

| EudraCT | Title | Phase | Status | Date |
|---|---|---|---|---|
| 2019-003807-37 | A Double-Blind, Randomized, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Odevixibat (A4250) in Children with Biliary Atresia Who Have Undergone a Kasai Hepatoportoenterostomy (BOLD) | Phase 3 | Ongoing | 2020-07-29 |
| 2015-001157-32 | An Exploratory Phase II Study to demonstrate the Safety and Efficacy of A4250 | Phase 2 | Completed | 2015-05-13 |
| 2014-004070-42 | An Exploratory, Phase IIa Cross-Over Study to Demonstrate the Efficacy | Phase 2 | Ongoing | 2014-12-09 |
| 2017-002325-38 | An Open-label Extension Study to Evaluate Long-term Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 2) | Phase 3 | Ongoing | |
| 2017-002338-21 | A Double-Blind, Randomized, Placebo-Controlled, Phase 3 Study to Demonstrate Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 1) | Phase 3 | Ongoing, Completed |
Home » Posts tagged 'APPROVALS 2021' (Page 6)
Tag Archives: APPROVALS 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Inclisiran
Inclisiran
CAS 1639324-58-5
- ALN-60212
- ALN-PCSsc
US FDA APPROVED
12/22/2021 |
To treat heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease as an add-on therapy, Leqvio
Inclisiran was first developed by Alnylam Pharmaceuticals, Inc. (Cambridge, Massachusetts, US). Development has now been assumed by The Medicines Company (Parsippany, New Jersey, US). One phase I and two phase II trials have been completed. Topline results of two phase III trials were also recently presented while other phase III trials are still ongoing as part of the ORION clinical development program. …..https://www.ncbi.nlm.nih.gov/books/NBK555477/
Inclisiran is a long-acting, synthetic small interfering RNA (siRNA) directed against proprotein convertase subtilisin-kexin type 9 (PCSK9), which is a serine protease that regulates plasma low-density lipoprotein cholesterol (LDL-C) levels. By binding to PCSK9 messenger RNA, inclisiran prevents protein translation of PCSK9, leading to decreased concentrations of PCSK9 and plasma concentrations of LDL cholesterol.1,2 Lowering circulating plasma LDL-C levels offers an additional benefit of reducing the risk of cardiovascular disease (CVD) and improving cardiovascular outcomes, as hypercholesterolemia is a major known risk factor for CVD.1,2
On December 11, 2020, the European Commission (EC) granted authorization for marketing inclisiran as the first and only approved siRNA for the treatment of adults with primary hypercholesterolemia (heterozygous familial and non-familial) or mixed dyslipidemia, alone or in combination with other lipid-lowering therapies. It is marketed under the trade name Leqvio 8 and is also currently under review by the FDA.
Inclisiran, sold under the brand name Leqvio, is a medication for the treatment of people with atherosclerotic cardiovascular disease (ASCVD), ASCVD risk equivalents and heterozygous familial hypercholesterolemia (HeFH). It is a small interfering RNA that inhibits translation of the protein PCSK9.[2][3][4] It is being developed by The Medicines Company which licensed the rights to inclisiran from Alnylam Pharmaceuticals.[5]
On 15 October 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Leqvio, intended for the treatment for primary hypercholesterolaemia or mixed dyslipidaemia.[6] Inclisiran was approved for use in the European Union in December 2020.[1]
History
In 2019 The Medicines Company announced positive results from pivotal phase III study (all primary and secondary endpoints were met with efficacy consistent with Phase I and II studies). The company anticipates regulatory submissions in the U.S. in the fourth quarter of 2019, and in Europe in the first quarter of 2020.[7] The Medicines Company is being acquired by Novartis.[8]

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b “Leqvio EPAR”. European Medicines Agency. 13 October 2020. Retrieved 6 January 2021.
- ^ Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. (January 2017). “A Highly Durable RNAi Therapeutic Inhibitor of PCSK9”. The New England Journal of Medicine. 376 (1): 41–51. doi:10.1056/NEJMoa1609243. PMC 5778873. PMID 27959715.
- ^ Spreitzer H (11 September 2017). “Neue Wirkstoffe: Inclisiran”. Österreichische Apotheker-Zeitung (in German) (19/2017).
- ^ “Proposed INN: List 114” (PDF). WHO Drug Information. WHO. 29 (4): 531f. 2015.
- ^ Taylor NP (26 August 2019). “Medicines Company’s PCSK9 drug hits phase 3 efficacy goals”. FierceBiotech.
- ^ “Leqvio: Pending EC decision”. European Medicines Agency (EMA). 16 October 2020. Retrieved 16 October 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “The Medicines Company Announces Positive Topline Results from First Pivotal Phase 3 Trial of Inclisiran”. The Medicines Company. Retrieved 29 August 2019.
- ^ “Novartis acquires medicines company”. Novartis. Retrieved 15 January 2020.
Further reading
- Ray KK, Landmesser U, Leiter LA, et al. (April 2017). “Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol” (PDF). N. Engl. J. Med. 376 (15): 1430–1440. doi:10.1056/NEJMoa1615758. hdl:10044/1/45416. PMID 28306389. S2CID 205101529.
- Ray KK, Wright RS, Kallend D, et al. (March 2020). “Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol”. N. Engl. J. Med. 382 (16): 1507–1519. doi:10.1056/NEJMoa1912387. PMID 32187462.
External links
- “Inclisiran”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03399370 for “Inclisiran for Participants With Atherosclerotic Cardiovascular Disease and Elevated Low-density Lipoprotein Cholesterol (ORION-10)” at ClinicalTrials.gov
- Clinical trial number NCT03400800 for “Inclisiran for Subjects With ACSVD or ACSVD-Risk Equivalents and Elevated Low-density Lipoprotein Cholesterol (ORION-11)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Leqvio |
| Other names | ALN-PCSsc, ALN-60212 |
| Routes of administration | Subcutaneous injection |
| ATC code | C10AX16 (WHO) |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1639324-58-5 |
| DrugBank | DB14901 |
| UNII | UOW2C71PG5 |
| KEGG | D11931 |
| Chemical and physical data | |
| Formula | C520H679F21N175O309P43S6 |
| Molar mass | 16248.27 g·mol−1 |
General References
- Kosmas CE, Munoz Estrella A, Sourlas A, Silverio D, Hilario E, Montan PD, Guzman E: Inclisiran: A New Promising Agent in the Management of Hypercholesterolemia. Diseases. 2018 Jul 13;6(3). pii: diseases6030063. doi: 10.3390/diseases6030063. [PubMed:30011788]
- German CA, Shapiro MD: Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs. 2020 Feb;34(1):1-9. doi: 10.1007/s40259-019-00399-6. [PubMed:31782112]
- Doggrell SA: Inclisiran, the billion-dollar drug, to lower LDL cholesterol – is it worth it? Expert Opin Pharmacother. 2020 Nov;21(16):1971-1974. doi: 10.1080/14656566.2020.1799978. Epub 2020 Aug 4. [PubMed:32749892]
- Goldstein JL, Brown MS: Regulation of low-density lipoprotein receptors: implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis. Circulation. 1987 Sep;76(3):504-7. doi: 10.1161/01.cir.76.3.504. [PubMed:3621516]
- Pratt AJ, MacRae IJ: The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem. 2009 Jul 3;284(27):17897-901. doi: 10.1074/jbc.R900012200. Epub 2009 Apr 1. [PubMed:19342379]
- Leiter LA, Teoh H, Kallend D, Wright RS, Landmesser U, Wijngaard PLJ, Kastelein JJP, Ray KK: Inclisiran Lowers LDL-C and PCSK9 Irrespective of Diabetes Status: The ORION-1 Randomized Clinical Trial. Diabetes Care. 2019 Jan;42(1):173-176. doi: 10.2337/dc18-1491. Epub 2018 Nov 28. [PubMed:30487231]
- Cupido AJ, Kastelein JJP: Inclisiran for the treatment of hypercholesterolaemia: implications and unanswered questions from the ORION trials. Cardiovasc Res. 2020 Sep 1;116(11):e136-e139. doi: 10.1093/cvr/cvaa212. [PubMed:32766688]
- Novartis: Novartis receives EU approval for Leqvio (inclisiran), a first-in-class siRNA to lower cholesterol with two doses a year [Link]
- Summary of Product Characteristics: Leqvio (inclisiran), solution for subcutaneous injection [Link]
Summary
- Atherosclerotic cardiovascular disease (ASCVD) remains one of the leading causes of death in Canada. Cholesterol, specifically low-density lipoprotein cholesterol (LDL-C), is a major risk factor for cardiovascular disease (CVD) and is thereby targeted to reduce the likelihood of a cardiovascular event, such as a myocardial infarction (MI) and stroke.
- Inclisiran, first developed by Alnylam Pharmaceuticals, Inc. (Cambridge, Massachusetts, US) then by The Medicines Company (Parsippany, New Jersey, US), is a small interfering ribonucleic acid (siRNA) molecule being investigated for the treatment of hypercholesterolemia.
- ORION-1 was a phase II, double-blind, placebo-controlled, multi-centre, randomized controlled trial of 501 patients. Patients were included in the trial if they had a history of ASCVD or were at high risk of ASCVD. The treatment arms were administered 200 mg, 300 mg, or 500 mg of inclisiran on day 1, or 100 mg, 200 mg, or 300 mg of inclisiran on days 1 and 90. The comparator was either placebo on day 1 or placebo on days 1 and 90. The primary end point was percentage change in LDL-C at day 180 from baseline.
- The ORION-1 study demonstrated that inclisiran, administered at various doses and intervals, compared with placebo, resulted in a statistically significant reduction in LDL-C levels (P < 0.001 for all comparisons versus placebo). The greatest reduction in LDL-C levels was obtained with the 300 mg dose of inclisiran given at days 1 and 90 with a 52.6% (95% confidence interval [CI]: −57.1 to −48.1) reduction at day 180 compared with baseline, and a mean absolute reduction in LDL-C levels of 1.66 (standard deviation 0.54) mmol/L. Results from the ORION-1 trial provided the necessary data to make a decision regarding the dosing regimen to be used in subsequent phase III trials, in particular the ORION-11 phase III trial.
- The ORION-11 study was a phase III international, multi-centre, and double-blind trial which randomized 1,617 participants (87% with established ASCVD) to inclisiran 300 mg (n = 810) or placebo (n = 807). An initial inclisiran dose of 300 mg given subcutaneously was administered at day 1, day 90, and then every six months for two doses, that is at days 270 and 450. The mean baseline LDL-C level was 2.8 mmol/L (inclisiran) and 2.7 mmol/L (placebo); 96% of participants were on high-dose statin therapy. There was a 50% time-averaged reduction in LDL-C levels from day 90 to day 540 (P < 0.00001). Pre-specified exploratory cardiovascular composite end point (cardiac death, cardiac arrest, MI, or stroke) occurred in 7.8% of inclisiran treated patients versus 10.3% of patients on placebo; this lower rate was mainly driven by a reduction in MI and stroke. With respect to adverse effects, 4.69% of patients on inclisiran reported an injection site reaction, compared with 0.5% of patients on placebo. All reactions were transient. There was no evidence of liver, kidney, muscle, or platelet toxicity.
- Inclisiran may be an option in the future as a cholesterol-lowering medication, where it would likely be used in patients who are unable to achieve their LDL-C targets despite maximally tolerated statin therapy or who are intolerant to statin therapy. However, results from the inclisiran cardiovascular outcome trial (ORION-4), are needed to confirm its efficacy in reducing CVD and its long-term safety.
- Inclisiran is not yet approved by any regulatory authority, but its ORION clinical development program identifies the year 2021 as the goal to reach worldwide markets.
///////////Inclisiran, LEQVIO, ALN 60212, ALN PCSsc , NOVARTIS, Leqvio, APPROVALS 2021, FDA 2021

NEW DRUG APPROVALS
ONE TIME
$10.00
Setmelanotide

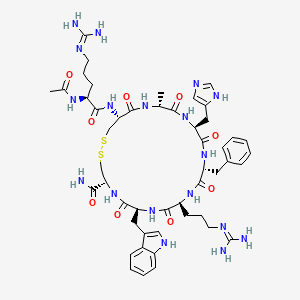

Setmelanotide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
- Molecular FormulaC49H68N18O9S2
- Average mass1117.309 Da
- N-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide (2->8)-disulfide
1,2-Dithia-5,8,11,14,17,20-hexaazacyclotricosane-4-carboxamide, 22-[[(2S)-2-(acetylamino)-5-[(diaminomethylene)amino]-1-oxopentyl]amino]-10-[3-[(diaminomethylene)amino]propyl]-16-(1H-imidazol-5-ylmeth yl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-13-(phenylmethyl)-, (4R,7S,10S,13R,16S,19R,22R)- [ACD/Index Name]10011920014-72-8[RN]Imcivree [Trade name]N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-Ltryptophyl- L-cysteinamide, cyclic (2-8)-disulfideN7T15V1FUYRM-493, BIM-22493UNII-N7T15V1FUYсетмеланотид [Russian] [INN]سيتميلانوتيد [Arabic] [INN]司美诺肽 [Chinese] [INN](4R,7S,10S,13R,16S,19R,22R)-22-[[(2S)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-13-benzyl-10-[3-(diaminomethylideneamino)propyl]-16-(1H-imidazol-5-ylmethyl)-7-(1H-indol-3-ylmethyl)-19-methyl-6,9,12,15,18,21-hexaoxo-1,2-dithia-5,8,11,14,17,20-hexazacyclotricosane-4-carboxamide
FDA 11/25/2020, Imcivree, To treat obesity and the control of hunger associated with pro-opiomelanocortin deficiency, a rare disorder that causes severe obesity that begins at an early age
Drug Trials Snapshot, 10MG/ML, SOLUTION;SUBCUTANEOUS, Orphan


update Imcivree EMA APPROVED 2021/7/16
DESCRIPTION
IMCIVREE contains setmelanotide acetate, a melanocortin 4 (MC4) receptor agonist. Setmelanotide is an 8 amino acid cyclic peptide analog of endogenous melanocortin peptide α-MSH (alpha-melanocyte stimulating hormone).
The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-Lhistidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C49H68N18O9S2 (anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide is:
 |
IMCIVREE injection is a sterile clear to slightly opalescent, colorless to slightly yellow solution. Each 1 mL of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with 2 to 4 molar equivalents of acetate, and the following inactive ingredients: 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-glycero-3phosphoethanolamine sodium salt, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 11 mg mannitol, 5 mg phenol, 10 mg benzyl alcohol, 1 mg edetate disodium dihydrate, and Water for Injection. The pH of IMCIVREE is 5 to 6.
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor. Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects. Setmelanotide increases resting energy expenditure in both obese animals and humans. Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).
Setmelanotide, sold under the brand name Imcivree, is a medication for the treatment of obesity.[1]
The most common side effects include injection site reactions, skin hyperpigmentation (skin patches that are darker than surrounding skin), headache and gastrointestinal side effects (such as nausea, diarrhea, and abdominal pain), among others.[1] Spontaneous penile erections in males and adverse sexual reactions in females have occurred with treatment.[1] Depression and suicidal ideation have also occurred with setmelanotide.[1]
SYN
WO 2011060355



Medical uses
Setmelanotide is indicated for chronic weight management (weight loss and weight maintenance for at least one year) in people six years and older with obesity due to three rare genetic conditions: pro-opiomelanocortin (POMC) deficiency, proprotein subtilisin/kexin type 1 (PCSK1) deficiency, and leptin receptor (LEPR) deficiency confirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes considered pathogenic (causing disease), likely pathogenic, or of uncertain significance.[1] Setmelanotide is the first FDA-approved treatment for these genetic conditions.[1]
Setmelanotide is not approved for obesity due to suspected POMC, PCSK1, or LEPR deficiency with variants classified as benign (not causing disease) or likely benign or other types of obesity, including obesity associated with other genetic syndromes and general (polygenic) obesity.[1]
Setmelanotide binds to and activates MC4 receptors in the paraventricular nucleus (PVN) of the hypothalamus and in the lateral hypothalamic area (LHA), areas involved in the regulation of appetite, and this action is thought to underlie its appetite suppressant effects.[2] In addition to reducing appetite, setmelanotide increases resting energy expenditure in both obese animals and humans.[3] Importantly, unlike certain other MC4 receptor agonists, such as LY-2112688, setmelanotide has not been found to produce increases in heart rate or blood pressure.[4]
Setmelanotide has been reported to possess the following activity profile (cAMP, EC50): MC4 (0.27 nM) > MC3 (5.3 nM) ≈ MC1 (5.8 nM) > MC5 (1600 nM) ≟ MC2 (>1000 nM).[5] (19.6-fold selectivity for MC4 over MC3, the second target of highest activity.)
History
Setmelanotide was evaluated in two one-year studies.[1] The first study enrolled participants with obesity and confirmed or suspected POMC or PCSK1 deficiency while the second study enrolled participants with obesity and confirmed or suspected LEPR deficiency; all participants were six years or older.[1] The effectiveness of setmelanotide was determined by the number of participants who lost more than ten percent of their body weight after a year of treatment.[1]
The effectiveness of setmelanotide was assessed in 21 participants, ten in the first study and eleven in the second.[1] In the first study, 80 percent of participants with POMC or PCSK1 deficiency lost ten percent or more of their body weight.[1] In the second study, 46 percent of participants with LEPR deficiency lost ten percent or more of their body weight.[1]
The study also assessed the maximal (greatest) hunger in sixteen participants over the previous 24 hours using an eleven-point scale in participants twelve years and older.[1] In both studies, some, but not all, of participants’ weekly average maximal hunger scores decreased substantially from their scores at the beginning of the study.[1] The degree of change was highly variable among participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for setmelanotide orphan disease designation, breakthrough therapy designation, and priority review.[1] The FDA granted the approval of Imcivree to Rhythm Pharmaceutical, Inc.[1]
Research
Setmelanotide is a peptide drug and investigational anti-obesity medication which acts as a selective agonist of the MC4 receptor.[6][4] Its peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It was first discovered at Ipsen and is being developed by Rhythm Pharmaceuticals for the treatment of obesity and diabetes.[6] In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity.[7] As of December 2014, the drug is in phase II clinical trials for obesity and PWS.[6][8][9][needs update] So far, preliminary data has shown no benefit of Setmelanotide in Prader-Willi syndrome.[10]
PATENT
WO 2007008704
WO 2011060355
WO 2011060352
US 20120225816
PAPER
Journal of Medicinal Chemistry, 61(8), 3674-3684; 2018
PATENT
https://patents.google.com/patent/US9314509
Synthesis of Example 1i.e., Ac-Arg-cyclo(Cys-D-Ala-His-D-Phe-Arg-Trp-Cys)-NH2

The title peptide having the above structure was assembled using Fmoc chemistry on an Apex peptide synthesizer (Aapptec; Louisville, Ky., USA). 220 mg of 0.91 mmol/g (0.20 mmoles) Rink Amide MBHA resin (Polymer Laboratories; Amherst, Mass., USA) was placed in a reaction well and pre-swollen in 3.0 mL of DMF prior to synthesis. For cycle 1, the resin was treated with two 3-mL portions of 25% piperidine in DMF for 5 and 10 minutes respectively, followed by 4 washes of 3-mL DMF—each wash consisting of adding 3 mL of solvent, mixing for 1 minute, and emptying for 1 minute. Amino acids stocks were prepared in NMP as 0.45M solutions containing 0.45M HOBT. HBTU was prepared as a 0.45M solution in NMP and DIPEA was prepared as a 2.73M solution in NMP. To the resin, 2 mL of the first amino acid (0 9 mmoles, Fmoc-Cys(Trt)-OH) (Novabiochem; San Diego, Calif., USA) was added along with 2 mL (0.9 mmoles) of HBTU and 1.5 mL (4.1 mmoles) of DIPEA. After one hour of constant mixing, the coupling reagents were drained from the resin and the coupling step was repeated. Following amino acid acylation, the resin was washed with two 3-mL aliquots of DMF for 1 minute. The process of assembling the peptide (deblock/wash/acylate/wash) was repeated for cycles 2-9 identical to that as described for cycle 1. The following amino acids were used: cycle 2) Fmoc-Trp(Boc)-OH (Genzyme; Cambridge, Mass., USA); cycle 3) Fmoc-Arg(Pbf)-OH (Novabiochem); cycle 4) Fmoc-DPhe-OH (Genzyme); cycle 5) Fmoc-His(Trt)-OH (Novabiochem); cycle 6) Fmoc-D-Ala-OH (Genzyme); cycle 7) Fmoc-Cys(Trt)-OH, (Novabiochem); and cycle 8) Fmoc-Arg(Pbf)-OH (Genzyme). The N-terminal Fmoc was removed with 25% piperidine in DMF as described above, followed by four 3-mL DMF washes for 1 minute. Acetylation of the N-terminus was performed by adding 0.5 mL of 3M DIPEA in NMP to the resin along with 1.45 mL of 0.45M acetic anhydride in NMP. The resin was mixed for 30 minutes and acetylation was repeated. The resin was washed with 3 mL of DMF for a total of 5 times followed with 5 washes with 5 mL of DCM each.
To cleave and deprotect the peptide, 5mL of the following reagent was added to the resin: 2% TIS/5% water/5% (w/v) DTT/88% TFA. The solution was allowed to mix for 3.5 hours. The filtrate was collected into 40 mL of cold anhydrous ethyl ether. The precipitate was pelleted for 10 minutes at 3500 rpm in a refrigerated centrifuge. The ether was decanted and the peptide was re-suspended in fresh ether. The ether workup was performed three times. Following the last ether wash, the peptide was allowed to air dry to remove residual ether.
The peptide was dissolved in 10% acetonitrile and analyzed by mass spectrometry and reverse-phase HPLC employing a 30×4.6 cm C18 column (Vydac; Hesperia, Calif., USA) with a gradient of 2-60% acetonitrile (0.1% TFA) over 30 minutes. This analysis identified a product with ˜53% purity. Mass analysis employing electrospray ionization identified a main product containing a mass of 1118.4 corresponding to the desired linear product. The crude product (˜100 mg) was diluted to a concentration of 2 mg/mL in 5% acetic acid. To this solution, 0.5M iodine/methanol was added dropwise with vigorous stirring until a pale yellow color was achieved. The solution was vigorously stirred for another 10 minutes. Excess iodine was then quenched by adding 1.0M sodium thiosulfate under continuous mixing until the mixture was rendered colorless. The peptide was re-examined by mass spectrometry analysis and HPLC. Mass spectrometry analysis identified a main species with a mass of 1116.4 which indicated successful oxidation to form the cyclic peptide. The peptide solution was purified on a preparative HPLC equipped with a C18 column using a similar elution gradient. The purified product was re-analyzed by HPLC for purity (>95%) and mass spectrometry (1116.9 which is in agreement with the expected mass of 1117.3) and subsequently lyophilized. Following lyophilization, 28 mg of purified product was obtained representing a 24% yield.
The other exemplified peptides were synthesized substantially according to the procedure described for the above-described synthetic process. Physical data for select exemplified peptides are given in Table 1.
TABLE 1 Example Mol. Wt. Mol. Wt. Purity Number (calculated) (ES-MS) (HPLC) 1 1117.3 1116.9 95.1% 2 1117.3 1116.8 99.2% 3 1280.5 1280.6 98.0% 5 1216.37 1216.20 99.9%
Preparation of Pamoate Salt of Example 1
The acetate salt of Example 1 (200 mg, 0.18 mmole) was dissolved in 10 mL of water. Sodium pamoate (155 mg, 0.36 mmole) was dissolved in 10 mL of water. The two solutions were combined and mixed well. The precipitates were collected by centrifugation at 3000 rpm for 20 minutes, washed for three times with water, and dried by lyophilization.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment for weight management for people with certain rare genetic conditions”. U.S. Food and Drug Administration (FDA) (Press release). 27 November 2020. Retrieved 27 November 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Kim GW, Lin JE, Blomain ES, Waldman SA (January 2014). “Antiobesity pharmacotherapy: new drugs and emerging targets”. Clinical Pharmacology and Therapeutics. 95 (1): 53–66. doi:10.1038/clpt.2013.204. PMC 4054704. PMID 24105257.
- ^ Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, et al. (April 2015). “RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals”. The Journal of Clinical Endocrinology and Metabolism. 100 (4): 1639–45. doi:10.1210/jc.2014-4024. PMC 4399297. PMID 25675384.
- ^ Jump up to:a b Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, et al. (February 2013). “Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques”. Diabetes. 62 (2): 490–7. doi:10.2337/db12-0598. PMC 3554387. PMID 23048186.
- ^ Muniyappa R, Chen K, Brychta R, Abel B, Mullins K, Staker P, et al. (June 2014). “A Randomized, Double-Blind, Placebo-Controlled, Crossover Study to Evaluate the Effect of a Melanocortin Receptor 4 (MC4R) Agonist, RM-493, on Resting Energy Expenditure (REE) in Obese Subjects” (PDF). Endocrine Reviews. Rhythm Pharmaceuticals. 35 (3). Retrieved 2015-05-21.
- ^ Jump up to:a b c Lee EC, Carpino PA (2015). “Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014)”. Pharmaceutical Patent Analyst. 4 (2): 95–107. doi:10.4155/ppa.15.1. PMID 25853469.
- ^ “Obesity and Diabetes Caused by Genetic Deficiencies in the MC4 Pathway”. Rhythm Pharmaceuticals. Retrieved 2015-05-21.
- ^ Jackson VM, Price DA, Carpino PA (August 2014). “Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies”. Expert Opinion on Investigational Drugs. 23 (8): 1055–66. doi:10.1517/13543784.2014.918952. PMID 25000213. S2CID 23198484.
- ^ “RM-493: A First-in-Class, Phase 2-Ready MC4 Agonist: A New Drug Class for the Treatment of Obesity and Diabetes”. Rhythm Pharmaceuticals. Archived from the original on 2015-06-14. Retrieved 2015-05-21.
- ^ Duis J, van Wattum PJ, Scheimann A, Salehi P, Brokamp E, Fairbrother L, et al. (March 2019). “A multidisciplinary approach to the clinical management of Prader-Willi syndrome”. Molecular Genetics & Genomic Medicine. 7 (3): e514. doi:10.1002/mgg3.514. PMC 6418440. PMID 30697974.
ADDITIONAL INFORMATION
The peptide sequence is Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2. It is being researched by Rhythm Pharmaceuticals for the treatment of obesity and diabetes. In addition, Rhythm Pharmaceuticals is conducting trials of setmelanotide for the treatment of Prader–Willi syndrome (PWS), a genetic disorder which includes MC4 receptor deficiency and associated symptoms such as excessive appetite and obesity. As of December 2014, the drug is in phase II clinical trials for obesity and PWS.
L-Cysteinamide, N2-acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-, cyclic (2->8)-disulfide
Ac-Arg-Cys(1)-D-Ala-His-D-Phe-Arg-Trp-Cys(1)-NH2
REFERENCES
1: Lee EC, Carpino PA. Melanocortin-4 receptor modulators for the treatment of obesity: a patent analysis (2008-2014). Pharm Pat Anal. 2015;4(2):95-107. doi: 10.4155/ppa.15.1. PubMed PMID: 25853469.
2: Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LH, Reitman ML, Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab. 2015 Apr;100(4):1639-45. doi: 10.1210/jc.2014-4024. Epub 2015 Feb 12. PubMed PMID: 25675384; PubMed Central PMCID: PMC4399297.
3: Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, Hofmann SM, Tschöp MH, Van der Ploeg LH, Müller TD. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015 Feb 4;7(3):288-98. doi: 10.15252/emmm.201404508. PubMed PMID: 25652173; PubMed Central PMCID: PMC4364946.
4: Jackson VM, Price DA, Carpino PA. Investigational drugs in Phase II clinical trials for the treatment of obesity: implications for future development of novel therapies. Expert Opin Investig Drugs. 2014 Aug;23(8):1055-66. doi: 10.1517/13543784.2014.918952. Epub 2014 Jul 7. Review. PubMed PMID: 25000213.
5: Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013 Feb;62(2):490-7. doi: 10.2337/db12-0598. Epub 2012 Oct 9. PubMed PMID: 23048186; PubMed Central PMCID: PMC3554387.
6: Kumar KG, Sutton GM, Dong JZ, Roubert P, Plas P, Halem HA, Culler MD, Yang H, Dixit VD, Butler AA. Analysis of the therapeutic functions of novel melanocortin receptor agonists in MC3R- and MC4R-deficient C57BL/6J mice. Peptides. 2009 Oct;30(10):1892-900. doi: 10.1016/j.peptides.2009.07.012. Epub 2009 Jul 29. PubMed PMID: 19646498; PubMed Central PMCID: PMC2755620.
External links
- “Setmelanotide”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Imcivree |
| Other names | RM-493; BIM-22493; IRC-022493; N2-Acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidyl-D-phenylalanyl-L-arginyl-L-tryptophyl-L-cysteinamide, cyclic (2-8)-disulfide |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 920014-72-8 |
| PubChem CID | 11993702 |
| ChemSpider | 10166169 |
| UNII | N7T15V1FUY |
| KEGG | D11927 |
| Chemical and physical data | |
| Formula | C49H68N18O9S2 |
| Molar mass | 1117.32 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]C[C@@H]1C(=O)N[C@H](C(=O)N[C@@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CCCN=C(N)N)NC(=O)C)C(=O)N)Cc2c[nH]c3c2cccc3)CCCN=C(N)N)Cc4ccccc4)Cc5cnc[nH]5 | |
| InChI[hide]InChI=1S/C49H68N18O9S2/c1-26-41(70)63-37(20-30-22-55-25-59-30)46(75)64-35(18-28-10-4-3-5-11-28)44(73)62-34(15-9-17-57-49(53)54)43(72)65-36(19-29-21-58-32-13-7-6-12-31(29)32)45(74)66-38(40(50)69)23-77-78-24-39(47(76)60-26)67-42(71)33(61-27(2)68)14-8-16-56-48(51)52/h3-7,10-13,21-22,25-26,33-39,58H,8-9,14-20,23-24H2,1-2H3,(H2,50,69)(H,55,59)(H,60,76)(H,61,68)(H,62,73)(H,63,70)(H,64,75)(H,65,72)(H,66,74)(H,67,71)(H4,51,52,56)(H4,53,54,57)/t26-,33+,34+,35-,36+,37+,38+,39+/m1/s1Key:HDHDTKMUACZDAA-PHNIDTBTSA-N |
///////////Setmelanotide, FDA 2020, 2020 APPROVALS, Imcivree, Orphan, PEPTIDE, ANTIOBESITY, UNII-N7T15V1FUY, сетмеланотид , سيتميلانوتيد , 司美诺肽 , BIM 22493, RM 493
CC1C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(C(=O)NC(CSSCC(C(=O)N1)NC(=O)C(CCCN=C(N)N)NC(=O)C)C(=O)N)CC2=CNC3=CC=CC=C32)CCCN=C(N)N)CC4=CC=CC=C4)CC5=CN=CN5
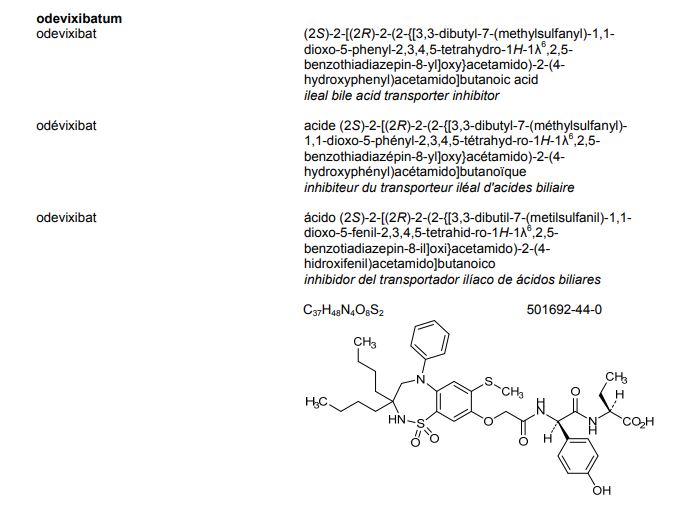
Odevixibat

Odevixibat
A-4250, AR-H 064974
CAS 501692-44-0
BUTANOIC ACID, 2-(((2R)-2-((2-((3,3-DIBUTYL-2,3,4,5-TETRAHYDRO-7-(METHYLTHIO)-1,1-DIOXIDO-5-PHENYL-1,2,5-BENZOTHIADIAZEPIN-8-YL)OXY)ACETYL)AMINO)-2-(4-HYDROXYPHENYL)ACETYL)AMINO)-, (2S)-
(2S)-2-[[(2R)-2-[[2-[(3,3-dibutyl-7-methylsulfanyl-1,1-dioxo-5-phenyl-2,4-dihydro-1λ6,2,5-benzothiadiazepin-8-yl)oxy]acetyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]butanoic acid
| Molecular Formula | C37H48N4O8S2 |
| Molecular Weight | 740.929 |
-
-
-
- UPDATE 7/20/2021FDA APPROVED, To treat pruritus,
-
- New Drug Application (NDA): 215498
Company: ALBIREO PHARMA INC
-
- AZD8294WHO 10706AR-H064974HY-109120CS-0078340D11716US9694018, 5Originator Albireo AB
- Developer Albireo AB; Albireo Pharma
- ClassAcetamides; Butyric acids; Hepatoprotectants; Small molecules; Sulfones; Thiazepines
- Mechanism of Action Sodium-bile acid cotransporter inhibitors
- Orphan Drug Status Yes – Primary biliary cirrhosis; Biliary atresia; Intrahepatic cholestasis; Alagille syndrome
- New Molecular Entity Yes
- Phase III Biliary atresia; Intrahepatic cholestasis
- Phase II Alagille syndrome; Cholestasis; Primary biliary cirrhosis
- No development reported Non-alcoholic steatohepatitis
- 22 Jul 2020 Albireo initiates an expanded-access programme for Intrahepatic cholestasis in USA, Canada, Australia and Europe
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Belgium (PO) after July 2020 (EudraCT2019-003807-37)
- 14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Germany, France, United Kingdom, Hungary (PO) (EudraCT2019-003807-37)
UPDATE Bylvay, FDA APPROVED2021/7/20 AND EMA 2021/7/16
Odevixibat, sold under the trade name Bylvay, is a medication for the treatment of progressive familial intrahepatic cholestasis (PFIC).[1]
The most common side effects include diarrhea, abdominal pain, hemorrhagic diarrhea, soft feces, and hepatomegaly (enlarged liver).[1]
Odevixibat is a reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT).[1][2]
In May 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for odevixibat for the treatment of PFIC in people aged six months or older.[1][3]
A-4250 (odevixibat) is a selective inhibitor of the ileal bile acid transporter (IBAT) that acts locally in the gut. Ileum absorbs glyco-and taurine-conjugated forms of the bile salts. IBAT is the first step in absorption at the brush-border membrane. A-4250 works by decreasing the re-absorption of bile acids from the small intestine to the liver, whichreduces the toxic levels of bile acids during the progression of the disease. It exhibits therapeutic intervention by checking the transport of bile acids. Studies show that A-4250 has the potential to decrease the damage in the liver cells and the development of fibrosis/cirrhosis of the liver known to occur in progressive familial intrahepatic cholestasis. A-4250 is a designated orphan drug in the USA for October 2012. A-4250 is a designated orphan drug in the EU for October 2016. A-4250 was awarded PRIME status for PFIC by EMA in October 2016. A-4250 is in phase II clinical trials by Albireo for the treatment of primary biliary cirrhosis (PBC) and cholestatic pruritus. In an open label Phase 2 study in children with cholestatic liver disease and pruritus, odevixibat showed reductions in serum bile acids and pruritus in most patients and exhibited a favorable overall tolerability profile.
![]()
Odevixibat is a highly potent, non-systemic ileal bile acid transport inhibitor (IBATi) that has has minimal systemic exposure and acts locally in the small intestine. Albireo is developing odevixibat to treat rare pediatric cholestatic liver diseases, including progressive familial intrahepatic cholestasis, biliary atresia and Alagille syndrome.
With normal function, approximately 95 percent of bile acids released from the liver into the bile ducts to aid in liver function are recirculated to the liver via the IBAT in a process called enterohepatic circulation. In people with cholestatic liver diseases, the bile flow is interrupted, resulting in elevated levels of toxic bile acids accumulating in the liver and serum. Accordingly, a product capable of inhibiting the IBAT could lead to a reduction in bile acids returning to the liver and may represent a promising approach for treating cholestatic liver diseases.
The randomized, double-blind, placebo-controlled, global multicenter PEDFIC 1 Phase 3 clinical trial of odevixibat in 62 patients, ages 6 months to 15.9 years, with PFIC type 1 or type 2 met its two primary endpoints demonstrating that odevixibat reduced serum bile acids (sBAs) (p=0.003) and improved pruritus (p=0.004), and was well tolerated with a low single digit diarrhea rate. These topline data substantiate the potential for odevixibat to be first drug for PFIC patients. The Company intends to complete regulatory filings in the EU and U.S. no later than early 2021, in anticipation of regulatory approval, issuance of a rare pediatric disease priority review voucher and launch in the second half of 2021.
Odevixibat is being evaluated in the ongoing PEDFIC 2 open-label trial (NCT03659916) designed to assess long-term safety and durability of response in a cohort of patients rolled over from PEDFIC 1 and a second cohort of PFIC patients who are not eligible for PEDFIC 1.
Odevixibat is also currently being evaluated in a second Phase 3 clinical trial, BOLD (NCT04336722), in patients with biliary atresia. BOLD, the largest prospective intervention trial ever conducted in biliary atresia, is a double-blind, randomized, placebo-controlled trial which will enroll approximately 200 patients at up to 75 sites globally to evaluate the efficacy and safety of odevixibat in children with biliary atresia who have undergone a Kasai procedure before age three months. The company also anticipates initiating a pivotal trial of odevixibat for Alagille syndrome by the end of 2020.
For more information about the PEDFIC 2 or BOLD studies, please visit ClinicalTrials.gov or contact medinfo@albireopharma.com.
The odevixibat PFIC program, or elements of it, have received fast track, rare pediatric disease and orphan drug designations in the United States. In addition, the FDA has granted orphan drug designation to odevixibat for the treatment of Alagille syndrome, biliary atresia and primary biliary cholangitis. The EMA has granted odevixibat orphan designation, as well as access to the PRIority MEdicines (PRIME) scheme for the treatment of PFIC. Its Paediatric Committee has agreed to Albireo’s odevixibat Pediatric Investigation Plan for PFIC. EMA has also granted orphan designation to odevixibat for the treatment of biliary atresia, Alagille syndrome and primary biliary cholangitis.

