
Home » Posts tagged 'Antineoplastic' (Page 4)
Tag Archives: Antineoplastic
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sendegobresib
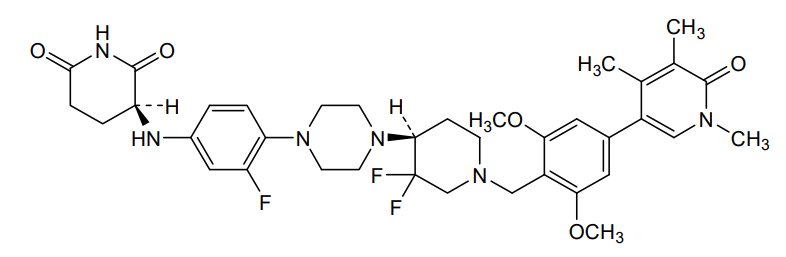
Sendegobresib
CAS 2704617-96-7
MFC37H45F3N6O5, 710.79
2,6-PIPERIDINEDIONE, 3-((4-(4-((4S)-1-((4-(1,6-DIHYDRO-1,4,5-TRIMETHYL-6-OXO-3-PYRIDINYL)-2,6-DIMETHOXYPHENYL)METHYL)-3,3-DIFLUORO-4-PIPERIDINYL)-1-PIPERAZINYL)-3-FLUOROPHENYL)AMINO)-, (3S)-
(3S)-3-[4-[4-[(4S)-1-[[2,6-dimethoxy-4-(1,4,5-trimethyl-6-oxo-3-pyridinyl)phenyl]methyl]-3,3-difluoropiperidin-4-yl]piperazin-1-yl]-3-fluoroanilino]piperidine-2,6-dione

bromodomain-containing protein 9 (BRD9) degradation inducer, antineoplastic, AW8PEP3VZ3, CFT 8634, ORPHAN DRUG
Sendegobresib is an orally bioavailable heterobifunctional protein degrader of bromodomain-containing protein 9 (BRD9; sarcoma antigen NY-SAR-29; rhabdomyosarcoma antigen MU-RMS-40.8), with potential antineoplastic activity. Sendegobresib is comprised of an E3 ligase-binding moiety and a BRD9-binding moiety. Upon oral administration, sendegobresib targets and binds to BRD9 with its BRD9-binding moiety. Upon BRD9 binding, the E3 ligase-binding moiety binds to cereblon (CRBN), a component of the CRL4-CRBN E3 ubiquitin ligase complex, which directs proteins for destruction, resulting in the proteasome-mediated degradation of BRD9. This leads to an inhibition of the growth of tumor cells that rely on BRD9 for survival. BRD9, a component of one form of the Brg/Brahma-Associated Factor (BAF) complex, is needed for the survival of certain cancer cells due to mutations.
A Study to Assess the Safety and Tolerability of CFT8634 in Locally Advanced or Metastatic SMARCB1-Perturbed Cancers, Including Synovial Sarcoma and SMARCB1-Null Tumors
CTID: NCT05355753
Phase: Phase 1
Status: Terminated
Date: 2024-12-17
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US355912448&_cid=P11-MINYJY-62955-1
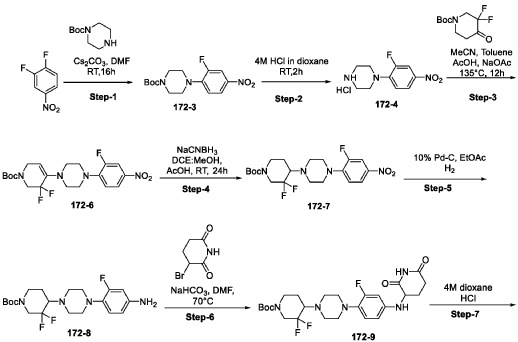
Synthesis of Compound 172
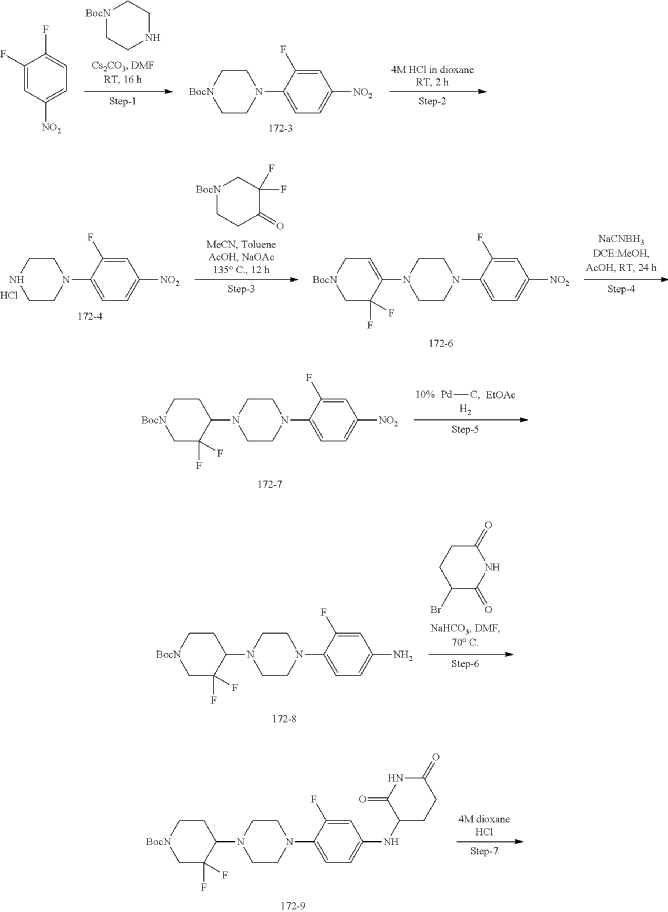
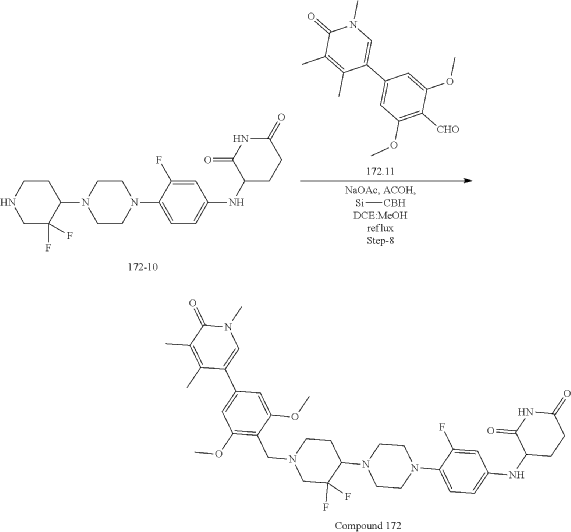
| Step-1: To a stirred solution of compound tert-butyl piperazine-1-carboxylate (85.40 g, 536.82 mmol) in DMF (500 mL) was added cesium carbonate (262.4 g, 805.4 mmol) and stirred for 15 min before adding 1,2-difluoro-4-nitro-benzene (100 g, 536.82 mmol). The reaction mixture stirred at RT for 16 h while monitoring by TLC. After completion, the reaction mass was quenched with ice flakes and the precipitated solid was filtered, dried under vacuum to afford tert-butyl 4-(2-fluoro-4-nitro-phenyl) piperazine-1-carboxylate 172-3 (152 g, 88.85% yield, 97.94% purity) as a yellow solid. |
PAT
PAT
PAT
- Enhanced hyt-induced protein degradation using lipid nanoparticle deliveryPublication Number: US-2023414723-A1Priority Date: 2020-10-26
- Compounds for targeted degradation of brd9Publication Number: WO-2021178920-A1Priority Date: 2020-03-05
- Compounds for targeted degradation of brd9Publication Number: US-2022098194-A1Priority Date: 2020-03-05
- Compounds for targeted degradation of brd9Publication Number: US-2023060334-A1Priority Date: 2020-03-05
- Compounds for targeted degradation of BRD9Publication Number: US-11691972-B2Priority Date: 2020-03-05Grant Date: 2023-07-04
- Selected compounds for targeted degradation of brd9Publication Number: US-2024245677-A1Priority Date: 2021-09-09
- Exosome-based cancer assaysPublication Number: US-11938164-B2Priority Date: 2021-04-07Grant Date: 2024-03-26
- Exosome-based cancer assaysPublication Number: US-2022331390-A1Priority Date: 2021-04-07
- Exosome-based cancer assaysPublication Number: WO-2022216765-A1Priority Date: 2021-04-07
- Enhanced hyt-induced protein degradation using lipid nanoparticle deliveryPublication Number: WO-2022093809-A1Priority Date: 2020-10-26
- Directed degron molecules and applications thereofPublication Number: WO-2023081400-A1Priority Date: 2021-11-04
- Directed degron molecules and applications thereofPublication Number: WO-2023081400-A9Priority Date: 2021-11-04
- Directed degron molecules and applications thereofPublication Number: EP-4426687-A1Priority Date: 2021-11-04
- Selected compounds for targeted degradation of brd9Publication Number: WO-2023039208-A1Priority Date: 2021-09-09
- Selected compounds for targeted degradation of brd9Publication Number: EP-4398904-A1Priority Date: 2021-09-09



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
/////////Sendegobresib, antineoplastic, AW8PEP3VZ3, CFT 8634, ORPHAN DRUG
Segigratinib, Ratangratinib
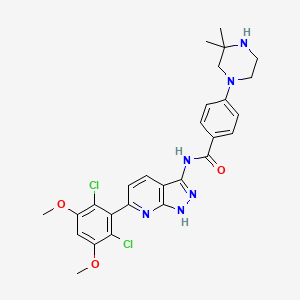
Segigratinib, Ratangratinib
CAS 1882873-93-9
MF C27H28Cl2N6O3 MW 555.5 g/mol
N-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-1H-pyrazolo[3,4-b]pyridin-3-yl]-4-(3,3-dimethylpiperazin-1-yl)benzamide
N-[6-(2,6-dichloro-3,5-dimethoxyphenyl)-1H-pyrazolo[5,4-b]pyridin-3-yl]-4-(3,3-dimethylpiperazin-1-yl)benzamide
fibroblast growth factor receptor tyrosine kinase inhibitor, antineoplastic, 3D 185, Ratangratinib, 3D-185, G0Z5E4YTB4, HH 185
Ratangratinib is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) types 1, 2, and 3 (FGFR1/2/3) and colony stimulating factor 1 receptor (CSF1R; CSF-1R; CD115; M-CSFR), with potential immunomodulatory and antineoplastic activities. Upon administration, ratangratinib binds to and inhibits FGFR1/2/3, which may result in the inhibition of FGFR1/2/3-mediated signal transduction pathways. This inhibits proliferation in FGFR1/2/3-overexpressing tumor cells. 3D185 also targets and binds to CSF1R, thereby blocking CSF1R activation and CSF1R-mediated signaling. This inhibits the activities of tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), and prevents immune suppression in the tumor microenvironment (TME). This enhances antitumor T-cell immune responses and inhibits the proliferation of tumor cells. FGFR, a family of receptor tyrosine kinases (RTKs) upregulated in many tumor cell types, plays a key role in cellular proliferation, migration and survival. CSF1R, also known as macrophage colony-stimulating factor receptor (M-CSFR) and CD115 (cluster of differentiation 115), is a cell-surface receptor that plays major roles in tumor cell proliferation and metastasis.
Efficacy and Safety of 3D185 Monotherapy in Subjects With Previously Treated Locally Advanced or Metastatic Cholangiocarcinoma
CTID: NCT05039892
Phase: Phase 2
Status: Not yet recruiting
Date: 2025-05-20
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016026445&_cid=P20-MIMK7T-68502-1
N-(6-(2,6-dichloro-3,5-dimethoxyphenyl)-1H-pyrazolo[3,4-b]pyridin-3-yl)-6-(3,3-dimethylpiperazin-1-yl)nicotinamide
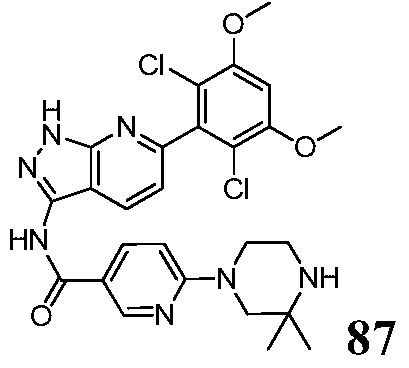
1H NMR(DMSO-d6,400MHz)δppm 13.39(s,1H),10.86(s,1H),8.81(d,1H,J=2.0Hz),8.40(d,1H,J=8.0Hz),8.16(dd,1H,J 1=2.4Hz,J 2=2.4Hz),7.08(t,2H,J=8.4Hz),6.90(d,1H,J=9.2Hz),3.99(s,6H),3.60(t,2H,J=4.0Hz),3.43(s,2H),2.82(t,2H,J=4.4Hz),1.04(s,6H).LCMS:556.2[M+H] +,RT=1.21min。
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US204149576&_cid=P20-MIMK4B-66027-1
1H NMR (d-MeOD, 400 MHz) δ ppm 8.52 (d, J=8.0 Hz, 1H), 8.03 (d, J=8.0 Hz, 2H), 7.15-7.13 (m, 3H), 6.94 (s, 1H), 3.99 (s, 6H), 3.58-3.57 (m, 2H), 3.44-3.40 (m, 4H), 10.50 (s, 6H).
