
Home » Posts tagged 'Antifungal'
Tag Archives: Antifungal
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Liranaftate
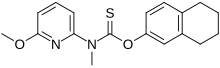
Liranaftate
リラナフタート
88678-31-3
(6-Methoxy-2-pyridinyl)methylcarbamothioic Acid O-(5,6,7,8-Tetrahydro-2-naphthalenyl) Ester
O-(5,6,7,8-Tetrahydronaphthalen-2-yl) (6-methoxypyridin-2-yl)methylcarbamothioate
Zefnart;Piritetrate;M-732
лиранафтат
ليرانافتات
利拉萘酯
| Formula | C18H20N2O2S |
|---|---|
| CAS | 88678-31-3 |
| Mol weight | 328.4286 |
| Efficacy | Antifungal, Ergosterol biosynthesis inhibitor |
|---|---|
| Comment | Thiocarbamate |
Liranaftate (trade name Zefnart) is a topical antifungal drug.[1] It is used as a 2% cream used to treat tinea pedis (athlete’s foot), tinea corporis (ringworm), and tinea cruris (jock itch).[2] It was approved for use in Japan in August 2000.[3][4]
Liranaftate works by inhibiting the fungal enzyme squalene epoxidase that is necessary for the fungus to synthesize sterols which are essential for cell membrane integrity.[5]
SYN
IN 2010MU02699
PAPER
Journal of Chemical and Pharmaceutical Research (2013), 5(11), 219-222,
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007010744
Conventionally, 0-aryl N- (6-alkoxy-2-pyridyl) -N-alkylthio-force rubamate has generally been produced by a method using thiophosgen. For example, in Patent Document 1, 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N- represented by the following reaction formula 0 or ii) A method for producing methylthiolbamate (4) is disclosed.
(Example 1)
1) Sodium 5, 6, 7, 8-Tetrahydro-2-naphthoside synthesis
[hua 6]
,She
To methanol (10 ml), 0.54 g (10.0 mmol) of sodium methoxide was added, and the mixture was stirred at room temperature. There, 1.50 g (10.0 mmol) of 5,6,7,8-tetrahydro-2-naphthol was added and he stirred for 1 hour at room temperature. The solvent was distilled off under reduced pressure to obtain 3.75 g ( q uant.) Of white powder. I left it overnight in a desiccator.
2) 2- [Ν- (1-imidazolithiocarbol) -Ν’-methyl] amino-6-methoxypyridin compound
[hua 7]
To ethyl acetate (30 ml), 2.07 g (15.0 mmol) of 6-methoxy-2-methylaminoviridin and 2.67 g (15.0 mmol) of 1,1, -thiocarboldiimidazole were added, and the mixture was heated under reflux for 2 hours. After allowing to cool, the solvent was distilled off under reduced pressure to obtain 3.70 g of brown oil. (Yield 99.3%). If necessary, further purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain pale yellow crystals.
Melting point: 58.0~60.0°C
NMR(CDC1 3 ) δ ppm:3.86(3H,s), 3.87(3H,s), 6.38 (lH’dd, J=7.5Hz, 0.7Hz), 6.61 (1H
,dd, J=8.3Hz, 0.7Hz), 6.82 (lH,t, J=1.0Hz) , 7.03 (lH,t, J=1.0Hz) , 7.46 (lH’dd, J= 8.3Hz, 7.5Hz), 7.72 (lH,t, J=1.0Hz)
IR(KBr)cm_1: 1604, 1590, 1571, 1465, 1359, 1303, 1120, 1013, 986, 822, 798 MS m/z: 248(M+)
3) Synthesis of 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) -N- (6-methoxy-2-pyridyl) -N-methylthiocarbamate
Dissolve 2- [N- (1-imidazolithiocarbol) -N-methyl] amino-6-methoxypyridin 250 mg (1.0 mmol) in N, N-dimethylformamide (4 ml), and then dissolve. At room temperature, Natrium 5, 6, 7, 8-tetrahydro-2-naphthoside 360 mg (2.0 mmol) was added. -After stirring at room temperature, the reaction solution was extracted with ethyl acetate (10 mlx2), and the insoluble material was filtered off on the way. The organic layer was washed with saturated brine, dried over magnesium sulfate, filtered off magnesium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) gave the title compound 266.6 mg (yield 81.3%).
Melting point: 99~100°C
NMR(CDCl 3) δ ppm:1.77(4H,bs), 2.75(4H,bs), 3.75(3H,s), 3.93(3H,s), 6.65(lH,d, J
=8.0Hz), 6.78-7.08(4H,m), 7.64(lH,t,J=8.0Hz)
IR(KBr) cm_1 : 1603, 1460, 1413, 1369, 1325, 1262, 1175, 1035, 808, 785
MS m/z: 328(M+)
(Example 2)
0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N-methylthio force Rubamate synthesis
[Chemical 9]
1.34 g (33.6 mmol) of 60% sodium hydride was added to N, N-dimethylformamide (20 ml), followed by the addition of 5, 6, 7, 8-tetrahydro-2-naphthol 4.65 g (30.5 mmol). After gas generation is complete, add 2- [N- (1-imidazolthiocarbonyl) -N-methyl] amino-6-methoxypyridin 7.45 g (30.0 mmol) and zinc chloride 2.05 g (15.0 mmol). rice field. After heating and stirring at 60 ° C for 3 hours and allowing to cool, the reaction solution was extracted with ethyl acetate (150 mlx2), and the insoluble material was filtered off on the way. The organic layer is washed with saturated brine, dried over magnesium sulfate, and filtered through magnesium sulfate.
Separately, the solvent was distilled off under reduced pressure. The obtained crystals were purified by one of the following methods.
[0028] A) Purification was performed by silica gel column chromatography (eco-gel C 200, hexane: ethyl silicate = 10: 1) to obtain 9.80 g of the indicated compound (yield 99.5%).
B) Suspended in hexane (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 9.65 g of crystals. Further, the mixture was suspended in methanol (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 8.62 g (yield 87.5%) of the indicated compound.
The physics and physics data of the obtained compound were consistent with the compounds obtained in the examples.
(Example 3)
1) Synthesis of 2- [N- [1-2 (1H) -pyridonylthiocarbol] -N-methyl] amino-6-methoxypyridine
[Chemical 10]
OMe
Add 6-methoxy-2-methylaminoviridin 690 mg (5.0 mmol) and 1, 1, -thiocarbol-di-2 (1H) -pyridone 1.16 g (5.0 mmol) to ethyl acetate (15 ml). Heated and refluxed for 1 hour. After allowing to cool, the solvent was distilled off under reduced pressure, and purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1)! ヽ, 297.4 mg of brown oil was obtained. (Yield 21.6%).
NMR(CDC1 3 ) δ ppm:3.77(3H,s), 3.93(3H,s), 6.66 (lH’dd, J=8.0Hz, 0.7Hz), 7.07 ( lH,d, J=8.0Hz), 7.14 (lH,d, J=7.5Hz) , 7.25 (lH’dd, J=8.0Hz, 4.0Hz) , 7.62 (lH’dd , J=8.0Hz, 7.5Hz), 7.78 (lH’dd, J=2.0Hz, 0.7Hz) , 8.43 (lH’dd, J=4.0Hz, 0.7Hz)
MS m/z: 275(M+)
[0031] 2) Synthesis of 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N-methylthiocarbamate
[Chemical 11]
OMe
N, N-dimethylformamide (2 ml), 2- [N- [1-2 (1H) -pyridonylthiocarbol] –N-methyl] amino-6-methoxypyridin 297 mg (1.08 mmol) and sodium 5 , 6, 7, 8-Tetrahydro-2-naphthoside 390 mg (2.16 mmol) was added and stirred overnight at room temperature. The reaction mixture was extracted with ethyl acetate (50 mlx2), the organic layer was washed with saturated brine, dried over magnesium sulfate, magnesium sulfate was filtered off, and the solvent was distilled off under reduced pressure. The obtained crystals were purified by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) to obtain the title compound 288.2 mg (81.4%).
SYN
CN 104725302
| Liranafate is a new-generation antifungal drug, a squalene cyclooxygenase inhibitor and a cell wall synthesis inhibitor, with the chemical name of 6-methoxy-2-N-methyl-pyridylamino-thio Formic acid-(5,6,7,8-tetrahydro)-β-naphthyl ester. A new type of antifungal drug jointly developed by Tosoh Corporation of Japan and Zenyaku Kogyo Corporation was first listed in Japan by Torii Corporation in August 2000. The antifungal drug exerts antifungal activity by inhibiting the squalene epoxidation reaction of fungal cells and inhibiting the synthesis of ergosterol, a component of cell membranes. effect is particularly evident. Today, with the increasing concern of the world about environmental pollution, the development of new green and effective drug synthesis methods is an important task faced by the research of drug synthesis. In recent years, room temperature ionic liquids have been widely used in various organic synthesis reactions as a new type of environmentally friendly reaction media. Compared with traditional organic solvents, ionic liquids have many advantages, such as extremely low vapor pressure, non-flammability, good thermal stability and recyclability. |
| At present, the main synthetic route of liranaftate is as follows: |
| |
| Among the four synthetic routes, the pyridine derivative intermediates of routes C and D need to be prepared through multi-step reactions, the routes are long, the steps are cumbersome, the actual operation is cumbersome, the cost is high, and they are not suitable for industrialized large-scale production. Although route A has simple steps, the yield of pyridine derivatives is low. Each intermediate structure in route B is relatively simple and easy to prepare, but this route uses 6-methoxy-2-methylaminopyridine and 5,6,7,8-tetrahydro-2-naphthoxysulfuryl chloride as raw materials to synthesize the In the process of lanaphthalate, isopropanol-water is used as the reaction medium, and the experiment shows that with the progress of the reaction, the reaction solution becomes viscous, and the reaction is difficult to complete. |
| Example 1 |
| (1) Ionic liquid [bmim]BF 4 Synthesis |
| |
| Add N-methylimidazole (14.8g, 0.18mol) and trichloroethane (80mL) to a dry 250mL three-neck flask, stir to make the mixture uniform, add 20.4mL of freshly distilled n-bromine to the dropping funnel Butane (26.03g, 0.19mol) was added dropwise for about 30min, and the reaction was refluxed for 4-5h (the reflux temperature was about 78±1℃). With the progress of the reaction, the reaction solution changed from colorless and transparent to white turbidity, light yellow turbidity, and the color gradually became darker until brownish red. After the reaction is completed, the liquids are separated into layers, the upper layer is lighter in color, which is the trichloroethane layer, and the lower layer is darker in color (brown red), which is the ionic liquid [bmim]Br layer. The prepared ionic liquid [bmim]Br and trichloroethane were separated, and the ionic liquid [bmim]Br was washed twice with trichloroethane, and then the trichloroethane in the ionic liquid [bmim]Br was washed with a water pump. The alkane was pumped away until the ionic liquid [bmim]Br liquid was no longer turbid, and then dried in a vacuum drying oven at 90 °C for 10-12 h to obtain relatively pure ionic liquid [bmim]Br. |
| |
