


Topical Premature Ejaculation Treatment
Take control.
Last longer.
<$3 per use for most men!!

PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Posts tagged 'AYURVEDA' (Page 7)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
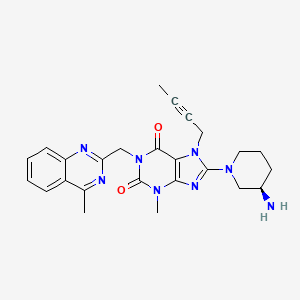
linagliptin
C25H28N8O2
CAS : 668270-12-0
Molecular Weight: 472.54
Purity: > 98%
(R)-8-(3-aminopiperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-1H-purine-2,6(3H,7H)-dione
8-(3R)-3-aminopiperidinyl)-7-butyn-2-yl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione
Solubility: Up to 25 mM in DMSO
Synonyms: BI-1356, BI1356, Linagliptin, Tradjenta, Trajenta
BI-1356 (Linagliptin) is a highly potent and selective dipeptidyl peptidase 4 (DPP-4) inhibitor (IC50 = 1 nM) for treatment of type II diabetes. [1] BI-1356 can increase incretin levels (GLP-1 and GIP), which increases insulin secretion and inhibits glucagon release, decreases gastric emptying, and decreases blood glucose levels. BI-1356 shows 10,000-fold more selectivity for DPP-4 against other protease/peptidases, including DPP-8, DPP-9, trypsin, plasmin, and thrombin, It is a DPP-4 inhibitor developed by Boehringer Ingelheim for the treatment of type II diabetes.
Linagliptin is a highly potent, selective DPP-4 inhibitor with IC50 of 1 nM.
“This study provides much-needed data on glucose-lowering treatment of elderly people with Type 2 Diabetes, inadequately controlled with common anti-hyperglycaemic agents”
Data published in The Lancet showed that elderly people with Type 2 Diabetes (T2D) treated for 24 weeks with the dipeptidyl peptidase-4 (DPP-4) inhibitor linagliptin, marketed by Boehringer Ingelheim and Eli Lilly and Company, experienced significant reductions in blood glucose levels (HbA1c) compared with those receiving placebo. In addition, the overall safety and tolerability profile of linagliptin was similar to placebo, with no significant difference in hypoglycaemia
Linagliptin (BI-1356, trade names Tradjenta and Trajenta) is a DPP-4 inhibitor developed by Boehringer Ingelheim for treatment of type II diabetes.
Linagliptin (once-daily) was approved by the US FDA on 2 May 2011 for treatment of type II diabetes.[1] It is being marketed by Boehringer Ingelheim and Lilly.
Linagliptin, namely 8-(3R)-3-aminopiperidinyl)-7-butyn-2-yl-3-methyl-1-(4-methylquinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione, of formula (A), is a long acting inhibitor of dipeptidylpeptidase-IV (DPP-IV) activity, at present under development for the treatment of type II diabetes mellitus.

The synthesis of Linagliptin is reported in US 7,407,955 , according to the scheme below, where 8-bromo xanthine of formula (B) is condensed with 3-(R)-Boc-aminopiperidine of formula (C) to obtain a compound of formula (D), which is converted to Linagliptin (A) by deprotection of the amine function

Optically active 3-aminopiperidine protected as the tert-butylcarbamate (Boc), compound (C), although commercially available, is very expensive and difficult to prepare; moreover in this process impurities are very difficult to remove, particularly on an industrial scale, in particular because of the Boc protective group. For this reason,US 2009/0192314 discloses a novel process for the preparation of Linagliptin (A) which makes use of a 3-(R)-aminopiperidine protected as a phthalimide of formula (E).

US ‘955 is schematically represented in scheme

U.S. Patent No. 7,820,815 (“US ‘815) discloses a process for preparation of Linagliptin wherein it is prepared by deprotecting 1 -[(4-methyl-quinazolin-2-yl)methyl]-3- methyl-7-(2-butyn-1 -yl)-8-(3-(R)-phthalimidopiperidin-1 -yl)-xanthine of formula Ilia in presence of ethanolamine. The 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2- butyn-1 -yl)-8-(3-(R)phthalimidopiperidin-1 -yl)-xanthine is prepared by condensing 1 -[(4- l methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromo xanthine of formula III with (R)-3-phthalimidopiperidine of formula I la. The process disclosed in US ‘815 is schematically represented in scheme-ll.

Scherre
PCT Publications WO 2004/018468 and WO 2006/048427 describe synthesis of Linagliptin. Crystalline forms of Linagliptin, Forms A, B, C, D, and E are described in the PCT Publication No. WO 2007/128721. According to WO 2007/128721, Linagliptin prepared according to Publication No.
WO 2004/018468 is present in ambient temperature as a mixture of two enantiotropic polymorphs. The temperature at which the two polymorphs transform into one another is 25±15° C. The pure high temperature form (polymorph A), can be obtained by heating the mixture to temperatures>40° C. The low temperature form (polymorph B) is obtained by cooling to temperatures<10° C.”.
According to WO 2007/128721, the transition point between forms A and B is at room temperature, such that they exist as a polymorphic mixture. In addition, WO 2007/128721 teaches that form D “is obtained if polymorph C is heated to a temperature of 30-100° C. or dried at this temperature”. Since the procedure to obtain form C according to this application includes drying at 70° C., the dried form C is expected to be obtained in admixture with form D.
WO 2007/128721 teaches that Form E is obtained only at high temperatures (after melting of form D at 150±3° C.), and therefore is not relevant industrially.







Example 1: Preparation of a compound of formula (II) with X=OEt
Example 2: Preparation of a compound of formula (II) with X=OH
Example 3: Preparation of a compound of formula (IV) with R = OCH(CH3)2
Example 4: Preparation of Linagliptin
Example 5: Preparation of a compound of formula (IV) with R = S(CH2)11CH3
Example 6: Preparation of Linagliptin
Example 7: Preparation of a compound of formula (IV) with R=C7H5N2S (2-mercaptobenzoimidazole)
Example 8: Preparation of Linagliptin
http://pubs.rsc.org/en/content/articlelanding/2015/ob/c5ob01111f#!divAbstract
By employing a rhodium–Duanphos complex as the catalyst, β-alkyl (Z)-N-acetyldehydroamino esters were smoothly hydrogenated in a highly efficient and enantioselective way. Excellent enantioselectivities together with excellent yields were achieved for a series of substrates. An efficient approach for the synthesis of the intermediate of the orally administered anti-diabetic drugs Alogliptin and Linagliptin in the DPP-4 inhibitor class was also developed.


