
Home » Posts tagged 'DIABETES' (Page 5)
Tag Archives: DIABETES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Whey beneficially affects diabetes and cardiovascular disease risk factors in obese adults
View original post 296 more words
(Z)-5-((1-(4-Chloro-2-(trifluoromethyl)benzyl)-1H-indazol-5-yl)methylene)-3-((3R,4R)-3-fluoro-1-methylpiperidin-4-yl)thiazolidine-2,4-dione for the treatment of hyperglycemia in patients with type 2 diabetes mellitus.


Estrogen Related Receptor alpha (ERR-a) modulators useful for treating, ameliorating, or inhibiting the progression of disease states, disorders, and
conditions mediated by ERR-a activity. BACKGROUND OF THE INVENTION
Nuclear receptors are members of a superfamily of transcription factors.
The members of this family share structural similarities and regulate a diverse set of biological effects (Olefsky, J. M. J. Biol. Chem. 2001 , 276(40), 36863-36864). Ligands activate or repress these transcription factors that control genes involved in metabolism, differentiation and reproduction (Laudet, V. and H. Gronmeyer. The Nuclear Receptor Factbooks. 2002, San Diego: Academic Press). Presently, the human genome project has identified about 48 members for this family and cognate ligands have been identified for about 28 of them (Giguere, V. Endocrine Rev. 1999, 20(5), 689-725). This protein family is composed of modular structural domains that can be interchanged within the members of the family without loss of function. A typical nuclear receptor contains a hypervariable N-terminus, a conserved DNA binding domain (DBD), a hinge region, and a conserved ligand- binding domain (LBD). The function of the DBD is targeting of the receptor to specific DNA sequences (Nuclear Hormone Receptor (NHR) response elements or NREs), and the function of the LBD is recognition of its cognate ligand. Within the sequence of the nuclear receptor there are regions involved in transcriptional activation. The Activation Function 1 (AF-1 ) domain is situated at the N-terminus and constitutively activates transcription (Rochette-Egly, C. et al. Cell 1997, 90, 97-107; Rochette-Egly, C. et al. Mol. Endocrinol. 1992, 6, 2197-2209), while the Activation Function 2 (AF-2) domain is embedded within the LBD and its transcriptional activation is ligand dependent (Wurtz, J.M. et al. Nat. Struct. Biol. 1996, 3, 87-94). Nuclear receptors can exist as monomers, homodimers or heterodimers and bind to direct or inverted nucleotide repeats (Laudet and
Gronmeyer, 2002; Aranda, A. and A. Pascual. Physiol. Rev. 2001 , 81 (3), 1269- 1304).
The members of this family exist either in an activated or repressed basal biological state. The basic mechanism of gene activation involves ligand dependent exchange of co-regulatory proteins. These co-regulatory proteins are referred to as co-activators or co-repressors (McKenna, L.J. et al. Endocrine Rev. 1999, 20, 321 -344). A nuclear receptor in the repressed state is bound to its DNA response element and is associated with co-repressor proteins that recruit histone de-acetylases (HDACs) (Jones, P.L. and Y.B. Shi. Curr. Top. Microbiol. Immunol. 2003, 274, 237-268). In the presence of an agonist there is an exchange of co- repressors with co-activators that in turn recruit transcription factors that assemble into an ATP dependent chromatin-remodeling complex. Histones are hyper- acetylated, causing the nucleosome to unfold, and repression is alleviated. The AF-2 domain acts as the ligand dependent molecular switch for the exchange of co-regulatory proteins. In the presence of an agonist the AF-2 domain undergoes a conformational transition and presents a surface on the LBD for interaction with co-activator proteins. In the absence of an agonist or in the presence of an antagonist the AF-2 domain presents a surface that promotes interactions with co- repressor proteins. The interaction surfaces on the LBD for both co-activators, and co-repressors overlap and provide a conserved molecular mechanism for gene activation or repression that is shared by the members of this family of transcription factors (Xu, H.E. et al. Nature 2002, 415 (6873), 813-817).
Natural ligands that modulate the biological activity of nuclear receptors have been identified for only approximately one half of known nuclear receptors. Receptors for which no natural ligand has been identified are termed “orphan receptors.” The discovery of ligands or compounds that interact with an orphan receptor will accelerate the understanding of the role of the nuclear receptors in physiology and disease and facilitate the pursuit of new therapeutic approaches. Estrogen related receptors (ERRs) constitutes a sub-class of these receptors where no ligand has been identified.
ERR-a (also known as ERR-1 ), an orphan receptor, is the first of the three identified members of the estrogen receptor related subfamily of orphan nuclear receptors (ERR-a, β, γ). The ERR subfamily is closely related to the estrogen receptors (ER-a and ER-β). ERR-a and ERR-β were first isolated by a low stringency hybridization screen (Giguere, V. et al. Nature 1988, 331 , 91 -94) followed later with the discovery of ERR-γ (Hong, H. et al. J. Biol. Chem. 1999, 274, 22618-22626). The ERRs and ERs share sequence similarity with the highest homology observed in their DBDs, approximately 60%, and all interact with the classical DNA estrogen response element. Recent biochemical evidence suggested that the ERRs and ERs share target genes, including pS2, lactoferin, aromatase and osteopontin, and share co-regulator proteins (Giguere, V. Trends in Endocrinol. Metab. 2002, 13, 220-225; Vanacker, J.M. et al. EMBO J. 1999, 18, 4270-4279; Kraus, R.J. et al. J. Biol. Chem. 2002, 272, 24286-24834; Hong et al., 1999; Zhang, Z. and C.T. Teng. J. Biol. Chem. 2000, 275, 20387-20846).
Therefore, one of the main functions of ERR is to regulate the response of estrogen responsive genes. The effect of the steroid hormone estrogen is primarily mediated in the breast, bone and endometrium. Thus, the identification of compounds that will interact with ERRs should provide a benefit for the treatment of bone related disease, breast cancer and reproduction.
ERR-a is shown to be present both in normal and breast cancer tissue (Ariazi, E.A. et al. Cancer Res. 2002, 62, 6510-6518). It has been reported that the main function of ERR-a in normal breast tissue is that of a repressor for estrogen responsive genes. In breast cancers or cell lines that are non-estrogen responsive (ER-a negative), ERR-a has been reported to be in an activated state (Ariazi et al., 2002). Therefore, compounds that will interact with ERR-a may be useful agents for the treatment of breast cancer that is ER-a negative and non- responsive to classical anti-estrogenic therapy, or may be used as an adjunct agent for anti-estrogen responsive breast cancers. These agents may act as antagonists by reducing the biological activity of ERR-a in these particular tissues.
Many post-menopausal women experience osteoporosis, a condition that is a result of the reduction of estrogen production. Reduction of estrogen levels results in an increase of bone loss (Turner, R.T. et al. Endocrine Rev. 1994, 15(3), 275-300). An anabolic effect on bone development has been observed on the administration of estrogens to postmenopausal patients with osteoporosis (Pacifici, R. J. Bone Miner. Res. 1996, 1 1 (8), 1043-1051 ) but the molecular mechanism is unknown since ER-a and ER-β knock-out animals have minor skeletal defects, where the action of estrogens is typically mediated (Korach, K. S. Science 1994, 266, 1524-1527; Windahl, S.H. et al. J. Clin. Invest. 1999, 104(7), 895-901 ). Expression of ERR-a in bone is regulated by estrogen (Bonnelye, E. et al. Mol. Endocrin. 1997, 1 1 , 905-916; Bonnelye, E. et al. J. Cell Biol. 2001 , 153, 971 -984). ERR-a is maintained throughout osteoblast differentiation stages.
Over-expression of ERR-a in rat calvaria osteoblasts, an accepted model of bone differentiation, results in an increase of bone nodule formation, while treatment of rat calvaria osteoblasts with ERR-a antisense results in a decrease of bone nodule formation. ERR-a also regulates osteopontin, a protein believed to be involved in bone matrix formation. Therefore compounds that will modulate ERR-a by increasing its activity can have an anabolic effect for the regeneration of bone density and provide a benefit over current approaches that prevent bone loss, but have no anabolic effect. Such compounds can enhance the activity of the receptor by two possible mechanisms: i) enhancing the association of the receptor with proteins that enhance its activity or improve the stability of the receptor; and ii) increasing the intracellular concentrations of the receptor and consequently increasing its activity. Conversely, with respect to bone diseases that are a result of abnormal bone growth, compounds that will interact with ERR-a and decrease its biological activity may provide a benefit for the treatment of these diseases by retarding bone growth. Antagonism of the association of the receptor with co- activator proteins decreases the activity of the receptor.
ERR-a is also present in cardiac, adipose, and muscle tissue and forms a transcriptional active complex with the PGC-1 co-activator family, co-activators implicated with energy homeostasis, mitochondria biogenesis, hepatic
gluconeogenesis and in the regulation of genes involved in fatty acid beta- oxidation (Kamei, Y. et al. Proc. Natl. Acad. Sci. USA 2003, 100(21 ), 12378- 12383). ERR-a regulates the expression of the medium chain acyl-CoA
dehydrogenase promoter (MCAD). Medium chain acyl-CoA dehydrogenase is a gene involved in the initial reaction in fatty acid beta-oxidation. It is believed that in the adipose tissue ERR-a regulates energy expenditure through the regulation of MCAD (Sladek, R. et al. Mol. Cell. Biol. 1997, 17, 5400-5409; Vega, R.B. and D.P. Kelly. J. Biol. Chem. 1997, 272, 31693-31699). In antisense experiments in rat calvaria osteoblasts, in addition to the inhibition of bone nodule formation, there was an increase in adipocyte differentiation markers including aP2 and PPAR-γ (Bonnelye, E. et al. Endocrinology 2002, 143, 3658-3670). Recently an ERR-a knockout model has been described that exhibited reduced fat mass relative to the wild type and DNA chip analysis data indicated alteration of the expression levels of genes involved in adipogenesis and energy metabolism (Luo, J. et al. Mol. Cell. Biol. 2003, 23(22), 7947-7956). More recently it has been shown that ERR-a regulates the expression of endothelial nitric oxide synthase, a gene that has a protective mechanism against arteriosclerosis (Sumi, D. and L.J. Ignarro. Proc Natl. Acad. Sci. 2003, 100, 14451 -14456). The biochemical evidence supports the involvement of ERR-a in metabolic homeostasis and differentiation of cells into adipocytes. Therefore, compounds interacting with ERR-a can affect energy homeostasis and may therefore provide a benefit for the treatment of obesity and metabolic syndrome related disease indications, including arteriosclerosis and diabetes (Grundy, S.M. et al. Circulation 2004, 109(3), 433-438).
There is a continuing need for new ERR-a inverse agonists. There is also a need for ERR-a inverse agonists useful for the treatment of conditions including but not limited to ankylosing spondylitis, artherosclerosis, arthritis (such as rheumatoid arthritis, infectious arthritis, childhood arthritis, psoriatic arthritis, reactive arthritis), bone-related diseases (including those related to bone formation), breast cancer (including those unresponsive to anti-estrogen therapy), cardiovascular disorders, cartilage-related disease (such as cartilage injury/loss, cartilage degeneration, and those related to cartilage formation),
chondrodysplasia, chondrosarcoma, chronic back injury, chronic bronchitis, chronic inflammatory airway disease, chronic obstructive pulmonary disease, diabetes, disorders of energy homeostasis, gout, pseudogout, lipid disorders, metabolic syndrome, multiple myeloma, obesity, osteoarthritis, osteogenesis imperfecta, osteolytic bone metastasis, osteomalacia, osteoporosis, Paget’s disease, periodontal disease, polymyalgia rheumatica, Reiter’s syndrome, repetitive stress injury, hyperglycemia, elevated blood glucose level, and insulin resistance.
Scheme 1



Scheme 2


Scheme 3


Scheme 4


Scheme 5



Scheme 6

Scheme 7

Scheme 8

Scheme 9

without methyl
Example 199
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3-(c/‘s- 4-fluoropiperidin-3-yl)-1 ,3-thiazolidine-2,4-dione
 note………..this is without methyl
note………..this is without methyl(A) 1 ,1 -Dimethylethyl c/‘s-3-[(5Z)-5-[(1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H- indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl]-4-fluoropiperidine- 1 -carboxylate was prepared from (5Z)-5-({1 -[2-chloro-4-
(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ) and 1 ,1 -dimethylethyl frans-3-hydroxy-4- fluoropiperidine-1 -carboxylate (prepared as described in US 2007/249589) following General Procedure W.
(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yljmethylidene)- 3-(c/s-4-fluoropiperidin-3-yl)-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl c/s-3-[(5Z)-5-[(1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl]-4-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.21 (s, 1 H), 7.95 (s, 1 H), 7.72 (d, 1 H), 7.65 (s, 1 H), 7.45 – 7.50 (m, 1 H), 7.30 – 7.38 (m, 2H), 6.66 (d, 1 H), 5.80 (s, 2H), 4.83 – 5.04 (m, 2H), 4.08 – 4.20 (m, 2H), 3.99 – 4.08 (m, 1 H), 3.81 – 3.91 (m, 1 H), 2.27 – 2.40 (m, 1 H), 2.02 – 2.13 (m, 1 H).
LC/MS: mass calcd. for C24Hi9CIF4N4O2S: 538.08, found 539.5 [M+1 ]+
Example 201
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3-(c/‘s- 3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
 note. this is without methyl
note. this is without methyl(A) 1 ,1 -Dimethylethyl c/‘s-4-[(5Z)-5-[(1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H- indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl]-3-fluoropiperidine- 1 -carboxylate was prepared from (5Z)-5-({1 -[2-chloro-4- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ) and 1 ,1 -dimethylethyl frans-4-hydroxy-3- fluoropiperidine-1 -carboxylate (prepared as described in US 2007/249589) following General Procedure J.(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-(c/s-3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl c/‘s-4-[(5Z)-5-[(1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl]-3-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.00 (s, 1 H), 7.96 (s, 1 H), 7.72 (d, 1 H), 7.48 – 7.54 (m, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 4.57 – 4.75 (m, 1 H), 4.40 – 4.56 (m, 1 H), 3.25 – 3.46 (m, 2H), 3.18 (qd, 1 H), 2.83 – 3.03 (m, 1 H), 2.72 (t, 1 H), 1 .88 (br. s., 1 H), 1 .72 (d, 1 H).
LC/MS: mass calcd. for C2 H19CIF4N4O2S: 538.08, found 539.5 [M+1 ]+
Example 273
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3- (frans-3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
 note——- this is without methyl but precursor to desired compd
note——- this is without methyl but precursor to desired compdPreparation 1 :
(A) To the solution of 1 ,1 -dimethylethyl frans-4-(2,4-dioxo-1 ,3-thiazolidin-3-yl)- 3-hydroxypiperidine-1 -carboxylate (from Example 270, 0.68 mmol) in DCM (5 ml_) in a plastic bottle was added bis(2-methoxyethyl)aminosulfur trifluoride (3 equiv) and a drop of ethanol. After stirring at rt for 3 h, the reaction was concentrated and the resultant residue was purified by silica gel chromatography (hexane/EtOAc) to provide 1 ,1 -dimethylethyl trans-4- (2,4-dioxo-1 ,3-thiazolidin-3-yl)-3-fluoropiperidine-1 -carboxylate as a pale yellow solid.
(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5-yl}methylidene)- 3-[frans-3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione was prepared from [4-chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5-carbaldehyde (from
Example 1 ) and 1 ,1 -dimethylethyl frans-4-(2,4-dioxo-1 ,3-thiazolidin-3-yl)-3- fluoropiperidine-1 -carboxylate following General Procedure F.
Preparation 2:
(A) A mixture of 1 ,1 -dimethylethyl 7-oxa-3-azabicyclo[4.1 .0]heptane-3- carboxylate (from Example 270; 47.7 mmol), [(5Z)-5-({1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ; 31 .8 mmol) and magnesium perchlorate (23.9 mmol) in DMF (70 mL) was heated at 1 15 °C for 2-4 h. After cooling to rt, the mixture was slowly poured into water (300 mL) with vigorous stirring, and the resultant precipitate was filtered, thoroughly washed with water and dried to afford a mixture of 1 ,1 -dimethylethyl frans-4-{(5Z)-5-[(1 –
{[4-chloro-2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5-yl)methylidene]- 2,4-dioxo-1 ,3-thiazolidin-3-yl}-3-hydroxypiperidine-1 -carboxylate and the corresponding regioisomer, 1 ,1 -dimethylethyl frans-3-{(5Z)-5-[(1 -{[4-chloro- 2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo- 1 ,3-thiazolidin-3-yl}-4-hydroxypiperidine-1 -carboxylate in ratio of ~ 3.3 : 1 .
(B) To an ice-cooled solution of the above mixture of 1 ,1 -dimethylethyl frans- 4-{(5Z)-5-[(1 -{[4-chloro-2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5- yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl}-3-hydroxypiperidine-1 – carboxylate and the regioisomer, 1 ,1 -dimethylethyl frans-3-{(5Z)-5-[(1 -{[4- chloro-2-(trifluoromethyl)phenyl]methyl}-1 H-indazol-5-yl)methylidene]-2,4- dioxo-1 ,3-thiazolidin-3-yl}-4-hydroxypiperidine-1 -carboxylate in DCM (350 mL) was slowly added bis(2-methoxyethyl)aminosulfur trifluoride (47.7 mmol). After stirring for 1 h, the solution was allowed to warm to rt and stir overnight. The reaction was then quenched with sat’d aq. NaHCO3 and after separating phases, the organic phase was dried (Na2SO4) and concentrated to ~ 40 mL. The solution was loaded onto a silica gel column (Analogix, 200g) and eluted with heptanes/DCM/EtOAc (40:57:3).
Product-containing fractions were combined and concentrated to afford a crude product mixture as a pale yellow foam. Treatment of this foam with ether (~ 20 mL) led to product precipitation; additional ether (200 mL) was added portionwise with stirring and after cooling to ~ 5 °C, the mixture was filtered through a glass fiber filter and washed with cold ether to afford 1 ,1 – dimethylethyl frans-4-{(5Z)-5-[(1 -{[4-chloro-2-(trifluoromethyl)phenyl]- methyl}-1 H-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl}-3- fluoropiperidine-1 -carboxylate as an essentially white powder. (C) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-[frans-3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl frans-4-{(5Z)-5-[(1 -{[4-chloro-2- (trifluoromethyl)phenyl]methyl}-1 H-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl}-3-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.02 (s, 1 H), 7.96 (s, 1 H), 7.72 (d, 1 H), 7.47 – 7.56 (m, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 5.10 – 5.33 (m, 1 H), 4.40 – 4.55 (m, 1 H), 3.52 (d, 1 H), 3.14 (d, 1 H), 2.68 (br. s., 2H), 2.43 (qd, 1 H), 1 .70 – 1 .90 (m, 2H).
LC/MS: mass calcd. for C2 H2oCIF4N4O2S: 538.09, found 539.3 [M+1 ]+
main compd
Example 277
(5Z)-5-({1-[4-Chloro-2-(trifluoromethyl)benzyl]-1H-indazol-5-yl}methylidene)-3- (frans-3-fluoro-1-methylpiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
 desired compd
desired compd(5Z)-5-({1-[4-Chloro-2-(trifluoromethyl)benzyl]-1H-indazol-5-yl}methylidene)- 3-[ trans -3-fluoro-1-methylpiperidin-4-yl]-1,3-thiazolidine-2,4-dione was prepared from (5Z)-5-({1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-[ trans -3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione (Example 273) and formaldehyde following General Procedure R.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.01 (s, 1 H), 7.96 (s, 1 H), 7.72 (s, 1 H), 7.51 (d, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 5.25 – 5.48 (m, 1 H), 4.28 – 4.42 (m, 1 H), 3.24 – 3.36 (m, 1 H), 2.85 – 2.96 (m,
1 H), 2.56 (qd, 1 H), 2.37 (s, 3H), 2.07 – 2.17 (m, 2H), 1 .77 (dd, 1 H).
LC/MS: mass calcd. for C25H2iCIF4N4O2S: 552.1 , found 553.3 [M+1 ]+