| Then prepare 0.03mol NaBF 4 of aqueous solution. Add 6.58g (about 0.03mol) ionic liquid [bmim]Br and 5-10mL water to a 100mL round-bottomed single diameter flask, stir, ice-water bath, and dropwise add NaBF 4 The solution (completed dropwise addition in about 5min), continue to stir for 10-20min, the solution is yellow and transparent, pour it into a separatory funnel, extract twice with dichloromethane, combine the dichloromethane layers, and wash the dichloromethane layer 2 with 50 mL of water times, and then the dichloromethane layer was washed with anhydrous MgSO 4 Dry, filter, evaporate the dichloromethane under normal pressure in a water bath (50-52°C), and dry the remaining dark yellow viscous liquid in a vacuum drying oven at 90°C for 10-12h to obtain the ionic liquid [bmim]BF 4 。 |
| |
| (2) Synthesis of 6-methoxy-2-chloropyridine 2 |
| 2,6-dichloropyridine (10g, 0.068mol) and sodium methoxide (24.5g, 0.136mol) were put into the reaction flask, heated under reflux for 4-5h, and the reaction was completed by TLC (ethyl acetate: petroleum ether=1 : 15), concentrated to remove methanol, added 100 mL of water, extracted with ethyl acetate, combined the organic phases, washed with saturated brine, dried, filtered, and the filtrate was concentrated to obtain 9 g of a crude colorless oily product with a yield of 92.5%. used for the next reaction. |
| (3) Synthesis of 6-methoxy-2-methylaminopyridine 3 |
| Take 6-methoxy-2-chloropyridine 2 (9g, 0.127mol), cuprous chloride (1.72g, 0.0017mol) and methylamine aqueous solution (29mL, mass concentration is 25%-30%) and add it to the autoclave , sealed and heated to 120 °C for 7 h, the reaction was stopped, ethyl acetate was added for extraction, the organic phases were combined, washed with saturated brine, dried, and the filtrate was concentrated to obtain 6.18 g of brown oil, the yield was 71.2%, and the HPLC purity was 98% . |
| (4) Synthesis of 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride 4 |
| Mix 50 mL of ethyl acetate, thiophosgene (4.25 mL, 0.056 mol) and 5,6,7,8-tetrahydro-2-naphthol (6.3 g, 0.0425 mol), and cool it in an ice-salt bath to below 0 °C. Add 10 mL of potassium carbonate (3 g, 0.022 mol) solution, continue to stir the reaction after the dropwise addition, and check by TLC (developing solvent: petroleum ether) that the reaction is complete, add 100 mL of water, extract with ethyl acetate, wash the organic phase with saturated brine, Dry, filter, and concentrate the filtrate to obtain 8.7 g of yellow oil with a yield of 90.4%, which can be directly used in the next reaction without purification. |
| (5) Synthesis of Liranaftate 1 |
| The prepared ionic liquid [bmim]BF 4 (100mL), 6-methoxy-2-methylaminopyridine 3 (5.7g, 0.0413mol) and potassium carbonate (5.7g, 0.0413mol) were mixed, cooled with ice water, and slowly added dropwise 5,6,7,8 -Tetrahydro-2-naphthyloxysulfuryl chloride 4 (8.7g, 0.0385mol) was added dropwise for 4h, slowly added 150mL of water under full stirring, continued to stir for 20min, filtered, washed with deionized water to obtain 12.2g of crude product, collected The yield was 96.81%, and acetone was recrystallized to obtain 11 g of white crystalline powder, the yield was 90%, and the HPLC purity was 99.7%. mp: 98.8-99.5°C, IR (2973cm -1 , 2930cm -1 , 2852cm -1 , 1416cm -1 , 1264cm -1 , 1037cm -1 ), 1 HNMR: 1.8 (m, 4H); 6.68(d, 1H) ;6.86(dd,1H);3.78(s,3H);3.98(s,3H);6.68(d,1H);6.86(dd,1H);7.05(d,1H);7.10(d.1H); 7.65 (dd, 1H), MS (m/z: 328, 181, 165, 108). |
| Example 2 |
| Under the same conditions, the ionic liquid 1-n-butyl-3-methylimidazolium tetrafluoroborate ([bmim]BF 4 ), N-ethylpyridine tetrafluoroborate ([EPy]BF 4 ), 1-n-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF 6 ), 1-hydroxyethyl-2,3-dimethylimidazolium chloride (LOH), 1-cyanopropyl-3-methylimidazolium chloride (LCN), 1-carboxyethyl-3-methylimidazole Chloride salt (LOOH), [Hnmp]HSO 4 The effects of and [bmim]OH on the synthesis of liranaftate are shown in Table 1. The results show that different ionic liquids have little effect on the yield of the synthesis and the yields are relatively high. |
| Table 1 Effects of different ionic liquids on the reaction yield |
| ionic liquidYield/%[bmim] BF 496.81[EPy]BF 496.83[bmim]PF 696.82LOH96.75LCN96.67LOOH96.05[Hnmp]HSO 496.06[bmim]OH95.98 |
| Example 3 |
| Whether the reaction medium used can be recovered and reused is an important content of “green chemistry”. This example specifically examines the reuse of ionic liquid for synthesizing liranaftate. After 5 times of use of ionic liquid, the product yield It just started to decrease, which shows that the ionic liquid can be recovered and reused effectively, and the reuse performance is good. It is a recyclable green solvent. |
SYN
| Comparative Example 1: |
| Put 10 g of 2,6-dichloropyridine, 100 ml of methanol, and 15 g of sodium methoxide into a reaction flask, heat under reflux for about 4 to 5 hours, concentrate to remove methanol, add 150 ml of water, extract with ethyl acetate, and concentrate under reduced pressure to remove ethyl acetate. 6-Methoxy2-chloropyridine was obtained as a colorless oil. |
| 9 g of 6-methoxy 2-chloropyridine, 1.72 g of cuprous chloride, and 29 ml of 30% methylamine aqueous solution were put into the reaction flask, heated and added with a mass fraction of 11.6 g of cuprous chloride, and the temperature was kept at 120 ° C for the reaction 8h, extracted three times with 150 ml of ethyl acetate, washed with saturated brine, concentrated under reduced pressure to remove the ethyl acetate to obtain 6.18 g of 6-methoxy-2-methylaminopyridine as a brown oily product. The two-step yield was 71.2%. |
| 50ml of carbon tetrachloride, 4.25g of thiophosgene, 6.3g of 5,6,7,8-tetrahydro-2-naphthol were added to the reaction flask, the ice-salt bath was lowered to below 0°C, and 10ml of 3g potassium carbonate aqueous solution was added dropwise. , Continue the reaction at 0°C after the dropwise addition, and detect by TLC (developing solvent: petroleum ether) after the reaction is completed, separate the organic phase, wash three times with saturated brine, and concentrate under reduced pressure to obtain red oily products 5, 6, 7 , 8.7g of 8-tetrahydro-2-naphthyloxysulfuryl chloride was directly used in the next reaction. |
| 100ml of acetone, 5.7g of 6-methoxy-2-methylaminopyridine and 5.7g of potassium carbonate were added to the reaction flask, cooled with ice water, and 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride was added dropwise 8.7g, continue to stir and react for 4h after dropping, add 150ml of water, continue to stir for 30min, and filter to obtain the crude product. The crude product was recrystallized with acetone to obtain 11 g of off-white crystalline powder. The weight yield was 174.6% based on 5,6,7,8-tetrahydro-2-naphthol. The maximum single impurity content determined by HPLC was 1.5%, which did not meet the requirements of the Pharmacopoeia. |
SYN
CN 106632018
| Example 1 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours; |
| Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, and the consumption of purified water was 25.0 g each time; |
| Drying: put the wet product into a drying box, control the temperature to 45 ℃ and dry for 4 hours, to obtain 24 g of the crude product of lira naphthate; |
| The synthesis yield is 81%; |
| (2) preparation of Liranaftate fine product: |
| Impurity removal: put 23g of Liranaftate crude product and 115g of absolute ethanol into the reaction flask, add 1.38g of medicinal charcoal, decolorize at 55°C under temperature control, remove impurities for 30 minutes, filter, transfer the filtrate to the reaction flask, control the temperature Crystallize at 55°C, centrifuge, dry, pulverize, and pack to obtain 22g of Lira naphthate fine product. |
| The purification yield was 92%. |
| Example 2 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 500g of absolute ethanol was added to the reaction flask, 25g of 2-methoxy-6-methylaminopyridine, 17.6g of anhydrous sodium carbonate and 62.6g of purified water were added to the reaction flask in turn, stirred for 30 minutes, and slowly added 2-(5,6,7,8-tetrahydronaphthyloxy) chlorothioformate 37.6g, added in 2.5 hours; |
| Reaction: control the temperature at 25°C for 2.5 hours, add 250 g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 50 g each time; |
| Drying: put the wet product into a drying box, control the temperature to 55 ℃ and dry for 4 hours to obtain 49 g of the crude product of lira naphthate; |
| The synthesis yield is 82%; |
| (2) preparation of Liranaftate fine product: |
| Impurity removal: put 49g of Liranaftate crude product and 245g of absolute ethanol into the reaction flask, add 2.9g of medicinal charcoal, decolorize at 55~65 ℃ of temperature, remove impurities for 30 minutes, filter, and transfer the filtrate to the reaction flask, The temperature was controlled at 65°C for crystallization, centrifugation, drying, pulverization, and packaging to obtain 45g of fine lanaftate. |
| The purification yield was 92%. |
| Example 3 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours; |
| Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 25.0 g each time; |
| Drying: put the wet product into a drying oven, control the temperature to 45~55 ℃ and dry for 4 hours, to obtain the crude product, 23.3 g of the crude liranaftate; |
| The synthesis yield is 82%; |
| (2) preparation of Liranaftate fine product: |
| Removal of impurities: 140g of absolute ethanol was added to the reaction flask, 23.3g of crude liranaftate was added, the temperature was controlled at 50°C and stirred for 30 minutes, 1.5g of medicinal charcoal was added, the temperature was controlled at 60°C for decolorization for 30 minutes, filtered, and the temperature was controlled Crystallize at 60°C, centrifuge, dry, pulverize, and package to obtain 23g of Lira naphthate fines. |
| The purification yield was 92%. |
SYN
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Koga H, Nanjoh Y, Makimura K, Tsuboi R (2009). “In vitro antifungal activities of luliconazole, a new topical imidazole”. Medical Mycology. 47 (6): 640–7. doi:10.1080/13693780802541518. PMID 19115136.
- ^ “Torii Pharmaceutical to Launch Antifungal Agent for External Use, “ZEFNART SOLUTION 2%”, in Japan” (Press release). Torii Pharmaceutical Co. Retrieved June 27, 2021.
- ^ “Liranaftate”. ncats.io. Retrieved June 27, 2021.
- ^ “Liranaftate”. Adis Insight. Retrieved June 27, 2021.
- ^ “Liranaftate”. targetmol.com. Retrieved June 27, 2021.
///////////////////Liranaftate , リラナフタート , Zefnart, Piritetrate, M-732, лиранафтат , ليرانافتات , 利拉萘酯 , ANTIFUNGAL, JAPAN 2000