Linagliptin is an inhibitor of DPP-4, an enzyme that degrades the incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). Both GLP-1 and GIP increase insulin biosynthesis and secretion from pancreatic beta cells in the presence of normal and elevated blood glucose levels. GLP-1 also reduces glucagon secretion from pancreatic alpha cells, resulting in a reduction in hepatic glucose output. Thus, linagliptin stimulates the release of insulin in a glucose-dependent manner and decreases the levels of glucagon in the circulation.


http://www.google.com/patents/WO2013098775A1?cl=en
In one aspect, the application provides a process for preparation of Linagliptin comprising reacting (R)-piperidine-3-amine of formula II or an acid addition salt thereof with 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine of formula III in the presence of a suitable base in an inert organic solvent.

In another aspect, the application provides Linagliptin or a pharmaceutically acceptable salt thereof, having less than about 0.15 area % of potential process related impurities viz., regio-impurity of the formula la, bromo-impurity of the formula lb and S- isomer as measured by HPLC.

L nag pt n S- somer
Example 1 : Preparation of Linagliptin
a) Preparation of 3-methyl-7-(2-butyn-l-yl)-8-bromo-xanthine (compound of formula IV)
3-Methyl-8-bromo-xanthine (30 gm) and N,N-dimethylformamide (170 ml_) were charged into a 1000 ml_ round bottomed flask equipped with a mechanical stirrer. Diisopropylethylamine (DIPEA, 1 5.9 gm) and 1 -bromo-2-butyne (16.2 gm) were added at 30°C. The reaction mixture was heated to 85 °C and maintained the temperature for 4 hours. The reaction mixture was cooled to 30°C and pre cooled water (300 ml_) was added. The solid formed was collected by filtration and washed with pre cooled water (150 ml_) and diethyl ether (30 ml_). The solid was dried in oven under vacuum at 50°C to get 30.9 gm of the title compound.
(b) Preparation of 1 -[(4-methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8- bromoxanthine (compound of formula III) 3-Methyl-7-(2-butyn-l-yl)-8-bromo-xanthine (10 gm) and Ν,Ν-dimethylacetamide (150 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (9.3 gm) and 2-(chloromethyl)-4- methylquinazoline (6.8 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 90 °C and maintained the temperature for 8 hours. The reaction mixture was cooled to 30°C and water (450 mL) was added and the mixture was stirred for 1 hour at 30°C. The solid formed was collected by filtration and washed with water (150 mL). The wet cake was charged into 500 mL round bottomed flask and toluene (220 mL) was added and the mixture was heated to reflux temperature and maintained for 1 hour. The mixture was cooled to 10°C and maintained for 2 hours. The solid was collected by filtration and washed with toluene (50 mL). The solid was dried in oven under vacuum at 80°C to get 10.8 gm of the title compound. Purity by HPLC: 99.59%
(c) Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (5 gm) and Ν,Ν-dimethylformamide (DMF, 50 mL) were charged into a 500 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (4.57 gm) and (R)-piperidine-3-amine dihydrochloride (2.86 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 80 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to room temperature and DMF was evaporated under vacuum, then dichloromethane (DCM, 50 mL) was added, and stirred for 15 minutes. The reaction mixture was filtered to separate out the non- dissolved material and the non-dissolved material was washed with 15 mL of dichloromethane. The dichloromethane was evaporated under vacuum to give 4 gm of crude Linagliptin.
Example 2: One pot process for preparation of Linagliptin
3-Methyl-8-bromo-xanthine (5 gm) and Ν,Ν-dimethylformamide (DMF, 28.5 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Diisopropylethylamine (DIPEA, 2.6 gm) and 1 -bromo-2-butyne (2.7 gm) were added at 30 °C. The reaction mixture was heated to 85 °C and maintained at this temperature for 4 hours. The reaction mixture is cooled to 30°C and Ν,Ν-dimethylformamide (DMF, 100 ml_) was added. Potassium carbonate (4.4 gm) and 2-(chloromethyl)-4- methylquinazoline (4.2 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 85 °C and maintained at this temperature for 4 hours. The reaction mixture was cooled to 30°C and Ν,Ν-dimethylformamide (DMF, 90 ml_) was added. Potassium carbonate (8.3 gm) and (R)-piperidine-3-amine dihydrochloride (5.2 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 80 °C and maintained at this temperature for 8 hours. The reaction mixture was cooled to 30 °C and DMF was evaporated under vacuum. Dichloromethane (DCM, 30 ml_) was added and stirred for 15 minutes. The reaction mixture was filtered to separate out the undissolved material and the undissolved material was washed with dichloromethane (30 ml_). The dichloromethane was evaporated under vacuum and 10% acetic acid (100 ml_) was added. The resulted solution was stirred for 30 minutes and washed with dichloromethane (25 ml_x3). The pH of the aqueous layer was adjusted to 8.5 with 10% aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (25 ml_x2) and the dichloromethane was evaporated under vacuum to get 1 .2 gm of Linagliptin.
