
Home » Posts tagged 'CHINA 2024' (Page 2)
Tag Archives: CHINA 2024
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Envonalkib
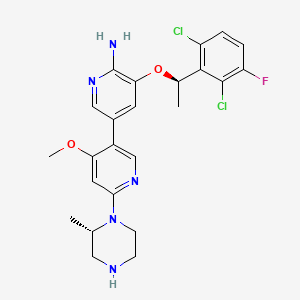
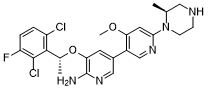
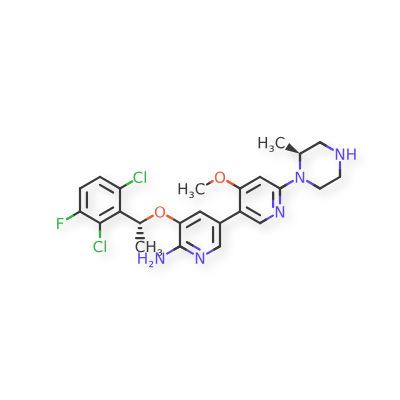
Envonalkib
- CAS 1621519-26-3
- QB7KTQ7VW9
- 5-((1R)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((2S)-2-methyl-1-piperazinyl)(3,3′-bipyridin)-6-amine
- 506.4 g/mol, C24H26Cl2FN5O2
TQ-B3139, Chia Tai Tianqing, Anluoqing, cancer
ENVONALKIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
SYN
https://patentscope.wipo.int/search/en/WO2014117718
Example 27: 5-[(2,6-dichloro-3-fluorophenyl)ethoxy-4′-methoxy-6′ …
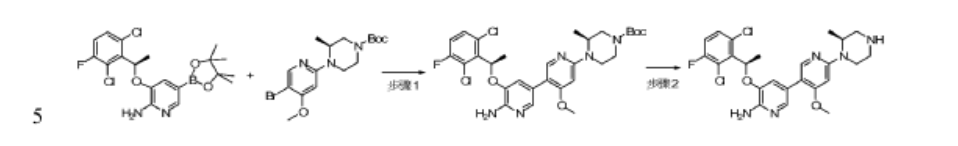
Step 1: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methyl-4-tert-butoxycarbonylpiperazin-1-yl)-3,3′-bipyridin-6-amine
To dioxane (10 mL) and water (1.5 mL) were added tert-butyl (S)-4-(5-bromo-4-methoxypyridin-2-yl)-3-methylpiperidin-1-carboxylate (106 mg, 0.275 mmol), (R)-3-(1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-aminopyridine (140 mg, 0.33 mmol), tetrakis(triphenylphosphine)palladium (32 mg, 0.0275 mmol) and cesium carbonate (179 mg, 0.55 mmol), the atmosphere was replaced with nitrogen, and the reaction was carried out at 100 ° C. overnight. After cooling, the mixture was separated by silica gel column chromatography to give 5-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6-(5-(2-methyl-4-tert-butoxycarbonylpiperidin-1-yl)-3,3′-bipyridin-6-amine) (70 mg) in a yield of 42%. MS m/z [ESI]: 606.2 [M+1].
Step 2: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-3,3′-bipyridin-6-amine
To a stirred dichloromethane solution (10 mL) of 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methyl-4-tert-butoxycarbonylpiperidin-1-yl)-3,3′-bipyridin-6-amine (67 mg, 0.11 mmol) was added trifluoroacetic acid (1 mL) and stirred for 1 hour. The pH was adjusted to greater than 13 with sodium hydroxide solution, and the mixture was extracted with dichloromethane. The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated. The product was separated and purified by column chromatography (with dichloromethane:methanol = 8:1 as eluent) to give 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperidin-1-yl)-3,3′-bipyridin-6-amine (30 mg). Yield: 55%, MS m/z [ESI]: 506.1[M+1]. 1H-NM (400 MHz, CDC1 3 ):5= 7.94(1H, s), 7.71(1H, s), 7.28-7.32(lH, m), 7.07(1H, t, J=8.4Hz), 6.97(1H, s), 6.04-6.13(2H, m), 4.86 (2H : s), 4.57-4.59(lH, m), 4.03 (1H, d, J=14Hz), 3.76(3H, s), 3.07-3.33(4H, m), 2.88-3.00(lH, m), 1.84(3H, d, J=6.8Hz), 1.34 (3H, d, J=6.8Hz).
SYN
CN107949560
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US154015806&_cid=P11-MEF9W1-27198-1
Example 27: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-[3,3′-bipyridin]-6-amine
General Synthetic Methods:
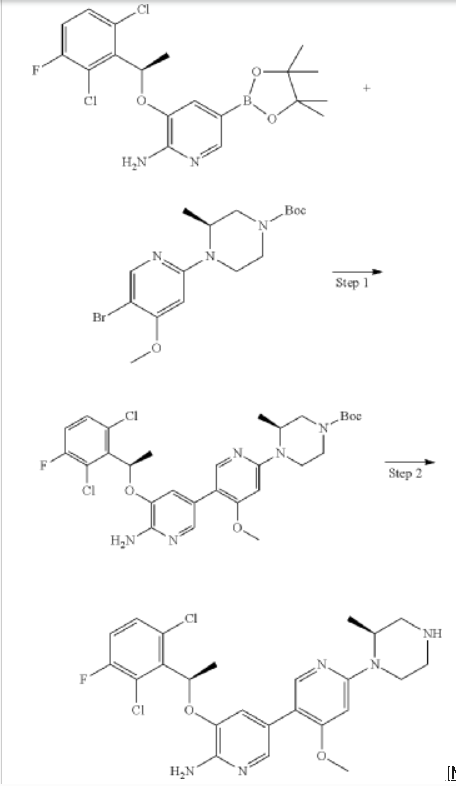
Step 1: (S)-tert-butyl 4-(6′-amino-5′-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4-methoxy-[3,3′-bipyridin]-6-yl)-3-methylpiperazine-1-carboxylate
Step 2: 5-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-4′-methoxy-6′-((S)-2-methylpiperazin-1-yl)-[3,3′-bipyridin]-6-amine
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Envonalkib, also known as TQ-B3139, is a novel small-molecule TKI, developed by Chia Tai Tianqing Pharmaceutical Group. It targets ALK, ROS1, and c-Met kinases, exhibiting potent antitumor activity against cancers harboring these genetic alterations. In 2024, the NMPA approved Envonalkib under the brand name Anluoqing for the treatment of adult patients with ALK-positive locally advanced or metastatic NSCLC who have not received prior ALK inhibitor therapy [24]. Envonalkib exerts its therapeutic effects through selective inhibition of the kinase activities of ALK, ROS1, and c-Met, thereby interrupting the downstream signaling pathways that are crucial for tumor cell proliferation and survival [25]. The inhibition of these targets results in cell cycle arrest and apoptosis in cancer cells。The clinical efficacy of Envonalkib was evidenced in a Phase III randomized, open-label, multicenter clinical trial (NCT04009317), which compared Envonalkib with crizotinib in treatment-naïve patients with ALK-positive advanced NSCLC [25,26]. In the reported study, Envonalkib demonstrated a me dian PFS of 24.87 months, which was markedly superior to the 11.60 months achieved with crizotinib (hazard ratio [HR] = 0.47, p < 0.0001). Notably, in patients harboring brain metastases, Envonalkib exhibited a
central nervous system objective response rate (CNS-ORR) of 78.95 %, a substantial improvement over the 23.81 % observed with crizotinib. In terms of safety profile, Envonalkib was generally well-tolerated. Treat ment-related adverse events (TRAEs) of Grade ≥3 were noted in 55.73 % of patients receiving Envonalkib, contrasting with the 42.86 % incidence in the crizotinib cohort. The predominant TRAEs encompassed elevated liver enzymes, neutropenia, and gastrointestinal symptoms, all of which
were amenable to effective management through appropriate support ive care measures. The regulatory approval of Envonalkib thus in troduces a novel therapeutic modality for patients with ALK-positive NSCLC, effectively addressing a significant unmet medical need within this patient population [25].
The synthesis of Envonalkib, illustrated in Scheme 6, initiates with Mitsunobu coupling of Envo-001 and Envo-002, affording Envo-003 [27]. Sequential reduction and NBS-bromination converts Envo-003 to
Envo-005 via Envo-004. Miyaura borylation of Envo-005 constructs Envo-006, which undergoes Suzuki-Miyaura cross-coupling with Envo-007 followed by deprotection to deliver Envonalkib. In parallel,
Envo-009 reacts with Envo-010 through Buchwald-Hartwig cross coupling to form Envo-011. This intermediate is brominated to produce Envo-007, which is used in the Suzuki-Miyaura coupling with Envo-006
[24] X. Li, Y. Xia, C. Wang, S. Huang, Q. Chu, Efficacy of ALK inhibitors in Asian
patients with ALK inhibitor-naïve advanced ALK-Positive non-small cell lung
cancer: a systematic review and network meta-analysis, Transl. Lung Cancer Res.
13 (2024) 2015–2022.
[25] Y. Yang, J. Min, N. Yang, Q. Yu, Y. Cheng, Y. Zhao, M. Li, H. Chen, S. Ren, J. Zhou,
W. Zhuang, X. Qin, L. Cao, Y. Yu, J. Zhang, J. He, J. Feng, H. Yu, L. Zhang, W. Fang,
Envonalkib versus crizotinib for treatment-naive ALK-Positive non-small cell lung
cancer: a randomized, multicenter, open-label, phase III trial, Signal Transduct
Target Ther 8 (2023) 301.
[26] R. Garcia-Carbonero, A. Carnero, L. Paz-Ares, Inhibition of HSP90 molecular
chaperones: moving into the clinic, Lancet Oncol. 14 (2013) e358–e369.
[27] F. Gong, X. Li, R. Zhao, X. Zhang, X. Xu, X. Liu, D. Xiao, Y. Han, Process for
Preparation of Pyridine Substituted 2-aminopyridine Protein Kinase Inhibitor
Crystal, 2017. CN107949560B.




AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Efficacy of ALK inhibitors in Asian patients with ALK inhibitor-naïve advanced ALK-positive non-small cell lung cancer: a systematic review and network meta-analysisPublication Name: Translational Lung Cancer ResearchPublication Date: 2024-08-31PMCID: PMC11384493PMID: 39263024DOI: 10.21037/tlcr-24-604
- Envonalkib versus crizotinib for treatment-naive ALK-positive non-small cell lung cancer: a randomized, multicenter, open-label, phase III trialPublication Name: Signal Transduction and Targeted TherapyPublication Date: 2023-08-14PMCID: PMC10423717PMID: 37574511DOI: 10.1038/s41392-023-01538-w
- Pharmacokinetic, pharmacodynamic, and behavioural studies of deschloroketamine in Wistar ratsPublication Name: British Journal of PharmacologyPublication Date: 2021-10-31PMID: 34519023DOI: 10.1111/bph.15680
//////////Envonalkib, china 2024, approvals 2024, TQ-B3139, TQ B3139, Chia Tai Tianqing, Anluoqing, cancer, QB7KTQ7VW9
Tunlametinib
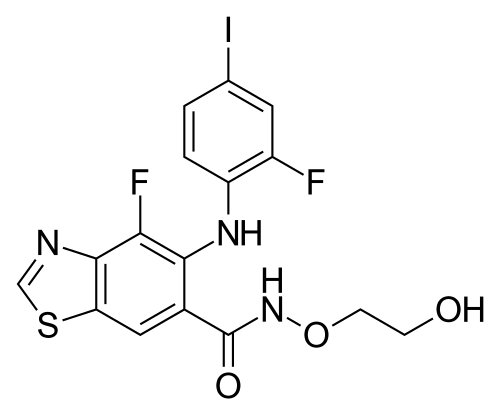
Tunlametinib
- CAS 1801756-06-8
- IF25NR1PV3
- HL085
- C16H12F2IN3O3S
491.3 g/mol
4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,3-benzothiazole-6-carboxamide
- 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)-6-benzothiazolecarboxamide
- 4-fluoro-5-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,3-benzothiazole-6-carboxamide
- 6-Benzothiazolecarboxamide, 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)-
Tunlametinib, an oral selective inhibitor of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), was developed by Shanghai KeChow Pharmaceuticals Co., Ltd. Marketed under the brand name
Keluping,
Tunlametinib is a pharmaceutical drug for the treatment of cancer. It is an inhbitor of mitogen-activated protein kinase kinase.[1]
In China, tunlametinib was approved in 2024 for the treatment of patients with NRAS-mutated advanced melanoma who were previously treated with a PD-1/PD-L1 targeting agent.[2][3]
It is also being studied for use in combination with vemurafenib in patients with advanced BRAF V600-mutant solid tumors.[4]
PAT
US9937158
PAT
https://patents.google.com/patent/WO2013107283A1/en
Step 1:

[0435] To a solution of 2,3,4-trifluorobromobenzene in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), sulfolane, HMPA, DMPU, prefer anhydrous THF, ethyl ether and dioxane) was added strong base (such as LDA, nBuLi,
LiHDMS) at low temperature (-50 °C 80 °C, prefer -78 °C) under nitrogen atmosphere. The reaction is kept stirring for some time (0.5-12 h, prefer 0.5-2 h) and is added dry ice. After several hours (3-12 h, prefer 5-10 h), 5-bromo-2,3,4-trifluorobenzoic acid is obtained after conventional workup.
Step 2:

[0436] 5-Bromo-2,3,4-trifluorobenzoic acid can be reacted with halogenated aniline (such as o-fluoroaniline, o-chloroaniline, o-bromoaniline, o-iodoaniline) in the presence of base (such as LDA, n-BuLi, LiHDMS) in appropriate solvent (include aliphatic and aromatic
hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2- methoxyethyl ether, tetrahydrofuran, dioxane), sulfolane, HMPA, DMPU, prefer anhydrous THF, ethyl ether and dioxane) at low temperature (-50 °C— -80 °C, prefer -78 °C) for some time (such as 3-12 h, prefer 5-10 h). 5-Bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid is obtained after conventional workup.
Step 3:

[0437] 5-Bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid can be reacted with MeOH in the presence of SOCl2 in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile(such as acetonitrile, propiononitrile), amide(such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methanol and ethanol). The reaction proceeds for several hours (3-12 h, prefer 5-10 h). Methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate is obtained after conventional workup.
Step 4:

[0438] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ester(such as ethyl acetate, methyl acetate), nitrile(such as acetonitrile, propiononitrile), amide(such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dioxane) was added base (such as aliphatic and aromatic amine(such as, but not limited to, N-ethyl-N-isopropylpropan-2-amine, triethylamine, diethylamine, DBU, t-butylamine, cyclopropanamine, dibutylamine, diisopropylamine, 1,2- dimethylpropanamine), inorganic base(such as Na2C03, K2C03, NaHC03, KHC03, t-BuONa, t- BuOK), prefer N-ethyl-N-isopropylpropan-2-amine) at ambient temperature under nitrogen atmosphere, followed by Pd catalyst (such as tris(dibenzylideneacetone)dipalladium,
bis(dibenzylideneacetone) palladium, bis(triphenylphosphine)palladium(II) chloride, palladium diacetate, tetrakis(triphenylphosphine)palladium, bis(triphenylphosphinepalladium)acetate, prefer tris(dibenzylideneacetone) dipalladium) and phosphine ligand (such as
dimethylbisdiphenylphosphinoxanthene, tri-tert-butylphosphine, tri-p-tolylphosphine, tris(4- chlorophenyl)phosphine, triisopropylphosphine, tris(2,6-dimethoxyphenyl)phosphine, 1, 1 ‘- bis(diphenylphosphino)ferrocene, prefer dimethylbisdiphenylphosphinoxanthene). The reaction is kept stirring at high temperature (80-130 °C, prefer 90-110 °C) for some time (8-24 h, prefer 12-18 h). Methyl 3,4-difluoro-2- ((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup. Step 5:

[0439] Methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxy benzyl)thio)benzoate can be reacted with azide (such as NaN3, KN3) at high temperature (60-120 °C, prefer 80-100 °C) in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer N,N-dimethylformamide and N,N-dimethylacetamide) for some time (1-12 h, prefer 3-10 h). Methyl 4-azido-3-fluoro-2-((2-fluorophenyl) amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup.
Step 6:

[0440] Methyl 4-azido-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxy
benzyl)thio)benzoate can be hydrogenated catalyzed by appropriate catalyst (such as Pd/C, Pt, Ni) in the solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ester(such as ethyl acetate, methyl acetate), amide (such as N,N-dimethylformamide, N,N- dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methanol, ethanol, propan-l-ol and water) for some time (1-12 h, prefer 3-10 h). Methyl 4- amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate is obtained after conventional workup. Step 7:

[0441] 4-Amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate can be deprotected in the presence of acid (such as CF3COOH, HCOOH, CH3COOH and n- C5H11COOH, prefer CF3COOH) at certain temperature (20-75 °C, prefer 25-75 °C) in
appropriate aromatic aliphatic ether (such as anisole and phenetole, prefer anisole) for some time (1-12 h, prefer 3-10 h). Methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate is obtained after conventional workup.
Step 8:

[0442] Methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercapto benzoate can be cyclized in the presence of acid (such as ^-toluenesulfonic acid, pyridinium toluene-4- sulphonate, formic acid, acetic acid, sulfuric acid) in appropriate solvent (include aliphatic and aromatic hydrocarbon (such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as
dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N- methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer methyl acetate, ethyl acetate and trimethoxymethane) for some time (0.2-12 h, prefer 0.5-10 h). Methyl 4-fluoro-5-((2- fluorophenyl)amino) benzo[d]thiazole-6-carboxylate is obtained after conventional workup. Step 9:

