
Home » Posts tagged 'APPROVALS 2025' (Page 3)
Tag Archives: APPROVALS 2025
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Taletrectinib
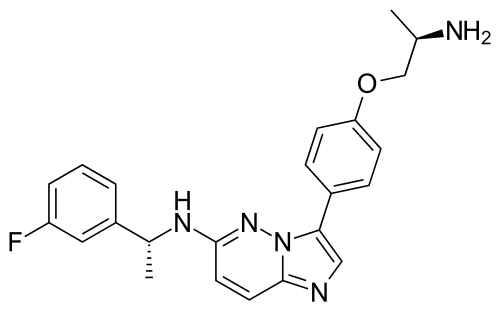
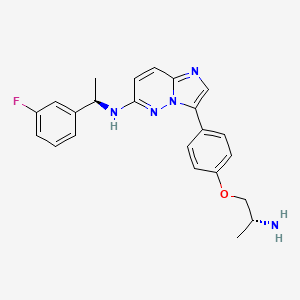
Taletrectinib
CAS 1505514-27-1
as salt: 1505515-69-4, Taletrectinib adipate
FDA 6/11/2025, Ibtrozi, To treat locally advanced or metastatic ROS1-positive non-small cell lung cancer ALSO CHINA 2024 APPROVED |
405.5 g/mol, C23H24FN5O, UNII-W4141180YD
3-[4-[(2R)-2-aminopropoxy]phenyl]-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine
Taletrectinib adipate
WeightAverage: 551.619
Monoisotopic: 551.254397378
Chemical FormulaC29H34FN5O5
DS-6051B, CAS 1505515-69-4,
6KLL51GNBG, 3-{4-[(2R)-2-aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo[1,2-b]pyridazin-6-amine; hexanedioic acid
Taletrectinib, sold under the brand name Ibtrozi, is an anti-cancer medication used for the treatment of non-small cell lung cancer.[1][2] It is used as the salt, taletrectinib adipate.[1] Taletrectinib is a kinase inhibitor.[1] It is taken by mouth.[1]
Taletrectinib was approved for medical use in the United States in June 2025.[3]
SYN
US20200062765
https://patentscope.wipo.int/search/en/detail.jsf?docId=US289038418&_cid=P12-MCIHV1-02369-1
Example 1
tert-Butyl [(2R)-1-(4-bromophenoxy)propan-2-yl]carbamate (1)
Example 2
6-Fluoroimidazo[1,2-b]pyridazine methanesulfonate (2)
Example 3
tert-Butyl {(2R)-1-[4-(6-fluoroimidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate (3)
Example 4
tert-Butyl {(2R)-1-[4-(6-{[(1R)-1-(3-fluorophenyl)ethyl]amino}imidazo[1,2-b]pyridazin-3-yl)phenoxy]propan-2-yl}carbamate hydrochloride (4)
Example 5
3-{4-[(2R)-2-Aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethylimidazo[1,2-b]pyridazin-6-amine dihydrochloride (5)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023272701&_cid=P12-MCIHPU-95869-1
The NMR data for the crystalline form A of Compound 1 adipate are as follows: 1H NMR (500 MHz, DMSO) δ 1.13-1.14 (d, J=5.0 Hz, 3H) , 1.47-1.48 (d, J=5.0 Hz, 7H) , 2.15-2.18 (t, J=5.0 Hz, J=10.0 Hz, 4H) , 3.25-3.29 (m, 1H) , 3.79-3.83 (m, 2H) , 4.80-4.85 (m, 1H) , 6.76-6.77 (d, J=5.0 Hz, 1H) , 6.92-6.94 (d, J=10.0 Hz, 2H) , 7.01-7.05 (t, J=10.0 Hz, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.64-7.65 (d, J=5.0 Hz, 1H) , 7.72-7.76 (t, J=10.0 Hz, 4H) .
[0148]
The IR data for the crystalline form A of Compound 1 adipate are as follows: IR (cm -1) : 1701, 1628, 1612, 1586, 1463, 1333, 1246, 1110, 829, 821.
Example 5: Preparation and Characterization of Crystalline Form A of Compound 1 Free Base
[0212]
Compound 1 HCl (75.5 g) (e.g., obtained by using the method described in Example 5 of U.S. Application Publication No. 2020/0062765) was dissolved in ethanol (604 mL) at 50℃. Sodium hydroxide (68.1 g) was added to the above solution. The mixture was cooled to 1℃ in 1.5 hours and stirred for 18.5 hours. The mixture was then filtered, and the solid thus obtained was washed with a cooled mixture of ethanol (151 mL) and water (151 mL) and dried. The solid thus obtained was confirmed to be the crystalline form A of Compound 1 free base.
[0213]
The NMR data for the crystalline form A of Compound 1 free base are as follows: 1H NMR (500 MHz, DMSO) δ 1.09-1.10 (d, J=5.0 Hz, 3H) , 1.48-1.49 (d, J=5.0 Hz, 3H) , 3.16-3.20 (m, 1H) , 3.75-3.79 (m, 2H) , 4.82-4.86 (m, 1H) , 6.76-6.78 (d, J=10.0 Hz, 1H) , 6.92-6.94 (m, 2H) , 7.01-7.05 (m, 1H) , 7.23-7.28 (m, 2H) , 7.37-7.42 (m, 1H) , 7.62-7.63 (d, J=5.0 Hz, 1H) , 7.72-7.75 (m, 4H) .
[0214]
The IR data for the crystalline form A of Compound 1 free base are as follows: IR (cm -1) : 3350, 3247, 3055, 2961, 2923, 2864, 1611, 1586, 1349, 829, 819.
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Taletrectinib is an oral, next-generation ROS1 TKI developed by Nuvation Bio Inc. for the treatment of ROS1-positive NSCLC. In 2024, the NMPA approved taletrectinib for adult patients with locally advanced or metastatic ROS1-positive NSCLC, regardless of prior ROS1TKI treatment [47]. Under an exclusive license agreement, Innovent Biologics will commercialize taletrectinib in China under the brand
name DOVBLERON®. Taletrectinib exerts its pharmacological action through the mechanism of selectively impeding the ROS1 receptor tyrosine kinase, which effectively disrupts the signaling cascades which are responsible for facilitating the growth and survival of cancer cells in ROS1-positive NSCLC. This inhibition of the ROS1 receptor tyrosine kinase is a key event in the drug’s mode of action, as it specifically targets the molecular processes that drive the progression of the disease in ROS1-positive NSCLC cases [48]. The NMPA granted approval founded on the data sourced from the crucial Phase 2 TRUST – I study. This study substantiated that patients administered with taletrectinib achieved sustained responses and extended PFS. Regarding safety, taletrectinib boasted a generally good tolerability. It presented an advantageous safety profile and favorable tolerability characteristics, as evidenced by the low incidences of dose reduction and treatment discontinuation triggered by adverse effects. [49]. Overall, taletrectinib represents a promising therapeutic option for patients with advanced ROS1-positive NSCLC, offering efficacy in both TKI-naïve and TKI-pretreated populations, including those with CNS metastases [50–52].
The synthesis of Taletrectinib, illustrated in Scheme 12, commences with Mitsunobu coupling of Tale-001 and Tale-002 to afford Tale-003, which then undergoes Suzuki coupling with Tale-004 constructing
Tale-005 [53]. Sequential acidolysis/deprotection of Tale-005 ultimately delivers Taletrectinib
[47] M. P´ erol, N. Yang, C.M. Choi, Y. Ohe, S. Sugawara, N. Yanagitani, G. Liu, F.G.M.
D. Braud, J. Nieva, M. Nagasaka, 1373P efficacy and safety of taletrectinib in
patients (pts) with ROS1+ non-small cell lung cancer (NSCLC): interim analysis of
global TRUST-II study, Ann. Oncol. 34 (2023) S788–S789.
[48] G. Harada, F.C. Santini, C. Wilhelm, A. Drilon, NTRK fusions in lung cancer: from
biology to therapy, Lung Cancer 161 (2021) 108–113.
[49] W. Li, A. Xiong, N. Yang, H. Fan, Q. Yu, Y. Zhao, Y. Wang, X. Meng, J. Wu, Z. Wang,
Y. Liu, X. Wang, X. Qin, K. Lu, W. Zhuang, Y. Ren, X. Zhang, B. Yan, C.M. Lovly,
C. Zhou, Efficacy and safety of taletrectinib in Chinese patients with ROS1+ non-
small cell lung cancer: the phase II TRUST-I study, J. Clin. Oncol. 42 (2024)
2660–2670.
[50] M. Nagasaka, D. Brazel, S.I. Ou, Taletrectinib for the treatment of ROS-1 positive
non-small cell lung cancer: a drug evaluation of phase I and II data, Expert Opin
Investig Drugs 33 (2024) 79–84.
[51] S. Waliany, J.J. Lin, Taletrectinib: TRUST in the continued evolution of treatments
for ROS1 fusion-positive lung cancer, J. Clin. Oncol. 42 (2024) 2622–2627.
[52] M. Nagasaka, Y. Ohe, C. Zhou, C.M. Choi, N. Yang, G. Liu, E. Felip, M. P´ erol,
B. Besse, J. Nieva, L. Raez, N.A. Pennell, A. Dimou, F. Marinis, F. Ciardiello,
T. Seto, Z. Hu, M. Pan, W. Wang, S. Li, S.I. Ou, TRUST-II: a global phase II study of
taletrectinib in ROS1-positive non-small-cell lung cancer and other solid tumors,
Future Oncol. 19 (2023) 123–135.
[53] Y. Takeda, K. Yoshikawa, Y. Kagoshima, Y. Yamamoto, R. Tanaka, Y. Tominaga,
M. Kiga, Y. Hamada, Preparation of imidazo[1,2-b]pyridazine Derivatives as
Potent Inhibitors of ROS1 Kinase and NTRK Kinase, 2013. WO2013183578A1.

Medical uses
Taletrectinib is indicated for the treatment of adults with locally advanced or metastatic ROS1-positive non-small cell lung cancer.[1][2]
Adverse effects
The FDA prescribing information for taletrectinib includes warnings and precautions for hepatotoxicity, interstitial lung disease/pneumonitis, QTc interval prolongation, hyperuricemia, myalgia with creatine phosphokinase elevation, skeletal fractures, and embryo-fetal toxicity.[1][3]
History
The efficacy of taletrectinib to treat ROS1-positive non-small cell lung cancer was evaluated in participants with locally advanced or metastatic, ROS1-positive non-small cell lung cancer enrolled in two multi-center, single-arm, open-label clinical trials, TRUST-I (NCT04395677) and TRUST-II (NCT04919811).[3] The efficacy population included 157 participants (103 in TRUST-I; 54 in TRUST-II) who were naïve to treatment with a ROS1 tyrosine kinase inhibitor (TKI) and 113 participants (66 in TRUST-I; 47 in TRUST-II) who had received one prior ROS1 tyrosine kinase inhibitor.[3] Participants may have received prior chemotherapy for advanced disease.[3] The US Food and Drug Administration (FDA) granted the application for taletrectinib priority review, breakthrough therapy, and orphan drug designations.[3]
Society and culture
Legal status
Taletrectinib was approved for medical use in the United States in June 2025.[3][4]
Names
Taletrectinib is the international nonproprietary name.[5]
Taletrectinib is sold under the brand name Ibtrozi.[3][4]
References
- ^ Jump up to:a b c d e f g “Prescribing Information for NDA 219713, Supplement 000” (PDF). Drugs@FDA. U.S. Food and Drug Administration. April 2025. Retrieved 14 June 2025.
- ^ Jump up to:a b Khan I, Sahar A, Numra S, Saha N, Nidhi, Parveen R (April 2025). “Efficacy and safety of taletrectinib for treatment of ROS1 positive non-small cell lung cancer: A systematic review”. Expert Opinion on Pharmacotherapy. 26 (6): 765–772. doi:10.1080/14656566.2025.2487150. PMID 40170301.
- ^ Jump up to:a b c d e f g h “FDA approves taletrectinib for ROS1-positive non-small cell lung cancer”. U.S. Food and Drug Administration (FDA). 11 June 2025. Retrieved 13 June 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “U.S. Food and Drug Administration Approves Nuvation Bio’s Ibtrozi (taletrectinib), a Next-Generation Oral Treatment for Advanced ROS1-Positive Non-Small Cell Lung Cancer”. Nuvation Bio (Press release). 12 June 2025. Retrieved 13 June 2025.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
- Clinical trial number NCT04395677 for “A Study of AB-106 in Subjects With Advanced NSCLC Harboring ROS1 Fusion Gene” at ClinicalTrials.gov
- Clinical trial number NCT04919811 for “Taletrectinib Phase 2 Global Study in ROS1 Positive NSCLC (TRUST-II)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Ibtrozi |
| License data | US DailyMed: Taletrectinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1505514-27-1as salt: 1505515-69-4 |
| PubChem CID | 72202474as salt: 72694302 |
| DrugBank | DB18711 |
| ChemSpider | 114934673as salt: 88297530 |
| UNII | W4141180YDas salt: 6KLL51GNBG |
| KEGG | D12363as salt: D12364 |
| ChEMBL | ChEMBL4650989as salt: ChEMBL4650361 |
| Chemical and physical data | |
| Formula | C23H24FN5O |
| Molar mass | 405.477 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Taletrectinib, FDA 2025, APPROVALS 2025, Ibtrozi, CANCER, AB-106, DS-6051a, UNII-W4141180YD, DS 6051B, APPROVALS 2024, CHINA 2024, Nuvation Bio Inc
Acoltremon
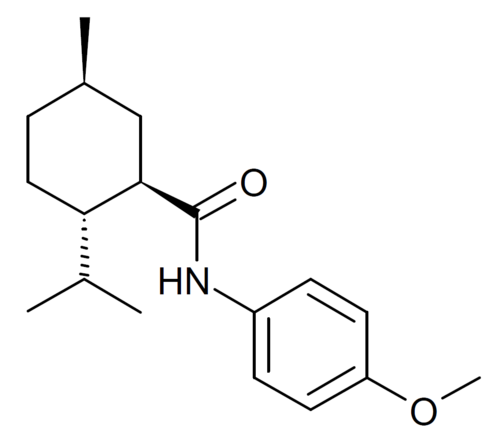
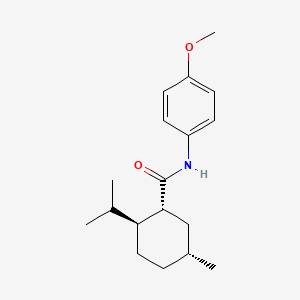
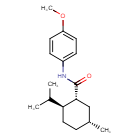
Acoltremon
CAS 68489-09-8
WeightAverage: 289.419
Monoisotopic: 289.204179113
Chemical FormulaC18H27NO2
FDA 2025, 5/28/2025, To treat the signs and symptoms of dry eye disease
Tryptyr |
WS 12
(1R,2S,5R)-N-(4-methoxyphenyl)-5-methyl-2-(propan-2-yl)cyclohexane-1-carboxamide
Fema No. 4681
N-(4-methoxyphenyl)-p-menthanecarboxamide
- OriginatorInstituto de Neurociencias de Alicante
- DeveloperAlcon; AVX Pharma
- ClassCyclohexanes; Ethers; Eye disorder therapies; Small molecules
- Mechanism of ActionTRPM8 protein stimulants
- RegisteredDry eyes
- 30 May 2025Alcon plans to launch Acoltremon for Dry eyes in USA in the third quarter of 2025
- 28 May 2025Registered for Dry eyes in USA (Ophthalmic) – First global approval
- 05 May 2025FDA assigns PDUFA action date of 30/05/2025 for Acoltremon for Dry eyes
Acoltremon sold under the brand name Tryptyr, is a medication used for the treatment of dry eye syndrome.[1]
PATENT
US 217370
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023114986&_fid=RU437402572
https://patentscope.wipo.int/search/en/detail.jsf?docId=US193167995&_cid=P11-MCE7BB-27500-1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012032209&_fid=US193167995
Medical uses
Acoltremon was approved for medical use in the United States in May 2025, for the treatment of signs and symptoms associated with dry eye disease.[2]
Pharmacology
Acoltremon acts as a potent and selective activator (opener) of the TRPM8 calcium channel, which is responsible for the sensation of coldness produced by menthol.[3] It is slightly less potent as a TRPM8 activator compared to icilin, but is a much more selective TRPM8 ligand when compared to menthol.[4]
Society and culture
Legal status
Acoltremon was approved for medical use in the United States in May 2025.[5]
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/217370s000lbl.pdf
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 29 May 2025. Archived from the original on 3 March 2025. Retrieved 29 May 2025.
- ^ Ma S, Gisselmann G, Vogt-Eisele AK, Doerner JF, Hatt H (October 2008). “Menthol derivative WS-12 selectively activates transient receptor potential melastatin-8 (TRPM8) ion channels”. Pakistan Journal of Pharmaceutical Sciences. 21 (4): 370–378. PMID 18930858.
- ^ Kühn FJ, Kühn C, Lückhoff A (February 2009). “Inhibition of TRPM8 by icilin distinct from desensitization induced by menthol and menthol derivatives”. The Journal of Biological Chemistry. 284 (7): 4102–4111. doi:10.1074/jbc.M806651200. PMID 19095656.
- ^ “Alcon Announces FDA Approval of Tryptyr (acoltremon ophthalmic solution) 0.003% for the Treatment of the Signs and Symptoms of Dry Eye Disease” (Press release). Alcon. 28 May 2025. Archived from the original on 29 May 2025. Retrieved 29 May 2025 – via Business Wire.
External links
- Clinical trial number NCT05285644 for “Study Evaluating the Safety and Efficacy of AR-15512 (COMET-2)” at ClinicalTrials.gov
- Clinical trial number NCT05360966 for “Study Evaluating the Safety and Efficacy of AR-15512 (COMET-3)” at ClinicalTrials.gov
| molecular structure | |
| 3D representation | |
| Clinical data | |
|---|---|
| Trade names | Tryptyr |
| Other names | AVX-012, WS-12 |
| License data | US DailyMed: Acoltremon |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 68489-09-8 |
| PubChem CID | 11266244 |
| DrugBank | DB19202 |
| ChemSpider | 9441255 |
| UNII | 1L7BVT4Z4Z |
| KEGG | D13125 |
| ChEMBL | ChEMBL2441929 |
| CompTox Dashboard (EPA) | DTXSID10460636 |
| Chemical and physical data | |
| Formula | C18H27NO2 |
| Molar mass | 289.419 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- [1]. Beck B, et al. Prospects for prostate cancer imaging and therapy using high-affinity TRPM8 activators. Cell Calcium. 2007 Mar;41(3):285-94. [Content Brief][2]. Ma S, et al. Menthol derivative WS-12 selectively activates transient receptor potential melastatin-8 (TRPM8) ion channels. Pak J Pharm Sci. 2008 Oct;21(4):370-8. [Content Brief]
///////Acoltremon, FDA 2025, APPROVALS 2025, WS-12, WS 12, Fema No. 4681, Tryptyr, 1L7BVT4Z4Z, AR-15512
Nerandomilast



