
Home » Posts tagged 'Celgene'
Tag Archives: Celgene
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
Today, the U.S. Food and Drug Administration approved Inrebic (fedratinib) capsules to treat adult patients with certain types of myelofibrosis.
“Prior to today, there was one FDA-approved drug to treat patients with myelofibrosis, a rare bone marrow disorder. Our approval today provides another option for patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The FDA is committed to encouraging the development of treatments for patients with rare diseases and providing alternative options, as not all patients respond in the same way.”
Myelofibrosis is a chronic disorder where scar tissue forms in the bone marrow and the production of the blood cells moves from the bone marrow to the spleen and liver, causing organ enlargement. It can cause extreme fatigue, shortness of breath, pain below the ribs, fever, night sweats, itching and bone pain. When myelofibrosis occurs on its own, it is called primary myelofibrosis. Secondary myelofibrosis occurs when there is excessive red blood cell production (polycythemia vera) or excessive platelet production (essential thrombocythemia) that evolves into myelofibrosis.
Jakafi (ruxolitinib) was approved by the FDA in 2011. The approval of Inrebic for intermediate-2 or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis was based on the results of a clinical trial where 289 patients with myelofibrosis were randomized to receive two different doses (400 mg or 500 mg daily by mouth) of fedratinib or placebo. The clinical trial showed that 35 of 96 patients treated with the fedratinib 400 mg daily dose (the dose recommended in the approved label) experienced a significant therapeutic effect (measured by greater than or equal to a 35% reduction from baseline in spleen volume at the end of cycle 6 (week 24) as measured by an MRI or CT scan with a follow-up scan four weeks later). As a result of treatment with Inrebic, 36 patients experienced greater than or equal to a 50% reduction in myelofibrosis-related symptoms, such as night sweats, itching, abdominal discomfort, feeling full sooner than normal, pain under ribs on left side, and bone or muscle pain.
The prescribing information for Inrebic includes a Boxed Warning to advise health care professionals and patients about the risk of serious and fatal encephalopathy (brain damage or malfunction), including Wernicke’s, which is a neurologic emergency related to a deficiency in thiamine. Health care professionals are advised to assess thiamine levels in all patients prior to starting Inrebic, during treatment and as clinically indicated. If encephalopathy is suspected, Inrebic should be immediately discontinued.
Common side effects for patients taking Inrebic are diarrhea, nausea, vomiting, fatigue and muscle spasms. Health care professionals are cautioned that patients may experience severe anemia (low iron levels) and thrombocytopenia (low level of platelets in the blood). Patients should be monitored for gastrointestinal toxicity and for hepatic toxicity (liver damage). The dose should be reduced or stopped if a patient develops severe diarrhea, nausea or vomiting. Treatment with anti-diarrhea medications may be recommended. Patients may develop high levels of amylase and lipase in their blood and should be managed by dose reduction or stopping the mediation. Inrebic must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
The FDA granted this application Priority Review designation. Inrebic also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Inrebic to Impact Biomedicines, Inc., a wholly-owned subsidiary of Celgene Corporation.
LINK
///////Inrebic , fedratinib, FDA 2019, Priority Review , Orphan Drug, Biomedicines, Celgene , bone marrow disorder
Tanzisertib
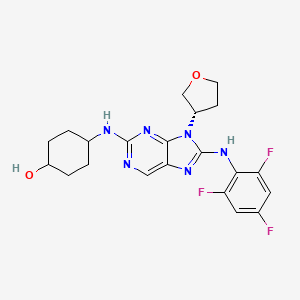
Tanzisertib
CAS 899805-25-5
trans-4-((9-((3S)-Tetrahydrofuran-3-yl)-8-((2,4,6-trifluorophenyl)amino)-9H-purin-2-yl)amino)cyclohexanol
4-[[9-[(3S)-oxolan-3-yl]-8-(2,4,6-trifluoroanilino)purin-2-yl]amino]cyclohexan-1-ol
C21-H23-F3-N6-O2, 448.4467
9557
Cyclohexanol, 4-[[9-[(3S)-tetrahydro-3-furanyl]-8-[(2,4,6-trifluorophenyl)amino]-9H-purin-2-yl]amino]-, trans-
- CC 930
- CC-930
- Tanzisertib
- UNII-M5O06306UO
- A c-Jun amino-terminal kinase inhibitor.UNII, M5O06306UO
Treatment of Idiopathic Pulmonary Fibrosis (IPF)
- Originator Celgene Corporation
- Class Antifibrotics; Small molecules
- Mechanism of ActionJ NK mitogen-activated protein kinase inhibitors
- Orphan Drug Status Yes – Idiopathic pulmonary fibrosis
- Discontinued Discoid lupus erythematosus; Idiopathic pulmonary fibrosis
- 16 Jul 2012 Celgene Corporation terminates a phase II trial in Discoid lupus erythematosus in USA (NCT01466725)
- 23 Feb 2012 Celgene initiates enrolment in a phase II trial for Discoid lupus erythematosus in the USA (NCT01466725)
- 08 Nov 2011The Committee for Orphan Medicinal Products (COMP) recommends orphan drug designation for tanzisertib in European Union for Idiopathic pulmonary fibrosis
Tanzisertib has been granted orphan drug status by the FDA for the treatment of idiopathic pulmonary fibrosis. A positive opinion has been received from the EU Committee for Orphan Medicinal Products (COMP
Tanzisertib has been used in trials studying the treatment of Fibrosis, Discoid Lupus, Pulmonary Fibrosis, Interstitial Lung Disease, and Lung Diseases, Interstitial, among others.