[0443] Methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate can be reacted with halogenations reagent (such as NIS) in the presence of acid (such as trifluoroacetic acid, trifluoromethanesulfonic acid, methanesulfonic acid, formic acid, acetic acid) at ambient temperature in appropriate solvent (include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N- dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer N,N-dimethylformamide and N,N-dimethylacetamide) for some time (1- 12 h, prefer 3-10 h). Methyl 4-fluoro-5-((2-fluoro-4-iodophenyl) amino)benzo[d]thiazole-6- carboxylate is obtained after conventional workup.
Step 10:

[0444] 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6-carboxylic acid can be reacted with O-(2-(vinyloxy)ethyl)hydroxylamine in the presence of coupling reagent(such as HOBt, EDCI, HATU, TBTU) at ambient temperature in appropriate solvent(include aliphatic and aromatic hydrocarbon(such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N- methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dichloromethane, 1,2- dichloroethane and N,N-dimethylformamide) for some time (1-12 h, prefer 3-10 h). 4-Fluoro-5- ((2-fluoro-4-iodophenyl) amino)-N-(2-(vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide is obtained after conventional workup. Step 11:

[0445] 4-Fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy)ethoxy)benzo[d]thiazole- 6-carboxamide can be reacted in the presence of acid (such as HCl, H2S04, trifluoroacetic acid) in appropriate solvent (include aliphatic and aromatic hydrocarbon (such as pentane, hexane, heptane, cyclohexane, petroleum ether, petrol, gasoline, benzene, toluene, xylene), aliphatic and aromatic halo-hydrocarbon (such as dichloromethane, 1,2-dichloroethane, chloroform, phenixin, chlorobenzene, o-dichlorobenzene), ether (such as diethyl ether, dibutyl ether, glycol dimethyl ether, 2-methoxyethyl ether, tetrahydrofuran, dioxane), ketone(such as acetone, methyl ethyl ketone, methyl isopropyl ketone, methyl isobutyl ketone), ester(such as ethyl acetate, methyl acetate), nitrile (such as acetonitrile, propiononitrile), amide (such as N,N-dimethylformamide, N,N-dimethylacetamide and N-methylpyrrolidin-2-one), DMSO, sulfolane, HMPA, DMPU, prefer dichloromethane and 1,2-dichloroethane) for some time (1-12 h, prefer 3-10 h). 4-Fluoro- 5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxy ethoxy)benzo[d]oxazole-6-carboxamide is obtained after conventional workup.
Example 9: Preparation of 4-fluoro-5-((2-fluoro-4-iodophenyDamino)-N-(2- hydroxyethoxy)benzo[d]thiazole-6-carboxamide (Compound 9)

Step 1: 5-bromo-2,3,4-trifluorobenzoic acid
[0510] To a solution of diisopropylamine (10.14 g, 100.20 mmol) in THF (100 mL) was added «-BuLi (40.08 mL, 2.5 M in hexane, 100.20 mmol) at -78 °C under nitrogen atmosphere. The stirring was maintained at this temperature for 1 h. Then a solution of l-bromo-2,3,4- trifluorobenzene (17.62 g, 83.50 mmol) in THF (120 mL) was added. After stirring for 1 h at -78 °C, the mixture was transferred to a bottle with dry ice. The mixture was stirred overnight at room temperature. The reaction was quenched with 10% aqueous HCl and pH was adjusted to 1- 2. The mixture was extracted with ethyl acetate (100 mL x 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated under reduced pressure to afford the desired product (20.12 g, 94.5% yield). 1H NMR (400 MHz, DMSO-d6): δ 13.95 (s, 1H), 7.97 (m, 1H).
Step 2: 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid
[0511] To a solution of 2-fluoroaniline (17.54 g, 157.80 mmol) and 5-bromo-2,3,4- trifluorobenzoic acid (20.12 g, 78.90 mmol) in THF (120 mL) was added LiHMDS (236.7 mL, 1 M in THF, 236.7 mmol) dropwisely at -78 °C under nitrogen atmosphere. The mixture was allowed to slowly warm to room temperature and stirred at this temperature overnight. The reaction was quenched with water (100 mL) and acidified to pH 2-3 with 10% HCl (aq.). The mixture was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the desired product (pale yellow solid, 24.24 g, 88.8% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.22 (s, 1H), 8.01 (dd, J= 7.4, 2.1 Hz, 1H), 7.25 (m, 1H), 7.10 (m, 3H).
Step 3: methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoate
[0512] To a solution of 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino) benzoic acid (24.24 g, 70.04 mmol) in MeOH (300 mL) was added thionyl chloride (20 mL). After stirring at 85 °C overnight, most MeOH was removed in vacuo. The residue was neutralized with saturated sodium bicarbonate (aq.) and extracted with ethyl acetate (100 mL χ 3). The combined organic layer was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. After purification by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v), the corresponding product was obtained as a white solid (22.33 g, 88.5% yield). 1H NMR (400 MHz, CDC13): δ 9.06 (s, 1H), 8.01 (dd, J= 7.1, 2.3 Hz, 1H), 7.04 (m, 4H), 3.92 (s, 3H).
Step 4: methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate
[0513] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl) amino)benzoate (22.33 g, 62.01 mmol) in anhydrous 1,4-dioxane (200 mL) was added N,N- diisopropylethylamine (16.03 g, 124.04 mmol). Then Pd2(dba)3 (2.84 g, 3.10 mmol) followed by Xantphos (3.59 g, 6.20 mmol) and 4-methoxy-a-toluenethiol (10.27 g, 65.11 mmol) was added under nitrogen atmosphere. The mixture was stirred overnight at 100 °C under N2 atmosphere and then allowed to warm to ambient temperature. The insoluble matter was filtered off and the filter cake was washed ethyl acetate. The filtrate was diluted with water (300 mL) and extracted with ethyl acetate (100 mL x 3). The combined organic layers were washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v) to give the desired product (pale yellow solid, 24.35 g, 90.6% yield). 1H NMR (400 MHz, CDC13): δ 9.12 (s, 1H), 7.78 (d, 1H), 7.25 (m, 6H), 6.85 (m, 2H), 4.03 (s, 2H), 3.90 (s, 3H), 3.80 (s, 3H). Step 5: methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate
[0514] To a solution of methyl 5-(4-methoxybenzylthio)-3,4-difluoro-2- ((2- fluorophenyl)amino)benzoate (24.35 g, 56.18 mmol) in DMF (200 mL) was added NaN3 (4.38 g, 67.41 mmol) at ambient temperature. The mixture was stirred at 90 °C for 3 h. Then water (200 mL) was added. The solution was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL) and brine (100 mL), dried over Na2S04 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v) and gave the desired product (white solid, 21.04 g, 82.1% yield). 1H NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 7.75 (s, 1H), 7.10 (m, 6H), 6.84 (m, 2H), 4.03 (s, 2H), 3.92 (s, 3H), 3.81 (s, 3H). Step 6: methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate To a solution of methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (21.04 g, 46.09 mmol) in MeOH (500 mL) was added and 10% palladium on carbon (3.40 g) under nitrogen atmosphere. Then the nitrogen atmosphere was completely changed to hydrogen atmosphere. The mixture was stirred for 2 h at ambient temperature. After the insoluble matter was filtered off, the solvent was evaporated in vacuo to give the desired product (19.46 g, 98.1% yield). 1H NMR (400 MHz, CDC13): δ 9.07 (s, 1H), 7.77 (s, 1H), 7.06 (m, 4H), 6.95 (m, 2H), 6.81 (d, J = 8.3 Hz, 2H), 4.68 (s, 2H), 3.85 (s, 5H), 3.81 (s, 3H).
Step 7: dimethyl 5,5′-disulfanediylbis(4-amino-3-fluoro-2-((2-fluorophenyl)amino)benzoate)
[0515] To a solution of methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (19.46 g, 45.21 mmol) in CH2C12 (180 mL) was added DDQ (11.29 g, 49.73 mmol) followed by water (20 mL). After stirring at ambient temperature for 10 h, the reaction was quenched by saturated sodium bicarbonate (aq., 100 mL). The aqueous layer was extracted by CH2C12 (100 mL χ 3). The combined organic phase was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 5: 1, v/v) to give the desired product (pale yellow solid, 9.81 g, 35.1% yield). 1H NMR (400 MHz, CDC13): δ 9.34 (s, 2H), 7.46 (s, 2H), 7.06 (m, 8H), 4.89 (br, 4H), 3.75 (s, 6H). Step 8: methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercaptobenzoate
[0516] To a solution of dimethyl 5,5′-disulfanediylbis(4-amino-3-fluoro-2-((2- fluorophenyl)amino)benzoate) (9.81 g, 15.86 mmol) in THF/MeOH (100 mL, 10: 1, v/v) was added NaBH4 (3.00 g, 79.29 mmol) portion-wise in 1 h. After stirring at ambient temperature for 1 h, the reaction was quenched with 10% HCl (aq.) and pH was adjusted to 1-2. The aqueous layer was extracted with CH2C12 (50 mL χ 3). The combined organic phase was washed with water (50 mL) and brine (50 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. The crude product was used directly in the next step without further purification.
Step 9: methyl 4-fluoro-5-((2-fluorophenyl)amino)benzofdJthiazole-6-carboxylate
[0517] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate in trimethyl orthoformate (50 mL) was added p-TsOU (0.61 g, 3.17 mmol). The reaction mixture was stirred for 1 h and treated with water (100 mL). The precipitate was filtered off and the filter cake was washed with water to afford the desired product (pale yellow solid, 8.64 g, 85.1% yield for two steps). 1H MR (400 MHz, CDC13): δ 9.13 (s, 1H), 8.68 (s, 1H), 8.46 (s, 1H), 7.10 (m, 1H), 7.01 (m, 1H), 6.92 (s, 2H), 3.97 (s, 3H).
Step 10: methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzofdJthiazole-6-carboxylate
[0518] To a solution of methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate (8.64 g, 26.97 mmol) in DMF (100 mL) was added NIS (6.68 g, 29.67 mmol) followed by trifluoroacetic acid (0.5 mL). After stirring for 5 h at ambient temperature, the reaction was treated by water (150 mL). The precipitate was filtered off and the filter cake was washed with water. The desired product was obtained as a yellow solid (10.34 g, 86.0% yield). 1H NMR (400 MHz, CDC13): δ 9.14 (s, 1H), 8.66 (s, 1H), 8.46 (s, 1H), 7.42 (d, J= 10.4 Hz, 1H), 7.31 (d, J= 8.8 Hz, 1H), 6.63 (dd, J= 15.0, 8.7 Hz, 1H), 3.97 (s, 3H).
Step 11: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6-carboxylic acid
[0519] To a solution of methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino) benzo[d]thiazole-6- carboxylate (10.34 g, 23.17 mmol) in THF and MeOH (20 mL, 4: 1, v/v) was added 5.0 M LiOH (aq., 2 mL, 10 mmol). After stirring at ambient temperature for 2 h, the reaction was treated with 1.0 M HCl (aq.) till the solution was acidic. The aqueous layer was extracted with ethyl acetate (50 mL x 3). The combined organic phase was washed with water (100 mL) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated to give the desired product (9.51 g, 95.0% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.10 (s, 1H), 9.18 (s, 1H), 8.68 (s, 1H), 8.45 (s, 1H), 7.41 (m, 1H), 7.30 (m, 1H), 6.65 (m, 1H). Step 12: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy)etho
carboxamide
[0520] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d]thiazole-6- carboxylic acid (519 mg, 1.20 mmol) in CH2C12 (10 mL) was added HOBt (254 mg, 1.63 mmol) and EDCI (314 mg, 1.63 mmol). The mixture was stirred for 1 h and O-(2-
(vinyloxy)ethyl)hydroxyl -amine (172 mg, 1.62 mmol) was added. After stirring for 4 h at ambient temperature, the reaction was treated with saturated H4C1 (aq.). The resultant mixture was extracted with CH2C12 (30 mL χ 3). The combined organic extracts were washed with water (30 mL) and brine (30 mL), dried over Na2S04 filtered, and concentrated in vacuo. The crude product (492 mg) was used directly in the next step without further purification.
Step 13: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)benzo[d]thiazole-6- carboxamide
[0521] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- (vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide (492 mg, 1.00 mmol) in CH2C12 (10 mL) was added 1.0 N HCl (aq., 5 mL, 5 mmol). After stirring for 1 h, the reaction mixture was neutralized with saturated NaHC03 (aq.). The aqueous layer was washed with CH2C12 (30 mL). The combined organic layer was washed with water (30 mL x 2) and brine (30 mL), dried over Na2S04, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH, 50: 1, v/v) and gave the desired product as a white solid (446 mg, 75.9% yield for the two steps). 1H MR (400 MHz, DMSO-d6): δ 11.80 (s, 1H), 9.55 (s, 1H), 8.22 (s, 1H), 8.12 (s, 1H), 7.55 (d, J= 11.0 Hz, 1H), 7.31 (d, J= 8.5 Hz, 1H), 6.48 (d, J= 9.2 Hz, 1H), 4.72 (s, 1H), 3.84 (m, 2H), 3.57 (m, 2H). MS APCI(+)m/z: 491.8, [M+H].
Example 9A: Preparation of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- hydroxyethoxy)benzo[d]thiazole-6-carboxamide (Compound 9)

Step 1: 5-bromo-2,3,4-trifluorobenzoic aci

[0522] To a solution of l-bromo-2,3,4-trifluorobenzene (13.64 g, 64.6 mmol) in THF (120 mL) was added lithium diisopropylamide (2.0 M in THF, 33.9 mL, 67.8 mmol) at -78 °C under nitrogen atmosphere. After stirring for 1 h at -78 °C, the mixture was transferred to a bottle with dry ice. The mixture was stirred overnight at room temperature. The reaction was quenched with 10% aqueous HC1 (300 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic extracts were washed with 5% sodium hydroxide (300 mL). The aqueous layer was acidized to pH 1 and extracted with ethyl acetate (200 mL χ 3). The combined organic extract was dried over Na2S04, filtered and concentrated under reduced pressure to afford the desired product (white solid, 13.51 g, 82% yield). 1H MR (400 MHz, CDC13): δ 13.94 (s, 1H), 7.95 (m,
1H).
Step 2: 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic

[0523] To a solution of 2-fluoroaniline (10.2 mL, 105.8 mmol) and 5-bromo-2,3,4- trifluorobenzoic acid (13.51 g, 52.9 mmol) in THF (120 mL) was added LiHMDS (158.7 mL, 1 M in THF, 158.7 mmol) dropwisely at -78 °C under nitrogen atmosphere. The mixture was allowed to slowly warm to room temperature and stirred at this temperature overnight. The reaction was quenched with 10% HC1 (aq., 100 mL) and extracted with ethyl acetate (200 mL x 3). The combined organic extracts were washed with water (200 mL x 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the desired product (pale yellow solid, 13.73 g, 75% yield). 1H MR (400 MHz, DMSO-d6): δ 9.21 (s, 1H), 8.01 (d, 1H), 7.26 (m, 1H), 7.01-7.16 (m, 3H).
Step 3: methyl 5-bromo-3,4-difluoro-2- -fluorophenyl)amino)benzoate

[0524] To a solution of 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoic acid (13.73 g, 39.6 mmol) in MeOH (300 mL) was added SOCl2 (60 mL). After stirring at 85 °C overnight, most MeOH was removed in vacuo. The residue was neutralized with saturated sodium bicarbonate (aq.) and extracted with ethyl acetate (300 mL χ 3). The combined organic extract was washed with water (200 mL x 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo to afford the corresponding product (gray solid, 12.58 g, 90% yield). 1H MR (400 MHz, CDC13): δ 9.09 (s, 1H), 8.05 (d, 1H), 7.00-7.14 (m, 4H), 3.94 (s, 3H).
Step 4: methyl 3,4-difluoro-2-((2-fluorophenyl)amino)-5-((4-methoxybenzyl)thio)benzoate

[0525] To a solution of methyl 5-bromo-3,4-difluoro-2-((2-fluorophenyl)amino)benzoate (12.85 g, 35.6 mmol) in anhydrous 1,4-dioxane (30 mL) was added N,N-diisopropylethylamine (9.21 g, 71.2 mmol). Then Pd2(dba)3 (1.63 g, 1.78 mmol) followed by Xantphos (2.06 g, 3.56 mmol) and 4-methoxy-a-toluenethiol (5.48 g, 35.6 mmol) was added under nitrogen atmosphere. The mixture was stirred overnight at 100 °C under N2 atmosphere and then allowed to cool to ambient temperature. The reaction was quenched with water (150 mL) and extracted with ethyl acetate (200 mL χ 3). The combined organic extract was washed with water (200 mL χ 3) and brine (200 mL) sequentially, dried over Na2S04, filtered and concentrated. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 50: 1, v/v) to give the desired product (pale yellow solid, 12.64 g, 82% yield). 1H NMR (400 MHz, CDC13): δ 9.12 (s, 1H), 7.78 (d, 1H), 7.06-7.44 (m, 6H), 6.82-6.88 (m, 2H), 4.03 (s, 2H), 3.90 (s, 3H), 3.80 (s, 3H).
Step 5: methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate

[0526] To a solution of methyl 5-(4-methoxybenzylthio)-3,4-difluoro-2-((2- fluorophenyl)amino)benzoate (12.64 g, 29.2 mmol) in DMF (30 mL) was added NaN3 (2.28 g, 35.0 mmol) at ambient temperature. The mixture was stirred at 90 °C for 3 h. Then water (150 mL) was added. The solution was extracted with ethyl acetate (100 mL χ 3). The combined organic extracts were washed with water (100 mL χ 3) and brine (100 mL), dried over Na2S04 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v) and gave the desired product (white solid, 10.38 g, 78% yield). 1H NMR (400 MHz, CDC13): δ 8.98 (s, 1H), 7.75 (s, 1H), 7.02-7.28 (m, 6H), 6.83- 6.85 (m, 2H), 4.03 (s, 2H), 3.92 (s, 3H), 3.81 (s, 3H).
Step 6: methyl 4-amino-5-(4-methoxybenzylthio)-3-fluoro-2-((2-fluorophenyl)amino)benzoate

[0527] To a solution of methyl 4-azido-5-(4-methoxybenzylthio)-3-fluoro-2-((2- fluorophenyl)amino)benzoate (10.38 g, 22.7 mmol) in MeOH (100 mL) was added and 10% palladium on carbon (1.55 g) under nitrogen atmosphere. Then the nitrogen atmosphere was completely changed to hydrogen atmosphere. The mixture was stirred at ambient temperature for 6 h. After the insoluble matter was filtered off, the solvent was evaporated in vacuo to give the desired product (9.79 g, 100% yield).1H MR (400 MHz, CDC13): δ 9.08 (s, 1H), 7.78 (s, 1H), 6.93-7.28 (m, 8H), 4.65 (s, 2H), 4.00 (s, 2H), 3.89 (s, 3H), 3.75 (s, 3H).
Step 7: methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-mercaptobenzoate

[0528] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5-((4- methoxybenzyl)thio)benzoate (9.79 g, 22.7 mmol) in anisole (12 mL) was added CF3COOH (20 mL). After stirring at ambient temperature for 23 h, the solvent was removed in vacuo. To the residue was added water (30 mL). The mixture was neutralized with 25% aqueous ammonia and extracted with ethyl acetate (100 mL χ 3). The combined organic layer was washed with water (100 mL x 3) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated to give the desired product (white solid, 5.28 g, 75% yield). The product was used directly in the next step without further purification.
Step 8: methyl 4-fluoro-5-((2-fluorophenyl)amino)benzofdJthiazole-6-carboxylate

[0529] To a solution of methyl 4-amino-3-fluoro-2-((2-fluorophenyl)amino)-5- mercaptobenzoate (2.07 g, 6.67 mmol) in trimethyl orthoformate (20 mL) was added p-TsOU (166 mg, 0.65 mmol). The reaction mixture was stirred for 1 h and treated with water (100 mL). The precipitate was filtered off and the filter cake was washed with water to afford the desired product (white solid, 1.963 g, 92% yield for two steps). 1H NMR (400 MHz, DMSO-d6): δ 9.01 (s, 1H), 8.08 (s, 1H), 7.90 (s, 1H), 7.15-6.78 (m, 4H), 3.91 (s, 3H).
Step 9: methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzofdJthiazole-6-carboxylate

[0530] To a solution of methyl 4-fluoro-5-((2-fluorophenyl)amino)benzo[d]thiazole-6- carboxylate (1.963 g, 6.14 mmol) in DMF (10 mL) was added NIS (1.5 g, 6.5 mmol) followed by trifluoroacetic acid (0.5 mL). After stirring for 4 h at ambient temperature, the reaction was treated by saturated H4C1 (aq.). The aqueous layer was extracted with ethyl acetate (150 mL χ 3). The combined organic layer was washed with water (100 mL x 3) and brine (100 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. After purification by flash column chromatography on silica gel (petroleum ether/ethyl acetate, 10: 1, v/v), the desired product was obtained as white solid (1.889 g, 69% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.03 (s, 1H), 8.10 (s, 1H), 7.93 (s, 1H), 7.18-6.72 (m, 3H), 3.91 (s, 3H).
Step 10: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-(vinyloxy
carboxamide

[0531] To a solution of O-(2-(vinyloxy)ethyl)hydroxyl-amine (172 mg, 1.62 mmol) in THF (6 mL) was added LiHMDS (2.5 mL, 1 M in THF, 2.5 mmol) at -78 °C. After stirring at this temperature for 10 min, a solution of methyl 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)benzo[d] thiazole-6-carboxylate (360 mg, 0.81 mmol) in THF was syringed dropwisely. Then the mixture was allowed to warm to ambient temperature, quenched with saturated NH4C1 (aq., 20 mL) and extracted with ethyl acetate (15 mL χ 3). The combined organic extract was washed with water (10 mL x 3) and brine (10 mL), dried over Na2S04, filtered and concentrated in vacuo. After purification by flash chromatography (petroleum ether/ethyl acetate, 10: 1, v/v), the desired product was obtained (410 mg, 98% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.85 (s, 1H),
8.98 (s, 1H), 8.04 (s, 1H), 7.89 (s, 1H), 7.55 (d, J= 10.8 Hz, 1H), 7.31 (d, J = 8.1 Hz, 1H), 6.53 (dd, J= 13.9, 6.6 Hz, 1H), 6.42 (d, J= 6.0 Hz, 1H), 4.21 (d, J= 14.5 Hz, 1H), 4.01 (m, 3H), 3.83 (m, 2H).
Step 11: 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2-hydroxyethoxy)benzofdJthiazole-6- carboxamide

[0532] To a solution of 4-fluoro-5-((2-fluoro-4-iodophenyl)amino)-N-(2- (vinyloxy)ethoxy)benzo[d]thiazole-6-carboxamide (410 mg, 0.8 mmol) in CH2C12 (5 mL) was added 1.0 N HCl (aq., 5 mL, 5 mmol) dropwise. After stirring for 1 h, the reaction mixture was neutralized with saturated NaHC03 (aq.). The organic layer was separated, washed with water (30 mL x 2) and brine (30 mL) sequentially, dried over Na2S04, filtered and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH, 15: 1, v/v) and the desired product was obtained as a white solid (290 mg, 52 % yield). 1H MR (400 MHz, DMSO-de): δ 11.83 (s, 1H), 8.92 (s, 1H), 8.03 (s, 1H), 7.90 (s, 1H), 7.56 (d, J= 9.4 Hz, 1H), 7.30 (d, J= 8.7 Hz, 1H), 6.41 (m, 1H), 4.72 (m, 1H), 3.85 (m, 2H), 3.59 (m, 2H). MS (ES+): m/z 492.35 [MH+].
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Tunlametinib, an oral selective inhibitor of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2), was developed by Shanghai KeChow Pharmaceuticals Co., Ltd. Marketed under the brand name
Keluping, it received conditional approval from the NMP in 2024 for the treatment of patients with advanced melanoma harboring NRAS mutations, particularly those who have not responded to anti-PD-1/PD-L1therapies [1]. Tunlametinib exerts its antitumor effects by targeting the MEK1/2 kinases within the RAS-RAF-MEK-ERK signaling pathway, thereby disrupting downstream signaling cascades and inhibiting tumor cell growth and proliferation [2]. Its clinical efficacy was demonstrated in a Phase II pivotal registration study (NCT05217303) involving patients with advanced NRAS-mutant melanoma [3]. The study reported a confirmed objective response rate (ORR) of 34.8 % and a median progression-free survival (mPFS) of 4.2 months. These findings suggest that Tunlametinib holds promise as a treatment option for NRAS-mutant melanoma, including in patients who have failed immunotherapy. In terms of safety, Tunlametinib has been generally well-tolerated [4]. Adverse events frequently encountered during treatment primarily consist of increased blood creatine phosphokinase (CPK) levels, diarrhea, and edema. Additionally, warnings and precautions pertinent to Tunlametinib therapy encompass decreased left ventricular ejection fraction (LVEF), skin toxicity, ocular toxicity, interstitial lung disease,
gastrointestinal reactions, and elevated CPK levels [5].
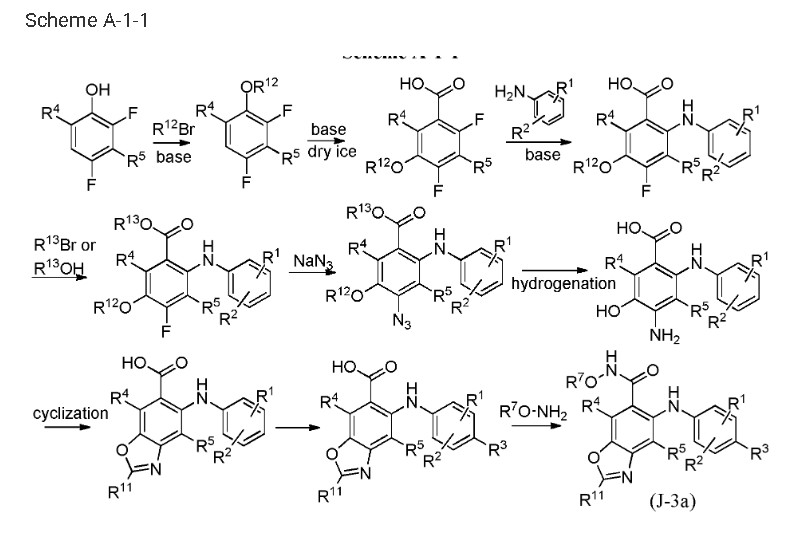
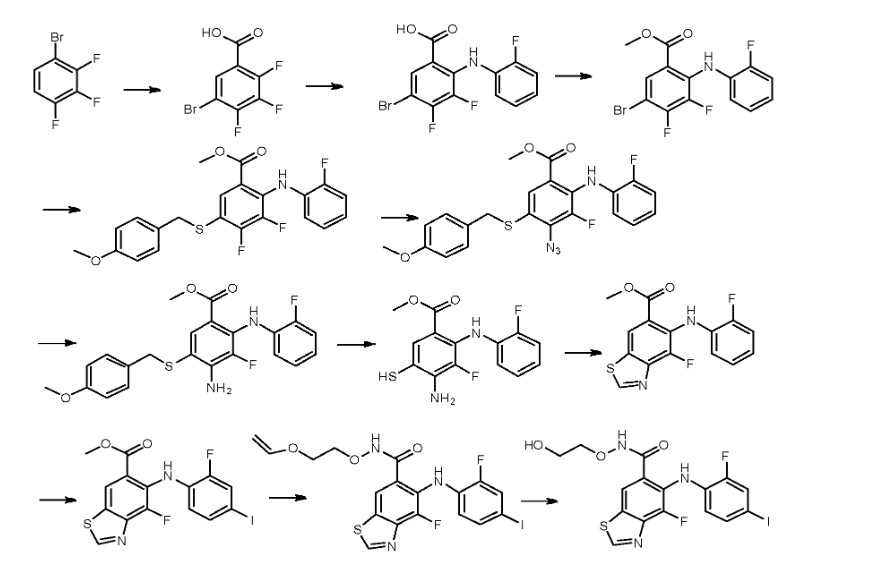
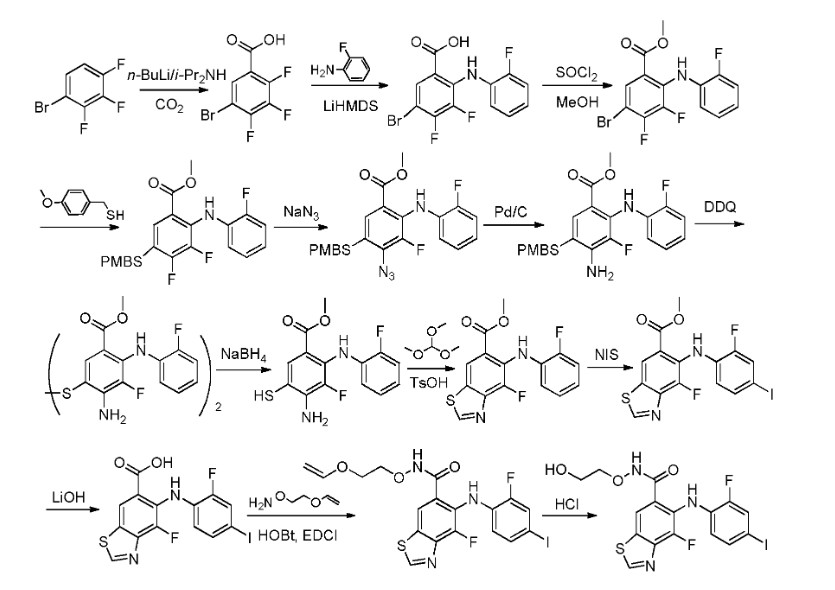
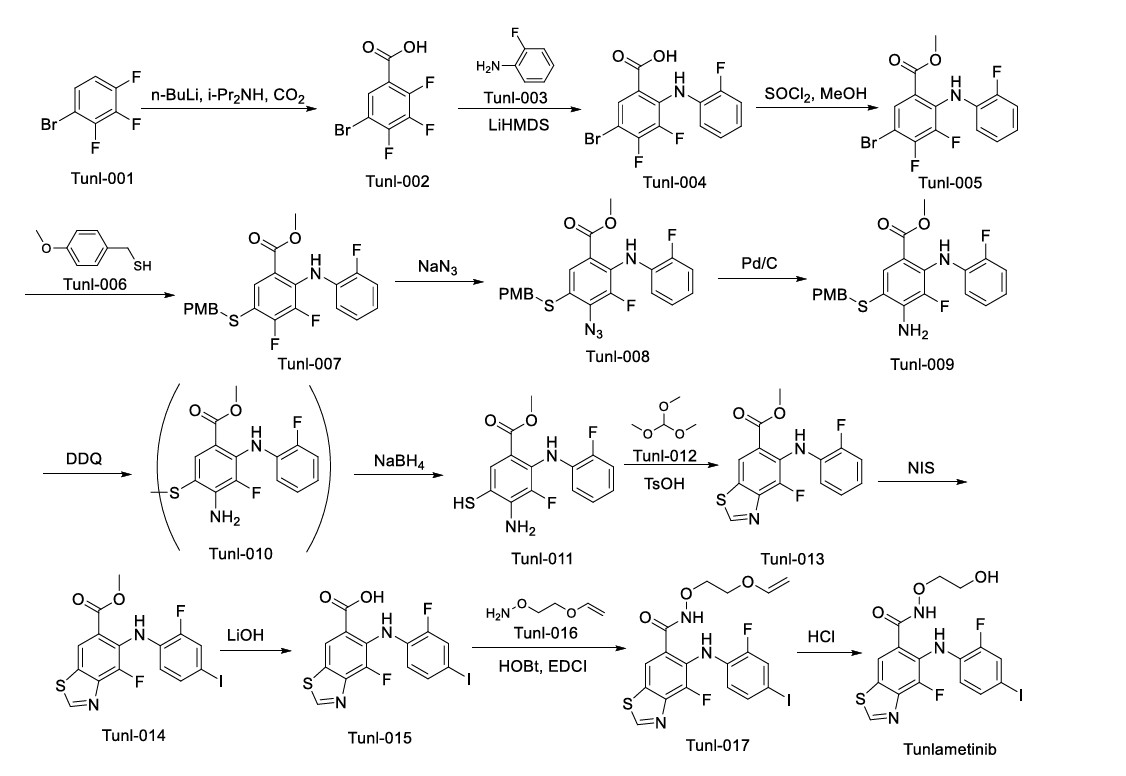
The synthetic pathway of Tunlametinib, illustrated in Scheme 1, begins with carboxylation of Tunl-001 to yield Tunl-002 [6]. Nucleophilic substitution of Tunl-002 with Tunl-003 then produces Tunl-004,
which undergoes esterification to form Tunl-005. Subsequent nucleophilic substitution between Tunl-05 and Tunl-006 generates Tunl-007. This intermediate undergoes azidation to afford Tunl-008, followed by
reduction to Tunl-009. Treatment of Tunl-009 with DDQ converts it to Tunl-010, which is deprotected to yield Tunl-011. Cycloaddition of Tunl-011 with Tunl-012 forms Tunl-013. Iodination of Tunl-013 gives
Tunl-014, which is hydrolyzed to produce Tunl-015. Amidation of Tunl-015 with Tunl-016 yields Tunl-017, and its subsequent acidolysis completes the synthesis of Tunlametinib.
[1] Y. Liu, Y. Cheng, G. Huang, X. Xia, X. Wang, H. Tian, Preclinical characterization of
tunlametinib, a novel, potent, and selective MEK inhibitor, Front. Pharmacol. 14
(2023) 1271268.
[2] S.J. Keam, Tunlametinib: first approval, Drugs 84 (2024) 1005–1010.
[3] X. Wei, Z. Zou, W. Zhang, M. Fang, X. Zhang, Z. Luo, J. Chen, G. Huang, P. Zhang,
Y. Cheng, J. Liu, J. Liu, J. Zhang, D. Wu, Y. Chen, X. Ma, H. Pan, R. Jiang, X. Liu,
X. Ren, H. Tian, Z. Jia, J. Guo, L. Si, A phase II study of efficacy and safety of the MEK inhibitor tunlametinib in patients with advanced NRAS-Mutant melanoma,
Eur. J. Cancer 202 (2024) 114008.
[4] Q. Zhao, T. Wang, H. Wang, C. Cui, W. Zhong, D. Fu, W. Xi, L. Si, J. Guo, Y. Cheng,
H. Tian, P. Hu, Phase I pharmacokinetic study of an oral, small-molecule MEK
inhibitor tunlametinib in patients with advanced NRAS mutant melanoma, Front.
Pharmacol. 13 (2022) 1039416.
[5] Y. Shi, X. Han, Q. Zhao, Y. Zheng, J. Chen, X. Yu, J. Fang, Y. Liu, D. Huang, T. Liu,
H. Shen, S. Luo, H. Yu, Y. Cao, X. Zhang, P. Hu, Tunlametinib (HL-085) plus
vemurafenib in patients with advanced BRAF V600-mutant solid tumors: an open-
label, single-arm, multicenter, phase I study, Exp. Hematol. Oncol. 13 (2024) 60.
[6] H. Tian, C. Ji, C. Liu, L. Kong, Y. Cheng, G. Huang, Benzoheterocyclic Compounds
and Use Thereof, 2014. US9937158B2.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Tunlametinib”. NCI Drug Dictionary. National Cancer Institute.
- “Tunlametinib Wins Approval in China for NRAS+ Advanced Melanoma After PD-1/PD-L1 Therapy”. 18 March 2024.
- Keam SJ (2024). “Tunlametinib: First Approval”. Drugs. 84 (8): 1005–1010. doi:10.1007/s40265-024-02072-x. PMID 39034326.
- Shi Y, Han X, Zhao Q, Zheng Y, Chen J, Yu X, et al. (2024). “Tunlametinib (HL-085) plus vemurafenib in patients with advanced BRAF V600-mutant solid tumors: An open-label, single-arm, multicenter, phase I study”. Experimental Hematology & Oncology. 13 (1): 60. doi:10.1186/s40164-024-00528-0. PMC 11167782. PMID 38867257.
| Clinical data | |
|---|---|
| Other names | HL-085 |
| ATC code | None |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1801756-06-8 |
| PubChem CID | 71621329 |
| ChemSpider | 115006753 |
| UNII | IF25NR1PV3 |
| ChEMBL | ChEMBL5095241 |
| Chemical and physical data | |
| Formula | C16H12F2IN3O3S |
| Molar mass | 491.25 g·mol−1 |
/////////Tunlametinib, CHINA 2024, APPROVALS 2024, Shanghai KeChow, Keluping,1801756-06-8, IF25NR1PV3, HL 085
Taletrectinib
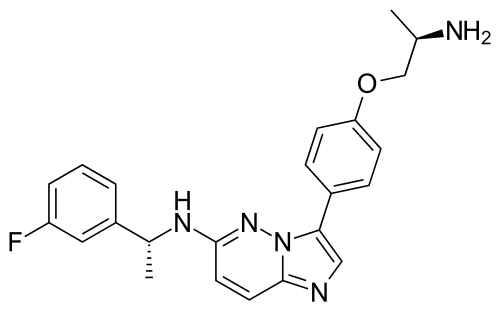
Taletrectinib
CAS 1505514-27-1
as salt: 1505515-69-4, Taletrectinib adipate
FDA 6/11/2025, Ibtrozi, To treat locally advanced or metastatic ROS1-positive non-small cell lung cancer ALSO CHINA 2024 APPROVED |
405.5 g/mol, C23H24FN5O, UNII-W4141180YD
3-[4-[(2R)-2-aminopropoxy]phenyl]-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine
Taletrectinib adipate
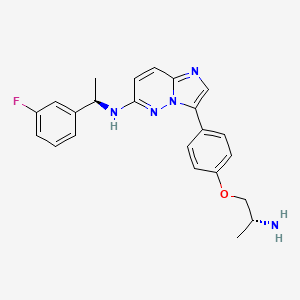
WeightAverage: 551.619
Monoisotopic: 551.254397378
Chemical FormulaC29H34FN5O5
DS-6051B, CAS 1505515-69-4,
6KLL51GNBG, 3-{4-[(2R)-2-aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine; hexanedioic acid
Taletrectinib, sold under the brand name Ibtrozi, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1][2] It is used as the salt, taletrectinib adipate.[1] Taletrectinib is a kinase inhibitor.[1] It is taken by mouth.[1]
Taletrectinib was approved for medical use in the United States in June 2025.[3]
SYN
US20200062765
https://patentscope.wipo.int/search/en/detail.jsf?docId=US289038418&_cid=P12-MCIHV1-02369-1
Example 1
tert-Butyl [(2R)-1-(4-bromophenoxy)propan-2-yl]carbamate (1)
Example 2
6-Fluoroimidazo[1,2-b]pyridazine methanesulfonate (2)
Example 3
tert-Butyl {(2R)-1-[4-(6-fluoroimidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate (3)
Example 4
tert-Butyl {(2R)-1-[4-(6-{[(1R)-1-(3-fluorophenyl)ethyl]amino}imidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate hydrochloride (4)
Example 5
3-{4-[(2R)-2-Aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethylimidazo[1,2-b]pyridazin-6-amine dihydrochloride (5)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023272701&_cid=P12-MCIHPU-95869-1
The NMR data for the crystalline form A of Compound 1 adipate are as follows: 1H NMR (500 MHz, DMSO) δ 1.13-1.14 (d, J=5.0 Hz, 3H) , 1.47-1.48 (d, J=5.0 Hz, 7H) , 2.15-2.18 (t, J=5.0 Hz, J=10.0 Hz, 4H) , 3.25-3.29 (m, 1H) , 3.79-3.83 (m, 2H) , 4.80-4.85 (m, 1H) , 6.76-6.77 (d, J=5.0 Hz, 1H) , 6.92-6.94 (d, J=10.0 Hz, 2H) , 7.01-7.05 (t, J=10.0 Hz, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.64-7.65 (d, J=5.0 Hz, 1H) , 7.72-7.76 (t, J=10.0 Hz, 4H) .
[0148]
The IR data for the crystalline form A of Compound 1 adipate are as follows: IR (cm -1) : 1701, 1628, 1612, 1586, 1463, 1333, 1246, 1110, 829, 821.
Example 5: Preparation and Characterization of Crystalline Form A of Compound 1 Free Base
[0212]
Compound 1 HCl (75.5 g) (e.g., obtained by using the method described in Example 5 of U.S. Application Publication No. 2020/0062765) was dissolved in ethanol (604 mL) at 50℃. Sodium hydroxide (68.1 g) was added to the above solution. The mixture was cooled to 1℃ in 1.5 hours and stirred for 18.5 hours. The mixture was then filtered, and the solid thus obtained was washed with a cooled mixture of ethanol (151 mL) and water (151 mL) and dried. The solid thus obtained was confirmed to be the crystalline form A of Compound 1 free base.
[0213]
The NMR data for the crystalline form A of Compound 1 free base are as follows: 1H NMR (500 MHz, DMSO) δ 1.09-1.10 (d, J=5.0 Hz, 3H) , 1.48-1.49 (d, J=5.0 Hz, 3H) , 3.16-3.20 (m, 1H) , 3.75-3.79 (m, 2H) , 4.82-4.86 (m, 1H) , 6.76-6.78 (d, J=10.0 Hz, 1H) , 6.92-6.94 (m, 2H) , 7.01-7.05 (m, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.62-7.63 (d, J=5.0 Hz, 1H) , 7.72-7.75 (m, 4H) .
[0214]
The IR data for the crystalline form A of Compound 1 free base are as follows: IR (cm -1) : 3350, 3247, 3055, 2961, 2923, 2864, 1611, 1586, 1349, 829, 819.
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Taletrectinib is an oral, next-generation ROS1 TKI developed by Nuvation Bio Inc. for the treatment of ROS1-positive NSCLC. In 2024, the NMPA approved taletrectinib for adult patients with locally advanced or metastatic ROS1-positive NSCLC, regardless of prior ROS1TKI treatment [47]. Under an exclusive license agreement, Innovent Biologics will commercialize taletrectinib in China under the brand
name DOVBLERON®. Taletrectinib exerts its pharmacological action through the mechanism of selectively impeding the ROS1 receptor tyrosine kinase, which effectively disrupts the signaling cascades which are responsible for facilitating the growth and survival of cancer cells in ROS1-positive NSCLC. This inhibition of the ROS1 receptor tyrosine kinase is a key event in the drug’s mode of action, as it specifically targets the molecular processes that drive the progression of the disease in ROS1-positive NSCLC cases [48]. The NMPA granted approval founded on the data sourced from the crucial Phase 2 TRUST – I study. This study substantiated that patients administered with taletrectinib achieved sustained responses and extended PFS. Regarding safety, taletrectinib boasted a generally good tolerability. It presented an advantageous safety profile and favorable tolerability characteristics, as evidenced by the low incidences of dose reduction and treatment discontinuation triggered by adverse effects. [49]. Overall, taletrectinib represents a promising therapeutic option for patients with advanced ROS1-positive NSCLC, offering efficacy in both TKI-naïve and TKI-pretreated populations, including those with CNS metastases [50–52].
The synthesis of Taletrectinib, illustrated in Scheme 12, commences with Mitsunobu coupling of Tale-001 and Tale-002 to afford Tale-003, which then undergoes Suzuki coupling with Tale-004 constructing
Tale-005 [53]. Sequential acidolysis/deprotection of Tale-005 ultimately delivers Taletrectinib
[47] M. P´ erol, N. Yang, C.M. Choi, Y. Ohe, S. Sugawara, N. Yanagitani, G. Liu, F.G.M.
D. Braud, J. Nieva, M. Nagasaka, 1373P efficacy and safety of taletrectinib in
patients (pts) with ROS1+ non-small cell lung cancer (NSCLC): interim analysis of
global TRUST-II study, Ann. Oncol. 34 (2023) S788–S789.
[48] G. Harada, F.C. Santini, C. Wilhelm, A. Drilon, NTRK fusions in lung cancer: from
biology to therapy, Lung Cancer 161 (2021) 108–113.
[49] W. Li, A. Xiong, N. Yang, H. Fan, Q. Yu, Y. Zhao, Y. Wang, X. Meng, J. Wu, Z. Wang,
Y. Liu, X. Wang, X. Qin, K. Lu, W. Zhuang, Y. Ren, X. Zhang, B. Yan, C.M. Lovly,
C. Zhou, Efficacy and safety of taletrectinib in Chinese patients with ROS1+ non-
small cell lung cancer: the phase II TRUST-I study, J. Clin. Oncol. 42 (2024)
2660–2670.
[50] M. Nagasaka, D. Brazel, S.I. Ou, Taletrectinib for the treatment of ROS-1 positive
non-small cell lung cancer: a drug evaluation of phase I and II data, Expert Opin
Investig Drugs 33 (2024) 79–84.
[51] S. Waliany, J.J. Lin, Taletrectinib: TRUST in the continued evolution of treatments
for ROS1 fusion-positive lung cancer, J. Clin. Oncol. 42 (2024) 2622–2627.
[52] M. Nagasaka, Y. Ohe, C. Zhou, C.M. Choi, N. Yang, G. Liu, E. Felip, M. P´ erol,
B. Besse, J. Nieva, L. Raez, N.A. Pennell, A. Dimou, F. Marinis, F. Ciardiello,
T. Seto, Z. Hu, M. Pan, W. Wang, S. Li, S.I. Ou, TRUST-II: a global phase II study of
taletrectinib in ROS1-positive non-small-cell lung cancer and other solid tumors,
Future Oncol. 19 (2023) 123–135.
[53] Y. Takeda, K. Yoshikawa, Y. Kagoshima, Y. Yamamoto, R. Tanaka, Y. Tominaga,
M. Kiga, Y. Hamada, Preparation of imidazo[1,2-b]pyridazine Derivatives as
Potent Inhibitors of ROS1 Kinase and NTRK Kinase, 2013. WO2013183578A1.