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
SULCONAZOLE

SULCONAZOLEсульконазол , سولكونازول , 硫康唑
- Molecular FormulaC18H15Cl3N2S
- Average mass397.749 Da
1H-Imidazole, 1-[2-[[(4-chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]- [ACD/Index Name]
4332
5D9HAA5Q5S
61318-90-9[RN]
(±)-1-[2,4-Dichloro-b-[(p-chlorobenzyl)thio]phenethyl]imidazole
1-[2-[[(4-Chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole: SulconazoleCAS Registry Number: 61318-90-9
CAS Name: 1-[2-[[(4-Chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole
Additional Names: (±)-1-[2,4-dichloro-b-[(p-chlorobenzyl)thio]phenethyl]imidazole
Molecular Formula: C18H15Cl3N2S
Molecular Weight: 397.75
Percent Composition: C 54.35%, H 3.80%, Cl 26.74%, N 7.04%, S 8.06%
Literature References: Prepn: K. A. M. Walker, DE2541833; idem,US4055652 (1976, 1977 both to Syntex). HPLC determn in plasma: M. Fass et al.,J. Pharm. Sci.70, 1338 (1981). Mechanism of action study: W. H. Beggs, Biochem. Arch.10, 117 (1994). Clinical trial in tinea pedis: W. A. Akers et al.,J. Am. Acad. Dermatol.21, 686 (1989). Review of pharmacology and clinical efficacy: P. Benfield, S. P. Clissold, Drugs35, 143-153 (1988).
Derivative Type: Nitrate
CAS Registry Number: 61318-91-0
Manufacturers’ Codes: RS-44872
Trademarks: Exelderm (Syntex); Myk (Cassenne); Sulcosyn (Syntex)
Molecular Formula: C18H15Cl3N2S.HNO3
Molecular Weight: 460.76
Percent Composition: C 46.92%, H 3.50%, Cl 23.08%, N 9.12%, S 6.96%, O 10.42%
Properties: Colorless crystals from acetone, mp 130.5-132°.
Melting point: mp 130.5-132°
Therap-Cat: Antifungal.
Keywords: Antifungal (Synthetic); Imidazoles.
Sulconazole (trade name Exelderm) is an antifungal medication of the imidazole class. It is available as a cream or solution to treat skin infections such as athlete’s foot, ringworm, jock itch, and sun fungus.[1][2] Although not used commercially for insect control, sulconazole nitrate exhibits a strong anti-feeding effect on the keratin-digesting Australian carpet beetle larvae Anthrenocerus australis.[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

DE 2541833 US 4038409
(Read example 5 and 9 in US patent.)
https://patents.google.com/patent/US4038409A/en
EXAMPLE 5Alternative Route to 1-[β-(R-carbonylthio)phenethyl]imidazolesA. Preparation of 1-[2,4-dichloro-β-(methylcarbonylthio)-phenethyl]imidazole, oxalate.1-(β,2,4-Trichlorophenethylimidazole (1.19g) in 5 ml of dry tetrahydrofuran was added to preformed sodium thioacetate, generated in situ from 720 mg thioacetic acid and sodium hydride (480 mg 57% dispersion in mineral oil) in 20 ml. tetrahydrofuran and the mixture stirred and refluxed under nitrogen for 18 hours. The solvent was removed under reduced pressure, water (20 ml) added and the product extracted with ether. The extracts were washed with water, dried (MgSO4), evaporated and the residue chromatographed on silica gel eluting with 10-20% acetone in dichloromethane. The pure product in ether was treated dropwise with ethereal oxalic acid until precipitation was complete, and the thus obtained oxalate salt of 1-[2,4-dichloro-β-(methylcarbonylthio)phenethyl]imidazole recrystallized from acetone/ethyl acetate with mpBy substituting other available sodium thioacids for sodium thioacetate, other compounds of this invention may be prepared.
EXAMPLE 9A. Preparation of 1-[2,4-dichloro-β-(4-chlorobenzylthio)-phenethyl]imidazoleTo a stirred solution of 330 mg sodium hydroxide in 30 ml methanol under nitrogen is added 810 mg of 1-[2,4-dichloro-β-(methylcarbonylthio)phenethyl]imidazole oxalate and the mixture is stirred at room temperature for ca. 30 minutes (until thin layer chromatography shows the disappearance of the ester). α,p-dichlorotoluene (350 mg) is then added, the solution stirred a further 15 minutes and the solvent removed under reduced pressure. Ether and water are then added to the residue and the ether extract washed with water, dried (MgSO4) and concentrated. Dropwise addition of nitric acid (d = 1.42) until precipitation is complete gives the nitrate salt of 1-[2,4-dichloro-β-(4-chlorobenzylthio)phenethyl]imidazole, recrystallized from acetone, mp 130.5°-132° C.B. By using other compounds of this invention exemplified by those set forth in Examples 2 and 4 and other suitable (substituted) hydrocarbyl halides (or mesylates, tosylates), other compounds may be prepared.
SYN
https://www.sciencedirect.com/science/article/pii/S2095177917301405
SYN
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 6258-66-8 | C7H7ClS | 4-chlorobenzyl mercaptan | Benzenemethanethiol, 4-chloro- |
| 24155-42-8 | C11H10Cl2N2O | 1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethanol | 1H-Imidazole-1-ethanol, α-(2,4-dichlorophenyl)- |
References
- ^ Drugs.com: sulconazole topical
- ^ Fromtling RA (April 1988). “Overview of medically important antifungal azole derivatives”. Clinical Microbiology Reviews. 1 (2): 187–217. doi:10.1128/CMR.1.2.187. PMC 358042. PMID 3069196.
- ^ Sunderland MR, Cruickshank RH, Leighs SJ (2014). “The efficacy of antifungal azole and antiprotozoal compounds in protection of wool from keratin-digesting insect larvae”. Textile Research Journal. 84 (9): 924–931. doi:10.1177/0040517513515312.
| Clinical data | |
|---|---|
| Trade names | Exelderm |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a698018 |
| Routes of administration | Topical |
| ATC code | D01AC09 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 61318-90-9 |
| PubChem CID | 5318 |
| ChemSpider | 5127 |
| UNII | 5D9HAA5Q5S |
| KEGG | D08535 |
| ChEBI | CHEBI:9325 |
| ChEMBL | ChEMBL1221 |
| CompTox Dashboard (EPA) | DTXSID8044129 |
| Chemical and physical data | |
| Formula | C18H15Cl3N2S |
| Molar mass | 397.74 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////SULCONAZOLE, сульконазол , سولكونازول , 硫康唑 , Antifungal,

NEW DRUG APPROVALS
ONE TIME
$10.00
Saperconazole

Saperconazole

The most common systemic fungal infections in humans are blastomycosis, candidosis, aspergillosis, histoplasmosis, coccidioidomycosis, paracoccidioidomycosis, and cryptococcosis.
Fungal diseases are often confined to typical anatomic sites, and many involve a primary focus in the lung, with more characteristic manifestations of specific fungal infections appearing once the infection spreads from a primary site. For example, blastomycosis primarily involves the lungs, and occasionally spreads to the skin. Similarly, the primary form of coccidioidomycosis occurs as an acute, benign, self-limiting respiratory disease, which can then progress to a chronic, often-fatal infection of the skin, lymph glands, liver, and spleen. Other infectious diseases such as paracoccidioidomycosis and candidiasis present in different manners, and depending on the etiology, may exhibit several forms involving internal organs, the lymph nodes, skin, and mucous membranes. Diagnosis of specific fungal diseases can be made by isolation of the causative fungus from various specimens, such as sputum, urine, blood, or the bone marrow, or with certain fungus types, through evidence of tissue invasion.
Many patients suffering from severe systemic fungal infections are hardly, or not at all, able to receive medication via oral administration, as such patients are often in a coma or suffering from severe gastroparesis. As a result, the use of insoluble or sparingly soluble antifungals such as itraconazole free base, which are difficult to administer intravenously to treat such patients, is significantly impeded.
Local or superficial fungal infections are caused by dermatophytes or fungi that involve the outer layers of the skin, nails, or hair. Such infections may present as a mild inflammation, and can cause alternating remissions and eruptions of a gradually extending, scaling, raised lesion. Yeast infections, such as candidiasis and oral candidiasis (thrush), are usually localized to the skin and mucous membranes, with the symptoms varying depending on the site of infection. In many instances, such infections appear as erythematous, often itchy, exudative patches in the groin, axillas, umbilicus, between toes, and on finger-webs. Oral thrush involves an inflamed tongue or buccal mucosa, typically accompanied by white patches of exudate. Chronic mucocutaneous candidiasis is manifested in the form of red, pustular, crusted, thickened lesions on the forehead or nose.Itraconazole or (±)-£is-4-[4-[4-[4-[[2-(2,4-dichlorophenyl)-2-(lH-l-2,4-triazol-l- ylmethyl)- 1 ,3-dioxolan-4-yl]methoxy]phenyl]- 1 -ρiperazinyl]phenyl]-2,4-dihydro-2-( 1 – methyl-propyl)-3H-l,2,4-triazol-3-one, is a broadspectrum antifungal compound developed for oral, parenteral and topical use and is disclosed in US-4,267,179.
The development of effϊcaceous pharmaceutical compositions of itraconazole and saperconazole is hampered considerably by the fact that said compounds are only very sparingly soluble in water. The solubility of both compounds can be increased by complexation with cyclodextrins or derivatives thereof as described in WO 85/02767 and US-4,764,604.
Unexpectedly, it has now been found that each of the individual stereoisomers of itraconazole and saperconazole have greater water solubility than the diastereomeric mixtures of said compounds, in particular when complexed with cyclodextrin or its derivatives. As a result, pharmaceutical compositions having good bioavailability, yet comprising less cyclodextrin as a complexing agent, can be prepared. The present invention is concemced with the stereoisomeric forms of itraconazole (X = CI) and saperconazole (X = F), which may be represented by the formula

