
Home » Posts tagged 'DAPAGLIFLOZIN'
Tag Archives: DAPAGLIFLOZIN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DAPAGLIFLOZIN
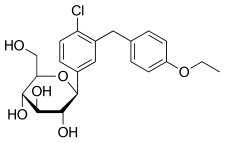

DAPAGLIFLOZIN, BMS-512148
ダパグリフロジン;
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
Cas 461432-26-8
| Molecular Formula: C21H25ClO6 |
| Molecular Weight: 408.87 |
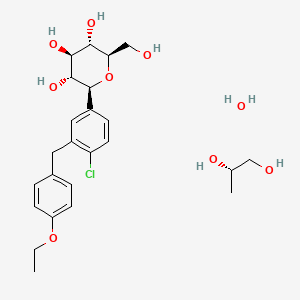
Dapagliflozin propandiol monohydrate; 960404-48-2
| Molecular Weight | 502.98 |
| Formula | C21H25ClO6•C3H8O2•H2O |
Bristol-Myers Squibb (Originator)
AstraZeneca
TYPE 2 DIABETES,SGLT-2 Inhibitors
launched 2012, as forxiga in EU, FDA 2014, JAPAN PMDA 2014
Dapagliflozin propanediol monohydrate was first approved by European Medicine Agency (EMA) on November 12, 2012, then approved by the U.S. Food and Drug Administration (FDA) on January 8, 2014, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on March 24, 2014. It was co-developed and co-marketed as Forxiga® by Bristol-Myers Squibb and AstraZeneca in EU.
Dapagliflozin propanediol monohydrate is a sodium-glucose co-transporter 2 (SGLT2) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Forxiga® is available as tablet for oral use, containing 5 mg or 10 mg of free Dapagliflozin. The recommended starting dose is 5 mg once daily in the morning.

Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
WILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013-- USFDA



Sales:$518.7 Million (Y2015); 
$235.8 Million (Y2014);
$33 Million (Y2013);ATC Code:A10BX09
Approved Countries or AreaUpdate Date:2015-07-29
- US
- EU
- JP
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-01-08 | Marketing approval | Farxiga | Type 2 diabetes | Tablet | 5 mg/10 mg | AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-11-12 | Marketing approval | Forxiga | Type 2 diabetes | Tablet, Film coated | Eq. 5 mg/10 mg Dapagliflozin | Bristol-Myers Squibb, AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-03-24 | Marketing approval | Forxiga | Type 2 diabetes | Tablet, Film coated | 5 mg/10 mg | Bristol-Myers Squibb, AstraZeneca, Ono |
MoreChemical Structure
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.

Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
http://online.wsj.com/article/PR-CO-20131212-910828.html?dsk=y
Reference:1. WO03099836A1 / US6515117B2.
2. WO2010048358.
3. J. Med. Chem. 2008, 51, 1145–1149.
4. WO2004063209A2 / US7375213B2.
5. WO2008002824A1 / US7919598B2.Route 2
Reference:1. WO2010022313 / US8283454B2.Route 3
Reference:1. WO2013068850.Route 4
Reference:1. Org. Lett. 2012, 14, 1480-1483.
PAPER
https://www.future-science.com/doi/10.4155/fmc-2020-0154
Patent
https://patents.google.com/patent/WO2016178148A1/en
1. A process for the preparation of dapagliflozin in amorphous form, the process comprising:
(a) reducing a compound of formula II to a compound of formula ΠΙ in the presence of a Lewis acid;

(b) silylating a compound of formula IV with hexamethyldisilazane to form a compound of formula V;

(c) reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;

(d) converting the compound of formula VII to dapagliflozin;
(e) acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;

(f) optionally, purifying the compound of formula VIII with a solvent selected from halogenated hydrocarbons, alcohols, ethers, or mixtures thereof; (g) hydrolyzing the compound of formula VIII obtained in step (f) to give dapagliflozin;
EXAMPLE 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
To a suspension of 5-bromo-2-chiorobenzoic acid (lOg) in methylene di chloride (40niL), dimethylformamide (0.2g) and thionyl chloride were added and the reaction mixture was refluxed for about 2h. After completion of reaction, the solvent was distilled out. The mass obtained was degassed under vacuum followed by stripping with cyclohexane to give crude 5-bromo-2-chlorobenzoyl chloride (10.8g).
[0189] EXAMPLE 2: Preparation of 5-bromo-2-chloro-4′-ethoxybenzophenone (compound of Formula II)
5-bromo-2-chlorobenzoyl chloride (10.7g) was dissolved in methylene dichloride (40mL) and the reaction mixture was cooled to about -8°C to about -12°C under inert atmosphere. Aluminum chloride (5.65g) was added to the reaction mixture followed by addition of a solution of ethoxybenzene in methylene dichloride. The reaction mixture was stirred for about lh at about -8°C to -12°C and then quenched in dilute hydrochloric acid followed by extraction with methylene dichloride. The organic layer was washed with sodium bicarbonate solution and concentrated. The residue obtained was crystallized from methanol to give 5-bromo-2-chloro-4′-ethoxybenzophenone (8.5g). HPLC purity: 99.34%
[0190] EXAMPLE 3: Preparation of 5-bromo-2-chloro-4′-ethoxydiphenylmethane (compound of formula III)
To a mixture of 5-bromo-2-chloro-4′-ethoxybenzophenone (lOg) and methylene dichloride (50mL), cooled to about 0°C to about 5°C, triethylsilane (11.98g) and titanium chloride (22.3g) were added. The reaction mixture was stirred for about 3h at about 10°C to about 15°C. The reaction mixture was quenched into chilled water. The organic layer was separated, washed with water and sodium bicarbonate solution and concentrated under vacuum followed by stripping with toluene. The residue obtained was stirred with methanol, filtered and dried to give 5-bromo-2-chloro-4′-ethoxydiphenylmethane (9g). HPLC purity: 99.4%
[0191] EXAMPLE 4: Preparation of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5- lactone (compound of Formula V)
To a mixture of D-glucono- 1,5 -lactone (lOg) and iodine (0.28g) in methylene dichloride (80mL), hexamethyldisilazane (36.1g) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed to give 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5-lactone as liquid (25g). HPLC purity: 95%
[0192] EXAMPLE 5: Preparation of D-glucopyranoside, methyl l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (compound of Formula VII wherein R is methyl)
To a mixture of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l ,5-lactone (25g) and 5- bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) in tetrahydrofuran (174mL), cooled to about -75°C to about -88 °C under nitrogen atmosphere, n-butyl lithium in hexane (50mL) was slowly added. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. The residue was purified with a mixture of toluene and cyclohexane. Yield: 1 lg as thick mass with 80-85% HPLC purity.
reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;
[0193] EXAMPLE 6: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl) methyl] phenyl]
To a mixture of D-glucopyranoside, methyl l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] in methylene di chloride (40mL) and acetonitrile (40mL), cooled to about -40°C to about -45°C, triethylsilane (8.74g) was added followed by addition of boron trifluoride etherate (10.67g) maintaining the temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum to give title compound (1 lg) as thick residue with 80-85% HPLC purity.
[0194] EXAMPLE 7: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (lS)-
To a cooled solution of D-glucitol, l,5-anhydro-l -C-[4-chloro-3-[(4-ethoxyphenyl) methyl] phenyl]- (l lg) in methylene dichloride (55mL) at about 0°C to about 5°C, diisopropylethylamme, dmiethylaminopyridine and acetic anhydride were added and the reaction mixture was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol. The residue was purified with methanol and charcoal, followed by diisopropyl ether and methanol crystallization. Yield: lOg; HPLC purity: 99.6%
acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;
[0195] EXAMPLE 8: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (Dapagliflozin)
To a stirred solution of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, (lOg) in THF: methanol: water mixture (50mL: 50mL:30mL), sodium hydroxide was added and the reaction mixture was stirred. After completion of reaction, the solvents were distilled out under vacuum and the residue obtained was dissolved in methylene dichloride and washed with water and brine and dried over sodium sulfate. The reaction mixture was concentrated and degassed to give off- white to white solids of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]- (dapagliflozin) Yield: 7g (XRD matches with amorphous form) HPLC purity: 99.8%
[0197] EXAMPLE 10: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (IS)- from D-glucono-1,5- lactone (One-pot Synthesis)
To a mixture of D-glucono-l,5-lactone (lOg) in methylene dichloride (80mL), hexamethyldisilazane (36. lg) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed. The residue obtained was dissolved in tetrahydrofuran. 5-Bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) was added to the reaction mixture which was cooled to about -75°C to about-85°C under nitrogen atmosphere. n-Butyl lithium in hexane (50mL) was slowly added to the reaction mixture maintaining the temperature between -75°C to about -85°C. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. This residue was purified by a mixture of toluene and cyclohexane. To the product obtained, methylene dichloride and acetonitrile were added and the reaction mixture was cooled to about -40°C to about -45°C. Triethylsilane (8.74g) was added to the reaction mixture followed by addition of boron trifluoride etherate (10.67g) maintaining temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution. The aqueous layer was separated and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum. The thick residue obtained was dissolved in methylene dichloride and cooled to about 0°C to about 5°C. Diisopropylethylamine, dimethylaminopyridine and acetic anhydride were added to the reaction mixture which was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol The residue obtained was recrystallized with methanol and charcoal to give title compound (iOg) with 99.7% HPLC purity.
PAPER
Bioorganic & Medicinal Chemistry, 26(14), 3947-3952; 2018
https://www.sciencedirect.com/science/article/abs/pii/S0968089618309386?
Abstract
The cardiovascular complications were highly prevalent in type 2 diabetes mellitus (T2DM), even at the early stage of T2DM or the state of intensive glycemic control. Therefore, there is an urgent need for the intervention of cardiovascular complications in T2DM. Herein, the new hybrids of NO donor and SGLT2 inhibitor were design to achieve dual effects of anti-hyperglycemic and anti-thrombosis. As expected, the preferred hybrid 2 exhibited moderate SGLT2 inhibitory effects and anti-platelet aggregation activities, and its anti-platelet effect mediated by NO was also confirmed in the presence of NO scavenger. Moreover, compound 2 revealed significantly hypoglycemic effects and excretion of urinary glucose during an oral glucose tolerance test in mice. Potent and multifunctional hybrid, such as compound 2, is expected as a potential candidate for the intervention of cardiovascular complications in T2DM.
Graphical abstract