Medical uses
Taletrectinib is indicated for the treatment of adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer.[1][2]
Adverse effects
The FDA prescribing information for taletrectinib includes warnings and precautions for hepatotoxicity, interstitial lung disease/pneumonitis, QTc interval prolongation, hyperuricemia, myalgia with creatine phosphokinase elevation, skeletal fractures, and embryo-fetal toxicity.[1][3]
History
The efficacy of taletrectinib to treat ROS1-positive non-small cell lung cancer was evaluated in participants with locally advanced or metastatic, ROS1-positive non-small cell lung cancer enrolled in two multi-center, single-arm, open-label clinical trials, TRUST-I (NCT04395677) and TRUST-II (NCT04919811).[3] The efficacy population included 157 participants (103 in TRUST-I; 54 in TRUST-II) who were naïve to treatment with a ROS1 tyrosine kinase inhibitor (TKI) and 113 participants (66 in TRUST-I; 47 in TRUST-II) who had received one prior ROS1 tyrosine kinase inhibitor.[3] Participants may have received prior chemotherapy for advanced disease.[3] The US Food and Drug Administration (FDA) granted the application for taletrectinib priority review, breakthrough therapy, and orphan drug designations.[3]
Society and culture
Legal status
Taletrectinib was approved for medical use in the United States in June 2025.[3][4]
Names
Taletrectinib is the international nonproprietary name.[5]
Taletrectinib is sold under the brand name Ibtrozi.[3][4]
References
- ^ Jump up to:a b c d e f g “Prescribing Information for NDA 219713, Supplement 000” (PDF). Drugs@FDA. U.S. Food and Drug Administration. April 2025. Retrieved 14 June 2025.
- ^ Jump up to:a b Khan I, Sahar A, Numra S, Saha N, Nidhi, Parveen R (April 2025). “Efficacy and safety of taletrectinib for treatment of ROS1 positive non-small cell lung cancer: A systematic review”. Expert Opinion on Pharmacotherapy. 26 (6): 765–772. doi:10.1080/14656566.2025.2487150. PMID 40170301.
- ^ Jump up to:a b c d e f g h “FDA approves taletrectinib for ROS1-positive non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 11 June 2025. Retrieved 13 June 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “U.S. Food and Drug Administration Approves Nuvation Bio’s Ibtrozi (taletrectinib), a Next-Generation Oral Treatment for Advanced ROS1-Positive Non-Small Cell Lung Cancer”. Nuvation Bio (Press release). 12 June 2025. Retrieved 13 June 2025.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
- Clinical trial number NCT04395677 for “A Study of AB-106 in Subjects With Advanced NSCLC Harboring ROS1 Fusion Gene” at ClinicalTrials.gov
- Clinical trial number NCT04919811 for “Taletrectinib Phase 2 Global Study in ROS1 Positive NSCLC (TRUST-II)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Ibtrozi |
| License data | US DailyMed: Taletrectinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1505514-27-1as salt: 1505515-69-4 |
| PubChem CID | 72202474as salt: 72694302 |
| DrugBank | DB18711 |
| ChemSpider | 114934673as salt: 88297530 |
| UNII | W4141180YDas salt: 6KLL51GNBG |
| KEGG | D12363as salt: D12364 |
| ChEMBL | ChEMBL4650989as salt: ChEMBL4650361 |
| Chemical and physical data | |
| Formula | C23H24FN5O |
| Molar mass | 405.477 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Taletrectinib, FDA 2025, APPROVALS 2025, Ibtrozi, CANCER, AB-106, DS-6051a, UNII-W4141180YD, DS 6051B, APPROVALS 2024, CHINA 2024, Nuvation Bio Inc
Iptacopan


Iptacopan
1644670-37-0
422.525, C25H30N2O4
- 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl) benzoic acid
- BENZOIC ACID, 4-((2S,4S)-4-ETHOXY-1-((5-METHOXY-7-METHYL-1H-INDOL-4-YL)METHYL)-2-PIPERIDINYL)-
- Iptacopan
- LNP 023
- LNP-023
- LNP023
- NVP-LNP023
- NVP-LNP023-NX
Fda approved, To treat paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta ‘CHINA 2024
Iptacopan is a small-molecule factor B inhibitor previously investigated as a potential treatment for the rare blood disease paroxysmal nocturnal hemoglobinuria (PNH) by inhibiting the complement factor B.1 Factor B is a positive regulator of the alternative complement pathway, where it activates C3 convertase and subsequently C5 convertase.2 This is of particular importance to PNH, where one of the disease hallmarks is the mutation of the PIGA gene. Due to this mutation, all progeny erythrocytes will lack the glycosyl phosphatidylinositol–anchored proteins that normally anchor 2 membrane proteins, CD55 and CD59, that protect blood cells against the alternative complement pathway.3 Additionally, iptacopan has the benefit of targeting factor B, which only affect the alternative complement pathway, leaving the classic and lectin pathway untouched for the body to still mount adequate immune responses against pathogens.2
On December 6th, 2023, Iptacopan under the brand name Fabhalta was approved by the FDA for the treatment of adults with PNH. This approval was based on favorable results obtained from the phase III APPL-PNH and APPOINT-PNH studies, where 82.3% and 77.5% of patients experienced a sustained hemoglobin improvement without transfusions respectively.5
Iptacopan , sold under the brand name Fabhalta, is a medication used for the treatment of paroxysmal nocturnal hemoglobinuria.[1] It is a complement factor B inhibitor that was developed by Novartis.[1] It is taken by mouth.[1]
Iptacopan was approved by the US Food and Drug Administration (FDA) for the treatment of adults with paroxysmal nocturnal hemoglobinuria in December 2023.[2][3]
Medical uses
Iptacopan is indicated for the treatment of adults with paroxysmal nocturnal hemoglobinuria.[1][4]
Side effects
The FDA label for iptacopan contains a black box warning for the risk of serious and life-threatening infections caused by encapsulated bacteria, including Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B.[1]
Research
In a clinical study with twelve participants, iptacopan as a single drug led to the normalization of hemolytic markers in most patients, and no serious adverse events occurred during the 12-week study.[5][6]
Iptacopan is also investigated as a drug in other complement-mediated diseases, like age-related macular degeneration and some types of glomerulopathies.[7]
PATENT
https://patents.google.com/patent/US9682968B2/en
Example-26Example-26a4-((2S,4S)-(4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl))benzoic acid ((+) as TFA Salt)