cis-©,and the pharmaceutically acceptable acid addition salt forms thereof. The three asterisks indicate the three chiral centers, and ‘cis’ means that the (lH-l,2,4-triazol-l-ylmethyl) moiety and the substituted phenoxy moiety are located at the same side of the plane defined by the 1,3-dioxolane ring.
The four possible stereoisomeric cis forms can be described using various rules of nomenclature. The following tables show the correlation among the C. A. stereochemical descriptor, the absolute configuration at each of the chiral centers and the specific optical
20 rotation [α]jj in 1% methanol (itraconazole; table I) (saperconazole; table H).
Table I

itraconazole

Table π

saperconazole

Itraconazole is a broad-spectrum antifungal agent developed for oral, parenteral and topical use, and is disclosed in U.S. Patent No. 4,267,179. Itraconazole is a synthetic triazole derivative that disrupts the synthesis of ergosterol, the primary sterol of fungal cell membranes. This disruption appears to result in increased permeability and leakage of intracellular content, and at high concentration, cellular internal organelles involute, peroxisomes increase, and necrosis occurs.
As set forth in the USP Dictionary of Drug Names and USAN, itraconazole is defined as 4-[4-[4-[4- [[2-(2,4-dichlorophenyl)-2-(lH-l,2,4-triazol-l-ylmethyl)-l,3-dioxolan-4-yl] methoxy]phenyl]-l-piperazinyl]phenyl]- 2,4-dihydro-2-(l-methylpropyl)-3H-l,2,4-triazol-3-one, or alternatively, as (±)-l-5ec-butyl-4-[/7-[4-[/7-[[(2R*,4S*)-2-(2,4-dichlorophenyl)-2-(lH-l,2,4-triazol-l-ylmethyl)-l,3-dioxolan-4-yl]methoxy]phenyl]-l-piperazinyl]phenyl]-Δ2-l,2,4-triazolin-5-one. There are three asymmetric carbons in itraconazole: one in the sec-butyl side chain on the triazolone and two in the dioxolane ring. As a result, eight possible stereoisomers of itraconazole exist: (R,R,R), (S,S,S), (R,R,S), (S,S,R), (R,S,S), (R,S,R), (S,R,S), and (S,R,R).
(±)Cz‘s-Itraconazole comprises a mixture of only those isomers that describe a “cis” relationship in the dioxolane ring, i.e., the (1Η-1, 2, 4-triazol-l-ylmethyl) moiety and the substituted phenoxy moiety are located on the same side of a plane defined by the 1, 3-dioxolane ring. By convention, the first represented chiral center is at the C-2 position of the dioxolane ring, the second is at the C-4 position of the dioxolane ring, and the third is in the sec-butyl group. Hence, (±)c.s-itraconazole is a mixture of (R,S,S), (R,S,R), (S,R,S) and (S,R,R) isomers.
The four possible stereoisomeric cis forms of itraconazole, and
diastereomeric pairs thereof, are described in more detail in U.S. Patent Nos. 5,474,997 and 5,998,413. In general, the individual stereoisomeric forms of c s-itraconazole have antifungal properties, and contribute to the overall activity of (±)cw-itraconazole.
(±)Ciy-Itraconazole free base is only very sparingly soluble in water, and thus it is extremely difficult to prepare effective pharmaceutical compositions containing the same. A number of means have been used to increase the solubility of itraconazole free base, including complexing or co-formulation with cyclodextrins or derivatives thereof, as described in U.S. Patent No. 4,764,604, U.S. Patent No.5,998,413, and U.S. Patent No. 5,707,975, and coating beads with a film comprising a hydrophilic polymer and itraconazole, as described in U.S. Patent No. 5,633,015.
[0014] Another approach to increase solubility of itraconazole focuses on preparation of the stereoisomers of c s-itraconazole, and in particular (2R, 4S) itraconazole, which may comprise a mixture of two diastereomers ((R,S,S) and
(R,S,R)), as described in U.S. Patent Nos. 5,414,997 and 5,998,413.
Commercially available itraconazole (SPORANOX® brand (±)cis-itraconazole, Janssen Pharmaceutica Products, L.P., Titusville, NJ, U.S.A.) is a free base and a racemic mixture of the cis isomer in the dioxolane ring and is represented by structural formula (I):

