives |
| US20040053908 | 18 Apr 2003 | 18 Mar 2004 | Yasuhiro Funahashi | Nitrogen-containing aromatic derivatives |
| US20040242506 | 9 Aug 2002 | 2 Dec 2004 | Barges Causeret Nathalie Claude Marianne | Formed from paroxetine hydrochloride and ammonium glycyrrhyzinate by precipitation, spray, vacuum or freeze drying, or evaporation to glass; solid or oil; masks the bitter taste of paroxetine and has a distinctive licorice flavor; antidepressants; Parkinson’s disease |
| US20040253205 | 10 Mar 2004 | 16 Dec 2004 | Yuji Yamamoto | c-Kit kinase inhibitor |
| US20070004773 * | 22 Jun 2006 | 4 Jan 2007 | Eisai R&D Management Co., Ltd. | Amorphous salt of 4-(3-chiloro-4-(cycloproplylaminocarbonyl)aminophenoxy)-7-method-6-quinolinecarboxamide and process for preparing the same |
| US20070078159 | 22 Dec 2004 | 5 Apr 2007 | Tomohiro Matsushima | Has excellent characteristics in terms of physical properties (particularly, dissolution rate) and pharmacokinetics (particularly, bioavailability), and is extremely useful as an angiogenesis inhibitor or c-Kit kinase inhibitor |
| US20070117842 * | 22 Apr 2004 | 24 May 2007 | Itaru Arimoto | Polymorph of 4-[3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy]-7-methoxy-6- quinolinecarboxamide and a process for the preparation of the same |
| EP0297580A1 | 30 Jun 1988 | 4 Jan 1989 | E.R. SQUIBB & SONS, INC. | Amorphous form of aztreonam |
| JP2001131071A | Title not available | |||
| JP2005501074A | Title not available | |||
| JPS6422874U | Title not available | |||
| WO2002032872A1 | 19 Oct 2001 | 25 Apr 2002 | Itaru Arimoto | Nitrogenous aromatic ring compounds |
| WO2003013529A1 | 9 Aug 2002 | 20 Feb 2003 | Barges Causeret Nathalie Claud | Paroxetine glycyrrhizinate |
| WO2004039782A1 | 29 Oct 2003 | 13 May 2004 | Hirai Naoko | QUINOLINE DERIVATIVES AND QUINAZOLINE DERIVATIVES INHIBITING AUTOPHOSPHORYLATION OF Flt3 AND MEDICINAL COMPOSITIONS CONTAINING THE SAME |
| WO2004080462A1 | 10 Mar 2004 | 23 Sep 2004 | Eisai Co Ltd | c-Kit KINASE INHIBITOR |
| WO2004101526A1 | 22 Apr 2004 | 25 Nov 2004 | Itaru Arimoto | Polymorphous crystal of 4-(3-chloro-4-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-qunolinecarboxamide and method for preparation thereof |
| WO2005044788A1 | 8 Nov 2004 | 19 May 2005 | Eisai Co Ltd | Urea derivative and process for producing the same |
| WO2005063713A1 | 22 Dec 2004 | 14 Jul 2005 | Itaru Arimoto | Crystal of salt of 4-(3-chloro-4-(cyclopropylaminocarbonyl)amino-phenoxy)-7-methoxy-6-quinolinecarboxamide or of solvate thereof and processes for producing these |
| WO2006030826A1 | 14 Sep 2005 | 23 Mar 2006 | Eisai Co Ltd | Medicinal composition |
UPDATE………….
1H NMR PREDICT OF LENVATINIB BASE




ACIPHEX, RABEPRAZOLE SODIUM, patent exp 8 th Nov 2013

AS SODIUM SALT

Bottle of rabeprazole 20 mg tablets
ACIPHEX, RABEPRAZOLE SODIUM
Drug Patent Expiration and Exclusivity
US 5045552 – Uspto – United States Patent and Trademark Office
| Active Ingredient | Form | Dosage | Drug Type | Application | Product | |
|---|---|---|---|---|---|---|
| RABEPRAZOLE SODIUM | TABLET, DELAYED RELEASE; ORAL | 10MG **Federal Register determination that product was not discontinued or withdrawn for safety or efficacy reasons** | DISCN | 020973 | 001 | |
| RABEPRAZOLE SODIUM | TABLET, DELAYED RELEASE; ORAL | 20MG | RX | 020973 | 002 |
EISAI INC’s ACIPHEX.

