PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
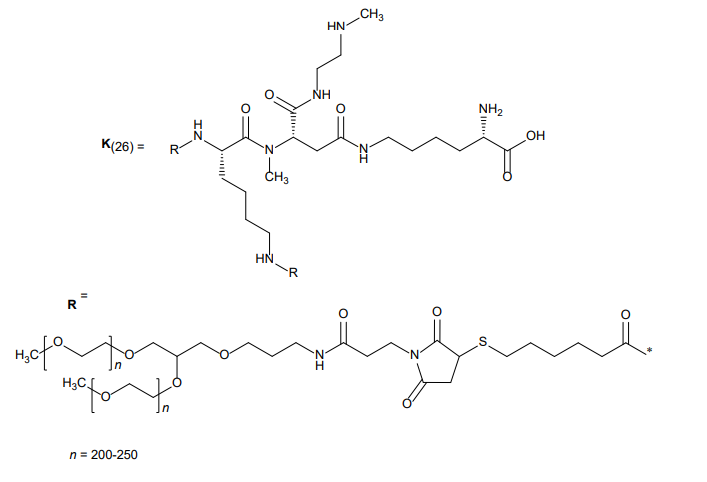
MOLECULAR FORMULA C231H386N64O67S5 + (C2H4O)4n MOLECULAR WEIGHT approx. 45 kDa
The structure of navepegritide (YUVIWEL®) is built using a “prodrug” design. It is not a simple small molecule, but rather a complex conjugate consisting of three distinct components designed to release the active drug slowly over time.
1. The Active Part: C-Type Natriuretic Peptide (CNP)
The core of the molecule is a synthetic 38-amino acid peptide (CNP-38).
Sequence: This peptide mimics the natural human C-type natriuretic peptide, which is essential for bone growth.
Function: Once released, this peptide binds to the natriuretic peptide receptor B (NPR-B) on the surface of chondrocytes (cartilage cells) in the growth plates, stimulating bone formation.
2. The Carrier: Polyethylene Glycol (PEG)
To prevent the body from clearing the small peptide too quickly, it is attached to a large, inert carrier.
Type: It uses a multi-arm, branched 40 kDa Polyethylene Glycol (PEG) molecule.
Purpose: The PEG carrier acts as a shield and a “weight,” making the molecule too large to be filtered out rapidly by the kidneys. This is what allows for once-weekly dosing instead of daily injections.
3. The Linker: TransCon™ Technology
This is the most critical part of the structure. The peptide is attached to the PEG carrier via a cleavable linker.
Mechanism: This linker is designed to break down spontaneously at a predictable rate under physiological conditions (neutral pH and body temperature).
The Result: As the linker slowly breaks, it releases the unmodified, active CNP-38 into the bloodstream. Because the peptide is released in its natural state, it retains its full biological activity.
Summary Table: Structural Components
Component
Description
Role
Peptide
CNP-38 (38 amino acids)
The “payload” that stimulates bone growth.
Linker
pH-sensitive cleavable bond
Controls the slow release of the peptide.
Carrier
40 kDa PEG
Increases the half-life and prevents rapid clearance.
Note: This structure is technically a prodrug because the large PEG-bound version is inactive; only the released CNP-38 peptide performs the therapeutic work.
C-Type natriuretic peptide (CNP), human, (89-126)-fragment (1-38) (CNP-38), conjugated at N6 of Lys26 with four O-methylpoly(ethylene glycol) chains (approx. 10 kDa each) via a cleavable tetra-antennary linker; L-leucyl-L-glutaminyl-L-?-glutamyl-L-histid
Poly(oxy-1,2-ethanediyl), ?-hydro-?-methoxy-, 26,26,26,26-tetraether with L-leucyl-L-glutaminyl-L-?-glutamyl-L-histidyl-L-prolyl-L-asparaginyl-L-alanyl-L-arginyl-L-lysyl-L-tyrosyl-L-lysylglycyl-L-alanyl-L-asparaginyl-L-lysyl-L-lysylglycyl-L-leucyl-L-sery
FDA 2026, APPROVALS 2026, 2/27/2026, Yuviwel, Y3BH8M899D, MN-266, TRANSCON CNP, PA (224-233), Influenza, DA-66438, ACP-015, WHO 11981,
To increase linear growth in pediatric patients 2 years and older with achondroplasia with open epiphyses
Navepegritide is a prodrug consisting of a 38-amino acid C-type natriuretic peptide (CNP) moiety conjugated to a multi-arm polyethylene glycol (PEG) carrier via a cleavable linker. This structure allows for the once-weekly dosing approved by the FDA for children with achondroplasia.