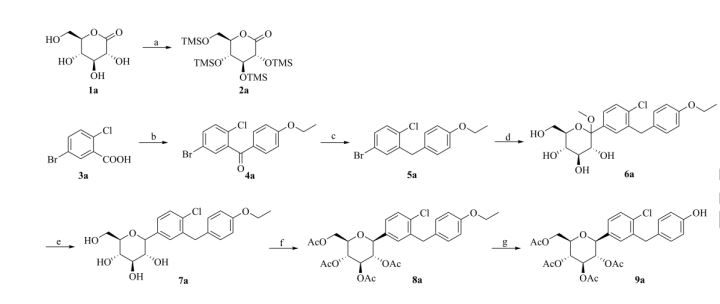
Scheme 1. Synthesis of target compounds 1-3. Reagents and conditions: (a) TMSCl, NMM, THF, 35 °C; (b) (COCl)2, CH2Cl2, DMF, then phenetole, AlCl3, 0 °C; (c) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, 25 °C; (d) n-BuLi, THF, toluene, -78 °C, then 2a followed by MeOH, CH3SO3H; (e) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, -10 °C; (f) Ac2O, pyridine, CH2Cl2, DMAP;
PATENT
Indian Pat. Appl., 2014MU03972,
PATENT
| Dapagliflozin, also known as SGLT2 inhibitor, chemical name is (2S,3R,4R,5S,6R)-2-[3-(4-ethoxybenzyl)-4-chlorophenyl]-6- Hydroxymethyltetrahydro-2H-pyran-3,4,5-triol, a sodium-glucose cotransporter 2 inhibitor, announced by the U.S. Food and Drug Administration (FDA) on January 8, 2014 , approved the use of dapagliflozin for the treatment of type 2 diabetes, the specific structural formula is as follows: |
| |
| Dapagliflozin works by inhibiting sodium-glucose transporter 2 (SGLT2), a protein in the kidney that allows glucose to be reabsorbed into the blood. This allows excess glucose to be excreted through the urine, thereby improving blood sugar control without increasing insulin secretion. |
| At present, there are two main methods for synthesizing dapagliflozin. One uses 5-bromo-2-chlorobenzoic acid as the starting material, which is chlorinated, Falk acylated, reduced, and then combined with 2,3,4 ,6-tetra-0-trimethylsilyl-D-glucopyranosic acid 1,5-lactone is condensed, methyl etherified, and demethoxylated to obtain dapagliflozin. The specific process route is as follows: |
| |
| The method has expensive starting materials and too many process steps, so it is not suitable for industrial production, and dangerous n-butyllithium needs to be used in the reaction process, so the requirements for experimental conditions are too high; |
| Another method is to use o-toluidine as the starting material, undergo bromination, diazotization, chlorination, and alkylation reactions, and then react with 2,3,4,6-tetra-0-trimethylsilyl -D-glucopyranosic acid 1,5-lactone is condensed, then methyl etherified and demethoxylated to obtain dapagliflozin. The specific process route is as follows: |
| |
| AIBN will be used in this reaction, which will produce highly toxic cyanide, which will seriously pollute the environment, and also requires the use of n-butyllithium, which requires high experimental conditions and is dangerous to operate, and is not suitable for large-scale production. |
| Example 1 |
| Weigh 16g (0.8mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodobenzene to it 2000mL of THF solution (total 2000mL: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsided, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene. |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 423.6 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 93%. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 2500mL of toluene, then add 5.6g of europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 259.49 g of solid with a yield of 85%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:3 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 147.1 g of white rice-like crystals with a yield of 80.2% and a purity of 99.2% (determined by high performance liquid chromatography, external standard method). |
| IR(cm-1):1689,1612,1588,1523,1455,1253,1089,820。1H NMR(500MHz,CDCl 3 ):δ:7.36(d,J=8.2Hz,1H),7.32(d,J=1.9Hz,1H),7.23(dd,J=8.3,2.0Hz,1H),7.09(d,J=8.6Hz,2H),6.82(d,J=8.6Hz,2H),4.85(s,1H),4.41(s,3H),3.93~4.02(m,5H),3.70(dd,J=11.7,1.3Hz,1H),3.44(dd,J=11.7,5.6Hz,1H),3.26~3.28(m,1H)。LC-MS,m/z:[M+Na]+=431。 |
| Example 2 |
| Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 272.75mL of 33% hydrogen bromide in acetic acid solution (1.1mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 381.5 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 83.8%. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , dried to obtain solid 281.2g, yield 92.1%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 150.9 g of white rice-like crystals with a yield of 88.2% and a purity of 99.5% (determined by high performance liquid chromatography, external standard method). |
| Example 3 |
| Weigh 37.4g (1.862mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 818.25mL of 33% hydrogen bromide in acetic acid solution (3.3mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 375.3 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 82.4%. |
| Weigh 217.83g 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 7500mL toluene, then add 5.6g europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 273.25 g of solid with a yield of 89.5%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:5 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 152.7 g of white rice-like crystals with a yield of 82.9% and a purity of 99.4% (determined by high performance liquid chromatography, external standard method). |
| Example 4 |
| Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 425.1 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose in 94% yield. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, control the temperature at -20~-15°C, after the dropwise addition, raise the temperature to 5°C for 2 hours, concentrate in vacuo to an oily substance, then add to it 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, then 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 278.6 g of solid with a yield of 91.2%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was completed, and 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 155.62 g of white rice-like crystals with a yield of 84.5% and a purity of 99.7% (determined by high performance liquid chromatography, external standard method). |
Patent
Sodium-glucose co-transporter-2 (SGLT2) inhibitors are a group of oral medicines used for treating diabetes that have been approved since 2013. SGLT2 inhibitors prevent the kidneys from re-absorbing glucose back into the blood by passing into the bladder. Glucose is re-absorbed back into the blood via the renal proximal tubules. SGLT2 is a protein predominantly expressed in the renal proximal tubules and is likely to be major transporter responsible for this uptake. Glucose-lowering effect of SGLT-2 inhibitors occurs via an insulin-independent mechanism mostly through glucosuria by increasing the urinary excretion of glucose.
It has been shown that the treatment with SGLT2 inhibitors in patients with type II diabetes lowers HbAlc, reduces body weight, lowers systemic blood pressure (BP) and induces a small increase in LDL-C and HDL-C levels.
SGLT2 inhibitors inhibit the reabsorption of sodium and glucose from the tubule and hence, more sodium is delivered in the macula densa causing arteriole dilation, reduced intraglomerular pressure and decreased hyperfiltration. SGLT2 inhibitors cause natriuresis and volume depletion, and an increase in circulating levels of renin, angiotensin and aldosterone. They also reduce albuminuria and slow GFR loss through mechanisms that appear independent of glycemia.
Dapagliflozin is a highly potent and reversible SGLT2 inhibitor, which increases the amount of glucose excreted in the urine and improves both fasting and post-prandial plasma glucose levels in patients with type 2 diabetes. Dapagliflozin has also been shown to tend to reduce liver fat content in some studies in a diabetic population.
Dapagliflozin is available on the market in the form of dapagliflozin propanediol monohydrate and is sold under trade name Forxiga or Farxiga in the form of fdm-coated tablets. Further it is available on the market as a combination product with metformin hydrochloride which is sold under trade name Xigduo IR or Xigduo XR in the form of film-coated tablets. In addition, it is available on the market as a combination product with saxagliptin hydrochloride which is sold under trade name Qtem in the form of film-coated tablets. Moreover, it is available on the market as a combination product with saxagliptin hydrochloride and metformin hydrochloride which is sold under trade name Qtemmet XR in the form of film-coated tablets.
Dapagliflozin as a monotherapy and in a combination with other active substances has demonstrated its efficacy in improving glycaemic control and reducing body weight and blood pressure in a broad spectrum of patients with type II diabetes, including those with high baseline HbAlc and the elderly. A sustained reduction in serum uric acid concentration was also observed. Dapagliflozin provides significant improvement in HbAlc, reduction in insulin dose and reduction in body weight in patients with type 1 diabetes as adjunct therapy to adjustable insulin.
Dapagliflozin can be in its free form or any stereoisomer or any pharmaceutically acceptable salt or co crystal complex or a hydrate or a solvate thereof and in any polymorphic forms and any mixtures thereof.
Dapagliflozin as a substance was first disclosed in US 6,515,117. The process for the preparation of dapagliflozin involves the reaction of 4-bromo- 1 -chloro-2-(4-ethoxybenyl)benzene with 2,3,4,6-tetra-O-trimethyl silyl -D-gluconolactone, the obtained compound 3 on demethoxylation yields diastereomeric mixture of Dapagliflozin. Hie diastereomeric mixture of dapagliflozin is further acetylated with acetic anhydride in the presence of pyridine and dimethylaminopyridine yields, then recrystallized from absolute ethanol to yield the desired tetra-acetyJated b-C-glucoside as a white solid. Compound tetra-acetylated b-C-glucoside is treated with lithium hydroxide hydrate which undergoes deprotection to yield the compound dapagliflozin.
Several other documents, patents and applications disclose the process for the preparation of dapagliflozin such as for example WOO 127128, WO03099836, W02004063209, W02006034489,
W02010022313, WO2012019496, W02013064909, W02013068850, W02013079501, WO2014094544, WO2014159151, WO2014206299, W02015040571, WO2015044849, WO2015063726, WO2015132803, WO2015155739, W02016098016, WO2016128995, WO2016178148, WO2017042683, WO2017063617, W02018029611, WO2018029264, WO2018142422.
Prior art documents already provided some compositions of SGLT2 inhibitor dapagliflozin.
W02008116179 discloses immediate release formulation in the form of a stock granulation or in the form of a capsule or a tablet which comprises dapagliflozin propylene glycol hydrate, one or more bulking agent, one or more binder and one or more disintegrant.
WO2011060256 describes the bilayer tablet comprising dapagliflozin having sustained release profde in one layer and metformin in another layer while WO2011060290 describes immediate release formulation of dapagliflozin and metformin.
WO2012163546 discloses the pharmaceutical composition comprising cyclodextrin and dapagliflozin.
Co-crystals of dapagliflozin with lactose are described in WO2014178040.
Solid dispersion compositions comprising amorphous dapagliflozin and at least one polymer are disclosed in W02015011113 and in WO2015128853.
CN103721261 discloses the combination of SGLT2 inhibitor with vitamins such as vitamin B.
Pharmaceutical composition preparation comprising dapagliflozin L-proline and metformin and/or DPP-IV inhibitor is disclosed in WO2018124497.
EP2252289A1 provides a combination of SGLT inhibitor with DPP4 inhibitor showing synergistic effect in increasing plasma active GLP-1 level in a patient over that provided by administration of the SGLT inhibitor or the DPP4 inhibitor alone.
EP2395983A1 relates to a pharmaceutical composition comprising a SGLT2 inhibitor, a DPP4 inhibitor and a third antidiabetic agent which is suitable in the treatment or prevention of one or more conditions selected from type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance and hyperglycemia.
Example A: HPLC method
The purity of Dapagliflozine in general may be determined with the following HPLC method: column: XBridge C18, 150×4.6mm, 3.5; flow-rate: 0.9ml/min; column temperature: 50°C, wavelength: UV 225 nm; mobile phase: eluent A: 0.1% H3PO4, Eluent B: methanol; gradient:
Sample preparation: Accurately weigh about 40mg of sample and dissolve in 50 ml of solvent. Calculation: Use area per cent method. Do not integrate solvent peaks.
Example 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
5-bromo-2-chlorobenzoic acid (450 g) was suspended in dichloromethane (2.25 L) and dimethylformamide (0.74 ml). At 15 – 30°C oxalyl chloride (180.3 ml) was slowly added. During addition gas evolution of HC1 and CO2 occurred. The reaction was performed at 20-30°C. The reaction was considered to be complete if 2-chloro-5-bromobenzoic acid was below 1% (area percent purity). The mixture was concentrated at elevated temperature until oily residue was obtained.
Example 2: Preparation of (5-bromo-2-chlorophenyl)(4-ethoxyphenyl)methanone
Dichloromethane (900 ml) was charged into reactor and then aluminum chloride (267.6 g) was added. The reaction mixture was cooled below 5°C and ethoxybenzene (256.1 ml) was slowly added. After complete addition, the mixture was gradually cooled below -5°C. In a separate reactor, 5-bromo-2-chlorobenzoyl chloride (485g) was dissolved in dichloromethane (900 ml). This solution was slowly added to the mixture of aluminum chloride and ethoxybenzene with such rate that temperature was kept below -5°C. After complete addition the mixture was stirred below -5°C until reaction was finished. The reaction was considered to be complete if methyl ester was below 1 % (the reaction mixture is sampled in methanol). After reaction was completed the reaction mixture was slowly added into cooled 1M HC1 solution and flushed with of dichloromethane (450 ml). The organic phase was separated and water phase was washed again with dichloromethane. Organic phases were combined and washed with water and NaHC03 solution. So obtained organic phase was concentrated to oily residue and dissolved in methanol ethyl acetate mixture in 10 to 1 ratio at reflux temperature. The clear solution was gradually cooled down to 35-45 °C and seeded with pure 5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone. The reaction mass was gradually cooled down to 0-10°C and stirred at that temperature up to 4 hours. The precipitate was isolated and washed with precooled methanol. The product was dried to a final LOD (Loss on drying) content of less than 1.0% with a yield of 564g (87% mass yield).
Example 3: Preparation of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene
5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone (400g) was dissolved in 1.62L tetrahydrofuran. Into solution NaBTL (53.5g ) was added. After addition, the mixture was stirred at ambient temperature for 30-60 min followed by cooling of reaction mixture below -5°C. Aluminum chloride (314g) was added in portion and reaction mixture maintained below 5°C. After addition, the reaction mixture was gradually heated to reflux temperature and stirred until reaction was complete. Reaction mixture was cooled to ambient temperature and mixture of THF/water was slowly added into reaction mixture followed by addition of water and stirred at ambient temperature. Organic phase was collected and washed with saturated NaCl solution. Organic phase was concentrated to oily residue and dissolved in ethanol (800ml) at elevated temperature. Solution was cooled to 25-30°C and seeded with pure 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene. The reaction mass was gradually cooled to -2 to 10°C and stirred at that temperature. The product was isolated and washed with precooled ethanol and dried until final LOD (Loss on drying) content was less than 1.0%. Yield was 322 g (89%).
Example 4: Preparation of 3R,4S,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (97.5g ) and toluene (1.46L) was charge into reactor. Solution was heated to reflux temperature and approximately half of the solvent was distilled out. Tetrahydrofuran (195 mL) was charged into the solution and mixture was cooled below -70°C. Solution of 15% «-Buli in hexane (227.5 ml) was slowly added and temperature was kept below -70°C. After complete addition solution was stirred at temperature below -70°C to complete reaction. Solution of 2,3,4,6-tetra-O-trimethylsilyl-D-gluconolactone (182 g) in toluene (243 mL) was added into reaction mixture at temperature below -70°C. After complete addition, the mixture was stirred below -70°C, warmed to approximately -65°C and then mixture of 57.6 g methanesulfonic acid in 488 ml methanol was added. After addition, the mixture was gradually warmed to ambient temperature and stirred until reaction was complete. After reaction was finished reaction mixture was slowly added into saturated NaHCCL solution (630ml) and stirred. Into quenched mixture 975 ml of heptane and 585 ml methanol was added. The mixture was stirred for additional 15 min. Organic phase was washed with water/methanol mixture several times. Water phases were combined and distilled to remove organic solvents. Into the residual water phase, toluene was added to perform extraction. Organic phases were combined and washed with water. Organic phase was distillated at elevated temperature until oily residue was obtained.
Example 5: Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl) tetrahydro2H-pyran-3,4,5-triol – Dapagliflozin
Dichloromethane (656 mL) was charged into oily residue from step 4 and stirred at ambient temperature until clear solution was obtained. Triethysilane (122 mL) was added into the so obtained solution. Reaction mixture was cooled below -30°C and 94.2 mL of boron trifluoride etherate was slowly added at temperatures below -30°C. After complete addition, the mixture was stirred below -30°C for one hour and gradually warmed to -5 to 0°C until reaction was completed. After reaction was finished saturated NaHCCL solution (468 mL) was slowly added. Reaction mixture was distilled to remove organic solvents followed by addition ethyl acetate into the residue. Organic phase was collected and washed again with saturated NaHC03 and water. So obtained organic phase was distillated at elevated temperature until oily residue is obtained.
Example 6: Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate.
Oily residual from example 5 was dissolved in dichloromethane (602 mL) at ambient temperature followed by addition of DMAP (6.22g). Reaction mixture was cooled to 0 – 10°C and 144.3 mL of acetic anhydride was added at temperatures below 10°C. Reaction mixture was gradually warmed to ambient temperature and stirred until reaction was completed. Reaction mass was washed with water with saturated NaHC03. Organic phase was collected and concentrated to oily residue to which ethanol (1.68L) was charged and approximately 300 ml ethanol was removed by distillation. The clear solution was gradually cooled to 60-65 °C and seeded. The reaction mass was gradually cooled to 20-25 °C and product was isolated. The product was dried at 50°C in vacuum until LOD (Loss on drying) below 1.0% 123g of product was obtained with yield 71%. HPLC purity: 99.97 %.
Formula 9 Formula 2
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (740 g) as prepared according to process described in examples 1 to 6 was charged into solution of methanol (2.27L), water (0.74L) and NaOH (23 lg) at 35-45°C and stirred at 35-45°C until reaction was completed. After reaction was finished 1M HC1 (1.63L) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. Combined organic phases were washed with water and concentrated at elevated temperature until oily residue was obtained. Content of impurity IMP A was below 0.02%.
Example 7a: Preparation of amorphous dapagliflozin.
Oily residue as prepared according to example 7 comprising approximately 262 g of dapagliflozin was dissolved in toluene (2.5L) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (5.3L) at temperature between 10 to 15°C and stirring rate with P/V at 4 W/m3. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was 14 · 104 m/s. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of impurity IMP A was below 0.02% and residual heptane and toluene were 1673 ppm and below 89 ppm.
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (30 g) of 4 different qualities obtained by the process known in the prior art was charged into solution of methanol (90 mL), water (30 mL) and NaOH (9.36 g) and stirred at 35-45°C until reaction was completed and sampled for HPLC analysis (Sample 1).
After reaction was finished 1M HC1 (66 mL) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. To the combined organic phases 68ml of 1M NaOH was added and pH was set to 12.5 to 13.5. Phases were separated and organic phase is washed again with 68ml of water without pH correction. So obtained organic phase was sampled for HPLC analysis (Sample 2) and concentrated at elevated temperature until oily residue was obtained.
Oily residue comprising approximately 21 g of dapagliflozin was dissolved in toluene (210 mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (420 mL) at temperature between 10 to 15°C and stirring rate with P/V as defined in Table 1. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was as defined in Table 1. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Amorphous dapagliflozin with content of impurity IMP A as shown in Table 1 and residual heptane and toluene as shown in Table 1 was obtained for each cases.
Table 1 : Process parameters used in the preparation of four different starting materials (cases).
As it is evident from Table 2 the final amorphous dapagliflozin prepared by the extraction process according to the present invention contains less than 0.02% of impurity IMP A irrespective of the level of impurity IMP A present in the starting material.
Table 2: Content of impurity IMP A in the final amorphous dapagliflozin obtained with and without extraction.
Example 9
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 25 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 10
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 40 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 11
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -10°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1480 ppm and 732 ppm.
Example 12
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 20°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 5°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled hcptanc Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2873 ppm and 639 ppm.
Comparative Example 1
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at -5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1940 ppm and 1557 ppm.
Comparative Example 2
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 25°C and stirring rate with P V at 16 W/m3. After complete addition, the suspension was cooled to 20°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 3663 ppm and 2047 ppm.
Comparative Example 3
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 30°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2425 ppm and 1812 ppm.
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
https://patents.google.com/patent/WO2017206808A1/en
Daggliflozin (English name: Dapagliflozin) is a new Sodium glucose co-transporters 2 (SGLT-2) inhibitor developed by Bristol-Myers Squibb and AstraZeneca. Approved by the European Commission on November 14, 2012, and marketed in the United States on January 8, 2014, to improve glycemic control in adult patients with type 2 diabetes by combining diet and exercise; the trade name is Farxiga, currently offering 5 mg and 10 mg tablets. At the same time, a combination of dapagliflozin and metformin hydrochloride has also been marketed.The chemical name of dapagliflozin is (2S,3R,4R,5S,6R)-2-(3-(4-ethoxybenzyl)-4-chlorophenyl)-6-hydroxymethyltetrahydro-2H – pyran-3,4,5-triol, the chemical formula is C 21 H 25 ClO 6 , CAS No. 461432-26-8, the structural formula is shown as 2, clinically used as a pharmaceutical for dapagliflozin (S) -1,2-propanediol monohydrate, the structural formula is as shown in 1.

