
Home » Posts tagged 'Alzheimer’s'
Tag Archives: Alzheimer’s
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
VALILTRAMIPROSATE


VALILTRAMIPROSATE
1034190-08-3
- (S)-3-(2-Amino-3-methylbutanamido)propane-1-sulfonic acid
- BLU8499
- WHO 11912
| Molecular Weight | 238.30 |
|---|---|
| Formula | C8H18N2O4S |
| CAS No. | 1034190-08-3 |
ALZ-801
Synonyms: valiltramiprosate, NRM-8499, homotaurine prodrug, 3-APS
This is a prodrug of homotaurine, a modified amino acid previously developed under the names tramiprosate and Alzhemed™. ALZ-801 is converted to homotaurine in vivo, but is more easily absorbed and lasts longer in the blood than tramiprosate.
Tramiprosate was reported to inhibit Aβ42 aggregation into toxic oligomers (Gervais et al., 2007; Kocis et al., 2017). Both ALZ-801 and tramiprosate are metabolized to 3-sulfopranpanoic acid (3-SPA), which is normally found in brain and also inhibits Aβ42 aggregation (Hey et al., 2018). A more recent study found that homotaurine activates GABA receptors, and suggests an alternative mechanism of action for ALZ-801 (Meera et al., 2023).
After tramiprosate failed in Phase 3, its maker, NeuroChem, marketed it as a nutritional supplement. Years later, a subgroup analysis of the trial data indicated a potential positive effect in participants who carried two copies of ApoE4 (Abushakra et al., 2016; Abushakra et al., 2017). Alzheon licensed ALZ-801 from NeuroChem and is developing it for Alzheimer’s disease.
ALZ-801 is a potent and orally available small-molecule β-amyloid (Aβ) anti-oligomer and aggregation inhibitor, valine-conjugated proagent of Tramiprosate with substantially improved PK properties and gastrointestinal tolerability compared with the parent compound. ALZ-801 is an advanced and markedly improved candidate for the treatment of alzheimer’s disease.
SCHEME

REF 1
US20080146642
https://patents.google.com/patent/US20080146642A1/en
HCL WATER, Dowex™ Marathon™ C ion-exchange column
General/Typical Procedure: [0311] (i) The solid material was dissolved in water (25 mL). The solution was passed through a Dowex™ Marathon™ C ion-exchange column (strongly acidic, 110 g (5 eq), prewashed). The strong acidic fractions were combined and treated with concentrated HCl (10 mL). The mixture was stirred at 50° C. for 30 minutes, and then was concentrated to dryness. The residual material was co-evaporated with EtOH (ethanol) to completely remove water. EtOH (100 mL) was added to the residue. The mixture was stirred at reflux for 1 h, and then cooled to room temperature. The solid material was collected by filtration. The solid material was dissolved in water (10 mL). The solution was added drop wise to EtOH (100 mL). The product slowly crystallized. The suspension was stirred at room temperature for 30 minutes. The solid material was collected by filtration and it was dried in a vacuum oven (60° C.). ID A2. 1H NMR (D2O).δ. 0.87-0.90 (m, 6H), 1.83 (qt, J = 7.2 Hz, 2H), 2.02-2.09 (m, 1H), 2.79 (t, J = 7.8 Hz, 2H), 3.20-3.29 (m, 2H), 3.60 (d, J = 6.3 Hz, 2H); 13C NMR (D2O).δ. 17.20, 17.77, 24.11, 30.00, 38.29, 48.63, 58.96, 169.35; m/z 237 (M-1).
- [1]. John A. Hey, et al. Discovery and Identification of an Endogenous Metabolite of Tramiprosate and Its Prodrug ALZ-801 that Inhibits Beta Amyloid Oligomer Formation in the Human Brain. CNS Drugs. 2018; 32(9): 849–861.[2]. Hey JA, et al. Clinical Pharmacokinetics and Safety of ALZ-801, a Novel Prodrug of Tramiprosate in Development for the Treatment of Alzheimer’s Disease. Clin Pharmacokinet. 2018 Mar;57(3):315-333. [Content Brief]
////////VALILTRAMIPROSATE, ALZ-801, ALZ 801, BLU 8499, WHO 11912
ATUZAGINSTAT