PATENT
https://patents.google.com/patent/US9694018B1/en
Example 5
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N—{(R)-α-[N—((S)-1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine, Mw. 740.94.
This compound is prepared as described in Example 29 of WO3022286.
PATENT
https://patents.google.com/patent/WO2003022286A1/sv
Example 29
1,1-Dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(N-((R)-α-[N-((S)- 1-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine
A solution of 1,1-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-[N-((R)-α-carboxy-4-hydroxybenzyl)carbamoylmethoxy]-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine (Example 18; 0.075 g, 0.114 mmol), butanoic acid, 2-amino-, 1,1-dimethylethyl ester, hydrochloride, (2S)-(0.031 g, 0.160 mmol) and Ν-methylmorpholine (0.050 ml, 0.457 mmol) in DMF (4 ml) was stirred at RT for 10 min, after which TBTU (0.048 g, 0.149 mmol) was added. After 1h, the conversion to the ester was complete. M/z: 797.4. The solution was diluted with toluene and then concentrated. The residue was dissolved in a mixture of DCM (5 ml) and TFA (2 ml) and the mixture was stirred for 7h. The solvent was removed under reduced pressure. The residue was purified by preparative HPLC using a gradient of 20-60% MeCΝ in 0.1M ammonium acetate buffer as eluent. The title compound was obtained in 0.056 g (66 %) as a white solid. ΝMR (400 MHz, DMSO-d6): 0.70 (3H, t), 0.70-0.80 (6H, m), 0.85-1.75 (14H, m), 2.10 (3H, s), 3.80 (2H, brs), 4.00-4.15 (1H, m), 4.65 (1H, d(AB)), 4.70 (1H, d(AB)), 5.50 (1H, d), 6.60 (1H, s), 6.65-7.40 (11H, m), 8.35 (1H, d), 8.50 (1H, d) 9.40 (1H, brs).
PATENT
https://patents.google.com/patent/US20140323412A1/en
PATENT
https://patents.google.com/patent/WO2013063526A1/e
PATENT
https://patents.google.com/patent/WO2019245448A1/en
The compound l,l-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(A/-{(R)-a-[A/-((S)-l-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-l,2,5-benzothiadiazepine (odevixibat; also known as A4250) is disclosed in WO 03/022286. The structure of odevixibat is shown below.

As an inhibitor of the ileal bile acid transporter (IBAT) mechanism, odevixibat inhibits the natural reabsorption of bile acids from the ileum into the hepatic portal circulation. Bile acids that are not reabsorbed from the ileum are instead excreted into the faeces. The overall removal of bile acids from the enterohepatic circulation leads to a decrease in the level of bile acids in serum and the liver. Odevixibat, or a pharmaceutically acceptable salt thereof, is therefore useful in the treatment or prevention of diseases such as dyslipidemia, constipation, diabetes and liver diseases, and especially liver diseases that are associated with elevated bile acid levels.
According to the experimental section of WO 03/022286, the last step in the preparation of odevixibat involves the hydrolysis of a tert-butyl ester under acidic conditions. The crude compound was obtained by evaporation of the solvent under reduced pressure followed by purification of the residue by preparative HPLC (Example 29). No crystalline material was identified.
Amorphous materials may contain high levels of residual solvents, which is highly undesirable for materials that should be used as pharmaceuticals. Also, because of their lower chemical and physical stability, as compared with crystalline material, amorphous materials may display faster
decomposition and may spontaneously form crystals with a variable degree of crystallinity. This may result in unreproducible solubility rates and difficulties in storing and handling the material. In pharmaceutical preparations, the active pharmaceutical ingredient (API) is for that reason preferably used in a highly crystalline state. Thus, there is a need for crystal modifications of odevixibat having improved properties with respect to stability, bulk handling and solubility. In particular, it is an object of the present invention to provide a stable crystal modification of odevixibat that does not contain high levels of residual solvents, that has improved chemical stability and can be obtained in high levels of crystallinity.
Example 1
Preparation of crystal modification 1
Absolute alcohol (100.42 kg) and crude odevixibat (18.16 kg) were charged to a 250-L GLR with stirring under nitrogen atmosphere. Purified water (12.71 kg) was added and the reaction mass was stirred under nitrogen atmosphere at 25 ± 5 °C for 15 minutes. Stirring was continued at 25 ± 5 °C for 3 to 60 minutes, until a clear solution had formed. The solution was filtered through a 5.0 m SS cartridge filter, followed by a 0.2 m PP cartridge filter and then transferred to a clean reactor.
Purified water (63.56 kg) was added slowly over a period of 2 to 3 hours at 25 ± 5 °C, and the solution was seeded with crystal modification 1 of odevixibat. The solution was stirred at 25 ± 5 °C for 12 hours. During this time, the solution turned turbid. The precipitated solids were filtered through centrifuge and the material was spin dried for 30 minutes. The material was thereafter vacuum dried in a Nutsche filter for 12 hours. The material was then dried in a vacuum tray drier at 25 ± 5 °C under vacuum (550 mm Hg) for 10 hours and then at 30 ± 5 °C under vacuum (550 mm Hg) for 16 hours. The material was isolated as an off-white crystalline solid. The isolated crystalline material was milled and stored in LDPE bags.
An overhydrated sample was analyzed with XRPD and the diffractogram is shown in Figure 2.
Another sample was dried at 50 °C in vacuum and thereafter analysed with XRPD. The diffractogram of the dried sample is shown in Figure 1.
The diffractograms for the drying of the sample are shown in Figures 3 and 4 for 2Q ranges 5 – 13 ° and 18 – 25 °, respectively (overhydrated sample at the bottom and dry sample at the top).
References
- ^ Jump up to:a b c d “First treatment for rare liver disease”. European Medicines Agency (EMA) (Press release). 21 May 2021. Retrieved 21 May 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Odevixibat”. Albireo Pharma. Retrieved 21 May 2021.
- ^ “Bylvay: Pending EC decision”. European Medicines Agency (EMA). 19 May 2021. Retrieved 21 May 2021.
External links
- “Odevixibat”. Drug Information Portal. U.S. National Library of Medicine.
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04336722 | Efficacy and Safety of Odevixibat in Children With Biliary Atresia Who Have Undergone a Kasai HPE (BOLD) | Phase 3 | Recruiting | 2020-09-02 |
| NCT04483531 | Odevixibat for the Treatment of Progressive Familial Intrahepatic Cholestasis | Available | 2020-08-25 | |
| NCT03566238 | This Study Will Investigate the Efficacy and Safety of A4250 in Children With PFIC 1 or 2 | Phase 3 | Active, not recruiting | 2020-03-05 |
| NCT03659916 | Long Term Safety & Efficacy Study Evaluating The Effect of A4250 in Children With PFIC | Phase 3 | Recruiting | 2020-01-21 |
| NCT03608319 | Study of A4250 in Healthy Volunteers Under Fasting, Fed and Sprinkled Conditions | Phase 1 | Completed | 2018-09-19 |
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT02630875 | A4250, an IBAT Inhibitor in Pediatric Cholestasis | Phase 2 | Completed | 2018-03-29 |
| NCT02360852 | IBAT Inhibitor A4250 for Cholestatic Pruritus | Phase 2 | Terminated | 2017-02-23 |
| NCT02963077 | A Safety and Pharmakokinetic Study of A4250 Alone or in Combination With A3384 | Phase 1 | Completed | 2016-11-16 |
EU Clinical Trials Register
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Bylvay |
| Routes of administration |
By mouth |
| ATC code |
|
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C37H48N4O8S2 |
| Molar mass | 740.93 g·mol−1 |
| 3D model (JSmol) | |
////////////odevixibat, Orphan Drug Status, phase 3, Albireo, A-4250, A 4250, AR-H 064974
CCCCC1(CN(C2=CC(=C(C=C2S(=O)(=O)N1)OCC(=O)NC(C3=CC=C(C=C3)O)C(=O)NC(CC)C(=O)O)SC)C4=CC=CC=C4)CCCC
| publicationnumber | |||||||
| US-2020046635-A1 | |||||||
| US-2020046636-A1 | US-2020046757-A1 | US-2020046758-A1 | US-2020002299-A1 | WO-2019245448-A1 | WO-2019245449-A1 | US-2019046451-A1 | US-2019070217-A1 |
| US-10441605-B2 | |||||||
| US-2017224720-A1 | |||||||
| US-2017224721-A1 | |||||||
| US-2018264029-A1 | |||||||
| US-2018360869-A1 | |||||||
| US-2018360870-A1 | |||||||
| US-2018360871-A1 | |||||||
| WO-2017133517-A1 | |||||||
| US-2017143738-A1 | |||||||
| US-2017143783-A1 | |||||||
| EP-2968230-A2 | |||||||
| EP-2968262-A1 | |||||||
| US-2014271734-A1 | |||||||
| US-2014275090-A1 | |||||||
| WO-2014144485-A1 | |||||||
| WO-2014144485-A9 | |||||||
| WO-2014144650-A2 | |||||||
| EP-2770990-A1 | |||||||
| EP-2771003-A1 | |||||||
| EP-2771003-B1 | |||||||
| EP-3266457-A1 | |||||||
| EP-3278796-A1 | |||||||
| US-10512657-B2 | |||||||
| US-2013108573-A1 | |||||||
| US-2013109671-A1 | |||||||
| US-2013338093-A1 | |||||||
| US-2014243281-A1 | |||||||
| US-2014323412-A1 | |||||||
| US-2016310518-A1 | |||||||
| US-2017368085-A1 | |||||||
| US-2019169217-A1 | |||||||
| US-2020069715-A1 | |||||||
| WO-2013063512-A1 | |||||||
| WO-2013063526-A1 | |||||||
| EP-2739286-A2 | |||||||
| WO-2013020108-A2 | |||||||
| EP-2637646-B1 | |||||||
| EP-2637668-B1 | |||||||
| EP-3023102-A1 | |||||||
| EP-3023102-B1 | |||||||
| EP-3400944-A1 | |||||||
| US-10000528-B2 | |||||||
| US-10011633-B2 | |||||||
| US-10093697-B2 | |||||||
| US-10221212-B2 | |||||||
| US-2012114588-A1 | |||||||
| US-2013225511-A1 | |||||||
| US-2013236541-A1 | |||||||
| US-2015031636-A1 | |||||||
| US-2015031637-A1 | |||||||
| US-2016193277-A1 | |||||||
| US-2016194353-A1 | |||||||
| US-2017182059-A1 | |||||||
| US-2017182115-A1 | |||||||
| US-2018022776-A1 | |||||||
| US-2018030088-A1 | |||||||
| US-2018030089-A1 | |||||||
| US-2018162904-A1 | |||||||
| US-2018362577-A1 | |||||||
| US-9688720-B2 | |||||||
| US-9694018-B1 | |||||||
| US-10555950-B2 | |||||||
| US-2016220577-A1 | |||||||
| US-9339480-B2 | |||||||
| WO-2008039829-A2 | |||||||
| US-2009069285-A1 | |||||||
| US-7842684-B2 | |||||||
| WO-2007051995-A2 | |||||||
| EP-1896408-A1 | |||||||
| EP-1896409-A1 | |||||||
| EP-1896457-A1 | |||||||
| US-2010048529-A1 | |||||||
| US-2010048530-A1 | |||||||
| US-2010137273-A1 | |||||||
| US-2010152156-A1 | |||||||
| US-2010168039-A1 | |||||||
| US-2010168075-A1 | |||||||
| US-7893048-B2 | |||||||
| US-7906502-B2 | |||||||
| WO-2006137792-A1 | |||||||
| WO-2006137794-A1 | |||||||
| WO-2006137795-A1 | |||||||
| US-2010216759-A1 | |||||||
| US-7863265-B2 | |||||||
| US-2008194494-A1 | |||||||
| US-2009186834-A1 | |||||||
| WO-2006102674-A2 | |||||||
| US-2009005321-A1 | |||||||
| EP-1831151-A1 | |||||||
| US-2008114064-A1 | |||||||
| WO-2006065214-A1 | |||||||
| EP-1699759-A1 | |||||||
| US-2007142304-A1 | |||||||
| US-2008064676-A1 | |||||||
| US-2010099657-A2 | |||||||
| US-7871998-B2 | |||||||
| WO-2005061452-A1 | |||||||
| EP-1638922-A1 | |||||||
| EP-1638926-A1 | |||||||
| EP-1638930-A1 | |||||||
| EP-1675820-A2 | |||||||
| EP-1676833-A1 | |||||||
| US-2005148656-A1 | |||||||
| US-2006142389-A1 | |||||||
| US-2006178432-A1 | |||||||
| US-2006194879-A1 | |||||||
| US-2006258866-A1 | |||||||
| US-2007099928-A1 | |||||||
| US-2007099997-A1 | |||||||
| US-2007244198-A1 | |||||||
| US-7309720-B2 | |||||||
| WO-2004110984-A1 | |||||||
| WO-2004113270-A2 | |||||||
| WO-2004113276-A1 | |||||||
| WO-2004113283-A1 | |||||||
| EP-1610770-A1 | |||||||
| EP-1610770-B1 | |||||||
| EP-1894564-A2 | |||||||
| US-2006199797-A1 | |||||||
| US-7514421-B2 | |||||||
| WO-2004089350-A1 | |||||||
| EP-1572626-A1 | |||||||
| US-2005131068-A1 | |||||||
| WO-2004056748-A1 | |||||||
| EP-1539120-A1 | |||||||
| US-2006083790-A1 | |||||||
| WO-2004006899-A1 | |||||||
| EP-1521742-A1 | |||||||
| US-2005239766-A1 | |||||||
| US-7470678-B2 | |||||||
| WO-2004005247-A1 | |||||||
| EP-1517679-A1 | |||||||
| EP-1517679-B1 | |||||||
| EP-1517883-A1 | |||||||
| EP-1517883-B1 | |||||||
| EP-1517883-B8 | |||||||
| US-2005222261-A1 | |||||||
| US-2005256198-A1 | |||||||
| US-2005267149-A1 | |||||||
| US-7351858-B2 | |||||||
| US-7355069-B2 | |||||||
| US-7521461-B2 | |||||||
| WO-2004000294-A1 | |||||||
| WO-2004000790-A1 | |||||||
| EP-1478368-A1 | |||||||
| US-2005124557-A1 | |||||||
| WO-03061663-A1 | |||||||
| EP-1458672-A1 | |||||||
| EP-1458672-B1 | |||||||
| EP-1458673-A1 | |||||||
| EP-1458673-B1 | |||||||
| EP-1458677-A1 | |||||||
| EP-1458677-B1 | |||||||
| US-2005113362-A1 | |||||||
| US-2005171204-A1 | |||||||
| US-2005215630-A1 | |||||||
| US-2005282822-A1 | |||||||
| US-7256307-B2 | |||||||
| US-7276539-B2 | |||||||
| US-7488844-B2 | |||||||
| US-7514471-B2 | |||||||
| WO-03051821-A1 | |||||||
| WO-03051822-A1 | |||||||
| WO-03051826-A1 | |||||||
| EP-1427423-B1 | |||||||
| EP-1427423-B9 | |||||||
| US-2005038009-A1 | |||||||
| US-7132416-B2 |
Viloxazine, ヴィロキサジン;

Viloxazine
- Molecular FormulaC13H19NO3
- Average mass237.295 Da
update FDA APPROVED 2021/4/2, Qelbree, Viloxazine hydrochloride
| Formula |
C13H19NO3. HCl
|
|---|---|
| CAS |
35604-67-2
|
| Mol weight |
273.7558
|
2-[(2-Ethoxyphenoxy)methyl]morpholine
256-281-7 [EINECS]
3489
46817-91-8 free [RN], Hcl 35604-67-2
5I5Y2789ZF
Emovit [Wiki]
Morpholine, 2-((2-ethoxyphenoxy)methyl)-
Morpholine, 2-[(2-ethoxyphenoxy)methyl]-
UNII:5I5Y2789ZF

NEW DRUG APPROVALS
ONE TIME
$10.00
Viloxazine (trade names Vivalan, Emovit, Vivarint and Vicilan) is a morpholine derivative and is a selective norepinephrine reuptake inhibitor (NRI). It was used as an antidepressant in some European countries, and produced a stimulant effect that is similar to the amphetamines, except without any signs of dependence. It was discovered and brought to market in 1976 by Imperial Chemical Industries and was withdrawn from the market in the early 2000s for business reasons.

Clip
https://www.sciencedirect.com/science/article/pii/S0040402015302659

Patent
US 20180265482
| Viloxazine ((R,S)-2-[(2-ethoxyphenoxy)methyl]morpholine]) is a bicyclic morpholine derivative, assigned CAS No. 46817-91-8 (CAS No. 35604-67-2 for the HCl salt). It is characterized by the formula C 13H 19NO 3, with a molecular mass of 237.295 g/mol. Viloxazine has two stereoisomers, (S)-(−)- and (R)-(+)-isomer, which have the following chemical structures: |
| Viloxazine is known to have several desirable pharmacologic uses, including treatment of depression, nocturnal enuresis, narcolepsy, sleep disorders, and alcoholism, among others. In vivo, viloxazine acts as a selective norepinephrine reuptake inhibitor (“NRI”). |
| Between the two stereoisomers, the (S)-(−)-isomer is known to be five times as pharmacologically active as the (R)-(+)-isomer. See, e.g., “Optical Isomers of 2-(2-ethoxyphenoxymethyl)tetrahydro-1,4 oxazine (viloxazine) and Related Compounds” (Journal of Medicinal Chemistry, Jan. 9, 1976, 19(8); 1074) in which it is disclosed that optical isomers of 2-(2-ethoxyphenoxymethyl)tetrahydro-1,4-oxazine (viloxazine) and 2-(3-methoxyphenoxymethyl)tetrahydro-1,4-oxazine were prepared and absolute configurations assigned. The synthesis of optical isomers of viloxazine analogs of known configuration was accomplished by resolution of the intermediate 4-benzyl-2-(p-toluenesulfonyloxymethyl)tetrahydro-1,4-oxazine isomers. |
| Some unsatisfactory methods of synthesizing viloxazine are known in the art. For example, as disclosed in U.S. Pat. No. 3,714,161, viloxazine is prepared by reacting ethoxyphenol with epichlorohydrin to afford the epoxide intermediate 1-(2-ethoxyphenoxy)-2,3-epoxypropane. This epoxide intermediate is then treated with benzylamine followed with chloroacetyl chloride. The resulting morpholinone is then reduced by lithium aluminum hydride and then by Pd/C-catalyzed hydrogenation to yield viloxazine free base. |
| Yet another unsatisfactory synthesis of viloxazine is disclosed in U.S. Pat. No. 3,712,890, which describes a process to prepare viloxazine HCl, wherein the epoxide intermediate, 1-(2-ethoxyphenoxy)-2,3-epoxypropane, is reacted with 2-aminoethyl hydrogen sulfate in ethanol in the presence of sodium hydroxide to form viloxazine free base. The product is extracted with diethyl ether from the aqueous solution obtained by evaporating the solvent in the reaction mixture then adding water to the residue. The ethereal extract is dried over a drying agent and the solvent is removed. Viloxazine HCl salt is finally obtained by dissolving the previous residue in isopropanol, concentrated aqueous HCl, and ethyl acetate followed by filtration. |
| The foregoing methods of synthesizing viloxazine suffer from a number of deficiencies, such as low reaction yield and unacceptably large amount of impurities in the resulting product. Effective elimination or removal of impurities, especially those impurities possessing genotoxicity or other toxicities, is critical to render safe pharmaceutical products. For example, certain reagents traditionally utilized in viloxazine HCl preparation, such as epichlorohydrin and 2-aminoethyl hydrogen sulfate, present a special problem due to their toxicity. There is a need for effective methods to remove or limit harmful impurities down to a level that is appropriate and safe according to contemporary sound medical standards and judgment. Accordingly, a continuing and unmet need exists for new and improved methods of manufacturing viloxazine and its various salts to yield adequate quantities of pharmacologically desirable API with predictable and reliable control of impurities. |
| Polymorph control is also an important aspect of producing APIs and their associated salts that are used in pharmaceutical products. However, no polymorphs of viloxazine HCl have previously been disclosed. A need therefore exists for new polymorphic forms of viloxazine that have improved pharmacological properties. |
PATENT
WO 2011130194
For the sake of convenience and without putting any limitations thereof, the methods of manufacture of viloxazine have been separated into several steps, each step being disclosed herein in a multiplicity of non-limiting embodiments. These steps comprise Step 1, during which 2-ethoxyphenol and epichlorhydrin are reacted to produce l-(2-ethoxyphenoxy)-2,3-epoxypropane (Epoxide 1); Step 2, during which l-(2-ethoxyphenoxy)-2,3-epoxypropane (Epoxide 1) is converted into viloxazine base which is further converted into viloxazine salt, and Step 3, during which viloxazine salt is purified/recrystallized, and various polymorphic forms of viloxazine salt are prepared.
The above-mentioned steps will be considered below in more details.
[0031] The process of the Step 1 may be advantageously carried out in the presence of a phase-transfer catalyst to afford near quantitative yield of l-(2-ethoxyphenoxy)-2,3-epoxypropane. Alternatively, the process may make use of a Finkelstein catalyst described in more details below. Additionally, the reaction may take place without the use of the catalyst.
FIG. 1, depicted below, schematically illustrates the preparation of l-(2-ethoxyphenoxy)-2,3-epoxypropane (“Epoxide 1”) in accordance with Step I of an exemplary synthesis of viloxazine:
STEP I:
Epoxide 1
In one embodiment of the Step 1, the preparation of l-(2-ethoxyphenoxy)-2,3-epoxypropane (epoxide 1) can be effected by the use of a phase transfer catalyst in the presence of a solid or liquid base with a solution of a corresponding phenol and epichlorohydrin in one or more solvents (Fig. 1). The phase transfer catalyst can be selected from ammonium salts, such as benzyltriethylammonium salts, benzyltrimethylammonium salts, and tetrabutylammonium salts, phosphonium salts, guanidinium salts, crown ether, polyethylene glycol, polyethylene glycol ether, or polyethylene glycol ester, or other phase transfer catalysts know in the art. The solid or liquid base can be a carbonate such as alkali carbonate, NaOH, KOH, LiOH, LiOH/LiCl, amines such as mono-, di- or tri-substituted amines (such as diethylamine, triethylamine, dibutylamine, tributylamine), DMAP, or other appropriate base. The solvents used in the solution of a corresponding phenol and epichlorohydrin include but are not limited to ethers such as methyl t-butyl ether, ketones, non-substituted or substituted aromatic solvents (xylene), halo-substituted hydrocarbons (e.g. CH2C12, CHC13), THF, DMF, dioxanes, non-substituted and substituted pyridines, acetonitrile, pyrrolidones, nitromethane , or other appropriate solvent. Additional catalyst, such as, for example, Finkelstein catalyst, can also be used in the process of this embodiment. This reaction preferably takes place at an elevated temperature. In one variation of the embodiment, the temperature is above 50°C. In another variation, epichlorohydrin, potassium carbonate, and a phase transfer catalyst are mixed with a solution of 2-ethoxyphenol in a solvent at an elevated temperature, such as 50 – 60°C. After the reaction is complete, the reaction mixture can be washed with water, followed by work-up procedures known in the art. Variations of this embodiment of the invention are further disclosed in Examples 1-8.
[0033] In one variation of the above embodiment of the Step 1 , Epoxide 1 is prepared by reacting 2-ethoxyphenol and epichlorohydrin in a solvent in the presence of two different catalysts, and a base in a solid state. The first catalyst is a phase transfer catalyst as described above; the second catalyst is a Finkelstein reaction catalyst. Without putting any limitation
hereon, metal iodide and metal bromide salts, such as potassium iodide, may be used as an example of a Finkelstein catalyst. The phase transfer catalyst and a solvent may be selected from any phase transfer catalysts and solvents known in the art. Potassium carbonate may be used as a non-limiting example of a solid base. Using the solid base in a powdered form may be highly beneficial due to the greatly enhanced interface and limiting the side reactions. This variation of the embodiment is further illustrated by Example 9. In another variation of the embodiment, liquid base such as triethylamine can be used to replace the solid base.
[0034] In a different embodiment of Step 1 , 2-ethoxyphenol and epichlorohydrin are reacted in a solvent-free system that comprises a solid or liquid base, a phase transfer catalyst as listed above and a Finkelstein catalyst.
[0035] FIG. 2, depicted below, schematically illustrates the preparation of l-(2-ethoxyphenoxy)-2,3-epoxypropane (“Epoxide 1”) in accordance with the Step I of another exemplary synthesis of viloxazine ( biphasic):
STEP I (alternative embodiment):
In this embodiment of Step 1, illustrated in Fig. 2, Epoxide 1 can be prepared by reacting epichlorohydrin with 2-ethoxyphenol in the presence of a catalytic amount of a phase transfer catalyst without the use of solvents at elevated temperatures in a two-stage process to afford near quantitative yield of l-(2-ethoxyphenoxy)-2,3-epoxypropane with very few side products. This embodiment of the invention is further illustrated by a non-limiting Example 12. The phase transfer catalyst for this embodiment can be selected from ammonium salts such as benzyltriethylammonium salts, benzyltrimethylammonium salts, tetrabutylammonium salts, etc; phosphonium salts, guanidinium salts, crown ether, polyethylene glycol, polyethylene glycol ether, or polyethylene glycol ester, or other phase transfer catalysts know in the art. The first stage of the process of this embodiment may take place without a solvent in a presence of a large excess of epichlorohydrin. This stage is followed by a de-chlorination stage, before or after
removal of excess epichlorohydrin, using a base and a solvent. The reaction produces l-(2-ethoxyphenoxy)-2,3-epoxypropane in high yield. Example of the bases used herein include but are not limited to NaOH, KOH, LiOH, LiOH/LiCl, K2C03, Na2C03, amines such as mono-, di-or tri-substituted amines (such as diethylamine, triethylamine, dibutylamine, tributylamine etc.), DMAP. In one variation of this embodiment of Step 1, the phase transfer catalyst may be used only at the de-chlorination stage of the process. The de-chlorination stage can be carried out in a biphasic system or in a single phase system. For a biphasic system, it can be an organic-aqueous liquid biphasic system, or a liquid-solid biphasic system. Solvents that are useful for the process include but are not limited to non-substituted and substituted aromatic solvents (e.g. toluene, benzene, chlorobenzene, dimethylbenzene, xylene), halo-substituted hydrocarbons (e.g. CH2C12, CHC13), THF, dioxanes, DMF, DMSO, non-substituted and substituted pyridines, ketones, pyrrolidones, ethers, acetonitrile, nitromethane. As mentioned above, this process takes place at the elevated temperature. In one variation of the embodiment, the temperature is above 60°C. In another variation, 2-ethoxyphenol and epichlorohydrin are heated to 60 – 90°C for a period of time in the presence of phase transfer catalyst. Excess of epichlorohydrin is removed and the residue is dissolved in a solvent such as toluene or benzene treated with an aqueous base solution, such as NaOH, KOH, LiOH, LiOH/LiCl. In yet another variation of the embodiment, the residue after epichlorohydrin removal can be dissolved in one or more of the said solvent and treated with a base (solid or liquid but not an aqueous solution) and optionally a second phase transfer catalyst, optionally at elevated temperatures.
[0036] In yet another embodiment of Step 1 , Epoxide 1 can also be prepared by using a catalyst for a so-called Finkelstein reaction in the presence of a Finkelstein catalyst but without the need to use a phase transfer catalyst. Finkelstein catalysts useful herein include metal iodide salts and metal bromide salts, among others. In one variation of this embodiment, 2-ethoxyphenol and epichlorohydrin are dissolved in a polar aprotic solvent such as DMF, and a catalytic amount of an iodide such as potassium iodide and a base, as solid or liquid, are used. Preferably, the base is used as a solid, such as potassium carbonate powder. This embodiment is further illustrated by the Example 11.
[0037] In the alternative embodiment of Step 1 , Epoxide 1 can also be prepared by a different method that comprises reacting epichlorohydrin and the corresponding phenol in the presence of a base at a temperature lower than the ambient temperature, especially when a base solution is used, and without the use of a phase transfer catalyst. This embodiment is illustrated by the Example 10.
[0038] A very high, almost quantitative, yield of 1 -(2-ethoxyphenoxy)-2,3-epoxypropane can be obtained through realizing the above-described embodiments of Step 1 , with less impurities generated in Epoxide 1.
[0039] Epoxide 1 , produced in Step 1 as described above, is used to prepare viloxazine base (viloxazine), which is further converted into viloxazine salt through the processes of Step 2.
[0040] FIG. 3, depicted below, schematically illustrates the preparation of viloxazine
(“Step Ila”) and the preparation of viloxazine hydrochloride (“Step lib”), as well as their purification (“Step III”) in accordance with another example embodiment hereof:
STEP Ila:
Hydrogen Sulfate
STEP lib:
Step III:
Conversion
Viloxazine free base ► Viloxazine salt
Wash/ raction
Recrystallization
Purified viloxazine salt
In the embodiment of Step 2, illustrated in Fig. 3, the preparation of viloxazine base is achieved by reacting the Epoxide 1 intermediate prepared in Step 1 and aminoethyl hydrogen sulfate in presence of a large excess of a base as illustrated by the Examples 5-7 and 14. The base may be present as a solid or in a solution. Preferably, the molar ratio of the base to Epoxide 1 is more than 10. More preferably the ratio is more than 12. Even more preferably, the ratio is between 15 and 40. It was unexpectedly discovered that the use of a higher ratio of a base results in a faster reaction, less impurities, and lower reaction temperature.
[0041] Further advantages may be offered by a specific variation of this embodiment, wherein the base is added to the reaction mixture in several separate steps. For example, a third of the base is added to the reaction mixture, and the mixture is stirred for a period of time. Then the rest of the base is added followed by additional stirring. Alternatively, half of the base is added initially followed by the second half after some period of time, or the base is added in three different parts separated by periods of time. The bases used herein include but are not limited to NaOH, KOH, LiOH, LiOH/LiCl, K2C03, Na2C03, amines such as mono-, di- or tri-substituted amines (such as diethylamine, triethylamine, dibutylamine, tributylamine), DMAP, and combinations thereof. . In one embodiment of the invention, the base is KOH. In another embodiment, the base is NaOH. In a further embodiment, the base is K2C03 powder. In yet further embodiment, the base is triethylamine. This embodiment is illustrated further by
Examples 13,15 and 16.
[0042] In another exemplary embodiment of Step 2, viloxazine is produced by cyclization of novel intermediate compound “Diol 1 ,” which is made from Epoxide 1 and N-benzyl-aminoethanol. This method allows one to drastically reduce the use of potentially toxic materials in the manufacturing process, completely eliminating some of them such as aminoethyl hydrogen sulfate. The first stage of the reaction results in the formation of an intermediate of Formula 3 (Diol 1), which is a new, previously unidentified compound.
[0043] Formula 3
Diol 1
FIG. 4, depicted below, schematically illustrates the preparation of viloxazine and its salts via “Diol 1” in accordance with another exemplary embodiment hereof (Bn = benzyl, Et = ethyl):
Viloxazine HCI
As illustrated in Fig. 4, Diol 1 is turned into N-benzyl viloxazine by cyclization. Removal of the benzyl protective group yields viloxazine base. Similarly, FIG. 5, depicted below, schematically illustrates the cyclization of Diol 1, as well as some side-reactions thereof.
Uses
Viloxazine hydrochloride was used in some European countries for the treatment of clinical depression.[4][5]
Side effects
Side effects included nausea, vomiting, insomnia, loss of appetite, increased erythrocyte sedimentation, EKG and EEG anomalies, epigastric pain, diarrhea, constipation, vertigo, orthostatic hypotension, edema of the lower extremities, dysarthria, tremor, psychomotor agitation, mental confusion, inappropriate secretion of antidiuretic hormone, increased transaminases, seizure, (there were three cases worldwide, and most animal studies (and clinical trials that included epilepsy patients) indicated the presence of anticonvulsant properties, so was not completely contraindicated in epilepsy,[6]) and increased libido.[7]
Drug interactions
Viloxazine increased plasma levels of phenytoin by an average of 37%.[8] It also was known to significantly increase plasma levels of theophylline and decrease its clearance from the body,[9] sometimes resulting in accidental overdose of theophylline.[10]
Mechanism of action
Viloxazine, like imipramine, inhibited norepinephrine reuptake in the hearts of rats and mice; unlike imipramine, it did not block reuptake of norepinephrine in either the medullae or the hypothalami of rats. As for serotonin, while its reuptake inhibition was comparable to that of desipramine (i.e., very weak), viloxazine did potentiate serotonin-mediated brain functions in a manner similar to amitriptyline and imipramine, which are relatively potent inhibitors of serotonin reuptake.[11] Unlike any of the other drugs tested, it did not exhibit any anticholinergic effects.[11]
It was also found to up-regulate GABAB receptors in the frontal cortex of rats.[12]
Chemical properties
It is a racemic compound with two stereoisomers, the (S)-(–)-isomer being five times as pharmacologically active as the (R)-(+)-isomer.[13]
History
Viloxazine was discovered by scientists at Imperial Chemical Industries when they recognized that some beta blockers inhibited serotonin reuptake inhibitor activity in the brain at high doses. To improve the ability of their compounds to cross the blood brain barrier, they changed the ethanolamine side chain of beta blockers to a morpholine ring, leading to the synthesis of viloxazine.[14]:610[15]:9 The drug was first marketed in 1976.[16] It was never approved by the FDA,[5] but the FDA granted it an orphan designation (but not approval) for cataplexy and narcolepsy in 1984.[17] It was withdrawn from markets worldwide in 2002 for business reasons.[14][18]
As of 2015, Supernus Pharmaceuticals was developing formulations of viloxazine as a treatment for ADHD and major depressive disorder under the names SPN-809 and SPN-812.[19][20]
Research
Viloxazine has undergone two randomized controlled trials for nocturnal enuresis (bedwetting) in children, both of those times versus imipramine.[21][22] By 1990, it was seen as a less cardiotoxic alternative to imipramine, and to be especially effective in heavy sleepers.[23]
In narcolepsy, viloxazine has been shown to suppress auxiliary symptoms such as cataplexy and also abnormal sleep-onset REM[24] without really improving daytime somnolence.[25]
In a cross-over trial (56 participants) viloxazine significantly reduced EDS and cataplexy.[18]
Viloxazine has also been studied for the treatment of alcoholism, with some success.[26]
While viloxazine may have been effective in clinical depression, it did relatively poorly in a double-blind randomized controlled trial versus amisulpride in the treatment of dysthymia.[27]
It is also under investigation as a treatment for attention deficit hyperactivity disorder.[28]
REFERNCES
- ^ Bouchard JM, Strub N, Nil R (October 1997). “Citalopram and viloxazine in the treatment of depression by means of slow drop infusion. A double-blind comparative trial”. Journal of Affective Disorders. 46 (1): 51–8. doi:10.1016/S0165-0327(97)00078-5. PMID 9387086.
- ^ Case DE, Reeves PR (February 1975). “The disposition and metabolism of I.C.I. 58,834 (viloxazine) in humans”. Xenobiotica. 5 (2): 113–29. doi:10.3109/00498257509056097. PMID 1154799.
- ^ “SID 180462– PubChem Substance Summary”. Retrieved 5 November 2005.
- ^ Pinder, RM; Brogden, RN; Speight, ™; Avery, GS (June 1977). “Viloxazine: a review of its pharmacological properties and therapeutic efficacy in depressive illness”. Drugs. 13 (6): 401–21. doi:10.2165/00003495-197713060-00001. PMID 324751.
- ^ Jump up to:a b Dahmen, MM, Lincoln, J, and Preskorn, S. NARI Antidepressants, pp 816-822 in Encyclopedia of Psychopharmacology, Ed. Ian P. Stolerman. Springer-Verlag Berlin Heidelberg, 2010. ISBN 9783540687061
- ^ Edwards JG, Glen-Bott M (September 1984). “Does viloxazine have epileptogenic properties?”. Journal of Neurology, Neurosurgery, and Psychiatry. 47 (9): 960–4. doi:10.1136/jnnp.47.9.960. PMC 1027998. PMID 6434699.
- ^ Chebili S, Abaoub A, Mezouane B, Le Goff JF (1998). “Antidepressants and sexual stimulation: the correlation” [Antidepressants and sexual stimulation: the correlation]. L’Encéphale (in French). 24 (3): 180–4. PMID 9696909.
- ^ Pisani F, Fazio A, Artesi C, et al. (February 1992). “Elevation of plasma phenytoin by viloxazine in epileptic patients: a clinically significant drug interaction”. Journal of Neurology, Neurosurgery, and Psychiatry. 55 (2): 126–7. doi:10.1136/jnnp.55.2.126. PMC 488975. PMID 1538217.
- ^ Perault MC, Griesemann E, Bouquet S, Lavoisy J, Vandel B (September 1989). “A study of the interaction of viloxazine with theophylline”. Therapeutic Drug Monitoring. 11 (5): 520–2. doi:10.1097/00007691-198909000-00005. PMID 2815226.
- ^ Laaban JP, Dupeyron JP, Lafay M, Sofeir M, Rochemaure J, Fabiani P (1986). “Theophylline intoxication following viloxazine induced decrease in clearance”. European Journal of Clinical Pharmacology. 30 (3): 351–3. doi:10.1007/BF00541543. PMID 3732375.
- ^ Jump up to:a b Lippman W, Pugsley TA (August 1976). “Effects of viloxazine, an antidepressant agent, on biogenic amine uptake mechanisms and related activities”. Canadian Journal of Physiology and Pharmacology. 54 (4): 494–509. doi:10.1139/y76-069. PMID 974878.
- ^ Lloyd KG, Thuret F, Pilc A (October 1985). “Upregulation of gamma-aminobutyric acid (GABA) B binding sites in rat frontal cortex: a common action of repeated administration of different classes of antidepressants and electroshock”. The Journal of Pharmacology and Experimental Therapeutics. 235 (1): 191–9. PMID 2995646.
- ^ Danchev ND, Rozhanets VV, Zhmurenko LA, Glozman OM, Zagorevskiĭ VA (May 1984). “Behavioral and radioreceptor analysis of viloxazine stereoisomers” [Behavioral and radioreceptor analysis of viloxazine stereoisomers]. Biulleten’ Eksperimental’noĭ Biologii i Meditsiny (in Russian). 97 (5): 576–8. PMID 6326891.
- ^ Jump up to:a b Williams DA. Antidepressants. Chapter 18 in Foye’s Principles of Medicinal Chemistry, Eds. Lemke TL and Williams DA. Lippincott Williams & Wilkins, 2012. ISBN 9781609133450
- ^ Wermuth, CG. Analogs as a Means of Discovering New Drugs. Chapter 1 in Analogue-based Drug Discovery. Eds.IUPAC, Fischer, J., and Ganellin CR. John Wiley & Sons, 2006. ISBN 9783527607495
- ^ Olivier B, Soudijn W, van Wijngaarden I. Serotonin, dopamine and norepinephrine transporters in the central nervous system and their inhibitors. Prog Drug Res. 2000;54:59-119. PMID 10857386
- ^ FDA. Orphan Drug Designations and Approvals: Viloxazine Page accessed August 1, 2-15
- ^ Jump up to:a b Vignatelli L, D’Alessandro R, Candelise L. Antidepressant drugs for narcolepsy. Cochrane Database Syst Rev. 2008 Jan 23;(1):CD003724. Review. PMID 18254030
- ^ Bloomberg Supernus profile Page accessed August 1, 2015
- ^ Supernus. Psychiatry portfolio Page accessed August 1, 2015
- ^ Attenburrow AA, Stanley TV, Holland RP (January 1984). “Nocturnal enuresis: a study”. The Practitioner. 228 (1387): 99–102. PMID 6364124.
- ^ ^ Yurdakök M, Kinik E, Güvenç H, Bedük Y (1987). “Viloxazine versus imipramine in the treatment of enuresis”. The Turkish Journal of Pediatrics. 29 (4): 227–30. PMID 3332732.
- ^ Libert MH (1990). “The use of viloxazine in the treatment of primary enuresis” [The use of viloxazine in the treatment of primary enuresis]. Acta Urologica Belgica (in French). 58 (1): 117–22. PMID 2371930.
- ^ Guilleminault C, Mancuso J, Salva MA, et al. (1986). “Viloxazine hydrochloride in narcolepsy: a preliminary report”. Sleep. 9 (1 Pt 2): 275–9. PMID 3704453.
- ^ Mitler MM, Hajdukovic R, Erman M, Koziol JA (January 1990). “Narcolepsy”. Journal of Clinical Neurophysiology. 7 (1): 93–118. doi:10.1097/00004691-199001000-00008. PMC 2254143. PMID 1968069.
- ^ Altamura AC, Mauri MC, Girardi T, Panetta B (1990). “Alcoholism and depression: a placebo controlled study with viloxazine”. International Journal of Clinical Pharmacology Research. 10 (5): 293–8. PMID 2079386.
- ^ León CA, Vigoya J, Conde S, Campo G, Castrillón E, León A (March 1994). “Comparison of the effect of amisulpride and viloxazine in the treatment of dysthymia” [Comparison of the effect of amisulpride and viloxazine in the treatment of dysthymia]. Acta Psiquiátrica Y Psicológica de América Latina (in Spanish). 40 (1): 41–9. PMID 8053353.
- ^ Mattingly, GW; Anderson, RH (December 2016). “Optimizing outcomes in ADHD treatment: from clinical targets to novel delivery systems”. CNS Spectrums. 21 (S1): 45–59. doi:10.1017/S1092852916000808. PMID 28044946.
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2004229942 | Analeptic and antidepressant combinations | 2004-05-12 | 2004-11-18 |
| US2004242698 | Analeptic and antidepressant combinations | 2004-05-12 | 2004-12-02 |
| US2002192302 | Transdermal and topical administration of antidepressant drugs using basic enhancers | 2002-06-19 | 2002-12-19 |
| US2018105877 | GENETIC POLYMORPHISMS ASSOCIATED WITH DEPRESSION | 2017-05-19 | |
| US2013244990 | GENETIC POLYMORPHISMS ASSOCIATED WITH DEPRESSION | 2012-11-30 | 2013-09-19 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2009197849 | TRANSDERMAL AND TOPICAL ADMINISTRATION OF DRUGS USING BASIC PERMEATION ENHANCERS | 2008-09-03 | 2009-08-06 |
| US2006150989 | Method of diagnosing, treating and educating individuals with and/or about depression | 2005-01-12 | 2006-07-13 |
| US8153159 | Modafinil modified release pharmaceutical compositions | 2004-09-17 | 2012-04-10 |
| US2004229940 | Analeptic and antidepressant combinations | 2004-05-12 | 2004-11-18 |
| US2004229941 | Analeptic and antidepressant combinations | 2004-05-12 | 2004-11-18 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9434703 | METHODS FOR PRODUCING VILOXAZINE SALTS AND NOVEL POLYMORPHS THEREOF | 2014-12-19 | 2015-05-07 |
| US2012220962 | TRANSDERMAL AND TOPICAL ADMINISTRATION OF VITAMINS USING BASIC PERMEATION ENHANCERS | 2012-05-10 | 2012-08-30 |
| US9358204 | FORMULATIONS OF VILOXAZINE | 2013-02-07 | 2013-08-08 |
| US2012289515 | Combination therapy for cognitive distortions, resisting relapse of unipolar non-psychotic depression | 2011-05-25 | 2012-11-15 |
| US2009317453 | TRANSDERMAL AND TOPICAL ADMINISTRATION OF DRUGS USING BASIC PERMEATION ENHANCERS | 2008-12-23 | 2009-12-24 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2006013834 | Room temperature stable aqueous liquid pharmaceutical composition | 2005-02-25 | 2006-01-19 |
| US2004220262 | Transdermal and topical administration of drugs using basic permeation enhancers | 2004-06-03 | 2004-11-04 |
| US2003124176 | Transdermal and topical administration of drugs using basic permeation enhancers | 2002-06-21 | 2003-07-03 |
| US6713089 | Quick release pharmaceutical compositions of drug substances | 2001-07-10 | 2004-03-30 |
| WO0015195 | QUICK RELEASE PHARMACEUTICAL COMPOSITIONS OF DRUG SUBSTANCES | 2000-03-23 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2014315720 | POLYSACCHARIDE ESTER MICROSPHERES AND METHODS AND ARTICLES RELATING THERETO | 2014-04-04 | 2014-10-23 |
| US2014113826 | POLYSACCHARIDE ESTER MICROSPHERES AND METHODS AND ARTICLES RELATING THERETO | 2013-10-23 | 2014-04-24 |
| US2014052264 | Porous, Stabilized Craniomaxillofacial Implants and Methods and Kits Relating Thereto | 2013-08-12 | 2014-02-20 |
| US2014348936 | GASTRORETENTIVE CONTROLLED RELEASE VEHICLES THAT INCLUDE ETHYLENE COPOLYMERS, ETHYL CELLULOSES, AND/OR THERMOPLASTIC POLYURETHANES | 2012-12-17 | 2014-11-27 |
| US2016304475 | METHODS FOR PRODUCING VILOXAZINE SALTS AND NOVEL POLYMORPHS THEREOF | 2016-06-24 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017319698 | GASTRORETENTIVE GEL FORMULATIONS | 2015-10-26 | |
| US8231899 | Quick release pharmaceutical compositions of drug substances | 2004-01-13 | 2012-07-31 |
| US9662338 | FORMULATIONS OF VILOXAZINE | 2016-06-03 | 2016-09-29 |
| US9603853 | FORMULATIONS OF VILOXAZINE | 2016-05-18 | 2016-09-08 |
| US2015306230 | DRUG DELIVERY VEHICLES COMPRISING CELLULOSE DERIVATIVES, STARCH DERIVATIVES, AND COMBINATIONS THEREOF | 2014-06-12 | 2015-10-29 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US3959273 | Morpholine derivatives | 1976-05-25 | |
| US8802596 | Multi-functional ionic liquid compositions for overcoming polymorphism and imparting improved properties for active pharmaceutical, biological, nutritional, and energetic ingredients | 2012-06-07 | 2014-08-12 |
| US6060516 | N1-propargylhydrazines, N2-propargylhydrazines and their analogs for the treatment of depression, anxiety and neurodegeneration | 2000-05-09 | |
| US6479491 | Disubstituted morpholine, oxazepine or thiazepine derivatives, their preparation and their use as dopamine d4 receptor antagonists | 2002-11-12 | |
| US2017258801 | FORMULATIONS OF VILOXAZINE | 2017-05-25 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US6034117 | Methods of treating and diagnosing sleep disordered breathing and means for carrying out the method | 2000-03-07 | |
| EP0923370 | ACETYL CHOLINE ESTERASE INHIBITORS FOR TREATING AND DIAGNOSING SLEEP DISORDERED BREATHING | 1999-06-23 | 2005-11-16 |
| US2014328884 | CONTROLLED RELEASE VEHICLES HAVING DESIRED VOID VOLUME ARCHITECTURES | 2012-12-17 | 2014-11-06 |
| US9096743 | PROCESS FOR FORMING FILMS, FIBERS, AND BEADS FROM CHITINOUS BIOMASS | 2010-06-01 | 2012-05-10 |
| US2003104041 | Transdermal and topical administration of drugs using basic permeation enhancers | 2002-06-20 | 2003-06-05 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| WO9807710 | DISUBSTITUTED MORPHOLINE, OXAZEPINE OR THIAZEPINE DERIVATIVES, THEIR PREPARATION AND THEIR USE AS DOPAMINE D4 RECEPTOR ANTAGONISTS | 1998-02-26 | |
| US2014051771 | Biomedical Devices Comprising Molded Polyethylene Components | 2013-08-12 | 2014-02-20 |
| US9278134 | DUAL FUNCTIONING IONIC LIQUIDS AND SALTS THEREOF | 2009-12-29 | 2012-02-23 |
| US8232265 | Multi-functional ionic liquid compositions for overcoming polymorphism and imparting improved properties for active pharmaceutical, biological, nutritional, and energetic ingredients | 2007-04-26 | 2012-07-31 |
| US2005074487 | Transdermal and topical administration of drugs using basic permeation enhancers | 2004-06-07 | 2005-04-07 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9199918 | SMALL MOLECULE INHIBITORS OF AGBL2 | 2012-02-14 | 2013-12-12 |
| US9403783 | METHODS FOR PRODUCING VILOXAZINE SALTS AND NOVEL POLYMORPHS THEREOF | 2011-10-13 | |
| US7973043 | Combination therapy for depression, prevention of suicide, and various medical and psychiatric conditions | 2003-07-25 | 2011-07-05 |
| US6207662 | Disubstituted morpholine, oxazepine or thiazepine derivatives, their preparation and their use as dopamine D4 receptor antagonists | 2001-03-27 | |
| EP0920423 | DISUBSTITUTED MORPHOLINE, OXAZEPINE OR THIAZEPINE DERIVATIVES, THEIR PREPARATION AND THEIR USE AS DOPAMINE D4 RECEPTOR ANTAGONISTS | 1999-06-09 | 2005-01-26 |
| Clinical data | |
|---|---|
| Routes of administration | By mouth, intravenous infusion[1] |
| ATC code | N06AX09 (WHO) |
| Legal status | |
| Legal status | In general: uncontrolled |
| Pharmacokinetic data | |
| Elimination half-life | 2–5 hours |
| Excretion | Renal[2] |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 46817-91-8 35604-67-2 (HCl salt) |
| PubChem CID | 5666 |
| ChemSpider | 5464 |
| UNII | 5I5Y2789ZF |
| KEGG | D08673 |
| ChEMBL | ChEMBL306700 |
| ECHA InfoCard | 100.051.148 |
| Chemical and physical data | |
| Formula | C13H19NO3 |
| Molar mass | 237.295 g/mol g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| SMILES[hide]O(c1ccccc1OCC)CC2OCCNC2 | |
| InChI[hide]InChI=1S/C13H19NO3/c1-2-15-12-5-3-4-6-13(12)17-10-11-9-14-7-8-16-11/h3-6,11,14H,2,7-10H2,1H3 Key:YWPHCCPCQOJSGZ-UHFFFAOYSA-N |
/////////////////Viloxazine, ヴィロキサジン , Emovit, Vivalan, Emovit, Vivarint, Vicilan
Iobenguane I 131
Iobenguane I 131