PATENT
https://patents.google.com/patent/US20090048275A1/de

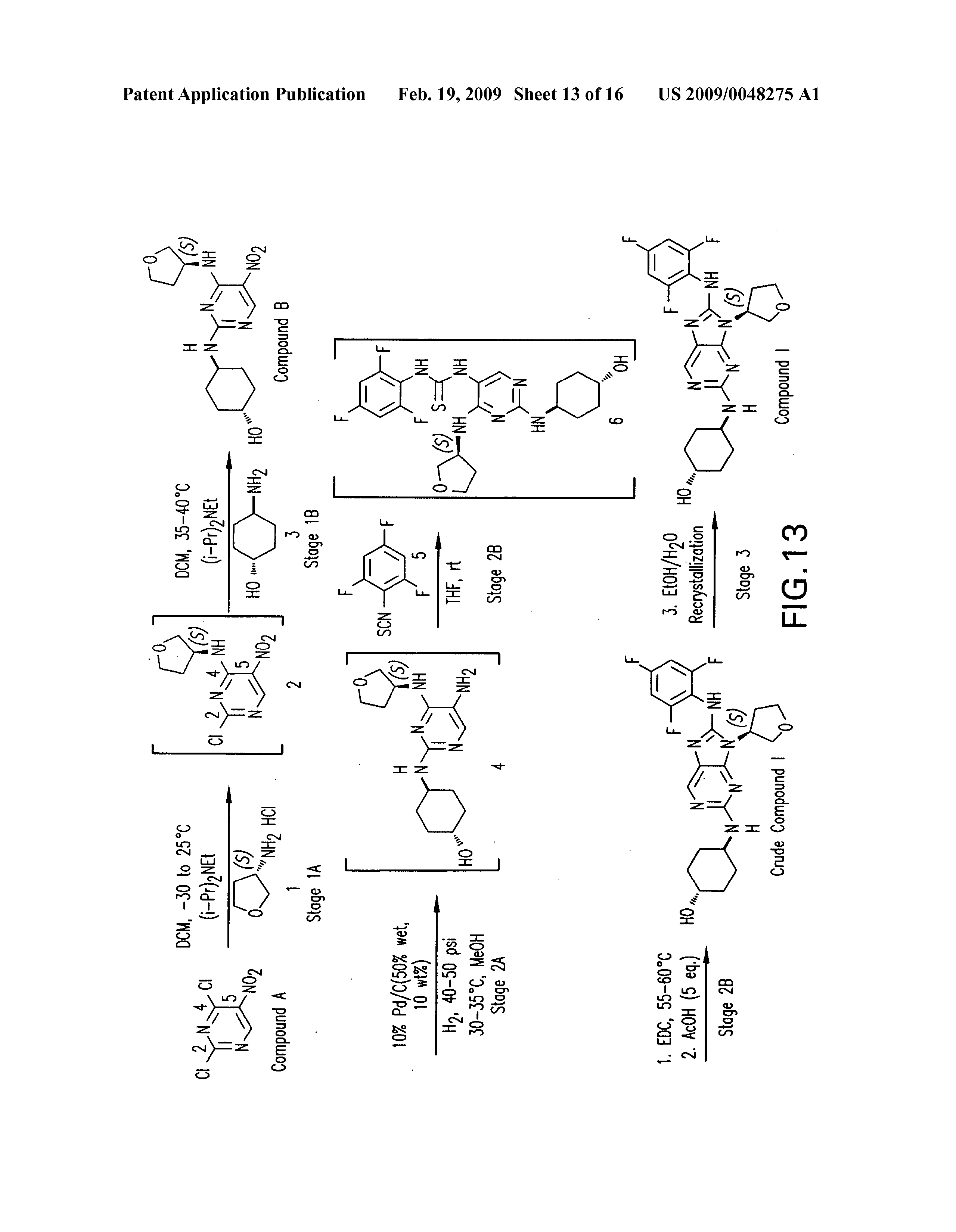

PATENT
WO 2006076595
US 20070060598
WO 2008057252
US 20080021048
US 20140094456
WO 2014055548
PATENT
WO 2015153683
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015153683
/////////Tanzisertib, CC 930, Idiopathic Pulmonary Fibrosis, Orphan Drug, phase II, CELGENE
c1c(c(c(cc1F)F)Nc2n(c3nc(ncc3n2)N[C@H]4CC[C@@H](CC4)O)[C@@H]5COCC5)F
Celgene oral Crohn’s drug GED-0301, Mongersen impresses in Phase II

Nogra制药,Celgene
公司寡核苷酸(Oligonucleotides)
克罗恩病(Crohn’s disease)
Shares in Celgene Corp have risen steadily following promising mid-stage data of its closely-watched Crohn’s disease drug mongersen.
Read more at: http://www.pharmatimes.com/Article/14-10-20/Celgene_oral_Crohn_s_drug_impresses_in_Phase_II.aspx#ixzz3Gr7C3e6u
| Company | Nogra Pharma Ltd. |
| Description | Antisense oligonucleotide targeting SMAD family member 7 (MADH7; SMAD7) |
| Molecular Target | SMAD family member 7 (MADH7) (SMAD7) |
| Mechanism of Action | |
| Therapeutic Modality | Nucleic acid: Linear RNA: Antisense |
| Latest Stage of Development | Phase II |
| Standard Indication | Crohn’s disease |
| Indication Details | Treat moderate to severe Crohn’s disease |
| Regulatory Designation | |
| Partner |
Mongersen (GED-0301) from Celgene Corp. (NASDAQ:CELG) produced clinical remission rates as high as 65.1% in a Phase II trial in 166 patients with moderate to severe Crohn’s disease, according to an abstract published in advance of the United European Gastroenterology’s meeting in Vienna.
In the trial, 55% of patients receiving 40 mg/day of mongersen and 65.1% of those receiving 160 mg/day achieved clinical remission compared with 9.5% of placebo patients (p<0.0001 for both). A cohort receiving 10 mg/day achieved a clinical remission rate of 12.2%, which was not significantly better than placebo.
The study’s primary outcomes were clinical remission, defined by a CDAI score less than 150 at day 15 and maintained for more than two weeks, and safety. Mongersen was well-tolerated, and toxicities associated with systemically active antisense therapies were not observed.
The study’s secondary endpoint is clinical response, defined as a CDAI score reduction of 100 points at day 28. Those rates were dose-dependent: 36.6%, 57.5% and 72.1% for the low, medium and high doses compared with 16.7% for placebo.
Celgene said it plans to start Phase III testing of mongersen shortly. The company paid $710 million up front to obtain exclusive, worldwide rights to the antisense oligonucleotide targeting SMAD family member 7 (MADH7; SMAD7) from Nogra Pharma Ltd. (Dublin, Ireland) in April. Nogra is eligible for $1.9 billion in milestones, plus tiered single-digit royalties.