PAT
- Indazole compounds as fgfr kinase inhibitor, preparation and use thereofPublication Number: EP-3184521-A1Priority Date: 2014-08-19
- Indazole compounds as FGFR kinase inhibitor, preparation and use thereofPublication Number: US-10562900-B2Priority Date: 2014-08-19Grant Date: 2020-02-18
- Indazole compounds as fgfr kinase inhibitor, preparation and use thereofPublication Number: US-2017275291-A1Priority Date: 2014-08-19
- Indazole compounds as fgfr kinase inhibitor, preparation and use thereofPublication Number: WO-2016026445-A1Priority Date: 2014-08-19
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////segigratinib, antineoplastic, 3D 185, Ratangratinib, 3D-185, G0Z5E4YTB4, HH 185
Rupitasertib
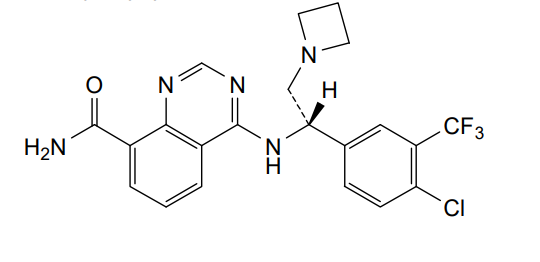
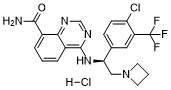
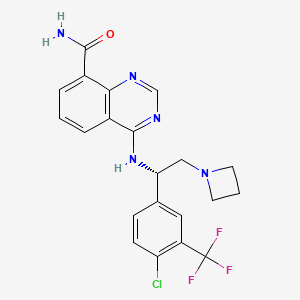
Rupitasertib
CAS 1379545-95-5
MF C21H19ClF3N5O 449.9 g/mol
4-({(1S)-2-(azetidin-1-yl)-1-[4-chloro-3-(trifluoromethyl)phenyl]ethyl}amino)quinazoline-8-carboxamide
4-[[(1S)-2-(azetidin-1-yl)-1-[4-chloro-3-(trifluoromethyl)phenyl]ethyl]amino]quinazoline-8-carboxamide
serine/ threonine kinase inhibitor, antineoplastic, EMD SERONO, Gastric cancer; HER2 positive breast cancer; Solid tumours, M2698 HCl, M2698 hydrochloride, MSC2363318A, MSC 2363318A, MSC-2363318A, M2698, M-269, M 2698. Rupitasertib HCl, 0DXG50I4WD
- OriginatorEMD Serono
- DeveloperEMD Serono; Evexta Bio
- ClassAntineoplastics; Small molecules
- Mechanism of Action70 kDa ribosomal protein S6 kinase inhibitors; Proto-oncogene protein c-akt inhibitors
- PreclinicalGlioblastoma; HER2 negative breast cancer
- No development reportedGastric cancer; HER2 positive breast cancer; Solid tumours
- 28 Oct 2025No recent reports of development identified for preclinical development in Gastric-cancer in France (PO)
- 28 Jun 2025No recent reports of development identified for phase-I development in HER2-positive-breast-cancer(Combination therapy, Late-stage disease, Metastatic disease) in USA (PO)
- 28 Jun 2025No recent reports of development identified for phase-I development in Solid-tumours(Combination therapy, Late-stage disease) in USA (PO)
- First-in-Human Dose Escalation Trial in Subjects With Advanced Malignancies
- CTID: NCT01971515
- Phase: Phase 1
- Status: Completed
- Date: 2018-09-19
Rupitasertib is an orally available inhibitor of the serine/threonine protein kinases ribosomal protein S6 Kinase (p70S6K) and Akt (protein kinase B), with potential antineoplastic activity. Upon administration, rupitasertib binds to and inhibits the activity of p70S6K and Akt. This prevents the activation of the PI3K/Akt/p70S6K signaling pathway and inhibits tumor cell proliferation in cancer cells that have an overactivated PI3K/Akt/p70S6K signaling pathway. Constitutive activation and dysregulated signaling of the PI3K/Akt/p70S6K pathway are frequently associated with tumorigenesis of many tumor types; targeting multiple kinases in this pathway is more efficacious than targeting a single kinase.
An optimized S6K inhibitor to overcome limitations of PAM pathway inhibitors
In just over 20 years, protein kinase inhibitors have changed the face of oncology and opened the new eras of targeted therapies and precision medicine. However, with few exceptions, no patient can be cured by one of these drugs alone. Today, scientists seek to develop novel kinase inhibitors[1] with improved efficacy and the potential to overcome resistances. The dual S6K AKT1/3 inhibitor rupitasertib (formerly DIACC3010, acquired from Merck KGaA, Darmstadt, Germany) has both of these characteristics and reaches brain metastases. After successfully completing a Phase I trial in patients with advanced/refractory solid tumors, including breast cancer, the drug candidate will be evaluated in a Phase 2/3 trial in ER+ HER2 breast cancer, which is expected to start in 2024.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012069146&_cid=P10-MIJPKI-12294-1
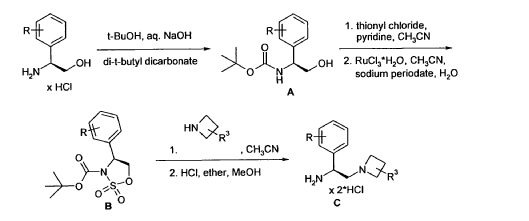
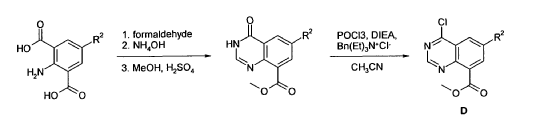
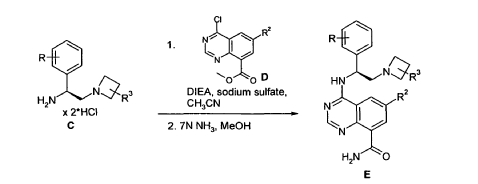
Example 4 was prepared following the general synthesis of A-E starting with (S)-2- amino-2-(3,4-di-fluoro-phenyl)-ethanol.LCMS [384.20 (M+1)]. 1H NMR (DMSO-d6, ppm) 1.92 (2H), 2.75 (1H), 2.93 (1H), 3.15 (4H), 5.43 (1H), 7.34 (2H), 7.53 (1H), 7.68 (1H), 7.81 (1H), 8.58 (4H), 10.30 (1H).
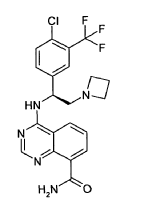
4-[(S)-2-Azetidin-1-yl-1-(4-chloro-3-trifluoromethylphenyl)-ethylamino]-guinazoline-8- carboxylic acid amide (5)
IC50 P70S6K [nM]: 0.9
pS6 MDA-MB-468 [nM]: 11
Akt1 IC50 [nM]: 1.4
Aurora B IC50 [nM]: 100
PAT
- Quinazoline carboxamide azetidinesPublication Number: SG-190318-A1Priority Date: 2010-11-24
- Quinazoline carboxamide azetidinesPublication Number: US-2013252942-A1Priority Date: 2010-11-24
- Quinazoline carboxamide azetidinesPublication Number: US-8946247-B2Priority Date: 2010-11-24Grant Date: 2015-02-03
- SMAC Mimetic for Treating Myelodysplastic SyndromesPublication Number: US-2015158908-A1Priority Date: 2009-07-02
- Methods of treating a ras protein-related disease or disorderPublication Number: US-2025049810-A1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- p70S6K/Akt dual inhibitor DIACC3010 is efficacious in preclinical models of gastric cancer alone and in combination with trastuzumabPublication Name: Scientific ReportsPublication Date: 2023-09-25PMCID: PMC10520030PMID: 37749105DOI: 10.1038/s41598-023-40612-9
- TTD: Therapeutic Target Database describing target druggability informationPublication Name: Nucleic Acids ResearchPublication Date: 2023-09-15PMCID: PMC10767903PMID: 37713619DOI: 10.1093/nar/gkad751
- Identification of Clinical Candidate M2698, a Dual p70S6K and Akt Inhibitor, for Treatment of PAM Pathway-Altered CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-10-01PMID: 34596404DOI: 10.1021/acs.jmedchem.1c01087
- Phase 1 study of M2698, a p70S6K/AKT dual inhibitor, in patients with advanced cancerPublication Name: Journal of Hematology & OncologyPublication Date: 2021-08-18PMCID: PMC8371902PMID: 34407844DOI: 10.1186/s13045-021-01132-z
- M2698 is a potent dual-inhibitor of p70S6K and Akt that affects tumor growth in mouse models of cancer and crosses the blood-brain barrierPublication Name: American journal of cancer researchPublication Date: 2016PMCID: PMC4859885PMID: 27186432
////////////Rupitasertib, antineoplastic, EMD SERONO, Gastric cancer; HER2 positive breast cancer; Solid tumours, M2698 HCl, M2698 hydrochloride, MSC2363318A, MSC 2363318A, MSC-2363318A, M2698, M-269, M 2698. Rupitasertib HCl, 0DXG50I4WD
Potrasertib

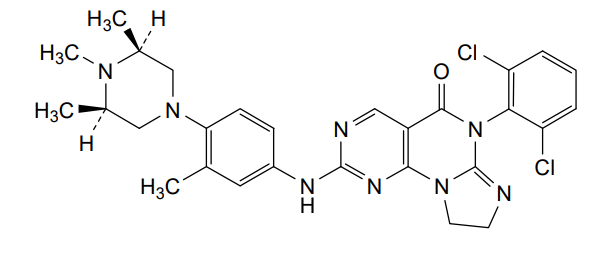
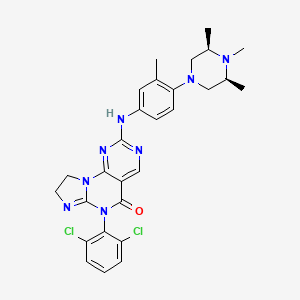
Potrasertib
CAS 2226938-19-6
MFC28H30Cl2N8O MW 565.5 g/mol
6-(2,6-dichlorophenyl)-2-{3-methyl-4-[(3R,5S)-3,4,5-trimethylpiperazin-1-yl]anilino}-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one
7-(2,6-dichlorophenyl)-12-[3-methyl-4-[(3S,5R)-3,4,5-trimethylpiperazin-1-yl]anilino]-2,5,7,11,13-pentazatricyclo[7.4.0.02,6]trideca-1(13),5,9,11-tetraen-8-one
serine/ threonine kinase inhibitor, antineoplastic, IMP 7068, WEE1-IN-10, orb2664172, 621K13UG4B, Phase 1, Solid tumours
- OriginatorIMPACT Therapeutics
- ClassAntineoplastics; Small molecules
- Mechanism of ActionWEE1 protein inhibitors
- Phase ISolid tumours
- 28 Mar 2024No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Monotherapy) in Taiwan (PO)
- 28 Mar 2024No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease, Monotherapy) in USA (PO)
- 20 Oct 2023Efficacy, adverse events, pharmacodynamics and pharmacokinetics data from the phase I WEE1 trial in Solid tumours presented at the 48th European Society for Medical Oncology Congress (ESMO-2023)
Potrasertib is an investigational drug that is a selective inhibitor of WEE1 kinase, a protein crucial for the cell cycle. It is being studied for the treatment of various advanced solid tumors, including small cell lung cancer, ovarian, and colorectal cancers. By blocking the WEE1 kinase, potrasertib causes cancer cells with DNA damage to undergo premature, error-prone mitosis, which leads to cell death.
How it works
- Potrasertib is a serine/threonine kinase inhibitor.
- It works by targeting WEE1 kinase, which regulates the cell’s response to DNA damage.
- By inhibiting WEE1, it prevents cancer cells from repairing DNA damage before dividing, forcing them into a state that leads to cell death.
- This mechanism is particularly effective in tumors with a defective p53 gene, as these tumors rely more heavily on the WEE1 checkpoint for survival.
Potential uses
- Combination therapy: It is being explored in combination with chemotherapy (like gemcitabine and cisplatin) or radiotherapy to enhance their effectiveness against cancer.
- Monotherapy: It is also being studied as a standalone treatment for certain cancers, including ovarian, colorectal, and non-small cell lung cancer, especially those with high replication stress or WEE1 dependency.
Current status
- Potrasertib is still an investigational drug and is not yet approved for widespread clinical use.
- It is undergoing clinical trials to evaluate its safety and effectiveness in treating advanced cancers.