Key Details
Purpose: It is designed to increase linear growth by providing continuous exposure to C-type natriuretic peptide (CNP), a protein that helps regulate bone growth.
Mechanism: As a prodrug, it uses Ascendis Pharma’s TransCon technology to release active CNP slowly into the body over a week, maintaining steady levels and avoiding high peaks.
Clinical Benefits: In the pivotal ApproaCH trial, patients treated with navepegritide showed a significant improvement in annualized growth velocity (AGV) compared to those on a placebo. It also showed potential improvements in body proportionality and lower-limb alignment.
Administration: It is administered via a once-weekly subcutaneous injection, offering a less frequent alternative to daily treatments like vosoritide.
Safety: Most common side effects include injection site reactions (redness, itching, or swelling) and a risk of low blood pressure (hypotension).
25 Feb 2026Vanda Pharmaceuticals has patent protection for an improved method of treatment with milsaperidone in USA
25 Feb 2026Vanda Pharmaceuticals has patents pending for an improved method of treatment with milsaperidone in China, Australia, Israel, Mexico and worldwide
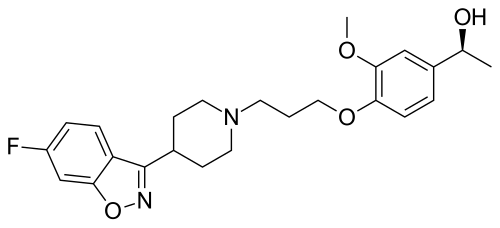
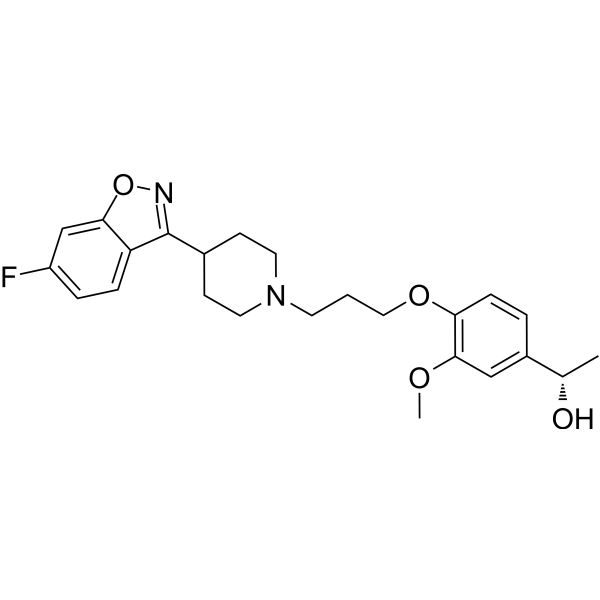
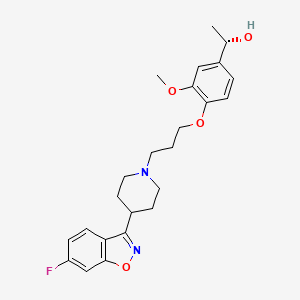
56.36 g of boran complex of (3aR, 7R)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1 ,3,2]oxazaborole (1 equivalent) is dissolved under nitrogen in methylenchloride, and the solution is cooled to 0°C. A 1M solution of 1-(4-{3-[4-(6-fluoro-benzo[d]isoxazol-3-yl)-piperidin-1-yl]-propoxy}-3-methoxy-phenyl)-ethanone (iloperidone; 1 equivalent) in methylenchloride is added via a dropping funnel over 90 minutes while the internal temperature is maintained at 0°C ± 2°C. After the addition is complete, the mixture is stirred at 0°C for 20 hours. The reaction mixture is then poured into precooled methanol (0-5°C) during 1 hour. The solution is warmed to room temperature and stirred until the H2 evolution ceases. The solution is concentrated by distillation and the residue dried in vacuum, treated with methanol and stirred for about 1 hour at 50°C and an additional hour at 0CC. The product is isolated by filtration and dried under reduced pressure for 3 hours at 50°C. The title compound is obtained (white crystals).