| Patent | Expiration | |
|---|---|---|
| US 5045552*PED | 2013-11-8 | |
| US 5045552 | Pyridine derivatives having anti-ulcerative activity
Pyridine derivatives useful for preventing or treating peptic ulcers, pharmaceutical preparations and methods of treating peptic ulcers are described.
|
2013-5-8(expired) |
Exclusivity
Exclusivity is marketing rights granted by the FDA to the EISAI INC.
Eisai Co. Ltd. announced that Halaven (eribulin mesylate), an anti-cancer agent, has now been launched in Russia

Eribulin
Eribulin mesylate
Eisai R&D Management Co., Ltd.
13/9/2013
Halaven is a novel anticancer agent discovered and developed in-house by Eisai and is currently approved in more than 50 countries, including Japan, the United States and in Europe. In Russia, Halaven was approved in July 2012 for the treatment of locally advanced or metastatic breast cancer previously treated with at least two chemotherapy regimens including an anthracycline and a taxane. Approximately 50,000 women in Russia are newly diagnosed with breast cancer each year, with this type of cancer being the leading cause of death in women aged 45 to 55 years. read all at…………………….
http://www.dddmag.com/news/2013/09/eisai-launches-halaven-cancer-drug-russia

Eribulin mesylate (Halaven; Eisai) — a synthetic analogue of the marine natural product halichondrin B that interferes with microtubule dynamics — was approved in November 2010 by the US Food and Drug Administration for the treatment of metastatic breast cancer.
Family members of the product patent, WO9965894, have SPC protection in the EU until 2024 and one of its Orange Book listed filings, US8097648, has US154 extension till January 2021.
The drug also has NCE exclusivity till November 2015.
Halichondrin B, a large polyether macrolide, was isolated 25 years ago from the marine sponge Halichondria okadai
Eribulin is an anticancer drug marketed by Eisai Co. under the trade name Halaven. Eribulin mesylate was approved by the U.S. Food and Drug Administration on November 15, 2010, to treat patients with metastatic breast cancer who have received at least two prior chemotherapy regimens for late-stage disease, including both anthracycline– and taxane-based chemotherapies.[1] It was approved by Health Canada on December 14, 2011 for treatment of patients with metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease. [2]
Eribulin is also being investigated by Eisai Co. for use in a variety of other solid tumors, including non-small cell lung cancer, prostate cancer and sarcoma.[3]
Eribulin has been previously known as E7389 and ER-086526, and also carries the US NCI designation NSC-707389.
Eribulin mesylate is an analogue of halichondrin B, which in 1986 was isolated from the marine sponge Halichondria okadai toxic Pacific.Halichondrin B has a significant anti-tumor activity. The Eribulin synthetically obtained has a simpler but still complex molecular structure.Taxanes such as to inhibit the spindle apparatus of the cell, but it is engaged in other ways.
Patent Data
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
Delist Requested |
|---|---|---|---|---|---|---|---|
| N201532 | 001 | 6214865 | Jul 20, 2023 | Y | |||
| N201532 | 001 | 6469182 | Jun 16, 2019 | U – 1096 | |||
| N201532 | 001 | 7470720 | Jun 16, 2019 | Y | |||
| N201532 | 001 | 8097648 | Jan 22, 2021 | U – 1096 |
Exclusivity Data
| Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N201532 | 001 | NCE | Nov 15, 2015 |
The substance inhibits the polymerization of tubulin into microtubules and encapsulates tubulin molecules in non-productive aggregates from. The lack of training of the spindle apparatus blocks the mitosis and ultimately induces apoptosis of the cell. Eribulin differs from known microtubule inhibitors such as taxanes and vinca alkaloids by the binding site on microtubules, also it does not affect the shortening. This explains the effectiveness of the new cytostatic agent in taxane-resistant tumor cell lines with specific tubulin mutations.

