
Home » Posts tagged 'CDK Inhibitor'
Tag Archives: CDK Inhibitor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SNS-032, BMS-387032 A potent and selective Cdk inhibitor
SNS 032, BMS-387032
N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide
Cas 345627-80-7, MP 165-167° C
M.Wt:380.53, Formula:C17H24N4O2S2
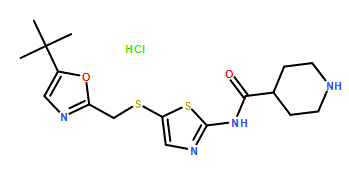
SNS 032, BMS-387032 HYDROCHLORIDE
| Formula | C17H24N4O2S2 . HCl |
|---|---|
| MW | 380.5 . 36.5 |
| CAS | 345627-90-9 |
A potent and selective Cdk inhibitor
Potent inhibitor of cyclin-dependent kinases (cdks) 9, 2 and 7 (IC50 values are 4, 38 and 62 nM respectively). Displays no activity against 190 additional kinases (IC50 >1000 nM). Arrests the cell cycle at G2/M; inhibits transcription, proliferation and colony formation, and induces apoptosis in RPMI-8226 multiple myeloma cells. Prevents tumor cell-induced VEGF secretion and in vitro angiogenesis. SNS-032 (BMS-387032) has firstly been described as a selective inhibitor of CDK2 with IC50 of 48 nM in cell-free assays and is 10- and 20-fold selective over CDK1/CDK4. It is also found to be sensitive to CDK7/9 with IC50 of 62 nM/4 nM, with little effect on CDK6. Phase 1.
Quality Control & MSDS
COA NMR HPLC Datasheet SDS/MSDS
- COA (Certificate Of Analysis)
- HPLC
- NMR (Nuclear Magnetic Resonance)
- MSDS (Material Safety Data Sheet)
SNS-032 (BMS-387032) is a potent and selective inhibitor of cyclin-dependent kinases (CDKs) 2, 7, and 9 [1], with IC50 values of 38 nM, 62 nM and 4 nM, respectively [2].
CDKs mean a family of serine/threonine kinases regulating cell cycle process. Some CDKs are related to transcription control and are often perturbed in cancer cells [3].
Decrease in the phosphorylation at Ser5 and Ser2 in the C-terminal domain (CTD) of RNA Pol II can indicate the inhibition to CDK9 and CDK7 [1]. Chronic lymphocytic leukemia (CLL) cells treated with SNS-032 for 6 or 24 hours showed a decrease in the phosphorylation of Ser2 and Ser5 of the CTD of RNA Pol II, this appeared to be both time- and concentration- dependent, and remarkably consistent among samples. For the phosphorylation of Ser2, the inhibition of SNS-032 was greater than that for the phosphorylation of Ser5, this was consistent with the fact that IC50 for the inhibition of CDK9 was lower compared with that for the inhibition of CDK7 (4 nM vs 62 nM). After 6 hours of SNS-032 exposure, protein levels of CDK7 and CDK9 were stable, but declined at 24 hours [4].
In patients with chronic lymphocytic leukemia (CLL), infusion of SNS-032 in a total dose of 75 mg/m2 resulted in a decrease in the phosphorylation at Ser5 and Ser2 in the C-terminal domain of RNA Pol II. This indicated the inhibition to Cdk9 and Cdk7 by SNS-032. This inhibition was first seen 2 hours after the beginning of the infusion with SNS-032, was pronounced after 6 hours and returned to baseline after 24 hours [1].