(i)
SPORANOX has been approved for use as an antifungal agent for treating immunocompromised and non-immunocompromised patients having: blastomycosis (pulmonary and extrapulmonary); histoplasmosis, including chronic cavitary pulmonary disease and disseminated non-meningeal histoplasmosis; and aspergillosis. In addition, in non-immunocompromised patients, it has been approved for treatment of onychomycosis. See generally, Physician ‘s Desk Reference, 56th ed. (2002). The compound has also been investigated for use in coccidioidomycosis, cryptococcosis, dermatophyte, and candidiasis infections.
Adverse effects associated with the administration of (±)cts-itraconazole free base include nausea, vomiting, anorexia, headache, dizziness, hepatotoxicity, and inhibition of drug metabolism in the liver, leading to numerous, clinically significant, adverse drug interactions. See, Physician ‘s Desk Reference, 56th ed. (2002); Honig et al., J. Clin. Pharmacol. 33:1201-1206 (1993) (terfenadine interaction); Gascon and Dayer, Eur. J. Clin. Pharmacol., 41_:573-578 (1991) (midazolam interaction); and Neuvonen et al, Clin. Pharmacol. Therap., 60:54-61 (1996) (lovastatin interaction). Reactions associated with hypersensitivity, such as urticaria and serum liver enzymes elevation, are also associated with the administration of the drug. A more serious, though less common, adverse effect is hepatotoxicity. See, e.g., Lavrijsen et al., Lancet, 340:251-252 (1992).
In addition, as discussed herein, c/s-itraconazole free base is only very sparingly soluble in water. Thus, due to its relative non-polarity and insolubility, itraconazole free base suffers from two other drawbacks: it cannot be readily formulated in parenteral solution, and it does not effectively penetrate the blood-brain barrier. The latter problem is exacerbated by drug interactions, such as one observed between itraconazole free base and valproate, as described in Villa et al. , Rev. Inst. Med. Trop., Sao Paulo, pp. 231-234 (Jul-Aug 2000), which is incorporated by reference herein in its entirety. In another case of CNS fungal infection, extremely high doses of itraconazole free base were used to treat residual aspergillus infection, as reported by Imai et al., Intern. Med, 38(10):829-832 (1999), which is incorporated by reference herein in its entirety. As a result, numerous therapeutic indications that require rapid achievement of effective blood levels or access to the CNS are difficult to treat or beyond treatment with itraconazole free base.
Furthermore, the emergence of antifungal resistance (e.g., in Aspergillus fumigatus isolates as described by Dannaoui et al., J. Antimicrob. Chemother., 47:333-340 (2001), which is incorporated by reference herein in its entirety) presents an added challenge to the efficacy of itraconazole free base. For those strains of fungi that show resistance, high and relatively constant levels of itraconazole free base must be produced in the target organs of infected patients.
Over the years, a number of formulation routes have been used in order to enhance the adsorption and bioavailability of itraconazole. For example, the currently marketed SPORANOX® solid dosage capsule form of itraconazole free base utilizes sugar-based beads coated with a hydrophilic polymer and an amorphous film of itraconazole. See Physicians Desk Reference, 56th ed., pp.1800- 1804 (2002); and U.S. Patent No. 5,633,015. This dosage form requires up to two capsules three times daily depending on the condition being treated.
Even with the various formulation routes, the dosage amounts and dose frequency for itraconazole can be burdensome to patients. In addition, administration of existing dosage forms of itraconazole have shown significant variability in bioavailability and adsorption, which likely results from food effects. See, Physician ‘s
Desk Reference, 56th ed., pp. 1800-1804 (2002). Thus, it would be desirable to increase bioavailability and adsorption and decrease the per-dose pill count and decrease dosing frequency (e.g., twice a day to once a day) associated with administration of itraconazole in order to provide an improvement over current therapy, particularly with regard to patient compliance, convenience, ease of ingestion, especially with regard to immunocompromized polypharmacy patients (e.g., AIDS or cancer patients).
Posaconazole and Saperconazole Chemistry and Uses
Other related conazoles have also been discovered and used as antifungals. Two of these conazoles that are closely structurally related to itraconazole are posaconazole and saperconazole. Posaconazole (CAS Registry Number: 171228-49-2; CAS Name: 2,5-Anhydro-l ,3,4-trideoxy-2-C-(2,4-difluorophenyl)-4-[[4-[4-[4-[l -[(1 S,2S)- 1 -ethyl-2-hydroxypropyl]- 1 ,5-dihydro-5-oxo-4H- 1 ,2,4-triazol-4-yl]phenyl]- 1 -piperazinyl]phenoxy]methyl]- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)-D-t/Veo-pentitol; Additional Name: (3R-c s)-4-[4-[4-[4-[5-(2,4-difluorophenyl)-5-(l,2,4-triazol-l-ylmethyl)tetrahydrofuran-3-ylmethoxy]phenyl]piperazin- 1 -yl]phenyl]-2-[l (S)-ethyl-2(S)-hydroxypropyl]-3,4-dihydro-2H-l,2,4-triazol-3-one) is represented by structural formula (II):

(II)
Saperconazole (CAS Registry Number: 110588-57-3; CAS Name: 4-[4-[4-[4-[[2-(2,4-Difluorophenyl)-2-(lH-l,2,4-triazol-l-ylmethyl)-l,3-dioxolan-4-yl]methoxy]phenyl]- 1 -piperazinyl]phenyl]-2,4-dihydro-2-(l -methylpropyl)-3H- 1 ,2,4-triazol-3-one; Additional Name: (±)-l-sec-butyl-4-[ -[4-| -[[(2R* 4S*)-2-(2,4- difluorophenyl)-2-( 1 H- 1 ,2,4-triazol- 1 -ylmethyl)- 1 ,3 -dioxolan-4-yl]methoxy]phenyl]- 1 -piperazinyl]phenyl]-Δ2-l,2,4-triazolin-5-one) is represented by structural formula (III):