Example 3: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 ml_) were charged into a 1000 ml_ round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine dihydrochloride (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95°C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 ml_). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at room temperature. The aqueous layer was separated and washed with 60 ml_ of dichloromethane. The aqueous layer was charged into another flask and 200 ml_ of dichloromethane and 100 ml_ of aqueous sodium hydroxide solution was added drop-wise at 30 °C. The mixture was stirred for one hour at 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45°C. Isopropyl alcohol (100 mL) was added to the residue and stirred for 3 hours at room temperature. Filtered the compound and washed with isopropyl alcohol (20 mL) and dried the compound at below 60 °C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 99.0%
Example 4: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at room temperature. The reaction mixture was heated to 95 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to room temperature and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at room temperature. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at room temperature. The mixture was stirred for one hour at room temperature and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Hexane (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with Hexane (40 mL) and dried the compound at below 60°C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 98.92%
Example 5: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95°C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at 30 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at 30°C. The mixture was stirred for one hour at 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Toluene (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with Toluene (40 mL) and dried the compound at below 60 °C under vacuum to give 16.8 gm of Linagliptin. Purity: 98.91 %, PXRD pattern: Fig. 2.
Example 6: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine (1 1 .5 gm) were added to the reaction mixture at 30°C. The reaction mixture was heated to 95 °C and maintained at that temperature for 8 hours. The reaction mixture was cooled to 30°C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 10% aqueous acetic acid solution and stirred for one hour at 30 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) was added drop-wise at room temperature (pH is > 10). The mixture was stirred for one hour 30 °C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Ethyl acetate (100 mL) was added to the residue and stirred for 3 hours at 30 °C. Filtered the compound and washed with ethyl acetate (40 mL) and dried the compound at below 60 °C under vacuum to give 17.6 gm of Linagliptin. PXRD pattern: Fig. 2, Purity: 98.72%
Example 7: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (4 gm) and methyl isobutyl ketone (MIBK 100 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (3.7 gm) and (R)-piperidine-3-amine dibenzoyl-D-tartrate (6.1 gm) were added to the reaction mixture at 26°C. The reaction mixture was heated to 100°C and maintained at that temperature for 6 hours. The reaction mixture was cooled to 30 °C and filtered, and the salt was washed with MIBK (8 mL). The filtrate was charged into another flask and added slowly 10% aqueous acetic acid solution (40 mL) and stirred for one hour at 26°C. The aqueous layer was separated and washed with 12 mL of dichloromethane. The aqueous layer was charged into another flask and 40 mL of dichloromethane and 20 mL of 16 % aqueous sodium hydroxide solution was added drop-wise at 26°C. The mixture was stirred for one hour at 26 °C and the organic layer was separated and the aqueous layer was extracted with 20 ml of dichloromethane. Combined the organic layers and evaporated under vacuum at below 45 °C. Isopropyl alcohol (8 mL) was added to the residue and evaporated under vacuum at below 45 °C. Isopropyl alcohol (16 mL) was added to the residue and stirred for 2 hours at 2Q°C. Filtered the compound and washed with isopropyl alcohol (4 mL) and dried the compound at 60 °C under vacuum to give 3.2 gm of Linagliptin. PXRD pattern: Fig. 2, Chemical Purity: 98.68%, Chiral Purity: 99.82%, S-isomer content: 0.12%, Regio impurity: 0.57%, Bromo impurity: 0.28%
Example 8: Preparation of Linagliptin
1 -[(4-Methyl-quinazolin-2-yl)methyl]-3-methyl-7-(2-butyn-1 -yl)-8-bromoxanthine (20 gm) and methyl isobutyl ketone (MIBK 200 mL) were charged into a 1000 mL round bottomed flask equipped with a mechanical stirrer. Potassium carbonate (18.3 gm) and (R)-piperidine-3-amine dihydrochloride (8.4 gm) were added to the reaction mixture at 26°C. The reaction mixture was heated to ‘\ 00 °C and maintained at that temperature for 4 hours. The reaction mixture was cooled to 30 °C and filtered and washed with MIBK (40 mL). The filtrate was charged into another flask and added 200 mL of 10% aqueous acetic acid solution and stirred for 30 minutes at 28 °C. The aqueous layer was separated and washed with 60 mL of dichloromethane. The aqueous layer was charged into another flask and 200 mL of dichloromethane and 100 mL of aqueous sodium hydroxide solution (16 gm of sodium hydroxide in 100 mL of water) were added drop- wise at 28°C (pH is > 10). The mixture was stirred for one hour at 28°C and the organic layer was separated and the aqueous layer was extracted with 100 ml of dichloromethane. Combined the organic layers and divided into 5 equal parts.
Part 1 : The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (16 mL) was added to the residue stirred for 30 minutes at 28 °C and 48 mL of MTBE was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 8 mL of MTBE and dried the compound at 65 °C under vacuum to give 3.0 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.46%, Regio impurity: 0.37%, Bromo impurity: 0.03%