FDA approves first treatment for rare adrenal tumors
The U.S. Food and Drug Administration today approved Azedra (iobenguane I 131) injection for intravenous use for the treatment of adults and adolescents age 12 and older with rare tumors of the adrenal gland (pheochromocytoma or paraganglioma) that cannot be surgically removed (unresectable), have spread beyond the original tumor site and require systemic anticancer therapy. This is the first FDA-approved drug for this use.
update………APPROVED JAPAN 2021, 2021/9/27, Raiatt MIBG-I 131
July 30, 2018
Release
The U.S. Food and Drug Administration today approved Azedra (iobenguane I 131) injection for intravenous use for the treatment of adults and adolescents age 12 and older with rare tumors of the adrenal gland (pheochromocytoma or paraganglioma) that cannot be surgically removed (unresectable), have spread beyond the original tumor site and require systemic anticancer therapy. This is the first FDA-approved drug for this use.
“Many patients with these ultra-rare cancers can be treated with surgery or local therapies, but there are no effective systemic treatments for patients who experience tumor-related symptoms such as high blood pressure,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Patients will now have an approved therapy that has been shown to decrease the need for blood pressure medication and reduce tumor size in some patients.”
Pheochromocytomas are rare tumors of the adrenal glands. These glands are located right above the kidneys and make hormones including stress hormones called epinephrines and norepinephrines. Pheochromocytomas increase the production of these hormones, leading to hypertension (high blood pressure) and symptoms such as headaches, irritability, sweating, rapid heart rate, nausea, vomiting, weight loss, weakness, chest pain or anxiety. When this type of tumor occurs outside the adrenal gland, it is called a paraganglioma.
The efficacy of Azedra was shown in a single-arm, open-label, clinical trial in 68 patients that measured the number of patients who experienced a 50 percent or greater reduction of all antihypertensive medications lasting for at least six months. This endpoint was supported by the secondary endpoint, overall tumor response measured by traditional imaging criteria. The study met the primary endpoint, with 17 (25 percent) of the 68 evaluable patients experiencing a 50 percent or greater reduction of all antihypertensive medication for at least six months. Overall tumor response was achieved in 15 (22 percent) of the patients studied.
The most common severe side effects reported by patients receiving Azedra in clinical trials included low levels of white blood cells (lymphopenia), abnormally low count of a type of white blood cells (neutropenia), low blood platelet count (thrombocytopenia), fatigue, anemia, increased international normalized ratio (a laboratory test which measures blood clotting), nausea, dizziness, hypertension and vomiting.
As it is a radioactive therapeutic agent, Azedra includes a warning about radiation exposure to patients and family members, which should be minimized while the patient is receiving Azedra. The risk of radiation exposure is greater in pediatric patients. Other warnings and precautions include a risk of lower levels of blood cells (myelosuppression), underactive thyroid, elevations in blood pressure, renal failure or kidney injury and inflammation of lung tissue (pneumonitis). Myelodysplastic syndrome and acute leukemias, which are cancers of the blood and bone marrow, were observed in patients who received Azedra, and the magnitude of this risk will continue to be studied. Azedra can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception after receiving Azedra. Radiation exposure associated with Azedra may cause infertility in males and females.
The FDA granted this application Fast Track, Breakthrough Therapy and Priority Review designations. Azedra also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Azedra to Progenics Pharmaceuticals, Inc.
Iobenguane (131I); Iobenguane I 131; Iobeguane I 131; 3-Iodobenzylguanidine; 131I-MIBG; Azedra
77679-27-7 CAS NUMBER
PATENT US 4584187
Guanidine, [[3-(iodo-131I)phenyl]methyl]-
- [[3-(Iodo-131I)phenyl]methyl]guanidine
- 131I-MIBG
- Azedra
- Iobenguane (131I)
- Iobenguane I 131
- Ultratrace Iobenguane 131I
- [131I]-m-Iodobenzylguanidine
- [131I]-m-Iodobenzylguanidine
- m-Iodobenzylguanidine-131I
- m-[131I]Iodobenzylguanidine
| Molecular Formula: | C8H10IN3 |
|---|---|
| Molecular Weight: | 279.095 g/mol |

(I 131-meta-iodobenzylguanidine sulfate)
Iobenguane sulfate; M-Iodobenzylguanidine hemisulfate; MIBG; 87862-25-7; 3-Iodobenzylguanidine hemisulfate; 3-Iodobenzyl-guanidine hemisulfate
| Molecular Formula: | C16H22I2N6O4S |
|---|---|
| Molecular Weight: | 648.259 g/mol |
AdreView
(iobenguane I 123) Injection for Intravenous Use
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
SYN
CN 106187824
DESCRIPTION
AdreView (iobenguane I 123 Injection) is a sterile, pyrogen-free radiopharmaceutical for intravenous injection. Each mL contains 0.08 mg iobenguane sulfate, 74 MBq (2 mCi) of I 123 (as iobenguane sulfate I 123) at calibration date and time on the label, 23 mg sodium dihydrogen phosphate dihydrate, 2.8 mg disodium hydrogen phosphate dihydrate and 10.3 mg (1% v/v) benzyl alcohol with a pH of 5.0 – 6.5. Iobenguane sulfate I 123 is also known as I 123 meta-iodobenzlyguanidine sulfate and has the following structural formula:
 |
Physical Characteristics
Iodine 123 is a cyclotron-produced radionuclide that decays to Te 123 by electron capture and has a physical half-life of 13.2 hours.
Iobenguane I-131 is a guanidine analog with specific affinity for tissues of the sympathetic nervous system and related tumors. The radiolabeled forms are used as antineoplastic agents and radioactive imaging agents. (Merck Index, 12th ed) MIBG serves as a neuron-blocking agent which has a strong affinity for, and retention in, the adrenal medulla and also inhibits ADP-ribosyltransferase.
Iobenguane i-131 is a Radioactive Diagnostic Agent. The mechanism of action of iobenguane i-131 is as a Radiopharmaceutical Activity.
Iobenguane I-131 is an I 131 radioiodinated synthetic analogue of the neurotransmitter norepinephrine. Iobenguane localizes to adrenergic tissue and, in radioiodinated forms, may be used to image or eradicate tumor cells that take up and metabolize norepinephrine.
Iobenguane, also known as metaiodobenzylguanidine or mIBG, or MIBG (tradename Adreview) is a radiopharmaceutical,[1] used in a scintigraphy method called MIBG scan. Iobenguane is a radiolabeled molecule similar to noradrenaline.
The radioisotope of iodine used for the label can be iodine-123 (for imaging purposes only) or iodine-131 (which must be used when tissue destruction is desired, but is sometimes used for imaging also).

Pheochromocytoma seen as dark sphere in center of the body (it is in the left adrenal gland). Image is by MIBG scintigraphy, with radiation from radioiodine in the MIBG. Two images are seen of the same patient from front and back. Note dark image of the thyroid due to unwanted uptake of iodide radioiodine from breakdown of the pharmaceutical, by the thyroid gland in the neck. Uptake at the side of the head are from the salivary glands. Radioactivity is also seen in the bladder, from normal renal excretion of iodide.
It localizes to adrenergic tissue and thus can be used to identify the location of tumors[2] such as pheochromocytomas and neuroblastomas. With I-131 it can also be used to eradicate tumor cells that take up and metabolize norepinephrine.
Thyroid precautions
Thyroid blockade with (nonradioactive) potassium iodide is indicated for nuclear medicine scintigraphy with iobenguane/mIBG. This competitively inhibits radioiodine uptake, preventing excessive radioiodine levels in the thyroid and minimizing the risk of thyroid ablation ( in the case of I-131). The minimal risk of thyroid carcinogenesis is also reduced as a result.
The FDA-approved dosing of potassium iodide for this purpose are as follows: infants less than 1 month old, 16 mg; children 1 month to 3 years, 32 mg; children 3 years to 18 years, 65 mg; adults 130 mg.[3] However, some sources recommend alternative dosing regimens.[4]
Not all sources are in agreement on the necessary duration of thyroid blockade, although agreement appears to have been reached about the necessity of blockade for both scintigraphic and therapeutic applications of iobenguane. Commercially available iobenguane is labeled with iodine-123, and product labeling recommends administration of potassium iodide 1 hour prior to administration of the radiopharmaceutical for all age groups,[5] while the European Associated of Nuclear Medicine recommends (for iobenguane labeled with either I-131 or I-123,) that potassium iodide administration begin one day prior to radiopharmaceutical administration, and continue until the day following the injection, with the exception of newborns, who do not require potassium iodide doses following radiopharmaceutical injection.[4]
Product labeling for diagnostic iodine-131 iobenguane recommends potassium iodide administration one day before injection and continuing 5 to 7 days following.[6] Iodine-131 iobenguane used for therapeutic purposes requires a different pre-medication duration, beginning 24–48 hours prior to iobenguane injection and continuing 10–15 days following injection.[7]
Alternative imaging modality for pheochromocytoma
The FDOPA PET/CT scan has proven to be nearly 100% sensitive for detection of pheochromocytomas, vs. 90% for MIBG scans.[8][9][10] Centers which offer FDOPA PET/CT, however, are rare.
Clinical trials
Iobenguane I 131 for cancers
Iobenguane I 131 (as Azedra) has had a clinical trial as a treatment for malignant, recurrent or unresectable pheochromocytoma and paraganglioma, and the US FDA has granted it a Priority Review.[11]

2-(3-Iodobenzyl)guanidine
35MRW7B4AD
80663-95-2 [RN]
Guanidine, ((3-iodophenyl)methyl)-
Guanidine, N”-[(3-iodophenyl)methyl]-
iobenguane
|
|
| Clinical data | |
|---|---|
| Synonyms | meta-iodobenzylguanidine mIBG, MIBG |
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C8H10IN3 |
| Molar mass | 275.09 g/mol |
| 3D model (JSmol) | |

Title: Iobenguane
CAS Registry Number: 80663-95-2
CAS Name: [(3-Iodophenyl)methyl]guanidine
Additional Names: m-iodobenzylguanidine; MIBG
Molecular Formula: C8H10IN3
Molecular Weight: 275.09
Percent Composition: C 34.93%, H 3.66%, I 46.13%, N 15.28%
Literature References: Norepinephrine analog with specific affinity for tissues of sympathetic nervous system and related tumors; prepd as 123I and 131I labeled forms. Prepn and imaging studies: D. M. Wieland et al., J. Nucl. Med. 21, 349 (1980); eidem, US4584187 (1986). Improved synthesis: P. A. P. M. van Doremalen, A. G. M. Janssen, J. Radioanal. Nucl. Chem. Lett. 96, 97 (1985). Metabolism in man: T. J. Mangner et al., J. Nucl. Med. 27, 37 (1986). HPLC determn in serum and urine: D. Schwabe et al., J. Chromatogr. 487, 177 (1989). Radiopharmacokinetics: S. Ertl et al., Nucl. Med. Commun. 8, 643 (1987). Clinical evaluation of myocardial imaging: D. Fagret et al., Eur. J. Nucl. Med. 15, 624 (1989). Diagnostic use in pheochromocytoma: B. Shapiro et al., J. Nucl. Med. 26, 576 (1985); therapeutic use: M. Krempf et al., J. Clin. Endocrinol. Metab. 72, 455 (1991). Symposia on therapeutic and diagnostic use in neuroblastoma: Advances in Neuroblastoma Research 2, A. E. Evans et al., Eds. (Alan R. Liss, Inc., New York, 1988) p 643-726; Med. Pediatr. Oncol. 15, 157-228 (1987). Review of pharmacology: J. C. Sisson, D. M. Weiland, Am. J. Physiol. Imaging 1, 96-103 (1986); of biodistribution and clinical studies: A. R. Wafelman et al., Eur. J. Nucl. Med. 21, 545-559 (1994); of therapeutic use in neural crest tumors: L. Troncone, V. Rufini, Anticancer Res. 17, 1823-1832 (1997).
Derivative Type: Sulfate
Molecular Formula: (C8H10IN3)2.H2SO4
Molecular Weight: 648.26
Percent Composition: C 29.64%, H 3.42%, I 39.15%, N 12.96%, S 4.95%, O 9.87%
Properties: Colorless crystals from water + ethanol, mp 166-167°.
Melting point: mp 166-167°
Therap-Cat: Radiolabeled forms as antineoplastic; diagnostic aid (radioactive imaging agent).
References
- Jump up^ “Iobenguane I 131 Sulfate (Intravenous Route) – MayoClinic.com”. Retrieved 2007-12-15.
- Jump up^ Scarsbrook AF, Ganeshan A, Statham J, et al. (2007). “Anatomic and functional imaging of metastatic carcinoid tumors”. Radiographics. 27 (2): 455–77. doi:10.1148/rg.272065058. PMID 17374863.
- Jump up^ Kowalsky RJ, Falen, SW. Radiopharmaceuticals in Nuclear Pharmacy and Nuclear Medicine. 2nd ed. Washington DC: American Pharmacists Association; 2004.
- ^ Jump up to:a b “Archived copy” (PDF). Archived from the original (PDF) on 2011-10-07. Retrieved 2011-10-07.
- Jump up^ http://nuclearpharmacy.uams.edu/resources/adreview.pdf
- Jump up^ Iobenguane Sulfate I 131 Injection Diagnostic package insert. Bedford, MA: CIS-US, Inc. July 1999.
- Jump up^ “Archived copy” (PDF). Archived from the original (PDF) on 2011-10-07. Retrieved 2011-10-07.
- Jump up^ 6-[18FFluorodopamine Positron Emission Tomographic (PET) Scanning for Diagnostic Localization of Pheochromocytoma. Pacek et al. 2001] full text
- Jump up^ Pheochromocytoma Imaging Updated May 2017
- Jump up^ Luster M, Karges W, Zeich K, Pauls S, Verburg FA, Dralle H; et al. (2010). “Clinical value of (18)F-fluorodihydroxyphenylalanine positron emission tomography/computed tomography ((18)F-DOPA PET/CT) for detecting pheochromocytoma”. European journal of nuclear medicine and molecular imaging. 37 (3): 484–93. doi:10.1007/s00259-009-1294-7. PMID 19862519.
- Jump up^ FDA grants priority review to Azedra for rare neuroendocrine tumors Jan 2018
External links
- Iodine 131-metaiodobenzylguanidine at the NCI Drug Dictionary
/////////////// Azedra, iobenguane I 131, fda 2018, Progenics Pharmaceuticals, Fast Track, Breakthrough Therapy, Priority Review, orphan drug, Iobenguane (131I), Iobenguane I 131, Iobeguane I 131, 3-Iodobenzylguanidine, 131I-MIBG, Azedra
C1=CC(=CC(=C1)I)CN=C(N)N

NEW DRUG APPROVALS
ONE TIME
$10.00
ABL 001, Asciminib
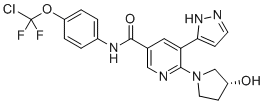
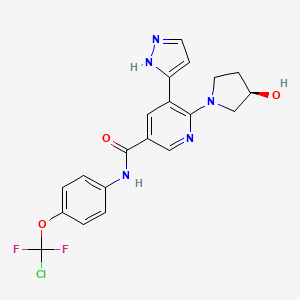
ABL001 / Asciminib
Cas 1492952-76-7
Chemical Formula: C20H18ClF2N5O3
Molecular Weight: 449.8428
Elemental Analysis: C, 53.40; H, 4.03; Cl, 7.88; F, 8.45; N, 15.57; O, 10.67
N-[4-[Chloro(difluoro)methoxy]phenyl]-6-[(3R)-3-hydroxypyrrolidin-1-yl]-5-(1H-pyrazol-5-yl)pyridine-3-carboxamide
3-Pyridinecarboxamide, N-[4-(chlorodifluoromethoxy)phenyl]-6-[(3R)-3-hydroxy-1-pyrrolidinyl]-5-(1H-pyrazol-3-yl)-
PHASE 3, Chronic Myeloid Leukemia, NOVARTIS
UPDATE FDA APPROVED 10/29/2021,
| Scemblix |
To treat Philadelphia chromosome-positive chronic myeloid leukemia with disease that meets certain criteria
Asciminib, sold under the brand name Scemblix, is a medication used to treat Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML).[1][2][3] Asciminib is a protein kinase inhibitor.[1]
The most common adverse reactions include upper respiratory tract infections, musculoskeletal pain, fatigue, nausea, rash, and diarrhea.[2]
Asciminib was approved for medical use in the United States in October 2021.[1][4][5]
The U.S. Food and Drug Administration (FDA) granted the application for asciminib priority review, fast track, orphan drug, and breakthrough therapy designations.[2][6][7]
Asciminib is an orally bioavailable, allosteric Bcr-Abl tyrosine kinase inhibitor with potential antineoplastic activity. Designed to overcome resistance, ABL001 binds to the Abl portion of the Bcr-Abl fusion protein at a location that is distinct from the ATP-binding domain. This binding results in the inhibition of Bcr-Abl-mediated proliferation and enhanced apoptosis of Philadelphia chromosome-positive (Ph+) hematological malignancies. The Bcr-Abl fusion protein tyrosine kinase is an abnormal enzyme produced by leukemia cells that contain the Philadelphia chromosome.
ABL001 has been used in trials studying the health services research of Chronic Myelogenous Leukemia and Philadelphia Chromosome-positive Acute Lymphoblastic Leukemia.