Potrasertib is an investigational new drug that is being evaluated by IMPACT Therapeutics for the treatment of advanced solid tumors. It is oral inhibitor of WEE1 kinase, a key regulator of cell cycle checkpoints.[1][2]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018090939&_cid=P21-MI6TEY-70275-1

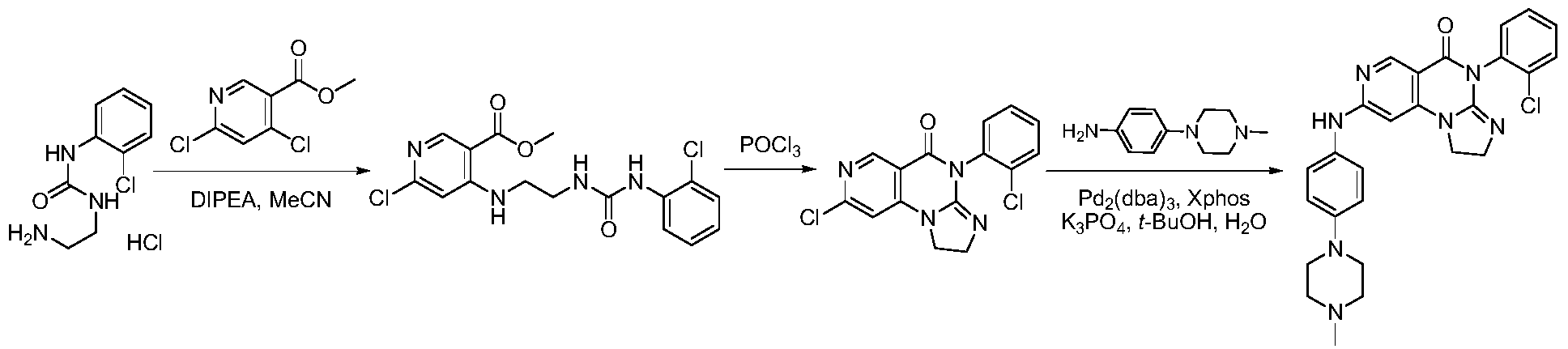
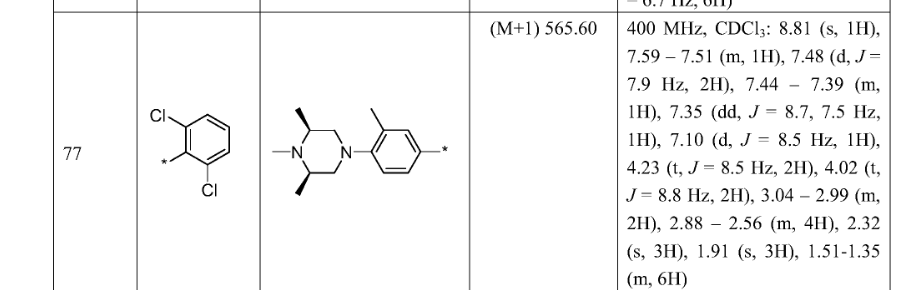
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021073491&_cid=P21-MI6TF3-70349-1
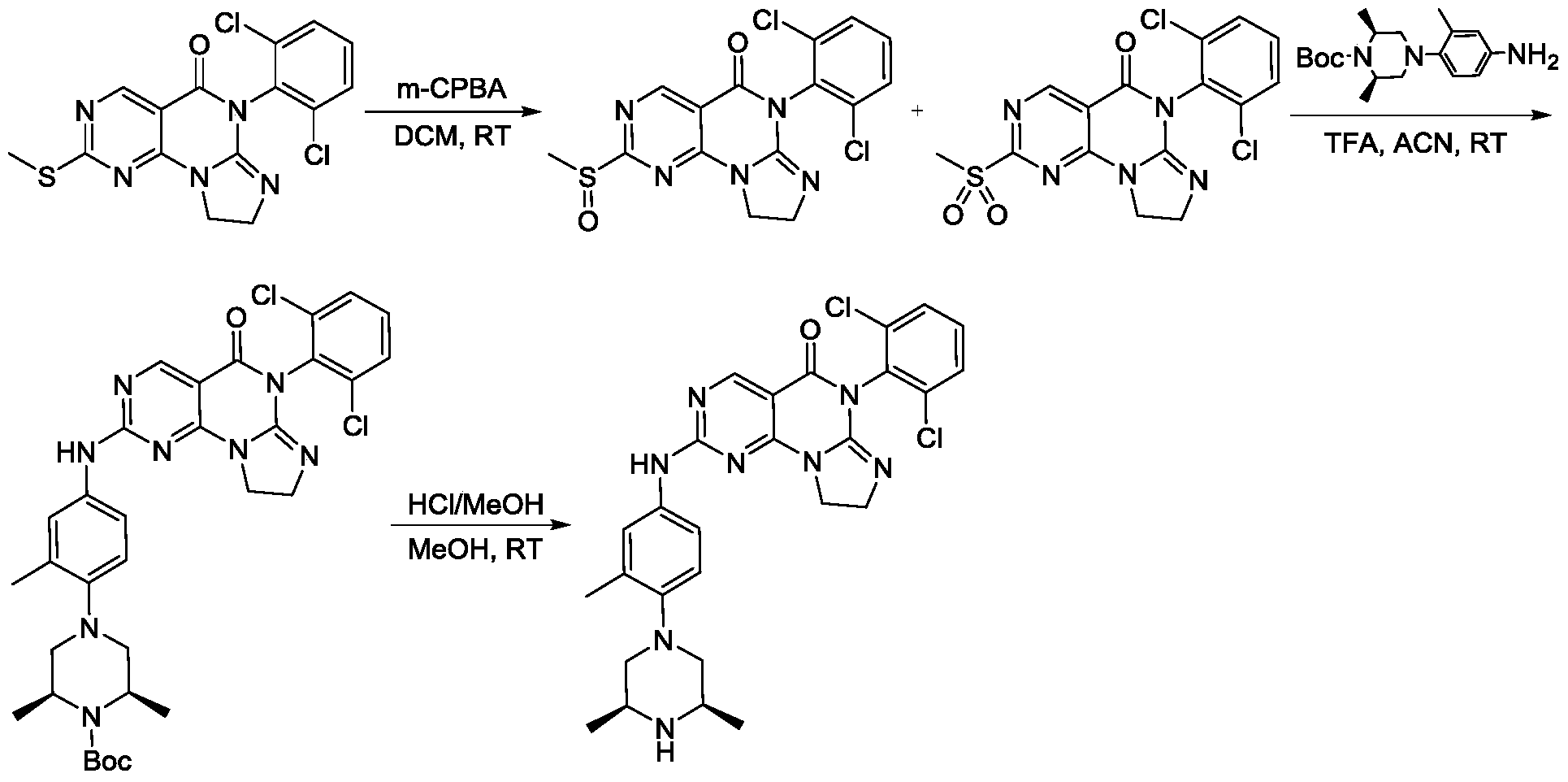
Example 1
SIMILAR NOT SAME
[0117]6-(2,6-dichlorophenyl)-2-((4-((3S,5R)-3,5-dimethylpiperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one
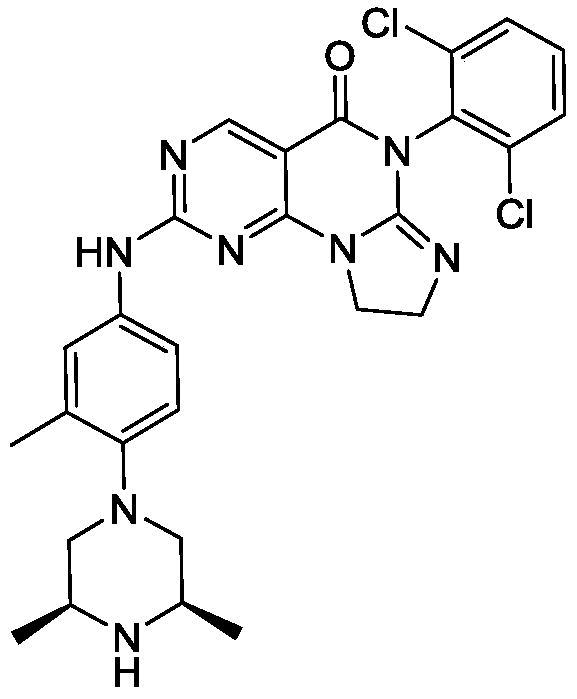
SIMILAR NOT SAME
[0128]6-(2,6-dichlorophenyl)-2-((4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one
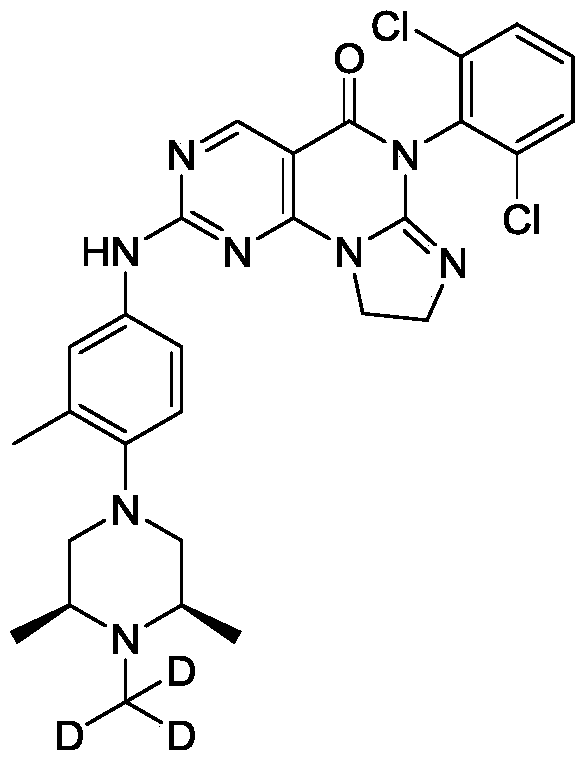
[0130]a) Preparation of (2S,6R)-2,6-dimethyl-1-(methyl-d3)-4-(2-methyl-4-nitro)piperazine: Sodium hydride (385.03 mg, 9.63 mmol, 60% purity) was added to a solution of (3S,5R)-3,5-dimethyl-1-(2-methyl-4-nitro)piperazine (2 g, 8.02 mmol) in N,N-dimethylformamide (15 mL). The mixture was stirred at 0 °C for 25 hours, then trideuterated iodomethane (1.16 g, 8.02 mmol, 499.09 μL) was added, and the mixture was stirred at 0 °C for 2 hours. The reaction was quenched by adding an aqueous sodium bicarbonate solution (30 mL) at 0 °C, extracted with ethyl acetate (50 mL × 3), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to obtain the target crude product (1.5 g, yellow-green solid). LC-MS(ESI): m/z(M+1) + 267.1. 1 H NMR (400MHz, CDCl
3 ): δ8.04-8.01 (m, 2H), 6.96 (d, J = 12.0Hz, 1H), 3.10 (d, J = 12Hz, 2H), 2.65 (t , J=12Hz, 2H), 2.45-2.43 (m, 2H), 2.36 (s, 3H), 1.16-1.15 (d, J=4.0Hz, 6H).
[0131]b) Preparation of 4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylaniline: Under nitrogen protection, palladium on carbon (281.58 μmol, 10% purity) was added to a methanol (5 mL) solution of (2S,6R)-2,6-dimethyl-1-(methyl-d3)-4-(2-methyl-4-nitro)piperazine (1.5 g, 5.63 mmol). The resulting suspension was purified multiple times under vacuum with hydrogen. The mixture was stirred at 25 °C for 12 hours under a hydrogen atmosphere (15 psi). The reaction mixture was filtered, and the filtrate was concentrated under reduced pressure to give the target crude product (1.3 g, black solid). LC-MS (ESI): m/z (M+1) + 237.1.
[0132]c) Preparation of 6-(2,6-dichlorophenyl)-2-((4-(((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylphenyl)amino)-8,9-dihydroimidazo[1,2-a]pyrimidino[5,4-e]pyrimidin-5(6H)-one: 4-((3S,5R)-3,5-dimethyl-4-(methyl-d3)piperazin-1-yl)-3-methylaniline (459.32 mg, 1.94 mmol) and the prepared 6-(2,6-dichlorophenyl)-2- A mixture (700 mg, crude) of crude (methanesulfonyl)-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one and 6-(2,6-dichlorophenyl)-2-(methanesulfonyl)-8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-one was dissolved in acetonitrile (5 mL) and trifluoroacetic acid (20.14 mg, 0.177 mmol, 13.08 μL) was added. The mixture was stirred at 20–25 °C for 2 hours, filtered, and the filtrate was concentrated under reduced pressure to give the crude product. The crude product was purified by reversed-phase HPLC to give the target compound (56.89 mg, 100.00 μmol, yellow solid, 5.66% yield). LC-MS (ESI): m/z (M+1) + 568.0.
1 H NMR (400MHz, CDCl 3 ): δ8.81 (s, 1H), 7.49 (d, J=3.8Hz, 3H), 7.41-7.34 (m, 3H), 7.02 (d, J=4.2Hz, 1H), 4.25-4.21 (m, 2H), 4.02 (t, J=8.0Hz, 2H), 2.95 (d, J=6.0Hz 2H), 2.62 (t, J=6.0Hz, 2H), 2.46-2.41 (m, 2H), 2.34 (s, 6H), 1.15 (d, J=6.4Hz, 6H).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022188802&_cid=P21-MI6TVM-79837-1
PAT
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: US-11345711-B2Priority Date: 2016-11-16Grant Date: 2022-05-31
- 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidine-5(6h)-ketone compoundPublication Number: EP-3543242-B1Priority Date: 2016-11-16Grant Date: 2024-01-03
- Compound 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-ketonePublication Number: ES-2968252-T3Priority Date: 2016-11-16Grant Date: 2024-05-08
- 8,9-dihydroimidazole[1,2-a]pyrimido[5,4-e]pyrimidine-5(6h)-ketone compoundPublication Number: EP-3543242-A1Priority Date: 2016-11-16
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: US-10703759-B2Priority Date: 2016-11-16Grant Date: 2020-07-07
- 8,9-DIHYDROIMIDAZO[1,2-a]PYRIMIDO[5,4-e]PYRIMIDIN-5(6H)-ONESPublication Number: US-2019308984-A1Priority Date: 2016-11-16
- 8,9-DIHYDROIMIDAZO[1,2-a]PYRIMIDO[5,4-e]PYRIMIDIN-5(6H)-ONESPublication Number: US-2020385394-A1Priority Date: 2016-11-16
- 8,9-Dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6H)-onesPublication Number: CN-109906227-BPriority Date: 2016-11-16Grant Date: 2022-03-11
- Dihydroimidazo pyrimido pyrimidinone compoundPublication Number: WO-2021073491-A1Priority Date: 2019-10-16
- DihydroimidazopyrimidopyrimidinonesPublication Number: CN-114502559-APriority Date: 2019-10-16
- Dihydroimidazopyrimidopyrimidinone compoundsPublication Number: CN-114502559-BPriority Date: 2019-10-16Grant Date: 2024-02-02
- Dihydroimidazo pyrimido pyrimidinone compoundPublication Number: US-2024010655-A1Priority Date: 2019-10-16
- 8,9-dihydroimidazo[1,2-a]pyrimido[5,4-e]pyrimidin-5(6h)-onesPublication Number: CA-3043945-A1Priority Date: 2016-11-16
- Use of Wee1 kinase inhibitors in the treatment of cancerPublication Number: CN-118338905-APriority Date: 2021-11-26
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: WO-2023093840-A1Priority Date: 2021-11-26
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: WO-2022188802-A1Priority Date: 2021-03-10
- The use of Wee1 kinase inhibitors in the treatment of cancer diseasesPublication Number: CN-117202908-APriority Date: 2021-03-10
- Use of wee1 kinase inhibitors in the treatment of cancerPublication Number: US-2024091233-A1Priority Date: 2021-03-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | IMP7068 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2226938-19-6 |
| PubChem CID | 139503236 |
| UNII | 621K13UG4B |
| Chemical and physical data | |
| Formula | C28H30Cl2N8O |
| Molar mass | 565.50 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- “IMP 7068”. AdisInsight. Springer Nature Switzerland AG.
- Wang Z, Li W, Li F, Xiao R (January 2024). “An update of predictive biomarkers related to WEE1 inhibition in cancer therapy”. Journal of Cancer Research and Clinical Oncology. 150 (1): 13. doi:10.1007/s00432-023-05527-y. PMC 10794259. PMID 38231277.