The cell cycle-regulated cyclin-dependent kinases (CDKs), CDK1, 2, and 4 have been extensively studied as potential therapeutic targets in cancer. Recent research has additionally underscored the potential role of several constitutively active CDKs including CDK7 and 9 as cancer targets. Phosphorylation of the c-terminal domain (CTD) of RNA Polymerase II by CDK7 and 9 are critical steps in transcriptional regulation. Inhibition of these kinases is predicted to have the greatest effect on the expression of proteins with short t½ and short-lived mRNA, including proteins involved in apoptotic regulation. CDK7 also activates cell-cycle CDKs 1, 2, 4 and 6. SNS-032 (formerly BMS-387032) has previously been described as a selective inhibitor of CDK2 with potent antitumor activity in animal models. Here we show that in addition to inhibition of CDK2, SNS-032 also inhibits CDK7/cyclinH and CDK9/cyclinT at low nanomolar concentrations in biochemical assays. The compound is highly selective for CDK inhibition; in a panel of 208 kinases, only four non-CDK proteins were inhibited by >50% at 1 μM SNS-032. The cellular pharmacology of SNS- 032 mirrors the biochemical data. Cells treated with SNS-032 show a rapid cell cycle arrest and onset of cell death that corresponds with inhibition of multiple substrates of CDK2, 7, and 9. For instance, inhibition of Rb phosphorylation, accumulation of cyclin E protein and cell-cycle arrest at GI and G2 are observed in multiple cell lines in a time and dose-dependent manner, consistent with inhibition of CDK2 and CDK7. Furthermore, SNS-032 inhibits CDK9-mediated phosphorylation of Ser2 in the CTD with an IC50 = 200 nM. Corresponding with inhibition of RNA polymerase II, the short half-life, anti-apoptotic protein Mcl-1 is rapidly depleted from cells, coincident with the phosphorylation of p53. Expression of Mcl-1 is a candidate predictor of aggressive disease and resistance to chemotherapy in CLL and is essential for survival of B-cell lymphoma and multiple myelomas, supporting the use of SNS-032 as a treatment for these diseases. SNS-032, a selective inhibitor of multiple CDKs involved in apoptosis and cell cycle regulation, has potential for antitumor activity in both solid and hematological cancers. SNS-032 is currently in phase 1 clinical studies.
SNS-032, was designed as a selective CDK2 inhibitor. Here, we show that in addition to CDK2, CDK 7 and 9 inhibitory activities also contribute to the biological activity of the molecule. The CDK2/cyclin E complex regulates entry of cells into S phase by phosphorylating Rb, a negative regulator of the transcription factor E2F. CDK2 phosphorylates a number of additional substrates, including cyclin E, signaling its degradation. Inhibiting CDK2 should therefore arrest cells in G1 and stabilize cyclin E. The cellcycle CDKs (CDK1, 2 4 and 6) are activated by phosphorylation by CDK7/cyclin H (also called CAK). Inhibition of CDK7 would therefore also result in cell-cycle arrest at multiple points in the cell cycle due to failure to activate the cell cycle CDKs. CDK 7 and 9 activate transcription by phosphorylating the CTD of RNA pol II. Inhibition of CTD phosphorylation has been shown to inhibit transcription and reduce expression of short lived proteins, including those involved in apoptosis regulation. Stalling of RNA polymerase has also been shown to activate p53, leading to apoptosis. Thus, the CDK7 and 9 inhibitory activities of SNS-032 are expected to cause cytotoxicity via induction of apoptosis.
SNS-032 is a selective CDK inhibitor, preferentially targeting CDK2, CDK7 and CDK9 in vitro. • In cell models, SNS-032 shows dual activity, targeting both cell cycle progression and apoptosis pathway proteins. • SNS-032 Inhibited CDK9 and 7-mediated phosphorylation of ser 2 and ser 5 of the CTD of RNA pol II and in turn downregulates the antiapoptotic protein Mcl-1. • SNS-032 induced a cell cycle arrest, and increased cyclin E levels are consistent with inhibition of cell cycle CDKs • Mcl-1 is a key survival factor in many B-cell malignancies. SNS-032 is being pursed as treatment for these diseases.
| Biological Activity | ||||||
|---|---|---|---|---|---|---|
| Description | SNS-032 is a novel, potent and selective CDK inhibitor of CDK2, CDK7 and CDK9 with IC50 of 38 nM, 62 nM and 4 nM, respectively. | |||||
| Targets | CDK2 | CDK7 | CDK9 | |||
| IC50 | 38 nM | 62 nM | 4 nM [1] | |||
| In Vitro | SNS-032 has low sensitivity to CDK1 and CDK4 with IC50 of 480 nM and 925 nM, respectively. SNS-032 effectively kills chronic lymphocytic leukemia cells in vitro regardless of prognostic indicators and treatment history. Compared with flavopiridol and roscovitine, SNS-032 is more potent, both in inhibition of RNA synthesis and at induction of apoptosis. SNS-032 activity is readily reversible; removal of SNS-032 reactivates RNA polymerase II, which led to resynthesis of Mcl-1 and cell survival. [1] SNS-032 inhibits three dimensional capillary network formations of endothelial cells. SNS-032 completely prevents U87MG cell–mediated capillary formation of HUVECs. In addition, SNS-032 significantly prevents the production of VEGF in both cell lines, SNS-032 prevents in vitro angiogenesis, and this action is attributable to blocking of VEGF. Preclinical studies have shown that SNS-032 induces cell cycle arrest and apoptosis across multiple cell lines. [2] SNS-032 blocks the cell cycle via inhibition of CDKs 2 and 7, and transcription via inhibition of CDKs 7 and 9. SNS-032 activity is unaffected by human serum. [3]SNS-032 induces a dose-dependent increase in annexin V staining and caspase-3 activation. At the molecular level, SNS-032 induces a marked dephosphorylation of serine 2 and 5 of RNA polymerase (RNA Pol) II and inhibits the expression of CDK2 and CDK9 and dephosphorylated CDK7. [4] | |||||
| In Vivo | SNS-032 prevents tumor cell-induced VEGF secretion in a tumor coculture model. [2] SNS-032, a new CDK inhibitor, is more selective and less cytotoxic and has been shown to prolong stable disease in solid tumors. [4] | |||||
| Clinical Trials | SNS-032 currently in phase I clinical trial for chronic lymphocytic leukemia (CLL) and multiple myeloma (MM). | |||||
| Description | SNS-032 is a selective inhibitor of CDK2 with IC50 of 48 nM. | |||||
| Targets | CDK2 | CDK7 | CDK9 | |||
| IC50 | 48 nM | 62 nM | 4 nM | |||
CLIP
http://www.mdpi.com/1420-3049/19/9/14366/htm#B39-molecules-19-14366
SNS032, previously called BMS-387032, has been developed by Sunesis. This compound, which contains a thiazole unit, selectively inhibits CDK2 (IC50: 38 nM), CDK7 (IC50: 62 nM) and CDK9 (IC50: 4 nM) [39]. Preclinical studies demonstrated that SNS032 was able to inhibit cell cycle activity along with transcription [20].
SNS032 is in phase I clinical trials for the treatment of chronic lymphoid leukemia along with multiple myeloma, and the mode of administration is intravenous [39]. The purpose is to evaluate the dose-escalation of SNS-032 along with its safety, pharmacokinetics, pharmacodynamic activity and clinical efficacy. Biomarker analyses demonstrated mechanism-based pharmacodynamic activity with inhibition of CDK7 and CDK9, although limited clinical activity in heavily pretreated patients was observed [39].
Tong, W.G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective CDK2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. ASCO Annual Meeting. J. Clin. Oncol. 2010, 28, 3015–3022.