ATUZAGINSTAT, COR388
cas 2211981-76-7
Cyclopentanecarboxamide, N-[(1S)-5-amino-1-[2-(2,3,6-trifluorophenoxy)acetyl]pentyl]-
Cyclopentanecarboxamide, n-((1s)-5-amino-1-(2-(2,3,6-trifluorophenoxy)acetyl)pentyl)-N-((3s)-7-amino-2-oxo-1-(2,3,6- trifluorophenoxy)heptan-3-yl)cyclopentanecarboxamide
C19H25F3N2O3
386.415
UNII-DGN7ROZ8EN
- OriginatorCortexyme
- DeveloperQuince Therapeutics
- ClassAnti-inflammatories; Antibacterials; Antidementias; Antineoplastics; Antiparkinsonians; Neuroprotectants; Small molecules
- Mechanism of ActionPeptide hydrolase inhibitors
- Phase II/IIIAlzheimer’s disease
- Phase IIPeriodontal disorders
- PreclinicalParkinson’s disease; Squamous cell cancer
- 27 Jan 2023COR 388 licensed to Lighthouse Pharmaceuticals in the US
- 01 Aug 2022Atuzaginstat is available for licensing as of 01 Aug 2022. http://www.quincetx.com
- 01 Aug 2022Cortexyme is now called Quince Therapeutics
You need to be a logged in or subscribed to view this content
This small molecule is an orally available protease inhibitor targeting the lysine proteases of the periodontal pathogen Porphyromonas gingivalis. Known as gingipains, these proteases penetrate gingival tissue and cause inflammation at the site of periodontitis (O’Brien-Simpson et al., 2009). Periodontitis has been linked epidemiologically to cognitive impairment, and P. gingivalis bacterial lipopolysaccharide has been detected in postmortem brain tissue of people with AD (Poole et al., 2013). Oral P. gingivalis has been called a risk factor for Alzheimer’s disease (Kanagasingam et al., 2020).
Cortexyme’s approach is based on the theory that P. gingivalis invades the brain, where gingipains contribute to Alzheimer’s pathology (see Sabbagh and Decourt, 2022). The company reported elevated gingipain in brain tissue from people with AD, and a correlation between levels of gingipain and tau proteins in postmortem middle temporal gyrus from AD and healthy control tissue. P. gingivalis DNA was detected in postmortem cortices from people with AD and healthy controls, and in CSF of AD patients (Jan 2019 news on Dominy et al., 2019). In the same study, they show that in mice, oral P. gingivalis infection led to the appearance of bacterial DNA in the brain, increased brain Aβ42 production, neuroinflammation, and hippocampal degeneration. The first three findings were reported to be reduced by atuzaginstat; results for hippocampal cell death were not reported.
In preclinical work from other labs, infection with P. gingivalis was reported to worsen AD pathology and cognitive impairment in AD transgenic mice, and to cause neuroinflammation, memory impairment, neurodegeneration, micro- and astrogliosis, increased brain Aβ and phospho-tau, and neurofibrillary tangles in wild-type C57Bl6 mice (Ishida et al., 2017; Ilievski et al., 2018; Ding et al., 2018). For a review of the preclinical literature, see Costa et al., 2021.
In human neurons grown in culture, P. gingivalis infection led to tau phosphorylation and degradation, synapse loss, and cell death (Haditsch et al., 2020).
P. gingivalis is associated with cardiovascular disease. In rabbits, oral infection was reported to increase arterial plaque and levels of the inflammatory marker CRP. Both were reversed by treatment with COR388 (2020 AAIC abstract). In aged dogs with periodontal disease, ninety days of COR388 reduced oral bacterial load and gum pathology (Arastu-Kapur et al., 2020). In addition, older dogs had bacterial antigens and ribosomal RNA in their brains, consistent with systemic infection seen in humans.
Findings
Two Phase 1 trials of atuzaginstat were completed by June 2019. In a single-dose study of 5 to 250 mg capsules in 34 healthy adults, the compound was safe and well-tolerated. A multiple-dose study assessed safety and tolerability in 24 healthy older adults (mean age of 60 years) and nine with AD (mean age 72). According to a company press release and a poster presentation at the 2018 CTAD conference, healthy adults received 25, 50, or 100 mg COR388 or placebo every 12 hours for 10 days; AD patients took 50 mg or placebo every 12 hours for 28 days. The pharmacokinetic profiles of COR388 in AD and controls were reported to be similar. All volunteers with AD had P. gingivalis DNA fragments in their CSF at baseline. COR388 caused no serious adverse reactions, and no one withdrew. Gingipains also were reported to degrade ApoE, and 28 days of treatment with COR388 was claimed to reduce CSF ApoE fragments (2020 AAIC abstract).
A Phase 2/3 trial (GAIN) evaluating a 48-week course of COR388 in 643 people with mild to moderate AD began in April 2019. Participants took either 40 mg, 80 mg, or placebo twice daily. The primary endpoint was to be ADAS-Cog11 score, and the ADCS-ADL was added later as a co-primary functional endpoint. Further outcomes included CDR-SB, MMSE, NPI, the Winterlight Speech Assessment, MRI brain scans, and change in periodontal disease status. Investigators assessed CSF Aβ and tau, plus P. gingivalis DNA and gingipains in CSF, blood, and saliva, before and after treatment. A dental substudy of 228 participants is assessing effects of COR388 on periodontal disease. This trial involves 93 sites in the U.S. and Europe. The U.S. sites are offering a 48-week open-label extension.
According to a presentation at the 2020 CTAD, GAIN was fully enrolled. At baseline, more than 80 percent of participants had CSF Aβ and tau levels consistent with amyloid positivity or an AD diagnosis. All had detectable antibodies to P. gingivalis in their blood. In the dental substudy, 90 percent had periodontal disease. In December 2020, an independent data-monitoring committee recommended continuing the trial after a planned futility analysis of 300 patients treated for six months (press release).
In February 2021, the FDA placed a partial clinical hold on GAIN because of liver abnormalities in some participants (press release). Dosing in the open-label extension was stopped, but the placebo-controlled portion of GAIN continued. Cortexyme characterized the liver effects as reversible and showing no risk of long-term effects.
In October 2021, Cortexyme announced top-line results indicating the trial had missed its co-primary endpoints of ADAS-Cog11 and ADCS-ADL (press release). The company reported a statistically significant 57 percent slowing of decline on the ADAS-Cog11 in a subgroup with detectable saliva P. gingivalis DNA at baseline who took the higher dose; a 42 percent slowing on the lower dose did not reach statistical significance. This prespecified subgroup analysis included 242 participants; it found no effect on the ADCS-ADL. Improvements in ADAS-Cog and other cognitive endpoints correlated with reductions in saliva P. gingivalis DNA, according to data presented at CTAD 2021 in November. The most common treatment-related adverse events were gastrointestinal, occurring in 12 to 15 percent of treated participants. The treatment groups had dose-related liver enzyme elevations greater than three times the upper limit of normal, in 7 and 15 percent of participants on low and high doses, respectively, with bilirubin elevation reported in two participants on the high dose. The elevations occurred mainly in the first six weeks of treatment, and all resolved without long-term effects. Discontinuations due to transaminase elevations numbered one on placebo, and five and 17 in the 40 mg and 80 mg groups, respectively. The overall dropout rate was 25 percent in the placebo group, and 40 percent in atuzaginstat groups. There were five deaths in the high dose arm, and one in the low dose. All were deemed unrelated to drug. There was no evidence of ARIA or other imaging abnormalities.
At CTAD, the company announced plans for a confirmatory trial, pending discussions with regulators. The plan was to test atuzaginstat in people with mild to moderate AD and evidence of P. gingivalis infection, at the lower dose of 40 mg twice daily, reached by titration to minimize liver effects. The company was also planning a trial in Parkinson’s disease to begin in 2022. These trials were never registered.
In September 2021, Cortexyme began a Phase 1 trial of a second-generation lysin-gingipain inhibitor, COR588 (press release). This compound is expected to require only once-daily dosing. Results were expected in May 2022.
In January 2022, the company announced that the FDA had placed a full clinical hold on atuzaginstat due to concerns about liver toxicity (press release). The company said it intended to develop its backup compound, COR588, for Alzheimer’s disease, pending Phase 1 results. In July 2022, Cortexyme announced that COR588 had met safety and tolerability endpoints in a single- and multiple-ascending dose study in healthy adults (press release).
In August 2022, Cortexyme discontinued the gingipain inhibitor program, and offered it for external licensing (press release). The company changed its name to Quince, and its focus to bone disease. In January 2023, Quince put out word that it had sold Cortexyme’s legacy small molecule protease inhibitor portfolio to Lighthouse Pharmaceuticals, a company co-founded by a former Cortexyme CEO (press release).
For all trials of atuzaginstat, see clinicaltrials.gov.
SCHEME