///////potrasertib, antineoplastic, IMP 7068, WEE1-IN-10, orb2664172, 621K13UG4B, Phase 1, Solid tumours
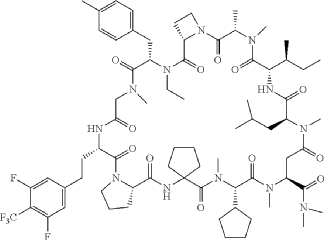
Paluratide
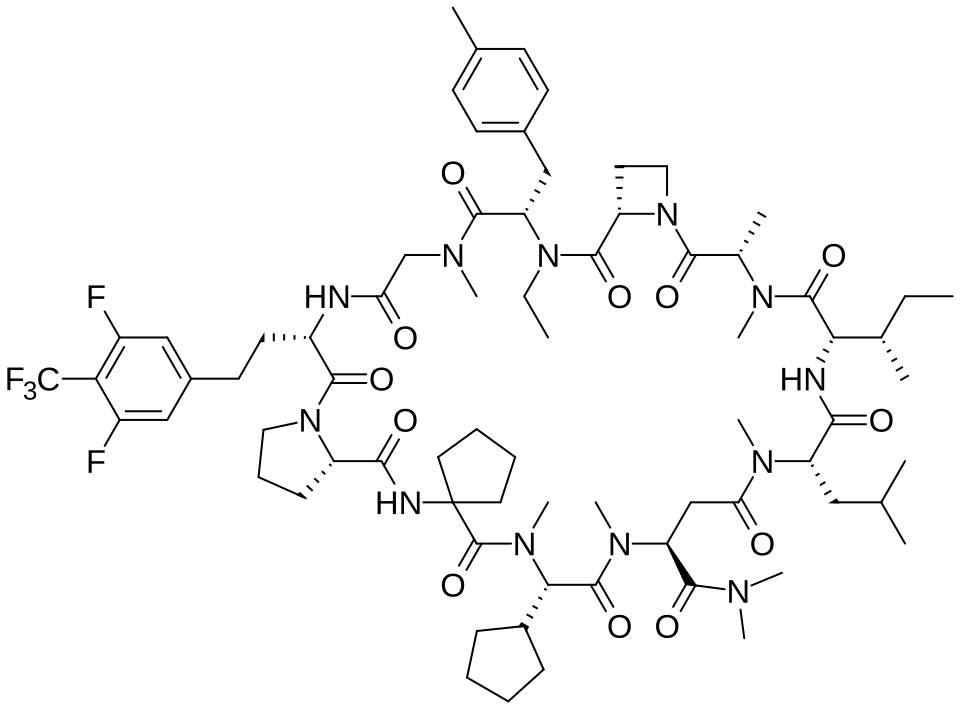


Paluratide
CAS 2676177-63-0
MFC73H105F5N12O12 MW 1437.7 g/mol
1,11-anhydro[N-methyl-L-alanyl-(2S)-azetidine-2-carbonyl-N-ethyl-4-methyl-L-phenylalanyl-N-methylglycyl-3-{[3,5-difluoro-4-(trifluoromethyl)phenyl]methyl}-L-alanyl-L-prolyl-2-
aminocyclopentane-1-carbonyl-(2S)-N-methyl-3-cyclopentylglycyl-1-
(dimethylamino)-N-methyl-L-aspart-4-yl-N-methyl-L-leucyl-Lisoleucine]
(3S,9S,12S,17S,20S,23S,27S,30S,36S)-20-[(2S)-butan-2-yl]-30-cyclopentyl-3-[2-[3,5-difluoro-4-(trifluoromethyl)phenyl]ethyl]-10-ethyl-N,N,7,17,18,24,28,31-octamethyl-9-[(4-methylphenyl)methyl]-23-(2-methylpropyl)-2,5,8,11,16,19,22,25,29,32,35-undecaoxospiro[1,4,7,10,15,18,21,24,28,31,34-undecazatricyclo[34.3.0.012,15]nonatriacontane-33,1′-cyclopentane]-27-carboxamide
G-protein Ras (rat sarcoma virus) inhibitor, antineoplastic, LUNA 18, CHUGAI, AW3YP3CD9X
Paluratide (development code LUNA18) was an investigational cyclic peptide KRAS inhibitor developed by Chugai Pharmaceutical, a member of the Roche Group, for the treatment of cancers with KRAS mutations.[1] The compound was notable as an orally bioavailable macrocyclic peptide that could target intracellular protein-protein interactions, a class of targets traditionally considered “undruggable.”[2]
Development was discontinued in July 2025 due to a narrow therapeutic window compared to competing KRAS inhibitors.[3]
Ras Inhibitor LUNA18 is an orally bioavailable cyclic peptide and Ras inhibitor, with potential antineoplastic activity. Upon oral administration, Ras inhibitor LUNA18 selectively targets, binds to and inhibits Ras, thereby inhibiting Ras-dependent signaling and inhibits proliferation of tumor cells in which Ras is overexpressed and/or mutated. Ras serves an important role in cell signaling, division and differentiation. Mutations of Ras may induce constitutive signal transduction leading to tumor cell growth, proliferation, invasion, and metastasis.
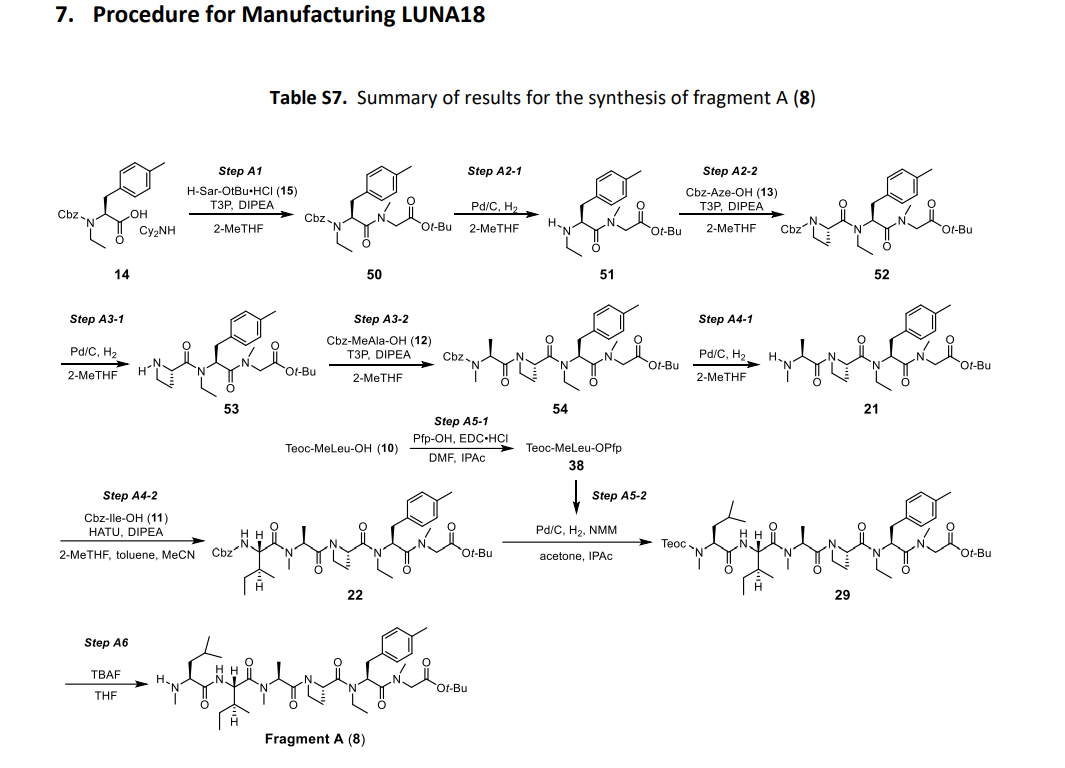
Paluratide (LUNA18 is synthesized using a novel liquid-phase peptide synthesis (LPPS) method, not traditional solid-phase methods, to overcome challenges with N-alkylated cyclic peptides. This process involves a convergent route of 24 telescoped chemical transformations, a final crystallization step, and a focus on specific strategies to manage side reactions like diketopiperazine formation and low reactivity of sterically hindered amino acids.
Key aspects of the synthesis
- Liquid-phase synthesis: A novel, high-yielding LPPS process was developed to enable the large-scale production of paluratide. This is a departure from traditional solid-phase methods, which have limitations with solubility and waste.
- Convergent synthetic route: The synthesis uses a convergent approach, meaning smaller fragments of the peptide are synthesized separately and then joined together. The overall process includes 24 telescoped chemical transformations followed by a final crystallization step.
- Addressing synthesis challenges: Specific strategies were employed to overcome key difficulties:
- Low reactivity: Amino acids with N-alkylation are sterically hindered, so more reactive and stable protecting groups were used to ensure efficient coupling.
- Side reactions: The method was designed to prevent side reactions like diketopiperazine formation in intermediates and incomplete hydrolysis of active esters.
- Instability: The peptide backbone is sensitive to acidic conditions, so a mildly acidic aqueous medium was chosen for workup and purification to maintain stability.
- Protecting group selection: Cbz-protected amino acid active esters were preferred over Boc-protected ones because they are less prone to forming N-carboxyanhydrides (NCA) under activating conditions, which can reduce yield and purity.
- Purification: A final crystallization step is used for purification.
PAT
- Method for producing eutectic of cyclic peptidePublication Number: WO-2024195801-A1Priority Date: 2023-03-20
- Method for producing cyclic peptide crystalsPublication Number: WO-2024085235-A1Priority Date: 2022-10-20
- Composition containing peptide, surfactant, and polymerPublication Number: WO-2024080308-A1Priority Date: 2022-10-12
- Methods for producing cyclic compounds comprising n-substituted amino acid residuesPublication Number: EP-4086272-A1Priority Date: 2021-05-07
- Methods for producing cyclic compounds comprising n-substituted amino acid residuesPublication Number: US-2022411462-A1Priority Date: 2021-05-07
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.5c00260?ref=PDF



PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US383248369&_cid=P20-MI3YXS-80609-1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Mechanism of action
Paluratide functions as a pan-RAS inhibitor, targeting multiple RAS isoforms including KRAS, NRAS, and HRAS.[1] The compound binds with high affinity to KRASG12D, with a dissociation constant (Kd) of 0.043 nM, and blocks the interaction between KRASG12D and the guanine nucleotide exchange factor SOS1 with an IC50 of less than 2.2 nM.[4]
Unlike covalent KRAS inhibitors that target specific mutations (such as sotorasib for KRASG12C), paluratide was designed to inhibit RAS proteins through disruption of protein-protein interactions with guanine nucleotide exchange factors (GEFs).[1] This mechanism allows the drug to affect RAS signalling regardless of the specific mutation, theoretically providing broader applicability across different KRAS-mutant cancers. The compound also demonstrates activity against downstream signalling pathways, affecting ERK and AKT phosphorylation.[4]
Medical uses
Paluratide was being developed for the treatment of locally advanced or metastatic solid tumors harbouring RAS gene alterations.[5] The drug demonstrated significant cellular activity against multiple cancer types with KRAS mutations in preclinical studies, including colorectal cancer, gastric cancer, non-small cell lung cancer, and pancreatic cancer.[1]
Chemistry
Paluratide is an 11-member (11-mer) cyclic peptide with a molecular weight in the range of 1000–2000 g/mol, classified as a “middle-size” cyclic peptide.[1] The compound features extensive N-alkylation, a modification that reduces hydrogen bond donors and improves oral absorption while maintaining cellular permeability.[2] Its structure allows it to navigate the challenging boundary between small molecules and biologics, achieving properties of both classes. The compound demonstrated oral bioavailability ranging from 21% to 47% in preclinical animal studies without requiring special formulations.[1]
Discovery
Paluratide was discovered through Chugai Pharmaceutical’s cyclic peptide platform using an mRNA display library screening approach.[1] The initial hit compound, designated AP8747, was identified from the mRNA display library and subsequently underwent extensive chemical optimization without scaffold hopping (maintaining the basic cyclic peptide structure).[1] The optimization focused on increasing plasma stability, improving absorption, reducing clearance, and reducing hydrogen bond donors to achieve oral bioavailability.
The final clinical compound, LUNA18, emerged after modifications to four amino acid positions (positions 5, 7, 10, and 11) from an intermediate compound (compound 40). Key structure-activity relationship findings included: the side chain at position 5 preferring aromatic over aliphatic groups; physicochemical properties being adjustable at position 11; and biological activity enhancement through modifications at positions 7 and 10.[1]
Chugai also developed a novel synthetic methodology that enabled the broadly applicable synthesis of highly N-alkylated cyclic peptide-like drugs.[6] This method overcame three major technical challenges: formation of diketopiperazine, insufficient reactivity of amidation due to steric hindrance, and instability of cyclic peptides under acidic conditions. Using this approach, more than 4,000 cyclic peptides were synthesized with a process yield of 31% and final product purity of 97%.[6]
Clinical trials
A Phase 1 dose-escalation and cohort expansion study (NCT05012618) was initiated in August 2021 to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary activity of paluratide administered as a single agent or in combination with other anti-cancer drugs.[5] The study, in the United States and Japan, was designed to enrol approximately 195 patients with locally advanced or metastatic solid tumors positive for documented RAS alterations.[5]
Paluratide was administered orally as capsules.[5] The study also evaluated combination therapy with cetuximab, an EGFR inhibitor.[5]
References
- Tanada M, Tamiya M, Matsuo A, Chiyoda A, Takano K, Ito T, et al. (August 2023). “Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor”. Journal of the American Chemical Society. 145 (30): 16610–16620. Bibcode:2023JAChS.14516610T. doi:10.1021/jacs.3c03886. PMID 37463267.
- Ohta A, Tanada M, Shinohara S, Morita Y, Nakano K, Yamagishi Y, et al. (November 2023). “Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets”. Journal of the American Chemical Society. 145 (44): 24035–24051. Bibcode:2023JAChS.14524035O. doi:10.1021/jacs.3c07145. PMID 37874670.
- Taylor NP (24 October 2025). “Roche axes 4 Chugai solid tumor assets in early-phase clear-out”. Fierce Biotech.
- “LUNA18 (Paluratide) – KRAS Inhibitor, ERK Inhibitor, RAS Inhibitor”. MedChemExpress.
- “A Dose-escalation Study of LUNA18 in Patients With Locally Advanced or Metastatic Solid Tumors (With Expansion)”. ClinicalTrials.gov. 29 July 2025. NCT05012618.
- Nomura K, Hashimoto S, Takeyama R, Tamiya M, Kato T, Muraoka T, et al. (October 2022). “Broadly Applicable and Comprehensive Synthetic Method for N-Alkyl-Rich Drug-like Cyclic Peptides”. Journal of Medicinal Chemistry. 65 (19): 13401–13412. doi:10.1021/acs.jmedchem.2c01296. PMID 36109865.