GED-0301, an antisense oligonucleotide targeting the SMAD7 gene, is in phase II clinical trials at Nogra Pharma for the oral treatment of moderate to severe Crohn’s disease.
生物技术公司新基(Celgene)从爱尔兰制药商Nogra制药手中获得了一种处于后期临床开发的克罗恩病(Crohn’s disease)药物GED-0301。GED-0301是一种口服反义药物,靶向于Smad7信使RNA(mRNA),该药开发用于中度至重度克罗恩病 的治疗。反义药物是一种合成的核酸拷贝,旨在结合导致疾病的基因的mRNA,关闭基因的表达;口服;【Celgene签署$26亿协议获克罗恩病反义药物 GED-0301】http://www.hfoom.com/product/20140425/8311.html
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract suffered by approximately one million patients in the United States. The two most common forms of IBD are Crohn’s disease (CD) and ulcerative colitis (UC). Although CD can affect the entire gastrointestinal tract, it primarily affects the ilieum (the distal or lower portion of the small intestine) and the large intestine. UC primarily affects the colon and the rectum. Current treatment for both CD and UC include aminosalicylates (e.g., 5- aminosalicylic acid, sulfasalazine and mesalamine), antibiotics (e.g., ciprofloxacin and metronidazole), corticosteroids (e.g., budesonide or prednisone), immunosuppressants (e.g., azathioprine or methotrexate) and tumor necrosis factor (TNF) antagonists (e.g., infliximab (Remicade®)). Patient response to these therapies varies with disease severity and it can vary over cycles of active inflammation and remission. Moreover, many of the current therapies for IBD are associated with undesirable side effects.
Although the etiologies of CD and UC are unknown, both are considered inflammatory diseases of the intestinal mucosa. Recent studies have demonstrated that TGF-β 1 acts as a potent immunoregulator able to control mucosal intestinal inflammation. TGF-βΙ binds a heterodimeric transmembrane serine/threonine kinase receptor containing two subunits, TGF-βΙ Rl and TGF-βΙ R2. Upon ligand binding, the TGF-βΙ Rl receptor is phosphorylated by the constitutively active TGF-βΙ R2 receptor and signal is propagated to the nucleus by proteins belonging to the SMAD family. Activated TGF-β Ι Rl directly phosphorylates SMAD2 and SMAD3 proteins, which then interact with SMAD4. The complex of SMAD2/SMAD3/SMAD4 translocates to the nucleus and modulates the transcription of certain genes.
Additional studies have demonstrated that another SMAD protein, SMAD7, also plays a role in inflammation. SMAD7, an intracellular protein, has been shown to interfere with binding of SMAD2/SMAD3 to the TGF-βΙ Rl preventing phosphorylation and activation of these proteins. Further, increased expression of SMAD7 protein is associated with an inhibition of TGF-βΙ mediated-signaling. Mucosal samples from IBD patients are characterized by high levels of SMAD7 and reduced levels of phosphorylated-SMAD3 indicating that TGF-βΙ -mediated signaling is compromised in these patients.
Recent studies have focused on SMAD7 as a target for treating patients suffering from IBD.
Such therapies include anti-SMAD7 antisense therapies. As such, there is a need for methods based on predictive biomarkers that can be used to identify patients that are likely (or unlikely) to respond to treatment with anti- SMAD7 therapies.
GTCGCCCCTTCTCCCCGCAGC
GED-0301, Mongersen
Phosphorothioate antisense oligonucleotide targeting human mothers against decapentaplegic homolog 7 (SMAD7) gene, whose sequence is 5′-GTCGCCCCTTCTCCCCGCAGC-3′, wherein ‘C’ at postions 3 and 16 is 5-methyl 2′-deoxycytidine 5′-monophosphate
WO 2004087920
http://www.google.com/patents/WO2004087920A1?cl=en
…………………………
WO 2013037970
http://www.google.com/patents/WO2013037970A1?cl=en
…………………
WO 2013158868
http://www.google.com/patents/WO2013158868A1?cl=en
……………………………………………
http://www.google.com/patents/WO2014140333A1?cl=en
5*-GTCGCCCCTTCTCCCCGCAGC-3* (SEQ ID NO: 3).
| Reference | ||
|---|---|---|
| 1 | BADARU, A.; PIHOKER, C.: ‘Type 2 diabetes in childhood: clinical characteristics and role of beta-cell autoimmunity‘ CURR. DIAB. REP. vol. 12, 2012, pages 75 – 81 | |
| 2 | * | BHAT ET AL: “Antisense inhibition of 11betahydroxysteroid dehydrogenase type 1 improves diabetes in a novel cortisone-induced diabetic KK mouse model“, BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 365, no. 4, 20 November 2007 (2007-11-20), pages 740-745, XP022384861, ISSN: 0006-291X, DOI: 10.1016/J.BBRC.2007.11.032 |
| 3 | * | GUTIERREZ-AGUILAR ET AL: “Minor contribution of SMAD7 and KLF10 variants to genetic susceptibility of type 2 diabetes“, DIABETES & METABOLISM, PARIS, AMSTERDAM, NL, vol. 33, no. 5, 10 October 2007 (2007-10-10), pages 372-378, XP022327080, ISSN: 1262-3636, DOI: 10.1016/J.DIABET.2007.06.002 |
| 4 | * | H. Y. CHEN ET AL: “The Protective Role of Smad7 in Diabetic Kidney Disease: Mechanism and Therapeutic Potential“, DIABETES, vol. 60, no. 2, 27 October 2010 (2010-10-27), pages 590-601, XP55071874, ISSN: 0012-1797, DOI: 10.2337/db10-0403 |
| 5 | HONG, S. ET AL.: ‘Smad7 sensitizes tumor necrosis factor induced apoptosis through the inhibition of antiapoptotic gene expression by suppressing activation of the nuclear factor-kappaB pathway‘ CANCER RES. vol. 67, 2007, pages 9577 – 9583 | |
| 6 | HOOK, S. M. ET AL.: ‘Smad2: A candidate gene for the murine autoimmune diabetes locus Idd21.1‘ 1. CLIN. ENDOCRINOL. METAB. vol. 96, 2011, pages E2072 – E2077 | |