The development of a reproducible process for multihundred gram production of (Z)-5-((1-(4-chloro-2-(trifluoromethyl)benzyl)-1H-indazol-5-yl)methylene)-3-((3R,4R)-3-fluoro-1-methylpiperidin-4-yl)thiazolidine-2,4-dione (26), a potent and selective inhibitor of estrogen-related receptor 1 (ERR1), is described. This multihundred gram synthesis was achieved via magnesium perchlorate-catalyzed regioselective epoxide ring-opening of tert-butyl 7-oxa-3-azabicyclo[4.1.0]heptane-3-carboxylate (9) with thiazolidine-2,4-dione (6, TZD) to form a diastereomeric mixture tert-butyl 4-(2,4-dioxothiazolidin-3-yl)-3-hydroxypiperidine-1-carboxylate (17), of which the 3-hydroxyl group was functionally transformed to 3-fluoro derivative 19 after treatment with Deoxo-Fluor. Chiral separation of 19 provided the desired diastereomer (3R,4R)-21 that was converted to the secondary amine 23 TFA salt. Reductive amination of 23 produced the key intermediate N-methyl 24. Knoevenagel condensation of24 with 1-(4-chloro-2-(trifluoromethyl)benzyl)-1H-indazole-5-carbaldehyde (5) produced the final product 26 in 10% overall yield (99.7% HPLC area% with ≥99.5% de) after a convergent eight synthetic steps with the only column purification being the chiral HPLC separation of 3R,4R–21 from 3S,4S–22.
Citations
- Bignan, G; WO 2011149841 2011
- Li, X; 246th American Chemical Society National Meeting 2013
- Slade, D; J Org Chem 2009, 74, 6331
- Collot, V; Tetrahedron 1999, 55, 6917
- Patta, S; Indian J Chem 2005, 2404
- Maccari, R; Bioorg Med Chem 2005, 13, 2809
- Corona, J; Org Process Res Dev 2010, 14, 712
- Chen, S; Bioorg Med Chem Lett 2007, 17, 2134
- Boto, A; Eur J Org Chem 2005, 673
- Saavedra, J; J Org Chem 1979, 44, 4516
- Bosmans, J; WO 2005000838 2005
- Kratzel, M; Heterocycles 1995, 41, 897
- Daly, A; Tetrahedron Lett 1999, 40, 3617
- Cresswell, A; Org Lett 2010, 12, 2936
- Ready, J; Angew Chem, Int Ed 2002, 41, 1394
- Tandon, V; Tetrahedron Lett 1993, 34, 4403
- Zhao, S; Heterocycles 1994, 39, 163
- Imanishi, T; Synth Comm 1978, 8, 99
- White, J; J Org Chem 2004, 69, 2573
- Lal, G; Chem Commun 1999, 215
- Singh, R; Synthesis 2002, 17, 2561
- Singh, R; J Fluorine Chem 2002, 116, 23
- Shaw, S; J Org Chem 2013, 78, 8892
- Grunewald, G; J Med Chem 2001, 44, 2849
Sotagliflozin, LX 4211 in phase 2 For type 1, 2 diabetes
LX 4211, Sotagliflozin, LP-802034 , lex 1287
UNII-6B4ZBS263Y
Methyl (5S)-5-[4-chloro-3-(4-ethoxybenzyl)phenyl]-1-thio-beta-L-xylopyranoside
β-L-Xylopyranoside, methyl 5-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-thio-, (5S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol,
(5S)-Methyl 5-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-thio-beta-L-xylopyranoside
1018899-04-1
C21H25ClO5S, 424.94, LP-802034
LX-4211 is a dual SGLT2/1 inhibitor; Antidiabetic agents.
LX-4211 is a SGLT-2 inhibitor being evaluated in phase II clinical studies at Lexicon Pharmaceuticals for the oral treatment of type 2 diabetes.



Summary
- Co-administration of LX4211 led to a nearly one-third reduction in mealtime insulin for Type 1 diabetics.
- Although there was no reduction in basal insulin use, the LX4211 group saw better glucose control, lower HbA1c, and weight loss.
- Partnering LX4211 is still management’s top priority but independent development in Type 1 diabetes is at least an option.
Lexicon Pharmaceuticals (LXRX) continues to generate data on its SGLT-1/2 inhibitor LX4211 that suggest this is an effective and promising medication for treating not only Type 2 diabetes (the common target for non-insulin medications for diabetes), but also Type 1 as well. Lexicon’s most recent update, a small short-term Phase II study in Type 1 diabetics is certainly a positive update, but it’s not what investors really want to see. Lexicon still needs to find a development partner for LX4211 and the ongoing delays don’t help sentiment or the long-term prospects for the drug.
A Potentially Meaningful Addition To Type 1 Care
On Monday morning, Lexicon released top-line data from a small (33-patient) Phase II study of LX4211 in Type 1 diabetics on insulin. The results support the notion that SGLT inhibition can play a valuable role in improving glucose control for Type 1 diabetics.
This small study enrolled generally well-controlled patients (HbA1c levels ranging from 7 to 9, with an average of 7.9) and the addition of LX4211 led to 32% reduction in bolus (mealtime) insulin versus a 6% reduction in the placebo group. Even with the lower bolus insulin, patients in the LX4211 group showed a 0.55% reduction in HbA1c versus a 0.06% reduction in the placebo group. Patients taking LX4211 demonstrated better glucose control (more time spent in the target range of 70-180 mg/dL) and saw a 1.7kg weight loss versus a 0.5kg weight gain in the placebo group

……………………..
Scheme 1 :




3(a) 3(b)


4(a) 4(b)

Scheme 3:

…………………
http://www.google.com/patents/EP2332947A1?cl=en
EXAMPLES
-
Aspects of this invention can be understood from the following examples.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2.3-d][13]dioxol-5-yl)(morpholino)methanone
-
To a 12L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(-)-xylose (504.40 g, 3.360 mol), acetone (5L, reagent grade) and anhydrous MgSO4 powder (811.23g, 6.740 mol / 2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol / 0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24°C over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH = 8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt% water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23g, 86% yield, 96.7 area% pure by GC). 1H NMR (400 MHz, DMSO-d6)δ1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93 – 4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 13C NMR (101MHz, DMSO-d6) δ26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73.
-
To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0g, 131 mmol) in acetone (375 mL, 15X) and H2O (125 mL, 5X) was added NaHC03 (33.0g, 3.0 equiv), NaBr (2.8g, 20 mol%) and TEMPO (0.40g, 2 mol%) at 20°C. The mixture was cooled to 0-5°C and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20°C for 24h. Methanol (20 mL) was added and the mixture was stirred at 20°C for 1h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2X). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12X x3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5X) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0g, 79%). 1H NMR (methanol-d4), δ 6.00 (d, J= 3.2 Hz, 1H), 4.72 d, J= 3.2 Hz, 1H), 4.53 (d, J= 3.2 Hz, 1H), 4.38 (d, J= 3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H).
-
To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0g, 24.5 mmol) in THF (100 mL, 20X) was added TBTU (11.8g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20°C for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20°C for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2X x2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4:1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), 8 6.02 (d, J= 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J= 3.2 Hz, 1H), 4.58 (d, J= 3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahvdrofuro[2.3-d][1,3]dioxol-5-yl)(morpholino)methanone
-
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2 solution (1.6 L) and bleach/K2HPO4 solution (400 mL),∼1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25°C. Additional charges of TEMPO (14.3g, 0.5 mol%) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to < 10°C and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15°C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L x 3). The combined organic layer was concentrated under vacuo (∼100-120 torr) at < 35°C (28-32°C vapor, 45-50°C bath) to low volume (- 6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached < 1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5°C and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ5.96 (d, J = 3.6 Hz, 1H), 4.5 8 (d, J = 3.6 Hz, 1H), 4.53 (d, J =3.2Hz,1H), 4.30 (d, J= 3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) 8 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25. 1.
-
The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (-80 L) to remove excess morpholine. When final volume reached -12 L, heptane (14 L) was added slowly at 60-70°C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136°C (DSC). 1H NMR (CDCl3), δ 6.02 (d, J = 3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene:

6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene
-
A 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF = 0.003 wt% water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step.
-
A jacketed 2L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF = 0.003 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 6°C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9°C. The resulting orange solution was diluted with dichloromethane (75mL) and was cooled to -7°C. Then a solution of 2-chloro-5-iodobenzoyl chloride (≤ 0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3°C. The reaction mixture was warmed slightly and held at +5°C for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450mL pre-cooled (∼5°C) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10min with internal temperature remaining below 28°C. The quenched biphasic mixture was stirred at 20°C for 45min and the lower organic phase was washed with 1N aq. HCl (200mL), twice with saturated aq sodium bicarbonate (200mL per wash), and with saturated aq sodium chloride (200mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93g, 99.0 area% by HPLC at 220nm, 1.0 area% regioisomer at 200nm, 98.5 % “as-is” yield).
-
A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300mL, KF = 0.004 wt% water) and the suspension was set stirring under nitrogen and was cooled to about 5°C.Then triethylsilane (28mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24mL, 194.46mmo1,2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150mL) followed by saturated aq sodium bicarbonate (150mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq sodium bicarbonate (100 mL), and with saturated aq sodium chloride (50mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45°C was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45°C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone
-
To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500mag, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5°C, and the mixture was stirred for 1.5 h at 0-5°C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5°C and the mixture was kept stirring for 1h, warmed to 20°C and stirred at 20°C for 2 hours. The reaction was quenched with saturated aq NH4CI, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7. 88 (dd, J= 8.4, 2.0 Hz, 1H), 7.82 (d, J= 2.0 Hz, 1H), 7.50 (d, J= 8.4 Hz, 1H), 7.12 (d, J= 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.07 (d, J = 3.2 Hz, 1H), 5.21 (d, J = 3.2 Hz, 1H), 4.58 (d, J = 3.2 Hz, 1H), 4.56 (d, J = 3.2 Hz, 1H), 4.16 (d, J = 7.2 Hz, 2H), 4.03 (q, J = 7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone
-
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4X to the morpholinoamide) at room temperature and cooled to -5°C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at -5°C over 3 hours. This Grignard solution was used in the ketone formation below.
-
[0055]To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity = 97 wt%, 2.01 kg, 7.34 mol) and THF (11 L, 5.5X) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30°C. The homogeneous solution was then cooled to – 25°C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at -25°C over 3 hours. Then the above Grignard solution was added to this solution at -20 over 41 minutes. The resulting solution was further stirred at -20°C before quench. The reaction mixture was added to 10 wt% aqueous NH4Cl (10 L, 5X) at 0°C with vigorous stirring, and stirred for 30 minutes at 0°C. To this mixture was added slowly 6 N HCl (4 L, 2X) at 0°C to obtain a clear solution and stirred for 30 minutes at 10°C. After phase split, the organic layer was washed with 25 wt% aq NaCl (5 L, 2.5X). Then the organic layer was concentrated to a 3X solution under the conditions (200 mbar, bath temp 50°C). EtOAc (24 L, 12X) was added, and evaporated to a 3X solution under the conditions (150 mbar, bath temp 50°C). After removed solids by a polish filtration, EtOAc (4 L, 2X) was added and concentrated to dryness (150 mbar, bath temp 50°C). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70°C to obtain a 2.5X homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5X) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5X) was added slowly to a little cloudy solution at 70°C. After stirred for 0.5 h at 70°C, the suspension was slowly cooled to 60°C and stirred for 1 h at 60°C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4X), dried under vacuum at 45°C to give the desired ketone as fluffy solids (2.57 kg, 100 wt% by HPLC, purity-adjusted yield: 81%).
(2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate:

6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
-
To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)-((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17X) was added CeCl3.7H2O (118.5g, 1.2 equiv) and the mixture was stirred at 20°C until all solids were dissolved. The mixture was then cooled to -78°C and NaBH4 (12.03g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed -70°C. The mixture was stirred at – 78°C for 1 hour, slowly warmed to 0°C and quenched with saturated aq NH4Cl (550 mL, 5X). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1L, 10X x2) and washed with brine (550 mL, 5X). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100°C and stirred for 15 hours. The mixture was then cooled to room temperature (20°C) and concentrated under vacuum to give a yellow oil (crude, ∼118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0°C. Then, Ac2O (195 mL, -8.0 equiv) was added and the mixture was warmed to 20°C and stirred at 20°C for 2h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, x2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (-133g).
-
To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4X) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80°C for 3.5 hours. The mixture was cooled to 20°C and Mel (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20°C for 3h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1.3 L, 10X) and washed with H2O (650 mL, 5X x2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5X) and the mixture was reslurried at 60°C for 2h and then cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 70 mL, x3). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) δ 7.37 (d, J= 8.0 Hz, 1H), 7.20 (dd, J= 8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J = 9.6 Hz, 1H), 5.20 (t, J = 9.6 Hz, 1H), 5.05 (t, J= 9.6 Hz, 1H), 4.51 (d, J=9.6Hz, 1H), 4.38 (d, J= 9.6Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2.11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J= 7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
-
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19X). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25°C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt% aq NH4Cl (2.5 L, 1X) and the mixture was concentrated under vacuum to 5X, diluted with water (10 L, 4X) and MTBE (12.5L, 5X). The mixture was cooled to 10°C and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2X). The combined aqueous layer was extracted with MTBE (12.5 L, 5X). The combined organic layers were washed with brine (2.5 L, 1X) and concentrated under vacuum to 3X. MeCN (15 L, 6X) was added. The mixture was concentrated again to 10 L (4X) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN.
-
The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol% aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80°C for 2 hours and then cooled to 20°C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2X) and diluted with MTBE (15 L, 6X). The organic layer was separated, washed with brine (5 L, 2X) and concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the mixture was concentrated to 7.5 L (3X).
-
The above MeCN solution of (3S,4R,SR,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10°C, added with dimethylaminopyridine (17.53 g, 2.5 mol%), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2X, 6.0 equiv) so that the temperature of the mixture was kept below 20°C. The reaction was then warmed to 20°C and stirred for 1 hour and diluted with MTBE (15 L, 6X). The mixture was slowly quenched with water (7.5 L, 3X). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2X), 1 N NaHSO4 (5 L, 2X), and brine (5 L, 2X) in sequence.
-
The organic layer was then concentrated under vacuum to 5 L (2X). MeCN (12.5 L, 5X) was added and the solution was concentrated to 7.5 L (3X) (KF = 0.08%). Dioxane (12.5 L, 5X) was added and the solution was concentrated to 7.50 L (3X) (KF = 0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL).
-
To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80°C for 3 hours (>97% conversion). The mixture was cooled to 20°C and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20°C for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20°C for 1 hours. The mixture was then diluted with MTBE (25 L, 10X) and washed with water (12.5 L, 5X x2). The organic layer was separated and concentrated under vacuum to -5 L (2X). MeOH (12.5 L, 5X) was added and the mixture was concentrated to 5X to afford a slurry. The mixture was then heated at 60°C for 1 hour and cooled to 0°C and stirred at 0°C for 1 hour. The mixture was filtered and the cake was washed with MeOH (0°C, 2.5 L, 1X x2, 1.0 L, 0.4X). The cake was dried under vacuum at 45°C overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol
-
To a slurry of (2S,3S,4R,SS,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mo1) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 20°C and the mixture was stirred at 20°C for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.0g, 95%). 1H NMR (CDCl3) δ 7.38 (d, J = 8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, 1H), 4.15 (d, J = 9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, 1H), 3.50 (m, 2H), 3.42 (br s, 1H), 2.95 (br s, 1H), 2.57 (br s, 1H), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
…………
http://www.google.com/patents/WO2010009197A1?cl=en
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H- pyran-3,4,5-triol:

