
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

TL 487
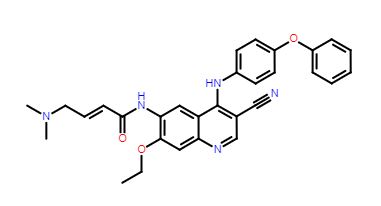
TL-487
- Molecular Weight, 507.58, MF C30 H29 N5 O3
Teligene Inc(2E)-N-[3-Cyano-7-ethoxy-4-[(4-phenoxyphenyl)amino]-6-quinolinyl]-4-(dimethylamino)-2-butenamide
(E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide
Maleate in anhydrous or monohydrate CAS, 2326561-36-6, AND 2326561-38-8 form are BTK and HER-2 kinase inhibitor useful for treating cancer
Useful for treating breast cancer, ovary cancer and colon cancer. are BTK and HER-2 kinase inhibitor useful for treating cancer.
Anticancer protein kinase inhibitor
The compound was originally claimed in WO2013152135 , and may provide the structure of TL-487 , a small molecule inhibitor to HERs, being investigated by Teligene for the treatment of breast cancer; in July 2016, the company intended to develop the product as a class 1.1 chemical drug in China.
PATENT
US 20150057312
PATENT
WO2013152135

PATENT
WO-2019096327
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019096327&redirectedID=true
Novel crystalline maleate salt of (E)-N-(3-cyano-7-ethoxy-4-((4-phenoxyphenyl)amino)quinolin-6-yl)-4-(dimethylamino)but-2-enamide (first disclosed in WO2013152135) and its hydrates (monohydrate) and anhydrates, process for its preparation, composition comprising it and its use for treating cancers such as breast cancer, ovary cancer, colon cancer, prostate cancer, kidney cancer, bladder cancer, stomach cancer, lung cancer, mantle cell lymphoma and multiple myeloma are claimed. The compound is disclosed to be an irreversible inhibitor to BTK and Her-2 (also known as Erb-2 or neu).
///////////////TL-487, PRECLINICAL, CHINA, breast cancer, ovary cancer, olon cancer, BTK, HER-2 kinase inhibitor,
CN(C)C\C=C\C(=O)Nc3cc4c(Nc2ccc(Oc1ccccc1)cc2)c(cnc4cc3OCC)C#N
TENAPANOR
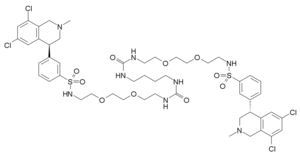


Tenapanor
Molecular FormulaC50H66Cl4N8O10S2
Average mass1145.049 Da
1234423-95-0 [RN]
1234423-95-0 (free base) 1234365-97-9 (2HCl)
9652
3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)-N-(26-((3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)phenyl)sulfonamido)-10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosyl)benzenesulfonamide
Benzenesulfonamide, N,N’-(10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosane-1,26-diyl)bis[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]-
12,15-Dioxa-2,7,9-triazaheptadecanamide, 17-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-
1-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethyl]-3-[4-[2-[2-[2-[[3-[(4S)-6,8-dichloro-2-methyl-3,4-dihydro-1H-isoquinolin-4-yl]phenyl]sulfonylamino]ethoxy]ethoxy]ethylcarbamoylamino]butyl]urea
17-[[[3-[(4S)-6,8-Dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-1,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulfonyl]amino]ethoxy]ethoxy]ethyl]-8-oxo-12,15-dioxa-2,7,9-triazaheptadecanamide
Tenapanor, also known as AZD-1722 and RDX 5791, is an inhibitor of the sodium-proton (Na(+)/H(+)) exchanger NHE3, which plays a prominent role in sodium handling in the gastrointestinal tract and kidney. Tenapanor possesses an excellent preclinical safety profile and, as of now, there are no serious concerns about its side effects.
Tenapanor is a drug developed by Ardelyx, which acts as an inhibitor of the sodium-proton exchanger NHE3. This antiporterprotein is found in the kidney and intestines, and normally acts to regulate the levels of sodium absorbed and secreted by the body. When administered orally, tenapanor selectively inhibits sodium uptake in the intestines, limiting the amount absorbed from food, and thereby reduces levels of sodium in the body.[1] This may make it useful in the treatment of chronic kidney disease and hypertension, both of which are exacerbated by excess sodium in the diet.[2]
Ardelyx and licensees Kyowa Hakko Kirin and Fosun Pharma are developing tenapanor, an NHE3 (Na+/H+ exchange-3) inhibitor that increases fluid content in the GI tract and which also reduces GI tract pain via an unknown TRPV-1-dependent pathway, for treating constipation-predominant irritable bowel syndrome (IBS-C) and hyperphosphatemia in patients with end stage renal disease (ESRD).
Syn

PATENT
WO2010078449
PATENT
WO-2019091503
A novel crystalline form of tenapanor free base, process for its preparation, composition comprising it and its use for the preparation of tenapanor with chemical purity >98.8% is claimed. Also claimed are salt forms of tenapanor, preferably tenapanor phosphate and their use for treating irritable bowel syndrome, constipation, hyperphosphatemia, final stage renal failure, chronic kidney disease and preventing excess sodium in patients with kidney and heart conditions. Further claimed are processes for the preparation of tenapanor comprising the steps of reaction of a diamine compound with 1,4-diisocyanatobutane, followed by deprotection and condensation to obtain tenapanor. Novel intermediates of tenapanor and their use for the preparation of tenapanor are claimed. Tenapanor is known to be a sodium hydrogen exchanger 3 inhibitor and analgesic.
enapanor, having the chemical name 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazaheptadecaneamide, is a selective inhibitor of the sodium protonic NHE3 antiporter. Orally administered tenapanor selectively inhibits the absorption of sodium in the intestine. This leads to an increase of water content in the digestive tract, improved bowel flow and normalization of the frequency of bowel movement and stool consistency. At the same time it exhibits antinociceptive activity and ability to lower serum phosphate levels. Because of these properties, it is clinically tested for the treatment of irritable bowel syndrome, especially when accompanied by constipation, treatment of hyperphosphatemia, especially in patients with dialysis with final stage renal failure, treatment of chronic kidney disease, and prevention of excess sodium in patients with kidney and heart conditions. The tenapanor molecule, which was first described in the international patent application WO 2010/078449, has the following structural formula:
In this document, tenapanor was prepared as bishydrochloride salt. The bishydrochloride salt was prepared only in the form of an amorphous foam, which, after solidification, required grinding for further processing. However, the thus obtained particles are of varying sizes, while a narrow particle size distribution is required for pharmaceutical use in order to ensure uniform behavior. The amorphous foam obtained in the said document is essentially a thickened reaction mixture or a slightly purified reaction mixture containing, in addition to tenapanor, various impurities. The possibilities to purify the reaction mixtures are limited. Moreover, amorphous foams tend to adsorb solvents, and it is usually difficult to remove (or dry out) the residual solvents from the amorphous foam. This is undesirable for pharmaceutical use. A typical feature of amorphous foams is a large specific surface, resulting in a greater interaction of the substance with the surrounding environment. This significantly increases the risk of decomposition of the substance, for example through air oxygen, moisture or light. The present invention aims at overcoming these problems.
It would be advantageous to provide tenapanor solid forms (tenapanor free base or tenapanor salts) which are precipitated in solid forms, thus allowing to filter off the liquid reaction mixture containing the impurities. This results in a significantly improved purity.
The process used in WO 2010/078449 for the preparation of bishydrochloride salt of tenapanor was based on the preparation of 3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzene-l-sulfonyl chloride of formula III from 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l, 2,3,4-te
Scheme 1
The said document also discloses resolution of the starting tetrahydroisoquinoline of formula II by L-or D-dibenzoylt
(II) (S-II) (R-II)
Scheme 2
WO 2010/078449 discloses further steps of preparation of tenapanor, as shown in Scheme 3.
(V) (I)
Scheme 3
Individual synthetic steps described in Scheme 3 result in low yields: 42% for the reaction of the chloride of formula III with 2-(2-(2-aminoethoxy)ethoxy)ethylamine of formula IV, and 59% for the subsequent reaction with 1,4-diisocyanatobutane of formula V. The products of both synthetic steps are isolated by preparative chromatography which is technologically an unsuitable isolation and purification technique. The low yields and the need to use preparative chromatography for the isolation are caused by an abundance of side products and impurities and by the inability of the intermediates as well as of the product to provide a crystalline form.
Thus present invention thus further aims at providing a method of preparation of tenapanor which would be economically effective, in particular in relation to the expensive starting compound 4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline, and which would also enable industrial scale production, in particular by removing steps which cannot be scaled up effectively or which cannot be scaled up at all. Furthermore, the method of preparation of tenapanor should provide tenapanor in a form which is useful for use in pharmaceutical forms and does not have the disadvantages of an amorphous foam.
Tenapanor free base in the form of an amorphous solid foam was prepared by the procedure disclosed in patent application WO 2010/078449, Example 202. The chemical purity of the tenapanor prepared by this procedure was 96.5% (HPLC). The structure of tenapanor was verified by MS and H and 13C NMR spectra.
Step A
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline
Potassium carbonate (9.30 g) and anhydrous xylene (500 ml) were added to the reaction vessel. Benzyl mercaptane (25 g) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
(S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline 50 g in anhydrous xylene (500 ml), Pd2(dba)3 (3 g) and Xantphos (3 g). The resulting solution was stirred at 25 °C for 30 minutes and then added to a solution of benzyl mercaptane. The resulting reaction mixture was maintained at 140 °C for 16 h. The mixture was then concentrated and the residue was subjected to preparative chromatography on silica gel with the mobile phase ethyl acetate / petroleum ether (1: 100-1 :50). 20 g of product are obtained as a yellow oil (36% yield).
Ste B
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochloride
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (16 g) was dissolved in the reaction vessel in acetic acid/water (160 mL: 16 mL) mixture. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the well stirred mixture. After disappearance of the starting material, the reaction mixture was purged with nitrogen and concentrated in vacuo. A product (10 g, 66.6%) was obtained as a colorless substance.
Step C
Preparation of (S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l, 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
2-(2-(2-Aminoethoxy)ethoxy)ethylamine HC1 (30 g; 0.2 mol) and triethylamine (5.2 g; 52 mmol) were dissolved in dichloromethane (500 ml) and the mixture was chilled in an ice bath. (S)-3-(6,8-Dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (10 g; 26 mmol) was added in parts during 40 minutes to the chilled reaction mixture. The ice bath was removed and the reaction mixture was stirred at laboratory temperature for additional 30 minutes.
The dichloromethane solution was extracted three times by brine (2x 250 ml), dried over sodium sulphate, and concentrated in vacuo. The residue was purified using preparative chromatography on silica gel with dichloromethane-methanol mobile phase.
Yield 7.2 g. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Step D
Preparation of 17-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl]sulphonyl]amino]-N-[2-[2-[2-[[[3-[(4S)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl -4-isoquinolinyl] phenyl] sulphonyl] amino] ethoxy] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7,9-triazah
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide (5g; 10 mmol) prepared in step A was dissolved in dichloromethane (50 ml). Triethylamine (1.5 g; 14.9 mmol) and 1 ,4-diisocyanatobutane (0.48 g; 3.4 mmol) were added to the solution. The reaction mixture was cooled using ice and stirred overnight. The resulting fine suspension was filtered off, the filtrate was concentrated and the obtained product was purified by preparative chromatography on on silica gel with dichloromethane-methanol mixture as a mobile phase
Yield: 2 g of tenapanor in the form of amorphous solid foam. HPLC purity 96.5 %.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. *H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H).
Ste E
Preparation of bishydrochloride salt of tenapanor
Tenapanor free base (1 g; 0.85 mmol) prepared in step B was dissolved in a mixture of methanol (10 ml) and 4M aqueous HCl (0.5 ml; 2 mmol) under mild reflux. The solution was concentrated on rotary vacuum evaporator, and the title product was obtained in the yield of 1 g of amorphous solid foam.
Example 1
Preparation of tenapanor, crystalline form I
Tenapanor free base (200 mg, 0.17 mmol), prepared as in step D of the comparative example, was dissolved in 0.4 ml acetonitrile under mild reflux. The clear solution was cooled at the rate of 1 °C/min with stirring to laboratory temperature (i.e., range from 22 °C to 26 °C) and then stirred for additional 2 hours at this temperature. The resulting crystals were isolated by filtration on sintered glass filter and dried for 6 hours in a vacuum oven at 40 °C. Crystallization yield was 170 mg of crystalline form I of tenapanor. HPLC showed a purity of 99.5%.
Examples 4 to 9 illustrate the inventive method of preparation of crystalline tenapanor.
Example 4
Preparation of (5)- -(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinoline
DIPEA (9.6 mL) and anhydrous dioxane (100 mL) were added to a reaction vessel. Benzyl mercaptan (8.1 ml) was added dropwise to the stirred mixture under ice -cooling. The resulting mixture was stirred at 25 °C for lh.
In a second reaction vessel, (S)-4-(3-bromophenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline (21.2 g) in anhydrous dioxane (140 mL), Pd2(dba)3 (835 mg)and Xantphos (835 mg) were mixed. The resulting solution was stirred at 25 °C for 30 minutes and then added to the solution of benzyl mercaptan. The resulting reaction mixture was maintained at gentle reflux for 3 hours.
After cooling, the suspension obtained was filtered through a thin layer of celite. HC1 was added to the filtrate. The precipitated hydrochloride was isolated by filtration, washed well and dried. 21 g of pinkish product were obtained (81.6% yield).
Example 5
Preparation of (5) -3 -(6 , 8 -dichloro-2 -methyl- 1,2,3 ,4-tetr ahydroisoquinolin-4-yl)benzenesulf onyl chloride hydrochlorid
(S)-4-(3-(benzylthio)phenyl)-6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinoline hydrochloride (11.1 g) was stirred in DCM/2M HC1 (70 mL:6 mL) mixture in a reaction vessel. The mixture was cooled in an ice bath and then gaseous Cl2 was introduced into the vigorously stirred mixture. After disappearance of the starting material, the resulting suspension was bubbled through by nitrogen and the product was filtered off and washed with DCM. 9.2 g of white product was obtained (82.7% yield).
Example 6
In the reaction vessel, t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (21.8 g) was stirred in DCM. The mixture was cooled in an ice bath under an inert atmosphere. To the cooled solution was
added 1 ,4-diisocyanatobutane (6.14 g) and TEA (0.1 mL). The cooling bath was removed and the reaction mixture was further stirred for 2 h.
35% HCl was added to the reaction mixture and the mixture was stirred under gentle reflux overnight.
After cooling, the precipitated product was filtered off and washed with DCM.
The product was recrystallized from propan-2-ol. 22.3 g of white product was obtained (80% yield).
Example 7
Preparation of (5)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l , 2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonamide
(S)-3-(6,8-dichloro-2-methyl-l ,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (11.7 g) prepared in Example 2 was stirred in dichloromethane (100 ml) and the suspension was cooled in an ice bath. To the cooled suspension was added a solution of t-butyl 2-(2-(2-amionoethoxy)ethoxy)ethylcarbamate (6.8 g) and DIPEA (14 ml) in DCM (50 ml). The resulting solution was stirred for 2 hours in an ice bath. The reaction mixture was extracted twice with water. Concentrated HCl (15 mL) was added to the dichloromethane solution and the mixture heated at gentle reflux for 2 h.
The precipitated product, after cooling, was extracted into water. The aqueous phase was separated and basified with Na2C03. The product as the free base was extracted into DCM and the dichloromethane solution was dried over sodium sulfate and concentrated in vacuo. 12.9 g of product were obtained.
Yield 93.4%. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S.
Example 8
Preparation of 17-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l ,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-triazaheptadecanamide (tenapanor free base)
(S)-N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-3-(6,8-dichloro-2-methyl-l,2,3,4- tetrahydroisoquinolin- 4-yl)benzenesulfonamide (12.9 g) prepared in Example 4 was dissolved in dichloromethane (150 ml). To the solution was added triethylamine (0.3 ml) and 1,4-diisocyanatobutane (1.7 g). The reaction mixture was stirred at 25 °C for 2 h. The resulting reaction mixture was extracted with water and aqueous Na2C03. The dichloromethane solution of the product was dried over sodium sulfate and concentrated to a solid foam. Yield 13.9 g. The crude product was taken up in acetone (100 ml) and then recrystallized from methanol (80 ml). 7.3 g of white crystalline product was obtained. Yield 49.8%.
HRMS 1143.3186 [M+H]+, C5oH66Cl4N8010S2. !H NMR (500MHz, DMSO, ppm):7.69-7.66 (m, 6H), 7.54-7.50 (m, 6H), 6.89 (bs, 2H), 5.9 (t, 2H), 5.79 (t, 2H), 4.4 (dd, 2H), 3.7 (dd, 4H), 3.44-3.44 (m, 8H), 3.35 (dd, 8H), 3.12 (dd, 4H), 2.96-2.64 (m, 12H), 2.37 (s, 6H), 1.31 (bs, 4H)
Example 9
Preparation of 17-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl]phenyl] sulfonyl]amino]-N-[2-[2-[2-[[[3-[(45)-6,8-dichloro-l,2,3,4-tetrahydro-2-methyl-4-isoquinolinyl] phenyl] sulf onyl] amino] ethoxy ] ethoxy ] ethyl] – 8 -oxo- 12,15 -dioxa-2 ,7 ,9-
(S)-3-(6,8-dichloro-2-methyl-l,2,3,4-tetrahydroisoquinolin-4-yl)benzenesulfonyl chloride hydrochloride (0.81 g) prepared in Example 2 and l,l’-(butane-l,4-diyl)bis(3-(2-(2-(2-aminoethoxy)ethoxy)ethyl)urea) dihydrochloride prepared according to Example 3 (0.48 g) were stirred in anhydrous ΝΜΡ (10 ml). To the suspension was added DIPEA (2 mL) and the resulting solution was stirred at 60 °C for 1.5 h. Water (10 mL) was added dropwise to the reaction mixture and the mixture was cooled to 5 °C. The precipitated product was isolated and stirred in acetone at 5 °C overnight. The beige product was filtered off (0.67 g) and recrystallized from methanol (12 ml).
0.53 g of a colorless crystalline product was obtained.
Yield 78.7 %. HRMS 502.1247 [M+H]+, C22H29CI2N3O4S. DSC analysis showed the melting temperature of 130.5 °C.
Example 10
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 11
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of tetrahydrofurane (THF). From the thus prepared solution, 1 ml is taken and hydrobromic acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with hydrobromic acid precipitated from the solution in solid stable form, the salt was filtered off, washed with THF and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 12
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and phosphoric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with phosphoric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Example 13
Tenapanor (1.48 g, 1.3 mmol) is dissolved in 10 ml of acetone. From the thus prepared solution, 1 ml is taken and citric acid (0.4 mmol) is added. The mixture is stirred at room temperature for 24 hours. Salt of tenapanor with citric acid precipitated from the solution in solid stable form, the salt was filtered off, washed with acetone and dried by stream of inert gas. XRPD confirmed amorphousness of the product.
Other pharmaceutically acceptable acids were tested by the procedures shown in Examples 10-13, but did not yield salts which would precipitate in amorphous stable solid form from the solution. The tested acids were: methanesulfonic acid, benzenesulfonic acid, oxalic acid, maleinic acid, tartaric acid, fumaric acid, trichloroacetic acid.
Example 14
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, a solution of phosphoric acid in THF (50 μ1/5 ml) is added dropwise during 10 minutes. The resulting suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with phosph (79 %) oric is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 430 mg of colourless salt of tenapanor with phosphoric acid. XRPD showed amorphousness of the product.
Example 15
Tenapanor (500 mg, 0.44 mmol) is dissolved in 20 ml of THF at 45 °C. To this clear solution, hydrobromic acid (48%; 100 μΐ) is added dropwise during 10 minutes. A fine precipitate forms already during the dropwise addition of HBr, and the suspension is stirred at room temperature for 30 minutes. The precipitated salt of tenapanor with HBr is filtered off, washed with 3 ml of THF and dried by stream of inert gas. Yield: 397 mg (69 %) of colourless salt of tenapanor with HBr (1 :2). XRPD showed amorphousness of the product.
References
- ^ Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D (2014). “Intestinal inhibition of the na+/h+ exchanger 3 prevents cardiorenal damage in rats and inhibits na+ uptake in humans”. Sci Transl Med. 6 (227): 227ra36. doi:10.1126/scitranslmed.3007790. PMID 24622516.
- ^ Salt-buster drug cuts sodium absorbed from food. New Scientist, 14 March 2014
REFERENCES
1: Johansson SA, Knutsson M, Leonsson-Zachrisson M, Rosenbaum DP. Effect of Food Intake on the Pharmacodynamics of Tenapanor: A Phase 1 Study. Clin Pharmacol Drug Dev. 2017 Mar 24. doi: 10.1002/cpdd.341. [Epub ahead of print] PubMed PMID: 28339149.
2: Johansson S, Rosenbaum DP, Ahlqvist M, Rollison H, Knutsson M, Stefansson B, Elebring M. Effects of Tenapanor on Cytochrome P450-Mediated Drug-Drug Interactions. Clin Pharmacol Drug Dev. 2017 Mar 16. doi: 10.1002/cpdd.346. [Epub ahead of print] PubMed PMID: 28301096.
3: Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor Treatment of Patients With Constipation-Predominant Irritable Bowel Syndrome: A Phase 2, Randomized, Placebo-Controlled Efficacy and Safety Trial. Am J Gastroenterol. 2017 Feb 28. doi: 10.1038/ajg.2017.41. [Epub ahead of print] PubMed PMID: 28244495.
4: Carney EF. Dialysis: Efficacy of tenapanor in hyperphosphataemia. Nat Rev Nephrol. 2017 Apr;13(4):194. doi: 10.1038/nrneph.2017.27. PubMed PMID: 28239171.
5: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Åstrand M, Johansson S, Knutsson M, Langkilde AM, Chertow GM. Effect of Tenapanor on Serum Phosphate in Patients Receiving Hemodialysis. J Am Soc Nephrol. 2017 Feb 3. pii: ASN.2016080855. doi: 10.1681/ASN.2016080855. [Epub ahead of print] PubMed PMID: 28159782.
6: Koliani-Pace J, Lacy BE. Update on the Management of Chronic Constipation. Curr Treat Options Gastroenterol. 2017 Mar;15(1):126-134. doi: 10.1007/s11938-017-0118-2. Review. PubMed PMID: 28116695.
7: Charoenphandhu N, Kraidith K, Lertsuwan K, Sripong C, Suntornsaratoon P, Svasti S, Krishnamra N, Wongdee K. Na(+)/H(+) exchanger 3 inhibitor diminishes hepcidin-enhanced duodenal calcium transport in hemizygous β-globin knockout thalassemic mice. Mol Cell Biochem. 2017 Mar;427(1-2):201-208. doi: 10.1007/s11010-016-2911-y. PubMed PMID: 27995414.
8: Thammayon N, Wongdee K, Lertsuwan K, Suntornsaratoon P, Thongbunchoo J, Krishnamra N, Charoenphandhu N. Na(+)/H(+) exchanger 3 inhibitor diminishes the amino-acid-enhanced transepithelial calcium transport across the rat duodenum. Amino Acids. 2017 Apr;49(4):725-734. doi: 10.1007/s00726-016-2374-1. PubMed PMID: 27981415.
9: Afsar B, Vaziri ND, Aslan G, Tarim K, Kanbay M. Gut hormones and gut microbiota: implications for kidney function and hypertension. J Am Soc Hypertens. 2016 Dec;10(12):954-961. doi: 10.1016/j.jash.2016.10.007. Review. PubMed PMID: 27865823.
10: Johansson S, Leonsson-Zachrisson M, Knutsson M, Spencer AG, Labonté ED, Deshpande D, Kohler J, Kozuka K, Charmot D, Rosenbaum DP. Preclinical and Healthy Volunteer Studies of Potential Drug-Drug Interactions Between Tenapanor and Phosphate Binders. Clin Pharmacol Drug Dev. 2016 Sep 22. doi: 10.1002/cpdd.307. [Epub ahead of print] PubMed PMID: 27654985.
11: Ketteler M, Liangos O, Biggar PH. Treating hyperphosphatemia – current and advancing drugs. Expert Opin Pharmacother. 2016 Oct;17(14):1873-9. doi: 10.1080/14656566.2016.1220538. Review. PubMed PMID: 27643443.
12: Johansson S, Rosenbaum DP, Knutsson M, Leonsson-Zachrisson M. A phase 1 study of the safety, tolerability, pharmacodynamics, and pharmacokinetics of tenapanor in healthy Japanese volunteers. Clin Exp Nephrol. 2016 Jul 1. [Epub ahead of print] PubMed PMID: 27368672.
13: Block GA, Rosenbaum DP, Leonsson-Zachrisson M, Stefansson BV, Rydén-Bergsten T, Greasley PJ, Johansson SA, Knutsson M, Carlsson BC. Effect of Tenapanor on Interdialytic Weight Gain in Patients on Hemodialysis. Clin J Am Soc Nephrol. 2016 Sep 7;11(9):1597-605. doi: 10.2215/CJN.09050815. PubMed PMID: 27340281; PubMed Central PMCID: PMC5012484.
14: Nusrat S, Miner PB Jr. New pharmacological treatment options for irritable bowel syndrome with constipation. Expert Opin Emerg Drugs. 2015;20(4):625-36. doi: 10.1517/14728214.2015.1105215. Review. PubMed PMID: 26548544.
15: Spencer AG, Greasley PJ. Pharmacologic inhibition of intestinal sodium uptake: a gut centric approach to sodium management. Curr Opin Nephrol Hypertens. 2015 Sep;24(5):410-6. doi: 10.1097/MNH.0000000000000154. Review. PubMed PMID: 26197202.
16: Zielińska M, Wasilewski A, Fichna J. Tenapanor hydrochloride for the treatment of constipation-predominant irritable bowel syndrome. Expert Opin Investig Drugs. 2015;24(8):1093-9. doi: 10.1517/13543784.2015.1054480. Review. PubMed PMID: 26065434.
17: Thomas RH, Luthin DR. Current and emerging treatments for irritable bowel syndrome with constipation and chronic idiopathic constipation: focus on prosecretory agents. Pharmacotherapy. 2015 Jun;35(6):613-30. doi: 10.1002/phar.1594. Review. PubMed PMID: 26016701.
18: Gerritsen KG, Boer WH, Joles JA. The importance of intake: a gut feeling. Ann Transl Med. 2015 Mar;3(4):49. doi: 10.3978/j.issn.2305-5839.2015.03.21. PubMed PMID: 25861604; PubMed Central PMCID: PMC4381464.
19: Labonté ED, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Dy E, Black D, Zhong Z, Langsetmo I, Spencer AG, Bell N, Deshpande D, Navre M, Lewis JG, Jacobs JW, Charmot D. Gastrointestinal Inhibition of Sodium-Hydrogen Exchanger 3 Reduces Phosphorus Absorption and Protects against Vascular Calcification in CKD. J Am Soc Nephrol. 2015 May;26(5):1138-49. doi: 10.1681/ASN.2014030317. PubMed PMID: 25404658; PubMed Central PMCID: PMC4413764.
20: Spencer AG, Labonte ED, Rosenbaum DP, Plato CF, Carreras CW, Leadbetter MR, Kozuka K, Kohler J, Koo-McCoy S, He L, Bell N, Tabora J, Joly KM, Navre M, Jacobs JW, Charmot D. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med. 2014 Mar 12;6(227):227ra36. doi: 10.1126/scitranslmed.3007790. PubMed PMID: 24622516.