- “Chugai Announces 2025 2nd Quarter Results” (Press release). Chugai Pharmaceutical. 24 July 2025.
External links
- Phase 1 Clinical Trial Information at ClinicalTrials.gov
- Development of LUNA18 at Journal of the American Chemical Society
| Clinical data | |
|---|---|
| Other names | LUNA18 |
| Routes of administration | Oral administration |
| Legal status | |
| Legal status | Development discontinued |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2676177-63-0 |
| PubChem CID | 166509683 |
| ChemSpider | 129321315 |
| UNII | AW3YP3CD9X |
| Chemical and physical data | |
| Formula | C73H105F5N12O12 |
| Molar mass | 1437.707 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////Paluratide, antineoplastic, LUNA 18, CHUGAI, AW3YP3CD9X
Nuvisertib
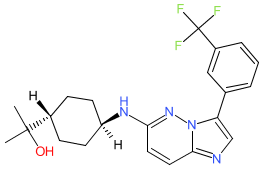
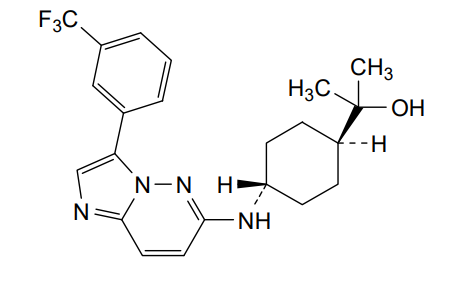

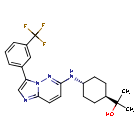
Nuvisertib
CAS 1361951-15-6
MF C22H26ClF3N4O MW418.5 g/mol
2-[(1r,4r)-4-({3-[3-(trifluoromethyl)phenyl]imidazo[1,2-b]pyridazin-6-yl}amino)cyclohexyl]propan-2-ol
serine/ threonine kinase inhibitor, antineoplastic, Orphan Drug, myelofibrosis, SGI-9481, SGI 9481, TP-3654, TP 3654, EOB0N7BOY4
The chemical structure for nuvisertib was obtained from proposed INN list 130 (Feb. 2024), in which the compound is described as a serine/ threonine kinase inhibitor with antineoplastic action. A structure match to clinical lead TP-3654 was made via PubChem. TP-3654 is declared as an orally available, second-generation pan-PIM kinase inhibitor [1-2].
| References |
| 1. Foulks JM, Carpenter KJ, Luo B, Xu Y, Senina A, Nix R, Chan A, Clifford A, Wilkes M, Vollmer D et al.. (2014) A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia, 16 (5): 403-12. [PMID:24953177] |
| 2. Wu CP, Li YQ, Chi YC, Huang YH, Hung TH, Wu YS. (2021) The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer Drugs. Int J Mol Sci, 22 (17). [PMID:34502348] |
Nuvisertib is an orally available, second-generation and selective ATP-competitive inhibitor of proviral integration site for Moloney murine leukemia virus (PIM) kinases, with potential antineoplastic activity. Upon oral administration, nuvisertib selectively binds to and prevents the activation of the PIM kinases. This prevents the activation of PIM-mediated signaling pathways and inhibits proliferation in cells that overexpress PIM. PIMs, constitutively active proto-oncogenic serine/threonine kinases, are upregulated in various types of cancers and play key roles in tumor cell proliferation and survival.
Nuvisertib, also known as TP-3654, is an oral, investigational, and highly selective PIM1 kinase inhibitor being studied in a Phase 1/2 clinical trial for intermediate- or high-risk myelofibrosis (MF). It is not currently an approved medication.
Key Information
- Mechanism of Action: Nuvisertib targets the PIM1 kinase pathway, which is often overactive in myelofibrosis and can promote cancer cell growth. By inhibiting this pathway, nuvisertib is being investigated for its potential to manage symptoms, reduce spleen size, improve blood counts, and slow the progression of bone marrow fibrosis.
- Current Status: Nuvisertib is in ongoing Phase 1/2 clinical trials (NCT04176198) as a monotherapy and in combination with JAK inhibitors like ruxolitinib and momelotinib.
- Designations: Nuvisertib has received Orphan Drug Designation for myelofibrosis
Study of TP-3654 in Patients With Advanced Solid Tumors
CTID: NCT03715504
Phase: Phase 1
Status: Completed
Date: 2023-11-14
SYN
WO2013013188
Example 31
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US427659372&_cid=P10-MHWTVL-76212-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US130491286&_cid=P10-MHWU33-81462-1
31. 4-((3-(3-(Trifluoromethyl)phenyl)imidazo[1,2-b]pyridazin-6-yl)amino)-trans-cyclohexyl)propan-2-ol (EX. 8-31)
| EX. 8-31 was prepared by similar procedures as in EX. 8-1 using 2-(trans-4-aminocyclohexyl)propan-2-ol. |

| 1H-NMR (CD 3OD/400 MHz): δ 8.82 (s, 1H), 8.19 (m, 1H), 7.88 (s, 1H), 7.62 (m, 3H), 6.70 (d, J=9.6 Hz, 1H), 3.71 (m, 1H), 2.26 (m, 2H), 1.95 (m, 2H), 1.36 (m, 1H), 1.27 (m, 4H), 1.21 (s, 6H). MS (ES +, m/z): (M+H) +: 419.6. |
| To a solution of trans-4-((tert-butoxycarbonyl)amino)cyclohexanecarboxylic acid (823 g, 3.38 mol) in EtOAc (4000 mL) was added EA/HCl (2500 mL). The mixture was stirred at 0° C. overnight. The reaction mixture was filtered and dried in vacuo to give a product of hydrochloride salt of trans-4-aminocyclohexanecarboxylic acid as white solid (604 g, 99.42% yield). |
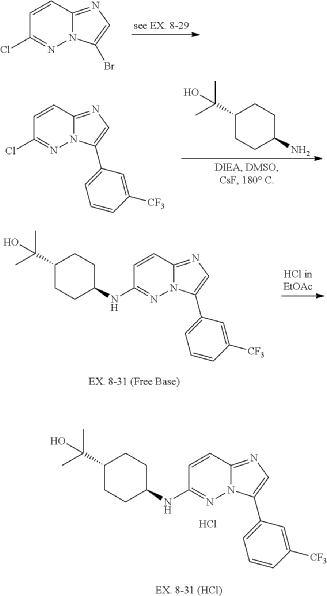
| 6-chloro-3-(3-(trifluoromethyl)phenyl)imidazo[1,2-b]pyridazine was prepared according to procedure in EX. 8-29. |
PAT
- Heterocyclic protein kinase inhibitorsPublication Number: ES-2834093-T3Priority Date: 2011-07-21Grant Date: 2021-06-16
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2021238183-A1Priority Date: 2011-07-21
- Imidazo[1,2-b]pyridazine and pyrazolo[1,5-a]pyrimidine derivatives and their use as protein kinase inhibitorsPublication Number: US-2012058997-A1Priority Date: 2006-11-06
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-9416132-B2Priority Date: 2011-07-21Grant Date: 2016-08-16
- Heterocyclic protein kinase inhibitorsPublication Number: WO-2013013188-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3409278-B1Priority Date: 2011-07-21Grant Date: 2020-09-16
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10875864-B2Priority Date: 2011-07-21Grant Date: 2020-12-29
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3812387-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10392392-B2Priority Date: 2011-07-21Grant Date: 2019-08-27
- Heterocyclic protein kinase inhibitorsPublication Number: US-2014329807-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2017002014-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2019071446-A1Priority Date: 2011-07-21
- Substituted imidazo[1,2-b]pyridazines as protein kinase inhibitorsPublication Number: US-2020102313-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-2734205-B1Priority Date: 2011-07-21Grant Date: 2018-03-21
- Heterocyclic protein kinase inhibitorsPublication Number: EP-3409278-A1Priority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: JP-2014520898-APriority Date: 2011-07-21
- Heterocyclic protein kinase inhibitorsPublication Number: JP-6105578-B2Priority Date: 2011-07-21Grant Date: 2017-03-29
- Substituted imidazo[1,2-B]pyridazines as protein kinase inhibitorsPublication Number: US-10047093-B2Priority Date: 2011-07-21Grant Date: 2018-08-14
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
REF
– Nuvisertib (TP-3654), an investigational highly selective oral PIM1 kinase inhibitor, is being evaluated in patients with relapsed or refractory myelofibrosis (MF) –
– Nuvisertib demonstrated symptom and spleen responses correlating with cytokine modulation in the preliminary Phase 1/2 data recently presented at the European Hematology Association (EHA) 2025 Congress –
MARLBOROUGH, Mass., June 12, 2025 /PRNewswire/ — Sumitomo Pharma America, Inc. (SMPA) today announced that the U.S. Food and Drug Administration (FDA) granted Fast Track Designation to nuvisertib (TP-3654) for the treatment of patients with intermediate or high-risk myelofibrosis (MF). The FDA Fast Track Designation is granted to investigational therapies being developed to treat serious or life-threatening conditions that demonstrate the potential to address unmet medical needs. Nuvisertib is an oral, investigational, highly selective inhibitor of PIM1 kinase, which demonstrated clinical activity including symptom and spleen responses correlating with cytokine modulation in the updated preliminary Phase 1/2 data presented at the European Hematology Association (EHA) 2025 Congress in Milan, Italy.
MF, a serious and rare type of blood cancer, is characterized by the buildup of fibrous tissues in the bone marrow which is caused by dysregulation in the Janus-associated kinase (JAK) signaling pathway. The clinical manifestations of MF include an enlarged spleen, debilitating symptoms and reduction in hemoglobin and/or platelets. MF affects 1 in 500,000 people worldwide.1
“This positive momentum for nuvisertib signals strong promise in our pipeline and reflects our dedication to addressing unmet medical needs on behalf of patients with myelofibrosis and their families,” said Tsutomu Nakagawa, Ph.D, President and Chief Executive Officer of SMPA. “Receiving FDA Fast Track Designation for nuvisertib in the treatment of myelofibrosis reinforces our confidence in its potential as a treatment option for patients facing a poor prognosis with limited treatment options. We are committed to working closely with the FDA to progress the clinical development of nuvisertib and bring an alternative treatment option to patients with myelofibrosis.”
Updated data from the ongoing Phase 1/2 study of nuvisertib in patients with relapsed/refractory MF were presented at the EHA Congress on June 12, 2025. Preliminary data showed that nuvisertib monotherapy appears to be well tolerated with no dose-limiting toxicities (DLTs). Evaluable patients showed clinical activity including a ≥25% spleen volume reduction (SVR25) in 22.2% of patients and a ≥50% reduction in total symptom score (TSS50) of 44.4% of patients, as well as improvement of bone marrow fibrosis (42.9% patients), hemoglobin (24% patients) and platelet count (26.7% patients). Data also showed that nuvisertib treatment led to significant cytokine modulation [reduction of pro-inflammatory cytokines (e.g. EN-RAGE, MIP-1β) and increase of anti-inflammatory cytokines (e.g. adiponectin)], which demonstrated significant (p<0.001) correlation with symptom and spleen responses. Preclinical2 and emerging clinical data support the development of nuvisertib in combination with JAK inhibitors for the treatment of patients with MF.
“The data observed to date demonstrate promising clinical activity for nuvisertib and the strong potential for selective PIM1 inhibition to slow the progression of myelofibrosis,” said Jatin Shah, MD, Chief Medical Officer, Oncology. “Patients with myelofibrosis are in need of new therapeutic approaches, including combination treatment options, that can provide increased and durable response rates with limited hematologic adverse events. The FDA Fast Track Designation reinforces the potential of nuvisertib to provide clinical benefits for patients with myelofibrosis, an unmet medical need.”
About Nuvisertib (TP-3654)
Nuvisertib (TP-3654) is an oral investigational selective inhibitor of PIM1 kinase, which has shown potential antitumor and antifibrotic activity through multiple pathways, including induction of apoptosis in preclinical models.2,3 Nuvisertib was observed to inhibit proliferation and increase apoptosis in murine and human hematopoietic cells expressing the clinically relevant JAK2 V617F mutation.3 Nuvisertib alone and in combination with ruxolitinib showed white blood cell and neutrophil count normalization, and also reduced spleen size and bone marrow fibrosis in JAK2 V617F and MPLW515L murine models of myelofibrosis.2 The safety and efficacy of nuvisertib is currently being clinically evaluated in a Phase 1/2 study in patients with intermediate and high-risk myelofibrosis (NCT04176198). The U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation to nuvisertib for the indication of myelofibrosis in May 2022. The Japan Ministry of Health, Labour and Welfare (MHLW) granted Orphan Drug Designation to nuvisertib for the treatment of myelofibrosis in November 2024.
About Sumitomo Pharma
Sumitomo Pharma Co., Ltd., is a global pharmaceutical company based in Japan with key operations in the U.S. (Sumitomo Pharma America, Inc.), Canada (Sumitomo Pharma Canada, Inc.), and Europe (Sumitomo Pharma Switzerland GmbH) focused on addressing patient needs in oncology, urology, women’s health, rare diseases, psychiatry & neurology, and cell & gene therapies. With several marketed products in the U.S., Canada, and Europe, a diverse pipeline of early- to late-stage assets, we aim to accelerate discovery, research, and development to bring novel therapies to patients sooner. For more information on SMPA, visit our website https://www.us.sumitomo-pharma.com or follow us on LinkedIn.
The Sumitomo corporate symbol mark is a trademark of Sumitomo Pharma Co., Ltd., used under license. SUMITOMO PHARMA is a trademark of Sumitomo Pharma Co., Ltd., used under license. SUMITOMO is a registered trademark of Sumitomo Chemical Co., Ltd., used under license. Sumitomo Pharma America, Inc. is a U.S. subsidiary of Sumitomo Pharma Co., Ltd.
©2025 Sumitomo Pharma America, Inc. All rights reserved.
References
- U.S. National Library of Medicine. (n.d.). Primary myelofibrosis: Medlineplus Genetics. MedlinePlus. https://medlineplus.gov/genetics/condition/primary-myelofibrosis/
- Dutta A., Nath D, Yang Y, et al. Genetic ablation of Pim1 or pharmacologic inhibition with TP-3654 ameliorates myelofibrosis in murine models. Leukemia. 2022; 36 (3): 746-759. doi: 10.1038/s41375-021-01464-2.