LEX-1287 The compound is an inhibitor of the sodium glucose co-transporter 2, and may be useful in the treatment of diabetes and a variety of other diseases and conditions. See U.S. patent application no. 11/862,690, filed September 28, 2007.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4- ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran-3,4,5-triol To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-
(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0.164mol) in MeOH (900 mL, 10X) was added NaOMe in MeOH (25 wt%, 18 mL, 0.2X) at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. The mixture was then
18
LEX-1287 concentrated to 300 mL, added to H2O (IL) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, x3) and the cake was dried under vacuum at 45°C overnight to afford the desired methyl thiolate (67.Og, 95%). IH NMR (CDC13) δ 7.38 (d, J = 8.4 Hz, IH), 7.22 (m, 2H), 7.11 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 4.35 (d, J = 9.6 Hz, IH), 4.15 (d, J = 9.6 Hz, IH), 4.10-3.95 (m, 3H), 3.64 (t, J = 8.8 Hz, IH), 3.50 (m, 2H), 3.42 (br s, IH), 2.95 (br s, IH), 2.57 (br s, IH), 2.17 (s, 3H), 1.40 (t, J = 7.2 Hz, 3H).
6.9. Preparation of Crystalline Anhydrous (2S,3R,4R,5S,6R)-2-(4-chloro-
3-f4-ethoxybenzyl)phenyl)-6-fmethylthio)tetrahydro-2H-pyran- 3,4,5-triol Form 1
Under slightly positive nitrogen pressure, to a 50 L reactor was charged MeOH (12 L) and the triacetate (1.70 Kg, 3.09 mol). Methanol (5L) was added as a rinse. The slurry was then added NaOMe in MeOH (25 wt%, 340 mL, 0.2X) in 15 minutes at 200C and the mixture was stirred at 200C for 2 hours until all solids disappeared. To the mixture was added slowly water (25.5 L, 15X) in 45 minutes with 5 g seeding (DSC123°C). Solids crashed out and the mixture was stirred at 200C for 1 hour, cooled to 00C and stirred for 30 minutes. The solid was filtered and washed with water (1.7 L, IX, x2) and the cake was dried under vacuum at 45°C overnight to afford the title compound (m.p. ~ 123 0C by DSC peak; 1.28 Kg, 97.7% yield).
…………..
http://www.google.com/patents/US20090030198

EXAMPLES
Aspects of this invention can be understood from the following examples, which do not limit its scope.
6.1. Synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone

To a 12 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged L-(−)-xylose (504.40 g, 3.360 mol), acetone (5 L, reagent grade) and anhydrous MgSO4 powder (811.23 g, 6.740 mol/2.0 equiv). The suspension was set stirring at ambient and then concentrated H2SO4 (50 mL, 0.938 mol/0.28 equiv) was added. A slow mild exotherm was noticed (temperature rose to 24° C. over about 1 hr) and the reaction was allowed to stir at ambient overnight. After 16.25 hours, TLC suggested all L-xylose had been consumed, with the major product being the bis-acetonide along with some (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction mixture was filtered and the collected solids were washed twice with acetone (500 mL per wash). The stirring yellow filtrate was neutralized with concentrated NH4OH solution (39 mL) to pH =8.7. After stirring for 10 min, the suspended solids were removed by filtration. The filtrate was concentrated to afford crude bis-acetonide intermediate as a yellow oil (725.23 g). The yellow oil was suspended in 2.5 L water stirring in a 5 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler. The pH was adjusted from 9 to 2 with 1N aq. HCl (142 mL) and stirred at room temperature for 6 h until GC showed sufficient conversion of the bis-acetonide intermediate to (3aS,5 S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol. The reaction was neutralized by the addition of 50% w/w aq. K2HPO4 until pH=7. The solvent was then evaporated and ethyl acetate (1.25 L) was added to give a white suspension which was filtered. The filtrate was concentrated in vacuo to afford an orange oil which was dissolved in 1 L methyl tert-butyl ether. This solution had KF 0.23 wt % water and was concentrated to afford (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol as an orange oil (551.23 g, 86% yield, 96.7 area % pure by GC). 1H NMR (400 MHz, DMSO-d6) δ 1.22 (s, 3 H) 1.37 (s, 3 H) 3.51 (dd, J=11.12, 5.81 Hz, 1 H) 3.61 (dd, J=11.12, 5.05 Hz, 1 H) 3.93-4.00 (m, 1 H) 3.96 (s, 1 H) 4.36 (d, J=3.79 Hz, 1 H) 4.86 (br. s., 2 H) 5.79 (d, J=3.54 Hz, 1 H). 3C NMR (101 MHz, DMSO-d6) δ 26.48, 27.02, 59.30, 73.88, 81.71, 85.48, 104.69, 110.73. To a solution of (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol (25.0 g, 131 mmol) in acetone (375 mL, 15×) and H2O (125 mL, 5×) was added NaHCO3 (33.0 g, 3.0 equiv), NaBr (2.8 g, 20 mol %) and TEMPO (0.40 g, 2 mol %) at 20° C. The mixture was cooled to 0-5° C. and solid trichloroisocyanuric acid (TCCA, 30.5 g, 1.0 equiv) was then added in portions. The suspension was stirred at 20° C. for 24h. Methanol (20 mL) was added and the mixture was stirred at 20° C. for 1 h. A white suspension was formed at this point. The mixture was filtered, washed with acetone (50 mL, 2×). The organic solvent was removed under vacuum and the aqueous layer was extracted with EtOAc (300 mL, 12× ×3) and the combined organic layers were concentrated to afford an oily mixture with some solid residue. Acetone (125 mL, 5×) was added and the mixture was filtered. The acetone solution was then concentrated to afford the desired acid ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid) as a yellow solid (21.0 g, 79%).1H NMR (methanol-d4), δ 6.00 (d, J=3.2 Hz, 1H), 4.72 d, J=3.2 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.38 (d, J=3.2 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H). To a solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (5.0 g, 24.5 mmol) in THF (100 ML, 20×) was added TBTU (11.8 g, 1.5 equiv), N-methylmorpholine (NMM, 4.1 mL, 1.5 equiv) and the mixture was stirred at 20° C. for 30 min. Morpholine (3.2 mL, 1.5 equiv) was then added, and the reaction mixture was stirred at 20° C. for an additional 6h. The solid was filtered off by filtration and the cake was washed with THF (10 mL, 2× ×2). The organic solution was concentrated under vacuum and the residue was purified by silica gel column chromatography (hexanes:EtOAc, from 1:4 to 4: 1) to afford 4.3 g of the desired morpholine amide (64%) as a white solid. 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H).
6.2. Alternative synthesis of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone
A solution of the diol (3aS,5S,6R,6aS)-5-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-6-ol in acetonitrile (5.38 kg, 65% w/w, 3.50 kg active, 18.40 mol), acetonitrile (10.5 L) and TEMPO (28.4 g, 1 mol %) were added to a solution of K2HPO4 (0.32 kg, 1.84 mol) and KH2PO4 (1.25 kg, 9.20 mol) in water (10.5 L). A solution of NaClO2 (3.12 kg, 80% w/w, 27.6 mole, 1.50 eq) in water (7.0 L) and a solution of K2HPO4 (2.89 kg, 0.90 eq) in water (3.0 L) were prepared with cooling. Bleach (3.0 L, approximate 6% household grade) was mixed with the K2HPO4 solution. Approximately 20% of the NaClO2solution (1.6 L) and bleach/K2HPO4 solution (400 mL, 1 mol %) were added. The remainders of the two solutions were added simultaneously. The reaction mixture turned dark red brown and slow exotherm was observed. The addition rate of the NaClO2 solution was about 40 mL/min (3-4 h addition) and the addition rate for the bleach/K2HPO4 solution was about 10-12 mL/min (10 hr addition) while maintaining the batch at 15-25° C. Additional charges of TEMPO (14.3 g, 0.5 mol %) were performed every 5-6 hr until the reaction went to completion (usually two charges are sufficient). Nitrogen sweep of the headspace to a scrubber with aqueous was performed to keep the green-yellowish gas from accumulating in the vessel. The reaction mixture was cooled to <10° C. and quenched with Na2SO3 (1.4 kg, 0.6 eq) in three portions over 1 hr. The reaction mixture was then acidified with H3PO4 until pH reached 2.0-2.1 (2.5-2.7 L) at 5-15° C. The layers were separated and the aqueous layer was extracted with acetonitrile (10.5 L ×3). The combined organic layer was concentrated under vacuo (˜100-120 torr) at <35° C. (28-32° C. vapor, 45-50° C. bath) to low volume (˜6-7 L) and then flushed with acetonitrile (40 L) until KF of the solution reached <1% when diluted to volume of about 12-15Lwith acetonitrile. Morpholine (1.61 L, 18.4 mol, 1.0 eq) was added over 4-6 h and the slurry was aged overnight under nitrogen. The mixture was cooled to 0-5° C. and aged for 3 hours then filtered. The filter cake was washed with acetonitrile (10 L). Drying under flowing nitrogen gave 4.13 kg of the morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid as a white solid (92-94% pure based on 1H NMR with 1,4-dimethoxybenzene as the internal standard), 72-75% yield corrected for purity. 1H NMR (D2O) δ 5.96 (d, J=3.6 Hz, 1H), 4.58 (d, J=3.6 Hz, 1H), 4.53 (d, J=3.2 Hz, 1H), 4.30 (d, J=3.2 Hz, 1H), 3.84 (m, 2H), 3.18 (m, 2H), 1.40 (s, 1H), 1.25 (s, 1H). 13H NMR (D2O) δ 174.5, 112.5, 104.6, 84.2, 81.7, 75.0, 63.6, 43.1, 25.6, 25.1. The morpholine salt of ((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylic acid (7.85 kg, 26.9 mol), morpholine (2.40 L, 27.5 mol) and boric acid (340 g, 5.49 mol, 0.2 eq) were added to toluene (31 L). The resulting slurry was degassed and heated at reflux with a Dean-Stark trap under nitrogen for 12 h and then cooled to room temperature. The mixture was filtered to remove insolubles and the filter cake washed with toluene (5 L). The filtrate was concentrated to about 14 L and flushed with toluene (˜80 L) to remove excess morpholine. When final volume reached 12 L, heptane (14 L) was added slowly at 60-70° C. The resulting slurry was cooled gradually to room temperature and aged for 3 h. It was then filtered and washed with heptane (12 L) and dry under nitrogen gave a slightly pink solid (6.26 kg, 97% pure, 98% yield). m.p.: 136° C. (DSC). 1H NMR (CDCl3), δ 6.02 (d, J=3.2 Hz, 1H), 5.11 (br s, 1H), 4.62 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 3.9-3.5 (m, 8H), 1.51 (s, 3H), 1.35 (s, 3H). 13C NMR (methanol-d4) δ 26.84, 27.61, 44.24, 47.45, 68.16, 77.14, 81.14, 86.80, 106.87, 113.68, 169.05.
6.3. Synthesis of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene

A 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with 2-chloro-5-iodobenzoic acid (199.41 g, 0.706 mol), dichloromethane (1.2L, KF=0.003 wt % water) and the suspension was set stirring at ambient temperature. Then N,N-dimethylformamide (0.6 mL, 1.1 mol %) was added followed by oxalyl chloride (63 mL, 0.722 mol, 1.02 equiv) which was added over 11 min. The reaction was allowed to stir at ambient overnight and became a solution. After 18.75hours, additional oxalyl chloride (6 mL, 0.069 mol, 0.10 equiv) was added to consume unreacted starting material. After 2 hours, the reaction mixture was concentrated in vacuo to afford crude 2-chloro-5-iodobenzoyl chloride as a pale yellow foam which will be carried forward to the next step. A jacketed 2 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and pressure-equalized addition funnel with gas bubbler was charged with aluminum chloride (97.68 g, 0.733 mol, 1.04 equiv), dichloromethane (0.65 L, KF=0.003 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 6° C. Then ethoxybenzene (90 mL, 0.712 mol, 1.01 equiv) was added over 7 minutes keeping internal temperature below 9° C. The resulting orange solution was diluted with dichloromethane (75 mL) and was cooled to −7° C. Then a solution of 2-chloro-5-iodobenzoyl chloride (<0.706 mol) in 350 mL dichloromethane was added over 13 minutes keeping the internal temperature below +3° C. The reaction mixture was warmed slightly and held at +5° C. for 2 hours. HPLC analysis suggested the reaction was complete and the reaction was quenched into 450 mL pre-cooled (˜5° C.) 2N aq. HCl with stirring in a jacketed round bottom flask. This quench was done in portions over 10 min with internal temperature remaining below 28° C. The quenched biphasic mixture was stirred at 20° C. for 45 min and the lower organic phase was washed with 1N aq. HCl (200 mL), twice with saturated aq. sodium bicarbonate (200 mL per wash), and with saturated aq. sodium chloride (200 mL). The washed extract was concentrated on a rotary evaporator to afford crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone as an off-white solid (268.93 g, 99.0 area % by HPLC at 220 nm, 1.0 area % regioisomer at 200 nm, 98.5 % “as-is” yield). A jacketed 1 L three-necked round bottom flask with mechanical stirrer, rubber septum with temperature probe and gas bubbler was charged with crude (2-chloro-5-iodophenyl)(4-ethoxyphenyl)methanone (30.13 g, 77.93 mmol), acetonitrile (300 mL, KF=0.004 wt % water) and the suspension was set stirring under nitrogen and was cooled to about 5° C. Then triethylsilane (28 mL, 175.30 mmol, 2.25 equiv) was added followed by boron trifluoride-diethyletherate (24 mL, 194.46 mmol, 2.50 equiv) which was added over about 30 seconds. The reaction was warmed to ambient over 30 min and was stirred for 17 hours. The reaction was diluted with methyl tert-butyl ether (150 mL) followed by saturated aq sodium bicarbonate (150 mL) which was added over about 1 minutes. Mild gas evolution was noticed and the biphasic solution was stirred at ambient for 45 minutes. The upper organic phase was washed with saturated aq. sodium bicarbonate (100 mL), and with saturated aq. sodium chloride (50 mL). The washed extract was concentrated on a rotary evaporator to about one half of its original volume and was diluted with water (70 mL). Further concentration in vacuo at 45° C. was done until white prills formed which were allowed to cool to ambient while stirring. After about 30 minutes at ambient, the suspended solids were isolated by filtration, washed with water (30 mL), and were dried in vacuo at 45° C. After about 2.5 hours, this afforded 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene as a slightly waxy white granular powder (28.28 g, 98.2 area % by HPLC at 220 nm, 97.4 % “as-is” yield).
6.4. Synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanone

To a solution of 1-chloro-2-(4-ethoxybenzyl)-4-iodobenzene (500 mg, 1.34 mmol) in THF (5.0 mL) was added i-PrMgCl (2.0M in THF, 1.0 mL, 2.00 mmol) at 0-5° C., and the mixture was stirred for 1.5 h at 0-5° C. A solution of (3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)(morpholino)methanone (146.5 mg, 0.536 mmol) in THF (1.0 mL) was added dropwise at 0-5° C. and the mixture was kept stirring for 1 h, warmed to 20° C. and stirred at 20° C. for 2 hours. The reaction was quenched with saturated aq NH4Cl, extracted with MTBE, washed with brine. The organic layer was concentrated and the residue was purified by silica gel column chromatography to afford the desired ketone (178 mg, 76%) as a white solid. 1H NMR (CDCl3) δ 7.88 (dd, J=8.4, 2.0 Hz, 1H), 7.82 (d, J=2.0 Hz, 1H), 7.50 (d, J=8.4 Hz, 1H), 7.12 (d, J=8.4 Hz, 2H), 6.86 (d, J=8.4 Hz, 2H), 6.07 (d, J=3.2 Hz, 1H), 5.21 (d, J=3.2 Hz, 1H), 4.58 (d, J=3.2 Hz, 1H), 4.56 (d, J=3.2 Hz, 1H), 4.16 (d, J=7.2 Hz, 2H), 4.03 (q, J=7.2 Hz, 2H), 1.54 (s, 3H), 1.42 (t, J=7.2 Hz, 3H), 1.37 (s, 3H).
6.5. Alternative synthesis of (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d]1,3]dioxol-5-yl)methanone
To a 20 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet was charged with the iodide (3.00 kg, 8.05 mol) and THF (8 L, 4× to the morpholinoamide) at room temperature and cooled to −5° C. To the above solution was added dropwise a solution of i-PrMgCl in THF (Aldrich 2 M, 4.39 L, 8.82 mol) at −5° C. over 3 hours. This Grignard solution was used in the ketone formation below. To a 50 L reactor equipped with a mechanical stirrer, a temperature controller, and a nitrogen inlet was charged the morpholinoamide (HPLC purity=97 wt %, 2.01 kg, 7.34 mol) and THF (11 L, 5.5×) at room temperature and stirred for 45 minutes at room temperature and for 15 minutes at 30° C. The homogeneous solution was then cooled to −25° C. To this solution was added a solution of t-BuMgCl in THF (Aldrich 1 M, 7.32 L, 7.91 mol) at −25° C. over 3 hours. Then the above Grignard solution was added to this solution at −20 over 41 minutes. The resulting solution was further stirred at −20° C. before quench. The reaction mixture was added to 10 wt % aqueous NH4Cl (10 L, 5×) at 0° C. with vigorous stirring, and stirred for 30 minutes at 0° C. To this mixture was added slowly 6 N HCl (4 L, 2×) at 0° C. to obtain a clear solution and stirred for 30 minutes at 10° C. After phase split, the organic layer was washed with 25 wt % aq NaCl (5 L, 2.5×). Then the organic layer was concentrated to a 3× solution under the conditions (200 mbar, bath temp 50° C.). EtOAc (24 L, 12×) was added, and evaporated to a 3× solution under the conditions (150 mbar, bath temp 50° C.). After removed solids by a polish filtration, EtOAc (4 L, 2×) was added and concentrated to dryness (150 mbar, bath temp 50° C.). The wet cake was then transferred to a 50 L reactor equipped with a mechanical stirrer, a temperature controller and a nitrogen inlet. After EtOAc was added, the suspension was heated at 70° C. to obtain a 2.5× homogeneous solution. To the resulting homogeneous solution was added slowly heptane (5 L, 2.5×) at the same temperature. A homogeneous solution was seeded and heptane (15 L, 7.5×) was added slowly to a little cloudy solution at 70° C. After stirred for 0.5 h at 70° C., the suspension was slowly cooled to 60° C. and stirred for 1 h at 60° C. The suspension was then slowly cool to room temperature and stirred for 14 h at the same temperature. The crystals were collected and washed with heptane (8 L, 4×), dried under vacuum at 45° C. to give the desired ketone as fluffy solids (2.57 kg, 100 wt % by HPLC, purity-adjusted yield: 81%).
6.6. Synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate

To a solution of the ketone (4-chloro-3-(4-ethoxybenzyl)phenyl)((3aS,5R,6S,6aS)-6-hydroxy-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methanone (114.7 g, 0.265 mol) in MeOH (2 L, 17×) was added CeCl3.7H2O (118.5 g, 1.2 equiv) and the mixture was stirred at 20° C. until all solids were dissolved. The mixture was then cooled to −78° C. and NaBH4 (12.03 g, 1.2 equiv) was added in portions so that the temperature of the reaction did not exceed −70° C. The mixture was stirred at −78° C. for 1 hour, slowly warmed to 0° C. and quenched with saturated aq NH4Cl (550 mL, 5×). The mixture was concentrated under vacuum to remove MeOH and then extracted with EtOAc (1.1 L, 10× ×2) and washed with brine (550 mL, 5×). The combined organics were concentrated under vacuum to afford the desired alcohol as a colorless oil (crude, 115 g). To this colorless oil was added AcOH (650 mL) and H2O (450 mL) and the mixture was heated to 100° C. and stirred for 15 hours. The mixture was then cooled to room temperature (20° C.) and concentrated under vacuum to give a yellow oil (crude, 118 g). To this crude oil was added pyridine (500 mL) and the mixture was cooled to 0° C. Then, Ac2O (195 mL, ˜8.0 equiv) was added and the mixture was warmed to 20° C. and stirred at 20° C. for 2 h. The reaction was quenched with H2O (500 mL) and diluted with EtOAc (1000 mL). The organic layer was separated and concentrated under vacuum to remove EtOAc and pyridine. The residue was diluted with EtOAc (1000 mL) and washed with aq NaHSO4 (1N, 500 mL, ×2) and brine (300 mL). The organic layer was concentrated to afford the desired tetraacetate intermediate as a yellow foam (˜133 g). To a solution of tetraacetate (133 g, 0.237 mol assuming pure) and thiourea (36.1, 2.0 equiv) in dioxane (530 mL, 4×) was added trimethylsilyl trifluoromethanesulfonate (TMSOTf) (64.5 mL, 1.5 equiv) and the reaction mixture was heated to 80° C. for 3.5 hours. The mixture was cooled to 20° C. and MeI (37 mL, 2.5 equiv) and N,N-diisopropylethylamine (DiPEA) (207 mL, 5.0 equiv) was added and the mixture was stirred at 20° C. for 3 h. The mixture was then diluted with methyl tertiary-butyl ether (MTBE) (1. 3 L, 10×) and washed with H2O (650 mL, 5× ×2). The organic layer was separated and concentrated under vacuum to give a yellow solid. To this yellow solid was added MeOH (650 mL, 5×) and the mixture was reslurried at 60° C. for 2 h and then cooled to 0C and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 70 mL, ×3). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (88 g, 60% over 4 steps) as a pale yellow solid. 1H NMR (CDCl3) 6 7.37 (d, J=8.0 Hz, 1H), 7.20 (dd, J=8.0, 2.0 Hz, 1H), 7.07 (m, 2H), 6.85 (m, 2H), 5.32 (t, J=9.6 Hz, 1H), 5.20 (t, J=9.6 Hz, 1H), 5.05 (t, J =9.6 Hz, 1H), 4.51 (d, J =9.6 Hz, 1H), 4.38 (d, J=9.6 Hz, 1h), 4.04 (m, 2H), 2.17 (s, 3H), 2. 11 (s, 3H), 2.02 (s, 3H), 1.73 (s, 3H), 1.42 (t, J=7.2 Hz, 3H).
6.7. Alternative synthesis of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate
To a 50 L reactor under nitrogen atmosphere, 40 L MeOH was charged, followed with the ketone (2.50 kg, 5.78 mol) and CeCl3.7H2O (2.16 kg, 1.0 equiv). Methanol (7.5 L) was added as rinse (totally 47.5 L, 19×). A freshly prepared solution of NaBH4 (87.5 g, 0.4 equiv) in aqueous 1 N NaOH (250 mL) was added slowly (35 min) at 15-25° C. The mixture was then stirred for 15 min. HPLC analysis of the reaction mixture showed approximately 90:10 diastereomeric ratio. The reaction was quenched with 10 wt % aq NH4Cl (2.5 L, 1×) and the mixture was concentrated under vacuum to 5×, diluted with water (10 L, 4×) and MTBE (12.5 L, 5×). The mixture was cooled to 10° C. and 6 N aq HCl was added until the pH of the mixture reached 2.0. Stirring was continued for 10 minutes and the layers were separated. The organic layer was washed with H2O (5L, 2×). The combined aqueous layer was extracted with MTBE (12.5 L, 5×). The combined organic layers were washed with brine (2.5 L, 1×) and concentrated under vacuum to 3×. MeCN (15 L, 6×) was added. The mixture was concentrated again to 10 L (4×) and any solid residue was removed by a polish filtration. The cake was washed with minimal amount of MeCN. The organic filtrate was transferred to 50 L reactor, and a pre-prepared 20 mol % aqueous H2SO4 solution (61.8 mL 98% concentrated H2SO4 and 5 L H2O) was added. The mixture was heated to 80° C. for 2 hours and then cooled to 20° C. The reaction was quenched with a solution of saturated aqueous K2CO3 (5 L, 2×) and diluted with MTBE (15 L, 6×). The organic layer was separated, washed with brine (5 L, 2×) and concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the mixture was concentrated to 7.5 L (3×). The above MeCN solution of (3S,4R,5R,6S)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl)tetrahydro-2H-pyran-2,3,4,5-tetraol was cooled to 10° C., added with dimethylaminopyridine (17.53 g, 2.5 mol %), followed by slow addition of acetic anhydride (3.23 L, 6.0 equiv) and triethylamine (5 L, 2×, 6.0 equiv) so that the temperature of the mixture was kept below 20° C. The reaction was then warmed to 20° C. and stirred for 1 hour and diluted with MTBE (15 L, 6×). The mixture was slowly quenched with water (7.5 L, 3×). The organic layer was separated and washed with saturated aqueous KHCO3 (5L, 2×), 1 N NaHSO4 (5 L, 2×), and brine (5 L, 2×) in sequence. The organic layer was then concentrated under vacuum to 5 L (2×). MeCN (12.5 L, 5×) was added and the solution was concentrated to 7.5 L (3×) (KF=0.08%). Dioxane (12.5 L, 5×) was added and the solution was concentrated to 7.50 L (3×) (KF=0.02%). Any residual solid was removed by a polish filtration and the cake was washed with minimal amount of dioxane (500 mL). To the above filtrate was added thiourea (880 g, 2.0 equiv) and TMSOTf (1.57 L, 1.5 equiv). The reaction mixture was heated to 80° C. for 3 hours (>97% conversion). The mixture was cooled to 20° C. and methyl iodide (541 mL, 1.5 equiv) and diethylisopropylamine (3.02 L, 3.0 equiv) were added and the mixture was stirred at 20° C. for 18 hours. An extra methyl iodide charge (90 mL, 0.25 equiv) was added and the mixture was stirred at 20° C. for 1 hours. The mixture was then diluted with MTBE (25 L, 10×) and washed with water (12.5 L, 5× ×2). The organic layer was separated and concentrated under vacuum to ˜5 L (2×). MeOH (12.5 L, 5×) was added and the mixture was concentrated to 5× to afford a slurry. The mixture was then heated at 60° C. for 1 hour and cooled to 0° C. and stirred at 0° C. for 1 hour. The mixture was filtered and the cake was washed with MeOH (0° C., 2.5 L, 1× ×2, 1.0 L, 0.4×). The cake was dried under vacuum at 45° C. overnight to afford the desired triacetate (1.49 kg, 47% over 4 steps) as a pale yellow/off-white solid.
6.8. Synthesis of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triol

To a slurry of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H-pyran-3,4,5-triyl triacetate (90.0 g, 0. 164 mol) in MeOH (900 mL, 10×) was added NaOMe in MeOH (25 wt %, 18 mL, 0.2×) at 20° C. and the mixture was stirred at 20° C. for 2 hours until all solids disappeared. The mixture was then concentrated to 300 mL, added to H2O (1 L) and stirred for 1 hour. The solid was filtered and washed with H2O (100 mL, ×3) and the cake was dried under vacuum at 45° C. overnight to afford the desired methyl thiolate (67.0 g, 95%). 1H NMR (CDCl3) 6 7.38 (d, J=8.4 Hz, 1H), 7.22 (m, 2H), 7.11 (d, J=8.8 Hz, 2H), 6.83 (d, J=8.8 Hz, 2H), 4.35 (d, J=9.6 Hz, 1H), 4.15 (d, J=9.6 Hz, 1H), 4.10-3.95 (m, 3H), 3.64 (t, J=8.8 Hz, 1H), 3.50 (m, 2H), 2.73 (br s, 3H), 2.17 (s, 3H), 1.40 (t, J=7.2 Hz, 3H).
…………..
SGLT inhibitors: a novel target for diabetes.
Kanwal A, Banerjee SK.
Pharm Pat Anal. 2013 Jan;2(1):77-91. doi: 10.4155/ppa.12.78.
clinical trials………..http://clinicaltrials.gov/search/intervention=LX-4211+OR+LX4211
On the importance of synthetic organic chemistry in drug discovery: reflections on the discovery of antidiabetic agent ertugliflozin. , Med. Chem. Commun., 2013, 4, 101
Carbohydrate Derivatives and Glycomimetic Compounds in Established and Investigational Therapies of Type 2 Diabetes Mellitus

A PART IS PASTED
Carbohydrate Derivatives and Glycomimetic Compounds in Established and Investigational Therapies of Type 2 Diabetes Mellitus
László Somsák, Éva Bokor, Katalin Czifrák, László Juhász and Marietta Tóth (2011). Carbohydrate Derivatives and Glycomimetic Compounds in Established and Investigational Therapies of Type 2 Diabetes Mellitus, Topics in the Prevention, Treatment and Complications of Type 2 Diabetes, Prof. Mark Zimering (Ed.), ISBN: 978-953-307-590-7, InTech, DOI: 10.5772/23463. Available from: http://www.intechopen.com/books/topics-in-the-prevention-treatment-and-complications-of-type-2-diabetes/carbohydrate-derivatives-and-glycomimetic-compounds-in-established-and-investigational-therapies-of-
1. Introduction
Diabetes mellitus is characterized by chronically elevated serum glucose levels resulting in damage of several tissues (e. g. retina, kidney, nerves) due to higher protein glycation, retardation of wound healing, impaired insulin secretion, enhanced insulin resistance, cell apoptosis, and increased oxidative stress. Type 2 diabetes (T2DM), representing 90-95 % of all diabetic cases, is a multifactorial disease where impaired insulin secretion and the development of insulin resistance ultimately leads to hyperglycemia (Hengesh, 1995). The end of the 20th century has witnessed a dramatic increase in the number of patients diagnosed with diabetes worldwide. The predicted number for the year 2025 is well over 300 million representing a 4-5 % yearly increase of the population above 20 years of age (Treadway et al., 2001). This striking prevalence can even be an underestimate due to methodological uncertainties as well as undiagnosed cases (Green et al., 2003). The highest increases are expected in the developing countries of Africa, Asia, and South America, while European populations seem to be less affected (Diamond, 2003). T2DM has been considered as the adult- or late-onset variant, however, the recent decade has seen the appearance and spreading of the disease among young people including children: this forecasts severe economic and health service burdens in the near future (Alberti et al., 2004; Ehtisham & Barrett, 2004).
The epidemic of T2DM is in conjunction with genetic susceptibility: evidence for a genetic component to the disease are accumulating, and the potential of these factors in the treatment and prevention of diabetes has been reviewed (Barroso, 2005; Bonnefond et al., 2010; Sladek et al., 2007; Toye & Gauguier, 2003). A similarly high contribution to this epidemic may originate from behavioral factors such as sedentary lifestyle, overly rich nutrition, and obesity (Bloomgarden, 2004).
Especially due to its long term complications (Brownlee, 2001) like retinopathy, neuropathy, nephropathy, and in particular cardiovascular diseases, as well as significantly higher risk of myocardial infarction, stroke, gangrene, and limb amputation diabetes has become one of the largest contributors to disability and mortality. Although several pathomechanisms (Lowell & Shulman, 2005;Panunti et al., 2004; Stumvoll et al., 2005) are under investigation, no firm understanding of the molecular origins (Ross et al., 2004) of the disease exists. Thereby, all available and investigational treatments are symptomatic. As the complications can first of all be attributed to the high blood glucose levels, current antidiabetic therapies (Table 1) aim at reaching normoglycemia. However, most of the applied oral hypoglycemic agents (Cheng & Fantus, 2005; Krentz & Bailey, 2005; Mizuno et al., 2008; Padwal et al., 2005; Rendell, 2004; Uwaifo & Ratner, 2005) have several side effects and are inadequate for 30-40 % of the patients (Wagman & Nuss, 2001). On the other hand, their efficacy is lost over the time, and several concerns exist regarding their safety (Israili, 2011).
TABLE 1.
Main types of current therapeutic agents for T2DM and their major side effects (Israili, 2011;Moller, 2001)
The complexity of T2DM offers many potential points of intervention for pharmacotherapy for which the main molecular targets and strategies such as insulin secretagogues, insulin sensitizers, hormones, inhibitors of PTP-1B, GSK3, and hepatic glucose production, methods for altering lipid metabolism, combination therapies, etc. have been reviewed in details (Israili, 2011; Morral, 2003; Nourparvar et al., 2004; Wagman et al., 2004).
Among the numerous methods used to treat type 2 diabetes and investigated to find new therapeutic possibilities there are several approaches which apply carbohydrate (especially glucose) derivatives as well as compounds mimicking the properties of sugars. Based on our experience in the chemistry of carbohydrates and glycomimetics, in this survey we summarize the roles of such compounds in combatting type 2 diabetes relying on the review literature and very recent primary scientific papers.
2. Inhibitors of α-glucosidase enzymes
Starch and sucrose are the most important dietary carbohydrates but they are not directly available for the cells. They are digested in the gastrointestinal tract to monosaccharides which can be absorbed to the circulation to raise the serum concentration (Hanhineva et al., 2010). The normal blood glucose level (3.6–5.8 mM) fluctuates throughout the day, is usually lowest in the morning, before the first meal of the day, and rises after meals for an hour or two.
A medically applied treatment of diabetes is to retard the absorption of glucose by inhibition of the carbohydrate hydrolyzing enzymes α-amylase and α-glucosidase in the digestive tract. In humans the digestion of starch, maltodextrins, and maltooligosaccharides includes several stages: degradation of the polymeric substrates results in shorter oligomers which are than cleaved by α-amylase into smaller oligosaccharides. This mixture is broken down to monosaccharides by α-glucosidase from the non-reducing end of the oligosaccharides. By inhibition of these enzymes the rate of glucose production can be reduced that contributes to diminishing the blood glucose levels, too (Tundis et al., 2010). Such inhibitors decrease postprandial hyperglycaemia and hyperinsulinaemia, thereby may improve sensitivity to insulin and release the stress on β-cells (Scheen, 2003).
Glycosidases are a long known and studied class of glycoenzymes for which an enormous number of compounds have been tested as inhibitors (El Ashry et al., 2000a; El Ashry et al., 2000b; El Ashry et al., 2000c; Lillelund et al., 2002). Analogues of monosaccharides in which the ring oxygen is replaced by a nitrogen atom are known as iminosugars (or less properly azasugars) comprising both natural and synthetic molecules (Table 2) which, as the most potent inhibitors of glycosidases, have high pharmacological potential not only in the context of T2DM (Asano, 2009; Compain & Martin, 2007).
The naturally occurring salacinol and analogous sugar mimics with a 4-thiofuranoid type ring (Table 2) belong to a growing class of zwitterionic glycosidase inhibitors, which attract great interest both as synthetic targets and applications for α-glucosidase inhibition (Praly & Vidal, 2010).
The positive charge on the sulfur atom in the thiosugar derivatives and in the iminosugar-based glycosidase inhibitors at physiological pH is facilitating the binding in the active sites of glycosidase enzymes as a mimicry of the charge of the oxocarbeniumion-like transition state formed during hydrolysis of the natural enzyme substrate (Zechel & Withers, 2000). The stabilizing electrostatic interactions between the ammonium (protonated nitrogen) or sulfonium (positively charged sulfur) moieties and an active-site carboxylate residue are considered to be a possible mechanism of action of these inhibitors (Mohan & Pinto, 2007).
Three competitive inhibitors of α-glucosidases: acarbose, miglitol, and voglibose (de Melo et al., 2006) (Table 3) are used as drugs in the treatment of T2DM under various brand names. These compounds are known to inhibit a wide range of glycosidases. In the absence of specificity and because of the known serious side effects, the applications of these first generation iminosugar drugs are limited. Current investigations aim at discovering safer, more specific, and effective iminosugar based derivatives not only as hypoglycemic agents but for several other purposes among others in oncology, as antivirals, and against cystic fibrosis as reviewed in (Home et al., 2011).