Nerandomilast
CAS 1423719-30-5
C20H25ClN6O2S
| Molecular Weight | 448.97 |
|---|---|
| Formula | C20H25ClN6O2S |
fda 2025, approvals 2025, Jascayd,10/7/2025, To treat idiopathic pulmonary fibrosis
[1-[[(5R)-2-[4-(5-chloropyrimidin-2-yl)piperidin-1-yl]-5-oxo-6,7-dihydrothieno[3,2-d]pyrimidin-4-yl]amino]cyclobutyl]methanol
Cyclobutanemethanol, 1-[[(5R)-2-[4-(5-chloro-2-pyrimidinyl)-1-piperidinyl]-6,7-dihydro-5-oxidothieno[3,2-d]pyrimidin-4-yl]amino]-
1-[[(5R)-2-[4-(5-Chloro-2-pyrimidinyl)-1-piperidinyl]-6,7-dihydro-5-oxidothieno[3,2-d]pyrimidin-4-yl]amino]cyclobutanemethanol
Nerandomilast (BI 1015550) is an investigational oral medication being studied for the treatment of idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF). It is a preferential inhibitor of phosphodiesterase 4B (PDE4B) and has shown potential in slowing lung function decline in patients with IPF.
Key points about nerandomilast:
- Mechanism of Action:Nerandomilast inhibits PDE4B, an enzyme that plays a role in inflammation and fibrosis.
- Clinical Trials:Phase 3 clinical trials have shown that nerandomilast can slow lung function decline in patients with IPF and PPF.
- Efficacy:The trials demonstrated that nerandomilast led to a smaller decline in forced vital capacity (FVC), a measure of lung function, compared to placebo.
- Safety:Diarrhea was the most frequent adverse event, but serious adverse events were balanced across treatment groups.
- Progressive Fibrosing ILDs:Nerandomilast is also being investigated in other progressive fibrosing interstitial lung diseases (ILDs) beyond IPF.
- FDA Designation:Nerandomilast received Breakthrough Therapy Designation from the FDA for the treatment of IPF.
- Not a Cure:While nerandomilast can slow disease progression, it does not cure pulmonary fibrosis.
- Not Yet Approved:Nerandomilast is still an investigational drug and is not yet approved for use.
Nerandomilast (BI 1015550) is an orally active inhibitor of PDE4B with an IC50 value of 7.2 nM. Nerandomilast has good safety and potential applications in inflammation, allergic diseases, pulmonary fibrosis, and chronic obstructive pulmonary disease (COPD).
SCHEME


1H NMR (400 MHz, DMSO-D6) 1.57–1.84 (m, 2H), 1.96 (br d, J = 12.5 Hz, 2H), 2.10–2.21 (m, 2H), 2.24–
2.41 (m, 2H), 2.82–2.98 (m, 2H), 3.06 (br t, J = 11.7 Hz, 2H), 3.13–3.27 (m, 2H), 3.36–3.47 (m, 1H), 3.71 (d, J =
5.64 Hz, 2H), 4.70 (br d, J = 12.5 Hz, 2H), 4.84 (t, J = 5.7 Hz, 1H), 7.35 (s, 1H), 8.85 (s, 2H).


1H NMR (DMSO-d6, 400 MHz) 1.87–1.92 (m, 2H), 2.12–2.17 (m, 2H), 3.08 (ddd, J = 12.8, 12.8, 2.8 Hz,
2H), 3.21 (m, 1H), 3.34–3.42 (m, 2H), 8.47 (br, 2H), 8.19 (s, 2H).
PATENT
US20150045376
WO2013026797
PAPER
https://pubs.acs.org/doi/10.1021/acs.oprd.4c00309

A robust and scalable synthesis process for Nerandomilast (1, BI 1015550), a selective PDE4B inhibitor with potential therapeutic properties for the treatment of respiratory diseases, was developed and implemented at a pilot plant on a multikilogram scale. Key aspects of the process include the efficient synthesis of intermediate (1-((2-chloro-6,7-dihydrothieno[3,2-d]pyrimidin-4-yl)amino)cyclobutyl)methanol (4) by means of a regioselective SNAr reaction between (1-aminocyclobutyl)methanol (6) and 2,4-dichloro-6,7-dihydrothieno[3,2-d]pyrimidine (5), a new convergent synthesis of 5-chloro-2-(piperidin-4-yl)pyrimidine (3) by means of a Suzuki coupling, and a highly enantioselective sulfide oxidation to give chiral nonracemic (R)-2-chloro-4-((1-(hydroxymethyl)cyclobutyl)amino)-6,7-dihydrothieno[3,2-d]pyrimidine 5-oxide (2).

- [1]. Pouzet P A, et al. Piperidino-dihydrothienopyrimidine sulfoxides and their use for treating COPD and asthma. United States. US9150586.[2]. Herrmann FE, et al. BI 1015550 is a PDE4B Inhibitor and a Clinical Drug Candidate for the Oral Treatment of Idiopathic Pulmonary Fibrosis. Front Pharmacol. 2022 Apr 20;13:838449. [Content Brief]
//////////Nerandomilast, BI 1015550, I5DGT51IB8, fda 2025, approvals 2025, Jascayd,
ETRIPAMIL



ETRIPAMIL
CAS 1593673-23-4
AS ACETATE 512.64 CAS 2891832-59-8
HCL SALT 2560549-35-9
WeightAverage: 452.595
Monoisotopic: 452.267507647
Chemical FormulaC27H36N2O4
12/12/2025, FDA 2025, APPROVALS 2025
Benzoic acid, 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]methylamino]ethyl]-, methyl ester
methyl 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]-methylamino]ethyl]benzoate
- Methyl 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]methylamino]ethyl]benzoate
- (-)-MSP 2017
- MSP 2017
- OriginatorMilestone Pharmaceuticals
- DeveloperCorxel Pharmaceuticals; Milestone Pharmaceuticals
- ClassAmines; Antiarrhythmics; Benzoates; Esters; Ischaemic heart disorder therapies; Small molecules
- Mechanism of ActionCalcium channel antagonists
- PreregistrationParoxysmal supraventricular tachycardia
- Phase IIAtrial fibrillation
- Phase IUnspecified
- No development reportedAngina pectoris
- 14 May 2025Milestone Pharmaceuticals has patent protection for etripamil in the USA
- 28 Mar 2025Milestone pharmaceuticals plans to request a Type A meeting with USFDA to discuss the issues raised in the complete response letter
- 28 Mar 2025USFDA has issued a Complete Response Letter (CRL) regarding New Drug Application (NDA) for Etripamil for Paroxysmal supraventricular tachycardia
Etripamil has been used in trials studying the treatment of Paroxysmal Supraventricular Tachycardia (PSVT).
Etripamil (MSP-2017) is a short-acting, L-type calcium-channel antagonist. Etripamil inhibits calcium influx through slow calcium channels, thereby slowing AV node conduction and prolonging the AV node refractory period. Etripamil increases heart rate and decreases systolic blood pressure. Etripamil can be used in the study of paroxysmal supraventricular tachycardia (PSVT).
To treat episodes of paroxysmal supraventricular tachycardia
SCHEME
SIDE CHAIN

MAIN

SYN
US20180110752/ U.S. Patent No. 10,117,848,
EXAMPLES
Example 1: Synthesis methyl 3-(2-((4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl)(methyl)amino)ethyl)benzoate
Part I: Synthesis of 5-Bromo-2-(3,4-dimethoxyphenyl)-2-isopropylpentanenitrile
Part II: Synthesis of methyl 3-(2-(methylamino)ethyl)benzoate
Part III: Reaction of Compound II with Compound III Produced Compound I
| Analysis of the product by mass spectrometry revealed a peak with a mass-to-charge ratio (m/z) of 453, corresponding to the M+H molecular ion of compound I. |
Example 2: Concentrated Solution of Acetate Salt of Compound I
| A concentrated aqueous solution of the acetate salt of compound I is formed according to the following protocol: |
| This protocol readily can be adapted to provide a concentrated solution of the methanesulfonate salt of compound I. |
PRED BY CHIRAL SEPERATION
US20230065401
WO2016165014
EP4119137 chiral sepn done
[0034] In one embodiment the present invention is a kit for treating a cardiac arrhythmia (e.g., PSVT or atrial fibrillation), angina, or a migraine in a subject in need thereof wherein the kit comprises a nasal delivery system comprising two doses of a therapeutically effective amount of compound I having a structure according to the formula:
and instructions for nasally administering to the subject (i) a first dose, and, optionally, (ii) a second dose of an aqueous composition comprising a pharmaceutically acceptable acetate or methanesulfonate salt of compound I, or a racemate or enantiomer thereof, wherein the acetate or methanesulfonate salt of compound I, or the racemate or enantiomer thereof, is dissolved in the aqueous composition at a concentration of 350 mg/mL± 50 mg/mL, and wherein the second dose of the compound is to be administered between 5 minutes and 60 minutes after the first dose.
Cross ref U.S. Patent No. 10,117,848,
[0336]
- 1. A method of treating a cardiac arrhythmia in a subject in need thereof with a therapeutically effective amount of compound I having a structure according to the formula:
the method comprising nasally administering to the subject (i) a first dose, and (ii) a second dose of an aqueous composition comprising a pharmaceutically acceptable acetate or methanesulfonate salt of compound I, or a racemate or enantiomer thereof, wherein the acetate or methanesulfonate salt of compound I, or the racemate or enantiomer thereof, is dissolved in the aqueous composition at a concentration of 350 mg/mL ± 50 mg/mL, and wherein the second dose of the compound is administered between 5 minutes and 25 minutes after the first dose.
PATENT
Journal of the American College of Cardiology (2018), 72(5), 489-497
American Heart Journal (2022), 253, 20-29
Expert Opinion on Investigational Drugs (2020), 29(1), 1-4
EP4119137 WO2016165014
EP-2170050-B1
US-9737503-B2
US-4968717-A
EP-0231003-A2
- [1]. Stambler BS, et al. Etripamil Nasal Spray for Rapid Conversion of Supraventricular Tachycardia to Sinus Rhythm. J Am Coll Cardiol. 2018 Jul 31;72(5):489-497. [Content Brief][2]. Milestone Pharmaceuticals Announces USAN Approval of Generic Name “Etripamil” for its Phase 2 Clinical Development Product for the Treatment of Paroxysmal Supraventricular Tachycardia.[3]. Ascah A, et al. Cardiovascular and Pharmacokinetic Profiles of Intravenous Etripamil in Conscious Telemetered Cynomolgus Monkeys. Int J Toxicol. 2025 Apr 1:10915818251327963. [Content Brief][4]. Pion J, et al. Preclinical Safety Evaluation of Etripamil Nasal Spray in Cynomolgus Macaques (Macaca fascicularis) to Assess for Safety in Patients With Paroxysmal Supraventricular Tachycardia. Int J Toxicol. 2024 Sep-Oct;43(5):503-510. [Content Brief]
//////////ETRIPAMIL, (-)-MSP 2017, MSP 2017, FDA 2025, APPROVALS 2025
Olezarsen


Olezarsen
Olezarsen is an ASO directed inhibitor of Apolipoprotein C-III (apoC-III) mRNA, conjugated to a ligand containing three N-acetyl galactosamine (GalNAc) residues to enable delivery of the ASO to hepatocytes.
TRYNGOLZA contains olezarsen sodium as the active ingredient. Olezarsen sodium is a white to yellow solid and it is freely soluble in water and in phosphate buffer. The molecular formula of olezarsen sodium is C 296H 419N 71O 154P 20S 19Na 20and the molecular weight is 9124.48 daltons. The chemical name of olezarsen sodium is DNA, d(P-thio) ([2′- O-(2-methoxyethyl)] rA-[2′- O-(2-methoxyethyl)] rG-[2′- O-(2-methoxyethyl)] m5rC-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-m5C-T-T-G-T-m5C-m5C-A-G-m5C-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] m5rU-[2′- O-(2-methoxyethyl)] rA-[2′- O-(2-methoxyethyl)]m5rU), 5′-[26-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]-14,14-bis[[3-[[6-[[2-(acetylamino)-2-deoxy-β-D-galactopyranosyl]oxy]hexyl]amino]-3-oxopropoxy]methyl]-8,12,19-trioxo-16-oxa-7,13,20-triazahexacos-1-yl hydrogen phosphate], sodium salt (1:20).