![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://journals.prous.com/journals/dof/20083311/html/df330932/images/sch01.gif)
SNS-032 (formerly BMS-387032) is a small-molecule cyclin-dependent kinase (CDK) inhibitor currently in phase I clinical trials for the treatment of B-cell malignancies and advanced solid tumors. Preclinical studies have shown that SNS-032 is a specific and potent inhibitor of CDK2, 7 and 9 which induces cell cycle arrest and apoptosis in tumor cell lines. It was shown to inhibit in vitro angiogenesis and prostaglandin E2 (PGE2) production, both strongly associated with tumorigenesis. Phase I clinical trials support the safety and tolerability of SNS-032 as evaluated in dose-escalation studies. The compound is currently administered by i.v. infusion but has shown promising potential for oral delivery.
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://journals.prous.com/journals/dof/20083311/html/df330932/images/sch02.gif)
 NMR
NMR
CLIP
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://i0.wp.com/www.rcsb.org/pdb/images/56H_600.gif)
![Image result for N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,](https://i0.wp.com/www.mdpi.com/ijms/ijms-14-21805/article_deploy/html/images/ijms-14-21805f2-1024.png)
The structures of representative protein kinases inhibitors based on the aminopyrazole scaffold.http://www.mdpi.com/1422-0067/14/11/21805/htm
CLIP
N-(Cycloalkylamino)acyl-2-aminothiazole Inhibitors of Cyclin-Dependent Kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a Highly Efficacious and Selective Antitumor Agent,

N-Acyl-2-aminothiazoles with nonaromatic acyl side chains containing a basic amine were found to be potent, selective inhibitors of CDK2/cycE which exhibit antitumor activity in mice. In particular, compound 21 {N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4-piperidinecarboxamide, BMS-387032}, has been identified as an ATP-competitive and CDK2-selective inhibitor which has been selected to enter Phase 1 human clinical trials as an antitumor agent. In a cell-free enzyme assay, 21 showed a CDK2/cycE IC50 = 48 nM and was 10- and 20-fold selective over CDK1/cycB and CDK4/cycD, respectively. It was also highly selective over a panel of 12 unrelated kinases. Antiproliferative activity was established in an A2780 cellular cytotoxicity assay in which 21 showed an IC50 = 95 nM. Metabolism and pharmacokinetic studies showed that 21 exhibited a plasma half-life of 5−7 h in three species and moderately low protein binding in both mouse (69%) and human (63%) serum. Dosed orally to mouse, rat, and dog, 21showed 100%, 31%, and 28% bioavailability, respectively. As an antitumor agent in mice, 21administered at its maximum-tolerated dose exhibited a clearly superior efficacy profile when compared to flavopiridol in both an ip/ip P388 murine tumor model and in a sc/ip A2780 human ovarian carcinoma xenograft model.
CLIP