A mixture of methyl 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoate, Intermediate 6-2b peak-1 (tr=1.9 min), (84 mg, 0.192 mmol) and LiOH in H2O (1 mL, 1 mmol) in THF (1 mL)/MeOH (2 mL) was stirred at room temperature for 16 h, and then concentrated. The resulting residue was purified by RP-HPLC (HC-A) to afford the title compound. Absolute stereochemistry was determined by comparison with enantiopure synthesis in Example-26c. 1H NMR (TFA salt, 400 MHz, D2O) δ 8.12 (d, J=8.19 Hz, 2H), 7.66 (br. d, J=8.20 Hz, 2H), 7.35 (d, J=3.06 Hz, 1H), 6.67 (s, 1H), 6.25 (d, J=3.06 Hz, 1H), 4.65 (dd, J=4.28, 11.49 Hz, 1H), 4.04 (d, J=13.00 Hz, 1H), 3.87-3.98 (m, 2H), 3.53-3.69 (m, 5H), 3.38-3.50 (m, 1H), 3.20-3.35 (m, 1H), 2.40 (s, 3H), 2.17-2.33 (m, 2H), 2.08 (br. d, J=15.70 Hz, 1H), 1.82-1.99 (m, 1H), 1.28 (t, J=7.03 Hz, 3H); HRMS calcd. for C26H31N2O3 (M+H)+ 423.2284, found 423.2263.
PATENT
Example 1
PAPER
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.9b01870
The alternative pathway (AP) of the complement system is a key contributor to the pathogenesis of several human diseases including age-related macular degeneration, paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and various glomerular diseases. The serine protease factor B (FB) is a key node in the AP and is integral to the formation of C3 and C5 convertase. Despite the prominent role of FB in the AP, selective orally bioavailable inhibitors, beyond our own efforts, have not been reported previously. Herein we describe in more detail our efforts to identify FB inhibitors by high-throughput screening (HTS) and leveraging insights from several X-ray cocrystal structures during optimization efforts. This work culminated in the discovery of LNP023 (41), which is currently being evaluated clinically in several diverse AP mediated indications.



a Reagents and conditions: (a) i PrMgCl·LiCl, Cbz-Cl, THF; (b) Zn, AcOH; (c) LiBH4, THF; (d) TBDPS-Cl, imidazole, DMF; (e) separation of diastereomers by flash chromatography; (f) TBAF, THF; (g) NaH, EtI, DMF; (h) Ba(OH)2, i PrOH, H2O; (i) K2CO3, MeI, DMF; (j) H2, Pd/C, MeOH; (k) (±)-50, DIPEA, DMA; (l) K2CO3, MeOH; then TMS-diazomethane, toluene, MeOH; (m) chiral SFC; (n) LiOH, H2O, MeOH, THF; (o) (2S,4S)-50, NaBH(OAc)3, DCE.
4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). Step 1: tert-Butyl 4-(((2S,4S)-4-Ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58). To a solution of tert-butyl 4-formyl-5-methoxy-7-methyl1H-indole-1-carboxylate (57) (1.5 g, 5.18 mmol) and methyl 4- ((2S,4S)-4-ethoxypiperidin-2-yl)benzoate ((2S,4S)-50) (1.185 g, 4.50 mmol) in DCE (20 mL) was added NaBH(OAc)3 (3 g, 14.1 mmol), and this was stirred at rt for 21.5h. Additional tert-butyl 4-formyl-5- methoxy-7-methyl-1H-indole-1-carboxylate (57) (500 mg, 1.90 mmol) was added, and this was stirred for 20 h. The reaction was diluted with EtOAc, washed successively with 5% aqueous NaHCO3, H2O, and brine, dried over Na2SO4, filtered, and concentrated to provide the title compound (2.415 g, quant) which was used without further purification. MS (ESI+) m/z 537.4 (M + H). The absolutestereochemistry was ultimately determined via cocrystallization of 41 with the catalytic domain of FB. Step 2: 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid (41, LNP023). To a solution of tert-butyl 4-(((2S,4S)-4-ethoxy-2-(4-(methoxycarbonyl)phenyl)- piperidin-1-yl)methyl)-5-methoxy-7-methyl-1H-indole-1-carboxylate (58) (2.415 g, 4.50 mmol) in THF (10 mL) and MeOH (20 mL) was added 1 M LiOH in H2O (15 mL, 15 mmol), and this was stirred at 70 °C for 8 h. The reaction was cooled to rt, diluted with H2O, half saturated aqueous KHSO4 and citric acid, saturated with sodium chloride, then extracted with 9:1 DCM/TFE, dried with Na2SO4, filtered, and concentrated. RP-HPLC-B purification provided the title compound (730 mg, 38% for 2 steps). 1 H NMR (400 MHz, D2O) δ 7.96 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 3.2 Hz, 1H), 6.66 (s, 1H), 6.20 (s, 1H), 4.62−4.47 (m, 1H), 4.06 (d, J = 13.2 Hz, 1H), 3.97−3.76 (m, 2H), 3.66−3.48 (m, 5H), 3.43−3.29 (m, 1H), 3.26−3.15 (m, 1H), 2.35 (s, 3H), 2.31−2.11 (m, 2H), 2.00 (d, J = 15.4 Hz, 1H), 1.93−1.74 (m, 1H), 1.25−1.07 (m, 3H). HRMS calcd for C25H31N2O4 (M + H)+ 423.2284, found 423.2263. 4-((2S,4S)-(4-Ethoxy-1-((5-methoxy-7-methyl-1H-indol-4- yl)methyl)piperidin-2-yl))benzoic Acid Hydrochloride (41· HCl). To a solution of 41 (620 mg, 1.47 mmol) in H2O (10 mL) and acetonitrile (3 mL) was added 5 M aqueous HCl (0.5 mL, 2.5 mmol). The mixture was then lyophilized, and the resulting solid was suspended in i PrOH and heated to 70 °C. The mixture turned into a solution after 1.5 h and was then cooled to rt with stirring. After about 5 h, the mixture turned into a suspension and the solid was collected by filtration and dried under high vacuum at 50 °C to provide the title compound as the hydrochloride salt (450 mg, 65%). 1 H NMR (400 MHz, methanol-d4) δ 10.73 (s, 1H), 8.23 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3 Hz, 2H), 7.36−7.31 (m, 1H), 6.77 (s, 1H), 6.42−6.31 (m, 1H), 4.40−4.19 (m, 2H), 3.87−3.80 (m, 1H), 3.76 (s, 3H), 3.68− 3.50 (m, 4H), 3.45−3.38 (m, 1H), 2.51 (s, 3H), 2.30−2.18 (m, 2H), 2.13−1.89 (m, 2H), 1.31 (t, J = 7.0 Hz, 3H). MS (ESI+) m/z 423.3 (M + H).
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Iptacopan (Fabhalta®), a first-in-class oral therapeutic agent discovered by Novartis, specifically targets the complement Factor B protein within the alternative complement system. NMPA granted
marketing authorization in 2024, indicated for complement inhibitor-naïve adult patients diagnosed with paroxysmal nocturnal hemoglobinuria (PNH) [75]. By competitively binding to the catalytic domain of
Factor B, the drug effectively blocks C3 convertase assembly, thereby suppressing downstream cleavage of C3 into its active fragments. This dual inhibitory action addresses both intravascular erythrocyte
destruction and extravascular hemolytic processes characteristic of PNHpathogenesis [76]. Clinical validation emerged from the multinational APPOINT-PNH study (ClinicalTrials.gov identifier NCT04820530), where treatment-naïve participants exhibited sustained hemoglobin
stabilization (≥12 g/dL) in 79.6 % of cases, achieving transfusion in dependence over 24 weeks. Secondary endpoints revealed significant improvements in fatigue scores and health-related quality metrics [77]. Safety monitoring identified encapsulated bacterial infection as critical risks, necessitating mandatory vaccination ≥2 weeks pre-treatment. Common treatment-emergent adverse events comprised transient gastrointestinal disturbances (nausea 18.3 %, diarrhea 14.7 %) and mild
cephalgia (22.1 %), with resolution typically occurring within 4 weeks [78].
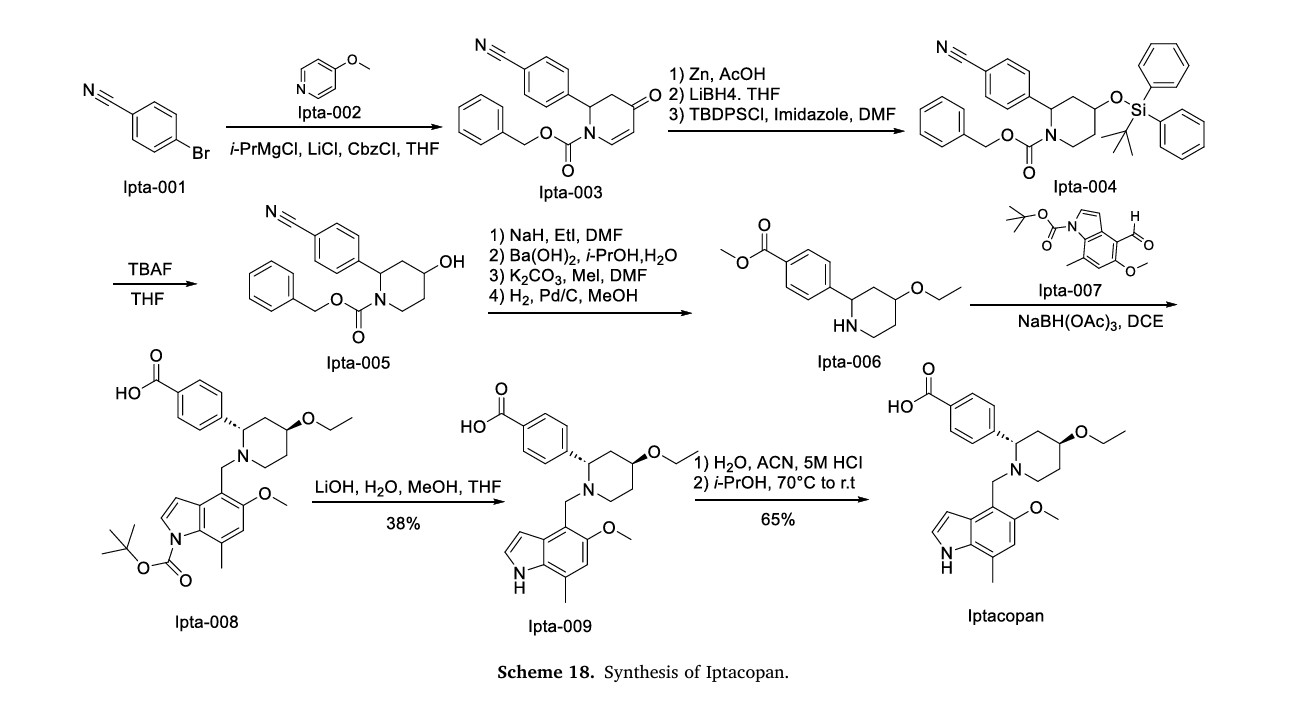
The synthetic pathway of Iptacopan, delineated in Scheme 18, initiates with nucleophilic substitution between Ipta-001 and Ipta-002, followed by Grignard coupling yielding Ipta-003 [79]. This intermedi
ate undergoes NaBH4-mediated reduction and TMSCl-induced silanization to afford Ipta-004. Acid-catalyzed TMS deprotection (HCl/MeOH) delivers Ipta-005, which progresses through sequential alkylation (methyl iodide/K2CO3 catalytic hydrogenation (H)/Pd–C), transesterification (EtONa), and to construct Ipta-006. Condensation with Ipta-007 and subsequent reduction forms Ipta-008. Strategic TFA-mediated Boc cleavage in DCM followed by HCl-induced salt formation in dioxane ultimately furnishes Iptacopan hydrochloride.
75-79
[75] Iptacopan, Drugs and Lactation Database (Lactmed®), National Institute of Child
Health and Human Development, Bethesda (MD), 2006.
[76] J.H. Jang, L. Wong, B.S. Ko, S.S. Yoon, K. Li, I. Baltcheva, P.K. Nidamarthy,
R. Chawla, G. Junge, E.S. Yap, Iptacopan monotherapy in patients with paroxysmal
nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study, Blood
Adv 6 (2022) 4450–4460.
[77] A.M. Risitano, C. de Castro, B. Han, A.G. Kulasekararaj, J.P. Maciejewski,
P. Scheinberg, Y. Ueda, S. Vallow, G. Bermann, M. Dahlke, R. Kumar, R. Peffault de
Latour, Patient-reported improvements in patients with PNH treated with
iptacopan from two phase 3 studies, Blood Adv 9 (2025) 1816–1826.
[78] C.M. de Castro, B.J. Patel, Iptacopan for the treatment of paroxysmal nocturnal
hemoglobinuria, Expert Opin Pharmacother 25 (2024) 2331–2339.
[79] N. Mainolfi, T. Ehara, R.G. Karki, K. Anderson, A. Mac Sweeney, S.M. Liao, U.
A. Argikar, K. Jendza, C. Zhang, J. Powers, D.W. Klosowski, M. Crowley,
T. Kawanami, J. Ding, M. April, C. Forster, M. Serrano-Wu, M. Capparelli,
R. Ramqaj, C. Solovay, F. Cumin, T.M. Smith, L. Ferrara, W. Lee, D. Long,
M. Prentiss, A. De Erkenez, L. Yang, F. Liu, H. Sellner, F. Sirockin, E. Valeur,
P. Erbel, D. Ostermeier, P. Ramage, B. Gerhartz, A. Schubart, S. Flohr, N. Gradoux,
R. Feifel, B. Vogg, C. Wiesmann, J. Maibaum, J. Eder, R. Sedrani, R.A. Harrison,
M. Mogi, B.D. Jaffee, C.M. Adams, Discovery of 4-((2S,4S)-4-Ethoxy-1-((5-
methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (LNP023), a
factor B inhibitor specifically designed to be applicable to treating a diverse array
of complement mediated diseases, J. Med. Chem. 63 (2020) 5697–5722.
.
//////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
| Clinical data | |
|---|---|
| Trade names | Fabhalta |
| Other names | LNP023 |
| AHFS/Drugs.com | Fabhalta |
| License data | US DailyMed: Iptacopan |
| Routes of administration | By mouth |
| Drug class | Complement factor B inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1644670-37-0 |
| PubChem CID | 90467622 |
| DrugBank | DB16200 |
| ChemSpider | 75533872 |
| UNII | 8E05T07Z6W |
| KEGG | D12251D12252 |
| ChEMBL | ChEMBL4594448 |
| PDB ligand | JGQ (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C25H30N2O4 |
| Molar mass | 422.525 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f “Fabhalta- iptacopan capsule”. DailyMed. 5 December 2023. Archived from the original on 10 December 2023. Retrieved 10 December 2023.
- ^ “Novartis receives FDA approval for Fabhalta (iptacopan), offering superior hemoglobin improvement in the absence of transfusions as the first oral monotherapy for adults with PNH”. Novartis (Press release). Archived from the original on 12 December 2023. Retrieved 6 December 2023.
- ^ “Novel Drug Approvals for 2023”. U.S. Food and Drug Administration (FDA). 6 December 2023. Archived from the original on 21 January 2023. Retrieved 10 December 2023.
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2023/218276Orig1s000ltr.pdf Archived 10 December 2023 at the Wayback Machine
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jang JH, Wong L, Ko BS, Yoon SS, Li K, Baltcheva I, et al. (August 2022). “Iptacopan monotherapy in patients with paroxysmal nocturnal hemoglobinuria: a 2-cohort open-label proof-of-concept study”. Blood Advances. 6 (15): 4450–4460. doi:10.1182/bloodadvances.2022006960. PMC 9636331. PMID 35561315.
- ^ “Novartis Phase III APPOINT-PNH trial shows investigational oral monotherapy iptacopan improves hemoglobin to near-normal levels, leading to transfusion independence in all treatment-naïve PNH patients”. Novartis (Press release). Archived from the original on 12 December 2023. Retrieved 6 September 2023.
- ^ Schubart A, Anderson K, Mainolfi N, Sellner H, Ehara T, Adams CM, et al. (April 2019). “Small-molecule factor B inhibitor for the treatment of complement-mediated diseases”. Proceedings of the National Academy of Sciences of the United States of America. 116 (16): 7926–7931. Bibcode:2019PNAS..116.7926S. doi:10.1073/pnas.1820892116. PMC 6475383. PMID 30926668.
External links
- Clinical trial number NCT04558918 for “Study of Efficacy and Safety of Twice Daily Oral LNP023 in Adult PNH Patients With Residual Anemia Despite Anti-C5 Antibody Treatment (APPLY-PNH)” at ClinicalTrials.gov
- Clinical trial number NCT04820530 for “Study of Efficacy and Safety of Twice Daily Oral Iptacopan (LNP023) in Adult PNH Patients Who Are Naive to Complement Inhibitor Therapy (APPOINT-PNH)” at ClinicalTrials.gov
///////Iptacopan, fda 2023, approvals, 2023, paroxysmal nocturnal hemoglobinuria, 12/5/2023, Fabhalta , LNP 023, LNP-023, LNP023, NVP-LNP023, NVP-LNP023-NX