Olezarsen
FDA APPROVED 12/19/2024, Tryngolza, To treat familial chylomicronemia syndrome
Drug Trials Snapshot
- AKCEA-APOCIII-LRX
- ALL-P-AMBO-5′-O-(((6-(5-((TRIS(3-(6-(2-ACETAMIDO-2-DEOXY-.BETA.-D-GALACTOPYRANOSYLOXY)HEXYLAMINO)-3-OXOPROPOXYMETHYL))METHYL)AMINO-5-OXOPENTANAMIDO)HEXYL))PHOSPHO)-2′-O-(2-METHOXYETHYL)-P-THIOADENYLYL-(3′-O->5′-O)-2′-O-(2-METHOXYETHYL)-P-THIOGUANYLYL-(3
- DNA, D(P-THIO)((2′-O-(2-METHOXYETHYL))RA-(2′-O-(2-METHOXYETHYL))RG-(2′-O-(2-METHOXYETHYL))M5RC-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETHYL))M5RU-M5C-T-T-G-T-M5C-M5C-A-G-M5C-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETHYL))M5RU-(2′-O-(2-METHOXYETH
- IONIS-APOCIII-LRX
- ISIS-APOCIII-LRX
- ISIS-678354
Olezarsen, sold under the brand name Tryngolza, is a medication used in the treatment of familial chylomicronemia syndrome.[1][2] It is given by injection under the skin.[1]
Olezarsen was approved for medical use in the United States in December 2024.[1][3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9127276 | No | 2015-09-08 | 2034-05-01 |  |
| US9181549 | No | 2015-11-10 | 2034-05-01 | |
| US9593333 | No | 2014-02-14 | 2034-02-14 | |
| US9157082 | No | 2012-04-27 | 2032-04-27 | |
| US9163239 | No | 2014-05-01 | 2034-05-01 | |
Medical uses
Olezarsen is indicated as an adjunct to diet to reduce triglycerides in adults with familial chylomicronemia syndrome.[1]
Pharmacology
Olezarsen is an apolipoprotein C-III-directed antisense oligonucleotide.[1] By binding to apolipoprotein C-III mRNA, it causes its degradation, which in turn increases clearance of plasma triglycerides and very low-density lipoprotein (VLDL).[5]
Adverse effects
In a 66-patient trial, olezarsen was demonstrated to cause following side effects:[5][6]
- injection site reactions
- hypersensitivity reactions (due to immunogenic potential of the medication)
- arthralgia
- thrombocytopenia
- hyperglycemia
- elevation of liver enzymes
History
The US Food and Drug Administration (FDA) granted the application of olezarsen orphan drug designation in February 2024.[7] In August 2024, European Medicines Agency also granted olezarsen this designation.[8]
Society and culture
Legal status
Olezarsen was approved for medical use in the United States in December 2024.[3][9]
Names
Olezarsen is the international nonproprietary name.[10]
Olezarsen is sold under the brand name Tryngolza.[1]
References
^ Jump up to:a b c d e f g “Tryngolza- olezarsen sodium injection, solution”. DailyMed. 19 December 2024. Retrieved 25 January 2025.
- ^ Spagnuolo, Catherine M; Hegele, Robert A (2023). “Recent advances in treating hypertriglyceridemia in patients at high risk of cardiovascular disease with apolipoprotein C-III inhibitors”. Expert Opinion on Pharmacotherapy. 24 (9): 1013–1020. doi:10.1080/14656566.2023.2206015. PMID 37114828.
- ^ Jump up to:a b “Novel Drug Approvals for 2024”. U.S. Food and Drug Administration (FDA). 1 October 2024. Retrieved 20 December 2024.
- ^ New Drug Therapy Approvals 2024 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2025. Archived from the original on 21 January 2025. Retrieved 21 January 2025.
- ^ Jump up to:a b Stroes, Erik S.G.; Alexander, Veronica J.; Karwatowska-Prokopczuk, Ewa; Hegele, Robert A.; Arca, Marcello; Ballantyne, Christie M.; et al. (16 May 2024). “Olezarsen, Acute Pancreatitis, and Familial Chylomicronemia Syndrome”. New England Journal of Medicine. 390 (19): 1781–1792. doi:10.1056/NEJMoa2400201. ISSN 0028-4793.
- ^ Ionis Pharmaceuticals, Inc. (11 December 2024). A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study of AKCEA-APOCIII-LRx Administered Subcutaneously to Patients With Familial Chylomicronemia Syndrome (FCS) (Report). clinicaltrials.gov.
- ^ “Olezarsen Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). Retrieved 20 December 2024.
- ^ “EU/3/24/2973 – orphan designation for treatment of familial chylomicronaemia syndrome | European Medicines Agency (EMA)”. http://www.ema.europa.eu. 21 August 2024. Retrieved 22 February 2025.
- ^ “Tryngolza (olezarsen) approved in U.S. as first-ever treatment for adults living with familial chylomicronemia syndrome as an adjunct to diet” (Press release). Ionis Pharmaceuticals. 19 December 2024. Retrieved 20 December 2024 – via PR Newswire.
- ^ World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 87”. WHO Drug Information. 36 (1). hdl:10665/352794.
Further reading
Karwatowska-Prokopczuk, Ewa; Tardif, Jean-Claude; Gaudet, Daniel; Ballantyne, Christie M.; Shapiro, Michael D.; Moriarty, Patrick M.; et al. (2022). “Effect of olezarsen targeting APOC-III on lipoprotein size and particle number measured by NMR in patients with hypertriglyceridemia”. Journal of Clinical Lipidology. 16 (5): 617–625. doi:10.1016/j.jacl.2022.06.005. PMID 35902351.
- Tardif, Jean-Claude; Karwatowska-Prokopczuk, Ewa; Amour, Eric St; Ballantyne, Christie M; Shapiro, Michael D; Moriarty, Patrick M; et al. (6 April 2022). “Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk”. European Heart Journal. 43 (14): 1401–1412. doi:10.1093/eurheartj/ehab820. PMC 8986458. PMID 35025993.
External links
“Olezarsen (Code C180652)”. NCI Thesaurus.
- Clinical trial number NCT04568434 for “A Study of Olezarsen (Formerly Known as AKCEA-APOCIII-LRx) Administered to Patients With Familial Chylomicronemia Syndrome (FCS) (BALANCE)” at ClinicalTrials.gov
- Tardif JC, Karwatowska-Prokopczuk E, Amour ES, Ballantyne CM, Shapiro MD, Moriarty PM, Baum SJ, Hurh E, Bartlett VJ, Kingsbury J, Figueroa AL, Alexander VJ, Tami J, Witztum JL, Geary RS, O’Dea LSL, Tsimikas S, Gaudet D: Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur Heart J. 2022 Apr 6;43(14):1401-1412. doi: 10.1093/eurheartj/ehab820. [Article]
- Karwatowska-Prokopczuk E, Tardif JC, Gaudet D, Ballantyne CM, Shapiro MD, Moriarty PM, Baum SJ, Amour ES, Alexander VJ, Xia S, Otvos JD, Witztum JL, Tsimikas S: Effect of olezarsen targeting APOC-III on lipoprotein size and particle number measured by NMR in patients with hypertriglyceridemia. J Clin Lipidol. 2022 Sep-Oct;16(5):617-625. doi: 10.1016/j.jacl.2022.06.005. Epub 2022 Jun 23. [Article]
- Hooper AJ, Bell DA, Burnett JR: Olezarsen, a liver-directed APOC3 ASO therapy for hypertriglyceridemia. Expert Opin Pharmacother. 2024 Oct;25(14):1861-1866. doi: 10.1080/14656566.2024.2408369. Epub 2024 Sep 26. [Article]
- Bergmark BA, Marston NA, Prohaska TA, Alexander VJ, Zimerman A, Moura FA, Murphy SA, Goodrich EL, Zhang S, Gaudet D, Karwatowska-Prokopczuk E, Tsimikas S, Giugliano RP, Sabatine MS: Olezarsen for Hypertriglyceridemia in Patients at High Cardiovascular Risk. N Engl J Med. 2024 May 16;390(19):1770-1780. doi: 10.1056/NEJMoa2402309. Epub 2024 Apr 7. [Article]
- FDA News: FDA approves drug to reduce triglycerides in adult patients with familial chylomicronemia syndrome [Link]
- FDA Approved Drug Products: TRYNGOLZA (olezarsen) injection, for subcutaneous use [Link]
| Clinical data | |
|---|---|
| Trade names | Tryngolza |
| Other names | IONIS-APOCIII-LRX |
| License data | US DailyMed: Olezarsen |
| Routes of administration | Subcutaneous |
| Drug class | Antisense oligonucleotide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2097587-83-02298451-31-5 |
| DrugBank | DB18728 |
| UNII | S3RS2SA30LNSY2BY6PSB |
| KEGG | D13023 |
////Olezarsen, FDA 2024, APPROVALS 2025, Tryngolza, ISIS-678354, ISIS 678354, familial chylomicronemia syndrome
Fitusiran
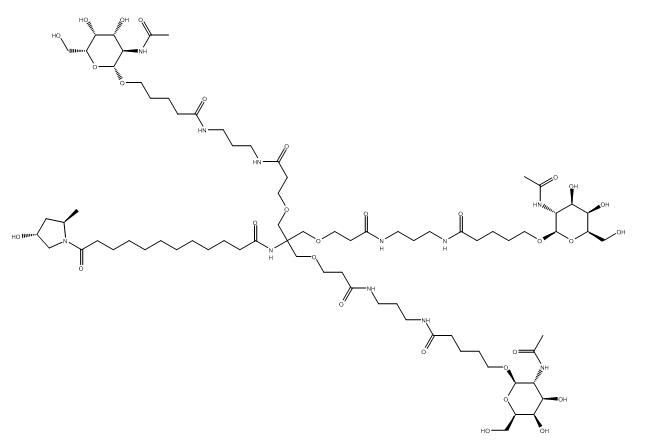
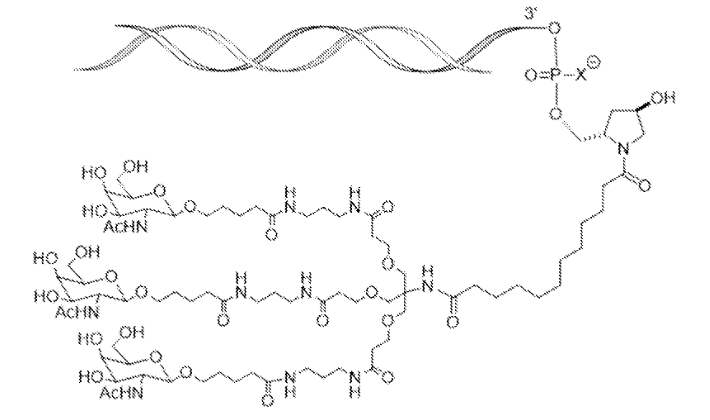
Fitusiran
1711.0 g/mol, C78H139N11O30
FDA APPROVED 3/28/2025, Qfitlia, To prevent or reduce the frequency of bleeding episodes in hemophilia A or B
Press Release
- CAS 1499251-18-1
- EX-A12034
- DA-53206
- N-[1,3-Bis[3-[3-[5-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxypentanoylamino]propylamino]-3-oxopropoxy]-2-[[3-[3-[5-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxypentanoylamino]propylamino]-3-oxopropoxy]methyl]propan-2-yl]-12-[(2R,4R)-4-hydroxy-2-methylpyrrolidin-1-yl]-12-oxododecanamide
Fitusiran Sodium

43 Sodium salt of duplex of [(2S,4R)-1-{1-[(2-acetamido-2-deoxy-β-D-galactopyranosyl)oxy]-16,16-bis({3-[(3-{5-[(2-acetamido-2-deoxy-β-D-galactopyranosyl)oxy]pentanamido}propyl)amino]-3-oxopropoxy}methyl)-5,11,18-trioxo-14-oxa-6,10,17-triazanonacosan-29-oyl}-4-hydroxypyrrolidin-2-yl]methyl hydrogen all–P–ambo-2′-deoxy-2′-fluoro-P-thioguanylyl-(3’→5′)-2′-O-methyl-P-thioguanylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methylcytidylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluoro-3′-adenylate and all–P–ambo-2′-O-methyl-P-thiouridylyl-(3’→5′)-2′-deoxy-2′-fluoro-P-thiouridylyl-(3’→5′)-2′-O-methylguanylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluoroguanylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-O-methylguanylyl-(3’→5′)-2′-O-methylguanylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methylguanylyl-(3’→5′)-2′-deoxy-2′-fluorouridylyl-(3’→5′)-2′-O-methyluridylyl-(3’→5′)-2′-deoxy-2′-fluoroadenylyl-(3’→5′)-2′-O-methyladenylyl-(3’→5′)-2′-deoxy-2′-fluorocytidylyl-(3’→5′)-2′-O-methyl-P-thiocytidylyl-(3’→5′)-2′-O-methyl-P-thioadenylyl-(3’→5′)-2′-O-methylguanosine
C520H636F21N175Na43O309P43S6 : 17193.39
[1609016-97-8]
Fitusiran, sold under the brand name Qfitlia, is a medication used for the treatment of hemophilia.[1] It is an antithrombin-directed small interfering ribonucleic acid.[1] It is given by subcutaneous injection.[1] Fitusiran reduces the amount of a protein called antithrombin.[2]
The most common side effects include viral infection, common cold symptoms (nasopharyngitis) and bacterial infection.[2]
Fitusiran was approved for medical use in the United States in March 2025.[2]
PATENT
https://patents.google.com/patent/WO2023240199A2/en
Medical uses
Fitusiran is indicated for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in people aged twelve years of age and older with hemophilia A or hemophilia B, with or without factor VIII or IX inhibitors (neutralizing antibodies).[1][2]
Adverse effects
The US Food and Drug Administration prescription label for fitusiran contains a boxed warning for thrombotic events (blood clotting) and gallbladder disease (with some recipients requiring gallbladder removal).[2] The label also has a warning about liver toxicity and the need to monitor liver blood tests at baseline and then monthly for at least six months after initiating treatment with fitusiran or after a dose increase of fitusiran.[2]
History
The efficacy and safety of fitusiran were assessed in two multicenter, randomized clinical trials which enrolled a total of 177 adult and pediatric male participants with either hemophilia A or hemophilia B.[2] In one study, participants had inhibitory antibodies to coagulation factor VIII or coagulation factor IX and previously received on-demand treatment with medicines known as “bypassing agents” for bleeding.[2] In the second study, participants did not have inhibitory antibodies to coagulation factor VIII or coagulation factor IX and previously received on-demand treatment with clotting factor concentrates.[2] In the two randomized trials, participants received either a fixed dose of fitusiran monthly or their usual on-demand treatment (bypassing agents or clotting factor concentrates) as needed for nine months.[2] The fixed dose of fitusiran is not approved because it led to excessive clotting in some participants.[2]
The US Food and Drug Administration (FDA) granted the application for fitusiran orphan drug and fast track designations. The FDA granted the approval of Qfitlia to Sanofi.
Society and culture
Legal status
Fitusiran was approved for medical use in the United States in March 2025.[2][3]
Names
Fitusiran is the international nonproprietary name.[4]
Fitusiran is sold under the brand name Qfitlia.[1][2]
References
^ Jump up to:a b c d e f “Qfitlia- fitusiran injection, solution”. DailyMed. 26 March 2025. Retrieved 2 April 2025.
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves Novel Treatment for Hemophilia A or B, with or without Factor Inhibitors”. U.S. Food and Drug Administration. 28 March 2025. Retrieved 29 March 2025. This article incorporates text from this source, which is in the public domain.
- ^ “Qfitlia approved as the first therapy in the US to treat hemophilia A or B with or without inhibitors”. Sanofi (Press release). 28 March 2025. Retrieved 29 March 2025.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1). hdl:10665/331046.
Further reading
Srivastava A, Rangarajan S, Kavakli K, Klamroth R, Kenet G, Khoo L, et al. (May 2023). “Fitusiran prophylaxis in people with severe haemophilia A or haemophilia B without inhibitors (ATLAS-A/B): a multicentre, open-label, randomised, phase 3 trial”. The Lancet. Haematology. 10 (5): e322 – e332. doi:10.1016/S2352-3026(23)00037-6. PMID 37003278.
- Young G, Kavakli K, Klamroth R, Matsushita T, Peyvandi F, Pipe SW, et al. (March 2025). “Safety and efficacy of a fitusiran antithrombin-based dose regimen in people with hemophilia A or B: the ATLAS-OLE study”. Blood. doi:10.1182/blood.2024027008. PMID 40053895.
- Young G, Srivastava A, Kavakli K, Ross C, Sathar J, You CW, et al. (April 2023). “Efficacy and safety of fitusiran prophylaxis in people with haemophilia A or haemophilia B with inhibitors (ATLAS-INH): a multicentre, open-label, randomised phase 3 trial”. Lancet (London, England). 401 (10386): 1427–1437. doi:10.1016/S0140-6736(23)00284-2. PMID 37003287.
External links
- Clinical trial number NCT03417102 for “A Study of Fitusiran (ALN-AT3SC) in Severe Hemophilia A and B Patients With Inhibitors (ATLAS-INH)” at ClinicalTrials.gov
- Clinical trial number NCT03417245 for “A Study of Fitusiran (ALN-AT3SC) in Severe Hemophilia A and B Patients Without Inhibitors” at ClinicalTrials.gov
- Clinical trial number NCT03754790 for “Long-term Safety and Efficacy Study of Fitusiran in Patients With Hemophilia A or B, With or Without Inhibitory Antibodies to Factor VIII or IX (ATLAS-OLE)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Qfitlia |
| Other names | ALN-AT3SC |
| License data | US DailyMed: Fitusiran |
| Routes of administration | Subcutaneous |
| Drug class | Anthithrombin production inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 1499251–18–1 |
| DrugBank | DB15002 |
| UNII | SV9W47ZLE1 |
| KEGG | D11810 |
| Chemical and physical data | |
| Formula | C520H636F21N175Na43O309P43S6 |
| Molar mass | 17193.48 g·mol−1 |
////////Fitusiran, Qfitlia, FDA 2025, APPROVALS 2025, EX-A12034, DA-53206
Gepotidacin




Gepotidacin
CAS
1075236-89-3 |
GSK2140944
WeightAverage: 448.527
Monoisotopic: 448.222288786 Chemical FormulaC24H28N6O3
(3R)-3-({4-[({2H,3H,4H-pyrano[2,3-c]pyridin-6-yl}methyl)amino]piperidin-1-yl}methyl)-1,4,7-triazatricyclo[6.3.1.0^{4,12}]dodeca-6,8(12),9-triene-5,11-dione
FDA APPROVED 3/25/2025,Blujepa, To treat uncomplicated urinary tract infections
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Gepotidacin hydrochloride | 30Z5B7ACV6 | 1075235-46-9 | DPAHPKBTWARMFG-FSRHSHDFSA-N |
| Gepotidacin mesylate | 5P7X0H2O6B | 1624306-20-2 | MTLHHQWYERWLIX-RGFWRHHQSA-N |
Gepotidacin, sold under the brand name Blujepa, is an antibiotic medication used for the treatment of urinary tract infection.[1] Gepotidacin is a triazaacenaphthylene bacterial type II topoisomerase inhibitor.[1][2] It is used as the salt gepotidacin mesylate, and is taken by mouth.[1]
Gepotidacin was approved for medical use in the United States in March 2025.[1][3]
SYNTHESIS
Gepotidacin
Gepotidacin (GSK2140944) is a triazaacenaphtylene developed by GSK and belongs to the class of Novel Bacterial Topoisomerase Inhibitors (NBTI). This new antibiotic is currently being investigated in three phase 3 clinical trials.
Gepotidacin is derived from the analogue GSK299423 described by Bax et al. [9], which results from a medicinal chemistry program initiated after an unbiased antibacterial screening [10].
2.2.1 Chemical synthesis
The synthesis of gepotidacin has been described in two patents in 2008 and 2016 and comprises 11 steps (Fig. 2) [11,12]. First, 2-chloro-6-methoxy-3-nitro-pyridine reacts with 2-amino-propane-1,3-diol through nucleophilic aromatic substitution (SNAr). The resulting diol is then protected with 2,2-dimethoxypropane in presence of p-toluenesulfonic acid (PTSA) followed by the reduction of the nitro group with hydrogen and 10% Pd/C. The aniline thus formed is then alkylated with ethyl bromoacetate. Cyclization is performed in basic conditions using sodium hydride, followed by oxidation using manganese dioxide. The acetal is then cleaved and the released diol reacts with methanesulfonic anhydride to form the third cycle of the triazaacenaphtylene core. Substitution with Boc-amino-piperidine, followed by deprotection and subsequent purification by chiral chromatography affords the primary amine derivative, which can be condensed by reductive amination with the corresponding aldehyde to give the free base of gepotidacin. The mono-hydrochloride salt is obtained by reaction with one equivalent of HCl 1 M in diethylether [13].