Structure and mechanism
Structurally, eribulin is a fully synthetic macrocyclic ketone analogue of the marine sponge natural product halichondrin B,[4][5] the latter being a potent naturally-occurring mitotic inhibitor with a unique mechanism of action found in the Halichondria genus of sponges.[6][7] Eribulin is a mechanistically-unique inhibitor of microtubule dynamics,[8][9] binding predominantly to a small number of high affinity sites at the plus ends of existing microtubules.[10] Eribulin exerts its anticancer effects by triggering apoptosis of cancer cells following prolonged and irreversible mitotic blockade.[11][12]
A new synthetic route to E7389 was published in 2009.[13]
References
- ^“FDA approves new treatment option for late-stage breast cancer” (Press release). USFDA. 2010-11-15. Retrieved November 15, 2010.
- ^Notice of Decision for HALAVEN
- ^http://www.clinicaltrials.gov/ct2/results?term=eribulin+OR+E7389
- ^ Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsky BM, Palme MH, Habgood GJ, Singer LA, Dipietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ, Littlefield BA (February 2001). “In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B”. Cancer Res.61 (3): 1013–21. PMID11221827.
- ^ Yu MJ, Kishi Y, Littlefield BA (2005). “Discovery of E7389, a fully synthetic macrocyclic ketone analogue of halichondrin B”. In Newman DJ, Kingston DGI, Cragg, GM. Anticancer agents from natural products. Washington, DC: Taylor & Francis. ISBN0-8493-1863-7.
- ^ Hirata Y, Uemura D (1986). “Halichondrins – antitumor polyether macrolides from a marine sponge”. Pure Appl. Chem.58 (5): 701–710. doi:10.1351/pac198658050701.
- ^ Bai RL, Paull KD, Herald CL, Malspeis L, Pettit GR, Hamel E (August 1991). “Halichondrin B and homohalichondrin B, marine natural products binding in the vinca domain of tubulin. Discovery of tubulin-based mechanism of action by analysis of differential cytotoxicity data”. J. Biol. Chem.266 (24): 15882–9. PMID1874739.
- Jordan MA, Kamath K, Manna T, Okouneva T, Miller HP, Davis C, Littlefield BA, Wilson L (July 2005). “The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth”. Mol. Cancer Ther.4 (7): 1086–95. doi:10.1158/1535-7163.MCT-04-0345. PMID16020666.
- Okouneva T, Azarenko O, Wilson L, Littlefield BA, Jordan MA (July 2008). “Inhibition of Centromere Dynamics by Eribulin (E7389) during Mitotic Metaphase”. Mol. Cancer Ther.7 (7): 2003–11. doi:10.1158/1535-7163.MCT-08-0095. PMC2562299. PMID18645010.
- Smith JA, Wilson L, Azarenko O, Zhu X, Lewis BM, Littlefield BA, Jordan MA (February 2010). “Eribulin Binds at Microtubule Ends to a Single Site on Tubulin to Suppress Dynamic Instability”. Biochemistry49 (6): 1331–7. doi:10.1021/bi901810u. PMC2846717. PMID20030375.
- Kuznetsov G, Towle MJ, Cheng H, Kawamura T, TenDyke K, Liu D, Kishi Y, Yu MJ, Littlefield BA (August 2004). “Induction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin B macrocyclic ketone analog E7389”. Cancer Res.64 (16): 5760–6. doi:10.1158/0008-5472.CAN-04-1169. PMID15313917.
- ^ Towle MJ, Salvato KA, Wels BF, Aalfs KK, Zheng W, Seletsky BM, Zhu X, Lewis BM, Kishi Y, Yu MJ, Littlefield BA (January 2011). “Eribulin induces irreversible mitotic blockade: implications of cell-based pharmacodynamics for in vivo efficacy under intermittent dosing conditions”. Cancer Res.71 (2): 496–505. doi:10.1158/0008-5472.CAN-10-1874. PMID21127197.
- ^ Kim DS, Dong CG, Kim JT, Guo H, Huang J, Tiseni PS, Kishi Y (November 2009). “New syntheses of E7389 C14-C35 and halichondrin C14-C38 building blocks: double-inversion approach”. J. Am. Chem. Soc.131 (43): 15636–41. doi:10.1021/ja9058475. PMID19807076.