Part 2: The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (24 mL) was added to the residue stirred for 30 minutes at 28 °C and the resulted solution was cooled to 5°C and stirred for 1 hour. Filtered the compound and washed with 5 mL of chilled methanol and dried the compound at 65°C under vacuum to give 3.0 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.41 %, Regio impurity: 0.38%, Bromo impurity: 0.03%
Part 3: The organic layer was distilled off completely under vacuum at 45 °C. Methanol (8 mL) was added to the residue and distilled off completely under vacuum at 45°C. Methanol (20 mL) was added to the residue stirred for 30 minutes at 28 °C and 20 mL of MTBE was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 8 mL of MTBE and dried the compound at 65 °C under vacuum to give 2.8 gm of Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.47%, Regio impurity: 0.36%, Bromo impurity: 0.03%.
Part 4: The organic layer was distilled off completely under vacuum at 45 °C. Isopropyl alcohol (8 mL) was added to the residue and distilled off completely under vacuum at 45 °C. Methanol (16 mL) was added to the residue stirred for 30 minutes at 28 °C and 16 mL of isopropyl alcohol was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 4 mL of isopropyl alcohol and dried the compound at 65 °C under vacuum to give 2.9 gm of Linagliptin. PXRD pattern: Fig. 1 .
Chemical Purity: 99.44%, Regio impurity: 0.38%, Bromo impurity: 0.02%.
Part 5: The organic layer was distilled off completely under vacuum at 45 °C. Ethyl acetate (8 mL) was added to the residue and distilled off completely under vacuum at 45 °C. Ethyl acetate (16 mL) was added to the residue stirred for 30 minutes at 28°C and 16 mL of methanol was added over a period of 30 minutes to the resulted solution at 27°C and stirred for 1 hour. Filtered the compound and washed with 4 mL of ethyl acetate and dried the compound at 65 °C under vacuum to give 0.7 gm of Linagliptin. PXRD pattern: Fig. 2.
Chemical Purity: 99.57%, Regio impurity: 0.29%, Bromo impurity: 0.02%
Example 9: Purification of Linagliptin
Linagliptin (3.5 gm) was dissolved in 10% aqueous acetic acid and stirred for 15 minutes. Dichloromethane (50 mL) was added to the solution and stirred for 30 minutes. The aqueous layer was separated and the pH of this layer was adjusted to 8.5 using 10% aqueous sodium bicarbonate solution. The aqueous layer was extracted with dichloromethane (50 mLx2). The dichloromethane was evaporated under vacuum to give 3 gm of Linagliptin.
Example 10: Purification of Linagliptin
Linagliptin (31 gm) and methanol (124 mL) were charged into 500 mL round bottomed flask and the solution was heated to 40 °C and stirred for 60 minutes. Charcoal (3 gm) was added to the clear solution and stirred for 30 minutes. The solution was filtered through Hy-flow and the Hy-flow bed was washed with methanol (30 mL). Filtrate was charged into 1000 mL round bottomed flask and methyl tertiary butyl ether was added drop-wise to the solution and stirred for 2 hours at 30 °C. The precipitate so formed was filtered and the wet cake was washed with methyl tertiary butyl ether (30 mL) to get 25.6 gm of pure Linagliptin. PXRD pattern: Fig. 3. Chemical Purity: 99.57%, Chiral purity: 99.73%, Regio impurity: 0.10%, Bromo impurity: 0.1 %
Example 1 1 : Purification of Linagliptin
Linagliptin (4 gm) and methanol (24 mL) were charged into 100 mL round bottomed flask and the solution is heated to 50 °C and stirred for 60 minutes. Methyl tertiary butyl ether (MTBE, 80mL) was charged into 500 mL round bottomed flask and the methanol solution containing linagliptin was added drop-wise at 27 °C and stirred for 2 hours at same temperature. The precipitate formed was filtered and the wet cake was washed with methyl tertiary butyl ether (8 mL) to get 2.6 gm of pure Linagliptin. PXRD pattern: Fig. 2, Bromo impurity content: 0.04%.
Example 12: Purification of Linagliptin
a) Preparation of linagliptin-(D)-tartrate
Linagliptin (10 gm) and methanol (300 mL) were charged into 1000 mL round bottomed flask and (D)-tartaric acid solution (3.3 gm of (D)-tartaric acid in 100 mL of methanol) was added at 26 °C. The solution was heated to 65 °C and stirred for 60 minutes. The solution was cooled to 28 °C and stirred for 2 hours at 27 °C. The precipitate formed was filtered and the wet cake was washed with methanol (20 mL) and the solid was dried under vacuum at 55°C to get 8.3 gm of Linagliptin-(D)-tartrate. PXRD pattern: Fig. 4. Chemical Purity: 99.72%, Chiral purity: 99.89%, Regio impurity: 0.08%, Bromo impurity: 0.05%, S-isomer: 0.1 1%.
b) Isolation of pure Linagliptin
Linagliptin-(D)-tartrate (8 gm) and water (100 mL) were charged into 1000 mL round bottomed flask and stirred for 30 minutes at 26 °C. Dichloromethane (80 mL) was added to the solution and cooled to 5°C. Aqueous sodium hydroxide solution (0.6 gm of NaOH is added to 20 mL of water) was added to the mixture at 5°C and maintained for 1 hour. Layers were separated and aqueous layer was extracted with dichloromethane (20 mL). Combined both organic layers and dried over sodium sulphate and distilled off the organic layer under vacuum at 45 °C. Hexane (20 mL) was added to the crude and stirred for 1 hour at 26°C. The precipitate was filtered and washed with 4 mL of hexane and dried the compound at 60°C under vacuum to give 6 gm of pure Linagliptin. PXRD pattern: Fig. 2, Chemical Purity: 99.67%, Chiral purity: 99.85%, (S)-isomer content: 0.1 5%, Regio impurity: 0.09%, Bromo impurity: 0.07%.
PATENT
http://www.google.com/patents/US20130123282
Example 35Preparation of 8-bromo-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione
Example 36Preparation of (R)-8-(3-Amino-piperidin-1-yl)-7-(but-2-ynyl)-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (Form-XXII)
Results in 2010 from a Phase III clinical trial of linagliptin showed that the drug can effectively reduce blood sugar.[2]