- Originator Novartis
- Developer Novartis; Novartis Oncology
- Class Antineoplastics; Pyrazoles; Pyrrolidines; Small molecules
- Mechanism of Action Bcr-abl tyrosine kinase inhibitors
Highest Development Phases
- Phase III Chronic myeloid leukaemia
- No development reported Precursor cell lymphoblastic leukaemia-lymphoma
Most Recent Events
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in Australia (PO)
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in France (PO)
- 04 Nov 2017 No recent reports of development identified for phase-I development in Acute-lymphoblastic-leukaemia(Second-line therapy or greater) in Germany (PO)
- The tyrosine kinase activity of the ABLl protein is normally tightly regulated, with the N-terminal cap region of the SH3 domain playing an important role. One regulatory mechanism involves the N-terminal cap glycine-2 residue being myristoylated and then interacting with a myristate binding site within the SHI catalytic domain. A hallmark of chronic myeloid leukemia (CML) is the Philadelphia chromosome (Ph), formed by the t(9,22) reciprocal chromosome translocation in a haematopoietic stem cell. This chromosome carries the BCR-ABL1 oncogene which encodes the chimeric BCR-ABL1 protein, that lacks the N-terminal cap and has a constitutively active tyrosine kinase domain.Although drugs that inhibit the tyrosine kinase activity of BCR-ABL1 via an ATP-competitive mechanism, such as Gleevec® / Glivec® (imatinib), Tasigna® (nilotinib) and Sprycel® (dasatinib), are effective in the treatment of CML, some patients relapse due to the emergence of drug-resistant clones, in which mutations in the SHI domain compromise inhibitor binding. Although Tasigna® and Sprycel® maintain efficacy towards many Gleevec-resistant mutant forms of BCR-ABLl, the mutation in which the threonine-315 residue is replaced by an isoleucine (T315I) remains insensitive to all three drugs and can result in CML patients developing resistance to therapy. Therefore, inhibiting BCR-ABLl mutations, such as T315I, remains an unmet medical need. In addition to CML, BCR-ABLl fusion proteins are causative in a percentage of acute lymphocytic leukemias, and drugs targeting ABL kinase activity also have utility in this indication.Agents targeting the myristoyl binding site (so-called allosteric inhibitors) have potential for the treatment of BCR-ABLl disorders (J. Zhang, F. J. Adrian, W. Jahnke, S. W. Cowan- Jacob, A. G. Li, R. E. Iacob4, T. Sim, J. Powers, C. Dierks, F. Sun, G.-R. Guo, Q. Ding, B. Okram, Y. Choi, A. Wojciechowski, X. Deng, G. Liu, G. Fendrich, A. Strauss, N. Vajpai, S. Grzesiek, T. Tuntland, Y. Liu, B. Bursulaya, M. Azam, P. W. Manley, J. R. Engen, G. Q. Daley, M. Warmuth., N. S. Gray. Targeting BCR-ABL by combining allosteric with ATP -binding-site inhibitors. Nature 2010;463:501-6). To prevent the emergence of drug resistance from ATP inhibitor and/or allosteric inhibitor use, a combination treatment using both types of inhibitor can be developed for the treatment of BCR-ABLl related disorders. In particular, the need exists for small molecules, or combinations thereof, that inhibit the activity of BCR-ABLl and BCR-ABLl mutations via the ATP binding site, the myristoyl binding site or a combination of both sites.Further, inhibitors of ABL 1 kinase activity have the potential to be used as therapies for the treatment of metastatic invasive carcinomas and viral infections such as pox and Ebola viruses.The compounds from the present invention also have the potential to treat or prevent diseases or disorders associated with abnormally activated kinase activity of wild-type ABL1, including non-malignant diseases or disorders, such as CNS diseases in particular neurodegenerative diseases (for example Alzheimer’s, Parkinson’s diseases), motoneuroneuron diseases (amyotophic lateral sclerosis), muscular dystrophies, autoimmune and inflammatory diseases (diabetes and pulmonary fibrosis), viral infections, prion diseases.
Asciminib is an allosteric inhibitor of BCR-ABL kinase in phase III clinical development at Novartis for the treatment of patients with chronic myelogenous leukemia (CML) in chronic phase who have been previously treated with ATP-binding site tyrosine kinase inhibitors. Early clinical trials are also under way in patients with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) and as first-line threapy of CML.
PATENT
To illustrate tautomerism with the following specific examples, (R)-N-(4- (chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)-5-(lH-pyrazol-5-yl)nicotinamide
(right structure, below) is a tautomer of (R)-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)-5-(lH-pyrazol-3-yl)nicotinamide (left structure, below) and vice versa:

[0045] Where the plural form (e.g. compounds, salts) is used, this includes the singular
Example 9
(R)-N-(4-(Chlorodifluoromethoxy)phenyl)-6-(3-hvdroxypyrrolidin-l-yl)-5-(lH-pyrazol-5- vDnicotinamide

[00365] A mixture of (R)-5-Bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)nicotinamide (Stage 9.2, 100 mg, 0.216 mmol) and 5-(4 ,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- 1 -((2-(trimethylsilyl)ethoxy)methyl)- IH-pyrazole (215 mg, 0.663 mmol), Pd(PPh3)2Cl2 (17 mg, 0.024 mmol), Na2C03 (115 mg, 1.081 mmol), DME (917 μί), water (262 μΕ) and EtOH (131 μί) in a MW vial was sealed, evacuated / purged 3 times with argon and subjected to MW irradiation at 125°C for 20 min. The RM was diluted with 2 mL
of DME, stirred with Si-Thiol (Silicycle 1.44 mmol/g, 90 mg, 0.130 mmol) for 3 h. The mixture was centrifuged and the supernatant was filtered through a 0.45 μηι PTFE filter and the solvent was evaporated off under reduced pressure. The crude product was purified by flash
chromatography (RediSep® Silica gel column, 12 g, cyclohexane / EtOAc from 40% to 100% EtOAc) to afford the protected intermediate as a colorless oil. Ethylene diamine (96 μί, 1.428 mmol) and TBAF 1 M in THF (1.428 mL, 1.428 mmol) were then added and the RM was stirred at 80-85°C for 5 days. The solvent was evaporated off under reduced pressure and the residue was dissolved in EtOAc (40 mL), washed 3 times with sat. aq. NaHCC and brine, dried over Na2S04 and The solvent was evaporated off under reduced pressure to give a residue which was purified by preparative SFC (Column DEAP, from 25% to 30% in 6 min) to yield the title compound as a white solid.
[00366] Alternatively, Example 9 was prepared by adding TFA (168 mL, 2182 mmol) to a solution of N-(4-(chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinamide (Stage 9.1, 31.3 g, 54.6 mmol) in DCM (600 mL). The mixture was stirred at RT for 2.5 h. The solvent was evaporated off under reduced pressure and the residue was dissolved in EtOAc (1.5 L),washed with a sat. solution of NaHC03 (3 x 500 mL) and brine (500 mL), dried over Na2S04 and the solvent was evaporated off under reduced pressure to give a residue which was suspended in DCM (300 mL), stirred at RT for 15 min, filtered, washed with DCM (200 mL), dried and purified by chromatography (Silica gel, 1 kg, DCM / MeOH 95:5). The residue was dissolved in MeOH (500 mL) and treated with Si-Thiol (Biotage, 5.0 g , 6.5 mmol) for 16 h at 25°C. The resin was filtered off, the solvent was evaporated off under reduced pressure and the residue was crystallized from MeCN to afford the title compound as a white crystalline solid.
[00367] Alternatively, Example 9 was prepared by the dropwise addition of aqueous HC1
(7.7 mL of 6M) to a solution of N-(4-(chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-( 1 -(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinamide (Stage 9.1, 3.8 g, 7.12 mmol) in MeOH (20 mL) and THF (10 mL) with cooling (below 35°C). The mixture was stirred at 22°C for 2 h and then added to cooled (10°C) 1.2 M NaOH (22 mL).
Throughout the addition the temperature was kept below 30°C and pH was kept in the range of 9-10. The RM was then stirred for 30 min at 30°C. The solvent was evaporated off under reduced pressure, until the desired compound precipitated. The precipitate was filtered and dried to give the title compound as a yellow solid.
[00368] Analytical data for Example 9: HPLC (Condition 5) tR = 5.54 min, HPLC Chiral
(CHIRALCEL® OD-H, 250 x 4.6 mm, eluent : n-heptane/EtOH/MeOH (85: 10:5), 1 mL/min, UV 210 nm) tR = 10.17 min, UPLC-MS (condition 3) tR = 0.93 min, m/z = 450.3 [M+H]+, m/z = 494.1 [M+formic acid-H]“; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.65 – 1.76 (m, 1 H) 1.76 – 1.87 (m, 1 H) 2.93 (d, J=l 1.73 Hz, 1 H) 3.19 – 3.29 (m, 2 H) 3.35 – 3.51 (m, 1 H) 4.10 – 4.25 (m, 1 H) 4.89 (br. s, 1 H) 6.41 (br. s, 1 H) 7.33 (d, J=8.50 Hz, 2 H) 7.57/7.83 (br. s, 1 H) 7.90 (d, J=8.50 Hz, 2 H) 8.07 (br. s, 1 H) 8.77 (br. s, 1 H) 10.23 (s, 1 H) 12.97/13.15 (br. s, 1 H).
[00369] Stage 9.1 : N-(4-(Chlorodifluoromethoxy)phenyl)-6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-( 1 -(tetrahydro-2H-pyran-2- l)- 1 H-pyrazol-5-yl)nicotinamide

[00370] l-(Tetrahydro-2H-pyran-2-yl)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (29.6 g, 102 mmol), K3P04 (51.6 g, 236 mmol) and Pd(PPh3)4 (4.55 g, 3.93 mmol) were added to a suspension of (R)-5-bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin-l-yl)nicotinamide (Stage 9.2, 36.4 g, 79 mmol) in toluene (360 mL) under an argon atmosphere and the mixture was stirred at 110°C for 4 h. The RM was poured into brine (500 mL) and extracted with EtOAc (2 x 1 L). The combined extracts were washed with brine (500 mL), dried over Na2S04, and the solvent was evaporated off under reduced pressure to give a residue which was purified by chromatography (Silica gel column, 1.5 kg, DCM / MeOH 95:5) to afford a dark yellow foam, that was dissolved in MeOH / DCM (1 L of 3: l) and treated with Si-Thiol (Biotage, 35 g , 45.5 mmol) for 17 h at 30°C. The resin was filtered off, and solvent was evaporated off under reduced pressure, until the desired compound crystallized. The product was filtered washed with MeOH and dried to afford the title compound.
[00371] Alternatively, Stage 9.1 was prepared by adding 4-(chlorodifluoromethoxy)aniline
(16.6 g, 84.9 mmol), NMM (21.7 g, 212.1 mmol), hydroxybenzotriazole hydrate (HOBt H20, 11.9 g, 77.77 mmol) and l-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCIHCl, 20.9 g, 109.0 mmol) to a solution of 6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinic acid (Stage 9.4, 29.83 g, 70.7 mmol) in THF (271 mL). The mixture was stirred for 1.5 h at 25°C and then at 65°C for 16 h. After cooling the RM to 35 °C, further EDCIHCl (13.3 g, 69.4 mmol) was added and the RM was stirred for 1.5 h at 35°C then again at 65°C for 16 h. After cooling the RM to 35°C, water (150 mL) was added, the THF was removed under reduced pressure, EtOAc (180 mL) was added and the mixture was stirred for at 35 °C fori h. The two layers were separated and the aq. phase was then extracted with EtOAc (60 mL). The combined organic layers were washed with water (90 mL), brine (90 mL). The solvent was evaporated off under reduced pressure to give a brown solid which was purified by column chromatography (Silica gel, DCM / MeOH 40: 1 to 20: 1) to afford the title compound as a yellow solid.
[00372] Analytical data for Stage 9.1: HPLC (Condition 5) tR = 6.12 min, UPLC-MS
(Condition 3) tR = 1.06 min, m/z = 533.2 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.36 -2.02 (m, 7 H) 2.23 – 2.38 (m, 1 H) 3.08 – 3.29 (m, 2 H) 3.32 – 3.52 (m, 2 H) 3.73 – 3.93 (m, 1 H) 4.13 – 4.25 (m, 1 H) 4.80 – 4.90 (m, 1 H) 4.95 – 5.17 (m, 1 H) 6.33 – 6.50 (m, 1 H) 7.33 (d, J=8.99 Hz, 2 H) 7.61 (d, J=1.56 Hz, 1 H) 7.86 (d, J=8.99 Hz, 2 H) 7.97 – 8.11 (m, 1 H) 8.82 (s, 1 H) 10.13 – 10.25 (m, 1 H).
[00373] Stage 9.2: (R)-5-Bromo-N-(4-(chlorodifluoromethoxy)phenyl)-6-(3-hydroxypyrrolidin- 1 -yl)nicotinamide

[00374] (R)-Pyrrolidin-3-ol (9.55 g, 109.6 mmol) and DIPEA (35.1 ml, 201.3 mmol) were added to a suspension of 5-bromo-6-chloro-N-(4-(chlorodifluoromethoxy)phenyl)nicotinamide (Stage 9.3, 37.7 g, 91.5 mmol) in iPrOH (65 mL) and stirred at 140°C for 1 h. EtOAc (700 mL) was added and the solution was washed IN HC1 (2 x 200 mL), sat. NaHCC (200 mL) and brine (2 x 200 mL), dried over Na2S04, and the solution was concentrated under reduced pressure until crystallization commenced. n-Heptane (1 L) were added and the mixture was stirred at RT for 30 min, filtered and washed with ΪΡΓ20 (500 mL) to afford the title compound as a white crystalline solid. HPLC (Condition 5) tR = 6.68 min, UPLC-MS (Condition 3) tR = 1.10 min, m/z =
462.2/464.2 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.78 – 2.01 (m, 2 H) 3.55 (d, J=l 1.34 Hz, 1 H) 3.66 – 3.75 (m, 1 H) 3.79 – 3.93 (m, 2 H) 4.34 (br. s, 1 H) 4.98 (d, =3.13 Hz, 1 H) 7.32 (d, J=8.99 Hz, 2 H) 7.84 (d, J=8.99 Hz, 2 H) 8.33 (d, J=1.96 Hz, 1 H) 8.66 (d, J=1.96 Hz, 1 H) 10.21 (s, 1 H).
[00375] Stage 9.3: 5-Bromo-6-chloro-N- 4-(chlorodifluoromethoxy)phenyl)nicotinamide

[00376] DMF (2.55 mL, 33.0 mmol) and SOCl2 (24.08 ml, 330 mmol) were added to a suspension of 5-bromo-6-chloro-nicotinic acid (26 g, 110 mmol) in toluene (220 mL) and the RM was stirred at 80°C for 1 h. The solvent was evaporated off under reduced pressure and the residue was dissolved in THF (220 mL) and cooled to -16°C. DIPEA (38.4 mL, 220 mmol) was added, followed by dropwise addition of a solution of 4-(chlorodifluoromethoxy)aniline (22.35 g, 115 mmol) in THF (220 mL) over 15 min. The suspension was stirred for 1 h at RT. The solvent was evaporated off under reduced pressure and the residue was dissolved in TBME (700 mL), washed with IN HC1 (2 x 200 mL), sat. NaHC03 (200 mL) and brine (2 x 200 mL), dried over Na2S04, and the solvent was evaporated off under reduced pressure to give the product which was crystallized from EtOAc – n-heptane to afford the title compound as a white crystalline solid. HPLC (Condition 5) tR = 7.77 min, UPLC-MS (Condition 3) tR = 1.24 min, m/z =
409.1/411.1/413.1 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 7.38 (d, =8.99 Hz, 2 H) 7.85 (d, =8.99 Hz, 2 H) 8.72 (br. s, 1 H) 8.92 (br. s, 1 H) 10.68 (s, 1 H).
[00377] Stage 9.4: 6-((R)-3-Hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazol-5-yl)nicotinic acid

[00378] Aq. NaOH (180 niL of 2.6 M) was added to a solution of methyl 6-((R)-3-hydroxypyrrolidin- 1 -yl)-5-(l -(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinate (Stage 9.5, 11 lg, 299 mmol) in MeOH (270 mL) and the RM was stirred at RT for 14 h. The MeOH was evaporated off under reduced pressure and the aq. residue was treated with brine (90 mL), extracted with MeTHF twice (540 mL + 360 mL) and the combined organic layers were washed with water (90 mL). MeTHF was added to the combined aq. layers, the biphasic mixture was cooled to 0 °C and acidified (pH = 4-4.5) with aq. HC1 solution (18%) and extracted with
MeTHF. The combined organic extracts were washed with brine and the solvent was evaporated off under reduced pressure to give a residue which was recrystallized from a EtOAc / TBME (1 : 1) to afford the title compound as a white solid. HPLC (Condition 7) tR = 4.74 min, LC-MS
(Condition 8) tR = 3.37 min, m/z = 359.0 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.44 (br. s, 2 H), 1.51 (d, J=11.54 Hz, 2 H), 1.64 – 1.86 (m, 4 H), 1.90 (br. s, 1 H), 2.31 (d, J=9.29 Hz, 1 H), 2.77 (br. s, 1 H), 3.10 (br. s, 1 H), 3.21 (d, J=8.78 Hz, 2 H), 3.27 – 3.51 (m, 4 H), 3.87 (d, J=11.54 Hz, 1 H), 4.16 (br. s, 1 H), 4.75 – 4.93 (m, 1 H), 5.04 (br. s, 1 H), 6.35 (d, J=17.32 Hz, 1 H), 7.51 – 7.64 (m, 1 H), 7.64 – 7.82 (m, 1 H), 8.67 (d, J=2.26 Hz, 1 H), 12.58 (br. s, 1 H).
[00379] Stage 9.5: Methyl 6-((R)-3-hydroxypyrrolidin-l-yl)-5-(l-(tetrahydro-2H-pyran-2-yl)- 1 H-pyrazol-5-yl)nicotinate

[00380] A mixture of (R)-methyl 5-bromo-6-(3-hydroxypyrrolidin-l-yl)nicotinate (Stage
9.6, 90 g, 299 mmol), l-(tetrahydro-2H-pyran-2-yl)-lH-pyrazole-5-boronic acid pinacol ester (103.9 g, 373.6 mmol), K3P04 (126.9 g, 597.7 mmol), Pd(PPh3)2Cl2 (6.29 g, 8.97 mmol) in toluene (900 mL) was stirred at 92°C and for 16 h. After cooling the mixture to RT, the solution was washed with water (450 mL), 5% NaHCC solution (430 mL) and the solvent was evaporated off under reduced pressure to give a residue which was used without further purifications in the next step. HPLC (Condition 7) tR = 6.929 min, LC-MS (Condition 8) tR = 4.30 min, m/z = 373.0 [M+H ; XH-NMR (400 MHz, DMSO-d6) δ ppm 1.19 – 1.28 (m, 1 H), 1.35 – 1.63 (m, 4 H), 1.63 -1.86 (m, 3 H), 1.89 (br. s, 1 H), 2.12 – 2.39 (m, 1 H), 3.11 (br. s, 1 H), 3.18 – 3.48 (m, 4 H), 3.78 (s, 4 H), 3.88 (d, J=11.54 Hz, 1 H), 4.08 – 4.24 (m, 1 H), 4.86 (dd, J=18.20, 2.89 Hz, 1 H), 5.02 (d, J=8.28 Hz, 1 H), 6.39 (br. s, 1 H), 7.58 (d, J=1.25 Hz, 1 H), 7.78 (br. s, 1 H), 8.69 (t, J=2.01 Hz, 1 H).
[00381] Stage 9.6: (R)-methyl 5-bromo-6-(3-hydroxypyrrolidin-l-yl)nicotinate

[00382] DIPEA (105.3 g, 142.2 mL, 814.4 mmol) was added to a solution of methyl-5-bromo-6-chroronicotinate (85 g, 339.5 mmol) and (R)-pyrrolidin-3-ol (54.2 g, 441.2 mmol) in isopropyl acetate and the RM was stirred at 70°C for 14 h . The solvent was evaporated off under reduced pressure to give a the residue which was dissolved in toluene (850 mL), washed with water (127 mL) and brine (127 mL)and concentrated under reduced pressure until precipitation commenced. n-Heptane (340 mL) was slowly added to the stirred mixture at 22 °C, which was then cooled to 0 °C and the product was filtered, washed with a toluene / n-heptane mixture
(1 : 1.5) and dried to give the title compound as a yellow solid. HPLC (Condition 7) tR = 8.54 min, LC-MS (Condition 8) tR = 4.62 min, m/z = 300.9/302.9 [M+H]+; XH-NMR (400 MHz, DMSO-d6) δ ρριη 1.77 – 1.99 (m, 2 H), 3.57 (d, J=11.54 Hz, 1 H), 3.72 (ddd, J=l 1.11, 7.97, 3.26 Hz, 1 H), 3.78 (s, 3 H), 3.81 -3.90 (m, 2 H), 4.26 – 4.39 (m, 1 H), 4.99 (br. s, 1 H), 8.11 (d, J=2.01 Hz, 1 H), 8.56 (d, J=1.76 Hz, 1 H).
PAPER
- By Wylie, Andrew A.; Schoepfer, Joseph; Jahnke, Wolfgang; Cowan-Jacob, Sandra W.; Loo, Alice; Furet, Pascal; Marzinzik, Andreas L.; Pelle, Xavier; Donovan, Jerry; Zhu, Wenjing; et al
- From Nature (London, United Kingdom) (2017), 543(7647), 733-737.
By Wylie, Andrew A. et alFrom Nature (London, United Kingdom), 543(7647), 733-737; 2017
PAPER
- By Molica, Matteo; Massaro, Fulvio; Breccia, Massimo
- From Expert Opinion on Pharmacotherapy (2017), 18(1), 57-65.
PATENT
US 20170216289
PAPER
- By El Rashedy, Ahmed A.; Olotu, Fisayo A.; Soliman, Mahmoud E. S.
- From Chemistry & Biodiversity (2018), 15(3), n/a.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
 |
|
| Clinical data | |
|---|---|
| Trade names | Scemblix |
| Other names | ABL001 |
| Routes of administration |
By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII |
|
| KEGG | |
| ChEMBL |
|
| PDB ligand | |
| Chemical and physical data | |
| Formula | C20H18ClF2N5O3 |
| Molar mass | 449.84 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Jump up to:a b c d “Scemblix- asciminib tablet, film coated”. DailyMed. Retrieved 4 November 2021.
- ^ Jump up to:a b c “FDA approves asciminib for Philadelphia chromosome-positive chronic myeloid leukemia”. U.S. Food and Drug Administration (FDA) (Press release). 29 October 2021. Retrieved 4 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Breccia M, Colafigli G, Scalzulli E, Martelli M (August 2021). “Asciminib: an investigational agent for the treatment of chronic myeloid leukemia”. Expert Opinion on Investigational Drugs. 30 (8): 803–811. doi:10.1080/13543784.2021.1941863. PMID 34130563.
- ^ “Scemblix: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 29 October 2021.
- ^ “FDA approves Novartis Scemblix (asciminib), with novel mechanism of action for the treatment of chronic myeloid leukemia”. Novartis (Press release). Retrieved 29 October 2021.
- ^ “Asciminib Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 27 February 2017. Retrieved 29 October 2021.
- ^ “Novartis receives FDA Breakthrough Therapy designations for investigational STAMP inhibitor asciminib (ABL001) in chronic myeloid leukemia”. Novartis (Press release). 8 February 2020. Retrieved 29 October 2021.
External links
- “Asciminib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02081378 for “A Phase I Study of Oral ABL001 in Patients With CML or Ph+ ALL” at ClinicalTrials.gov
- Clinical trial number NCT03106779 for “Study of Efficacy of CML-CP Patients Treated With ABL001 Versus Bosutinib, Previously Treated With 2 or More TKIs” at ClinicalTrials.gov
////////////////ABL001, Asciminib, ABL 001, ABL-001, PHASE 3, Chronic Myeloid Leukemia, NOVARTIS
O=C(NC1=CC=C(OC(F)(Cl)F)C=C1)C2=CN=C(N3C[C@H](O)CC3)C(C4=CC=NN4)=C2

NEW DRUG APPROVALS
one time
$10.00
TRILACICLIB, G1T28
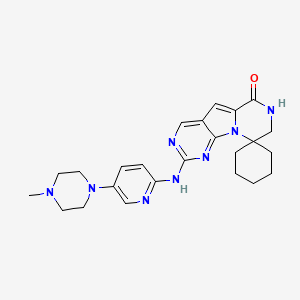
Trilaciclib
update 2021/2/12 US FDA APPROVED COSELA
- Molecular FormulaC24H30N8O
- Average mass446.548 Da
- G1T 28
CAS 1374743-00-6
2′-{[5-(4-Methyl-1-piperazinyl)-2-pyridinyl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
G1T28, SHR 6390
Spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one, 7′,8′-dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]-
- 7′,8′-Dihydro-2′-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]spiro[cyclohexane-1,9′(6’H)-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
- 2′-[[5-(4-Methylpiperazin-1-yl)pyridin-2-yl]amino}-7′,8′-dihydro-6’H-spiro[cyclohexane-1,9′-pyrazino[1′,2′:1,5]pyrrolo[2,3-d]pyrimidin]-6′-one
UNII:U6072DO9XG
Reduction of Chemotherapy-Induced Myelosuppression
Trilaciclib dihydrochloride
1977495-97-8
In phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin
![]()
PATENT, WO 2014144326, Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
| Norman Sharpless | |
|---|---|
 |
|
| Born | Norman Edward Sharpless September 20, 1966 Greensboro, North Carolina |
| Nationality | American |
| Other names | Ned Sharpless |
| Occupation | Director, Lineberger Comprehensive Cancer Center Founder, G1 Therapeutics ($GTHX) |
| Notable work | Wellcome Distinguished Professor, American Society of Clinical Investigation Member, Association of American Cancer Institute board of directors, |
NCI Director Dr. Norman E. Sharpless

NCI Director Dr. Norman E. Sharpless, Credit: National Institutes of Health
Norman E. “Ned” Sharpless, M.D., was officially sworn in as the 15th director of the National Cancer Institute (NCI) on October 17, 2017. Prior to his appointment, Dr. Sharpless served as the director of the University of North Carolina (UNC) Lineberger Comprehensive Cancer Center, a position he held since January 2014.
Dr. Sharpless was a Morehead Scholar at UNC–Chapel Hill and received his undergraduate degree in mathematics. He went on to pursue his medical degree from the UNC School of Medicine, graduating with honors and distinction in 1993. He then completed his internal medicine residency at the Massachusetts General Hospital and a hematology/oncology fellowship at Dana-Farber/Partners Cancer Care, both of Harvard Medical School in Boston.
After 2 years on the faculty at Harvard Medical School, he joined the faculty of the UNC School of Medicine in the Departments of Medicine and Genetics in 2002. He became the Wellcome Professor of Cancer Research at UNC in 2012.
Dr. Sharpless is a member of the Association of American Physicians as well as the American Society for Clinical Investigation (ASCI), the nation’s oldest honor society for physician–scientists, and served on the ASCI council from 2011 to 2014. Dr. Sharpless was an associate editor of Aging Cell and deputy editor of the Journal of Clinical Investigation. He has authored more than 150 original scientific papers, reviews, and book chapters, and is an inventor on 10 patents. He cofounded two clinical-stage biotechnology companies: G1 Therapeutics and HealthSpan Diagnostics.
In addition to serving as director of NCI, Dr. Sharpless continues his research in understanding the biology of the aging process that promotes the conversion of normal self-renewing cells into dysfunctional cancer cells. Dr. Sharpless has made seminal contributions to the understanding of the relationship between aging and cancer, and in the preclinical development of novel therapeutics for melanoma, lung cancer, and breast cancer.
| Record ID | Title | Status | Phase |
|---|---|---|---|
| NCT03041311 | Carboplatin, Etoposide, and Atezolizumab With or Without Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Extensive Stage Small Cell Lung Cancer (SCLC) | Recruiting | 2 |
| NCT02978716 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Gemcitabineand Carboplatin in Metastatic Triple Negative Breast Cancer (mTNBC) | Recruiting | 2 |
| NCT02514447 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Patients With Previously Treated Extensive Stage SCLC Receiving Topotecan Chemotherapy | Recruiting | 2 |
| NCT02499770 | Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Etoposide and Carboplatin in Extensive Stage Small Cell Lung Cancer (SCLC) | Active, not recruiting | 2 |
Synthesis
WO 2016040858


Trilaciclib (G1T28)
Trilaciclib is a potential first-in-class short-acting CDK4/6 inhibitor in development to preserve hematopoietic stem cells and enhance immune system function during chemotherapy. Trilaciclib is administered intravenously prior to chemotherapy and has the potential to significantly improve treatment outcomes.
G1 is currently evaluating trilaciclib in four Phase 2 clinical trials: three studies in patients with small-cell lung cancer (SCLC), and one study in patients with triple-negative breast cancer (TNBC). Preliminary data from the SCLC trials were presented at the American Society of Clinical Oncology 2017 Annual Meeting and at the 2016 World Conference on Lung Cancer.
Data from a Phase 1 trial in healthy volunteers were presented at the American Society of Clinical Oncology 2015 Annual Meeting and published in Science Translational Medicine. Trilacicilib has been extensively studied in animals; these preclinical data have been presented at several scientific meetings and published in Molecular Cancer Therapeutics, Science Translational Medicine, and Cancer Discovery.
Trilaciclib is a small molecule, competitive inhibitor of cyclin dependent kinases 4 and 6 (CDK4/6), with potential antineoplastic and chemoprotective activities. Upon intravenous administration, trilaciclib binds to and inhibits the activity of CDK4/6, thereby blocking the phosphorylation of the retinoblastoma protein (Rb) in early G1. This prevents G1/S phase transition, causes cell cycle arrest in the G1 phase, induces apoptosis, and inhibits the proliferation of CDK4/6-overexpressing tumor cells. In patients with CDK4/6-independent tumor cells, G1T28 may protect against multi-lineage chemotherapy-induced myelosuppression (CIM) by transiently and reversibly inducing G1 cell cycle arrest in hematopoietic stem and progenitor cells (HSPCs) and preventing transition to the S phase. This protects all hematopoietic lineages, including red blood cells, platelets, neutrophils and lymphocytes, from the DNA-damaging effects of certain chemotherapeutics and preserves the function of the bone marrow and the immune system. CDKs are serine/threonine kinases involved in the regulation of the cell cycle and may be overexpressed in certain cancer cell types. HSPCs are dependent upon CDK4/6 for proliferation.
Trilaciclib (G1T28) is a CDK4/6 inhibitor in phase II clinical development as a chemoprotectant at G1 Therapeutics for first- or second-line treatment in patients with metastatic triple negative breast cancer, in combination with gemcitabine and carboplatin. Also, phase II trials are ongoing in newly diagnosed, treatment-naive small-cell lung cancer patients, in combination with carboplatin, etoposide, and atezolizumab and phase I trials in previously treated small-cell lung cancer patients, in combination with topotecan.
U.S. Patent Nos. 8,822,683; 8,598,197; 8,598,186, 8,691,830, 8,829,102, 8,822,683, 9, 102,682, 9,499,564, 9,481,591, and 9,260,442, filed by Tavares and Strum and assigned to Gl Therapeutics describe a class of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amine cyclin dependent kinase inhibitors including those of the formula with variables as defined therein):

U.S. Patent Nos. 9,464,092, 9,487,530, and 9,527,857 which are also assigned to Gl Therapeutics describe the use of the above pyrimidine-based agents in the treatment of cancer.
These patents provide a general synthesis of the compounds that is based on a coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. Such coupling reactions are sometimes referred to as Buchwald coupling (see WO Ί56 paragraph 127; reference WO 2010/020675). The lactam of the fused chloropyrimidine, for example, a 2-chloro-spirocyclo-pyrrolo[2,3-d]pyrimidine-one such as Intermediate K as shown below can be prepared by dehydration of the corresponding carboxylic acid. The reported process to prepare intermediate IK requires seven steps.

(Intermediate IK; page 60, paragraph 215 of WO Ί56)
WO 2013/148748 (U.S. S.N. 61/617,657) entitled “Lactam Kinase Inhibitors” filed by Tavares, and also assigned to Gl Therapeutics likewise describes the synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines via the coupling reaction of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine.
WO 2013/163239 (U.S. S.N. 61/638,491) “Synthesis of Lactams” describes a method for the synthesis of this class of compounds with the variation that in the lactam preparation step, a carboxylic acid can be cyclized with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. The purported improvement is that cyclization can occur without losing the protecting group on the amine before cyclization. The typical leaving group is “tBOC” (t-butoxycarbonyl). The application teaches (page 2 of WO 2013/163239) that the strong acid is, for example, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride or mixed anhydrides. An additional step may be necessary to take off the N-protecting group. The dehydrating agent can be a carbodiimide-based compound such as DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-dimethylaminopropyl)carbodiimide, or DIC (Ν,Ν-diisopropylcarbodiimide). DCC and DIC are in the same class of reagents-carbodiimides. DIC is sometimes considered better because it is a liquid at room temperature, which facilitates reactions.
WO 2015/061407 filed by Tavares and licensed to Gl Therapeutics also describes the synthesis of these compounds via the coupling of a fused chloropyrimidine with a heteroaryl amine to form the central disubstituted amine. WO ‘407 focuses on the lactam production step and in particular describes that the fused lactams of these compounds can be prepared by treating the carboxylic acid with an acid and a dehydrating agent in a manner that a leaving group on the amine is not removed during the amide-forming ring closing step.
Other publications that describe compounds of this general class include the following. WO 2014/144326 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of normal cells during chemotherapy using pyrimidine based CDK4/6 inhibitors. WO 2014/144596 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for protection of hematopoietic stem and progenitor cells against ionizing radiation using pyrimidine based CDK4/6 inhibitors. WO 2014/144847 filed by Strum et al. and assigned to Gl Therapeutics describes HSPC-sparing treatments of abnormal cellular proliferation using pyrimidine based CDK4/6 inhibitors. WO2014/144740 filed by Strum et al. and assigned to Gl Therapeutics describes highly active anti -neoplastic and anti-proliferative pyrimidine based CDK 4/6 inhibitors. WO 2015/161285 filed by Strum et al. and assigned to Gl Therapeutics describes tricyclic pyrimidine based CDK inhibitors for use in radioprotection. WO 2015/161287 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for the protection of cells during chemotherapy. WO 2015/161283 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use in HSPC-sparing treatments of RB-positive abnormal cellular proliferation. WO 2015/161288 filed by Strum et al. and assigned to Gl Therapeutics describes analogous tricyclic pyrimidine based CDK inhibitors for use as anti -neoplastic and anti-proliferative agents. WO 2016/040858 filed by Strum et al. and assigned to Gl Therapeutics describes the use of combinations of pyrimidine based CDK4/6 inhibitors with other anti-neoplastic agents. WO 2016/040848 filed by Strum et al. and assigned to Gl Therapeutics describes compounds and methods for treating certain Rb-negative cancers with CDK4/6 inhibitors and topoisomerase inhibitors.
Other biologically active fused spirolactams and their syntheses are described, for example, in the following publications. Griffith, D. A., et al. (2013). “Spirolactam-Based Acetyl-CoA Carboxylase Inhibitors: Toward Improved Metabolic Stability of a Chromanone Lead Structure.” Journal of Medicinal Chemistry 56(17): 7110-7119, describes metabolically stable spirolactams wherein the lactam resides on the fused ring for the inhibition of acetyl-CoA carboxylase. WO 2013/169574 filed by Bell et al. describes aliphatic spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2007/061677 filed by Bell et al. describes aryl spirolactams as CGRP receptor antagonists wherein the lactam resides on the spiro ring. WO 2008/073251 filed by Bell et al. describes constrained spirolactam compounds wherein the lactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031606 filed by Bell et al. describes carboxamide spirolactam compounds wherein the spirolactam resides on the spiro ring as CGRP receptor antagonists. WO 2006/031610, WO 2006/031491, and WO 2006/029153 filed by Bell et al. describe anilide spirolactam compounds wherein the spirolactam resides on the spiro ring; WO 2008/109464 filed by Bhunai et al. describes spirolactam compounds wherein the lactam resides on the spiro ring which is optionally further fused.
Given the therapeutic activity of selected N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines, it would be useful to have additional methods for their preparation. It would also be useful to have new intermediates that can be used to prepare this class of compounds.
PATENT
WO 2014144596
PATENT
Compound 89 (also referred to as Compound T)
| WO2014144847A3 | |
| Inventors | Norman E. Sharpless, Jay Copeland Strum, John Emerson Bisi, Patrick Joseph Roberts, Francis Xavier Tavares |
| Applicant | G1 Therapeutics, Inc. |
EXAMPLES
Intermediates B, E, K, L, 1A, IF and 1CA were synthesized according to US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C..
The patents WO 2013/148748 entitled Lactam Kinase Inhibitors to Tavares, F.X., WO 2013/163239 entitled Synthesis of Lactams to Tavares, F.X., and US 8,598,186 entitled CDK Inhibitors to Tavares, F.X. and Strum, J.C. are incorporated by reference herein in their entirety. Example 1
Synthesis of tert-butyl N- [2- [(5-bromo-2-chloro-pyrimidin-4yl)amino] ethyl] carbamate, Compound 1

To a solution of 5-bromo-2,4-dichloropyrimidine (3.2 g, 0.0135 mol) in ethanol (80 mL) was added Hunig’s base (3.0 mL) followed by the addition of a solution of N-(tert- butoxycarbonyl)-l,2-diaminoethane (2.5 g, 0.0156 mole) in ethanol (20 mL). The contents were stirred overnight for 20 hrs. The solvent was evaporated under vacuum. Ethyl acetate (200 mL) and water (100 mL) were added and the layers separated. The organic layer was dried with magnesium sulfate and then concentrated under vacuum. Column chromatography on silica gel using hexane/ethyl acetate (0- 60%) afforded tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4- yl)amino]ethyl]carbamate. 1HNMR (d6-DMSO) δ ppm 8.21 (s, 1H), 7.62 (brs, 1H), 7.27 (brs, 1H), 3.39 (m, 2H), 3.12 (m, 2H), 1.34 (s, 9H). LCMS (ESI) 351 (M + H).
Example 2
Synthesis of tert-butyl N-[2-[[2-chloro-5-(3,3-diethoxyprop-l-ynyl)pyrimidin-4- yl] amino] ethyl] carbamate, Compound 2