- Foulks JM, Carpenter KJ, Luo B, et al. A small-molecule inhibitor of PIM kinases as a potential treatment for urothelial carcinomas. Neoplasia. 2014;16(5):403-412.
SOURCE Sumitomo Pharma America
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer DrugsPublication Name: International Journal of Molecular SciencesPublication Date: 2021-08-30PMCID: PMC8431370PMID: 34502348DOI: 10.3390/ijms22179440
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- PIM kinase inhibitors: Structural and pharmacological perspectivesPublication Name: European Journal of Medicinal ChemistryPublication Date: 2019-06-15PMID: 30954777DOI: 10.1016/j.ejmech.2019.03.050
- A Small-Molecule Inhibitor of PIM Kinases as a Potential Treatment for Urothelial CarcinomasPublication Name: Neoplasia (New York, N.Y.)Publication Date: 2014-05PMCID: PMC4198696PMID: 24953177DOI: 10.1016/j.neo.2014.05.004
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- The Second-Generation PIM Kinase Inhibitor TP-3654 Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Cytotoxic Anticancer DrugsPublication Name: International Journal of Molecular SciencesPublication Date: 2021-08-30PMCID: PMC8431370PMID: 34502348DOI: 10.3390/ijms22179440
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- PIM kinase inhibitors: Structural and pharmacological perspectivesPublication Name: European Journal of Medicinal ChemistryPublication Date: 2019-06-15PMID: 30954777DOI: 10.1016/j.ejmech.2019.03.050
- A Small-Molecule Inhibitor of PIM Kinases as a Potential Treatment for Urothelial CarcinomasPublication Name: Neoplasia (New York, N.Y.)Publication Date: 2014-05PMCID: PMC4198696PMID: 24953177DOI: 10.1016/j.neo.2014.05.004
///////Nuvisertib, serine/ threonine kinase inhibitor, antineoplastic, Orphan Drug, myelofibrosis, SGI-9481, SGI 9481, TP-3654, TP 3654, EOB0N7BOY4
Neladalkib
Neladalkib
CAS 2739866-40-9
MF C23H22ClFN6O MW 452.9 g/mol
(19R)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(25),2(6),4,8,11,13(18),14,16,21,23-decaen-22-amine

anaplastic lymphoma kinase (ALK) inhibitor, antineoplastic, NVL-655, NVL 655, J32P26A6BC, ALK-IN-27
Neladalkib is a small molecule drug. The usage of the INN stem ‘-alkib’ in the name indicates that Neladalkib is a ALK (anaplastic lymphoma kinase) inhibitor. Neladalkib is under investigation in clinical trial NCT06765109 (Neladalkib (NVL-655) for TKI-naive Patients With Advanced ALK-Positive NSCLC). Neladalkib has a monoisotopic molecular weight of 452.15 Da.
ALK Inhibitor NVL-655 is an orally bioavailable, brain-penetrant, selective small molecule inhibitor of the receptor tyrosine kinase (RTK) anaplastic lymphoma kinase (ALK), with potential antineoplastic activity. Upon oral administration, ALK inhibitor NVL-655 specifically targets, binds to and inhibits ALK fusion proteins and activating mutations, including the acquired resistance mutations solvent front mutation (SFM) G1202R and the compound mutations G1202R/L1196M and G1202R/G1269A. The inhibition of ALK leads to the disruption of ALK-mediated signaling and the inhibition of cell growth in ALK-expressing tumor cells. ALK belongs to the insulin receptor superfamily and plays an important role in nervous system development. ALK is not expressed in healthy adult human tissue but ALK dysregulation and gene rearrangements are associated with a variety of tumor cell types. NVL-655 is able to penetrate the blood-brain-barrier (BBB) and may therefore exert its activity against EGFR-driven central nervous system (CNS) primary tumors and CNS metastases.
- Expanded Access Program of Neladalkib (NVL-655) for Patients With Advanced ALK+ NSCLC or Other ALK+ Solid TumorsCTID: NCT06834074Status: AvailableDate: 2025-09-22
- Neladalkib (NVL-655) for TKI-naive Patients With Advanced ALK-Positive NSCLCCTID: NCT06765109Phase: Phase 3Status: RecruitingDate: 2025-08-29
- A Study of Neladalkib (NVL-655) in Patients With Advanced NSCLC and Other Solid Tumors Harboring ALK Rearrangement or Activating ALK Mutation (ALKOVE-1)CTID: NCT05384626Phase: Phase 1/Phase 2Status: RecruitingDate: 2025-07-24
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023196910&_cid=P20-MHSIQF-58684-1
SYN
PAT
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2022098212-A1Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2022340586-A9Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-2023076627-A1Priority Date: 2020-05-05
- Heteroaromatic macrocyclic ether chemotherapeutic agentsPublication Number: US-11667649-B2Priority Date: 2020-05-05Grant Date: 2023-06-06
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: US-2023322797-A1Priority Date: 2022-04-07
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: WO-2023196900-A1Priority Date: 2022-04-07
- Solid forms, pharmaceutical compositions and preparation of heteroaromatic macrocyclic ether compoundsPublication Number: WO-2023196900-A9Priority Date: 2022-04-07
- Methods of treating solid tumor using (19r)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentaazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(24),2(6),4,8,11,13,15,17,21(25),22-decaen-22-aminePublication Number: WO-2023196910-A1Priority Date: 2022-04-07
- Methods of treating solid tumor using (19r)-5-chloro-3-ethyl-16-fluoro-10,19-dimethyl-20-oxa-3,4,10,11,23-pentaazapentacyclo[19.3.1.02,6.08,12.013,18]pentacosa-1(24),2(6),4,8,11,13,15,17,21(25),22-decaen-22-aminePublication Number: EP-4504189-A1Priority Date: 2022-04-07
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////neladalkib, antineoplastic, NVL-655, NVL 655, J32P26A6BC, ALK-IN-27
Nefextinib
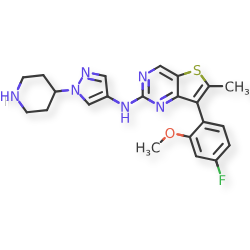
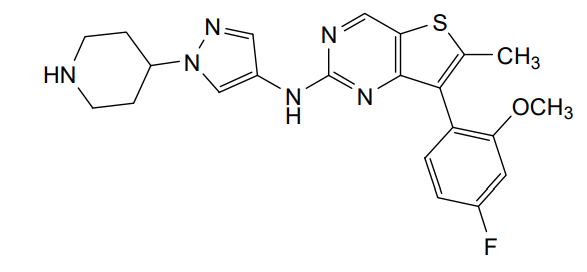
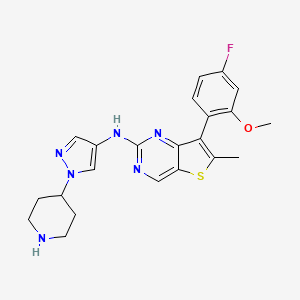
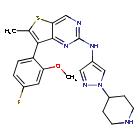
Nefextinib
CAS 2070931-57-4
MF C22H23FN6OS MW 438.52
7-(4-fluoro-2-methoxyphenyl)-6-methyl-N-[1-(piperidin4-yl)-1H-pyrazol-4-yl]thieno[3,2-d]pyrimidin-2-amine
7-(4-FLUORO-2-METHOXYPHENYL)-6-METHYL-N-(1-(PIPERIDIN-4-YL)-1H-PYRAZOL-4-YL) THIENO (3,2-D)PYRIMIDIN-2-AMINE
tyrosine kinase inhibitor, antineoplastic, DL772G3NN7, MAX-40279, MAX 40279
Nefextinib is an orally bioavailable inhibitor of the fibroblast growth factor receptor (FGFR) and FMS-like tyrosine kinase 3 (FLT3; CD135; STK1; FLK2), with potential antineoplastic activity. Upon oral administration, nefextinib binds to and inhibits both FGFR and FLT3, including FLT3 mutant forms, which results in the inhibition of FGFR/FLT3-mediated signal transduction pathways. This inhibits proliferation in FGFR/FLT3-overexpressing tumor cells. FGFR, a family of receptor tyrosine kinases, is upregulated in many tumor cell types. FLT3, a class III receptor tyrosine kinase (RTK), is overexpressed or mutated in most B-lineage neoplasms and in acute myeloid leukemias. They both play key roles in cellular proliferation and survival.
- A Phase 2 Study to Evaluate the Safety and Efficacy of Max-40279-01 in Patients With Advanced Gastric Cancer or Gastroesophageal Junction CancerCTID: NCT05395780Phase: Phase 2Status: Unknown statusDate: 2022-06-02
- MAX-40279 in Subjects With Acute Myelogenous Leukemia (AML)CTID: NCT03412292Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- MAX-40279-01 in Patients With Advanced Solid TumorsCTID: NCT04183764Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- Study of MAX-40279 in Patients With Relapsed or Refractory Acute Myelogenous Leukemia (AML)CTID: NCT04187495Phase: Phase 1Status: Unknown statusDate: 2022-01-19
- A Clincal Study of Max-40279-01 in Patients With Advanced Colorectal CancerCTID: NCT05130021Phase: Phase 2Status: Unknown statusDate: 2021-12-06
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017012559&_cid=P22-MHRG1L-67142-1
[0488]N-[7-(4-fluoro-2-methoxyphenyl)-6-methylthieno[3,2-d]pyrimidin-2-yl]-1-(piperidin-4-yl)-1H-pyrazol-4-amine (compound 31)
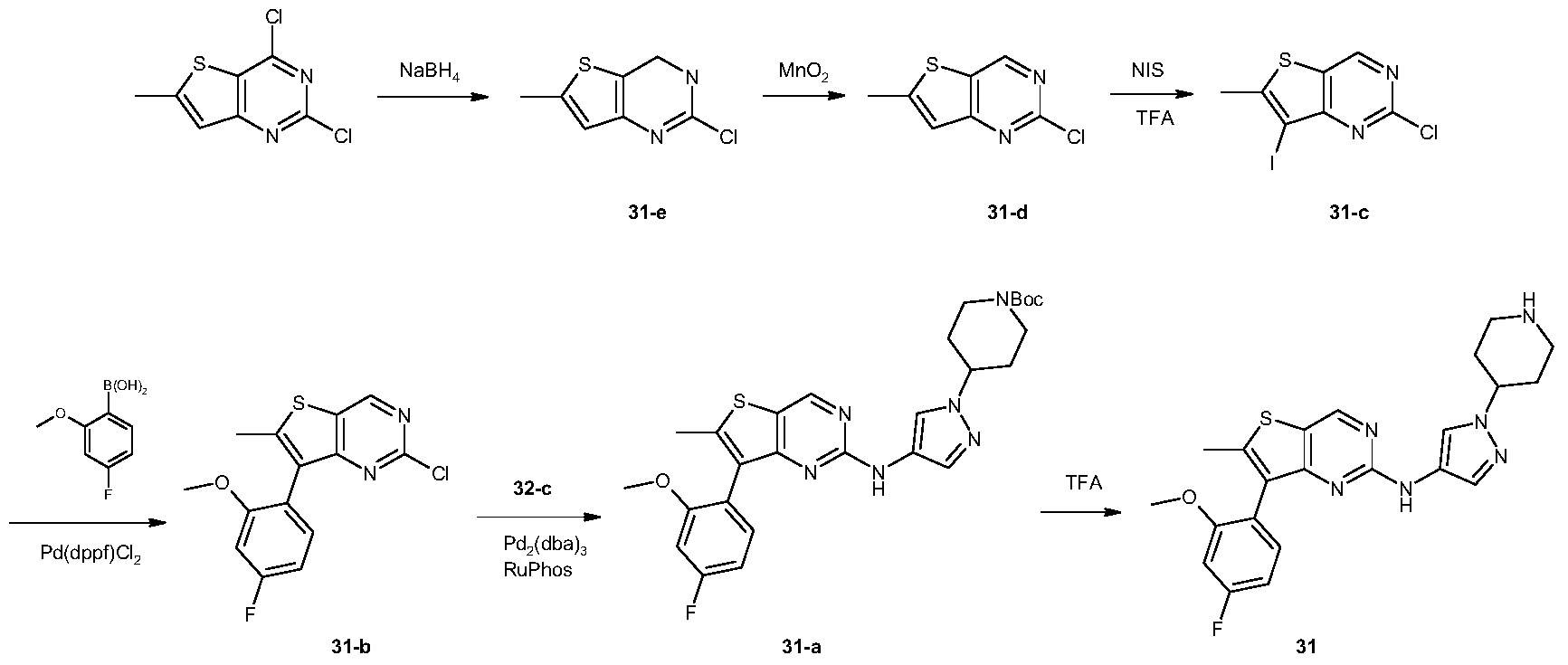
[0491]2,4-Dichloro-6-methylthiophene[3,2-d]pyrimidine (10 g, 45.6 mmol) was dissolved in tetrahydrofuran (100 mL) and ethanol (100 mL). The reaction mixture was cooled to 0 °C, and sodium borohydride (12.5 g, 198 mmol) was added in portions. The reaction mixture was brought to room temperature and stirred for 16 hours. It was then diluted with water (500 mL) and adjusted to pH 7 with 1 N hydrochloric acid solution. The aqueous phase was extracted with ethyl acetate (150 mL × 3). The organic phase was washed successively with water (100 mL × 3) and saturated brine (100 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a white solid 31-e (7.5 g, yield: 88%). This product required no further purification. LC-MS (ESI): m/z = 187 [M+H] + .
[0492]Synthesis of compound 31-d
[0493]Compound 31-e (7.5 g, 40 mmol) was dissolved in chloroform (300 mL) at 0 °C, and activated manganese dioxide (35 g, 400 mmol) was added. The reaction mixture was brought to room temperature and stirred for 16 hours. The reaction mixture was filtered through diatomaceous earth, and the filter cake was washed with chloroform (100 mL × 3). The combined filtrates were concentrated under reduced pressure to give a white solid 31-d (6.6 g, yield: 89%), which did not require further purification. LC-MS (ESI): m/z = 185 [M + H]+.
[0494]Synthesis of compound 31-c
[0495]Compound 31-d (3.1 g, 16.8 mmol) was dissolved in trifluoroacetic acid (30 mL) at 0 °C. N-iodosuccinimide (5.7 g, 25.3 mmol) was added in portions. The reaction mixture was brought to room temperature and stirred for 1 hour. The reaction was quenched with water (50 mL) and extracted with dichloromethane (50 mL × 3). The organic phase was washed successively with water (50 mL × 3) and saturated brine (50 mL), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a white solid 31-c (4.9 g, yield: 94%). This product required no further purification. LC-MS (ESI): m/z = 311 [M + H] + .