(III)
Consequently, there is a need for soluble forms of conazoles including cis itraconazole, posaconazole and saperconazole that can be readily formulated for use in various modes of administration, including parenteral and oral administration.
A. Preparation of intermediates: Example 1a) utilizing water separator, by 200 parts of glycerin, 90 parts of 1- (2,4-difluorophenyl) -2- (1H-1,2,4- three mixture of 1-yl) ethanone, 600 parts of methanesulfonic acid, 190 parts of benzene was stirred first at reflux for 3 hours, then stirred at room temperature overnight. The reaction mixture was added dropwise a solution of sodium bicarbonate. The product was extracted with chloroform, the extract was washed with water, dried, filtered and evaporated. With 4-methyl-2-pentanone and the residue triturated product was filtered off and dried, yielding 80 parts (67.2%) (cis + trans) -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolane-4-methanol (intermediate 1).
b) by 69 parts of 3,5-dinitrobenzoyl chloride, 80 parts of (cis + trans) -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolane-4-methanol, 400 parts of pyridine and 520 parts of dichloromethane was stirred at room temperature for 3 hours. The reaction mixture was evaporated, and the residue was dissolved in water. The product was extracted with chloroform. The extract was dried, filtered and evaporated. The residue was subjected to silica gel column chromatography, eluting with chloroform / methanol (99:1v / v). Pure fractions were collected, the eluent was evaporated, to give 90 parts (70.4%) of cis -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1 ylmethyl) -1,3-dioxolane-4-methanol 3,5-dinitrobenzoate (residue) (intermediate 2).
c) by 90 parts of (cis) -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxo- dioxolan-4-methanol 3,5-dinitrobenzoate, 16 parts of 50% sodium hydroxide solution, 800 parts of 1,4-dioxane, 400 parts of water and the mixture was stirred at room temperature overnight. The reaction mixture was poured into water and the product was extracted with dichloromethane, extracts washed with water, dried, filtered and evaporated. With 4-methyl-2-pentanone and the residue triturated product was filtered off and dried, yielding 30 parts (56.0%) of cis -2- (2,4-difluorophenyl) -2- (1H-1, 2,4-triazol-1-ylmethyl) -1,3-dioxolane-4-methanol (residue) (intermediate 3).
d) by 11.4 parts of methanesulfonyl chloride, 25 parts of cis -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1, mixture of 1,3-dioxolane-4-methanol, 300 parts of pyridine, 390 parts of dichloromethane was stirred at room temperature for 3 hours. The reaction mixture was evaporated, and the residue was dissolved in chloroform. The organic phase was dried, filtered and evaporated. The residue was triturated with dipropyl ether. The product was filtered off and dried, yielding 29.4 parts (93.2%) of cis -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) – 1,3-dioxolane-4-methanol methanesulfonate (residue) of intermediate 4).
In a similar manner there were also prepared: cis-2- (2,4-difluorophenyl) -2- (1H- imidazol-1-ylmethyl) -1,3-dioxolane-4-methanol mesylate ethanedioate (1/1) (interm. 5).
Example 2a) over 2 hours, dissolved in 100 parts of pyridine 121.2 parts of 2-naphthalenesulfonyl chloride was added dropwise to a stirred, was dissolved in 1300 parts of dichloromethane, and 122.0 parts of (cis + trans ) -2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolane-4-methanol and 1.0 parts of N, N- dimethyl-4-pyridin-amine solution. Upon completion, stirring was continued at room temperature overnight. The reaction mixture was washed twice with water, and evaporated under reduced pressure. The residue was purified by silica gel column chromatography, eluting with chloroform. Pure fractions were collected, the eluent was evaporated. The residue was crystallized from 4-methyl-2-pentanone. The product was filtered off and dried, yielding 102.3 parts (51.0%) of cis – [[2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-yl-methyl ) -1,3-dioxolan-4-yl] methyl] -2-naphthalene sulfonate; mp139.5 ℃ (intermediate 6).
Example 3a) at 70 ℃, under nitrogen atmosphere, by 9.0 parts of 4- [4- (4-nitrophenyl) -1-piperazinyl] phenol, 13.6 parts of cis-2- [2,4- difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolane-4-methanol methanesulfonate ester, 6.0 parts of potassium hydroxide and 90 parts of a mixture of DMF was stirred overnight. After cooling, the reaction mixture was diluted with water. The precipitated product was filtered off and purified by silica gel column chromatography, the chloroform / ethyl acetate / hexane / methanol (500:300:200:0.5v / v / v / v) mixture as eluent. Pure fractions were collected, the eluent was evaporated. The residue was crystallized 4-methyl-2-pentanone. The product was filtered off and dried, yielding 6.69 parts (38.5%) of cis -1- [4 – [[2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol – 1- ylmethyl) -1,3-dioxolan-4-yl] methoxy) phenyl] -4- (4-nitrophenyl) piperazine; mp169.8 ℃ (Intermediate 7) .
b) at atmospheric pressure, 50 ℃, with 2 parts of 5% palladium – on-charcoal catalyst by 38.3 parts of cis -1- [4 – [[2- (2,4-difluorophenyl) -2- (1H -1,2,4-triazol-1-ylmethyl) -1,3-dioxolan-4-yl] methoxy] phenyl] -4- (4-nitrophenyl) piperazine, 2 parts of a solution of thiophene (4% solution in methanol) and 600 parts of 2-methoxy-ethanol mixture. After absorption of the calculated amount of hydrogen finished, hot filtered to remove the catalyst, and the filtrate was saturated with water. After cooling, the precipitated product was filtered off, washed with water and 2-propanol and crystallized from 1,4-dioxane. The product was filtered off and dried, yielding 22.7 parts (62.6%) of cis-4- [4- [4 – [[2- (2,4-difluorophenyl) -2- (1H-1,2,4- triazol-1-ylmethyl) -1,3-dioxolan-4-yl] methoxy] phenyl] -1-piperazinyl] aniline; mp193.0 ℃ (interm. 8).
Example 4a) by 10 parts of 2,4-dihydro-4- [4- [4- [4-methoxyphenyl) -1-piperazinyl] phenyl] -3H-1,2,4- triazol-3-one (U.S. Patent No. 4,267,179 in the implementation of the method in Example ⅩⅦ obtained), 1.5 parts of sodium hydride (50% dispersion), 300 parts of the mixture consisting of dimethyl sulfoxide, at 60 ℃ under a nitrogen atmosphere begging, stirring, until no bubble up. Was then added 5.24 parts of 2-bromopropane, and at 60 ℃, stirring was continued for 1 hour. Further added 1.5 parts of sodium hydride (50% dispersion) and stirring was continued until no more bubble up. Then 5.24 parts of 2-bromopropane was added, and the whole was stirred for 1 hour at 60 ℃. The reaction mixture was cooled, poured into water and the product was extracted with chloroform. The extract was washed with water, dried, filtered and evaporated. The residue was purified by silica gel column chromatography, eluting with chloroform / methanol (99:1v / v). Pure fractions were collected, the eluent was evaporated, the residue was crystallized in 1-butanol, yielding 5.2 parts (47% (2,4-dihydro-4- [4- [4- (4-methoxyphenyl ) -1-piperazinyl] phenyl] -2- (1-methylethyl) -3H-1,2,4- triazol-3-one; mp209.5 ℃ (intermediate 9).