To tert-butyl N-[2-[(5-bromo-2-chloro-pyrimidin-4-yl)amino]ethyl]carbamate (1.265 g, 6 mmol) in THF (10 mL) was added the acetal (0.778 mL, 5.43 mmol), Pd(dppf)CH2Cl2 (148 g), and triethylamine (0.757 mL, 5.43 mmol). The contents were degassed and then purged with nitrogen. To this was then added Cul (29 mg). The reaction mixture was heated at reflux for 48 hrs. After cooling, the contents were filtered over CELITE™ and concentrated. Column chromatography of the resulting residue using hexane/ethyl acetate (0- 30%) afforded tert-butyl N- [2- [ [2-chloro-5 -(3 ,3 -diethoxyprop- 1 -ynyl)pyrimidin-4-yl]amino] ethyl] carbamate. 1HNMR (d6-DMSO) δ ppm 8.18 (s, 1H), 7.63 (brs, 1H), 7.40 (brs, 1H), 5.55 (s, 1H), 3.70 (m, 2H), 3.60 (m, 2H), 3.42 (m, 2H), 3.15 (m, 2H), 1.19 – 1.16 (m, 15H). LCMS (ESI) 399 (M + H).
Example 3
Synthesis of tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7- yl] ethyl] carbamate, Compound 3

To a solution of the coupled product (2.1 g, 0.00526 mole) in THF (30 mL) was added TBAF solid (7.0 g). The contents were heated to and maintained at 65 degrees for 2 hrs. Concentration followed by column chromatography using ethyl acetate/hexane (0-50%) afforded tert-butyl N-[2-[2-chloro-6-(diethoxymethyl)pyrrolo[2,3-d]pyrimidin-7-yl]ethyl]carbamate as a pale brown liquid (1.1 g). 1FiNMR (d6-DMSO) δ ppm 8.88 (s, 1H), 6.95 (brs, 1H), 6.69 (s, 1H), 5.79 (s, 1H), 4.29 (m, 2H), 3.59 (m, 4H), 3.34 (m, 1H), 3.18 (m, 1H), 1.19 (m, 9H), 1.17 (m, 6H). LCMS (ESI) 399 (M + H).
Example 4
Synthesis of tert-buty\ N-[2-(2-chloro-6-formyl-pyrrolo [2,3-d] pyrimidin-7- yl)ethyl] carbamate, Compound 4

To the acetal (900 mg) from the preceeding step was added AcOH (8.0 mL) and water
(1.0 mL). The reaction was stirred at room temperature for 16 hrs. Cone, and column chromatography over silica gel using ethyl acetate/hexanes (0- 60%) afforded tert-butyl N-[2-(2- chloro-6-formyl-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl]carbamate as a foam (0.510 g). 1HNMR (d6-DMSO) δ ppm 9.98 (s, 1H), 9.18 (s, 1H), 7.66 (s, 1H), 6.80 (brs, 1H), 4.52 (m, 2H), 4.36 (m, 2H), 1.14 (s, 9H). LCMS (ESI) 325 (M + H).
Example 5
Synthesis of 7- [2-(teri-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3-d] pyrimidine-6- carboxylic acid, Compound 5

To the aldehyde (0.940 g) from the preceeding step in DMF (4 mL) was added oxone (1.95 g, 1.1 eq). The contents were stirred at room temp for 7 hrs. Silica gel column chromatography using hexane/ethyl acetate (0- 100%) afforded l-\2-(tert- butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g). 1HNMR (d6-DMSO) δ ppm 9.11 (s, 1H), 7.39 (s, 1H), 4.38 (m, 2H), 4.15 (m, 2H), 1.48 (m, 9H). LCMS (ESI) 341(M + H).
Example 6
Synthesis of methyl 7-[2-(teri-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate, Compound 6

To a solution of 2-chloro-7-propyl-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid (0.545 g, 0.00156 mole) from the preceeding step in toluene (3.5 mL) and MeOH (1 mL) was added TMS- diazomethane (1.2 mL). After stirring overnight at room temperature, the excess of TMS- diazomethane was quenched with acetic acid (3 mL) and the reaction was concentrated under vacuum. The residue was purified by silica gel column chromatography with hexane/ethyl acetate (0- 70%) to afford methyl 7-[2-(tert-butoxycarbonylamino)ethyl]-2-chloro-pyrrolo[2,3- d]pyrimidine-6-carboxylate as an off white solid (0.52 g). 1HNMR (d6-DMSO) δ ppm 9.10 (s, 1H), 7.45 (s, 1H), 6.81 (brs, 1H) 4.60 (m, 2H), 3.91 (s, 3H), 3.29 (m, 2H), 1.18 (m, 9H) LCMS (ESI) 355 (M + H).
Example 7
Synthesis of Chloro tricyclic amide, Compound 7

To methyl 7- [2-(tert-butoxycarbonylamino)ethyl] -2-chloro-pyrrolo [2,3 -d]pyrimidine-6- carboxylate (0.50 g, 0.0014 mole) from the preceeding step in dichloromethane (2.0 mL) was added TFA (0.830 mL). The contents were stirred at room temperature for 1 hr. Concentration under vacuum afforded the crude amino ester which was suspended in toluene (5 mL) and Hunig’s base (0.5 mL). The contents were heated at reflux for 2 hrs. Concentration followed by silica gel column chromatography using hexane/ethyl acetate (0- 50%) afforded the desired chloro tricyclic amide (0.260 g). 1HNMR (d6-DMSO) δ ppm 9.08 (s, 1H), 8.48 (brs, 1H), 7.21 (s, 1H) 4.33 (m, 2H), 3.64 (m, 2H). LCMS (ESI) 223 (M + H).
Example 8
Synthesis of chloro-N-methyltricyclic amide, Compound 8

To a solution of the chloro tricycliclactam, Compound 7, (185 mg, 0.00083 mole) in DMF (2.0 mL) was added sodium hydride (55% dispersion in oil, 52 mg). After stirring for 15 mins, methyl iodide (62 μί, 1.2 eq). The contents were stirred at room temperature for 30 mins. After the addition of methanol (5 mL), sat NaHCOs was added followed by the addition of ethyl acetate. Separation of the organic layer followed by drying with magnesium sulfate and concentration under vacuum afforded the N-methylated amide in quantitative yield. 1FiNMR (d6-DMSO) δ ppm 9.05 (s, 1H), 7.17 (s, 1H) 4.38 (m, 2H), 3.80 (m, 2H), 3.05 (s, 3H). LCMS (ESI) 237 (M + H). Example 9
Synthesis of l-methyl-4-(6-nitro-3-pyridyl)piperazine, Compound 9

To 5-bromo-2-nitropyridine (4.93 g, 24.3 mmole) in DMF (20 mL) was added N- methylpiperazine (2.96 g, 1.1 eq) followed by the addition of DIPEA (4.65 mL, 26.7 mmole). The contents were heated at 90 degrees for 24 hrs. After addition of ethyl acetate (200 mL), water (100 mL) was added and the layers separated. Drying followed by concentration afforded the crude product which was purified by silica gel column chromatography using (0-10%) DCM/Methanol. 1HNMR (d6-DMSO) δ ppm 8.26 (s, 1H), 8.15 (1H, d, J = 9.3 Hz), 7.49 (1H, d, J = 9.4 Hz), 3.50 (m, 4H), 2.49 (m, 4H), 2.22 (s, 3H).
Example 10
Synthesis of 5-(4-methylpiperazin-l-yl)pyridin-2-amine, Compound 10

To l-methyl-4-(6-nitro-3-pyridyl)piperazine (3.4 g) in ethyl acetate (100 mL) and ethanol (100 mL) was added 10%> Pd/C (400 mg) and then the reaction was stirred under hydrogen (10 psi) overnight. After filtration through CELITE™, the solvents were evaporated and the crude product was purified by silica gel column chromatography using DCM/ 7N ammonia in MeOH (0- 5%) to afford 5-(4-methylpiperazin-l-yl)pyridin-2-amine (2.2 g). 1HNMR (d6-DMSO) δ ppm 7.56 (1H, d, J = 3 Hz), 7.13 (1H, m), 6.36 (1H, d, J = 8.8 Hz), 5.33 (brs, 2H), 2.88 (m, 4H), 2.47 (m, 4H), 2.16 (s, 3H).
Example 11
Synthesis of tert-butyl 4-(6-amino-3-pyridyl)piperazine-l-carboxylate, Compound 11

This compound was prepared as described in WO 2010/020675 Al .
Synthesis of Compound 89 (also referred to as Compound T)

Compound 89 was synthesized in a similar manner to that described for compound 78 and was converted to an HCl salt. 1HNMR (600 MHz, DMSO-d6) δ ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS (ESI) 447 (M + H)
PATENT
WO 2014144740
PATENT
Preparation of Active Compounds
Syntheses
The disclosed compounds can be made by the following general schemes:

Scheme 1
In Scheme 1, Ref-1 is WO 2010/020675 Al; Ref-2 is White, J. D.; et al. J. Org. Chem. 1995, 60, 3600; and Ref-3 Presser, A. and Hufher, A. Monatshefte fir Chemie 2004, 135, 1015.

Scheme 2
In Scheme 2, Ref-1 is WO 2010/020675 Al; Ref-4 is WO 2005/040166 Al; and Ref-5 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.

92


93 
3) Pd/C/H2 ![]()
Scheme 6

Scheme 7
NHfOH

Scheme 8
In Scheme 8, Ref-1 is WO 2010/020675 Al; Ref-2 is WO 2005/040166 Al; and Ref-3 is Schoenauer, K and Zbiral, E. Tetrahedron Letters 1983, 24, 573.
Alternatively, the lactam can be generated by reacting the carboxylic acid with a protected amine in the presence of a strong acid and a dehydrating agent, which can be together in one moiety as a strong acid anhydride. Examples of strong acid anhydrides include, but are not limited to, trifluoroacetic acid anhydride, tribromoacetic acid anhydride, trichloroacetic acid anhydride, or mixed anhydrides. The dehydrating agent can be a carbodiimide based compound such as but not limited to DCC (Ν,Ν-dicyclohexylcarbodiimide), EDC (l-ethyl-3-(3-
dimethylaminopropyl)carbodiimide or DIC (Ν,Ν-diisopropylcarbodiimide). An additional step may be necessary to take off the N-protecting group and the methodologies are known to those skilled in the art.
Alternatively, the halogen moiety bonded to the pyrimidine ring can be substituted with any leaving group that can be displaced by a primary amine, for example to create an intermediate for a final product such as Br, I, F, SMe, SO2Me, SOalkyl, SO2alkyl. See, for Exmaple PCT /US2013/037878 to Tavares.
Other amine intermediates and final amine compounds can be synthesized by those skilled in the art. It will be appreciated that the chemistry can employ reagents that comprise reactive functionalities that can be protected and de-protected and will be known to those skilled in the art at the time of the invention. See for example, Greene, T.W. and Wuts, P.G.M., Greene’s Protective Groups in Organic Synthesis, 4th edition, John Wiley and Sons.

Scheme 9
CDK4/6 Inhibitors of the present invention can be synthesized according to the generalized Scheme 9. Specific synthesis and characterization of the Substituted 2-aminopyrmidines can be found in, for instance, WO2012/061156.
Compounds T, Q, GG, and U were prepared as above and were characterized by mass spectrometry and NMR as shown below:
Compound T
1H NMR (600 MHz, DMSO- d6) ppm 1.47 (br. s., 6 H) 1.72 (br. s., 2 H) 1.92 (br. s., 2 H) 2.77 (br. s., 3 H) 3.18 (br. s., 2 H) 3.46 (br. s., 2 H) 3.63 (br. s., 2 H) 3.66 (d, J=6.15 Hz, 2 H) 3.80 (br. s., 2 H) 7.25 (s, 1 H) 7.63 (br. s., 2 H) 7.94 (br. s., 1 H) 8.10 (br. s., 1 H) 8.39 (br. s., 1 H) 9.08 (br. s., 1 H) 11.59 (br. s., 1 H). LCMS ESI (M + H) 447.
PATENT
Synthesis of N-(heteroaryl)-pyrrolo[3,2-d]pyrimidin-2-amines. The application appears to be particularly focused on methods for the preparation of trilaciclib and an analog of it. Trilaciclib is the company’s lead CDK4/6 inhibitor presently in phase II trials against small-cell lung cancer and triple negative breast cancer. Interestingly, the company is working on a second CDK4/6 inhibitor, G1T38 , which is in a phase II trial against breast cancer.
GENERAL METHODS
The structure of starting materials, intermediates, and final products was confirmed by standard analytical techniques, including NMR spectroscopy and mass spectrometry. Unless otherwise noted, reagents and solvents were used as received from commercial suppliers. Proton nuclear magnetic resonance spectra were obtained on a Bruker AVANCE 500 at 500 MHz in DMSO-dis. HPLC analyses were performed on a Waters HPLC using the below HPLC method.
HPLC Method
Column: Atlantis T3 (150 χ 4.6, 3 μιη)
Column Temperature: 40°C
Flow Rate: 1 mL/min
Detection: UV @ 275 nm
Analysis Time: 36 min
Mobile Phase A: Water (with 0.1% Trifluoroacetic Acid)
Mobile Phase B : Acetonitrile (with 0.1% Trifluoroacetic Acid)
Sample preparation: dissolve PC sample, wet or dry solid (~1 mg of active compound) in acetonitrile/water (1/1) to achieve complete dissolution.
HPLC Method Gradient

Example 1. General Routes of Synthesis

Scheme 1-1 : Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3, Step 4, Step 5, or Step 6. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the hydroxyl group of the fused spirolactam is converted to a leaving group.
In Step 5 the leaving group is dehydrated to afford a compound of Formula IV. In Step 6 the compound of Formula IV is optionally deprotected.

Scheme 1-2: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the appropriately substituted spirolactam is protected with a group selected from R2. In Step 3 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 3 or Step 4. Oxidation prior to Step 3 results in undesired byproducts. In Step 4 the compound of Formula IV is optionally deprotected.

Scheme 1-3 : Starting from an appropriately substituted alkyl glycinate, compounds of the present invention can be prepared. In Step 1 the appropriately substituted alkyl glycinate is subjected to cyclohexanone and TMSCN in the presence of base to afford a cyano species. In Step 2 the appropriately substituted cyanospecies is reduced and subsequently cyclized to afford a compound of Formula I.
Scheme 1-4

Scheme 1-4: Starting from an appropriately substituted l-(aminomethyl)cyclohexan-l-amine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted l-(aminomethyl)cyclohexan-l -amine is reductively aminated with an aldehyde. In Step 2 the appropriately substituted cyclohexane amine is optionally deprotected (i.e.: the group selected from R2 if not H is optionally replaced by H). In Step 3 the cyclohexane amine is cyclized to afford a compound of Formula I. In Step 4 the compound of Formula I is optionally protected.
1-5

Conversion

Scheme 1-5: Starting from an appropriately substituted halo pyrimidine, compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a
substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2, Step 3, Step 4, or Step 5. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the hydroxyl group of the fused spirolactam is converted to a leaving group. In Step 4 the leaving group is dehydrated to afford a compound of Formula IV. In Step 5 the compound of Formula IV is optionally deprotected.
S

Scheme 1-6: Starting from an appropriately substituted halo pyrimidine compounds of the present invention can be prepared. In Step 1 the appropriately substituted halo pyrimidine is subjected to l,4-diazaspiro[5.5]undecan-3-one in the presence of base and heat to afford a substituted spirolactam. In Step 2 the protected spirolactam is cyclized in the presence of base to afford a fused spirolactam of Formula IV. The fused spirolactam can be optionally oxidized to a sulfoxide or sulfone after Step 2 or Step 3. Oxidation prior to Step 2 results in undesired byproducts. In Step 3 the compound of Formula IV is optionally deprotected.

Scheme 1-7: Starting from compound of Formula IV a CDK4/6 inhibitor can be prepared. In Step 1 a heteroaryl amine is subjected to a base and a compound of Formula IV is added slowly under chilled conditions to afford a nucleophilic substitution reaction. The compound of Formula IV can previously be prepared as described in the schemes herein.
Example 2. Representative Routes of Synthesis
Scheme 2-1

quant, yield 2 steps
isolated

70% yield 2 steps 75% yield 95% yield
isolated isolated isolated
Scheme 2-1 : An ester route is one embodiment, of the present invention. Ideally, the best synthesis scheme would afford crystalline intermediates to provide material of consistent purity without column chromatography, and high yielding steps while using safe and cost effective reagents when possible.
The first step in the ester route is a SNAr nucleophilic substitution of CI group in commercially available ester 3 using spirolactam 4. Due to low reactivity of 4, a reaction temperature of 85-95 °C was required. Because of the temperature requirements, DIPEA and dimethylacetamide were selected as the base and solvent, respectively. The reaction follows second-order kinetics and usually stalls after -85% conversion. Therefore, the reaction was typically stopped after 60 hours by first cooling it to room temperature at which point solid formation was observed. The mixture was then partitioned between MTBE and water and product was filtered with excellent purity with -53% yield of the desired product 5. The obtained
compound 5 was protected with a Boc group using Boc anhydride and DMAP as the catalyst and dichloromethane as the solvent. The intermediate 6 was obtained in a quantitative yield. Due to the semi-solid nature of compound 6, the material was taken to the next step without further purification. The Dieckmann condensation was initially performed with strong bases such as LiHMDS and tBuOK. A similar result to the aldehyde route (Scheme 2-2) was obtained: a partial deprotection of Boc group was observed that required column chromatography. However, the best results were obtained when DBU was used as base and THF as solvent. The reaction outcome was complete, clean conversion of 6 to 7. Moreover, the product crystallized from the reaction mixture upon seeding, and a quantitative yield was obtained for the two steps.
The hydroxyl group of 7 was removed via a two-step procedure. First, compound 7 was converted completely into triflate 8 using triflic anhydride and triethylamine in dichloromethane. The reaction was found to proceed well at 0°C. Due to the potential instability of the triflate intermediate, it was not isolated. It was immediately taken to the next step and reduced with triethylsilane and palladium tetrakis to afford the product 9 after ethyl acetate crystallization in -70% yield. The Boc group of 9 was removed using trifluoroacetic acid in dichloromethane to afford 10. Intermediate 10 was converted into the final sulfone 11 using Oxone™ in acetonitrile/water solvent system.
The obtained sulfone 11 was use-tested in the coupling step and was found to perform well. In conclusion, the route to sulfone 11 was developed which eliminated the use of column chromatography with good to excellent yields on all steps.
Scheme 2-2

Molecular Weight: 421 
Scheme 2-2: The first step of Scheme 2-2 consistently afforded product 13 contaminated with one major impurity found in substantial amount. Thorough evaluation of the reaction impurity profile by LC-MS and 2D MR was performed, which showed the impurity was structurally the condensation of two aldehyde 12 molecules and one molecule of lactam 4. Therefore, column chromatography was required to purify compound 13, which consistently resulted in a modest 30% yield. A solvent screen revealed that sec-butanol, amyl alcohol, dioxane, and tert-butanol can all be used in the reaction but a similar conversion was observed in each case. However, tert-butanol provided the cleanest reaction profile, so it was selected as a solvent for the reaction. Assessing the impact of varying the stoichiometric ratio of 4 and 12 on the reaction outcome was also investigated. The reaction was performed with 4 equivalents of amine 4 in an attempt to disrupt the 2: 1 aldehyde/amine composition of the impurity. The result was only a marginal increase in product 13 formation. The temperature impact on the reaction outcome was evaluated next. The coupling of aldehyde 12 and 4 was investigated at two different temperatures: 50 °C and 40 °C with 1 : 1 ratio of aldehyde/amine. Reactions were checked at 2 and 4 hours and then every 12 hours. The reaction progress was slow at 50°C and was accompanied by growth of other impurities. The reaction at 40°C was much cleaner; however the conversion was lower in the same time period. The mode of addition of the reagents was investigated as well at 80°C with a slow addition (over 6 hours) of either aldehyde 12 or amine 4 to the reaction mixture. The product distribution did not change and an about 1 to 1 ratio was observed between product and impurity when amine 4 was added slowly to the reaction mixture containing aldehyde 12 and
DIPEA at reflux. The product distribution did change when aldehyde 12 was added slowly to the mixture of amine 4 and DIPEA. However, the major product of the reaction was the undesired impurity. Other organic bases were tried as well as different ratios of DIPEA. No product was observed when potassium carbonate was used as a base. The results of the experiments are presented in Table 1 below.
Table 1

Compound 13 was successfully formed in three cases: triethylamine, 2,6-lutidine and DIPEA, with the DIPEA result being the best. The use of Boc protected spirolactam 4 had no effect on the impurity formation as well. Its utilization was speculated to be beneficial in performing the coupling step together with the following step, preparation of compound 14.
The major impurity formed during Step 1 of Scheme 2-2 is:

Chemical Formula:€2)Η;Μ(¾ 6( 2ί>2
Molecular Weight: 527.4903
The second step (Boc protection of the free lactam) proceeded well using DMAP as a catalyst in dichloromethane at room temperature. The product 14 is a thick oil, and, therefore, cannot be purified by crystallization. The Boc protected intermediate 14 was cyclized successfully into the desired pentacyclic structure 10 upon treatment with a strong base such as LiHMDS or tBuOK. Surprisingly, the Boc group was partially removed during the reaction. The level of deprotection was independent from the internal reaction temperature and was positively correlated with excess of base used. Therefore the mixture of the desired product 10 and 10-Boc compound was treated with acid to completely deprotect Boc group. The conversion of methyl sulfide into the final sulfone 11 was carried out with Oxone™. Initially a mixture of methanol and water was used for the reaction. As the result, a partial displacement of sulfone by methoxy group was detected. The methanol was replaced with acetonitrile and the sulfone displacement was eliminated.
In summary, the ester route (Scheme 2-1) is preferred because:
1. Formation of the impurity during the first step of Scheme 2-2 was unavoidable and resulted in yields of < 35%.
2. Column purification was required to isolate intermediate 14.
3. The aldehyde starting material was not commercially available and required two synthetic steps from the corresponding ester.

Scheme 2-3 : Starting with cyclohexanone, compounds of the present invention can be prepared. In Step 1 the methyl glycinate is subjected to cyclohexanone and TMSCN in the presence of tri ethyl amine in DCM to afford 15. In Step 2 15 hydrogenated with hydrogen gas in the presence of catalytic platinum oxide and subsequently undergoes an intramolecular cyclization to afford compound 16 which is used in the schemes above.

Scheme 2-4: Starting with compound 17, compounds of the present invention can be prepared. In Step 1 compound 17 is subjected to ethyl 2-oxoacetate in the presence platinum on carbon and hydrogen gas to afford compound 18. In Step 2 compound 18 is Boc-deprotected with hydrochloric acid. In Step 3 compound 18 is cyclized to afford compound 16 which is used in the schemes above.
Scheme 2-5

11 19
Scheme 2-5: Starting from compound 11 the CDK 4/6 inhibitor 19 can be prepared. In Step 1 5-(4-methylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 19. Compound 11 can be prepared as described in the schemes herein.

Scheme 2-6: Starting from compound 11 the CDK 4/6 inhibitor 20 can be prepared. In Step 1 5-(4-isopropylpiperazin-l-yl)pyridin-2-amine is subjected to LiHMDS and compound 11 is added slowly under chilled conditions to afford a nucleophilic substitution reaction and compound 20. Compound 11 can be prepared as described in the schemes herein.
Preparation of Compound 5:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate 3 (49.2 g, 0.21 mol, 1.00 equiv.), spirolactam 4 (39.2 g, 0.23 mol, 1.10 equiv.), DIPEA (54.7 g, 0.42 mol, 2.00 equiv.), and DMAc (147.6 mL, 3 vol). The batch was heated to 90-95 °C, and after 60 h, IPC confirmed -14% (AUC) of ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate remained. The batch was cooled to RT, and precipitate formation was observed. The suspension was diluted with MTBE (100 mL, 2 vol) and water (442 mL, 9 vol) and stirred for 2 h at RT. The product was isolated by vacuum filtration and washed with MTBE (49 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford compound 5 [41.0 g, 53% yield] as an off-white solid with a purity of >99% AUC. ¾ MR (CDCh): δ 8.76 (d, J = 2.0 Hz, 1H), 6.51-6.29 (br, 1H), 4.33 (q, J = 7.0 Hz, 2H), 3.78 (s, 2H), 3.58 (s, 2H), 2.92 (s, 2H), 2.53 (s, 3H), 1.63-1.37 (m, 12H). LCMS (ESI, m/z = 365.3 [M+H]).
Preparation of Compound 6:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 5 [41.0 g, 0.11 mol, 1.00 equiv.], Boc-anhydride (36.8 g, 0.17 mol, 1.50 equiv.), DMAP (1.37 g, 0.01 mol, 0.10 equiv.), and dichloromethane (287 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained (AUC). The batch was concentrated into a residue under reduced pressure and taken to the next step (a quantitative yield is assumed for this step). An aliquot (200 mg) was purified by column chromatography (heptanes/ethyl acetate 0 to 100%) to afford compound 6. 1H MR (CDCh): δ 8.64 (s, 1H), 4.31 (q, J = 7.0 Hz, 2H), 4.07 (s, 2H), 3.83 (S, 2H), 3.15 (m, 2H), 2.56 (s, 3H), 172 (m, 3H), 1.59 (m, 15H), 1.42 (t, J= 7.0 Hz, 3H). LCMS (ESI, m/z = 465.2 [M+H]).
Preparation of Compound 7:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 6 [residue from a previous step, quantitative yield assumed, 52.2 g, 0.11 mol, 1.00 equiv.], and THF (261 mL, 5 vol). The batch was cooled to 0°C and 1,8-diazabicyclo[5.4.0]un-dec-7-ene (17.1 g, 0.11 mmol, 1.00 equiv.) was added keeping the internal temperature in 0-10°C range. After the addition was complete, the cooling bath was removed and the reaction mixture was allowed to warm up to RT and after 2 h, IPC confirmed no starting material remained. The batch was seeded with the product (1.0 g) and was cooled to 0°C. The slurry was stirred at 0°C for 2 h. The product was isolated by vacuum filtration and washed with cold (0°C) THF (50 mL, 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 7 [47 g, quantitative yield] as a light orange solid with a purity of >99% AUC. The color of the product changed into yellow once the batch was exposed to air for an extended period of time (~ 1 day). Material was isolated with substantial amount DBU, according to proton NMR. However, it did not interfere with the next step. 1H MR (CDCh): δ 8.71 (s, 1H), 4.03 (s, 2H), 2.57 (s, 3H), 1.85 (m, 10H), 1.51 (s, 9H). LCMS (ESI, m/z = 419.2 [M+H]).
Preparation of Compound 8:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 7 [40.8 g, 0.10 mol, 1.00 equiv.], triethylamine (31.5 g, 0.31 mol, 3.20 equiv.), and dichloromethane (408 mL, 10 vol). The batch was purged with N2 for 15 min and was cooled to 0°C. Triflic anhydride (44.0 g, 0.16 mol, 1.60 equiv.) was added keeping the
internal temperature in 0-10°C range. The batch was stirred at 0°C and after 3 h, IPC confirmed -7.0% (AUC) of 7 remained. [It was speculated that the product was hydrolyzing back into starting material during the analysis.] Once the reaction was deemed complete, the batch was transferred to a 1 L, separatory funnel and was washed with 50% saturated sodium bicarbonate (200 mL, 5 vol). [It was prepared by mixing saturated sodium bicarbonate (100 mL) with water (100 mL)).] The aqueous layer was separated and was extracted with DCM (2×40 mL, 1 vol). The organic layers were combined and concentrated into a residue under reduced pressure and taken to the next step. LCMS (ESI, m/z = 551.6 [M+H]).
Preparation of Compound 9:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with compound 8 [residue from a previous step, quantitative yield assumed, 53.7 g, 0.10 mol, 1.00 equiv.], and THF (110 mL, 2 vol). The solvent was removed under vacuum distillation and the procedure was repeated two times. The flask was charged with triethylsilane (22.7 g, 0.20 mol, 2.00 equiv.), and DMF (268 mL, 5 vol). The batch was degassed by five cycles of evacuation, followed by backfilling with nitrogen. The flask was charged with tetrakis(triphenylphosphine)palladium(0) (11.3 g, 0.01 mol, 0.1 equiv.). The batch was heated to 45-50°C, and after 14 h, IPC confirmed no starting material remained. The batch was transferred to a 500 mL, separatory funnel while still warm. The reaction was partitioned between water (5 vol) and ethyl acetate (5 vol). The aqueous layer was extracted with ethyl acetate (3 x3 vol). The organic layers were combined and concentrated down to 2 volumes. The precipitate was filtered and washed with ethyl acetate (2x 1 vol). The solid cake was conditioned for 1 h and dried under vacuum at 40°C for 16 h to afford 9 [27.5 g, 70% yield] as a yellow solid with a purity of -98% AUC. Proton NMR showed some triphenylphosphine oxide present. ¾ NMR (DMSO-i¾):5 9.01 (s, 1H), 7.40 (s, 1H), 4.30 (s, 2H), 2.58 (m, 2H), 2.58 (s, 3H), 1.81 (m, 5H), 1.51 (s, 9H). LCMS (ESI, m/z = 403.4 [M+H]).
Preparation of Compound 10 from the Scheme 2-1 route:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged 9 (12.8 g, 31.8 mmol, 1.00 equiv.) and dichloromethane (64 mL, 5 vol). Trifluoroacetic acid (18.2 g, 159 mmol, 5.00 equiv.) was added over 20 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (200 mL). The aqueous layer was extracted with dichlorom ethane (3 x3 vol). The organic layers were combined and concentrated down to 1 volume. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [6.72 g, 70% yield] as an off-white solid with a purity of 99.1% AUC. ¾ NMR (DMSO-dis): δ 8.95 (s, 1H), 8.32 (s, 1H), 7.15 (s, 1H), 3.68 (d, J = 1.0 Hz, 2H), 2.86 (m, 2H), 2.57 (s, 3H), 1.92 (m, 2H), 1.73 (m, 3H), 1.39 (m, 3H). LCMS, ESI, m/z = 303.2 [M+H]).
Preparation of Compound 10 from Scheme 2-2 route:
A 50 mL, three-neck flask equipped with a magnetic stirring bar, thermocouple, N2 inlet was charged 14 (680 mg, 1.62 mmol, 1.00 equiv.) and THF (6.8 mL, 10 vol). A I M solution of potassium tert-butoxide (3.2 mL, 3.24 mmol, 2.00 equiv.) in THF was added over 10 min and the solution was stirred for 2 h at RT. IPC confirmed reaction was complete. The batch was acidified with 4 N hydrogen chloride solution in dioxane (2.4 mL, 9.72 mmol, 6.00 equiv.) and stirred for additional 1 h. The batch was transferred to a 500 mL, separatory funnel and washed with saturated sodium bicarbonate (100 mL). The aqueous layer was extracted with ethyl acetate (3 x20 vol). The organic layers were combined and concentrated down to 3volumes and product precipitated. The precipitate was filtered and conditioned for 1 h and dried under vacuum at 40 °C for 16 h to afford 9 [489 mg, quantitative yield] as an off-white solid.
Preparation of Compound 11 :
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 10 (9.00 g, 29.8 mmol, 1.00 equiv.), and acetonitrile (180 mL, 20 vol). A solution of Oxone™ (45.9 g, 0.15 mol, 5.00 equiv.) in water (180 mL, 20 vol) was added to the batch over 20 min. The batch was stirred for 2 h and IPC confirmed the reaction was complete. The batch was concentrated down to ½ of the original volume and was extracted with dichloromethane DCM (4x 10 vol). The organic layers were combined; polish filtered and concentrated down to -10 vol of DCM. The product was slowly crystallized out by addition of heptanes (-30 vol). The mixture was cooled to 0°C and the product was filtered and dried under vacuum at 40 °C for 16 h to afford 11 [9.45 g, 95% yield] as an off-white solid with a purity of >99% AUC. ¾ NMR (CDCb): 5 9.24 (s, 1H), 7.78 (s, 1H), 7.46 (s, 1H), 3.89 (d, J= 2.0 Hz, 2H), 3.43 (s, 3H), 2.98 (m, 2H), 2.10 (m, 2H), 1.86 (m, 3H), 1.50 (m, 3H). LCMS (ESI, m/z = 335.2 [M+H]).
Preparation of Compound 13:
A 250 mL, single-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet, and reflux condenser was charged with 4-chloro-2-(methylthio)pyrimidine-5-carbaldehyde (2.00 g, 10.6 mmol, 1.00 equiv.), spirolactam 4 (1.96 g, 11.7 mmol, 1.10 equiv.), DIPEA (2.74 g, 21.2 mmol, 2.00 equiv.), and fert-butanol (20 mL, 10 vol). The batch was heated to 80-85 °C, and after 24 h, IPC confirmed no aldehyde 12 remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 13 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 14:
A 500 mL, three-neck flask equipped with a mechanical overhead stirrer, thermocouple, N2 inlet was charged with 13 [0.98 g, 3.00 mmol, 1.00 equiv.], Boc-anhydride (4.90 g, 21.5 mmol, 7.00 equiv.), DMAP (36 mg, 0.30 mmol, 0.10 equiv.), and dichloromethane (7 mL, 7 vol). The batch was stirred for 3 h at RT. IPC confirmed no starting material remained. The batch was cool to RT and concentrated into a residue, which was loaded on silica gel column. The product was eluted with ethyl acetate/heptanes (0% to 100%). The product containing fractions were pulled out and concentrated to afford 14 [0.98 g, 29% yield] as an off-white solid.
Preparation of Compound 15:
To a suspension of methyl glycinate (500 g, 3.98 mol, 1 eq) in DCM (10 L) was added
TEA dropwise at rt under nitrogen atmosphere, followed by the addition of cyclohexanone (781 g, 7.96 mol, 2 eq) dropwise over 15 min. To the resulting mixture was added TMSCN (591 g, 5.97 mol, 1.5 eq) dropwise over 1 hour while maintaining the internal reaction temperature below 35
°C. After stirred at rt for 2 hrs, the suspension became a clear solution. The progress of the reaction was monitored by H- MR.
When the methyl glycinate was consumed completely as indicated by H-NMR analysis, the reaction was quenched by water (5 L). The layers were separated. The aqueous layer was extracted with DCM (1 L). The combined organic phase was washed with water (5 L X 2) and
dried over Na2S04 (1.5 Kg). After filtration and concentration, 1.24 Kg of crude 15 was obtained as oil.
The crude 15 was dissolved in IPA (4 L). The solution was treated with HC1/IPA solution (4.4 mol/L, 1.1L) at RT. A large amount of solid was precipitated during the addition. The resulting suspension was stirred for 2 hrs. The solid product was collected by vacuum filtration and rinsed with MTBE (800 mL). 819 g of pure 15 was obtained as a white solid. The yield was 88.4%. ¾- MR (300 MHz, CD3OD) 4.20 (s, 2H), 3.88 (s, 3H), 2.30-2.40 (d, J = 12 Hz, 2H), 1.95-2.02 (d, J = 12 Hz, 2H), 1.55-1.85 (m, 5H), 1.20-1.40 (m, 1H).
Preparation of Compound 16:
To a solution of 15 (10 g, 43 mmol) in MeOH (100 mL) was added methanolic hydrochloride solution (2 .44 mol/L, 35.3 mL, 2 eq) and Pt02 (0.5 g, 5 wt %). The reaction suspension was stirred with hydrogen bubble at 40 °C for 6 hours. H- MR analysis showed consumption of 15. To the reaction mixture was added K2CO3 (15 g, 108 mmol, 2.5 eq) and the mixture was stirred for 3 hrs. The suspension was filtered and the filtrate was concentrated to dryness. The residual oil was diluted with DCM (100 mL) and resulting suspension was stirred for 3 hrs. After filtration, the filtrate was concentrated to provide crude 16 (6.6 g) as an oil. The crude 16 was diluted with EtOAc/hexane (1 : 1, 18 mL) at rt for 2 hrs. After filtration, 16 (4 g) was isolated. The obtained 16 was dissolved in DCM (16.7 mL) and hexane (100 mL) was added dropwise to precipitate the product. After further stirred for 1 h, 2.8 g of the pure 16 was isolated as a white solid. The yield was 39%. HPLC purity was 98.3%; MS (ESI): 169.2 (MH+); 1 H-NMR (300 MHz, D2O) 3.23 (s, 3H), 3.07 (s, 3H), 1.37-1.49 (m, 10H).
Preparation of compound 19:
5-(4-methylpiperazin-l-yl)pyridin-2-amine (803.1 g; 3.0 equivalents based on sulfone 11) was charged to a 22 L flask. The flask was blanketed with N2 and anhydrous THF added (12.4 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (4.7 L; 1.2 equivalents based on sulfone 11) was added via an addition funnel in three equal additions to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C. The sulfone 11 (455.1 g; 1.00 equivalents) was added in five additions. Reaction proceeded until HPLC analysis of an IPC sample indicated less than 3% of sulfone 11 remained.
To quench the reaction, the contents of the 22L flask were transferred to a 100 L flask containing water. After stirring for 30 minutes at <30°C, the crude product was collected by filtration, washed with water and dried to afford 19 (387 g, 99.1% purity, 63.7% yield).
Preparation of compound 20:
5-(4-isopropylpiperazin-l-yl)pyridin-2-amine (1976.2 g; 3.0 equivalents based on sulfone 11) was charged to a 50 L flask. The flask was blanketed with N2 and anhydrous THF added (10.7 kg). The resulting black-purple solution was cooled in an ice bath to < 5°C. 1M LiHMDS (9.6 kg; 3.6 equivalents based on sulfone) was added via an addition funnel at a rate to keep the temperature below 10°C. Upon the completion of the addition, the reaction mixture was warmed to 16°C over 120 minutes by removing the ice bath. The sulfone (1000 g; 1.00 mol) was added in five additions. The reaction proceeded until HPLC analysis of an IPC sample indicated less than 1% of sulfone 11 remained. After completion of the reaction, ammonium chloride was added to the reaction mixture. The mixture stirred at < 32°C for at least 30 minutes and the solids collected by filtration to afford 20 (900 g, 99.1% purity, 64.2% yield).
Alternate Route to Spirolactam via cyclohexanone:
Scheme 2-7

26
In one embodiment the spirolactam is made via the synthetic scheme above. By reducing the nitrile group before addition of the glycinate group the reaction sequence proceeds in higher yield. The chemistry used in Step 1 is described in the literature (J. Org. Chem. 2005, 70,8027-8034), and was performed on a kilogram scale. The chemistry to convert Compound 24 into the
spirolactam was also demonstrated on kilogram scale. The Boc protection of Compound 23, is carried out at -70°C in order to limit formation of the di-Boc protected product. Experimental details of a 200 g pilot run are described below.
Step 1

200 g of cyclohexanone 21 was converted to 22 using Ti(Oi-Pr)4 /TMSCN/NH3. After work-up, 213 g of 22 was obtained. The H- MR was clean. The yield was 84%. The titanium salts were removed by vacuum filtration. In one embodiment, the titanium salts are removed by centrifugation or Celite filtration.
Step 2

190 g of 22 was mixed with LAH (2 eq) in MTBE for 30 minutes at 45°C. After work-up, 148 g of crude 23 was obtained.
Step 3

136 g of the crude 23 from step 2 was converted to 24 with 0.9 eq of B0C2O at -70°C. The reaction was completed and worked up. After concentration, 188 g of 24 was obtained. The yield was 86%. The H-NMR and C-NMR spectra confirmed that the compound was pure.
Step 4

188 g of 24 was subjected to methyl 2-bromoacetate and K2CO3 in acetonitrile to afford 25. 247 g of crude 25 was obtained.
Step 5

247 g of 25 was subjected to TFA in DCE heated to reflux to afford 26. After work-up, 112 g of 6 as TFA salt was obtained. H- MR was clean.
Step 6

26 27
Compound 26 was stirred in EtOH in the presence at room temperature overnight to afford 27. In one embodiment DCM is used as the solvent instead of EtOH.
Example 3. Purge of residual palladium from Step 5 Scheme 2-1:
Since palladium was used in Step 5 of Scheme 2-1, the levels of residual Pd present in the subsequent synthetic steps was determined. Table 2 below and Figure 3 show the palladium levels in the isolated solids.
Table 2

The material after Step 5 was isolated containing 1.47% (14700 ppm) of residual palladium. This data represents the highest level of palladium in the worst case scenario. The workup conditions of the latter steps purged nearly all of the palladium and the final product, 19 bis HC1 salt, contained 14 ppm of Pd, which is below the standard 20 ppm guidline. The Pd levels will likely be even lower once the catal st loading is optimized in Step 5.