The synthesis of β-type C-aryl glycosidic bonds is a key point in the synthetic route during the preparation of dapagliflozin. At present, there are four synthetic methods for the synthesis of dapagliflozin reported in the literature and patents.Route 1: The synthetic route of dapagliflozin reported in patent WO03099836A1 is as follows:

The route uses 2-chloro-5-bromobenzoic acid (12) as raw material to react with phenethyl ether to form intermediate 11 and then triethylsilane to obtain intermediate 10; intermediate 10 and n-butyl The lithium is reacted at -78 ° C, and then subjected to a nucleophilic addition reaction with the intermediate 9, and then methoxylated to obtain the intermediate 8; the intermediate 8 is subjected to acylation reduction and deprotection to obtain the intermediate 2. The disadvantage of this method is that the β-type C-aryl glycosidic bond synthesis of the compound is carried out at a low temperature of -78 ° C, which is obviously difficult to meet the needs of industrial production; and, through nucleophilic addition, methoxylation, The five-step reaction of acetylation, reduction and hydrolysis can synthesize the β-type C-aryl glycosidic bond. The procedure is relatively long, and the purity of the intermediate 2 is only 94%.Route 2: The synthetic route of dapagliflozin reported in the literature OrgLett.2012, 14, 1480 is as follows:

The intermediate 14 of the route is reacted with di-n-butyl-n-hexylmagnesium for 48 hours at 0 ° C, and then reacted with zinc bromide to prepare an organozinc reagent by Br/Mg/Zn exchange reaction, and then with intermediate 4 Intermediate 3 was prepared by nucleophilic substitution reaction; finally, intermediate 2 was obtained by deprotection with sodium methoxide. The synthesis method is relatively novel, and the synthesis step is short. However, the research experiment is conducted only as a synthesis method, and the post treatment of the intermediate 3 is performed by column chromatography. The purity of the intermediate 2 produced was not reported. Moreover, the di-n-butyl-n-hexylmagnesium reagent used in the route is not a commonly used reagent, and is not commercially available in China. It can only be prepared by reacting dibutylmagnesium with n-hexyllithium reagent before the test, and the operation is cumbersome and difficult to mass. use.Route 3: The synthetic route of dapagliflozin reported in patent WO2013068850A2 is as follows:

The route uses 1,6-anhydroglucose (20) as a raw material, protects the 2,4-hydroxyl group by tert-butyldiphenylchlorosilane, and then protects the 3-position hydroxyl group with phenylmagnesium bromide. Intermediate 18. The intermediate 14 is subjected to an Br/Mg/Al exchange reaction to prepare an organoaluminum reagent 16, which is reacted with an intermediate 18 to form an intermediate 15, and finally, deprotected to obtain an intermediate 2. The synthesis method is very novel and is also used as a synthetic methodological study. The purification of the intermediates is carried out by column chromatography. The 1,6-anhydroglucose (20) used in the route is very expensive; and the multi-step reaction in the route uses a format reagent, a preparation format reagent or an organoaluminum reagent, which is cumbersome and cumbersome to perform, and is difficult to scale synthesis. The purity of the intermediate 2 produced was not reported.Route 4: The synthetic route of dapagliflozin reported in patent WO2013152476A1 is as follows:

The route uses 2-chloro-5-iodobenzoic acid (24) as raw material to form intermediate 22 by Friedel acylation and reduction reaction, and exchange with I-Mg at -5 ° C with isopropyl magnesium chloride lithium chloride. The intermediate 8 is obtained by nucleophilic addition and methoxylation with the intermediate 9, and then the intermediate 2 is obtained by reduction with triethylsilane, and the intermediate 2 is further purified by co-crystallizing with L-valine. Finally, The pure intermediate 2 was obtained by removing L-valine. This route is a modified route of Route 1, which replaces n-butyllithium with isopropylmagnesium chloride chloride to raise the reaction temperature of the reaction from -78 °C to -5 °C. However, the problem of a long step of synthesizing a β-type C-aryl glycosidic bond still exists. The obtained intermediate 2 is not optically pure, and needs to be purified by co-crystallizing with L-valine, and the work amount of post-treatment is increased, and finally the purity of the intermediate 2 is 99.3%.Among the four synthetic routes described above for dapagliflozin, route one and route four are commonly used synthetic methods for β-type C-aryl glycosidic bonds, and the route is long, and the optical purity of the obtained product is not high, and further purification is required. Post processing is cumbersome. Moreover, the reaction required at -78 °C in Route 1 requires high equipment and high energy consumption, which undoubtedly increases the cost. Although both Route 2 and Route 3 are new methods, most of the purification of intermediates used is column chromatography. Such a process is not suitable for scale production in factories; and some of the synthetic routes are used. Reagents are not commercially available or expensive, and there is no advantage in such route costs. Therefore, there is an urgent need to find a new method for the synthesis of dapagliflozin, and to enable industrial production, and the route has a cost advantage.Repeating the procedure reported in the literature in Equation 2, the yield of Intermediate 3 was only 46%. The organic zinc reagent is prepared by Br/Mg/Zn exchange reaction, and the exchange reaction yield is 78%; and the raw material is prepared by X/Li/Zn exchange reaction to prepare an organic zinc reagent, and the exchange reaction yield is 98.5%, which is also the two Different reaction pathways lead to the essential reason for the different yields of intermediate 3. Moreover, the price of commercially available 1.0 mol/L di-n-butyl magnesium n-heptane solution 500 mL is 1380 yuan, and the price of 1.6 mol/L n-hexyl lithium n-hexane solution 500 mL is 950 yuan, and 2.5 mol/L n-butyl lithium. The price of 500 mL of n-hexane solution is only 145 yuan. Therefore, the method for preparing dapagliflozin by preparing an organozinc reagent by X/Li/Zn and then synthesizing the β-type C-aryl glycosidic bond designed by the invention has the advantages of cost, ease of operation and industrialization. Very obvious advantage.In order to solve this problem, the original compound company uses a eutectic method in the production of dapagliflozin to make dapagliflozin together with a solvent or an amino acid compound, since the compound 2 sugar ring structure contains four hydroxyl groups and is easy to absorb moisture and deteriorate. The crystal is made into a relatively stable solid, easy to store, stable and controllable in quality, and easy to prepare. Among them, the marketed dapagliflozin forms a stable eutectic with (S)-1,2-propanediol and water (1). The original crystal form patent (CN101479287B, CN103145773B) reported that all 11 crystal forms are dapagliflozin solvate or dapagliflozin. Crystal. Among them, there are two preparation methods for the da forme (S)-1,2-propanediol monohydrate (1) having a crystal structure of type Ia:Method 1: The preparation method is as follows:

Compound 7 is deprotected with sodium hydroxide to obtain compound 2, then compound 2 is extracted with isopropyl acetate, (S)-1,2-propanediol ((S)-PG) is added, and seed crystal of compound 1 is added. Then, cyclohexane was added to crystallize and separated to obtain a eutectic of the compound (1) of the type Ia.Method 2: The preparation method is as follows:

Compound 8 is subjected to reduction of methoxy group by triethylsilane and boron trifluoride diethyl ether complex, and then the reaction solution is extracted with methyl tert-butyl ether (MTBE), and (S)-1,2-propanediol ( (S)-PG), a seed crystal of the compound 1 is added, and then cyclohexane is added to crystallize, and the mixture is separated and dried to obtain a eutectic of the compound (1) of the type Ia.The above two methods for preparing the eutectic are all used in the cyclohexane solvent, which is listed in the appendix of the 2015 edition of the Pharmacopoeia (four parts) as the second type of solvent that should be restricted, with a residual limit of 0.388%. The solvent residue of the final product obtained must reach the specified limit, and the post-treatment process is complicated, time-consuming and labor-intensive, and the production cost is correspondingly increased. The invention finds a suitable solvent on the basis of the synthetic route to prepare a medicinal crystal form, and has obvious advantages in both the method and the process operation steps.The synthetic route is as follows:

Comparative Example 1, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4- Preparation of chlorophenyl]glucosamine (Compound 3)Under nitrogen protection, 1.0 mol/L di-n-butylmagnesium-n-heptane solution (16 mL) was cooled to 0 ° C, and 1.6 mol/L n-hexane lithium n-hexane solution (10 mL) was slowly added dropwise. After the addition was completed, 0 ° C After stirring for 15 h, dry n-butyl ether (2.5 mL) was added to prepare a solution of di-n-butyl-n-hexylmagnesium lithium solution, which was calibrated with iodine and stored for use.Zinc bromide (2.7 g) and lithium bromide (1.04 g) were added with n-butyl ether (20 mL), heated to 50 ° C for 4 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (8 mL) and n-butyl ether (5 mL) under nitrogen, cooled to 0 ° C, and 0.61 mol/L was added dropwise. n-Butyl-n-hexylmagnesium lithium solution (13.1 mL), after the addition is completed, the reaction was kept at 0 ° C for 48 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution were added, and the reaction was kept at 0 ° C for 1 h, and added 2 , 3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (25 mL), heated to 100 ° C to stir the reaction, after TLC detection reaction, add 1 mol / L diluted hydrochloric acid (60 mL), taken after stirring extraction, the organic phase was washed with water (40 mL), then washed with saturated brine (40 mL), dried over anhydrous Na 2 SO 4, concentrated under reduced pressure, column chromatography (petroleum ether / Ethyl acetate = 20:1) 10.38 g of Compound 3 as a pale yellow oil. Yield: 46%. Purity: 99.02%. The organozinc reagent prepared by the method has an iodine calibration yield of 78%.The calibration method of the concentration of the prepared organic zinc reagent: accurately weighed iodine (1 mmol), placed in a three-necked flask, replaced nitrogen, and added anhydrous 0.5 mol/L LiCl tetrahydrofuran solution (5 mL), stirred and dissolved, and cooled to 0 ° C. The prepared organozinc reagent was slowly added dropwise until the color of the brownish yellow solution disappeared.Example 2 (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (2.25 g) and lithium bromide (0.87 g) were added with n-butyl ether (30 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and n-butyl ether (10 mL) under nitrogen, cooled to -20 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (14mL), control the internal temperature does not exceed -10 ° C, after the completion of the addition, the temperature is incubated at -20 ° C for 0.5 h, adding the above-mentioned spare zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at 20 ° C for 3 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (11.59g) toluene (50mL) solution, heat to 120 ° C and stir the reaction for 4h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (40 mL), water (20 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.8 g of Compound 3 as a white solid was obtained in a yield: 72.42%. Purity: 99.47%. Melting point: 99.5 to 101.6 °C. (The organic zinc reagent prepared by this method was iodine-calibrated in a yield of 98.5%.) ESI-MS (m/z): 767.30 [M+Na] + . 1 H-NMR (400 MHz, CDCl 3 ): δ 7.33 (1H, d), 7.14-7.17 (2H, m), 7.05 (2H, d), 6.79-6.81 (2H, dd), 5.39 (1H, t ), 5.21-5.31 (2H, m), 4.33 (1H, d), 4.17-4.20 (1H, dd), 3.94-4.11 (5H, m), 3.79-3.83 (1H, m), 1.39 (3H, t ), 1.20 (9H, s), 1.16 (9H, s), 1.11 (9H, s), 0.86 (9H, s).Example 3, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3) PrepareZinc bromide (3.38 g) and lithium bromide (1.3 g) were added with n-butyl ether (40 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (20 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -50 ° C, and slowly added dropwise 2.5 mol / L-butyllithium hexane solution (8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -50 ° C for 10 h, adding the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at -20 ° C for 10 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (34.77g) toluene (80mL) solution, heat to 100 ° C and stir the reaction for 24h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (60 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.854 g of Compound 3 as a white solid. Yield: 72.81%. Purity: 99.53%.Example 4, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)N-butyl ether (50 mL) was added to zinc iodide (3.19 g) and lithium iodide (1.34 g), and the mixture was heated to 50 ° C for 1.5 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -60 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -60 ° C for 5 h, and the above-mentioned alternate zinc iodide and lithium iodide n-butyl ether solution is added. The reaction was stirred at 25 ° C for 1 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (23.2g) toluene (50mL) solution, heat to 140 ° C reflux reaction for 0.5h, after TLC detection reaction was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous SO 4 Na 2, concentrated by weight of n-heptane (15mL) and methanol (60 mL) Crystallization gave 10.51 g of Compound 3 as a white solid, yield 70.5%. Purity: 99.41%.Example 5, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)To the zinc bromide (2.25 g) and lithium bromide (0.87 g), cyclopentyl methyl ether (30 mL) was added, and the mixture was heated to 50 ° C for 3 hours, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and cyclopentyl methyl ether (10 mL) under nitrogen, cooled to -5 ° C, and slowly added dropwise. Mol / L n-hexyl lithium n-hexane solution (12.5mL), control the internal temperature does not exceed 0 ° C, after the addition is completed, the reaction is kept at -5 ° C for 3 h, adding the above-mentioned spare zinc bromide and lithium bromide cyclopentyl methyl ether The solution was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (40 mL) was added and heated to 80 ℃ reaction was stirred 6h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous 2 SO 4 Na, and concentrated under reduced pressure, Recrystallization of n-heptane (15 mL) and methanol (60 mL) gave 8.15 g of Compound 3 as a white solid. Purity: 99.39%.Example 6, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (4.5 g) and lithium bromide (1.74 g) were added with n-butyl ether (60 mL), heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -30 ° C, and slowly added dropwise 2.5 mol / L-butyllithium n-hexane solution (8.4mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -30 ° C for 3 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution is added. The reaction was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (50 mL) was added and heated to 120 ° C for stirring. the reaction 4h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure, n-heptyl Recrystallization of the alkane (15 mL) and methanol (60 mL) gave 10.38 g of Compound 3 as a white solid. Purity: 99.54%.Example 7, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Methyl bromide (40 mL) was added to zinc bromide (2.25 g) and lithium bromide (0.87 g), and the mixture was heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL), methyl tert-butyl ether (15 mL), cooled to -40 ° C, and slowly added dropwise. 1.6mol/L n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -40 ° C for 4 h, and the above-mentioned alternate zinc bromide and lithium bromide are added. The butyl ether solution was incubated at 5 ° C for 7 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (50 mL) was added and heated. to 90 deg.] C the reaction was stirred 8h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (40 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure Recrystallization from n-heptane (15 mL) and methanol (60 mL) gave 9.41 g of Compound 3 as a white solid. Purity: 99.42%. Example 8. Preparation of dapagliflozin (S)-1,2-propanediol monohydrate eutectic (Compound 1)To the compound 3 (37.27 g), methanol (190 mL) was added, and sodium methoxide (10.8 g) was added thereto, and the mixture was heated under reflux for 3 hours. After the TLC reaction was completed, methanol was concentrated, and isopropyl acetate (100 mL) was added to the residue, and water was added. (60 mL), extracted with stirring and the organic phase washed with water (50 mL). (S)-1,2-propanediol (3.8g) and water (0.9g) were added to the organic phase, stirred until it was dissolved, and n-heptane (200 mL) was added, and the mixture was stirred for 2 hours under ice-cooling, suction filtration, filter cake Washing with n-heptane and drying at 30 ° C gave 23.89 g of Compound 1 as a white solid. Yield: 95%. Purity: 99.79%. Melting point: 69.1 to 75.6 °C. The product obtained was subjected to KF = 3.74% (theoretical value: 3.58%). ESI-MS (m/z): 431.22 [M+Na] + . 1 H-NMR (400 MHz, CD 3 OD): δ 7.33 – 7.37 (2H, m), 7.28-7.30 (1H, dd), 7.11 (2H, d), 6.80-6.83 (2H, dd), 4.1 ( 1H, d), 3.98-4.05 (4H, m), 3.88-3.91 (1H, dd), 3.74-3.82 (1H, m), 3.68-3.73 (1H, m), 3.37-3.49 (5H, m), 3.28-3.34 (1H, m), 1.37 (1H, t), 1.15 (3H, d).The crystal form of the obtained product was subjected to thermogravimetric analysis (TGA) by a Universal V4.7A TA instrument, and the TGA curve (Fig. 1) showed a weight loss of about 18.52% from about room temperature to about 240 ° C. The original form Ia crystal form The TGA plot shows a value of 18.7%.The crystal form of the obtained product was subjected to differential scanning calorimetry (DSC) by a Universal V4.7A TA instrument, and the DSC curve (Fig. 2) showed endotherm in the range of about 60 ° C to 85 ° C. The DSC plot shows a range of approximately 50 ° C to 78 ° C.
The crystal form of the obtained product was examined by a Bruker D8advance instrument for powder X-ray diffraction (PXRD), and the 2X value of the PXRD pattern (Fig. 3) (CuKα).