[α]D20– 19.3° (c=1 in chloroform) Mp: 138.2 – 138.8°C
The boran complex used as starting material can be obtained as follows:
200 ml of a solution of (3aR, 7R)-1-methyl-3,3-diphenyl-tetrahydro-pyrrolo[1,2-c][1,3,2]oxazaborole (1M in toluene) is stirred at room temperature under nitrogen. 1.2 equivalent borane-dimethylsulfide complex is added with a syringe. The solution is stirred for 2 further hours at room temperature. The borane complex is then crystallised by addition of 4 vol dry hexane and cooling to -12°C for 1.5 hour. The product is isolated by filtration in a sintered glass funnel and dried in vacuum at 40°C. The boran complex is obtained /white crystals).
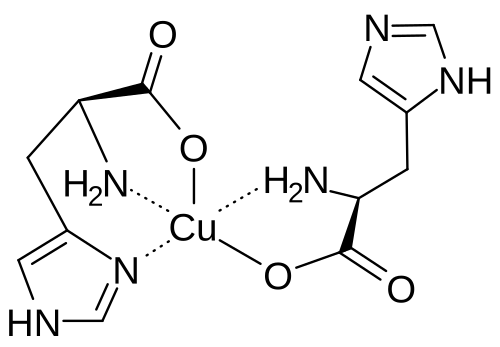
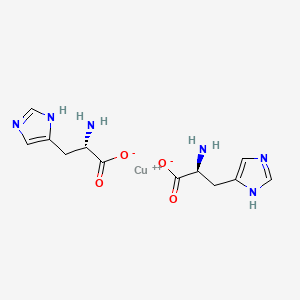
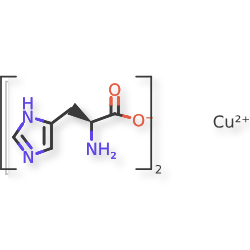
Copper histidinate, sold under the brand name Zycubo, is a medication used for the treatment of Menkes disease.[1] Copper histidinate is a copper replacement therapy given by subcutaneous injection.[1][2]
The most common side effects include infections, respiratory problems, seizures, vomiting, fever, anemia and injection site reactions.[2]
Copper histidinate was approved for medical use in the United States in January 2026.[2]
Menkes disease is a neurodegenerative disorder caused by a genetic defect that impairs a child’s ability to absorb copper.[2] The disease is characterized by seizures, failure to gain weight and grow, developmental delays, and intellectual disability.[2] It leads to abnormalities of the vascular system, bladder, bowel, bones, muscles, and nervous system.[2]
SYN
A275388 — Flores-Pulido AA, Jimenez-Perez VM, Garcia-Chong NR: Sintesis y uso de histidinato de cobre en ninos con enfermedad de Menkes en Mexico. Gac Med Mex. 2019;155(2):191-195. doi: 10.24875/GMM.18004310. [PubMed:31056589]
World Health Organization (2025). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 94”. WHO Drug Information. 39 (3). hdl:10665/383022.