NEW DRUG APPROVALS
ONE TIME
$10.00
Rezivertinib
BPI-7711, Rezivertinib
1835667-12-3
C27H30N6O3, 486.576
N-[2-[2-(dimethylamino)ethoxy]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide
Beta Pharma in collaboration Chinese licensee CSPC Pharmaceuticals Group , is developing BPI-7711
In June 2021, this drug was reported to be in phase 3 clinical development.
APPROVALS 2024, CHINA 2024
- OriginatorBeta Pharma
- ClassAmides; Amines; Antineoplastics; Indoles; Phenyl ethers; Pyrimidines; Small molecules
- Mechanism of ActionEpidermal growth factor receptor antagonists
- Phase IIINon-small cell lung cancer
- 30 Dec 2020Chemical structure information added
- 09 Apr 2020Beta Pharma initiates a phase I trial for Non-small cell lung cancer (In volunteers) in China (PO) (NCT04135833)
- 25 Mar 2020Beta Pharma completes a phase I pharmacokinetic trial for Non-small cell lung cancer (In volunteers) in China (NCT04135820)
N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl)amino)phenyl)-2-propenamideThe epidermal growth factor receptor (EGFR, Herl, ErbB l) is a principal member of the ErbB family of four structurally-related cell surface receptors with the other members being Her2 (Neu, ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR exerts its primary cellular functions though its intrinsic catalytic tyrosine protein kinase activity. The receptor is activated by binding with growth factor ligands, such as epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-a), which transform the catalytically inactive EGFR monomer into catalytically active homo- and hetero- dimers. These catalytically active dimers then initiate intracellular tyrosine kinase activity, which leads to the autophosphorylation of specific EGFR tyrosine residues and elicits the downstream activation of signaling proteins. Subsequently, the signaling proteins initiate multiple signal transduction cascades (MAPK, Akt and JNK), which ultimately mediate the essential biological processes of cell growth, proliferation, motility and survival.EGFR is found at abnormally high levels on the surface of many types of cancer cells and increased levels of EGFR have been associated with advanced disease, cancer spread and poor clinical prognosis. Mutations in EGFR can lead to receptor overexpression, perpetual activation or sustained hyperactivity and result in uncontrolled cell growth, i.e. cancer. Consequently, EGFR mutations have been identified in several types of malignant tumors, including metastatic lung, head and neck, colorectal and pancreatic cancers. In lung cancer, mutations mainly occur in exons 18 to 21, which encode the adenosine triphosphate (ATP)-binding pocket of the kinase domain. The most clinically relevant drug- sensitive EGFR mutations are deletions in exon 19 that eliminate a common amino acid motif (LREA) and point mutations in exon 21, which lead to a substitution of arginine for leucine at position 858 (L858R). Together, these two mutations account for nearly 85% of the EGFR mutations observed in lung cancer. Both mutations have perpetual tyrosine kinase activity and as a result they are oncogenic. Biochemical studies have demonstrated that these mutated EGFRs bind preferentially to tyrosine kinase inhibitor drugs such as erlotinib and gefitinib over adenosine triphosphate (ATP).Erlotinib and gefitinib are oral EGFR tyrosine kinase inhibitors that are first line monotherapies for non-small cell lung cancer (NSCLC) patients having activating mutations in EGFR. Around 70% of these patients respond initially, but unfortunately they develop resistance with a median time to progression of 10-16 months. In at least 50% of these initially responsive patients, disease progression is associated with the development of a secondary mutation, T790M in exon 20 of EGFR (referred to as the gatekeeper mutation). The additional T790M mutation increases the affinity of the EGFR kinase domain for ATP, thereby reducing the inhibitory activity of ATP- competitive inhibitors like gefitinib and erlotinib.Recently, irreversible EGFR tyrosine kinase inhibitors have been developed that effectively inhibit the kinase domain of the T790M double mutant and therefore overcome the resistance observed with reversible inhibitors in the clinic. These inhibitors possess reactive electrophilic functional groups that react with the nucleophilic thiol of an active-site cysteine. Highly selective irreversible inhibitors can be achieved by exploiting the inherent non-covalent selectivity of a given scaffold along with the location of a particular cysteine residue within the ATP binding site. The acrylamide moieties of these inhibitors both undergo a Michael reaction with Cys797 in the ATP binding site of EGFRT790M to form a covalent bond. This covalent mechanism is thought to overcome the increase in ATP affinity of the T790M EGRF double mutant and give rise to effective inhibition. However, these inhibitors may cause various undesired toxicities. Therefore, development of new inhibitors for treatment of various EGFR-related cancers is still in high demand.
PatentCN201580067776) N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H- Indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (compound of formula I) can be prepared by the following synthetic route:
PATENT
https://patents.google.com/patent/WO2016094821A2/enExample 1N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)amino)phenyl)acrylamide (1) Sche

N-(4-(2-(Dimethylamino)ethoxy)-2-methoxy-5-nitrophenyl)-4-(l-methyl-lH- indol-3-yl)pyrimidin-2-amine (Scheme 1, Intermediate B). To a slurry of NaH (30 mmol, 60% oil dispersion prewashed with hexanes) and 50 mL of 1,4-dioxane was added 2-dimethylaminoethanol (27 mmol, 2.7 mL) dropwise with stirring under N2. After stirring for 1 h, a slurry of A (5.4 mmol) in 50 mL of 1,4-dioxane was added portion-wise over 15 min under a stream of N2. The resulting mixture was stirred overnight, then poured into water and the solid was collected, rinsed with water, and dried under vacuum to yield 2.6 g of product as a yellow solid. A purified sample was obtained from chromatography (silica gel; CH2C12-CH30H gradient). 1H NMR (300 MHz, DMSO) δ 2.26 (s, 6H), 2.70 (t, 2H, J = 6 Hz), 3.87 (s, 3H), 4.01 (s, 3H), 4.32 (t, 2H, J = 6 Hz), 7.00-7.53 (m, 5H), 8.18-8.78 (m, 5H); C24H26N604 m/z MH+ 463.4-(2-(Dimethylamino)ethoxy)-6-methoxy-Nl-(4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)benzene-l,3-diamine (Scheme 1, Intermediate C). A suspension of 2.6 g of Intermediate B, 1.6 g of Fe°, 30 mL of ethanol, 15 mL of water, and 20 mL of cone. HC1 was heated to 78 °C for 3 h. The solution was cooled to room temperature, adjusted to pH 10 with 10% NaOH (aq) and diluted with CH2C12. The mixture was filtered through Dicalite, and the filtrate layers were separated. The aqueous phase was extracted with CH2C12 twice, and the combined organic extracts were dried over Na2S04 and concentrated. Column chromatography (silica gel, CH2Cl2-MeOH gradient) afforded 1.2 g of Intermediate C as a solid. C24H28N602 m/z MH+ 433.N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(l-methyl-lH-indol-3- yl)pyrimidin-2-yl)amino)phenyl)acrylamide (1). To a solution of Intermediate C (2.8 mmol) in 50 mL of THF and 10 mL of water was added 3-chloropropionychloride (2.8 mmol) dropwise with stirring. After 5 h of stirring, NaOH (28 mmol) was added and the mixture was heated at 65°C for 18 h. After cooling to room temperature, THF was partially removed under reduced pressure, and the mixture was extracted with CH2C12, dried over Na2S04, and concentrated. Chromatography of the crude product (silica gel, CH2Cl2-MeOH) afforded 0.583 g of Example 1 as a beige solid. 1H NMR (300 MHz, DMSO) δ 2.28 (s, 6H), 2.50-2.60 (m, 2H), 3.86 (s, 3H), 3.90 (s, 3H), 4.19 (t, 2H, = 5.5 Hz), 5.73-5.77 (m, IH), 6.21-6.27 (m, IH), 6.44-6.50 (m, IH), 6.95 (s, IH), 7.11-7.53 (overlapping m, 3H), 7.90 (s, IH), 8.27-8.30 (overlapping m, 3H), 8.55 (s, IH), 8.84 (s, IH), 9.84 (s, IH) ppm; C27H30N6O3 m/z MH+ 487
PATENT WO2021115425
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021115425&tab=FULLTEXT&_cid=P20-KQN9F3-73566-1Epidermal growth factor receptors (EGFR, Her1, ErbB1) are the main members of the ErbB family of four structurally related cell surface receptors, and the other members are Her2 (Neu, ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR exerts its main cellular functions through its inherent catalytic tyrosine protein kinase activity. The receptor is activated by binding to growth factor ligands, such as epidermal growth factor (EGF) and transforming growth factor-α (TGF-α). The catalytically inactive EGFR monomer is transformed into a catalytically active homopolymer and Heterodimer. These catalytically active dimers then initiate intracellular tyrosine kinase activity, which leads to autophosphorylation of specific EGFR tyrosine residues and elicits downstream activation of signaling proteins. Subsequently, the signal protein initiates multiple signal transduction cascades (MAPK, Akt, and JNK), which ultimately regulate the basic biological processes of cell growth, proliferation, motility, and survival.
EGFR has been found to have abnormally high levels on the surface of many types of cancer cells, and elevated EGFR levels have been associated with advanced disease, cancer spread, and poor clinical prognosis. Mutations in EGFR can lead to overexpression of the receptor, permanent activation or continuous hyperactivity, leading to uncontrolled cell growth, which is cancer. Therefore, EGFR mutations have been identified in several types of malignant tumors, including metastatic lung cancer, head and neck cancer, colorectal cancer, and pancreatic cancer. In brain cancer, mutations mainly occur in exons 18-21, which encode the adenosine triphosphate (ATP)-binding pocket of the kinase domain. The most clinically relevant drug-sensitive EGFR mutations are deletions in exon 19 and point mutations in exon 21. The former eliminates a common amino acid motif (LREA), and the latter results in position 858 (L858R). The arginine is replaced by leucine. Together, these two mutations account for nearly 85% of the EGFR mutations observed in lung cancer. Both mutations have permanent tyrosine kinase activity, so they are carcinogenic. In at least 50% of patients who initially responded to current therapies, the progression of the disease is related to the development of a secondary mutation, T790M (also known as the goalkeeper mutation) in exon 20 of EGFR.
BPI-7711 is a third-generation EGFR-TKI compound developed by Beida Pharmaceuticals and disclosed in International Patent No. WO2017/218892. It is the N-(2-(2-(dimethylamino) )Ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide methanesulfonic acid salt:
Need to develop improved properties containing N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indole-3 -Yl)pyrimidin-2-yl)amino)phenyl)acrylamide pharmaceutically acceptable salt, in particular the pharmaceutical composition of BPI-7711 and its use, and the preparation of said pharmaceutical composition suitable for large-scale production method.
PATENT
WO2021061695 , for another filing, assigned to Beta Pharma, claiming a combination of an EGFR inhibitor (eg BPI-7711) and a CDK4/6 inhibitor, useful for treating cancer.
PATENT
WO-2021121146
Novel crystalline polymorphic form A of rezivertinib – presumed to be BPI-7711 – useful for treating diseases mediated by EGFR mutations eg lung cancer, preferably non-small cell lung cancer (NSCLC).Epidermal growth factor receptor (EGFR) is a type of transmembrane receptor tyrosine kinase in the human body. The activation (ie phosphorylation) of this kinase is of great significance to the inhibition of tumor cell proliferation, angiogenesis, tumor invasion, metastasis and apoptosis. EGFR kinase is involved in the disease process of most cancers, and these receptors are overexpressed in many major human tumors. Overexpression, mutations, or high expression of ligands associated with these family members can lead to some tumor diseases, such as non-small cell lung cancer, colorectal cancer, breast cancer, head and neck cancer, cervical cancer, bladder cancer, and thyroid. Cancer, stomach cancer, kidney cancer, etc.
In recent years, epidermal growth factor receptor tyrosine kinase has become one of the most attractive targets in current anti-tumor drug research. In 2003, the US FDA approved the first epidermal growth receptor tyrosine kinase inhibitor (EGFR-TKI) drug (gefitinib) for the treatment of advanced non-small cell lung cancer (NSCLC). Development of a generation of EGFR inhibitors. Numerous clinical trials have confirmed that for patients with EGFR-positive non-small cell lung cancer, the therapeutic effect of molecular targeted drugs is significantly better than traditional chemotherapy.
Although the first-generation EGFR-inhibiting targeted drugs responded well to the initial treatment of many non-small cell lung cancer (NSCLC) patients, most patients will eventually develop disease progression due to drug resistance (such as EGFR secondary T790M mutation). The emergence of drug resistance is caused by various mechanisms based on the mutations in the original EGFR pathway activity. In the drug resistance research on the first generation of EGFR inhibitors, the research frontier is the irreversible third generation EFGR inhibitor.
But so far, the third-generation EGFR inhibitors worldwide, in addition to AstraZeneca O’Higgins imatinib developed, there is no other effective against T790M resistance mutations in patients with drug approved for clinical use; Several drug candidates for the T790M mutation are in clinical development. The chemical structure of this third-generation EGFR inhibitor is completely different from that of the first-generation. The main difference from the first-generation EGFR inhibitors is that they both use a highly selective core structure to replace the low-selective aminoquinoline core structure of the first and second-generation EGFR-TKIs. Compared with wild-type EGFR, these third-generation compounds are highly specific and selective for the T790M mutation after EGFR positive resistance.
Chinese Patent Application No. CN201580067776.8 discloses a compound of the following formula I, which also belongs to the third-generation EGFR-TKI class of small molecule targeted drugs. The compound has a high inhibitory effect on non-small cell lung cancer (NSCLC) cells with single-activity mutation and T790M double-mutant EGFR, and its effective inhibitory concentration is significantly lower than the concentration required to inhibit the activity of wild-type EGFR tyrosine kinase. It has good properties, low side effects and good safety.
Chinese Patent Application No. CN201780050034.3 also discloses various salts and corresponding crystal forms of the compound of the above formula I. Example 2 discloses two crystal forms of the methanesulfonate of the compound of formula I, 2A and 2B, respectively.In the following examples, the “room temperature” can be 15-25°C.[0041](1) N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidine -2-yl)amino)phenyl)acrylamide (compound of formula I)[0042]
[0043]Known (for example, see CN201580067776.8) N-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H- Indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide (compound of formula I) can be prepared by the following synthetic route:[0044]
[0045]Step 1-Preparation of Intermediate J:[0046]
[0047]Preparation: In a 10L reaction flask, add 6L of anhydrous tetrahydrofuran solvent, protected by nitrogen, and cool to 0°C. While stirring, slowly add 101 g of sodium hydride (101 g, 2.52 mol), and the internal temperature does not exceed 10° C., and add 234 g of dimethylaminoethanol (234 g, 2.62 mol). After the addition, the temperature is adjusted to room temperature to prepare a sodium alkoxide solution.[0048]In a 30L reaction flask, add N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(1-methyl-1H-indol-3-yl)-2-pyrimidinamine ( Starting material B) (430g, 1.10mol), then add 9L of tetrahydrofuran, start stirring, dissolve it, control the temperature at 10±10°C, slowly add the prepared sodium alkoxide solution dropwise. Control the temperature at 10±10℃ and keep it for 5.0h. When the raw material content is ≤0.5%, the reaction ends. Control the temperature at 10±10°C, slowly add 3% hydrochloric acid solution dropwise, adjust the pH of the solution to 6-7, stir for 1.5h and then stand for stratification, separate the organic phase, and concentrate to 15-20L. After cooling to 20±5°C, 4.3 kg of water was slowly added dropwise, filtered, and dried to obtain 497 g of yellow powder intermediate J with a yield of 98.0% and an HPLC purity of 99.3%. MS m/z: 463.2 [M+1].[0049]Nuclear magnetic data: 1 HNMR (d 6 -DMSO): δ ppm: 8.78 (s, 1H); 8.42-8.28 (m, 3H); 8.16 (s, 1H); 7.53 (d, 1H, J = 8.28); 7.29- 7.20 (m, 2H); 7.13-7.07 (m, 1H); 7.01 (s, 1H); 4.33 (t, 2H, J = 5.65); 4.02 (s, 3H); 3.88 (s, 3H); 2.71 ( t, 2H, J = 5.77); 2.27 (s, 6H).[0050]Step 2-Preparation of Intermediate K:[0051]
[0052]Preparation: Add 5L of tetrahydrofuran and Intermediate J (350g, 108mmol) to a 10L hydrogenation reactor, add 17.5g of wet palladium charcoal, replace the hydrogenation reactor with hydrogen, adjust the pressure value to 0.2MPa, control the temperature at 25°C, and keep the temperature for reaction. At 9h, HPLC monitors the progress of the reaction, and stops the reaction when the substrate is ≤0.5%. Filter, concentrate the filtrate under reduced pressure until the solvent volume is about 2L, adjust the internal temperature to room temperature, slowly add 4L n-heptane dropwise within 4-7 hours, filter and dry the solid under reduced pressure to obtain 285g of white powder intermediate K The yield was 86%, and the HPLC purity was 99.60%. MS m/z: 433.3 [M+1].
Nuclear magnetic data: 1 HNMR (CDCl 3 ): δ ppm: 8.42 (d, 1H, J = 7.78), 8.28 (s, 1H), 8.26-8.23 (m, 1H), 7.78 (s, 1H), 7.51 (d, 1H,J=8.28),7.41(s,1H),7.26-7.23(m,1H),7.19- 7.11(m,2H),6.72(s,1H), 4.38(br,2H),4.06(t, 2H,J=5.77), 3.88(s,3H), 3.75(s,3H), 2.63(t,2H,J=5.77), 2.26(s,6H).
Step 3-Preparation of compound of formula I:
Add 250 mL of anhydrous tetrahydrofuran solvent and Intermediate K (14 g, 32 mmol) to the reaction flask and stir, cool to 0-5° C., add 10% hydrochloric acid (12 ml), and stir for 20 minutes. At 0-5°C, slowly drop 3-chloropropionyl chloride (5.6 g, 45 mmol) into the reaction flask. Stir for 3 hours, after sampling test (K/(U+K)≤0.5%) is qualified, add 36% potassium hydroxide aqueous solution (75ml, 480mmol), heat to 23-25°C, and stir for 12 hours. Raise the temperature to 50-60°C and stir for 4 hours. After the sampling test (U/(U+L)≤0.1%) is qualified, stand still for liquid separation. Separate the organic phase, wash with 10% brine three times, dry, filter, and concentrate the organic phase to 150 ml. The temperature was raised to 40° C., 150 ml of n-heptane was slowly added dropwise, and the temperature was lowered to room temperature to precipitate crystals. Filtered and dried to obtain 10.71 g of light brown solid (compound of formula I), yield 68%, HPLC purity: 99.8% (all single impurities do not exceed 0.15%). MS m/z: 487.3 [M+1].[0057]Nuclear magnetic data (Figure 1): 1 HNMR (d 6 -DMSO): δppm: 9.84 (s, 1H), 8.90 ~ 8.82 (m, 1H), 8.32-8.25 (m, 2H), 7.89 (s, 1H) ,7.51(d,1H,J=8.25), 7.27~7.10(m,1H), 6.94(s,1H), 6.49(dd,1H,J=16.88,10.13), 6.25(dd,1H,J=16.95 ,1.81),5.80~5.75(m,1H),4.19(t,2H,J=5.57),3.88(d,6H,J=14.63,6H),3.34(s,3H),2.58(d,2H, J=5.5), 2.28 (s, 6H).
(2) N-(2-(2-(Dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1H-indol-3-yl)pyrimidine -2-yl)amino)phenyl)acrylamide methanesulfonate (Form A) preparation
Example 1
The compound of formula I (3 g, 6.1 mmol) was dissolved in 24 ml of dimethyl sulfoxide DMSO solvent, the temperature was raised to 65° C., and the mixture was stirred and dissolved. Add an equivalent amount of methanesulfonic acid (0.59 g, 6.1 mmol) to the system. The temperature was lowered to 50°C, and 12ml of isopropyl acetate IPAc was slowly added. Stir at 50°C for 1 hour, then lower the temperature to 15°C. 21ml IPAc was added in 4 hours. The solution was stirred and crystallized at 15°C, filtered under reduced pressure, the filter cake was washed with isopropyl acetate, and washed with acetone to reduce the residual DMSO solvent. Blow drying at 50°C (or vacuum drying at 50°C) to obtain 3.16 g of a pale yellow solid (crystal form A). HPLC purity is 100%, yield is 88%, DMSO: <100ppm; IPAc: <100ppm. MS m/z: 487.2 [M+1-MsOH]. Melting point: 242-244°C.
Nuclear magnetic data (figure 2): 1 HNMR(d 6 -DMSO): δppm: 9.57(brs,1H), 9.40(s,1H), 8.71(s,1H), 8.48(s,1H), 8.32(d ,1H,J=7.9),8.29(d,1H,J=5.3),7.96(s,1H),7.51(d,1H,J=8.2),7.23(ddd,1H,J=7.9,7.1,0.8 ), 7.19 (d, 1H, J = 5.4), 7.15 (ddd, 1H, J = 7.8, 7.3, 0.5), 6.94 (s, 1H), 6.67 (dd, 1H, J = 16.9, 10.2), 6.27 ( dd, 1H, J = 16.9, 1.8), 5.57 (dd, 1H, J = 16.9, 1.7), 4.44 (t, 2H, J = 4.6), 3.89 (s, 3H), 3.88 (s, 3H), 3.58 (t, 2H, J=4.6), 2.93 (s, 6H), 2.39 (s, 3H).
After testing, the powder X-ray diffraction pattern of crystal form A obtained in this example has diffraction angle 2θ values of 11.06±0.2°, 12.57±0.2°, 13.74±0.2°, 14.65±0.2°, 15.48±0.2°, 16.58±0.2°, 17.83±0.2°, 19.20±0.2°, 19.79±0.2°, 20.88±0.2°, 22.05±0.2°, 23.06±0.2°, 24.23±0.2°, 25.10±0.2°, 25.71±0.2°, 26.15±0.2°, 27.37±0.2°, 27.42±0.2° has a characteristic peak; its XRPD spectrum is shown in Figure 3 and the attached table, DSC diagram is shown in Figure 4, TGA diagram is shown in Figure 5, and infrared spectrum IR diagram is shown in Figure 6. Show.
Example 2
[0066]The compound of formula I (28.25 g, 58.1 mmol) was dissolved in 224 ml of dimethyl sulfoxide DMSO solvent, the temperature was raised to 15-35° C., and the mixture was stirred to clear. 0.97 equivalents of methanesulfonic acid (5.4 g, 0.97 mmol) were added to the system in batches. Slowly add 448 ml of methyl isobutyl ketone (MIBK). Stir for 1 hour, then lower the temperature to 10-15°C. The solution was reacted with salt formation at 10-15°C, sampled, and HPLC detected the residue of the compound of formula I in the mother liquor (≤0.4%). After the reaction was completed, vacuum filtration was performed to obtain 32 g of the crude methanesulfonate of the compound of formula I.Add 3g of the crude methanesulfonate of the compound of formula I into 24ml of dimethyl sulfoxide DMSO solvent, stir to clear at 65°C, cool down, slowly add 48ml of methyl isobutyl ketone (MIBK) dropwise, stir and crystallize 6-8 After hours, vacuum filtration, drying at 60° C. (or 60° C. vacuum drying) to obtain the target crystal form A. Melting point: 242-244°C. The XRPD pattern of the crystal form is consistent with Figure 3 (Figure 7), and all characteristic peaks are within the error range.
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Rezivertinib, also known as BPI-7711, is a third-generation epidermal growth factor receptor (EGFR) TKI, developed by Beta Pharm. Rezivertinib selectively targets both EGFR-sensitizing mutations
and the T790 M resistance mutation, thereby addressing resistance mechanisms associated with first- and second-generation EGFR-tyrosine kinase inhibitors. In 2024, the NMPA approved Rezivertinib mesylate capsules (trade name: Ruibida) for the treatment of adult patients with locally advanced or metastatic NSCLC who have progressed during or after EGFR-TKI therapy and have confirmed EGFR T790 M mutation-positive status. Rezivertinib exerts its antitumor activity by forming covalent bonds with mutant EGFR, particularly the T790 M mutation, which effectively blocks the downstream signaling pathways responsible for promoting tumor cell proliferation and survival [21]. The mechanism of Rezivertinib effectively inhibits tumor growth in patients harboring T790M-mediated resistance to first- and second-generation EGFR-TKIs. In a Phase IIb clinical trial (NCT03812809), Rezivertinib demonstrated significant clinical efficacy among patients with EGFR T790 M mutation-positive NSCLC who had experienced disease progression following prior EGFR-TKI therapy. The trial reported an ORR of
64.6 % and a median PFS of 12.2 months, highlighting its potent antitumor activity in this specific patient cohort. In terms of safety, Rezivertinib exhibited a favorable tolerability profile [22]. The most
frequently observed treatment-related adverse events were rash, diarrhea, and elevated liver enzymes, predominantly of mild to moderate severity (grade 1 or 2). No dose-limiting toxicities were noted, and its safety profile aligned with those of other third-generation EGFR-TKIs.
The synthesis of Rezivertinib, illustrated in Scheme 5, initiates with nucleophilic substitution reaction between Rezi-001 and Rezi-002,affording Rezi-003 [23]. Fe-mediated reduction of Rezi-003 yields
Rezi-004, followed by amidation with Rezi-005 to deliver Rezivertinib [20] J.J. Cui, E.W. Rogers, Preparation of Fluorodimethyltetrahydroethenopyrazolobenzoxatriazacyclotridecinone
Derivatives for Use as Antitumor Agents, 2017. US20180194777A1.
[21] Y. Shi, Y. Zhao, S. Yang, J. Zhou, L. Zhang, G. Chen, J. Fang, B. Zhu, X. Li, Y. Shu,
J. Shi, R. Zheng, D. Wang, H. Yu, J. Huang, Z. Zhuang, G. Wu, L. Zhang, Z. Guo,
M. Greco, X. Li, Y. Zhang, Safety, efficacy, and pharmacokinetics of rezivertinib
(BPI-7711) in patients with advanced NSCLC with EGFR T790M mutation: a phase
1 dose-escalation and dose-expansion study, J. Thorac. Oncol. 17 (2022) 708–717.