There are characteristic peaks at 3.749°, 7.52°, 7.995°, 8.664°, 15.134°, 15.708°, 17.069°, 18.946°, 20.049°, which are completely consistent with the characteristic peaks of the PXRD pattern of the Ia crystal form in the original patent.In combination with the nuclear magnetic data and melting point of the prepared crystal form, the crystal form of the product (Compound 1) obtained by the present invention is consistent with the pharmaceutically acceptable crystalline form Ia reported in the original patent.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleCN101479287A *2006-06-282009-07-08布里斯托尔-迈尔斯斯奎布公司Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetesCN104496952A *2014-11-282015-04-08深圳翰宇药业股份有限公司Synthesis method of dapagliflozinCN105153137A *2015-09-172015-12-16上海应用技术学院Preparation method of empagliflozinFamily To Family CitationsCN104829572B *2014-02-102019-01-04江苏豪森药业集团有限公司Dapagliflozin novel crystal forms and preparation method thereofCN105399735A *2015-12-292016-03-16上海应用技术学院Empagliflozin intermediate, and preparation method and application thereof* Cited by examiner, † Cited by third party
Non-Patent Citations
TitleCHEN DEJIN ET AL., CHINA MASTER’S THESES FULL-TEXT DATABASE, ENGINEERING TECHNOLOGY I, vol. B016-731, no. 3, 15 March 2016 (2016-03-15) *LEMAIRE S. ET AL.: “Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents”, PURE APPL. CHEM., vol. 86, no. 3, 4 March 2014 (2014-03-04), pages 329 – 333 *LEMAIRE S. ET AL.: “Stereoselective C-Glycosylation Reactions with Arylzinc Reagents”, ORGANIC LETTERS, vol. 14, no. 6, 2 March 2012 (2012-03-02), pages 1480 – 1483, XP055069093 ** Cited by examiner, † Cited by third partyCLIP
Chemical Synthesis
Dapagliflozin propanediol hydrate, an orally active sodium glucose cotransporter type 2 (SGLT-2) inhibitor, was developed by Bristol-Myers Squibb (BMS) and AstraZeneca for the once-daily treatment of type 2 diabetes. As opposed to competitor SGLT-2 inhibitors, dapagliflozin was not associated with renal toxicity or long-term deterioration of renal function in phase III clinical trials. The drug exhibits excellent SGLT2 potency with more than 1200 fold selectivity over the SGLT1 enzyme.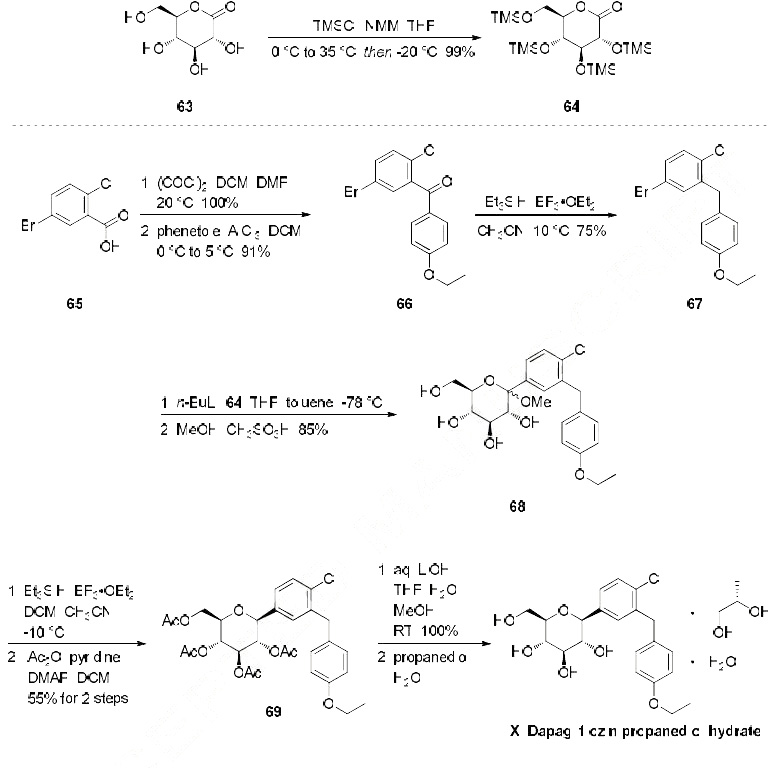
Dapagliflozin propanediol monohydrate
PAPER
https://link.springer.com/article/10.1007/s12039-020-1747-x

PATENTS
WO 2010138535
WO 2011060256
WO 2012041898
WO 2012163990
WO 2013068850
WO 2012163546
WO 2013068850
WO 2013079501

The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7

dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .

formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2

formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
PATENT
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
http://www.google.com/patents/US20120282336
http://www.tga.gov.au/pdf/auspar/auspar-dapagliflozin-propanediol-monohydrate-130114.pdf
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
PAPER
In Vitro Characterization and Pharmacokinetics of Dapagliflozin …
dmd.aspetjournals.org/content/…/DMD29165_supplemental_data_.doc
Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)
-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33
(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),
6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,
1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,
140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,
64.5, 63.1, 39.2, 15.2.
HRMS calculated for C21H25ClNaO6 (M+Na)+
For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
: 431.1237; found 431.1234. Anal. Calcd
SECOND SETJ. Med. Chem., 2008, 51 (5), pp 1145–1149DOI: 10.1021/jm701272q
1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);
13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
HPLC
- HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
– Eluent A: 0.1 % formic acid in water
– Eluent B: 0.1 % formic acid in acetonitrile
– Injection volume: 3 mL
– Flow: 0.7 mL/min
– Gradient:Time [min][%] B0.02525.06526.07029.07029.52535.025……………………..
PATENT
http://www.google.com/patents/WO2013068850A2?cl=en
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))

[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.56-7.54 (m, 2H), 7.43-7.31 (m, 13H), 7.29-7.22 (m, 6H), 7.07- 7.04 (m, 2H), 7.00 (d, J= 2.0 Hz, IH), 6.87 (dd, J= 8.4, 2.0 Hz, IH), 6.83-6.81 (m, 2H), 4.18 (d, J= 9.6 Hz, IH), 4.02 (q, J= 6.9 Hz, 2H), 3.96 (d, J= 10.8 Hz, 2H), 3.86 (ddd, J= 11.3, 7.7, 1.1 Hz, IH), 3.76 (ddd, J= 8.4, 8.4, 4.8 Hz, IH), 3.56 (ddd, J= 9.0, 6.4, 2.4 Hz, IH), 3.50 (dd, J=
I I.4, 5.4 Hz, IH), 3.44 (dd, J= 9.4, 8.6 Hz, IH), 3.38 (dd, J= 8.8, 8.8 Hz, IH), 1.70 (dd, J= 7.8, 5.4 Hz, IH, OH), 1.42 (t, J= 6.8 Hz, 3H), 1.21 (d, J= 5.2 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 157.4 (C), 138.8 (C), 137.4 (C), 136.3 (CH x2), 136.1 (CH x2), 135.2 (CH x2), 135.0 (C), 134.9 (CH x2), 134.8 (C), 134.2 (C), 132.8 (C), 132.0 (C), 131.6 (CH), 131.1 (C), 129.9 (CH x2), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.58 (CH x2), 127.57 (CH x2), 127.54 (CH x2), 127.31 (CH), 127.28 (CH x2), 114.4 (CH x2), 82.2 (CH), 80.5 (CH), 79.3 (CH), 76.3 (CH), 72.7 (CH), 63.4 (CH2), 62.7 (CH2), 38.2 (CH2), 27.2 (CH3 x3), 26.6 (CH3 x3), 19.6 (C), 19.2 (C), 14.9 (CH3). EXAMPLE 25 -Synthesis of dapagliflozin ((25,3R,4R,55,6/?)-2-[4-chloro-3-(4- ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; (Ij))

IVj’ U
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
1H NMR (400 MHz, CD3OD) δ 7.37-7.34 (m, 2H), 7.29 (dd, J= 8.2, 2.2 Hz, 1H), 7.12-7.10 (m, 2H), 6.82-6.80 (m, 2H), 4.10 (d, J= 9.6 Hz, 2H), 4.04 (d, J= 9.2 Hz, 2H), 4.00 (q, J= 7.1 Hz, 2H), 3.91-3.87 (m, 1H), 3.73-3.67(m, 1H), 3.47-3.40 (m, 3H), 3.31-3.23 (m, 2H), 1.37 (t, J= 7.0 Hz, 3H);
13C NMR (100 MHz, CD3OD) δ 157.4 (C), 138.6 (C), 138.5 (C), 133.1 (C), 131.5 (C), 130.5 (CH), 129.4 (CH x2), 128.7 (CH), 126.8 (CH), 114.0 (CH x2), 80.5 (CH), 80.8 (CH), 78.3 (CH), 75.0 (CH), 70.4 (CH), 63.0 (CH2), 61.7 (CH2), 37.8 (CH2), 13.8 (CH3);
LCMS (ESI) m/z 426 (100, [M+NH4]+), 428 (36, [M+NH4+2]+), 447 (33, [M+K]+).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)

III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- “D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
PATENT
WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
LINK>>> (WO2016147197) A NOVEL PROCESS FOR PREPARING (2S,3R,4R,5S,6R)-2-[4-CHLORO-3-(4-ETHOXYBENZYL)PHENY 1] -6-(HY DROXY METHYL)TETRAHYDRO-2H-PY RAN-3,4,5-TRIOL AND ITS AMORPHOUS FORM
PATENT
PATENT
WO2016018024, CRYSTALLINE COMPOSITE COMPRISING DAPAGLIFLOZIN AND METHOD FOR PREPARING SAME
HANMI FINE CHEMICAL CO., LTD. [KR/KR]; 59, Gyeongje-ro, Siheung-si, Gyeonggi-do 429-848 (KR)
Dapagliflozin, sold under the brand name Farxiga among others, is a medication used to treat type 2 diabetes and, with certain restrictions, type 1 diabetes.[2] It is also used to treat adults with certain kinds of heart failure.[3][4][5]
Common side effects include hypoglycaemia (low blood sugar), urinary tract infections, genital infections, and volume depletion (reduced amount of water in the body).[6] Diabetic ketoacidosis is a common side effect in type 1 diabetic patients.[7] Serious but rare side effects include Fournier gangrene.[8] Dapagliflozin is a sodium-glucose co-transporter-2 (SGLT-2) inhibitor and works by removing sugar from the body with the urine.[9]
It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. In 2018, it was the 227th most commonly prescribed medication in the United States, with more than 2 million prescriptions.[10][11]
Medical uses
Dapagliflozin is used along with diet and exercise to improve glycemic control in adults with type 2 diabetes and to reduce the risk of hospitalization for heart failure among adults with type 2 diabetes and known cardiovascular disease or other risk factors.[12][3] It appears more useful than empagliflozin.[13][verification needed]
In addition, dapagliflozin is indicated for the treatment of adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3][4][5] It is also indicated to reduce the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14]
In the European Union it is indicated in adults:
- for the treatment of insufficiently controlled type 2 diabetes mellitus as an adjunct to diet and exercise:
- as monotherapy when metformin is considered inappropriate due to intolerance;
- in addition to other medicinal products for the treatment of type 2 diabetes;
- for the treatment of insufficiently controlled type 1 diabetes mellitus as an adjunct to insulin in patients with BMI ≥ 27 kg/m2, when insulin alone does not provide adequate glycaemic control despite optimal insulin therapy; and
- for the treatment of heart failure with reduced ejection fraction.[5]
Adverse effects
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Rarely, use of an SGLT2 drug, including dapagliflozin, is associated with necrotizing fasciitis of the perineum, also called Fournier gangrene.[15]
Dapagliflozin is also associated with hypotensive reactions. There are concerns it may increase the risk of diabetic ketoacidosis.[16]
Dapagliflozin can cause dehydration, serious urinary tract infections and genital yeast infections.[3] Elderly people, people with kidney problems, those with low blood pressure, and people on diuretics should be assessed for their volume status and kidney function.[3] People with signs and symptoms of metabolic acidosis or ketoacidosis (acid buildup in the blood) should also be assessed.[3] Dapagliflozin can cause serious cases of necrotizing fasciitis of the perineum (Fournier gangrene) in people with diabetes and low blood sugar when combined with insulin.[3]
To lessen the risk of developing ketoacidosis (a serious condition in which the body produces high levels of blood acids called ketones) after surgery, the FDA has approved changes to the prescribing information for SGLT2 inhibitor diabetes medicines to recommend they be stopped temporarily before scheduled surgery. Canagliflozin, dapagliflozin, and empagliflozin should each be stopped at least three days before, and ertugliflozin should be stopped at least four days before scheduled surgery.[17]
Symptoms of ketoacidosis include nausea, vomiting, abdominal pain, tiredness, and trouble breathing.[17]
Use is not recommended in patients with eGFR < 45ml/min/1.73m2, though data from 2021 shows the reduction in the kidney failure risks in people with chronic kidney disease using dapagliflozin.[18]
Mechanism of action
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2) which are responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter mechanism causes blood glucose to be eliminated through the urine.[19] In clinical trials, dapagliflozin lowered HbA1c by 0.6 versus placebo percentage points when added to metformin.[20]
Regarding its protective effects in heart failure, this is attributed primarily to haemodynamic effects, where SGLT2 inhibitors potently reduce intravascular volume through osmotic diuresis and natriuresis. This consequently may lead to a reduction in preload and afterload, thereby alleviating cardiac workload and improving left ventricular function.[21]
Selectivity
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/L), so that the drug does not interfere with intestinal glucose absorption.[22]
Names
Dapagliflozin is the International nonproprietary name (INN),[23] and the United States Adopted Name (USAN).[24]
There is a fixed-dose combination product dapagliflozin/metformin extended-release, called Xigduo XR.[25][26][27]
In July 2016, the fixed-dose combination of saxagliptin and dapagliflozin was approved for medical use in the European Union and is sold under the brand name Qtern.[28] The combination drug was approved for medical use in the United States in February 2017, where it is sold under the brand name Qtern.[29][30]
In May 2019, the fixed-dose combination of dapagliflozin, saxagliptin, and metformin hydrochloride as extended-release tablets was approved in the United States to improve glycemic control in adults with type 2 diabetes when used in combination with diet and exercise. The FDA granted the approval of Qternmet XR to AstraZeneca.[31] The combination drug was approved for use in the European Union in November 2019, and is sold under the brand name Qtrilmet.[32]
History
In 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued a positive opinion on the drug.[5]
Dapagliflozin was found effective in several studies in participants with type 2 and type 1 diabetes.[5] The main measure of effectiveness was the level of glycosylated haemoglobin (HbA1c), which gives an indication of how well blood glucose is controlled.[5]
In two studies involving 840 participants with type 2 diabetes, dapagliflozin when used alone decreased HbA1c levels by 0.66 percentage points more than placebo (a dummy treatment) after 24 weeks.[5] In four other studies involving 2,370 participants, adding dapagliflozin to other diabetes medicines decreased HbA1c levels by 0.54-0.68 percentage points more than adding placebo after 24 weeks.[5]
In a study involving 814 participants with type 2 diabetes, dapagliflozin used in combination with metformin was at least as effective as a sulphonylurea (another type of diabetes medicines) used with metformin.[5] Both combinations reduced HbA1c levels by 0.52 percentage points after 52 weeks.[5]
A long-term study, involving over 17,000 participants with type 2 diabetes, looked at the effects of dapagliflozin on cardiovascular (heart and circulation) disease.[5] The study indicated that dapagliflozin’s effects were in line with those of other diabetes medicines that also work by blocking SGLT2.[5]
In two studies involving 1,648 participants with type 1 diabetes whose blood sugar was not controlled well enough on insulin alone, adding dapagliflozin 5 mg decreased HbA1c levels after 24 hours by 0.37% and by 0.42% more than adding placebo.[5]
Dapagliflozin was approved for medical use in the European Union in November 2012.[5] It is marketed in a number of European countries.[33]
Dapagliflozin was approved for medical use in the United States in January 2014.[34][14]
In 2020, the U.S. Food and Drug Administration (FDA) expanded the indications for dapagliflozin to include treatment for adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3] It is the first in this particular drug class, sodium-glucose co-transporter 2 (SGLT2) inhibitors, to be approved to treat adults with New York Heart Association’s functional class II-IV heart failure with reduced ejection fraction.[3]
Dapagliflozin was shown in a clinical trial to improve survival and reduce the need for hospitalization in adults with heart failure with reduced ejection fraction.[3] The safety and effectiveness of dapagliflozin were evaluated in a randomized, double-blind, placebo-controlled study of 4,744 participants.[3] The average age of participants was 66 years and more participants were male (77%) than female.[3] To determine the drug’s effectiveness, investigators examined the occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visits.[3] Participants were randomly assigned to receive a once-daily dose of either 10 milligrams of dapagliflozin or a placebo (inactive treatment).[3] After about 18 months, people who received dapagliflozin had fewer cardiovascular deaths, hospitalizations for heart failure, and urgent heart failure visits than those receiving the placebo.[3]
In July 2020, the FDA granted AstraZeneca a Fast Track Designation in the US for the development of dapagliflozin to reduce the risk of hospitalisation for heart failure or cardiovascular death in adults following a heart attack.[35]
In August 2020, it was reported that detailed results from the Phase III DAPA-CKD trial showed that AstraZeneca’s FARXIGA® (dapagliflozin) on top of standard of care reduced the composite measure of worsening of renal function or risk of cardiovascular (CV) or renal death by 39% compared to placebo (p<0.0001) in patients with chronic kidney disease (CKD) Stages 2-4 and elevated urinary albumin excretion. The results were consistent in patients both with and without type 2 diabetes (T2D)[36]
In April 2021, the FDA expanded the indications for dapagliflozin (Farxiga) to include reducing the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14] The efficacy of dapagliflozin to improve kidney outcomes and reduce cardiovascular death in people with chronic kidney disease was evaluated in a multicenter, double-blind study of 4,304 participants.[14]
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN
https://pharmacia.pensoft.net/article/70626/
Synthesis
Dapagliflozin is an approved drug by U.S. Food and Drug Administration (FDA). Dapagliflozin is a representative of SGLT-2 inhibitors, actively considered to cure diabetes type 2. Thus, methodology of dapagliflozin synthesis has rarely published (Ellsworth et al. 2002; Meng 2008). Scheme 1 have shown the general synthetic route for the synthesis of dapagliflozin. Gluconolactone 3 which was protected by trimethylsilyl TMS was treated with aryl lithium. Aryl lithium was obtained by reacting aryl bromide 2 (exchange of Li/Br) with n-BuLi. Methyl C-aryl glucoside 4 was produced by treatment of resulting mixture with methane sulfonic acid in the presence of methanol. Compound 4 was subjected to acetylation in the presence of Ac2O, resulted in the formation of 5 followed by reduction of 5 to 6 with the help of Et3SiH and BF3.OEt2. Finally, dapagliflozin 1 was produced via hydrolysis of 6 by LiOH (Deshpande et al. 2008; Meng 2008).
Ellsworth B, Washburn WN, Sher PM, Wu G, Meng W (2002) C-Aryl glucoside SGLT2 inhibitors and method, Google Patents. https://patents.google.com/patent/US6515117B2/en
Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN (2008) “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. ” Journal of Medicinal Chemistry 51(5): 1145–1149. https://doi.org/10.1021/jm701272q
Deshpande PP, Ellsworth BA, Singh J, Denzel TW, Lai C, Crispino G, Randazzo ME, Gougoutas JZ (2008) Methods of producing C-aryl glucoside SGLT2 inhibitors, Google Patents. https://patents.google.com/patent/US7375213B2/en