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.243.471 |
| Chemical and physical data | |
| Formula | C50H66Cl4N8O10S2 |
| Molar mass | 1145.046 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////////Tenapanor, AZD 1722, RDX 5791, chronic kidney disease, hypertension
CN1CC(C2=CC(=CC(=C2C1)Cl)Cl)C3=CC(=CC=C3)S(=O)(=O)NCCOCCOCCNC(=O)NCCCCNC(=O)NCCOCCOCCNS(=O)(=O)C4=CC=CC(=C4)C5CN(CC6=C(C=C(C=C56)Cl)Cl)C
HM04 or H0900
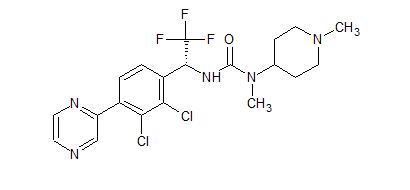
![3-[(1R)-1-(2,3-Dichloro-4-pyrazin-2-ylphenyl)-2,2,2-trifluoroethyl]-1-methyl-1-(1-methylpiperidin-4-yl)urea.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=91884613&t=l)
HM04 or H0900
Cas 1808913-24-7
(R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea
The compound was disclosed in WO2015134839 . Helsinn under license from Novo Nordisk , is investigating ghrelin antagonists for treating obesity, Prader-Willi syndrome and other metabolic disorders; in May 2015, the program was listed as being in preclinical development
Helps reducing ghrelin signaling activity and treating disorder associated with an increase in ghrelin level (eg food abuse, alcohol addiction, and Prader-Willi syndrome).
Ghrelin, a growth hormone-releasing peptide produced by ghrelinergic cells in the gastrointestinal tract, is understood to function as a neuropeptide that regulates energy metabolism by stimulating appetite. The modulation, for example inhibition, of ghrelin signaling, through the ghrelin/growth hormone secretagogue receptor (GHS-Rla), is an attractive target for pharmacological treatment of disorders associated with high ghrelin level. Potential disorders for treatment using ghrelin modulators include food abuse (such as binge eating, obesity, hyperphagia (or uncontrollable appetite), post-dieting body weight rebound (including post-dieting hyperphagia), alcohol addiction, and genetic diseases associated with increased ghrelin level (e.g., Prader-Willi syndrome (PWS)).
PATENT
US 20150252021

PATENT
WO2015134839
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015134839
Example 1
nthesis of Intermediate lk
Intermediate k
Step 1:
To a solution of la (100 g, 0.62 mol) in DMF (1.2 L) was added N-bromosuccinimide (110 g, 0.62 mol) at 0 °C. The mixture was stirred at room temperature for 4 h, then water (800 mL) was added and the resulting mixture was extracted with EtOAc (3 x 500 mL). The combined organic layers were dried over anhydrous Na2S04 and concentrated under reduced pressure. The residue was triturated with petroleum ether to provide lb (133.7 g, 89% yield) as a brown solid. !H-NMR (CDC13, 300 MHz): δ= 7.30 (d, 1 H), 6.59 (d, 1 H), 4.22 (br, 2 H). LC-MS: 241 [M+l]+.
Step 2:
To a solution of lb (133.7 g, 0.55 mol) in dry CH2C12 (1.5 L) was added acetic anhydride (110 g, 0.62 mol) dropwise over a period of 20 minutes at room temperature. The mixture was stirred at room temperature overnight, then diluted with CH2C12 (300 mL) and washed with water (150 mL) and brine (200 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was triturated with petroleum ether (300 mL) to provide compound lc (143.0 g, 91% yield) as a white solid. ¾-NMR (CDC13, 400 MHz): δ= 8.26 (d, 1 H), 7.63 (br, 1 H), 7.54 (d, 1 H), 2.26 (s, 3 H). LC-MS: 280 [M-l]“.
Step 3:
A mixture of compound lc (50.0 g, 0.18 mol), butyl vinyl ether (Id, 89.0 g, 0.89 mol), bis(l,3-diphenylphosphino)propane (DPPP, 22.0 g, 0.053 mol), TEA (100 mL, 0.71 mol) and Pd(OAc)2 (6.4 g, 0.027 mol) in DMSO (1.2 L) was heated at 130 °C under N2 overnight. After the reaction was completed, the mixture was cooled to 0 °C and 2N HC1 (480 mL) was added dropwise over a period of 30 minutes. Then, the mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous a2S04 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide le (19.5 g, 45% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 8.46 (d, 1 H), 7.82 (br, 1 H), 7.51 (d, 1 H), 2.63 (s, 3 H), 2.29 (s, 3 H). LC-MS: 244 [M-l]“.
Step 4:
To a solution of le (21.9 g, 89.4 mmol) in MeOH (350 mL) was added 2N NaOH solution (350 mL) at room temperature. The mixture was heated at 50 °C overnight, then cooled and concentrated under reduced pressure. The resulting solid was triturated with water (100 mL) for 30 min and filtered to provide If (18.0 g, 98% yield) as a brown solid. ¾-NMR (CDC13, 400 MHz): 3= 7.48 (d, 1 H), 6.68 (d, 1 H), 4.56 (br, 2 H), 2.62 (s, 3 H). LC-MS: 202[M-1]\
Step 5:
To a mixture of compound If (18.0 g, 89.2 mmol) and ice (360 g) in cone. HC1 (180 mL) was added a solution of NaN02 (9.2 g, 133.7 mmol) in water (20 mL) dropwise over a period of 30 minutes, and the resulting mixture stirred in an ice bath for 30 min. A solution of KI (74.0 g, 446 mmol) in water (360 mL) was added dropwise over 45 min at 0 °C. The mixture was stirred for 30 min and then extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 :40) to provide lg (23.9 g, 86% yield) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 7.6 (d, 1 H), 7.06 (d, 1 H), 2.62 (s, 3 H).
Step 6:
To a solution of lg (23.9 g, 76.1 mmol) in MeOH (100 mL)/THF (100 mL) was slowly added NaB¾ (2.9 g, 76.1 mmol) at 0 °C. The mixture was stirred at room temperature for 5 min, and then quenched with water (100 mL). The mixture was extracted with EtOAc (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 10) to provide lh (22.4 g, 93% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.26 (d, 1 H), 5.23 (q, 1 H), 2.17 (br, 1 H), 1.47 (d, 3 H).
Step 7:
To a mixture of lh (22.4 g, 70.9 mmol), phthalimide (12.5 g, 85.0 mmol) and PPh3 (22.3 g, 85.0 mmol) in dry THF (450 mL) was added DIAD (21.5 g, 106.3 mmol) at room temperature under N2 protection. The mixture was stirred at room temperature overnight and then concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc: PE=1 : 15) to provide li (18.5 g, 58% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.78-7.84 (m, 3 H), 7.70-7.73 (m, 2 H), 7.41-7.43 (d, 1 H), 5.76-5.81 (q, 1 H), 1.84 (d, 3 H).
Step 8:
A solution of li (7.2 g, 16.2 mmol) and hydrazine hydrate (98%, 4.0 g, 80.9 mmol) in MeOH (150 mL) was heated under reflux for 2 h, then cooled and concentrated under reduced pressure. The residue was diluted with water (100 mL) and extracted with CH2C12 (3 x 100 mL). The combined organic layers were dried over anhydrous Na2SC>4 and concentrated under reduced pressure to give lj (3.8 g, 75% yield) as a white solid. 1H-NMR (CDC13, 400 MHz): 3= 7.81 (d, 1 H), 7.25 (d, 1 H), 4.55 (q, 1 H), 1.36-1.38 (d, 3 H). LC-MS: 316 [M+l]+.
Step 9:
To a solution of lj (41. Og, 0.13 mol) in methyl tert-butyl ether (750 mL) was added slowly a solution of D-mandelic acid (7.8 g, 0.052 mol) in methyl tert-butyl ether (1 10 mL) at 45°C. The mixture was stirred at this temperature for 30 min then cooled and filtered. White solid obtained was partitioned between 5% NaOH solution (300 mL) and methyl tert-butyl ether (300 mL). The bi -phases were separated and the aqueous phase was extracted with methyl tert-butyl ether (300 mL). The combined organic layer was concentrated to provide Intermediate lk (12 g, 58.5% yield) as a white solid (ee%=98.0%, Chiralpak AD-H, 5 μπι, 4.6*250mm, mobile phase: Hex: EtOH : DEA=80 : 20 : 0.2), retention time = 6.408 min).
Example 2
Synthesis of Compoun
A suspension of N-methyl-4-piperidone 2a (13.3 g, 58.6 mmol), NH2Me (30% in MeOH, 100 mL) and Pd/C (0.66 g) in MeOH (200 mL) was heated at 60 °C under H2 atmosphere (50 psi) overnight, then cooled and filtered. The filtrate was concentrated under reduced pressure and the residue was dissolved in HC1 in dioxane (3N, 100 mL) and stirred for 30 min. The precipitate was filtered and washed with EtOAc (50 mL) to provide 2b (7.7g, 54% yield) as white powder. 1H-NMR (DMSO, 400 MHz): δ= 9.50 (br, 2 H), 3.48 (d, 2 H), 3.15-3.16 (m, 1 H), 2.96-3.01 (m, 2 H), 2.70 (s, 3 H), 2.51 (s, 3 H), 2.22-2.28 (m, 2 H), 1.94-2.02 (m, 2 H), LC-MS: 129 [M+l]+ .
Example 20
Synthesis of H0900
Step 1:
To a mixture of 16d (32 g, 120 mmol) in dry CH2CI2 (800 mL) was added Dess-Martin peroxide reagent (76 g, 180 mmol) portion- wise at 0 °C. The mixture was stirred at room temperature for 1 h, then diluted with DCM (800 mL), washed with aqueous NaHC03 solution (300 mL) and brine (300 mL). The organic phase was separated, dried over anhydrous Na2S04 and
concentrated under reduced pressure to afford crude 18a (31.4 g) which was used directly in the next step without further purification.
Step 2:
To a solution of 18a (12 g, 40 mmol) and 3b (22.2 g, 60 mmol) in DME (560 mL) were added Pd(PPh3)4 (9.25 g, 8 mmol) and Cul (1.52 g, 8 mmol) at room temperature. The mixture was stirred at 90 °C overnight, then concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE = 1 :5) to provide 18b (8.0 g, 79.3%) as a white solid. LC-MS: 253 [M+l]+.
Step 3:
To a solution of 18b (7 g, 27.7 mmol) and (¾)-tert-butylsulfinamide (7.27 g, 30.56 mmol) in dry THF (200 mL) was added Ti(i-OPr)4 (15.7 g, 55.4 mmol) dropwise at room temperature. The mixture was stirred at 80 °C overnight, and then cooled. Ethyl acetate (40 mL) was added, the resulting mixture was filtered and the filtrate was concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA:PE =1 :5) to provide 18c (6.8 g, 69%) as a yellow solid. 1H-NMR (CDC13, 400 MHz): 3= 9.10 (s, 1H), 8.97 (s, 1H), 8.72 (s, 1H), 8.64 (d, 1H),8.12 (d, 1H), 7.59 (d, 1H), 1.30 (s, 9H).LC-MS: 356 [M+l]+.
Step 4:
To a stirred solution of 18c (6.8 g, 19 mmol) and Tetrabutylammonium difluorotnphenylsilicate (15.8 g, 29 mmol) in dry THF (250 mL) was added a solution of TMSCF3 (11 g, 77 mmol) in anhydrous THF (50 mL) at -65 °C. The mixture was then stirred at -65 °C for 2 h, and at that point aqueous NH4CI solution (250 mL) was added. The mixture was diluted with ethyl acetate (250 mL), washed with brine (250 mL), dried over anhydrous Na2SC>4 and concentrated under reduced pressure. The residue was purified with silica gel column chromatography (silica, EA : PE=1 :2) to provide 18d (4.3 g, 52%) as a yellow solid. LC-MS: 426 [M+l]+.
Step 5:
To a stirred solution of 18d (4.3 g, 10.1 mmol) in MeOH (40 mL) was added a solution of HCl/MeOH (4N, 40 mL) at room temperature. The mixture was stirred for 1 h, then concentrated under reduced pressure. The residue was triturated with ethyl acetate (40 mL) to afford crude 18e (4.3g) which was directly in the next step without further purification. LC-MS: 322 [M+l]+.
Step 6:
To a solution of 18e (2.7 g, 7.1 mmol), 2b (3.4 g, 21.3 mmol) and TEA (80 mL) in DCM (220 mL) was added thiphosgene (3.15 g, 10.6 mmol) in DCM (40 mL) dropwise at 0 °C. The solution was warmed to ambient temperature and stirred for 1 h, then diluted with DCM ( 100 mL) and washed with aqueous Na2C03 solution (100 mL) and brine (100 mL). The organic layer was separated, dried over anhydrous Na2SC>4 and concentrated. The residue was purified with silica gel column chromatography (silica, DCM : CH3OH=10 : 1) to provide crude H0900 (2.13 g, ee%=92.5%) which was further purified through chiral separation to afford H0900 (1.6 g, 49% yield) as a white solid. (ee%=98.5%, Chiralpak IC 5um, 4.6*250mm, Phase: Hex: EtOH:
DEA=90: 10:0.2), retention tine =12.829 min. 1H-NMR (CDC13, 400 MHz): δ= 8.86 (d, 1H), 8.63 (dd, 1H), 8.55 (d, 1H), 7.47 (d, 1H), 7.40 (d, 1H), 6.28 (m, 1H), 5.18 (d, 1H), 4.12 (m, 1H), 2.88 (t, 2H), 2.77 (s, 3H), 2.22 (s, 3H), 2.05 (m, 2H), 2.48 (m, 2H), 1.52 (m, 2H), 1.73-1.49 (m, 4H). LC-MS: 476 [M+l]+.
PATENT
WO-2019118298
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019118298&tab=PCTDESCRIPTION&maxRec=1000
Novel crystalline fumarate salt forms of (R)-3-(1-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2,2,2-trifluoroethyl)-1-methyl-1-(1-methylpiperidin-4-yl) urea (also referred to as HM04 or H0900; designated as Forms 1-4), process for their preparation and compositions comprising them are claimed.
PWS occurs in approximately 1 in 10,000 births and is associated with deletion or lack of expression of region 15ql 1.2 of the paternal chromosome 15.
Characteristics of PWS include short stature, low muscle tone, and hyperphagia. Growth hormone replacement is frequently used to treat growth deficiencies and hypotonia. However, treatment for the insatiable appetite is lacking and PWS children can mature into adults suffering from obesity and type 2 diabetes. Levels of ghrelin are generally elevated in PWS; however, the relationship with ghrelin signaling and food intake in PWS remains unclear. See Purtell L., et ah, In adults with Prader-Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-l levels. Neuropeptides. 201 l;45(4):30l-7; Cummings D.E., et ah, Elevated plasma ghrelin levels in Prader Willi syndrome. Nature Medicine . 2002;8(7):643-4; DelParigi A., et ah, High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. The Journal of Clinical Endocrinology and Metabolism. 2002;87(l2):546l-4.
[005] Accordingly, it is desirable to find treatments that effectively inhibit GHSRla, that are tolerable to the patient, and that do not interfere with other functions of the growth hormones. GHSRla modulators, including inhibitors such as (R)-3-(l-(2,3-dichloro-4-(pyrazin-2-yl)phenyl)-2, 2, 2-trifluoroethyl)-l -methyl- l-(l-m ethylpiperidin-4-yl) urea (HM04, H0900) depicted below, are reported in LT.S. Patent No. 9,546,157.
Step 1 : Synthesis of compound 2A
[00106] 2,2,6,6-tetramethylpiperidine (7.20 kg, 51.1 mol, 3.0 eq.,
KF=0.30%) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer and mixed at RT under nitrogen protection. THF (50 L) was added into the reactor and stirred. The vessel was purged with nitrogen three times and cooled to 0 °C. n-BuLi (20.4 L, 3.0 eq.; 2.5 M hexane solution) was added to the mixture dropwise while keeping the temperature at about 0 °C to about 5 °C for over one hour. The color of the solution turned yellow. The mixture was stirred at about 0 °C to about 5 °C for 30 minutes. The mixture was cooled to about -78 °C to about -70 °C to form Solution A.
[00107] Compound 1 (3.25 kg, 17.0 mol. 1.0 eq., KF=0.03%) was dissolved in 15 L of THF to form Solution B.
[00108] Solution B was added to solution A dropwise at a temperature of about -70 °C to about -78 °C over one hour and then stirred for 30 minutes to form solution C. Tri-isopropyl borate ((i-PrO)3B) (3.52 kg, 18.7 mol., 1.1 eq.) was added dropwise into solution C over 10 minutes. The reaction mixture was stirred at a
temperature of about -70 °C to about -78 °C for one hour. HC1 (40 L, 3M, 7.0 eq.) was added over 30 minutes to quench the reaction. A 10 degree rise in temperature was noted.
[00109] The resulting aqueous layer was separated and extracted with EtOAc (40 L). The aqueous layer was separated and extracted twice again with EtOAc (35 L, 30 L). The organic layers were combined resulting in about 160 L of liquid. The combined organic layer was washed twice with 50 L of a 1M aqueous HC1 solution saturated with NaCl. The organic layer was concentrated to about 5 L in a 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 8 hours.
[00110] The residual EtOAc was swapped with DME for 3 times (10 L x 3). The organic layer was concentrated in the 50 L rotavapor at a temperature of about 50 °C to about 55 °C under 30-40 mmHg for about 6 hours. Each time about 5 L of residual remained. DME (20 L) was added to the residual to obtain a deep brown solution of 14.2% compound 2A (3.55 kg in 25 kg of solution; 88.8% yield; 97.4% purity (AETC by HPLC, retention time = 1.6 minutes); 0.24% residual ethyl acetate). 1H-NMR (400 MHz, DMSO): 5=8.55 (s, 2H), 7.36 (d, 1H), 7.69 (d, 1H). A second batch of compound 2A was prepared by the same method to produce 3.29 kg (95.4% purity, 82.3% yield, 0.11% residual ethyl acetate).
[00111] Step 2: Synthesis of Compound 3A
C! , N
M
K2CO3 (I .O equiv)
2A OH
DME/H20 3:1 (20 vol), 50 e C 3A N
[00112] Compound 2 A (2.91 kg in 20.5 kg solution) was added into a 100
L reactor at room temperature under nitrogen. DME (45 mL), 2-chloropyrazin (1.42 kg,
12.4 mol., 1.0 eq.), and Pd(dppf)Cl2 (10% w/w, 291 g) were added sequentially, and each
mixed at room temperature under nitrogen. Nitrogen was bubbled into the mixture for 20
minutes and the resulting mixture was purged and filled with nitrogen (3 times). The
mixture was heated to 48-52°C over 60 minutes. K2CO3 (2.57 kg, 18.6 mol, 1.5 eq.) was
added to 22 L of water in another reactor at room temperature and then added dropwise to
the compound 2 A mixture over 10 minutes. The mixture was stirred at 48-52°C for 16
hours and then cooled to room temperature. This procedure was repeated twice and all
three batches were combined.
[00113] An aqueous solution of K2CO3 (1.0 kg) was dissolved in 22 L of
water and added to the combined mixture to adjust the pH to 9. TBME (50 L) was added
into the mixture and filtered (PET filter, 3-5 pm, 205g/m2) to remove about 50 g of
sticky, brown solid material (catalyst analog). The aqueous layer was twice separated and
extracted with TBME (40 L, 40L).
[00114] The aqueous layer was combined with the aqueous layer of a
fourth batch prepared according to the above method. The pH of the combined aqueous layers was adjusted to pH<3 with HC1 (2N, 48 L). The solid precipitated out slowly as
the mixture was stirred at room temperature for 1 hour. The mixture was filtered (PET
filter, 3-5 pm, 205g/m2) over 30 minutes to obtain 20 kg of wet product. ACN (40 L) was
added into a 100 L reactor equipped with an overhead stirrer at room temperature. The 20
kg of wet product was added into the reactor and the reaction mixture heated to reflux
and stirred at reflux for 4 hours. The reaction mixture was cooled to room temperature
over 3 hours (around 15 °C/hour) and filtered to obtain 8.5 kg of wet solid. The wet solid
was dried under vacuum (20-30 mmHg) at 50-55 °C for 15 hours to obtain compound 3 A
as a pale white solid (6.1 kg; 97.4% purity (AUC by HPLC, retention time = 3.7
minutes); 83.8% yield). 1H-NMR (400 MHz, DMSO): 5=7.67 (d, 1H), 7.82 (d, 1H), 8.75
(d, 1H), 8.82 (t, 1H), 8.98 (d, 1H), 13.89 (bs, 1H).
[00115] Step 3: Synthesis of compound 6A
3A 6A N
N
[00116] Compound 3 A (6.1 kg, 22.7 mol, 1.0 eq.) was added into a 100 L
reactor equipped with a temperature probe, overhead stirrer, and condenser. Methanol
(92 L) was added into the reactor at room temperature. The mixture was cooled to
0-10 °C and added with SOCk (5.4 kg, 45.3 mol, 2.0 eq.) dropwise at 0-10 °C over 30
minutes. The reaction mixture was heated to reflux (65 °C) and stirred at reflux for 15
hours. A suspension was formed. Most of the solvent and SOCk was removed under
vacuum distillation until about 30 L remained. The mixture was concentrated under
vacuum (30-40 mmHg) at 50-55 °C for about 6 hours. Water (10 L) was added to the residual at -5 to 15 °C. The pH was adjusted to 8-9 with an aqueous solution of K2CO3 (200 g, dissolved in 2L of water) at -5 to 15 °C. The resulting aqueous layer was extracted twice with isopropyl acetate (25 L, 25 L). The combination of organic layers (about 50 kg) was washed with 20 L of NaHCCb aqueous layer. The organic layer was separated and washed with 10 L of of an aqueous solution of NaHCCb. All the aqueous layers were combined (55.8 kg). The organic layer was filtered through a silica pad (30 cm) and the pad washed with extra isopropyl acetate until the compound 6 A was filtered from the silica gel (about 3 hours). The organic layer was concentrated to about 5 L. THF (10 L) was added to the residual and concentrated to about 5 L (3 times) under vacuum (30-40 mmHg) at 50-55 °C for about 3 hours. Another 10 L of THF was added to the residual concentrate, giving a concentrated solution of compound 6A (15.8 kg; 32.83%,
5.19 kg compound 6A in solution; 97.9% purity (AUC by HPLC, retention time = 8.5 min); 80.8% yield). 1H-NMR (400 MHz, DMSO): 5=3.98 (s, 3H), 7.54 (d, 1H), 7.78 (d, 1H), 8.63 (d, 1H), 8.72 (t, 1H), 8.94 (d, 1H).
[00117] Step 4: Synthesis of compound 6B
[00118] THF (26 L) was added into a 100 L reactor equipped with a temperature probe and overhead stirrer under nitrogen. DIBAL-H (26 kg, 46 mol, 5.0 eq.) was added and the system purged and filled with nitrogen three times. The mixture was cooled to -78 to -70 °C to form solution A. A room temperature solution of compound 6A (2.6 kg, 9.2 mol, 1.0 eq.) in 52 L of THF was added dropwise at -78 to -70 °C over 30 minutes under nitrogen. The mixture was warmed to -30 °C over about 5-6 hours. The reaction mixture was stirred at -40 to -30 °C for 30 minutes. The mixture was slowly added to 42 L of 2N HCL over 1 hour reaching a maximum temperature of 35 °C. The mixture was extracted with 26 L of isopropyl acetate. The organic layer was separated and washed with 30 L of brine. This procedure was repeated and both batches of organic layer were combined and concentrated from about 100 L to about 5-10 L under vacuum.
A solid slowly formed during concentration. The mixture was cooled to 5-15 °C and stirred for 1 hour. The mixture was filtered (30-50 pm) over 30 minutes. The solid was dried under vacuum at 50 °C for 6 hours to obtain compound 6B as a brown solid (2.1 kg; 97.5% purity (AUC by HPLC, retention time = 8.6 min); 45.7% yield). 1H-NMR (400 MHz, DMSO): d = 4.65 (d, 2H), 5.68 (t, 1H), 7.62 (d, 1H), 7.68 (d, 1H), 8.72 (d, 1H),
8.80 (t, 1H), 8.94 (d, 1H).
[00119] Step 5: Synthesis of compound 7
[00120] DMSO (10 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 6B (2.05 kg, 8.04 mol, 1.0 eq.) was added under nitrogen at room temperature. Et3N (8 L) was added under nitrogen at RT and the mixture was then cooled to 15-20 °C.
SCb. pyridine (5.1 kg, 32.08 mol, 4.0 eq.) was dissolved into 10 L of DMSO at 5-15 °C in a separate flask and added to the mixture dropwise over 3.5 hours at about 20 °C. The reaction mixture was transferred to 70 L of ice-water. The suspension mixture was stirred at 0-10 °C for 1 hour and filtered (PET, 3-5 pm, 205 g/m2) by centrifuge over 1.5 hours to obtain compound 7 as a brown solid. The solid was dissolved in 35 L of DCM at room temperature. The resulting DCM layer was washed with 5 L of brine. The organic layer was separated and concentrated under vacuum at 40-45 °C to dryness to obtain compound 7 as a brown solid (2.33 kg; 96.3% purity (AEiC by HPLC, retention time = 9.2 minutes); 93.5% yield). 1H-NMR (400 MHz, DMSO): d = 7.67 (d, 1H), 7.99 (d, 1H), 8.67 (d, 1H), 8.75 (s, 1H), 8.99 (d, 1H), 10.56 (s, 1H).
[00121] Step 6: Synthesis of compound 8
[00122] THF (23 L) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at room temperature. Compound 7 (2.3 kg, 9.1 mol, 1.0 eq.) and (S)-2-methylpropane-2-sulfmamide (1.21 kg, 10 mol, 1.1 eq.) were added sequentially to the flask under nitrogen. Ti(OEt)4 (6.22 kg, 27.3 mol, 3.0 eq.) was added dropwise to the flask over 1 hour at 30-35 °C under nitrogen. The system was purged with nitrogen three times and then the mixture was stirred at room temperature for 2 hours. Isopropyl acetate (40 L) was added to the reaction mixture. The entire reaction mixture was then charged to 20 L of brine while stirring slowly at RT. A lot of solid was formed and no heat release was observed. The solid (about 18 kg) was filtered using centrifuge, and then the solid was slurried with 20 L of isopropyl acetate again for 20 minutes, and filtered again, resulting is slightly less solid (17.3 kg). The filtrates were then combined and washed with 20 L of brine. The organic layer was separated and concentrated in a rotavapor under vacuum (30-40 mmHg) at 40-50 °C for about 4 hours to remove the solvents and obtain a brown oil (compound 8). The oil was dissolved in DMF to obtain a black solution (7.36 kg; 40.1%; 3.0 kg compound 8 in solution; 92.1% purity (AUC by HPLC, retention time = 9.7 minutes); >100% yield). 1H-NMR (400 MHz, CDCb): d = 1.30 (s, 9H), 7.59 (d, 1H), 8.11 (d, 1H), 8.64 (s, 1H), 8.73 (m, 1H), 8.97 (s, 1H), 9.10 (s, 1H).
[00123] Step 7: Synthesis of compound 11
O
S
10 s C
8
11 N
[00124] DMF (26 L, 10 v/w) was added to a 50 L flask equipped with a temperature probe and overhead stirrer under nitrogen at 15 °C. Compound 8 (7.3 kg of
DMF solution, containing 2.9 kg, 8.1 mol, 1.0 eq.) and TBAA (2.44 kg, 8.1 mol, 1.0 eq.) were added sequentially to the flask under nitrogen. The mixture was cooled to 0-10 °C.
TMSCF3 (2.88 kg, 20.3 mol, 2.5 eq.) was then added to the flask over 60 min at 0-10 °C.
The reaction mixture was stirred at 0-5 °C under nitrogen protection for 3 hours.