PATENT
WO2021219637A1
https://patents.google.com/patent/WO2021219637A1/en
Gepotidacin mesylate dihydrate (Form 1)
Example la – Preparation Method 1
Acetone (5 ml) was added to gepotidacin (294.14 mg). To the slurry, methanesulfonic acid (3M solution in water, 1 equivalent) was added over a period of 60 minutes. The slurry was heated to 50°C for 3 hours, cooled slowly to 20°C, left stirring at 20°C for 5 hours and cooled further to 5°C. The slurry was stirred at 5°C overnight. The crystalline solids were filtered under vacuum, washed with acetone and dried in a vacuum oven at 60°C to give crystalline gepotidacin mesylate dihydrate (Form 1) in 72.9% yield.
References:
GLAXO GROUP LIMITED WO2008/128942, 2008, A1Yield:-
Steps:
Multi-step reaction with 12 steps
1.1: ethanol; water / 4 h / 0 °C / Heating / reflux
2.1: toluene-4-sulfonic acid / 20 °C
2.2: 0.33 h
3.1: hydrogen / palladium 10% on activated carbon / 1,4-dioxane / 20 °C / 760.05 Torr
4.1: potassium carbonate / N,N-dimethyl-formamide / 20 °C
5.1: sodium hydride / tetrahydrofuran / 3.25 h / 0 – 20 °C
6.1: manganese(IV) oxide / dichloromethane / 2 h / 20 °C
7.1: hydrogenchloride; water / tetrahydrofuran / 1 h / 20 °C
7.2: pH ~ 8
8.1: triethylamine / chloroform / 4.5 h / Heating / reflux
9.1: pyridine / acetonitrile / 5 h / 50 – 90 °C
10.1: hydrogenchloride / 1,4-dioxane; dichloromethane / 1 h / 20 °C
11.1: isopropylamine / methanol; acetonitrile / Resolution of racemate
12.1: methanol; chloroform / 20 °C
12.2: 0.5 h / 20 °C
Example 10 (lR)-l-({4-[(3,4-Dihydro-2H-pyrano[2,3-c]pyridin-6-ylmethyl)amino]- l-piperidinyl}methyl)-l,2-dihydro-4H,9H-imidazo[l,2,3-//]-l,8-naphthyridine-4,9- dione hydrochloride

A suspension of (\R)- 1 -[(4-amino- 1 -piperidinyl)methyl]- 1 ,2-dihydro-4Η,9Η- imidazo[l,2,3-ij]-l,8-naphthyridine-4,9-dione (for a preparation see Example 5(j)) (51 mg, 0.14 mmol) in chloroform:methanol (9:1, 3 ml) at rt under argon was treated with triethylamine (0.06ml) and stirred at rt for 10 min. The solution was then treated with 1,3- dihydrofuro[3,4-c]pyridine-6-carbaldehyde (for a synthesis see WO2004058144,
Example 126(e)) (21mg, 0.133mmol) and stirred for a further 2h. The solution was then treated with NaBH(OAc)3 (87mg) and stirred at rt for 2h. The reaction was then treated with saturated aqueous NaHCO (10ml) and extracted with 20% methanol/DCM (3 x 50ml). The combined organic extracts were dried (MgSO ), filtered, evaporated and chromatographed (0-20% methanol/DCM) to give the free base of the title compound as a light brown solid (20mg, 32%) MS (ES+) m/z 448 (MH+). δH (CDCl3, 400MHz) 1.15-1.49 (2H, m), 1.61-1.95 (2H, m), 1.99-2.09 (2H, m) 2.20-2.38 (IH, m), 2.45-2.85 (6H, m), 2.92-3.02(1H, m), 3.05-3.15 (IH, m), 3.78 (2H, s), 4.20 (2H, t), 4.30-4.42 (IH, m), 4.52-4.61 (IH, m), 4.95-5.05 (IH, m), 6.23-6.32 (2H, m), 7.00 (IH, s), 7.47-7.50 (2H, m), 8.07 (IH, s).
The free base in DCM was treated with one equivalent IM HCl in diethyl ether and then evaporated to give the title monohydrochloride salt.
PATENT
WO2004058144
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004058144&_cid=P20-M9AS9E-95245-1
Medical uses
Gepotidacin is indicated for the treatment of females aged twelve years of age and older weighing at least 40 kilograms (88 lb) with uncomplicated urinary tract infections (uUTI) caused by Escherichia coli, Klebsiella pneumoniae, Citrobacter freundii complex, Staphylococcus saprophyticus, and Enterococcus faecalis.[1]
Society and culture
Legal status
In October 2024, gepotidacin was granted priority review by the US Food and Drug Administration (FDA) for the treatment of uncomplicated urinary tract infections.[4]
Gepotidacin was approved for medical use in the United States in March 2025.[1][5]
Names
Gepotidacin is the international nonproprietary name.[6]
Gepotidacin is sold under the brand name Blujepa.[1][5]
Research
Gepotidacin is being studied for the treatment of Neisseria gonorrhoeae (gonorrhea) infection, including multidrug resistant strains.[7][8]
References
- ^ Jump up to:a b c d e f g h “Blujepa- gepotidacin tablet, film coated”. DailyMed. 25 March 2025. Retrieved 2 April 2025.
- ^ Biedenbach DJ, Bouchillon SK, Hackel M, Miller LA, Scangarella-Oman NE, Jakielaszek C, et al. (January 2016). “In Vitro Activity of Gepotidacin, a Novel Triazaacenaphthylene Bacterial Topoisomerase Inhibitor, against a Broad Spectrum of Bacterial Pathogens”. Antimicrobial Agents and Chemotherapy. 60 (3): 1918–1923. doi:10.1128/aac.02820-15. PMC 4776004. PMID 26729499.
- ^ Fick M, Sneha SK, Sunny ME (2025). “FDA approval”. Reuters.
- ^ “GSK’s investigational antibiotic granted FDA priority review for urinary tract infections”. PMLiVE. 18 October 2024. Retrieved 21 October 2024.
- ^ Jump up to:a b “Blujepa (gepotidacin) approved by US FDA for treatment of uncomplicated urinary tract infections (uUTIs) in female adults and pediatric patients 12 years of age and older”. GSK (Press release). 25 March 2025. Retrieved 28 March 2025.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 74”. WHO Drug Information. 29 (3). hdl:10665/331070.
- ^ Scangarella-Oman NE, Hossain M, Dixon PB, Ingraham K, Min S, Tiffany CA, et al. (December 2018). “Microbiological Analysis from a Phase 2 Randomized Study in Adults Evaluating Single Oral Doses of Gepotidacin in the Treatment of Uncomplicated Urogenital Gonorrhea Caused by Neisseria gonorrhoeae“. Antimicrobial Agents and Chemotherapy. 62 (12). doi:10.1128/AAC.01221-18. PMC 6256812. PMID 30249694.
- ^ Jacobsson S, Golparian D, Scangarella-Oman N, Unemo M (August 2018). “In vitro activity of the novel triazaacenaphthylene gepotidacin (GSK2140944) against MDR Neisseria gonorrhoeae“. The Journal of Antimicrobial Chemotherapy. 73 (8): 2072–2077. doi:10.1093/jac/dky162. PMC 6927889. PMID 29796611.
Further reading
- Wagenlehner F, Perry CR, Hooton TM, Scangarella-Oman NE, Millns H, Powell M, et al. (February 2024). “Oral gepotidacin versus nitrofurantoin in patients with uncomplicated urinary tract infection (EAGLE-2 and EAGLE-3): two randomised, controlled, double-blind, double-dummy, phase 3, non-inferiority trials”. Lancet. 403 (10428): 741–755. doi:10.1016/S0140-6736(23)02196-7. PMID 38342126. S2CID 267548740.
External links
- Clinical trial number NCT04020341 for “A Study to Evaluate Efficacy and Safety of Gepotidacin in the Treatment of Uncomplicated Urinary Tract Infection (UTI)” at ClinicalTrials.gov
- Clinical trial number NCT04187144 for “Comparative Study to Evaluate Efficacy and Safety of Gepotidacin to Nitrofurantoin in Treatment of Uncomplicated Urinary Tract Infection (UTI)” at ClinicalTrials.gov
- Ross JE, Scangarella-Oman NE, Flamm RK, Jones RN: Determination of disk diffusion and MIC quality control guidelines for GSK2140944, a novel bacterial type II topoisomerase inhibitor antimicrobial agent. J Clin Microbiol. 2014 Jul;52(7):2629-32. doi: 10.1128/JCM.00656-14. Epub 2014 Apr 23. [Article]
- Oviatt AA, Gibson EG, Huang J, Mattern K, Neuman KC, Chan PF, Osheroff N: Interactions between Gepotidacin and Escherichia coli Gyrase and Topoisomerase IV: Genetic and Biochemical Evidence for Well-Balanced Dual-Targeting. ACS Infect Dis. 2024 Apr 12;10(4):1137-1151. doi: 10.1021/acsinfecdis.3c00346. Epub 2024 Mar 5. [Article]
- GSK Press Release: Blujepa (gepotidacin) approved by US FDA for treatment of uncomplicated urinary tract infections (uUTIs) in female adults and paediatric patients 12 years of age and older [Link]
- FDA Approved Drug Products: Blujepa (gepotidacin) tablets for oral use (March 2025) [Link]
| Clinical data | |
|---|---|
| Trade names | Blujepa |
| Other names | GSK2140944 |
| AHFS/Drugs.com | Blujepa |
| License data | US DailyMed: Gepotidacin |
| Routes of administration | By mouth |
| ATC code | J01XX13 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1075236-89-3 |
| DrugBank | DB12134 |
| ChemSpider | 34982930 |
| UNII | DVF0PR037D5P7X0H2O6B |
| KEGG | D10878D10879 |
| ECHA InfoCard | 100.249.088 |
| Chemical and physical data | |
| Formula | C24H28N6O3 |
| Molar mass | 448.527 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
///////Gepotidacin, FDA 2025, APPROVALS 2025, Blujepa, GSK-2140944, GSK2140944
Vimseltinib
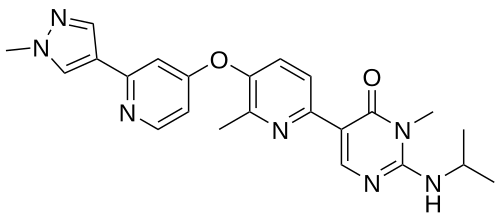


Vimseltinib
1628606-05-2 |
2/14/2025 FDA APPROVED, Romvimza
3-methyl-5-[6-methyl-5-[2-(1-methylpyrazol-4-yl)pyridin-4-yl]oxypyridin-2-yl]-2-(propan-2-ylamino)pyrimidin-4-one
C23H25N7O2, 431.5
- DP-6865
- PX9FTM69BF
- DCC3014
- UNII-PX9FTM69BF
- WHO 11443
DCC-3014- DP-6865
To treat symptomatic tenosynovial giant cell tumor for which surgical resection will potentially cause worsening functional limitation or severe morbidity
Vimseltinib is an orally bioavailable inhibitor of the tyrosine kinase receptor colony stimulating factor 1 receptor (CSF1R; CSF-1R; C-FMS; CD115; M-CSFR), with potential antineoplastic, macrophage checkpoint-inhibitory and immunomodulating activities. Upon administration, vimseltinib targets and binds to CSF1R expressed on monocytes, macrophages, and osteoclasts and inhibits the binding of the CSF1R ligands colony-stimulating factor-1 (CSF-1) and interleukin-34 (IL-34), to CSF1R. This prevents CSF1R activation and CSF1R-mediated signaling in these cells. This blocks the production of inflammatory mediators by macrophages and monocytes and reduces inflammation. By blocking the recruitment to the tumor microenvironment (TME) and activity of CSF1R-dependent tumor-associated macrophages (TAMs), vimseltinib inhibits the immunomodulating activity by macrophages and enhances T-cell infiltration and anti-tumor T-cell immune responses, which inhibits the proliferation of tumor cells. TAMs play key roles in the TME and allow for immune suppression; TAMs promote inflammation, tumor cell proliferation, angiogenesis, invasiveness and survival.
Vimseltinib, sold under the brand name Romvimza, is an anti-cancer medication used for the treatment of tenosynovial giant cell tumor.[1][2] Vimseltinib is a kinase inhibitor.[1][2] Vimseltinib is a macrophage colony-stimulating factor receptor antagonist.[3]
The most common adverse reactions, including laboratory abnormalities, include increased aspartate aminotransferase, periorbital edema, fatigue, rash, increased cholesterol, peripheral edema, face edema, decreased neutrophils, decreased leukocytes, pruritus, and increased alanine aminotransferase.[2]
Vimseltinib was approved for medical use in the United States in February 2025.[2][4]
PATENT
vimseltinib is a c-FMS (CSF-IR) and c-KIT dual inhibitor with anticancer and antiproliferative activities, can excite tyrosine protein kinase activity, influence protooncogene transcription, and is widely applied to research of anticancer drugs as an active molecule.
CN105120864B discloses heating the reaction mixture in a sealed tube at 100 ℃ for 2 days. The mixture was then cooled to room temperature, the solids were removed by filtration and the filtrate was concentrated to dryness and purified by silica gel chromatography to give 2- (isopropylamino) -3-methyl-5- (6-methyl-5- ((2- (1-methyl-1H-pyrazol-4-yl) pyridin-4-yl) oxy) pyridin-2-yl) pyrimidin-4 (3H) -one, amorphous form described.
CN113880812a reports another preparation method of Vimseltinib, and a small amount of target product meeting the requirement is finally obtained through a column chromatography purification process. The preparation method has complicated process and is not beneficial to industrialized mass production. There is no mention in this patent of reports on solid or crystalline forms of the compound of formula (I), and the purification process of column chromatography (EA/meoh=120:1 to 100:1) was repeated to give form a.
CN116283919A
https://patents.google.com/patent/CN116283919A/en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014145025&_cid=P21-M98JKR-94364-1
Example 2: A solution of Example C2 (0.13 g, 0.309 mmol) in DCM (5 mL) was treated portion-wise with mCPBA (0.09 g, 0.37 mmol), stirred at RT overnight, treated with TEA (0.5 mL) and Ν,Ν-dimethylamine HCl salt (500 mg) and stirred at RT for 2 h. The mixture was treated with satd. NaHCO3, extracted with DCM (2x) and the combined organics were dried over Na2SO4, concentrated to dryness and purified via silica gel chromatography (MeOH/DCM) to obtain 4-methoxy-N,N-dimethyl-5-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)pyrimidin-2-amine (60 mg, 47%). MS (ESI) m/z: 418.2 (M+H+).
[0199] A solution of 4-methoxy-N,N-dimethyl-5-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)pyrimidin-2-amine (0.060 g, 0.144 mmol) in acetic acid (5 mL) was treated with HBr (0.065 mL, 0.575 mmol), heated at 90°C for 6 h, cooled to RT and quenched with ice water. The solution was treated with NaHCO3 and NaCl, extracted with 1 : 1 THF/EtOAc (3x) and the combined organics were dried over Na2SO4 and concentrated to dryness. The material was treated with MeCN (1 mL), allowed to stand at RT and the
resulting solid was collected via filtration to afford 2-(dimethylamino)-5-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)pyrimidin-4(3H)-one (43 mg, 71%). 1H NMR (400 MHz, DMSO-d6): δ 11.23 (s, 1 H), 8.73 (s, 1 H), 8.36 (d, J = 5.7 Hz, 1 H), 8.30 (m, 1H), 8.26 (s, 1 H), 7.97 (s, 1 H), 7.51 (m, 1H), 7.23 (d, J = 2.4 Hz, 1 H), 6.62 (br s, 1 H), 3.85 (s, 3 H), 3.12 (s, 6 H), 2.35 (s, 3 H); MS (ESI) m/z: 404.2 (M+H+).
Example 3: A solution of Example C2 (0.13 g, 0.309 mmol) in DCM (5 mL) was treated portion-wise with mCPBA (0.09 g, 0.37 mmol), stirred at RT overnight, treated with isopropyl amine (0.5 mL) and stirred at RT overnight. The mixture was treated with satd. NaHCO3, extracted with DCM (2x) and the combined organics were dried over Na2SO4, concentrated to dryness and purified via silica gel chromatography (MeOH/DCM) to obtain N-isopropyl-4-methoxy-5-(6-methyl-5-((2-(1-methyl-1H-pyrazol-4-yl)pyridin-4-yl)oxy)pyridin-2-yl)pyrimidin-2-amine (63 mg, 47%). MS (ESI) m/z: 432.2 (M+H+).
PAPER
Discovery of vimseltinib (DCC-3014), a highly selective CSF1R switch-control kinase inhibitor, in clinical development for the treatment of Tenosynovial Giant Cell Tumor (TGCT)
https://www.sciencedirect.com/science/article/pii/S0960894X22004048