Jun et al. has reported a few improvements to the scheme 1. In the improved methodology scheme 2, trimethylsilyl chloride was added to gluconolactone 7 in the presence of N-methylmorpholine and tetrahydrofuran THF (Horton et al. 1981), followed by the formation of persilyated lactone 3. After completing the reaction of aryl bromide 2 with n-BuLi, added to persilyated lactone 3. Intermediate lactol 8 was produced by treating resulting reaction mixture with trifluoroacetic acid in aqueous form. Then ethyl C-aryl glycoside 9 yielded when subsequently compound 8 was subjected to methane-sulfonic acid in ethyl alcohol. Crude product 9 in the form of oil was secured after the screening of solvents. Jun et al. proposed that more than 98% pure 9 was collected as crystalline solvate after the crystallization of crude oil from n-propanol and n-heptane mixture (Yu et al. 2019). Moreover, Wang et al. proposed that a high extent of diastereoselectivity obtained after the reduction of tetra-O-unprotected methyl C-aryl glucoside by utilizing Et3SiH and BF3.Et2O (Wang et al. 2014). The nature of active pharmaceutical ingredient is amorphous foam which is isolated after the reduction of 9. Production of cocrystalline complex facilitate the isolation and purification of API (Deng et al. 2017). It is concluded that more than 99.7% pure dapagliflozin produced in overall 79% yield, after the crystallization of a mixture consists of n-heptane and ethyl acetate (Yu et al. 2019).
Zheng et al. designed the production methodology of dapagliflozin by introducing NO donor group at the last steps of general route of dapagliflozin synthesis (scheme 1). Novel hybrids achieved by the combination of dapagliflozin and NO donor, having excellent dual characteristics of anti-hyperglycemic and anti-thrombosis. The figure 2 represent the modifiable site (4-position) of dapagliflozin.
Horton D, Priebe W (1981) “Synthetic routes to higher-carbon sugars. Reaction of lactones with 2-lithio-, 3-dithiane. ” Carbohydrate Research 94(1): 27–41. https://doi.org/10.1016/S0008-6215(00)85593-7
Yu J, Cao Y, Yu H, Wang JJ (2019) “A Concise and Efficient Synthesis of Dapagliflozin. ” Organic Process Research & Development 23(7): 1458–1461. https://doi.org/10.1021/acs.oprd.9b00141
Wang X-j, Zhang L, Byrne D, Nummy L, Weber D, Krishnamurthy D, Yee N, Senanayake CH (2014) “Efficient synthesis of empagliflozin, an inhibitor of SGLT-2, utilizing an AlCl3-promoted silane reduction of a β-glycopyranoside. ” Organic Letters 16(16): 4090–4093. https://doi.org/10.1021/ol501755h
Deng J-H, Lu T-B, Sun CC, J-M Chen (2017) “Dapagliflozin-citric acid cocrystal showing better solid state properties than dapagliflozin. ” European Journal of Pharmaceutical Sciences 104: 255–261. https://doi.org/10.1016/j.ejps.2017.04.008

Scheme 3 has shown the formation strategy of new hybrids of nitric oxide with dapagliflozin. During the synthesis process, the compound 6 was treated with BBr3 that result in the formation of phenol 10, which was further subjected to condensation with bromoalkane and then undergo hydrolysis and produce 11 intermediates. Target compound was obtained by the reacting 11 with silver nitrate in acetonitrile (Li et al. 2018).
Li Z, Xu X, Deng L, Liao R, Liang R, Zhang B, Zhang LJB (2018) Design, synthesis and biological evaluation of nitric oxide releasing derivatives of dapagliflozin as potential anti-diabetic and anti-thrombotic agents. Bioorganic & Medicinal Chemistry 26(14): 3947–3952. https://doi.org/10.1016/j.bmc.2018.06.017

Lin et al. fabricated green route (scheme 4) for the production of dapagliflozin. 5-bromo-2-chlorobenzoic acid 12 and gluconolactone were utilized to initiate the synthesis. By taking BF3.Et2O in catalytic amount to produce 13, overall yield of 76% was obtained in one-pot way via considering the Friedel-Craft acylation and ketallization. There was no need to do work-up operations to separate the diaryl ketal 13 as it was easily crystallized from the mixture. Compound 14 was produce as a result of condensation between 13 and 3. Overall yield of 68% of compound 15 was produced by the deprotection of silyl group in ethyl alcohol media. In THF presence, single crystals of 15 was achieved and characterized by XRD-analysis. High yield of dapagliflozin was obtained after the reduction of 15 that was carried out by triethylsilane in the presence of boron trifluoride diethyl etherate in dichloromethane. Upon crystallization from the mixture having heptane and ethyl acetate, greater than 98% pure dapagliflozin was produced by green synthetic pathway (Hu et al. 2019).
Hu L, Zou P, Wei W, Yuan X-M, Qiu X-L, Gou S-H (2019) “Facile and green synthesis of dapagliflozin. ” Synthetic Communications 49(23): 3373–3379. https://doi.org/10.1080/00397911.2019.1666283
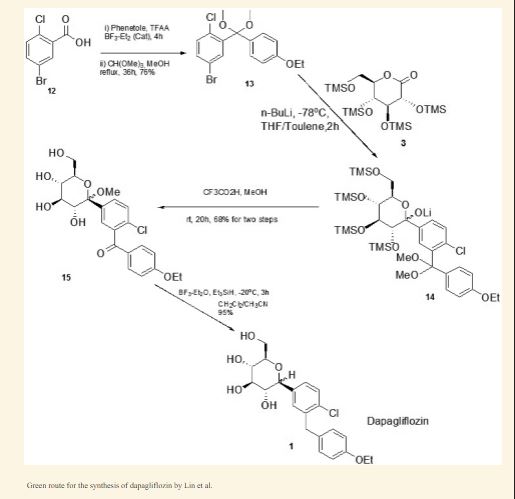
PAPER
A Concise and Efficient Synthesis of Dapagliflozin
Cite this: Org. Process Res. Dev.2019, 23, 7, 1458–1461
Publication Date:June 27, 2019
https://doi.org/10.1021/acs.oprd.9b00141
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00141
file:///C:/Users/Inspiron/Downloads/op9b00141_si_001.pdf SUPP
Abstract

A concise and efficient synthesis of the SGLT-2 inhibitor dapagliflozin (1) has been developed. This route involves ethyl C-aryl glycoside 9 as the key intermediate, which is easily crystallized and purified as the crystalline n-propanol solvate with high purity (>98.5%). The tetra-O-unprotected compound 9 could be directly reduced to crude dapagliflozin with high diastereoselectivity. The final pure API product 1 was isolated and purified with high purity (>99.7%). The process has been implemented on a multikilogram scale.
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetra-hydro-2Hpyran-3,4,5-triol (1)
Compound 1 has a melting point of 88.3oC.
The MsOH solution should be used immediately to avoid sulfonate formation and we have no data on the presence of sulfonates in the API.
1HNMR (400 MHz, DMSO-d6) δ (ppm) 7.32-7.37 (m, 2H), 7.22-7.24 (m, 1H), 7.08-7.10 (m, 2H), 6.80-6.84 (m, 2H), 4.94-4.96 (m, 2H), 4.82-4.83 (d, J=5.6 Hz, 1H), 4.43-4.46 (m, 1H), 3.93-4.02 (m, 5H), 3.68-3.73 (m, 1H), 3.42-3.48 (m, 1H), 3.10-3.28 (m, 4H), 1.27-1.30 (m, 3H).
13C NMR (100 MHz, DMSOd6) δ(ppm) 157.38, 140.14, 138.27, 132.40, 131.69, 131.27, 130.03, 129.13, 127.81, 114.78, 81.67, 81.18, 78.80, 75.19, 70.80, 63.37, 61.86, 38.14, 15.15.
IR(KBr): 3415, 2979, 2918, 1617, 1512, 1475, 1391, 1242, 1103, 1045, 913, 825 cm-1.
MS(m/z):431.12[M + Na]+ .
1H NMR and 13C NMR spectra for Compound
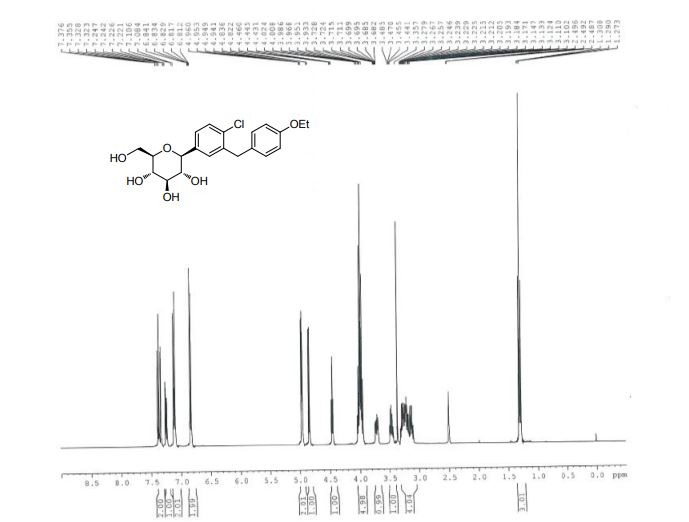
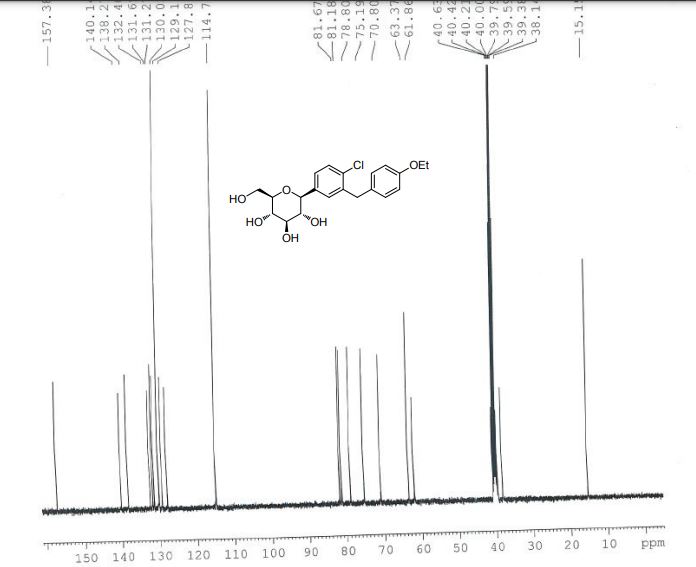
IR and Mass spectra for Compound
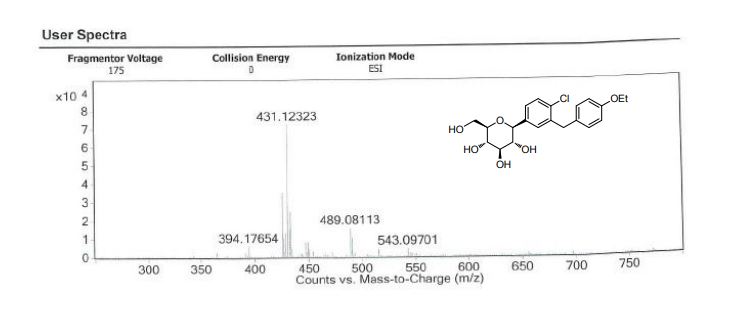
DSC spectra for Compound
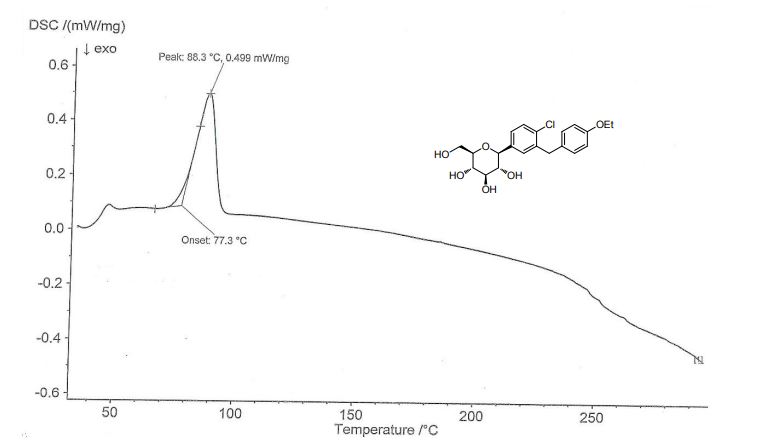
PAPER
https://pubs.acs.org/doi/10.1021/ol300220p
rg. Lett.2012, 14, 6, 1480–1483
Publication Date:March 2, 2012
https://doi.org/10.1021/ol300220p
Abstract