Patent
- US10730826, Compound 1a-racemic
- US10730826, Compound 1a-non-racemic
- Ketone inhibitors of lysine gingipainPublication Number: EP-3512846-A1Priority Date: 2016-09-16
- Ketone inhibitors of lysine gingipainPublication Number: US-2019210960-A1Priority Date: 2016-09-16
- Ketone inhibitors of lysine gingipainPublication Number: WO-2018053353-A1Priority Date: 2016-09-16
- Ketone inhibitors of lysine gingipainPublication Number: US-10730826-B2Priority Date: 2016-09-16Grant Date: 2020-08-04
- Ketone inhibitors of lysine gingipainPublication Number: US-2021053908-A1Priority Date: 2016-09-16
PATENT
WO2018053353
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018053353&_cid=P10-M1OFBK-46119-1



VIII. Examples
Example 1. Preparation of (S)-N-(7-amino-2-oxo-1-(2,3,6-trifluorophenoxy)heptan-3- yl)cyclopentanecarboxamide(1)hydrochloride
[0224] To a mixture of compound 1.4 (23.0 g, 67.2 mmol, 1.00 eq) in THF (200 mL) was added NMM (6.79 g, 67.2 mmol, 7.38 mL, 1.00 eq), isobutyl carbonochloridate (9.17 g, 67.2 mmol, 8.82 mL, 1.00 eq), and diazomethane (5.65 g, 134 mmol, 2.00 eq) at -40 °C under N2 (15 psi). The mixture was stirred at 0 °C for 30 min. LCMS showed the reaction was completed. FLO (200 mL) was added to the reaction and extracted with two 300-mL portions of ethyl acetate. The combined organic phase was washed with two 200-mL portions of brine (200, dried with anhydrous Na2SO4, filtered and concentrated under vacuum to provide crude compound 1.3 (30.0 g, crude) as a yellow oil.
[0225] To a mixture of compound 1.3 (20.0 g, 54.6 mmol, 1.00 eq) in EtOAc (300 mL) was
added hydrogen bromide(29.8 g, 121.7 mmol, 20.0 mL, 33% purity, 2.23 eq) at -20 °C under
N2 (15 psi). The mixture was stirred at -20 °C for 10 min. TLC (petroleum ether : ethyl
acetate = 0:1) showed the reaction was completed. The reaction was basified by addition of
saturated NaHCO3 until the pH of the mixture reached 8, and the mixture was extracted with
three 500-mL portions of EtOAc. The combined organic phase was washed with two 200-mL portions of brine, dried over anhydrous Na2SO4, filtered and concentrated under vacuum
to afford crude compound 1.2 (15.0 g, crude) as a yellow solid.
[0226] To a mixture of compound 1.2 (4.00 g, 9.54 mmol, 1.00 eq) in DMF (40.0 mL) was
added 2,6-difluorophenol (1.49 g, 11.4 mmol, 1.20 eq) and KF (1.66 g, 28.6 mmol, 670 μL,
3.00 eq) at 25 °C. The mixture was stirred at 25 °C for 3 h. TLC (petroleum ether: ethyl
acetate = 1:1) showed the reaction was completed. H2O (150 mL) was added to the mixture
and extracted with two 200-mL portions of ethyl acetate. The combined organic phase was
washed with two 100-mL portions of brine, dried with anhydrous Na2SO4, filtered, and
concentrated under vacuum. The residue was purified by silica gel chromatography
(petroleum ether: ethyl acetate = 100:1, 5:1) to afford compound 1.1 (2.50 g, 5.35 mmol,
56.1 % yield) as a yellow solid.
[0227] To a mixture of compound 1.1 (4.00 g, 8.22 mmol, 1.00 eq) in EtOAc (3.00 mL) was added HCl/EtOAc (40.0 mL) at 25 °C. The mixture was stirred at 25 °C for 2 h. TLC (petroleum ether : ethyl acetate=2:1) showed the reaction was completed. The mixture was concentrated in reduced pressure to provide (.S)-N-(7-amino-2-oxo-1-(2,3,6-trifluorophenoxy)heptan-3-yl)cyclopentanecarboxamide 1 hydrochloride salt (1.34 g, 3.16 mmol) as a light yellow solid. LCMS (ESI): m/z: [M + H] calcd for C19H25N2F3O3: 387.2; found 387.1; RT=2.508 min. 1HNMR (400 MHz, DMSO-d6) δ ppm 1.21 – 1.83 (m, 15 H) 2.60 – 2.81 (m, 3 H) 4.30 (ddd, J=9.70, 7.17, 4.52 Hz, 1 H) 5.02 – 5.22 (m, 2 H) 7.12 – 7.24 (m, 2 H) 7.98 (br s, 3 H) 8.32 (d, J=7.28 Hz, 1 H).
Paper Citations
- Raha D, Broce S, Haditsch U, Rodriguez L, Ermini F, Detke M, Kapur S, Hennings D, Roth T, Nguyen M, Holsinger LJ, Lynch CC, Dominy S. COR388, a novel gingipain inhibitor, decreases fragmentation of APOE in the central nervous system of Alzheimer’s disease patients: Abstract. Alzheimer’s & Dementia, 07 December 2020
- O’Brien-Simpson NM, Pathirana RD, Walker GD, Reynolds EC. Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect Immun. 2009 Mar;77(3):1246-61. Epub 2008 Dec 29 PubMed.
- Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013 Jan 1;36(4):665-77. PubMed.
- Kanagasingam S, Chukkapalli SS, Welbury R, Singhrao SK. Porphyromonas gingivalis is a Strong Risk Factor for Alzheimer’s Disease. J Alzheimers Dis Rep. 2020 Dec 14;4(1):501-511. PubMed.
- Sabbagh MN, Decourt B. COR388 (atuzaginstat): an investigational gingipain inhibitor for the treatment of Alzheimer disease. Expert Opin Investig Drugs. 2022 Oct;31(10):987-993. Epub 2022 Sep 1 PubMed.
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RL, Curtis MA, Dragunow M, Potempa J. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019 Jan;5(1):eaau3333. Epub 2019 Jan 23 PubMed.
- Ishida N, Ishihara Y, Ishida K, Tada H, Funaki-Kato Y, Hagiwara M, Ferdous T, Abdullah M, Mitani A, Michikawa M, Matsushita K. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech Dis. 2017;3:15. Epub 2017 Nov 6 PubMed.
- Ilievski V, Zuchowska PK, Green SJ, Toth PT, Ragozzino ME, Le K, Aljewari HW, O’Brien-Simpson NM, Reynolds EC, Watanabe K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One. 2018;13(10):e0204941. Epub 2018 Oct 3 PubMed.
- Ding Y, Ren J, Yu H, Yu W, Zhou Y. Porphyromonas gingivalis , a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing. 2018;15:6. Epub 2018 Jan 30 PubMed.
- Costa MJ, de Araújo ID, da Rocha Alves L, da Silva RL, Dos Santos Calderon P, Borges BC, de Aquino Martins AR, de Vasconcelos Gurgel BC, Lins RD. Relationship of Porphyromonas gingivalis and Alzheimer’s disease: a systematic review of pre-clinical studies. Clin Oral Investig. 2021 Mar;25(3):797-806. Epub 2021 Jan 20 PubMed.
- Haditsch U, Roth T, Rodriguez L, Hancock S, Cecere T, Nguyen M, Arastu-Kapur S, Broce S, Raha D, Lynch CC, Holsinger LJ, Dominy SS, Ermini F. Alzheimer’s Disease-Like Neurodegeneration in Porphyromonas gingivalis Infected Neurons with Persistent Expression of Active Gingipains. J Alzheimers Dis. 2020;75(4):1361-1376. PubMed.
- Ermini F, Rojas P, Dean A, Stephens D, Patel M, Haditsch U, Roth T, Rodriguez L, Broce S, Raha D, Nguyen M, Kapur S, Lynch CC, Dominy SS, Holsinger LJ, Hasturk H. Targeting porphyromonas gingivalis to treat Alzheimer’s disease and comorbid cardiovascular disease: abstract. Alzheimer’s & Dementia, 07 December 2020
- Arastu-Kapur S, Nguyen M, Raha D, Ermini F, Haditsch U, Araujo J, De Lannoy IA, Ryder MI, Dominy SS, Lynch C, Holsinger LJ. Treatment of Porphyromonas gulae infection and downstream pathology in the aged dog by lysine-gingipain inhibitor COR388. Pharmacol Res Perspect. 2020 Feb;8(1):e00562. PubMed.
///////ATUZAGINSTAT, COR388, COR 388, Cortexyme, Quince Therapeutics
CT 1812
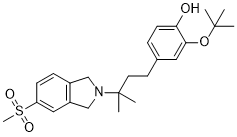
CT-1812
Elayta
Condition(s): Alzheimer’s Disease
U.S. FDA Status: Alzheimer’s Disease (Phase 2)
Company: Cognition Therapeutics Inc.
CAS: 1802632-22-9
Chemical Formula: C24H33NO4S
Molecular Weight: 431.591
2-(tert-butoxy)-4-(3-methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol
Phenol, 4-[3-[1,3-dihydro-5-(methylsulfonyl)-2H-isoindol-2-yl]-3-methylbutyl]-2-(1,1-dimethylethoxy)-
- Originator Cognition Therapeutics
- Class Antidementias; Neuroprotectants; Nootropics; Small molecules
- Mechanism of Action Sigma-2 receptor antagonists
- Phase II Alzheimer’s disease
- Phase I Cognition disorders
- 21 Feb 2019 Cognition Therapeutics receives patent for a composition of matter patent covering Elayta™ in Europe
- 19 Feb 2019 Pharmacokinetics and adverse events data from a phase I trial in Cognition disorders released by Cognition Therapeutics
- 22 Oct 2018 CTP push 289675: Updated KDM, forwarded USA line from PI/II to PII
CT-1812 is a first-in-class, orally available sigma-2/PGRMC1 antagonist (alpha beta oligomer receptor antagonist), is being developed by Cognition. sCT-1812 is a novel therapeutic candidate for Alzheimer’s disease
SYN