| 7 | KAWAMOTO, K. ET AL.: ‘Transforming growth factor beta 1 (TGF-?1) and rapamycin synergize to effectively suppress human T cell responses via upregulation of FoxP3+ Tregs‘ TRANSPL. IMMUNOL. vol. 23, 2010, pages 28 – 33 | |
| 8 | LI, M. O.; FLAVELL, R. A.: ‘TGF-beta: a master of all T cell trades‘ CELL vol. 134, 2008, pages 392 – 404 | |
| 9 | * | LIANG Y ET AL: “Reduction in Glucagon Receptor Expression by an Antisense Oligonucleotide Ameliorates Diabetic Syndrome in db/db Mice“, DIABETES, AMERICAN DIABETES ASSOCIATION, US, vol. 53, February 2004 (2004-02), pages 410-417, XP002995165, ISSN: 0012-1797, DOI: 10.2337/DIABETES.53.2.410 |
| 10 | * | LU ZHU ET AL: “Unraveling the biological functions of Smad7 with mouse models“, CELL & BIOSCIENCE, BIOMED CENTRAL LTD, LONDON, UK, vol. 1, no. 1, 28 December 2011 (2011-12-28), page 44, XP021132085, ISSN: 2045-3701, DOI: 10.1186/2045-3701-1-44 |
| 11 | LUO, X. ET AL.: ‘Systemic transforming growth factor-?1 gene therapy induces Foxp3+ regulatory cells, restores self-tolerance, and facilitates regeneration of beta cell function in overtly diabetic nonobese diabetic mice‘ TRANSPLANTATION vol. 79, 2005, pages 1091 – 1096 | |
| 12 | MARGOLLES-CLARK, E. ET AL.: ‘Small molecule costimulatory blockade: organic dye inhibitors of the CD40-CD154 interaction‘ J. MOL. MED. vol. 87, 2009, pages 1133 – 1143 | |
| 13 | * | MIZOBUCHI TERUAKI ET AL: “Differential expression of Smad7 transcripts identifies the CD4+CD45RChigh regulatory T cells that mediate type V collagen-induced tolerance to lung allografts“, THE JOURNAL OF IMMUNOLOGY, THE AMERICAN ASSOCIATION OF IMMUNOLOGISTS, US, vol. 171, no. 3, 1 August 2003 (2003-08-01), pages 1140-1147, XP002430371, ISSN: 0022-1767 |
| 14 | MONTELEONE, G. ET AL.: ‘A failure of transforming growth factor-?1 negative regulation maintains sustained NF-KB activation in gut inflammation‘ J. BIOL. CHEM. vol. 279, 2004, pages 3925 – 3932 | |
| 15 | MORITANI, M. ET AL.: ‘Abrogation of autoimmune diabetes in nonobese diabetic mice and protection against effector lymphocytes by transgenic paracrine TGF-?1‘ J. CLIN. INVEST. vol. 102, 1998, pages 499 – 506 | |
| 16 | * | NORA G. SMART ET AL: “Conditional Expression of Smad7 in Pancreatic [beta] Cells Disrupts TGF-[beta] Signaling and Induces Reversible Diabetes Mellitus“, CELL, vol. 88, no. 2, 31 January 2006 (2006-01-31), page 561, XP55071875, ISSN: 0092-8674, DOI: 10.1371/journal.pbio.0040039 |
| 17 | OLIVIERI, A. ET AL.: ‘Serum transforming growth factor ?1 during diabetes development in non-obese diabetic mice and humans‘ CLIN. EXP. IMMUNOL. vol. 162, 2010, pages 407 – 414 | |
| 18 | ‘Remington’s Pharmaceutical Sciences‘, 1990, MACK PUBLISHING COMPANY | |
| 19 | ROEP, B. O. ET AL.: ‘Satisfaction (not) guaranteed: re-evaluating the use of animal models of type 1 diabetes‘ NAT. REV. IMMUNOL. vol. 4, 2004, pages 989 – 997 | |
| 20 | * | S. M. HOOK ET AL: “Smad2: A Candidate Gene for the Murine Autoimmune Diabetes Locus Idd21.1“, JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM, vol. 96, no. 12, 5 October 2011 (2011-10-05), pages E2072-E2077, XP55071877, ISSN: 0021-972X, DOI: 10.1210/jc.2011-0463 |
| 21 | SHODA, L. K. ET AL.: ‘A comprehensive review of interventions in the NOD mouse and implications for translation‘ IMMUNITY vol. 23, 2005, pages 115 – 126 | |
| 22 | SMART, N. G. ET AL.: ‘Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus‘ PLOS BIOL. vol. 4, 2006, page E39 | |
| 23 | WALLBERG, M. ET AL.: ‘An islet-specific pulse of TGF-? abrogates CTL function and promotes ? cell survival independent of Foxp3+ T cells‘ J. IMMUNOL. vol. 186, 2011, pages 2543 – 2551 | |
| 24 | YAN, X.; CHEN, Y. G.: ‘Smad7: not only a regulator, but also a cross-talk mediator of TGF-beta signalling‘ BIOCHEM. J. vol. 434, 2011, pages 1 – 10 | |
| WO2003037368A2 * | Oct 31, 2002 | May 8, 2003 | Andreas Steinbrecher | Smad7 inhibitors for the treatment of cns diseases |
| WO2009129544A1 * | Apr 20, 2009 | Oct 22, 2009 | Baxter International Inc. | Microsphere-based composition for preventing and/or reversing new-onset autoimmune diabetes |
| WO2010054826A1 | Nov 13, 2009 | May 20, 2010 | Giuliani International Limited | Antisense compositions and methods of making and using same |
Oral Anti-Cancer Therapy Pomalidomide Now Approved by European Commission as Treatment for Patients with Relapsed/Refractory Multiple Myeloma – a Rare Form of Blood Cancer

POMALIDOMIDE
4-amino-2-(2,6-dixopiperidin-3- yl)isoindoline-l,3-dione; 3-(4-amino-l,3-dioxo-l,3-dihydro-isoindol-2-yl)-piperidine- 2,6-dione; 3-(4-amino-l ,3-dioxoisoindolin-2-yl)piperidine-2,6-dione; 1 ,3-dioxo-2-(2,6- dioxopiperidin-3-yl)-4-aminoisoindoline; 3-(l,3-dioxo-4-aminoisoindolin-2-yl)- piperidine-2,6-dione;
BOUDRY, Switzerland–(BUSINESS WIRE)–Aug. 9, 2013–Celgene International Sàrl, a wholly-owned subsidiary of Celgene Corporation (NASDAQ: CELG) today announced that the European Commission (EC) has granted approval for Pomalidomide Celgene®▼(pomalidomide),
in combination with dexamethasone, for the treatment of relapsed and refractory multiple myeloma (rrMM) in adult patients who have received at least two prior therapies including both lenalidomide and bortezomib and have demonstrated disease progression on the last therapy.1 Celgene intends to launch Pomalidomide Celgene in the EU under the trade name “IMNOVID®”, following submission of a regulatory notification to the European Medicines Agency (EMA) to change the trade name.