http://pubs.rsc.org/en/content/articlehtml/2016/md/c6md90040b
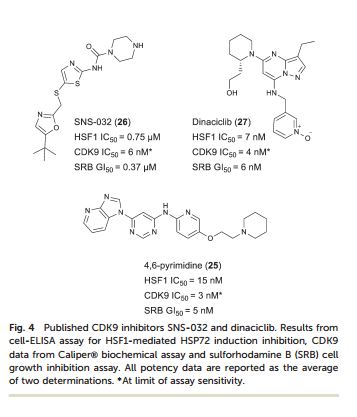
Heat shock factor 1 (HSF1) is a transcription factor that plays key roles in cancer, including providing a mechanism for cell survival under proteotoxic stress. Therefore, inhibition of the HSF1-stress pathway represents an exciting new opportunity in cancer treatment. We employed an unbiased phenotypic screen to discover inhibitors of the HSF1-stress pathway. Using this approach we identified an initial hit (1) based on a 4,6-pyrimidine scaffold (2.00 μM). Optimisation of cellular SAR led to an inhibitor with improved potency (25, 15 nM) in the HSF1 phenotypic assay. The 4,6-pyrimidine 25 was also shown to have high potency against the CDK9 enzyme (3 nM).

Discovery of 4,6-disubstituted pyrimidines as potent inhibitors of the heat shock factor 1 (HSF1) stress pathway and CDK9
E-mail: Paul.Workman@icr.ac.uk, Keith.Jones@icr.ac.uk
DOI: 10.1039/C6MD00159A
COMPD 25
1H NMR (500 MHz, DMSO-d6) δ 10.38 (s, 1H), 9.21 (s, 1H), 8.74 (d, J = 0.9 Hz, 1H), 8.62 (dd, J = 8.2, 1.5 Hz, 1H), 8.56 (dd, J = 4.7, 1.5 Hz, 1H), 8.16-8.13 (m, 2H), 7.64 (br d, J = 8.6 Hz, 1H), 7.52-7.47 (m, 2H), 4.14 (t, J = 5.9 Hz, 2H), 2.66 (t, J = 5.9 Hz, 2H), 2.47-2.42 (m, 4H), 1.53-1.47 (m, 4H), 1.42 – 1.33 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 160.74, 158.32, 156.72, 154.88, 150.74, 146.47, 145.38, 143.74, 134.21, 125.02, 124.16, 122.29, 119.60, 114.32, 94.06, 66.49, 57.35, 54.35, 25.54, 23.88. HRMS (ESI+ ): calcd for C22H25N8O (M + H)+ , 417.2146; found 417.2163.
NOTE, THERE IS ERROR IN STRUCTURE ABOVE OF SNS 032
References
References:
[1]. Tong W.G., Chen R., Plunkett W., et al. Phase I and Pharmacologic Study of SNS-032, a Potent and Selective Cdk2, 7, and 9 Inhibitor, in Patients With Advanced Chronic Lymphocytic Leukemia and Multiple Myeloma. Journal of Clinical Oncology, 2010, 28(18):3015- 3022.
[2]. Chipumuro E., Marco E., Christensen C.L., et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell, 2014, 159:1-14.
[3]. Meng H., Jin Y.M., Liu H., et al. SNS-032 inhibits mTORC1/mTORC2 activity in acute myeloid leukemia cells and has synergistic activity with perifosine against Akt. Journal of Hematology & Oncology, 2013, 6:18.
[4]. Chen R., Wierda W.G., Chubb S., et al. Mechanism of action of SNS032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood, 2009, 113(19):4637-4645.Chen et al (2010) Responses in mantle cell lymphoma cells to SNS-032 depend on the biological context of each cell line. Cancer Res. 70 6587. PMID: 20663900.
Conroy et al (2009) SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemother.Pharmacol. 64 723. PMID: 19169685.
Ali et al (2007) SNS-032 prevents tumor cell-induced angiogenesis by inhibiting vascular endothelial growth factor. Neoplasia 9 370. PMID: 17534442.
Misra et al (2004) N-(Cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J.Med.Chem. 47 1719. PMID: 15027863.
Abstract
SNS-032, a CDK inhibitor, exhibited modest to high anti-neuroblastoma activity against a panel of 109 neuroblastoma cell lines in the range of the therapeutic plasma levels reported for SNS-032 through a mechanism involving CDK7 and CDK9 inhibition-mediated down-regulation of XIAP, Mcl-1, BIRC2, cIAP-1 and surviving.
Abstract
The anti-AML mechanism of SNS-032, a cyclin-dependent kinase inhibitor, has been identified though characterizing in vitro effects of SNS-032 alone or in combination with perifosine.
Abstract
Although it induces apoptosis in cancer cells, SNS-032 has no significant effects on normal HSC and HPC in terms of self-renewal inhibition, differentiation suppression and apoptosis induction.
Abstract
The CDK7/9 inhibitor SNS-032-induced down-regulation of FIP1L1-PDGFRα and Bcr-Abl has the potential to be used to decrease the acquired resistant to imatinib.
Abstract
SNS-032, a CDK inhibitor, alone or in combination with Ara-C exhibited potent anti-AML activity, where down-regulation of antiapoptotic genes, cluding BCL2, XIAP amd MCL1, was associated with the synergistic anti-AML effect of the combination treatment.
CC(C)(C)C1=CN=C(O1)CSC2=CN=C(S2)NC(=O)C3CCNCC3
CDK Inhibitor, MK 7965, DINACICLIB, SCH 727965