Clinical trial number NCT00811785 for “Molecular Bases of Response to Copper Treatment in Menkes Disease, Related Phenotypes, and Unexplained Copper Deficiency” at ClinicalTrials.gov
Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
RegisteredAtopic dermatitis
27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)
Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
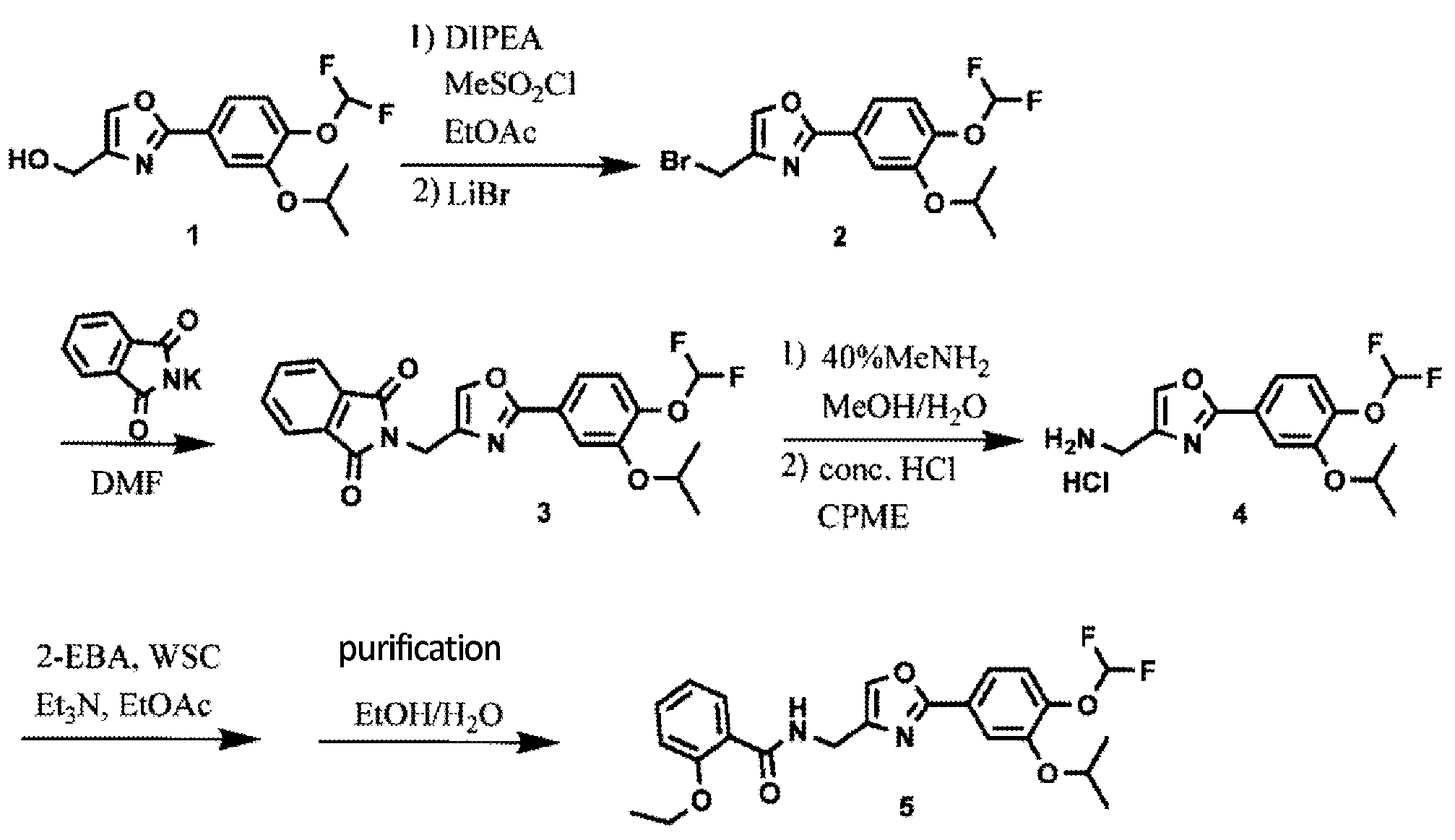
Synthesis of Oxazole Compound (Type A Crystal)
Compound (5) (white powder) was prepared in accordance with the method disclosed in Example 352 of PTL 1 (WO2007/058338).