//////////// BPI-7711, BPI 7711, rezivertinib, phase 3, CHINA 2024, APPROVALS 2024
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
CN1C=C(C2=CC=CC=C21)C3=NC(=NC=C3)NC4=CC(=C(C=C4OC)OCCN(C)C)NC(=O)C=C
NEW DRUG APPROVALS
one time
$10.00
Entinostat


Entinostat
- BAY 86-5274
- BAY86-5274
CAS 209783-80-2
209784-80-5 (HCl)
Bayer Schering Pharma Aktiengesellschaft
Pyridin-3-ylmethyl N-[[4-[(2-aminophenyl)carbamoyl]phenyl]methyl]carbamate
Entinostat, developed by Syndax Pharmaceuticals, is an oral selective histone deacetylase (HDAC) inhibitor primarily targeting class IHDACs (HDAC1, HDAC2, and HDAC3) . It was later licensed to
Jiangsu Hengrui Medicine Co., Ltd., for development and commercialization in China. In 2024, Entinostat has been approved by the NMPA for use in combination with exemestane to treat advanced breast cancer that is HR-positive and HER2-negative.
KHK and Syndax partner for breast cancer treatment entinostat in Japan and Korea
Japan-based Kyowa Hakko Kirin (KHK) has signed a license agreement with US-based Syndax Pharmaceuticals for the exclusive rights to develop and commercialise entinostat in Japan and Korea.
TOKYO and WALTHAM, Mass., Jan. 7, 2015 /PRNewswire/ — Kyowa Hakko Kirin Co., Ltd., (Headquarters: Chiyoda-ku, Tokyo; president and CEO: Nobuo Hanai, “Kyowa Hakko Kirin”) and Syndax Pharmaceuticals, Inc., (Waltham, Mass.; president and CEO:Arlene M. Morris, “Syndax”) today jointly announced that the companies have entered into a license agreement for the exclusive rights to develop and commercialize entinostat in Japan and Korea. Entinostat is a Class I selective histone deacetylase (HDAC) inhibitor being developed by Syndax in the United States and Europe in combination with hormone therapy for advanced breast cancer and immune therapy combinations in solid tumors.

Entinostat, also known as SNDX-275 and MS-275, is a benzamide histone deacetylase inhibitor undergoing clinical trials for treatment of various cancers.[1]
Entinostat inhibits class I HDAC1 and HDAC3 with IC50 of 0.51 μM and 1.7 μM, respectively.[2]
Entinostat (formerly known as MS-275) is a histone deacetylase (HDAC) inhibitor in phase III clincal trials at Syndax in combination with exemestane for the treatment of advanced HR-positive breast cancer.
Entinostat (MS-275) preferentially inhibits HDAC1 (IC50=300nM) over HDAC3 (IC50=8µM) and has no inhibitory activity towards HDAC8 (IC50>100µM). MS-275 induces cyclin-dependent kinase inhibitor 1A (p21/CIP1/WAF1), slowing cell growth, differentiation, and tumor development in vivo. Recent studies suggest that MS-275 may be particularly useful as an antineoplastic agent when combined with other drugs, like adriamycin.
In September 2013, Syndax Pharmaceuticals entered into a licensing, development and commercialization agreement with Eddingpharm in China and other asian countries. In 2013, a Breakthrough Therapy Designation was assigned to the compound for the treatment of locally recurrent or metastatic estrogen receptor-positive (ER+) breast cancer when added to exemestane in postmenopausal women whose disease has progressed following non-steroidal aromatase inhibitor therapy.
Clinical trials
There is an ongoing phase II trial studying the effect of entinostat on Hodgkin’s lymphoma.[3] It is in other phase II trials for advanced breast cancer (in combination with aromatase inhibitors)[4] and for metastatic lung cancer (in combination with erlotinib).[5] As of September 2013, the Food and Drug Administration is working with the industry to design phase III clinical trials. They seek to evaluate the application of Entinostat for the reduction, or prevention of, treatment resistance to aromatase inhibitors in hormone receptor positive breast cancer.[6] Syndax pharmaceuticals currently holds the rights to Entinostat and recently received $26.6 million in funds to advance treatments of resistant cancers using epigenetic tools.[7]
PHASE 3………..SYNDAX, BREAST CANCER
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Entinostat, developed by Syndax Pharmaceuticals, is an oral selec
tive histone deacetylase (HDAC) inhibitor primarily targeting class I
HDACs (HDAC1, HDAC2, and HDAC3) [7]. It was later licensed to
Jiangsu Hengrui Medicine Co., Ltd., for development and commercial
ization in China. In 2024, Entinostat has been approved by the NMPA for
use in combination with exemestane to treat advanced breast cancer that
is HR-positive and HER2-negative. This approval is specifically for pa
tients whose disease has progressed following prior endocrine therapy
[8]. Entinostat inhibits HDACs, increasing histone acetylation and
reactivating tumor suppressor genes. This mechanism restores sensi
tivity to endocrine therapy and prevents cancer cell proliferation [9].
The therapeutic agent exerts its effects by modulating the tumor
microenvironment through the suppression of immune regulatory cells,
thereby augmenting the immune response. Its clinical efficacy was
confirmed in the E2112 trial (NCT02115282), a global Phase III study.
When used in combination with exemestane, Entinostat demonstrated
the ability to extend PFS in patients with HR-positive, HER2-negative
breast cancer [10]. The median PFS was significantly extended to 6.32
months, contrasting with the 3.72 months observed in the control
cohort. In terms of safety profile, Entinostat demonstrated favorable
tolerability. The frequently encountered adverse events were primarily
neutropenia, fatigue, and nausea. Severe neutropenia occurred in 43 %
of patients but was manageable with supportive care. Liver function
abnormalities were reported but manageable with dose adjustments
[11].
The synthetic route of Entinostat is shown in Scheme 2 [12].
Enti-001 is first treated with trifluoroacetic anhydride to afford
Enti-002. Reaction of Enti-002 with oxalyl chloride yields the acyl
chloride intermediate, which undergoes condensation with Enti-003 to
form Enti-004. Subsequent alkaline hydrolysis of Enti-004 produces
Enti-005. This compound is activated with CDI followed by reaction
with Enti-006 to generate Enti-007. The synthesis concludes with acidic removal of the Boc protecting group from Enti-007, yielding Entinostat
[8] W. Li, Z. Sun, Mechanism of action for HDAC inhibitors-insights from omics
approaches, Int. J. Mol. Sci. 20 (2019) 1616.
[9] N. Bharathy, N.E. Berlow, E. Wang, J. Abraham, T.P. Settelmeyer, J.E. Hooper, M.
N. Svalina, Z. Bajwa, M.W. Goros, B.S. Hernandez, J.E. Wolff, R. Pal, A.M. Davies,
A. Ashok, D. Bushby, M. Mancini, C. Noakes, N.C. Goodwin, P. Ordentlich, J. Keck,
D.S. Hawkins, E.R. Rudzinski, A. Mansoor, T.J. Perkins, C.R. Vakoc, J.E. Michalek,
C. Keller, Preclinical rationale for entinostat in embryonal rhabdomyosarcoma,
Skelet Muscle 9 (2019) 12.
[10] B. Xu, Q. Zhang, X. Hu, Q. Li, T. Sun, W. Li, Q. Ouyang, J. Wang, Z. Tong, M. Yan,
H. Li, X. Zeng, C. Shan, X. Wang, X. Yan, J. Zhang, Y. Zhang, J. Wang, L. Zhang,
Y. Lin, J. Feng, Q. Chen, J. Huang, L. Zhang, L. Yang, Y. Tian, H. Shang, Entinostat,
a class I selective histone deacetylase inhibitor, plus exemestane for Chinese
patients with hormone receptor-positive advanced breast cancer: a multicenter,
randomized, double-blind, placebo-controlled, phase 3 trial, Acta Pharm. Sin. B 13
(2023) 2250–2258.
[11] E.T. Roussos Torres, W.J. Ho, L. Danilova, J.A. Tandurella, J. Leatherman, C. Rafie,
C. Wang, A. Brufsky, P. LoRusso, V. Chung, Y. Yuan, M. Downs, A. O’Connor, S.
M. Shin, A. Hernandez, E.L. Engle, R. Piekarz, H. Streicher, Z. Talebi, M.A. Rudek,
Q. Zhu, R.A. Anders, A. Cimino-Mathews, E.J. Fertig, E.M. Jaffee, V. Stearns, R.
M. Connolly, Entinostat, nivolumab and ipilimumab for women with advanced
HER2-negative breast cancer: a phase Ib trial, Nat Cancer 5 (2024) 866–879.
[12] T. Suzuki, T. Ando, K. Tsuchiya, T. Nakanishi, A. Saito, S. Yamashita, G. Shiraishi,
E. Tanaka, Preparation of Benzamide Derivatives as Anticancer Agents, 1998
JP10152462
SEE SCHEME AT END