Isopropyl acetate (60 L) was added to the mixture, followed by the addition of 45 L of
NaHCCb under stirring at 5-25 °C. The organic layer was separated, washed three times with NaHC03 (30 L x 3), and concentrated from 60 kg to 2.5 kg of brown oil. The oil product was dissolved in 20 L of TBME and filtered through a pad of silica gel (about 40 cm high, 30 cm diameter) over 2 hours to obtain 2.14 kg of compound 1 1 in TBME solution. The solution was concentrated at 45-50 °C to dryness to obtain compound 1 1 as a black oil (1.85 kg; 85.2% purity (AETC by HPLC, retention time = 9.1 minutes, 9.6 minutes for diastereoisomer); 53.6% yield). 1H-NMR (400 MHz, CDCh): d = 1.33 (s, 9H), 3.82-3.85 (d, 1H), 5.61-5.66 (m, 1H), 7.53-7.60 (m, 2H), 8.63-8.64 (d, 1H), 8.71-8.72 (m, 1H), 8.95 (s, 1H).
[00125] Step 8: Synthesis of compound 12 (free base)
[00126] Compound 1 1 (1.8 kg, 4.23 mol, 1.0 eq., crude) was added to a 50 L reactor equipped with a temperature probe and overhead stirrer under nitrogen at 25 °C. Anhydrous MeOH (18 L) was added to dissolve compound 1 1. Then MeOH/HCl (18 L, 1 N) was added dropwise at 25-30 °C over 10 minutes and the mixture was stirred at 25-30 °C for 1 hour. Water (15 L) was added to the reaction and the mixture concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 4 hours to remove the solvent. The pH of the mixture was adjusted to 10 with 5 L of K2CO3 solution. 20 L of EtOAc was then added to the mixture and the organic layer was separated and the aqueous layer extracted twice with EtOAc (15 L x 2). The organic layers were combined and washed with 10 L of brine. The combined organic layers contained 996 g of
compound 12 in 40 kg of EtOAc solution (84% purity (AUC by HPLC, retention time =
2.8 minutes). The organic layers were concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to a 7.5 kg volume of compound 12 in EtOAc solution (83% purity (AETC by HPLC, retention time = 2.7 minutes).
[00127] In a separate 50 L reactor equipped with a temperature probe and overhead stirrer, D-CSA was added (930 g, 4.0 mol, 1.0 eq. to 1.26 kg compound 12) and stirred at room temperature under nitrogen. EtOAc (10 L) and then the EtOAc solution of compound 12 (1.26 kg, 3.9 mol, 1.0 eq.) were each sequentially added to the reactor. The mixture was stirred at room temperature for 1 hour and slowly became a suspension. The mixture was filtered by centrifuge and washed with EtOAc to produce 2.3 kg of compound 12 as an off-white solid (96.0% purity).
[00128] The solid product, 20 L of EtOAc, and 10 L of 10% aqueous K2CO3 were added sequentially to a 50 L flask and stirred at room temperature until no solid remained (pH = 9-10). The organic layer was separated and the aqueous layer extracted twice with EtOAc (10 L x 2). The organic layers were combined (about 32 kg) and washed with 10 L of brine. The organic layer contained 716 g of compound 12 in
31.8 kg of solution.
[00129] The organic layer was concentrated under vacuum at 45-50 °C to about 8 L. Activated carbon (200 g) was added to the organic layer and the mixture stirred at 60-70 °C for 1 hour, cooled to room temperature, and filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to remove the activated carbon. The mixture was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to yield 710 g of compound 12 as a yellow solid (99.4% purity). [00130] D-CSA (410 g, 1.77 mol, 1.0 eq. to 680 g compound 12), 3.4 L iPrOH, and 68 mL of water were added sequentially to a 10 L reactor equipped with a temperature probe and overhead stirrer and stirred at room temperature under nitrogen. The mixture was heated to reflux (84 °C) to form solution A after 1 hour. Compound 12 (680 g) was dissolved in 3.4 L of iPrOH and added into solution A for one partition. A clear solution was formed and the temperature decreased to 65 °C. The mixture was stirred at 65 °C for about 15 minutes after which a solid appeared. The mixture was cooled to 10 °C over 2 hours, stirred at 10 °C for an additional 30 minutes, and filtered through a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to collect the 1.1 kg of white solid.
[00131] EtOAc (10 L), 1.1 kg of white solid product, and 5 L of 10% K2CO3 were added sequentially to a 20 L flask and mixed for 5 minutes. The solid dissolved (pH = 9-10). The EtOAc layer was separated and the aqueous layer extracted twice with EtOAc (5 L each). The organic layers were combined (about 20 L), washed with 5 L of brine, and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-55 °C for about 3 hours to remove most of the solution and until the residue weight reached 1 kg. Heptanes (1 L) was added to the mixture and stirred at room temperature for 30 minutes. The mixture was filtered using a Buchner funnel and filter paper (pore size: 30-50 pm) over 30 minutes to obtain 419 g of compound 12 base as a white solid (99.7% purity). The filtrate was concentrated to 135 g of compound 12 as a yellow solid (98.7% purity). 1H-NMR (400 MHz, CDCh): d = 1.85 (bs, 2H), 5.17 (m, 1H), 7.56 (d, 1H), 7.68 (d, 1H), 8.62 (d, 1H), 8.70-8.71 (m, 1H), 8.93 (s, 1H). Combined, the products resulted in a 40.7% yield of compound 12.
[00132] Step 9: Synthesis of compound 10
10A 10
[00133] Pd/C (40 g, 5% w/w) was added into a 10 L autoclave reactor at room temperature under nitrogen. THF (2 L), 2 L of methylamine (27%-30% alcoholic solution, 2.1 eq.), and 800 g of compound 10A (7 mol, 1.0 eq.) were sequentially added into the reactor. The system was purged with hydrogen three times. The mixture was stirred at hydrogen pressure (50 psi) at 70-75 °C overnight and was then filtered using a Biichner funnel and filter paper (pore size: 30-50 pm) over 10 minutes to remove the Pd/C. The filtrate was concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 3 hours to obtain 933 g of yellow oil. The mixture was distilled without a column at atmospheric pressure and the 140-170 °C portion was collected to obtain 763 g of compound 10 as a colorless oil (98.6% purity (AUC by HPLC, retention time = 4.8 minutes); 84.2% yield; 8000 ppm residual ethanol). A portion of the oil (563 g) was distilled using a 3 cm column at atmospheric pressure and the 140-170 °C portion was collected to obtain 510 g of compound 10 (75.8% yield; 134 ppm residual ethanol). 1H-NMR (400 MHz, CDCb): d = 0.82 (bs, 1H), 1.10-1.12 (q, 2H), 1.66 (d, 2H), 1.73-1.81 (t, 2H), 2.05 (s, 3H), 2.08-2.19 (m, 1H), 2.22 (s, 3H), 2.60 (d, 2H).
[00134] Step 10: Synthesis of HM04 fumarate salt
[00135] DCM (1L), 200 g CDI (1.23 mol, 2.0 eq.), and 35 g DABCO (0.31 mol, 0.5 eq.) were sequentially added into a 3 L reactor equipped with a temperature probe and overhead stirrer, and stirred at room temperature under nitrogen. The mixture was cooled to -10 to -5 °C. Compound 12 (200 g) was dissolved in 1 L of DCM and added into the mixture dropwise over 1 hour, followed by stirring for 16 hours at -10 to -5 °C. Compound 10 (159 g, 1.24 mol, 2.0 eq.) was added at -10 to 0 °C over 10 minutes. The mixture was then warmed to 0 to 5 °C and held for 2 hours. The mixture was concentrated under vacuum at 40-45 °C to about 1 L. HC1 (1 L of 1 N) was added to the residual and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to remove the DCM. Another 3 L of 1N HC1 was added to the residual and extracted three times with TBME (4 L, 2 L, 2 L). The aqueous layer was slowly adjusted to pH = 9-10 with 20% aqueous K2CO3 (about 1.5 L) and extracted with DCM (2 L x 3). The organic layers were combined (about 4 L) and washed three times with 0.25 N KH2PO4 (1.2 L x 3). The organic layer was washed with 2 L of brine to bring the pH to neutral and concentrated in a rotavapor under vacuum (30-40 mmHg) at 45-50 °C for about 2 hours to 450 g (335 mL). MTBE (1.5 L) was added to the residual and distilled until 500 mL of liquid was collected. This step was repeated four times with the addition of 500 mL of TBME and collection of 500 mL of distillate, with the exception that 330 mL of liquid was collected at the final distillation. About 1 to 1.2 L of residual remained in the flask. The residual was slowly cooled to room temperature and stirred at room temperature overnight. The mixture was filtered, washed twice with TBME (400 mL x 2), and dried to obtain 192 g of HM04 free base a light yellow solid (99.3% purity (AUC by HPLC, retention time = 11.0 minutes). The product on the wall was dissolved in DCM and concentrated under vacuum to obtain 22 g of HM04 free base as a brown sticky oil (97.6% purity). The filtrate was concentrated under vacuum to obtain 22.5 g of yellow solid (94.0% purity).
[00136] HM04 free base (187 g, 0.39 mol, 1.0 eq., 99.3% purity) and 1.9 L of ACN were sequentially added to a 3 L flask equipped with a temperature probe and overhead stirrer and stirred at 15 °C under nitrogen to obtain a light-yellow suspension. Fumaric acid (45.6 g, 0.39 mol, 1.0 eq.) was added to the flask and generated a white suspension after 1 minute. The reaction suspension was stirred overnight at room temperature, filtered (15-20 pm, ash<0.l5), washed twice with ACN (50 mL x 2), and dried under vacuum at 50 °C for 6 hours to obtain 207 g of HM04 fumarate salt as a light yellow solid (99.4% purity (AUC by HPLC, retention time = 11.1 minutes); 57.8% yield; 3100 ppm residual ACN). The filtrate was concentrated under vacuum to obtain 20.1 g of HM04 fumarate salt as a light yellow solid (97.3% purity).
[00137] A portion of the product (117 g) was further dried in a vacuum oven (20-40 mmHg) to lower the residual acetonitrile content. After drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours, the residual acetonitrile content was measured as 3100 ppm, 2570 ppm, 1300 ppm, and 256 ppm, respectively. After the drying process, 98 g of HM04 fumarate salt was isolated (99.4% purity (AUC by HPLC, retention time = 11.0 minutes); 1H-NMR (400 MHz, DMSO): d = 1.49-1.58 (m, 2H),
1.81-1.92 (m, 2H), 2.44-2.53 (m, 5H), 2.78 (s, 3H), 3.12 (m, 2H), 4.06-4.13 (m, 1H), 6.36-6.41 (m, 1H), 6.55 (s, 2H), 7.47 (d, 1H), 7.73 (d, 1H), 8.11 (d, 1H), 8.75 (d, 1H),
8.81-8.82 (m, 1H), 8.99 (d, 1H). The yield of 98g of HM04 fumarate salt isolated after drying the partial batch was extrapolated over the whole batch to calculate an
approximate yield of 48% for step 10.
[00138] XRPD analysis of HM04 fumarate salt products obtained after drying at 60 °C for 6 hours, 15 hours, and 72 hours; and at 65 °C for 18 hours was performed (see Figures 6-9, respectively). The XRPD profile showed that the HM04 fumarate salt product was consistent with Form 1.
Example 6. Streamlined Synthesis of HM04 Fumarate Salt Form 1
[00139] The overall yield of HM04 fumarate salt produced using Step 10 of Example 5 was calculated as approximately 48%. In order to increase the overall yield, a streamlined synthesis was investigated that eliminated the step of isolating HM04 free base. In particular, step 10 of the method of Example 5 shown in Figure 5 was changed. An overview of the streamlined synthesis beginning after step 9 of Example 5 is shown in Figure 10.
[00140] Streamlined HM04 Fumarate Salt Trial 1 : PCM (121.4 g). CPI (20.0 g, 123 mmol, 2 eq.) and DABCO (3.5 g, 31 mmol) were sequentially added into an inertized 1 L reactor. The mixture was cooled to -10 °C. Separately, a solution of DCM (132.5 g) and compound 12 (20.0 g, 62.1 mmol) were charged into a vessel and stirred until a solution was obtained. This solution was dropped into the 1 L reactor over 33 minutes by keeping the internal temperature at -10 to -5 °C. At the end of the addition, the vessel was rinsed with DCM (7.0 g), which was then added to the reaction mixture.
After stirring overnight (19 hours) and positive IPC, compound 10 (15.9 g, 124 mmol, 2 eq.) was added over 15 minutes and the vessel rinsed with DCM (9.0 g). After heating at 0 °C, 1 hour of stirring, positive IPC, and a further 1.5 hours of stirring, the mixture was heated at room temperature and charged with water (200.1 g). The aqueous layer was separated and the organic layer extracted twice with 1 N HC1 (201, 200 g). The combined aqueous layers containing the product were washed with TBME (148 g). After removal of the organic layer, the aqueous layer was charged with DCM (265.0 g) and 50% K2CO3 solution (about 240 ml) until reaching pH 9.61.
[00141] Meanwhile, a solution of KH2PO4 (8.2 g) in water (240 g) was prepared. The organic layer containing the product was charged with the KH2PO4 solution until reaching pH 7.12 (142.2 g). After separation of the aqueous layer, the organic layer was washed with water (200 g). After separation of the aqueous layer, the organic layer was evaporated at 50 °C. ACN (314.4 g) was added and the solvent distilled again at 70-75 °C under vacuum. ACN (235.8 g) was added and the solvent distilled again under vacuum. ACN (141.5 g) was added, the resulting solution polish filtered and the filter washed with ACN (16 g). After heating at 60 °C, fumaric acid (7.2 g, 62 mmol) was added to the solution, causing a white precipitate. After cooling to 20 °C over 1 hour, the suspension was filtered and washed twice with TBME (2 x 30 g). After drying on the filter with nitrogen flow, 70.7 g of wet raw product was obtained. This was slurried with TBME (177.0 g) for 1 hour, filtered, and washed with TBME (70 g). After drying on the filter under nitrogen flow, 33.0 g of wet product was obtained. Heating at 50 °C under vacuum afforded the dry product as a white powder of HM04 fumarate salt (21.1 g;
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US9926337 | SUBSTITUTED ASYMMETRIC UREAS AND MEDICAL USES THEREOF | 2016-12-02 | |
| US9546157 | p-Substituted Asymmetric Ureas and Medical Uses Thereof | 2015-03-06 | 2015-09-10 |
////////////HM04, H0900, Helsinn, Novo Nordisk, PRECLINICAL, obesity, Prader-Willi syndrome, ghrelin
CN(C1CCN(C)CC1)C(=O)N[C@H](c3ccc(c2cnccn2)c(Cl)c3Cl)C(F)(F)F
GFH 018
![(E)-3-[6-[2-(6-Methylpyridin-2-yl)-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl]-[1,2,4]triazolo[1,5-a]pyridin-5-yl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=132246793&t=l)
GFH-018
CAS 2169299-67-4
GenFleet Therapeutics
Advanced solid tumor; Cancer
TGF-beta Receptor Type-1 (TGFBR1; ALK5; SKR4; TbetaR-I) Inhibitors
Signal Transduction Modulators
GFH-018 , a TGFBR1 inhibitor, being investigated by GenFleet as an oral tablet formulation, for the treatment of cancer, including advanced solid tumors and hepatocellular carcinoma, in March 2019, the company was developing GFH-018 as a class 1 chemical drug in China, with a clinical trial expected to begin in the second half of 2019.


PATENT
WO2017215506
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017215506
PATENT
WO-2019114792
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019114792&tab=FULLTEXT&maxRec=1000
Novel crystalline and salt (hydrochloride, sulfate and mesylate) forms of a TGF-βRI inhibitor, designated as Forms A and B, processes for their preparation and compositions comprising them are claimed for treating cancers. The compound was originally claimed in WO2017215506 , assigned to Medshine Discovery Inc alone.
Step C: 1-9 (4.50 g, 15.20 mmol), 1-6 (4.43 g, 18.24 mmol), sodium carbonate (4.83 g, 45.60 mmol), [1,1′-bis (diphenyl) Phosphine) ferrocene] palladium dichloride (556.07 mg, 759.96 μmol), 2-biscyclohexylphosphine-2′, 6′-dimethoxybiphenyl (311.98 mg, 759.96 μmol) and [2-( 2-Aminophenyl)phenyl]-chloro-palladium-cyclohexyl-[2-(2,6-dimethoxyphenyl)phenyl]phosphine (547.64 mg, 759.96 μmol) was added to the dioxane (100 ml) and water (20 ml) in a mixed solvent. It was replaced with nitrogen three times and then heated to 90 to 100 ° C and stirred for 2 hours. The reaction mixture was poured into water (200 ml) and evaporated and evaporated. The combined organic layers were washed with EtOAc EtOAc m. The residue was purified on a silica gel column (eluent: methylene chloride/methanol, v/v=30/1) to afford crude crude product in petroleum ether/ethyl acetate (v/v=5/1) After stirring for 12 hours, the solid was collected by filtration, and the solid was concentrated and dried under reduced pressure to give 1-10. . 1 H NMR (400 MHz, CDCl3 . 3 ) [delta] 8.49 (S, IH), 7.82-7.74 (m, 2H), 7.59-7.46 (m, 4H), 6.99 (dd, J = 2.6,6.1Hz, IH), 4.39 (d, J = 6.3 Hz, 2H), 2.90 – 2.70 (m, 4H), 2.20 (s, 3H).
192 mg of the compound of formula (I) was weighed into a glass bottle. 10 ml of a tetrahydrofuran:acetic acid (v/v=9/1) mixed solvent was added, and after ultrasonic assisted for 30 minutes, the sample was dissolved into a clear solution. Stir on a magnetic stirrer (40 ° C). After 1.05 equivalents of p-toluenesulfonic acid monohydrate was slowly added, the sample was stirred overnight. After naturally cooling to room temperature, the supernatant was discarded by centrifugation, stirred for 10 hours by adding 10 ml of tetrahydrofuran, and the supernatant was discarded by centrifugation, and the same procedure was repeated twice more. The obtained solid was dried in a vacuum oven at 40 ° C for 1 hour, and after milling, it was further dried in a vacuum oven at 30 ° C for 16 hours to obtain a crystal form B of the compound of the formula (II).
.///////////////////GFH-018, GFH 018, GenFleet Therapeutics, Advanced solid tumor, Cancer, PRECLINICAL
NC(=O)/C=C/c4n5ncnc5ccc4c2c3CCCn3nc2c1cccc(C)n1
FDA approves new treatment Vyleesi (Bremelanotide) for hypoactive sexual desire disorder in premenopausal women

Bremelanotide
SYNTHESIS……. https://newdrugapprovals.org/2015/02/18/palatins-bremelanotide-under-clinical-trials-female-libido-enhancer/
The U.S. Food and Drug Administration today approved Vyleesi (bremelanotide) to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women.
“There are women who, for no known reason, have reduced sexual desire that causes marked distress, and who can benefit from safe and effective pharmacologic treatment. Today’s approval provides women with another treatment option for this condition,” said Hylton V. Joffe, M.D., M.M.Sc., director of the Center for Drug Evaluation and Research’s Division of Bone, Reproductive and Urologic Products. “As part of the FDA’s commitment to protect and advance the health of women, we’ll continue to support the development of safe and effective treatments for female sexual dysfunction.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not due to a co-existing medical or psychiatric condition, problems within the relationship or the effects of a medication or other drug substance. Acquired HSDD develops in a patient who previously experienced no problems with sexual desire. Generalized HSDD refers to …
- June 21, 2019
The U.S. Food and Drug Administration today approved Vyleesi (bremelanotide) to treat acquired, generalized hypoactive sexual desire disorder (HSDD) in premenopausal women.
“There are women who, for no known reason, have reduced sexual desire that causes marked distress, and who can benefit from safe and effective pharmacologic treatment. Today’s approval provides women with another treatment option for this condition,” said Hylton V. Joffe, M.D., M.M.Sc., director of the Center for Drug Evaluation and Research’s Division of Bone, Reproductive and Urologic Products. “As part of the FDA’s commitment to protect and advance the health of women, we’ll continue to support the development of safe and effective treatments for female sexual dysfunction.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not due to a co-existing medical or psychiatric condition, problems within the relationship or the effects of a medication or other drug substance. Acquired HSDD develops in a patient who previously experienced no problems with sexual desire. Generalized HSDD refers to HSDD that occurs regardless of the type of sexual activity, situation or partner.
Vyleesi activates melanocortin receptors, but the mechanism by which it improves sexual desire and related distress is unknown. Patients inject Vyleesi under the skin of the abdomen or thigh at least 45 minutes before anticipated sexual activity and may decide the optimal time to use Vyleesi based on how they experience the duration of benefit and any side effects, such as nausea. Patients should not use more than one dose within 24 hours or more than eight doses per month. Patients should discontinue treatment after eight weeks if they do not report an improvement in sexual desire and associated distress.
The effectiveness and safety of Vyleesi were studied in two 24-week, randomized, double-blind, placebo-controlled trials in 1,247 premenopausal women with acquired, generalized HSDD. Most patients used Vyleesi two or three times per month and no more than once a week. In these trials, about 25% of patients treated with Vyleesi had an increase of 1.2 or more in their sexual desire score (scored on a range of 1.2 to 6.0, with higher scores indicating greater sexual desire) compared to about 17% of those who took placebo. Additionally, about 35% of the patients treated with Vyleesi had a decrease of one or more in their distress score (scored on a range of zero to four, with higher scores indicating greater distress from low sexual desire) compared to about 31% of those who took placebo. There was no difference between treatment groups in the change from the start of the study to end of the study in the number of satisfying sexual events. Vyleesi does not enhance sexual performance.
The most common side effects of Vyleesi are nausea and vomiting, flushing, injection site reactions and headache. About 40% of patients in the clinical trials experienced nausea, most commonly with the first Vyleesi injection, and 13% needed medications for the treatment of nausea. About 1% of patients treated with Vyleesi in the clinical trials reported darkening of the gums and parts of the skin, including the face and breasts, which did not go away in about half the patients after stopping treatment. Patients with dark skin were more likely to develop this side effect.
In the clinical trials, Vyleesi increased blood pressure after dosing, which usually resolved within 12 hours. Because of this effect, Vyleesi should not be used in patients with high blood pressure that is uncontrolled or in those with known cardiovascular disease. Vyleesi is also not recommended in patients at high risk for cardiovascular disease.
When naltrexone is taken by mouth, Vyleesi may significantly decrease the levels of naltrexone in the blood. Patients who take a naltrexone-containing medication by mouth to treat alcohol or opioid dependence should not use Vyleesi because it could lead to naltrexone treatment failure.
In 2012, the FDA identified female sexual dysfunction as one of 20 disease areas of high priority and focused attention. The FDA held a two-day meeting in October 2014 to advance the agency’s understanding of female sexual dysfunction. During the first day of the meeting, the FDA solicited perspectives directly from patients about their condition and its impact on daily life. In 2016, the FDA published a draft guidance titled “Low Sexual Interest Desire and/or Arousal in Women: Developing Drugs for Treatment,” to assist companies developing drugs for the treatment of these conditions. The FDA is committed to continuing to work with companies to develop safe and effective treatments for female sexual dysfunction.
The FDA granted approval of Vyleesi to AMAG Pharmaceuticals.
REF
//////////////Vyleesi, bremelanotide, FDA 2019, HSDD, female sexual dysfunction, AMAG Pharmaceuticals, PT 141, SEX AROUSAL, LIBIDO ENHANCER,
Piclidenoson, иклиденозон , بيكليدينوسون , 匹利诺生 ,

CF 101, Piclidenoson
ALB-7208
CAS 152918-18-8
Chemical Formula: C18H19IN6O4
Molecular Weight: 510.28
(2S,3S,4R,5R)-3,4-Dihydroxy-5-{6-[(3-iodobenzyl)amino]-9H-purin-9-yl}-N-methyltetrahydro-2-furancarboxamide
N6-(3-Iodobenzyl)adenosine-5′-N-methyluronamide
β-D-Ribofuranuronamide, 1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-
1-Deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-β-D-ribofuranuronamide
CF 101 (known generically as IB-MECA) is an anti-inflammatory drug for rheumatoid arthritis patients. Its novel mechanism of action relies on antagonism of adenoside A3 receptors. CF101 is supplied as an oral drug and has an excellent safety profile. It is also being considered for the treatment of other autoimmune-inflammatory disorders, such as Crohn’s disease, psorasis and dry eye syndrome.

- Originator Can-Fite BioPharma
- Class Amides; Anti-inflammatories; Antineoplastics; Antipsoriatics; Antirheumatics; Eye disorder therapies; Iodobenzenes; Neuroprotectants; Purine nucleosides; Ribonucleosides; Small molecules
- Mechanism of Action Adenosine A3 receptor agonists; Immunosuppressants; Interleukin 23 inhibitors; Interleukin-17 inhibitors
- Phase III Plaque psoriasis; Rheumatoid arthritis
- Phase II Glaucoma; Ocular hypertension
- Phase I Uveitis
- Preclinical Osteoarthritis
- Discontinued Colorectal cancer; Dry eyes; Solid tumours
- 05 Feb 2019 Can-Fite BioPharma receives patent allowance for A3 adenosine receptor (A3AR) agonists in USA
- 05 Feb 2019 Can-Fite BioPharma receives patent allowance for A3 adenosine receptor (A3AR) agonists in North America, South America, Europe and Asia
- 21 Aug 2018 Phase-III clinical trials in Plaque psoriasis (Monotherapy) in Israel (PO)
Piclidenoson, also known as CF101, is a specific agonist to the A3 adenosine receptor, which inhibits the development of colon carcinoma growth in cell cultures and xenograft murine models. CF101 has been shown to downregulate PKB/Akt and NF-κB protein expression level. CF101 potentiates the cytotoxic effect of 5-FU, thus preventing drug resistance. The myeloprotective effect of CF101 suggests its development as an add-on treatment to 5-FU.
Piclidenoson is known to be a TNF-α synthesis inhibitor and a neuroprotectant. use as an A3 adenosine receptor agonist, useful for treating rheumatoid arthritis (RA), psoriasis, osteoarthritis and glaucoma.
Can-Fite BioPharma , under license from the National Institutes of Health (NIH), is developing a tablet formulation of CF-101, an adenosine A3 receptor-targeting, TNF alpha-suppressing low molecular weight molecule for the potential treatment of psoriasis, RA and liver cancer. The company is also investigating a capsule formulation of apoptosis-inducing namodenoson, the lead from a program of adenosine A3 receptor agonist, for treating liver diseases, including hepatocellular carcinoma (HCC). In January 2019, preclinical data for the treatment of obesity were reported. Also, see WO2019105217 , WO2019105359 and WO2019105082 , published alongside.
PATENT
WO-2019105388
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019105388&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of CF-101 (also known as piclidenoson; designated as Forms CS1, CS2 and CS3), processes for their preparation, compositions comprising them and their use as an A3 adenosine receptor agonist for treating rheumatoid arthritis, psoriasis, osteoarthritis and glaucoma are claimed
PAPER
Journal of medicinal chemistry (1994), 37(5), 636-46
https://pubs.acs.org/doi/pdf/10.1021/jm00031a014
PAPER
Journal of medicinal chemistry (1998), 41(10), 1708-15
https://pubs.acs.org/doi/abs/10.1021/jm9707737
PAPER
Bioorganic & Medicinal Chemistry (2006), 14(5), 1618-1629
PATENT
WO 2015009008
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015009008


PATENT
WO 2008111082
REFERENCES
1: Avni I, Garzozi HJ, Barequet IS, Segev F, Varssano D, Sartani G, Chetrit N, Bakshi E, Zadok D, Tomkins O, Litvin G, Jacobson KA, Fishman S, Harpaz Z, Farbstein M, Yehuda SB, Silverman MH, Kerns WD, Bristol DR, Cohn I, Fishman P. Treatment of Dry Eye Syndrome with Orally Administered CF101 Data from a Phase 2 Clinical Trial. Ophthalmology. 2010 Mar 19. [Epub ahead of print] PubMed PMID: 20304499.
2: Bar-Yehuda S, Rath-Wolfson L, Del Valle L, Ochaion A, Cohen S, Patoka R, Zozulya G, Barer F, Atar E, Piña-Oviedo S, Perez-Liz G, Castel D, Fishman P. Induction of an antiinflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum. 2009 Oct;60(10):3061-71. PubMed PMID: 19790055.