Scheme:
. J. Med Chem 2009, 52, 6433..
J. Med Chem 2007, 50, 6450…

| CN101735218A * | Dec 17, 2009 | Jun 16, 2010 | 廖国超 | Piperidine carbamic acid ester derivative and application thereof |
| US7407955 | Aug 12, 2003 | Aug 5, 2008 | Boehringer Ingelheim Pharma Gmbh & Co., Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US20040097510 * | Aug 12, 2003 | May 20, 2004 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | 8-[3-amino-piperidin-1-yl]-xanthines, the preparation thereof and their use as pharmaceutical compositions |
| US20090192314 | Mar 30, 2009 | Jul 30, 2009 | Boehringer Ingelheim International Gmbh | Process for the preparation of chiral 8-(3-aminopiperidin-1yl)-xanthines |
| WO2005085246A1 * | Feb 12, 2005 | Sep 15, 2005 | Boehringer Ingelheim Int | 8-[3-amino-piperidin-1-yl]-xanthine, the production thereof and the use in the form of a dpp inhibitor |
| Reference | ||
|---|---|---|
| 1 | CHIRALITY vol. 7, 1995, pages 90 – 95 | |
| 2 | * | JEAN L ET AL: “A convenient route to 1-benzyl 3-aminopyrrolidine and 3-aminopiperidine“, TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 42, no. 33, 13 August 2001 (2001-08-13), pages 5645-5649, XP004295831, ISSN: 0040-4039, DOI: DOI:10.1016/S0040-4039(01)00985-6 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014033746A2 * | Aug 6, 2013 | Mar 6, 2014 | Glenmark Pharmaceuticals Limited; Glenmark Generics Limited | Process for the preparation of dipeptidylpeptidase inhibitors |
| WO2014059938A1 * | Oct 17, 2013 | Apr 24, 2014 | 2Y-Chem, Ltd. | Method for preparing important intermediate of linagliptin |
| WO2014097314A1 * | Dec 16, 2013 | Jun 26, 2014 | Mylan Laboratories Ltd | An improved process for the preparation of linagliptin |
| WO2010072776A1 * | Dec 22, 2009 | Jul 1, 2010 | Boehringer Ingelheim International Gmbh | Salt forms of organic compound |
| CN101784270A * | Aug 15, 2008 | Jul 21, 2010 | 贝林格尔.英格海姆国际有限公司 | Pharmaceutical composition comprising a glucopyranosyl-substituted benzene derivative |
| CN102127080A * | Nov 2, 2005 | Jul 20, 2011 | 贝林格尔.英格海姆国际有限公司 | Method for producing chiral 8-(3-amino-piperidin-1-yl)-xanthines |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2015067539A1 * | Oct 31, 2014 | May 14, 2015 | Chemelectiva S.R.L. | Process and intermediates for the preparation of linagliptin |
| WO2015087240A1 | Dec 9, 2014 | Jun 18, 2015 | Ranbaxy Laboratories Limited | Process for the preparation of linagliptin and an intermediate thereof |
| WO2015107533A1 * | Sep 1, 2014 | Jul 23, 2015 | Harman Finochem Limited | A process for preparation of 1h-purine-2,6-dione, 8-[(3r)-3-amino-1-piperidinyl]-7 (2-butyn-1-yl)-3,7-dihydro-3-methyl-1-[(4-methyl-2quinazolinyl) methyl] and its pharmaceutically acceptable salts |
| Eckhardt M, et al. 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydropurine-2,6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J Med Chem. 2007; 50(26):6450-3. Pubmed ID: 18052023 | |
| 2. | Thomas L, et al. (R)-8-(3-amino-piperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3,7-dihydro-purine-2,6-dione (BI 1356), a novel xanthine-based dipeptidyl peptidase 4 inhibitor, has a superior potency and longer duration of action compared with other dipeptidyl peptidase-4 inhibitors. J Pharmacol Exp Ther. 2008; 325(1):175-82. Pubmed ID: 18223196 |
//////////BI-1356, BI1356, Linagliptin, Tradjenta, Trajenta, DPP-IV, DPP-4 inhibitor
VISIT
Take control.
Last longer.

Onion is a commonly known plant. The part used is the bulb, which grows underground. It is an essential part of our kitchen herbs and spices. Onion is used in lots of recipes all over the world particularly in South Asia. It is both herb and vegetable used in all seasons. It has been part of many herbal preparations particularly sexual medicines since ages due to its medical properties. Apart from the chemistry of onion, it has dry-warm characteristics, which are used to treat various ailments. It is a boon for those having wet-cold constitutions. Many diseases associated with wet-cold group can be treated with onion. In homeopathy it comes under the name Allium Cepa and is used for a multitude of symptoms. Following are some of the common uses: –
– See more at:
http://www.homeopathy.com.pk/kitchen-herbs/onion-health-benefits-medical-uses.php
webmd info
Onion is a plant. The bulb (rounded underground part) of the onion is used to make medicine.
Onion is used for treating digestion problems including loss of appetite, upsetstomach, and gallbladder disorders; for treating heart and blood vessel problems including chest pain (angina) and high blood pressure; and for preventing “hardening of the arteries” (atherosclerosis). It is also used for treating sore mouth and throat,whooping cough, bronchitis, asthma, dehydration, intestinal gas, parasitic worms, and diabetes. Some people use it as a diuretic to increase urine output.
Onion is applied directly to the skin for insect bites, wounds, light burns, boils, warts, and bruises.
In foods, onion is used in many recipes.
In manufacturing, the oil is used to flavor foods.
Onion might help reduce cholesterol levels, a risk factor for hardening of the arteries. There is some evidence that onion might also reduce lung tightness in people with asthma.
MEDICINAL QUALITIES OF ONION
In investigating the use of onion in medicinal terms, the onion is found to be a remedy for conditions with symptoms like those which are caused by exposure to onions, such as watering eyes, and a burning and running nose. When looking at the symptoms of cold, it is ironic that we would treat this ailment with an almost like-with-like therapy. Alliums are antibacterial and anti-fungal, so they can help ward off colds and treat colds with sinus congestion that shifts from side to side in the head. Onion will relieve coughs that cause a ripping or tearing pain in the throat or a cough that is merely an irritating dry tickle. The watery and inflamed eyes due to sinus con-gestion and hay fever will be greatly relieved with onion.
The onions ability to relieve congestions especially in the lungs and bronchial tract, is hard to believe until you have actually witnessed the results. The drawing of infection, congestion and colds out of the ear is also remarkable.
The onion will relieve stomach upset and other gastrointestinal disorders and it will also strengthen the appetite.
Onions help prevent thrombosis and reduce hypertension, according to the American Heart Association. 8 The natural constituents of yellow or white onions can “…raise HDL cholesterol by 30% over time”, according to Dr. Victor Gurewich of Tufts University.9
The onion is being used for compresses to be applied to the skin for acne, arthritis, and congestion, and used internally for worms. The onion is also known for its diuretic properties.
There have been cases in which the onion has been proven to be so effective as an antiviral that a cut piece of onion placed in a closed off room will prevent the person in the room to be safe from viruses.
Onion will relieve headache centered behind the forehead; earache in children and adults; stuffed up nose with discharge that makes nostrils and upper lip sore or stuffed up nose with discharge from the alternate nostril; toothache, especially in the molar area or the shifting from side to side or from one tooth to another; hoarseness and the early stages of laryngitis; abdominal colic in babies.
Keeping cooler rooms in the home, and getting plenty of fresh air may prevent the symptoms of a stuffy nose and hoarseness
Never mind the tears they bring on—onions are an ace ally in your fight against disease. A prized member of the lily family, they lavish you with health benefits while adding oodles of taste to your food.
A quick glimpse at their incredible health benefits:
My favorite way to enjoy onions is to slice them really thin, squeeze some lemon juice on top and add a little salt. Sprinkling a few freshly washed cilantro leaves adds fragrance and flavor to this simple, quick salad, without which no dinner of mine is complete.
…
…