BACKGROUND
CT1812 is a small-molecule antagonist of the sigma2 receptor, also known as the progesterone receptor membrane component 1. The rationale behind this therapeutic approach is that ligands for the sigma2/PGRMC1 receptor will compete with oligomeric Aβ binding to this receptor and thus interfere with Aβ-induced synaptic toxicity. CT1812 grew out of screening programs at Cognition Therapeutics. Company scientists have reported that compounds in this series not only block binding of a range of different Aβ species to this receptor but also displace it when applied after Aβ has bound (Dec 2014 conference news).
The structure of CT1812 has not been disclosed, but similar compounds in the series have been reported to enter the brain, occupy up to 80 percent of sigma2/PGRMC1 receptors, and restore behavioral deficits in APP transgenic mice (Izzo et al., 2014; Izzo et al., 2014).
FINDINGS
From September 2015 to May 2016, Cognition Therapeutics ran a Phase 1 trial in 80 healthy volunteers aged 18 to 75 in Melbourne, Australia; target enrollment was originally listed as 114. Single-ascending-dose administration was followed by multiple ascending doses given once daily for two weeks. The dose range in this trial spanned 10 to 650 mg; if this would not generate data to set a maximum tolerated dose, doses up to 1,350 mg were to be tried. Outcome measures included safety, tolerability, plasma pharmacokinetics, and CSF CT1812 concentration. At the 2016 and 2017 AAIC conferences, company scientists reported that single doses up to 1,120 mg were given, as were multiple doses of up to 840 mg in young and up to 560 mg in elderly volunteers. The drug was reported to be well-tolerated, with suitable pharmacokinetics, sufficient brain penetrance and target exposure, and minimal drug-drug interactions affecting cytochrome P450 activity (Catalano et al., 2016; Catalano et al., 2017).
From September 2016 to August 2017, Cognition Therapeutics ran a Phase 1/2 trial at four sites in Australia, enrolling 19 participants with mild to moderate Alzheimer’s disease supported by a recent MRI. It compared a four-week course of 90, 280, or 560 mg of CT 1812 to placebo, taken once daily, on safety and tolerability parameters. At the subsequent CTAD conference, Elayta was reported to have been generally safe and well tolerated, though there were four cases of lymphocytopenia. Exploratory measures such as ADAS-Cog14, verbal or category fluency tests recorded no difference between groups, but exploratory biomarker analyses yielded possible signals of synapse protection (Dec 2017 conference news).
In April 2018, a Phase 1/2 study started enrolling 21 people whose mild to moderate AD was confirmed by amyloid PET or CSF testing. Conducted at Yale University School of Medicine and dubbed COG0105 or SPARC, this trial will compare a six-month course of 100 or 300 mg of Elayta, or placebo. The primary outcome is cognition as assessed by the Alzheimer’s Disease Clinical Study Activities of Daily Living (ADCS-ADL), but the trial will also use the investigational PET tracer UCB-J, which binds to the synaptic vesicle glycoprotein 2A, in an attempt to monitor synapse density before and after treatment (see company press release; Jul 2016 news).
In summer 2018, a Phase 1b target engagement study at the University of Pennsylvania will start enrolling 18 people whose mild to moderate AD is confirmed by amyloid PET. Called COG0104 or SNAP, it will compare single injections of 90, 280, or 560 mg of Elayta or placebo for their ability to displace Aβ oligomers and clear them into the CSF, as measured by a CSF Aβ oligomer assay.
Also in summer 2018, a Phase 2 multi-center study is expected to begin enrolling 24 people with mild to moderate AD as confirmed by amyloid PET for a six-month course of 100 or 300 mg of Elayta, or placebo. As of May 22, 2018, this trial lists CT1812 pharmacodynamic effects on CSF biomarkers, specifically as assessed by CSF neurogranin levels, as primary outcome.
For all trials of this compound, see clinicaltrials.gov.
PATENT
WO 2015116923
https://patents.google.com/patent/WO2015116923A1
There are only five medications currently FDA-approved for the treatment of Alzheimer’s Disease (AD). Four are cholinesterase inhibitors: tacrine (COGNEX®; Sciele), donepezil (ARICEPT®; Pfizer), rivastigmine (EXELON®; Novartis), and galantamine (RAZADYNE®; Ortho-McNeil-Janssen). Donepezil, rivastigmine, and galantamine are successors to tacrine, a first generation compound rarely prescribed because of the potential for hepatotoxicity; they are roughly equally efficacious at providing symptomatic improvement of cognition and function at all stages of AD. The fifth approved medication is memantine (NAMENDA®; Forest), a low-affinity, use dependent N-methyl-D-aspartate glutamate receptor antagonist that offers similar benefits, but only in moderate to severe AD. The clinical effects of these compounds are small and impermanent, and currently available data are inconclusive to support their use as disease modifying agents. See, e.g., Kerchner et al, 2010, Bapineuzumab, Expert Opin Biol Ther., 10(7): 1121-1130. Clearly, alternative approaches to treatment of AD are required.
[004] Certain isoindoline compounds are provided that act as sigma-2 receptor functional antagonists and inhibit the deleterious effects of soluble Αβ oligomers. In some embodiments, isoindoline sigma-2 receptor antagonist compounds and compositions are used to treat or prevent synaptic dysfunction in a subject.
Example 21 illustrates representative preparation of 2-(Tert-butoxy)-
4-(3-methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol, Example Compound 62, as shown in Scheme 17.