3: Borea PA, Gessi S, Bar-Yehuda S, Fishman P. A3 adenosine receptor: pharmacology and role in disease. Handb Exp Pharmacol. 2009;(193):297-327. Review. PubMed PMID: 19639286.
4: Moral MA, Tomillero A. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2008 Mar;30(2):149-71. PubMed PMID: 18560631.
5: Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J Rheumatol. 2008 Jan;35(1):41-8. Epub 2007 Nov 15. PubMed PMID: 18050382
/////////////CF 101, Piclidenoson, CF101, CF-101, CF 101, ALB-7208, ALB 7208, ALB7208, IB MECA, Phase III, Plaque psoriasis, Rheumatoid arthritis, UNII-30679UMI0N, Пиклиденозон , بيكليدينوسون , 匹利诺生 , Can-Fite BioPharma
CNC(=O)[C@H]1O[C@H]([C@H](O)[C@@H]1O)N1C=NC2=C(NCC3=CC(I)=CC=C3)N=CN=C12
SHR-0532
SHR-0532
CAS 2166329-09-3
C24 H26 N4 O5 . C4 H6 O6
2-Pyridinecarboxamide, 5-cyano-N-[1-[(2R)-2-(1,3-dihydro-4-methyl-1-oxo-5-isobenzofuranyl)-2-hydroxyethyl]-4-piperidinyl]-4-methoxy-, (2R,3R)-2,3-dihydroxybutanedioate (1:1)
FREE FORM
1945997-37-4
5-Cyano-N-[1-[(2R)-2-(1,3-dihydro-4-methyl-1-oxo-5-isobenzofuranyl)-2-hydroxyethyl]-4-piperidinyl]-4-methoxy-2-pyridinecarboxamide
KCNJ potassium channel-1 inhibitor, Hypertension; Renal insufficiency
- Originator Jiangsu Hengrui Medicine Co.
- Class Antihypertensives
- Mechanism of Action Undefined mechanism
- Preclinical Hypertension
- 03 Jun 2019 Jiangsu Hengrui Medicine Co. plans a phase I trial for Hypertension (PO) in June 2019 (NCT03971929)
- 26 Aug 2018 Jiangsu HengRui Medicine plans a phase I trial for Hypertension (In volunteers) (PO) in August 2018 (NCT03645278)
Jiangsu Hengrui Medicine is developing an oral tablet formulation of SHR-0532, a small molecule specific inhibitor of ROMK (renal outer medullary potassium channel), for use as a diuretic to treat hypertension and renal insufficiency inducing water and sodium retention. In January 2019 a phase trial was completed, and in June 2019, another phase I trial for mild hypertension was planned.
PATENT
WO2016091042
WO 2017211271
CN 108113988
PATENT
WO2019011200
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019011200&redirectedID=true
Clinically, traditional diuretics have a risk of causing hypokalemia. ROMK antihypertensive diuretic development of new targets, as ROMK of inward rectifier K + channel (inwardly rectifying K channels, Kir) a family, belong Kir1 type, the maintenance of renal potassium ions play a crucial balance effect. In the rat kidney, there are at least three subtypes of ROMK channels: ROMK1, ROMK2, and ROMK3. Most of ROMK2 is distributed in the ascending limb of Henle (TALH); ROMK1 and ROMK3 are mainly expressed on the cortical collecting duct (CCD). Expressed in the TALH and ROMK of Na + / K + / 2Cl – transporter with regulating the secretion of potassium ions and sodium reabsorption, and expressed in the CCD ROMK of Na + / K + secretion was adjusted with potassium transporter. Therefore, blocking the ROMK site can be a good diuretic research direction by inhibiting the reabsorption of Na + by diuretic and reducing blood potassium and causing hypokalemia.
The second step, the synthesis of intermediate (III-1)
The third step, the synthesis of the compound of formula (I)
PATENT
WO-2019109935
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019109935&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of a renal outer medullary potassium channel inhibitor and their salts, preferably Form III, for treating hypertension or heart failure.
/////////////SHR-0532, SHR0532, SHR 0532, Jiangsu Hengrui Medicine Co, phase I, Antihypertensives
COc1cc(ncc1C#N)C(=O)NC2CCN(CC2)C[C@H](O)c4ccc3C(=O)OCc3c4C
SEVITERONEL, севитеронел , سيفيتيرونيل , 赛维罗奈 ,
SEVITERONEL
CAS Registry Number 1610537-15-9
Molecular formulaC18 H17 F4 N3 O3, MW 399.34
1H-1,2,3-Triazole-5-methanol, α-[6,7-bis(difluoromethoxy)-2-naphthalenyl]-α-(1-methylethyl)-, (αS)-
(αS)-α-[6,7-Bis(difluoromethoxy)-2-naphthalenyl]-α-(1-methylethyl)-1H-1,2,3-triazole-5-methanol
8S5OIN36X4
- Mechanism of ActionAndrogen receptor antagonists; Estrogen receptor antagonists; Steroid 17-alpha-hydroxylase inhibitors; Steroid 17-alpha-hydroxylase modulators
- WHO ATC codeL01 (Antineoplastic Agents)L01X-X (Other antineoplastic agents)
- EPhMRA codeL1 (Antineoplastics)L1X9 (All other antineoplastics)
1H-1,2,3-Triazole-5-methanol, alpha-(6,7-bis(difluoromethoxy)-2-naphthalenyl)-alpha-(1-methylethyl)-, (alphaS)-
Seviteronel (developmental codes VT-464 and, formerly, INO-464) is an experimental cancer medication which is under development by Viamet Pharmaceuticals and Innocrin Pharmaceuticals for the treatment of prostate cancer and breast cancer.[1] It is a nonsteroidalCYP17A1 inhibitor and works by inhibiting the production of androgens and estrogens in the body.[1] As of July 2017, seviteronel is in phase II clinical trials for both prostate cancer and breast cancer.[1] In January 2016, it was designated fast-track status by the United States Food and Drug Administration for prostate cancer.[1][2] In April 2017, seviteronel received fast-track designation for breast cancer as well.[1]
- Originator Viamet Pharmaceuticals
- Developer Innocrin Pharmaceuticals
- Clas sAntiandrogens; Antineoplastics; Fluorine compounds; Naphthalenes; Propanols; Small molecules; Triazoles
- Mechanism of Action Androgen receptor antagonists; Estrogen receptor antagonists; Steroid 17-alpha-hydroxylase inhibitors; Steroid 17-alpha-hydroxylase modulators
- Phase II Breast cancer; Prostate cancer; Solid tumours
- 31 Jan 2019 Innocrin Pharmaceutical completes a phase II trial in Prostate Cancer (Second-line therapy or greater, Hormone refractory) in the US (NCT02445976)
- 31 Jan 2019 Innocrin Pharmaceutical completes a phase II trial for Prostate Cancer (Hormone refractory) in the US, UK, Switzerland and Greece (NCT02012920)
- 31 Jan 2019 Innocrin Pharmaceuticals completes the phase I/II CLARITY-01 trial for Breast cancer (Late stage disease) in USA (NCT02580448)
- CYP-17 useful for treating fungal infections, prostate cancer, and polycystic ovary syndrome, assigned to Viamet Pharmaceuticals Inc , naming Hoekstra and Rafferty. Innocrin Pharmaceuticals , a spin-out of Viamet is developing oral seviteronel, the lead dual selective inhibitors of the 17,20-lyase activity of P450c17 (CYP17) and androgen receptor antagonist, which also includes VT-478 and VT-489, developed using the company’s Metallophile technology, for treating castration-resistant prostate cancer (CRPC) in men, breast cancer and androgen (AR) related cancers.
Pharmacology
Pharmacodynamics
Seviteronel is a nonsteroidal antiandrogen, acting specifically as an androgen synthesis inhibitor via inhibition of the enzyme CYP17A1, for the treatment of castration-resistant prostate cancer.[3][4][5][6][7][8] It has approximately 10-fold selectivity for the inhibition of 17,20-lyase (IC50 = 69 nM) over 17α-hydroxylase (IC50 = 670 nM), which results in less interference with corticosteroid production relative to the approved CYP17A1 inhibitor abiraterone acetate (which must be administered in combination with prednisone to avoid glucocorticoid deficiency and mineralocorticoid excess due to 17α-hydroxylase inhibition) and hence may be administerable without a concomitant exogenous glucocorticoid.[4][5][6][7][8] Seviteronel is 58-fold more selective for inhibition of 17,20-lyase than abiraterone (the active metabolite of abiraterone acetate), which has IC50 values for inhibition of 17,20-lyase and 17α-hydroxylase of 15 nM and 2.5 nM, respectively.[7] In addition, in in vitro models, seviteronel appears to possess greater efficacy as an antiandrogen relative to abiraterone.[6] Similarly to abiraterone acetate, seviteronel has also been found to act to some extent as an antagonist of the androgen receptor.[6]
Society and culture
Generic names
Seviteronel is the generic name of the drug and its INN.[9]
PATENT
WO2012064943
PATENT
WO-2019113312
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019113312&redirectedID=true
The present invention relates to a process for preparing compound 1 that is useful as an anticancer agent. In particular, the invention seeks to provide a new methodology for preparing compound 1 and substituted derivatives thereof.
Living organisms have developed tightly regulated processes that specifically import metals, transport them to intracellular storage sites and ultimately transport them to sites of use. One of the most important functions of metals such as zinc and iron in biological systems is to enable the activity of metalloenzymes. Metalloenzymes are enzymes that incorporate metal ions into the enzyme active site and utilize the metal as a part of the catalytic process. More than one-third of all characterized enzymes are metalloenzymes.
The function of metalloenzymes is highly dependent on the presence of the metal ion in the active site of the enzyme. It is well recognized that agents which bind to and inactivate the active site metal ion dramatically decrease the activity of the enzyme. Nature employs this same strategy to decrease the activity of certain metalloenzymes during periods in which the enzymatic activity is undesirable. For example, the protein TIMP (tissue inhibitor of metalloproteases) binds to the zinc ion in the active site of various matrix metalloprotease enzymes and thereby arrests the enzymatic activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain a l-( 1,2, 4-triazole) group that binds to the heme iron present in the active site of the target enzyme lanosterol demethylase and thereby inactivates the enzyme.
In the design of clinically safe and effective metalloenzyme inhibitors, use of the most appropriate metal-binding group for the particular target and clinical indication is critical. If a weakly binding metal-binding group is utilized, potency may be suboptimal. On the other hand, if a very tightly binding metal-binding group is utilized, selectivity for the target enzyme versus related metalloenzymes may be suboptimal. The lack of optimal selectivity can be a cause for clinical toxicity due to unintended inhibition of these off-target metalloenzymes.
One example of such clinical toxicity is the unintended inhibition of human drug metabolizing enzymes such as CYP2C9, CYP2C19 and CYP3A4 by the currently-available azole antifungal agents such as fluconazole and voriconazole. It is believed that this off-target inhibition is caused primarily by the indiscriminate binding of the currently utilized l-(l,2,4-triazole) to iron in the active site of CYP2C9, CYP2C19 and CYP3A4. Another example of this is the joint pain that has been observed in many clinical trials of matrix metalloproteinase inhibitors. This toxicity is considered to be related to inhibition of off-target metalloenzymes due to indiscriminate binding of the hydroxamic acid group to zinc in the off-target active sites.
Therefore, the search for metal-binding groups that can achieve a better balance of potency and selectivity remains an important goal and would be significant in the realization of therapeutic agents and methods to address currently unmet needs in treating and preventing diseases, disorders and symptoms thereof. Similarly, methods of synthesizing such therapeutic agents on the laboratory and, ultimately, commercial scale is needed. Addition of metal-based nucleophiles (Zn, Zr, Ce, Ti, Mg, Mn, Li) to azole-methyl substituted ketones have been effected in the synthesis of voriconazole (M. Butters, Org. Process Res. Dev. 2001, 5, 28-36). The nucleophile in these examples was an ethyl-pyrimidine substrate. Similarly, optically active azole-methyl epoxide has been prepared as precursor electrophile toward the synthesis of ravuconazole (A. Tsuruoka, Chem. Pharm. Bull. 1998, 46, 623-630). Despite this, the development of methodology with improved efficiency and selectivity is desirable
Preparation of Compound 4:
de
Acetone (850 L), 2,3-dihydroxynaphthalene (85.00 kg, 530.7 moles), and potassium carbonate (219.3 kg, 1,586.7 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 20 – 35 °C. Dimethyl sulfate (200.6 kg, 2131.09) was added to the stirred reaction at a rate that maintains the internal temperature of the exothermic reaction below 60 °C. This addition typically requires about 3 hours. At the end of the dimethyl sulfate addition, the reaction is continued to allow to stir while maintaining the internal temperature at 50 – 60 °C. After about 3 hours, the reaction was analyzed by HPLC. The reaction was concentrated by atmospheric pressure distillation of acetone. The distillation was continued until 340 – 425 L of distillate was collected. This represents 40 – 50 % of the initial charge of acetone. At the end of the distillation, the reaction mass is present as a thick suspension. While maintaining the internal temperature below 60 °C, the reactor contents were slowly diluted with water (850 L). When the addition is complete, the reaction was cooled to an internal temperature of 25 – 35 °C and stirring was continued for 1 – 2 hours after the designated internal temperature was reached. Compound 2 was isolated by filtration and the cake was washed with water (at least 3 X 85 L). Compound 2 was dried at 40 – 45 °C and full vacuum until the water content by Karl Fisher titration is found to be NMT 2.0 %. Typically, greater than 90 kg of dry product is obtained with an assay of >99.5% AUC by HPLC.
Dichloromethane (with a water content by Karl Fisher Titration of NMT 0.50%) (928 L) and 2,3-dimethoxynaphthalene (2, 116.00 kg, 616.3 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 20 – 35 °C. The reactor contents were cooled to an internal temperature of -5 to 0 °C. Aluminum chloride (164.72 kg, 1235.3 moles, 2.00 molar equivalents) was carefully added in portions to the reaction, while maintaining the internal temperature at -5 to +5 °C. This addition typically requires 5 – 6 hours. At the end of the addition, the reactor contents were cooled to an internal temperature of -15 to -5 °C. Isobutyryl chloride (102.08 kg, 958.05 moles, 1.55 molar equivalents) was slowly added to the reaction while maintaining the internal temperature at -15 to -5 °C. The addition typically requires about 3 hours. At the end of the isobutyryl chloride addition, the reaction was warmed to an internal temperature of 20 – 35 °C. When the temperature was reached, these conditions were maintained for 2 – 3 hours until the IPC indicated a level of residual starting material of NMT 2.0 % AUC by HPLC. The reactor contents were then cooled to 0 – 5 °C. The reaction was quenched by adding the reaction to a precooled (0 – 5 °C) 3M aqueous solution of hydrochloric hcid (Water, 754 L: cone. HC1, 406 L). The mixture was vigorously stirred for 15 – 20 minutes then the layers were allowed to settle. The lower, dichloromethane, product-containing layer was washed sequentially with 10 % aqueous sodium bicarbonate (1044 L), water (1160 L), then 10 % aqueous sodium chloride (1044 L). The reaction was concentrated by distillation under full vacuum and at an internal temperature of NMT 40 °C. The reaction concentrate was cooled to 20 – 35 °C and diluted with hexanes (812 L). The resultant slurry was warmed to 45 – 50 °C and these conditions were maintained for 1 – 2 hours. The reactor contents were cooled to 20 – 35 °C for 1 – 2 hours. Compound 3 was isolated by filtration. The cake was washed with fresh hexanes (232 L) twice, the filter was cooled, and the cake was washed an additional two times with hexanes. Compound 3 was dried under full vacuum at a jacket temperature of 45 °C. Typically, about 95 kg of dry product was isolated with a product purity of >90% by HPLC.
Acetic acid (212.5 L L) and l-(6,7-dimethoxynaphthalene-2-yl)-2-methylpropane-l- one (42.5 kg, 164.5 moles) were charged to a clean, fixed reactor with stirring and with the temperature maintained at 25 – 45 °C. Concentrated hydrochloric acid (425.0 L) was added carefully to the stirring reactor contents while maintaining reactor contents at an internal temperature of 25 – 45 °C. When the addition was complete, the internal temperature of the reaction was raised to 100 – 105 °C. Note that the reaction is a heterogeneous mixture. The reaction was stirred under these conditions for 6 – 8 hours. The reaction was cooled to 85 – 90 °C to which was carefully added a fresh portion of hydrochloric acid (127.5 L). The reaction was warmed to 100 – 105 °C and stirred for another 6 – 8 hours. The reaction was cooled to 85 – 90 °C. The reaction was cooled further to 70 – 80 °C. Water (212.5 L) was added to the well stirred reaction and the reactor contents were cooled to an internal temperature of 35 – 45 °C and stirred for 3 – 4 hours. Compound 4 was collected by filtration. The wet cake was washed with water (212.5 L). The wet cake was added to a clean reactor with a 5% aqueous sodium bicarbonate solution and stirred at an internal temperature of 35 – 45 °C for 1 – 2 hours.
Compound 4 was collected by filtration and washed with water (212.5 L). Compound 4 was dried under full vacuum and a temperature of < 50 °C until the water content of the dried material was found to be NMT 5.0% by Karl Fisher Titration. The yield is typically >31 kg with a purity >99.5 %.
Preparation of Compound 5:
The following difluoromethylation conditions listed in Table 1 were investigated:
Preparation 1:
The reaction flask was dried under an argon flow at 120 °C. (lS,2R)-l-Phenyl-2-(l- pyrrolidinyl)propan-l-ol (ligand 45) (196.6 g, 0.96 mol, 2.2 eq.) was added into the flask and then toluene (195 mL) was added. The solution was cooled to <12 °C. A solution of diethyl zinc (716.4 g, 0.87 mol, 15 wt%, 2 eq.) in toluene was added through a septum over 30 min at 0-10 °C. Further, a solution of ((Trimethylsilyl)ethynyl)-magnesium bromide in THF (1.81 kg; 0.87 mol, 9.7 wt%, 2 eq.) was added over 30 min at 0-10 °C. Finally, trifluoroethanol (87.0 g; 0.87 mol; 2 eq.) was added over 10 min at 0-10 °C. The reaction solution was stirred at 10-12 °C for 3 h. Compound 5 (143.4 g; 0.434 mol; 1 eq.) was added (as a solid) at room
temperature. The reaction mixture was stirred at room temperature for 1 h and at 55 °C for 17 h. The reaction solution was cooled to room temperature and dosed with aqueous HC1 (3600 mL; 7.5 wt%) within 20 min. The temperature of the mixture was kept below 25 °C. Toluene (1250 mL) was added and the mixture was stirred at room temperature for 5 min. The aqueous phase was separated and stored for the recycling of ligand 45. The organic phases were washed with water (638 mL) and concentrated via distillation under reduced pressure (50 mbar). The residue (approx. 184 g) was treated with heptane (200 mL), which was removed
via distillation. The residue was dissolved in heptane (2050 mL) at 50 °C. The mixture was cooled to room temperature and subsequently to -8 °C within 2 hours. The obtained suspension was stirred at -8 °C for 1 h. Crystallized compound 5 (20.0 g; 14%) was isolated via filtration, washed twice with cold (0 °C) heptane (2×20 mL) and dried under vacuum at 50 °C for 12 hours. The combined heptane phases were concentrated under reduced pressure to obtain a 48 wt% solution of compound 18b in heptane (yield: 83.0%). The solution was directly used for the next step.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.23 (s, 9H), 0.77 (d, J = 6.7 Hz, 3H), 0.93 (d, 7 = 6.7 Hz, 3H), 2.04 (sept., 7 = 6.7 Hz, 1H), 6.11 (s, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.35 (t, 27H,F = 73.4 Hz, 1H), 7.68 (dd, 7 = 8.6, 1.5 Hz, 1H), 7.84 (s, 1H), 7.87 (s, 1H), 7.93 (d, 7 = 8.6 Hz, 1H), 8.03 (s (broad), 1H);
HPLC (purity): 94%;
chiral HPLC: e.r. = 18:82.
Preparation 2:
(7S,2R)-l-Phenyl-2-(l-pyrrolidinyl)propan-l-ol (ligand 45) (13.0 kg, 63.3 mol, 2.2 eq.) was charged into the reactor and toluene (60 L) was added. The solution was cooled to < 12 °C. A solution of diethyl zinc (35.6 kg, 57.3 mol, 20 wt%, 2 eq.) in toluene was added via mass flow controller at 8-16 °C. Further, a solution of ((trimethylsilyl)ethynyl)-magnesium bromide in THF (11.5 kg; 57.3 mol, 9.7 wt%, 2 eq.) was added at 8-16 °C. Finally, trifluoroethanol (5.7 kg; 57.3 mol; 2 eq.) was added over 10 min at 8-16 °C.The reaction solution was stirred at 22-25 °C for 3 h. A solution of compound 5 (9.5 kg; 28.7 mol; 1 eq.) in toluene (20 L) was added at room temperature. The reaction mixture was stirred at 25 °C for 1 h and at 55 °C for 17 h. The reaction solution was cooled to room temperature and dosed in aqueous HC1 (225L; 7.5 wt%) within 20 min. The temperature of the mixture should be kept below 25 °C. Toluene (80 L) was added and the mixture was stirred at room temperature for 5 min. The organic phases was washed with water (50 L) and concentrated via distillation under reduced pressure (50 mbar). The residue was treated with heptane (100 L), which was removed via distillation. The residue was dissolved in heptane (100 L) at 50°C, which was removed via distillation. The residue was dissolved in heptane (25 L). Heptane (110 L) was added, the mixture was cooled to room temperature and subsequently to 0-5 °C and seeded with compound 5 (0.15 kg). The obtained suspension was cooled to -8 °C within 1 h and stirred at this temperature for 2 h. Crystallized compound 5 was removed via filtration. The filtrate was concentrated under reduced pressure to obtain a 48 wt% solution of compound 18b in heptane (calculated 8.8 kg, 71.6%). This solution was directly used for the next step.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.23 (s, 9H), 0.77 (d, J = 6.7 Hz, 3H), 0.93 (d, 7 = 6.7 Hz, 3H), 2.04 (sept., 7 = 6.7 Hz, 1H), 6.11 (s, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.35 (t, 27H,F = 73.4 Hz, 1H), 7.68 (dd, 7 = 8.6, 1.5 Hz, 1H), 7.84 (s, 1H), 7.87 (s, 1H), 7.93 (d, 7 = 8.6 Hz, 1H), 8.03 (s (broad), 1H);
HPLC (purity): 94%;
chiral HPLC: e.r. = 18:82.
Recovery of the chiral ligand ( lS,2R)-l-Phenvl-2-
-l-ol from the
Preparation 1:
The above acidic aqueous phase was diluted with toluene (1000 mL) and the mixture was treated with sodium hydroxide (50 wt% solution) to adjust the pH to 12. The mixture was warmed to 50 °C and sodium chloride (100 g) was added. The aqueous phase was separated and washed with toluene (1000 mL). The combined organic phases were washed with water (200 mL). The combined toluene phases were treated with water (1000 mL) and the pH was adjusted to 2 by the addition of a cone. HC1 solution. The aqueous phase was separated and the mixture was treated with sodium hydroxide (50 wt% solution) at 5 °C to adjust the pH to 12. After seeding, the suspension was stirred at 5 °C for 30 min. The solids were isolated, washed with cold (0 °C) water (4×100 mL) and dried under vacuum at 30 °C for 24 hours. Ligand 45 (178.9g; 91%) was obtained as slightly yellow crystalline solid.
HPLC (purity): 99%.
Preparation 2:
The acidic aqueous phase containing ligand 45 (500 L) was diluted with toluene (125 L) and treated with“Kieselgur” (20 L). The mixture was treated with sodium hydroxide (40 L; 50 wt% solution) to adjust the pH to 12 whereas the temperature was kept <55 °C. The suspension was stirred for 15-20 min and filtered to remove all solids. Toluene (80 L) was added and the aqueous phase was separated. The organic phase was treated with water (150 mL) and the pH was adjusted to 1.5-2 by the addition of an aqueous HC1 solution (10 L; 32 wt%). The aqueous phase was separated, toluene (150 L) was added, and the mixture was treated with sodium hydroxide (5 L; 50 wt% solution) at 5 °C to adjust the pH to 12-12.5. The organic phase was separated, washed with water (30 L), and concentrated under reduced
pressure at 50 °C. Approx. 100L of distillate was removed. A sample of the solution of ligand 45 in toluene was analyzed:
The NMR results indicated a 21.6 wt% solution of ligand 45 in toluene which corresponds to a calculated amount of 118.4 kg (83.6%) of ligand 45.
Preparation of Compound 18a
Preparation 1:
A solution of tertiary alcohol 18b (320 g; 48 wt%; 0.36 mol; 1 eq.) in heptane was dissolved in methanol (800 mL). Potassium carbonate (219 g; 1.58 mol; 4.4 eq.) was added (temperature was kept < 30 °C) and the suspension was stirred at room temperature for 3 h. Water (1250 mL) was added and the mixture was treated with a cone. HC1 solution (approx. 130 mL) to adjust the pH to 7.8. The reaction mixture was extracted twice with methyl- /-butyl ether (MTBE; 2×465 mL). The combined MTBE phases were washed with water (155 mL). Water (190 mL) was added to the MTBE phase and the organic solvent was distilled off under reduced pressure (50 mbar). The obtained emulsion of compound 18a (yield: 99%) was directly used for the next step.
1H-NMR (600.6 MHz, CDC13) d: 0.87 (d, J = 6.8 Hz, 3H), 1.09 (d, / = 6.8 Hz, 3H), 2.20 (sept. / = 6.8 Hz, 1H), 2.47 (s, 1H), 2.77 (s, 1H), 6.63 (t, 27H,F = 73.5 Hz, 1H), 6.63 (t, 2/H,F = 73.5 Hz, 1H), 7.65 (s, 1H), 7.69 (s, 1H), 7.74 (dd, 7 = 8.6, 1.7 Hz, 1H), 7.79 (d, / =
8.6 Hz, 1H), 8.06 (s (broad), 1H);
HPLC (purity): 95%.
Preparation 2:
The solution of tertiary alcohol 18b (48 wt%; 57.5 mol; 1 eq.) in heptane was dissolved in methanol (128 L). Potassium carbonate (35.0 kg; 253 mol; 4.4 eq.) was added (temperature was kept < 30 °C) and the suspension was stirred at 20-30 °C for 3 h. Water (200 L) was added and the mixture was treated with an aqueous HC1 solution (approx. 25 L; 32 wt%) to adjust the pH to 7.5 – 7.8. The reaction mixture was extracted twice with MTBE
(2×66.6 L). The combined MTBE phases were washed with water (25 L). Water (30 L) was added to the MTBE phase and the organic solvent was distilled off under reduced pressure (<80 mbar; 55°C). The residue was dissolved in tert-butanol (25 L). The resulting 18a was cooled to <30°C and used directly in the next step.
^-NMR (600.6 MHz, CDC13) d: 0.87 (d, / = 6.8 Hz, 3H), 1.09 (d, / = 6.8 Hz, 3H), 2.20 (sept. / = 6.8 Hz, 1H), 2.47 (s, 1H), 2.77 (s, 1H), 6.63 (t, 27H,F = 73.5 Hz, 1H), 6.63 (t, 2/H,F = 73.5 Hz, 1H), 7.65 (s, 1H), 7.69 (s, 1H), 7.74 (dd, 7 = 8.6, 1.7 Hz, 1H), 7.79 (d, / = 8.6 Hz, 1H), 8.06 (s (broad), 1H);
HPLC (purity): 95%.