A general, transition-metal-free, highly stereoselective cross-coupling reaction between glycosyl bromides and various arylzinc reagents leading to β-arylated glycosides is reported. The stereoselectivity of the reaction is explained by invoking anchimeric assistance via a bicyclic intermediate. Stereochemical probes confirm the participation of the 2-pivaloyloxy group. Finally, this new method was applied to a short and efficient stereoselective synthesis of Dapagliflozin and Canagliflozin.
CROSS REF J. Med. Chem 2008, 51, 1145
H NMR (360 MHz, MeOD): δ 7.35‐7.28 (m, 3H); 7.09 (d, J=8.4Hz, 2H), 6.80 (d, J=8.8Hz, 2H), 4.09 (d, J=9.5Hz, 1H); 4.03‐3.96 (m, 4H); 3.89‐3.85 (m, 1H); 3.71‐3.66 (m, 1H); 3.45‐3.36 (m, 4H), 3.28‐3.26 (m, 2H); 1.36 (t, J=6.9Hz, 3H).
13CNMR (90 MHz, MeOD): δ 14.80, 38.31, 61.80, 63.38, 69.88, 74.67, 77.93, 79.35, 81.08, 114.48, 126.35, 128.20, 129.01, 129.72, 130.62, 131.14, 134.15, 137.04, 139.01, 157.31.
PAPER
https://pubs.acs.org/doi/10.1021/ja00199a028
Research
One study found that it had no benefit on heart disease risk or overall risk of death in people with diabetes.[37] Another study found that in heart failure with a reduced ejection fraction, dapagliflozin reduced the risk of worsening of heart failure or progression to death from cardiovascular causes, irrespective of diabetic status.[38]
References
- ^ Jump up to:a b “Dapagliflozin (Farxiga) Use During Pregnancy”. Drugs.com. 30 August 2018. Retrieved 5 May 2020.
- ^ Jump up to:a b “Farxiga- dapagliflozin tablet, film coated”. DailyMed. 3 February 2020. Retrieved 5 May 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves new treatment for a type of heart failure”. U.S. Food and Drug Administration (FDA) (Press release). 5 May 2020. Retrieved 5 May 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b National Institute for Health and Care Excellence (24 February 2021). “Dapagliflozin for treating chronic heart failure with reduced ejection fraction”. NICE Technology Appraisal Auidance [TA679]. NICE. Retrieved 9 May 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “Forxiga EPAR”. European Medicines Agency (EMA). Retrieved 17 February 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Ptaszynska, Agata; Johnsson, Kristina M.; Parikh, Shamik J.; De Bruin, Tjerk W. A.; Apanovitch, Anne Marie; List, James F. (2014). “Safety Profile of Dapagliflozin for Type 2 Diabetes: Pooled Analysis of Clinical Studies for Overall Safety and Rare Events”. Drug Safety. 37 (10): 815–829. doi:10.1007/s40264-014-0213-4. PMID 25096959. S2CID 24064402.
- ^ Dandona, Paresh; Mathieu, Chantal; Phillip, Moshe; Hansen, Lars; Tschöpe, Diethelm; Thorén, Fredrik; Xu, John; Langkilde, Anna Maria; DEPICT-1 Investigators (2018). “Efficacy and Safety of Dapagliflozin in Patients with Inadequately Controlled Type 1 Diabetes: The DEPICT-1 52-Week Study”. Diabetes Care. 41(12): 2552–2559. doi:10.2337/dc18-1087. PMID 30352894. S2CID 53027785.
- ^ Hu, Yang; Bai, Ziyu; Tang, Yan; Liu, Rongji; Zhao, Bin; Gong, Jian; Mei, Dan (2020). “Fournier Gangrene Associated with Sodium-Glucose Cotransporter-2 Inhibitors: A Pharmacovigilance Study with Data from the U.S. FDA Adverse Event Reporting System”. Journal of Diabetes Research. 2020: 1–8. doi:10.1155/2020/3695101. PMC 7368210. PMID 32695827.
- ^ FARXIGA- dapagliflozin tablet, film coated. DailyMed. Retrieved 6 May 2021.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Dapagliflozin – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ “FDA Approves Farxiga to Treat Type 2 Diabetes” (Press release). U.S. Food and Drug Administration (FDA). 8 January 2014. Archived from the original on 9 January 2014. Retrieved 15 November 2016. This article incorporates text from this source, which is in the public domain.
- ^ Zelniker TA, Wiviott SD, Raz I, et al. (January 2019). “SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials”. Lancet. 393(10166): 31–9. doi:10.1016/S0140-6736(18)32590-X. PMID 30424892. S2CID 53277899.
However, in patients with atherosclerotic cardiovascular disease, the effect of empagliflozin on cardiovascular death was more pro-nounced than that of canagliflozin or dapagliflozin
- ^ Jump up to:a b c d “FDA Approves Treatment for Chronic Kidney Disease”. U.S. Food and Drug Administration (FDA) (Press release). 30 April 2021. Retrieved 30 April 2021. This article incorporates text from this source, which is in the public domain.
- ^ “FDA warns about rare occurrences of a serious infection of the genital area with SGLT2 inhibitors for diabetes”. U.S. Food and Drug Administration (FDA). 9 February 2019. This article incorporates text from this source, which is in the public domain.
- ^ “SGLT2 inhibitors: Drug Safety Communication – FDA Warns Medicines May Result in a Serious Condition of Too Much Acid in the Blood”. U.S. Food and Drug Administration (FDA). 15 May 2015. Archived from the original on 27 October 2016. Retrieved 15 November 2016. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “FDA revises labels of SGLT2 inhibitors for diabetes to include warning”. U.S. Food and Drug Administration. 19 March 2020. Retrieved 6 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ McMurray, John J.V.; Wheeler, David C.; Stefánsson, Bergur V.; Jongs, Niels; Postmus, Douwe; Correa-Rotter, Ricardo; Chertow, Glenn M.; Greene, Tom; Held, Claes; Hou, Fan-Fan; Mann, Johannes F.E.; Rossing, Peter; Sjöström, C. David; Toto, Roberto D.; Langkilde, Anna Maria; Heerspink, Hiddo J.L.; DAPA-CKD Trial Committees Investigators (2021). “Effect of Dapagliflozin on Clinical Outcomes in Patients with Chronic Kidney Disease, with and Without Cardiovascular Disease” (PDF). Circulation. 143 (5): 438–448. doi:10.1161/CIRCULATIONAHA.120.051675. PMID 33186054. S2CID 226948086.
- ^ “Life Sciences – Clarivate”. Clarivate. Archived from the original on 5 November 2007.
- ^ “UEndocrine: Internet Endocrinology Community”. uendocrine.com. Archived from the original on 5 February 2013.
- ^ Lan NS, Fegan PG, Yeap BB, Dwivedi G (October 2019). “The effects of sodium-glucose cotransporter 2 inhibitors on left ventricular function: current evidence and future directions”. ESC Heart Fail. 6 (5): 927–935. doi:10.1002/ehf2.12505. PMC 6816235. PMID 31400090.
- ^ Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59” (PDF). World Health Organization. 2008. p. 50. Retrieved 15 November 2016.
- ^ “Statement on a Nonproprietary Name Adopted by the USAN Council” (PDF). American Medical Association. Archived from the original (PDF) on 7 February 2012. Retrieved 15 November2016.
- ^ “US FDA Approves Once-Daily Xigduo XR Tablets for Adults with Type 2 Diabetes”. AstraZeneca. 30 October 2014.
- ^ “Drug Approval Package: Xigduo XR (dapagliflozin and metformin HCl) Extended-Release Tablets”. U.S. Food and Drug Administration (FDA). 7 April 2015. Retrieved 5 May 2020.
- ^ “Xigduo XR- dapagliflozin and metformin hydrochloride tablet, film coated, extended release”. DailyMed. 3 February 2020. Retrieved 5 May 2020.
- ^ “Qtern EPAR”. European Medicines Agency (EMA). Retrieved 7 May 2020.
- ^ “Drug Approval Package: Qtern (dapagliflozin and saxagliptin)”. U.S. Food and Drug Administration (FDA). 10 October 2018. Retrieved 8 May 2020.
- ^ “Qtern- dapagliflozin and saxagliptin tablet, film coated”. DailyMed. 24 January 2020. Retrieved 17 February 2020.
- ^ “Drug Approval Package: Qternmet XR”. U.S. Food and Drug Administration (FDA). 27 January 2020. Retrieved 17 February2020.
- ^ “Qtrilmet EPAR”. European Medicines Agency (EMA). Retrieved 30 March 2020.
- ^ “Forxiga”. Drugs.com. 4 May 2020. Retrieved 5 May 2020.
- ^ “Drug Approval Package: Farxiga (dapagliflozin) Tablets NDA #202293”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 5 May 2020.
- ^ “FARXIGA Granted Fast Track Designation in the US for Heart Failure Following Acute Myocardial Infarction Leveraging an Innovative Registry-Based Trial Design”. http://www.businesswire.com. 16 July 2020. Retrieved 20 July 2020.
- ^https://www.businesswire.com/news/home/20200830005009/en/FARXIGA-Demonstrated-Unprecedented-Reduction-Risk-Kidney-Failure
- ^ “Type 2 diabetes. Cardiovascular assessment of dapagliflozin: no advance”. Prescrire International. 29 (211): 23. January 2020. Retrieved 2 February 2020.
- ^ McMurray JJ, Solomon SD, Inzucchi SE, et al. (November 2019). “Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction”. New England Journal of Medicine. 381 (21): 1995–2008. doi:10.1056/NEJMoa1911303. PMID 31535829.
External links
- “Dapagliflozin”. Drug Information Portal. U.S. National Library of Medicine.
- “Dapagliflozin mixture with metformin hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
- “Dapagliflozin mixture with saxagliptin”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trials
- Clinical trial number NCT00528372 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled With Diet and Exercise” at ClinicalTrials.gov
- Clinical trial number NCT00643851 for “An Efficacy & Safety Study of BMS-512148 in Combination With Metformin Extended Release Tablets” at ClinicalTrials.gov
- Clinical trial number NCT00859898 for “Study of Dapagliflozin in Combination With Metformin XR to Initiate the Treatment of Type 2 Diabetes” at ClinicalTrials.gov
- Clinical trial number NCT00528879 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled on Metformin Alone” at ClinicalTrials.gov
- Clinical trial number NCT00660907 for “Efficacy and Safety of Dapagliflozin in Combination With Metformin in Type 2 Diabetes Patients” at ClinicalTrials.gov
- Clinical trial number NCT00680745 for “Efficacy and Safety of Dapagliflozin in Combination With Glimepiride (a Sulphonylurea) in Type 2 Diabetes Patients” at ClinicalTrials.gov
- Clinical trial number NCT01392677 for “Evaluation of Safety and Efficacy of Dapagliflozin in Subjects With Type 2 Diabetes Who Have Inadequate Glycaemic Control on Background Combination of Metformin and Sulfonylurea” at ClinicalTrials.gov
- Clinical trial number NCT00683878 for “Add-on to Thiazolidinedione (TZD) Failures” at ClinicalTrials.gov
- Clinical trial number NCT00984867 for “Dapagliflozin DPPIV Inhibitor add-on Study” at ClinicalTrials.gov
- Clinical trial number NCT00673231 for “Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin” at ClinicalTrials.gov
- Clinical trial number NCT02229396 for “Phase 3 28-Week Study With 24-Week and 52-week Extension Phases to Evaluate Efficacy and Safety of Exenatide Once Weekly and Dapagliflozin Versus Exenatide and Dapagliflozin Matching Placebo” at ClinicalTrials.gov
- Clinical trial number NCT02413398 for “A Study to Evaluate the Effect of Dapagliflozin on Blood Glucose Level and Renal Safety in Patients With Type 2 Diabetes (DERIVE)” at ClinicalTrials.gov
- Clinical trial number NCT01730534 for “Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58)” at ClinicalTrials.gov
- Clinical trial number NCT03036124 for “Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients With Chronic Heart Failure (DAPA-HF)” at ClinicalTrials.gov
///////////DAPAGLIFLOZIN, ダパグリフロジン, BMS 512148, TYPE 2 DIABETES, SGLT-2 Inhibitors, EU 2012, forxiga, FDA 2014, JAPAN 2014, DIABETES
- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- “FDA panel advises against approval of dapagliflozin”. 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;
- Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;
- Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ;
- Hecheng Huaxue , 18 (3), 389-392; 20105) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,.
- WO20100223136) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,.
- WO20100092437) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,.
- WO20090265378) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ;
- Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,.
- WO200800282410) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;..
- U.S. Pat Appl Publ,. 20,040,138,439
NEW DRUG APPROVALS
one time
$10.00
WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED


WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
LINK>>> (WO2016147197) A NOVEL PROCESS FOR PREPARING (2S,3R,4R,5S,6R)-2-[4-CHLORO-3-(4-ETHOXYBENZYL)PHENY 1] -6-(HY DROXY METHYL)TETRAHYDRO-2H-PY RAN-3,4,5-TRIOL AND ITS AMORPHOUS FORM
HARMAN FINOCHEM LIMITED [IN/IN]; 107, Vinay Bhavya Complex 159-A, C.S.T. Road Kalina, Mumbai 400098 Maharashtra (IN)
![]()
KADAM, Vijay Trimbak; (IN).
SAIKRISHNA; (IN).
CHOUDHARE, Tukaram Sarjerao; (IN).
MINHAS, Harpreet Singh; (IN).
MINHAS, Gurpreet Singh; (IN)

CHAIRMAN

HARPREET SINGH MINHAS
Owner, HARMAN FINOCHEM LIMITED

(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is sodium dependent glucose transporter (SGLT) which is currently under investigation for the treatment of type-2 diabetes. (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is marketed under the tradename Farxiga or Forxiga.
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is also known as D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4ethoxyphenyl)methyl]phenyl]-, (I S). (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3, 4,5 -triol is a white to off-white powder with a molecular formula of C2iH25C106 and a molecular weight of 408.87

Formula-I
US 6,515,117 B2 discloses (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol and its pharmaceutically acceptable salts. US 6,515,117 B2 also describes process for preparation of (2S,3R,4R,5S,6R)-2-[4- chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol which comprises reaction of 5-bromo-2-chloro-4′-ethoxydiphenylmethane with 2,3,4,6-tetra-O-trimethylsilyl- -D-glucolactone in presence of THF/Toluene, methansulfonic acid to yield o-methylglucoside product which further reacts with Et3SiH, BF3Et20 in presence of MDC and acetonitrile to yield yellow solidified foam which is dissolved in MDC, pyridine and followed by acetylation with acetic anhydride, DMAP to yield tetra acetylated- β-C-glucoside as a white solid which is further deprotected with LiOH H20 in presence of THF/MeOH/H20 to get (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
The drawback of said prior art is having multiple process steps which makes the process very lengthy and tedious. Moreover the process discloses use of hazardous chemicals like pyridine which is not applicable to industry.
Process for preparation of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenylJ-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol is disclosed in US 7,375,213 B2 and J.Med.Chem.2008, 51, 1145-1149. The preparation process is depicted in Scheme-I.