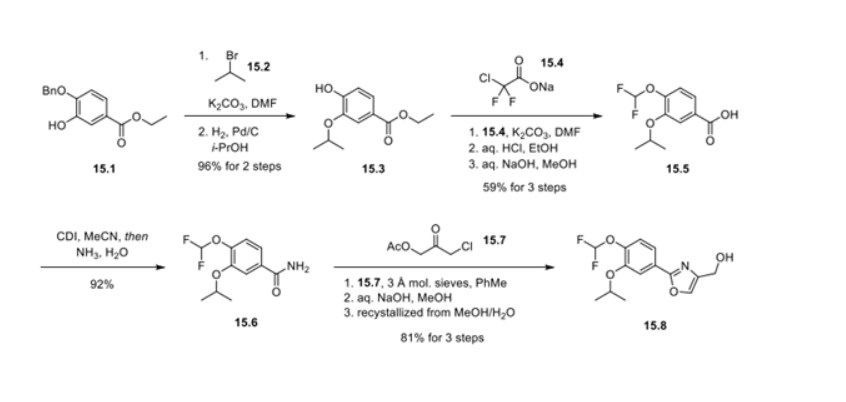
Synthesis of difamilast commenced with the monobenzylated protocatechuic acid ethyl ester 15.1. Phenol 15.1 was first converted into the corresponding isopropyl ether, which was subsequently debenzylated under palladium-catalyzed hydrogenation conditions to generate the phenolic intermediate 15.3. Difluoromethylation of 15.3 was accomplished by introducing sodium chlorodifluoroacetate 15.4 in the presence of potassium carbonate at an elevated temperature. The decarboxylative C− O bond-forming reaction presumably proceeded via a difluorocarbene species. The difluoromethylated product was treated with acid followed by ester hydrolysis under a basic medium to furnish benzoic acid derivative 15.5. Benzoic acid 15.5 was subsequently transformed into benzamide 15.6 via a benzoyl imidazole intermediate. Condensation of benzamide 15.6 with 1-acetoxy-3-chloroacetone 15.7 produced an oxazole derivative, which was subsequently saponified and recrystallized from 50% aqueous MeOH to generate alcohol 15.8.
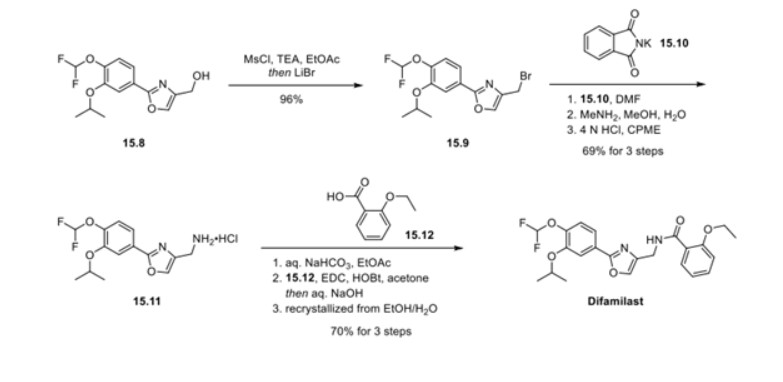
First, an activation−displacement process transformed alcohol 15.8 into bromide 15.9 via a mesylate intermediate. Alkyl bromide 15.9 was then treated with potassium phthalimide to incorporate the nitrogen center via an SN2-type displacement. Methylamine-mediated phthalimide deprotection and subsequent salt formation produced amine 15.11 as a hydrochloride salt in 69% yield over 3 steps. Finally, hydrochloride salt 15.11 was treated with aqueous sodium bicarbonate to generate a free amine, which was subjected to amide bond formation with 2-ethoxybenzoic acid 15.12 to deliver difamilast after recrystallization from aqueous EtOH.
Patent Documents 1 and 2 report an oxazole compound having a specific inhibitory action on phosphodiesterase 4 (PDE4) and a method for producing the same. PDE4 is the predominant PDE in inflammatory cells, inhibition of PDE4 increases intracellular cAMP concentration, and the increase in this concentration downregulates the inflammatory response through regulation of the expression of TNF-α, IL-23, and other inflammatory cytokines. .. Elevated cAMP levels also increase anti-inflammatory cytokines such as IL-10. Therefore, it is considered that the oxazole compound is suitable for use as an anti-inflammatory agent. For example, it may be useful for controlling skin eczema and dermatitis, including atopic dermatitis. Patent Document 3 describes an ointment that stably contains an oxazole compound having a specific inhibitory effect on PDE4 and can be efficiently absorbed into the skin. The contents of Patent Documents 1 to 3 are incorporated in the present specification by reference.