HALAVEN (eribulin mesylate) Injection is a non-taxane microtubule dynamics inhibitor. Eribulin mesylate is a synthetic analogue of halichondrin B, a product isolated from the marine sponge Halichondria okadai. The chemical name for eribulin mesylate is 11,15:18,21:24,28-Triepoxy-7,9-ethano12,15-methano-9H,15H-furo[3,2-i]furo[2′,3′:5,6]pyrano[4,3-b][1,4]dioxacyclopentacosin-5(4H)-one, 2[(2S)-3-amino-2-hydroxypropyl]hexacosahydro-3-methoxy-26-methyl-20,27-bis(methylene)-, (2R,3R,3aS,7R,8aS,9S,10aR,11S,12R,13aR,13bS,15S,18S,21S,24S,26R,28R,29aS)-, methanesulfonate (salt).
It has a molecular weight of 826.0 (729.9 for free base). The empirical formula is C40H59NO11 •CH4O3S. Eribulin mesylate has the following structural formula:
 |
HALAVEN is a clear, colorless, sterile solution for intravenous administration. Each vial contains 1 mg of eribulin mesylate as a 0.5 mg/mL solution in ethanol: water (5:95).


complete syn is available here
http://www.sciencedirect.com/science/article/pii/S0968089611010674

http://www.drugdevelopment-technology.com/projects/halaven-cancer/halaven-cancer1.html

 |
Nitrogen: dark blue, oxygen: red, hydrogen: light blue
graphics: Wurglics, Frankfurt am Main |
……………….
Macrocyclization process for preparing a macrocyclic intermediate of halichondrin B analogs, in particular eribulin, from a non-macrocyclic compound, using a carbon-carbon bond-forming reaction.

http://www.pnas.org/content/108/17/6699/F1.expansion.html

http://www.nature.com/nrd/journal/v8/n1/fig_tab/nrd2487_F6.html
UPDATED
Eisai has developed and launched eribulin mesylate for treating breast cancer. Follows on from WO2014208774, claiming use of a combination comprising eribulin mesylate and lenvatinib mesylate, for treating cancer.
Macrocyclization reactions and intermediates useful in the synthesis of analogs of halichondrin B
By: Fang, Francis G.; Kim, Dae-Shik; Choi, Hyeong-Wook; Chase, Charles E.; Lee, Jaemoon
Assignee: Eisai R&D Management Co., Ltd., Japan
The invention provides methods for the synthesis of eribulin or a pharmaceutically acceptable salt thereof (e.g., eribulin mesylate) through a macrocyclization strategy. The macrocyclization strategy of the present invention involves subjecting a non-macrocyclic intermediate to a carbon-carbon bond-forming reaction (e.g., an olefination reaction (e.g., Horner-Wadsworth-Emmons olefination), Dieckmann reaction, catalytic Ring-Closing Olefin Metathesis, or Nozaki-Hiyama-Kishi reaction) to afford a macrocyclic intermediate. The invention also provides compds. useful as intermediates in the synthesis of eribulin or a pharmaceutically acceptable salt thereof and methods for prepg. the same.
| WO2012129100A1 * | Mar 16, 2012 | Sep 27, 2012 | Eisai R&D Management Co., Ltd. | Methods and compositions for predicting response to eribulin |
| WO2012166899A2 * | May 31, 2012 | Dec 6, 2012 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of thyroid and kidney cancer subjects to lenvatinib compounds |
| CA2828946A1 * | Apr 16, 2012 | Oct 26, 2012 | Eisai R&D Management Co., Ltd. | Therapeutic agent for tumor |
| US7982060 * | Jun 3, 2005 | Jul 19, 2011 | Eisai R&D Management Co., Ltd. | Intermediates for the preparation of analogs of Halichondrin B |

 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com
JALGAON, MAHARASHTRA, INDIA

.