Cdiffense trial to evaluate vaccine against a leading cause of life-threatening, healthcare-associated infections worldwide
SWIFTWATER, Pa., Aug. 5, 2013 /PRNewswire/ — Sanofi Pasteur, the vaccines division of Sanofi (EURONEXT: SAN and NYSE: SNY), announced today the initiation of its Phase III clinical program called Cdiffense to evaluate the safety, immunogenicity and efficacy of an investigational vaccine for the prevention of primary symptomatic Clostridium difficile infection (CDI). Clostridium difficile (C. diff) is a potentially life-threatening, spore-forming bacterium that causes intestinal disease. The risk of C. diff increases with age, antibiotic treatment and time spent in hospitals or nursing homes, where multiple cases can lead to outbreaks. The investigational vaccine is designed to help protect at-risk individuals from C. diff, which is emerging as a leading cause of life-threatening, healthcare-associated infections (HAIs)worldwide, read all at…………….
http://www.pharmalive.com/sanofi-starts-phase-iii-trial-for-clostridium-difficile-vaccine
..
…

Bristol, UK (Scicasts) – New research into the fight against Dengue, an insect-borne tropical disease that infects up to 390 million people worldwide annually, may influence the development of anti-viral therapies that are effective against all four types of the virus.
The findings, led by researchers at the University of Bristol and published in the Journal of Biological Chemistry August 2 show that there may be significant differences in specific properties of the viral proteins for the four dengue virus types.
read all at scicasts
..
…………
…
….
…
 |
|
| Kokum fruits, seeds, pulp and rinds.jpg |
The rind of the Vrikshamla (Garcinia/Garcinia indica) fruit contains an active component called hydroxycitric acid (HCA), which supports normal fat and carbohydrate metabolism, a healthy appetite level and optimum body weight. The Department of Physiology researchers at the Georgetown University Medical Center in Washington DC, called Vrikshamla a safe, natural supplement for normal weight support. In an eight-week study of 60 volunteers, they reported Vrikshamla’s effectiveness in supporting normal weight, cholesterol, triglyceride and serum lipid levels.

hydroxycitric acid (HCA)

Vrikshamla grows in India the evergreen forests of the Western Ghats, the Southern Konkan region and in Goa. It is also cultivated in the Southern districts of Maharashtra and on the lower slopes of the Nilgiris mountains.
The active constituent in this herb, HCA, helps weight loss by promoting normal appetite levels and by reducing the body’s ability to form adipose (fatty) tissue. HCA also reduces blood lipid levels and naturally lowers cholesterol. Other useful phytochemicals in Vrikshamla are garcinol, isogarcinol, xanthochymol and isoxanthochymol.
Weight management: The herb known to slow down the body’s ability to store fat, potentially enabling more fat from foods to pass through the body without being stored. Additionally, HCA blocks the production and storage of fat and cholesterol when calorie consumption exceeds healthy levels. The herb is also known to reduce blood lipid levels and to naturally lower cholesterol.
read at
http://www.himalayahealthcare.com/products/pharmaceuticals/vrikshamla.htm

Garcinia indica, a plant in the mangosteen family (Clusiaceae), commonly known askokum, punar puli (tulu language), is a fruit-bearing tree that has culinary, pharmaceutical, and industrial uses.
The genus Garcinia, belonging to the family Clusiaceae, includes about 200 species found in the Old World tropics, mostly in Asia and Africa. Garcinia indica is indigenous to theWestern Ghats region of India located along the western coast of the country. Of the 35 species found in India, 17 are endemic. Of these, seven are endemic to the Western Ghats, six in the Andaman and Nicobar Islands and four in the northeastern region of India.
Garcinia indica is found in forest lands, riversides and wastelands. These plants preferevergreen forests, but sometimes they also thrive in areas with relatively low rainfall. It is also cultivated on a small scale. It does not require irrigation, spraying of pesticides or fertilizers.
Garcinia indica is known by various names across India, including aamsol, aamsul, bindin, biran, bhirand,bhrinda, brinda, bin’na, kokum (alternate spellings kokam and cocum), katambi, looikya, sour apple, panarpuli, ratamba, thekera (in Assam) and many others.

The dried skin of kokum fruits
The outer cover of fruit is dried in the sun to get aamsul or kokam. It is used as a slightly sour spice in recipes from Maharashtra. Kokum yields a peculiar flavour and blackish red colour. It is a preferred substitute for tamarind in curries and other dishes from the Konkanregion. It is also used in cuisine from Gujarat, where it is frequently used to add flavor and tartness to dal (lentil soup) for flavor balance, and parts of South India.

The vessel on the left contains syrup which is obtained from the vessel containing kokum rinds, on the right. The syrup is used to make kokum sherbet
Kokum squash or kokum concentrate is used in preparing a drink (sherbet) which is bright red in colour. Kokum sherbet improves digestion and cools the body during summers[citation needed].
Further, the extract/ concentrate of this fruit is called aagal in Konkani and Marathi. It is to added during the preparation of solkadhi, along with coconut milk.
The seed of Garcinia indica contains 23–26% oil, which remains solid at room temperature. It is used in the preparation of confectionery, medicines and cosmetics.
Recently, industries have started extracting hydroxycitric acid (HCA) from the rind of the fruit.[citation needed]
FDA Grants Priority Review To New Drug Application For MNK-795 Submitted By Depomed Licensee Mallinckrodt
Controlled Substance Analgesic Combination Product Uses Depomed’s Proprietary Acuform® Technology
NEWARK, Calif., July 29, 2013 /PRNewswire/ — Depomed, Inc. (NASDAQ:DEPO) announced today that the U. S. Food and Drug Administration (FDA) has accepted for filing a New Drug Application (NDA) from Mallinckrodt (NYSE: MNK) for MNK-795. MNK-795 is a controlled-release oral formulation of oxycodone and acetaminophen that has been studied for the management of moderate to severe acute pain where the use of an opioid analgesic is appropriate. MNK-795 is formulated with Depomed’s Acuform® drug delivery technology.
http://www.pharmalive.com/fda-grants-priority-review-to-new-drug-application-for-mnk-795