[Synthesis of Oxazole Compound (Type A Crystal)]
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
[Preparation of B-type crystal 2]
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
[0072]
[Chem. 6]
[0073]
Compound (1) 20.00 g (66.8 mmol) and 17.28 g (134 mmol) of diisopropylethylamine were added to 300 mL of ethyl acetate to cool the mixture, and 11.48 g (100 mmol) of methanesulfonyl chloride was introduced into the compound (1) at 10 to 30 ° C. Stir for hours. Subsequently, 17.41 g (200 mmol) of lithium bromide was added, and the mixture was stirred at 20 to 35 ° C. for 1 hour. 100 mL of water was added to the reaction solution to separate the layers, and the organic layer was concentrated under reduced pressure. 300 mL of ethyl acetate was added to the concentrated residue to dissolve it, and the mixture was concentrated again under reduced pressure. 200 mL of N, N-dimethylformamide and 17.33 g (93.6 mmol) of phthalimide potassium were added to the concentrated residue, and the mixture was reacted at 75 to 85 ° C. for 1 hour. 200 mL of water was added to the reaction solution to precipitate crystals, and the precipitated crystals were collected by filtration and dried at 80 ° C. to obtain 27.20 g (yield 95.01%) of compound (3).
[0074]
[Chem. 7]
[0075]
Compound (3) 20.00 g (46.7 mmol), 40 mL of a 40% aqueous methylamine solution, 40 mL of methanol, and 100 mL of water were mixed and reacted under reflux for 30 minutes. 200 mL of cyclopentyl methyl ether (CPME) and 20 mL of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75 ° C. to separate the liquids. A mixed solution of 100 mL of water and 20.00 g of sodium chloride was added to the organic layer, and the temperature was adjusted again to 65 to 75 ° C. to separate the liquids. 5 mL of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. Precipitated crystals were collected by filtration to obtain 27.58 g of wet crystals of compound (4).
[0076]
Wet crystals (46.7 mmol) of compound (4) were mixed with 120 mL of ethyl acetate and 7.1 mL (51.4 mmol) of triethylamine, and the mixture was stirred at 20 to 30 ° C. for 1 hour. To the reaction solution, 10.09 g (60.7 mmol) of 2-ethoxybenzoic acid and 11.63 g (60.7 mmol) of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC) were added, and 20 to 30 were added. The reaction was carried out at ° C. for 1 hour. 60 mL of water and 6 mL of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50 ° C. to separate the solutions. 60 mL of water and 6 mL of a 25%
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
Using the compound obtained in Example 347 and 2-bromopropane, white powdery N-[2-(4-difluoromethoxy-3-isopropoxyphenyl)oxazol-4-ylmethyl]-2-ethoxybenzamide was obtained following the procedure of Example 348.
Production Example 1: Production 1 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
[0146]
[Chem. 11]
[0147]
10.00 g (55.5 mmol) of compound (1a) and 9.20 g (66.6 mmol) of potassium carbonate were added to 40 ml of N,N-dimethylformamide and 6 ml of water, and the mixture was stirred until exotherm subsided. 16.92 g (111 mmol) of sodium chlorodifluoroacetate was added thereto, and the mixture was reacted at 95 to 110°C for 3 hours. 80 ml of butyl acetate and 80 ml of water were added to the reaction solution, and the solution was partitioned. 80 ml of water was added again to the organic layer, followed by partitioning. 3 ml of concentrated hydrochloric acid was added to the organic layer, and the mixture was stirred at 60 to 70°C for 30 minutes. 40 ml of water and 10 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the mixture was partitioned. 5.93 g (61.1 mmol) of sulfamic acid and 10 ml of water were added to the organic layer, and 22.08 g (61.0 mmol) of a 25% sodium chlorite aqueous solution was added dropwise thereto at a temperature of 20°C or below. The mixture was reacted at 20°C or below for 15 minutes, and 10 ml of a 25% sodium hydroxide aqueous solution was added dropwise thereto at a temperature of 20°C or below, followed by pouring in 83.95 g (66.6 mmol) of a 10% sodium sulfite aqueous solution. Additionally, 2 ml of concentrated hydrochloric acid was added and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 40 ml of methanol, 80 ml of water, and 10 ml of a 25% sodium hydroxide aqueous solution were added to the concentrated residue to dissolve the residue, and 5 ml of concentrated hydrochloric acid was added dropwise thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 11.81 g (yield: 86.4%) of compound (3) as a white powder.