Scheme-1
Prior art US’213 describes reaction of 2-chloro-5-bromo-4′-ethoxy-diphenylmethane with 2,3,4,6-tetra-O-trimethylsilyl-D-gluconolactone, n-BuLi in presence of THF and Heptane. After basification with TEA, the oily residue of methyl- l-C-(2-chloro-4′- ethoxy-diphenylmethan-3-yl)-a-D-glucopyranose obtained as solid compound after workup. This compound reacts with acetic anhydride in presence of THF, DIPEA and DMAP to get oily residue of methyl-2,3,4,6 tetra-0-acetyl-l-C-(2-chloro-4′-ethoxydiphenylmethan-3-yl)-a-D-glucopyranose which further undergoes reduction reaction in presence of acetonitirle, t riethylsilane, boron trifluoride etherate to yield 2,3,4,6-tetra-0-acetyl-l-C-(2-chloro-4′-ethoxydi henylmethan-3-yl)-β-D-glucopyranose which is further deprotected by reacting with LiOH monohydrate in presence of THF/MeOH/H20 to get (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
The said prior art describes multiple, time consuming process steps which involves getting the intermediate products as oily residue at various stages of the process, which is difficult to purify and handle for further process step. More over the workup involves multiple evaporation of product which may result in decomposition. Another drawback of the process is that the process describes n-BuLi reaction with two pot reaction. It is very difficult to transfer the material from one reactor to second reactor at -78 °C at industrial level with highly moisture sensitive reaction mass. This makes process uneconomical, cumbersome and commercially not viable. Further when practically the said method followed, a-Isomer of the final product is formed in the range of 6-8% along ith Des-bromo impurity formed in the range of 7-9 %, which increases after addition of n-butyllithium and kept the mass for overnight reaction. Moreover lactone ring cleavage is also observed in the range of 3-4% after addition of Methanesulphonic Acid/Methanol and maintained overnight for reaction completion, the removal of which is difficult from the final product.
WO 2008002824 A 1 discloses crystalline forms of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol comprising (S)-propylene glycol (PG), (R)-PG, EtOH, ethylene glycol (EG), 1 :2 L-proline, 1 : 1 L-proline, 1 : 1 L-proline hemihydrate, 1 : 1 L-phenylalanine and its preparation process.
In the light of the above drawbacks, it is necessitated to provide economical, robust, safe and commercially viable process for preparing (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
Accordingly, it is an objective of the present invention to provide a commercially viable process for the preparation of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxyb.enzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, prepared via riovel intermediates which gives higher yield and purity and facilitates easy recovery of the final compound. The purification process does not involve any costly technique/equipment, however, carried out with solvents which are industrially feasible. More over the present invention discloses the n-BuLi insitu reaction that makes the present invention cost-effective over the teachings of prior art.



Scheme-II

Formula-Ill Formula-IV
Formula-V where R1= allyl, prop-2-ynyl,isopropyl
Scheme-Ill

where R = allyl, prop-2-ynyl
Scheme-IV

Scheme-V
Examples:
Example-1: Preparation of 3,4,5-Tris-trimethylsiIanyloxy-6-trimethylsiIanyloxymethyl-tetrahydro-pyran-2-one
To 750 cc of dry THF added 1.12 mole 3,4,5-Trihydroxy-6-hydroxymethyl-tetrahydro-pyran-2-one at ambient temperature and stirred for 20 min. To the reaction mass added 9.0 mole N-Methyl morpholine and stirred for another 30.0 min at ambient temperature. Reaction mass was cooled to -5 °C to 0 °C and stirred for 30.0 min. Added 18.0 mole Trimethyl sillyl chloride at the temp -5 °C to 0 °C and stirred for 30.0 min. Temperature was raised to 25 °C to 30 °C and maintained for 18-20hrs. After reaction complies by GC, the reaction mass was cooled to -5 deg to 0 deg. Added Sat.Sodium bicarbonate solution to obtain the pH 7-8 and stirred for 1 hr at 0 °C. Added 500 cc toluene and stirred for lhr. Reaction mass was settled down for 30.0 min and layers were separated. To the Aqueous layer added 250 cc of toluene and stirred for 30.0 min. Layers separated and both the organic layers mixed and back washed with sat.brine solution. Organic layer was distilled under reduced pressure at a temperature of about 40 – 48 deg. Unload the oily mass . Purity: 92-96 %
Example-2: Preparation of 2-Allyloxy-2-[4-chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyI-tetrahydro-pyran-3,4,5-triol
To the mixture of 10 cc THF and 10 cc Toluene added 0.138 mole 4-(5-bromo-2-chlorobenzyl)phenyl ethyl ether at ambient temperature and stirred for 15 min. Cooled to -70 to -80°C in dry ice /acetone bath and stirred for 15 min. Added a solution of 0.014 mole n-Butyl lithium (1.9M in hexanes) at -70 to -80°C. and stirred for lhr. Added solution of 3, 4, 5-Tris-trimethylsilanyloxy-6-trimethylsilanyloxymethyl-tetrahydro-pyran-2-one in 5 cc of Toluene at -70 to -80°C and stirred for 2 to 3hrs. After the compliance of the reaction, reaction mass was quenched with Methane sulphonic acid and Allyl alcohol mixture at -70 to -80°C. Temperature was raised to ambient temperature and stirred overnight. Reaction mass was quenched with 30 cc sat.sodiumbicarbonate solution to bring the pH neutral to alkaline and stirred for 30.0 min. Layers separated and aqueous layer was extracted with 10 cc of Toluene. Organic layer was combined and washed with 30cc water and 50 cc sat. brine solution. Organic layer was distilled under reduced pressure to recover toluene. Solid compound was dissolved in 50cc of toluene and quenched in n-Hexane to obtain 83 % the compound as crystalline solid.
HPLC purity: 88 – 91 %
I R data:
Anomeric C-0 stretching: 1242 cm“1
Allylic C- O stretching: 1 177 cm“1
Allylic C- H stretching: 3010 – 3120 cm“1
Aromatic C- CI stretching: 820 cm“1
Lactones O – H stretching: 3240 – 3380 cm“1
Lactones C – 0 stretching: 1045 – 1092 cm“1
Aromatic C=C stretching: 1510 , 1548 , 1603 , 1703 cm“1
Alkane C – H stretching: 2877,2866, 2956, 2958, 2962 cm“1
Aromatic C – H stretching: 3050 – 3090 cm“1
Dip-Mass
(M+Na) 487.19 m/z
(M+K) 503.17 m/z
Example 3: Preparation of 2-prop-2ynyl-2-[4-Chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
To the mixture of 10 cc THF and 10 cc Toluene added 0.138 mole 4-(5-bromo-2-chlorobenzyl)phenyl ethyl ether at ambient temperature and stirred for 15 min. Cooled to -70 to -80°C in dry ice /acetone bath and stirred for 15 min. Added a solution of 0.014 mole n-Butyl lithium (1.9M in hexanes) at -70 to -80°C. and stirred for lhr. Added solution of 3, 4, 5-Tris-trimethylsilanyloxy-6-trimethylsilanyloxymethyl-tetrahydro-pyran-2-one in 5 cc of Toluene at -70 to -80°C and stirred for 2 to 3hrs. After the compliance of the reaction, the reaction mass was quenched with Methane sulphonic acid and propargyl alcohol mixture at -70 to -80°C. Temperature was raised to ambient temperature and stirred overnight. Reaction mass was quenched with 30 cc sat.sodiumbicarbonate solution to bring the pH neutral to alkaline. Reaction mass stirred for 30.0 min. Layers separated and aqueous layer was extracted with 10 cc of Toluene. Organic layer were combined and washed with 30cc water and 50 cc sat. brine solution. Organic layer was distilled under reduced pressure to recover toluene. Solid compound dissolved in 50cc of toluene and quenched in n-Hexane to obtain 75 – 80 % the compound as crystalline solid.
HPLC purity: 88 – 93 %
IR data:
Anomeric C-0 stretching: 1242 cm“1
Propargyl ~c CH stretching: 2125 cm“1
Propargyl C- H stretching : 3010 – 3120 cm“1
Aromatic C- CI stretching: 820 cm“1
Lactones O – H stretching: 3240 – 3380 cm“1
Lactones C – 0 stretching: 1045 – 1092 cm“1
Aromatic C=C stretching: 1510 , 1548 , 1603 , 1703 cm“1
Alkane C – H stretching: 2877, 2866,2956,2958,2962 cm“1
Aromatic C – H stretching: 3050 – 3090 cm“1
Dip-Mass
(M+Na) 485.25 m/z
(M+K) 501.25 m/z
Example-4: Preparation of 2-[4-Chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyI-tetrahydro-pyran-3,4,5-trioI
To the mixture of 20 cc (1 : 1 MDC + ACN) added 0.1 1 mole 2-Allyloxy-2-[4-chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol under argon atmosphere, and stirred the reaction mass for 30.0 min. Cooled the reaction mass to -40 to -55°C in a dry ice/acetone bath under argon atmosphere. Charged 3 mole Triethylsilane at -40 to -55°C and stirred the reaction mass for 30.0 min at -50 to -55°C. Slowly added Borontrifloride in diethyl ether solution at -40 to -55°C and stirred the reaction mass for 2 hrs. Quenched the reaction mass with 50 cc sat. sodium bicarbonate solution at -40 to -55°C . and stirred the reaction mass for 30.0 min. Slowly raised the temperature to 25 to 30°C. Settled down the reaction mass and separated the layers, extracted the aqueous layer with 100 cc of MDC. Combined the organic layer and wash with 500 cc water. Washed the organic layer with 500 cc of sat. Brine solution. Distilled out the MDC under reduced pressure below 40°C. to get 85 %the light yellow solid.
HPLC purity: 92-95 %
Example 5: Preparation of 2-[4-Chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
To the mixture of 20 cc (1 :1 MDC + ACN) added 0.11 mole 2-prop-2-ynyl-2-[4-Chloro-3-(4-ethoxy-benzyl)-phenyl]-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol under argon
atmosphere. Stirred the reaction mass for 30.0 min. Cooled the reaction mass to -40 to -55°C in a dry ice/acetone bath under argon atmosphere. Charged 3 mole Triethylsilane at -40 to -55°C and stirred the reaction mass for 30.0 min at -50 to -55°C. Slowly added Borontrifloride in diethyl ether solution at -40 to -55°C and stirred the reaction mass for 2 hrs. Quenched the reaction mass with 50 cc sat. sodium bicarbonate solution at -40 to -55°C and Stirred the reaction mass for 30.0 min. Slowly raised the temperature to 25 to 30°C. Settled down the reaction mass and separated the layers, extracted the aqueous layer with 100 cc of MDC. Combined the organic layer and washed with 500 cc water. Washed the organic layer with 500 cc of sat. Brine solution. Distilled out the MDC under reduced pressure below 40°C. to get 85%the light yellow solid.
HPLC purity: 90%
Example 6: Preparation of amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
To the solid obtained from example 4 charged 500cc of n-heptane and stirred for ½hrs at ambient temperature. Heated the reaction mass to 55-60°C and stirred it for 2-3 hrs.; cooled to room temperature and maintained for 4-5 hrs. Filtered the solid and washed the, cake with 100 cc n-heptane. Dried at 40-45°C under vacuum to get 85% amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
HPLC purity: 91-93%
Example 7: Preparation of amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
To the solid obtained from example 5 charged 500cc of n-heptane and stirred for ½ hrs at ambient temperature. Heated the reaction mass to 55-60°C and stirred it for 2-3 hrs., cooled to room temperature and maintained for 4-5 hrs. Filtered the solid and washed the cake with 100 cc n-heptane. Dried at 40-45 °C under vacuum to get 85-88% amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol.
HPLC purity: 89-91%
Example 8: Preparation of L-proline – (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyI]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol co crystal
To the 10 cc of Ethyl acetate charged 1.0 mole (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol under argon atmosphere at ambient temperature and stirred for 30.0 min to get clear solution. Slowly heated the reaction mass to 60 – 65°C and stirred for 1 hr. Slowly added L-proline at 60 -65°C and maintained for 1 hr. Slowly added 15 cc n-Heptane to the reaction mass at 60 -65°C and stirred the mass for 2.5 hrs. Cooled the mass to ambient temperature for 3-4 hrs and maintained for 5 hrs. Filtered the mass under argon atmosphere. Washed the cake with 10 cc n-Heptane. Dried the cake at 50-55°C under reduced pressure to get 92% titled compound.
HPLC purity: 99%
Example 9: Preparation of L-proline – (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triolco crystal
To the 10 cc of acetone charged 1.0 mole (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol under argon atmosphere at ambient temperature and stirred for 30.0 min to get clear solution. Slowly heated the reaction mass to 60 – 65°C and stirred for 1 hr. Slowly added“ proline at 60 -65°C and maintained for 1 hr. Slowly added 15 cc n-Heptane to the reaction mass at 60 -65°C and stirred the mass for 2.5 hrs. Cooled the mass to ambient temperature for 3-4 hrs and maintained for 5 hrs. Filtered the mass under argon atmosphere. Washed the cake with 10 cc n-Heptane. Dried the cake at 50-55°C under reduced pressure to get 93-95% titled compound.
HPLC purity: 98-99%
Example 10: Preparation of amorphous form of (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
To the 15 cc ethyl acetate added (2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol at ambient temperature and stirred for 30.0 min. Slowly added 5- 8 cc sat. sodium bicarbonate solution at ambient temperature and stirred for 1.5 hr to get the clear solution. Settled down and separated layers. Extracted the aqueous layer with 25 cc ethyl acetate.
Combined the organic layers and washed the ethyl acetate layer with 50 cc sat. Sodium chloride solution. Distilled out ethyl acetate under reduced pressure at 40 – 45°C to get ■fluffy solid. Charged 50 cc n-Heptane and stirred for 5 hrs to get 70-78% the title compound as Amorphous soild.
HPLC purity: 99.8-99.95 %
Example 11: Preparation of 2-chloro -4′- ethoxydiphenylmethane (impurity)
To the 20 cc THF and 20 cc Toluene added 0.25 mole 2-ehloro-5-bromo-4′- ethoxydiphenylmethane under argon atmosphere. Cooled the reaction mass to – 78° C. Slowly added n-Butyl lithium (1.9 M in hexane) at – 78° C and stirred for 30 min. Slowly added 20 % Ammonium chloride solution to the reaction mass. Brought the reaction mass to ambient temperature and stirred for 30 min. Settled and separated layers. Extracted the aqueous layer with 50 cc toluene. Washed the combined organic layer with 500 cc brine solution. Distilled out the toluene and charged heptanes, stirred for 2 – 3 hrs at ambient temperature. Filtered the product and dried the product at 45 – 50°C under reduced pressure to get 93 % titled compound.
Mass: (m+1) 247 m/z found 247.1 1
HPLC purity: 96.33 %
SHENDRA AURANGABAD, MAHARASHTRA, INDIA

Bhupinder Singh Manhas


////////WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
WO 2016018024, DAPAGLIFLOZIN, HANMI FINE CHEMICAL CO., LTD, NEW PATENT
![]()
(S) – propylene glycol and water, 1: 1 crystalline complex
PATENT
WO2016018024, CRYSTALLINE COMPOSITE COMPRISING DAPAGLIFLOZIN AND METHOD FOR PREPARING SAME
HANMI FINE CHEMICAL CO., LTD. [KR/KR]; 59, Gyeongje-ro, Siheung-si, Gyeonggi-do 429-848 (KR)
KIM, Ki Lim; (KR).
PARK, Chulhyun; (KR).
LEE, Jaeheon; (KR).
CHANG, Young-kil; (KR)

The present invention relates to a crystalline composite comprising dapagliflozin and a method for preparing the same. More specifically, the present invention provides a novel crystalline composite comprising dapagliflozin, which is an SGLT2 inhibitor, and a preparing method capable of economically preparing the novel crystalline composite at high purity.