19
The process developed in this route was a significant improvement over the one used for the first generation synthesis. Overall, the scheme consists of seven steps with five isolations, all by crystallization. No silica column chromatography is employed in the synthesis, which makes the process highly scalable. The process workup conditions can successfully purge the 1.47% of residual palladium after step 5 of Scheme 2-1.
///////////////TRILACICLIB, G1T28, G1T 28, SHR 6390, PHASE 2, G1 Therapeutics, Inc.
CN1CCN(CC1)C2=CN=C(C=C2)NC3=NC=C4C=C5C(=O)NCC6(N5C4=N3)CCCCC6
TRIENTINE HYDROCHLORIDE, 塩酸トリエンチン , 曲恩汀
TRIENTINE
- Molecular Formula C6H18N4
- Average mass 146.234 Da
112-24-3 CAS
曲恩汀, KD-034, MK-0681, MK-681, TECZA, TETA, TJA-250
1,2-Ethanediamine, N1,N2-bis(2-aminoethyl)-
1,8-diamino-3,6-diazaoctane

TRIENTINE HYDROCHLORIDE
- Molecular Formula C6H19ClN4
- Average mass 182.695 Da
38260-01-4 CAS
Launched – 1986 VALEANT, WILSONS DISEASE

塩酸トリエンチン
Trientine Hydrochloride

C6H18N4 2HCl : 219.16
2HCl : 219.16
[38260-01-4]
UPDATE CDSCO INDIA Trientine 08.06.2021 APPROVED
Trientine Tetrahydrochloride bulk and
Trientine Tetrahydrochloride capsules 333 mg
(Each capsule contains Trientine
tetrahydrochloride 333mg equivalent to
Trientine 167mg base)
For the treatment of Wilson’s disease
(hepatolenticular degeneration) in patients
intolerant to Penicillamine. It should be
used when continued treatment with
Penicillamine is no longer possible because
of intolerable or life endangering side
effects.
Aton Pharma, a subsidiary of Valeant Pharmaceuticals, has developed and launched Syprine, a capsule formulation of trientine hydrochloride, for treating Wilson disease.

Triethylenetetramine, abbreviated TETA and trien and also called trientine (INN), is an organic compound with the formula [CH2NHCH2CH2NH2]2. This oily liquid is colorless but, like many amines, assumes a yellowish color due to impurities resulting from air-oxidation. It is soluble in polar solvents. The branched isomer tris(2-aminoethyl)amine and piperazine derivatives may also be present in commercial samples of TETA.[1]
Trientine hydrochloride is a metal antagonist that was first launched by Merck, Sharp & Dohme in the U.S. in 1986 under the brand name Syprine for the oral treatment of Wilson’s disease.
Orphan drug designation has also been assigned in the U.S. for the treatment of patients with Wilson’s disease who are intolerant or inadequately responsive to penicillamine and in the E.U. by Univar for the treatment of Wilson’s disease

By condensation of ethylenediamine (I) with 1,2-dichloroethane (II)
Trientine hydrochloride is N,N’-bis (2-aminoethyl)-1,2-ethanediamine dihydrochloride. It is a white to pale yellow crystalline hygroscopic powder. It is freely soluble in water, soluble in methanol, slightly soluble in ethanol, and insoluble in chloroform and ether.
The empirical formula is C6H18N4·2HCI with a molecular weight of 219.2. The structural formula is:
NH2(CH2)2NH(CH2)2NH(CH2)2NH2•2HCI
Trientine hydrochloride is a chelating compound for removal of excess copper from the body. SYPRINE (Trientine Hydrochloride) is available as 250 mg capsules for oral administration. Capsules SYPRINE contain gelatin, iron oxides, stearic acid, and titanium dioxide as inactive ingredients.

Production
TETA is prepared by heating ethylenediamine or ethanolamine/ammonia mixtures over an oxide catalyst. This process gives a variety of amines, which are separated by distillation and sublimation.[2]
Uses
The reactivity and uses of TETA are similar to those for the related polyamines ethylenediamine and diethylenetriamine. It was primarily used as a crosslinker (“hardener”) in epoxy curing.[2]
The hydrochloride salt of TETA, referred to as trientine hydrochloride, is a chelating agent that is used to bind and remove copper in the body to treat Wilson’s disease, particularly in those who are intolerant to penicillamine. Some recommend trientine as first-line treatment, but experience with penicillamine is more extensive.[3]
Coordination chemistry
TETA is a tetradentate ligand in coordination chemistry, where it is referred to as trien.[4] Octahedral complexes of the type M(trien)Cl3 can adopt several diastereomeric structures, most of which are chiral.[5]
Trientine, chemically known as triethylenetetramine or N,N’-bis(2-aminoethyl)-l,2-ethanediamine belongs to the class of polyethylene polyamines. Trientine dihydrochloride is a chelating agent which is used to bind and remove copper in the body in the treatment of Wilson’s disease.
Trientine dihydrochloride (1)
Trientine dihydrochloride formulation, developed by Aton with the proprietary name SYPRINE, was approved by USFDA on November 8, 1985 for the treatment of patients with Wilson’s disease, who are intolerant to penicillamine. Trientine dihydrochloride, due to its activity on copper homeostasis, is being studied for various potential applications in the treatment of internal organs damage in diabetics, Alzheimer’s disease and cancer.
Various synthetic methods for preparation of triethylenetetramine (TETA) and the corresponding dihydrochloride salt have been disclosed in the prior art.
U.S. 4,806,517 discloses the synthesis of triethylenetetramine from ethylenediamine and monoethanolamine using Titania supported phosphorous catalyst while U.S. 4,550,209 and U.S. 5,225,599 disclose catalytic condensation of ethylenediamine and ethylene glycol for the synthesis of linear triethylenetetramine using catalysts like zirconium trimethylene diphosphonate, or metatungstate composites of titanium dioxide and zirconium dioxide.
U.S. 4,503,253 discloses the preparation of triethylenetetramine by reaction of an alkanolamine compound with ammonia and an alkyleneamine having two primary amino groups in the presence of a catalyst, such as supported phosphoric acid wherein the support is comprised of silica, alumina or carbon.
The methods described above for preparation of triethylenetetramine require high temperatures and pressure. Further, due to the various possible side reactions and consequent associated impurities, it is difficult to control the purity of the desired amine.
CN 102924289 discloses a process for trientine dihydrochloride comprising reduction of Ν,Ν’-dibenzyl-,N,N’-bis[2-(l,3-dioxo-2H-isoindolyl)ethyl]ethanediamine using hydrazine hydrate to give N,N’-dibenzyl-,N,N’-bis(2-aminoethyl)ethanediamine, which, upon condensation with benzyl chloroformate gave N,N’-dibenzyl-,N,N’-bis[2-(Cbz-amino)ethyl]ethanediamine, and further reductive deprotection to give the desired compound.
CS 197,093 discloses a process comprising reaction of triethylenetetramine with concentrated hydrochloric acid to obtain the crystalline tetrahydrochlonde salt. Further reaction of the salt with sodium ethoxide in solvent ethanol, filtration of the solid sodium chloride which is generated in the process, followed by slow cooling and crystallization of the filtrate provided the dihydrochloride salt. Optionally, aqueous solution of the tetrahydrochloride salt was passed through a column of an anion exchanger and the eluate containing free base was treated with a calculated amount of the tetrahydrochloride, evaporated, and the residue was crystallized from aqueous ethanol to yield the dihydrochloride salt.
The process is quite circuitous and cumbersome, requiring use of strong bases, filtration of sodium chloride and results in yields as low as 60%.
US 8,394,992 discloses a method for preparation of triethylenetetramine dihydrochloride wherein tertiary butoxycarbonyl (boc) protected triethylenetetramine is first converted to its tetrahydrochloride salt using large excess of hydrochloric acid in solvent isopropanol, followed by treatment of the resulting tetrahydrochloride salt with a strong base like sodium alkoxide to produce the amine free base (TETA) and sodium chloride salt in anhydrous conditions. The free amine is extracted with tertiary butyl methyl ether (TBME), followed by removal of sodium chloride salt and finally the amine free base TETA is treated with hydrochloric acid in solvent ethanol to give trientine hydrochloride salt.
PATENT
WO-2017046695
EXAMPLES
Example 1: Preparation of 2-([2-[cyanomethyl]-t-butyloxycarbonylamino]ethyl- 1-butyloxy carbonylamino)acetonitrile (5)
Potassium carbonate (481.9 g) was added to a stirred mixture of ethylenediamine (100.0 g) in acetonitrile (800 ml) and cooled to around 10°C. Chloroacetonitrile (263.8 g) was gradually added at same temperature and stirred at 25-30°C, till completion of the reaction, as monitored by HPLC. The mixture was cooled to 5-15°C and Boc-anhydride (762. lg) was added to it, followed by stirring at the same temperature. The temperature was raised to 25-30°C and the mass was stirred till completion of the reaction, as monitored by HPLC.
The reaction mass was filtered and the filtrate was concentrated. Toluene was added to the residue, and the mixture was heated to around 70°C followed by cooling and filtration to give 2-([2-[cyanomethyl)-t-butyloxycarbonylamino]ethyl-t-butyloxycarbonylamino) acetonitrile (5).
Yield: 506.8 g
% Yield: 89.9 %
Example 2: Preparation of t-butyl( N-2-aminoethyl)N-([2-[(2-aminoethyl)t-butyloxy)carbonylamino] ethyl) carbamate (6)
Raney nickel (120.0 g) in isopropanol (100 ml) was charged into an autoclave, followed by a mixture of Compound 5 (200 g) in isopropanol (400 ml). Cooled ammonia solution prepared by purging ammonia gas in 1400 ml isopropanol, equivalent to 125 g ammonia was gradually charged to the autoclave and the reaction was carried out around 15-25°C under hydrogen pressure of 2-5 Kg/cm2.
After completion of the reaction, as monitored by HPLC, the mass was filtered, concentrated, and methyl tertiary butyl ether was added to the residue. The mixture was heated to around 50°C, followed by cooling of the mass, stirring, optional seeding with compound 6 and filtration to give tertiary butyl-(N-2-aminoethyl)N-([2-[(2-aminoethyl)-(tert-butyloxy) carbonylamino] ethyl) carbamate.
Yield: 174 g
%Yield: 85 %
Example 3: Preparation of triethylenetetramine dihydrochloride (1)
Concentrated hydrochloric acid (121.5 g) was gradually added to a stirred mixture of tertiary-butyl-N-(2-aminoethyl)-N-2-[(2-aminoethyl)-(tert-butoxy) carbonyl] amino] ethyl} carbamate (Compound 6, 200.0 g) and water (1400 ml) at 20-30°C. The reaction mixture was heated in the temperature range of 100-105°C till completion of the reaction, as monitored by HPLC, with optionally distilling out water, if so required.
The reaction mass was concentrated and ethanol (600 ml) was added to the residue, followed by heating till a clear solution was obtained. The reaction mixture was gradually cooled with stirring, filtered and dried to provide triethylenetetramine dihydrochloride (1).
Yield: 88.9 g, (70 %)
Purity : > 99%
Patent
https://www.google.com/patents/US8394992
Trientine was said to be used in the synthesis of benzylidene-(2-{3-[2-(benzylidene-amino)-ethyl]-2-phenyl-imidazolidin-1-yl}-ethyl)-amine in French Patent No. FR2810035 to Guilard et al. Cetinkaya, E., et al., “Synthesis and characterization of unusual tetraminoalkenes,” J. Chem. Soc. 5:561-7 (1992), is said to be directed to synthesis of benzylidene-(2-{3-[2-(benzylidene-amino)-ethyl]-2-phenyl-imidazolidin-1-yl}-ethyl)-amine from trientine, as is Araki T., et al., “Site-selective derivatization of oligoethyleneimines using five-membered-ring protection method,” Macromol., 21:1995-2001 (1988). Triethylenetetramine may reportedly also be used in the synthesis of N-methylated triethylenetetramine, as reported in U.S. Pat. No. 2,390,766, to Zellhoefer et al.
Synthesis of polyethylenepolyamines, including triethylenetetramines, from ethylenediamine and monoethanolamine using pelleted group IVb metal oxide-phosphate type catalysts was reported by Vanderpool et al. in U.S. Pat. No. 4,806,517. Synthesis of triethylenetetramine from ethylenediamine and ethanolamine was also proposed in U.S. Pat. No. 4,550,209, to Unvert et al. U.S. Pat. No. 5,225,599, to King et al. is said to be directed to the synthesis of linear triethylene tetramine by condensation of ethylenediamine and ethylene glycol in the presence of a catalyst. Joint production of triethylenetetramine and 1-(2-aminoethyl)-aminoethyl-piperazine was proposed by Borisenko et al. in U.S.S.R. Patent No. SU1541204. U.S. Pat. No. 4,766,247 and European Patent No. EP262562, both to Ford et al., reported the preparation of triethylenetetramine by reaction of an alkanolamine compound, an alkaline amine and optionally either a primary or secondary amine in the presence of a phosphorous containing catalyst, for example phosphoric acid on silica-alumina or Group IIIB metal acid phosphate, at a temperature from about 175° C. to 400° C. under pressure. These patents indicate that the synthetic method used therein was as set forth in U.S. Pat. No. 4,463,193, to Johnson. The Ford et al. ‘247 patent is also said to be directed to color reduction of polyamines by reaction at elevated temperature and pressure in the presence of a hydrogenation catalyst and a hydrogen atmosphere. European Patent No. EP450709 to King et al. is said to be directed to a process for the preparation of triethylenetetramine and N-(2-aminoethyl)ethanolamine by condensation of an alkylenamine and an alkylene glycol in the presence of a condensation catalyst and a catalyst promoter at a temperature in excess of 260° C.
Russian Patent No. RU2186761, to Zagidullin, proposed synthesis of diethylenetriamine by reaction of dichloroethane with ethylenediamine. Ethylenediamine has previously been said to have been used in the synthesis of N-carboxylic acid esters as reported in U.S. Pat. No. 1,527,868, to Hartmann et al.
Japanese Patent No. 06065161 to Hara et al. is said to be directed to the synthesis of polyethylenepolyamines by reacting ethylenediamine with ethanolamine in the presence of silica-treated Nb205 supported on a carrier. Japanese Patent No. JP03047154 to Watanabe et al., is said to be directed to production of noncyclic polyethylenepolyamines by reaction of ammonia with monoethanolamine and ethylenediamine. Production of non-cyclic polyethylenepolyamines by reaction of ethylenediamine and monoethanolamine in the presence of hydrogen or a phosphorous-containing substance was said to be reported in Japanese Patent No. JP03048644. Regenerative preparation of linear polyethylenepolyamines using a phosphorous-bonded catalyst was proposed in European Patent No. EP115,138, to Larkin et al.
A process for preparation of alkyleneamines in the presence of a niobium catalyst was said to be provided in European Patent No. 256,516, to Tsutsumi et al. U.S. Pat. No. 4,584,405, to Vanderpool, reported the continuous synthesis of essentially noncyclic polyethylenepolyamines by reaction of monoethanolamine with ethylenediamine in the presence of an activated carbon catalyst under a pressure between about 500 to about 3000 psig., and at a temperature of between about 200° C. to about 400° C. Templeton, et al., reported on the preparation of linear polyethylenepolyamides asserted to result from reactions employing silica-alumina catalysts in European Patent No. EP150,558.
Production of triethylenetetramine dihydrochloride was said to have been reported in Kuhr et al., Czech Patent No. 197,093, via conversion of triethylenetetramine to crystalline tetrahydrochloride and subsequently to triethylenetetramine dihydrochloride. “A study of efficient preparation of triethylenetetramine dihydrochloride for the treatment of Wilson’s disease and hygroscopicity of its capsule,” Fujito, et al., Yakuzaigaku, 50:402-8 (1990), is also said to be directed to production of triethylenetetramine.
Preparation of triethylenetetramine salts used for the treatment of Wilson’s disease was said to be reported in “Treatment of Wilson’s Disease with Triethylene Tetramine Hydrochloride (Trientine),” Dubois, et al., J. Pediatric Gastro. & Nutrition, 10:77-81 (1990); “Preparation of Triethylenetetramine Dihydrochloride for the Treatment of Wilson’s Disease,” Dixon, et al., Lancet, 1(1775):853 (1972); “Determination of Triethylenetetramine in Plasma of Patients by High-Performance Liquid Chromatography,” Miyazaki, et al., Chem. Pharm. Bull., 38(4):1035-1038 (1990); “Preparation of and Clinical Experiences with Trien for the Treatment of Wilson’s Disease in Absolute Intolerance of D-penicillamine,” Harders, et al., Proc. Roy. Soc. Med., 70:10-12 (1977); “Tetramine cupruretic agents: A comparison in dogs,” Allen, et al., Am. J. Vet. Res., 48(1):28-30 (1987); and “Potentiometric and Spectroscopic Study of the Equilibria in the Aqueous Copper(II)-3,6-Diazaoctane-1,8-diamine System,” Laurie, et al., J.C.S. Dalton, 1882 (1976).
Preparation of Triethylenetetramine Salts by Reaction of Alcohol Solutions of Amines and acids was said to be reported in Polish Patent No. 105793, to Witek. Preparation of triethylenetetramine salts was also asserted in “Polycondensation of polyethylene polyamines with aliphatic dicarboxylic acids,” Witek, et al., Polimery, 20(3):118-119 (1975).
Baganz, H., and Peissker, H., Chem. Ber., 1957; 90:2944-2949; Haydock, D. B., and Mulholland, T. P. C., J. Chem. Soc., 1971; 2389-2395; and Rehse, K., et al., Arch. Pharm., 1994; 393-398, report on Strecker syntheses. Use of Boc and other protecting groups has been described. See, for example, Spicer, J. A. et al., Bioorganic & Medicinal Chemistry, 2002; 10: 19-29; Klenke, B. and Gilbert, I. H., J. Org. Chem., 2001; 66: 2480-2483.
FIG. 6 shows an 1H-NMR spectrum of a triethylenetetramine hydrochloride salt in D2O, as synthesized in Example 3. NMR values include a frequency of 400.13 Mhz, a 1H nucleus, number of transients is 16, points count of 32768, pulse sequence of zg30, and sweep width of 8278.15 H
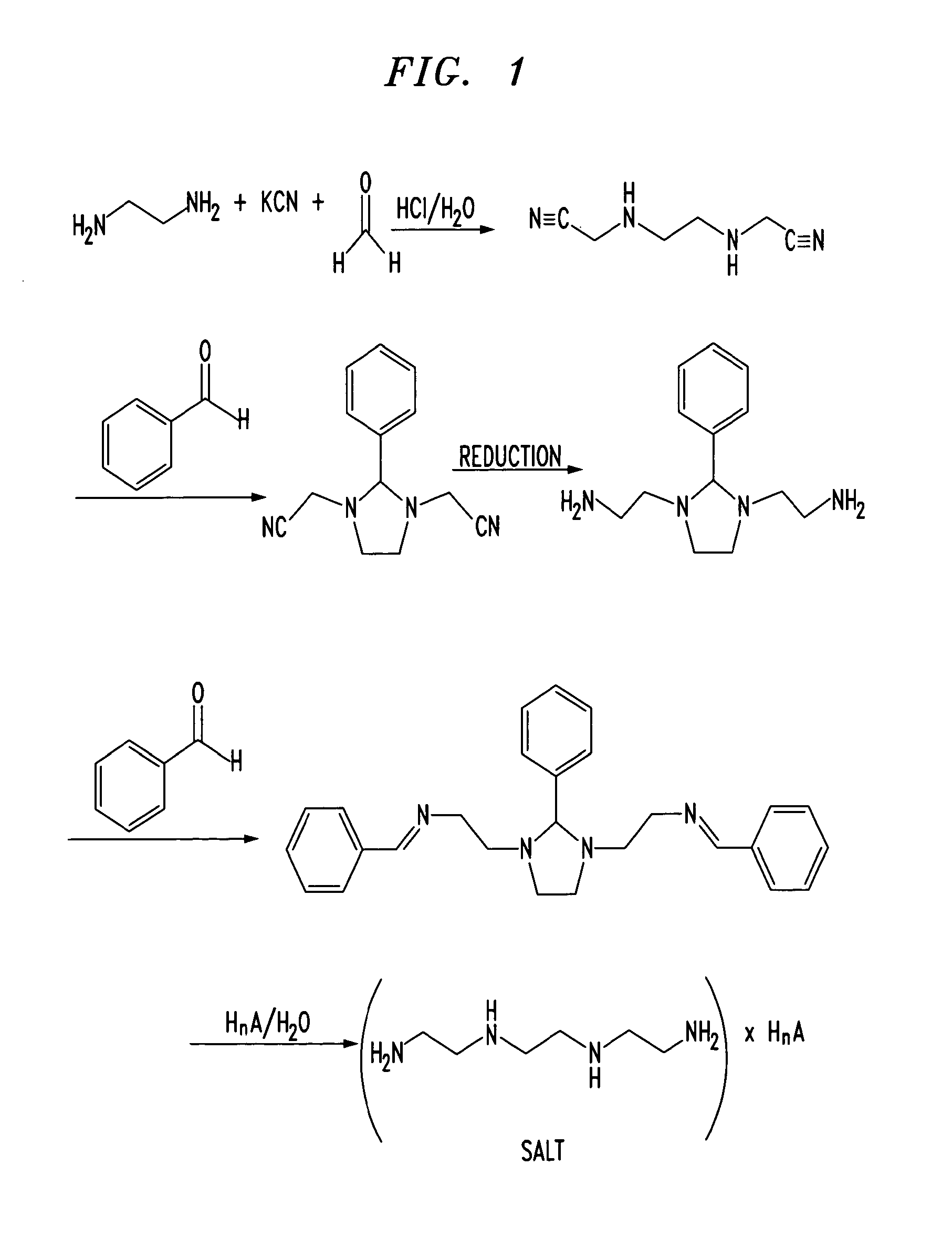
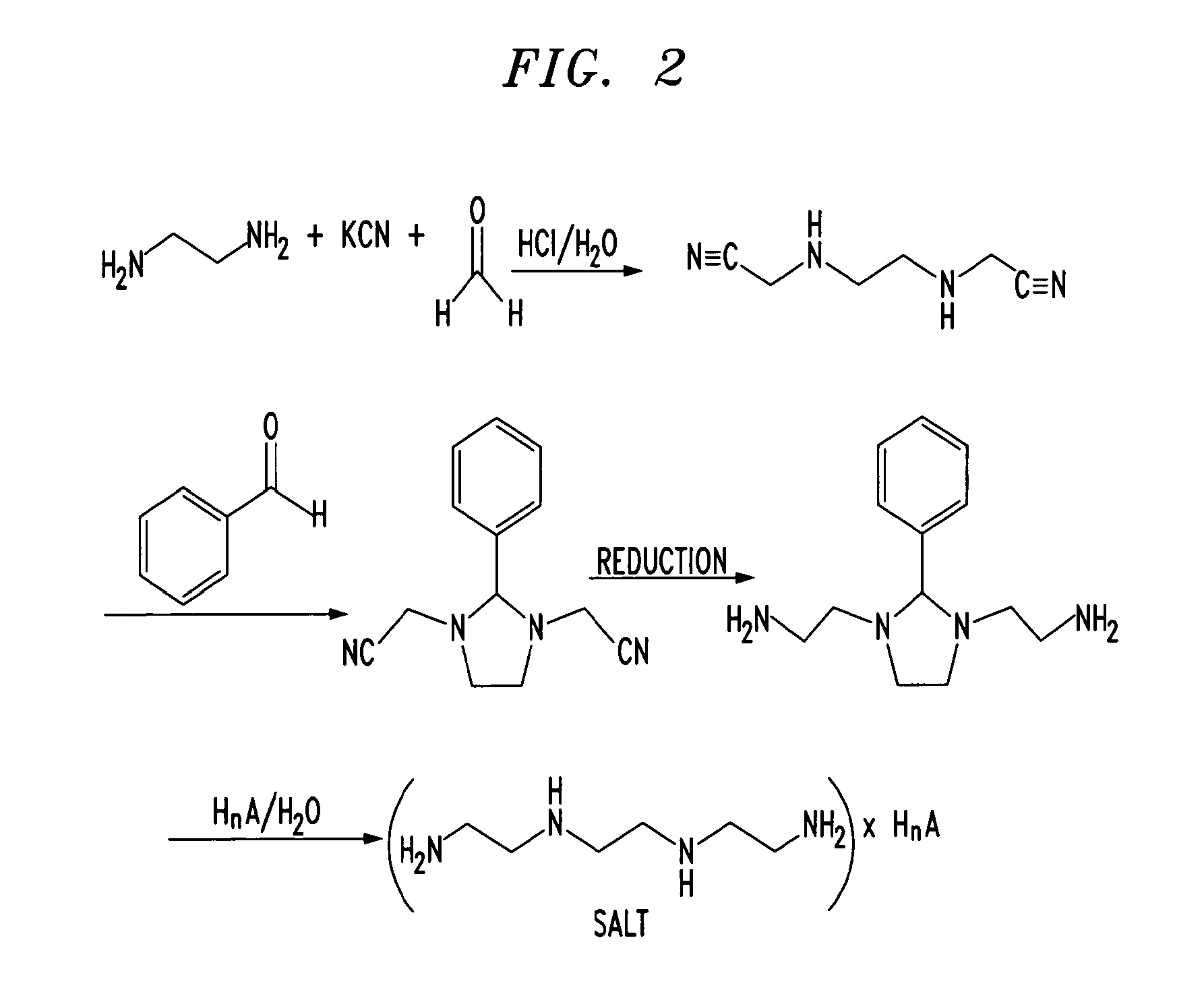

CLIP
Method of purification: Dissolve Trientine Hydrochloride in water while warming, and recrystallize by addition of ethanol (99.5). Or dissolve Trientine Hydrochloride in water while warming, allow to stand after addition of activated charcoal in a cool and dark place for one night, and filter. To the filtrate add ethanol (99.5), allow to stand in a cool and dark place, and recrystallize. Dry the crystals under reduced pressure not exceeding 0.67 kPa at 409C until ethanol odor disappears.
References
- “Ethyleneamines” (PDF). Huntsman. 2007.
- ^ Jump up to:a b Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. (2005). “Amines, Aliphatic”. Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a02_001.
- Jump up^ Roberts, E. A.; Schilsky, M. L. (2003). “A practice guideline on Wilson disease” (pdf). Hepatology. 37 (6): 1475–1492. doi:10.1053/jhep.2003.50252. PMID 12774027.
- Jump up^ von Zelewsky, A. (1995). Stereochemistry of Coordination Compounds. Chichester: John Wiley. ISBN 047195599X.
- Utsuno, S.; Sakai, Y.; Yoshikawa, Y.; Yamatera, H. (1985). “Three Isomers of the Trans-Diammine-[N,N′-bis(2-Aminoethyl)-1,2-Ethanediamine]-Cobalt(III) Complex Cation”. Inorganic Syntheses. 23: 79–82. doi:10.1002/9780470132548.ch16.
 |
|
 |
|
 |
|
| Names | |
|---|---|
| Other names
N,N’-Bis(2-aminoethyl)ethane-1,2-diamine; TETA; Trien; Trientine (INN); Syprine (brand name)
|
|
| Identifiers | |
|
3D model (Jmol)
|
|
| 605448 | |
| ChEBI | |
| ChemSpider | |
| ECHA InfoCard | 100.003.591 |
| EC Number | 203-950-6 |
| 27008 | |
| KEGG | |
| MeSH | Trientine |
|
PubChem CID
|
|
| RTECS number | YE6650000 |
| UNII | |
| UN number | 2259 |
| Properties | |
| C6H18N4 | |
| Molar mass | 146.24 g·mol−1 |
| Appearance | Colorless liquid |
| Odor | Fishy, ammoniacal |
| Density | 982 mg mL−1 |
| Melting point | −34.6 °C; −30.4 °F; 238.5 K |
| Boiling point | 266.6 °C; 511.8 °F; 539.7 K |
| Miscible | |
| log P | 1.985 |
| Vapor pressure | <1 Pa (at 20 °C) |
|
Refractive index (nD)
|
1.496 |
| Thermochemistry | |
| 376 J K−1 mol−1 (at 60 °C) | |
| Pharmacology | |
| A16AX12 (WHO) | |
| Hazards | |
| GHS pictograms |   |
| GHS signal word | DANGER |
| H312, H314, H317, H412 | |
| P273, P280, P305+351+338, P310 | |
|
EU classification (DSD)
|
|
| R-phrases | R21, R34, R43, R52/53 |
| S-phrases | (S1/2), S26, S36/37/39, S45 |
| Flash point | 129 °C (264 °F; 402 K) |
| Lethal dose or concentration (LD, LC): | |
|
LD50 (median dose)
|
|
| Related compounds | |
|
Related amines
|
|
|
Related compounds
|
|
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////////////TRIENTINE, 112-24-3, 曲恩汀 , KD-034 , MK-0681, MK-681, TECZA, TETA, TJA-250, Orphan drug
NCCNCCNCCN
Cetilistat, セチリスタット
Cetilistat, セチリスタット
- Molecular FormulaC25H39NO3
- Average mass401.582 Da
CAS 282526-98-1
2-(Hexadecyloxy)-6-methyl-4H-3,1-benzoxazin-4-one
282526-98-1 [RN]
2-Hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one
2-hexadecyl-oxy-6-methyl-4H-3,1-benzoxazin-4-one
4H-3,1-Benzoxazin-4-one, 2-(hexadecyloxy)-6-methyl
[282526-98-1]
2-(Hexadecycloxy)-6-methyl-4H-3,1-benzoxazin-4-one
ATL-962; ATL962;ATL 962
Trade Name:Oblean®
MOA:Pancreatic lipase inhibitor
Indication:Obesity
Status:Approved, 2013-09-20 JAPAN, Japan PMDA.
Company:Norgine (Originator) , Takeda

UPDATE 09.07.2021 INDIA CDSCO For the treatment of Obseity (limited to
patients with both type 2 diabetes mellitus
and dyslipidaemia, and with a BMI ≥ 25
kg/m2 inspite of dietary treatment and /or
excersise therapy)…………Cetilistat bulk and Cetilistat 120 mg tablets
patients with both type 2 diabetes mellitus
and dyslipidaemia, and with a BMI ≥ 25
kg/m2 inspite of dietary treatment and /or
excersise therapy)…………Cetilistat bulk and Cetilistat 120 mg tablets
Cetilistat was approved by Pharmaceuticals Medical Devices Agency of Japan (PMDA) on September 20, 2013. It was developed by Norgine and Takeda, then marketed as Oblean® by Takeda in Japan.
Cetilistat is a pancreatic lipase inhibitor, and it acts in the same way as the older drug orlistat (Xenical) by inhibiting pancreatic lipase, an enzyme that breaks down triglycerides in the intestine. Without this enzyme, triglycerides from the diet are prevented from being hydrolyzed into absorbable free fatty acids and are excreted undigested. It is usually used for the treatment of obesity (limited to patients with both type 2 diabetes mellitus and dyslipidemia, and with a BMI≥25 kg/m2 in spite of dietary treatment and/or exercise therapy).
Oblean® is available as tablet for oral use, containing 120 mg of free Cetilistat. The recommended dose is 120 mg three times a day immediately after each meal.
Cetilistat is a drug designed to treat obesity. It acts in the same way as the older drug orlistat (Xenical) by inhibitingpancreatic lipase, an enzyme that breaks down triglycerides in the intestine. Without this enzyme, triglycerides from the diet are prevented from being hydrolyzed into absorbable free fatty acids and are excreted undigested.[1]
In human trials, cetilistat was shown to produce similar weight loss to orlistat, but also produced similar side effects such as oily, loose stools, fecal incontinence, frequent bowel movements, and flatulence.[2][3] It is likely that the same precautions would apply in that absorption of fat-soluble vitamins and other fat-soluble nutrients may be inhibited, requiring vitamin supplements to be used to avoid deficiencies.
Central obesity have an important impact on the development of risk factors for coronary heart disease, including dislipidemia, glucose intolerance, insulin resistance and hypertension. These factors contribute to building cardiovascular (CV) disease as a major cause of death. The approach to obesity therapy should be designed to reduce CV risk and mortality. Diet and lifestyle changes remain the cornerstones of therapy for obesity, but the resultant weight loss is often small and long-term success is uncommon and disappointing. Drug therapy is considered for individuals with a body mass index greater than 30 kg/m2 or ranging from 25 to 30 kg/m2 if they have comorbid conditions. Antiobesity agents can be helpful to some patients in achieving and maintaining meaningful weight loss, but yet our pharmaceutical tools are of limited effectiveness considering the magnitude of the problem. At the present, only two drugs, orlistat and sibutramine, are approved for long-term treatment of obesity and promote no more than 5 to 10% of weight loss.
Rimonabant, a cannabinoid-1 receptor antagonist, was withdrawn from the market because of concerns about its safety, including risk of suicidal and seizures, although very effective in promoting clinically meaningful weight loss, reduction in waist circumference, and improvements in several metabolic risk factors, rimonabant, a cannabinoid-1 receptor antagonist was withdrawn from the market because it concerns about its safety, including risk of suicidal and seizures. Fortunately, recent fundamental insights into the neuroendocrine mechanisms regulating body weight provide an expanding list of molecular targets for novel, rationally designed antiobesity drugs. In this review, the therapeutic potential of some antiobesity molecules in the development will be analyzed based on an understanding of energy homeostasis.