[0496]Synthesis of compound 31-b
[0497]Compound 31-c (615 mg, 1.98 mmol), 2-methoxy-4-fluorophenylboronic acid (405 mg, 2.38 mmol), and sodium carbonate (630 mg, 5.94 mmol) were suspended in dioxane (5 mL) and water (5 mL). A [1,1′-bis(diphenylphosphine)ferrocene]palladium dichloride dichloromethane complex (163 mg, 0.2 mmol) was added. The mixture was purged three times with nitrogen and heated to 80 °C for 16 hours. After cooling to room temperature, the reaction solution was concentrated under reduced pressure. The residue was separated into layers by dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated and purified by silica gel column chromatography (petroleum ether:dichloromethane = 1:1) to give a white solid 31-b (240 mg, yield: 39%). LC-MS (ESI): m/z = 309 [M+H] + .
[0498]Synthesis of compound 31-a
[0499]Compound 31-b (240 mg, 0.78 mmol) and compound 32-c (208 mg, 0.78 mmol) were dissolved in N,N-dimethylformamide (3 mL), and potassium carbonate (323 mg, 2.34 mmol), 2-dicyclohexylphosphine-2′,6′-diisopropoxy-1,1′-biphenyl (112 mg, 0.24 mmol), and tris(dibenzylacetone)palladium (134 mg, 0.24 mmol) were added. The reaction was carried out under nitrogen protection at 110 °C for 16 hours. After cooling to room temperature, the reaction mixture was separated into layers by dichloromethane (50 mL) and water (50 mL). The organic phase was dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel thin-layer chromatography (petroleum ether: ethyl acetate = 1:1) to give a yellow viscous oil 31-a (190 mg, yield: 45%). LC-MS(ESI): m/z = 539[M+H] + .
[0500]Synthesis of Compound 31
[0501]31-a (190 mg, 0.35 mmol) was dissolved in dichloromethane (3 mL), and trifluoroacetic acid (3 mL) was added. The mixture was stirred at room temperature for 3 hours. The reaction solution was concentrated under reduced pressure, and the residue was separated into layers by ethyl acetate (50 mL) and 1N hydrochloric acid aqueous solution (50 mL). The aqueous phase was adjusted to pH = 10 with saturated potassium carbonate aqueous solution, and a solid precipitated. The solid was filtered, and the filter cake was washed with water (20 mL × 3). The solid was dried under vacuum to give a light yellow solid 31 (22 mg, yield: 14%). LC-MS (ESI): m/z = 439 [M+H] + .
[0502]
1H-NMR(400MHz,MeOD)δ:8.78(d,J=5Hz,1H),7.87(s,1H),7.48(s,1H),7.35(m,1H),7.05(dd,J=11Hz,J=2Hz,1H),6.91(m,1H),4.10(m,1H),3.79(s,3H),3.22(m,2H),2.77(m,2H),2.47(s,3H),2.03(m,2H),1.73(m,2H)ppm
PAT
- Condensed ring pyrimidine compound, intermediate, its preparation method, composition and applicationPublication Number: CN-106366093-BPriority Date: 2015-07-21Grant Date: 2020-08-18
- Condensation ring pyrimidine compounds, intermediates, methods for producing them, compositions and applicationsPublication Number: JP-6875372-B2Priority Date: 2015-07-21Grant Date: 2021-05-26
- Condensed ring pyrimidine compounds, intermediates, preparation methods, compositions and applications thereofPublication Number: KR-102591886-B1Priority Date: 2015-07-21Grant Date: 2023-10-20
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: EP-3354653-B1Priority Date: 2015-07-21Grant Date: 2019-09-04
- Fused ring pyrimidine compounds, intermediates, production methods, compositions and applications thereofPublication Number: JP-2018520202-APriority Date: 2015-07-21
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: US-10494378-B2Priority Date: 2015-07-21Grant Date: 2019-12-03
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: US-2018208604-A1Priority Date: 2015-07-21
- Fused ring pyrimidine compound, intermediate, and preparation method, composition and use thereofPublication Number: WO-2017012559-A1Priority Date: 2015-07-21
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////nefextinib, tyrosine kinase inhibitor, antineoplastic, DL772G3NN7, MAX-40279, MAX 40279
Muvadenant
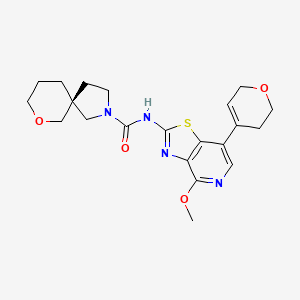
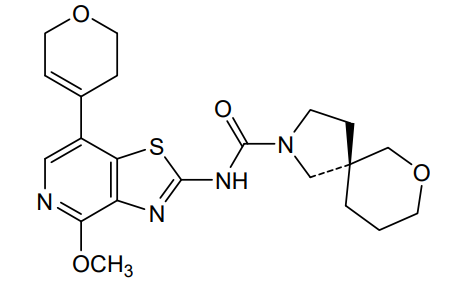
Muvadenant
CAS 2459881-03-7
MF C21H26N4O4S , 430.5 g/mol
(5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy[1,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5] decane-2-carboxamide
(5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide
adenosine receptor antagonist, antineoplastic, 6LSF69F6A8, M1069 , M 1069
Muvadenant is a small molecule drug. The usage of the INN stem ‘-adenant’ in the name indicates that Muvadenant is a adenosin receptor antagonist. Muvadenant has a monoisotopic molecular weight of 430.17 Da.
Adenosine is an ubiguitous modulator of numerous physiological activities, particularly within the cardiovascular, nervous and immune systems. Adenosine is related both structurally and metabolically to the bioactive nucleotides adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP) and cyclic adenosine monophosphate (cAMP), to the biochemical methylating agent S-adenosyl-L-methione (SAM) and structurally to the coenzymes NAD, FAD and coenzym A and to RNA.
Via cell surface receptors, adenosine modulates diverse physiological functions including induction of sedation, vasodilatation, suppression of cardiac rate and contractility, inhibition of platelet aggregability, stimulation of gluconeogenesis and inhibition of lipolysis. Studies show that adenosine is able to activate adenylate cyclases, open potassium channels, reduce flux through calcium channels, and inhibit or stimulate phosphoinositide turnover through receptor-mediated
mechanisms (Muller C. E. and Stein B., Current Pharmaceutical Design, 2: 501 , 1996; Muller C. E., Exp. Opin. Ther. Patents, 7(5): 419, 1997).
Adenosine receptors belong to the superfamily of G-protein-coupled receptors (GPCRs). Four major subtypes of adenosine receptors have been
pharmacologically, structurally and functionally characterized (Fredholm et al., Pharm. Rev., 46: 143-156, 1994) and referred to as A1, A2A, A2B and A3. Though the same adenosine receptor can couple to different G-proteins, adenosine A1 and A3 receptors usually couple to inhibitory G-proteins referred to as G, and Go which inhibit adenylate cyclase and down-regulate cellular cAMP levels. In contrast, the adenosine A2A and A2B receptors couple to stimulatory G-proteins referred to as Gs that activate adenylate cyclase and increase intracellular levels of cAMP (Linden J., Annu. Rev. Pharmacol. Toxicol., 41 : 775-87 2001).
PAT
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: US-2022119412-A1Priority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: EP-3914600-B1Priority Date: 2019-01-22Grant Date: 2024-08-07
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: CN-113939520-BPriority Date: 2019-01-22Grant Date: 2024-11-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: ES-2992081-T3Priority Date: 2019-01-22Grant Date: 2024-12-09
- Thiazolopyridine derivatives as adenosine receptor antagonists.Publication Number: JP-7600119-B2Priority Date: 2019-01-22Grant Date: 2024-12-16
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: AU-2020211697-A1Priority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: KR-20210116572-APriority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: CN-113939520-APriority Date: 2019-01-22
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: EP-3914600-A1Priority Date: 2019-01-22
- Thiazolopyridine derivative as an adenosine receptor antagonistPublication Number: JP-2022524914-APriority Date: 2019-01-22
- Combination therapy for cancerPublication Number: CN-117858723-APriority Date: 2021-06-07
- Combination treatment of cancerPublication Number: EP-4351640-A1Priority Date: 2021-06-07
- Combination Treatment of CancerPublication Number: JP-2024520764-APriority Date: 2021-06-07
- Combination Treatment of CancerPublication Number: US-2024279338-A1Priority Date: 2021-06-07
- Thiazolopyridine derivatives as adenosine receptor antagonistsPublication Number: WO-2020152132-A1Priority Date: 2019-01-22
- Novel crystalline forms of (s)-7-oxa-2-aza-spiro[4.5]decane-2-carboxylic acid [7-(3,6-dihydro-2h-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin-2-yl]-amide and co-crystal forms thereofPublication Number: TW-202415667-APriority Date: 2022-08-02
- Novel crystalline forms of (s)-7-oxa-2-aza-spiro[4.5]decane-2-carboxylic acid [7-(3,6-dihydro-2h-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin-2-yl]-amide and co-crystal forms thereofPublication Number: WO-2024028273-A1Priority Date: 2022-08-02
- Combination treatment of cancerPublication Number: WO-2022258622-A1Priority Date: 2021-06-07
- Combination treatment of cancerPublication Number: AU-2022288571-A1Priority Date: 2021-06-07
- Combination treatment of cancerPublication Number: CA-3220380-A1Priority Date: 2021-06-07
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020152132&_cid=P10-MHPOEV-06540-1
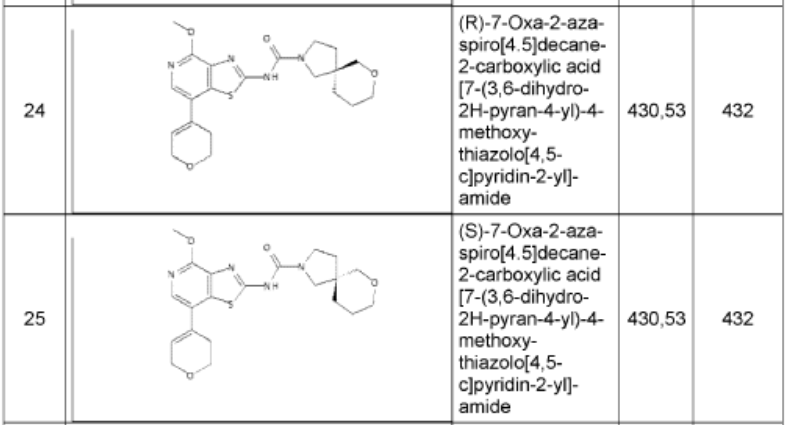
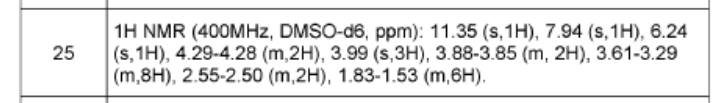
1. (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 24
and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5- c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 25
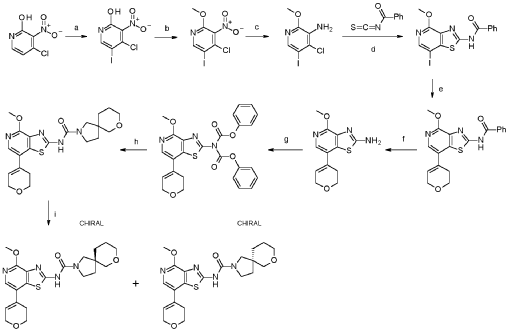
a. 4-chloro-5-iodo-3-nitropyridin-2-ol
Into a 250-mL round-bottom flask was placed 4-chloro-3-nitropyridin-2-ol (10.0 g, 54.4 mmol, 95%), N-lod-succinimid (NIS, 14.2 g, 59.9 mmol, 95%) in acetonitrile (115 mL). The solution was stirred for 1 h overnight at 80°C in an oil bath. The mixture was concentrated and the precipitate formed collected by filtration. The residue was washed with twice with petrol ether (500 mL) dried under vacuum at 60°C overnight. This resulted in 4-chloro-5-iodo-3-nitropyridin-2-ol (16.5 g, 97.9%, 97% purity) as a yellow solid. MS: m/z = 300.9 [M+H]+.
b. 4-chloro-5-iodo-2-methoxy-3-nitropyridine
Into a 500-mL round-bottom flask was placed 4-chloro-5-iodo-3-nitropyridin-2-ol (16.5 g, 53.3 mmol, 97%), Ag2CO3 (15.5 g, 53.3 mmol, 95%) in toluene (310 mL). To this suspension CH3I (15.9 g, 107 mmol, 95%) was added at 50°C and the mixture was stirred at 80°C for 4 h. The precipitate was collected by filtration and discarded. The filtrate was evaporated to dryness under vacuum and the residue purified by silica gel chromatography with ethyl acetate/petroleum ether (15:85).