Preparation of Compound 31
Preparation 1:
Benzyl bromide (39.4 g; 0.23 mol; 1 eq.) was dissolved in water (177 mL) and t-BuOH (200 mL). Diisopropylethylamine (DIPEA; 59.4 g; 0.46 mol; 2 eq.) and sodium azide (15.0 g; 0.23 mol; 1 eq.) were added. The suspension was stirred for 5 min at room temperature. A suspension of compound 18a (82 g; 0.23 mol; 1 eq.) in water (123 mL) was treated with t-BuOH (100 mL) and copper (I) iodide (8.8 g; 46 mmol; 0.2 eq.) was added and the temperature was kept below 30 °C. The yellow-brown suspension was stirred for 5 h at room temperature. Zinc powder (5.0 g; 76 mmol) and ammonium chloride (7.4 g; 0.14 mol) were added and the reaction mixture was stirred at room temperature for 3 hours. The mixture was diluted with MTBE (800 mL), water (280 mL), and an aqueous ammonia solution (120 g; 25 wt%). Solids were removed by filtration and additional MTBE (200 mL) and brine (200 mL) were added. The aqueous phase was separated and extracted with MTBE (400 mL). The combined organic phases were treated with water (150 mL) and MTBE was distilled off under reduced pressure (100 mbar). The obtained suspension of compound 31 (113 g; 50 wt%) in water (approx. 113 mL) was directly used for the next step.
Ή-NMEI (600.6 MHz, DMSO-D6) d: 0.66 (d, / = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 87%.
Preparation 2:
Benzyl bromide (11.0 kg g; 64.4 mol; 1,12 eq.) was dissolved in water (40 L) and t-BuOH (60 L). DIPEA (16.4 kg; 126.5 mol; 2,2 eq.) and sodium azide (4.12 kg; 63.3 mol; 1 eq.) were added. The suspension was stirred 5 min at room temperature. A mixture of compound 18a (20.5 kg; 57.5 mol; 1 eq.) in ieri-butanol (see previous step) was added together with water (5 L) and copper (I) iodide (2.2 kg; 11.5 mol; 0.2 eq.) at a temperature < 30 °C. The yellow-brown suspension was stirred for 5 h at room temperature. Zinc powder (1.25 kg; 19 mol, 0.33 eq.) and an aqueous solution of ammonium chloride (2.14 kg; 20 wt%; 40 mol; 0.7 eq.) were added and the reaction mixture was stirred at 20-30 °C for 2 hours. The reaction mixture was concentrated under vacuum (<200 mbar, 55 °C). The residue was diluted with MTBE (200 L), water (30 L), and an aqueous ammonia solution (30 kg; 25 wt%). Solids were removed by filtration over a pad of“Kieselgur NF” (2 kg). Brine (50 L) was added for a better phase separation. The aqueous phase was separated and washed with MTBE (200 L). The combined organic phases were washed with an aqueous HC1 solution (1 N, 52 L) and water (50 L). MTBE was distilled off under reduced pressure (<400 mbar, 55°C; distillate min. 230L). The oily residue was dissolved in ethanol (150 L), which was distilled off under reduced pressure (<300 mbar; 55°C; distillate min. 150-155L) and the residue was dissolved in additional ethanol (60 L). To the resulting solution of compound 31 was added water (24 L) and the mixture was warmed to 50-55 °C. The mixture was cooled to 30 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to <0 °C within 2 hours, and stirred at -5-0 °C for an additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (2 x 12 L). The wet product was dissolved in ethanol (115L) at 60 °C and water (24 L) was added. The mixture was cooled to 40 °C and the crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to <0 °C within 2 hours, and stirred at -5-0 °C for additional 2 hours. The solids were isolated and washed (without stirring) with ethanol/water (1/1; v/v) (3 x 8 L). Pure, wet compound 31 was isolated as a white solid, which was used for the next step without drying. 14.0 kg of wet 31 were obtained with a 31 content of 81.6 wt%. Based on the determined content, the calculated amount of pure 31 was 11.4 kg with a yield of 41% over two steps (from 18b).
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, J = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 HZ, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 87%.
Preparation 3: Synthesis of compound 31 directly from compound 18b
Benzyl bromide (1.64 g, 9.59 mmol, 1.12 eq) was dissolved in water (2.4 mL) and
MeOH (2.4 mL). K2CO3 (2.38 g, 17.2 mmol, 2.00 eq), sodium ascorbate (0.34 g, 1.72 mmol, 0.20 eq) and finally sodium azide (0.62 g, 9.40 mmol, 1.10 eq.) were added. The suspension was stirred for 5 min at room temperature. A suspension of 18b (3.08 g; 8.64 mmol, 1.00 eq) in water (2.5 mL) and MeOH (2.5 mL) and the resulting mixture was stirred for 10 min.
CuS04 (0.21 g, 1.30 mmol, 0.15 eq) were added (slightly exothermic reaction). The reaction mixture was stirred for 19 h and the conversion was determined by HPLC (conv. 100%, purity of compound 31 by HPLC: 83 area%). To the yellow-green suspension was added zinc powder (0.24 g, 4.13 mmol, 0.43 eq) and ammonium chloride (0.34 g, 6.36 mmol, 0.74 eq) were added and the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated under reduced pressure (150 mbar, 50 °C). The mixture was diluted with MTBE (40 mL), water (15 mL), and an aqueous ammonia solution (6.5 mL). Solids were removed by filtration and brine (5.5 mL) was added. The aqueous phase was separated and extracted with MTBE (20 mL). The combined organic phases were treated with water (10 mL) and the pH was adjusted to a pH of 1 by addition of cone. HC1. After phase separation, the organic layer was washed with water (10 mL). MTBE was distilled off under reduced pressure (100 mbar, 50°C) to give the crude compound 31 as an oil. Water (2.5 mL) and EtOH (30 mL) were added and the mixture was warmed to 50 °C. After cooling to 30 °C, the mixture was seeded with compound 31 and compound 31 started to precipitate. The mixture was kept for 1 h at 30 °C, then cooled to 0 °C over 2 h and kept at 0 °C for 2 h. The resulting product, 31, was collected by filtration and the filter cake was washed with small portions of EtOH/water (1:1). After drying, the product (2.97 g) was obtained as a pale yellow, crystalline solid with an HPLC purity of 79 area% and a NMR content of ca. 70 wt%.
Recrystallization of
31
Preparation 1:
To a suspension of compound 31 (96 g; 0.196 mol; 50 wt%) in water (96 mL) was added ethanol (480 mL) and the mixture was warmed to 50 °C. The mixture was cooled to 30 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to 0 °C within 2 hours and stirred at 0 °C for additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 40 mL). The wet product was dissolved in ethanol (280 mL) at 60 °C and water (56 mL) was added. The mixture was cooled to 40 °C and crystallization started. The suspension was stirred at 30 °C for 1 h, cooled to 0 °C within 2 hours, and stirred at 0 °C for an additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 28 mL). Pure, wet compound 31 (46.8 g on dried basis; 49 % over 2 steps) was isolated as a white solid, which was used for the next step without drying.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, J = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 HZ, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 99.5%;
chiral HPLC: e.r.: 0.2:99.8%.
mp of dried product: 110 °C.
Preparation 2:
14 kg of ethanol-wet 31 (content 81.6 wt%, calculated 11.4 kg, 23.7 mol) were suspended in ethanol (46 L) and the mixture was warmed to 50-55 °C, forming a homogenous solution at this temperature. Water (9 L) was added at 50-55 °C and the mixture was cooled to 40-45 °C. After the crystallization had started, the suspension was stirred at 40-45 °C for 1 h, cooled to 0 °C within 2 hours, and stirred at 0 °C for additional 2 hours. The solids were isolated and washed with ethanol/water (1/1; v/v) (3 x 8 L). Pure, wet compound 31 (14.5 kg) was isolated as a white solid, which was used for the next step without drying.
1H-NMR (600.6 MHz, DMSO-D6) d: 0.66 (d, / = 6.8 Hz, 3H), 0.83 (d, / = 6.7 Hz, 3H), 2.78 (sept. / = 6.8 Hz, 1H), 5.55 (s, 2H), 5.68 (s, 1H), 7.29 (t, 27H,F = 73.4 Hz, 1H), 7.32 (t, 27H,F = 73.4 Hz, 1H), 7.36 – 7.26 (m, 5H), 7.79 (s, 1H), 7.82 (s, 1H), 7.82 (dd, 7 = 8.8, 1.7 Hz, 1H), 7.86 (d, / = 8.8 Hz, 1H), 7.94 (s, 1H), 8.10 (s (broad), 1H);
HPLC (purity): 99.8%;
chiral HPLC: e.r.: 0.2:99.8%.
mp of dried product: 110 °C.
Preparation of Azidomethyl Pivalate Protected Triazole (6) from Compound 18a
1
Azidomethyl pivalate (1.42 g, 9.00 mmol, 1.05 eq) was suspended in water (6.0 mL) and t-BuOH (7.2 mL) and the suspension was stirred for 5 min. Compound 18a (theor. 3.08 g, 8.64 mmol, 1.00 eq), sodium ascorbate (0.48 g, 2.4 mmol, 0.30 eq), and CuS04 (0.08 g, 0.40 mmol, 0.05 eq.) were added. The reaction mixture was stirred for 19 h and conversion was determined by HPLC (conv. 98%, purity of the product by HPLC: 81 area%). To the green suspension was added MTBE (20 mL), water (10 mL), and an aqueous ammonia solution (2 g). A biphasic turbid mixture was formed. To improve phase separation, additional MTBE (20 mL) and water (10 mL) were added. The aqueous phase was separated and extracted with MTBE (20 mL). The combined organic phases were concentrated under reduced pressure (100 mbar, 50 °C) to give the crude product as a brown oil that solidified upon standing. HPLC purity: ca. 65 area%; NMR content of ca. 73 wt%.
1H-NMR (600.6 MHz, CDCL) d: 0.79 (d, 3H), 0.93 (d, 3H), 1.15 (s. 9H), 2.86 (sept, 1H), 3.12 (s, 1H), 6.20 (s, 2H), 6.59 (t/t, 27H,F = 73.5 Hz, 2H), 7.61 (1, 1H), 7.64 (s, 1H), 7.70 – 7.82 (m, 3H), 8.04 (s, 1H).
Preparation of Azidomethyl Pivalate Protected Triazole (6) from 18b
In a reaction flask, sodium ascorbate (277 mg, 1.4 mmol, 1.20 eq) and CuS04 (37 mg, 0.23 mmol, 0.20 eq.) were suspended in MeOH (11 mL). Azidomethyl pivalate (183 mg, 1.16 mmol, 1.00 eq) and 18b (183 mg, 1.16 mmol, 1.00 eq) were added and the mixture was warmed to 60 °C. The reaction mixture was stirred for 19 h and worked up. To the green suspension was added an aq NH4Cl solution (2 mL) and zinc powder, and the mixture was stirred for 2 h. MTBE (2 mL) was added and the aqueous phase was separated and extracted with MTBE (2 mL). The combined organic phases were concentrated under reduced pressure (100 mbar, 50 °C) to give 6 as a brown oil that solidified upon standing. HPLC purity: ca. 81 area%; NMR content of ca. 57 wt%.
1H-NMR (600.6 MHz, CDCL) d: 0.79 (d, 3H), 0.93 (d, 3H), 1.15 (s. 9H), 2.86 (sept, 1H), 3.12 (s, 1H), 6.20 (s, 2H), 6.59 (t/t, 27H,F = 73.5 Hz, 2H), 7.61 (1, 1H), 7.64 (s, 1H), 7.70 – 7.82 (m, 3H), 8.04 (s, 1H).
Preparation of Compound 1
Preparation 1:
Compound 31 (26 g; 53 mmol; 1 eq.) was dissolved in ethanol (260 mL) and Noblyst Pl 155 (2.2 g; 10 % Pd; 54 wt% water) was added. The autoclave was flushed with nitrogen and hydrogen (5 bar) was added. The reaction mixture was stirred at room temperature for 32 hours. The reaction mixture was treated with charcoal (2 g), stirred for 15 min, and the charcoal was filtered off. The filtrate was concentrated via distillation and the residue (approximately 42 g) was diluted with heptane (200 mL). The mixture was heated to reflux to
obtain a clear solution. The solution was cooled to room temperature within 1 h and the resulting suspension was cooled to 0 °C and stirred for 2 hours at 0 °C. The solids were isolated via filtration and washed with heptane/ethanol (10:1; v/v; 3×10 mL). Compound 1 (18.0 g; 85 %) was dried under vacuum at 60 °C for 24 hours and obtained as a white, crystalline solid.
1H-NMR (600 MHz) d: 0.80 (d, J = 6.8 Hz, 3H), 0.97 (d, / = 6.7 Hz, 3H), 2.83 (sept. / = 6.8 Hz, 1H), 6.60 (t, 27H,F = 73.5 Hz, 1H), 6.61 (t, 27H,F = 73.5 Hz, 1H), 7.61 (s, 1H), 7.65 (s, 1H), 7.68 (dd, / = 8.7, 1.6 Hz, 1H), 7.74 (s, 1H), 7.75 (d, / = 8.7 Hz, 1H), 8.02 (s (broad), 1H); HPLC (purity): 100%.
Preparation 2:
Compound 31 (26.5 kg; 53.5 mol; 1 eq.) was dissolved in ethanol (265 L) and Pd/C (2.0 kg; 10 % Pd; 54 wt% water) was added. The reactor was flushed with nitrogen, and hydrogen (4.5 bar) was added. The reaction mixture was stirred at 28-32 °C until the reaction was complete. The reaction mixture was treated with charcoal (1.3 kg) at a temperature of <
33 °C, stirred for 10 min, and the charcoal was filtered off, and the filter was washed with ethanol (10 L).The filtrates from two reactions were combined and concentrated via distillation under reduced pressure (max. 65 °C; distillate: min 480 L). The residue (approx. 50-60 L) was diluted with isopropylacetate (250 L). The mixture was again concentrated via distillation under reduced pressure (max. 65 °C; distillate: min 240-245 L). The residue (approx. 60-70 L) was cooled to 35-40 °C and isopropylacetate (125 L) and heptane (540 L) were added. The suspension was heated to reflux (approx. 88 °C) and stirred under reflux for 15-20 min. Subsequently, the mixture was cooled to 0-5 °C within 2 h and stirred at 0-5 °C for 2 hours. The solids were isolated via filtration and washed with heptane/isopropylacetate (5:1; v/v; 2×30 L; 0-5 °C). Wet 1 was dried under vacuum at 60 °C and was obtained as a white, crystalline solid (35.4 kg, 81.9%).
1H-NMR (600 MHz) d: 0.80 (d, / = 6.8 Hz, 3H), 0.97 (d, / = 6.7 Hz, 3H), 2.83 (sept. / = 6.8 Hz, 1H), 6.60 (t, 27H,F = 73.5 Hz, 1H), 6.61 (t, 27H,F = 73.5 Hz, 1H), 7.61 (s, 1H), 7.65 (s, 1H), 7.68 (dd, / = 8.7, 1.6 Hz, 1H), 7.74 (s, 1H), 7.75 (d, / = 8.7 Hz, 1H), 8.02 (s (broad), 1H); HPLC (purity): 100%.
Preparation 3: Preparation of Compound 1 from Compound 6
At room temperature, 6 (3.00 g, 5.84 mmol) was dissolved in MeOH (19.8 mL). NaOH (1.0 M, 19.8 mL) was added in one portion and the reaction mixture was stirred for 1 h at room temperature. The reaction progress was monitored by HPLC, which showed 98% conversion after 1 h. Aq. HC1 (19.8 mL) was added and the mixture was diluted with water (120 mL) and MTBE (60 mL), resulting in a clear biphasic solution. After phase separation, the organic phase was washed with aq NaHC03 (20 mL). The organic layer was concentrated under high vacuum (25 mbar, 45 °C) to yield 2.77 g of 1 as a greenish oil. The identity was confirmed by comparison of HPLC retention time with an authentic sample of 1 as well as by 1H NMR.
Recrystallization of Compound 1
Wet 1 (40 kg; isopropylacetate/heptane wet) was treated with isopropylacetate (110 L) and heptane (440 L). The suspension was heated to reflux (approx. 88 °C) and stirred under reflux for 15-20 min. Subsequently, the mixture was cooled to 0-5 °C within 2 h and stirred at 0-5 °C for 2 hours. The solids were isolated via filtration and washed with
heptane/isopropylacetate (5:1; v/v; 2×30 L; 0-5 °C). A sample was taken for analysis
(criterion: a) purity; NLT 99.0 A% by HPLC; b) single impurities, NMT 0.15 A% by HPLC; c) enantiomer VT-463, NMT 1.0 A% by HPLC). Wet 1 was dried under vacuum at 60 °C for not less than 12 h. A sample was taken for analysis: criterion: a) LOD; NMT 0.5 wt% by gravimetry; b) residual toluene, NMT 890 ppm by HS-GC. 1 was obtained as a white, crystalline solid (28.5 kg, 66.7% from 31).
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(11), 2444-2447.
https://www.sciencedirect.com/science/article/pii/S0960894X14003606

PATENT
WO 2016040896
https://patents.google.com/patent/WO2016040896A1/en
References
- ^ Jump up to:a b c d e http://adisinsight.springer.com/drugs/800035241
- ^ http://www.pharmaceutical-technology.com/news/newsfda-grants-fast-track-status-innocrins-seviteronel-treat-metastatic-crpc-4770025
- ^ Yin L, Hu Q, Hartmann RW (2013). “Recent progress in pharmaceutical therapies for castration-resistant prostate cancer”. Int J Mol Sci. 14 (7): 13958–78. doi:10.3390/ijms140713958. PMC 3742227. PMID 23880851.
- ^ Jump up to:a b Stein MN, Patel N, Bershadskiy A, Sokoloff A, Singer EA (2014). “Androgen synthesis inhibitors in the treatment of castration-resistant prostate cancer”. Asian J. Androl. 16 (3): 387–400. doi:10.4103/1008-682X.129133. PMC 4023364. PMID 24759590.
- ^ Jump up to:a b Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ (2014). “Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors”. Bioorg. Med. Chem. Lett. 24 (11): 2444–7. doi:10.1016/j.bmcl.2014.04.024. PMID 24775307.
- ^ Jump up to:a b c d Toren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns ES, Zoubeidi A, Moore W, Gleave ME (2015). “Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer”. Mol. Cancer Ther. 14 (1): 59–69. doi:10.1158/1535-7163.MCT-14-0521. PMID 25351916.
- ^ Jump up to:a b c Stephen Neidle (30 September 2013). Cancer Drug Design and Discovery. Academic Press. pp. 341–342. ISBN 978-0-12-397228-6.
- ^ Jump up to:a b Wm Kevin Kelly; Edouard J. Trabulsi, MD; Nicholas G. Zaorsky, MD (17 December 2014). Prostate Cancer: A Multidisciplinary Approach to Diagnosis and Management. Demos Medical Publishing. pp. 342–. ISBN 978-1-936287-59-8.
- ^ http://www.who.int/medicines/publications/druginformation/innlists/RL76.pdf
Further reading
- Gomez L, Kovac JR, Lamb DJ (2015). “CYP17A1 inhibitors in castration-resistant prostate cancer”. Steroids. 95: 80–7. doi:10.1016/j.steroids.2014.12.021. PMC 4323677. PMID 25560485.
- Bambury RM, Rathkopf DE (2015). “Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide”. Urol. Oncol. 34: 348–55. doi:10.1016/j.urolonc.2015.05.025. PMID 26162486.
External links[
 |
|
| Clinical data | |
|---|---|
| Synonyms | VT-464; INO-464 |
| Routes of administration |
By mouth |
| Drug class | Androgen biosynthesis inhibitor; Nonsteroidal antiandrogen |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C18H17F4N3O3 |
| Molar mass | 399.339 g/mol g·mol−1 |
| 3D model (JSmol) | |
References
-
Innocrin Pharmaceuticals Created as a Spin-out of the Prostate Cancer Program from Viamet Pharmaceuticals.
-
Viamet Pharmaceuticals and the Novartis Option Fund Enter Agreement for Development of Novel Metalloenzyme Inhibitors.
-
Innocrin Pharmaceuticals, Inc. Granted SME Status Designation by the European Medicines Agency.
-
A Single arm, open label, signal seeking, Phase II a trial of the activity of seviteronel in patients with androgen receptor (AR) positive solid tumours
-
Innocrin Pharmaceuticals and the Prostate Cancer Foundation (PCF) Join Forces for Innovative Phase 2 Clinical Study.
-
A Phase 2 Open-label Study to Evaluate the Efficacy and Safety of Seviteronel in Subjects With Castration-Resistant Prostate Cancer Progressing on Enzalutamide or Abiraterone
-
Innocrin Pharmaceuticals, Inc. Granted Fast Track Designation by FDA for VT-464 Treatment of Patients with Metastatic Castrate-resistant Prostate Cancer.
-
Innocrin Pharmaceuticals, Inc. Begins Phase 2 Study of Seviteronel in Women with Estrogen Receptor-positive or Triple-negative Breast Cancer and Expands Two Phase 2 Studies of Seviteronel in Men with Metastatic Castrate-resistant Prostate Cancer.
-
A Phase 2 Open-Label Study to Evaluate the Efficacy and Safety of VT-464 in Patients With Metastatic Castration Resistant Prostate Cancer Who Have Previously Been Treated With Enzalutamide, Androgen Receptor Positive Triple-Negative Breast Cancer Patients, and Men With ER Positive Breast Cancer
-
Innocrin Pharmaceuticals Inc. to Present Interim Results from Its Phase 1/2 Prostate Cancer Clinical Study and Preclinical Results That Demonstrate VT-464 Efficacy in a Clinically-Relevant Enzalutamide-Resistant Mouse Model.
-
A Phase 1/2 Open-Label Study to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamics of Seviteronel in Subjects With Castration-Resistant Prostate Cancer
-
A Phase 1/2 Open-Label, Multiple-Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Once-Daily VT-464 in Patients With Castration-Resistant Prostate Cancer
-
Viamet Pharmaceuticals Appoints Former Novartis Executive Marc Rudoltz, M.D. as Chief Medical Officer.
-
VIAMET PHARMACEUTICALS AND THE NATIONAL INSTITUTES OF HEALTH TO JOINTLY DEVELOP NOVEL VIAMET COMPOUND.
-
Viamet Pharmaceuticals Initiates Phase 1/2 Clinical Trial of Novel Prostate Cancer Therapy, VT-464.
-
Viamet Pharmaceuticals to Present at the 32nd Annual J.P. Morgan Healthcare Conference.
-
VIAMET PHARMACEUTICALS TO PRESENT AT THE 31st Annual J.P. MORGAN HEALTHCARE CONFERENCE.
-
Innocrin Pharmaceuticals, Inc. Initiates Phase 2 Castration-Resistant Prostate Cancer (CRPC) Study in Men Who Have Failed Enzalutmaide or Abiraterone.
-
Innocrin Pharmaceuticals Appoints Fred Eshelman, PharmD as CEO and is Granted Fast Track Designation by FDA for Seviteronel Treatment of Women with Triple-negative Breast Cancer and Women or Men with Estrogen Receptor-positive Breast Cancer.
-
Gucalp A, Bardia A, Gabrail N, DaCosta N, Danso M, Elias AD, et al. Phase 1/2 study of oral seviteronel (VT-464), a dual CYP17-lyase inhibitor and androgen receptor (AR) antagonist, in patients with advanced AR positive triple negative (TNBC) or estrogen receptor (ER) positive breast cancer (BC). SABCS-2016 2016; abstr. P2-08-04.
Available from: URL:http://www.abstracts2view.com/sabcs/view.php?nu=SABCS16L_1479
-
Innocrin Pharmaceuticals Presents Data from the Ongoing Phase 2 Trial of Seviteronel in Estrogen Receptor-positive or Triple-negative Breast Cancer (CLARITY-01) at the San Antonio Breast Cancer Symposium.
-
Innocrin Pharmaceuticals, Inc. Appoints Edwina Baskin-Bey, MD as Chief Medical Officer and Expands the Ongoing Phase 2 Study of Seviteronel in Women with Estrogen Receptor-positive or Triple-negative Breast Cancer (TNBC).
-
Innocrin Pharmaceuticals, Inc. Raises $28 Million in Series D Financing.
-
A Phase 1/2 Open-Label Study to Evaluate the Safety, Pharmacokinetics, Pharmacodynamics and Efficacy of Seviteronel in Subjects With Advanced Breast Cancer
-
Speers CW, Chandler B, Zhao S, Liu M, Wilder-Romans K, Olsen E, et al. Radiosensitization of androgen receptor (AR)-positive triple-negative breast cancer (TNBC) cells using seviteronel (SEVI), a selective CYP17 lyase and AR inhibitor. ASCO-2017 2017; abstr. e12102.
Available from: URL: http://abstracts.asco.org/199/AbstView_199_193240.html
-
Innocrin Pharmaceuticals, Inc. Appoints Charles F. Osborne Jr. as its Chief Financial Officer.
-
Viamet Pharmaceuticals Secures $18 Million Financing.
-
Viamet Pharmaceuticals Raises $4 Million Round of Financing.
///////////SEVITERONEL, VT-464, INO-464, VT 464, INO 464, Phase II, Breast cancer, Prostate cancer, Solid tumours, viamet, CANCER, севитеронел , سيفيتيرونيل , 赛维罗奈 ,
C1(=CN=NN1)C(C1=CC2=C(C=C1)C=C(C(=C2)OC(F)F)OC(F)F)(C(C)C)O
FDA approves new treatment Victoza (liraglutide) for pediatric patients with type 2 diabetes
The U.S. Food and Drug Administration today approved Victoza (liraglutide) injection for treatment of pediatric patients 10 years or older with type 2 diabetes. Victoza is the first non-insulin drug approved to treat type 2 diabetes in pediatric patients since metformin was approved for pediatric use in 2000. Victoza has been approved to treat adult patients with type 2 diabetes since 2010.
“The FDA encourages drugs to be made available to the widest number of patients possible when there is evidence of safety and efficacy,” said Lisa Yanoff, M.D, acting director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research. “Victoza has now been shown to improve blood sugar control in pediatric patients with type 2 diabetes. The expanded indication provides an additional treatment option at a time when
- June 17, 2019
The U.S. Food and Drug Administration today approved Victoza (liraglutide) injection for treatment of pediatric patients 10 years or older with type 2 diabetes. Victoza is the first non-insulin drug approved to treat type 2 diabetes in pediatric patients since metformin was approved for pediatric use in 2000. Victoza has been approved to treat adult patients with type 2 diabetes since 2010.
“The FDA encourages drugs to be made available to the widest number of patients possible when there is evidence of safety and efficacy,” said Lisa Yanoff, M.D, acting director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research. “Victoza has now been shown to improve blood sugar control in pediatric patients with type 2 diabetes. The expanded indication provides an additional treatment option at a time when an increasing number of children are being diagnosed with this disease.”
Type 2 diabetes is the most common form of diabetes, occurring when the pancreas cannot make enough insulin to keep blood sugar at normal levels. Although type 2 diabetes primarily occurs in patients over the age of 45, the prevalence rate among younger patients has been rising dramatically over the past couple of decades. The Diabetes Report Card published by the U.S. Centers for Disease Control and Prevention estimates that more than 5,000 new cases of type 2 diabetes are diagnosed each year among U.S. youth younger than age 20.
Victoza improves blood sugar levels by creating the same effects in the body as the glucagon-like peptide (GLP-1) receptor protein in the pancreas. GLP-1 is often found in insufficient levels in type 2 diabetes patients. Like GLP-1, Victoza slows digestion, prevents the liver from making too much glucose (a simple sugar), and helps the pancreas produce more insulin when needed. As noted on the label, Victoza is not a substitute for insulin and is not indicated for patients with type 1 diabetes or those with diabetic ketoacidosis, a condition associated with diabetes where the body breaks down fat too quickly because there is inadequate insulin or none at all. Victoza is also indicated to reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes and established cardiovascular disease; however, its effect on major adverse cardiovascular events in pediatrics was not studied and it is not indicated for this use in children.