Medical uses
Vimseltinib is indicated for the treatment of adults with symptomatic tenosynovial giant cell tumor for which surgical resection will potentially cause worsening functional limitation or severe morbidity.[1][2]
History
The efficacy of vimseltinib was evaluated in MOTION (NCT05059262), a double-blind, multicenter, randomized (2:1), placebo-controlled trial in participants with tenosynovial giant cell tumor for whom surgical resection may cause worsening functional limitation or severe morbidity.[2] Eligible participants had a confirmed diagnosis of tenosynovial giant cell tumor with measurable disease (RECIST v1.1) with at least one lesion having a minimum size of 2 cm.[2] Pp[-[p;articipants were randomized to placebo or vimseltinib, 30 mg twice weekly administered for 24 weeks, during the double-blind period (part 1).[2] During the open-label period (part 2), patients could continue vimseltinib and those receiving placebos could crossover to vimseltinib.[2] Randomization was stratified by tumor location (lower limb versus all other) and region (United States versus Non-US).[2] A total of 123 participants were randomized: 83 to the vimseltinib arm and 40 to placebo during part 1.[2]
The US. Food and Drug Administration (FDA) granted the application for vimseltinib priority review designation.[2]
Society and culture
Legal status
Vimseltinib was approved for medical use in the United States in February 2025.[2][5]
Names
Vimseltinib is the international nonproprietary name.[6]
Vimseltinib is sold under the brand name Romvimza.[1][2]
References
- ^ Jump up to:a b c d e “Romvimza- vimseltinib capsule”. DailyMed. 18 February 2025. Retrieved 3 March 2025.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves vimseltinib for symptomatic tenosynovial giant cell tumor”. U.S. Food and Drug Administration (FDA). 14 February 2025. Retrieved 16 February 2025. This article incorporates text from this source, which is in the public domain.
- ^ Caldwell TM, Ahn YM, Bulfer SL, Leary CB, Hood MM, Lu WP, et al. (October 2022). “Discovery of vimseltinib (DCC-3014), a highly selective CSF1R switch-control kinase inhibitor, in clinical development for the treatment of Tenosynovial Giant Cell Tumor (TGCT)”. Bioorganic & Medicinal Chemistry Letters. 74: 128928. doi:10.1016/j.bmcl.2022.128928. PMID 35961460.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ “U.S. FDA Grants Full Approval of Deciphera’s Romvimza (vimseltinib) for the Treatment of Symptomatic Tenosynovial Giant Cell Tumor (TGCT)” (Press release). Deciphera Pharmaceuticals. 14 February 2025. Retrieved 16 February 2025 – via Business Wire.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85”. WHO Drug Information. 35 (1). hdl:10665/340684.
External links
- Clinical trial number NCT05059262 for “Study of Vimseltinib for Tenosynovial Giant Cell Tumor (MOTION)” at ClinicalTrials.gov
- Caldwell TM, Ahn YM, Bulfer SL, Leary CB, Hood MM, Lu WP, Vogeti L, Vogeti S, Kaufman MD, Wise SC, Le Bourdonnec B, Smith BD, Flynn DL: Discovery of vimseltinib (DCC-3014), a highly selective CSF1R switch-control kinase inhibitor, in clinical development for the treatment of Tenosynovial Giant Cell Tumor (TGCT). Bioorg Med Chem Lett. 2022 Oct 15;74:128928. doi: 10.1016/j.bmcl.2022.128928. Epub 2022 Aug 10. [Article]
- Smith BD, Kaufman MD, Wise SC, Ahn YM, Caldwell TM, Leary CB, Lu WP, Tan G, Vogeti L, Vogeti S, Wilky BA, Davis LE, Sharma M, Ruiz-Soto R, Flynn DL: Vimseltinib: A Precision CSF1R Therapy for Tenosynovial Giant Cell Tumors and Diseases Promoted by Macrophages. Mol Cancer Ther. 2021 Nov;20(11):2098-2109. doi: 10.1158/1535-7163.MCT-21-0361. Epub 2021 Aug 25. [Article]
- FDA Approved Drug Products: Romvimza (vimseltinib) capsules for oral use (February 2025) [Link]
- FDA News Release: FDA approves vimseltinib for symptomatic tenosynovial giant cell tumor [Link]
| Clinical data | |
|---|---|
| Trade names | Romvimza |
| License data | US DailyMed: Vimseltinib |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1628606-05-2 |
| PubChem CID | 86267612 |
| IUPHAR/BPS | 11190 |
| DrugBank | DB17520 |
| ChemSpider | 95499700 |
| UNII | PX9FTM69BF |
| KEGG | D12238 |
| ChEMBL | ChEMBL5095202 |
| Chemical and physical data | |
| Formula | C23H25N7O2 |
| Molar mass | 431.500 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
//////Vimseltinib, FDA 2025, APPROVALS 2025, Romvimza, DCC-3014, DCC 3014, DP-6865, PX9FTM69BF, C3014, WHO 11443, DCC-3014, DP-6865,
TREOSULFAN