10 Compound 62
[0534] Scheme 17: Procedure for preparation of 2-(Tert-butoxy)-4-(3- methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol, Example Compound 62.
[0535] Preparation of compound l(Scheme 17): To a glass pressure -bottle at -30 °C containing a mixture of catechol (50.0 g, 454 mmol, 1.0 eq), concentrated sulfuric acid (0.3 mL) in dichloromethane (200 mL), isobutene (152.6 g, 2.72 mol, 6.0 eq) was condensed. After sealing the pressure-bottle with a threaded Teflon cap tipped with a Teflon-protected rubber O-ring, the mixture was heated at 35 °C for 3 h until a clear solution was obtained. After cooling (-30 °C), triethylamine (1.5 mL, 10.8 mmol) was added and the mixture was concentrated. The residue was suspended in 0.5 M NaOH (1 L) and stirred for 10 min. The dark-green colored solution was washed with petroleum ether (2x 100 mL) and the washing layers were reextracted with 0.5 M NaOH (3x 100 mL). The combined aqueous layers were brought to pH 7-8 with 2 N HCl (400 mL), and extracted with ethyl acetate (2* 1 L), dried over sodium sulfate and concentrated to afford product 1 (67.7 g, 90%) as a colorless oil, which was used directly for the next step reaction without further purification. TLC: PE/EA = 50/1 ; Rf (Catechol) = 0.1 ; Rf (Compound 1) = 0.6.
[0536] Preparation of compound 2 (Scheme 17): To a stirred solution of compound 1 (1 12.2 g, 676 mmol, 1.2 eq) and potassium iodide (1 12.2 g, 676 mmol, 1.0 eq) in methanol (2 L) at 0 °C was slowly added sodium hydroxide (27.0 g, 676 mmol, 1.0 eq), followed with aqueous sodium chlorite (7% aq., 718.8 mL, 710 mmol, 1.05 eq) dropwise over 3 h while keeping the reaction below 0 °C. The mixture was stirred at 0 °C for another 30 min and neutralized by adding 2 N HCl at 0 °C till pH 7, extracted with DCM (2 x 1 L). The organic layers were dried over sodium sulfate and concentrated to afford product 2 (179.8 g, 91%). TLC: PE/EA = 50/1; Rf(Compound 1) = 0.6 ; Rf (Compound 2) = 0.6.
[0537] Preparation of compound 3(Scheme 17): To a stirred solution of compound 2 (179.8 g, 616 mmol, 1.0 eq) and triethylamine (186.6 g, 1.85 mol, 3.0 eq) in dichloromethane (2 L) at 0 °C was slowly added acetyl chloride (53.2 g, 677 mmol, 1.1 eq). The mixture was stirred at 0 °C for another 30 min, and warmed up to rt, and stirred at rt for 3 h, water (1 L) was added into the reaction mixture and the organic layer was washed with brine, dried over sodium sulfate and concentrated to afford product 3 (206 g, 100%), which was used directly to the next step without further purification. TLC: PE/EA = 50/1; Rf (Compound 2) = 0.6; Rf (Compound 3) = 0.5.
[0538] Preparation of compound 4 (Scheme 17): To a stirred solution of compound 3 (206 g, 616 mmol, 1.0 eq) in triethylamine (4.0 L) was added 2- methylbut-3-yn-2-amine (102.5 g, 1.23 mol, 2.0 eq), Pd(PPh3)2Cl2 (15.1 g, 18.5 mmol, 0.03 eq) and copper(I) iodide (5.9 g, 31 mmol, 0.05 eq) and resulting mixture was stirred at rt for 17 h. The solvent was removed under reduced pressure and the crude product was purified by silica gel chromatography to afford the title compound 4 (132.7 g, 74%). TLC: PE/EA = 1/1; Rf (Compound 3) = 0.9; Rf (Compound 4) = 0.3. [0539] Preparation of compound 5(Scheme 17): To a stirred solution of compound 4 (104.5 g, 0.36 mol) in ethanol (1.5 L) was added Pd/C (10% wt, 10.5 g). The mixture was stirred under hydrogen (balloon) overnight, and filtered. The filtrate was evaporated to dryness to afford compound 5 (106.3 g, 100%), which was used directly to the next step without further purification. TLC: PE/EA = 1/1; Rf(Compound 4) = 0.3 ; Rf (Compound 5) = 0.3.
[0540] Preparation of compound 6 (Scheme 17): To a solution of o-xylene
(115.7 g, 1.09 mol, 1.0 eq) in chloroform (1.0 L) at 0 °C was added C1S03H (254 g, 2.18 mol, 2.0 eq) dropwise. After the addition, the reaction mixture was stirred at room temperature for 2 days, and poured into ice. The crude mixture was extracted with dichloromethane (3 x 1.0 L). The organic layers were combined, dried over anhydrous sodium sulfate, concentrated to afford the crude compound 6 (161.5 g, 80%) as a white solid, which was used directly to the next step without further purification. TLC: PE/EA = 5/1; Rf (Compound 6) = 0.7.
[0541] General procedure for the preparation of compound 7 (Scheme
17): To a stirred solution of compound 6 (161.5 g, 0.87 mol, 1.0 eq) in saturated sodium sulfite solution (273 g, 2.17 mol, 2.5 eq, in 2.0 L of water) was added dropwise 32% NaOH (69.4 g, 1.73 mol, 2.0 eq) till the solution reached pH 9. After stirring at rt overnight, the reaction mixture was acidified with cone. HC1 in ice- cooling bath till pH 1. The precipitate was filtered, and washed with ice-water (2x), dried in vacuo to afford the crude product 7 (131 g, 88%), which was used directly for next step without further purification. TLC: PE/EA = 5/1; Rf (Compound 6) = 0.7; Rf (Compound 7) = 0.6.