Select iminosugar and thiosugar type inhibitors and their effect againstα-glucosidases originating from mammalian gastrointestinal tract



TABLE 3.
α-Glucosidase inhibitors in the clinical practice against T2DM
CONTD……………………
Trelagliptin succinate (SYR-472) for the treatment of type 2 diabetes.

Trelagliptin succinate (SYR-472)
2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2, 4-dioxopyrimidin-1-yl]methyl]-4-fluorobenzonitrile; butanedioic acid
2-[6-[3(R)-Aminopiperidin-1-yl]-3-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-ylmethyl]-4-fluorobenzonitrile
2- [ [6- [ (3R) -3-amino-l-piperidinyl] -3, 4-dihydro-3- methyl-2, 4-dioxo-l (2H) -pyrimidinyl]methyl] -4-fluorobenzonitrile
succinic acid salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Sponsor/Developer: Takeda Pharmaceuticals and Furiex Pharmaceuticals
Mechanism of action: DPP-4 inhibitor
865759-25-7 cas FREE BASE
1029877-94-8 succinate
- SYR 111472 succinate
- SYR 472
- Syr-472
- Syr111472 succinate
- Trelagliptin succinate
- UNII-4118932Z90
- clinical trials….http://clinicaltrials.gov/search/intervention=SYR+472
Trelagliptin-succinate M. Wt: 475.47
Trelagliptin-succinate Formula: C22H26FN5O6
SYR-472 is an oral dipeptidyl peptidase IV inhibitor originated by Takeda. It is in phase III clinical trials for the treatment of type 2 diabetes.
- Diabetes affects 25.8 million people of all ages, or roughly 8.3 percent of the U.S. population.
- The World Health Organization predicts that there will be 366 million people worldwide affected by diabetes by the year 2030.
- The advent of trelagliptin succinate, a unique once weekly medication for patients with type 2 Diabetes is now the focus of clinical trials and exciting research and development.
- Phase III clinical trials of trelagliptin succinate commenced in September 2011, and are estimated to be complete by the second half of 2013.

Indication (Phase): Japan—Once-weekly oral treatment for type 2 diabetes (Phase III; study expected to be completed in second half of 2013)
trelagliptin succinate
Compound I, A, TRELAGLIPTIN which has the formula:

is a DPP-IV inhibitor that is described in U.S. patent application Ser. No. 11/080,992 filed Mar. 15, 2005 (see Compound 34). Its dosing, administration and biological activities are described in U.S. patent application Ser. No. 11/531,671 filed Sep. 13, 2006. U.S. patent application Ser. No. 11/080,992 and Ser. No. 11/531,671 are incorporated herein by reference in their entirety.
Dipeptidyl peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) (referred herein as “DPP-IV”) is a type II membrane protein and a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (e.g., intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of human disease states, including, but are not limit to, diabetes, particularly type II diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (IFG), metabolic acidosis, ketosis, appetite regulation and obesity; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; AIDS; and cancers.
DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV.
Compound (A) or a salt thereof has been reported as an inhibitor of dipeptidyl peptidase (DPP-IV) , which is an enzyme that decomposes glucagon-like peptide-1 (GLP-1) , a hormone increasing insulin secretion (patent document 1) .
In addition, a method including administering 1 – 250 mg of compound (A) or a salt thereof to a patient once per week (patent documents 2, 3), crystal polymorphs of compound (A) (patent documents 4, 5) , and a preparation of compound (A)
(patent documents 6, 7) have also been reported. Compound (A) and a salt thereof are recommended for oral administration in view of the easiness of self-administration, and a tablet, particularly a tablet in the dosage form for administration once per week, is desired. [0006]
The dosage form of once per week is expected to improve drug compliance of patients, whereas it requires supply of compound (A) or a salt thereof to patients in a high dose as compared to, for example, the dosage form of once per day. Since a solid preparation containing compound (A) or a salt thereof in a high dose increases its size, it may conversely degrade the drug compliance for patients, particularly infants and elderly patients having difficulty in swallowing
……………………..
SYNTHESIS
Compound 34 IS TRELAGLIPTIN

4-Fluoro-2-methylbenzonitrile (31).
A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32).
A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
Alternatively, 32 was made as follows.
4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AIBN (122 g) and heated to 75° C. A suspension of DBH (353 g) in DCE (500 mL) was added at 75° C. portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AIBN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10° C. over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20° C. over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (33).
A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows.
To a solution of 6-chloro-3-methyluracil (750 g) and N,N-diisopropylethylamine (998 mL) in NMP (3 L) was added (at <30° C. over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 60° C. for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5° C. for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2×2.25 L). The material was then dried in a vacuum oven at 40±5° C. for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
TFAsalt OF TRELAGLIPTIN
2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (34).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
FREE BASE NOF TRELAGLIPTIN
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 60° C. until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 60° C. and the mixture was allowed to cool to <25° C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2×3.6 L). The filtrate was concentrated at 45° C. under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
HCL salt OF TRELAGLIPTIN
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0° C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35° C. over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 50° C. (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
Succinate salt OF TRELAGLIPTIN

The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purified water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 30° C. over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 60° C. until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57° C. After stirring at 60° C. for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5° C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2×4.2 L). The material was then dried in a vacuum oven at 70±5° C. (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(−)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
Methanesulfonate salt
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75° C. and filtered through #3 Whatman filter paper. The solution was heated back to 75° C. and a 1M solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20° C./hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
…………………………
FORMULATION
COMPD A IS TRELAGLIPTIN
Examples (Comparative Example IA)
Succinate of compound (A) (26.6 mg) was weighed in a glass bottle and used as Comparative Example IA. (Comparative Example 2A)
The succinate of compound (A) and microcrystalline cellulose were uniformly mixed in a mortar at a ratio of 1:10, and the mixture (226.6 mg) was weighed in a glass bottle and used as Comparative Example 2A. (Comparative Example 3A)
The succinate of compound (A) and corn starch were uniformly mixed in a mortar at a ratio of 1:5, and the mixture (126.6 mg) was weighed in a glass bottle and used as Comparative Example 3A. (Example IA) Succinate of compound (A) , mannitol and corn starch according to the formulation of Table IA were uniformly mixed in a fluid bed granulator (LAB-I, POWREX CORPORATION) , and the mixture was granulated by spraying an aqueous solution of dissolved hypromellose 2910, and dried therein. The obtained granules were passed through a sieve -(16M) to give milled granules. To the milled granules were added croscarmellose sodium, microcrystalline cellulose and magnesium stearate, and they were mixed in a bag to give granules for tableting. The granules were punched by a rotary tableting machine (Correct 19K, Kikusui Seisakusho, Ltd.) with a 6.5 mmφ punch to give a plain tablet weighting 121 mg. On the other hand, titanium oxide, yellow ferric oxide and talc were dispersed in a hypromellose 2910 aqueous solution to prepare a film coating liquid. The aforementioned coating liquid was sprayed onto the above-mentioned plain tablet in a film coating machine (Hicoater HCP-75, Freund Corporation), to give 2500 film- coated tablets containing 3.125 mg of compound (A) (free form) per tablet. Table IA

………………………..
POLYMORPHS AND SYNTHESIS
FORM A
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared. For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days. The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE).
[0091] Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR.
[0092] Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1.
Table 1. Characteristic XRPD Peaks (CuKa) of Form A


Characterization Data of Form A of Compound I

8. Amorphous Form
[0137] The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis.
Table A. Approximate Solubilities of Compound I



Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool;CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation
…………………………
SYNTHESIS
EXAMPLES
1. Preparation of 2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro- 2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile and pharmaceutically acceptable salts


4-Fluoro-2-methylbenzonitrile (3)
[0166] A mixture of 2-bromo-5fluorotoluene ( 2) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was re fluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 3 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H). 2-Bromomethyl-4-fluorobenzonitrile (4)
[0167] A mixture of 4-fluoro-2-methylbenzonitrile (3) (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification.1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro- benzonitrile (6)
[0168] A mixture of crude 3-methyl-6-chlorouracil (5) (0.6 g, 3.8 mmol), 2- Bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO
(10 mL) was stirred at 60 C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=12, 8.4Hz, IH), 7.26 (d, J- 4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for Ci3H9ClFN3O2, 293.68; found 293.68.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, TFA salt (1) (TFA salt of Compound I)

[0169] 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4- fluoro-benzonitrile (5) (300 mg, 1.0 mmol), (i?)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 0C for 2 hrs. The final compound was obtained as a TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, HCl salt

[0170] The TFA salt of Compound I was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0 C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
General procedure for the preparation of salts of Compound I.
[0171] The benzonitrile product may be isolated as the free base if desired, but preferably, the product may be further converted to a corresponding acid addition salt. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of Compound I were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. [0172] The isolation and/or purification steps of the intermediate compounds in the above described process may optionally be avoided if the intermediates from the reaction mixture are obtained as relatively pure compounds and the by-products or impurities of the reaction mixture do not interfere with the subsequent reaction steps. Where feasible, one or more isolation steps may be eliminated to provide shorter processing times, and the elimination of further processing may also afford higher overall reaction yields.
…………………..
TABLET
2. Exemplary formulations comprising succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Provided are examples of tablet formulations that may be used to administer succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (Succinate salt of Compound I) according to the present invention. It is noted that the formulations provided herein may be varied as is known in the art.
The exemplary tablet formulations are as follows:
| 12.5 mg of Compound I (weight of free base form) per tablet | ||||
| Core Tablet Formulation | ||||
| (1) | 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4- | 17.0 | mg | |
| dioxo-3,4-dihydro-2H-pyrimidin-1- | ||||
| ylmethyl]-4-fluoro-benzonitrile (succinate salt) | ||||
| (2) | Lactose Monohydrate, NF, Ph, Eur | 224.6 | mg | |
| (FOREMOST 316 FAST FLO) | ||||
| (3) | Microcrystalline Cellulose, NF, Ph, Eur | 120.1 | mg | |
| (AVICEL PH 102) | ||||
| (4) | Croscarmellose Sodium, NF, Ph, Eur | 32.0 | mg | |
| (AC-DO-SOL) | ||||
| (5) | Colloidal Silicon Dioxide, NF, Ph, Eur | 3.2 | mg | |
| (CAB-O-SIL M-5P) | ||||
| (6) | Magnesium Stearate, NF, Ph, Eur | 3.2 | mg | |
| (MALLINCKRODT, Non-bovine Hyqual) | ||||
| TOTAL | 400.0 | mg | ||
| (per tablet) | ||||
…………..
US20080227798 AND US20120197018
POLYMORPHS AND SYNTHESIS
EXAMPLES Example 1 Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)

Compound I may be prepared by the follow synthetic route (Scheme 1)

A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)

Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)

Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)

Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)

Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile

The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent.
SUCCINATE SALT OF TRELAGLIPTIN
1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.
……………
patents
1. US 2013172377
2. WO 2011013639
3. WO 2009099172
4.WO 2009099171
5. WO 2008114807
6.WO 2008114800
7. WO 2008033851
8. WO 2007074884
9WO 2007035629
patent document 1: US2005/0261271
patent document 2: US2007/0060530
patent document 3: US2008/0287476
patent document 4: US2008/0227798
patent document 5: US2008/0280931
patent document 6: WO2008/114800
patent document 7: WO2011/013639
| US7906523 * | Oct 30, 2007 | Mar 15, 2011 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605 * | Nov 29, 2007 | Dec 27, 2011 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8188275 * | Oct 30, 2007 | May 29, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8222411 * | Sep 15, 2006 | Jul 17, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US20090275750 * | Sep 15, 2006 | Nov 5, 2009 | Jun Feng | Dipeptidyl peptidase inhibitors |
| WO2013183784A1 | Jun 4, 2013 | Dec 12, 2013 | Takeda Pharmaceutical Company Limited | Solid preparation |
| US20080227798 * | Nov 29, 2007 | Sep 18, 2008 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US20120197018 * | Feb 15, 2012 | Aug 2, 2012 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2007033265A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033266A2 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033350A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetes |
| EP1586571A1 * | Dec 21, 2004 | Oct 19, 2005 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
13 NMR TRELAGLIPTIN SUCCINATE

1H NMR TRELAGLIPTIN SUCCINATE

DAPAGLIFLOZIN SEES LIGHT

DAPAGLIFLOZIN, BMS-512148
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
cas 461432-26-8
| Molecular Formula: C21H25ClO6 |
| Molecular Weight: 408.87 |
Bristol-Myers Squibb (Originator)
AstraZeneca
TYPE 2 DIABETES,SGLT-2 Inhibitors
launched 2012, as forxiga in EU

Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
WILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013--
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.

Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
http://online.wsj.com/article/PR-CO-20131212-910828.html?dsk=y
………………………………………………………………..
PATENTS
WO 2010138535
WO 2011060256
WO 2012041898
WO 2012163990
WO 2013068850
WO 2012163546
WO 2013068850
WO 2013079501