TREOSULFAN
C6H14O8S2 MW 278.29
FDA APPROVED 1/21/2025 Grafapex
CAS
299-75-2 |
299-75-2
Treosulphan
Ovastat
Treosulfano
NSC-39069
- Dihydroxybusulfan
- L-threitol-1,4-dimethanesulfonate
[(2S,3S)-2,3-dihydroxy-4-methylsulfonyloxybutyl] methanesulfonate
Trecondi, Treosulfan was authorized for medical use in the European Union in June 2019
For use in combination with fludarabine as a preparative regimen for allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia and myelodysplastic syndrome
Treosulfan, sold under the brand name Trecondi among others, is an alkylating medication given to people before they have a bone marrow transplant from a donor known as allogeneic hematopoietic stem cell transplantation. It is used as a ‘conditioning’ treatment to clear the bone marrow and make room for the transplanted bone marrow cells, which can then produce healthy blood cells.[9][10] It is used together with another medicine called fludarabine in adults and children from one month of age with blood cancers as well as in adults with other severe disorders requiring a bone marrow transplant.[9] It belongs to the family of drugs called alkylating agents.[9] In the body, treosulfan is converted into other compounds called epoxides which kill cells, especially cells that develop rapidly such as bone marrow cells, by attaching to their DNA while they are dividing.[9]
The most common side effects include infections, nausea (feeling sick), stomatitis (inflammation of the lining of the mouth), vomiting, diarrhea, and abdominal pain (belly ache).[9] Tiredness, febrile neutropenia (low white blood cell counts with fever) and high blood levels of bilirubin (a breakdown product of red blood cells) are also seen in more than 1 in 10 adults, and rash also affects more than 1 in 10 children.[9] The most common adverse reactions include musculoskeletal pain, stomatitis, pyrexia, nausea, edema, infection, and vomiting.[7] Selected grade 3 or 4 nonhematological laboratory abnormalities include increased GGT, increased bilirubin, increased ALT, increased AST, and increased creatinine.[7]
Treosulfan was authorized for medical use in the European Union in June 2019,[9] and approved for medical use in the United States in January 2025.[7][11]
Medical Uses
Treosulfan in combination with fludarabine is indicated as part of conditioning treatment prior to allogeneic haematopoietic stem cell transplantation in adults with malignant and non malignant diseases, and in children older than one month with malignant diseases.[7][9]
History
Two main studies showed that treosulfan is at least as effective as busulfan, another medicine used to prepare people for haematopoietic stem cell transplantation.[9]
In one of the studies, involving 570 adults with acute myeloid leukaemia (a blood cancer) or myelodysplastic syndromes (conditions in which large numbers of abnormal blood cells are produced), 64% of patients given treosulfan (with fludarabine) had a successful transplant and were alive and disease-free after 2 years, compared with 51% of patients given busulfan (with fludarabine).[9]
In an additional study in 70 children with blood cancers, 99% of children given treosulfan (with fludarabine) were alive three months after their transplant.[9]
Efficacy was evaluated in MC-FludT.14/L Trial II (NCT00822393), a randomized active-controlled trial comparing treosulfan to busulfan with fludarabine as a preparative regimen for allogeneic transplantation. Eligible patients included adults 18 to 70 years old with AML or MDS, Karnofsky performance status ≥ 60%, and age ≥ 50 years or hematopoietic cell transplantation comorbidity index [HCTCI] score > 2. There were 570 patients randomized to treosulfan (n=280) or busulfan (n=290).
Society and culture
Legal status
Treosulfan was authorized for medical use in the European Union in June 2019,[9] and approved for medical use in the United States in January 2025.[11][12][13]
The US Food and Drug Administration granted orphan drug designation to treosulfan in 1994, for the treatment of ovarian cancer;[14] and in 2015, for conditioning treatment prior to hematopoietic stem cell transplantation in malignant and non-malignant diseases in adults and pediatric patients.[15]
In February 2004, orphan designation (EU/3/04/186) was granted by the European Commission to medac Gesellschaft fuer klinische Spezialpräparate mbH, Germany, for treosulfan for the conditioning treatment prior to haematopoietic progenitor cell transplantation.[16]
Names
Treosulfan is the international nonproprietary name.[17]
Treosulfan is sold under the brand names Trecondi[9] and Grafapex.[7]
SYN
Treosulfan is an active ingredient of the drug Ovastat . Treosulfan is indicated for the treatment of ovarian cancer and belongs to the class of alkylating agents, which prevents the growth and division of cancerous cells.
US3155702 discloses the preparation of Treosulfan by methanesulphonation of (2S,3S)- l,4-dibromobutane-2,3-diol with excess amount of silver methanesulphonate. The presence of free 2,3-diol in the starting material leads to side reactions and formation of undesired by-products which necessitates an additional purification step and thereby results in lower yields. Further, an additional filtration operation is also required to remove silver bromide salt generated during the process and un-reacted silver methanesulphonate, which makes the process less attractive for commercial manufacturing.
US3246012 discloses the preparation of Treosulfan by protection of hydroxyl group of dialkyl tartrates with corresponding aldehyde, ketone or a reactive derivatives to form corresponding cyclic 2,3-O-acetals and 2,3-O-ketals of butanetetrol esters followed by reduction using lithium aluminium hydride to obtain 2,3-O-acetal or ketal protected butanetetrol, which is further methanesulphonated and treated with acid. The use of highly pyrophoric and hazardous reducing agent renders the above process not ideal for industrial production. Organic Syntheses, Coll. Vol. 10, p. 297, 2004 discloses a similar reaction sequence followed by the final de-protection of methanesulphonated 2,3-O-diisopropylidene-L- threitol in methanesulfonic acid at reflux temperature, which leads to a sluggish reaction mixture and a higher number of impurities due to maintaining the reaction mixture for longer time at higher temperature.
IN 1568/MUM/2012 also discloses similar reaction sequence involving reduction of dimethyl-2,3-0-isopropylidene-L-tartrate by sodium-bis(2-methoxyethoxy) aluminium hydride followed by methanesulphonation and final deprotection with formic acid to yield Treosulfan.
KR101367641 describes reduction using lithium borohydride, which requires about 14 hours to complete the reaction and is further extended due to involvement of column chromatography purification. Tetrahedron, vol. 49, no. 30, p. 6645, 1993 describes reduction using sodium borohydride and lithium chloride, followed by flash chromatography purification. Reduction conditions as per Chem. Pharm. Bull. Vol. 42, No. 3, p. 68, 1994, are again not commercially feasible because of lithium aluminium hydride as reducing agent.
Haberland, M., Weber, S., Sharma, A. K., Upadhyay, S., Dua, H., Musmade, S., Singh, G., Lahiri, S., & Cabri, W. (2019). A process for the preparation of Treosulfan (Patent No. WO2019043587A2).
EXAMPLES Detailed experimental parameters suitable for the preparation of Treosulfan or intermediates according to the present invention are provided by the following examples, which are intended to be illustrative and not limiting.
Reference Example 1 (repetition of Tetrahedron, vol. 46, No. 12, p. 4165, 1990):
A reaction mixture of dimethyl-L-tartrate (10. Og), p-toluene sulfonic acid (0.013g) and p- anisaldehydedimethylacetal (l l.Og) in toluene (150ml) was refluxed and the azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture for 3-5 hours. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (50ml) and neutralised by addition of potassium carbonate (5.0g) followed by stirring for an hour . The reaction mixture was filtered and filtrate was evaporated to give yellow crude compound, which was further dissolved in dichloromethane (25ml) followed by addition of petroleum ether (100ml) and stirred for an hour at ambient temperature. The solid was filtered, washed with petroleum ether (20ml) and dried under vacuum at 35-40°C for 15-20 hours to obtain 16.63g (72.15%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-dicarboxylate having purity 98.4% by HPLC.
Reference Example 2 (repetition of Synthesis, No. 15, p. 2488-90, 2008):
A reaction mixture of dimethyl-L-tartrate (5.0g), p-toluene sulfonic acid (0.0064g) and p- anisaldehyde dimethylacetal (5.35g) in toluene (25ml) was refluxed and the azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture for 3-5 hours. The reaction mixture was cooled to ambient temperature, diluted with dichloromethane (25ml) and neutralised by addition of potassium carbonate (5.0g) followed by stirring for an hour. The reaction mixture was filtered and filtrate was evaporated to give yellow crude residues. The crude was further re-crystallized in petroleum ether (25ml), filtered the solid and washed with petroleum ether (15ml) followed by drying under vacuum at 35-40°C for 15-20 hours to obtain 7.4g (89.15%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-dicarboxylate having purity 98.8% by HPLC. Example-1: Preparation of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane- 4,5-dicarboxylate
A reaction mixture of dimethyl-L-tartrate (500g), p-toluene sulfonic acid (5.38g) and p- anisaldehyde dimethylacetal (665g) in toluene (2250ml) was refluxed to 110-115°C. The azeotropical mixture of toluene-methanol was continuously removed from the reaction mixture till the completion of the reaction. The reaction mixture was cooled to ambient temperature and quenched with aq. saturated sodium bicarbonate solution (2500ml), layers were separated. Resulting organic layer was washed with water (2500ml x 2) followed by evaporation of organic layer. Isopropyl alcohol (3500ml) was charged to the residue and heated to 60-70°C followed by cooling at ambient temperature. Reaction mixture was stirred at 0-5°C for 1-2 hours and filtered. The solid thus obtained was washed with pre- cooled isopropyl alcohol and dried under vacuum at 35-40°C for 15-20 hours to obtain 767.0g (92.93%) of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- dicarboxylate having purity 99.97% by HPLC.
Example-2: Preparation of (4S,5S)-2-(4-methoxyphenyl)-l 53-dioxo!ane-4,5- diyifdimethanol
Method-l :To a mixture of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- dicarboxylate (765g), Iodine (13. lg) in tetrahydrofuran (3750ml) and water (76ml), sodium borohydride (146.52g) was added at 0-15°C and stirred for 1 -2 hours at ambient temperature. The reaction was quenched with 30% aq. ammonium chloride (6100ml) solution and dichloromethane (7650ml). The layers were separated and the aqueous layer was extracted by dichloromethane (3800ml x 3) followed by washing of combined organic layers with water (3800ml), The resulting organic layer was evaporated at 35-65°C to obtain 525.0g (83.9%) of (4S,5S)-2-(4-methoxyphenyl)-l,3- dioxolane-4,5-diyl]dimethanol having purity 99.72% by HPLC. Method-2: To a mixture of dimethyl (4R,5R)-2-(4-methoxyphenyl)-l,3-dioxolane- 4,5-dicarboxylate (765g), Iodine (13.10g) in tetrahydrofuran (3750ml) and water (76.5ml), sodium borohydride (146.52g) was added at 0-10°C and stirred for Ihours at 0-5°C and stirred for 3-4 hours at ambient temperature. The reaction was quenched with 30% aq. ammonium chloride (6120ml) solution and dichloromethane (7650ml) at ambient temperature. The layers were separated and the aqueous layer was extracted by dichloromethane (3825m! x 3) followed by washing of combined organic layers with water (3825ml). The resulting organic layer was evaporated at 50-60°C to obtain 525 g (84.7%) of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-diyl]dirnethaiiol having purity 99.72% by HPLC. Example-3: Preparation of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene) dimethanesulfonate
Method-l:To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]dimethanol (145g) in dichloromethane (2175ml), pyridine (191g) and methanesulphonyl chloride (190. l g) was added at 0-5 °C. The reaction mixture was stirred for 2-3 hours at ambient temperature followed by quenching with water (1450ml). The organic layer was washed with water (1450ml x 4) and evaporated. The resulting residue was added to isopropanol (725ml) and stirred for 1-2 hours at ambient temperature and further for 1-2 hours at 0-5 C. The solid was filtered and washed with pre-cooled isopropanol (145ml). The resulting product was dissolved in acetone (1300ml) followed by addition of isopropanol (2610ml). Resulting reaction mixture was stirred for 1-2 hours at ambient temperature and then cooled at 0-5 °C. The solid thus obtained was filtered and washed with pre-cooled isopropanol (145ml x 2) and dried under vacuum at 30-35°C for 15-20 hours to give 190.8g (79.4%)of (4S,5S)-2-(4- methoxyphenyl)-l,3-dioxolane-4,5-diyl]bis(methylene) dimethanesulfonate. Method-2: To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]dimethanol (525, Og) in dichloromethane (7350ml), di-isopropylamine (663. Og) was added at ambient temperature followed by addition of methanesulphonyl chloride solution (624. Og in 525ml dichloromethane) at 0-10°C. The reaction mixture was stirred for 1-2 hours at 0-10 °C followed by stirring for 3-4 hours at ambient temperature. The organic layer was washed with water (2 x 5250ml) and evaporated. The residues were dissolved in acetone (4725ml) followed by addition of isopropanol (9450ml), stirred for about 1-2 hour at ambient temperature and then at 0-5 °C for 1-2 hours. The resulting solid was filtered, washed with pre-cooled isopropanol (525 x 2 ml)and dried under vacuum at 35-45°C for 15-20 hours to give 705.0g (81.45%) of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5-diyl]bis(methylene)
dimethanesulfonate having purity 99.92% by HPLC.
Example-4: Preparation of Treosulfan
Method-1: To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene) dimethanesulfonate (745. Og) in methanol (7450ml), concentrated hydrochloric acid (260ml) was added at 15-25°C followed by stirring for 10-15 hours at ambient temperature. The reaction mixture was cooled to 0-5°C and further stirred for 1-2 hours at 0-5°C followed by filtration and washing the solid with pre-cooled methanol (745ml). The solid thus obtained was dissolved in acetone (3725ml) followed by microne filtration. Di-isopropyl ether (7450ml) was added to the filtrate and stirred for 1-2 hours at ambient temperature and then cooled at 0-5°C. The solid thus obtained was filtered and washed with di-isopropyl ether (745ml x 2) followed by drying at 30-35°C for 15-20 hours to obtain 96.5g of Treosulfan having purity 99.9% by HPLC.
XRPD of Treosulfan obtained by above process is shown in Fig. 1. Method-2:To a solution of (4S,5S)-2-(4-methoxyphenyl)-l,3-dioxolane-4,5- diyl]bis(methylene)dimethanesulfonate (650. Og) in methanol (6500ml), 9N hydrochloric acid (227.5ml) was added at 0-10°C followed by stirring for 6-8 hours at ambient temperature. The reaction mixture was cooled to 0-5°C and further stirred for 1-2 hours followed by filtration and washing the solid with pre-cooled methanol (2 x 650ml). The solid thus obtained was dissolved in acetone (3250ml). Di-isopropyl ether (6500ml) was added to the resulting solution, stirred for 1-2 hours at ambient temperature and then cooled at 0-5°C. The solid thus obtained was filtered and washed with di- isopropyl ether (650ml x 2) followed by drying at 30-35°C for 15-20 hours to obtain 312g (68.4) of Treosulfan having purity 99.81% by HPLC.
PATENT
https://patents.google.com/patent/WO2020064815A1/en
Example 1 – Preparation of form B using water/isopropanol
99.8 mg treosulfan were weighed in a vial (volume 4.0 ml) which was equipped with a PTFE (Polytetrafluoroethylene) sealing and a stirrer. 1.5 ml of a mixture of 80 % by weight water and 20 % by weight isopropanol preheated to 65°C were then added. The resulting solution was completely taken up with a syringe (volume 5 ml) and filtered using a 0.2 pm filter into a second vial (volume 4.0 ml) . The syringe, second vial and filter had been tempered at 65°C before use. The solvents were allowed to evaporate from the open vial at room temperature to dryness which resulted in formation of crystals.
The XRPD pattern of the obtained crystals of form B according to the invention is shown in Figure 1.
PATENT
1568/MUM/2012
Abstract
Abstract: The present invention provides a convenient and cost-effective process for preparation of Treosulfan. The process comprises reduction of dimethyl 2,3-O-isopropylidene-L-tartrate with sodium-bis(2-methoxyethoxy)aluminum hydride to give the alcohol 2,3-O-isopropylidene-L-threitol (III), which on reaction with methanesulfonyl chloride led to 2,3-O-isopropylidene-L-threitol 1,4-bismethanesulfonate of formula (IV) and further treatment of compound (IV) with formic acid gave Treosulfan (I) having desired purity.
Treosulfan (I), chemically known as (2S,3S)-2,3-Dihydroxy-4-memylsidfonyIoxybutylj methanesulfonate is a drug commonly used for treating ovarian cancer. It belongs to the family of anti-cancer medicines called the alkylating agents, which prevent the growth and division of cancerous cells. Treosulfan has been used for bone-marrow ablation before stem-cell transplantation and in the treatment of malignant melanoma and breast cancer.
US 3,155,702 discloses synthesis of Treosulfan by replacement of the halogen function in L-Threitol-l,4-dibromobutane-2,3-diol, by treating with a large excess of an expensive reagent like silver methanesulfonate. Further, the presence of unprotected hydroxyl groups in the starting material inevitably leads to the formation of undesired impurities, which requires additional purification steps for removal of impurities as well for lowering the level of free silver in the active ingredient as per ICH guidelines, which results in lower yields and increases the costs substantially.
Another method reported in US 3,246,012 involves acetal formation of diethyl-L-tartrate with acetone to obtain 2,3-O-isopropylidene-diethyl-L-tartrate, which, when reduced with lithium aluminium hydride gives 2,3-0-methylene-L-threitol. The obtained alcohol was treated with methanesulfonyl chloride to yield the penultimate Treosulfan intermediate, 2,3-O-methylene-L-threitol-1,4-di-(methanesulfonate).
A similar approach which employs tartrate esters in the synthesis of Treosulfan, is disclosed in Organic Syntheses, (1993), Vol.8, p. 155 and Organic .Syntheses, (2004), Coll.Vol.10, p.297. L-tartaric acid is reacted with 2,2-dimethoxypropane in presence of methanol. The resulting methyl ester, dimethyl 2,3-O-isopropylidene-L-tartrate is reduced with lithium aluminium hydride to obtain 2,3-di-O-isopropylidene-L-threitol, which, upon reaction with methanesulfonyl chloride, followed by treatment with methanesulfonic acid yields Treosulfan.
Although these routes involve protection of the diol group and avoid impurities arising out of substitution at those alcohol functionalities, use of a highly pyrophoric, hazardous reagent such as lithium aluminium hydride severely limits their synthetic applicability, especially on commercial scale. Further, the final step involves reaction of 2,3-di-O-isopropylidene-L-threitol with methanesulfonic acid, which is quite sluggish and causes considerable rise in the total number of impurities due to long reaction time.
Thus, there is a need for a convenient, economical process for a commercial scale synthesis of Treosulfan (I), which overcomes the shortcomings of the prior art, does not involve use of hazardous, pyrophoric reagents and yields Treosulfan conforming to regulatory specifications.
The present inventors have developed a novel process for preparation of (2S,3S)-2,3-Dihydroxy-4-methylsulfonyloxybutyl] methanesulfonate (I). The scheme for synthesis comprises reaction of dimethyl 2,3-O-isopropylidene-L-tartrate of formula (II) with sodium-bis(2-methoxyethoxy) aluminum hydride to give the protected diol, 2,3-0-isopropylidene-L-threitoI (III), which on further treatment with methanesulfonyl chloride, followed by reaction of the resultant ester, 2,3-O-isopropyliden-L-threitol 1,4 bismethanesulfonate (IV) with formic acid, yields Treosulfan (I) having desired purity and with impurity levels conforming to ICH guidelines.
Scheme 1; Method embodied in the present invention for the preparation of Treosulfan (I)
In an embodiment, dimethyl 2,3 -O-isopropylidene-L-tartrate of formula (II) was treated with sodium-bis-(2-methoxyethoxy) aluminium hydride in presence of an organic solvent, and in the temperature range of 25 to 80°C, but preferably 60 to 75°C.
The organic solvent was selected from the group of toluene, xylenes, nitrobenzene, hexane, cyclohexane, heptane, N-methyl-2-pyrroIidone, ethers etc.
Upon completion of the reaction, as monitored by TLC, water was carefully added to the reaction mass and the mixture was extracted with a water immiscible organic solvent.
The organic solvent was selected from the group comprising of n-hexane, cyclohexane, heptane, methyl isobutyl ketone, 2-methyl tetrahydrofuran, cyclopentyl methyl ether etc.
The organic layer was separated and concentrated under reduced pressure to give 2,3-0-isopropylidene-L-threitol of formula (III) of desired purity.
It is pertinent to mention that the reaction was quite facile and the desired product was obtained with minimal formation of associated impurities and did not require any subsequent purification.
Further reaction of compound (III) with methanesulfonyl chloride was carried out at 25 to 35°C, in an organic solvent, in presence of an organic base.
The organic solvent was selected from the group comprising of chloroform, ethylene dichloride, dichloromethane, carbon tetrachloride etc., but preferably dichloromethane.
The organic base was selected from triethyl amine, tributyl amine and pyridine.
The reaction mixture was stirred at 25-35°C and after completion of the reaction as monitored by TLC, aqueous solution of sodium bicarbonate was added slowly to the reaction mass. The organic layer was separated, concentrated under reduced pressure and stirred with isopropyl alcohol to obtain the desired compound, 2,3-O-isopropylidene-L-threitol-l,4-bis(methanesulfonate) of formula (IV).
In a further embodiment, compound (TV) was hydrolyzed by treating with formic acid at 25 to 35°C based on TLC. After completion of the reaction, the reaction mass was concentrated and the product Treosulfan (I) was isolated by addition of isopropyl alcohol to the concentrated mass.
It is pertinent to mention that Organic Syntheses (2004), Coll.Vol. 10, p.297 discloses the hydrolysis reaction using methanesulfonic acid in ethanol at reflux temperature. However, the time taken for completion is about ten hours and the procedure is applicable only for laboratory scale reaction. The hydrolysis step disclosed in the present invention is easily scalable and so facile that it takes place at room temperature and within one to two hours. This reduces the time cycle for each batch run and also reduces the possibility of formation of undesired side products.
Dimethyl 2,3-O-isopropylidene-L-tartrate of formula (II) was prepared by the reaction of dimethyl -L-tartrate with acetone by following known synthetic procedures.
The following examples are meant to be illustrative of the present invention. These examples exemplify the invention and are not to be construed as limiting the scope of the invention.
EXAMPLES
Example 1: Synthesis of 2,3-O-isopropylidene-L-threitol (HI)
A solution of dimethyl-2,3-0-isopropylidene-L-tartrate (50.3 g) in toluene (50 ml) was gradually added to the stirred mixture of sodium-bis(2-methoxyethoxy) aluminum hydride (122.8 g) in toluene (50 ml) at 20-40°C. The reaction mixture was heated to 60-80°C, and the reaction was continued till completion, as monitored by TLC. When the reaction was complete, the mass was cooled to 25-3 5°C, quenched with careful addition of water (10ml) and concentrated. Treatment of the resulting residue with methyl tertiary butyl ether, followed by evaporation of the organic layer under reduced pressure afforded 2,3-0-isopropyliden -L-threitol ( III) as pale yellow oil. Yield: 29.8 g (81.2%) [α]D20 + 4.6.°(CHC13, c 5)
Example 2: Synthesis of 2,3-0-isopropylidene-L-threitol-l,4-bis(methanesulfonate)
(IV)
A stirred solution of 2,3-O-isopropylidene-L-threitol (100.2 g), methylene chloride (1250
ml) and pyridine (146.3 g) was cooled to 0-5°C and methanesulfonyl chloride (176.6 g)
was slowly added to it. Temperature of the reaction mixture was raised to 25-35°C and the
reaction was continued at the same temperature till completion of the reaction, as
monitored by HPLC. After completion of the reaction, aqueous sodium bicarbonate
solution was slowly added to the reaction mass and the organic layer was separated.
Aqueous layer from the reaction mixture was extracted with methylene chloride and the
organic layers were combined. Distillation of the organic solvent, optionally followed by
addition of isopropyl alcohol gave the product, 2,3-0-isopropylidene-L-threitol-l,4-
bis(methanesulfonate).
Yield: 160.7 g (79.7%)
[α]D20-21.6°(acetone,c2)
Example 3: Synthesis of Treosulfan (I)
A mixture of formic acid (98%, 1000 ml) and 2,3-0-isopropylidene-L-threitol-l,4-bis(methanesulfonate) (100.5 g) was stirred at room temperature until completion of the desired reaction, as monitored by TLC, When the reaction was complete, the reaction mass was concentrated under reduced pressure..
Treatment of the residue after evaporation with isopropanol yielded the final product Treosulfan, which was optionally subjected to further treatment with acetone and nexanes or petroleum ether, Yield: 74.3 g (85.0%) [α]D20 – 5.3°(acetone, c 2) Purity: > 99 %.
References
- ^ Jump up to:a b “Trecondi APMDS”. Therapeutic Goods Administration (TGA). 11 October 2022. Retrieved 25 January 2025.
- ^ “Updates to the Prescribing Medicines in Pregnancy database”. Therapeutic Goods Administration (TGA). 21 December 2022. Archived from the original on 3 April 2022. Retrieved 2 January 2023.
- ^ “Trecondi (Link Medical Products Pty Ltd T/A Link Pharmaceuticals)”. Therapeutic Goods Administration (TGA). 14 January 2025. Retrieved 25 January 2025.
- ^ “AusPAR: Trecondi”. Therapeutic Goods Administration (TGA). 4 July 2023. Retrieved 25 January 2025.
- ^ “Health product highlights 2021: Annexes of products approved in 2021”. Health Canada. 3 August 2022. Retrieved 25 March 2024.
- ^ “Treosulfan 5g Powder for Solution for Infusion – Summary of Product Characteristics (SmPC)”. (emc). Archived from the original on 20 May 2022. Retrieved 21 April 2020.
- ^ Jump up to:a b c d e f “Grafapex- treosulfan injection, powder, lyophilized, for solution”. DailyMed. 31 January 2025. Retrieved 2 April 2025.
- ^ “Trecondi Product Information” (PDF). European Medicines Agency (EMA). 21 April 2020.
- ^ Jump up to:a b c d e f g h i j k l m “Trecondi EPAR”. European Medicines Agency (EMA). 11 December 2018. Archived from the original on 16 March 2023. Retrieved 21 April 2020. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Romański M, Wachowiak J, Główka FK (October 2018). “Treosulfan Pharmacokinetics and its Variability in Pediatric and Adult Patients Undergoing Conditioning Prior to Hematopoietic Stem Cell Transplantation: Current State of the Art, In-Depth Analysis, and Perspectives”. Clinical Pharmacokinetics. 57 (10): 1255–1265. doi:10.1007/s40262-018-0647-4. PMC 6132445. PMID 29557088.
- ^ Jump up to:a b “FDA approves treosulfan with fludarabine as a preparative regimen for alloHSCT in adult and pediatric patients with AML or MDS”. U.S. Food and Drug Administration (FDA). 6 February 2025. Retrieved 8 March 2025. This article incorporates text from this source, which is in the public domain.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ “Medexus Announces FDA Approval of Grafapex (treosulfan) for Injection and Provides Business Update” (Press release). Medexus Pharmaceuticals. 22 January 2025. Retrieved 25 January 2025 – via Newsfile.
- ^ “Treosulfan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 16 May 1994. Retrieved 9 March 2025.
- ^ “Treosulfan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 8 April 2015. Retrieved 9 March 2025.
- ^ “EU/3/04/186”. European Medicines Agency (EMA). 17 September 2018. Archived from the original on 16 October 2019. Retrieved 21 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ World Health Organization (1972). “International nonproprietary names for pharmaceutical substances (INN). recommended INN: list 12”. WHO Chronicle. 26 (10).
External links
- “Treosulfan”. National Cancer Institute.
- [1]
- Clinical trial number NCT00822393 for “Clinical Phase III Trial Treosulfan-based Conditioning Versus Reduced-intensity Conditioning (RIC)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Trecondi, others |
| Other names | 1,2,3,4-Butanetetrol, 1,4-dimethanesulfonate, Threitol 1,4-dimethanesulfonate, Threitol 1,4-bismethanesulfonate; L-Threitol 1,4-bis(methanesulfonate); Threosulphan; Treosulphan; Tresulfan |
| AHFS/Drugs.com | International Drug Names |
| License data | US DailyMed: Treosulfan |
| Pregnancy category | AU: D[1][2] |
| Routes of administration | By mouth, intravenous |
| ATC code | L01AB02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)[1][3]<[4]CA: ℞-only[5]UK: POM (Prescription only)[6]US: ℞-only[7]EU: Rx-only[8]In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 299-75-2 |
| PubChem CID | 9882105 |
| DrugBank | DB11678 |
| ChemSpider | 8057780 |
| UNII | CO61ER3EPI |
| KEGG | C19557D07253 |
| ChEBI | CHEBI:82557 |
| CompTox Dashboard (EPA) | DTXSID0026173 |
| ECHA InfoCard | 100.005.529 |
| Chemical and physical data | |
| Formula | C6H14O8S2 |
| Molar mass | 278.29 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 101.5 to 105 °C (214.7 to 221.0 °F) |
| showSMILES | |
| showInChI | |
- Romanski M, Baumgart J, Bohm S, Glowka FK: Penetration of Treosulfan and its Active Monoepoxide Transformation Product into Central Nervous System of Juvenile and Young Adult Rats. Drug Metab Dispos. 2015 Dec;43(12):1946-54. doi: 10.1124/dmd.115.066050. Epub 2015 Oct 1. [Article]
- EMA Summary of Product Characteristics: Trecondi (treosulfan) powder for solution for infusion [Link]
- FDA Approved Drug Products: GRAFAPEX (treosulfan) for injection, for intravenous use [Link]
- EMC Summary of Product Characteristics: Treosulfan 5g Powder for Solution for Infusion [Link]
- NIH LiverTox: Alkylating Agents [Link]
- FDA News Release: FDA approves treosulfan with fludarabine as a preparative regimen for alloHSCT in adult and pediatric patients with AML or MDS [Link]
////////TREOSULFAN, Treosulphan, Ovastat, Treosulfano, Grafapex, acute myeloid leukemia, myelodysplastic syndrome, NSC-39069, Dihydroxybusulfan, L-threitol-1,4-dimethanesulfonate, Trecondi, FSA 2025, APPROVALS 2025, EMA 2019, EU 2019
CS(=O)(=O)OC[C@H](O)[C@@H](O)COS(C)(=O)=O
Suzetrigine