This resulted in 4-chloro-5-iodo-2-methoxy-3-nitropyridine (9.90 g, 52.6%, 89% purity) as a light yellow solid. MS: m/z = 315.5 [M+H]+.
c. 4-chloro-5-iodo-2-methoxypyridin-3-amine
Into a 250-mL 3-necked round-bottom flask was placed 4-chloro-5-iodo-2-methoxy-3-nitropyridine (9.90 g, 28.0 mmol, 89%), iron (16.5 g, 281 mmol, 95%) and NH 4C (9.40 g, 174 mmol, 99%) in ethanol (152 mL) and water (30 mL). The mixture was stirred for 2 h at 80°C in an oil bath. The reaction mixture was filtered over Celite, washed with ethanol and the mother liquor was concentrated to dryness. The residue was stirred for 30 min. with 100 ml water at 60°dried in vacuo. This resulted in 4-chloro-5-iodo-2-methoxypyridin-3-amine (7.20 g, 75%, 83% purity) as an off-white solid. It was used without further purification in the next step. MS: m/z = 285.9 [M+H]+.
d. N-[7-iodo-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2-yl]benzamide
Into a 500-mL round-bottom flask was placed 4-chloro-5-iodo-2-methoxypyridin-3-amine (7.20 g, 21.0 mmol, 83%) in acetone (150 mL) and benzoyl isothiocyanate (5.21 g, 31.5 mmol, 99%) was added dropwise at room temperature. The solution was stirred for 1 h at 50 °C in an oil bath. The solids were collected by filtration, washed with acetone and dried in vacuo to give N-[7-iodo-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (8.73 g, 91 %, 90% purity) as a white solid. MS: m/z = 412.2 [M+H]+.
e. N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin- 2-yl]benzamide
To a solution of N-[7-iodo-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (6.00 g, 13.1 mmol, 90%) and 2-(3,6-dihydro-2H-pyran-4-yl)-4,4,5,5-tetramethyl-1 ,3,2-dioxaborolane (6.13 g, 27.7 mmol, 95%) in dioxane (200 mL) and water (40.00 mL) were added NaOH (2.90 g, 68.9 mmol, 95%) and Pd(dppf)Cl2* dichloromethane (1.20 g, 1.40 mmol, 95%). After stirring for 1 h at 100°C under a nitrogen atmosphere, the mixture was concentrated to dryness under vacuo. The residue was purified by silica gel chromatography with ethyl acetate/hexane (95:5). This resulted in 3.32 g (62%, 90% purity) of N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide as colorless solid. MS: m/z = 368.1 [M+H]+.
f. 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin-2- amine
To a stirred mixture of N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]benzamide (3.27 g, 8.00 mmol, 90%) in water/methanol (1 :1 , 300 mL) was added NaOH (3.36 g, 80.0 mmol, 95%) at room temperature under nitrogen atmosphere. The mixture was stirred for overnight at 90°C under nitrogen atmosphere and evaporated to dryness. The residue was taken up in water and extracted 3 times with dichloromethane (100 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography, eluted with petrol ether/ethyl acetate (1 :1) to afford 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-amine (1.50 g, 68%, 96% purity) as a light brownish solid. MS: m/z = 264.1 [M+H]+.
g. phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy- [1,3]thiazolo[4,5c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate
To a stirred solution of 7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-amine (600 mg, 2.19 mmol, 96%) and phenyl chloroformate (1.81 g,
11.0 mmol, 95%) in THF (50 mL) was added K2CO3 (1.59 g, 11.0 mmol, 95%) and pyridine (913 mg, 11.0 mmol, 95%) at room temperature under nitrogen
atmosphere. The mixture was stirred for 6 h at 50° and then after re-cooling to room temperature quenched by the addition of water (300 mL). The mixture was extracted 3 times with dichloromethane (200 mL), the combined organic layers were washed once with brine (200 mL), dried over anhydrous Na2SO4, filtered, and evaporated to dryness under reduced pressure. This resulted in phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate (1.00 g, 69%, 76% purity) as a light yellow solid. The crude product was used in the next step directly without further purification. MS: m/z = 504.1 [M+H]+.
h. N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1,3]thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide
To a mixture of phenyl N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-N-(phenoxycarbonyl)carbamate (1.00 g, 1.52 mmol, 76.) and bis(7-oxa-2-azaspiro[4.5]decane), oxalic acid (1.19 g, 3.03 mmol, 95%) in THF (50 mL) was added diisopropylethyl amine (1.24 g, 9.09 mmol, 95%) at room temperature under nitrogen atmosphere. The mixture was stirred for 1 h at 60°. After re-cooling to room temperature, the mixture was extracted twice with dichloromethane (100 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated to dryness. The residue was purified by silica gel column chromatography, eluted with petrol ether/ethyl acetate (1 :1) to afford N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (600 mg, 92%) as a white solid. HPLC: 99.9 % purity, RT = 1.17 min. MS: m/z = 431.1 [M+H]+. 1 H NMR (300 MHz, DMSO-d6) d 1 1.37 (s, 1 H), 7.95 (s, 1 H), 6.25 (s, 1 H), 4.30-4.29 (m, 2H), 3.99 (s, 3H), 3.89 (t, J=5.4Hz, 2H), 3.61-3.29 (m, 8H), 2.55-2.51 (m, 2H), 1.82-1.54 (m, 6H).
i. (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5-c]pyridin- 2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 24
and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-thiazolo[4,5- c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide 25
N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (450 mg, 1.044 mmol, 1 equiv, 99.9%) was purified by chiral-preparative HPLC (Preparative HPLC-032, column: ChiralPak IA, 2*25cm, 5 mm; mobile phase, dichloromethane:ethanol (20:80); detector, UV). This resulted in (5R)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (178 mg, 39%) as a white solid. HPLC: 99.7 % purity, RT (chiral) = 3.86 min, 100% ee. MS: m/z = 431.2 [M+H]+. 1 H NMR (400 MHz, DMSO-d6) d 1 1.36 (s, 1 H), 7.94 (s, 1 H), 6.24 (s, 1 H), 4.29-4.27 (m, 2H), 3.97 (s,3H), 3.88 (t, J=5.2 Hz, 2H), 3.51-3.19 (m, 8H), 2.55-2.50 (m, 2H), 1.83-1.53 (m, 6H) and (5S)-N-[7-(3,6-dihydro-2H-pyran-4-yl)-4-methoxy-[1 ,3]thiazolo[4,5-c]pyridin-2-yl]-7-oxa-2-azaspiro[4.5]decane-2-carboxamide (171 mg, 38%) as a white solid. HPLC: 99.8 % purity, RT (chiral) = 5.23 min, 99.9% ee. MS: m/z = 431.2 [M+H]+. 1 H NMR (400 MHz, DMSO-d6) d 1 1.35 (s, 1 H), 7.94 (s, 1 H), 6.24 (s, 1 H), 4.29-4.28 (m, 2H), 3.99 (s, 3H), 3.88-3.85 (m, 2H), 3.61-3.29 (m, 8H), 2.55-2.50 (m,2H), 1.83-1.53 (m,6H).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024028273&_cid=P10-MHPOFP-06905-1
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////muvadenant, adenosine receptor antagonist, antineoplastic, 6LSF69F6A8, M1069 , M 1069
Lunresertib
Lunresertib
CAS 2719793-90-3
MF C18H20N4O2 MW 324.4 g/mol
(1P)-2-amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethyl1H-pyrrolo[2,3-b]pyridine-3-carboxamide
serine/ threonine kinase inhibitor, antineoplastic, N95U3A7N57, RP-6306, RP 6306
2-Amino-1-(3-hydroxy-2,6-dimethylphenyl)-5,6-dimethylpyrrolo[2,3-b]pyridine-3-carboxamide
Lunresertib is an investigational new drug that is being evaluated for the treatment of cancer. It is an oral small molecule inhibitor of PKMYT1, developed by Repare Therapeutics.[1] This drug targets cell cycle regulation in tumors with specific genetic alterations, including CCNE1 amplifications or FBXW7 and PPP2R1A loss of function mutations. It is currently in phase 1/2 clinical trials, both as monotherapy or in combination with camonsertib, an ATR inhibitor.[2]
Lunresertib is an orally bioavailable inhibitor of the human membrane-associated tyrosine– and threonine-specific cdc2-inhibitory kinase (PKMYT1), with potential antineoplastic activity. Upon oral administration, lunresertib targets, binds to and inhibits the activity of PKMYT1. This results in the inhibition of CDK1 phosphorylation, which may promote both premature mitosis and a prolonged mitotic arrest, and lead to the accumulation of unrepaired DNA damage and apoptosis in susceptible tumor cells, such as CCNE1-overexpressing tumor cells. PKMYT1 phosphorylates CDK1 specifically when CDK1 is complexed to cyclins, which blocks progression from G2 into mitosis.NCI Thesaurus (NCIt)
- Study of RP-6306 With FOLFIRI in Advanced Solid TumorsCTID: NCT05147350Phase: Phase 1Status: TerminatedDate: 2025-08-20
- Study of RP-6306 Alone or in Combination With RP-3500 or Debio 0123 in Patients With Advanced Solid TumorsCTID: NCT04855656Phase: Phase 1Status: RecruitingDate: 2025-08-06
- RP-6306 in Patients With Advanced CancerCTID: NCT05605509Phase: Phase 2Status: Active, not recruitingDate: 2025-07-14
- Study of RP-6306 With Gemcitabine in Advanced Solid TumorsCTID: NCT05147272Phase: Phase 1Status: TerminatedDate: 2025-06-17
- Liquid-biopsy Informed Platform Trial to Evaluate CDK4/6-inhibitor Resistant ER+/HER2- Metastatic Breast CancerCTID: NCT05601440Phase: Phase 2Status: RecruitingDate: 2025-01-14
- Phase 1 Study of RP-6306 With Carboplatin and Paclitaxel in TP53 Ovarian and Uterine Cancer
- CTID: NCT06107868
- Phase: Phase 1
- Status: Active, not recruiting
- Date: 2024-03-22
PAT
- Compounds, Pharmaceutical Compositions, and Methods of Preparing and Using CompoundsPublication Number: JP-2023521633-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: US-2023151014-A1Priority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: US-2023158022-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: EP-4126879-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: IL-296934-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of making the compounds and methods of using themPublication Number: KR-20230011279-APriority Date: 2020-04-01
- Compounds, pharmaceutical compositions and methods of making compounds and methods of their usePublication Number: CN-115916783-APriority Date: 2020-04-01
- Methods of using MYT1 inhibitorsPublication Number: JP-2023519430-APriority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: WO-2021195782-A1Priority Date: 2020-04-01
- Compounds, pharmaceutical compositions, and methods of preparing compounds and of their usePublication Number: AU-2021250744-A1Priority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: CA-3173955-A1Priority Date: 2020-04-01
- Methods of using MYT1 inhibitorsPublication Number: CN-115811976-APriority Date: 2020-04-01
- Methods of using myt1 inhibitorsPublication Number: EP-4125907-A1Priority Date: 2020-04-01
SYN
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021195781&_cid=P20-MHLE6P-37080-1
Step 9. To a suspension of 2-amino-1-(3-methoxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (2.22 g, 6.56 mmol, 77% purity) in DCM (25 mL) was added tribromoborane in DCM (1 M, 26 mmol, 26 mL) dropwise. The reaction mixture was stirred at RT for 45 min, then concentrated to dryness. The crude product was taken in DCM and placed in an ice bath and MeOH was added carefully (exotherm). The mixture was concentrated to dryness then co-evaporated twice with MeOH. The residue was triturated with saturated aqueous NaHCO3. The solids were collected by filtration on a Buchner funnel, washed with H2O and air-dried. The still wet solid was dissolved in DCM/MeOH, concentrated to dryness and triturated in 20% MeOH/DCM (50 mL). The solid was collected by filtration, washed with 20% MeOH/DCM, air-dried then dried in vacuo to afford 2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (1.60g, 75% yield) as a light beige solid. MS: [M+1]: 325.1. A different batch was purified by preparative HPLC to yield 2-amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (63% yield) as an off-white fluffy solid.
1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 7.82 (s, 1H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J =
8.2 Hz, 1H), 6.71 (br s, 2H), 6.64 (br s, 2H), 2.26 (s, 3H), 2.23 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
Chiral SFC separation of Compound 181 (1.60g, 4.93 mmol) (Instrument: Waters Prep 100 SFC-MS; Column: Phenomenex Lux Cellulose-2, 30 x 250 mm, 5 μm; Conditions: isocratic at 55% IPA + 10mM Ammonium Formate with 45% CO2 ; Flow Rate: 70 mL/min) provided
Compound 182 and Compound 183.
Compound 182 from SFC separation of 181. Peak 1 (retention time 3.94 min, 99.86%): (S)-2- amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (381 mg) was obtained as an off white fluffy solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1 H), 7.05 (d, J = 8.3 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.72 (s, 2H), 6.65 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
Compound 183 from SFC separation of 181. Peak 2 (retention time 4.35 min, 98.09%): (R)-2- amino-1-(3-hydroxy-2,6-dimethyl-phenyl)-5,6-dimethyl-pyrrolo[2,3-b]pyridine-3-carboxamide (495 mg) was obtained as an off white fluffy solid. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H), 7.83 (s, 1 H), 7.05 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.72 (s, 2H), 6.66 (s, 2H), 2.26 (s, 3H), 2.24 (s, 3H), 1.74 (s, 3H), 1.65 (s, 3H). MS: [M+1]: 325.1.
SYN
https://pubs.acs.org/doi/full/10.1021/acs.oprd.4c00493
REF
- The Science and Art of Structure-Based Virtual ScreeningPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2024-03-25PMCID: PMC11017385PMID: 38628791DOI: 10.1021/acsmedchemlett.4c00093
- Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306Publication Name: Journal of Medicinal ChemistryPublication Date: 2022-07-26PMCID: PMC9837800PMID: 35880755DOI: 10.1021/acs.jmedchem.2c00552
- CCNE1 amplification is synthetic lethal with PKMYT1 kinase inhibitionPublication Name: NaturePublication Date: 2022-04-20PMCID: PMC9046089PMID: 35444283DOI: 10.1038/s41586-022-04638-9
- Contributions in the domain of cancer research: Review¶Negative regulators of cyclin-dependent kinases and their roles in cancersPublication Name: Cellular and molecular life sciences : CMLSPublication Date: 2001-11PMCID: PMC11337304PMID: 11766887DOI: 10.1007/pl00000826
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | RP-6306 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2719793-90-3 |
| PubChem CID | 156869388 |
| ChemSpider | 115008046 |
| UNII | N95U3A7N57 |
| KEGG | D12736 |
| ChEMBL | ChEMBL5199076 |
| Chemical and physical data | |
| Formula | C18H20N4O2 |
| Molar mass | 324.384 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Szychowski J, Papp R, Dietrich E, Liu B, Vallée F, Leclaire ME, et al. (August 2022). “Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306”. Journal of Medicinal Chemistry. 65 (15): 10251–10284. doi:10.1021/acs.jmedchem.2c00552. PMC 9837800. PMID 35880755.
- Previtali V, Bagnolini G, Ciamarone A, Ferrandi G, Rinaldi F, Myers SH, et al. (July 2024). “New Horizons of Synthetic Lethality in Cancer: Current Development and Future Perspectives”. Journal of Medicinal Chemistry. 67 (14): 11488–11521. doi:10.1021/acs.jmedchem.4c00113. PMC 11284803. PMID 38955347.
///////lunresertib, Serine/ threonine kinase inhibitor, antineoplastic, N95U3A7N57, RP-6306, RP 6306

{kind=link}
{kind=link}
{kind=link}