Dapagliflozin (INN/USAN,[1] trade name Forxiga) is a drug used to treat type 2 diabetes. It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. Although dapagliflozin’s method of action would operate on both types of diabetes[1] and other conditions resulting inhyperglycemia, the current clinical trials specifically exclude participants with type 1 diabetes.[2][3]
In July 2011 an US Food and Drug Administration (FDA) committee recommended against approval until more data was available.[4] The Prescription Drug User Fee Act (PDUFA) date for dapagliflozin for the treatment of Type 2 diabetes was extended three months by the FDA to January 28, 2012.
In April 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency issued a positive opinion on the drug. It is now marketed in a number of European countries including the UK and Germany.
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[5] The efficacy of the this medication class has yet to be determined, but in initial clinical trials, dapagliflozin lowers HbA1c by 0.90 percentage points when added to metformin.[6]
Type II diabetes is the most common form of diabetes accounting for 90% of diabetes cases. Over 100 million people worldwide have type-2 diabetes (nearly 17 million in the U.S.) and the prevalence is increasing dramatically in both the developed and developing worlds. Type-II diabetes is a lifelong illness, which generally starts in middle age or later part of life, but can start at any age. Patients with type-2 diabetes do not respond properly to insulin, the hormone that normally allows the body to convert blood glucose into energy or store it in cells to be used later. The problem in type-2 diabetes is a condition called insulin resistance where the body produces insulin, in normal or even high amounts, but certain mechanisms prevent insulin from moving glucose into cells. Because the body does not use insulin properly, glucose rises to unsafe levels in the blood, the condition known as hyperglycemia.
Hyperglycemia, that is, elevated plasma glucose, is a hallmark of diabetes. Plasma glucose is normally filtered in the kidney in the glomerulus but is actively reabsorbed in the proximal tubule (kidney). Sodium-dependent glucose co-transporter SGLT2 appears to be the major transporter responsible for the reuptake of glucose at this site. The SGLT inhibitor phlorizin, and closely related analogs, inhibit this reuptake process in diabetic rodents and dogs, resulting in normalization of plasma glucose levels by promoting glucose excretion without hypoglycemic side effects. Long term (6 month) treatment of Zucker diabetic rats with an SGLT2 inhibitor has been reported to improve insulin response to glycemia, improve insulin sensitivity, and delay the onset of nephropathy and neuropathy in these animals, with no detectable pathology in the kidney and no electrolyte imbalance in plasma. Selective inhibition of SGLT2 in diabetic patients would be expected to normalize plasma glucose by enhancing the excretion of glucose in the urine, thereby improving insulin sensitivity and delaying the development of diabetic complications.
The treatment of diabetes is an important health concern and despite a wide range of available therapies, the epidemic continues. Type 2 diabetes (T2DM) is a progressive disease caused by insulin resistance and decreased pancreatic β-cell function. Insulin is produced by the pancreatic β-cell and mediates cellular glucose uptake and clearance. Insulin resistance is characterized by the lack of response to the actions of this hormone which results in decreased cellular clearance of glucose from the circulation and overproduction of glucose by the liver.
The currently available therapies to treat type 2 diabetes augment the action or delivery of insulin to lower blood glucose. However, despite therapy, many patients do not achieve control of their type 2 diabetes. According to the National Health and Nutrition Examination Survey (NHANES) III, only 36% of type 2 diabetics achieve glycemic control defined as a A1C<7.0% with current therapies. In an effort to treat type 2 diabetes, aggressive therapy with multiple pharmacologic agents may be prescribed. The use of insulin plus oral agents has increased from approximately 3 to 11% from NHANES II to III.
Thus, treatment of hyperglycemia in type 2 diabetes (T2DM) remains a major challenge, particularly in patients who require insulin as the disease progresses. Various combinations of insulin with oral anti-diabetic agents (OADs) have been investigated in recent years, and an increasing number of patients have been placed on these regimens. Poulsen, M. K. et al., “The combined effect of triple therapy with rosiglitazone, metformin, and insulin in type 2 diabetic patients”,Diabetes Care, 26 (12):3273-3279 (2003); Buse, J., “Combining insulin and oral agents”, Am. J. Med., 108 (Supp. 6a):23S-32S (2000). Often, these combination therapies become less effective in controlling hyperglycemia over time, particularly as weight gain and worsening insulin resistance impair insulin response pathways.
Hypoglycemia, weight gain, and subsequent increased insulin resistance are significant factors that limit optimal titration and effectiveness of insulin. (Holman, R. R. et al., “Addition of biphasic, prandial, or basal insulin to oral therapy in type 2 diabetes”, N. Engl. J. Med., 357 (17):1716-1730 (2007)). Weight gain with insulin therapy is predominantly a consequence of the reduction of glucosuria, and is thought to be proportional to the correction of glycemia. (Makimattila, S. et al., “Causes of weight gain during insulin therapy with and without metformin in patients with Type II diabetes mellitus”, Diabetologia, 42 (4):406-412 (1999)). Insulin drives weight gain when used alone or with OADs. (Buse, J., supra). In some cases, intensive insulin therapy may worsen lipid overload and complicate progression of the disease through a spiral of caloric surplus, hyperinsulinemia, increased lipogenesis, increased adipocity, increased insulin resistance, beta-cell toxicity, and hyperglycemia. (Unger, R. H., “Reinventing type 2 diabetes: pathogenesis, treatment, and prevention”, JAMA, 299 (10):1185-1187 (2008)). Among commonly used OADs, thiazolidinediones (TZDs) and sulfonylureas intrinsically contribute to weight gain as glucosuria dissipates with improved glycemic control. Weight gain is less prominent with metformin, acting through suppression of hepatic glucose output, or with incretin-based DPP-4 inhibitors. Overall, there is a pressing need for novel agents that can be safely added to insulin-dependent therapies to help achieve glycemic targets without increasing the risks of weight gain or hypoglycemia.
A novel approach to treating hyperglycemia involves targeting transporters for glucose reabsorption in the kidney. (Kanai, Y. et al., “The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose”, J. Clin. Invest., 93 (1):397-404 (1994)). Agents that selectively block the sodium-glucose cotransporter 2 (SGLT2) located in the proximal tubule of the kidney can inhibit reabsorption of glucose and induce its elimination through urinary excretion. (Brown, G. K., “Glucose transporters: structure, function and consequences of deficiency”, J. Inherit. Metab. Dis., 23 (3):237-246 (2000)). SGLT2 inhibition has been shown in pre-clinical models to lower blood glucose independently of insulin. (Han, S. et al., “Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats”, Diabetes, 57 (6):1723-1729 (2008); Katsuno, K. et al., “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”, J. Pharmacol. Exp. Ther., 320 (1):323-330 (2007)).
Dapagliflozin(BMS-512148) is a potent sodium-glucose transport proteins inhibitor with IC50 of 1.1 nM and 1.4uM for SGLT2 and SGLT1, respectively. Dapagliflozin (BMS-512148) inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine. Symptoms of hypoglycaemia occurred in similar proportions of patients in the dapagliflozin (2~4%) and placebo groups (3%). Signs, symptoms, and other reports suggestive of genital infections were more frequent in the dapagliflozin groups (2•5 mg, [8%]; 5 mg, [13%]; 10 mg, [9%]) than in the placebo group ( [5%]).
Dapagliflozin (which is disclosed in U.S. Pat. No. 6,515,117) is an inhibitor of sodium-glucose reabsorption by the kidney, by inhibiting SGLT2, which results in an increased excretion of glucose in the urine. This effect lowers plasma glucose in an insulin-independent manner.
Dapagliflozin is currently undergoing clinical development for treatment of type 2 diabetes. (Han, S. et al., supra; Meng, W. et al., “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes”, J. Med. Chem., 51 (5):1145-1149 (2008)). Phase 2a and 2b studies with dapagliflozin have demonstrated efficacy in reducing hyperglycemia either alone or in combination with metformin in patients with T2DM. (Komoroski, B. et al., “Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus”, Clin. Pharmacol. Ther., 85 (5):513-519 (2009); List, J. F. et al., “Dapagliflozin-induced glucosuria is accompanied by weight loss in type 2 diabetic patients”, 68th Scientific Sessions of the American Diabetes Association, San Francisco, Calif., Jun. 6-10, 2008, Presentation No. 0461P).
It has been found that dapagliflozin does not act through insulin signaling pathways and is effective in controlling blood sugar in patients whose insulin signaling pathways do not work well. This applies to extremes of insulin resistance, in type 2 diabetes as well as in insulin resistance syndromes, caused by, for example, mutations in the insulin receptor.
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Dapagliflozin is also associated with hypotensive reactions.

The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7]
- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- “FDA panel advises against approval of dapagliflozin”. 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ; Hecheng Huaxue , 18 (3), 389-392; 2010
5) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,. WO2010022313
6) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,. WO2010009243
7) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,. WO2009026537
8) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ; Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;
9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,. WO2008002824
10) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;.. U.S. Pat Appl Publ,. 20,040,138,439

dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .

formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2

formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
…..
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
http://www.google.com/patents/US20120282336
http://www.tga.gov.au/pdf/auspar/auspar-dapagliflozin-propanediol-monohydrate-130114.pdf
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
………………………………
In Vitro Characterization and Pharmacokinetics of Dapagliflozin …
dmd.aspetjournals.org/content/…/DMD29165_supplemental_data_.doc

Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)
-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33
(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),
6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,
1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,
140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,
64.5, 63.1, 39.2, 15.2.
HRMS calculated for C21H25ClNaO6 (M+Na)+
For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
: 431.1237; found 431.1234. Anal. Calcd
SECOND SET
1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);
13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
………………………
HPLC
-
HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
– Eluent A: 0.1 % formic acid in water
– Eluent B: 0.1 % formic acid in acetonitrile
– Injection volume: 3 mL
– Flow: 0.7 mL/min
– Gradient:Time [min] [%] B 0.0 25 25.0 65 26.0 70 29.0 70 29.5 25 35.0 25 ……………………..

……..
http://www.google.com/patents/WO2013068850A2?cl=en
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))

[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.56-7.54 (m, 2H), 7.43-7.31 (m, 13H), 7.29-7.22 (m, 6H), 7.07- 7.04 (m, 2H), 7.00 (d, J= 2.0 Hz, IH), 6.87 (dd, J= 8.4, 2.0 Hz, IH), 6.83-6.81 (m, 2H), 4.18 (d, J= 9.6 Hz, IH), 4.02 (q, J= 6.9 Hz, 2H), 3.96 (d, J= 10.8 Hz, 2H), 3.86 (ddd, J= 11.3, 7.7, 1.1 Hz, IH), 3.76 (ddd, J= 8.4, 8.4, 4.8 Hz, IH), 3.56 (ddd, J= 9.0, 6.4, 2.4 Hz, IH), 3.50 (dd, J=
I I.4, 5.4 Hz, IH), 3.44 (dd, J= 9.4, 8.6 Hz, IH), 3.38 (dd, J= 8.8, 8.8 Hz, IH), 1.70 (dd, J= 7.8, 5.4 Hz, IH, OH), 1.42 (t, J= 6.8 Hz, 3H), 1.21 (d, J= 5.2 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 157.4 (C), 138.8 (C), 137.4 (C), 136.3 (CH x2), 136.1 (CH x2), 135.2 (CH x2), 135.0 (C), 134.9 (CH x2), 134.8 (C), 134.2 (C), 132.8 (C), 132.0 (C), 131.6 (CH), 131.1 (C), 129.9 (CH x2), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.58 (CH x2), 127.57 (CH x2), 127.54 (CH x2), 127.31 (CH), 127.28 (CH x2), 114.4 (CH x2), 82.2 (CH), 80.5 (CH), 79.3 (CH), 76.3 (CH), 72.7 (CH), 63.4 (CH2), 62.7 (CH2), 38.2 (CH2), 27.2 (CH3 x3), 26.6 (CH3 x3), 19.6 (C), 19.2 (C), 14.9 (CH3). EXAMPLE 25 -Synthesis of dapagliflozin ((25,3R,4R,55,6/?)-2-[4-chloro-3-(4- ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; (Ij))

IVj’ U
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
1H NMR (400 MHz, CD3OD) δ 7.37-7.34 (m, 2H), 7.29 (dd, J= 8.2, 2.2 Hz, 1H), 7.12-7.10 (m, 2H), 6.82-6.80 (m, 2H), 4.10 (d, J= 9.6 Hz, 2H), 4.04 (d, J= 9.2 Hz, 2H), 4.00 (q, J= 7.1 Hz, 2H), 3.91-3.87 (m, 1H), 3.73-3.67(m, 1H), 3.47-3.40 (m, 3H), 3.31-3.23 (m, 2H), 1.37 (t, J= 7.0 Hz, 3H);
13C NMR (100 MHz, CD3OD) δ 157.4 (C), 138.6 (C), 138.5 (C), 133.1 (C), 131.5 (C), 130.5 (CH), 129.4 (CH x2), 128.7 (CH), 126.8 (CH), 114.0 (CH x2), 80.5 (CH), 80.8 (CH), 78.3 (CH), 75.0 (CH), 70.4 (CH), 63.0 (CH2), 61.7 (CH2), 37.8 (CH2), 13.8 (CH3);
LCMS (ESI) m/z 426 (100, [M+NH4]+), 428 (36, [M+NH4+2]+), 447 (33, [M+K]+).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)

III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- “D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
Pramlintide
Pramlintide (Symlin), a synthetic version of amylin, is a 37-amino acid peptide that is co-secreted with insulin by pancreatic β-cells. It was developed and approved in 2005 by the FDA for use in US patients with type I and II diabetes in conjunction with the administration of prandial insulin to improve postprandial glycemic control
 |
|---|
Pramlintide (Symlin) is a relatively new adjunct for diabetes (both type 1 and 2), developed by Amylin Pharmaceuticals (now a wholly owned subsidiary of Bristol Myers-Squibb). Pramlintide is delivered as an acetate salt.
Pramlintide is an analogue of amylin, a small peptide hormone that is released into the bloodstream by the β-cells of the pancreas along with insulin, after a meal.[1] Like insulin, amylin is completely absent in individuals with Type I diabetes.[2]
Reduction in glycated hemoglobin and weight loss have been shown in insulin-treated patients with type 2 diabetes taking pramlintide as an adjunctive therapy.[3]
By augmenting endogenous amylin, pramlintide aids in the absorption of glucose by slowing gastric emptying, promoting satiety via hypothalamic receptors (different receptors than for GLP-1), and inhibiting inappropriate secretion of glucagon, a catabolic hormone that opposes the effects of insulin and amylin. Pramlintide also has effects in raising the acute first-phase insulin response threshold following a meal.
Pramlintide has been approved by the FDA, for use by Type 1 and Type 2 Diabetics who use insulin.[4]Pramlintide allows patients to use less insulin, lowers average blood sugar levels, and substantially reduces what otherwise would be a large unhealthy rise in blood sugar that occurs in diabetics right after eating. Apart from insulin analogs, pramlintide is the only drug approved by the FDA to lower blood sugar in type 1 diabetics since insulin in the early 1920s.
Design and structure
Since native human amylin is highly amyloidogenic and potentially toxic, the strategy for designing pramlintide was to substitute residues from rat amylin, which is not amyloidogenic (but would presumably retain clinical activity). Proline residues are known to be structure-breaking residues, so these were directly grafted into the human sequence.
Amino acid sequences:
Pramlintide: KCNTATCATQRLANFLVHSSNNFGPILPPTNVGSNTY-(NH2)
Amylin: KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY-(NH2)
Rat amylin: KCNTATCATQRLANFLVRSSNNLGPVLPPTNVGSNTY-(NH2)
Pramlintide as protein is (positively charged).

- Jones MC (2007). “Therapies for diabetes: pramlintide and exenatide” (pdf). American Family Physician 75 (12): 1831–5. PMID 17619527.
- Edelman, Steve; Maier, Holly; Wilhelm, Ken (2008). “Pramlintide in the Treatment of Diabetes Mellitus”.BioDrugs 22 (6): 375–386. doi:10.2165/0063030-200822060-00004. ISSN 1173-8804.
- Hollander, Priscilla; Maggs, David G.; Ruggles, James A.; Fineman, Mark; Shen, Larry; Kolterman, Orville G.; Weyer, Christian (2004). “Effect of Pramlintide on Weight in Overweight and Obese Insulin-Treated Type 2 Diabetes Patients” (pdf). Obesity 12 (4): 661–668. doi:10.1038/oby.2004.76.ISSN 1930-7381.
- Ryan GJ, Jobe LJ, Martin R (2005). “Pramlintide in the treatment of type 1 and type 2 diabetes mellitus”. Clinical therapeutics 27 (10): 1500–12. doi:10.1016/j.clinthera.2005.10.009. PMID 16330288.

- www.symlin.com – product website
- www.amylin.com – Symlin page on the Amylin Pharmaceuticals website
| Pramlintide Acetate | |
| Pramlintide acetate is a relatively new adjunct treatment for diabetes (both type 1 and 2). |
Pramlintide Acetate, 196078-30-5,
| Synonym | Pramlintide Acetate,Pramlintide acetate hydrate |
| Molecular Formula | C171H267N51O53S2.X(C2H4O2).X(H2O) |
| Molecular Weight | 3949.39 |

LY2189265 (dulaglutide), a glucagon-like peptide-1 analog as once-weekly treatment for type 2 diabetes.

DULAGLUTIDE
PRONUNCIATION doo” la gloo’ tide
THERAPEUTIC CLAIM Treatment of type II diabetes
CHEMICAL NAMES
1. 7-37-Glucagon-like peptide I [8-glycine,22-glutamic acid,36-glycine] (synthetic
human) fusion protein with peptide (synthetic 16-amino acid linker) fusion protein with immunoglobulin G4 (synthetic human Fc fragment), dimer
2. [Gly8,Glu22,Gly36]human glucagon-like peptide 1-(7-37)-peptidyltetraglycyl-Lseryltetraglycyl-L-seryltetraglycyl-L-seryl-L-alanyldes-Lys229-[Pro10,Ala16,Ala17]human immunoglobulin heavy constant γ4 chain H-CH2-CH3 fragment, (55-55′:58-58′)-bisdisulfide dimer
STRUCTURAL FORMULA
Monomer
HGEGTFTSDV SSYLEEQAAK EFIAWLVKGG GGGGGSGGGG SGGGGSAESK 50
YGPPCPPCPA PEAAGGPSVF LFPPKPKDTL MISRTPEVTC VVVDVSQEDP 100
EVQFNWYVDG VEVHNAKTKP REEQFNSTYR VVSVLTVLHQ DWLNGKEYKC 150
KVSNKGLPSS IEKTISKAKG QPREPQVYTL PPSQEEMTKN QVSLTCLVKG 200
FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSRLT VDKSRWQEGN 250
VFSCSVMHEA LHNHYTQKSL SLSLG 275
Disulfide bridges location
55-55′ 58-58′ 90-150 90′-150′ 196-254 196′-254′
MOLECULAR FORMULA C2646H4044N704O836S18
MOLECULAR WEIGHT 59.67 kDa
MANUFACTURER Eli Lilly and Company
CODE DESIGNATION LY2189265
CAS REGISTRY NUMBER 923950-08-7
http://www.ama-assn.org/resources/doc/usan/dulaglutide.pdf
LY2189265 (dulaglutide), a glucagon-like peptide-1 analog, is a biologic entity being studied as a once-weekly treatment for type 2 diabetes.
Dulaglatuide works by stimulating cells to release insulin only when blood sugar levels are high.
Gwen Krivi, Ph.D., vice president, product development, Lilly Diabetes, said of the drug, “We believe dulaglutide, if approved, can bring significant benefits to people with type 2 diabetes.”
In fact, it might help to control both diabetics’ blood sugar and their high blood pressure.
Eli Lilly CEO John Lechleiter believes the drug has the potential to be a blockbuster. Lilly could be ready to seek approval by 2013.
For more information on dulaglutide clinical studies, click here.
PRESS RELEASES
Data Preseted at 49th EASD Annual Meeting Show Treatment with Lilly’s Investigational Dulaglutide Resulted in Improved Patient-Reported Health Outcomes – September 26, 2013
Lilly Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – April 16, 2013
Lilly Diabetes Announces Positive Results of Phase III Trials of Dulaglutide in Type 2 Diabetes – October 22, 2012
GLIPTINS: BETTER APPROACH FOR TYPE 2 DIABETES
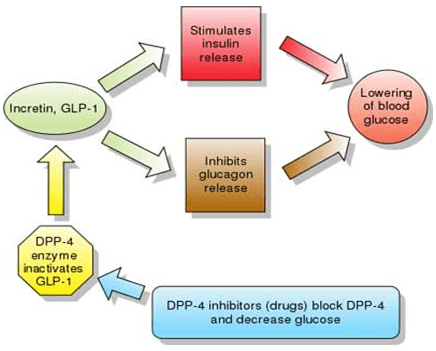
GLIPTINS: BETTER APPROACH FOR TYPE 2 DIABETES
Diabetes Mellitus is a metabolic disorder which results from defects in insulin secretion, insulin action, or both, further characterized by hyperglycemia, and causes long term damage and failure of various organs. It is estimated that 366 million people had Diabetes Mellitus in 2011; by 2030 this would have risen to 552 million. Many oral hypoglycaemic agents are…
read more: http://www.pharmatutor.org/articles/gliptins-better-approach-type-2-diabetes
Type 2 diabetic patients treated with DPP-4, Linagliptin experience reductions in blood glucose levels
linagliptin
C25H28N8O2
CAS : 668270-12-0
Molecular Weight: 472.54
Purity: > 98%
(R)-8-(3-aminopiperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-1H-purine-2,6(3H,7H)-dione
8-(3R)-3-aminopiperidinyl)-7-butyn-2-yl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione
Solubility: Up to 25 mM in DMSO
Synonyms: BI-1356, BI1356, Linagliptin, Tradjenta, Trajenta
BI-1356 (Linagliptin) is a highly potent and selective dipeptidyl peptidase 4 (DPP-4) inhibitor (IC50 = 1 nM) for treatment of type II diabetes. [1] BI-1356 can increase incretin levels (GLP-1 and GIP), which increases insulin secretion and inhibits glucagon release, decreases gastric emptying, and decreases blood glucose levels. BI-1356 shows 10,000-fold more selectivity for DPP-4 against other protease/peptidases, including DPP-8, DPP-9, trypsin, plasmin, and thrombin, It is a DPP-4 inhibitor developed by Boehringer Ingelheim for the treatment of type II diabetes.
Linagliptin is a highly potent, selective DPP-4 inhibitor with IC50 of 1 nM.
“This study provides much-needed data on glucose-lowering treatment of elderly people with Type 2 Diabetes, inadequately controlled with common anti-hyperglycaemic agents”