CDK Inhibitor, MK 7965, DINACICLIB, SCH 727965
REVIEW…….http://www.mdpi.com/2072-6694/6/4/2224/htm
One of the most popular CDK inhibitor in clinical trials in the recent years was dinaciclib (MK-7965, SCH 727965) (Figure 3), the inhibitor of CDK1, CDK2, CDK5, and CDK9. A Phase I trial on the effect of dinaciclib in combination with aprepitant was performed in patients with advanced malignancies [44]. Aprepitant is used for the prevention of chemotherapy-induced nausea and vomiting, is known as an inhibitor and inducer of CYP3A4, which metabolizes dinaciclib.
Coadministration of dinaciclib with aprepitant resulted in no clinically significant effect on the pharmacokinetics and did not alter the safety profile of dinaciclib. The first Phase I clinical trial on dinaciclib as a single agent was performed on patients with advanced malignancies [68]. Forty-eight patients with various solid tumors were treated and 10 of them achieved prolonged stable disease for at least four treatment cycles. Adverse effects were mild, the most common being nausea, anemia, decreased appetite and fatigue.
A phase II multi-center study of dinaciclib for relapsed and/or refractory AML was performed on 20 patients [69]. Temporary decrease in peripheral blood and/or bone marrow blasts was observed in 60% of patients. Four of 13 (31%) patients with circulating blasts had >50% decrease and 6 (46%) >80% decrease in the absolute blast count within 1–8 days of the first dinaciclib dose. Toxicities included diarrhea, fatigue, transaminitis, and manifestations of tumor lysis syndrome, with one patient who deceased of acute renal failure. Another Phase II study was performed of dinaciclib versus erlotinib in patients with non-small cell lung cancer [70].
Unfortunately, it was found that dinaciclib was not successful as monotherapy in non-small cell lung cancer. Most common toxicities included neutropenia, leukopenia, vomiting, and diarrhea. Yet another Phase II study was performed on dinaciclib versus capecitabine in patients with advanced breast cancer [71]. Dinaciclib treatment demonstrated antitumor activity in two of seven patients with ER-positive and ERBB 2-negative metastatic breast cancer, however efficacy was not superior to capecitabine (p = 0.991).
Toxicities included neutropenia, leukopenia, increase in aspartate aminotransferase, and febrile neutropenia. Phase I nonrandomized dose-escalation trial was performed, where patients with relapsed or refractory chronic lymphocytic leukemia were treated with dinaciclib and rituximab [72]. Four out of six patients achieved stable disease, and one patient achieved complete response. Drug-related adverse events were mostly hematological, digestive and metabolic and no dose-limiting toxicities were observed. Dinaciclib was also moved into Phase III development for refractory chronic lymphocytic leukemia [73]. Phase I/II clinical trial Dinaciclib in patients with relapsed multiple myeloma showed promise as single agent [74]. The overall confirmed response rate was 3 of 27 (11%). Adverse effects included leukopenia, thrombocytopenia, gastrointestinal symptoms, alopecia, and fatigue. –
FOR REF See more at: http://www.mdpi.com/2072-6694/6/4/2224/htm#sthash.amBuLwq1.dpuf

Dinaciclib (SCH-727965) is an experimental drug that inhibits cyclin-dependent kinases (CDKs.[1] It is being evaluated in clinical trials for various cancer indications.[2]
Mechanisms of action
- Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains.[3]
- Dinaciclib (SCH727665) inhibits the unfolded protein response (UPR) through a CDK1 and CDK5-dependent mechanism.[4]
Anti-tumoral action
- In chronic lymphocytic leukemia (CLL)
- In pancreatic cancer
- Dinaciclib inhibits pancreatic cancer growth and progression in murine xenograft models.[7]
- In osteosarcoma
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (S)-3-(((3-Ethyl-5-(2-(2-hydroxyethyl)piperidin-1-yl)pyrazolo[1,5-a]pyrimidin-7-yl)amino)methyl)pyridine 1-oxide | |
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS number | 779353-01-4 |
| ATC code | ? |
| PubChem | CID 46926350 |
| ChemSpider | 25027387 |
| ChEMBL | CHEMBL2103840 |
| Synonyms | SCH-727965 |
| Chemical data | |
| Formula | C21H28N6O2 |
Clinical trials

http://www.google.com.tr/patents/US8076479
One example of these inhibitors is the compound of Formula II.