b) by 4.7 parts of 2,4-dihydro-4- [4- [4- (4-methoxyphenyl) -1-piperazinyl] phenyl] -2- (1-methylethyl) -3H-1,2,4- triazol-3-one, a mixture of 75 parts of 48% aqueous hydrobromic acid was stirred at reflux for 3 hours. The reaction mixture was evaporated, and the residue was dissolved in a mixture of methanol and water. With sodium bicarbonate solution, and the whole was, and the product was extracted with chloroform. The extract was dried, filtered and evaporated. The residue was triturated with 2-propanol, yielding 3.9 parts (86%) of 2,4-dihydro-4- [4- [4- (4-hydroxyphenyl) -1-piperazinyl] phenyl] -2 – (1-methylethyl) -3H-1,2,4- triazol-3-one, mp208.4 ℃ (intermediate 10).
PATENT
| EP0006711A1 * | 13 Jun 1979 | 9 Jan 1980 | Janssen Pharmaceutica N.V. | Heterocyclic derivatives of (4-phenylpiperazin-1-yl-aryloxymethyl-1,3-dioxolan-2-yl)-methyl-1H-imidazoles and 1H-1,2,4-triazoles, processes for preparing them and compositions containing them |
| EP0118138A1 * | 24 Jan 1984 | 12 Sep 1984 | Janssen Pharmaceutica N.V. | ((4-(4-(4-Phenyl-1-piperazinyl)phenoxymethyl)-1,3-dioxolan-2-yl)methyl)-1H-imidazoles and 1H-1,2,4-triazoles |
| DE2804096A1 * | 31 Jan 1978 | 3 Aug 1978 | Janssen Pharmaceutica Nv | 1-(1,3-dioxolan-2-ylmethyl)-1h-imidazole und -1h-1,2,4-triazole und deren salze, verfahren zu ihrer herstellung und ihre verwendung bei der bekaempfung pathogener pilze und bakterien |
| Patent | Submitted | Granted |
|---|---|---|
| ORDERED MESOPOROUS SILICA MATERIAL [US2011081416] | 2010-10-15 | 2011-04-07 |
| BENZOYL PEROXIDE COMPOSITION FOR TREATING SKIN [US2011082216] | 2009-10-02 | 2011-04-07 |
| METHODS RELATED TO TIM 3, A TH1-SPECIFIC CELL SURFACE MOLECULE, FOR ACTIVATING ANTIGEN PRESENTING CELLS [US2015044229] | 2014-08-20 | 2015-02-12 |
| METHODS RELATED TO TIM 3, A TH1-SPECIFIC CELL SURFACE MOLECULE, FOR ACTIVATING ANTIGEN PRESENTING CELLS [US2015044230] | 2014-08-20 | 2015-02-12 |
| COSMETIC METHOD FOR INCREASING COLLAGEN EXPRESSION IN SKIN COMPRISING TOPICALLY APPLYING AN EXTRACT OF QUASSIA AMARA [US2015056310] | 2014-08-20 | 2015-02-26 |
| Flexible bone composite [US8771721] | 2013-03-15 | 2014-07-08 |
| Topical formulation [US8513304] | 2012-11-19 | 2013-08-20 |
| Prolonged release bioadhesive therapeutic systems [US8518442] | 2010-07-02 | 2013-08-27 |
| Preparation method for solid dispersions [US8216495] | 2009-03-25 | 2012-07-10 |
| Flexible bone composite [US8221782] | 2011-08-12 | 2012-07-17 |
| Patent | Submitted | Granted |
|---|---|---|
| Crystalline forms of conazoles and methods of making and using the same [US7446107] | 2005-03-31 | 2008-11-04 |
| CIS-itraconazole crystalline forms and related processes, pharmaceutical compositions and methods [US7078526] | 2004-01-29 | 2006-07-18 |
| Novel Saperconazole Crystalline Forms and Related Processes, Pharmaceutical Compositions and Methods [US2007293674] | 2007-12-20 | |
| NOVEL CRYSTALLINE FORMS OF CONAZOLES AND METHODS OF MAKING AND USING THE SAME [US2009088443] | 2009-04-02 | |
| CONTROLLED RELEASE VEHICLES HAVING DESIRED VOID VOLUME ARCHITECTURES [US2014328884] | 2012-12-17 | 2014-11-06 |
| MOLECULES WITH POTENT DHFR BINDING AFFINITY AND ANTIBACTERIAL ACTIVITY [US2014329840] | 2014-05-05 | 2014-11-06 |
| FUNCTIONALLY-MODIFIED OLIGONUCLEOTIDES AND SUBUNITS THEREOF [US2014330006] | 2012-11-15 | 2014-11-06 |
| ASPARTYL-TRNA SYNTHETASE-FC CONJUGATES [US2014335087] | 2012-12-27 | 2014-11-13 |
| GASTRORETENTIVE CONTROLLED RELEASE VEHICLES THAT INCLUDE ETHYLENE COPOLYMERS, ETHYL CELLULOSES, AND/OR THERMOPLASTIC POLYURETHANES [US2014348936] | 2012-12-17 | 2014-11-27 |
| HISTIDYL-TRNA SYNTHETASE-FC CONJUGATES [US2014349369] | 2014-03-14 | 2014-11-27 |
| ASPARTYL-TRNA SYNTHETASES [US2014302075] | 2012-12-06 | 2014-10-09 |
| Rhinosinusitis Prevention and Therapy with Proinflammatory Cytokine Inhibitors [US2014311482] | 2014-01-24 | 2014-10-23 |
| POLYSACCHARIDE ESTER MICROSPHERES AND METHODS AND ARTICLES RELATING THERETO [US2014315720] | 2014-04-04 | 2014-10-23 |
| MODIFIED GREEN TEA POLYPHENOL FORMULATIONS [US2014256616] | 2014-05-19 | 2014-09-11 |
| PLANT-BASED COMPOSITIONS AND USES THEREOF [US2014260466] | 2013-03-15 | 2014-09-18 |
| PLANT-BASED COMPOSITIONS AND USES THEREOF [US2014271928] | 2014-03-14 | 2014-09-18 |
| LIGHT AND ULTRASONIC TRANSDUCER DEVICE [US2014276247] | 2014-03-14 | 2014-09-18 |
| LIGHT AND/OR ULTRASONIC TRANSDUCER DEVICE WITH SENSOR FEEDBACK FOR DOSE CONTROL [US2014276248] | 2014-03-14 | 2014-09-18 |
| PHOTOPROTECTIVE COMPOSITION CONTAINING AN UNMODIFIED GELLING STARCH AND POLYAMIDE PARTICLES [US2014287005] | 2014-03-18 | 2014-09-25 |
| STABILIZED CHEMICAL DEHYDRATION OF BIOLOGICAL MATERIAL [US2014227686] | 2014-04-16 | 2014-08-14 |
| METHODS RELATED TO TIM 3, A TH1-SPECIFIC CELL SURFACE MOLECULE, FOR ACTIVATING ANTIGEN PRESENTING CELLS [US2014242094] | 2014-02-20 | 2014-08-28 |
| NOVEL ENCOCHLEATION METHODS, COCHLEATES AND METHODS OF USE [US2014242153] | 2014-01-30 | 2014-08-28 |
| METHODS OF REDUCING THE PROLIFERATION AND VIABILITY OF MICROBIAL AGENTS [US2010197621] | 2010-08-05 | |
| METHODS OF ADMINISTERING TOPICAL ANTIFUNGAL FORMULATIONS FOR THE TREATMENT OF FUNGAL INFECTIONS [US2010086504] | 2010-04-08 | |
| COMPOSITIONS AND METHODS FOR INCREASING ERYTHROPOIETIN (EPO) PRODUCTION [US2014024699] | 2011-12-09 | 2014-01-23 |
| PROLONGED RELEASE BIOADHESIVE THERAPEUTIC SYSTEMS [US2013310335] | 2013-07-26 | 2013-11-21 |
| Pharmaceutical Composition [US2013315988] | 2011-05-23 | 2013-11-28 |
| Topical Foam Composition [US2013315998] | 2013-08-05 | 2013-11-28 |
| ANTIFUNGAL NAIL COAT AND METHOD OF USE [US2013323189] | 2013-08-09 | 2013-12-05 |
| TOPICAL FORMULATIONS, SYSTEMS AND METHODS [US2013337031] | 2013-03-08 | 2013-12-19 |
LANOCONAZOLE
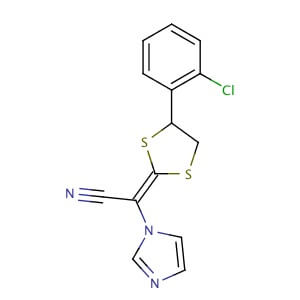
Lanoconazole
- Latoconazole, Lanoconazole, TJN-318, NND-318, Astat,
Nihon Nohyaku (Originator), Tsumura (Licensee)
| Synonym: | 2-[4-(2-Chlorophenyl)-1,3-dithiolan-2-ylidene]-2-imidazol-1-yl-acetonitrile |
| Application: | An antifungal compound |
| CAS Number: | 101530-10-3 |
| Molecular Weight: | 319.83 |
| Molecular Formula: | C14H10ClN3S2 |
Brief background information
| Appearance: | Crystalline |
| Physical State: | Solid |
| Solubility: | Soluble in chloroform, and methanol. Insoluble in water. |
| Storage: | Store at -20° C |
| Melting Point: | 129-132 °C |
| Boiling Point: | ~477.6 °C at 760 mmHg (Predicted) |
| Density: | ~1.4 g/cm3 (Predicted) |
| Refractive Index: | n20D 1.73 (Predicted) |
| pK Values: | pKb: 3.76 (Predicted) |
| WGK Germany: | 3 |
| RTECS: | NI3393500 |
| PubChem CID: | 3002820 |
| Merck Index: | 14: 5357 |
| MDL Number: | MFCD00865590 |
| Beilstein Registry: | 4819111 |
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | D01 | C 14 H 10 ClN 3 S 2 | 319.84 g / mol | 101530-10-3 |