Data published in The Lancet showed that elderly people with Type 2 Diabetes (T2D) treated for 24 weeks with the dipeptidyl peptidase-4 (DPP-4) inhibitor linagliptin, marketed by Boehringer Ingelheim and Eli Lilly and Company, experienced significant reductions in blood glucose levels (HbA1c) compared with those receiving placebo. In addition, the overall safety and tolerability profile of linagliptin was similar to placebo, with no significant difference in hypoglycaemia
INTRODUCTION
Linagliptin (BI-1356, trade names Tradjenta and Trajenta) is a DPP-4 inhibitor developed by Boehringer Ingelheim for treatment of type II diabetes.
Linagliptin (once-daily) was approved by the US FDA on 2 May 2011 for treatment of type II diabetes.[1] It is being marketed by Boehringer Ingelheim and Lilly.
-
Linagliptin, namely 8-(3R)-3-aminopiperidinyl)-7-butyn-2-yl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione, of formula (A), is a long acting inhibitor of dipeptidylpeptidase-IV (DPP-IV) activity, at present under development for the treatment of type II diabetes mellitus.

-
The synthesis of Linagliptin is reported in US 7,407,955 , according to the scheme below, where 8-bromo xanthine of formula (B) is condensed with 3-(R)-Boc-aminopiperidine of formula (C) to obtain a compound of formula (D), which is converted to Linagliptin (A) by deprotection of the amine function

-
Optically active 3-aminopiperidine protected as the tert-butylcarbamate (Boc), compound (C), although commercially available, is very expensive and difficult to prepare; moreover in this process impurities are very difficult to remove, particularly on an industrial scale, in particular because of the Boc protective group. For this reason,US 2009/0192314 discloses a novel process for the preparation of Linagliptin (A) which makes use of a 3-(R)-aminopiperidine protected as a phthalimide of formula (E).

-
Accordingly, a compound of formula (E) can be prepared starting from 3-aminopyridine by hydrogenation, reaction with phthalic anhydride, resolution through diastereoisomeric salts using expensive D-tartaric acid, and then cleavage of the tartrate salt.
-
This intermediate is, however, still expensive and its use in the substitution reaction of the bromine derivative of formula (B) is still poorly efficient, as it takes place under drastic reaction conditions.
-
As it can be noted, these processes make use of drastic reaction conditions, or expensive, difficult to prepare starting materials, thus negatively affecting costs. There is therefore the need for an alternative synthetic route to provide Linagliptin or a salt thereof with high enantiomeric and chemical purity, from low cost starting materials.



US ‘955 is schematically represented in scheme

U.S. Patent No. 7,820,815 (“US ‘815) discloses a process for preparation of Linagliptin wherein it is prepared by deprotecting 1 -[(4-methyl-quinazolin-2-yl)methyl]-3- methyl-7-(2-butyn-1 -yl)-8-(3-(R)-phthalimidopiperidin-1 -yl)-xanthine of formula Ilia in presence of ethanolamine. The 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2- butyn-1 -yl)-8-(3-(R)phthalimidopiperidin-1 -yl)-xanthine is prepared by condensing 1 -[(4- l methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromo xanthine of formula III with (R)-3-phthalimidopiperidine of formula I la. The process disclosed in US ‘815 is schematically represented in scheme-ll.

Scherre
PCT Publications WO 2004/018468 and WO 2006/048427 describe synthesis of Linagliptin. Crystalline forms of Linagliptin, Forms A, B, C, D, and E are described in the PCT Publication No. WO 2007/128721. According to WO 2007/128721, Linagliptin prepared according to Publication No.
WO 2004/018468 is present in ambient temperature as a mixture of two enantiotropic polymorphs. The temperature at which the two polymorphs transform into one another is 25±15° C. The pure high temperature form (polymorph A), can be obtained by heating the mixture to temperatures>40° C. The low temperature form (polymorph B) is obtained by cooling to temperatures<10° C.”.
According to WO 2007/128721, the transition point between forms A and B is at room temperature, such that they exist as a polymorphic mixture. In addition, WO 2007/128721 teaches that form D “is obtained if polymorph C is heated to a temperature of 30-100° C. or dried at this temperature”. Since the procedure to obtain form C according to this application includes drying at 70° C., the dried form C is expected to be obtained in admixture with form D.
WO 2007/128721 teaches that Form E is obtained only at high temperatures (after melting of form D at 150±3° C.), and therefore is not relevant industrially.
PATENT







Example 1: Preparation of a compound of formula (II) with X=OEt
-
The bromoxanthine of formula (B) prepared according to US 7,407, 955 (28.2 g, NMR title 90%, 56.0 mmols) and L-(+)-tartrate salt of (R)-ethylnipecotate (22.4 g, 72.8 mmols) are suspended in 50 mL of 1-methyl-2-pyrrolidone. The suspension is heated at 100° under stirring and, maintaining such temperature, diisopropylethylamine (38.3 ml, 224 mmols) is slowly dropwise added. The suspension is moderately refluxed for 2 hours. The mixture is cooled to 30°C and 400 mL of are dropwise added under vigorous stirring. The suspension is stirred for 30 minutes, then filtered off and the solid is washed with 100 mL of water. 27 g of solid product are obtained after drying with a 90% yield.
-
1H-NMR (300 MHz, CDCl3), δ 8.02 (d, 1H), 7.87 (d, 1H), 7.76 (t, 1H), 7.51 (t, 1H), 5.55 (s, 2H), 4.90 (s, 2H), 4.25 – 4.10 (m, 2H), 3.82 (dd, 1H), 3.65 – 3.51 (m, 4H), 3.33 (dd, 1H), 3.15 (m, 1H), 2.88 – 2.72 (m, 4H), 2.08 (m, 1H), 1.92 – 1.73 (m, 6H), 1.27 (t, 3H).
Example 2: Preparation of a compound of formula (II) with X=OH
-
The compound of formula (II) having X = OEt, prepared according to Example 1 (27 g, 51 mmols), is suspended in 270 mL of MeOH and 4.1 g of NaOH scales and 13.7 mL of water are added under stirring. The reaction mixture is maintained under stirring for 2 hours at reflux temperature and then cooled to 40°C and diluted with 400 ml of water.
-
[0080]The mixture is then acidified by adding 6.6 mL of acetic acid and the solid is filtered off and washed with water and dried under vacuum at 50°C, obtaining 21 g of product, with a yield of 82%.
-
1H-NMR (300 MHz, DMSO-d6), δ 8.11 (d, 1H), 7.85 (t, 1H), 7.80 (d, 1H), 7.62 (t, 1H), 5.30 (s, 2H), 4.87 (s, 2H), 3.79 (dd, 1H), 3.57 (m, 1H), 3.38 (s, 3H), 3.33 (dd, 1H), 3.10 (m, 1H), 2.85 (s, 3H), 2.62 (m, 1H), 1.95 (m, 1H), 1.78 – 1.60 (m, 6H).
Example 3: Preparation of a compound of formula (IV) with R = OCH(CH3)2
-
The compound of formula (II) with X=OH prepared according to Example 2 (0.5 g; 1 mmol), 5 ml of isopropanol and trietylamine (0.17 ml, 1.2 mmols) are mixed under stirring. 0.3 g of diphenylphosphorylazide (DPPA) are added in a sole portion. The mixture is heated at reflux temperature for 2 hours under stirring. The mixture is then cooled to room temperature and the solid is filtered off and washed with 2 ml of isopropyl alcohol. The solid is dried under vacuum at 50°C obtaining 0.4 g of product with a yield of 72%.
-
1H-NMR (300 MHz, DMSO-d6), δ 8.12 (d, 1H), 7.85 (t, 1H), 7.80 (d, 1H), 7.63 (t, 1H), 5.28 (s, 2H), 4.85 (s, 2H), 4.75 (ep, 1H), 4.27 (d, 1H), 3.78-3.55 (m, 4H), 3.35 (s, 3H), 2.85 (s, 3H), 1.85 – 1.60 (m, 6H). 1.42 (m, 1H), 1.02 (d, 6H).
Example 4: Preparation of Linagliptin
-
The carbamate of formula (IV), prepared according to Example 3 (400 mg, 0.72 mmols), is dissolved in 5 ml of 32% HCl in water. The reaction mixture is maintained under stirring at 65-70°C for 7 hours and then cooled to room temperature. The pH of the solution is brought to about 8-9 by treatment with 30% NaOH in water and the obtained suspension is stirred for 10 minutes and then filtered off. The solid is dissolved in 10 ml of AcOEt, the solution is filtered and the filtrate is evaporated under reduced pressure. 250 mg of Linagliptin are obtained with a yield of 73%.
Example 5: Preparation of a compound of formula (IV) with R = S(CH2)11CH3
-
The compound of formula (II) with X =OH, prepared according to Example 2 (3.0 g, 6 mmols), 30 ml of acetonitrile and triethylamine (1.09 ml, 7.8 mmols) are mixed together. Subsequently, 1.55 ml (7.2 mmols) of diphenylphosphorylazide (DPPA) are added. The reaction mixture is heated at reflux temperature for 1 hour under stirring and then cooled to 60°C and treated with dodecanethiol (1.87 ml, 7.8 mmols). The mixture is maintained under stirring at the same temperature for 30 minutes and then cooled to 25°C. The formed solid is filtered off and washed with 10 ml of acetonitrile. The solid is dried under vacuum at 60°C obtaining 3.5 g of product with a yield of 85%.
-
1H-NMR (300 MHz, DMSO-d6), δ 8.21 (d, 1H), 7.88 (t, 1H), 7.83 (d, 1H), 7.64 (t, 1H), 5.30 (s, 2H), 4.86 (s, 2H), 3.85 (m, 1H), 3.70 (d, 1H), 3.56 (d, 1H), 3.38 (s, 3H), 3.10-2.87 (m, 3H), 2.85 (s, 3H), 2.74 (t, 2H), 1.90-1.60 (m, 3H), 1.74 (s, 3H), 1.60-1.40 (m, 2H), 1.38-1.10 (m, 18H), 0.82 (t, 3H).
Example 6: Preparation of Linagliptin
-
The thiocarbamate of formula (IV) (10 g, 14,3 mmols), prepared according to Example 5, is dissolved in 100 mL of N-methylpyrrolidone (NMP) and treated with a 30% NaOH solution (7.6 g, 57.0 mmols). The reaction mixture is stirred for 3 hours and then diluted with water and acidified by adding concentrated H2SO4. The mixture is extracted with hexane and brought to pH 9.5 by adding 30% NaOH and repeatedly extracted with dichloromethane. The dichloromethane phases are collected and washed with water and then dried over Na2SO4, filtered and concentrated under reduced pressure. The so obtained oily residue is then dissolved in methyl tert-butyl ether (MTBE) and the mixture is maintained under stirring for 2 hours, then cooled to 0-5°C and the so obtained solid is filtered off, washed with MTBE and dried under vacuum at 50°C till constant weight. 4.2 g of Linagliptin with a yield of 63% are obtained.
Example 7: Preparation of a compound of formula (IV) with R=C7H5N2S (2-mercaptobenzoimidazole)
-
The compound of formula (II) with X =OH, prepared according to Example 2 (2.0 g, 4 mmols), 20 ml of acetonitrile and triethylamine (0.8 ml, 5.6 mmols) are mixed together. Subsequently, 1.43 g (5.2 mmols) of diphenylphosphorylazide (DPPA) are added. The reaction mixture is heated at reflux temperature for 1 hour under stirring and then cooled to 60°C and treated with 2-marcaptobenzimidazole (0.8 g, 5.2 mmols). The mixture is maintained under stirring at the same temperature for 30 minutes, then cooled to 25°C and evaporated under reduced pressure with Rotavapor®. The residue is treated with 50 ml of dichloromethane (CH2Cl2) and washed with 2X20 ml of 5% NaOH. The organic phase is dried over Na2SO4, filtered and concentrated under reduced pressure and the residue is triturated with 30 ml of MTBE. The so obtained solid is filtered off, dried under vacuum at 60°C till constant weight obtaining 2.5 g of light brown powder.
Example 8: Preparation of Linagliptin
-
Starting from the compound of formula (IV) as obtained in example 7 and following the procedure of example 6, product Linagliptin is obtained.
PAPER
DOI: 10.1039/C5OB01111F
http://pubs.rsc.org/en/content/articlelanding/2015/ob/c5ob01111f#!divAbstract
By employing a rhodium–Duanphos complex as the catalyst, β-alkyl (Z)-N-acetyldehydroamino esters were smoothly hydrogenated in a highly efficient and enantioselective way. Excellent enantioselectivities together with excellent yields were achieved for a series of substrates. An efficient approach for the synthesis of the intermediate of the orally administered anti-diabetic drugs Alogliptin and Linagliptin in the DPP-4 inhibitor class was also developed.


Mechanism of action
Linagliptin is an inhibitor of DPP-4, an enzyme that degrades the incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). Both GLP-1 and GIP increase insulin biosynthesis and secretion from pancreatic beta cells in the presence of normal and elevated blood glucose levels. GLP-1 also reduces glucagon secretion from pancreatic alpha cells, resulting in a reduction in hepatic glucose output. Thus, linagliptin stimulates the release of insulin in a glucose-dependent manner and decreases the levels of glucagon in the circulation.
PAPER

PATENT
http://www.google.com/patents/WO2013098775A1?cl=en
In one aspect, the application provides a process for preparation of Linagliptin comprising reacting (R)-piperidine-3-amine of formula II or an acid addition salt thereof with 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine of formula III in the presence of a suitable base in an inert organic solvent.

In another aspect, the application provides Linagliptin or a pharmaceutically acceptable salt thereof, having less than about 0.15 area % of potential process related impurities viz., regio-impurity of the formula la, bromo-impurity of the formula lb and S- isomer as measured by HPLC.