The synthesis of the compound of Formula II is described in the ‘878 publication according to Scheme II:
Scheme II:
Step 1—Amidization to Form Substituted Pyrazole

http://www.google.com.tr/patents/US8076479
Step 2—Formation and Dehalogenation of pyrazolo[1,5a]pyrimidine

Step 3—Amination (Two Separate, Sequential Reactions)

As described in the ‘878 publication, Synthetic Scheme II leading to the compound of Formula II has several disadvantages from the standpoint of commercial scale synthesis. In step 1, the starting material (compound “C”) used in the formation of compound “D” is a sticky, viscous oil which is difficult to process (weigh, transfer, and blend). Moreover, step 1, as described in the ‘878 publication, requires isolation and chromatographic purification of compounds C and D prior to carrying out each subsequent derivatization reaction. In addition, as described in the ‘878 publication, the reaction of compound C with malonate diester is carried out using the diester as a solvent. After isolation and purification of the resultant malonate adduct, compound D, ring closure to form diketone compound E is carried out in methanol. In accordance with the procedure described in the ‘878 publication, compound E is isolated and dried, then converted to the corresponding dichloride in N,N-dimethyl aniline by treatment with phosphorous oxychloride (POCl3). The dichloride thus formed was isolated and purified by chromatography prior to the sequential amination reactions. Additionally, the compounds of Formula G and of Formula II require chromatography purification and isolations, as described in the ‘878 publication.
As further described in the ‘878 publication, each of the amination reactions were run separately with isolation and chromatographic purification between amination reactions. Accordingly, the ‘878 publication describes the preparation of the compound of Formula II utilizing a scheme consisting of five separate reaction steps with intervening isolation and purification of the products, each sequential step being carried out in a different solvent system. The overall yield of the compound of Formula II reported for this synthesis, based on starting compound C (Scheme II) is about 20%.
Example 1Preparation of Diketone Compound E (Scheme VI) 3-Ethylpyrazolo[1,5-a]pyrimidine-5,7(4H,6H)-dione

To a 250 ml, three-necked flask equipped with a thermometer, a reflux condenser and mechanical stirrer was charged 3-amino-4-ethylpyrazole oxalate (10 g, 50 mmole), dimethylmalonate (10 ml, 88 mmole), methyl alcohol (80 ml) and sodium methoxide (50 ml, 245 mmole, 25% in methyl alcohol). The batch was heated at reflux for 16 hours then cooled to room temperature. Celite (5 g) and water (60 ml) were added to the batch and agitated for 10 minutes. The batch was filtered to remove the solid residue. The filtrate was pH adjusted to pH˜3 with aqueous HCl (10 ml) to effect precipitation. The precipitate (compound “E”) was filtered and washed with water (40 ml). The wet cake was dried for 18 hours in vacuum oven maintained in the range of oven at 45° C. to 55° C., to give a solid product (84.3%, 7.5 g). C8H9N3O3, Mp: 200-205° C.; NMR in DMSO-d6: 1.05 (t, 3H), 2.23 (q, 2H), 3.26 (bs, 1H), 3.89 (bs, 1H), 7.61 (s, 1H), 11.50(bs, 1H).
Example 2Preparation of Dichloride Compound F (Scheme VI) 5,7-Dichloro-3-Ethylpyrazolo[1,5-a]pyrimidine

Into a 3-neck flask fitted with an inert gas inlet, a reflux condenser and a mechanical stirring apparatus and containing 83 liters of acetonitrile was placed 3-Ethylpyrazolo[1,5-a]pyrimidine-5,7(4H,6H)-dione (E) prepared as described in Step 1 (11.0 kg, 61.5 mole), N,N-dimethylaniline (8.0 L, 63 mole) and POCl3 (7 kg, 430 mole). With stirring the mixture was brought to reflux and maintained under refluxing conditions for 15 hours. The reaction mixture was sampled periodically to monitor the amount of compound “E” present. After the conversion was complete, the solution was cooled to 15° C. Into the cooled reaction mixture was added water which had been cooled to a temperature of less than 20° C. The product is filtered and washed with 4 aliquots of acetonitrile-water (1:3) which had been cooled to a temperature of 20° C. followed by a wash with 10× water. The wet cake is dried in a vacuum oven maintained at 40° C. for at least 15 hours to yield the compound “F” (86.7%); 1H NMR (CDCl3): 1.32(t, 3H), 2.81 (q, 2H), 6.92 (s, 1H), 8.10 (s, 1H)
mp: 90-95° C.
Example 3Preparation of Compound G (Scheme VI) 5-Chloro-3-Ethyl-N-[(1-oxido-pyridinyl)methyl]pyrazolo-[1,5-a]pyrimidine-5.7(4H,6H)-dion-7-amine