Best Mode for Carrying out the Invention
Mode for the Invention
TABLE 1
| column | Ascentis Express RP-Amide 4.6mm × 150mm (diameter × height), 2.7㎛ (Aldrich) |
| The mobile phase | A: Formic acid 1mL/1000mL in H 2 OB: Formic acid 1mL/1000mL in Acetonitrile (ACN) |
| Test Solution | Acetonitrile Test specimen 5mg / 10mL in 50% (ACN) |
| Column temperature | 25 ℃ |
| Wavelength detector | UV, 220nm |
| Dose | 3 ㎕ |
| Flow rate | 0.7 mL / min |
| Operating hours | 40 min |
Table 2
| Gradient systems | ||
| Time (min) | Mobile phase A (%) | Mobile phase B (%) |
| 0 | 75 | 25 |
| 0-25 | 35 | 65 |
| 25-26 | 30 | 70 |
| 26-29 | 30 | 70 |
| 29-35 | 75 | 25 |
| 35-40 | 75 | 25 |
Claims
According to claim 1, wherein said crystalline complex is in the X- ray diffraction pattern of 9.7, 11.1, 13.7, 17.3, 18.7, 20.0, 20.4, 21.4, 27.5, 33.9, 36.2, 40.4, and the characteristic peaks at 2θ of 43.9 ± 0.2 ° containing crystalline complexes.
According to claim 4, wherein the mixing ratio by the spirit and mannitol dapa glyph is 1: 0.5 to 2 mole ratio, the method of producing a crystalline complex.
FIGURES
 CEO, YOUNG KIL CHANG
CEO, YOUNG KIL CHANG
/////////WO 2016018024, DAPAGLIFLOZIN, HANMI FINE CHEMICAL CO., LTD, New patent
AstraZeneca’s Forxiga receives positive advice from Scottish Medicines Consortium

DAPAGLIFLOZIN
SYNTHESIS https://newdrugapprovals.org/2013/12/18/dapagliflozin-sees-light/
AstraZeneca announced that the Scottish Medicines Consortium (SMC) has issued positive advice for use of its Forxiga, a selective and reversible inhibitor of sodium-glucose co-transporter-2, as part of a triple therapy regimen for type 2 diabetes.
ANTHONYFLOZIN………Find one if you can in this review

find here
http://medcheminternational.blogspot.in/p/flozin-series.html
1 TOFOGLIFLOZIN
2 SERGLIFLOZIN
3 DAPAGLIFLOZIN
4 IPRAGLIFLOZIN
5 EMPAGLIFLOZIN
6 LUSEOGLIFLOZIN
7 REMOGLIFLOZIN
8 ERTUGLIFLOZIN
9 SOTAGLIFLOZON


 DR ANTHONY
DR ANTHONY
BLOGS………





DAPAGLIFLOZIN…FDA approves AZ diabetes drug Farxiga
DAPAGLIFLOZIN, BMS-512148
The US Food and Drug Administration has finally approved AstraZeneca’s diabetes drug Farxiga but is insisting on six post-marketing studies, including a cardiovascular outcomes trial.
The approval was expected given that the agency’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1 last month that the benefits of Farxiga (dapagliflozin), already marketed in Europe as Forxiga, outweigh identified risks. The FDA rejected the drug in January 2012 due to concerns about possible liver damage and the potential link with breast and bladder cancer.

DAPAGLIFLOZIN SEES LIGHT
DAPAGLIFLOZIN, BMS-512148
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
cas 461432-26-8
| Molecular Formula: C21H25ClO6 |
| Molecular Weight: 408.87 |
Bristol-Myers Squibb (Originator)
AstraZeneca
TYPE 2 DIABETES,SGLT-2 Inhibitors
launched 2012, as forxiga in EU
Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
WILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013--
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.

Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
http://online.wsj.com/article/PR-CO-20131212-910828.html?dsk=y
………………………………………………………………..
PATENTS
WO 2010138535
WO 2011060256
WO 2012041898
WO 2012163990
WO 2013068850
WO 2012163546
WO 2013068850
WO 2013079501

Dapagliflozin (INN/USAN,[1] trade name Forxiga) is a drug used to treat type 2 diabetes. It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. Although dapagliflozin’s method of action would operate on both types of diabetes[1] and other conditions resulting inhyperglycemia, the current clinical trials specifically exclude participants with type 1 diabetes.[2][3]
In July 2011 an US Food and Drug Administration (FDA) committee recommended against approval until more data was available.[4] The Prescription Drug User Fee Act (PDUFA) date for dapagliflozin for the treatment of Type 2 diabetes was extended three months by the FDA to January 28, 2012.
In April 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency issued a positive opinion on the drug. It is now marketed in a number of European countries including the UK and Germany.
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine.[5] The efficacy of the this medication class has yet to be determined, but in initial clinical trials, dapagliflozin lowers HbA1c by 0.90 percentage points when added to metformin.[6]
Type II diabetes is the most common form of diabetes accounting for 90% of diabetes cases. Over 100 million people worldwide have type-2 diabetes (nearly 17 million in the U.S.) and the prevalence is increasing dramatically in both the developed and developing worlds. Type-II diabetes is a lifelong illness, which generally starts in middle age or later part of life, but can start at any age. Patients with type-2 diabetes do not respond properly to insulin, the hormone that normally allows the body to convert blood glucose into energy or store it in cells to be used later. The problem in type-2 diabetes is a condition called insulin resistance where the body produces insulin, in normal or even high amounts, but certain mechanisms prevent insulin from moving glucose into cells. Because the body does not use insulin properly, glucose rises to unsafe levels in the blood, the condition known as hyperglycemia.
Hyperglycemia, that is, elevated plasma glucose, is a hallmark of diabetes. Plasma glucose is normally filtered in the kidney in the glomerulus but is actively reabsorbed in the proximal tubule (kidney). Sodium-dependent glucose co-transporter SGLT2 appears to be the major transporter responsible for the reuptake of glucose at this site. The SGLT inhibitor phlorizin, and closely related analogs, inhibit this reuptake process in diabetic rodents and dogs, resulting in normalization of plasma glucose levels by promoting glucose excretion without hypoglycemic side effects. Long term (6 month) treatment of Zucker diabetic rats with an SGLT2 inhibitor has been reported to improve insulin response to glycemia, improve insulin sensitivity, and delay the onset of nephropathy and neuropathy in these animals, with no detectable pathology in the kidney and no electrolyte imbalance in plasma. Selective inhibition of SGLT2 in diabetic patients would be expected to normalize plasma glucose by enhancing the excretion of glucose in the urine, thereby improving insulin sensitivity and delaying the development of diabetic complications.
The treatment of diabetes is an important health concern and despite a wide range of available therapies, the epidemic continues. Type 2 diabetes (T2DM) is a progressive disease caused by insulin resistance and decreased pancreatic β-cell function. Insulin is produced by the pancreatic β-cell and mediates cellular glucose uptake and clearance. Insulin resistance is characterized by the lack of response to the actions of this hormone which results in decreased cellular clearance of glucose from the circulation and overproduction of glucose by the liver.
The currently available therapies to treat type 2 diabetes augment the action or delivery of insulin to lower blood glucose. However, despite therapy, many patients do not achieve control of their type 2 diabetes. According to the National Health and Nutrition Examination Survey (NHANES) III, only 36% of type 2 diabetics achieve glycemic control defined as a A1C<7.0% with current therapies. In an effort to treat type 2 diabetes, aggressive therapy with multiple pharmacologic agents may be prescribed. The use of insulin plus oral agents has increased from approximately 3 to 11% from NHANES II to III.
Thus, treatment of hyperglycemia in type 2 diabetes (T2DM) remains a major challenge, particularly in patients who require insulin as the disease progresses. Various combinations of insulin with oral anti-diabetic agents (OADs) have been investigated in recent years, and an increasing number of patients have been placed on these regimens. Poulsen, M. K. et al., “The combined effect of triple therapy with rosiglitazone, metformin, and insulin in type 2 diabetic patients”,Diabetes Care, 26 (12):3273-3279 (2003); Buse, J., “Combining insulin and oral agents”, Am. J. Med., 108 (Supp. 6a):23S-32S (2000). Often, these combination therapies become less effective in controlling hyperglycemia over time, particularly as weight gain and worsening insulin resistance impair insulin response pathways.
Hypoglycemia, weight gain, and subsequent increased insulin resistance are significant factors that limit optimal titration and effectiveness of insulin. (Holman, R. R. et al., “Addition of biphasic, prandial, or basal insulin to oral therapy in type 2 diabetes”, N. Engl. J. Med., 357 (17):1716-1730 (2007)). Weight gain with insulin therapy is predominantly a consequence of the reduction of glucosuria, and is thought to be proportional to the correction of glycemia. (Makimattila, S. et al., “Causes of weight gain during insulin therapy with and without metformin in patients with Type II diabetes mellitus”, Diabetologia, 42 (4):406-412 (1999)). Insulin drives weight gain when used alone or with OADs. (Buse, J., supra). In some cases, intensive insulin therapy may worsen lipid overload and complicate progression of the disease through a spiral of caloric surplus, hyperinsulinemia, increased lipogenesis, increased adipocity, increased insulin resistance, beta-cell toxicity, and hyperglycemia. (Unger, R. H., “Reinventing type 2 diabetes: pathogenesis, treatment, and prevention”, JAMA, 299 (10):1185-1187 (2008)). Among commonly used OADs, thiazolidinediones (TZDs) and sulfonylureas intrinsically contribute to weight gain as glucosuria dissipates with improved glycemic control. Weight gain is less prominent with metformin, acting through suppression of hepatic glucose output, or with incretin-based DPP-4 inhibitors. Overall, there is a pressing need for novel agents that can be safely added to insulin-dependent therapies to help achieve glycemic targets without increasing the risks of weight gain or hypoglycemia.
A novel approach to treating hyperglycemia involves targeting transporters for glucose reabsorption in the kidney. (Kanai, Y. et al., “The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose”, J. Clin. Invest., 93 (1):397-404 (1994)). Agents that selectively block the sodium-glucose cotransporter 2 (SGLT2) located in the proximal tubule of the kidney can inhibit reabsorption of glucose and induce its elimination through urinary excretion. (Brown, G. K., “Glucose transporters: structure, function and consequences of deficiency”, J. Inherit. Metab. Dis., 23 (3):237-246 (2000)). SGLT2 inhibition has been shown in pre-clinical models to lower blood glucose independently of insulin. (Han, S. et al., “Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats”, Diabetes, 57 (6):1723-1729 (2008); Katsuno, K. et al., “Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level”, J. Pharmacol. Exp. Ther., 320 (1):323-330 (2007)).
Dapagliflozin(BMS-512148) is a potent sodium-glucose transport proteins inhibitor with IC50 of 1.1 nM and 1.4uM for SGLT2 and SGLT1, respectively. Dapagliflozin (BMS-512148) inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter causes blood glucose to be eliminated through the urine. Symptoms of hypoglycaemia occurred in similar proportions of patients in the dapagliflozin (2~4%) and placebo groups (3%). Signs, symptoms, and other reports suggestive of genital infections were more frequent in the dapagliflozin groups (2•5 mg, [8%]; 5 mg, [13%]; 10 mg, [9%]) than in the placebo group ( [5%]).
Dapagliflozin (which is disclosed in U.S. Pat. No. 6,515,117) is an inhibitor of sodium-glucose reabsorption by the kidney, by inhibiting SGLT2, which results in an increased excretion of glucose in the urine. This effect lowers plasma glucose in an insulin-independent manner.
Dapagliflozin is currently undergoing clinical development for treatment of type 2 diabetes. (Han, S. et al., supra; Meng, W. et al., “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes”, J. Med. Chem., 51 (5):1145-1149 (2008)). Phase 2a and 2b studies with dapagliflozin have demonstrated efficacy in reducing hyperglycemia either alone or in combination with metformin in patients with T2DM. (Komoroski, B. et al., “Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus”, Clin. Pharmacol. Ther., 85 (5):513-519 (2009); List, J. F. et al., “Dapagliflozin-induced glucosuria is accompanied by weight loss in type 2 diabetic patients”, 68th Scientific Sessions of the American Diabetes Association, San Francisco, Calif., Jun. 6-10, 2008, Presentation No. 0461P).
It has been found that dapagliflozin does not act through insulin signaling pathways and is effective in controlling blood sugar in patients whose insulin signaling pathways do not work well. This applies to extremes of insulin resistance, in type 2 diabetes as well as in insulin resistance syndromes, caused by, for example, mutations in the insulin receptor.
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Dapagliflozin is also associated with hypotensive reactions.

The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7]
- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- “FDA panel advises against approval of dapagliflozin”. 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ; Hecheng Huaxue , 18 (3), 389-392; 2010
5) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,. WO2010022313
6) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,. WO2010009243
7) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,. WO2009026537
8) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ; Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;
9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,. WO2008002824
10) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;.. U.S. Pat Appl Publ,. 20,040,138,439

dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .
formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2
formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
…..
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
http://www.google.com/patents/US20120282336
http://www.tga.gov.au/pdf/auspar/auspar-dapagliflozin-propanediol-monohydrate-130114.pdf
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
………………………………
In Vitro Characterization and Pharmacokinetics of Dapagliflozin …
dmd.aspetjournals.org/content/…/DMD29165_supplemental_data_.doc

Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)
-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33
(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),
6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,
1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,
140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,
64.5, 63.1, 39.2, 15.2.
HRMS calculated for C21H25ClNaO6 (M+Na)+
For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
: 431.1237; found 431.1234. Anal. Calcd
SECOND SET
1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);
13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
………………………
HPLC
-
HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
– Eluent A: 0.1 % formic acid in water
– Eluent B: 0.1 % formic acid in acetonitrile
– Injection volume: 3 mL
– Flow: 0.7 mL/min
– Gradient:Time [min] [%] B 0.0 25 25.0 65 26.0 70 29.0 70 29.5 25 35.0 25 ……………………..

……..
http://www.google.com/patents/WO2013068850A2?cl=en
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))
[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.56-7.54 (m, 2H), 7.43-7.31 (m, 13H), 7.29-7.22 (m, 6H), 7.07- 7.04 (m, 2H), 7.00 (d, J= 2.0 Hz, IH), 6.87 (dd, J= 8.4, 2.0 Hz, IH), 6.83-6.81 (m, 2H), 4.18 (d, J= 9.6 Hz, IH), 4.02 (q, J= 6.9 Hz, 2H), 3.96 (d, J= 10.8 Hz, 2H), 3.86 (ddd, J= 11.3, 7.7, 1.1 Hz, IH), 3.76 (ddd, J= 8.4, 8.4, 4.8 Hz, IH), 3.56 (ddd, J= 9.0, 6.4, 2.4 Hz, IH), 3.50 (dd, J=
I I.4, 5.4 Hz, IH), 3.44 (dd, J= 9.4, 8.6 Hz, IH), 3.38 (dd, J= 8.8, 8.8 Hz, IH), 1.70 (dd, J= 7.8, 5.4 Hz, IH, OH), 1.42 (t, J= 6.8 Hz, 3H), 1.21 (d, J= 5.2 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 157.4 (C), 138.8 (C), 137.4 (C), 136.3 (CH x2), 136.1 (CH x2), 135.2 (CH x2), 135.0 (C), 134.9 (CH x2), 134.8 (C), 134.2 (C), 132.8 (C), 132.0 (C), 131.6 (CH), 131.1 (C), 129.9 (CH x2), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.58 (CH x2), 127.57 (CH x2), 127.54 (CH x2), 127.31 (CH), 127.28 (CH x2), 114.4 (CH x2), 82.2 (CH), 80.5 (CH), 79.3 (CH), 76.3 (CH), 72.7 (CH), 63.4 (CH2), 62.7 (CH2), 38.2 (CH2), 27.2 (CH3 x3), 26.6 (CH3 x3), 19.6 (C), 19.2 (C), 14.9 (CH3). EXAMPLE 25 -Synthesis of dapagliflozin ((25,3R,4R,55,6/?)-2-[4-chloro-3-(4- ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; (Ij))
IVj’ U
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
1H NMR (400 MHz, CD3OD) δ 7.37-7.34 (m, 2H), 7.29 (dd, J= 8.2, 2.2 Hz, 1H), 7.12-7.10 (m, 2H), 6.82-6.80 (m, 2H), 4.10 (d, J= 9.6 Hz, 2H), 4.04 (d, J= 9.2 Hz, 2H), 4.00 (q, J= 7.1 Hz, 2H), 3.91-3.87 (m, 1H), 3.73-3.67(m, 1H), 3.47-3.40 (m, 3H), 3.31-3.23 (m, 2H), 1.37 (t, J= 7.0 Hz, 3H);
13C NMR (100 MHz, CD3OD) δ 157.4 (C), 138.6 (C), 138.5 (C), 133.1 (C), 131.5 (C), 130.5 (CH), 129.4 (CH x2), 128.7 (CH), 126.8 (CH), 114.0 (CH x2), 80.5 (CH), 80.8 (CH), 78.3 (CH), 75.0 (CH), 70.4 (CH), 63.0 (CH2), 61.7 (CH2), 37.8 (CH2), 13.8 (CH3);
LCMS (ESI) m/z 426 (100, [M+NH4]+), 428 (36, [M+NH4+2]+), 447 (33, [M+K]+).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)
III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- “D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}