L nag pt n S- somer
Example 1 : Preparation of Linagliptin
a) Preparation of 3-methyl-7-(2-butyn-l-yl)-8-bromo-xanthine (compound of formula IV)
3-Methyl-8-bromo-xanthine (30 gm) and N,N-dimethylformamide (170 ml_) were charged into a 1000 ml_ round bottomed flask equipped with a mechanical stirrer. Diisopropylethylamine (DIPEA, 1 5.9 gm) and 1 -bromo-2-butyne (16.2 gm) were added at 30°C. The reaction mixture was heated to 85 °C and maintained the temperature for 4 hours. The reaction mixture was cooled to 30°C and pre cooled water (300 ml_) was added. The solid formed was collected by filtration and washed with pre cooled water (150 ml_) and diethyl ether (30 ml_). The solid was dried in oven under vacuum at 50°C to get 30.9 gm of the title compound.
(b) Preparation of 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8- bromoxanthine (compound of formula III) 3-Methyl-7-(2-butyn-l-yl)-8-bromo-xanthine (10 gm) and Ν,Ν-dimethylacetamide (150 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (9.3 gm) and 2-(chloromethyl)-4- methylquinazoline (6.8 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 90 °C and maintained the temperature for 8 hours. The reaction mixture was cooled to 30°C and water (450 mL) was added and the mixture was stirred for 1 hour at 30°C. The solid formed was collected by filtration and washed with water (150 mL). The wet cake was charged into 500 mL round bottomed flask and toluene (220 mL) was added and the mixture was heated to reflux temperature and maintained for 1 hour. The mixture was cooled to 10°C and maintained for 2 hours. The solid was collected by filtration and washed with toluene (50 mL). The solid was dried in oven under vacuum at 80°C to get 10.8 gm of the title compound. Purity by HPLC: 99.59%
(c) Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (5 gm) and Ν,Ν-dimethylformamide (DMF, 50 mL) were charged into a 500 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (4.57 gm) and (R)-piperidine-3-amine dihydrochloride (2.86 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 80 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to room temperature and DMF was evaporated under vacuum, then dichloromethane (DCM, 50 mL) was added, and stirred for 15 minutes. The reaction mixture was filtered to separate out the non- dissolved material and the non-dissolved material was washed with 15 mL of dichloromethane. The dichloromethane was evaporated under vacuum to give 4 gm of crude Linagliptin.
Example 2: One pot process for preparation of Linagliptin
3-Methyl-8-bromo-xanthine (5 gm) and Ν,Ν-dimethylformamide (DMF, 28.5 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Diisopropylethylamine (DIPEA, 2.6 gm) and 1 -bromo-2-butyne (2.7 gm) were added at 30 °C. The reaction mixture was heated to 85 °C and maintained at this temperature for 4 hours. The reaction mixture is cooled to 30°C and Ν,Ν-dimethylformamide (DMF, 100 ml_) was added. Potassium carbonate (4.4 gm) and 2-(chloromethyl)-4- methylquinazoline (4.2 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 85 °C and maintained at this temperature for 4 hours. The reaction mixture was cooled to 30°C and Ν,Ν-dimethylformamide (DMF, 90 ml_) was added. Potassium carbonate (8.3 gm) and (R)-piperidine-3-amine dihydrochloride (5.2 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 80 °C and maintained at this temperature for 8 hours. The reaction mixture was cooled to 30 °C and DMF was evaporated under vacuum. Dichloromethane (DCM, 30 ml_) was added and stirred for 15 minutes. The reaction mixture was filtered to separate out the undissolved material and the undissolved material was washed with dichloromethane (30 ml_). The dichloromethane was evaporated under vacuum and 10% acetic acid (100 ml_) was added. The resulted solution was stirred for 30 minutes and washed with dichloromethane (25 ml_x3). The pH of the aqueous layer was adjusted to 8.5 with 10% aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (25 ml_x2) and the dichloromethane was evaporated under vacuum to get 1 .2 gm of Linagliptin.
Example 3: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 ml_) were charged into a 1000 ml_ round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine dihydrochloride (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95°C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 ml_). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at room temperature. The aqueous layer was separated and washed with 60 ml_ of dichloromethane. The aqueous layer was charged into another flask and 200 ml_ of dichloromethane and 100 ml_ of aqueous sodium hydroxide solution was added drop-wise at 30 °C. The mixture was stirred for one hour at 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45°C. Isopropyl alcohol (100 mL) was added to the residue and stirred for 3 hours at room temperature. Filtered the compound and washed with isopropyl alcohol (20 mL) and dried the compound at below 60 °C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 99.0%
Example 4: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 95 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to room temperature and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at room temperature. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at room temperature. The mixture was stirred for one hour at room temperature and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Hexane (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with Hexane (40 mL) and dried the compound at below 60°C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 98.92%
Example 5: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95°C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at 30 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at 30°C. The mixture was stirred for one hour at 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Toluene (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with Toluene (40 mL) and dried the compound at below 60 °C under vacuum to give 16.8 gm of Linagliptin. Purity: 98.91 %, PXRD pattern: Fig. 2.
Example 6: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at 30 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at room temperature (pH is > 10). The mixture was stirred for one hour 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Ethyl acetate (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with ethyl acetate (40 mL) and dried the compound at below 60 °C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 98.72%
Example 7: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (4 gm) and methyl isobutyl ketone (MIBK 100 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (3.7 gm) and (R)-piperidine-3-amine dibenzoyl-D-tartrate (6.1 gm) were added to the reaction mixture at 26°C. The reaction mixture was heated to 100°C and maintained at that temperature for 6 hours. The reaction mixture was cooled to 30 °C and filtered, and the salt was washed with MIBK (8 mL). The filtrate was charged into another flask and added slowly 10% aqueous acetic acid solution (40 mL) and stirred for one hour at 26°C. The aqueous layer was separated and washed with 12 mL of dichloromethane. The aqueous layer was charged into another flask and 40 mL of dichloromethane and 20 mL of 16 % aqueous sodium hydroxide solution was added drop-wise at 26°C. The mixture was stirred for one hour at 26 °C and the organic layer was separated and the aqueous layer was extracted with 20 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Isopropyl alcohol (8 mL) was added to the residue and evaporated under vacuum at below 45 °C. Isopropyl alcohol (16 mL) was added to the residue and stirred for 2 hours at 2Q°C. Filtered the compound and washed with isopropyl alcohol (4 mL) and dried the compound at 60 °C under vacuum to give 3.2 gm of Linagliptin. PXRD pattern: Fig. 2, Chemical Purity: 98.68%, Chiral Purity: 99.82%, S-isomer content: 0.12%, Regio impurity: 0.57%, Bromo impurity: 0.28%
Example 8: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine dihydrochloride (8.4 gm) were added to the reaction mixture at 26°C. The reaction mixture was heated to ‘\ 00 °C and maintained at that temperature for 4 hours. The reaction mixture was cooled to 30 °C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 200 mL of 10% aqueous acetic acid solution and stirred for 30 minutes at 28 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) were added drop- wise at 28°C (pH is > 10). The mixture was stirred for one hour at 28°C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and divided into 5 equal parts.
Part 1 : The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (16 mL) was added to the residue stirred for 30 minutes at 28 °C and 48 mL of MTBE was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 8 mL of MTBE and dried the compound at 65 °C under vacuum to give 3.0 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.46%, Regio impurity: 0.37%, Bromo impurity: 0.03%
Part 2: The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (24 mL) was added to the residue stirred for 30 minutes at 28 °C and the resulted solution was cooled to 5°C and stirred for 1 hour. Filtered the compound and washed with 5 mL of chilled methanol and dried the compound at 65°C under vacuum to give 3.0 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.41 %, Regio impurity: 0.38%, Bromo impurity: 0.03%
Part 3: The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (20 mL) was added to the residue stirred for 30 minutes at 28 °C and 20 mL of MTBE was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 8 mL of MTBE and dried the compound at 65 °C under vacuum to give 2.8 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.47%, Regio impurity: 0.36%, Bromo impurity: 0.03%.
Part 4: The organic layer was distilled off completely under vacuum at 45 °C. Isopropyl alcohol (8 mL) was added to the residue and distilled off completely under vacuum at 45 °C. Methanol (16 mL) was added to the residue stirred for 30 minutes at 28 °C and 16 mL of isopropyl alcohol was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 4 mL of isopropyl alcohol and dried the compound at 65 °C under vacuum to give 2.9 gm of Linagliptin. PXRD pattern: Fig. 1 .
Chemical Purity: 99.44%, Regio impurity: 0.38%, Bromo impurity: 0.02%.
Part 5: The organic layer was distilled off completely under vacuum at 45 °C. Ethyl acetate (8 mL) was added to the residue and distilled off completely under vacuum at 45 °C. Ethyl acetate (16 mL) was added to the residue stirred for 30 minutes at 28°C and 16 mL of methanol was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 4 mL of ethyl acetate and dried the compound at 65 °C under vacuum to give 0.7 gm of Linagliptin. PXRD pattern: Fig. 2.
Chemical Purity: 99.57%, Regio impurity: 0.29%, Bromo impurity: 0.02%
Example 9: Purification of Linagliptin
Linagliptin (3.5 gm) was dissolved in 10% aqueous acetic acid and stirred for 15 minutes. Dichloromethane (50 mL) was added to the solution and stirred for 30 minutes. The aqueous layer was separated and the pH of this layer was adjusted to 8.5 using 10% aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (50 mLx2). The dichloromethane was evaporated under vacuum to give 3 gm of Linagliptin.
Example 10: Purification of Linagliptin
Linagliptin (31 gm) and methanol (124 mL) were charged into 500 mL round bottomed flask and the solution was heated to 40 °C and stirred for 60 minutes. Charcoal (3 gm) was added to the clear solution and stirred for 30 minutes. The solution was filtered through Hy-flow and the Hy-flow bed was washed with methanol (30 mL). Filtrate was charged into 1000 mL round bottomed flask and methyl tertiary butyl ether was added drop-wise to the solution and stirred for 2 hours at 30 °C. The precipitate so formed was filtered and the wet cake was washed with methyl tertiary butyl ether (30 mL) to get 25.6 gm of pure Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.57%, Chiral purity: 99.73%, Regio impurity: 0.10%, Bromo impurity: 0.1 %
Example 1 1 : Purification of Linagliptin
Linagliptin (4 gm) and methanol (24 mL) were charged into 100 mL round bottomed flask and the solution is heated to 50 °C and stirred for 60 minutes. Methyl tertiary butyl ether (MTBE, 80mL) was charged into 500 mL round bottomed flask and the methanol solution containing linagliptin was added drop-wise at 27 °C and stirred for 2 hours at same temperature. The precipitate formed was filtered and the wet cake was washed with methyl tertiary butyl ether (8 mL) to get 2.6 gm of pure Linagliptin. PXRD pattern: Fig. 2, Bromo impurity content: 0.04%.
Example 12: Purification of Linagliptin
a) Preparation of linagliptin-(D)-tartrate
Linagliptin (10 gm) and methanol (300 mL) were charged into 1000 mL round bottomed flask and (D)-tartaric acid solution (3.3 gm of (D)-tartaric acid in 100 mL of methanol) was added at 26 °C. The solution was heated to 65 °C and stirred for 60 minutes. The solution was cooled to 28 °C and stirred for 2 hours at 27 °C. The precipitate formed was filtered and the wet cake was washed with methanol (20 mL) and the solid was dried under vacuum at 55°C to get 8.3 gm of Linagliptin-(D)-tartrate. PXRD pattern: Fig. 4. Chemical Purity: 99.72%, Chiral purity: 99.89%, Regio impurity: 0.08%, Bromo impurity: 0.05%, S-isomer: 0.1 1%.
b) Isolation of pure Linagliptin
Linagliptin-(D)-tartrate (8 gm) and water (100 mL) were charged into 1000 mL round bottomed flask and stirred for 30 minutes at 26 °C. Dichloromethane (80 mL) was added to the solution and cooled to 5°C. Aqueous sodium hydroxide solution (0.6 gm of NaOH is added to 20 mL of water) was added to the mixture at 5°C and maintained for 1 hour. Layers were separated and aqueous layer was extracted with dichloromethane (20 mL). Combined both organic layers and dried over sodium sulphate and distilled off the organic layer under vacuum at 45 °C. Hexane (20 mL) was added to the crude and stirred for 1 hour at 26°C. The precipitate was filtered and washed with 4 mL of hexane and dried the compound at 60°C under vacuum to give 6 gm of pure Linagliptin. PXRD pattern: Fig. 2, Chemical Purity: 99.67%, Chiral purity: 99.85%, (S)-isomer content: 0.1 5%, Regio impurity: 0.09%, Bromo impurity: 0.07%.
PATENT
http://www.google.com/patents/US20130123282
-
[0181]8-Bromo-3-methylxanthine was reacted with 1-bromo-2-butyne in the presence of base in a mixture of N-methyl pyrrolidone and toluene mixture. The reaction mixture was heated overnight. The reaction completion was determined, and the mixture was then cooled to ambient temperature. A solid precipitate formed on cooling precipitation. The product, 3-Methyl-7-(2-butyne-1-yl)-8-bromoxanthine, having greater than 95% purity was isolated by filtration and washed with toluene.
- Example 34Preparation of (R)-8-(3-amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (Form-XXII): A. 3-Methyl-7-(2-butyne-1-yl)-8-bromoxanthine
Example 35Preparation of 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione
-
[0182]3-Methyl-7-(2-butine-1-yl)-8-bromoxanthine was reacted with 2-(chloromethyl)-4-methylquinazoline in the presence of base under phase transfer catalyst using a N-methyl pyrrolidone/toluene mixture as the reaction solvent. The reaction mixture was heated overnight. When the reaction was complete, the reaction mixture was cooled to ambient temperature. A solid precipitate formed and was separated by filtration and washed with toluene and then with water to provide the product, 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione having more than 97% purity.
Example 36Preparation of (R)-8-(3-Amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (Form-XXII)
-
[0183](R)-3-N-tert-Butoxycarbonylaminopiperidine was reacted with 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione in the presence of base. The reaction mixture was heated overnight. When the reaction was complete, the reaction mixture was cooled to ambient temperature. The cooled reaction mixture was washed several times with water and separated. The resulting 1-[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine organic solution was greater than 95%. Purified by HPLC. An excess of aqueous HCl solution was added to the obtained 1-[(4-methylquinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1-yl)-8-[(R)-3-(tert-butoxycarbonylamino)-piperidin-1-yl]-2,6-dioxo-2,3,6,7-tetrahydro-1H-purine organic solution. The resulting mixture was stirred under heating until complete conversion was observed. Aqueous base was added to the reaction. The resulting mixture was stirred and separated. The organic phase was washed with aqueous base and separated. A non-polar or moderately polar solvent was added to the resulting organic phase. The mixture was partially concentrated to achieve precipitation, and the concentrated mixture was cooled and filtered to provide the wet crude product. The crude product was re-crystallized from alcohol, filtered and dried in vacuum oven with heating to afford dry solid Form-XXII of (R)-8-(3-amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione having more than 98% purity.
Clinical trials
Results in 2010 from a Phase III clinical trial of linagliptin showed that the drug can effectively reduce blood sugar.[2]

Scheme:
. J. Med Chem 2009, 52, 6433..
J. Med Chem 2007, 50, 6450…

References
- H. Spreitzer (September 1, 2008). “Neue Wirkstoffe – BI-1356”. Österreichische Apothekerzeitung (in German) (18/2008): 918.
- Wang, Y, Serradell, N, Rosa, E, Castaner, R (2008). “BI-1356”. Drugs of the Future 33 (6): 473–477. doi:10.1358/dof.2008.033.06.1215244.
- ^ “FDA Approves Type 2 Diabetes Drug from Boehringer Ingelheim and Lilly”. 3 May 2011.
- “Four Phase III Trials Confirm Benefits of BI’s Oral, Once-Daily Type 2 Diabetes Therapy”. Genetic Engineering & Biotechnology News. 28 June 2010.
| CN101735218A * | Dec 17, 2009 | Jun 16, 2010 | 廖国超 | Piperidine carbamic acid ester derivative and application thereof |
| US7407955 | Aug 12, 2003 | Aug 5, 2008 | Boehringer Ingelheim Pharma Gmbh & Co., Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US20040097510 * | Aug 12, 2003 | May 20, 2004 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US20090192314 | Mar 30, 2009 | Jul 30, 2009 | Boehringer Ingelheim International Gmbh | Process for the preparation of chiral 8-(3-aminopiperidin-1yl)-xanthines |
| WO2005085246A1 * | Feb 12, 2005 | Sep 15, 2005 | Boehringer Ingelheim Int | 8-[3-amino-piperidin-1-yl]-xanthine, the production thereof and the use in the form of a dpp inhibitor |
| Reference | ||
|---|---|---|
| 1 | CHIRALITY vol. 7, 1995, pages 90 – 95 | |
| 2 | * | JEAN L ET AL: “A convenient route to 1-benzyl 3-aminopyrrolidine and 3-aminopiperidine“, TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 42, no. 33, 13 August 2001 (2001-08-13), pages 5645-5649, XP004295831, ISSN: 0040-4039, DOI: DOI:10.1016/S0040-4039(01)00985-6 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014033746A2 * | Aug 6, 2013 | Mar 6, 2014 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Process for the preparation of dipeptidylpeptidase inhibitors |
| WO2014059938A1 * | Oct 17, 2013 | Apr 24, 2014 | 2Y-Chem, Ltd. | Method for preparing important intermediate of linagliptin |
| WO2014097314A1 * | Dec 16, 2013 | Jun 26, 2014 | Mylan Laboratories Ltd | An improved process for the preparation of linagliptin |
| WO2010072776A1 * | Dec 22, 2009 | Jul 1, 2010 | Boehringer Ingelheim International Gmbh | Salt forms of organic compound |
| CN101784270A * | Aug 15, 2008 | Jul 21, 2010 | 贝林格尔.英格海姆国际有限公司 | Pharmaceutical composition comprising a glucopyranosyl-substituted benzene derivative |
| CN102127080A * | Nov 2, 2005 | Jul 20, 2011 | 贝林格尔.英格海姆国际有限公司 | Method for producing chiral 8-(3-amino-piperidin-1-yl)-xanthines |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015067539A1 * | Oct 31, 2014 | May 14, 2015 | Chemelectiva S.R.L. | Process and intermediates for the preparation of linagliptin |
| WO2015087240A1 | Dec 9, 2014 | Jun 18, 2015 | Ranbaxy Laboratories Limited | Process for the preparation of linagliptin and an intermediate thereof |
| WO2015107533A1 * | Sep 1, 2014 | Jul 23, 2015 | Harman Finochem Limited | A process for preparation of 1h-purine-2,6-dione, 8-[(3r)-3-amino-1-piperidinyl]-7 (2-butyn-1-yl)-3,7-dihydro-3-methyl-1-[(4-methyl-2quinazolinyl) methyl] and its pharmaceutically acceptable salts |
| Eckhardt M, et al. 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J Med Chem. 2007; 50(26):6450-3. Pubmed ID: 18052023 | |
| 2. | Thomas L, et al. (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J Pharmacol Exp Ther. 2008; 325(1):175-82. Pubmed ID: 18223196 |
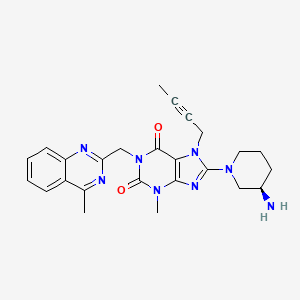
//////////BI-1356, BI1356, Linagliptin, Tradjenta, Trajenta, DPP-IV, DPP-4 inhibitor