READ ALL AT
http://www.pharmalive.com/ec-approves-celgene-blood-cancer-drug
CAS 19171-19-8
Pomalidomide, an analogue of thalidomide, is an immunomodulatory antineoplastic agent. FDA approved on February 8, 2013.
Pomalidomide is indicated for patients with multiple myeloma who have received at least two prior therapies including lenalidomide and bortezomib and have demonstrated disease progression on or within 60 days of completion of the last therapy.
Pomalidomide (INN, originally CC-4047 or 3-amino-thalidomide, trade name Pomalyst[1] in the US) is a derivative of thalidomidemarketed by Celgene. It is anti-angiogenic and also acts as an immunomodulator. Pomalidomide was approved in February 2013 by the U.S. Food and Drug Administration (FDA) as a treatment for relapsed and refractory multiple myeloma.[2] It received a similar approval from the European Commission in August 2013, and is expected to be marketed in Europe under the brand nameImnovid.[3]
Origin and development
The parent compound of pomalidomide, thalidomide, was originally discovered to inhibit angiogenesis in 1994.[4] Based upon this discovery, thalidomide was taken into clinical trials for cancer, leading to its ultimate FDA approval for multiple myeloma.[5] Further structure activity studies done in Dr. Robert D’Amato’s lab at Boston Children’s Hospital led to the first claim in 1995 that amino-thalidomide had antitumor activity.[6] Interestingly, the pronounced anti-tumor activity is due to its ability to directly inhibit both the tumor cell and vascular compartments of myeloma cancers.[7] This dual activity of pomalidomide makes it more efficacious than thalidomide in vitro and in vivo.[8]
Clinical trials
Phase I trial results showed tolerable side effects.[9]
Phase II clinical trials for multiple myeloma and myelofibrosis reported ‘promising results’.[10][11]
Phase III results were reported at ASH in 2012 and showed significant extension of progression-free survival (median 3.6 months vs. 1.8 months; P < 0.001), and overall survival in patients taking pomalidomide and dexamethasone v. dexamethasone alone.[12]
Mechanism
Pomalidomide directly inhibits angiogenesis and myeloma cell growth. This dual effect is central to its activity in myeloma, rather than other pathways such as TNF alpha inhibition, since potent TNF alpha inhibitors including rolipram and pentoxifylline do not inhibit myeloma cell growth nor angiogenesis.[7] Up regulation of Interferon gamma, IL-2 and IL-10 as well as down regulation of IL-6 have been reported for pomalidomide. These changes may contribute to pomalidomide’s anti-angiogenic and anti-myeloma activities.
Pregnancy and sexual contact warnings
Because Pomalyst can cause harm to unborn babies when administered during pregnancy, women taking Pomalyst must not become pregnant. Women must produce two negative pregnancy tests and use contraception methods before beginning Pomalyst. Women must commit either to abstain continuously from heterosexual sexual intercourse or to use two methods of reliable birth control, beginning 4 weeks prior to initiating treatment with Pomalyst, during therapy, during dose interruptions and continuing for 4 weeks following discontinuation of Pomalyst therapy. Pomalyst is present in the semen of patients receiving the drug. Therefore, males must always use a latex or synthetic condom during any sexual contact with females of reproductive potential while taking POMALYST and for up to 28 days after discontinuing Pomalyst, even if they have undergone a successful vasectomy. Male patients taking Pomalyst must not donate sperm.
Pomalidomide simple structure, synthesis is relatively easy. The glutamine ( 1 ), the compound 2 protected amino, thionyl chloride to ring palladium on carbon hydrogenation later deprotected to give compound 3 , 3 , and 4 direct condensation Pomalidomide.
PATENTS

| US PATENT No | Patent ExpirY | patent use code |
|---|---|---|
| 5635517 | Jul 24, 2016 | U-1359 |
| 6045501 | Aug 28, 2018 | U-1361 |
| 6315720 | Oct 23, 2020 | U-1361 |
| 6316471 | Aug 10, 2016 | U-1360 |
| 6476052 | Jul 24, 2016 | U-1360 |
| 6561976 | Aug 28, 2018 | U-1361 |
| 6561977 | Oct 23, 2020 | U-1361 |
| 6755784 | Oct 23, 2020 | U-1361 |
| 6908432 | Aug 28, 2018 | U-1361 |
| 8158653 | Aug 10, 2016 | |
| 8198262 | Oct 19, 2024 | U-1360 |
| 8204763 | Aug 28, 2018 | U-1361 |
| 8315886 | Oct 23, 2020 | U-1361 |
| Exclusivity Code | Exclusivity Date |
|---|---|
| ODE | Feb 8, 2020 |
| NCE | Feb 8, 2018 |
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 8198262 | 2013-02-08 | 2024-10-19 |
| United States | 8204763 | 2013-02-08 | 2018-08-28 |
| United States | 8315886 | 2013-02-08 | 2020-10-23 |
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 5635517 | 2013-02-08 | 2016-07-24 |
| United States | 6045501 | 2013-02-08 | 2018-08-28 |
| United States | 6315720 | 2013-02-08 | 2020-10-23 |
| United States | 6316471 | 2013-02-08 | 2016-08-10 |
| United States | 6476052 | 2013-02-08 | 2016-07-24 |
| United States | 6561976 | 2013-02-08 | 2018-08-28 |
| United States | 6561977 | 2013-02-08 | 2020-10-23 |
| United States | 6755784 | 2013-02-08 | 2020-10-23 |
| United States | 6908432 | 2013-02-08 | 2018-08-28 |
| United States | 8158653 | 2013-02-08 | 2016-08-10 |
Pomalidomide-2013, FDA approved anticancer drugs. Pomalidomide isthalidomide (thalidomide) derivative, for the treatment of multiple myeloma. Trade name Pomalyst, developed by Celgene.