[0542] Preparation of compound 8 (Scheme 17): To a stirred solution of compound 7 (130 g, 0.76 mol, 1.0 eq) and potassium carbonate (211 g, 1.53 mol, 2.0 eq) in DMF (300 mL) was added iodomethane (96 mL, 1.53 mol, 2.0 eq). The reaction was stirred at 40 °C overnight. The reaction mixture was evaporated to dryness, extracted with ethyl acetate. The organic layers were washed with water and brine, dried over sodium sulfate and concentrated, purified by flash column chromatography (PE: EA,10: 1 ~ 5: 1) to afford compound 8 (85.2 g, 61%). TLC: PE/EA = 5/1; Rf (Compound 7) = 0.6; Rf (Compound 8) = 0.3. [0543] Preparation of compound 9 (Scheme 17):To a stirred solution of compound 8 (78.2 g, 424 mmol, 1.0 eq) in 1 ,2-dichloroethane (1.2 L), were added N-bromosuccinimide (166 g, 934 mmol, 2.2 eq) and AIBN (6.9 g, 42.4 mmol, 0.1 eq). The reaction was stirred at reflux overnight. The reaction was diluted with water and dichloromethane. The organic layer was collected, and dried over sodium sulfate and concentrated, purified by flash column chromatography to afford compound 9, which was further recrystallized from hot methanol to afford the pure product 8 (75 g, 52%). TLC: PE/EA = 5/1; Rf (Compound 8) = 0.3; Rf (Compound 9) = 0.2.
[0544] Preparation of compound 10 (Scheme 17):To a stirred solution of compound 5 (46 g, 157 mmol, 1.0 eq) and compound 9 (53.5 g, 157 mmol, 1.0 eq) in THF (460 mL) was added triethylamine (47.7 g, 472 mmol, 3.0 eq). The reaction was stirred at 40 °C overnight, filtered and the filtrate was evaporated to dryness and purified by flash column chromatography to afford compound 10 (45 g, 63%). TLC: PE/EA = 1/1; Rf (Compound 5) = 0.3; Rf (Compound 9) = 1.0; Rf (Compound 10) = 0.4.
[0545] Preparation of Compound 62 (Scheme 17):To a stirred solution of compound 10 (45 g, 98.4 mmol) in methanol (300 mL) was added sodium methoxide (844 mg, 15.6 mmol, 0.16 eq) in one portion. The solution was stirred at rt overnight. Water (250 mL) was added dropwise into the reaction mixture over 1 h, the mixture was stirred at rt for 2 h, and filtered. The white solid was collected and dried on vacuum overnight to afford pure example Compound 62 base (38 g, 89%>). TLC: PE/EA = 1/1; Rf (Compound 10) = 0.4; Rf (Compound 62) = 0.4; ESI-MS: 432 (M+l)+; 1H NMR (400 MHz, CDC13) δ 7.80-7.78 (m, 2H). 7.40-7.38 (m, 1H), 6.87-6.79 (m, 3H), 5.58 (s, 1H), 4.11 (s, 4H), 3.05 (s, 3H), 2.61-2.57 (m, 2H), 1.76- 1.72 (m, 2H), 1.48 (s, 9H), 1.18 (s, 6H). Example 22: Preparation of (2-(4-(4-Hydroxy-3-methoxyphenyl)-2- methylbutan-2-yl)isoindolin-4-yl)(piperazin-l-yl)methanone,
REFERENCES
1: Grundman M, Morgan R, Lickliter JD, Schneider LS, DeKosky S, Izzo NJ,
Guttendorf R, Higgin M, Pribyl J, Mozzoni K, Safferstein H, Catalano SM. A phase
1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a
novel therapeutic candidate for Alzheimer’s disease. Alzheimers Dement (N Y).
2019 Jan 23;5:20-26. doi: 10.1016/j.trci.2018.11.001. eCollection 2019. PubMed
PMID: 30723776; PubMed Central PMCID: PMC6352291.
Paper Citations
- Catalano S, Grundman M, Schneider LS, DeKosky S, Morgan R, Higgin M, Pribyl J, Mozzoni K, Izzo NJ, Safferstein H, Lickliter J. A Two-Part, Double-Blind, Placebo-Controlled, Phase 1 Study of the Safety and Pharmacokinetics of Single and Multiple Ascending Doses of Ct1812 in Healthy Volunteers. Alzheimer’s & Dementia, July 2016, Volume 12, Issue 7, Supplement
- Catalano S, Grundman M, Schneider LS, DeKosky S, Morgan R, Guttendorf R, Higgin M, Pribyl J, Mozzoni K, Izzo NJ, Safferstein H. A Phase 1 Safety Trial of the aβ Oligomer Receptor Antagonist CT1812. Alzheimer’s & Dementia, July 2017, Volume 13, Issue 7
- Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, Wostein H, Walko T 3rd, Vaswani A, Wardius M, Syed Z, Ravenscroft J, Mozzoni K, Silky C, Rehak C, Yurko R, Finn P, Look G, Rishton G, Safferstein H, Miller M, Johanson C, Stopa E, Windisch M, Hutter-Paier B, Shamloo M, Arancio O, LeVine H 3rd, Catalano SM. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One. 2014;9(11):e111898. Epub 2014 Nov 12 PubMed.
- Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, Rehak C, Yurko R, Look G, Rishton G, Safferstein H, Cruchaga C, Goate A, Cahill MA, Arancio O, Mach RH, Craven R, Head E, LeVine H 3rd, Spires-Jones TL, Catalano SM. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS One. 2014;9(11):e111899. Epub 2014 Nov 12PubMed.
/////CT-1812, CT 1812, CT1812, Alzheimers , Cognition Therapeutics, Elayta, phase 2, Cognition disorders
OC1=CC=C(CCC(C)(N2CC3=C(C=C(S(=O)(C)=O)C=C3)C2)C)C=C1OC(C)(C)C
Polyphenols and Alzheimers
“Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease in the growing population of elderly people. A hallmark of AD is the accumulation of plaques in the brain of AD patients. The plaques predominantly consist of aggregates of amyloid-beta (Abeta), a peptide of 39-42 amino acids generated in vivo by specific, proteolytic cleavage of the amyloid precursor protein.” (Finder & Glockshuber)
Stefani & Rigacci review the evidence for polyphenols and their ability to reduce amyloid aggregation. Natural polyphenols are emerging as an increasingly attractive treatment for amyloid disease prevention and therapy. Evidence suggests that they can inhibit the production of amyloidogenic peptides, increase antioxidant enzyme activity and reduce inflammation. The researchers suggest that we should now be describing them as potentially multitargeting drugs.
Potential sources of polyphenols are found in, amongst others, vegetables, fruit and green tea.