Cetilistat has completed Phase 1 and 2 trials in the West and is currently in Phase 3 trials in Japan where it is partnered with Takeda.[4] Norgina BV has now acquired the full global rights to cetilistat from Alizyme after the latter went into administration.[5]
A published phase 2 trial found cetilistat significantly reduced weight with and was better tolerated than orlistat.[6
CLIP
Cetilistat (Oblean®)
Cetilistat is a selective pancreatic lipase inhibitor which was approved in Japan in September 2013
for the treatment of obesity. The drug was discovered by Alizyme PLC and later co-developed with
Takeda. Cetilistat demonstrated a lower incidence of adverse gastrointestinal events during a 12 week clinical trial, and the degree of weight loss associated with cetilistat is comparable to that of other approved antiobesity therapies.30 The most likely process-scale preparation of cetilistat is described below in Scheme. 4.31
Commercially available hexadecanol (21) was treated with phosgene in THF/toluene to give the
corresponding chloroformate (22), which was immediately subjected to commercial 2-amino-5-
methylbenzoic acid (23) in pyridine. Subsequent slow addition of methyl chloroformate at room
temperature resulted in the formation of cetilistat (IV), which was produced in 31% overall yield from
hexadecanol.31
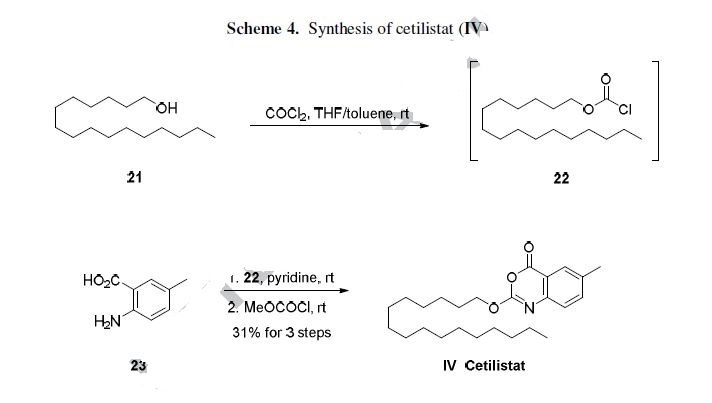
REF FOR ABOVE ONLY
30 Kopelman, P.; Groot, G. d. H.; Rissanen, A.; Rossner, S.; Toubro, S.; Palmer, R.; Hallam, R.;
Bryson, A.; Hickling, R. I. Obesity 2010, 18, 108.
31. Hodson, H. F.; Downham, R.; Mitchell, T. J.; Carr, B. J.; Dunk, C. R.; Palmer, R. M. J. US
Patent 20030027821A1, 2003.
SYNTHESIS
Route 1
WO2000040569
AND
http://www.google.co.in/patents/US6624161

WO0040569A1 / US6656934B2.2. WO0040247A1 / US6624161B2.
Route 2
http://www.google.co.in/patents/US7396952
Carbamic ester derivatives of the general formula (1) and especially (2-carboxy-4-methylphenyl)carbamic esters of the general formula (1′)

are suitable intermediates for active pharmaceutical ingredients.
Thus, for example, hexadecyl (2-carboxy-4-methylphenyl)carbamate as compound of the formula (1′) with R═C16H33 is disclosed as an intermediate in the preparation of 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one of the formula (3)

from the originally published version of WO-A 00/40569.
2-Hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one of the formula (3) is described therein as potential active ingredient for the treatment of obesity and type II diabetes.
In this originally published version of WO-A 00/40569, two synthetic routes 1 and 2 are described for preparing 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one (3), each of which starts from the 5-methyl-substituted anthranilic acid (4).
In the two-stage synthetic route 1, the 5-methyl-substituted anthranilic acid (4) is reacted with hexadecyl chloroformate (5) and subsequently with methyl chloroformate to give 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one (3), although the overall yield obtained is only 31%.
The one-stage synthetic route 2 with an excess of pyridine affords 2-hexadecyl-oxy-6-methyl-4H-3,1-benzoxazin-4-one (3) in an even lower yield of 15%.

The starting compound which is required for both the synthetic routes 1 and 2, the 5-methyl-substituted anthranilic acid (4), is not easily obtainable, however.
It is prepared by the method described in J. Org. Chem. 1952, 17, 141. This starts from p-toluidine, which is reacted with chloral hydrate and hydroxylamine hydrochloride. The resulting oxime is cyclized with acid catalysis, and subsequently the ring is cleaved again by oxidation under basic conditions.

The disadvantages of this synthesis are the low yields and the fact that only very low concentrations can be used. For this reason, this synthetic route is unattractive for an industrial reaction.
Further alternative routes known in principle for obtaining anthranilic acids are as follows:
J. Org. Chem. 1978, 43, 220 and Chem. Ber. 1909, 42, 430 disclose initial nitration of 3-cyanotoluene, then reduction of the nitro group and subsequent hydrolysis of the nitrile to the carboxylic acid.

A disadvantage of this synthesis is that the nitration of 3-cyanotoluene does not proceed selectively and therefore a further purification step is necessary. This requires additional effort and reduces the yield.
The synthesis which is described in J. Chem. Soc. Perkin I, 1973, 2940 and which starts from 3-toluic acid with subsequent nitration and reduction of the nitro group also has the same disadvantage.
The synthesis which is disclosed in Monatsh. Chem. 1920, 41, 155 and starts from 2,4-dimethyl-1-nitrobenzene is likewise unsuitable because oxidation of the methyl group next to the nitro group does not proceed selectively and therefore an elaborate separation of isomers is necessary.

EP-A 0 034 292 discloses a process for preparing optionally substituted anthranilic acids which includes a transition metal-catalysed carbonylation reaction with carbon monoxide to give an anthranilic acid derivative. This carbonylation reaction takes place in an aqueous reaction medium containing a trialkylamine and a catalyst formed from palladium and a tertiary phosphine. The anthranilic acid derivatives can be obtained by eliminating the protective group. The precursors employed for the carbonylation are obtained starting from optionally substituted anilines as shown in principle in the reaction scheme below:

EP-A 0 034 292 describes this reaction sequence of acetylation (a), halogenation (b), carbonylation (c) and subsequent elimination of the acetyl group (d) as affording the optionally substituted anthranilic acids in good yields (>80%). However, the introduction of the acetyl group is a disadvantage. This is necessary because the free anilines give only poor yields in transition metal-catalysed carbonylation reactions because of pronounced complexation [J. Org. Chem. 1981, 46, 4614-4617].
WO-A 97/28118 discloses a comparable process.
Because of the diverse difficulties, described above, associated with the known processes for preparing optionally substituted anthranilic acids and the yields, which are only unsatisfactory and thus limiting for the overall process, of the subsequent synthetic routes 1 and 2, the object of the present invention was to provide an improved process for preparing carbamic ester derivatives of the general formula (1).

US7396952B2.
EXAMPLES Example 1 Synthesis of hexadecyl 4-methylphenylcarbamate

91 g (375 mmol) of 1-hexadecanol were added to a solution of 50 g (375 mmol) of p-tolyl isocyanate in 50 ml of toluene, and the resulting solution was heated under reflux for 8 h. After cooling to room temperature and stirring at this temperature for 12 h, the precipitated solid was filtered off. The colourless solid was washed twice with 10 ml of toluene each time and then dried in vacuo. 80 g (213 mmol, 57%) of the desired carbamate were obtained in the form of a colourless solid with a melting point of 75° C. The melting point agreed with literature data (75-76° C., Microchem J. 1962, 6, 179).
1H-NMR (CDCl3, 400 MHz): δ=0.88 ppm (t, J=7.3 Hz, 3H), 1.25-1.40 (m, 26 H), 1.66 (sext, J=6.9 Hz, 2H), 2.30 (s, 3H), 4.14 (t, J=6.9 Hz, 2H), 6.53 (br, 1 H), 7.10 (d, J=7.8 Hz, 2H), 7.25 (d, J=8.3 Hz, 2H). Elemental Analysis Showed: Calculated: C 76.8%, H 11.0%, N 3.7% Found: C 76.9%, H 11.2%, N 3.7%.
Example 2 Synthesis of hexadecyl (2-bromo-4-methylphenyl)carbamate

19 g (119 mmol) of bromine were added dropwise to a solution of 45 g (119 mmol) of the carbamate in 225 ml (235 g) of glacial acetic acid at room temperature over the course of 1 h, and then the resulting solution was stirred at room temperature for 1 h. After addition of a further 25 ml (26 g, 437 mmol) of glacial acetic acid, the reaction mixture was stirred at 40° C. for 5 h and then cooled to room temperature. The precipitated solid was filtered off and washed with 20 ml of glacial acetic acid. Drying in vacuo resulted in 40 g (88 mmol, 74%) of the desired bromo compound in the form of a colourless solid with a melting point of 57° C.
1H-NMR (CDCl3, 400 MHz): δ=0.93 ppm (t, J=6.6 Hz, 3H), 1.25-1.43 (m, 26 H), 1.73 (sext, J=6.8 Hz, 2H), 2.33 (s, 3H), 4.21 (t, J=6.7 Hz, 2H), 7.04 (br, 1H), 7.14 (d, J=8.4 Hz, 1H), 7.37 (s, 1H), 8.02 (d, J=8.3 Hz, 1H). 13C-NMR (CDCl3, 100 MHz): δ=14.2 ppm, 20.4, 22.7. 25.9, 29.0, 29.3, 29.4, 29.6 (2C), 29.7 (2C), 29.8 (4C), 32.0, 65.7, 112.5, 120.3, 129.0, 132.5, 133.5, 134.1, 153.5. Elemental Analysis Showed: Calculated: C 63.4%, H 8.9%, N 3.1% Found: C 63.6%, H 8.9%, N 3.1%.
Example 3 Synthesis of 2-hexadecyloxycarbonylamino-5-methylbenzoic acid

217.5 g (478.5 mmol) of hexadecyl (2-bromo-4-methylphenyl)carbamate, 0.5 g (0.7 mmol) of bis(triphenylphosphine)palladium dichloride and 2.5 g (9.3 mmol) of triphenylphosphine were introduced into an autoclave. The autoclave was closed, flushed with nitrogen and an oxygen-free solution of 78.1 g (565.3 mmol) of potassium carbonate in 400 ml of water is added. The autoclave is evacuated and then 2 bar of carbon monoxide are injected and heated to 115° C. The pressure is subsequently adjusted to 8 bar. After CO uptake ceases, the mixture is cooled to RT and 200 ml of toluene are added. The pH is adjusted to 2 with 2M aqueous HCl solution, and the organic phase is separated off. The aqueous phase is extracted anew with 100 ml of toluene, the organic phase is separated off, and the two toluene extracts are combined. Removal of the solvent in vacuo results in 154.9 g (369.2 mmol, 77%) of 2-hexadecyloxycarbonylamino-5-methylbenzoic acid in the form of a pale yellow-coloured solid.
1H-NMR (CDCl3, 400 MHz): δ=0.88 ppm (t, J=6.7 Hz, 3H), 1.24-1.40 (m, 26 H), 1.70 (sext, J=6.8 Hz, 2H), 2.33 (s, 3H), 4.17 (t, J=6.8 Hz, 2H), 7.38 (d, J=8.7 Hz, 1H), 7.90 (s, 1H), 8.35 (d, J=8.6 Hz, 1H). Signal of the NH proton not identifiable.13C-NMR (CDCl3, 100 MHz): δ=14.1 ppm, 20.5, 22.7. 25.9, 29.0, 29.3, 29.4, 29.6 (2 C), 29.7 (6 C), 32.0, 65.5, 113.6, 119.0, 131.1, 131.8, 136.3, 140.1, 153.9, 172.5.
Example 4 Synthesis of 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one

4.0 g (10.0 mmol) of 2-hexadecyloxycarbonylamino-5-methylbenzoic acid are introduced into 20 ml of pyridine at 0° C. under a nitrogen atmosphere, and 4.93 g (45.4 mmol) of ethyl chloroformate are added dropwise to the resulting solution at 0° C. over the course of 20 min. After the reaction mixture has been stirred at 0° C. for 1 h and at room temperature for 2 h it is added to 30 ml of ice-water. The solid is filtered off and dried in vacuo. 3.3 g (8.2 mmol, 82%) of 2-hexadecyloxy-6-methyl-4H-3,1-benzoxazin-4-one are obtained in the form of a pale yellow coloured solid with a melting point of 67° C. (literature: 72-73° C., WO 00/40569).
1H-NMR (CDCl3, 400 MHz): δ=0.86 ppm (t, J=6.6 Hz, 3H), 1.24-1.42 (m, 26 H), 1.75-1.82 (m, 2H), 2.40 (s, 3H), 4.41 (t, J=6.8 Hz, 2H), 7.30 (d, J=8.3 Hz, 1H), 7.51 (dd, J=8.2, 1.9 Hz, 1H), 7.90 (d, J=0.9 Hz, 1H).
The 1H-NMR data agree with the literature data from WO-A 00/40569.

Patent
https://www.google.com/patents/CN104341370A?cl=en
cetirizine orlistat (2-methyl-6-firing sixteen -4H-3, 1- benzo ah winded -4- Korea, cetilistat) is a long-acting Alizyme developed and potent specific gastrointestinal lipase inhibitor, with the active serine site of the gastric and intestinal lumen gastric lipase and lipase membrane forms a covalent bond to inactivate the enzyme, and to reduce calorie intake, weight control therapeutic effect. The biggest advantage of the drug is not acting on the nervous system, does not affect other activity in the gastrointestinal tract, it is more secure than existing similar drugs orlistat. Its structural formula is as follows:

West Division for the benefit of his synthesis and intermediates have been described in U.S. Patent US2007232825 and US2003027821, domestic literature orlistat no cetirizine synthesis of relevant reports.
U.S. Patent US2007232825 2-amino-5-methyl-benzoic acid starting material, direct and vilify chloroformate cetyl alcohol vinegar into the ring, get cetirizine orlistat. The reaction byproducts and more difficult W purification needs over baby gel column, resulting in a low yield, suitable for mass industrialization. Directions are as follows:

Patent US2003027821 W toluene different acid vinegar as raw material to produce amino acid vinegar intermediate chloroformate, cetyl alcohol and vinegar reaction, after the desert generation essays glycosylation chloroformate caprolactone ring closure to give cetirizine orlistat. This method requires a great deal of glacial acetic acid, the presence of H waste discharge more harsh reaction conditions, equipment requirements, is not conducive to industrial production and other defects.

The present invention is a W under the technical program realization:






The following combination of embodiments of the present invention will be further described below.
(Sixteen essays firing oxygen-amino) -5- Preparation of 2-methyl benzoate desert vinegar; [0041] Example 1

4. 9g H phosgene will be added to 50 blood dichloromethane firing, the temperature was lowered to OC, a solution of 2-amino-5 Desert benzoic acid methyl ester (5g) and H hexylamine (13.8 blood) dichloro A firing (20 blood) solution, the addition was complete OC to maintain 15min, warmed to room temperature the reaction mix of football.

[0042] The 5. 26g cetyl alcohol was added to the reaction solution at room temperature the reaction of. After completion of the reaction, filtered and the filtrate was concentrated in vacuo spin dry, dry methanol residue fight starched coating, filtration, the filter cake is dried to constant weight. To give a white solid powder 9. Ig, namely 2- (sixteen essays firing oxygen-ylamino) -5-benzoic acid methyl ester desert; Yield; 85%.
2- (grilled oxygen sixteen essays) -5-methyl-benzoic acid methyl ester prepared; [0043] Example 2

Under nitrogen blanket IOg 2- (sixteen grilled oxygen essays) -5- desert benzoic acid methyl ester was dissolved in 1,4-dioxane (50mL) and water Qiao blood), and Ilg anhydrous carbonate Bell, 1.44g methacrylic acid test, 0. 731g Pd (dppf) 2Cl2, the mixture at 105C for 3 hours. Completion of the reaction, cool down, filtered and the filtrate spin dry, the residue of anhydrous methanol wash coating, the filter cake dried to give a gray solid 6. 5g, is 2- (xvi grilled oxygen essays) -5-methyl benzoic acid methyl ester in 75% yield.
2- (grilled oxygen sixteen essays) -5-methyl-benzoic acid; [0044] Example 3

The 7g 2- (sixteen grilled oxygen essays) -5-methyl-benzoic acid methyl ester was added to 35mL tetraammine clever furans and 7mL water mixture, adding ammonia oxidation in 20. Ig, 6 (TC reaction of the reaction is completed, the reaction mixture was concentrated, the residue was added 70mL of ice water, 6M hydrochloric suppression of 7, the filter cake was dried to constant weight to give a gray solid 6. 2g, namely 2- (sixteen firing oxygen-ylamino essays ) -5-methyl-benzoic acid, yield 92%.
Preparation of 2-methyl-6-firing sixteen -4H-3, 1- benzo Lai ah winded -4- (cetirizine Division him); 4 [0045] Example

The 66g 2- (XVI essays firing oxo-ylamino) -5-methylbenzoic acid in 330mL of information coincidence floating in an ice bath, was slowly added dropwise 45mL chloroformate caprolactone, after the addition was complete, naturally rise to room temperature The reaction of. After completion of the reaction, the reaction solution was poured into 700mL ice water, filtered, and the filter cake was dried to constant weight to give a gray solid 56g, that is, sixteen firing-6-methyl-2- -4H-3, 1- benzo Lai ah winded -4- (cetirizine orlistat), a yield of 85%. Mass spectrum shown in Figure 2, ESI-MS〇b / z): 402 [M + Tin +; X- ray diffraction as shown in (3 consistent with the data reported in FIG patent US2012101090), analyzed as shown in Table 1, Figure 1 FIG. 2 W and W Table 1 confirm that the product was obtained as cetirizine orlistat.
[0046] Table 1






CLIP

| Synthesis of Cetilistat |
| 1. Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050; 2. Beijing Union Pharmaceutical Factory, Beijing 102600 |
Cetilistat was synthesized from 2-amino-5-methylbenzoic acid and cetyl chloroformate via acylation to give 2-[[(hexadecyloxy)carbonyl]amino]-5-methylbenzoic acid, which was subjected to intramolecular dehydrationcyclization in the presence of POCl3 with an overall yield of 90% and purity over 99%. This one-pot method was simple and suitable for large-scale application.
CLIP
http://amogsobgy.com/downloads/AkumentisChechwt/CurrOpinInvestigDrugs.pdf

 CLICK ON IMAGE
CLICK ON IMAGE
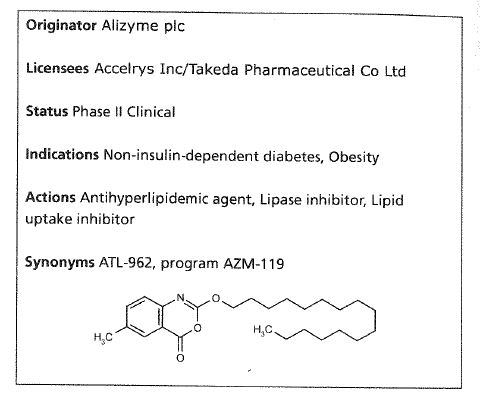
Cetilistat, a new lipase inhibitor for the treatment of obesity – AMOGS
amogsobgy.com/downloads/AkumentisChechwt/CurrOpinInvestigDrugs.pdf
by R Padwal – Cited by 26 – Related articles
clinical trials, and the above-mentioned lipase inhibitor cetilistat, which is the focus of this review.Synthesis and SAR. Cetilistat (2-hexadecyloxy-6-methyl-4H-3 …
PATENT
SEE
http://www.google.co.in/patents/WO1999016758A1?cl=en
CLIP
Taken from Ayurajan

https://ayurajan.blogspot.in/2016_01_01_archive.html
Cetilistat | Inhibitor of Gastrointestinal Lipases | Inhibitor of Pancreatic Lipases | Anti-Obesity Drug
Cetilistat [2-(Hexadecyloxy)-6-methyl-4H-3,1-benzoxazin-4-one] is a novel highly lipophilic benzoxazinone that inhibits gastrointestinal (GI) and pancreatic lipases, and is chemically distinct from Orlistat [1].
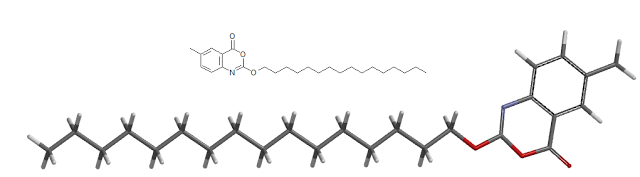 |
| Cetilistat: 2D and 3D Structure |
Pancreatic lipase is the enzyme that breaks down triglycerides in the intestine. Inhibition of this enzyme ensures that triglycerides from the diet are prevented from being hydrolyzed into absorbable free fatty acids and are excreted undigested.
In Phase I clinical trials in healthy volunteers, Cetilistat increased faecal fat excretion and was well tolerated. Cetilistat produced a clinically and statistically significant weight loss in obese patients in this short-term 12-week study. This was accompanied by significant improvements in other obesity-related parameters. Cetilistat treatment was well tolerated. The risk-benefit demonstrated in this study in terms of weight loss vs intolerable GI adverse effects shows that Cetilistat merits further evaluation for the pharmacotherapy of obesity and related disorders.
The NDA submission is based on the results of three Phase 3 clinical trials in obese patients with type 2 diabetes and dyslipidemia: a 52-week placebo-controlled, double-blind study to evaluate the efficacy and safety, and two open-label studies to evaluate safety, 24-week and 52-week respectively. The results of the 52-week placebo-controlled, double-blind study demonstrate that Cetilistat 120mg three times daily is superior to placebo in the primary endpoint, with a mean reduction in body weight from baseline of -2.776% with Cetilistat versus -1.103% with placebo (p=0.0020). Greater reduction in HbA1c and low-density lipoprotein cholesterol were also observed in patients treated with Cetilistat, compared to placebo. In all these three studies, Cetilistat showed a good safety profile and was well tolerated.
Cetilistat was approved in Japan in September 2013 for the treatment of obesity. Cetilistat (Tradename: Oblean) is approved for a dosage of 120 mg three times a day for the treatment of obesity with complications.
The drug was discovered by UK based Alizyme PLC and in 2003 Takeda acquired the rights for development and commercialisation for Japan. Norgine acquired all rights to the product from Alizyme in October 2009 [3].
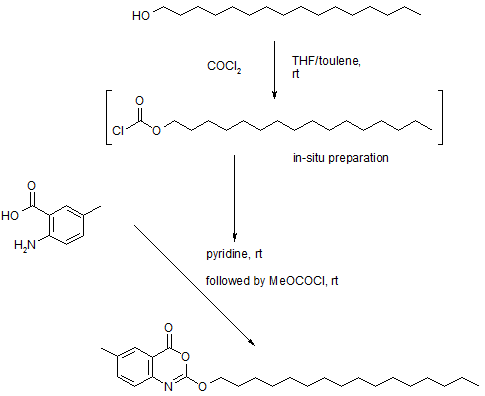
Cetilistat Synthesis
US20030027821A1: It appears to be the industrial process. The yields are in the range of 30-35%.
Identification:
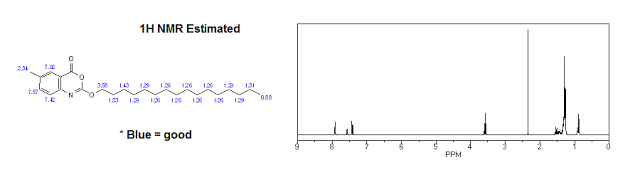 |
| 1H NMR (Estimated) for Cetilistat |
Experimental: 1H-NMR δH (400 MHz, CDCl3) 0.87 (3H, t, J 6.8, CH2CH3), 1.24-1.45 (26H, m, 13×CH2), 1.75-1.83 (2H, m, OCH2CH2), 2.41 (3H, s, ArCH3), 4.41 (2H, t, J 6.7, OCH2), 7.3 (1H, d, J 8.3, ArH), 7.51 (1H, dd, J 8.5, 2.0, ArH), 7.90 (1H, d, J 1.1, ArH); m/z (ES+) 402 (MH+); M Pt. 72-73° C.
Sideeffects: The most frequently experienced adverse events were those involving the gastrointestinal (GI) tract. The proportion of patients and the total number of GI adverse events reported in each of the active treatment groups were higher compared to the placebo group. However, GI adverse events were predominantly mild to moderate in intensity, with no evidence of a dose relationship.
The most frequently reported GI-related adverse events included increased defecation, soft stools, abdominal pain, flatulence and fatty/oily stool, which were all reported more frequently in the treatment arms compared to the placebo arm.
Faecal incontinence, flatus with discharge, oily evacuation and oily spotting occurred in only 1.8-2.8% of subjects in the active treatment arms and was not dose-related. Adverse events generally occurred on only one occasion and resolved rapidly.
Serum vitamin D, vitamin E and β-carotene levels were decreased significantly in the Cetilistat treatment arms. Generally, these reductions in vitamin levels did not take the levels outside the normal range and none required the use of vitamin supplements.
References FOR ABOVE ONLY
- Kopelman, P.; et. al. Cetilistat (ATL-962), a novel lipase inhibitor: a 12-week randomized, placebo-controlled study of weight reduction in obese patients. Int J Obes (Lond) 2007, 31(3), 494-499.
- Hodson, H.; et. al. 2-Oxy-benzoxazinone derivatives for the treatment of obesity.US20030027821A1
- Cetilistat Approval (here).

| CN1359378A * | Jan 6, 2000 | Jul 17, 2002 | 阿利茨默治疗学有限公司 | 2-oxy-benzoxazine derivatives for the treatment of obesity |
| CN1785967A * | Dec 12, 2005 | Jun 14, 2006 | 兰爱克谢斯德国有限责任公司 | Process for the preparation of carbamic acid derivatives |
| CN103936687A * | Mar 24, 2014 | Jul 23, 2014 | 重庆东得医药科技有限公司 | Method for preparing cetilistat |
| WO2013166037A1 * | Apr 30, 2013 | Nov 7, 2013 | The Trustees Of Columbia University In The City Of New York | Non-retinoid antagonists for treatment of eye disorders |
PATENT
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20030013707 | 6 Jul 2001 | 16 Jan 2003 | Hodson Harold Francis | 2-amino-benzoxazinone derivatives for the treatment of obesity |
| EP0034292A2 | 31 Jan 1981 | 26 Aug 1981 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Process for the preparation of anthranilic acid derivatives |
| WO1997028118A1 | 30 Jan 1997 | 7 Aug 1997 | Hoechst Celanese Corporation | Process for preparing anthranilic acids |
| Reference | ||
|---|---|---|
| 1 | Chem. Ber. 1909, 42, 430. | |
| 2 | J. Chem. Soc. Perkin I, 1973, 2940; Peter H. Gore et al. Friedel-Crafts Reactions, Part XXV.<SUP>1 </SUP>Acetylation and Benzoylation of Iodobenzene and of o-, m-, and p- Iodotoluenes. | |
| 3 | J. Org. Chem. 1952, 17, 141 B. R. Baker et al.; “An Antimalarial Alkaloid From Hydrangea, XIV, Synthesis of 5- ,6-,7-, and 8-Monosubstituted Derivatives“. | |
| 4 | J. Org. Chem. 1978, vol. 43, No. 2, 220 T.H. Fisher et al.; “Kinetic Study of the N-Bromosuccin-imide Bromination of Some 4-Substituted 3-Cyanotoluenes“. | |
| 5 | J. Org. Chem. 1981, 46, 4614-4617 Donald Valentine, Jr. et al; “Practical, Catalytic Synthesis of Anthranilic Acids“. | |
| 6 | Monatsch. Chem. 1920, 41, 155. | |
| 7 | Thomas G. Back et al.: “Conjugate Additions of o-Iodoanilines and Methyl Anthranilates to Acetylenic Sulfones. A New Route to Quinolones Including First Syntheses of Two Alkaloids from the Medical Herb Ruta chalepensis” Journal of Organic Chemistry., Bd. 68, 2003, Seiten 2223-2233, XP002371555 USAmerican Chemical Society, Easton. Seite 2227, Spalte 1, Reaktionsschema 4 und Spalte 2, Zeile 8-Zeile 9; Seite 2231, Spalte 2, Zeile 43-Zeile 54. | |
| 8 | * | Yadav et al., New Journal of Chemistry (2000), 24(8), 571-573. |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8883780 | 22 Apr 2010 | 11 Nov 2014 | Norgine B.V. | Crystal of a benzoxazinone compound |
References
- Yamada Y, Kato T, Ogino H, Ashina S, Kato K (2008). “Cetilistat (ATL-962), a novel pancreatic lipase inhibitor, ameliorates body weight gain and improves lipid profiles in rats”. Hormone and Metabolic Research. 40 (8): 539–43. doi:10.1055/s-2008-1076699. PMID 18500680.
- Kopelman, P; Bryson, A; Hickling, R; Rissanen, A; Rossner, S; Toubro, S; Valensi, P (2007). “Cetilistat (ATL-962), a novel lipase inhibitor: A 12-week randomized, placebo-controlled study of weight reduction in obese patients”. International journal of obesity (2005). 31 (3): 494–9. doi:10.1038/sj.ijo.0803446. PMID 16953261.
- Padwal, R (2008). “Cetilistat, a new lipase inhibitor for the treatment of obesity”. Current opinion in investigational drugs (London, England : 2000). 9 (4): 414–21. PMID 18393108.
- http://www.alizyme.com/alizyme/products/cetilistat/ Archived January 7, 2009, at the Wayback Machine.
- Norgine acquires cetilistat
- “Weight loss, HbA1c reduction, and tolerability of cetilistat in a randomized, placebo-controlled phase 2 trial in obese diabetics: comparison with orlistat (Xenical).”. Obesity. 18: 108–15. Jan 2010. doi:10.1038/oby.2009.155. PMID 19461584.
- Japan PMDA.
セチリスタット
Cetilistat

C25H39NO3 : 401.58
[282526-98-1]
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
2-(Hexadecyloxy)-6-methyl-4H-3,1-benzoxazin-4-one
|
|
| Identifiers | |
| CAS Number | 282526-98-1 |
| ATC code | none |
| PubChem | CID 9952916 |
| ChemSpider | 8128526 |
| UNII | LC5G1JUA39 |
| KEGG | D09208 |
| ChEMBL | CHEMBL2103825 |
| Chemical data | |
| Formula | C25H39NO3 |
| Molar mass | 401.582 g/mol |
///////////////Cetilistat, ATL-962, ATL962, ATL 962, 2013-09-20, JAPAN, APPROVED, Japan PMDA, 282526-98-1, セチリスタット
 SEE
SEE
Annual Reports in Medicinal Chemistry
2014 – Science
… versus vehicle-treated mice.34Noteworthy in the multistep synthesis of canagliflozin is …CETILISTAT (ANTIOBESITY)43–52 Class: Pancreatic lipase inhibitor …
Ponesimod

Ponesimod
Phase III
MW 460.97, C23 H25 Cl N2 O4 S
A sphingosine-1-phosphate receptor 1 (S1P1) agonist potentially for the treatment of multiple sclerosis.
(Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one
- (2Z,5Z)-5-[[3-Chloro-4-[(2R)-2,3-dihydroxypropoxy]phenyl]methylene]-3-(2-methylphenyl)-2-(propylimino)-4-thiazolidinone
- 5-[3-Chloro-4-[((2R)-2,3-dihydroxypropyl)oxy]benz-(Z)-ylidene]-2-((Z)-propylimino)-3-(o-tolyl)thiazolidin-4-one
- ACT 128800
ACT-128800; RG-3477; R-3477
CAS No. 854107-55-4
update 18/3/21 FDA APPROVEDAS PONVORY
SYNTHESIS
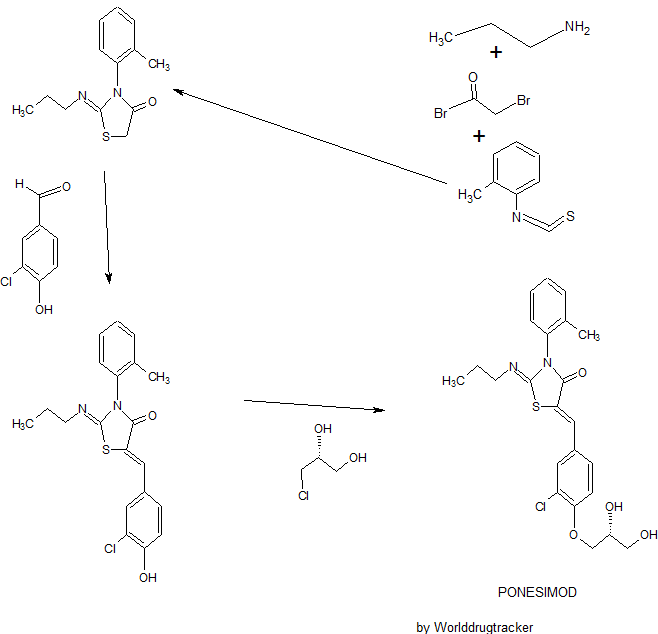
Ponesimod
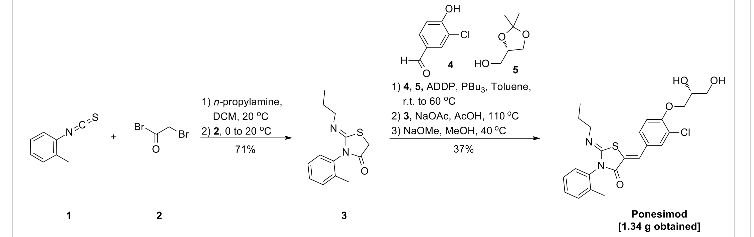