Pomalidomide simple structure, synthesis is relatively easy. (From glutamine 1 ), the compound 2 is protected amino, thionyl chloride off ring after deprotection to obtain a compound with palladium on carbon hydrogenation of 3 , 3 and 4 the direct condensation Pomalidomide.
……………………..
http://www.google.com/patents/WO2012177678A2?cl=en

…………………………


Figure 1: Chronological view of the history of thalidomide and its analogs
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-Amino-2-(2,6-dioxopiperidin-3-yl)isoindole-1,3-dione | |
| Clinical data | |
| Trade names | Imnovid, Pomalyst |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy cat. |
|
| Legal status | |
| Routes | Oral |
| Pharmacokinetic data | |
| Protein binding | 12–44% |
| Metabolism | Hepatic (mostly CYP1A2 andCYP3A4 mediated; some minor contributions by CYP2C19 andCYP2D6) |
| Half-life | 7.5 hours |
| Excretion | Urine (73%), faeces (15%) |
| Identifiers | |
| CAS number | 19171-19-8 |
| ATC code | L04AX06 |
| PubChem | CID 134780 |
| Chemical data | |
| Formula | C13H11N3O4 |
| Mol. mass | 273.24 g/mol |


Figure 2: The mechanism of TLP in multiple myeloma. TLP refers to thalidomide, lenalidomide and pomalidomide.
CLIP


HONORED
Celgene’s George Muller (left) and Roger Shen-Chu Chen celebrate at the Heroes of Chemistry banquet.
Credit: Linda Wang/C&EN
The satisfaction of helping patients is what drives George Muller as an industrial scientist. Muller is coinventor of Celgene’s Polamyst for multiple myeloma.
“It’s wonderful to be able to think that the work one did in the lab ended up helping patients,” he says. “Over my career, I’ve met patients who were taking drugs on which I had worked. It’s always amazing to see the positive effects on the lives of these patients. Some of them get their lives back.”
Muller says that during the course of developing Pomalyst, they made hundreds of compounds. “We worked on the project for probably 15-plus years,” he says. The drug was approved in 2014.
References
- “Pomalyst (Pomalidomide) Official Website”. Celgene Corporation. Retrieved 2013-08-10.
- “Pomalyst (Pomalidomide) Approved By FDA For Relapsed And Refractory Multiple Myeloma”. The Myeloma Beacon. Retrieved 2013-08-10.
- “Pomalidomide Approved In Europe For Relapsed And Refractory Multiple Myeloma”. The Myeloma Beacon. Retrieved 2013-08-10.
- D’Amato, Robert J.; Loughnan, Michael S.; Flynn, Evelyn; Folkman, Judah (1994). “Thalidomide is an inhibitor of angiogenesis”. Proceedings of the National Academy of Sciences of the United States of America 91 (9): 4082–5. Bibcode:1994PNAS…91.4082D. doi:10.1073/pnas.91.9.4082. JSTOR 2364596. PMC 43727. PMID 7513432.
- http://vectorblog.org/2013/04/from-thalidomide-to-pomalyst-better-living-through-chemistry/
- http://patft.uspto.gov/netacgi/nph-Parser?Sect1=PTO2&Sect2=HITOFF&p=1&u=%2Fnetahtml%2FPTO%2Fsearch-bool.html&r=1&f=G&l=50&co1=AND&d=PTXT&s1=5,712,291.PN.&OS=PN/5,712,291&RS=PN/5,712,291
- D’Amato, R; Lentzsch, S; Anderson, KC; Rogers, MS (2001). “Mechanism of action of thalidomide and 3-aminothalidomide in multiple myeloma”. Seminars in Oncology 28 (6): 597–601. doi:10.1016/S0093-7754(01)90031-4. PMID 11740816.
- Lentzsch, S; Rogers, MS; Leblanc, R; Birsner, AE; Shah, JH; Treston, AM; Anderson, KC; D’Amato, RJ (2002). “S-3-Amino-phthalimido-glutarimide inhibits angiogenesis and growth of B-cell neoplasias in mice”. Cancer Research 62 (8): 2300–5. PMID 11956087.
- Streetly, Matthew J.; Gyertson, Kylie; Daniel, Yvonne; Zeldis, Jerome B.; Kazmi, Majid; Schey, Stephen A. (2008). “Alternate day pomalidomide retains anti-myeloma effect with reduced adverse events and evidence of in vivo immunomodulation”. British Journal of Haematology 141 (1): 41–51. doi:10.1111/j.1365-2141.2008.07013.x. PMID 18324965.
- Jump up^ “Promising Results From 2 Trials Highlighting Pomalidomide Presented At ASH” (Press release). Celgene. December 11, 2008. Retrieved October 28, 2012.
- Jump up^ Tefferi, Ayalew (December 8, 2008). “Pomalidomide Therapy in Anemic Patients with Myelofibrosis: Results from a Phase-2 Randomized Multicenter Study”. 50th ASH Annual Meeting and Exposition. San Francisco. Retrieved October 28, 2012.
- Jump up^ “Phase III Study (MM-003) of Pomalidomide Plus Low-Dose Dexamethasone Demonstrates Significant Progression-Free and Overall Survival Improvement for Patients with Relapsed or Refractory Multiple Myeloma.”. 11 Dec 2012.
External links
POMALYST is an immunomodulatory antineoplastic agent. The chemical name is (RS)-4-Amino-2-(2,6-dioxo-piperidin-3-yl)-isoindoline-1,3dione and it has the following chemical structure:
 |
The empirical formula for pomalidomide is C13H11N3O4 and the gram molecular weight is 273.24.
Pomalidomide is a yellow solid powder. It has limited to low solubility into organic solvents and it has low solubility in all pH solutions (about 0.01 mg/mL). Pomalidomide has a chiral carbon atom which exists as a racemic mixture of the R(+) and S(-) enantiomers.