DONEPEZIL SYNTHESIS


Donepezil, marketed under the trade name Aricept by its developer Eisai and partnerPfizer, is a centrally acting reversible acetylcholinesterase inhibitor. Its main therapeutic use is in the palliative treatment of Alzheimer’s disease.Common side effects include gastrointestinal upset. It has an oral bioavailability of 100% and easily crosses the blood–brain barrier. Because it has a biological half-life of about 70 hours, it can be taken once a day.
Currently, no definitive proof shows the use of donepezil or other similar agents alters the course or progression of Alzheimer’s disease (AD). However, 6 to 12-month controlled studies have shown modest benefits in cognition and/or behavior.Pilot studies have reported donepezil therapy may potentially have effects on markers of disease progression, such as hippocampal volume. Therefore, many neurologists, psychiatrists, and primary-care physicians use donepezil in patients with Alzheimer’s disease. In 2005, the UK National Institute for Clinical Excellence (NICE) withdrew its recommendation for use of the drug for mild-to-moderate AD, on the basis of no significant improvement in functional outcome, quality of life, or behavioral symptoms. However, NICE revised its guidelines to suggest donepezil be used in moderate-stage patients for whom the evidence is strongest.

While the drug is currently indicated for mild to moderate Alzheimer’s, evidence from two clinical trials also indicates it may be effective for moderate to severe disease. An example of this is a Karolinska Institute paper published in The Lancet in early 2006, which states donepezil improves cognitive function even in patients with severe AD symptoms. In Oct. 2006 the U.S. Food and Drug Administration also approved Aricept for treatment of severe dementia.

【通用名】 Donepezil hydrochloride, BNAG, E-2020, Eranz, Memorit, Memac, Aricept
【化学名】 (?-1-Benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylmethyl)piperidine hydrochloride; (?-2-(1-Benzylpiperidin-4-ylmethyl)-5,6-dimethoxyindan-1-one hydrochloride
【CAS登记号】 120011-70-3, 123958-79-2 ([2-14C]-labeled), 142057-77-0 (deleted CAS), 120014-06-4 (free base)
【分子式】 C24-H29-N-O3.Cl-H
【分子量】 415.958
【化学活性】 Alzheimer’s Dementia, Treatment of , Analgesic and Anesthetic Drugs, Antimigraine Drugs, Attention Deficit Hyperactivity Disorder (ADHD), Treatment of, Autism, Treatment of, Cognition Disorders, Treatment of, Immunologic Neuromuscular Disorders, Treatment of, Migraine, Prophylactic Treatment of, Multiple Sclerosis, Agents for, Neurologic Drugs, Psychopharmacologic Drugs, Vascular Dementia, Treatment of, Acetylcholinesterase Inhibitors
【开发阶段】 Launched-1997
【研究机构】 Eisai (Originator), National Institute of Mental Health (Not Determined), Bracco (Licensee), Pfizer (Licensee)

Donepezil inhibiting Torpedo californicaacetylcholinesterase. See Proteopedia1eve.