The efficacy and safety of Victoza for reducing blood sugar in patients with type 2 diabetes was studied in several placebo-controlled trials in adults and one placebo-controlled trial with 134 pediatric patients 10 years and older for more than 26 weeks. Approximately 64% of patients in the pediatric study had a reduction in their hemoglobin A1c (HbA1c) below 7% while on Victoza, compared to only 37% who achieved these results with the placebo. HbA1c is a blood test that is routinely performed to evaluate how well a patient’s diabetes is controlled, and a lower number indicates better control of the disease. These results occurred regardless of whether the patient also took insulin at the same time. Adult patients who took Victoza with insulin or other drugs that increase the amount of insulin the body makes (e.g., sulfonylurea) may have an increased risk of hypoglycemia (low blood sugar). Meanwhile, pediatric patients 10 years and older taking Victoza had a higher risk of hypoglycemia regardless of whether they took other therapies for diabetes.
The prescribing information for Victoza includes a Boxed Warning to advise health care professionals and patients about the increased risk of thyroid C-cell tumors. For this reason, patients who have had, or have family members who have ever had medullary thyroid carcinoma (MTC) should not use Victoza, nor should patients who have an endocrine system condition called multiple endocrine neoplasia syndrome type 2 (MEN 2). In addition, people who have a prior serious hypersensitivity reaction to Victoza or any of the product components should not use Victoza. Victoza also carries warnings about pancreatitis, Victoza pen sharing, hypoglycemia when used in conjunction with certain other drugs known to cause hypoglycemia including insulin and sulfonylurea, renal impairment or kidney failure, hypersensitivity and acute gallbladder disease. The most common side effects are nausea, diarrhea, vomiting, decreased appetite, indigestion and constipation.
The FDA granted this application Priority Review. The approval of Victoza was granted to Novo Nordisk.
//////Victoza, liraglutide, FDA 2019, Priority Review, Novo Nordisk, DIABETES
VX-445, Elexacaftor, エレクサカフトル
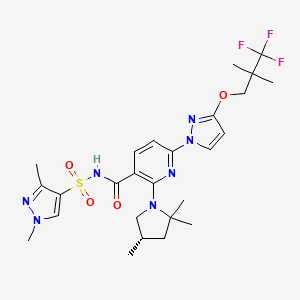

VX-445, Elexacaftor, エレクサカフトル
597.658 g/mol, C26H34F3N7O4S
N-[(1,3-Dimethyl-1H-pyrazol-4-yl)sulfonyl]-6-[3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethyl-1-pyrrolidinyl]-3-pyridinecarboxamide
3-Pyridinecarboxamide, N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)-2-((4S)-2,2,4-trimethyl-1-pyrrolidinyl)-
Cas 2216712-66-0
WHO 11180
Treatment of cystic fibrosis, CFTR modulator
Elexacaftor is under investigation in clinical trial NCT03525548 (A Study of VX-445 Combination Therapy in CF Subjects Homozygous for F508del (F/F)).
Cystic fibrosis transmembrane conductance regulator (CFTR) corrector designed to restore Phe508del CFTR protein function in patients with cystic fibrosis when administered with tezacaftor and ivacaftor.
VX-445 (elexacaftor), tezacaftor, and ivacaftor triple-drug combo
Vertex Pharmaceuticals (NASDAQ: VRTX) already claims a virtual monopoly in treating the underlying cause of cystic fibrosis (CF). The biotech’s current three CF drugs should generate combined sales of close to $3.5 billion this year. Another blockbuster is likely to join those three drugs on the market in 2020 — Vertex’s triple-drug CF combo featuring VX-445 (elexacaftor), tezacaftor, and ivacaftor.
EvaluatePharma projects that this triple-drug combo will rake in close to $4.3 billion by 2024. The market researcher pegs the net present value of the drug at nearly $20 billion, making it the most valuable pipeline asset in the biopharmaceutical industry right now.
PATENT
WO 2018107100
Also disclosed herein is Compound 1:
[0013] N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide.
Synthesis of Compound 1
[00256] Part A: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00257] Step 1: methyl-2,4-dimethyl-4-nitro-pentanoate
[00258] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature.2-Nitropropane (1.5 kg, 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl tert-butyl ether (MTBE) (14 L).2 M HCl (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible– a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HCl (3 L). After separation, the HCl washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. Crude product was treated with MgSO4 and filtered to afford methyl-2,4-dimethyl-4-nitro-pentanoate as a clear green oil (3.16 kg, 99% yield).
[00259] 1H NMR (400 MHz, Chloroform-d) δ 3.68 (s, 3H), 2.56– 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J = 6.8 Hz, 3H).
[00260] Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate
[00261] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00262] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CO3 (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
[00263] Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one
[00264] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at ~2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (~1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to ~1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white crystalline solid (2.042 kg, 16.1 mol, 87 %).1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
[00265] Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00266] A glass lined 120 L reactor was charged with lithium aluminum hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by
vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and the product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br d, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H).
[00267] Part B: Preparation of N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
[00268] Preparation of starting materials:
[00269] 3,3,3-Trifluoro-2,2-dimethyl-propan-1-ol
[00270] A 1 L 3 neck round bottom flask was fitted with a mechanical stirrer, a cooling bath, an addition funnel, and a J-Kem temperature probe. The vessel was charged with lithium aluminum hydride (LAH) pellets (6.3 g, 0.1665 mol) under a nitrogen atmosphere. The vessel was then charged with tetrahydrofuran (200 mL) under a nitrogen atmosphere. The mixture was allowed to stir at room temperature for 0.5 hours to allow the pellets to dissolve. The cooling bath was then charged with crushed ice in water and the reaction temperature was lowered to 0 oC. The addition funnel was charged with a solution of 3,3,3-trifluoro-2,2-dimethyl-propanoic acid (20 g, 0.1281 mol) in tetrahydrofuran (60 mL) and the clear pale yellow solution was added drop wise over 1 hour. After the addition was complete the mixture was allowed to slowly warm to room temperature and stirring was continued for 24 hours. The suspension was cooled to 0 oC with a crushed ice-water in the cooling bath and then quenched by the very slow and drop wise addition of water (6.3 ml), followed by sodium hydroxide solution (15 weight %; 6.3 mL) and then finally with water (18.9 mL). The reaction temperature of the resulting white suspension was recorded at 5 oC. The suspension was stirred at ~5 oC for 30 minutes and then filtered through a 20 mm layer of Celite. The filter cake was washed with tetrahydrofuran (2 x 100 mL). The filtrate was dried over sodium sulfate (150 g) and then filtered. The filtrate was concentrated under reduced pressure to provide a clear colorless oil (15 g) containing a mixture of the product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol in THF (73 % weight of product ~10.95g, and 27 wt.% THF as determined by 1H-NMR). The distillate from the rotary evaporation was distilled at atmospheric pressure using a 30 cm Vigreux column to provide 8.75 g of a residue containing 60 % weight of THF and 40 % weight of product (~3.5 g). The estimated total amount of product is 14.45 g (79% yield).1H NMR (400 MHz, DMSO-d6) δ 4.99 (t, J = 5.7 Hz, 1H), 3.38 (dd, J = 5.8, 0.9 Hz, 2H), 1.04 (d, J = 0.9 Hz, 6H).
[00271] tert-Butyl 3-oxo-2,3-dihydro-1H-pyrazole-1-carboxylate
[00272] A 50L Syrris controlled reactor was started and jacket set to 20 °C, stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added and the reactor was capped. The reaction was heated to an internal temperature of 40 °C and the system was set to hold jacket temp at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 ^C for 1 h. The reaction mixture was cooled to 20 ^C and triethyamine (2.483 kg, 3.420 L, 24.54 mol) was added portion wise (exothermic), maintaining reaction temp <30 °C.
A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L, 22.76 mol) in MeOH (2.860 L) was added portion wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 ^C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear light amber oil. The resulting oil was transferred to the 50L reactor, stirred and added water (7.150 L) and heptane (7.150 L). The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol), then began dropwise addition of acid. The jacket was set to 0 °C to absorb the quench exotherm. After addition (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L), and washed a second time with water (3.575 L) and pulled dry. The crystalline solid was scooped out of the filter into a 20L rotovap bulb and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and then distilled off 1-2 volumes of solvent. The slurry in the rotovap flask was filtered and the solids washed with heptane (3.575 L) and pulled dry. The solid was further dried in vacuo (50 °C , 15 mbar) to give tert-butyl 5-oxo-1H-pyrazole-2-carboxylate (2921 g, 71%) as coarse, crystalline solid.1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J = 2.9 Hz, 1H), 5.90 (d, J = 2.9 Hz, 1H), 1.54 (s, 9H).
[00273] Step A: tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate
[00274] A mixture of 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (10 g, 70.36 mmol) and tert-butyl 3-hydroxypyrazole-1-carboxylate (12.96 g, 70.36 mmol) in toluene (130 mL) was treated with triphenyl phosphine (20.30 g, 77.40 mmol) followed by isopropyl N-isopropoxycarbonyliminocarbamate (14.99 mL, 77.40 mmol) and the mixture was stirred at 110 °C for 16 hours. The yellow solution was concentrated under reduced
pressure, diluted with heptane (100mL) and the precipitated triphenylphosphine oxide was removed by filtration and washed with heptane/toluene 4:1 (100mL). The yellow filtrate was evaporated and the residue purified by silica gel chromatography with a linear gradient of ethyl acetate in hexane (0-40%) to give tert-butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (12.3 g, 57%) as an off white solid. ESI-MS m/z calc.308.13477, found 309.0 (M+1) +; Retention time: 1.84 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 3.0 Hz, 1H), 6.15 (d, J = 3.0 Hz, 1H), 4.18 (s, 2H), 1.55 (s, 9H), 1.21 (s, 6H).
[00275] Step B: 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
[00276] tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (13.5 g, 43.79 mmol) was treated with 4 M hydrogen chloride in dioxane (54.75 mL, 219.0 mmol) and the mixture was stirred at 45 °C for 1 hour. The reaction mixture was evaporated to dryness and the residue was extracted with 1 M aqueous NaOH (100ml) and methyl tert-butyl ether (100ml), washed with brine (50ml) and extracted with methyl tert-butyl ether (50ml). The combined organic phases were dried, filtered and evaporated to give 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 96%) as an off white waxy solid. ESI-MS m/z calc.208.08235, found 209.0 (M+1) +;
Retention time: 1.22 minutes.1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 7.52 (d, J = 2.2 Hz, 1H), 5.69 (t, J = 2.3 Hz, 1H), 4.06 (s, 2H), 1.19 (s, 6H).
[00277] Step C: tert-Butyl 2,6-dichloropyridine-3-carboxylate
[00278] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and left to stir overnight at room temperature. At this point, HCl 1N (400 mL) was added and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL) and the combined organics layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert-butyl 2,6-dichloropyridine-3-carboxylate as a colorless oil. ESI-MS m/z calc.247.01668, found 248.1 (M+1) +; Retention time: 2.27 minutes.1H NMR (300 MHz, CDCl3) ppm 1.60 (s, 9H), 7.30 (d, J=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
[00279] Step D: tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate
[00280] To a solution of tert-butyl 2,6-dichloropyridine-3-carboxylate (10.4 g, 41.9 mmol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 41.93 mmol) in DMF (110 mL) were added potassium carbonate (7.53 g, 54.5 mmol) and 1,4-diazabicyclo[2.2.2]octane (706 mg, 6.29 mmol) and he mixture was stirred at room temperature for 16 hours. The cream suspension was cooled in a cold water bath and cold water (130 mL) was slowly added. The thick suspension was stirred at room temperature for 1 hour, filtered and washed with plenty of water to give tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 99%) as an off white solid. ESI-MS m/z calc.419.12234, found 420.0 (M+1) +; Retention time: 2.36 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 2.9 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.9 Hz, 1H), 4.27 (s, 2H), 1.57 (s, 9H), 1.24 (s, 6H).
[00281] Step E: 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid
[00282] tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 40.25 mmol) was suspended in isopropanol (85 mL) treated with hydrochloric acid (34 mL of 6 M, 201 mmol) and heated to reflux for 3 hours (went almost complete into solution at reflux and started to precipitate again). The suspension was diluted with water (51 mL) at reflux and left to cool to room
temperature under stirring for 2.5 h. The solid was collected by filtration, washed with isopropanol/water 1:1 (50mL), plenty of water and dried in a drying cabinet under vacuum at 45-50 °C with a nitrogen bleed overnight to give 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (13.7 g, 91%) as an off white solid. ESI-MS m/z calc.363.05975, found 364.0 (M+1) +; Retention time: 1.79 minutes. 1H NMR (400 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.44 (d, J = 2.9 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 6.25 (d, J = 2.9 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).
[00283] Step F: 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide
[00284] 2-Chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (100 mg, 0.2667 mmol) and CDI (512 mg, 3.158 mmol) were combined in THF (582.0 µL) and the mixture was stirred at room temperature. Meanwhile, 1,3-dimethylpyrazole-4-sulfonyl chloride (62 mg, 0.3185 mmol) was combined with ammonia (in methanol) in a separate vial, instantly forming a white solid. After stirring for an additional 20 min, the volatiles were removed by evaporation, and 1 mL of dichloromethane was added to the solid residue, and was also evaporated. DBU (100 µL, 0.6687 mmol) was then added and the mixture stirred at 60 °C for 5 minutes, followed by addition of THF (1 mL) which was subsequently evaporated. The contents of the vial containing the CDI activated carboxylic acid in THF were then added to the vial containing the newly formed sulfonamide and DBU, and the reaction mixture was stirred for 4 hours at room temperature. The reaction mixture was diluted with 10 mL of ethyl acetate, and washed with 10 mL solution of citric acid (1 M). The aqueous layer was extracted with ethyl acetate (2x 10 mL) and the combined organics were washed with brine, dried over sodium sulfate, and concentrated to give the product as white solid (137 mg, 99%) that was used in the next step without further purification. ESI-MS m/z calc.520.09076, found 521.1 (M+1) +; Retention time: 0.68 minutes.
[00285] Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide
[00286] 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (137 mg, 0.2630 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (118 mg, 0.7884 mmol) , and potassium carbonate (219 mg, 1.585 mmol) were combined in DMSO (685.0 µL) and the mixture was heated at 130 ^C for 16 hours. The reaction was cooled to room temperature, and 1 mL of water was added. After stirring for 15 minutes, the contents of the vial were allowed to settle, and the liquid portion was removed via pipet and the remaining solids were dissolved with 20 mL of ethyl acetate and were washed with 1 M citric acid (15 mL). The layers were separated and the aqueous layer was extracted two additional times with 15 mL of ethyl acetate. The organics were combined, washed with brine, dried over sodium sulfate and concentrated. The resulting solid was further purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-10%) to give N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (72 mg, 41%) as a white solid. ESI-MS m/z calc.597.2345, found 598.3 (M+1) +; Retention time: 2.1 minutes.1H NMR (400 MHz, DMSO) δ 12.36 (s, 1H), 8.37 (s, 1H), 8.22 (d, J = 2.8 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 4.23 (s, 2H), 3.81 (s, 3H), 2.56 (d, J = 10.4 Hz, 1H), 2.41 (t, J = 8.7 Hz, 1H), 2.32 (s, 3H), 2.18 (dd, J = 12.4, 6.1 Hz, 1H), 1.87 (dd, J = 11.7, 5.5 Hz, 1H), 1.55 (d, J = 11.2 Hz, 6H), 1.42 (t, J = 12.0 Hz, 1H), 1.23 (s, 6H), 0.81 (d, J = 6.2 Hz, 3H).
[00287] Alternative Steps F and G:
[00288] Alternative Step F: 2-chloro-N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinamide
[00289]
[
[00291] To a suspension of 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (20.0 g, 53.89 mmol) in THF (78.40 mL) was added solid carbonyldiimidazole (approximately 10.49 g, 64.67 mmol) portion wise and the resulting solution was stirred at room temperature (slight exotherm from 18-21 °C was observed). After 1 h, solid 1,3-dimethylpyrazole-4-sulfonamide
(approximately 11.33 g, 64.67 mmol) was added, followed by DBU (approximately 9.845 g, 9.671 mL, 64.67 mmol) in two equal portions over 1 min (exotherm from 19 to 35 °C). The reaction mixture was stirred at room temperature for 16 h. The reaction mixture was diluted with ethyl acetate (118 mL) and then HCl (approximately 107.8 mL of 2 M, 215.6 mmol). The phases were separated and the aqueous phase was extracted
with ethyl aceate (78 mL). The combined organics were washed with water (39.2 mL), then brine (40 mL), dried over sodium sulfate and concentrated. The resulting foam was crystallized from a 1:1 isopropanol:heptane mixture (80 mL) to afford 2-chloro-N-((1,3-dimethyl-1H-pyrazol-4-yl)sulfonyl)-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinamide (26.1 g, 93%) as a white solid. ESI-MS m/z calc.520.0, found 520.9 (M+1) +; Retention time: 1.83 minutes.
[00292] Alternative Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide
[
[00294] 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (20.0 g, 38.39 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (approximately 14.36 g, 95.98 mmol), and K2CO3 (approximately 26.54 g, 192.0 mmol) were combined in DMSO (80.00 mL) and 1,2-diethoxyethane (20.00 mL) in a 500-mL flask with reflux condenser. The reaction mixture was heated at 120 °C for 16 h then cooled to room temperature. The reaction was diluted with DCM (200.0 mL) and HCl (approximately 172.8 mL of 2 M, 345.5 mmol); aqueous pH ~1. The phases were separated, and the aqueous phase was extracted with DCM (100.0 mL). The organic phases were combined, washed with water (100.0 mL) (3 x), and dried (Na2SO4) to afford an amber solution. The solution was filtered through a DCM-packed silica gel bed (80 g; 4 g/g) and washed with 20% EtOAc/DCM (5 x 200 mL). The combined filtrate/washes were concentrated to afford 22.2 g of an off-white powder. The powder was slurried in MTBE (140 mL) for 30 min. The solid was collected by filtration (paper/sintered-glass) to afford 24 g after air-drying. The solid was transferred to a drying dish and vacuum-dried (40 °C/200 torr/N2 bleed) overnight to afford 20.70 g (90%) of a white powder. ESI-MS m/z calc.
597.2345, found 598.0 (M+1)+; Retention time: 2.18 minutes.
[00295] 1H NMR (400 MHz, Chloroform-d) δ 13.85 (s, 1H), 8.30 (d, J = 8.6 Hz, 1H), 8.23 (d, J = 2.8 Hz, 1H), 8.08 (s, 1H), 7.55 (d, J = 8.5 Hz, 1H), 5.98 (d, J = 2.8 Hz, 1H), 4.24 (s, 2H), 3.86 (s, 3H), 3.44 (dd, J = 10.3, 8.4 Hz, 1H), 3.09 (dd, J = 10.3, 7.8 Hz, 1H), 2.67– 2.52 (m, 1H), 2.47 (s, 3H), 2.12 (dd, J = 12.3, 7.8 Hz, 1H), 1.70 (dd, J = 12.4, 9.6 Hz, 1H), 1.37 (s, 3H), 1.33 (s, 3H), 1.27 (s, 6H), 1.20 (d, 3H).
[00296] Alternative Synthesis of 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
Step 1: Preparation of 3,3,3-trifluoro-2,2-dimethylpropan-1-ol
A reactor was loaded with toluene (300 mL) and 3,3,3-trifluoro-2,2-dimethylpropanoic acid (30 g, 192.2 mmol), capped, purged under nitrogen. The reaction was set to control the internal temperature to 40 °C. A solution of Vitride (65% in toluene. approximately 119.6 g of 65 %w/w, 115.4 mL of 65 %w/w, 384.4 mmol) was set up for addition via syringe, and addition was begun at 40 °C, with the target addition temperature between 40 and 50 °C. The reaction was stirred at 40 °C for 90 min. The reaction was cooled to 10 °C then the remaining Vitride was quenched with slow addition of water (6 mL). A solution of 15 % aq NaOH (30 mL) was added in portions, and solids precipitated half way through the base addition. Water (60.00 mL) was added. The mixture was warmed to 30 °C and held for at least 15 mins. The mixture was then cooled to 20 °C. The
aqueous layer was removed. The organic layer was washed with water (60 mL x 3), and then washed with brine (60 mL). The washed organic layer was dried under Na2SO4, followed with MgSO4. The mix was filtered through Celite, and the cake washed with toluene (60.00 mL) and pulled dry. The product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (22.5 g, 82%) was obtained as clear colorless solution.
Step 2: Preparation of 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate
A reactor was charged with 3,3,3-trifluoro-2,2-dimethylpropan-1-ol (17.48 g, 123.0 mmol) solution in toluene (250g), 1-(tert-butyl) 4-ethyl 3-hydroxy-1H-pyrazole-1,4-dicarboxylate (30.0 g, 117.1 mmol), and PPh3 (35.33 g, 134.7 mmol). The reaction was heated to 40 °C. DIAD (26.09 mL, 134.7 mmol) was weighed and placed into a syringe and added over 10 minutes while maintaining an internal temperature ranging between 40 and 50 °C. The reaction was then heated to 100 °C over 30 minutes. After holding at 100 °C for 30 minutes, the reaction was complete, and the mixture was cooled to 70 °C over 15 minutes. Heptane (180.0 mL) was added, and the jacket was cooled to 15 °C over 1 hour. (TPPO began crystallizing at ~35 °C). The mixture stirring at 15 °C was filtered (fast), the cake was washed with a pre-mixed solution of toluene (60 mL) and heptane (60 mL) and then pulled dry. The clear solution was concentrated to a waxy solid (45 °C, vacuum, rotovap). Crude 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate (53.49g) was obtained as a waxy solid, (~120% of theoretical mass recovered).
Step 3: Preparation of 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid
A solution of 1-(tert-butyl) 4-ethyl 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-1,4-dicarboxylate (50.0 g, 131 mmol) in 2-methyltetrahydrofuran (500 mL) was prepared in a reactor and stirred at 40 °C. Portions of KOt-Bu (80.85 g, 720.5 mmol) were then added over 30 minutes. Addition was exothermic. After 2053.49g UPLC-MS showed complete removal of the Boc group, so water (3.53 g, 3.53 mL, 196 mmol) was added drop-wise addition via syringe over 20 min to keep the reaction temperature between 40-50 °C. The mixture was then stirred for 17 hours to complete the reaction. The mixture was then cooled to 20 °C and water (400 mL) was added. The stirring was stopped and the layers were separated. The desired product in the aqueous layer was returned to the reactor and the organic layer was discarded. The aqueous layer was washed with 2-Me-THF (200 mL). Isopropanol (50. mL) was added followed by dropwise addition of aqueous HCl (131 mL of 6.0 M, 786.0 mmol) to adjust the pH to ❤ while maintaining the temperature below 30 °C. The resulting solid was then isolated by filtration and the filter cake washer with water (100 mL) then pulled dry until a sticky cake was obtained. The solids were then dried under vacuum at 55 °C to afford 3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (23.25 g) as an off-white fine solid.
[00297] Step 4: Preparation of 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole
3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazole-4-carboxylic acid (1.0 equiv) was added to a reactor followed by DMF (6.0 vol, 2.6 wt equiv). The mixture was stirred at 18– 22 °C. DBU (0.2 equiv.) was charged to the reaction mixture at a rate of approximately 45 mL/min. The reaction temperature was then raised to 98– 102 °C over 45 minutes. The reaction mixture was stirred at 98– 102 °C for no less than 10 h. The reaction mixture was then cooled to -2°C to 2 °C over approximately 1 hour and was used without isolation to make ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate.
[00298] Alternate procedure for the preparation of 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid
[00299] Step 1. Ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate
[00300] A solution of ethyl 2,6-dichloronicotinate (256 g, 1.16 mol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (242 g, 1.16 mol) in DMF (1.53 L) was treated with potassium carbonate (209 g, 1.51 mol) and DABCO (19.6 g, 174 mmol). The resultant suspension was stirred allowed to exotherm from 14 to 25 °C and then maintained at 20– 25 °C with external cooling for 3 days. The suspension was cooled to below 10 °C when water (2.0 L) was added in a thin stream while maintaining the temperature below 25 °C. After the addition was complete, the suspension was stirred for an additional 1 h. The solid was collected by filtration (sintered-glass/polypad) and the filter-cake was washed with water (2 x 500-mL) and dried with suction for 2 h to afford water-damp ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate (512 g; 113% yield) as white powder which was used without further steps in the subsequent reaction.
[00301] Step 2.2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1h-pyrazol-1-yl)nicotinic acid
[00302] The water-damp ethyl 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl)nicotinate (455 g, 1.16 mol; assumed 100% yield from previous step) in EtOH (1.14 L) and THF (455 mL) was stirred at ambient temperature (17 °C) when 1 M NaOH (1.16 L, 1.16 mol) was added. The reaction mixture exothermed to 30 °C and was further warmed at 40 °C for 2 h. The solution was quenched with 1 M HCl (1.39 L, 1.39 mol) which resulted in an immediate precipitation which became thicker as the acid was added. The creamy suspension was allowed to cool to room temperature and was stirred overnight. The solid was collected by filtration (sintered-glass/poly pad). The filter-cake was washed with water (2 x 500-mL). The filter-cake was dried by suction for 1 h but remained wet. The damp solid was transferred to a 10-L Buchi flask for further drying (50 °C/20 torr), but was not effective. Further effort to dry by chasing with i-PrOH was also ineffective. Successful drying was accomplished after the damp solid was backfilled with i-PrOAc (3 L), the suspension was heated at 60 °C (homogenization), and re-concentrated to dryness (50 °C/20 torr) to afford dry 2-chloro-6-(3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1h-pyrazol-1-yl)nicotinic acid (408 g; 97% yield for two steps) as a fine, white powder. The product was further dried in a vacuum oven (50 °C/10 torr/N2 bleed) for 2 h but marginal weight loss was observed. 1H NMR (400 MHz, DMSO-d6) δ 13.64 (s, 1H), 8.49– 8.36 (m, 2H), 7.77 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.8 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).19F NMR (376 MHz, DMSO-d6) δ -75.2. KF analysis: 0.04% water.
2. Preparation of Form A of Compound 1
[00303] The crystalline Form A of Compound 1 was obtained as a result of the following synthesis. Combined 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide(108 g, 207.3 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (77.55 g, 518.2 mmol), was combined with K2CO3 (143.2 g, 1.036 mol) in DMSO (432.0 mL) and 1,2-
diethoxyethane (108.0 mL) in a 1-L RB flask with a reflux condenser. The resulting suspension was heated at 120°C and was stirred at temperature overnight. Then the reaction was diluted with DCM (1.080 L) and HCl (933.0 mL of 2 M, 1.866 mol) was slowly added. The liquid phases were separated, and the aqueous phase was extracted with DCM (540.0 mL).The organic phases were combined, washed with water (540.0 mL) (3 x), then dried with (Na2SO4) to afford an amber solution. Silica gel (25 g) was added and then the drying agent/silica gel was filtered off. The filter-bed was washed with DCM (3 x 50-mL). The organic phases were combined and concentrated (40 °C/40 torr) to afford crude N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (198.6 g, 160% theory) as an off-white solid. The solid was diluted with MTBE (750 mL), warmed at 60 °C (external temperature), and mixed to a homogenous suspension. The suspension was cooled to 30 °C with stirring and the solid was collected by filtration, air-dried, and vacuum-dried to afford Compound 1 (111.1 g; 90 %) as a fine, white powder.
[00304] The crystalline Form A of Compound 1 was also obtained through the following procedure. A suspension of Compound 1 (150.0 g, 228.1 mmol) in iPrOH (480 mL) and water (120 mL) was heated at 82 °C to obtain a solution. The solution was cooled with a J-Kem controller at a cooling rate of 10 °C/h. Once the temperature reached 74 °C, the solution was seeded with a sample of Compound 1 in crystalline Form A. Crystallization occurred immediately. The suspension was cooled to 20 °C. The solid was collected by filtration, washed with i-PrOH (2 x 75 mL), air-dried with suction, and vacuum-dried (55 °C/300 torr/N2 bleed) to afford Compound 1, Form A (103.3 g) as a white powder.. The sample was cooled to ~5 °C, let stir for 1 h, and then the solid was collected by filtration (sintered glass/paper). the filter-cake was washed with i-PrOH (75 mL) (2 x), air-dried with suction, air-dried in a drying dish (120.6 g mostly dried), vacuum-dried (55 °C/300 torr/N2 bleed) for 4 h, and then RT overnight. Overnight drying afforded 118.3 g (87% yield) of a white powder.