Suzetrigine
CAS
2649467-58-1 |
Average: 473.4
Monoisotopic: 473.137396951
Chemical Formula
C21H20F5N3O4
FDA 1/30/2025, Journavx
To treat moderate to severe acute pain
Press Release
- 2-Pyridinecarboxamide, 4-[[[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)tetrahydro-4,5-dimethyl-5-(trifluoromethyl)-2-furanyl]carbonyl]amino]-
- 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)-4,5- dimethyl-5-(trifluoromethyl)oxolane-2- carboxamido]pyridine-2-carboxamide
- 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)-4,5-dimethyl-5-(trifluoromethyl)oxolane-2-amido]pyridine2-carboxamide
- 4-[[[(2R,3S,4S,5R)-3-(3,4-Difluoro-2-methoxyphenyl)tetrahydro-4,5-dimethyl-5-(trifluoromethyl)-2-furanyl]carbonyl]amino]-2-pyridinecarboxamide
- CS-0641183
- HY-148800
- VX 548
- VX-548
- VX548
- Management of
Acute, moderate pain
Suzetrigine, sold under the brand name Journavx, is a medication used for the management of pain.[1][2] It is a non-opioid, small-molecule analgesic that works as a selective inhibitor of Nav1.8-dependent pain-signaling pathways in the peripheral nervous system,[3][4] avoiding the addictive potential of opioids. Suzetrigine is taken by mouth.[1]
The most common adverse reactions include itching, muscle spasms, increased blood level of creatine kinase, and rash.[1][2]
It was developed by Vertex Pharmaceuticals,[5] and was approved for medical use in the United States in January 2025.[2][6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines.[2]
Medical uses
Suzetrigine is indicated for the treatment of moderate to severe acute pain in adults.[1][2]
FDA Approves Novel Non-Opioid Treatment for Moderate to Severe Acute Pain
First Drug Approved in New Class of Non-Opioid Pain Medicines; Agency Continues to Take Steps to Support New Approaches for Pain Management
For Immediate Release:January 30, 2025
Today, the U.S. Food and Drug Administration approved Journavx (suzetrigine) 50 milligram oral tablets, a first-in-class non-opioid analgesic, to treat moderate to severe acute pain in adults. Journavx reduces pain by targeting a pain-signaling pathway involving sodium channels in the peripheral nervous system, before pain signals reach the brain.
Journavx is the first drug to be approved in this new class of pain management medicines.
Pain is a common medical problem and relief of pain is an important therapeutic goal. Acute pain is short-term pain that is typically in response to some form of tissue injury, such as trauma or surgery. Acute pain is often treated with analgesics that may or may not contain opioids.
The FDA has long supported development of non-opioid pain treatment. As part of the FDA Overdose Prevention Framework, the agency has issued draft guidance aimed at encouraging development of non-opioid analgesics for acute pain and awarded cooperative grants to support the development and dissemination of clinical practice guidelines for the management of acute pain conditions.
“Today’s approval is an important public health milestone in acute pain management,” said Jacqueline Corrigan-Curay, J.D., M.D., acting director of the FDA’s Center for Drug Evaluation and Research. “A new non-opioid analgesic therapeutic class for acute pain offers an opportunity to mitigate certain risks associated with using an opioid for pain and provides patients with another treatment option. This action and the agency’s designations to expedite the drug’s development and review underscore FDA’s commitment to approving safe and effective alternatives to opioids for pain management.”
The efficacy of Journavx was evaluated in two randomized, double-blind, placebo- and active-controlled trials of acute surgical pain, one following abdominoplasty and the other following bunionectomy. In addition to receiving the randomized treatment, all participants in the trials with inadequate pain control were permitted to use ibuprofen as needed for “rescue” pain medication. Both trials demonstrated a statistically significant superior reduction in pain with Journavx compared to placebo.
The safety profile of Journavx is primarily based on data from the pooled, double-blind, placebo- and active-controlled trials in 874 participants with moderate to severe acute pain following abdominoplasty and bunionectomy, with supportive safety data from one single-arm, open-label study in 256 participants with moderate to severe acute pain in a range of acute pain conditions.
The most common adverse reactions in study participants who received Journavx were itching, muscle spasms, increased blood level of creatine phosphokinase, and rash. Journavx is contraindicated for concomitant use with strong CYP3A inhibitors. Additionally, patients should avoid food or drink containing grapefruit when taking Journavx.
The application received Breakthrough Therapy, Fast Track and Priority Review designations by the FDA.
The FDA granted approval of Journavx to Vertex Pharmaceuticals Incorporated.
PATENTS
https://patentimages.storage.googleapis.com/08/4f/6e/4f104b27a3772f/US11919887.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=US407339565&_cid=P22-M90R90-47554-1



Step 1:
NEt₂ (7.7 mL, 55.2 mmol) was added to a solution of
ethyl 2-diazo-3-oxo-pentanoate (6.69 g, 39.3 mmol) in
DCM (80 mL) with stirring at 0° C. under nitrogen. Trimethylsilyl trifluoromethanesulfonate (8.5 mL, 47.0 mmol)
was added dropwise over 5 mins and the mixture was stirred
for a further 30 mins at 0° C. The reaction mixture was
diluted with pentane (100 mL), the layers separated and the
organic phase washed with dilute aqueous sodium bicarbonate (100 mL) and brine (100 mL). The organic layer was
dried (MgSO4), and concentrated in vacuo to give ethyl
(Z)-2-diazo-3-trimethylsilyloxy-pent-3-enoate (9.4 g, 99%)
as a red oil. H NMR (500 MHz, Chloroform-d) 8 5.33 (q,
J=7.0 Hz, 1H), 4.25 (q, J=7.1 Hz, 2H), 1.67 (d, J=7.0 Hz,
3H), 1.29 (t, J=7.1 Hz, 3H), 0.22 (s, 9H) ppm.
Step 2:
To a solution of 1,1,1-trifluoropropan-2-one (8 mL, 89.4
mmol) in DCM (80 mL) stirring at -78° C. was added TiCl
(70 mL of 1 M in DCM, 70.00 mmol) via cannula. To the
resulting solution, a solution of ethyl (Z)-2-diazo-3-trimethylsilyloxy-pent-3-enoate (36.1 g of 31.3% w/w, 46.6 mmol)
in 40 mL of DCM was added dropwise over 15 mins. After
100 mins the reaction was carefully quenched with water,
allowing the temperature to rise slowly, and then extracted
with DCM. The combined organic layers were dried
(MgSO), filtered, and concentrated in vacuo. Purification
by flash chromatography (330 g SiO₂, 0 to 20% EtOAc in
heptane) gave ethyl 2-diazo-6,6,6-trifluoro-5-hydroxy-4,5-
dimethyl-3-oxo-hexanoate (8.82 g, 67%), which was stored
as a solution in toluene. H NMR (500 MHz, Chloroform-d)
8 4.33 (q, J=7.1 Hz, 2H), 4.14 (q, J=7.0 Hz, 1H), 3.98 (s,
1H), 1.43 (q, J=1.2 Hz, 3H), 1.35 (t, J=7.1 Hz, 3H), 1.31 (dq.
J=7.0, 1.4 Hz, 3H) ppm. ESI-MS m/z calc. 282.08273, found
283.1 (M+1)*; 281.0 (M-1)-.
Step 3:
A solution of rhodium tetraacetate (245 mg, 0.55 mmol)
in benzene (32 mL) was heated at reflux for 10 min before
a solution of ethyl 2-diazo-6,6,6-trifluoro-5-hydroxy-4,5-
dimethyl-3-oxo-hexanoate (10 g, 35.4 mmol) in benzene (13
mL) was added slowly via addition funnel while refluxing
for 60 mins. The mixture was then concentrated in vacuo to
give ethyl rac-(4R, 5R)-4,5-dimethyl-3-oxo-5-(trifluoromethyl)tetrahydrofuran-2-carboxylate (9.0 g, 100%) as a
green coloured residue containing residual catalyst, and as a
mixture of epimers at the position next to the ester. This
material was used without further purification. H NMR
(500 MHz, Chloroform-d) 8 4.83-4.57 (m, 1H), 4.38-4.16
(m, 2H), 2.60 (dddd, J=9.3, 8.2, 5.6, 1.4 Hz, 1H), 1.73-1.63
(m, 3H), 1.30 (t, J=7.1 Hz, 3H), 1.24 (ddq, J=6.4, 4.1, 1.9
Hz, 3H) ppm.
Step 4:
To a stirred solution of ethyl rac-(4R,5R)-4,5-dimethyl- 5
3-oxo-5-(trifluoromethyl)tetrahydrofuran-2-carboxylate (48
g, 188.83 mmol) in DCM (400 mL) stirring at -78° C. was
added DIPEA (29.680 g, 40 mL, 229.64 mmol). A solution
of trifluoromethylsulfonyl trifluoromethanesulfonate
(53.440 g, 32 mL, 189.41 mmol) in DCM (200 mL) was 10
added to the reaction mixture at the same temperature over
1 h. The reaction mixture was stirred for 30 mins at 0° С.
before being quenched with 100 mL saturated aqueous
NaHCO3 solution. The organic layer was separated and
aqueous layer extracted with DCM (160 mL). The combined 15
organic layers were dried (MgSO) and concentrated in
vacuo to give ethyl rac-(4R,5R)-2,3-dimethyl-2-(trifluoromethyl)-4-(trifluoromethylsulfonyloxy)-3H-furan-5-carboxylate (71 g, 97%). H NMR (400 MHz, Chloroform-d) 8
4.38-4.32 (m, 2H), 3.29-3.23 (m, 1H), 1.64 (s, 3H), 1.37- 20
1.33 (m, 6H) ppm.
STEP 5
To stirred a solution of ethyl rac-(4R,5R)-2,3-dimethyl2-(trifluoromethyl)-4-(trifluoromethylsulfonyloxy)-3Hfuran-5-carboxylate (26 g, 67.311 mmol) in toluene (130.00
mL) was added (3,4-difluoro-2-methoxy-phenyl)boronic
acid (14 g, 74.5 mmol) followed by K3PO4 (100 mL of 2 M,
200.00 mmol) under an argon atmosphere. The reaction was
degassed before tetrakis(triphenylphosphine)palladium(0)
(4 g, 3.46 mmol) was added. After further degassing, the
reaction was heated at 100° C. for 2 hours. The reaction was
diluted in water and the aqueous layer extracted with EtOAc
(2×100 mL). The combined organic layers were concentrated in vacuo. Purification by flash chromatography (SiO.
0 to 10% EtOAc in heptane) gave ethyl 4-(3,4-difluoro-2- 35
methoxy-pheny1)-2,3-dimethyl-2-(trifluoromethyl)-3Hfuran-5-carboxylate (24.4 g, 93%) as a 6:1 diastereomeric
mixture, with the major isomer believed to be ethyl rac-(4R,
5R)-4-(3,4-difluoro-2-methoxy-phenyl)-2,3-dimethyl-2-
(trifluoromethyl)-3H-furan-5-carboxylate. Major isomer: H 40
NMR (400 MHz, Chloroform-d) 8 6.88-6.79 (m, 2H), 4.17-
4.09 (m, 2H), 3.90 (s, 3H), 3.46 (q, J=7.4 Hz, 1H), 1.67 (s,
3H), 1.12 (t, J=7.4 Hz, 3H), 1.06 (dd, J=5.4, 2.7 Hz, 3Н)
ppm. Minor isomer ¹H NMR (400 MHz, Chloroform-d) 8
6.88-6.79 (m, 2H), 4.17-4.09 (m, 2H), 3.88 (s, 3H), 3.76- 45
3.71 (m, 1H), 1.51 (s, 3H), 1.12 (t, J=7.4 Hz, 3H), 0.99 (dd,
J=5.4, 2.7 Hz, 3H) ppm. ESI-MS m/z calc. 380.1047, found
381.02 (M+1)+.
Step 6:
To an ice-cooled solution of ethyl 4-(3,4-difluoro-2- 50
methoxy-phenyl)-2,3-dimethyl-2-(trifluoromethyl)-3Hfuran-5-carboxylate (110 g, 243.0 mmol) in DCM (360 mL)
was added BBr, (370 mL of 1 M, 370.0 mmol) dropwise.
Upon completion the mixture was quenched by addition of
water and aqueous sodium bicarbonate solution, the aqueous 55
layer extracted with DCM and the combined organic layers
dried (MgSO) and concentrated in vacuo. The residue was
dissolved in DCM (430 mL) at ambient temperature and
TFA (40 mL, 519.2 mmol) was added, then the reaction was
heated to 45° C. Upon completion, the mixture was
quenched by addition of aqueous sodium bicarbonate solution and the aqueous layer extracted with DCM, dried
(MgSO) and concentrated in vacuo to give the desired
product in a 5:1 mixture of diastereomers. Recrystallization
was carried out by solubilizing the crude in the smallest
possible amount of DCM and adding a layer of heptane on
top of this solution (liquid-liquid diffusion). After approx. 1



https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021113627&_cid=P22-M90RUB-70989-1