POMALYST is available in 1 mg, 2 mg, 3 mg and 4 mg capsules for oral administration. Each capsule contains pomalidomide as the active ingredient and the following inactive ingredients: mannitol, pregelatinized starch and sodium stearyl fumarate. The 1 mg capsule shell contains gelatin, titanium dioxide, FD&C blue 2, yellow iron oxide, white ink and black ink. The 2 mg capsule shell contains gelatin, titanium dioxide, FD&C blue 2, yellow iron oxide, FD&C red 3 and white ink. The 3 mg capsule shell contains gelatin, titanium dioxide, FD&C blue 2, yellow iron oxide and white ink. The 4 mg capsule shell contains gelatin, titanium dioxide, FD&C blue 1, FD&C blue 2 and white ink.
NDA 204026
APPR..2013-02-08
Dosages/Routes/Forms
Celgene
| Strength | Form/Route | Marketing Status | RLD | TE Code |
|---|---|---|---|---|
| 1MG | CAPSULE;ORAL | 1 | 0 | |
| 2MG | CAPSULE;ORAL | 1 | 0 | |
| 3MG | CAPSULE;ORAL | 1 | 0 | |
| 4MG | CAPSULE;ORAL | 1 | 1 |
Approval History
2013-10-03
001
Manufacturing Change or Addition
Celgene makes good start to 2013 as Revlimid hits $1 billion
April 26, 2013
Celgene Corp has posted a healthy set of financials for the first quarter despite a 4.1% decline in net income to $410.2 million, as product sales increased 15% to $1.43 billion. read more at————http://www.pharmatimes.com/Article/13-04-26/Celgene_makes_good_start_to_2013_as_Revlimid_hits_1_billion.aspx

(RS)-3-(4-amino-1-oxo 1,3-dihydro-2H-isoindol- 2-yl)piperidine-2,6-dione
Lenalidomide
REVLIMID® is an oral immunomodulatory drug marketed in the United States and many international markets, in combination with dexamethasone, for treatment of patients with multiple myeloma who have received at least one prior therapy. It is also marketed in the United States and certain international markets for the treatment of transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes, or MDS, associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalitie.Revlimid Worldwide annual sales in 2011 was $3.2bLenalidomide (Revlimid) is a derivative of thalidomideintroduced in 2004.It was initially intended as a treatment for multiple myeloma, for which thalidomide is an accepted therapeutic treatment. Lenalidomide has also shown efficacy in the class of hematological disorders known as myelodysplastic syndromes (MDS). Lenalidomide has significantly improved overall survival in myeloma (which generally carries a poor prognosis), although toxicity remains an issue for users. [1]It costs $163,381 per year for the average patient.[2]
Lenalidomide has been used to successfully treat both inflammatory disorders and cancers in the past 10 years. There are multiple mechanisms of action, and they can be simplified by organizing them as mechanisms of action in vitro and in vivo.[3] In vitro, lenalidomide has three main activities: direct anti-tumor effect, inhibition of the microenvironment support for tumor cells, and immunomodulatory role. In vivo, lenalidomide induces tumor cell apoptosis directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects, and by immunomodulatory activity. Lenalidomide has a broad range of activities that can be exploited to treat many hematologic and solid cancers.
- McCarthy; Philip L. McCarthy, Kouros Owzar, Craig C. Hofmeister, et al. (May 10, 2012). “Lenalidomide after Stem-Cell Transplantation for Multiple Myeloma”. N Engl J Med 366 (19): 1770–1781. doi:10.1056/NEJMoa1114083. PMID 22571201.
- Badros, Ashraf Z. Badros (May 10, 2012). “Lenalidomide in Myeloma — A High-Maintenance Friend”. N Engl J Med 366 (19): 1836–1838. doi:10.1056/NEJMe1202819. PMID 22571206.
- Vallet S, Palumbo A, Raje N, Boccadoro M, Anderson KC (July 2008). “Thalidomide and lenalidomide: Mechanism-based potential drug combinations”. Leukemia & Lymphoma 49 (7): 1238–45. doi:10.1080/10428190802005191. PMID 18452080.
Celgene phase 3 – Oral Apremilast Achieves Statistical Significance for the Primary Endpoint of PASI-75 in the First Phase III Study in Patients with Psoriasis

APREMILAST, N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide
mar02,2013
Celgene International Sàrl, a subsidiary of Celgene Corporation (NASDAQ: CELG) today presented the results from ESTEEM 1, the Company’s first phase III study in psoriasis, at the American Academy of Dermatology annual meeting in Miami, Florida.
“I see this as a prime candidate for future management of psoriasis that allows us to treat a range of patients, including more moderate cases earlier on”
The company previously announced statistical significance for the primary and major secondary endpoint of PASI-75 at Week 16 and the Static Physician Global Assessment for patients receiving apremilast in the ESTEEM 1&2 phase III studies. ESTEEM 1&2 are the phase III registrational randomized, placebo-controlled studies evaluating the Company’s oral small-molecule inhibitor of phosphodiesterase-4 (PDE4) in patients with moderate-to-severe chronic plaque psoriasis.
ESTEEM 1, presented today, evaluated efficacy and safety in a range of patients. Approximately one-third of the study population was systemic and/or phototherapy treatment-naïve. Nearly 30 percent of the overall study population had prior biologic therapy, which included biologic-failures.
In the ESTEEM 1 study, a significantly higher percentage of apremilast-treated patients demonstrated PASI-75 at week 16 than did placebo patients (33.1% vs. 5.3%; P<0.0001). Significantly higher PASI-75 scores at week 16 were demonstrated across all patient segments enrolled in this study, including systemic-naïve and biologic-naïve patients receiving apremilast 30 mg BID compared with placebo (38.7% vs. 7.6%; P<0.0001 and 35.8% vs. 5.9%; P<0.0001 respectively). Apremilast demonstrated maintenance of effect over time, as measured by the Mean Percent Change from Baseline in PASI score over 32 weeks, with apremilast demonstrating a 54.9% reduction at week 16 and a 61.9% reduction at week 32.
Statistical significance at week 16 was also demonstrated in the major secondary endpoint, Static Physician Global Assessment (sPGA) of clear or almost clear (P<0.0001), and other key secondary endpoints (change in BSA, Pruritus VAS, DLQI), as well as in assessments of difficult to treat areas (nail and scalp psoriasis).