albiglutide,
Friday 2 August 2013, London UK
GlaxoSmithKline plc (LSE:GSK) today announced that the US Prescription Drug User Fee Act (PDUFA) goal date for albiglutide, an investigational once-weekly treatment for adult patients with type 2 diabetes, has been extended by three months to 15 April 2014 to provide time for a full review of information submitted by GSK in response to the Food and Drug Administration’s requests.
http://www.pharmalive.com/fda-delays-approval-decision-for-gsk-s-albiglutide
Albiglutide is a glucagon-like peptide-1 agonist (GLP-1 agonist) drug under investigation by GlaxoSmithKline for treatment oftype 2 diabetes. It is a dipeptidyl peptidase-4-resistant glucagon-like peptide-1 dimer fused to human albumin.
Albiglutide has a half-life of four to seven days, which is considerably longer than the other two GLP-1 analogs approved for market use, exenatide (Byetta) and liraglutide (Victoza). GLP-1 drugs are currently only available for subcutaneous administration on a daily basis, so a GLP-1 drug with a longer half-life is desirable. Such a drug would only need to be injected biweekly or weekly instead of daily, reducing the discomfort and inconvenience of GLP-1 administration considerably.
It has not yet been determined whether albiglutide is as effective an antidiabetic agent as GLP-1 drugs currently on the market, and final data remain to be published regarding the incidence of adverse effects related to the drug. To evaluate the efficacy and safety of the drug, albiglutide is undergoing eight Phase III clinical trials. Four of these trials should report useful data by end 2010
…………………………….
………..



Donepezil, marketed under the trade name Aricept by its developer Eisai and partnerPfizer, is a centrally acting reversible acetylcholinesterase inhibitor. Its main therapeutic use is in the palliative treatment of Alzheimer’s disease.Common side effects include gastrointestinal upset. It has an oral bioavailability of 100% and easily crosses the blood–brain barrier. Because it has a biological half-life of about 70 hours, it can be taken once a day.

While the drug is currently indicated for mild to moderate Alzheimer’s, evidence from two clinical trials also indicates it may be effective for moderate to severe disease. An example of this is a Karolinska Institute paper published in The Lancet in early 2006, which states donepezil improves cognitive function even in patients with severe AD symptoms. In Oct. 2006 the U.S. Food and Drug Administration also approved Aricept for treatment of severe dementia.

【通用名】 Donepezil hydrochloride, BNAG, E-2020, Eranz, Memorit, Memac, Aricept
【化学名】 (?-1-Benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylmethyl)piperidine hydrochloride; (?-2-(1-Benzylpiperidin-4-ylmethyl)-5,6-dimethoxyindan-1-one hydrochloride
【CAS登记号】 120011-70-3, 123958-79-2 ([2-14C]-labeled), 142057-77-0 (deleted CAS), 120014-06-4 (free base)
【分子式】 C24-H29-N-O3.Cl-H
【分子量】 415.958
【化学活性】 Alzheimer’s Dementia, Treatment of , Analgesic and Anesthetic Drugs, Antimigraine Drugs, Attention Deficit Hyperactivity Disorder (ADHD), Treatment of, Autism, Treatment of, Cognition Disorders, Treatment of, Immunologic Neuromuscular Disorders, Treatment of, Migraine, Prophylactic Treatment of, Multiple Sclerosis, Agents for, Neurologic Drugs, Psychopharmacologic Drugs, Vascular Dementia, Treatment of, Acetylcholinesterase Inhibitors
【开发阶段】 Launched-1997
【研究机构】 Eisai (Originator), National Institute of Mental Health (Not Determined), Bracco (Licensee), Pfizer (Licensee)

Donepezil inhibiting Torpedo californicaacetylcholinesterase. See Proteopedia1eve.