Into a 3-liter, three-necked flask equipped with a thermometer, a reflux condenser and mechanical stirrer was charged an aliquot of the dichloride compound “F” prepared in Step 2 (150 g, 0.69 mole), potassium phosphate tribasic monohydrate (338.0 g, 1.47 mole), the dihydrochloride salt of N-oxide-pyridin-3-yl-methylamine, compound F1a (142.5 g, 0.72 mole), water (1500 ml) and acetonitrile (300 ml). The batch was heated at reflux for 6 hours. At the end of the refluxing period the batch was cooled to room temperature over 2 hours and then held at room temperature for 4 hours. The resulting precipitate was filtered and washed with water (600 ml). The wet cake was returned to the flask with water (1500 ml) and acetonitrile (300 ml), and heated to reflux. Reflux was maintained for 6 hours additional. At the end of the second reflux period the reaction mixture was cooled to room temperature over a 2 hour period and left to stand at room temperature for 4 hours. The resulting precipitate was filtered and washed with water (600 ml). The wet cake was dried in an air draft oven at 50° C. for 18 hours to give the first amine adduct “G” material (179 g, 84.9%). mp: 187-189C; NMR in CDCl3, 1.26(t, 3H), 2.73(q, 2H), 4.60(d, 2H), 5.87(s, 1H), 6.83(bs, 1H), 7.33(t, 1H), 7.70(d, 1H), 7.84(s, 1H), 8.58(d, 1H), 8.64(d, 1H).
Example 4
Preparation of the Compound of Formula II (Scheme VI) 1-[3-Ethyl-7-[(1-oxido-3-pyridinyl)methyl]amino]pyrazolo[1,5-a]pyrimidin-5-yl]-2(s)-piperidinemethanol