Patent
http://www.google.im/patents/WO2010022988A1?cl=en
In EP 0 847 992 A1 (which co-patent is US 6,794,392) benzamide derivatives as medicament for the treatment of malignant tumors, autoimmune diseases, de- rmatological diseases and parasitism are described. In particular, these derivatives are highly effective as anticancer drugs, preferred for the haematological malignancy and solid tumors. The preparation of N-(2-aminophenyl)-4-[N- (pyridine-3-yl)methoxycarbonylaminomethyl]-benzamide is described on page 57, Example 48. The compound is neither purified by chromatography nor purified by treatment with charcoal. The final step of the process comprises the re- crystallization from ethanol.
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm“1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
In EP 0 974 576 B1 a method for the production of monoacylated phenylenediamine derivatives is described. The preparation of N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl] benzamide is described on pages 12 to 13, Example 6. The final step of the process comprises the purification of the compound via silica gel column chromatography.
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm‘1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form. In J. Med. Chem. 1999, 42, 3001-3003, the synthesis of new benzamide derivatives and the inhibition of histone deacetylase (HDAC) is described. The process for the production of N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth- oxycarbonylaminomethyl] benzamide is described. The final step of the process comprises the purification of the compound via silica gel column chromatography (ethyl acetate).
Said compound has a melting point (mp) of 159 – 160 0C.
The IR spectrum shows the following bands: IR(KBr) cm‘1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
In WO 01/12193 A1 a pharmaceutical formulation comprising N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl]benzamide is described.
In WO 01/16106 a formulation comprising N-(2-aminophenyl)-4-[N-(pyridine-3- yl)methoxycarbonylamino-methyl]benzamide, having an increased solubility and an improved oral absorption for benzamide derivatives, and pharmaceutically acceptable salts thereof are described.
In WO 2004/103369 a pharmaceutical composition is described which comprises histone deacetylase inhibitors. That application concerns the combined use of N-(2-aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino- methyl]benzamide together with different cancer active compounds. In fact that application is a later application, which is based on the above mentioned matter and thus concerns the Polymorph A form. Finally, JP 2001-131130 (11-317580) describes a process for the purification of monoacylphenylenediamine derivatives. In Reference Example 2, the process for the production of crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide is described. Said compound has a melting point (mp) of 159 – 160 0C,
The IR spectrum shows the following bands: IR(KBr) cm“1: 3295, 1648, 1541 , 1508, 1457, 1309, 1183, 742.
The data indicate the Polymorph A form.
Moreover, Working Example 1 describes the purification of crude N-(2- aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide in aqueous acid medium together with carbon The final crystallization is done under aqueous conditions at 40-500C.
Following the description to that example it can be seen from the Comparative Examples 1 – 3 that the crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth- oxycarbonylaminomethyl] benzamide is not purified by dissolution under reflux conditions in either ethanol, methanol or acetonithle followed by a recrystalliza- tion at 2°C. As a result, these recrystallisations do not yield any pure compound.
In addition a “purification” of crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide in ethanol under reflux conditions to- gether with carbon is dechbed. After filtering off the carbon the compound is re- crystallized at 2°C. The purification effect of this method is very limited. 1 ,1 % of an impurity remain in the N-(2-aminophenyl)-4-[N-(pyridine-3-yl) methoxycarbonylaminomethyl] benzamide. As a result, this procedure does not yield any pure compound.
None of the state of the art documents refer to a polymorph B of N-(2- aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylamino-methyl]benzamide and no physicochemical features of said compound are known. Several biological and clinical studies have been done with N-(2-aminophenyl)- 4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide. For example, Kummar et al., Clin Cancer Res. 13 (18), 2007, pp 5411-5417 describe a phase I trial of N-(2-aminophenyl)-4-[N-(pyridine-3-yl) meth-oxycarbonylaminomethyl] benzamide in refractory solid tumors. The compound was applied orally.
The crude N-(2-aminophenyl)-4-[N-(pyridine-3-yl)methoxycarbonylaminomethyl]- benzamide of step a) can be produced according to the method described in example 6 of EP 0974 576 B1.
PATENT
http://www.google.co.in/patents/EP0974576A2?cl=en
Example 6Synthesis of N-(2-aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide (an example in which after activation with N,N’-carbonyldiimidazole, an acid was added to carry out reaction)
-
[0082]7.78 g (48 mmole) of N,N’-carbonyldiimidazole were added to a 1,3-dimethyl-2-imidazolidinone (50 g) suspension including 11.45 g (40 mmole) of 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic acid. After stirring at room temperature for 2 hours, 17.30 g (0.16 mole) of 1,2-phenylenediamine were added to the solution. After cooling to 2°C, 9.60 g (0.1 mole) of methanesulfonic acid were added dropwise. After stirring for 2 hours, water was added, and the deposited solid was collected by filtration. Purification was then carried out through silica gel column chromatography to obtain 10.83 g (yield: 72%) of N-(2-aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide.
Reaction selectivity based on the result in HPLCRetention Time/min. Area % Benzoylimidazole as Active Intermediate 4.3 0.00 Monoacylated Phenylenediamine 4.7 98.91 Diacylated Phenylenediamine 11.7 1.09 Analysis data of the product
mp. 159-160°C
1H NMR (270MHz, DMSO-d6) δ ppm: 4.28 (2H, d, J=5.9Hz), 4.86 (2H, s), 5.10 (2H, s), 6.60 (1H, t, J=7.3Hz), 6.78 (1H, d, J=7Hz), 6.97 (1H, t, J=7Hz), 7.17 (1H, d, J=8Hz), 7.3-7.5 (3H, m), 7.78 (1H, d, J=8Hz), 7.93 (2H, d, J=8Hz), 8.53 (1H, d, J=3.7Hz), 8.59 (1H, s), 9.61 (1H, s).
IR (KBr) cm-1: 3295, 1648, 1541, 1508, 1457, 1309, 1183, 742
PATENT
WO 2009076206
http://www.google.com/patents/WO2009076206A1?cl=en
Suzuki et al (Suzuki et al Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives, J Med Chem 1999, 42, (15), 3001-3) discloses benzamide derivatives having histone deacetylase inhibitory activity and methods of making benzamide derivatives having histone deacetylase inhibitory activity. Suzuki et al is hereby incorporated herein by reference in its entirety.
[18] An example of the synthesis method of Suzuki et al to produce MS-275 via a three- step procedure in 50.96% overall yield is outlined in Scheme 3 below.
Scheme 3: Previous Procedure for Synthesis of MS-275 en rt, 4h
(used without purification)

[Overall yield: 0.91 x 0.56 x 100 = 50.96%;

MS-275 [19] In addition to the modest overall yield, the procedure of Suzuki et al has other disadvantages, such as a tedious method for the preparation of an acid chloride using oxalyl chloride and requiring the use of column chromatography for purification.
The synthesis of MS-275 is shown below in Scheme 4 as an example of Applicants invention of a two-step procedure: [37] Scheme 4: Preparation of MS-275
Scheme 4: New Synthesis of MS-275 (4)

Condensation of 3-(hydroxymethyl)pyridine (7) and 4-(aminomethyl)benzoic in the presence of CDI gave 4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic Acid (8) in 91.0% yield. In the previous method of Suzuki et ah, the carboxylic acid derivative 8 was first converted into acyl chloride hydrochloride by treatment of oxalyl chloride in toluene and then reacted with imidazole to form the acylimidazole intermediate. (Suzuki et al., Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives. J Med Chem 1999, 42, (15), 3001-3.). However, Applicants synthesized the imidazolide of intermediate 8 by treatment with CDI at about 55-60 0C in THF. The imidazolide was cooled to ambient and further reacted in situ with 1,2-phenylenediamine in the presence of TFA to afford MS-275
(4).
Experimental Section
[62] iV-(2-Aminophenyl)-4-[iV-(pyridin-3-ylmethoxycarbonyl) aminomethyl] benzamide (4, MS-275).
[63] To a suspension of 4-[N-(Pyridin-3-ylmethoxycarbonyl)aminomethyl]benzoic
Acid (5.0 g, 0.017 mol) in THF (100 mL) was added CDI (3.12 g, 0.019 mol), and the mixture stirred for 3 h at 60 0C. After formation of acylimidazole the clear solution was cooled to room temperature (rt). To this was added 1,2-phenylenediamine (15.11 g, 0.14 mmol) and trifluoroacetic acid (1.2 mL, 0.015 mol) and then stirred for 16 h. The reaction mixture was evaporated to remove THF and crude product was stirred in a mixture of hexane and water (2:5, v/v) for 1 h and filtered and dried. The residue was stirred in dichloromethane twice to afford pure MS-275 (4) as off white powder 5.25 g, 80% yield:
mp 159-160 * C; IR (KBr) 3295, 1648, 1541, 1508, 1457, 1309, 1183, 742 cm“1.
1H NMR (DMSO-J6) δ 4.28 (d, 2H, J = 5.9 Hz), 4.86 (s, 2H), 5.10 (s, 2H), 6.60 (t, IH, J = 7.3 Hz), 6.78 (d, IH, J = 7 Hz), 6.97 (t, IH, J= 7 Hz), 7.17 (d, IH, J= 8 Hz), 7.3-7.5(m, 3H), 7.78 (d, IH, J= 8 Hz), 7.93 (d, 2H, J = 8 Hz), 8.53 (d, IH, J = 3.7 Hz), 8.59 (s, IH), 9.61 (s, IH);
HRMS: calcd 376.1560 (C2iH2oN4θ3), found 376.1558. These spectral and analytical data are as previously reported in J Med Chem 1999, 42, (15), 3001-3.
[64] 4-[7V-(Pyridin-3-ylmethoxycarbonyI)aminomethyl] benzoic Acid (8) may be prepared as follows. To a suspension of l, l’-carbonyldiimidazole (CDI, 25.6 g, 158 mmol) in THF (120 mL) was added 3-pyridinemethanol (7, 17.3 g, 158 mmol) in THF (50 mL) at 10 0C, and the mixture stirred for 1 h at rt. The resulting solution was added to a suspension of 4-(aminomethyl)benzoic acid (22.6 g, 158 mmol), DBU (24.3 g, 158 mmol), and triethylamine (22.2 mL, 158 mmol) in THF (250 mL). After stirring for 5 h at rt, the mixture was evaporated to remove THF and then dissolved in water (300 mL). The solution was acidified with HCl (pH 5) to precipitate a white solid which was collected by filtration, washed with water (300 mL) and methanol (50 mL), respectively, and dried to yield pure 8 (41.1 g, 91% yield):
mp 207-208 0 C;
IR (KBr) 3043, 1718, 1568, 1434, 1266, 1 108, 1037, 984, 756 cm4; 1H NMR (DMSO-^6) δ 4.28 (d, 2H, J= 5.9 Hz), 5.10 (s, 2H), 7.3-7.5 (m, 3H), 7.7-8.1 (m, 4H), 8.5-8.7 (m, 2H). These spectral and analytical data are as previously reported in Suzuki et al, J Med Chem 1999, 42, (15), 3001-3.
PAPER
Volume 18, Issue 11, 1 June 2010, Pages 3925–3933
http://www.sciencedirect.com/science/article/pii/S0968089610003378
PAPER
see
Bioorg Med Chem 2008, 16(6): 3352
http://www.sciencedirect.com/science/article/pii/S0968089607010577
PAPER
see
Bioorganic and Medicinal Chemistry Letters, 2004 , vol. 14, 1 pg. 283 – 287
http://www.sciencedirect.com/science/article/pii/S0960894X03010539
PAPER
J Med Chem 1999, 42(15): 3001
http://pubs.acs.org/doi/abs/10.1021/jm980565u
N-(2-Aminophenyl)-4-[N-(pyridin-3-ylmethoxycarbonyl)aminomethyl]benzamide (1, MS-275). To a solution of imidazole (0.63 g, 9.2 mmol) in THF (20 mL) was added 3 (1 g, 2.9 mmol), and the mixture stirred for 1 h at room temperature. After imidazole hydrochloride was removed by filtration, 1,2-phenylenediamine (2.52 g, 23.2 mmol) and trifluoroacetic acid (0.2 mL, 2.6 mmol) were added to the filtrate and stirred for 15 h. The reaction mixture was evaporated to remove THF and partitioned between ethyl acetate (500 mL) and water (400 mL). The organic layer was washed with water and dried and then purified by silica gel column chromatography (ethyl acetate) to give 1 (0.62 g, 56% yield):
mp 159−160 °C;
1H NMR (DMSO-d6) δ 4.28 (d, 2H, J = 5.9 Hz), 4.86 (s, 2H), 5.10 (s, 2H), 6.60 (t, 1H, J = 7.3 Hz), 6.78 (d, 1H, J = 7 Hz), 6.97 (t, 1H, J = 7 Hz), 7.17 (d, 1H, J = 8 Hz), 7.3−7.5(m, 3H), 7.78 (d, 1H, J = 8 Hz), 7.93 (d, 2H, J = 8 Hz), 8.53 (d, 1H, J = 3.7 Hz), 8.59 (s, 1H), 9.61 (s, 1H);
IR (KBr) 3295, 1648, 1541, 1508, 1457, 1309, 1183, 742 cm-1.
Anal. (C21H20N4O3) C, H, N.
………………………………………………………………………..
see
Bulletin of the Korean Chemical Society, 2014 , vol. 35, 1 pg. 129 – 134
http://koreascience.or.kr/article/ArticleFullRecord.jsp?cn=JCGMCS_2014_v35n1_129
PAPER
see
ChemMedChem, 2013 , vol. 8, 5 pg. 800 – 811
PAPER
see
ACS Medicinal Chemistry Letters, 2013 , vol. 4, 10 pg. 994 – 999
http://pubs.acs.org/doi/full/10.1021/ml400289e
References
- Phase I trial of 5-azacitidine (5AC) and SNDX-275 in advanced lung cancer (NSCLC)
- Novel Sulphonylpyrroles as Inhibitors of Hdac S Novel Sulphonylpyrroles
- A Phase 2 Multi-Center Study of Entinostat (SNDX-275) in Patient With Relapsed or Refractory Hodgkin’s Lymphoma
- A Phase 2, Multicenter Study of the Effect of the Addition of SNDX-275 to Continued Aromatase Inhibitor (AI) Therapy in Postmenopausal Women With ER+ Breast Cancer Whose Disease is Progressing
- A Phase 2 Exploratory Study of Erlotinib and SNDX-275 in Patients With Non-small Cell Lung Carcinoma Who Are Progressing on Erlotinib
- Breakthrough Designation Granted to Entinostat for Advanced Breast Cancer Silas Inman Published Online: Wednesday, September 11, 2013 http://www.onclive.com/web-exclusives/Breakthrough-Designation-Granted-to-Entinostat-for-Advanced-Breast-Cancer
- http://www.syndax.com/assets/130827%20Syndax%20Series%20B%20news%20release.pdf
- References:
1. Saito, A. et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA 96 4592-4597 (1999).
2. Jaboin, J., et al. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res 62 6108-6115 (2002).
3. Rosato RR, et al. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res 2003; 63: 3637–3645.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0847992B1 * | Sep 30, 1997 | Jun 23, 2004 | Schering Aktiengesellschaft | Benzamide derivatives, useful as cell differentiation inducers |
| US7244751 * | Feb 2, 2004 | Jul 17, 2007 | Shenzhen Chipscreen Biosciences Ltd. | N-(2-amino-5-fluorophenyl)-4-[N-(Pyridn-3-ylacryloyl)aminomethyl]benzamide or other derivatives for treating cancer and psoriasis |
| Reference | ||
|---|---|---|
| 1 | * | MAI A: ‘Histone deacetylation in epigenetics: an attractive target for anticancer therapy‘ MED RES REV. vol. 25, no. 3, May 2005, pages 261 – 309 |
| 2 | * | SUZUKI T ET AL.: ‘Synthesis and histone deacetylase inhibitory activity of new benzamide derivatives‘ J MED CHEM. vol. 42, no. 15, 29 July 1999, pages 3001 – 3003 |

| Names | |
|---|---|
| Preferred IUPAC name(Pyridin-3-yl)methyl ({4-[(2-aminophenyl)carbamoyl]phenyl}methyl)carbamate | |
| Other namesSNDX-275; MS-275 | |
| Identifiers | |
| CAS Number | 209783-80-2 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:132082 |
| ChEMBL | ChEMBL27759 |
| ChemSpider | 4111 |
| ECHA InfoCard | 100.158.999 |
| IUPHAR/BPS | 7007 |
| KEGG | D09338 |
| PubChem CID | 4261 |
| UNII | 1ZNY4FKK9H |
| CompTox Dashboard (EPA) | DTXSID0041068 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C21H20N4O3 |
| Molar mass | 376.4085 g/mol |
| Pharmacology | |
| ATC code | L01XH05 (WHO) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
/////////////////approvals 2024, china 2024, Entinostat, SNDX 275, MS 275, Syndax, BAY 86-5274, BAY86-5274
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}