NMR CDCL3 FROM NET
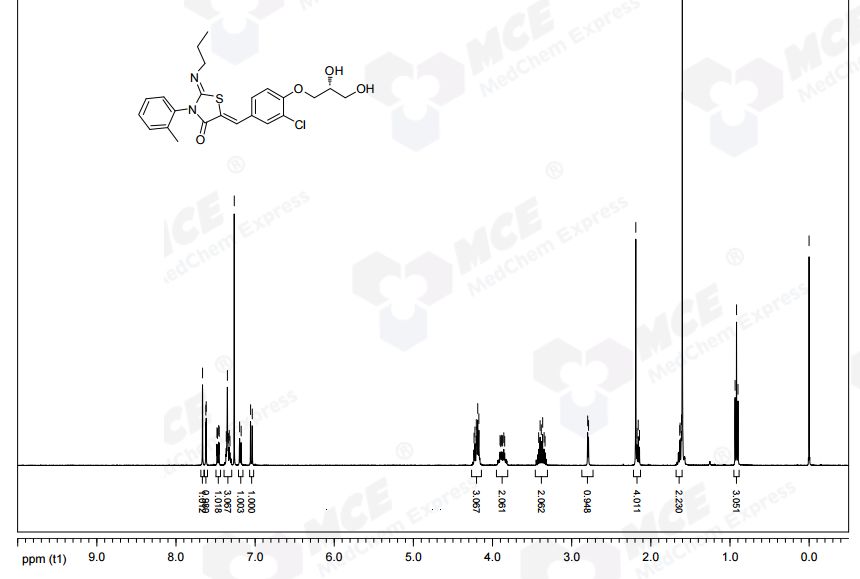
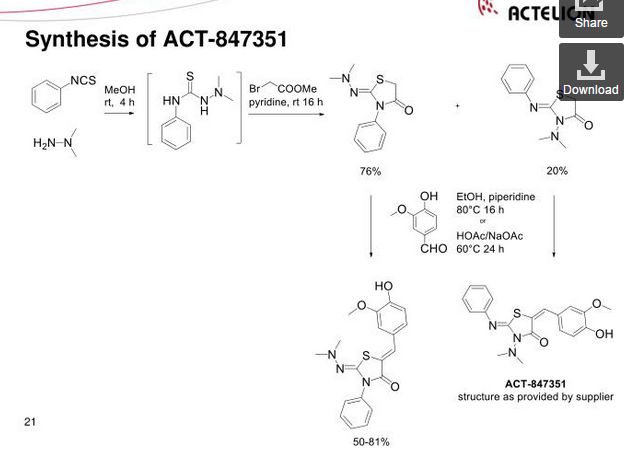
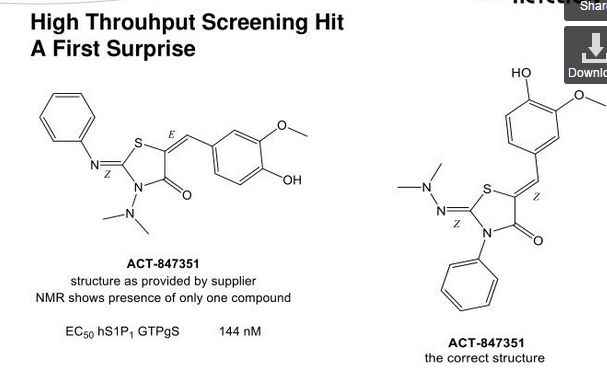
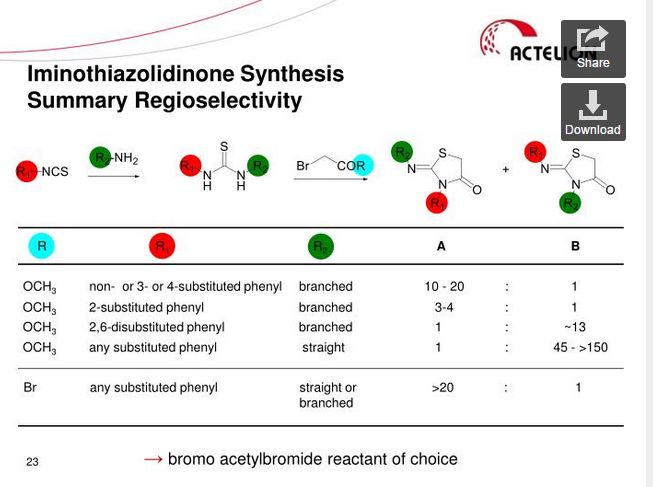
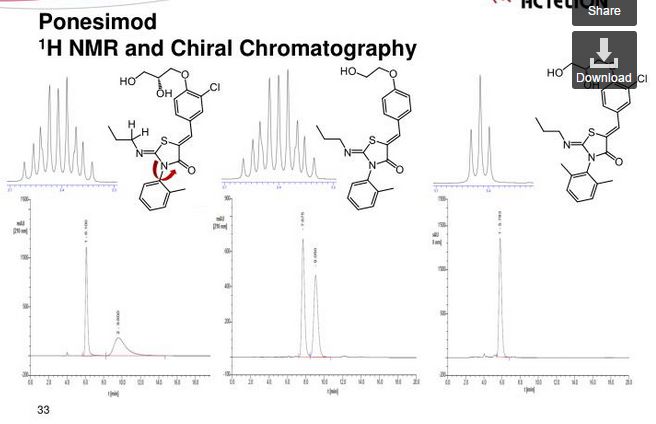
Ponesimod (INN, codenamed ACT-128800) is an experimental drug for the treatment of multiple sclerosis (MS) and psoriasis. It is being developed by Actelion.
The first oral treatment for relapsing multiple sclerosis, the nonselective sphingosine-1-phosphate receptor (S1PR) modulator fingolimod, led to identification of a pivotal role of sphingosine-1-phosphate and one of its five known receptors, S1P1R, in regulation of lymphocyte trafficking in multiple sclerosis. Modulation of S1P3R, initially thought to cause some of fingolimod’s side effects, prompted the search for novel compounds with high selectivity for S1P1R. Ponesimod is an orally active, selective S1P1R modulator that causes dose-dependent sequestration of lymphocytes in lymphoid organs. In contrast to the long half-life/slow elimination of fingolimod, ponesimod is eliminated within 1 week of discontinuation and its pharmacological effects are rapidly reversible. Clinical data in multiple sclerosis have shown a dose-dependent therapeutic effect of ponesimod and defined 20 mg as a daily dose with desired efficacy, and acceptable safety and tolerability. Phase II clinical data have also shown therapeutic efficacy of ponesimod in psoriasis. These findings have increased our understanding of psoriasis pathogenesis and suggest clinical utility of S1P1R modulation for treatment of various immune-mediated disorders. A gradual dose titration regimen was found to minimize the cardiac effects associated with initiation of ponesimod treatment. Selectivity for S1P1R, rapid onset and reversibility of pharmacological effects, and an optimized titration regimen differentiate ponesimod from fingolimod, and may lead to better safety and tolerability. Ponesimod is currently in phase III clinical development to assess efficacy and safety in relapsing multiple sclerosis. A phase II study is also ongoing to investigate the potential utility of ponesimod in chronic graft versus host disease.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
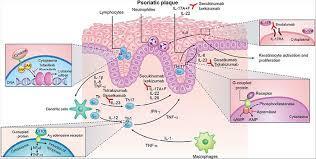
Biology and pharmacology of sphingosine-1-phosphate receptor 1
The past decades have witnessed major advances in the treatment of autoimmune and chronic inflammatory diseases. A plethora of novel therapies targeting specific molecules involved in the inflammatory or immune system activation cascades have become available. These have significantly increased our understanding of disease pathogenesis and improved the management of immune-mediated disorders. However, most of the targeted therapies are biological drugs which need to be injected, are eliminated slowly (e.g. over several weeks) and can lose efficacy or tolerability due to their potential immunogenicity. In an attempt to overcome these hurdles, pharmaceutical research has made considerable efforts to develop novel oral targeted therapies for autoimmune and chronic inflammatory diseases.
Sphingosine-1-phosphate receptor 1 (S1P1R) is one of five known G protein-coupled receptors with nanomolar affinity for the lysophospholipid sphingosine-1-phosphate (S1P), which is generated through physiologic metabolism of the cell membrane constituent sphingomyelin by all cells [Brinkmann, 2007]. S1P receptors, including S1P1R, are widely expressed in many tissues [Chun et al. 2010]. S1P1R expression on lymphocytes controls their egress from thymus and secondary lymphoid organs [Cyster and Schwab, 2012]. Lymphocyte egress requires a gradient of S1P concentration, which is established by a high S1P concentration in blood and lymph compared with a low concentration in the interstitial fluid of lymphoid organs [Grigorova et al. 2009].
Synthetic S1P1 receptor modulators disrupt the interaction of the physiologic S1P ligand with S1P1R by promoting initial activation followed by sustained internalization and desensitization of S1P1R [Hla and Brinkmann, 2011; Pinschewer et al. 2011]. Experiments conducted in animal models of transplant rejection, multiple sclerosis, lupus erythematosus, arthritis and inflammatory bowel disease with the first-generation, nonselective S1P receptor modulator, fingolimod, have demonstrated the potential efficacy of this mode of action across several immune-mediated chronic inflammatory conditions [Brinkmann, 2007]. Fingolimod is a structural analog of sphingosine that is phosphorylated in the body by a sphingosine kinase to generate the bioactive form of the drug, fingolimod phosphate, which binds to multiple S1P receptors [Brinkmann, 2007]. Clinical trials in multiple sclerosis (MS) have confirmed the efficacy of fingolimod in relapsing MS, but not in primary progressive disease, and led to the approval of the first oral medication for the treatment of relapsing forms of MS in 2010 [Kappos et al. 2010].
The mechanism of action of fingolimod has increased our understanding of MS pathogenesis. T and B cells, but not natural killer (NK) cells, express functional S1P1R and are affected by fingolimod [Cyster and Schwab, 2012]. Furthermore, S1P1R is differentially expressed and regulated in functionally distinct subsets of lymphocytes and fingolimod has been shown to predominantly affect naïve T cells and central memory T cells (TCM) while sparing effector memory T cells (TEM), and terminally differentiated effector T cells (TE) in patients with relapsing MS [Mehling et al. 2008, 2011]. This has raised the possibility that, at least in MS, retention of TCM cells, which include pro-inflammatory T helper 17 (Th17) cells, by fingolimod may prevent their accumulation in the cerebrospinal fluid (CSF) and subsequent differentiation to TE cells in the central nervous system (CNS) [Hla and Brinkmann, 2011]. The effects of S1P1R modulation on B cells are less well defined. Recent data from patients with relapsing MS have shown predominant reduction of memory B cells and recently activated memory B cells (CD38int-high) in peripheral blood after treatment with fingolimod [Claes et al. 2014; Nakamura et al. 2014]. As memory B cells are implicated in the pathogenesis of MS and other autoimmune diseases, these observations suggest another potential mechanism underlying the therapeutic effects of S1P1R modulators.
Astrocytes, microglia, oligodendrocytes and neurons express various S1P receptors including S1P1R, S1P3R and S1P5R. Fingolimod has been shown to penetrate the CNS tissues and in vitro studies have shown activation of astrocytes and oligodendrocytes by fingolimod [Foster et al. 2007]. Conditional deletion of S1P1R on neural cells in mice reduced the severity of experimental autoimmune encephalomyelitis (EAE) and reductions in the clinical scores were paralleled by decreased demyelination, axonal loss and astrogliosis [Choi et al. 2011]. Unfortunately, there was no beneficial effect in a recently completed, large study of fingolimod in patients with primary progressive MS [Lublin et al. 2015], suggesting that the direct effect on CNS cells alone may not be sufficient. Taken together, these data suggest the possibility of a direct beneficial effect of S1P1R modulation in the brain of patients with relapsing MS [Dev et al. 2008]; however, its contribution to efficacy relative to the immunological effects remains unclear.
Initial studies in rodents suggested that modulation of S1P3R on cardiac myocytes by fingolimod was associated with a reduction of heart rate (HR) by activation of G-protein-coupled inwardly rectifying potassium channels (GIRK) that regulate pacemaker frequency, and the shape and duration of action potentials [Koyrakh et al. 2005; Camm et al. 2014]. Modulation of S1P2R and S1P3R on myofibroblasts by fingolimod was also shown to stimulate extracellular matrix synthesis [Sobel et al. 2013]. Modulation of these receptors on vascular smooth muscle cells appeared to be associated with vasoconstriction, leading to the slight increase in blood pressure observed with fingolimod treatment [Salomone et al. 2003; Watterson et al. 2005; Hu et al. 2006; Lorenz et al. 2007; Kappos et al. 2010]. These observations raised the possibility that some side effects associated with fingolimod treatment could be avoided by more selective S1P1R modulators, thus triggering the search for novel compounds.
Currently, there are several selective S1P1R modulators in clinical development [Gonzalez-Cabrera et al.2014; Subei and Cohen, 2015]. Here we review data and the development status of ponesimod, a selective S1P1R modulator developed by Actelion Pharmaceuticals Ltd.http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Ponesimod, a selective, rapidly reversible, orally active, sphingosine-1-phosphate receptor modulator
Ponesimod (ACT-128800 (Z,Z)-5-[3-chloro-4-(2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one) is a selective, rapidly reversible, orally active, S1P1R modulator. Ponesimod emerged from the discovery of a novel class of S1P1R agonists based on the 2-imino-thiazolidin-4-one scaffold (Figure 1) [Bolli et al. 2010]. Ponesimod activates S1P1R with high potency [half maximal effective concentration (EC50) of 5.7 nM] and selectivity. Relative to the potency of S1P, the potency of ponesimod is 4.4 higher for S1P1R and 150-fold lower for S1P3R, resulting in an approximately 650-fold higher S1P1R selectivity compared with the natural ligand.

Chemical structure of ponesimod, C23H25N2O4CIS (molecular weight 460.98).http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4707431/
Clinical trials
In a 2009–2011 Phase II clinical trial including 464 MS patients, ponesimod treatment resulted in fewer new active brain lesions thanplacebo, measured during the course of 24 weeks.[3][4]
In a 2010–2012 Phase II clinical trial including 326 patients with psoriasis, 46 or 48% of patients (depending on dosage) had a reduction of at least 75% Psoriasis Area and Severity Index (PASI) score compared to placebo in 16 weeks.[3][5]
SEE https://clinicaltrials.gov/ct2/show/NCT02425644
Adverse effects
Common adverse effects in studies were temporary bradycardia (slow heartbeat), usually at the beginning of the treatment,dyspnoea (breathing difficulties), and increased liver enzymes (without symptoms). No significant increase of infections was observed under ponesimod therapy.[3] QT prolongation is detectable but was considered to be too low to be of clinical importance in a study.[6]
Mechanism of action
Like fingolimod, which is already approved for the treatment of MS, ponesimod blocks the sphingosine-1-phosphate receptor. This mechanism prevents lymphocytes (a type of white blood cells) from leaving lymph nodes.[3] Ponesimod is selective for subtype 1 of this receptor, S1P1.[7]
PAPER
Bolli, Martin H.; Journal of Medicinal Chemistry 2010, V53(10), P4198-4211 CAPLUS
2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1Receptor Agonists
Drug Discovery Chemistry, Actelion Pharmaceuticals Ltd., Gewerbestrasse 16, CH-4123 Allschwil, Switzerland
J. Med. Chem., 2010, 53 (10), pp 4198–4211
DOI: 10.1021/jm100181s
Publication Date (Web): May 06, 2010
Copyright © 2010 American Chemical Society
*To whom correspondence should be addressed. Phone: + 41 61 565 65 70. Fax: + 41 61 565 65 00. E-mail:martin.bolli@actelion.com.

Sphingosine-1-phosphate (S1P) is a widespread lysophospholipid which displays a wealth of biological effects. Extracellular S1P conveys its activity through five specific G-protein coupled receptors numbered S1P1 through S1P5. Agonists of the S1P1 receptor block the egress of T-lymphocytes from thymus and lymphoid organs and hold promise for the oral treatment of autoimmune disorders. Here, we report on the discovery and detailed structure−activity relationships of a novel class of S1P1 receptor agonists based on the 2-imino-thiazolidin-4-one scaffold. Compound 8bo (ACT-128800) emerged from this series and is a potent, selective, and orally active S1P1 receptor agonist selected for clinical development. In the rat, maximal reduction of circulating lymphocytes was reached at a dose of 3 mg/kg. The duration of lymphocyte sequestration was dose dependent. At a dose of 100 mg/kg, the effect on lymphocyte counts was fully reversible within less than 36 h. Pharmacokinetic investigation of8bo in beagle dogs suggests that the compound is suitable for once daily dosing in humans.
(Z,Z)-5-[3-Chloro-4-((2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolyl-thiazolidin-4-one (8bo)
…………..DELETED…………… column chromatography on silica gel eluting with heptane:ethyl acetate 1:4 to give the title compound (1.34 g, 37%) as a pale-yellow foam.
1H NMR (CDCl3): δ 0.94 (t, J = 7.3 Hz, 3 H), 1.58−1.70 (m, 2 H), 2.21 (s, 3 H), 3.32−3.48 (m, 2 H), 3.82−3.95 (m, 3 H), 4.12−4.27 (m, 4 H), 7.07 (d, J = 8.8 Hz, 1 H), 7.21 (d, J = 7.0 Hz, 1 H), 7.31−7.39 (m, 3 H), 7.49 (dd, J = 8.5, 2.0 Hz, 1 H), 7.64 (d, J= 2.0 Hz, 1 H), 7.69 (s, 1 H).
13C NMR (CDCl3): δ 11.83, 17.68, 23.74, 55.42, 63.46, 69.85, 70.78, 133.48, 120.75, 123.71, 127.05, 128.25, 128.60, 129.43, 130.06, 131.13, 131.50, 134.42, 136.19, 146.98, 154.75, 166.12. LC-MS (ES+): tR 0.96 min. m/z: 461 (M + H).
HPLC (ChiralPak AD-H, 4.6 mm × 250 mm, 0.8 mL/min, 70% hexane in ethanol): tR 11.8 min. Anal. (C23H25N2O4SCl): C, H, N, O, S, Cl.
PATENT
WO 2014027330
https://www.google.com/patents/WO2014027330A1?cl=3Den
The present invention relates inter alia to a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (hereinafter also referred to as the “COMPOUND” or “compound (2)”), especially in crystalline form C which form is described in WO 2010/046835. The preparation of COMPOUND and its activity as immunosuppressive agent is described in WO 2005/054215. Furthermore, WO 2008/062376 describes a new process for the preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one which can be used as an intermediate in the preparation of COMPOUND.
Example 1 a) below describes such a process of preparing (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one according to WO 2008/062376. According to WO 2008/062376 the obtained (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one can then be transformed into COMPOUND by using standard methods for the alkylation of phenols. Such an alkylation is described in Example 1 b) below. Unfortunately, this process leads to the impurity (2Z,5Z)-5-(3-chloro-4-((1 ,3-dihydroxypropan-2-yl)oxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one which is present in about 2% w/w in the crude product (see Table 1 ) and up to 6 recrystallisations are necessary in order to get this impurity below 0.4% w/w (see Tables 1 and 2) which is the specified limit based on its toxicological qualification.


the obtained (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one to form (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (2):

.
The reaction of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ) with 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one can be performed under conditions which are typical for a Knoevenagel condensation. Such conditions are described in the literature for example in Jones, G., Knoevenagel Condensation in Organic Reaction, Wiley: New York, 1967, Vol. 15, p 204; or Prout, F. S., Abdel-Latif, A. A., Kamal, M. R., J. Chem. Eng. Data, 2012, 57, 1881-1886.
2-[(Z)-Propylimino]-3-o-tolyl-thiazolidin-4-one can be prepared as described in WO 2008/062376, preferably without the isolation and/or purification of intermediates such as the thiourea intermediate that occurs after reacting o-tolyl-iso-thiocyanate with n-propylamine. Preferably 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one obtained according to WO 2008/062376 is also not isolated and/or purified before performing the Knoevenagel condensation, i.e. before reacting 2-[(Z)-propylimino]-3-o-tolyl-thiazolidin-4-one with (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde (1 ), i.e. in a preferred embodiment compound (2) is prepared in a one-pot procedure analogous to that described in WO 2008/062376.
Example 1 : (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one

a) Preparation of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one:

Acetic acid solution: To acetic acid (149.2 mL) are added sodium acetate (1 1 .1 1 g, 2.00 eq.) and 3-chloro-4-hydroxybenzaldehyde (10.60 g, 1.00 eq.) at 20 °C. The mixture is stirred at 20 °C until complete dissolution (2 to 3 h).
n-Propylamine (4.04 g, 1.00 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 40 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (13.53 g, 1.00 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). 70 mL of dichloromethane are distilled off under atmospheric pressure and jacket temperature of 60 °C. The temperature is adjusted to 42 °C and the acetic acid solution is added to the reaction mixture. The resulting solution is heated to 58 °C and stirred at this temperature for 15 h before IPC (conversion specification≥ 95 %). 25 mL of solvents are distilled off under vacuum 900 – 500 mbars and jacket temperature of 80 °C. The temperature is adjusted to 60 °C and water (80.1 mL) is added to the reaction mixture over 1 h. The resulting yellow suspension is stirred at 60 °C for 30 min. The suspension is cooled to 20 °C over 1 h and stirred at this temperature for 30 min.
The product is filtered and washed with a mixture of acetic acid (30 mL) and water (16 mL) and with water (50 mL) at 20 °C. The product is dried under vacuum at 50 °C for 40 h to afford a pale yellow solid; yield 25.93 g (78 %).
b) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
To a suspension of (2Z,5Z)-5-(3-chloro-4-hydroxy-benzylidene)-2-propylimino-3-o-tolyl-thiazolidin-4-one (10.00 g, 1.00 eq.) in ethanol (47.2 mL) is added (R)-3-chloro-1 ,2-
propanediol (3.37 g, 1.18 eq.) at 20 °C. Potassium tert-butoxide (3.39 g, 1.13 eq.) is added in portions at 20 °C. The resulting fine suspension is stirred at 20 °C for 25 min before being heated to reflux (88 °C). The reaction mixture is stirred at this temperature for 24 h before IPC (conversion specification≥ 96.0 %). After cooling down to 60 °C, acetonitrile (28.6 mL) and water (74.9 mL) are added. The resulting clear solution is cooled from 60 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.010 g, 0.001 eq.; crystalline form C can be prepared as described in WO 2010/046835) are added at 50 °C. The suspension is heated from 0 °C to 50 °C, cooled to 0 °C over 6 h and stirred at this temperature for 12 h.
The product is filtered and washed with a mixture of acetonitrile (23.4 mL) and water (23.4 mL) at 0 °C. The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield 1 1.91 g (84 %).
c) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
Recrystallisation I: The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in acetonitrile (30 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 12.8 mL).
Recrystallisation II: The wet product is dissolved in acetonitrile (27.0 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 1 1.3 mL).
Recrystallisation III: The wet product is dissolved in acetonitrile (24.3 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4- one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 10.1 mL).
Recrystallisation IV: The wet product is dissolved in acetonitrile (21.9 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 9.1 mL).
Recrystallisation V: The wet product is dissolved in acetonitrile (19.7 mL) at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h. During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed with acetonitrile at -10 °C (2 x 8.2 mL).
Recrystallisation VI: The wet product is dissolved in acetonitrile (23.9 mL) at 70 °C. Water (20 mL) is added at 70 °C. The reaction mixture is cooled from 70 °C to 0 °C over 2 h.
During the cooling ramp, (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2- (propylimino)-3-(o-tolyl)thiazolidin-4-one seeds of crystalline form C (0.0075 g, 0.00075 eq.) are added at 50 °C. The suspension is heated up to 52 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h. The product is filtered and washed twice with a mixture of acetonitrile (4.5 mL) and water (4.5 mL) at -10 °C.
The product is dried under vacuum at 45 °C for 24 h to afford a pale yellow solid; yield: 7.0 g (70 %).
Example 2: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde

Potassium tert-butoxide (1 18 g, 1.20 eq.) is added to n-propanol (963 mL) followed by 3-chloro-4-hydroxybenzaldehyde (137 g, 1.00 eq.). To the mixture is added (R)-3-chloro-1 ,2-propanediol (126 g, 1.30 eq.). The suspension is heated to 90 °C and stirred at this temperature for 17 h. Solvent (500 mL) is distilled off at 120 °C external temperature and reduced pressure. Water is added (1.1 L) and solvent (500 mL) is removed by distillation. The turbid solution is cooled to 20 °C. After stirring for one hour a white suspension is obtained. Water (500 mL) is added and the suspension is cooled to 10 °C. The suspension is filtered and the resulting filter cake is washed with water (500 mL). The product is dried at 50 °C and reduced pressure to yield 149 g of a white solid (73%), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 3: (R)-3-Chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde
Potassium tert-butoxide (8.60 g, 1.20 eq.) is added to n-propanol (70 mL) below 15 °C, the temperature is allowed to rise. After the addition the temperature is corrected again to below 15 °C before addition of 3-chloro-4-hydroxybenzaldehyde (10 g, 1 .00 eq.). The suspension is heated to 40 °C and stirred for 30 min. (R)-3-Chloro-1 ,2-propanediol (9.18 g, 1.30 eq.) is added at 40 °C. The resulting suspension is heated to 60 °C and stirred at this temperature for 15 h then heated to 94 °C till meeting the IPC-specification (specification conversion≥ 90.0 %). The mixture is cooled to 30 °C and n-propanol is partially distilled off (-50 mL are distilled off) under reduced pressure and a maximum temperature of 50 °C, the jacket temperature is not allowed to raise above 60 °C.
Water (81 mL) is added and a second distillation is performed under the same conditions (24 mL are distilled off). The mixture is heated till homogeneous (maximum 54 °C) and then cooled to 24 °C. At 24 °C the mixture is seeded with crystalline (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of form A (0.013 g, 0.00085 eq.). How to obtain the crystalline seeds is described in Examples 2 and 5. The reaction mixture is cooled to 0 °C over 7.5 h.
The product is filtered and washed with water (2 x 35 mL) and once with methyl tert-butyl ether (20 mL) at 5 °C. The product is dried under vacuum at 40 °C for 20 h to afford an off-white solid; yield: 10.6 g (72 %), which is (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde in crystalline form A.
Example 4: (2Z,5Z)-5-(3-Chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)- 3-(o-tolyl)thiazolidin-4-one
a) Preparation of crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
n-Propylamine (5.23 g, 1.32 eq.) is added to a solution of o-tolyl-iso-thiocyanate (10 g, 1.00 eq.) in dichloromethane (100 mL) at 20 °C. The resulting pale yellow solution is agitated for 15 min at 20 °C before IPC (conversion specification≥ 99.0 %). The reaction is cooled to -2 °C. Bromoacetyl bromide (14.88 g, 1.10 eq.) is added and the resulting solution is stirred for 15 min at -2 °C. Pyridine (10.92 g, 2.05 eq.) is then added slowly at -2 °C. The intensive yellow reaction mixture is stirred for 15 min at -2 °C before IPC (conversion specification≥ 93.0 %). Dichloromethane is partially distilled off (66 mL are distilled off) under atmospheric pressure and jacket temperature of 60 °C. Ethanol (1 1 1.4 mL), sodium acetate (12.75 g, 2.30 eq.) and (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde from Example 3 (14.38 g, 0.93 eq.) are added. The remaining dichloromethane and a part of ethanol are distilled off (49.50 mL are distilled off) under atmospheric pressure and jacket temperature up to 85 °C. The reaction mixture (orange suspension) is stirred for 3 – 5 h under reflux (78 °C) before IPC (conversion specification≥ 97.0 %).
Water (88.83 mL) is added and the temperature adjusted to 40 °C before seeding with micronized (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.075 g, 0.0024 eq.). The reaction mixture is cooled to 0 °C over 5 h, heated up to 40 °C, cooled to 0 °C over 6 h and stirred at this temperature for 2 h.
The product is filtered and washed with a 1 :1 ethanohwater mixture (2 x 48 mL) at 0 °C. The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 24.71 g (86 %).
b) Purification of (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one:
The crude (2Z,5Z)-5-(3-chloro-4-((R)-2,3-dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one (10 g) is dissolved in ethanol (40 mL) at 70 °C. The temperature is adjusted at 50 °C for seeding with micronised (2Z,5Z)-5-(3-chloro-4-((R)-2,3- dihydroxypropoxy)benzylidene)-2-(propylimino)-3-(o-tolyl)thiazolidin-4-one in crystalline form C (0.016 g, 0.0016 eq.). The reaction mixture is cooled from 50 °C to 0 °C over 4 h, heated up to 50 °C, cooled to 0 °C over 6 h and agitated at this temperature for 2 h.
The product is filtered and washed with ethanol at 0 °C (2 x 12.8 mL). The product is dried under vacuum at 45 °C for 10 h to afford a pale yellow solid; yield: 9.2 g (92 %).
Example 5: Preparation of crystalline seeds of (R)-3-chloro-4-(2,3-dihydroxypropoxy)- benzaldehyde
10 mg of (R)-3-chloro-4-(2,3-dihydroxypropoxy)-benzaldehyde of at least 99.5% purity by 1 H-NMR assay is dissolved in a 4 mL vial by adding 1 mL of pure ethanol (puriss p. a.). The solvent is allowed to evaporate through a small hole in the cap (approx. 2 mm of diameter) of the vial until complete dryness. The white solid residue is crystalline (R)-3-chloro-4-(2,3- dihydroxypropoxy)-benzaldehyde in crystalline form A. Alternatively, methanol or methylisobutylketone (both in puriss p. a. quality) is used. This procedure is repeated until sufficient seeds are made available.

PATENT
WO 2005054215
SEE https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2005054215
| WO2005054215A1 | Nov 16, 2004 | Jun 16, 2005 | Actelion Pharmaceuticals Ltd | 5-(benz- (z) -ylidene) -thiazolidin-4-one derivatives as immunosuppressant agents |
| WO2008062376A2 | Nov 22, 2007 | May 29, 2008 | Actelion Pharmaceuticals Ltd | New process for the preparation of 2-imino-thiazolidin-4-one derivatives |
| WO2010046835A1 | Oct 19, 2009 | Apr 29, 2010 | Actelion Pharmaceuticals Ltd | Crystalline forms of (r) -5- [3-chloro-4- ( 2, 3-dihydroxy-propoxy) -benz [z] ylidene] -2- ( [z] -propylimino) -3-0-tolyl-thiazolidin-4-one |
| Reference | ||
|---|---|---|
| 1 | * | BOLLI, M.H. ET AL.: “2-Imino-thiazolidin-4-one Derivatives as Potent, Orally Active S1P1 Receptor Agonists“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 53, no. 10, 2010, pages 4198-4211, XP55090073, ISSN: 0022-2623, DOI: 10.1021/jm100181s |
References
- “Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: Favorable impact of dose up-titration”. The Journal of Clinical Pharmacology 54: 179–88. Feb 2014. doi:10.1002/jcph.244. PMID 24408162.
- “Mass balance, pharmacokinetics and metabolism of the selective S1P1 receptor modulator ponesimod in humans”. Xenobiotica 45: 139–49. Feb 2015. doi:10.3109/00498254.2014.955832. PMID 25188442.
- H. Spreitzer (29 September 2014). “Neue Wirkstoffe – Ponesimod”. Österreichische Apothekerzeitung (in German) (20/2014): 42.
- “Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised phase II trial”. Journal of Neurology, Neurosurgery 85: 1198–208. Nov 2014. doi:10.1136/jnnp-2013-307282. PMC 4215282. PMID 24659797.
- “Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double-blind, placebo-controlled phase 2 trial”. The Lancet 384: 2036–45. Dec 2014. doi:10.1016/S0140-6736(14)60803-5. PMID 25127208.
- “Effect of Ponesimod, a selective S1P1 Receptor Modulator, on the QT Interval in Healthy Subjects”. Basic 116: 429–37. May 2015.doi:10.1111/bcpt.12336. PMID 25287214.
- “Ponesimod”. Actelion. Retrieved 31 October 2014.

ABOUT PONESIMOD
Ponesimod is a potent orally active, selective sphingosine-1-phosphate receptor 1 (S1P1) immunomodulator.
Ponesimod prevents lymphocytes from leaving lymph nodes, thereby reducing circulating blood lymphocyte counts and preventing infiltration of lymphocytes into target tissues. The lymphocyte count reduction is rapid, dose-dependent, sustained upon continued dosing, and quickly reversible upon discontinuation. Initial data suggest that ponesimod does not cause lymphotoxicity by destroying/depleting lymphocytes or interfering with their cellular function. Other blood cells e.g. cells of the innate immune system are largely unaffected. Ponesimod is therefore considered a promising new oral agent for the treatment of a variety of autoimmune disorders.
CURRENT STATUS
OPTIMUM (Oral Ponesimod versus Teriflunomide In relapsing MUltiple sclerosis) is a Phase III multi-center, randomized, double-blind, parallel-group, active-controlled superiority study to compare the efficacy and safety of ponesimod to teriflunomide in patients with relapsing multiple sclerosis (RMS). The study aims to determine whether ponesimod is more efficacious than teriflunomide in reducing relapses. The study is expected to enroll approximately 1’100 patients, randomized in 2 groups in a 1:1 ratio to receive ponesimod 20 mg/day or teriflunomide 14 mg/day, and is expected to last a little over 3 years. An additional study to further characterize the utility and differentiation of ponesimod in multiple sclerosis is being discussed with Health Authorities.
Ponesimod is also evaluated in a Phase II open-label, single-arm, intra-subject dose-escalation study to investigate the biological activity, safety, tolerability, and pharmacokinetics of ponesimod in patients suffering from moderate or severe chronic graft versus host disease (GvHD)inadequately responding to first- or second-line therapy. The study will also investigate the clinical response to ponesimod treatment in these patients. Approximately 30 patients will be enrolled to receive ponesimod in escalating doses of 5, 10, and 20 mg/day over the course of 24 weeks. The study is being conducted at approximately 10 sites in the US and is expected to last approximately 18 months.
AVAILABLE CLINICAL DATA
The decision to move into Phase III development was based on the Phase IIb dose-finding study with ponesimod in patients with relapsing-remitting multiple sclerosis. A total of 464 patients were randomized into this study and the efficacy, safety and tolerability of three ponesimod doses (10, 20, and 40 mg/day) versus placebo, administered once daily for 24 weeks.
The primary endpoint of this study was defined as the cumulative number of new gadolinium-enhancing lesions on T1-weighted magnetic resonance imaging (MRI) scans at weeks 12, 16, 20, and 24 after study drug initiation. A key secondary endpoint of this study was the annualized relapse rate over 24 weeks of treatment. Patients who completed 24 weeks of treatment were offered the opportunity to enter into an extension study. This ongoing trial is investigating the long-term safety, tolerability, and efficacy of 10 and 20 mg/day of ponesimod in patients with relapsing-remitting multiple sclerosis, in a double-blind fashion. The study continues to provide extensive safety and efficacy information for ponesimod in this indication, with some patients treated for more than 6 years.
The safety database from all studies with ponesimod now comprises more than 1,300 patients and healthy volunteers.
MILESTONES
2015 – Phase III program in multiple sclerosis initiated
2011 – Phase IIb dose-finding study in multiple sclerosis successfully completed
2006 – Entry-into-man
2004 – Preclinical development initiated
KEY SCIENTIFIC LITERATURE
Olsson T et al. J Neurol Neurosurg Psychiatr. 2014 Nov;85(11):1198-208. doi: 10.1136/jnnp-2013-307282. Epub 2014 Mar 21
Freedman M.S, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 420 (P923).
Fernández Ó, et al. Multiple Sclerosis Journal, 2012; 18 (4 suppl): 417 (P919).
Piali L, Froidevaux S, Hess P, et al. J Pharmacol Exp Ther 337(2):547-56, 2011
Bolli MH, Abele S, Binkert C, et al. J Med Chem. 53(10):4198-211, 2010
Kappos L et al. N Engl J Med. 362(5):387-401, 2010
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2Z,5Z)-5-{3-Chloro-4-[(2R)-2,3-dihydroxypropoxy]benzylidene}-3-(2-methylphenyl)-2-(propylimino)-1,3-thiazolidin-4-one
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | 2 main metabolites |
| Biological half-life | 31–34 hrs[1] |
| Excretion | Feces (57–80%, 26% unchanged), urine (10–18%)[2] |
| Identifiers | |
| CAS Number | 854107-55-4 |
| ATC code | none |
| PubChem | CID 11363176 |
| ChemSpider | 9538103 |
| ChEMBL | CHEMBL1096146 |
| Synonyms | ACT-128800 |
| Chemical data | |
| Formula | C23H25ClN2O4S |
| Molar mass | 460.974 g/mol |
////Ponesimod, Phase III , A sphingosine-1-phosphate receptor 1, S1P1 agonist, multiple sclerosis. ACT-128800; RG-3477; R-3477, autoimmune disease, lymphocyte migration, multiple sclerosis, psoriasis, transplantation
CCC/N=C\1/N(C(=O)/C(=C/C2=CC(=C(C=C2)OC[C@@H](CO)O)Cl)/S1)C3=CC=CC=C3C

{kind=link}
{kind=link}
{kind=link}