Into a three-neck flask fitted with a mechanical stirrer and a reflux condenser were placed the first amine adduct prepared in Step 3, compound “G”, (7 kg, 23 mole), amino-alcohol compound G1a (5.6 kg, 43.3 mole), sodium carbonate (3.5 kg, 33.0 mole), 110 ml of water and 1-methyl-2-pyrrolidinone (NMP) (11 L). The reaction mixture was heated to 150° C. for 4 days. After chromatography indicated that the reaction was complete (90-95% substrate consumed), the reaction mixture was cooled to room temperature and quenched by adding water. The mixture was then extracted with ethyl acetate. The batch was dried by distillation of the water azeotrope under atmospheric pressure and concentrated to about 28 L volume. THF was added and the solution was heated to reflux until all the solids dissolve. Ethyl acetate and trietylamine are added to the hot solution. The batch was cooled to ambient and then agitated with the temperature maintained in the range of from 20° C. to 25° C. for 12 hours. The solids were collected by filtration, washed first with ethyl acetate then water, and dried in the filter under vacuum for 24 hours with the temperature maintained at from 40° C. to 50° C., yielding 4.9 kg, 51.3% of the compound of Formula II.
DSC, 168.6° C.; Specific Rotation (10 mg/ml in MeOH, 20° C.), −117.8 °;
1HNMR (400 MHz, DMSO): 8.31 ppm (1H, s), 8.11-8.13 ppm (1H, td, J=5.7 Hz, J=1.4 Hz), 7.97 ppm (1H, t, J=6.7 Hz), 7.68 ppm (1H, s), 7.41 ppm (1H, s), 7.37-7.43 ppm (1H, dd), 5.55 ppm (1H, s), 4.85 ppm (1H, t, J=5.4 Hz), 4.49-4.59 ppm (3H, m), 4.24-4.28 ppm (1H, broad), 3.27-3.46 ppm (2H, m), 2.76-2.83 ppm (1H, t, J=13.0 Hz), 2.45-2.50 ppm (2H, q, J=7.5 Hz), 1.72-1.79 (1H, m), 1.54-1.68 ppm (6H, m), 1.30-1.34 ppm (1H, m), 1.16 ppm (3H, t, J=7.5 Hz)
References
- Parry, D; Guzi, T; Shanahan, F; Davis, N; Prabhavalkar, D; Wiswell, D; Seghezzi, W; Paruch, K; Dwyer, M. P.; Doll, R; Nomeir, A; Windsor, W; Fischmann, T; Wang, Y; Oft, M; Chen, T; Kirschmeier, P; Lees, E. M. (2010). “Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor”. Molecular Cancer Therapeutics 9 (8): 2344–53. doi:10.1158/1535-7163.MCT-10-0324. PMID 20663931.
- Jump up^ Bose P, Simmons GL, Grant S (2013). “Cyclin-dependent kinase inhibitor therapy for hematologic malignancies”. Expert Opin Investig Drugs 22 (6): 723–38.doi:10.1517/13543784.2013.789859. PMC 4039040. PMID 23647051.
- Martin, M. P.; Olesen, S. H.; Georg, G. I.; Schönbrunn, E (2013). “Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains”. ACS Chemical Biology 8 (11): 2360–5. doi:10.1021/cb4003283. PMC 3846258. PMID 24007471.
- Nguyen, T. K.; Grant, S (2013). “Dinaciclib (SCH727665) inhibits the unfolded protein response (UPR) through a CDK1 and CDK5-dependent mechanism”. Molecular Cancer Therapeutics 13(3): 662–74. doi:10.1158/1535-7163.MCT-13-0714. PMID 24362465.
- Jump up^ Desai, B. M.; Villanueva, J; Nguyen, T. T.; Lioni, M; Xiao, M; Kong, J; Krepler, C; Vultur, A; Flaherty, K. T.; Nathanson, K. L.; Smalley, K. S.; Herlyn, M (2013). “The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling”. PLoS ONE 8 (3): e59588. doi:10.1371/journal.pone.0059588. PMC 3601112. PMID 23527225.
- Jump up^ Johnson, A. J.; Yeh, Y. Y.; Smith, L. L.; Wagner, A. J.; Hessler, J; Gupta, S; Flynn, J; Jones, J; Zhang, X; Bannerji, R; Grever, M. R.; Byrd, J. C. (2012). “The novel cyclin-dependent kinase inhibitor dinaciclib (SCH727965) promotes apoptosis and abrogates microenvironmental cytokine protection in chronic lymphocytic leukemia cells”. Leukemia 26 (12): 2554–7.doi:10.1038/leu.2012.144. PMC 3645353. PMID 22791353.
- Jump up^ Feldmann, G; Mishra, A; Bisht, S; Karikari, C; Garrido-Laguna, I; Rasheed, Z; Ottenhof, N. A.; Dadon, T; Alvarez, H; Fendrich, V; Rajeshkumar, N. V.; Matsui, W; Brossart, P; Hidalgo, M; Bannerji, R; Maitra, A; Nelkin, B. D. (2011). “Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models”.Cancer biology & therapy 12 (7): 598–609. PMC 3218385. PMID 21768779.
- Jump up^ Fu, W; Ma, L; Chu, B; Wang, X; Bui, M. M.; Gemmer, J; Altiok, S; Pledger, W. J. (2011). “The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells”. Molecular Cancer Therapeutics 10 (6): 1018–27. doi:10.1158/1535-7163.MCT-11-0167. PMID 21490307.
- Jump up^ Fu, W; Sharma, S. S.; Ma, L; Chu, B; Bui, M. M.; Reed, D; Pledger, W. J. (2013). “Apoptosis of osteosarcoma cultures by the combination of the cyclin-dependent kinase inhibitor SCH727965 and a heat shock protein 90 inhibitor”. Cell Death and Disease 4 (3): e566. doi:10.1038/cddis.2013.101. PMC 3613821. PMID 23538447.
- Jump up^ Nemunaitis, J. J.; Small, K. A.; Kirschmeier, P; Zhang, D; Zhu, Y; Jou, Y. M.; Statkevich, P; Yao, S. L.; Bannerji, R (2013). “A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies”. Journal of Translational Medicine 11 (1): 259. doi:10.1186/1479-5876-11-259.PMC 3853718. PMID 24131779.
- Jump up^ Mita, M; Joy, A. A.; Mita, A; Sankhala, K; Jou, Y. M.; Zhang, D; Statkevich, P; Zhu, Y; Yao, S. L.; Small, K; Bannerji, R; Shapiro, C. L. (2013). “Randomized Phase II Trial of the Cyclin-Dependent Kinase Inhibitor Dinaciclib (MK-7965) Versus Capecitabine in Patients with Advanced Breast Cancer”. Clinical Breast Cancer 14 (3): 169–76. doi:10.1016/j.clbc.2013.10.016.PMID 24393852.
- Jump up^ Stephenson, J. J.; Nemunaitis, J; Joy, A. A.; Martin, J. C.; Jou, Y. M.; Zhang, D; Statkevich, P; Yao, S. L.; Zhu, Y; Zhou, H; Small, K; Bannerji, R; Edelman, M. J. (2014). “Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer”. Lung Cancer 83 (2): 219–23.doi:10.1016/j.lungcan.2013.11.020. PMID 24388167.
External links
- dinaciclib at the US National Library of Medicine Medical Subject Headings (MeSH)

Patent Submitted Granted
Process and intermediates for the synthesis of (3-alkyl-5-piperidin-1-yl-3,3a-dihydro-pyrazolo[1,5-a]pyrimidin-7-yl)-amino derivatives and intermediates [US8076479]2008-03-06 GRANT2011-12-13
Process for resolving chiral piperidine alcohol and process for synthesis of pyrazolo[1,5-a] pyrimidine derivatives using same [US7786306]2008-02-28 GRANT2010-08-31
Sequential Administration of Chemotherapeutic Agents for Treatment of Cancer [US2011129456]2011-06-02
TARGETING CDK4 AND CDK6 IN CANCER THERAPY [US2011009353]2011-01-13
Pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2007225270]2007-09-27
PYRAZOLO[1,5-a]PYRIMIDINES [US2007275963]2007-11-29
Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2007281951]2007-12-06
Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2008050384]2008-02-28
Novel pyrazolopyrimidines as cyclin dependent kinase inhibitors [US2007054925]2007-03-08