Research leading to the development of donepezil began in 1983 at Eisai, and the first Phase I clinical trial took place in 1989. In 1996, Eisai received approval from the United States Food and Drug Administration (USFDA) for donepezil under the brand Aricept, which it co-marketed with Pfizer. As of 2011, Aricept was the world’s best-selling Alzheimer’s disease treatment. The first generic donepezil became available in November 2010 with the USFDA approval of a formulation prepared by Ranbaxy Labs. In April 2011 a second generic formulation, from Wockhardt, received tentative USFDA marketing approval
| 标题: | Cyclic amine cpd., its use and pharmaceutical compsns. comprising it |
| 作者: | Sugimoto, H.; Tsuchiya, Y.; Higurashi, K.; Karibe, N.; Iimura, Y.; Sasaki, A.; Yamanashi, Y.; Ogura, H.; Araki, S.; Kosasa, T.; Kusota, A.; Kozasa, M.; Yamatsu, K. (Eisai Co., Ltd.) |
| 来源: | AU 8818216; EP 0296560; EP 0673927; EP 0742207; JP 1989079151; JP 1998067739; US 4895841; US 5100901 |
 |
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 标题: | Synthesis of 1-benzyl-4-[(5,6-dimethoxy[2-14C]-1-indanon)-2-yl]methylpiperidine hydrochloride (E-2020-14C) |
| 作者: | Sugimoto, H.; Mishima, M.; Iimura, Y. |
| 来源: | J Label Compd Radiopharm 1989,27(7),835-9 |
|
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 作者: | Casta馿r, J.; Prous, J. |
| 来源: | Drugs Fut 1991,16(1),16 |
|
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 标题: | Synthesis of 1-benzyl-4-[(5,[C-11]6-dimethoxy-1-oxoindan-2-yl)methyl]piperidine: A promising ligand for visualisation of acetylcholine esterase by PET |
| 作者: | Santens, P.; DeReuck, J.; Dierckx, R.A.; Siegers, G.; Vermeirsch, H.; De Vos, F. |
| 来源: | J Label Compd Radiopharm 2000,43(6),595 |
 |
|
| 合成路线图解说明:11C-Labeled donepezil was prepared by methylation of 2-(1-benzylpiperidin-4-ylmethyl)-6-hydroxy-5-methoxyindan-1-one (I) with 11CH3I by means of tetrabutylammonium hydroxide in DMF. | |
……………………………..
………………..
……………………….
……………………
……………………….
Donepezil hydrochloride is a useful memory enhancer introduced by the Japanese pharmaceutical company Eisai. Its preparation was described in patent no. EP 296560. In this patent Donepezil was produced by reaction of 5,6-dimethoxy-1- indanone with 1 -benzyl-4-formylpiperidine in the presence of a strong base, such as lithium diisopropylamide followed by reduction of the double bond. According to this method, Donepezil was obtained (Scheme 1). Patent application WO 99/36405 describes another process for the synthesis of Donepezil. According to this patent, 2-alkoxycarbonyl-1-indanones are reacted with (4-pyridinyl) methyl halide moiety followed by hydrolysis and decarboxylation to give the 2-(4-pyridinyl)methyl-1-indanone derivative. This is followed by reaction with benzyl halides to obtain the corresponding quaternary ammonium salt, and followed by hydrogenation of the pyridine ring to obtain Donepezil (Scheme 2).
Patent application WO 99/36405 describes another process for the synthesis of Donepezil. According to this patent, 2-alkoxycarbonyl-1-indanones are reacted with (4-pyridinyl) methyl halide moiety followed by hydrolysis and decarboxylation to give the 2-(4-pyridinyl)methyl-1-indanone derivative. This is followed by reaction with benzyl halides to obtain the corresponding quaternary ammonium salt, and followed by hydrogenation of the pyridine ring to obtain Donepezil (Scheme 2). Patent application WO 97/22584 describes the preparation of Donepezil by reaction of pyridine-4-carboxyaldehyde with malonic acid to give 3-(pyridin-4-yl)-2- propenoic acid, followed by hydrogenation of the double bond to give 3-(piperidin-4-yl)-2-propionic acid. Reaction of this intermediate with methyl chloroformate afforded 3-[N-(methyloxycarbonyl) piperidin-4-yl]propionic acid. This was followed by reaction with oxalyl chloride to give methyl 4-(2-chlorocarbonylethyl)piperidin-1-carboxylate. Reaction with 1,2-dimethoxybenzene in the presence of aluminum chloride afforded methyl 4-[3-(3,4-dimethoxyphenyl)-3-oxopropyl]piperidin-1 -carboxylate. Reaction with tetramethyldiaminomethane afforded 4-[2-(3,4-dimethoxybenzoyl)allyl] piperidin-1-carboxylate. Reaction with sulfuric acid afforded methyl 4-(5,6-dimethoxy-1-oxoindan-2-yl)methylpiperidin-1- carboxylate. This was followed by treatment with base to give 5,6-dimethoxy-2-(piperidin-4-ylmethyl) indan-1-one, then reaction with benzyl bromide afforded Donepezil (Scheme 3).
Patent application WO 97/22584 describes the preparation of Donepezil by reaction of pyridine-4-carboxyaldehyde with malonic acid to give 3-(pyridin-4-yl)-2- propenoic acid, followed by hydrogenation of the double bond to give 3-(piperidin-4-yl)-2-propionic acid. Reaction of this intermediate with methyl chloroformate afforded 3-[N-(methyloxycarbonyl) piperidin-4-yl]propionic acid. This was followed by reaction with oxalyl chloride to give methyl 4-(2-chlorocarbonylethyl)piperidin-1-carboxylate. Reaction with 1,2-dimethoxybenzene in the presence of aluminum chloride afforded methyl 4-[3-(3,4-dimethoxyphenyl)-3-oxopropyl]piperidin-1 -carboxylate. Reaction with tetramethyldiaminomethane afforded 4-[2-(3,4-dimethoxybenzoyl)allyl] piperidin-1-carboxylate. Reaction with sulfuric acid afforded methyl 4-(5,6-dimethoxy-1-oxoindan-2-yl)methylpiperidin-1- carboxylate. This was followed by treatment with base to give 5,6-dimethoxy-2-(piperidin-4-ylmethyl) indan-1-one, then reaction with benzyl bromide afforded Donepezil (Scheme 3).
 Patent application EP 711756 describes the preparation of Donepezil by reaction of 5,6-dimethoxy-1- indanone with pyridin-4-aldehyde to give 5,6-dimethoxy-2-(pyridin-4-yl)methylene indan-1-one. Reaction with benzyl bromide afforded 1-benzyl-4-(5,6-dimethoxyindan-1-on-2-ylidene)methylpyridinium bromide. Hydrogenation in the presence of platinum oxide afforded Donepezil (Scheme 4).
Patent application EP 711756 describes the preparation of Donepezil by reaction of 5,6-dimethoxy-1- indanone with pyridin-4-aldehyde to give 5,6-dimethoxy-2-(pyridin-4-yl)methylene indan-1-one. Reaction with benzyl bromide afforded 1-benzyl-4-(5,6-dimethoxyindan-1-on-2-ylidene)methylpyridinium bromide. Hydrogenation in the presence of platinum oxide afforded Donepezil (Scheme 4).

United States Patent 6844440

EP 1386607 A1

FDA Grants Priority Review To New Drug Application For MNK-795 Submitted By Depomed Licensee Mallinckrodt
Controlled Substance Analgesic Combination Product Uses Depomed’s Proprietary Acuform® Technology
NEWARK, Calif., July 29, 2013 /PRNewswire/ — Depomed, Inc. (NASDAQ:DEPO) announced today that the U. S. Food and Drug Administration (FDA) has accepted for filing a New Drug Application (NDA) from Mallinckrodt (NYSE: MNK) for MNK-795. MNK-795 is a controlled-release oral formulation of oxycodone and acetaminophen that has been studied for the management of moderate to severe acute pain where the use of an opioid analgesic is appropriate. MNK-795 is formulated with Depomed’s Acuform® drug delivery technology.
http://www.pharmalive.com/fda-grants-priority-review-to-new-drug-application-for-mnk-795
 In my practice this product
In my practice this product