Application
-
antifungal
Synthesis pathway
| Synthesis a) |
|---|
  |

Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| Japan | Astatine | Tsumura |
| Ukraine | No | No |
Formulations
-
1% cream;
-
1% ointment;
-
1% solution
Links
-
EP 218 736 (Nihon Nohyaku; EP-prior. 9.10.1985).
1. Oka, H., et al., 1992. Therapeutic efficacy of latoconazole in formulations of clinical use on experimental dermatophytosis in guinea pigs. Arzneimittel-Forschung. 42(3): 345-9. PMID: 1497697
2. Niwano, Y., et al., 1994. Therapeutic efficacy of lanoconazole, a new imidazole antimycotic agent, for experimental cutaneous candidiasis in guinea pigs. Antimicrobial agents and chemotherapy. 38(9): 2204-6. PMID: 7811048
3 http://aac.asm.org/content/38/9/2204.full.pdf
References 1. Seo, A., Kanno, H., Hasegawa, N. et al. (Nihon Nohyaku Co., Ltd.). Antimycotic agent and fungicidal agent. US 4738976. 2. Seo, A ., Sugano, H., Hasegawa, C., Ikeda, K., Munechica, Y., Konoe, T., Konaka, M. (Nihon Nohyaku Co., Ltd.). Antifungal agent. JP 87093227. 3. Seo , A., Sugano, H., Hasegawa, C., Ikeda, K., Nishimura, A., Miyashiro, Y. (Nihon Nohyaku Co., Ltd.). Non-medicinal bactericidal agents and method for their preparation. JP 87093204. 4. Seo, A., Sugano, H., Hasegawa, C., Miyashiro, Y., Nishimura, A., Ikeda, K. (Nihon Nohyaku Co., Ltd.). Ketene S, S-acetals. JP 85218387. 5. Seo, A., Kanno, H., Hasegawa, N. et al. (Nihon Nohyaku Co., Ltd.). A novel ketene S, S-acetal deriv., a process for manufacturing thereof and a method for curing mycosis by administering it. EP 218736.

MAKE IN INDIA
http://makeinindia.com/sector/pharmaceuticals/
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL 



{kind=link}
{kind=link}