Research leading to the development of donepezil began in 1983 at Eisai, and the first Phase I clinical trial took place in 1989. In 1996, Eisai received approval from the United States Food and Drug Administration (USFDA) for donepezil under the brand Aricept, which it co-marketed with Pfizer. As of 2011, Aricept was the world’s best-selling Alzheimer’s disease treatment. The first generic donepezil became available in November 2010 with the USFDA approval of a formulation prepared by Ranbaxy Labs. In April 2011 a second generic formulation, from Wockhardt, received tentative USFDA marketing approval
| 标题: | Cyclic amine cpd., its use and pharmaceutical compsns. comprising it |
| 作者: | Sugimoto, H.; Tsuchiya, Y.; Higurashi, K.; Karibe, N.; Iimura, Y.; Sasaki, A.; Yamanashi, Y.; Ogura, H.; Araki, S.; Kosasa, T.; Kusota, A.; Kozasa, M.; Yamatsu, K. (Eisai Co., Ltd.) |
| 来源: | AU 8818216; EP 0296560; EP 0673927; EP 0742207; JP 1989079151; JP 1998067739; US 4895841; US 5100901 |
 |
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 标题: | Synthesis of 1-benzyl-4-[(5,6-dimethoxy[2-14C]-1-indanon)-2-yl]methylpiperidine hydrochloride (E-2020-14C) |
| 作者: | Sugimoto, H.; Mishima, M.; Iimura, Y. |
| 来源: | J Label Compd Radiopharm 1989,27(7),835-9 |
|
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 作者: | Casta馿r, J.; Prous, J. |
| 来源: | Drugs Fut 1991,16(1),16 |
|
|
| 合成路线图解说明:The condensation of 5,6-dimethoxy-1-indanone (I) with 1-benzylpiperidine-4-carboxaldehyde (II) by means of butyllithium and diisopropylamine in THF gives 1-benzyl-4-(5,6-dimethoxy-1-oxoindan-2-ylidenemethyl)piperidine (III), which is reduced with H2 over Pd/C in THF and treated with HCl in dichloromethane – ethyl acetate. | |
| 标题: | Synthesis of 1-benzyl-4-[(5,[C-11]6-dimethoxy-1-oxoindan-2-yl)methyl]piperidine: A promising ligand for visualisation of acetylcholine esterase by PET |
| 作者: | Santens, P.; DeReuck, J.; Dierckx, R.A.; Siegers, G.; Vermeirsch, H.; De Vos, F. |
| 来源: | J Label Compd Radiopharm 2000,43(6),595 |
 |
|
| 合成路线图解说明:11C-Labeled donepezil was prepared by methylation of 2-(1-benzylpiperidin-4-ylmethyl)-6-hydroxy-5-methoxyindan-1-one (I) with 11CH3I by means of tetrabutylammonium hydroxide in DMF. | |
……………………………..
………………..
……………………….
……………………
……………………….
Donepezil hydrochloride is a useful memory enhancer introduced by the Japanese pharmaceutical company Eisai. Its preparation was described in patent no. EP 296560. In this patent Donepezil was produced by reaction of 5,6-dimethoxy-1- indanone with 1 -benzyl-4-formylpiperidine in the presence of a strong base, such as lithium diisopropylamide followed by reduction of the double bond. According to this method, Donepezil was obtained (Scheme 1). Patent application WO 99/36405 describes another process for the synthesis of Donepezil. According to this patent, 2-alkoxycarbonyl-1-indanones are reacted with (4-pyridinyl) methyl halide moiety followed by hydrolysis and decarboxylation to give the 2-(4-pyridinyl)methyl-1-indanone derivative. This is followed by reaction with benzyl halides to obtain the corresponding quaternary ammonium salt, and followed by hydrogenation of the pyridine ring to obtain Donepezil (Scheme 2).
Patent application WO 99/36405 describes another process for the synthesis of Donepezil. According to this patent, 2-alkoxycarbonyl-1-indanones are reacted with (4-pyridinyl) methyl halide moiety followed by hydrolysis and decarboxylation to give the 2-(4-pyridinyl)methyl-1-indanone derivative. This is followed by reaction with benzyl halides to obtain the corresponding quaternary ammonium salt, and followed by hydrogenation of the pyridine ring to obtain Donepezil (Scheme 2). Patent application WO 97/22584 describes the preparation of Donepezil by reaction of pyridine-4-carboxyaldehyde with malonic acid to give 3-(pyridin-4-yl)-2- propenoic acid, followed by hydrogenation of the double bond to give 3-(piperidin-4-yl)-2-propionic acid. Reaction of this intermediate with methyl chloroformate afforded 3-[N-(methyloxycarbonyl) piperidin-4-yl]propionic acid. This was followed by reaction with oxalyl chloride to give methyl 4-(2-chlorocarbonylethyl)piperidin-1-carboxylate. Reaction with 1,2-dimethoxybenzene in the presence of aluminum chloride afforded methyl 4-[3-(3,4-dimethoxyphenyl)-3-oxopropyl]piperidin-1 -carboxylate. Reaction with tetramethyldiaminomethane afforded 4-[2-(3,4-dimethoxybenzoyl)allyl] piperidin-1-carboxylate. Reaction with sulfuric acid afforded methyl 4-(5,6-dimethoxy-1-oxoindan-2-yl)methylpiperidin-1- carboxylate. This was followed by treatment with base to give 5,6-dimethoxy-2-(piperidin-4-ylmethyl) indan-1-one, then reaction with benzyl bromide afforded Donepezil (Scheme 3).
Patent application WO 97/22584 describes the preparation of Donepezil by reaction of pyridine-4-carboxyaldehyde with malonic acid to give 3-(pyridin-4-yl)-2- propenoic acid, followed by hydrogenation of the double bond to give 3-(piperidin-4-yl)-2-propionic acid. Reaction of this intermediate with methyl chloroformate afforded 3-[N-(methyloxycarbonyl) piperidin-4-yl]propionic acid. This was followed by reaction with oxalyl chloride to give methyl 4-(2-chlorocarbonylethyl)piperidin-1-carboxylate. Reaction with 1,2-dimethoxybenzene in the presence of aluminum chloride afforded methyl 4-[3-(3,4-dimethoxyphenyl)-3-oxopropyl]piperidin-1 -carboxylate. Reaction with tetramethyldiaminomethane afforded 4-[2-(3,4-dimethoxybenzoyl)allyl] piperidin-1-carboxylate. Reaction with sulfuric acid afforded methyl 4-(5,6-dimethoxy-1-oxoindan-2-yl)methylpiperidin-1- carboxylate. This was followed by treatment with base to give 5,6-dimethoxy-2-(piperidin-4-ylmethyl) indan-1-one, then reaction with benzyl bromide afforded Donepezil (Scheme 3).
 Patent application EP 711756 describes the preparation of Donepezil by reaction of 5,6-dimethoxy-1- indanone with pyridin-4-aldehyde to give 5,6-dimethoxy-2-(pyridin-4-yl)methylene indan-1-one. Reaction with benzyl bromide afforded 1-benzyl-4-(5,6-dimethoxyindan-1-on-2-ylidene)methylpyridinium bromide. Hydrogenation in the presence of platinum oxide afforded Donepezil (Scheme 4).
Patent application EP 711756 describes the preparation of Donepezil by reaction of 5,6-dimethoxy-1- indanone with pyridin-4-aldehyde to give 5,6-dimethoxy-2-(pyridin-4-yl)methylene indan-1-one. Reaction with benzyl bromide afforded 1-benzyl-4-(5,6-dimethoxyindan-1-on-2-ylidene)methylpyridinium bromide. Hydrogenation in the presence of platinum oxide afforded Donepezil (Scheme 4).

United States Patent 6844440

EP 1386607 A1