Example 6
rel-(2S,3R,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (20), (2S,3R,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)- 5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (21), rel- (2R,3S,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2- carbonyl]amino]pyridine-2-carboxamide (22), and (2R,3S,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy- phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (23)
[00676] Step 7:
[00677] (4-[[3-(3-Chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (420 mg, 0.8827 mmol) was separated by chiral SFC [(R,R)-Whelk-O1 column, 5 µm particle size, 25 cm x 21.2 mm from Regis Technologies, MeOH, 20 mM NH3], followed by further purification of one or more of the fractions by chiral SFC using a Chiralpak IC column, 5 µm particle size, 25 cm x 20 mm from Daicel or a Chiralpak ID column, 5 µum particle size, 25 cm x 20 mm from Daicel to give:
[00678] First Eluting Isomer: rel-(2S,3R,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (20, 30 mg, 7.1%) (further purified by chiral SFC using Chiralpak IC column). 1H NMR (500 MHz, Chloroform-d) δ 8.92 (s, 1H), 8.47 (d, J = 5.5 Hz, 1H), 8.21 (dd, J = 5.6, 2.1 Hz, 1H), 8.09 (d, J = 2.2 Hz, 1H), 7.87 (d, J = 4.1 Hz, 1H), 7.26 (dd, J = 8.8, 5.8 Hz, 1H), 7.03 (t, J = 8.4 Hz, 1H), 5.87 – 5.82 (m, 1H), 4.77 (d, J = 10.6 Hz, 1H), 3.98 (td, J = 11.2, 8.3 Hz, 1H), 3.88 (s, 3H), 2.51 (dd, J = 13.2, 11.7 Hz, 1H), 2.42 (dd, J = 13.2, 8.3 Hz, 1H), 1.69 (s, 3H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00679] Second Eluting Isomer: (2S,3R,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (21, 29 mg, 6.7%) (further purified by chiral SFC using Chiralpak ID column). 1H NMR (500 MHz, Chloroform-d) δ 8.56 (s, 1H), 8.48 (d, J = 5.5 Hz, 1H), 8.08 (dd, J = 5.5, 2.2 Hz, 1H), 7.98 (d, J = 2.1 Hz, 1H), 7.86 (d, J = 4.4 Hz, 1H), 7.23 (dd, J = 8.8, 5.8 Hz, 1H), 7.01 (t, J = 8.4 Hz, 1H), 5.86 (d, J = 4.2 Hz, 1H), 4.80 (d, J = 9.7 Hz, 1H), 4.10 – 4.00 (m, 1H), 3.93 (s, 3H), 3.52 – 3.48 (m, 1H), 2.86 (dd, J = 13.9, 8.4 Hz, 1H), 2.16 -2.07 (m, 1H), 1.64 (s, 2H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00680] Third Eluting Isomer: rel-(2R,3S,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (22, 42 mg, 9.5%).
1H NMR (500 MHz, Chloroform-d) δ 8.87 (s, 1H), 8.33 (d, J = 5.6 Hz, 1H), 8.08 (dd, J = 5.6, 2.2 Hz, 1H), 7.98 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 4.5 Hz, 1H), 7.12 (dd, J = 8.8, 5.8 Hz, 1H), 6.89 (t, J = 8.4 Hz, 1H), 5.79 (d, J = 4.5 Hz, 1H), 4.63 (d, J = 10.7 Hz, 1H), 3.85 (td, J = 11.2, 8.4 Hz, 1H), 3.74 (s, 3H), 2.37 (dd, J = 13.2, 11.7 Hz, 1H), 2.28 (dd, J = 13.1, 8.4 Hz, 1H), 1.55 (s, 3H) ppm. ESI-MS m/z calc.
475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00681] Fourth Eluting Isomer: (2R,3S,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (23, 40 mg, 8.8%).
1H NMR (500 MHz, Chloroform-d) δ 8.43 (s, 1H), 8.35 (d, J = 5.5 Hz, 1H), 7.95 (dd, J = 5.5, 2.2 Hz, 1H), 7.85 (d, J = 2.2 Hz, 1H), 7.73 (d, J = 4.3 Hz, 1H), 7.10 (dd, J = 8.8, 5.9 Hz, 1H), 6.87 (t, J = 8.4 Hz, 1H), 5.76 – 5.71 (m, 1H), 4.67 (d, J = 9.7 Hz, 1H), 3.97 – 3.87 (m, 1H), 3.80 (s, 3H), 2.73 (dd, J = 13.9, 8.4 Hz, 1H), 1.98 (dd, J = 13.9, 11.6 Hz, 1H), 1.51 (s, 3H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00682] Compound 22 – Solid Form A
Efficacy
When people used suzetrigine in clinical studies conducted through 2024, there was a reduction in pain typically from seven to four on the standard numerical scale used to rate pain.[7][8] Suzetrigine provided pain relief equal to a combination of hydrocodone and paracetamol (acetaminophen) (5 mg of hydrocodone bitartrate and 325 mg of acetaminophen).[8][9]
Suzetrigine suppresses pain at the same level as an opioid, but without the risks of addiction, sedation, or overdose.[10] An alternative to opioids, it is the first pain medication to be approved by the Food and Drug Administration in two decades.[10]
The efficacy of suzetrigine was evaluated in two randomized, double-blind, placebo- and active-controlled trials of acute surgical pain, one following abdominoplasty and the other following bunionectomy.[2] Both trials found that suzetrigine reduced pain more effectively than a placebo.[2]
Contraindications
Concomitant use of suzetrigine with strong CYP3A inhibitors is contraindicated.[1][2]
Adverse effects
Common adverse effects of suzetrigine may include itching, rash, muscle spasms, and increased levels of creatine kinase.[2] Mild side effects may include nausea, constipation, headache, and dizziness.[7][8] As of 2024, long-term safety and side effects remain undetermined.[8]
In preliminary research, suzetrigine had no serious neurological, behavioral, or cardiovascular effects.[3]
Interactions
Consuming grapefruit while using suzetrigine may cause an adverse grapefruit–drug interaction.[1][2]
Mechanism of action
Suzetrigine operates on peripheral nerves, avoiding the addictive potential of opioids which affect the central nervous system.[3][4][7] Unlike opioid medications, which reduce pain signals in the brain, suzetrigine works by closing sodium channels in peripheral nerves, inhibiting pain-signaling nerves from transmitting painful sensations to the brain.[3][4][7]
In pharmacological studies, suzetrigine selectively inhibited Nav1.8 channels, but not other voltage-gated sodium channels, and bound to a unique site on these sodium channels with a novel allosteric mechanism, by binding to the channel’s second voltage sensing domain, thereby stabilizing the closed state, causing tonic inhibition. It exerts its action on dorsal root ganglion.[3]
History
Vertex Pharmaceuticals announced in January 2024 that suzetrigine had successfully met several endpoints in its Phase III clinical trials.[5] The company announced in July 2024 that the FDA had accepted a new drug application for suzetrigine.[11] The FDA granted the application for suzetrigine priority review, fast track, and breakthrough therapy designations.[2][11] In January 2025, the FDA granted approval of Journavx to Vertex Pharmaceuticals.[2]
Society and culture
Legal status
Suzetrigine was approved for medical use in the United States in January 2025.[2]
Names
Suzetrigine is the international nonproprietary name.[12]
Suzetrigine is sold under the brand name Journavx.[1][2]




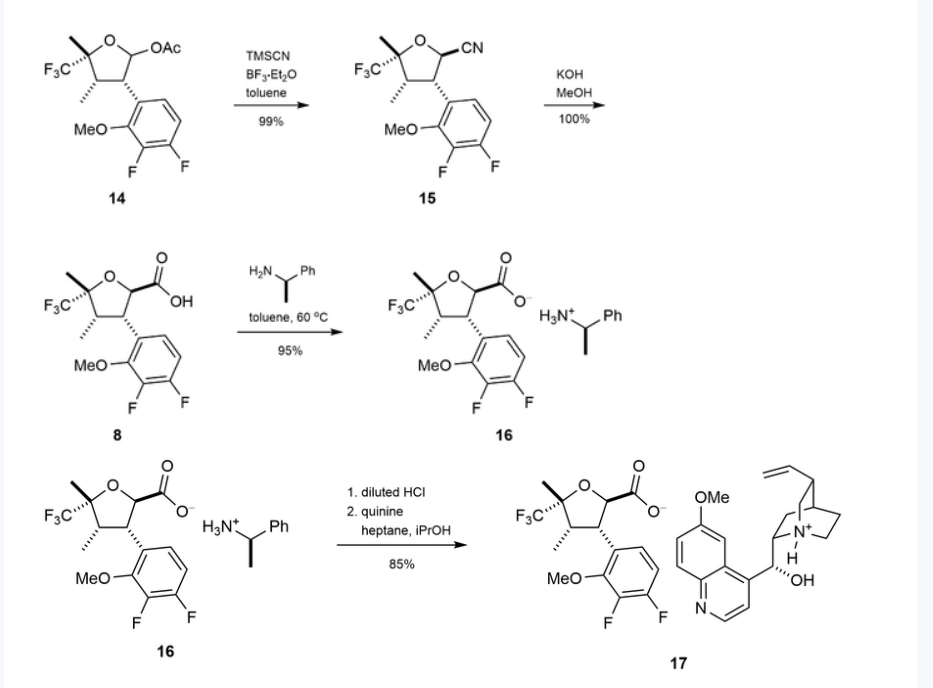
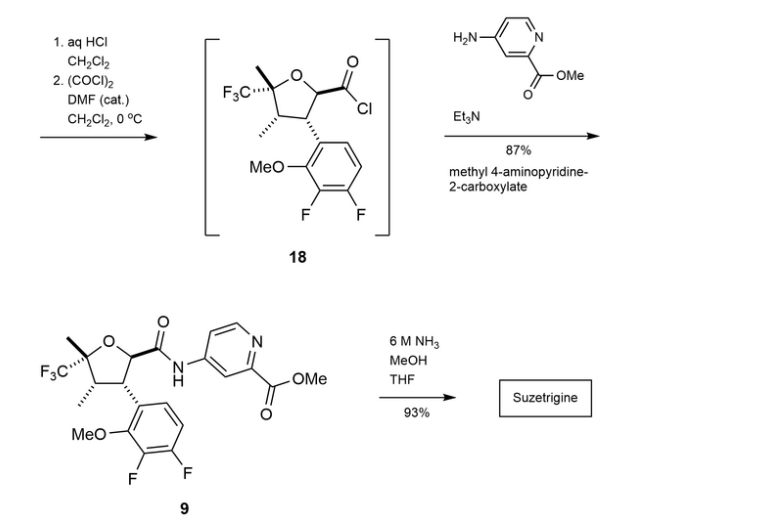

References
a) WO2021113627A1 (Vertex, 10.06.2021; USA-prior. 06.12.2019).
US11834441B2 (Vertex, 05.12.2023; USA-prior. 06.12.2019).
b) WO2022256660A1 (Vertex, 08.12.2022; USA-prior. 04.06.2021).
WO2024123815A1 (Vertex, 13.06.2024; USA-prior. 06.12.2022).
WO2022256708A1 (Vertex, 08.12.2022; USA-prior. 04.06.2021, 02.12.2021).
Source:
Suzetrigine, in Kleemann A., Kutscher B., Reichert D., Bossart M., Pharmaceutical Substances, Thieme. https://pharmaceutical-substances.thieme.com/lexicon/KD-19-0151, accessed: 05-29-2025
| Clinical data | |
|---|---|
| Pronunciation | /suˈzɛtrɪdʒiːn/ soo-ZE-tri-jeen |
| Trade names | Journavx |
| Other names | VX-548 |
| AHFS/Drugs.com | Journavx |
| License data | US DailyMed: Suzetrigine |
| Routes of administration | By mouth |
| Drug class | Nav1.8 sodium channel blocker; Analgesic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2649467-58-1 |
| PubChem CID | 156445116 |
| DrugBank | DB18927 |
| ChemSpider | 128942439 |
| UNII | LOG73M21H5 |
| KEGG | D12860 |
| ChEMBL | ChEMBL5314487 |
| Chemical and physical data | |
| Formula | C21H20F5N3O4 |
| Molar mass | 473.400 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f g h “Journavx- suzetrigine tablet, film coated”. DailyMed. 6 February 2025. Retrieved 2 April 2025.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves Novel Non-Opioid Treatment for Moderate to Severe Acute Pain” (Press release). U.S. Food and Drug Administration (FDA). 30 January 2025. Archived from the original on 7 February 2025. Retrieved 30 January 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e Osteen, Jeremiah D.; Immani, Swapna; Tapley, Tim L.; Indersmitten, Tim; Hurst, Nicole W.; Healey, Tiffany; et al. (January 2025). “Pharmacology and Mechanism of Action of Suzetrigine, a Potent and Selective NaV1.8 Pain Signal Inhibitor for the Treatment of Moderate to Severe Pain”. Pain and Therapy. doi:10.1007/s40122-024-00697-0. PMID 39775738.
- ^ Jump up to:a b c Jones, Jim; Correll, Darin J.; Lechner, Sandra M; Jazic, Ina; Miao, Xiaopeng; Shaw, David; et al. (August 2023). “Selective Inhibition of NaV1.8 with VX-548 for Acute Pain”. The New England Journal of Medicine. 389 (5): 393–405. doi:10.1056/NEJMoa2209870. PMID 37530822. S2CID 260377748.
- ^ Jump up to:a b “Vertex Announces Positive Results From the VX-548 Phase 3 Program for the Treatment of Moderate-to-Severe Acute Pain” (Press release). Vertex. 30 January 2024. Archived from the original on 25 December 2024. Retrieved 31 January 2025 – via Business Wire.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ Jump up to:a b c d Broadfoot, Marla (20 August 2024). “New Painkiller Could Bring Relief to Millions — without Addiction Risk”. Scientific American. Archived from the original on 30 December 2024. Retrieved 31 January 2025.
- ^ Jump up to:a b c d Hang Kong, Aaron Yik; Tan, Hon Sen; Habib, Ashraf S. (September 2024). “VX-548 in the Treatment of Acute Pain”. Pain Management. 14 (9): 477–486. doi:10.1080/17581869.2024.2421749. PMC 11721852. PMID 39552600.
- ^ Kingwell, Katie (December 2024). “NaV1.8 inhibitor poised to provide opioid-free pain relief”. Nature Reviews. Drug Discovery. 24 (1): 3–5. doi:10.1038/d41573-024-00203-3. PMID 39668193.
- ^ Jump up to:a b Dolgin, Elie (January 2025). “US drug agency approves potent painkiller – the first non-opioid in decades”. Nature. 638 (8050): 304–305. doi:10.1038/d41586-025-00274-1. PMID 39885357.
- ^ Jump up to:a b “Vertex Announces FDA Acceptance of New Drug Application for Suzetrigine for the Treatment of Moderate-to-Severe Acute Pain” (Press release). Vertex. 30 July 2024. Retrieved 31 January 2025 – via Business Wire.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 90”. WHO Drug Information. 37 (3). hdl:10665/373341.
Further reading
- Oliver, Brian; Devitt, Catherine; Park, Grace; Razak, Alina; Liu, Sun Mei; Bergese, Sergio D. (2025). “Drugs in Development to Manage Acute Pain”. Drugs. 85 (1): 11–19. doi:10.1007/s40265-024-02118-0. PMID 39560856.
External links
- “Suzetrigine (Code C199115)”. NCI Thesaurus.
- Clinical trial number NCT05661734 for “A Single-arm Study to Evaluate Safety and Effectiveness of VX-548 for Acute Pain” at ClinicalTrials.gov
- Clinical trial number NCT05558410 for “Evaluation of Efficacy and Safety of VX-548 for Acute Pain After an Abdominoplasty” at ClinicalTrials.gov
//////////Suzetrigine, Journavx, FDA 2025, APPROVALS 2025, CS-0641183, HY-148800, VX 548, VX-548, VX548, Breakthrough Therapy, Fast Track, Priority Review

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}