PATENT
WO-2019113476
Example 1: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
Step 1: methyl-2,4-dimethyl-4-nitro-pentanoate
[00110] Tetrahydrofuran (THF, 4.5 L) was added to a 20 L glass reactor and stirred under N2 at room temperature. 2-Nitropropane (1.5 kg, 16.83 mol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (1.282 kg, 8.42 mol) were then charged to the reactor, and the jacket temperature was increased to 50 °C. Once the reactor contents were close to 50 °C, methyl methacrylate (1.854 kg, 18.52 mol) was added slowly over 100 minutes. The reaction temperature was maintained at or close to 50 °C for 21 hours. The reaction mixture was concentrated in vacuo then transferred back to the reactor and diluted with methyl tert-butyl ether (MTBE) (14 L). 2 M HCl (7.5 L) was added, and this mixture was stirred for 5 minutes then allowed to settle. Two clear layers were visible– a lower yellow aqueous phase and an upper green organic phase. The aqueous layer was removed, and the organic layer was stirred again with 2 M HCl (3 L). After separation, the HCl washes were recombined and stirred with MTBE (3 L) for 5 minutes. The aqueous layer was removed, and all of the organic layers were combined in the reactor and stirred with water (3 L) for 5 minutes. After separation, the organic layers were concentrated in vacuo to afford a cloudy green oil. Crude product was treated with MgSO4 and filtered to afford methyl-2,4-dimethyl-4-nitro-pentanoate as a clear green oil (3.16 kg, 99% yield).
[00111] 1H NMR (400 MHz, Chloroform-d) δ 3.68 (s, 3H), 2.56– 2.35 (m, 2H), 2.11 – 2.00 (m, 1H), 1.57 (s, 3H), 1.55 (s, 3H), 1.19 (d, J = 6.8 Hz, 3H).
Step 2: Synthesis of methyl (2S)-2,4-dimethyl-4-nitro-pentanoate
[00112] A reactor was charged with purified water (2090 L; 10 vol) and then potassium phosphate monobasic (27 kg, 198.4 moles; 13 g/L for water charge). The pH of the reactor contents was adjusted to pH 6.5 (± 0.2) with 20% (w/v) potassium carbonate solution. The reactor was charged with racemic methyl-2,4-dimethyl-4-nitro-pentanoate (209 kg; 1104.6 moles), and Palatase 20000L lipase (13 L, 15.8 kg; 0.06 vol).
[00113] The reaction mixture was adjusted to 32 ± 2 °C and stirred for 15-21 hours, and pH 6.5 was maintained using a pH stat with the automatic addition of 20% potassium carbonate solution. When the racemic starting material was converted to >98% ee of the S-enantiomer, as determined by chiral GC, external heating was switched off. The reactor was then charged with MTBE (35 L; 5 vol), and the aqueous layer was extracted with MTBE (3 times, 400-1000L). The combined organic extracts were washed with aqueous Na2CO3 (4 times, 522 L, 18 % w/w 2.5 vol), water (523 L; 2.5 vol), and 10% aqueous NaCl (314 L, 1.5 vol). The organic layer was concentrated in vacuo to afford methyl (2S)-2,4-dimethyl-4-nitro-pentanoate as a mobile yellow oil (>98% ee, 94.4 kg; 45 % yield).
Step 3: Synthesis of (3S)-3,5,5-trimethylpyrrolidin-2-one
[00114] A 20 L reactor was purged with N2. The vessel was charged sequentially with DI water-rinsed, damp Raney® Ni (2800 grade, 250 g), methyl (2S)-2,4-dimethyl-4-nitro-pentanoate (1741g, 9.2 mol), and ethanol (13.9 L, 8 vol). The reaction was stirred at 900 rpm, and the reactor was flushed with H2 and maintained at ~2.5 bar. The reaction mixture was then warmed to 60 °C for 5 hours. The reaction mixture was cooled and filtered to remove Raney nickel, and the solid cake was rinsed with ethanol (3.5 L, 2 vol). The ethanolic solution of the product was combined with a second equal sized batch and concentrated in vacuo to reduce to a minimum volume of ethanol (~1.5 volumes). Heptane (2.5 L) was added, and the suspension was concentrated again to ~1.5 volumes. This was repeated 3 times; the resulting suspension was cooled to 0-5 °C, filtered under suction, and washed with heptane (2.5 L). The product was dried under vacuum for 20 minutes then transferred to drying trays and dried in a vacuum oven at 40 °C overnight to afford (3S)-3,5,5-trimethylpyrrolidin-2-one as a white solid (2.042 kg, 16.1 mol, 87 %). 1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (dd, J = 12.4, 8.6 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
Step 4: Synthesis of (4S)-2,2,4-trimethylpyrrolidine hydrochloride
[00115] A glass lined 120 L reactor was charged with lithium aluminum hydride pellets (2.5 kg, 66 mol) and dry THF (60 L) and warmed to 30 °C. The resulting suspension was charged with (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C, then cautiously quenched with the addition of ethyl acetate (EtOAc) (1.0 L, 10 moles), followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq), and then a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 equiv water with 1.4 equiv sodium hydroxide relative to aluminum), followed by 7.5 L water. After the addition was complete, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C. The resultant solution was concentrated by vacuum distillation to a slurry. Isopropanol (8 L) was added and the solution was concentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added, and the product was slurried by warming to about 50 °C. MTBE (6 L) was added, and the slurry was cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L MTBE and dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford (4S)-2,2,4-trimethylpyrrolidine•HCl as a white solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (br d, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H).
Example 2: Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
Example 2A
[00116] 2,2,6,6-tetramethylpiperidin-4-one (50.00 g, 305.983 mmol, 1.000 equiv), tributylmethyl ammonium chloride (2.89 g, 3.0 mL, 9.179 mmol, 0.030 equiv), chloroform (63.92 g, 43.2 mL, 535.470 mmol, 1.750 equiv), and DCM
(dichloromethane) (100.0 mL, 2.00 vol) were charged to a 1000 mL three-neck round bottom flask equipped with an overhead stirrer. The reaction mixture was stirred at 300 rpm, and 50 wt% NaOH (195.81 g, 133.2 mL, 2,447.863 mmol, 8.000 equiv) was added dropwise (via addition funnel) over 1.5 h while maintaining the temperature below 25 °C with intermittent ice/acetone bath. The reaction mixture was stirred at 500 rpm for 18 h, and monitored by GC (3% unreacted piperidinone after 18 h). The suspension was diluted with DCM (100.0 mL, 2.00 vol) and H2O (300.0 mL, 6.00 vol), and the phases were separated. The aqueous phase was extracted with DCM (100.0 mL, 2.00 vol). The organic phases were combined and 3 M hydrochloric acid (16.73 g, 153.0 mL, 458.974 mmol, 1.500 equiv) was added. The mixture was stirred at 500 rpm for 2 h. The conversion was complete after approximately 1 h. The aqueous phase was saturated with NaCl, H2O (100.0 mL, 2.00 vol) was added to help reduce the emulsion, and the phases were separated. The aqueous phase was extracted with DCM (100.0 mL, 2.00 vol) twice. H2O (100.0 mL, 2.00 vol) was added to help with emulsion separation. The organic phases were combined, dried (MgSO4), and concentrated to afford 32.6 g (85%) of crude 5,5-dimethyl-3-methylenepyrrolidin-2-one (19) as a pale orange clumpy solid. The crude was recrystallized from hot (90°C) iPrOAc (71.7 mL, 2.2 vol. of crude), cooled to 80 °C, and ~50 mg of crystalline 5,5-dimethyl-3-methylenepyrrolidin-2-one (19) was added for seeding. Crystallization started at 77 °C, the mixture was slowly cooled to ambient temperature, and aged for 2 h. The solid was collected by filtration, washed with 50/50 iPrOAc/heptane (20.0 mL, 0.40 vol) twice, and dried overnight in the vacuum oven at 40 °C to afford the desired product (23.70 g, 189.345 mmol, 62% yield) as a white sand colored crystalline solid.
1H NMR (400 MHz, CDCl3, 7.26 ppm) δ 7.33 (bs, 1H), 5.96– 5.95 (m, 1H), 5.31-5.30 (m, 1H), 2.6 (t, J = 2.5 Hz, 2H), 1.29 (s, 6H).
Example 2B
[00117] Step 1: Under a nitrogen atmosphere, 2,2,6,6-tetramethylpiperidin-4-one (257.4 kg, 1658.0 mol, 1.00 eq.), tri-butyl methyl ammonium chloride (14.86 kg, 63.0 mol, 0.038 eq.), chloroform (346.5 kg, 2901.5 mol, 1.75 eq.) and DCM (683.3 kg) were added to a 500 L enamel reactor. The reaction was stirred at 85 rpm and cooled to 15~17°C. The solution of 50wt% sodium hydroxide (1061.4 kg, 13264.0 mol, 8.00 eq.) was added dropwise over 40 h while maintaining the temperature between 15~25°C. The reaction mixture was stirred and monitored by GC.
[00118] Step 2: The suspension was diluted with DCM (683.3 kg) and water (1544.4 kg). The organic phase was separated. The aqueous phase was extracted with DCM (683.3 kg). The organic phases were combined, cooled to 10°C and then 3 M
hydrochloric acid (867.8 kg, 2559.0 mol, 1.5 eq.) was added. The mixture was stirred at 10~15 °C for 2 h. The organic phase was separated. The aqueous phase was extracted with DCM (683.3 kg x 2). The organic phases were combined, dried over Na2SO4 (145.0 kg) for 6 h. The solid was filtered off and washed with DCM (120.0 kg). The filtrate was stirred with active charcoal (55 kg) for 6 h. The resulting mixture was filtered and the filtrate was concentrated under reduced pressure (30~40°C, -0.1MPa). Then isopropyl acetate (338 kg) was added and the mixture was heated to 87~91°C, stirred for 1 h. Then the solution was cooled to 15 °C in 18 h and stirred for 1 h at 15 °C. The solid was collected by filtration, washed with 50% isopropyl acetate/hexane (80.0 kg x 2) and dried overnight in the vacuum oven at 50 °C to afford 5,5-dimethyl-3-methylenepyrrolidin-2-one as an off white solid, 55% yield.
Example 3: Synthesis of (S)-3,5,5-trimethyl-pyrrolidin-2-one from 5,5-dimethyl-3- methylenepyrrolidin-2-one
Example 3A – Use of Rh Catalyst
Step 1 – Preparation of Rh Catalyst Formation:
[00119] In a 3 L Schlenk flask, 1.0 l of tetrahydrofurn (THF) was degassed with an argon stream. Mandyphos Ligand SL-M004-1 (1.89 g) and [Rh(nbd)Cl]2 (98%, 0.35 g) (chloronorbornadiene rhodium(I) dimer) were added. The resulting orange catalyst solution was stirred for 30 min at room temperature to form a catalyst solution.
Step 2:
[00120] A 50 L stainless steel autoclave was charged with 5,5-dimethyl-3-methylenepyrrolidin-2-one (6.0 kg) and THF (29 L). The autoclave was sealed and the resulting suspension was flushed with nitrogen (3 cycles at 10 bar), and then released of pressure. Next the catalyst solution from Step 1 was added. The autoclave was flushed with nitrogen without stirring (3 cycles at 5 bar) and hydrogen (3 cycles at 5 bar). The pressure was set to 5 bar and a 50 L reservoir was connected. After 1.5 h with stirring at 1000 rpm and no hydrogen uptake the reactor was flushed again with nitrogen (3 cycles at 10 bar) with stirring and additional catalyst solution was added. The autoclave was again flushed to hydrogen with the above described procedure (3 x 5 bar N2, 3 x 5 bar H2) and adjusted to 5 bar. After 2 h, the pressure was released, the autoclave was flushed with nitrogen (3 cycles at 5 bar) and the product solution was discharged into a 60 L inline barrel. The autoclave was charged again with THF (5 L) and stirred with 1200 rpm for 5 min. The wash solution was added to the reaction mixture.
Step 3:
[00121] The combined solutions were transferred into a 60 L reactor. The inline barrel was washed with 1 L THF which was also added into the reactor. 20 L THF were removed by evaporation at 170 mbar and 40°C.15 L heptane were added. The distillation was continued and the removed solvent was continuously replaced by heptane until the THF content in the residue was 1% w/w (determined by NMR). The reaction mixture was heated to 89°C (turbid solution) and slowly cooled down again (ramp: 14°C/h). Several heating and cooling cycles around 55 to 65°C were made. The off-white suspension was transferred to a stirred pressure filter and filtered (ECTFE-pad, d = 414 mm, 60 my, Filtration time = 5 min). 10 L of the mother liquor was transferred back into the reactor to wash the crystals from the reactor walls and the obtained slurry was also added to the filter. The collected solid was washed with 2 x 2.5 l heptane, discharged and let dry on the rotovap at 40°C and 4 mbar to obtain the product, (S)-3,5,5-trimethyl-pyrrolidin-2-one; 5.48Kg (91%), 98.0% ee.
Example 3B – Use of Ru Catalyst
[00122] The reaction was performed in a similar manner as described above in Example 3A except the use of a Ru catalyst instead of a Rh catalyst.
[00123] Compound (15) (300 g) was dissolved in THF (2640 g, 10 Vol) in a vessel. In a separate vessel, a solution of [RuCl(p-cymene){(R)-segphos}]Cl (0.439g, 0.0002 eq) in THF (660 g, 2.5 Vol) was prepared. The solutions were premixed in situ and passed through a Plug-flow reactor (PFR). The flow rate for the Compound (15) solution was at 1.555 mL/min and the Ru catalyst solution was at 0.287 mL/min. Residence time in the PFR was 4 hours at 30 °C, with hydrogen pressure of 4.5 MPa. After completion of reaction, the THF solvent was distilled off to give a crude residue. Heptane (1026 g, 5 vol) was added and the resulting mixture was heated to 90 °C. The mixture was seeded with 0.001 eq. of Compound 16S seeds. The mixture was cooled to -15 °C at 20 °C/h. After cooling, heptane (410 g, 2 vol) was added and the solid product was recovered by filtration. The resulting product was dried in a vacuum oven at 35 °C to give (S)-3,5,5-trimethyl-pyrrolidin-2-one (281.77 g, 98.2 % ee, 92 % yield).
Example 3C – Analytical Measurements
[00124] Analytical chiral HPLC method for the determination of the conversion, chemoselectivity, and enantiomeric excess of the products from Example 3A and 3B was made under the following conditions
Instrument: Agilent Chemstation 1100
Column: Phenomenex Lux 5u Cellulose-2, 4.6 mm x 250 mm x 5 um, LHS6247 Solvent: Heptane/iPrOH (90:10)
Flow: 1.0 ml/min
Detection: UV (210 nm)
Temperature: 25°C
Sample concentration: 30 μl of reaction solution evaporated, dissolved in 1 mL heptane/iPrOH (80/20)
Injection volume: 10.0 μL, Run time 20 min
Retention times:
5,5–‐dimethyl–3–methylenepyrrolidin–‐2–‐one: 13.8 min (S)-3,5,5-trimethyl-pyrrolidin-2-one: 10.6 min
(R)-3,5,5-trimethyl-pyrrolidin-2-one: 12.4 min
Example 4: Synthesis of (S)-3,5,5-trimethyl-pyrrolidin-2-one from 5,5-dimethyl-3- methylenepyrrolidin-2-one
[00125] Mandyphos (0.00479 mmol, 0.12 eq) was weighed into a GC vial. In a separate vial Ru(Me-allyl)2(COD) (16.87 mg, 0.0528 mmol) was weighed and dissolved in DCM (1328 µL). In another vial HBF4•Et2O (6.6 µL) and BF3 ^Et2O (2.0 µL) were dissolved in DCM (240 µL). To the GC vial containing the ligand was added, under a flow of argon, the Ru(Me-allyl)2(COD) solution (100 µL; 0.00399 mmol, 0.1eq) and the HBF4•Et2O / BF3 ^Et2O solution (20 µL; 1 eq HBF4 ^Et2O and catalytic BF3 ^Et2O). The resulting mixtures were stirred under a flow of argon for 30 minutes.
[00126] 5,5-dimethyl-3-methylenepyrrolidin-2-one (5 mg, 0.0399 mmol) in EtOH (1 mL) was added. The vials were placed in the hydrogenation apparatus. The apparatus was flushed with H2 (3×) and charged with 5 bar H2. After standing for 45 minutes, the apparatus was placed in an oil bath at temperature of 45°C. The reaction mixtures were stirred overnight under H2.200 µL of the reaction mixture was diluted with MeOH (800 µL) and analyzed for conversion and ee.
1H NMR (400 MHz, Chloroform-d) δ 6.39 (s, 1H), 2.62 (ddq, J = 9.9, 8.6, 7.1 Hz, 1H), 2.17 (ddd, J = 12.4, 8.6, 0.8 Hz, 1H), 1.56 (dd, J = 12.5, 9.9 Hz, 1H), 1.31 (s, 3H), 1.25 (s, 3H), 1.20 (d, J = 7.1 Hz, 3H).
Table 1: IPC method for Asymmetric Hydrogenation
Example 5. Synthesis of (S)-2,2,4-trimethylpyrrolidine hydrochloride from (S)- 3,5,5-trimethyl-pyrrolidin-2-one
Example 5A
[00127] Anhydrous THF (100ml) was charged to a dry 750ml reactor and the jacket temperature was set to 50°C. Once the vessel contents were at 50°C LiAlH4pellets (10g, 263mmol, 1.34 eq.) were added. The mixture was stirred for 10 minutes, then a solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) (25g, 197mmol) in anhydrous THF (100ml) was added dropwise over 45 minutes, maintaining the temperature between 50-60°C. Once the addition was complete the jacket temperature was increased to 68°C and the reaction stirred for 18.5hrs. The reaction mixture was cooled to 30°C then saturated sodium sulfate solution (20.9ml) was added dropwise over 30 minutes, keeping the temperature below 40°C. Vigorous evolution of hydrogen was observed and the reaction mixture thickened but remained mixable. The mixture thinned towards the end of the addition. The mixture was cooled to 20°C, diluted with iPrOAc (100ml) and stirred for an additional 10 minutes. The suspension was then drained and collected through the lower outlet valve, washing through with additional iPrOAc (50ml). The collected suspension was filtered through a celite pad on a sintered glass funnel under suction and washed with iPrOAc (2x50ml).
[00128] The filtrate was transferred back to the cleaned reactor and cooled to 0°C under nitrogen. 4M HCl in dioxane (49.1ml, 197mmol, 1eq.) was then added dropwise over 15 minutes, maintaining the temperature below 20°C. A white precipitate formed. The reactor was then reconfigured for distillation, the jacket temperature was increased to 100 °C, and distillation of solvent was carried out. Additional i-PrOAc (100 mL) was added during concentration, after >100 mL distillate had been collected. Distillation was continued until ~250 mL total distillate was collected, then a Dean-Stark trap was attached and reflux continued for 1 hour. No water was observed to collect. The reaction mixture was cooled to 20 °C and filtered under suction under nitrogen. The filtered solid was washed with i-PrOAc (100 mL), dried under suction in nitrogen, then transferred to a glass dish and dried in a vacuum oven at 40 °C with a nitrogen bleed. (S)-2,2,4-Trimethylpyrrolidine hydrochloride (17S•HCl) was obtained as a white solid (24.2g, 82%).
GC analysis (purity): >99.5%
GC chiral purity: 99.5%
Water content (by KF): 0.074%
Residual solvent (by 1H-NMR): 0.41%
Example 5B
[00129] To a glass lined 120 L reactor was charged LiAlH4 pellets (2.5 kg 66 mol, 1.2 equiv.) and dry THF (60 L) and warmed to 30 °C. To the resulting suspension was
charged (S)-3,5,5-trimethylpyrrolidin-2-one (7.0 kg, 54 mol) in THF (25 L) over 2 hours while maintaining the reaction temperature at 30 to 40 °C. After complete addition, the reaction temperature was increased to 60 – 63 °C and maintained overnight. The reaction mixture was cooled to 22 °C and sampled to check for completion, then cautiously quenched with the addition of EtOAc (1.0 L, 10 moles, 0.16 eq) followed by a mixture of THF (3.4 L) and water (2.5 kg, 2.0 eq) then followed by a mixture of water (1.75 kg) with 50 % aqueous sodium hydroxide (750 g, 2 eq water with 1.4 eq sodium hydroxide relative to aluminum), followed by 7.5 L water (6 eq“Fieser” quench). After the addition was completed, the reaction mixture was cooled to room temperature, and the solid was removed by filtration and washed with THF (3 x 25 L). The filtrate and washings were combined and treated with 5.0 L (58 moles) of aqueous 37% HCl (1.05 equiv.) while maintaining the temperature below 30°C.
[00130] The resultant solution was concentrated by vacuum distillation to a slurry in two equal part lots on the 20 L Buchi evaporator. Isopropanol (8 L) was charged and the solution reconcentrated to near dryness by vacuum distillation. Isopropanol (4 L) was added and the product slurried by warming to about 50 °C. Distillation from Isopropanol continued until water content by KF is≤ 0.1 %. Methyl tertbutyl ether (6 L) was added and the slurry cooled to 2-5 °C. The product was collected by filtration and rinsed with 12 L methyl tert-butyl ether and pulled dry with a strong nitrogen flow and further dried in a vacuum oven (55 °C/300 torr/N2bleed) to afford (S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid (6.21 kg, 75% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, , 3H).
Example 5C
[00131] With efficient mechanical stirring, a suspension of LiAlH4 pellets (100 g 2.65 mol; 1.35 eq.) in THF (1 L; 4 vol. eq.) warmed at a temperature from 20 °C– 36 °C (heat of mixing). A solution of (S)-3,5,5-trimethylpyrrolidin-2-one (250 g; 1.97 mol) in THF (1 L; 4 vol. eq.) was added to the suspension over 30 min. while allowing the reaction temperature to rise to ~60 °C. The reaction temperature was increased to near reflux (~68 °C) and maintained for about 16 h. The reaction mixture was cooled to below 40 °C and cautiously quenched with drop-wise addition of a saturated aqueous solution of Na2SO4 (209 mL) over 2 h. After the addition was completed, the reaction mixture was cooled to ambient temperature, diluted with i-PrOAc (1 L), and mixed thoroughly. The solid was removed by filtration (Celite pad) and washed with i-PrOAc (2 x 500 mL). With external cooling and N2 blanket, the filtrate and washings were combined and treated with drop-wise addition of anhydrous 4 M HCl in dioxane (492 mL; 2.95 mol; 1 equiv.) while maintaining the temperature below 20 °C. After the addition was completed (20 min), the resultant suspension was concentrated by heating at reflux (74– 85 °C) and removing the distillate. The suspension was backfilled with i-PrOAc (1 L) during concentration. After about 2.5 L of distillate was collected, a Dean-Stark trap was attached and any residual water was azeotropically removed. The suspension was cooled to below 30 °C when the solid was collected by filtration under a N2 blanket. The solid is dried under N2 suction and further dried in a vacuum oven (55 °C/300 torr/N2 bleed) to afford 261 g (89% yield) of (S)-2,2,4-trimethylpyrrolidine•HCl as a white, crystalline solid. 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 2H), 3.33 (dd, J = 11.4, 8.4 Hz, 1H), 2.75 (dd, J = 11.4, 8.6 Hz, 1H), 2.50– 2.39 (m, 1H), 1.97 (dd, J = 12.7, 7.7 Hz, 1H), 1.42 (s, 3H), 1.38 (dd, J = 12.8, 10.1 Hz, 1H), 1.31 (s, 3H), 1.05 (d, J = 6.6 Hz, 3H). 1H NMR (400 MHz, CDCl3) δ 9.55 (d, J = 44.9 Hz, 2H), 3.52 (ddt, J = 12.1, 8.7, 4.3 Hz, 1H), 2.94 (dq, J = 11.9, 5.9 Hz, 1H), 2.70– 2.51 (m, 1H), 2.02 (dd, J = 13.0, 7.5 Hz, 1H), 1.62 (s, 3H), 1.58– 1.47 (m, 4H), 1.15 (d, J = 6.7 Hz, 3H).
Example 5D
[00132] A 1L four-neck round bottom flask was degassed three times. A 2M solution of LiAlH4 in THF (100 mL) was charged via cannula transfer. (S)-3,5,5-trimethylpyrrolidin-2-one (19.0 g) in THF (150 mL) was added dropwise via an addition funnel over 1.5 hours at 50-60 °C, washing in with THF (19 mL). Upon completion of the addition, the reaction was stirred at 60 °C for 8 hours and allowed to cool to room temperature overnight. GC analysis showed <1% starting material remained.
[00133] Deionized water (7.6 mL) was added slowly to the reaction flask at 10-15 °C, followed by 15% potassium hydroxide (7.6 mL). Isopropyl acetate (76 mL) was added, the mixture was stirred for 15 minutes and filtered, washing through with isopropyl acetate (76 mL).
[00134] The filtrate was charged to a clean and dry 500 mL four neck round bottom flask and cooled to 0-5 °C. 36% Hydrochloric acid (15.1 g, 1.0 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (190 mL), was carried out to leave a residual volume of ~85 mL. Karl Fischer analysis = 0.11% w/w H2O. MTBE (methyl tertiary butyl ether) (19 mL) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (25 mL) and drying under vacuum at 40-45 °C to give crude (S)-2,2,4-trimethylpyrrolidine hydrochloride as a white crystalline solid (17.4 g, 78% yield). GC purity = 99.5%. Water content = 0.20% w/w. Chiral GC gave an ee of 99.0% (S).
Ruthenium content = 0.004 ppm. Lithium content = 0.07 ppm.
[00135] A portion of the dried crude (S)-2,2,4-trimethylpyrrolidine hydrochloride (14.3g) was charged to a clean and dry 250 mL four-neck round bottom flask with isopropanol (14.3 mL) and the mixture held at 80-85 °C (reflux) for 1 hour to give a clear solution. The solution was allowed to cool to 50 °C (solids precipitated on cooling) then MTBE (43 mL) was added and the suspension held at 50-55 °C (reflux) for 3 hours. The solids were filtered off at 10 °C, washing with MTBE (14 mL) and dried under vacuum at 40 °C to give recrystallised (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystallised solid (13.5 g, 94% yield on recrystallisation, 73% yield). GC purity = 99.9%. Water content = 0.11% w/w. Chiral GC gave an ee of 99.6 (S). Ruthenium content = 0.001 ppm. Lithium content = 0.02 ppm.