 .
.



.
 MANUDEVI
MANUDEVI

The US FDA has cleared Eisai’s ACIPHEX Sprinkle (rabeprazole sodium) as 12-week gastroesophageal reflux disease (GERD) therapy for use in children between one to eleven years age
28 MAR 2013
The US FDA has cleared Eisai’s ACIPHEX Sprinkle (rabeprazole sodium) as 12-week gastroesophageal reflux disease (GERD) therapy for use in children between one to eleven years age.
The approval was based on positive data from multicenter, double-blind trial conducted in 127 pediatric patients between one to 11 years of age with GERD.
Eisai president and CEO Lonnel Coats said, “We are proud to offer a new treatment option for young children who suffer from GERD.”
The parallel-group study comprised a treatment period of 12 weeks and an extension period of two dose levels of rabeprazole for 24 weeks.
Healing was achieved in 81% of patients during the treatment period and 90% retained healing during the extension period.
The active ingredient in ACIPHEX Delayed-Release Tablets is rabeprazole sodium, a substituted benzimidazole that inhibits gastric acid secretion. Rabeprazole sodium is known chemically as 2-[[[4-(3methoxypropoxy)-3-methyl-2-pyridinyl]-methyl]sulfinyl]-1H–benzimidazole sodium salt. It has an empirical formula of C18H20N3NaO3S and a molecular weight of 381.43. Rabeprazole sodium is a white to slightly yellowish-white solid. It is very soluble in water and methanol, freely soluble in ethanol, chloroform and ethyl acetate and insoluble in ether and n-hexane. The stability of rabeprazole sodium is a function of pH; it is rapidly degraded in acid media, and is more stable under alkaline conditions. The structural formula is:
Figure 1
 |
ACIPHEX is available for oral administration as delayed-release, enteric-coated tablets containing 20 mg of rabeprazole sodium.
Inactive ingredients of the 20 mg tablet are carnauba wax, crospovidone, diacetylated monoglycerides, ethylcellulose, hydroxypropyl cellulose, hypromellose phthalate, magnesium stearate, mannitol, propylene glycol, sodium hydroxide, sodium stearyl fumarate, talc, and titanium dioxide. Iron oxide yellow is the coloring agent for the tablet coating. Iron oxide red is the ink pigment.
MHLW Japan approved Eisai’s Inovelon (rufinamide) as an adjunctive therapy to other antiepileptic drugs in the treatment of Lennox-Gastaut syndrome

Rufinamide
mar26, 2013
The agency approved Eisai’s Inovelon (rufinamide) as an adjunctive therapy to other antiepileptic drugs in the treatment of Lennox-Gastaut syndrome
Rufinamide is an anticonvulsant medication. It is used in combination with other medication and therapy to treat Lennox–Gastaut syndrome and various other seizure disorders. Rufinamide, a triazole derivative, was developed in 2004 by Novartis Pharma, AG, and is manufactured by Eisai.
Rufinamide was approved by the US Food and Drug Administration on November 14, 2008 as adjunctive treatment of seizures associated with Lennox-Gastaut syndrome in children 4 years and older and adults. Its official FDA-approved labeling does not mention use in the treatment of partial seizures inasmuch as clinical trials submitted to the FDA were marginal. However, several recent clinical trials suggest that the drug has efficacy for partial seizures It is marketed under the brand name Banzel. It is also marketed in the European Union under the brand name Inovelon.
The mechanism of action of rufinamide is unknown. However, it is presumed to involve stabilization of the sodium channel inactive state, effectively keeping these ion channels closed. Although the direct mechanism of action may be different, several other antiepileptic agents also stabilize a sodium channel inactive state including phenytoin, carbamazepine, and lacosamide (stabilizes the slow inactive state).