“I see this as a prime candidate for future management of psoriasis that allows us to treat a range of patients, including more moderate cases earlier on,” said Kristian Reich, M.D., SCIderm Research Institute and Dermatologikum Hamburg, Germany.
The overall safety and tolerability profile was consistent with results from previously reported phase III psoriatic arthritis trials. No cases of tuberculosis or lymphoma were observed through week 16, and there was no increased risk of cardiovascular events or serious opportunistic infection. Apremilast was generally well tolerated. The most common adverse events (AEs) greater than placebo were diarrhea, nausea and headache. Greater than 96% of patients in the study reported no AEs or mild to moderate AEs. A similar percentage of patients reported both serious AEs and severe AEs in the apremilast 30 mg BID treatment group compared to placebo (2.1% vs. 2.8% and 3.6% vs. 3.2%, respectively).
An NDA submission to the U.S. Food and Drug Administration, based on the combined ESTEEM 1&2 studies for psoriasis, is expected in the second half of 2013. The Company previously announced it expects to file a separate NDA for psoriatic arthritis in the first quarter of 2013. A combined PsA/psoriasis MAA submission in Europe is also planned for the second half of 2013.
Top-line positive results from the two pivotal, randomized, placebo-controlled phase III studies of apremilast in psoriasis (ESTEEM 1&2) were released in January 2013. The studies included more than 1,200 patients with moderate-to-severe psoriasis and are ongoing. Results from PSOR-005, a phase IIb dose-range study, were recently published in The Lancet (http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(12)60642-4/fulltext).
About ESTEEM 1 & 2
ESTEEM 1 & 2 are two pivotal phase III randomized, placebo-controlled studies evaluating apremilast in subjects with a diagnosis of moderate-to-severe chronic plaque psoriasis for at least 12 months prior to the screening, and at baseline, and who were also candidates for phototherapy and/or systemic therapy. Approximately 1,250 patients were randomized 2:1 to receive either apremilast 30 mg BID or placebo for the first 16 weeks, followed by a maintenance phase from weeks 16-32 in which placebo subjects were switched to apremilast 30 mg BID through week 32, and a randomized withdrawal phase for responders from Week 32-Week 52 based on their initial apremilast randomization and PASI response.
Apremilast, an oral small-molecule inhibitor of phosphodiesterase 4 (PDE4), works intracellularly to modulate a network of pro-inflammatory and anti-inflammatory mediators. PDE4 is a cyclic adenosine monophosphate (cAMP)-specific PDE and the dominant PDE in inflammatory cells (see http://discoverpde4.com/). PDE4 inhibition elevates intracellular cAMP levels, which in turn down-regulates the inflammatory response by modulating the expression of TNF-α, IL-23, and other inflammatory cytokines. Elevation of cAMP also increases anti-inflammatory cytokines such as IL-10. To learn more go to www.discoverpde4.com/.
Top-line positive results from three pivotal randomized, placebo-controlled phase III studies of apremilast in PsA (PALACE 1, 2 & 3) were released in September 2012. PALACE 1 was also presented as an oral presentation at the ACR annual meeting in November 2012. Taken together, the PALACE program comprises the most comprehensive psoriatic arthritis studies to date intended for regulatory submission.
Results from PSA-001, the phase II study of apremilast in psoriatic arthritis, were recently published online in the journal Arthritis & Rheumatism (http://onlinelibrary.wiley.com/doi/10.1002/art.34627/abstract).
A randomized, placebo-controlled phase III study (POSTURE) of apremilast in ankylosing spondylitis (AS) began enrolling patients in April 2012. AS, a debilitating disease, which may cause fusion of the spine, arthritis, inflammation of the eye and damage to the heart, affects approximately 1.5 million people in the U.S. and Europe. The trial will randomize approximately 450 patients to receive 20 mg or 30 mg apremilast BID, or placebo BID.
Psoriasis is an immune-mediated, non-contagious chronic inflammatory skin disorder of unknown cause. The disorder is a chronic recurring condition that varies in severity from minor localized patches to complete body coverage. Plaque psoriasis is the most common type of psoriasis. About 80 percent of people who develop psoriasis have plaque psoriasis, which appears as patches of raised, reddish skin covered by silvery-white scales. These patches, or plaques, frequently form on the elbows, knees, lower back, and scalp. Psoriasis occurs nearly equally in males and females. Recent studies show that there may be an ethnic link. Psoriasis is believed to be most common in Caucasians and slightly less common in other ethnic groups. Worldwide, psoriasis is most common in Scandinavia and other parts of northern Europe. About 10 percent to 30 percent of patients with psoriasis also develop a condition called psoriatic arthritis, which causes pain, stiffness and swelling in and around the joints.
Celgene International Sàrl, located in Boudry, in the Canton of Neuchâtel, Switzerland, is a wholly owned subsidiary and international headquarters of Celgene Corporation. Celgene Corporation, headquartered in Summit, New Jersey, is an integrated global pharmaceutical company engaged primarily in the discovery, development and commercialization of innovative therapies for the treatment of cancer and inflammatory diseases through gene and protein regulation. For more information, please visit the Company’s website at www.celgene.com.
Apremilast is an orally available small molecule inhibitor of PDE4 being developed by Celgene for ankylosing spondylitis, psoriasis, and psoriatic arthritis.[1][2] The drug is currently in phase III trials for the three indications. Apremilast, an anti-inflammatory drug, specifically inhibits phosphodiesterase 4. In general the drug works on an intra-cellular basis to moderate proinflammatory and anti-inflammatory mediator production.
Apremilast is being tested for its efficacy in treating “psoriasis, psoriatic arthritis and other chronic inflammatory diseases such as ankylosing spondylitis, Behcet’s disease, and rheutmatoid arthritis.”
- “Apremilast Palace Program Demonstrates Robust and Consistent Statistically Significant Clinical Benefit Across Three Pivotal Phase III Studies (PALACE-1, 2 & 3) in Psoriatic Arthritis” (Press release). Celgene Corporation. 6 September 2012. Retrieved 2012-09-10.
- “US HOT STOCKS: OCZ, VeriFone, Men’s Wearhouse, AK Steel, Celgene”. The Wall Street Journal. 6 September 2012. Retrieved 2012-09-06