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
[0149]
[Chem. 12]
[0150]
10.00 g (53.2 mmol) of compound (1b), 9.55 g (69.1 mmol) of potassium carbonate, and 8.50 g (69.1 mmol) of isopropyl bromide were added to 40 ml of N,N-dimethylformamide, and the mixture was reacted at 75 to 85°C for 2 hours. 80 ml of butyl acetate and 80 ml of water were added to the reaction solution, and the mixture was partitioned. 5.68 g (58.5 mmol) of sulfamic acid and 10 ml of water were added to the organic layer, and 21.15 g (58.5 mmol) of a 25% sodium chlorite aqueous solution was added dropwise thereto at 20°C or below, followed by reaction for 15 minutes. 10 ml of a 25% sodium hydroxide aqueous solution was added thereto at 20°C or below, and subsequently 80.41 g (63.8 mmol) of a 10% sodium sulfite aqueous solution was poured in. Additionally, 2 ml of concentrated hydrochloric acid was added, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 40 ml of methanol, 80 ml of water, and 10 ml of a 25% sodium hydroxide aqueous solution were added to the concentrated residue, and the residue was dissolved, followed by dropwise addition of 5 ml of concentrated hydrochloric acid to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 12.09 g (yield: 92.4%) of compound (3) as a white powder.
[0151]
Production Example 3: Production of Compound (7)
Compound (7) was produced in accordance with the following reaction scheme.
[0152]
[Chem. 13]
Production Example 4: Production of Compound (11)
Compound (11) was produced in accordance with the following reaction scheme.
[0160]
[Chem. 14]
[0161]
Synthesis of Compound (9)
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
Synthesis of Compound (10)
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
Synthesis of Compound (11)
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
Type B Crystal Preparation 2
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
[0072]
[0073]
20.00 g (66.8 mmol) of compound (1) and 17.28 g (134 mmol) of diisopropylethylamine were added to 300 mL of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto, and the mixture was stirred at 20 to 35°C for 1 hour. 100 mL of water was added to the reaction solution, and the mixture was separated, followed by concentration of the organic layer under reduced pressure. 300 mL of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 mL of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue, and reacted at 75 to 85°C for 1 hour. 200 mL of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 27.20 g (yield: 95.01%) of compound (3).
[0074]
[0075]
20.00 g (46.7 mmol) of compound (3), 40 mL of a 40% methylamine aqueous solution, 40 mL of methanol, and 100 mL of water were mixed and reacted for 30 minutes under reflux. 200 mL of cyclopentyl methyl ether (CPME) and 20 mL of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by separation. A mixture of 100 mL of water and 20.00 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by separation. 5 mL of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration, thereby obtaining 27.58 g of compound (4) as a wet crystal.
[0076]
The wet crystal (46.7 mmol) of compound (4) was mixed with 120 mL of ethyl acetate and 7.1 mL (51.4 mmol) of triethylamine, and stirred at 20 to 30°C for 1 hour. 10.09 g (60.7 mmol) of 2-ethoxybenzoic acid and 11.63 g (60.7 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 60 mL of water and 6 mL of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by separation. 60 mL of water and 6 mL of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% sodium hydroxide aqueous solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 12 mL of ethanol. The filtrate was cooled, and 10 mg of the type B crystal (a seed crystal) was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 18.38 g (88.18%) of compound (5).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader

















 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.



{kind=link}
{kind=link}