Example 5E:
[00136] A reactor was charged with lithium aluminum hydride (LAH) (1.20 equiv.) and 2-MeTHF (2-methyltetrahydrofuran) (4.0 vol), and heated to internal temperature of 60 °C while stirring to disperse the LAH. A solution of (S)-3,5,5-trimethylpyrrolidin-2-one (1.0 equiv) in 2-MeTHF (6.0 vol) was prepared and stirred at 25 °C to fully dissolve the (S)-3,5,5-trimethylpyrrolidin-2-one. The (S)-3,5,5-trimethylpyrrolidin-2-one solution was added slowly to the reactor while keeping the off-gassing manageable, followed by rinsing the addition funnel with 2-MeTHF (1.0 vol) and adding it to the reactor. The reaction was stirred at an internal temperature of 60 ± 5 °C for no longer than 6 h. The internal temperature was set to 5 ± 5 °C and the agitation rate was increased. A solution of water (1.35 equiv.) in 2-MeTHF (4.0v) was prepared and added slowly to the reactor while the internal temperature was maintained at or below 25 °C. Additional water (1.35 equiv.) was charged slowly to the reactor while the internal temperature was maintained at or below 25 °C. Potassium hydroxide (0.16 equiv.) in water (0.40 vol) was added to the reactor over no less than 20 min while the temperature was maintained at or below 25 °C. The resulting solids were removed by filtration, and the reactor and cake were washed with 2-MeTHF (2 x 2.5 vol). The filtrate was transferred back to a jacketed
vessel, agitated, and the temperature was adjusted to 15 ± 5 °C. Concentrated aqueous HCl (35-37%, 1.05 equiv.) was added slowly to the filtrate while maintaining the temperature at or below 25 °C and was stirred no less than 30 min. Vacuum was applied and the solution was distilled down to a total of 4.0 volumes while maintaining the internal temperature at or below 55 °C, then 2-MeTHF (6.00 vol) was added to the vessel. The distillation was repeated until Karl Fischer analysis (KF) < 0.20% w/w H2O. Isopropanol was added (3.00 vol), and the temperature was adjusted to 70 °C (65– 75 °C) to achieve a homogenous solution, and stirred for no less than 30 minutes at 70 °C. The solution was cooled to 50 °C (47– 53 °C) over 1 hour and stirred for no less than 1 h, while the temperature was maintained at 50°C (47– 53 °C). The resulting slurry was cooled to -10 °C (-15 to -5°C) linearly over no less than 12 h. The slurry was stirred at -10 °C for no less than 2 h. The solids were isolated via filtration or centrifugation and were washed with a solution of 2-MeTHF (2.25 vol) and IPA (isopropanol) (0.75 vol). The solids were dried under vacuum at 45 ± 5 °C for not less than 6 h to yield (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl).
Example 6: Phase Transfer Catalyst (PTC) Screens for the Synthesis of 5,5- dimethyl-3-methylenepyrrolidin-2-one
[00137] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), PTC (0.05 eq.), and chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added dropwise over 2 min. The reaction mixture was stirred until completion as assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion and assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the
organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. Reaction results are summarized in Table 2.
Table 2
Example 7: Solvent Screens for the Synthesis of 5,5-dimethyl-3- methylenepyrrolidin-2-one
[00138] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), tetrabutylammonium hydroxide (0.12 g, 0.153 mmol, 0.050 eq), chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.), and solvent (2 vol. or 4 vol., as shown in Table 3 below) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion and assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL,
2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC.
Reaction results are summarized in Table 3.
Table 3
Example 8: Base Screens for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin- 2-one
[00139] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), tetrabutylammonium hydroxide (0.12 g, 0.153 mmol, 0.050 eq), and chloroform (0.64 g, 0.4 mL, 5.36 mmol, 1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath, and a solution of an amount wt% sodium hydroxide as shown in Table 4 below in water (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion and assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase is extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as
an internal HPLC standard. Solution yield was assessed by HPLC. Reaction results are summarized in Table 4.
Table 4
Example 9: Various Amounts of Phase Transfer Catalyst (PTC) for the Synthesis of 5,5-dimethyl-3-methylenepyrrolidin-2-one
[00140] In this experiment, various amounts of PTCs were tested as described below: Tetrabutylammonium hydroxide (0.01 eq.), TBAB (0.01 eq.), Tributylmethylammonium chloride (0.01 eq.), Tetrabutylammonium hydroxide (0.02 eq.), TBAB (0.02 eq.), Tributylmethylammonium chloride (0.02 eq.), Tetrabutylammonium hydroxide (0.03 eq.), TBAB (0.03 eq.), Tributylmethylammonium chloride (0.03 eq.).
[00141] 2,2,6,6-tetramethylpiperidin-4-one (500.0 mg, 3.06 mmol, 1.0 eq.), PTC (0.12 g, 0.153 mmol, 0.050 eq), and chloroform (1.75 eq.) were charged into a vial equipped with a magnetic stir bar. The vial was cooled in an ice bath, and a solution of 50 wt% sodium hydroxide (0.98 g, 24.48 mmol, 8.0 eq.) was added drop wise over 2 min. The reaction mixture was stirred until completion, assessed by GC analysis. The reaction mixture was diluted with DCM (2.0 mL, 4.0v) and H2O (3.0 mL, 6.0v). The phases were separated and the aqueous phase was extracted with DCM (1.0 mL, 2.0v). The organic phases were combined and 2 M hydrochloric acid (0.17 g, 2.3 mL, 4.59 mmol, 1.5 eq.) was added. The reaction mixture was stirred until completion, assessed by
HPLC. The aqueous phase was saturated with NaCl and the phases were separated. The aqueous phase was extracted with DCM (1.0 mL, 2.0v) twice, the organic phases were combined, and 50 mg of biphenyl in 2 mL of MeCN was added as an internal HPLC standard. Solution yield was assessed by HPLC. The reaction results are summarized in Table 5.
Table 5
Example 10: Preparation of 2,2,6,6-tetramethylpiperidin-4-one hydrochloride (14•HCl)
[00142] 2,2,6,6-tetramethyl-4-piperidinone (14) (30 g, 193.2 mmol, 1.0 eq) was charged to a 500 mL nitrogen purged three necked round bottomed flask equipped with condenser. IPA (300 mL, 10 vol) was added to the flask and the mixture heated to 60 °C until dissolved.
[00143] To the solution at 60 °C was added 5-6 M HCl in IPA (40 mL, 214.7 mmol, 1.1 eq) over 10 min and the resulting suspension stirred at 60 °C for 30 min then allowed to cool to ambient temperature. The suspension was stirred at ambient temperature overnight, then filtered under vacuum and washed with IPA (3 x 60 mL, 3 x 2 vol). The cream colored solid was dried on the filter under vacuum for 10 min.
[00144] The wet cake was charged to a 1 L nitrogen purged three necked round bottomed flask equipped with condenser. IPA (450 mL, 15 vol) was added to the flask and the suspension heated to 80 °C until dissolved. The mixture was allowed to cool slowly to ambient temperature over 3 h and the resulting suspension stirred overnight at ambient temperature.
[00145] The suspension was filtered under vacuum, washed with IPA (60 mL, 2 vol) and dried on the filter under vacuum for 30 min. The resulting product was dried in a vacuum oven at 40 °C over the weekend to give 2,2,6,6-tetramethylpiperidin-4-one hydrochloride (14•HCl) a white crystalline solid, 21.4 g, 64% yield.
Example 11: Synthesis of (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) from (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S)
[00146] Each reactor was charged with (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF, H2, and the catalyst shown in the below table. The reactor was heated to 200 °C and pressurized to 60 bar, and allowed to react for 12 hours. GC analysis showed that (S)-2,2,4-trimethylpyrrolidine was produced in the columns denoted by“+.”
[00147] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 2% Pt-0.5%Sn/SiO2 catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 130 °C under 80 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (74.8% yield, 96.1% ee).
Alternate synthesis
[00148] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 4% Pt-2%Sn/TiO2catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 200 °C under 50 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (88.5% yield, 29.6% ee).
Alternate synthesis
[00149] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.05 mL/min into a packed bed reactor prepacked with 2% Pt-0.5%Sn/TiO2 catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 20 mL/min. The reaction was carried out at 150 °C under 50 bar pressure with a WHSV (Weigh Hourly Space Velocity) of 0.01-0.02 h-1. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (90.9% yield, 98.0% ee).
Alternate synthesis
[00150] A 2.5% solution of (S)-3,5,5-trimethyl-pyrrolidin-2-one (16S) in THF was flowed at 0.03 mL/min into a packed bed reactor prepacked with 2% Pt-8%Sn/TiO2catalyst immobilized on silica gel. H2 gas was also flowed into the packed bed reactor at 40 mL/min. The reaction was carried out at 180 °C under 55 bar pressure with a residence time of 6 min. The product feed was collected in a batch tank and converted to (S)-2,2,4-trimethylpyrrolidine HCl in batch mode: 36% Hydrochloric acid (1.1 eq.) was added keeping the temperature below 20 °C. Distillation of the solvent, backfilling with isopropyl acetate (4v), was carried out to leave a residual volume of 5v. Karl Fischer analysis < 0.2% w/w H2O. MTBE (methyl tertiary butyl ether) (1v) was added at 20-30 °C and the solids were filtered off under nitrogen at 15-20 °C, washing with isopropyl acetate (1.5v) and drying under vacuum at 40-45 °C to give (S)-2,2,4-trimethylpyrrolidine hydrochloride (17S•HCl) as a white crystalline solid (90.4% yield, 96.8% ee).
Example 12: Preparation of N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3- trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4- trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
Compound 1
I. Preparation of Starting Materials:
A. Synthesis of 3,3,3-Trifluoro-2,2-dimethylpropionic acid (31), morpholine salt:
Step 1: tert-Butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (28)
[00151] A 2L 3-necked round-bottom flask, equipped with a J-Kem thermocouple and an overhead stirrer, was purged with nitrogen for >20 minutes. Hexyllithium solution (2.3 M in hexanes; 1.05 equiv; 0.260 L, 597 mmol) was transferred into the flask via cannula. The flask was then cooled to–65°C in a dry ice/isopropyl alcohol bath and diisopropylamine (1.05 equiv; 0.842 L; 597mmol) was added via an addition funnel, and the internal temperature was maintained at–40 ±5 °C. Once the diisopropylamine addition was complete, tetrahydrofuran (THF) (0.423 L; 6.4 vol) was added to the reactor and the reaction was warmed to room temperature and stirred for 15 minutes. The solution was then cooled to–60 °C and ethyl isobutyrate (1.0 equiv; 0.754 L; 568 mmol) was added dropwise maintaining the temperature below–45 °C. 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) (0.9 equiv; 0.616 L; 511 mmol) was then added dropwise to the reaction flask and the temperature was maintained below–45 °C. In a separate flask, tert-butyldimethylsilyl chloride (TBSCl) (1.05 equiv; 89.9 g; 597 mmol) was dissolved in THF (2.2 vol w.r.t. TBSCl) and then added to the 2L reactor. The internal temperature was maintained at≤–30°C during the addition of the TBSCl solution. The resulting reaction mixture was allowed to warm to room
temperature and stirred overnight under inert atmosphere. The reaction solution was transferred to a 2L one-neck round-bottom flask. Additional THF (50 mL, x 2) was used to rinse and transfer. The solution was concentrated in vacuo to remove most of the THF. Hexanes were added to the concentrated tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (500 mL). The organic phase was washed with three times with water (500 mL x 3), to remove salts. The organic layer was dried over Na2SO4 (100 g). The solution was filtered and the waste cake washed with additional hexanes (100 mL). The resulting hexanes solution of tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was concentrated in vacuo. A quantitative 1H-NMR assay was performed with benzyl benzoate as an internal standard. The quantitative NMR assay indicated that 108.6 grams of tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane (83% yield) was present, and that 1.2 mol% of ethyl isobutyrate relative to tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was also present. The resulting tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane solution was used without further purification for the photochemical reaction of Step 2. Step 2: 3,3,3-Trifluoro-2,2-dimethylpropionic acid (31), morpholine salt
[00152] Stock solution A: The concentrated tert-butyl((1-ethoxy-2-methylprop-1-en-1- yl)oxy)dimethylsilane (198 g; 0.86 mol) was dissolved in acetonitrile (895 g; 1.14 L; 5.8 vol) to give a cloudy, yellow solution that was then filtered. The density of the clear, filtered solution was measured to be 0.81 g/mL and the molar concentration was calculated to be 0.6 M. This is referred to as stock solution A (substrate).
[00153] Stock solution B: The catalyst and reagent solution was prepared by dissolving Ru(bpy)3Cl2 hexahydrate in acetonitrile, followed by adding ethanol and pyrrolidine to give a red-colored solution (density measured: 0.810 g/mL). The molar concentration of the catalyst was calculated to be 0.00172 M. The molar concentration of the solution with respect to EtOH/pyrrolidine was calculated to be ~2.3 M. See Table 6.
Table 6
(i) Photochemical Trifluoromethylation
[00154] CF3I gas was delivered to the reactor directly from the lecture bottle using a regulator and mass flow controller. Stock solutions A and B were pumped at 6.7 g/min and 2.07 g/min, respectively, to mix in a static mixer. The resulting solution was then combined with CF3I in a static mixer. The CF3I was metered into the reactor via a mass flow controller at 2.00 g/min (2 equiv). Liquid chromatography (LC) assay indicated that 1.0% of the tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane was left unreacted. Details of the reaction parameters are shown in the table below. The reaction stream was passed through the 52 mL photoreactor while being irradiated with the 800 W 440-445 LED light source. The first 5 minutes of eluent was discarded. Thereafter the eluent was collected for a total of 3.05 hours. A total of ~2.3 L of solution was collected during the reaction (~1.06 mol). See Table 7.
Table 7
(ii) Saponification & Salt Formation
[00155] The saponication of the crude solution (4.1 L, from 1.60 mol tert-butyl((1-ethoxy-2-methylprop-1-en-1-yl)oxy)dimethylsilane) was carried out in a 5L 4-necked round-bottomed flask in 2 roughly equal size batches using 15 wt% NaOH (aq) (total ~320g NaOH) at 50 °C for 2-4 h. Upon completion of the reaction determined by gas chromatography (GC) analysis, the re-combined batches were cooled to room
temperature and hexanes (500 mL) and toluene (500 mL) were added to give a clear phase separation. The top organic layer was washed with half-brine (1 L) and combined with the first portion of the product-containing aqueous solution (4.5 L). The combined aqueous stream was washed with hexanes (500 mL) and concentrated to 2-3 L to remove a majority of volatile acetonitrile. To the aqueous phase was added concentrated HCl (1 L, 12 N) and the resulting mixture was extracted with hexanes (4 x 1 L). The combined hexanes extracts were washed with half brine (2 x 500 mL) and concentrated to give an oil (216 g). The oil was dissolved in THF (580 mL), and morpholine (120 mL, 1.0 equiv) was added slowly via an addition funnel. Upon completion of addition, the batch was seeded (0.5-1 g) with morpholine salt, and the seeds were held and allowed to thicken over 30 min. Hexanes (1660 mL) were added over ~ 2 h, and the mixture was aged for another 3 h. The batch was filtered, washed with hexanes (~500 mL) in portions and dried under vacuum/dry air flush to give 3,3,3-trifluoro-2,2-dimethylpropionic acid, morpholine salt as a white solid (283 g, 73%).1H NMR (400 MHz, CD3OD) δ 3.84-3.86 (m, 4H), 3.15-3.18 (m, 4H), 1.33 (s, 6H); 19F NMR (376 MHz, CD3OD): δ -75.90 (s, 3F).
B. Synthesis of 3,3,3-Trifluoro-2,2-dimethyl-propan-1-ol (5)
[00156] A 1 L 3 neck round bottom flask was fitted with a mechanical stirrer, a cooling bath, an addition funnel, and a J-Kem temperature probe. The vessel was charged with lithium aluminum hydride (LAH) pellets (6.3 g, 0.1665 mol) under a nitrogen atmosphere. The vessel was then charged with tetrahydrofuran (200 mL) under a nitrogen atmosphere. The mixture was allowed to stir at room temperature for 0.5 hours to allow the pellets to dissolve. The cooling bath was then charged with crushed ice in water and the reaction temperature was lowered to 0 oC. The addition funnel was charged with a solution of 3,3,3-trifluoro-2,2-dimethyl-propanoic acid (20 g, 0.1281 mol) in tetrahydrofuran (60 mL) and the clear pale yellow solution was added drop wise over 1 hour. After the addition was complete the mixture was allowed to slowly warm to room temperature and stirring was continued for 24 hours. The suspension was cooled to 0 oC with a crushed ice-water in the cooling bath and then quenched by the very slow and drop wise addition of water (6.3 ml), followed by sodium hydroxide solution (15 weight %; 6.3 mL) and then finally with water (18.9 mL). The reaction temperature of the resulting white suspension was recorded at 5 oC. The suspension was stirred at ~5 oC for 30 minutes and then filtered through a 20 mm layer of Celite. The filter cake was washed with tetrahydrofuran (2 x 100 mL). The filtrate was dried over sodium sulfate (150 g) and then filtered. The filtrate was concentrated under reduced pressure to provide a clear colorless oil (15 g) containing a mixture of the product 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol in THF (73 % weight of product ~10.95g, and 27 wt.% THF as determined by 1H-NMR). The distillate from the rotary evaporation was distilled at atmospheric pressure using a 30 cm Vigreux column to provide 8.75 g of a residue containing 60 % weight of THF and 40 % weight of product (~3.5 g), which corresponds to 14.45 g (79% yield).1H NMR (400 MHz, DMSO-d6) δ 4.99 (t, J = 5.7 Hz, 1H), 3.38 (dd, J = 5.8, 0.9 Hz, 2H), 1.04 (d, J = 0.9 Hz, 6H).
C. Synthesis of tert-Butyl 3-oxo-2,3-dihydro-1H-pyrazole-1-carboxylate (22)
[00157] A 50L Syrris controlled reactor was started and the jacket was set to 20 °C, stirring at 150 rpm, reflux condenser (10 °C) and nitrogen purge. MeOH (2.860 L) and methyl (E)-3-methoxyprop-2-enoate (2.643 kg, 22.76 mol) were added and the reactor was capped. The reaction was heated to an internal temperature of 40 °C and the system was set to hold jacket temp at 40 °C. Hydrazine hydrate (1300 g of 55 %w/w, 22.31 mol) was added portion wise via addition funnel over 30 min. The reaction was heated to 60 ^C for 1 h. The reaction mixture was cooled to 20 ^C and triethylamine (2.483 kg, 3.420 L, 24.54 mol) was added portion wise, maintaining reaction temp <30 °C. A solution of Boc anhydride (di-tert-butyl dicarbonate) (4.967 kg, 5.228 L, 22.76 mol) in MeOH (2.860 L) was added portion wise maintaining temperature <45 °C. The reaction mixture was stirred at 20 ^C for 16 h. The reaction solution was partially concentrated to remove MeOH, resulting in a clear light amber oil. The resulting oil was transferred to the 50L reactor, stirred and added water (7.150 L) and heptane (7.150 L). The additions caused a small amount of the product to precipitate. The aqueous layer was drained into a clean container and the interface and heptane layer were filtered to separate the solid (product). The aqueous layer was transferred back to the reactor, and the collected solid was placed back into the reactor and mixed with the aqueous layer. A dropping funnel was added to the reactor and loaded with acetic acid (1.474 kg, 1.396 L, 24.54 mol), then began dropwise addition of acid. The jacket was set to 0 °C to absorb the quench exotherm. After addition (pH=5), the reaction mixture was stirred for 1 h. The solid was collected by filtration and washed with water (7.150 L) and washed a second time with water (3.575 L) and pulled dry. The crystalline solid was scooped out of the filter into a 20L rotovap bulb and heptane (7.150 L) was added. The mixture was slurried at 45 °C for 30 mins, and then 1-2 volumes of solvent was distilled off. The slurry in the rotovap flask was filtered and the solids washed with heptane (3.575 L) and pulled dry. The solid was further dried in vacuo (50 °C, 15 mbar) to give tert-butyl 5-oxo-1H-pyrazole-2-carboxylate (2921 g, 71%) as coarse solid.1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 7.98 (d, J = 2.9 Hz, 1H), 5.90 (d, J = 2.9 Hz, 1H), 1.54 (s, 9H).
II. Preparation of Compound I
Step A: tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (23)
[00158] A mixture of 3,3,3-trifluoro-2,2-dimethyl-propan-1-ol (10 g, 70.36 mmol) and tert-butyl 3-hydroxypyrazole-1-carboxylate (12.96 g, 70.36 mmol) in toluene (130 mL) was treated with triphenyl phosphine (20.30 g, 77.40 mmol) followed by isopropyl N-
isopropoxycarbonyliminocarbamate (14.99 mL, 77.40 mmol) and the mixture was stirred at 110 °C for 16 hours. The yellow solution was concentrated under reduced pressure, diluted with heptane (100mL) and the precipitated triphenylphosphine oxide was removed by filtration and washed with heptane/toluene 4:1 (100mL). The yellow filtrate was evaporated and the residue purified by silica gel chromatography with a linear gradient of ethyl acetate in hexane (0-40%) to give tert-butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (12.3 g, 57%) as an off white solid. ESI-MS m/z calc.308.13477, found 309.0 (M+1) +; Retention time: 1.84 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 3.0 Hz, 1H), 6.15 (d, J = 3.0 Hz, 1H), 4.18 (s, 2H), 1.55 (s, 9H), 1.21 (s, 6H).
Step B: 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (7)
[00159] tert-Butyl 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazole-1-carboxylate (13.5 g, 43.79 mmol) was treated with 4 M hydrogen chloride in dioxane (54.75 mL, 219.0 mmol) and the mixture was stirred at 45 °C for 1 hour. The reaction mixture was evaporated to dryness and the residue was extracted with 1 M aqueous NaOH (100ml) and methyl tert-butyl ether (100ml), washed with brine (50ml) and extracted with methyl tert-butyl ether (50ml). The combined organic phases were dried, filtered and evaporated to give 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 96%) as an off white solid. ESI-MS m/z calc.208.08235, found 209.0 (M+1) +; Retention time: 1.22 minutes.1H NMR (400 MHz, DMSO-d6) δ 11.91 (s, 1H), 7.52 (d, J = 2.2 Hz, 1H), 5.69 (t, J = 2.3 Hz, 1H), 4.06 (s, 2H), 1.19 (s, 6H).
Step C: tert-Butyl 2,6-dichloropyridine-3-carboxylate (25)
[00160] A solution of 2,6-dichloropyridine-3-carboxylic acid (10 g, 52.08 mmol) in THF (210 mL) was treated successively with di-tert-butyl dicarbonate (17 g, 77.89 mmol) and 4-(dimethylamino)pyridine (3.2 g, 26.19 mmol) and left to stir overnight at room temperature. At this point, HCl 1N (400 mL) was added and the mixture was stirred vigorously for about 10 minutes. The product was extracted with ethyl acetate (2x300mL) and the combined organics layers were washed with water (300 mL) and brine (150 mL) and dried over sodium sulfate and concentrated under reduced pressure to give 12.94 g (96% yield) of tert-butyl 2,6-dichloropyridine-3-carboxylate as a colorless oil. ESI-MS m/z calc.247.01668, found 248.1 (M+1) +; Retention time: 2.27 minutes.1H NMR (300 MHz, CDCl3) ppm 1.60 (s, 9H), 7.30 (d, J=7.9 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H).
Step D: tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (26)
[00161] To a solution of tert-butyl 2,6-dichloropyridine-3-carboxylate (10.4 g, 41.9 mmol) and 3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)-1H-pyrazole (9.0 g, 41.93 mmol) in DMF (110 mL) were added potassium carbonate (7.53 g, 54.5 mmol) and 1,4-diazabicyclo[2.2.2]octane (706 mg, 6.29 mmol) and the mixture was stirred at room temperature for 16 hours. The cream suspension was cooled in a cold water bath and cold water (130 mL) was slowly added. The thick suspension was stirred at room temperature for 1 hour, filtered and washed with plenty of water to give tert-butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 99%) as an off white solid. ESI-MS m/z calc.419.12234, found 420.0 (M+1) +; Retention time: 2.36 minutes.1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 2.9 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 2.9 Hz, 1H), 4.27 (s, 2H), 1.57 (s, 9H), 1.24 (s, 6H).
Step E: 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (10)
[00162] tert-Butyl 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylate (17.6 g, 40.25 mmol) was suspended in isopropanol (85 mL) treated with hydrochloric acid (34 mL of 6 M, 201 mmol) and heated to reflux for 3 hours (went almost complete into solution at reflux and started to precipitate again). The suspension was diluted with water (51 mL) at reflux and left to cool to room temperature under stirring for 2.5 h. The solid was collected by filtration, washed with
isopropanol/water 1:1 (50mL), plenty of water and dried in a drying cabinet under vacuum at 45-50 °C with a nitrogen bleed overnight to give 2-chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (13.7 g, 91%) as an off white solid. ESI-MS m/z calc.363.05975, found 364.0 (M+1) +; Retention time: 1.79 minutes. 1H NMR (400 MHz, DMSO-d6) δ 13.61 (s, 1H), 8.44 (d, J = 2.9 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 6.25 (d, J = 2.9 Hz, 1H), 4.28 (s, 2H), 1.24 (s, 6H).
Step F: 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (13)
[00163] 2-Chloro-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxylic acid (100 mg, 0.2667 mmol) and CDI (512 mg, 3.158 mmol) were combined in THF (582.0 µL) and the mixture was stirred at room temperature. Meanwhile, 1,3-dimethylpyrazole-4-sulfonyl chloride (62 mg, 0.3185 mmol) was combined with ammonia (in methanol) in a separate vial, instantly forming a white solid. After stirring for an additional 20 min, the volatiles were removed by evaporation, and 1 mL of dichloromethane was added to the solid residue, and was also evaporated. DBU (100 µL, 0.6687 mmol) was then added and the mixture stirred at 60 °C for 5 minutes, followed by addition of THF (1 mL) which was subsequently evaporated. The contents of the vial containing the CDI activated carboxylic acid in THF were then added to the vial containing the newly formed sulfonamide and DBU, and the reaction mixture was stirred for 4 hours at room temperature. The reaction mixture was diluted with 10 mL of ethyl acetate, and washed with 10 mL solution of citric acid (1 M). The aqueous layer was extracted with ethyl acetate (2x 10 mL) and the combined organics were washed with brine, dried over sodium sulfate, and concentrated to give 2-chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide as white solid (137 mg, 99%) that was used in the next step without further purification. ESI-MS m/z calc.520.09076, found 521.1 (M+1) +;
Retention time: 0.68 minutes.
Step G: N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (Compound 1)
[00164] 2-Chloro-N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]pyridine-3-carboxamide (137 mg, 0.2630 mmol), (4S)-2,2,4-trimethylpyrrolidine (Hydrochloride salt) (118 mg, 0.7884 mmol), and potassium carbonate (219 mg, 1.585 mmol) were combined in DMSO (685.0 µL) and the mixture was heated at 130 ^C for 16 hours. The reaction was cooled to room temperature, and 1 mL of water was added. After stirring for 15 minutes, the contents of the vial were allowed to settle, and the liquid portion was removed via pipet and the remaining solids were dissolved with 20 mL of ethyl acetate and were washed with 1 M citric acid (15 mL). The layers were separated and the aqueous layer was extracted two additional times with 15 mL of ethyl acetate. The organics were combined, washed with brine, dried over sodium sulfate and concentrated. The resulting solid was further purified by silica gel chromatography eluting with a gradient of methanol in dichloromethane (0-10%) to give N-(1,3-dimethylpyrazol-4-yl)sulfonyl-6-[3-(3,3,3-trifluoro-2,2-dimethyl-propoxy)pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide (72 mg, 41%) as a white solid. ESI-MS m/z calc.597.2345, found 598.3 (M+1) +; Retention time: 2.1 minutes.1H NMR (400 MHz, DMSO) δ 12.36 (s, 1H), 8.37 (s, 1H), 8.22 (d, J = 2.8 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 6.93 (d, J = 8.2 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 4.23 (s, 2H), 3.81 (s, 3H), 2.56 (d, J = 10.4 Hz, 1H), 2.41 (t, J = 8.7 Hz, 1H), 2.32 (s, 3H), 2.18 (dd, J = 12.4, 6.1 Hz, 1H), 1.87 (dd, J = 11.7, 5.5 Hz, 1H), 1.55 (d, J = 11.2 Hz, 6H), 1.42 (t, J = 12.0 Hz, 1H), 1.23 (s, 6H), 0.81 (d, J = 6.2 Hz, 3H).
///////////VX-445, Elexacaftor, VX445, エレクサカフトル , PHASE 3, CYSTIC FIBRIOSIS, VX 445
C[C@@H]1CN(c2nc(ccc2C(=O)NS(=O)(=O)c3cn(C)nc3C)n4ccc(OCC(C)(C)C(F)(F)F)n4)C(C)(C)C1
CC1CC(N(C1)C2=C(C=CC(=N2)N3C=CC(=N3)OCC(C)(C)C(F)(F)F)C(=O)NS(=O)(=O)C4=CN(N=C4C)C)(C)C
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY

