
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

CLARITHROMYCIN


Clarithromycin
Synonyms:A-56268, TE-031, 6-O-methylerythromycin, ATC:J01FA09Use:macrolide antibioticChemical name:6-O-methylerythromycinFormula:C38H69NO13
- MW:747.96 g/mol
- CAS-RN:81103-11-9
- 81103-11-9
klacid XL / Klaricid XL / Macladin / Naxy / Veclam / Zeclar
(3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-6-{[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy}-14-ethyl-12,13-dihydroxy-4-{[(2R,4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy}-7-methoxy-3,5,7,9,11,13-hexamethyl-1-oxacyclotetradecane-2,10-dione
Synthesis Reference
Jih-Hua Liu, David A. Riley, “Preparation of crystal form II of clarithromycin.” U.S. Patent US5844105, issued May, 1997. US5844105
NEW DRUG APPROVALS
ONE TIME
$10.00
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Clarithromycin citrate | 16K08R7NG0 | 848130-51-8 | MDRWXDRMSKEMRE-AZFLODHXSA-N |
ClarithromycinCAS Registry Number: 81103-11-9CAS Name: 6-O-MethylerythromycinManufacturers’ Codes: A-56268; TE-031Trademarks: Biaxin (Abbott); Clarosip (Grñenthal); Clathromycin (Taisho); Cyllind (Abbott); Klacid (Abbott); Klaricid (Abbott); Macladin (Guidotti); Naxy (Sanofi Winthrop); Veclam (Zambon); Zeclar (Abbott)Molecular Formula: C38H69NO13Molecular Weight: 747.95Percent Composition: C 61.02%, H 9.30%, N 1.87%, O 27.81%Literature References: Semisynthetic macrolide antibiotic; derivative of erythromycin, q.v. Prepn: Y. Watanabe et al.,EP41355; eidem,US4331803 (1981, 1982 both to Taisho); and in vitro antibacterial activity: S. Morimoto et al.,J. Antibiot.37, 187 (1984). In vitro and in vivo antibacterial activity: P. B. Fernandes et al.,Antimicrob. Agents Chemother.30, 865 (1986). Comparative antibacterial spectrum in vitro: C. Benson et al.,Eur. J. Clin. Microbiol.6, 173 (1987); H. M. Wexler, S. M. Finegold, ibid. 492. HPLC determn in biological fluids: D. Croteau et al.,J. Chromatogr.419, 205 (1987); in plasma: H. Amini, A. Ahmadiani, J. Chromatogr. B817, 193 (2005). Acute toxicity study: S. Abe et al.,Chemotherapy (Tokyo)36, Suppl. 3, 274 (1988). Symposium on pharmacology and comparative clinical studies: J. Antimicrob. Chemother.27, Suppl. A, 1-124 (1991). Comprehensive description: I. I. Salem, Anal. Profiles Drug Subs. Excip.24, 45-85, (1996).Properties: Colorless needles from chloroform + diisopropyl ether (1:2), mp 217-220° (dec). Also reported as crystals from ethanol, mp 222-225° (Morimoto). uv max (CHCl3): 288 nm (e 27.9). uv max (CHCl3): 240, 288 nm; (methanol): 211, 288 nm. [a]D24 -90.4° (c = 1 in CHCl3). Stable at acidic pH. LD50 in male, female mice, male, female rats (mg/kg): 2740, 2700, 3470, 2700 orally, 1030, 850, 669, 753 i.p., >5000 all s.c. (Abe).Melting point: mp 217-220° (dec); mp 222-225° (Morimoto)Optical Rotation: [a]D24 -90.4° (c = 1 in CHCl3)Absorption maximum: uv max (CHCl3): 288 nm (e 27.9). uv max (CHCl3): 240, 288 nmToxicity data: LD50 in male, female mice, male, female rats (mg/kg): 2740, 2700, 3470, 2700 orally, 1030, 850, 669, 753 i.p., >5000 all s.c. (Abe)Therap-Cat: Antibacterial.Keywords: Antibacterial (Antibiotics); Macrolides.
Clarithromycin, a semisynthetic macrolide antibiotic derived from erythromycin, inhibits bacterial protein synthesis by binding to the bacterial 50S ribosomal subunit. Binding inhibits peptidyl transferase activity and interferes with amino acid translocation during the translation and protein assembly process. Clarithromycin may be bacteriostatic or bactericidal depending on the organism and drug concentration.
Clarithromycin, sold under the brand name Biaxin among others, is an antibiotic used to treat various bacterial infections.[2] This includes strep throat, pneumonia, skin infections, H. pylori infection, and Lyme disease, among others.[2] Clarithromycin can be taken by mouth as a pill or liquid.[2]
Common side effects include nausea, vomiting, headaches, and diarrhea.[2] Severe allergic reactions are rare.[2] Liver problems have been reported.[2] It may cause harm if taken during pregnancy.[2] It is in the macrolide class and works by decreasing protein production of some bacteria.[2]
Clarithromycin was developed in 1980 and approved for medical use in 1990.[3][4] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[5] Clarithromycin is available as a generic medication.[2] It is made from erythromycin and is chemically known as 6-O-methylerythromycin.[6]
Medical uses
Clarithromycin is primarily used to treat a number of bacterial infections including pneumonia, Helicobacter pylori, and as an alternative to penicillin in strep throat.[2] Other uses include cat scratch disease and other infections due to bartonella, cryptosporidiosis, as a second line agent in Lyme disease and toxoplasmosis.[2] It may also be used to prevent bacterial endocarditis in those who cannot take penicillin.[2] It is effective against upper and lower respiratory tract infections, skin and soft tissue infections and helicobacter pylori infections associated with duodenal ulcers.
Spectrum of bacterial susceptibility
Staphylococcus aureusAerobic Gram-positive bacteria
Aerobic Gram-negative bacteria
- Haemophilus parainfluenzae
- Haemophilus influenzae
- Moraxella catarrhalis
Helicobacter
Mycobacteria
Mycobacterium avium complex consisting of:
Other bacteria
Safety and effectiveness of clarithromycin in treating clinical infections due to the following bacteria have not been established in adequate and well-controlled clinical trials:[7]
Aerobic Gram-positive bacteria
- Streptococcus agalactiae
- Streptococcus (Groups C, F, G)
- Viridans group streptococci
Aerobic Gram-negative bacteria
Anaerobic Gram-positive bacteria
- Clostridium perfringens
- Peptococcus Niger
- Cutibacterium acnes
Anaerobic Gram-negative bacteria
- Prevotella melaninogenica (formerly Bacteroides melaninogenicus)
Contraindications
- Clarithromycin should not be taken by people who are allergic to other macrolides or inactive ingredients in the tablets, including microcrystalline cellulose, croscarmelose sodium, magnesium stearate, and povidone
- Clarithromycin should not be used by people with a history of cholestatic jaundice and/or liver dysfunction associated with prior clarithromycin use.[7]
- Clarithromycin should not be used in the setting of hypokalaemia (low blood potassium)
- Use of clarithromycin with the following medications: cisapride, pimozide, astemizole, terfenadine, ergotamine, ticagrelor, ranolazine or dihydroergotamine is not recommended.[7]
- It should not be used with colchicine in people with kidney or liver impairment.[7]
- Concomitant use with cholesterol medications such as lovastatin or simvastatin.[7]
- Hypersensitivity to clarithromycin or any component of the product, erythromycin, or any macrolide antibiotics.[7]
- QT prolongation or ventricular cardiac arrhythmias, including torsade de pointes.[7]
Side effects
The most common side effects are gastrointestinal: diarrhea (3%), nausea (3%), abdominal pain (3%), and vomiting (6%). It also can cause headaches, insomnia, and abnormal liver function tests. Allergic reactions include rashes and anaphylaxis. Less common side effects (<1%) include extreme irritability, hallucinations (auditory and visual), dizziness/motion sickness, and alteration in senses of smell and taste, including a metallic taste. Dry mouth, panic attacks, and nightmares have also been reported, albeit less frequently.[8]
Cardiac
In February 2018, the FDA issued a Safety Communication warning with respect to an increased risk for heart problems or death with the use of clarithromycin, and has recommended that alternative antibiotics be considered in those with heart disease.[9]
Clarithromycin can lead to a prolonged QT interval. In patients with long QT syndrome, cardiac disease, or patients taking other QT-prolonging medications, this can increase risk for life-threatening arrhythmias.[10]
In one trial, the use of short-term clarithromycin treatment was correlated with an increased incidence of deaths classified as sudden cardiac deaths in stable coronary heart disease patients not using statins.[11] Some case reports suspect it of causing liver disease.[12]
Liver and kidney
Clarithromycin has been known to cause jaundice, cirrhosis, and kidney problems, including kidney failure.[citation needed]
Central nervous system
Common adverse effects of clarithromycin in the central nervous system include dizziness, headaches. Rarely, it can cause ototoxicity, delirium and mania.
Infection
A risk of oral candidiasis and vaginal candidiasis, due to the elimination of the yeast’s natural bacterial competitors by the antibiotic, has also been noted.
Pregnancy and breastfeeding
Clarithromycin should not be used in pregnant women except in situations where no alternative therapy is appropriate.[7] Clarithromycin can cause potential hazard to the fetus hence should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.[7] For lactating mothers it is not known whether clarithromycin is excreted in human milk.[7]
Interactions
Clarithromycin inhibits a liver enzyme, CYP3A4, involved in the metabolism of many other commonly prescribed drugs. Taking clarithromycin with other medications that are metabolized by CYP3A4 may lead to unexpected increases or decreases in drug levels.
A few of the common interactions are listed below.
Colchicine
Clarithromycin has been observed to have a dangerous interaction with colchicine as the result of inhibition of CYP3A4 metabolism and P-glycoprotein transport. Combining these two drugs may lead to fatal colchicine toxicity, particularly in people with chronic kidney disease.[7]
Statins
Taking clarithromycin concurrently with certain statins (a class of drugs used to reduce blood serum cholesterol levels) increases the risk of side effects, such as muscle aches and muscle break down (rhabdomyolysis).[13]
Calcium channel blockers
Concurrent therapy with calcium channel blocker may increase risk of low blood pressure, kidney failure, and death, compared to pairing calcium channel blockers with azithromycin, a drug similar to clarithromycin but without CYP3A4 inhibition.[14] Administration of clarithromycin in combination with verapamil have been observed to cause low blood pressure, low heart rate, and lactic acidosis.[7]
Carbamazepine
Clarithromycin may double the level of carbamazepine in the body by reducing its clearance, which may lead to toxic symptoms of carbamazepine, such as double vision, loss of voluntary body movement, nausea, as well as hyponatremia.[15]
HIV medications
Depending on the combination of medications, clarithromycin therapy could be contraindicated, require changing doses of some medications, or be acceptable without dose adjustments.[16] For example, clarithromycin may lead to decreased zidovudine concentrations.[17]
Mechanism of action
Clarithromycin prevents bacteria from multiplying by acting as a protein synthesis inhibitor. It binds to 23S rRNA, a component of the 50S subunit of the bacterial ribosome, thus inhibiting the translation of peptides.[citation needed]
Pharmacokinetics
MetabolismUnlike erythromycin, clarithromycin is acid-stable, so can be taken orally without having to be protected from gastric acids. It is readily absorbed, and diffuses into most tissues and phagocytes. Due to the high concentration in phagocytes, clarithromycin is actively transported to the site of infection. During active phagocytosis, large concentrations of clarithromycin are released; its concentration in the tissues can be over 10 times higher than in plasma. Highest concentrations are found in liver, lung tissue, and stool.
Clarithromycin has a fairly rapid first-pass metabolism in the liver. Its major metabolites include an inactive metabolite, N-desmethylclarithromycin, and an active metabolite, 14-(R)-hydroxyclarithromycin. Compared to clarithromycin, 14-(R)-hydroxyclarithromycin is less potent against mycobacterial tuberculosis and the Mycobacterium avium complex. Clarithromycin (20%-40%) and its active metabolite (10%-15%) are excreted in urine. Of all the drugs in its class, clarithromycin has the best bioavailability at 50%, which makes it amenable to oral administration. Its elimination half-life is about 3 to 4 hours with 250 mg administered every 12 h, but increased to 5 to 7 h with 500 mg administered every 8 to 12 h. With any of these dosing regimens, the steady-state concentration of this metabolite is generally attained within 3 to 4 days.[18]
History
Clarithromycin was invented by researchers at the Japanese drug company Taisho Pharmaceutical in 1980.[3] The product emerged through efforts to develop a version of the antibiotic erythromycin that did not experience acid instability in the digestive tract, causing side effects, such as nausea and stomachache. Taisho filed for patent protection for the drug around 1980 and subsequently introduced a branded version of its drug, called Clarith, to the Japanese market in 1991. In 1985, Taisho partnered with the American company Abbott Laboratories for the international rights, and Abbott also gained FDA approval for Biaxin in October 1991. The drug went generic in Europe in 2004 and in the US in mid-2005.
Society and culture

A pack of Clarithromycin tablets manufactured by Taisho Pharmaceutical
Available forms
Clarithromycin is available as a generic medication.[2] In the United States, clarithromycin is available as immediate release tablets, extended release tablets, and granules for oral suspension.[2]
Brand names
Clarithromycin is available under several brand names in many different countries, for example Biaxin, Crixan, Claritron, Clarihexal, Clacid, Claritt, Clacee, Clarac, Clariwin, Claripen, Clarem, Claridar, Cloff, Fromilid, Infex, Kalixocin, Karicin, Klaricid, Klaridex, Klacid, Klaram, Klabax, MegaKlar, Monoclar, Resclar, Rithmo, Truclar, Vikrol and Zeclar.
Manufacturers
In the UK the drug product is manufactured in generic form by a number of manufacturers including Somex Pharma, Ranbaxy, Aptil and Sandoz.
SYN
CN 109705180

SYN
Indian Pat. Appl., 2014DE00731, 31 Aug 2016

SYN
Heterocycles, 31(12), 2121-4; 1990

SYN
https://patents.google.com/patent/WO2006064299A1/enErythromycin A is known to be a useful macrolide antibiotic having a strong activity against Gram-positive bacteria, this compound has an undesirable property that it loses rapidly the antibacterial activity by the acid in stomach when administered orally, where- upon its blood concentration remains at a low level. 6-0-Alkyl derivatives of Erythromycin- A are well known as an useful antibacterial agents. 6-O-Methyl-Erythromycin-A (Clarithromycin) and a pharmaceutically acceptable salt is a potent macrolide antibiotic as reported in US Patent No. 4,331 ,803. Clarithromycin is stable in acidic medium and also remarkable in vivo activity and has a strong antibacterial property against Gram-positive bacteria compared to Erythromycin- A. This compound shows excellent effect for the treatment of infections by oral administration.A number of synthetic processes have been reported for preparing 6-O-alkyl erythromycin. US Patent No. 4,331 ,803 discloses a method for the preparation of Clarithromycin by methylating 6-OH group of 2′-O-3′-N-benzyloxycarbonyl erythromycinFormula (III)

21,3′-O-Protected ErythromycinMethylation of 6-OH group of the 2′,3′-benzyloxycarbonyl erythromycin was carried out using methyl iodide in the presence of a suitable base in a solvent. Clarithromycin was obtained from the compound after removing benzyloxycarbonyl group by hydrogenolysis and then subjecting to the reductive methylation in the presence of excess amount of farmaldehyde. Clarithromycin can also be synthesized by the methylation of 6-OH position of Erythromycin-A-9-OximeFormula (II)

Erythromycin-9-OximeSynthesis of Clarithromycin using 9-oxime or its derivatives are well reported in US Patent Nos. 5,274,085; 4,680,386; 4,668,776; 4,670,549 and 4,672,109. In case of Erythromycin-9-Oxime derivatives, the oxime is protected before methylation step with 2- alkenyl group (US Patent Nos. 4,670,549; 4,668,776) or benzyl group (US Patent Nos. 4,680,386 and 4,670,549). However, it has been reported (Ref. Journal of Antibiotics 46, No. 6, Page No. 647, year 1993) that when the Erythromycin-A-9-Oxime is protected by trimethylsilyl group, which is very unstable under basic condition pose potential impurities formation during methylation. There are some methods reported in US Patent Nos., e.g. , 4,680,386; 4,670,549 and US Patent No. 4,311,803 for the synthesis of Clarithromycin by using chlorobenzyloxycarbonyl group for protection at 2′ and 3′ function of of Erythromycin-A-9-Oxime derivatives.For the protection of 2′-OH group (US Patent No. 4,311 ,803) requires large amounts of benzyl chloroformate which poses problems in handling because of its severe irritating and toxic properties. This protection step also leads to the formation of 3′ -N- demethylation, which requires an additional re-methylation step. The de-protection of chlorobenzyloxy carbonyl group leads to the formation of undesired side products. In earlier reported processes, e.g. , US Patent No. 4,990,602; EP 0,272,110 Al where the methylation has been done on Erythromycin-A-9-Oxime derivatives by the protection of 2′ and 4″ hydroxyl groups using DMSO and THF as a solvent at 0° to 50C or at room temperature, smooth methylation takes place with less side product formation. However, by using the above methylation processes the formation of 6, 11-O-dimethyl erythromycin- A (Compound- A) is always more than 1.0 % in Clarithromycin. Hence, there is a need for an efficient methylation process for the production of Clarithromycin with lesser amount of 6,11-O-dimethyl erythromycin-A than reported previously.




EXAMPLE 1Erythromycin-A-9-OximeTo a solution of 201 Ltr water in 561 Kg isopropyl alcohol is added 282 Kg (4057 mol) of hydroxyl amine hydrochloride under stirring and the reaction mixture is brought to 10 to 200C. Caustic flakes (162 Kg, 4050 mol) is added slowly to the reaction mixture by keeping temperature between 10° to 200C. After 15 minutes of completion of addition, pH of reaction mixture is adjusted to 6.5 to 7.0 by the slow addition of glacial acetic acid (96 Ltr, 100.8 Kg, 1678.6 mole). To the stirred reaction mass is added 300 Kg (408.8 mole) of Erythromycin-A base and reaction mixture is stirred at 55° C for 28 hours. After completion of the reaction, mixture is brought to ambient temperature and to it a mixture of ammonia solution (270 Kg) and water (600 Ltr) is added within 1 hour followed by 3000 Ltr of fresh water in next two hours and stirred the reaction mass for further 1 hour. White solid product obtained is centrifuged, wet cake is washed with water and dried at 6O0C for 12 hours to give 270 Kg of erythromycin-A Oxime. Melting point = 156° to 158°C.EXAMPLE 22′,4″-O-Bis(trimethylsilyl)-erythro?nycin-A-9[O-(l-methoxy-l-methyl ethyl)oximeTo a solution of 80 Kg (106.8 mole) of Erythromycin-A-9-Oxime in 400 Ltr of dichloromethane is added 38.50 Kg (534 mole) of 2-methoxy propene at 100C temperature 19.25 Kg (166.6 mole) of pyridine hydrochloride is added under stirring and the reaction mixture is stirred at 8 to 12° C for 6 hours then to it is added 19.30 Kg (119.5 mole) of HMDS and stirring is continued for 12 to 15 hours at 15° to 18°C temperature. After completion of reaction, 400 Ltr of saturated aqueous sodium carbonate solution is added and the mixture is stirred thoroughly at room temperature. Aqueous layer is further extracted with fresh DCM (100 Ltr). Both DCM extracts are mixed together and washed with water (200 Ltr) followed by brine solution (200 Ltr). The solvent is evaporated under reduced pressure. To the obtained crude solid mass is charged isopropyl alcohol (240 Ltr) and distilled out 80 Ltr of isopropyl alcohol. To the reaction mixture 160 Ltr of water is charged and stirring continued at room temperature for 1 hour. Solid crystalline product obtained is centrifuged and dried at 60° to 650C for 8 hours under vacuum to give 85 Kg of title compound. Melting point = 125° to 126°C. HPLC Purity = More than 90 % .EXAMPLE 3Clarithromycin-9- OximeTo a solution of 80 Kg (82.98 mole) of 2′,4″-O-bis(trimethylsilyl)-erythromycin-A- 9-[O-(l-methoxy methyl ethyl)Oxime] in 1200 Ltr of a mixture of dimethyl sulfoxide and diethylether (1 : 1) are added methyl iodide (20.62 Kg, 145.2 mole) and 6.48 Kg (98.35 mole) of 85 % potassium hydroxide powder and the reaction mixture is stirred for 90 minutes at room temperature. To the reaction mass is added 53 Kg of 40 % dimethylamine solution and stirring is continued for 1 hour diethylether layer is separated and DMSO layer is further extracted with fresh diethylether (200 Ltr). Combined ether layer is washed with water and concentrated in vacuum. To the obtained semi solid mass 330 Ltr of isopropyl alcohol is charged and then distilled out 165 Ltr of isopropyl alcohol. To the obtained slurry 165 Ltr of water and 21.71 Kg formic acid (99%) are added and the mixture is stirred at room temperature for 30 minutes. 622 Ltr of water is added to the reaction mixture and pH is adjusted between 10.5 and 11.5 with 25 % aqueous sodium hydroxide solution. Solid compound obtained is centrifuged and wet cake is kept as such for further reaction on the basis of moisture content. Wet weight = 95 Kg, Moisture Content = 33 %, Dried weight = 62 KgEXAMPLE 46-O-Methyl erythromycin- A (Clarithromycin)62 Kg of 6-O-Methyl erythromycin-9-Oxime is charged into a mixture of 434 Ltr of isopropyl alcohol and water (1: 1) and to it is added 38.80 Kg of sodium metabisulphite (203 mole) and then the mixture is heated to reflux for 6 to 8 hours. To the reaction mixture is charged water (620 Ltr) at ambient temperature and then the mixture is adjusted to pH about 10.5 to 11.5 by adding 25% aqueous sodium hydroxide solution and stirred for further 1 hour. White solid crude product is centrifuged, washed with water (300 Ltr), dried at 65° to 750C for 8 hours to give 40 Kg of crude Clarithromycin which on re- crystallization with chloroform isopropyl alcohol mixture provided 20 Kg of Clarithromycin (Form II).
SYNEP 0041355; US 4331803J Antibiot 1984,37(2),187-189
| EP 0147062 |
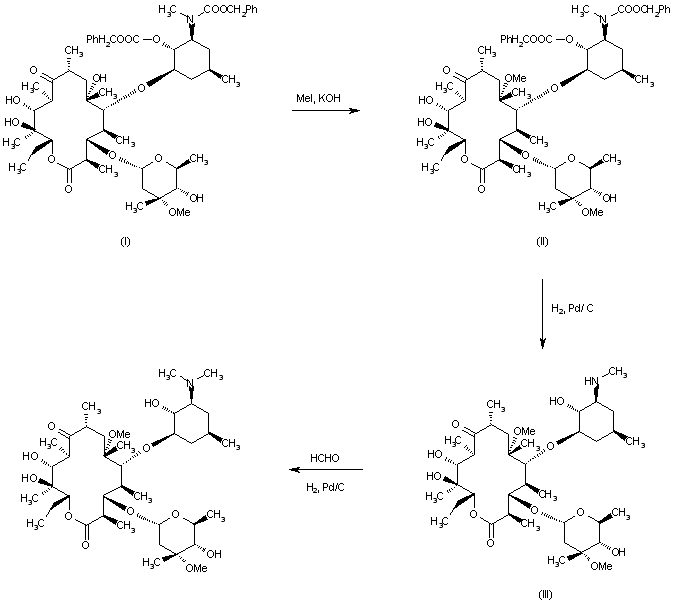
The methylation of 2′-O,N-bis(benzyloxycarbonyl)-N-demethylerythromycin A (I) with methyl iodide and KOH or NaHI in DMSO-dimethoxyethane gives the 6-O-methyl derivative (II), which is deprotected by hydrogenation with H2 over Pd/C in ethanol acetic acid affording 6-O-methyl-N-demethylerythromycin A (III). Finally, this compound is methylated with formaldehyde under reductive conditions (H2-Pd/C) in ethanol/acetic acid.
CLIP


2 Clarithromycin. Initial attempts of making clarithromycin (2) from erythromycin (1) by methylation of 8 gave approximately equal amounts of 2 and 10 by methylation at O-6 and O-11, respectively (Scheme 2, route A).[28–30] This allowed 2 to be obtained in approximately 39% yield, but it contained a small impurity of di-O-methylated 9. To improve the yields and obtain 2 in pure form, other alternatives were explored. During methylation of analogues of 8 it was observed that the conformation of the macrocyclic core plays an important role for the regioselectivity of the O-methylation.[31] As oximes are readilyhydrolysed and may have different conformations than ketone 8, oximes 11 and 13 were subjected to methylation. Interestingly, methylation of 13, but not of 11, proved to be highly selective for O-6 and provided 14 in 86% yield (Scheme2 route B); an observation which supports that 13 populates different conformations compared to 8 and 11 under the methylation conditions.[31] Compound 14 was then hydrogenated with Pd/C to deprotect the two benzyloxycarbonyl groups and the 2-chlorobenzyl group. The N-methylamine was then methylated by reductive amination and the oxime was deprotected by hydrolysis to provide clarithromycin (2). This procedure was further modified for process-scale synthesis so that clarithromycin (2) could be obtained in 70% yield starting from oxime 11 without the isolation of any intermediate.[32][28] M. Shigeo, T. Yoko, W. Yoshiaki, O. Sadafumi, J. Antibiot. 1984, 37, 187 – 189. [29] Y. Watanabe, T. Adachi, T. Asaka, M. Kashimura, S. Morimoto, Heterocycles 1990, 31, 2121 – 2124. [30] E. H. Flynn, H. W. Murphy, R. E. McMahon, J. Am. Chem. Soc. 1955, 77, 3104 – 3106. [31] Y. Watanabe, S. Morimoto, T. Adachi, M. Kashimura, T. Asaka, J. Antibiot. 1993, 46, 647 – 660.32] R. A. Dominguez, M. D. C. C. Rodriguez, L. . D. Tejo, R. N. Rib, J. S. Cebrin, J. I. B. Bilbao, 2003, US6642364B2.
References
- ^ https://www.ema.europa.eu/documents/psusa/clarithromycin-list-nationally-authorised-medicinal-products-psusa/00000788/202004_en.pdf
- ^ Jump up to:a b c d e f g h i j k l m n “Clarithromycin”. The American Society of Health-System Pharmacists. Archivedfrom the original on September 3, 2015. Retrieved September 4, 2015.
- ^ Jump up to:a b Greenwood D (2008). Antimicrobial drugs : chronicle of a twentieth century medical triumph (1 ed.). Oxford: Oxford University Press. p. 239. ISBN 9780199534845. Archived from the original on 2016-03-05.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 498. ISBN 9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Kirst HA (2012). Macrolide Antibiotics (2 ed.). Basel: Birkhäuser Basel. p. 53. ISBN 9783034881050. Archived from the original on 2016-03-05.
- ^ Jump up to:a b c d e f g h i j k l “BIAXIN® Filmtab® (clarithromycin tablets, USP) BIAXIN® XL Filmtab® (clarithromycin extended-release tablets) BIAXIN® Granules (clarithromycin for oral suspension, USP)” (PDF). November 2, 2015. Archived (PDF) from the original on August 24, 2015. Retrieved November 2, 2015.
- ^ “Clarithromycin Side Effects in Detail – Drugs.com”. Drugs.com. Archived from the original on 2017-08-19. Retrieved 2017-08-18.
- ^ “Safety Alerts for Human Medical Products – Clarithromycin (Biaxin): Drug Safety Communication – Potential Increased Risk of Heart Problems or Death in Patients With Heart Disease”. FDA. Retrieved 24 February 2018.
- ^ Yamaguchi S, Kaneko Y, Yamagishi T, et al. [Clarithromycin-induced torsades de pointes]. Nippon Naika Gakkai Zasshi. 2003;92(1):143–5.
- ^ Winkel P, Hilden J, Fischer Hansen J, Hildebrandt P, Kastrup J, Kolmos HJ, et al. (2011). “Excess sudden cardiac deaths after short-term clarithromycin administration in the CLARICOR trial: why is this so, and why are statins protective?”. Cardiology. 118 (1): 63–7. doi:10.1159/000324533. PMID 21447948. S2CID 11873791.
- ^ Tietz A, Heim MH, Eriksson U, Marsch S, Terracciano L, Krähenbühl S (January 2003). “Fulminant liver failure associated with clarithromycin”. The Annals of Pharmacotherapy. 37 (1): 57–60. doi:10.1345/1542-6270(2003)037<0057:flfawc>2.0.co;2. PMID 12503933.
- ^ Patel AM, Shariff S, Bailey DG, Juurlink DN, Gandhi S, Mamdani M, et al. (June 2013). “Statin toxicity from macrolide antibiotic coprescription: a population-based cohort study”. Annals of Internal Medicine. 158 (12): 869–76. doi:10.7326/0003-4819-158-12-201306180-00004. PMID 23778904. S2CID 21222679.
- ^ Gandhi S, Fleet JL, Bailey DG, McArthur E, Wald R, Rehman F, Garg AX (December 2013). “Calcium-channel blocker-clarithromycin drug interactions and acute kidney injury”. JAMA. 310 (23): 2544–53. doi:10.1001/jama.2013.282426. PMID 24346990.
- ^ Gélisse P, Hillaire-Buys D, Halaili E, Jean-Pastor MJ, Vespignan H, Coubes P, Crespel A (November 2007). “[Carbamazepine and clarithromycin: a clinically relevant drug interaction]”. Revue Neurologique. 163 (11): 1096–9. doi:10.1016/s0035-3787(07)74183-8. PMID 18033049.
- ^ Sekar VJ, Spinosa-Guzman S, De Paepe E, De Pauw M, Vangeneugden T, Lefebvre E, Hoetelmans RM (January 2008). “Darunavir/ritonavir pharmacokinetics following coadministration with clarithromycin in healthy volunteers”. Journal of Clinical Pharmacology. 48 (1): 60–5. doi:10.1177/0091270007309706. PMID 18094220. S2CID 38368595.
- ^ Polis MA, Piscitelli SC, Vogel S, Witebsky FG, Conville PS, Petty B, et al. (August 1997). “Clarithromycin lowers plasma zidovudine levels in persons with human immunodeficiency virus infection”. Antimicrobial Agents and Chemotherapy. 41 (8): 1709–14. doi:10.1128/AAC.41.8.1709. PMC 163990. PMID 9257746.
- ^ Ferrero JL, Bopp BA, Marsh KC, Quigley SC, Johnson MJ, Anderson DJ, et al. (1990). “Metabolism and disposition of clarithromycin in man”. Drug Metabolism and Disposition. 18 (4): 441–6. PMID 1976065.
- ReferencesAllevi, P. et al.: Bioorg. Med. Chem. (BMECEP) 7, 12, 2749 (1999)Watanabe, Y. et al.: Heterocycles (HTCYAM) 31, 12, 2121 (1990).EP 158 467 (Taisho Pharmaceutical Co.; 22.3.1985; J-prior. 6.4.1984).EP 272 110 (Taisho Pharmaceutical Co.; 16.12.1987; J-prior. 17.12.1986).US 2 001 037 015 (Teva Pharm.; 15.12.2000; USA-prior. 29.2.2000).KR 2 000 043 839 (Hanmi Pharm.; ROK-prior. 29.12.1998).EP 1 150 990 (Hanmi Pharm.; 7.11.2001; ROK-prior. 29.12.1998)EP 41 355 (Taisho Pharmaceutical Co.; 27.5.1981; J-prior. 4.6.1980).Preparation of O,N-dicarbobenzoxy-N-demethylerythromycin:Flynn, E. H. et al.: J. Am. Chem. Soc. (JACSAT) 77, 3104 (1955).Process for preparation of erythromycin A oxime:US 5 808 017 (Abbott; 15.9.1998; USA-prior. 10.4.1996).Alternative synthesis of clarithromycin:Liao, G.; Zhang, G.; He, T.: Zhongguo Kangshengsu Zazhi (ZKZAEY) 27, 3, 148 (2002) (in Chinese).EP 1 134 229 (Hanmi Pharmac. Co.; 19.9.2001; ROK-prior. 15.3.2000).Crystal form 0 of clarithromycin:The Merck Index, 13th Ed., 2362, p. 408.US 5 945 405 (Abbott; 31.8.1999; USA-prior. 17.1.1997).
External links
- “Clarithromycin”. Drug Information Portal. U.S. National Library of Medicine.
- US patent 4331803, Watanabe Y, Morimoto S, Omura S, “Novel erythromycin compounds”, issued 1981-05-19, assigned to Taisho Pharmaceutical
- “FDA review finds additional data supports the potential for increased long-term risks with antibiotic clarithromycin (Biaxin) in patients with heart disease” (PDF). FDA Drug Safety Communication.
| Clinical data | |
|---|---|
| Trade names | Biaxin, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a692005 |
| License data | EU EMA: by INNUS DailyMed: Clarithromycin |
| Pregnancy category | AU: B3 |
| Routes of administration | By mouth, intravenous |
| Drug class | Macrolides |
| ATC code | J01FA09 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-onlyEU: Rx-only [1]In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 50% |
| Protein binding | low binding |
| Metabolism | hepatic |
| Elimination half-life | 3–4 h |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 81103-11-9 |
| PubChem CID | 84029 |
| DrugBank | DB01211 |
| ChemSpider | 10342604 |
| UNII | H1250JIK0A |
| KEGG | D00276 |
| ChEMBL | ChEMBL1741 |
| CompTox Dashboard (EPA) | DTXSID3022829 |
| ECHA InfoCard | 100.119.644 |
| Chemical and physical data | |
| Formula | C38H69NO13 |
| Molar mass | 747.964 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCC[C@@H]1[C@@]([C@@H]([C@H](C(=O)[C@@H](C[C@@]([C@@H]([C@H]([C@@H]([C@H](C(=O)O1)C)O[C@H]2C[C@@]([C@H]([C@@H](O2)C)O)(C)OC)C)O[C@H]3[C@@H]([C@H](C[C@H](O3)C)N(C)C)O)(C)OC)C)C)O)(C)O | |
| hideInChIInChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21(4)28(40)19(2)17-37(9,47-14)33(52-35-29(41)25(39(11)12)16-20(3)48-35)22(5)30(23(6)34(44)50-26)51-27-18-36(8,46-13)32(43)24(7)49-27/h19-27,29-33,35,41-43,45H,15-18H2,1-14H3/t19-,20-,21+,22+,23-,24+,25+,26-,27+,29-,30+,31-,32+,33-,35+,36-,37-,38-/m1/s1 Key:AGOYDEPGAOXOCK-KCBOHYOISA-N | |
| (what is this?) (verify) |
////////////////////CLARITHROMYCIN, Antibacterial, Antibiotics, Macrolides, A-56268, TE-031,
#CLARITHROMYCIN, #Antibacterial, #Antibiotics, #Macrolides, #A-56268, #TE-031,
ERYTHROMYCIN

Erythromycin
NSC-55929
UNII63937KV33D
CAS number114-07-8
- Molecular FormulaC37H67NO13
- Average mass733.927 Da
- эритромицин [Russian] [INN]إيريثروميسين [Arabic] [INN]红霉素 [Chinese] [INN]
IUPAC Name(3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-6-{[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy}-14-ethyl-7,12,13-trihydroxy-4-{[(2R,4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy}-3,5,7,9,11,13-hexamethyl-1-oxacyclotetradecane-2,10-dione
Synthesis ReferenceTakehiro Amano, Masami Goi, Kazuto Sekiuchi, Tomomichi Yoshida, Masahiro Hasegawa, “Process for preparing erythromycin A oxime or a salt thereof.” U.S. Patent US5274085, issued October, 1966.
US5274085ErythromycinCAS Registry Number: 114-07-8Additional Names: E-Base; E-Mycin; Erythromycin ATrademarks: Aknemycin (Hermal); Aknin (Lichtenstein); Emgel (GSK); Ery-Derm (Abbott); Erymax (Merz); Ery-Tab (Abbott); Erythromid (Abbott); ERYC (Warner-Chilcott); Erycen (APS); Erycin (Nycomed); Erycinum (Cytochemia); Ermysin (Orion); Gallimycin (Bimeda); Ilotycin (Lilly); Inderm (Dermapharm); PCE (Abbott); Retcin (DDSA); Staticin (Westwood); Stiemycin (Stiefel)Molecular Formula: C37H67NO13Molecular Weight: 733.93Percent Composition: C 60.55%, H 9.20%, N 1.91%, O 28.34%Literature References: Antibiotic substance produced by a strain of Streptomyces erythreus (Waksman) Waksman & Henrici, found in a soil sample from the Philippine Archipelago. Isoln: McGuire et al.,Antibiot. Chemother.2, 281 (1952); Bunch, McGuire, US2653899 (1953 to Lilly); Clark, Jr., US2823203 (1958 to Abbott). Properties: Flynn et al.,J. Am. Chem. Soc.76, 3121 (1954). Solubility data: Weiss et al.,Antibiot. Chemother.7, 374 (1957). Structure: Wiley et al.,J. Am. Chem. Soc.79, 6062 (1957). Configuration: Hofheinz, Grisebach, Ber.96, 2867 (1963); Harris et al.,Tetrahedron Lett.1965, 679. There are three erythromycins produced during fermentation, designated A, B, and C; A is the major and most important component. Erythromycins A and B contain the same sugar moieties, desosamine, q.v., and cladinose (3-O-methylmycarose). They differ in position 12 of the aglycone, erythronolide, A having an hydroxyl substituent. Component C contains desosamine and the same aglycone present in A but differs by the presence of mycarose, q.v., instead of cladinose. Structure of B: P. F. Wiley et al.,J. Am. Chem. Soc.79, 6070 (1957); of C: eidem,ibid. 6074. Synthesis of the aglycone, erythronolide B: E. J. Corey et al.,ibid.100, 4618, 4620 (1978); of erythronolide A: eidem,ibid.101, 7131 (1979). Asymmetric total synthesis of erythromycin A: R. B. Woodward et al.,ibid.103, 3215 (1981). NMR spectrum of A: D. J. Ager, C. K. Sood, Magn. Reson. Chem.25, 948 (1987). HPLC determn in plasma: W. Xiao et al., J. Chromatogr. B817, 153 (2005). Biosynthesis: Martin, Goldstein, Prog. Antimicrob. Anticancer Chemother., Proc. 6th Int. Congr. Chemother.II, 1112 (1970); Martin et al.,Tetrahedron31, 1985 (1975). Cloning and expression of clustered biosynthetic genes: R. Stanzak et al.,Biotechnology4, 229 (1986). Reviews: T. J. Perun in Drug Action and Drug Resistance in Bacteria1, S. Mitsuhashi, Ed. (University Park Press, Baltimore, 1977) pp 123-152; Oleinick in Antibioticsvol. 3, J. W. Corcoran, F. E. Hahn, Eds. (Springer-Verlag, New York, 1975) pp 396-419; Infection10, Suppl. 2, S61-S118 (1982). Comprehensive description: W. L. Koch, Anal. Profiles Drug Subs.8, 159-177 (1979).Properties: Hydrated crystals from water, mp 135-140°, resolidifies with second mp 190-193°. Melting point taken after drying at 56° and 8 mm. [a]D25 -78° (c = 1.99 in ethanol). uv max (pH 6.3): 280 nm (e 50). pKa1 8.8. Basic reaction. Readily forms salts with acids. Soly in water: ~2 mg/ml. Freely sol in alcohols, acetone, chloroform, acetonitrile, ethyl acetate. Moderately sol in ether, ethylene dichloride, amyl acetate.Melting point: mp 135-140°, resolidifies with second mp 190-193°pKa: pKa1 8.8Optical Rotation: [a]D25 -78° (c = 1.99 in ethanol)Absorption maximum: uv max (pH 6.3): 280 nm (e 50) Derivative Type: EthylsuccinateCAS Registry Number: 41342-53-4Trademarks: Anamycin (Chephasaar); Arpimycin (Rosemont); E.E.S. (Abbott); Eritrocina (Abbott); Eryliquid (Linden); Eryped (Abbott); Erythroped (Abbott); Esinol (Toyama); Monomycin (Grñenthal); Paediathrocin (Abbott); Pediamycin (Abbott); Refkas (Maruko)Molecular Formula: C43H75NO16Molecular Weight: 862.05Percent Composition: C 59.91%, H 8.77%, N 1.62%, O 29.70%Literature References: Prepn: GB830846; R. K. Clark, US2967129 (1960, 1961 both to Abbott).Properties: Hydrated crystals from acetone + water, mp 109-110°. [a]D -42.5°.Melting point: mp 109-110°Optical Rotation: [a]D -42.5° Therap-Cat: Antibacterial.Therap-Cat-Vet: Antibacterial.Keywords: Antibacterial (Antibiotics); Macrolides.
NEW DRUG APPROVALS
one time
$10.00
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Erythromycin estolate | XRJ2P631HP | 3521-62-8 | AWMFUEJKWXESNL-JZBHMOKNSA-N |
| Erythromycin ethylsuccinate | 1014KSJ86F | 1264-62-6 | NSYZCCDSJNWWJL-YXOIYICCSA-N |
| Erythromycin gluceptate | 2AY21R0U64 | 23067-13-2 | ZXBDZLHAHGPXIG-VTXLJDRKSA-N |
| Erythromycin lactobionate | 33H58I7GLQ | 3847-29-8 | NNRXCKZMQLFUPL-WBMZRJHASA-N |
| Erythromycin phosphate | I8T8KU14X7 | 4501-00-2 | VUEMAFLGEMYXIH-YZPBMOCRSA-N |
| Erythromycin stearate | LXW024X05M | 643-22-1 | YAVZHCFFUATPRK-YZPBMOCRSA-N |
| Erythromycin sulfate | KVW9N83AME | 7184-72-7 | XTSSJGRRFMNXGO-YZPBMOCRSA-N |
| Erythromycin thiocyanate | Y7A95YRI88 | 7704-67-8 | WVRRTEYLDPNZHR-YZPBMOCRSA-N |
Erythromycin is an antibiotic which belongs to the group of macrolide antibiotics. The pharmaceutically distributed product consists of three components: Erythromycin A, B, and C where Erythromycin A represents the main component. Naturally this antibiotic is synthesized by the grampositive bacteria Streptomyces erythreus (Saccharopolyspora erythrea).
In 1949 Erythromycin was found for the first time in a soil sample in the Philippine region Iloilo. A research team, led by J. M. McGuire, was able to isolate Erythromycin which was part of the soil sample. Under the brand name Ilosone the product was launched commercially in 1952. They named the brand after the region where the antibiotic was found. Analogically the first product name was Ilotycin. Furthermore, in 1953 the U.S. patent was granted. Since 1957 the structure of Erythromycin is known and in 1965 the X-ray structure analysis gave awareness of the absolute configuration. In 1981, almost 30 years after the detection of Erythromycin, Robert B. Woodward, the Nobel prize laureate of chemistry in 1965, and his coworkers posthumously reported the first synthesis of Erythromycin A
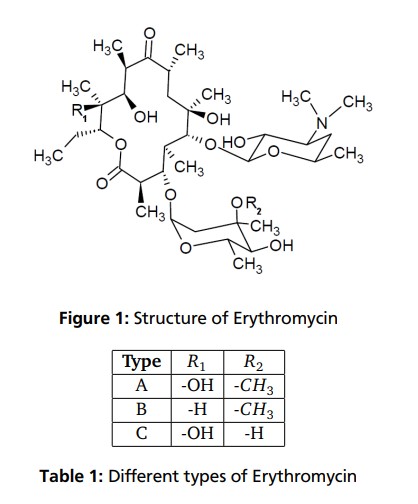
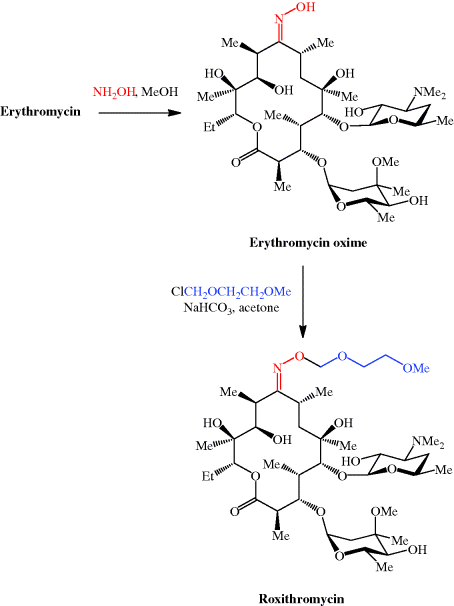
The structural characteristic of macrolides, to which Erythromycin affiliates, is a macrocyclic lactone ring of fourteen, fifteen or sixteen members. In case of Erythromycin the lactone ring consists of 14-members. Substituents on the mainchain are cladinose on C-3 and desosamine on C-5. Erythromycin is not a single compound but represents an alloy of structural very similar components. The main constituents are Erythromycin A, B and C. As shown in Table 1 and Figure 1 they only differ in two rests on the lactone ring or on the cladinose each case. In addition to the variants already mentioned, further variants, like Erythromycin D and E are known. They are pre- and post-stages in the biosynthesis and often do not have antibiotic effects
Chemical and Pharmacokinetic Properties Formula: C37H67NO13 CAS-Number: 114-07-8 Molar Mass: 733.93g/mol Half Hife 1.5 hours pkA: 8,6 – 8,8 Melting Point: 411K (hydrat) 463-466K (anhydrous)
Erythromycin is an antibiotic used for the treatment of a number of bacterial infections.[1] This includes respiratory tract infections, skin infections, chlamydia infections, pelvic inflammatory disease, and syphilis.[1] It may also be used during pregnancy to prevent Group B streptococcal infection in the newborn,[1] as well as to improve delayed stomach emptying.[3] It can be given intravenously and by mouth.[1] An eye ointment is routinely recommended after delivery to prevent eye infections in the newborn.[4]
Common side effects include abdominal cramps, vomiting, and diarrhea.[1] More serious side effects may include Clostridium difficile colitis, liver problems, prolonged QT, and allergic reactions.[1] It is generally safe in those who are allergic to penicillin.[1] Erythromycin also appears to be safe to use during pregnancy.[2] While generally regarded as safe during breastfeeding, its use by the mother during the first two weeks of life may increase the risk of pyloric stenosis in the baby.[5][6] This risk also applies if taken directly by the baby during this age.[7] It is in the macrolide family of antibiotics and works by decreasing bacterial protein production.[1]
Erythromycin was first isolated in 1952 from the bacteria Saccharopolyspora erythraea.[1][8] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[9] The World Health Organization classifies it as critically important for human medicine.[10] It is available as a generic medication.[5] In 2017, it was the 215th most commonly prescribed medication in the United States, with more than two million prescriptions.[11][12]
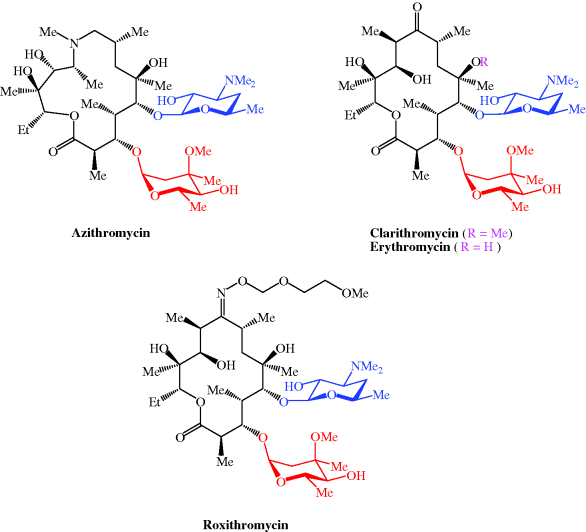
Table 4.2.1 Therapeutic indications for the macrolide antibiotics.
Medical uses
Erythromycin can be used to treat bacteria responsible for causing infections of the skin and upper respiratory tract, including Streptococcus, Staphylococcus, Haemophilus and Corynebacterium genera. The following represents MIC susceptibility data for a few medically significant bacteria:[13]
- Haemophilus influenzae: 0.015 to 256 μg/ml
- Staphylococcus aureus: 0.023 to 1024 μg/ml
- Streptococcus pyogenes: 0.004 to 256 μg/ml
- Corynebacterium minutissimum: 0.015 to 64 μg/ml
It may be useful in treating gastroparesis due to this promotility effect. It has been shown to improve feeding intolerances in those who are critically ill.[14] Intravenous erythromycin may also be used in endoscopy to help clear stomach contents.
Available forms

Enteric-coated erythromycin capsule from Abbott Labs
Erythromycin is available in enteric-coated tablets, slow-release capsules, oral suspensions, ophthalmic solutions, ointments, gels, enteric-coated capsules, non enteric-coated tablets, non enteric-coated capsules, and injections. The following erythromycin combinations are available for oral dosage:[15]
- erythromycin base (capsules, tablets)
- erythromycin estolate (capsules, oral suspension, tablets), contraindicated during pregnancy[16]
- erythromycin ethylsuccinate (oral suspension, tablets)
- erythromycin stearate (oral suspension, tablets)
For injection, the available combinations are:[15]
- erythromycin gluceptate
- erythromycin lactobionate
For ophthalmic use:
- erythromycin base (ointment)
Adverse effects
Gastrointestinal disturbances, such as diarrhea, nausea, abdominal pain, and vomiting, are very common because erythromycin is a motilin agonist.[17] Because of this, erythromycin tends not to be prescribed as a first-line drug.
More serious side effects include arrhythmia with prolonged QT intervals, including torsades de pointes, and reversible deafness. Allergic reactions range from urticaria to anaphylaxis. Cholestasis, Stevens–Johnson syndrome, and toxic epidermal necrolysis are some other rare side effects that may occur.
Studies have shown evidence both for and against the association of pyloric stenosis and exposure to erythromycin prenatally and postnatally.[18] Exposure to erythromycin (especially long courses at antimicrobial doses, and also through breastfeeding) has been linked to an increased probability of pyloric stenosis in young infants.[19][20] Erythromycin used for feeding intolerance in young infants has not been associated with hypertrophic pyloric stenosis.[19]
Erythromycin estolate has been associated with reversible hepatotoxicity in pregnant women in the form of elevated serum glutamic-oxaloacetic transaminase and is not recommended during pregnancy. Some evidence suggests similar hepatotoxicity in other populations.[21]
It can also affect the central nervous system, causing psychotic reactions, nightmares, and night sweats.[22]
Interactions
Erythromycin is metabolized by enzymes of the cytochrome P450 system, in particular, by isozymes of the CYP3A superfamily.[23] The activity of the CYP3A enzymes can be induced or inhibited by certain drugs (e.g., dexamethasone), which can cause it to affect the metabolism of many different drugs, including erythromycin. If other CYP3A substrates — drugs that are broken down by CYP3A — such as simvastatin (Zocor), lovastatin (Mevacor), or atorvastatin (Lipitor)—are taken concomitantly with erythromycin, levels of the substrates increase, often causing adverse effects. A noted drug interaction involves erythromycin and simvastatin, resulting in increased simvastatin levels and the potential for rhabdomyolysis. Another group of CYP3A4 substrates are drugs used for migraine such as ergotamine and dihydroergotamine; their adverse effects may be more pronounced if erythromycin is associated.[22] Earlier case reports on sudden death prompted a study on a large cohort that confirmed a link between erythromycin, ventricular tachycardia, and sudden cardiac death in patients also taking drugs that prolong the metabolism of erythromycin (like verapamil or diltiazem) by interfering with CYP3A4.[24] Hence, erythromycin should not be administered to people using these drugs, or drugs that also prolong the QT interval. Other examples include terfenadine (Seldane, Seldane-D), astemizole (Hismanal), cisapride (Propulsid, withdrawn in many countries for prolonging the QT time) and pimozide (Orap). Theophylline, which is used mostly in asthma, is also contraindicated.
Erythromycin and doxycycline can have a synergistic effect when combined and kill bacteria (E. coli) with a higher potency than the sum of the two drugs together. This synergistic relationship is only temporary. After approximately 72 hours, the relationship shifts to become antagonistic, whereby a 50/50 combination of the two drugs kills less bacteria than if the two drugs were administered separately.[25]
It may alter the effectiveness of combined oral contraceptive pills because of its effect on the gut flora. A review found that when erythromycin was given with certain oral contraceptives, there was an increase in the maximum serum concentrations and AUC of estradiol and dienogest.[26][27]
Erythromycin is an inhibitor of the cytochrome P450 system, which means it can have a rapid effect on levels of other drugs metabolised by this system, e.g., warfarin.
Pharmacology
Mechanism of action
Erythromycin displays bacteriostatic activity or inhibits growth of bacteria, especially at higher concentrations.[28] By binding to the 50s subunit of the bacterial rRNA complex, protein synthesis and subsequent structure and function processes critical for life or replication are inhibited.[28] Erythromycin interferes with aminoacyl translocation, preventing the transfer of the tRNA bound at the A site of the rRNA complex to the P site of the rRNA complex. Without this translocation, the A site remains occupied, thus the addition of an incoming tRNA and its attached amino acid to the nascent polypeptide chain is inhibited. This interferes with the production of functionally useful proteins, which is the basis of this antimicrobial action.
Erythromycin increases gut motility by binding to Motillin, thus it is a Motillin receptor agonist in addition to its antimicrobial properties.
Pharmacokinetics
Erythromycin is easily inactivated by gastric acid; therefore, all orally administered formulations are given as either enteric-coated or more-stable salts or esters, such as erythromycin ethylsuccinate. Erythromycin is very rapidly absorbed, and diffuses into most tissues and phagocytes. Due to the high concentration in phagocytes, erythromycin is actively transported to the site of infection, where, during active phagocytosis, large concentrations of erythromycin are released.
Metabolism
Most of erythromycin is metabolised by demethylation in the liver by the hepatic enzyme CYP3A4. Its main elimination route is in the bile with little renal excretion, 2%-15% unchanged drug. Erythromycin’s elimination half-life ranges between 1.5 and 2.0 hours and is between 5 and 6 hours in patients with end-stage renal disease. Erythromycin levels peak in the serum 4 hours after dosing; ethylsuccinate peaks 0.5-2.5 hours after dosing, but can be delayed if digested with food.[29]
Erythromycin crosses the placenta and enters breast milk. The American Association of Pediatrics determined erythromycin is safe to take while breastfeeding.[30] Absorption in pregnant patients has been shown to be variable, frequently resulting in levels lower than in nonpregnant patients.[29]
Chemistry
Composition
Standard-grade erythromycin is primarily composed of four related compounds known as erythromycins A, B, C, and D. Each of these compounds can be present in varying amounts and can differ by lot. Erythromycin A has been found to have the most antibacterial activity, followed by erythromycin B. Erythromycins C and D are about half as active as erythromycin A.[13][31] Some of these related compounds have been purified and can be studied and researched individually.
Synthesis
Over the three decades after the discovery of erythromycin A and its activity as an antimicrobial, many attempts were made to synthesize it in the laboratory. The presence of 10 stereogenic carbons and several points of distinct substitution has made the total synthesis of erythromycin A a formidable task.[32] Complete syntheses of erythromycins’ related structures and precursors such as 6-deoxyerythronolide B have been accomplished, giving way to possible syntheses of different erythromycins and other macrolide antimicrobials.[33] Woodward successfully completed the synthesis of erythromycin A.[34][35][36]
Erythromycin related compounds
History
In 1949 Abelardo B. Aguilar, a Filipino scientist, sent some soil samples to his employer Eli Lilly. Eli Lilly’s research team, led by J. M. McGuire, managed to isolate erythromycin from the metabolic products of a strain of Streptomyces erythreus (designation changed to Saccharopolyspora erythraea) found in the samples.[37]
Lilly filed for patent protection on the compound which was granted in 1953.[38] The product was launched commercially in 1952 under the brand name Ilosone (after the Philippine region of Iloilo where it was originally collected). Erythromycin was formerly also called Ilotycin.
The antibiotic clarithromycin was invented by scientists at the Japanese drug company Taisho Pharmaceutical in the 1970s as a result of their efforts to overcome the acid instability of erythromycin.
Scientists at Chugai Pharmaceuticals discovered an erythromycin-derived motilin agonist called mitemcinal that is believed to have strong prokinetic properties (similar to erythromycin) but lacking antibiotic properties. Erythromycin is commonly used off-label for gastric motility indications such as gastroparesis. If mitemcinal can be shown to be an effective prokinetic agent, it would represent a significant advance in the gastrointestinal field, as treatment with this drug would not carry the risk of unintentional selection for antibiotic-resistant bacteria.
Society and culture
Cost
It is available as a generic medication.[5]
In the United States in 2014 the price increased to seven dollars per tablet.[39]
The price of Erythromycin rose three times between 2010 and 2015, from 24 cents per tablet in 2010 to $8.96 in 2015.[40] In 2017, a Kaiser Health News study found that the per-unit cost of dozens of generics doubled or even tripled from 2015 to 2016, increasing spending by the Medicaid program. Due to price increases by drug manufacturers, Medicaid paid on average $2,685,330 more for Erythromycin in 2016 compared to 2015 (not including rebates).[41] By 2018, generic drug prices had climbed another 5% on average.[42]
Brand names
Brand names include Robimycin, E-Mycin, E.E.S. Granules, E.E.S.-200, E.E.S.-400, E.E.S.-400 Filmtab, Erymax, Ery-Tab, Eryc, Ranbaxy, Erypar, EryPed, Eryped 200, Eryped 400, Erythrocin Stearate Filmtab, Erythrocot, E-Base, Erythroped, Ilosone, MY-E, Pediamycin, Zineryt, Abboticin, Abboticin-ES, Erycin, PCE Dispertab, Stiemycine, Acnasol, and Tiloryth.
See also
Erythromycin/tretinoin, a combination of tretinoin and the antibiotic erythromycin
SYN
https://basicmedicalkey.com/macrolide-antibiotics/embed/#?secret=VMg8PBg4K9
Synthesis
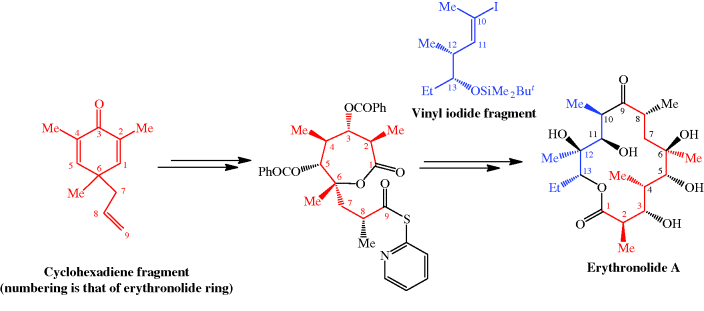
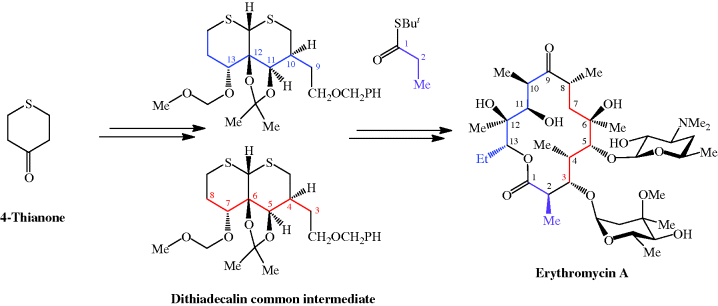
The total synthesis of the erythromycins (Figure 4.2.2) poses a supreme challenge and has attracted the attention of some of the world’s most eminent synthetic chemists, leading to many elegant examples of the total synthesis of complex natural products. The total synthesis of the erythronolide A aglycone (lacking the sugar units) was first reported by E. J. Corey (Nobel Prize in Chemistry in 1990) in a series of articles in the late 1970s (Scheme 4.2.2) (Corey et al., 1979 and references cited therein), and the total synthesis of erythromycin (known then as erythromycin A) by R. B. Woodward (Nobel Prize in Chemistry in 1965) in a series of articles in 1981, after his death (Scheme 4.2.3) (Woodward et al., 1981 and references cited therein). The Woodward synthesis is particularly elegant, as the dithiadecalin intermediate supplies both the C3-C8 and C9-C13 fragments (Scheme 4.2.3).
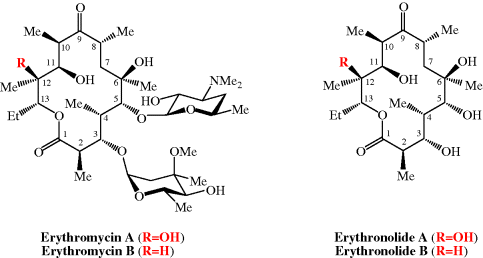
Figure 4.2.2 Erythromycins A and B and their aglycones, erythronolides A and B
Scheme 4.2.2 Corey’s total synthesis of erythronolide A (38 steps from the cyclohexadiene fragment; 0.04% overall yield)
Scheme 4.2.3 Woodward’s total synthesis of erythromycin (56 steps from 4-thianone; 0.01% overall yield)
Once again, erythromycin is such a complex antibiotic that its commercial production by total synthesis will never be feasible, and it is obtained from the submerged culture of free or immobilised Saccharopolyspora erythraea (El-Enshasy et al., 2008).
We have now seen a number of examples of how very complex semi-synthetic antibiotics can be prepared through the combination of fermentation (to give the complex natural product) and chemical modification, so you will no doubt already have spotted that both clarithromycin and roxithromycin are semi-synthetic macrolide antibiotics. Clarithromycin can be obtained in a five-step synthetic procedure, from erythromycin oxime (Brunet et al., 2007), while roxithromycin can also be prepared from this oxime (Massey et al., 1970) in a single step (Scheme 4.2.4) (Gouin d’Ambrieres et al., 1982). What is not so obvious is that azithromycin is also a semi-synthetic macrolide, having originally been produced by PLIVA Pharmaceuticals from erythromycin oxime via a sequence of reactions which included the well-known Beckmann rearrangement (Djoki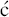 et al., 1986). For more on the synthesis of the erythromycins, see Paterson and Mansuri (1985).
et al., 1986). For more on the synthesis of the erythromycins, see Paterson and Mansuri (1985).
Scheme 4.2.4 Preparation of the semi-synthetic macrolide antibiotic roxithromycin
CLIP

Erythromycin. Erythromycin (1) was discovered in 1952 during the investigation of soil samples from Iloilo, Philippines for antibiotic activity[18, 19] and its molecular structure was assigned in 1957.[20] The microorganism that produced erythromycin was isolated and characterised as Streptomyces erythreus, strain NRRL 2338.[18, 19] Over the years, strain improvements and genetic engineering has allowed the yield of erythromycin to be increased so that 8–10 g L1 can now be produced from a tryptic soy broth.[21–25] Erythromycin forms anhydro-erythromycin 6 and 6:9, 9:12 spiroketal 7 under the acidic conditions in the stomach (Scheme 1), which results in the loss of its antibacterial activity and induction of abdominal pain.[26, 27] Generation of by-products 6 and 7 occurs through an acid-catalysed intramolecular reaction of the C-6 hydroxyl group with the C-9 keto moiety. To avoid this by-product formation several different semi-synthetic derivatives of erythromycin have been prepared in which either of these two functionalities are modified. They led to the discovery of clarithromycin (2) by O-6 methylation of erythromycin (Figure 3). Removal of the C-9 ketone by the formation of an oxime followed by Beckmann rearrangement and reduction led to azithromycin (3), which belongs to a new class of macrolides called “azalides”. Alternatively, conversion of the C-9 ketone to an amine, followed by reaction with an aldehyde, gave dirithromycin (4). Yet another approach involved the transformation of clarithromycin to the conformationally restricted telithromycin
SYN
Chemical Synthesis
Erythromycin, (3R,4S,5S,6R,7R,9R,11R,12R,13S,14R)-4-[(2,6-dideoxy-3-Cmethyl-3-O-methyl-α-L-ribo-hexopyranosyl)-oxy]-14-ethyl-7,12,13-trihydroxy- 3,5,7,9,11,13-hexamethyl-6-[[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy ]oxacyclotetradecan-2,10-dione (32.2.1), is more specifically called erythromycin A. It was first isolated in 1952 from the culture liquid of microorganisms of the type Streptomyces erytherus. Minor amounts of erythromycin B and C were also found in the culture fluid. Erythromycin B differs from A in that a hydrogen atom is located at position 12 in the place of a hydroxyl group, while erythromycin C differs from A in that the residue of a different carbohydrate, micarose (2-6-di-deoxy-3-C-methyl-L-ribohexose), is bound to the macrocycle in position 3 in the place of cladinose (4-methoxy-2,4-dimethyl-tetrahydropyran-3,6-diol).
Erythromycin A is produced only microbiologically using active strains of microorganisms of the type Saccharopolospora erythraea.
SYN

Erythromycin synthesis by modular polyketide synthases. The three genes EryAI-III encode three proteins of PKS: DEBS1 (the loading module, modules 1, 2) DEBS2 (modules 3, 4), DEBS3 (modules 5, 6, TE domain). Thus, PKS consists of the loading module, six extension modules, and TE domain. Each module includes from three to six domains: AT-acyl transferase, ACP-acyl carrier protein, KS-ketosynthase, KR-ketoreductase, DH-dehydratase, ERenoyl reductase.
CLIP
The chemical synthesis of Erythromycin poses a huge challenge. The molecule contains ten stereogenic centers of which five are arranged consecutively. R. B. Woodward and his research team first succeeded in synthesizing Erythromycin A. The reaction sequence, however, is so complicated that the yield was only about 0,02 % and, thus, the synthesis is not utilizable comercially. This is the reason for the preferred use of the biosynthesis of Erythromycin via fermentation of Streptomyces erythreus. Other scientists and research teams dealt with the synthesis of Erythromycin as well and developed very similar approaches. Most methods for the Erythromycin synthesis are based on the construction of the aglycon from secoic acid via glycosylation. Indeed the process is also possible inversely: first, a glycosylation, then a lactonization occurs. The yield, however, is considerably less. While earlier scientist mainly dealt with the production of the different secoic acids, the lactonization process is the major problem today because there is no fully developed method for it yet. A lot of side reactions such as dimerization and polymerization appear, because a 14 membered ring is hard to enclose. Even if the chemical synthesis of Erythromycin has no importance for the comercial fabrication of the antibiotic, it is still important for the development and fabrication of its derivatives.
References
- ^ Jump up to:a b c d e f g h i j k “Erythromycin”. The American Society of Health-System Pharmacists. Archived from the original on 2015-09-06. Retrieved Aug 1, 2015.
- ^ Jump up to:a b “Prescribing medicines in pregnancy database”. Australian Government. August 23, 2015. Archived from the original on April 8, 2014.
- ^ Camilleri M, Parkman HP, Shafi MA, Abell TL, Gerson L (January 2013). “Clinical guideline: management of gastroparesis”. The American Journal of Gastroenterology. 108 (1): 18–37, quiz 38. doi:10.1038/ajg.2012.373. PMC 3722580. PMID 23147521.
- ^ Matejcek A, Goldman RD (November 2013). “Treatment and prevention of ophthalmia neonatorum”. Canadian Family Physician. 59 (11): 1187–90. PMC 3828094. PMID 24235191.
- ^ Jump up to:a b c Hamilton RJ (2013). Tarascon pocket pharmacopoeia(2013 delux lab-coat ed., 14th ed.). [Sudbury, Mass.]: Jones & Bartlett Learning. p. 72. ISBN 9781449673611.
- ^ Kong YL, Tey HL (June 2013). “Treatment of acne vulgaris during pregnancy and lactation”. Drugs. 73 (8): 779–87. doi:10.1007/s40265-013-0060-0. PMID 23657872. S2CID 45531743.
- ^ Maheshwai N (March 2007). “Are young infants treated with erythromycin at risk for developing hypertrophic pyloric stenosis?”. Archives of Disease in Childhood. 92 (3): 271–3. doi:10.1136/adc.2006.110007. PMC 2083424. PMID 17337692. Archived from the original on 7 November 2012.
- ^ Vedas JC (2000). Biosynthesis : polyketides and vitamins. Berlin [u.a.]: Springer. p. 52. ISBN 9783540669692.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ World Health Organization (2019). Critically important antimicrobials for human medicine (6th revision ed.). Geneva: World Health Organization. hdl:10665/312266. ISBN 9789241515528. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2020”. ClinCalc. Retrieved 11 April 2020.
- ^ “Erythromycin – Drug Usage Statistics”. ClinCalc. Retrieved 11 April 2020.
- ^ Jump up to:a b “Erythromycin Susceptibility and Minimum Inhibitory Concentration (MIC) Data” (PDF). TOKU-E.
- ^ Lewis K, Alqahtani Z, Mcintyre L, Almenawer S, Alshamsi F, Rhodes A, et al. (August 2016). “The efficacy and safety of prokinetic agents in critically ill patients receiving enteral nutrition: a systematic review and meta-analysis of randomized trials”. Critical Care. 20 (1): 259. doi:10.1186/s13054-016-1441-z. PMC 4986344. PMID 27527069.
- ^ Jump up to:a b “Erythromycin Oral, Parenteral Advanced Patient Information”. Drugs.com. Archived from the original on 2009-11-30.
- ^ Sexually Transmitted Diseases Treatment Guidelines 2006Archived 2010-02-11 at the Wayback Machine Centers for Disease Control and Prevention. MMWR 2006;55
- ^ Weber FH, Richards RD, McCallum RW (April 1993). “Erythromycin: a motilin agonist and gastrointestinal prokinetic agent”. The American Journal of Gastroenterology. 88 (4): 485–90. PMID 8470625.
- ^ “Pregnancy and lactation”. Archived from the original on 2014-04-20.
- ^ Jump up to:a b Maheshwai N (March 2007). “Are young infants treated with erythromycin at risk for developing hypertrophic pyloric stenosis?”. Archives of Disease in Childhood. 92 (3): 271–3. doi:10.1136/adc.2006.110007. PMC 2083424. PMID 17337692.
- ^ Lund M, Pasternak B, Davidsen RB, Feenstra B, Krogh C, Diaz LJ, et al. (March 2014). “Use of macrolides in mother and child and risk of infantile hypertrophic pyloric stenosis: nationwide cohort study”. BMJ. 348: g1908. doi:10.1136/bmj.g1908. PMC 3949411. PMID 24618148.
- ^ McCormack WM, George H, Donner A, Kodgis LF, Alpert S, Lowe EW, Kass EH (November 1977). “Hepatotoxicity of erythromycin estolate during pregnancy”. Antimicrobial Agents and Chemotherapy. 12 (5): 630–5. doi:10.1128/AAC.12.5.630. PMC 429989. PMID 21610.
- ^ Jump up to:a b “Erythromycine”. Belgisch Centrum voor Farmacotherapeutische Informatie. Archived from the original on 2015-10-06.
- ^ Hunt CM, Watkins PB, Saenger P, Stave GM, Barlascini N, Watlington CO, et al. (January 1992). “Heterogeneity of CYP3A isoforms metabolizing erythromycin and cortisol” (PDF). Clinical Pharmacology and Therapeutics. 51 (1): 18–23. doi:10.1038/clpt.1992.3. hdl:2027.42/109905. PMID 1732074. S2CID 28056649.
- ^ Ray WA, Murray KT, Meredith S, Narasimhulu SS, Hall K, Stein CM (September 2004). “Oral erythromycin and the risk of sudden death from cardiac causes”. The New England Journal of Medicine. 351 (11): 1089–96. doi:10.1056/NEJMoa040582. PMID 15356306.
- ^ Pena-Miller R, Laehnemann D, Jansen G, Fuentes-Hernandez A, Rosenstiel P, Schulenburg H, Beardmore R (April 23, 2013). “When the most potent combination of antibiotics selects for the greatest bacterial load: the smile-frown transition”. PLOS Biology. 11 (4): e1001540. doi:10.1371/journal.pbio.1001540. PMC 3635860. PMID 23630452.
- ^ Blode H, Zeun S, Parke S, Zimmermann T, Rohde B, Mellinger U, Kunz M (October 2012). “Evaluation of the effects of rifampicin, ketoconazole and erythromycin on the steady-state pharmacokinetics of the components of a novel oral contraceptive containing estradiol valerate and dienogest in healthy postmenopausal women”. Contraception. 86 (4): 337–44. doi:10.1016/j.contraception.2012.01.010. PMID 22445438.
- ^ Simmons KB, Haddad LB, Nanda K, Curtis KM (January 2018). “Drug interactions between non-rifamycin antibiotics and hormonal contraception: a systematic review”. American Journal of Obstetrics and Gynecology. 218 (1): 88–97.e14. doi:10.1016/j.ajog.2017.07.003. PMID 28694152. S2CID 36567820.
- ^ Jump up to:a b Katzung PHARMACOLOGY, 9e Section VIII. Chemotherapeutic Drugs Chapter 44. Chloramphenicol, Tetracyclines, Macrolides, Clindamycin, & Streptogramins
- ^ Jump up to:a b “unknown”. Archived from the original on 2014-04-19. Cite uses generic title (help)
- ^ American Academy of Pediatrics Committee on Drugs (September 2001). “Transfer of drugs and other chemicals into human milk” (PDF). Pediatrics. 108 (3): 776–89. doi:10.1542/peds.108.3.776. PMID 11533352. Archived from the original (PDF) on 2016-03-05.
- ^ Kibwage IO, Hoogmartens J, Roets E, Vanderhaeghe H, Verbist L, Dubost M, et al. (November 1985). “Antibacterial activities of erythromycins A, B, C, and D and some of their derivatives”. Antimicrobial Agents and Chemotherapy. 28 (5): 630–3. doi:10.1128/aac.28.5.630. PMC 176346. PMID 4091529.
- ^ Pal S (2006). “A journey across the sequential development of macrolides and ketolides related to erythromycin”. Tetrahedron. 62(14): 3171–3200. doi:10.1016/j.tet.2005.11.064.
- ^ Evans DA, Kim AS (1997). “Synthesis of 6-Deoxyerythronolide B. Implementation of a General Strategy for the Synthesis of Macrolide Antibiotics”. Tetrahedron Lett. 38: 53–56. doi:10.1016/S0040-4039(96)02258-7.
- ^ Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung, Balaram P, Browne LJ, Card PJ, et al. (1981). “Asymmetric Total Synthesis of Erythromycin. 1. Synthesis of an Erythronolide A Seco Acid Derivative via Asymmetric Induction”. J. Am. Chem. Soc. 103 (11): 3210–3213. doi:10.1021/ja00401a049.
- ^ Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung, Balaram P, Browne LJ, Card PJ, et al. (1981). “Asymmetric Total Synthesis of Erythromycin. 2. Synthesis of an Erythronolide A Lactone System”. J. Am. Chem. Soc. 103 (11): 3213–3215. doi:10.1021/ja00401a050.
- ^ Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung, Balaram P, Browne LJ, Card PJ, et al. (1981). “Asymmetric Total Synthesis of Erythromycin. 3. Total Synthesis of Erythromycin”. J. Am. Chem. Soc. 103 (11): 3215–3217. doi:10.1021/ja00401a051.
- ^ Hibionada, F. Remembering the battle of Dr. Abelardo Aguilar: Cure for millions, deprived of millions. The News Today. Retrieved 22 September 2015
- ^ U.S. Patent 2,653,899
- ^ Stahl, Stephanie (September 26, 2014) Health: Generic Drugs Prices Increasing Archived 2016-04-09 at the Wayback Machine CBS Philadelphia. Retrieved March 24, 2016.
- ^ “Some Generic Drugs See Huge Price Increases”. http://www.medscape.com. Retrieved 29 June 2018.
- ^ “Climbing cost of decades-old drugs threatens to break Medicaid bank”. Philly.com. Retrieved 29 June 2018.
- ^ “Are Drugs Really Getting More Expensive? Yes”. The GoodRx Prescription Savings Blog. 27 February 2018. Retrieved 29 June2018.
External links
- “Erythromycin”. Drug Information Portal. U.S. National Library of Medicine.
- U.S. Patent 2,653,899
| Clinical data | |
|---|---|
| Trade names | Eryc, Erythrocin, others[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682381 |
| License data | US DailyMed: Erythromycin |
| Pregnancy category | AU: A[2] |
| Routes of administration | By mouth, intravenous (IV), intramuscular (IM), topical, eye drops |
| Drug class | Macrolide antibiotic |
| ATC code | D10AF02 (WHO) J01FA01 (WHO) S01AA17 (WHO) QJ51FA01 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | Depends on the ester type between 30% – 65% |
| Protein binding | 90% |
| Metabolism | liver (under 5% excreted unchanged) |
| Elimination half-life | 1.5 hours |
| Excretion | bile |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 114-07-8 |
| PubChem CID | 12560 |
| IUPHAR/BPS | 1456 |
| DrugBank | DB00199 |
| ChemSpider | 12041 |
| UNII | 63937KV33D |
| KEGG | D00140 |
| ChEBI | CHEBI:42355 |
| ChEMBL | ChEMBL532 |
| PDB ligand | ERY (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID4022991 |
| ECHA InfoCard | 100.003.673 |
| Chemical and physical data | |
| Formula | C37H67NO13 |
| Molar mass | 733.937 g·mol−1 |
| hideSMILESCC[C@@H]1[C@@]([C@@H]([C@H](C(=O)[C@@H](C[C@@]([C@@H]([C@H]([C@@H]([C@H](C(=O)O1)C)O[C@H]2C[C@@]([C@H]([C@@H](O2)C)O)(C)OC)C)O[C@H]3[C@@H]([C@H](C[C@H](O3)C)N(C)C)O)(C)O)C)C)O)(C)O | |
| hideInChIInChI=1S/C37H67NO13/c1-14-25-37(10,45)30(41)20(4)27(39)18(2)16-35(8,44)32(51-34-28(40)24(38(11)12)15-19(3)47-34)21(5)29(22(6)33(43)49-25)50-26-17-36(9,46-13)31(42)23(7)48-26/h18-26,28-32,34,40-42,44-45H,14-17H2,1-13H3/t18-,19-,20+,21+,22-,23+,24+,25-,26+,28-,29+,30-,31+,32-,34+,35-,36-,37-/m1/s1 Key:ULGZDMOVFRHVEP-RWJQBGPGSA-N | |
| (verify) |
//////////erythromycin, NSC-55929, NSC 55929, эритромицин , إيريثروميسين , 红霉素 , ANTIBACTERIAL, MACROLIDES, ANTIBIOTICS
#erythromycin, #NSC-55929, #NSC 55929, #эритромицин , #إيريثروميسين , #红霉素 , #ANTIBACTERIAL, #MACROLIDES, #ANTIBIOTICS
Dirithromycin

Dirithromycin
LY 237216
- LY-237216
(1R,2R,3R,6R,7S,8S,9R,10R,12R,13S,15R,17S)-9-{[(2S,3R,4S,6R)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy}-3-ethyl-2,10-dihydroxy-7-{[(2R,4R,5S,6S)-5-hydroxy-4-methoxy-4,6-dimethyloxan-2-yl]oxy}-15-[(2-methoxyethoxy)methyl]-2,6,8,10,12,17-hexamethyl-4,16-dioxa-14-azabicyclo[11.3.1]heptadecan-5-one
UNII1801D76STL
CAS number62013-04-1
Synthesis Reference
Counter FT, Ensminger PW, Preston DA, Wu CY, Greene JM, Felty-Duckworth AM, Paschal JW, Kirst HA: Synthesis and antimicrobial evaluation of dirithromycin (AS-E 136; LY237216), a new macrolide antibiotic derived from erythromycin. Antimicrob Agents Chemother. 1991 Jun;35(6):1116-26. Pubmed.DirithromycinCAS Registry Number: 62013-04-1CAS Name: (1R,2R,3R,6R,7S,8S,9R,10R,12R,13S,15R,17S)-7-[(2,6-Dideoxy-3-C-methyl-3-O-methyl-a-L-ribo-hexopyranosyl)oxy]-3-ethyl-2,10-dihydroxy-15-[(2-methoxyethoxy)methyl]-2,6,8,10,12,17-hexamethyl-9-[[3,4,6-trideoxy-3-(dimethylamino)-b-D-xylo-hexopyranosyl]oxy]-4,16-dioxa-14-azabicyclo[11.3.1]heptadecan-5-oneAdditional Names: [9S(R)]-9-deoxo-11-deoxy-9,11-[imino[2-(2-methoxyethoxy)ethylidene]oxy]erythromycinManufacturers’ Codes: LY-237216; AS-E 136Trademarks: Dynabac (Lilly); Noriclan (Lilly); Nortron (Lilly); Valodin (Ferrer)Molecular Formula: C42H78N2O14Molecular Weight: 835.07Percent Composition: C 60.41%, H 9.41%, N 3.35%, O 26.82%Literature References: Semi-synthetic derivative of erythromycin, q.v. Prepn: BE840431 (1976 to Thomae); R. Maier et al.,US4048306 (1977 to Boehringer, Ing.). Synthesis, 1H- and 13C-NMR, and antimicrobial evaluation: F. T. Counter et al.,Antimicrob. Agents Chemother.35, 1116 (1991). X-ray structure determn: P. Luger, R. Maier, J. Cryst. Mol. Struct.9, 329 (1979). HPLC determn in plasma: G. W. Whitaker, T. D. Lindstrom, J. Liq. Chromatogr.11, 3011 (1988). Symposium on antibacterial activity, pharmacology, and clinical experience: J. Antimicrob. Chemother.31, Suppl. C, 1-185 (1993).Properties: Crystals from ethanol/water, mp 186-189° (dec) (Counter). pKa 9.0 in 66% aq dimethyl fluoride. LD50 in mice (g/kg): >1 s.c.; >1 orally (Maier).Melting point: mp 186-189° (dec) (Counter)pKa: pKa 9.0 in 66% aq dimethyl fluorideToxicity data: LD50 in mice (g/kg): >1 s.c.; >1 orally (Maier)Therap-Cat: Antibacterial.Keywords: Antibacterial (Antibiotics); Macrolides.
Dirithromycin is a macrolide glycopeptide antibiotic.[1]
For the treatment of the following mild-to-moderate infections caused by susceptible strains of microorganisms: acute bacterial exacerbations of chronic bronchitis, secondary bacterial infection of acute bronchitis, community-acquired pneumonia, pharyngitis/tonsilitis, and uncomplicated skin and skin structure infections.
Dirithromycin (Dynabac) is a more lipid-soluble prodrug derivative of 9S-erythromycyclamine prepared by condensation of the latter with 2-(2-methoxyethoxy)acetaldehyde. The 9N, 11O-oxazine ring thus formed is a hemi-aminal that is unstable under both acidic and alkaline aqueous conditions and undergoes spontaneous hydrolysis to form erythromycyclamine. Erythromycyclamine is a semisynthetic derivative of erythromycin in which the 9-ketogroup of the erythronolide ring has been converted to an amino group. Erythromycyclamine retains the antibacterial properties of erythromycin oral administration. The prodrug, dirithromycin, is provided as enteric coated tablets to protect it from acid catalyzed hydrolysis in the stomach. Orally administered dirithromycin is absorbed rapidly into the plasma, largely from the small intestine. Spontaneous hydrolysis to erythromycyclamine occurs in the plasma. Oral bioavailability is estimated to be about 10%, but food does not affect absorption of the prodrug.
NEW DRUG APPROVALS
one time
$10.00
Discontinuation
Dirithromycin is no longer available in the United States.[2] Since the production of dirithromycin is discontinued in the U.S, National Institutes of Health recommend that people taking dirithromycin should consult their physicians to discuss switching to another treatment.[3] However, dirithromycin is still available in many European countries.
Clip
https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/chem.201902716

In attempts to modify the C-9 keto moiety of erythromycin, (9S)-erythromycinylamine (21) was prepared by the reduction of oxime 17 with sodium borohydride (Scheme 4).[13] Amine 21 displayed good in vitro antimicrobial activity against Staphylococcus aureus, [38–44] but had poor bioavailability due to the polar primary amine. In search of compounds in this class with better oral bioavailability, efforts were directed towards masking the amine in 21 as an imine with aromatic and aliphatic aldehydes.[40] These efforts were based on the idea that such imines would be hydrolysed at physiological pH after absorption from the intestine, but somewhat unexpectedly, lead to the discovery of dirithromycin (4) when 21 was treated with aldehyde 22. In this reaction, 9- N-11-O-oxazine epi-dirithromycin (23) is first formed as the kinetic product, which then undergoes conversion into the thermodynamically stable dirithromycin (4).[45–47] Due to issues with the stability of aldehyde 22 on process-scale synthesis, this procedure was later modified so that dimethyl acetal 24 was used for commercial production.[48]
13] S. Djokic´, Z. Tamburasˇev, Tetrahedron Lett. 1967, 8, 1645 – 1647.
[38] R. Maier, E. Woitun, B. Wetzel, W. Reuter, H. Goeth, U. Lechner, 1977, US4048306A. [39] E. Wildsmith, 1974, US3780019A. [40] E. H. Massey, B. S. Kitchell, L. D. Martin, K. Gerzon, J. Med. Chem. 1974, 17, 105 – 107. [41] E. Wildsmith, Tetrahedron Lett. 1972, 13, 29 – 30. [42] K. Gerzon, M. H. William, DPMA Deutsches Patent, 1972, DE1966310A1. [43] G. H. Timms, E. Wildsmith, Tetrahedron Lett. 1971, 12, 195 – 198. [44] E. H. Massey, B. Kitchell, L. D. Martin, K. Gerzon, H. W. Murphy, Tetrahedron Lett. 1970, 11, 157 – 160. [45] P. Luger, R. Maier, J. Cryst. Mol. Struct. 1979, 9, 329 – 338. [46] F. T. Counter, P. W. Ensminger, D. A. Preston, C. Y. Wu, J. M. Greene, A. M. Felty-Duckworth, J. W. Paschal, H. A. Kirst, Antimicrob. Agents Chemother. 1991, 35, 1116 – 1126. [47] J. Firl, A. Prox, P. Luger, R. Maier, E. Woitun, K. Daneck, J. Antibiot. 1990, 43, 1271 – 1277. [48] J. M. Mcgill, Synthesis 1993, 11, 1089 – 1091.
Clip
Dirithromycin is the second-generation erythromycin macrocyclic (fourteen member ring) lactone antibiotics; made from the condensation reaction between 2-methoxyethoxy acetaldehyde and erythromycylamine. It has similar structure to erythromycin. It can subject to in vivo non-enzymatic hydrolysis into erythromycin cyclic amines. It takes effect through targeting the 50S ribosomal subunit of sensitive pathogenic microorganisms, blocking the bacterial peptide bond formation, which further inhibits protein synthesis to play antibacterial activity.
Compared with erythromycin and other new macrocyclic lactone antibiotics, this drug has the following characteristics: (1) antibacterial effect: in addition to retaining the antibacterial effect against gram positive bacteria; it also has strong effect on a variety of G- bacteria, Anaerobic bacteria and other pathogens, such as Mycoplasma, Chlamydia and spirochete. Dirithromycin has stronger effect than erythromycin on Staphylococcus aureus and Staphylococcus epidermidis. (2) Pharmacokinetics: compared with other macrolide antibiotics in the vine, the half-life of erythromycin is longer with the plasma elimination tl/2 being longer than 24h. Its tissue permeability is strong. It can be administered once a day. So it will also be competitive in the market with characteristics that are different from other antibiotics.
Lilly’s products in the United States was listed in Spain in September 1993, listed in 1996 in US after the approval of FDA and had been included in Pharmacopoeia USP 23; it was listed in 2005 in the domestic market. At present, there are a number of domestic dysthromycin enteric-coated tablets and enteric-coated capsules approved for clinical use.
Synthetic route
Route 1: erythromycin is first reacted with hydrazine hydrate to generate erythromycin hydrazone (2), erythromycin hydrazone is used for synthesizing erythromycylamine (3), and finally reacted with 2-methoxyethoxy acetaldehyde (5) to generate dysthromycin (1), as shown in the figure:
Route 2: Erythromycin is reacted with hydroxylamine to generate erythromycin oxime; erythromycin oxime can be reduced to obtain erythromycin amine, and is then condensed with 2- (2- methoxyethoxy) acetaldehyde ethylene glycol to generate dysthromycin (DRM), the specific reaction route is as follows:

Clip
https://link.springer.com/article/10.1007/s00894-003-0172-7
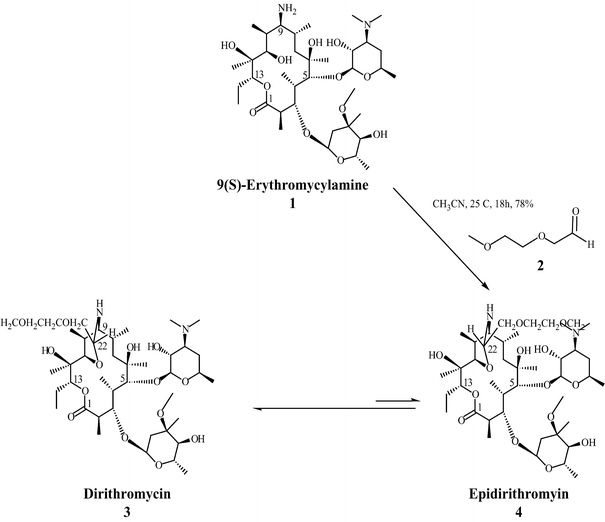
References
- ^ McConnell SA, Amsden GW (April 1999). “Review and comparison of advanced-generation macrolides clarithromycin and dirithromycin”. Pharmacotherapy. 19 (4): 404–15. doi:10.1592/phco.19.6.404.31054. PMID 10212011.
- ^ “Dynabac Drug Details”. U.S. Food and Drug Administration. Retrieved 2007-05-25.
- ^ “Dirithromycin”. MedlinePlus. U.S. National Library of Medicine. January 1, 2006. Archived from the original on 2007-03-29. Retrieved 2007-05-25.
| Clinical data | |
|---|---|
| Trade names | Dynabac |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a604026 |
| License data | US FDA: Clarithromycin |
| Pregnancy category | B |
| Routes of administration | Oral |
| ATC code | J01FA13 (WHO) |
| Pharmacokinetic data | |
| Bioavailability | 10% |
| Protein binding | 15 to 30% |
| Metabolism | Hyrolized to erythromycyclamine in 1.5 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 62013-04-1 |
| PubChem CID | 6917067 |
| DrugBank | DB00954 |
| ChemSpider | 5292341 |
| UNII | 1801D76STL |
| KEGG | D03865 |
| ChEBI | CHEBI:474014 |
| ChEMBL | ChEMBL3039471 |
| CompTox Dashboard (EPA) | DTXSID7048956 |
| ECHA InfoCard | 100.152.704 |
| Chemical and physical data | |
| Formula | C42H78N2O14 |
| Molar mass | 835.086 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 186 to 189 °C (367 to 372 °F) (dec.) |
| hideSMILESO=C4O[C@@H]([C@](O)(C)[C@H]1O[C@@H](N[C@H]([C@@H]1C)[C@H](C)C[C@](O)(C)[C@H](O[C@@H]2O[C@H](C)C[C@H](N(C)C)[C@H]2O)[C@H]([C@H](O[C@@H]3O[C@@H](C)[C@H](O)[C@@](OC)(C)C3)[C@H]4C)C)COCCOC)CC | |
| hideInChIInChI=1S/C42H78N2O14/c1-15-29-42(10,49)37-24(4)32(43-30(56-37)21-52-17-16-50-13)22(2)19-40(8,48)36(58-39-33(45)28(44(11)12)18-23(3)53-39)25(5)34(26(6)38(47)55-29)57-31-20-41(9,51-14)35(46)27(7)54-31/h22-37,39,43,45-46,48-49H,15-21H2,1-14H3/t22-,23-,24+,25+,26-,27+,28+,29-,30-,31+,32+,33-,34+,35+,36-,37+,39+,40-,41-,42-/m1/s1 Key:WLOHNSSYAXHWNR-NXPDYKKBSA-N | |
| (what is this?) (verify) |
/////////// Dirithromycin, LY 237216, LY-237216, Antibacterial
#Dirithromycin, #LY 237216, #LY-237216, #Antibacterial
Brivudine
Brivudine
ブリブジン;
| Formula | C11H13BrN2O5 |
|---|---|
| CAS | 69304-47-8 |
| Mol weight | 333.1353 |
(E)-5-(2-Bromovinyl)-2′-deoxyuridine2M3055079H5-[(E)-2-bromoethenyl]-2′-deoxyuridine5-[(E)-2-Bromovinyl]-2′-deoxyuridine
626769304-47-8[RN]BrivudineCAS Registry Number: 69304-47-8CAS Name: 5-[(1E)-2-Bromoethenyl]-2¢-deoxyuridineAdditional Names: (E)-5-(2-bromovinyl)-2¢-deoxyuridine; brivudin; BVDUTrademarks: Brivex (Menarini); Brivirac (Menarini); Nervinex (Menarini); Zecovir (Guidotti); Zostex (Berlin-Chemie)Molecular Formula: C11H13BrN2O5Molecular Weight: 333.14Percent Composition: C 39.66%, H 3.93%, Br 23.99%, N 8.41%, O 24.01%Literature References: Analog of thymidine, q.v., with selective activity against herpes simplex virus type 1 and varicella-zoster virus. Prepn: A. S. Jones et al.,DE2915254; eidem, US4424211 (1979, 1984 both to University of Birmingham and Rega Institut); and antiviral activity: E. De Clercq et al,Proc. Natl. Acad. Sci. USA76, 2947 (1979). Mechanism of action studies: H. S. Allaudeen et al.,ibid.78, 2698 (1981); J. Balzarini, E. De Clercq, Methods Find. Exp. Clin. Pharmacol.11, 379 (1989). Cytotoxic properties vs viral tumor cells: C. Grignet-Debrus et al.,Cancer Gene Ther.7, 215 (2000). CE determn in plasma and urine: J. Olgemöller et al.,J. Chromatogr. B726, 261 (1999). Clinical evaluation in herpetic keratitis: P. C. Maudgal, E. De Clercq, Curr. Eye Res.10, Suppl., 193 (1991). Clinical comparison with acyclovir, q.v., in herpes zoster: S. W. Wassilew et al., Antiviral Res.59, 49, 57 (2003). Review of pharmacology and clinical efficacy in herpes zoster: S. J. Keam et al.,Drugs64, 2091-2097 (2004); of antiviral activity, mechanism of action, and clinical efficacy: E. De Clercq, Med. Res. Rev.25, 1-20 (2005).Properties: White needles from methanol-water, mp 123-125° (dec). uv max: 253, 295 nm (e 13100, 10300).Melting point: mp 123-125°Absorption maximum: uv max: 253, 295 nm (e 13100, 10300)Therap-Cat: Antiviral.Keywords: Antiviral; Purines/Pyrimidinones.
Brivudine (trade names Zostex, Mevir, Brivir, among others) is an antiviral drug used in the treatment of herpes zoster (“shingles”). Like other antivirals, it acts by inhibiting replication of the target virus.
Medical uses
Brivudine is used for the treatment of herpes zoster in adult patients. It is taken orally once daily, in contrast to aciclovir, valaciclovir and other antivirals.[1] A study has found that it is more effective than aciclovir, but this has been disputed because of a possible conflict of interest on part of the study authors.[2]
Contraindications
The drug is contraindicated in patients undergoing immunosuppression (for example because of an organ transplant) or cancer therapy, especially with fluorouracil (5-FU) and chemically related (pro)drugs such as capecitabine and tegafur, as well as the antimycotic drug flucytosine, which is also related to 5-FU. It has not been proven to be safe in children and pregnant or breastfeeding women.[1]
Adverse effects
The drug is generally well tolerated. The only common side effect is nausea (in 2% of patients). Less common side effects (<1%) include headache, increased or lowered blood cell counts (granulocytopenia, anaemia, lymphocytosis, monocytosis), increased liver enzymes, and allergic reactions.[1]
Interactions
Brivudine interacts strongly and in rare cases lethally with the anticancer drug fluorouracil (5-FU), its prodrugs and related substances. Even topically applied 5-FU can be dangerous in combination with brivudine. This is caused by the main metabolite, bromovinyluracil (BVU), irreversibly inhibiting the enzyme dihydropyrimidine dehydrogenase (DPD) which is necessary for inactivating 5-FU. After a standard brivudine therapy, DPD function can be compromised for up to 18 days. This interaction is shared with the closely related drug sorivudine which also has BVU as its main metabolite.[1][3]
There are no other relevant interactions. Brivudine does not significantly influence the cytochrome P450 enzymes in the liver.[1]
Pharmacology
Spectrum of activity
The drug inhibits replication of varicella zoster virus (VZV) – which causes herpes zoster – and herpes simplex virus type 1 (HSV-1), but not HSV-2 which typically causes genital herpes. In vitro, inhibitory concentrations against VZV are 200- to 1000-fold lower than those of aciclovir and penciclovir, theoretically indicating a much higher potency of brivudine. Clinically relevant VZV strains are particularly sensitive.[4]
Mechanism of action
Brivudine is an analogue of the nucleoside thymidine. The active compound is brivudine 5′-triphosphate, which is formed in subsequent phosphorylations by viral (but not human) thymidine kinase and presumably by nucleoside-diphosphate kinase. Brivudine 5′-triphosphate works because it is incorporated into the viral DNA, but then blocks the action of DNA polymerases, thus inhibiting viral replication.[1][4]
Pharmacokinetics
Brivudine is well and rapidly absorbed from the gut and undergoes first-pass metabolism in the liver, where the enzyme thymidine phosphorylase[5] quickly splits off the sugar component, leading to a bioavailability of 30%. The resulting metabolite is bromovinyluracil (BVU), which does not have antiviral activity. BVU is also the only metabolite that can be detected in the blood plasma.[1][6]
Highest blood plasma concentrations are reached after one hour. Brivudine is almost completely (>95%) bound to plasma proteins. Terminal half-life is 16 hours; 65% of the substance are found in the urine and 20% in the faeces, mainly in form of an acetic acid derivative (which is not detectable in the plasma), but also other water-soluble metabolites, which are urea derivatives. Less than 1% is excreted in form of the original compound.[1]
- Brivudine 5′-triphosphate, the active metabolite
- Bromovinyluracil (BVU), the main inactive metabolite
- The acetic acid derivative predominantly found in urine
Chemistry
The molecule has three chiral carbon atoms in the deoxyribose (sugar) part all of which have defined orientation; i.e. the drug is stereochemically pure.[1] The substance is a white powder.
Manufacturing
Main supplier is Berlin-Chemie, now part of Italy’s Menarini Group. In Central America is provided by Menarini Centro America and Wyeth.
History
The substance was first synthesized by scientists at the University of Birmingham in the UK in 1976. It was shown to be a potent inhibitor of HSV-1 and VZV by Erik De Clercq at the Rega Institute for Medical Research in Belgium in 1979. In the 1980s the drug became commercially available in East Germany, where it was marketed as Helpin by a pharmaceutical company called Berlin-Chemie. Only after the indication was changed to the treatment of herpes zoster in 2001 did it become more widely available in Europe.[7][8]
Brivudine is approved for use in a number of European countries including Austria, Belgium, Germany, Greece, Italy, Portugal, Spain and Switzerland.[9]
Etymology
The name brivudine derives from the chemical nomenclature bromo–vinyl–deoxyuridine or BVDU for short. It is sold under trade names such as Bridic, Brival, Brivex, Brivir, Brivirac, Brivox, Brivuzost, Zerpex, Zonavir, Zostex, and Zovudex.[9]
Research
A Cochrane Systematic Review examined the effectiveness of multiple antiviral drugs in the treatment of herpes simplex virus epithelial keratitis. Brivudine was found to be significantly more effective than idoxuridine in increasing the number of successfully healed eyes of participants.[10]
PATENT
EP-03792271
Process for preparing brivudine, useful for treating herpes zoster infection and cancer (eg pancreatic cancer). Also claims novel intermediate of brivudine. Brivudine is an antiviral drug approved under the brand name Zostex®. Represents the first patenting to be seen from Aurobindo that focuses on brivudine.
Brivudine is chemically known as 5-[(lE)-2-bromoethenyl]-2′-deoxyuridine. Brivudine is an analogue of the nucleoside thymidine with high and selective antiviral activity against varicella zoster virus and herpes simplex virus. Brivudine is an antiviral drug approved under the brand name Zostex® for treatment of herpes zoster. Brivudine is also useful to inhibit the upregulation of chemoresistance genes (Mdr1 and DHFR) during chemotherapy. Overall, the gene expression changes associated with Brivudine treatment result in the decrease or prevention of chemoresistance. In addition, it has been shown to enhance the cytolytic activity of NK-92 natural killer cells towards a pancreatic cancer cell line in vitro.
[0003] Brivudine (I) is disclosed first time in DE 2915254. This patent discloses a process for the preparation of Brivudine by coupling E-5-(2-bromovinyl) uracil with 1-chloro-2-deoxy-3,5-di-O-p-toluoyl-α-D-erythro-pentofuranose to obtain E-5-(2-bromovinyl)-3′,5′-di-O-p-toluoyl-2′-deoxyuridine as a mixture of α and β isomers. This mixture was subjected to chromatographic purification to obtain pure β-isomer. In the subsequent stage E-5-(2-bromovinyl)-3′,5′-di-O-p-toluoyl-2′-deoxyuridine was treated with sodium methoxide to yield Brivudine. The process is depicted in the below as Scheme I:
[0004] The major disadvantages associated with the process disclosed in DE 2915254 includes the use of expensive starting material, formation of unwanted excess of α-isomer. The undesired α-isomer result in a final product of low purity, making chromatographic purification methods not feasible at an industrial scale. Additionally, the process involves the use of bromine for the synthesis of E-5-(2-bromovinyl)uracil, which is a well known carcinogen.
[0005] GB 2125399 describe another process for the preparation of Brivudine involves the bromination and simultaneous dehydrohalogenation of 5-ethyl-2′-deoxyuridine in the presence of halogenated hydrocarbon solvent. The process is depicted in the below as Scheme – II:
[0006] The major disadvantages associated with the process disclosed in GB 2125399 includes the use of bromine for bromination, which make the process carcinogenic and the use of halogenated solvents like chloroform, carbon tetrachloride and dichloroethane for bromination makes the process environmentally hazardous.
[0007] US 2010298530 A1 discloses a process for the preparation of Brivudine by coupling 5-Iodo deoxyuridine with methyl acrylate in presence of palladium acetate to form (E)-5-(carbomethoxyvinyl)-2′-deoxyuridine, which is hydrolyzed with sodium hydroxide solution to obtain (E)-5-(carboxyvinyl)-2′-deoxyuridine, which undergoes bromination by using N-Bromo succinimide [NBS] to obtain Brivudine. The process is depicted in the below as Scheme – III:
[0008] The major disadvantages associated with the process disclosed in US 2010298530 A1 includes the use of expensive palladium acetate as catalyst and chromatographic purification method not feasible at an industrial scale. In addition, the above process involves the use of methyl acrylate and the process liberates iodine, which are highly carcinogenic. It makes the process environment unfriendly.
[0009] The inventors of the present invention found an alternative route to prepare Brivudine (I), which is industrial feasible, can avoid the use of potentially hazardous, expensive chemicals and to minimize the formation of undesired α-isomer and the other process related impurities. The present invention directed towards a process for the preparation of Brivudine of Formula – I with high purity and high yield.
EXAMPLE-1:
PREPARATION OF URIDINE ACRYLIC ACID
[0026] Trimethylchlorosilane (0.6 ml, 5 mmol) was added to the suspension of uracil acrylic acid (6.35g, 34.9 mmol) in hexamethyldisilazane (70 ml) and resulting mixture was refluxed till the clear solution was obtained. Hexamethyldisilazane was evaporated under vacuum and further co-evaporated with o-xylene to remove the traces of hexamethyldisilazane to yield viscous oily silylated uracil acrylic acid. The silylated uracil acrylic acid was dissolved in dichloromethane (100 ml) under nitrogen atmosphere, cooled at 0-10 °C. Anhydrous zinc chloride (0.63 grams, 4.6 mmol) and chloro-sugar (10 grams, 23.2 mmol) were added to the above solution. The reaction was monitored by qualitative HPLC and was essentially completed in 3 hours. After completion of the reaction dichloromethane was evaporated under vacuum. Methyl tert-butyl ether (100 ml) was added and stirred for 1 hour at 40-45 °C. The product was isolated after filtration at 25-30 °C. HPLC analysis showed the complete consumption of chloro-sugar and the ratio β/α = 98.
Yield: 75%
EXAMPLE-2:
PREPARATION OF DIBENZOYL BRIVUDINE
[0027] Uridine acrylic acid (5.0 grams, 8.69 mmol) was suspended in a mixture of tetrahydrofuran (45.0 ml) and water (5.0 ml). Potassium acetate (0.93 grams, 9.56 mmol) and N-bromosuccinamide (1.70 grams, 9.56 mmol) was added to the suspension and the resulting mixture was stirred for 2 hours at 25-30 °C. The solvent was removed under reduced pressure to the dryness and methanol (50 ml) was poured, suspension was stirred for 1 hour at 25-30 °C. The product was isolated after filtration.
Yield: 55%
EXAMPLE-3:
PREPARATION OF BRIVUDINE (FORMULA – I)
[0028] Dibenzoyl Brivudine (6 grams, 9.83 mmol) was suspended in methanol (30 ml) at 20-30 °C. A solution of 25 % w/w sodium methoxide (2.76 grams, 12.78 mmol) in methanol was added to the suspension and was allowed for 1hour at the same temp. The reaction was monitored by qualitative HPLC. Methanol was evaporated under vacuum and resulting residue was dissolved in water (25 ml). The aqueous mass was washed with methylene dichloride (2×20 ml) and the product was isolated from water at pH 2-3.
Yield: 85%
SYN
http://sioc-journal.cn/Jwk_yjhx/EN/10.6023/cjoc201410034

In this paper, a simple and practical method for the preparation of brivudine (BVDU) and its analog nucleoside derivatives via condensation of the easily obtainable 5-formyl pyrimidine nucleosides with carbon tetrabromide followed by an efficient and stereoselective debromination promoted by diethyl phosphite and triethylamine is presented.
Li Peiyuan, Zhang Jianrui, Guo Shenghai, Zhang Xinying, Fan Xuesen. New Synthesis of Brivudine and Its Analogs[J]. Chin. J. Org. Chem., 2015, 35(4): 910-916.
SYN 1
Tetrahedron Lett 1979,454415-8
J Carbohydates Nucleosides Nucleotides 1977,4(5),4415
5-Chloromercuri-2′-deoxyuridine (II) is prepared from 2′-deoxyuridine (I) by reaction with mercuriacetate and natrium chloride (1). Condensation of (II) with ethylacrylate (A) and lithium palladium chloride gives (E)-5-(2-carbethoxyvinyl)-2′-deoxyuridine (III), which is readily hydrozyled to (E)-5-(2-carboxyvinyl)-2′-deoxyuridine (IV) under basic conditions (0.5M NaOH). The final step involves the reaction of (IV) with N-bromosuccinimide to produce (E)-5-(2-bromovinyl)-2′-deoxyuridine.
SYN 2
he condensation of 2-deoxy-3,5-di-O-(phenylacetyl)-beta-D-erythro-pentofuranosyl chloride (I) with 2,4-bis-O-(trimethylsilyl)-5(E)-(2-bromovinyl)uracil (II) in acetonitrile (Lewis acid catalyst), or in CHCl3-pyridine (Bronsted acid catalyst), gives 3′,5′-di-O-(phenylacetyl)-5(E)-(2-bromovinyl)-2′-deoxyuridine (III) and its anomer that is eliminated by TLC (silicagel). Finally, (III) is treated with sodium methoxide in methanol.
| Int Symp: Basic Clin Approach Virus Chemother 1988,Poster M17 |
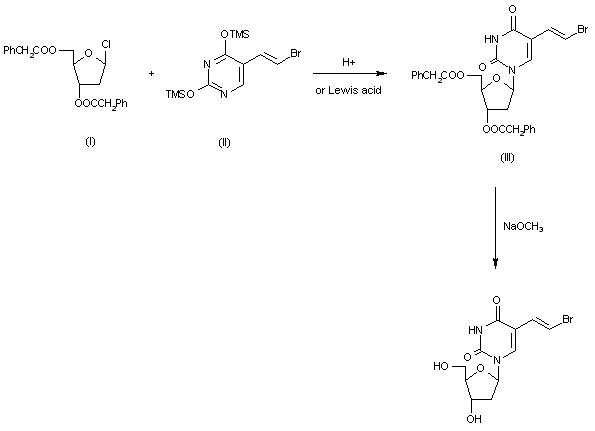
SYN
- Synthetic Method of Brivudine
- (CAS NO.: ), with its systematic name of (E)-5-(2-Bromovinyl)-2′-deoxyuridine, could be produced through many synthetic methods.Following is one of the reaction routes:
 2-Deoxy-3,5-di-O-(phenylacetyl)-beta-D-erythro-pentofuranosyl chloride (I) is condensed with 2,4-bis-O-(trimethylsilyl)-5(E)-(2-bromovinyl)uracil (II) in acetonitrile (Lewis acid catalyst), or in CHCl3-pyridine (Bronsted acid catalyst), to produce 3,5-di-O-(phenylacetyl)-5(E)-(2-bromovinyl)-2-deoxyuridine (III) and its anomer that is eliminated by TLC (silicagel). Finally, (III) is treated with sodium methoxide in methanol.
2-Deoxy-3,5-di-O-(phenylacetyl)-beta-D-erythro-pentofuranosyl chloride (I) is condensed with 2,4-bis-O-(trimethylsilyl)-5(E)-(2-bromovinyl)uracil (II) in acetonitrile (Lewis acid catalyst), or in CHCl3-pyridine (Bronsted acid catalyst), to produce 3,5-di-O-(phenylacetyl)-5(E)-(2-bromovinyl)-2-deoxyuridine (III) and its anomer that is eliminated by TLC (silicagel). Finally, (III) is treated with sodium methoxide in methanol.
References
- ^ Jump up to:a b c d e f g h i Jasek W, ed. (2007). Austria-Codex (in German) (62nd ed.). Vienna: Österreichischer Apothekerverlag. pp. 5246–8. ISBN 978-3-85200-181-4.
- ^ “Brivudin (Zostex) besser als Aciclovir (Zovirax a.a.)?”. Arznei-telegramm (in German). 5/2007.
- ^ “UAW – Aus Fehlern lernen – Potenziell tödlich verlaufende Wechselwirkung zwischen Brivudin (Zostex) und 5-Fluoropyrimidinen” (PDF). Deutsches Ärzteblatt (in German). 103 (27). 7 July 2006.
- ^ Jump up to:a b Steinhilber D, Schubert-Zsilavecz M, Roth HJ (2005). Medizinische Chemie (in German). Stuttgart: Deutscher Apotheker Verlag. pp. 581–2. ISBN 3-7692-3483-9.
- ^ Desgranges C, Razaka G, Rabaud M, Bricaud H, Balzarini J, De Clercq E (December 1983). “Phosphorolysis of (E)-5-(2-bromovinyl)-2′-deoxyuridine (BVDU) and other 5-substituted-2′-deoxyuridines by purified human thymidine phosphorylase and intact blood platelets”. Biochemical Pharmacology. 32 (23): 3583–90. doi:10.1016/0006-2952(83)90307-6. PMID 6651877.
- ^ Mutschler E, Schäfer-Korting M (2001). Arzneimittelwirkungen (in German) (8 ed.). Stuttgart: Wissenschaftliche Verlagsgesellschaft. p. 847. ISBN 3-8047-1763-2.
- ^ De Clercq E (December 2004). “Discovery and development of BVDU (brivudin) as a therapeutic for the treatment of herpes zoster”. Biochemical Pharmacology. 68 (12): 2301–15. doi:10.1016/j.bcp.2004.07.039. PMID 15548377.
- ^ Tringali C, ed. (2012). Bioactive Compounds from Natural Sources (2nd ed.). CRC Press. p. 170.
- ^ Jump up to:a b International Drug Names: Brivudine.
- ^ Wilhelmus KR (January 2015). “Antiviral treatment and other therapeutic interventions for herpes simplex virus epithelial keratitis”. The Cochrane Database of Systematic Reviews. 1: CD002898. doi:10.1002/14651858.CD002898.pub5. PMC 4443501. PMID 25879115.
| Clinical data | |
|---|---|
| Trade names | Zostex, Mevir, Brivir, many others |
| Other names | BVDU |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category | Contraindicated |
| Routes of administration | Oral |
| ATC code | J05AB15 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 30% |
| Protein binding | >95% |
| Metabolism | Thymidine phosphorylase |
| Metabolites | Bromovinyluracil |
| Elimination half-life | 16 hours |
| Excretion | 65% renal (mainly metabolites), 20% faeces |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 69304-47-8 |
| PubChem CID | 446727 |
| ChemSpider | 394011 |
| UNII | 2M3055079H |
| KEGG | D07249 |
| ChEMBL | ChEMBL31634 |
| CompTox Dashboard (EPA) | DTXSID0045755 |
| Chemical and physical data | |
| Formula | C11H13BrN2O5 |
| Molar mass | 333.138 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Specific rotation | +9°±1° |
| Density | 1.86 g/cm3 |
| Melting point | 165 to 166 °C (329 to 331 °F) (decomposes) |
| hideSMILESBr[C@H]=CC=1C(=O)NC(=O)N(C=1)[C@@H]2O[C@@H]([C@@H](O)C2)CO | |
| hideInChIInChI=1S/C11H13BrN2O5/c12-2-1-6-4-14(11(18)13-10(6)17)9-3-7(16)8(5-15)19-9/h1-2,4,7-9,15-16H,3,5H2,(H,13,17,18)/b2-1+/t7-,8+,9+/m0/s1 Key:ODZBBRURCPAEIQ-PIXDULNESA-N |
Sinovac COVID-19 vaccine, CoronaVac,

Sinovac COVID-19 vaccine, CoronaVac,
- PiCoVacc
CoronaVac, also known as the Sinovac COVID-19 vaccine,[1] is an inactivated virus COVID-19 vaccine developed by the Chinese company Sinovac Biotech.[2] It has been in Phase III clinical trials in Brazil,[3] Chile,[4] Indonesia,[5] the Philippines,[6] and Turkey.[7]
It relies on traditional technology similar to BBIBP-CorV and BBV152, other inactivated-virus COVID-19 vaccines in Phase III trials.[8] CoronaVac does not need to be frozen, and both the vaccine and raw material for formulating the new doses could be transported and refrigerated at 2–8 °C (36–46 °F), temperatures at which flu vaccines are kept.[9]
Brazil announced results on 13 January 2021 showing 50.4% effective at preventing symptomatic infections, 78% effective in preventing mild cases needing treatment, and 100% effective in preventing severe cases.[10] Final Phase III results from Turkey announced on 3 March 2021 showed an efficacy of 83.5%.[11] Interim results in Indonesia were announced on 11 January 2021 with an efficacy of 65.3%.[12] A detailed report containing confidence intervals, efficacy by age and side effects has not yet been released.
CoronaVac is being used in vaccination campaigns by certain countries in Asia,[13][14][15] South America,[16][17][18] North America,[19][20] and Europe.[21] In March, a Sinovac spokesman told Reuters production capacity for CoronaVac could reach 2 billion doses a year by June 2021.[22] As of March 21, 70 million doses of CoronaVac had been administered worldwide.[23
Technology
CoronaVac is an inactivated vaccine. It uses a similar, more traditional technology as in BBIBP-CorV and BBV152, other inactivated-virus vaccines for COVID-19 in Phase III trials.[24][25] CoronaVac does not need to be frozen, and both the vaccine and raw material for formulating the new doses could be transported and refrigerated at 2–8 °C (36–46 °F), temperatures at which flu vaccines are kept.[26] CoronaVac could remain stable for up to three years in storage, which might offer some advantage in vaccine distribution to regions where cold chains are not developed.[27]
NEW DRUG APPROVALS
one time
$10.00
Efficacy

Empty bottle of CoronaVac
On 7 January 2021, results from Phase III trials in Brazil among 13,000 volunteers revealed the vaccine was 78% effective in preventing symptomatic cases of COVID-19 requiring medical assistance (grade 3 on the WHO Clinical Progression Scale[28]) and 100% effective against moderate and severe infections.[29] After mounting pressure from scientists, Butantan said on 12 January that these rates only included volunteers who had mild to severe cases of COVID-19.[30] The overall efficacy, including asymptomatic cases and symptomatic cases not requiring medical assistance (WHO grade 2), was 50.38%.[31] Of the 220 participants infected, 160 cases were in the placebo group and 60 cases in the group that received CoronaVac.[32]
On 3 March 2021, final Phase III results from Turkey showed an efficacy of 83.5%. The final efficacy rate was based on 41 infections, 32 of which had received a placebo, said Murat Akova, head of the Phase III trials in Turkey. He added the vaccine prevented hospitalization and severe illness in 100% of cases, saying six people who were hospitalized were all in the placebo group. The final results were based on a 10,216 participants, 6,648 of whom received the vaccine as part of the Phase III study that began mid-September. Turkey had announced an interim result with 29 infections in December, which placed the efficacy at 91.25%.[33][34]
On 11 January, Indonesia released Phase III results from an interim analysis of 25 cases which showed an efficacy rate of 65.3% based on data of 1,600 participants in the trial.[35] The trial was conducted in the city of Bandung, and it was not clear how Indonesian scientists made their calculations.[30]
Variability in results
Officials said the lowered figure of 50.4% included “very light” cases of COVID-19 among participants omitted in the earlier analysis. Ricardo Palácios, Medical Director of Instituto Butantan said Sinovac’s relatively low efficacy rate of 50% was due to more rigorous standards for what counts as an infection among trial participants. The Institute included six types of cases in its results: asymptomatic, very mild, mild, two levels of moderate, and severe, while western vaccine makers generally included only mild, moderate, and severe categories. Brazil’s trial was also largely made up of frontline health care workers. “They are more exposed to the virus and may explain the relatively low efficacy rate,” said Yanzhong Huang, a senior fellow for global health at the Council on Foreign Relations.[36]
The release of more definitive data on CoronaVac’s efficacy was delayed because Sinovac needed to reconcile results from different trials using varying protocols.[32] According to Instituto Butantan director Dimas Covas, the Brazilian group was considered more vulnerable to infection and exposure to higher viral loads. In Turkish and Indonesian Phase III trials, the composition of volunteers was similar to that of the general population.[37]
COVID-19 variants
On March 10, Instituto Butantan Director Dimas Covas said CoronaVac was efficient against three variants of COVID-19 in the country; British B.1.1.7, South African 501.V2, and Brazil’s P.1, of which are derived variants P.1 from Manaus state, and P.2 from Rio de Janeiro.[38]
CoronaVac and other inactivated virus vaccines have all parts of the virus. Butantan said this may generate a more comprehensive immune response compared to other vaccines using only a part of the spike protein used by COVID-19 to infect cells. Tests run by Butantan used the serum of vaccinated people, which are placed in a cell culture and subsequently infected with the variants. The neutralization consists of determining whether antibodies generated from the vaccine will neutralize the virus in the culture.[38]
Clinical trials
For broader coverage of this topic, see COVID-19 vaccine.
Phase I–II
In a Phase II clinical trial completed in July 2020 and published in The Lancet, CoronaVac showed seroconversion of neutralising antibodies for 109 (92%) of 118 participants in the 3 μg group, 117 (98%) of 119 in the 6 μg group, after the days 0 and 14 schedule; whereas at day 28 after the days 0 and 28 schedule, seroconversion was seen in 114 (97%) of 117 in the 3 μg group, 118 (100%) of 118 in the 6 μg group.[39]
In May, CoronaVac began Phase I–II trials in China on adults over the age 60, and in September CoronaVac began Phase I–II trials in China on children ages 3–17.[40] Phase II results for older adults published in The Lancet showed CoronaVac was safe and well tolerated in older adults, with neutralising antibody induced by a 3 μg dose were similar to those of a 6 μg dose.[41]
Phase III
Latin America
In late July 2020, Sinovac began conducting a Phase III vaccine trial to evaluate efficacy and safety on 9,000 volunteer healthcare professionals in Brazil, collaborating with Butantan Institute.[42][43] On 19 October, São Paulo Governor João Doria said the first results of the clinical study conducted in Brazil proved that among the vaccines being tested in the country, CoronaVac is the safest, the one with the best and most promising immunization rates.[44] On 23 October, São Paulo announced the creation of six new centers for trials of CoronaVac, increasing the number of volunteers in the trials to 13,000.[45]
Brazil briefly paused Phase III trials on 10 November after the suicide of a volunteer before resuming on 11 November. Instituto Butantan said the suicide had no relation to the vaccine trial.[46][47]
In August, a Phase III trial was started in Chile, headed by Pontifical Catholic University of Chile, which was expected to include 3,000 volunteers between the ages of 18 and 65.[48]
Europe
In September, Turkey began Phase III trials with 13,000 volunteers on a two-dose 14-day interval.[49] The monitoring process for CoronaVac is underway at 25 centers in 12 cities across the country.[50]

The Governor of West Java Ridwan Kamil participating in phase 3 trial of the Sinovac COVID-19 vaccine in Indonesia.
Asia
In August, Sinovac began Phase III trials in Indonesia with Bio Farma in Bandung involving 1,620 volunteers.[51] In November, Padjadjaran University Medical School provided an update that the trials were running smoothly and that “at most, they found a slight body fever which disappeared within two days”.[52]
In October, Saudi Arabia signed an agreement with Sinovac to distribute CoronaVac to 7,000 healthcare workers, after conducting Phase III trials with the Saudi Arabian National Guard.[53]
Manufacturing
_2021_A.jpg)
Brazilian version of CoronaVac, manufactured by Butantan
In March, a Sinovac spokesman told Reuters production capacity for CoronaVac could reach 2 billion doses a year by June. The figure is double the capacity of 1 billion doses in bulk ingredients the firm said it could reach by February.[22]
After Indonesia’s Phase III trials, Bio Farma plans to ramp up production to 250 million doses a year.[54]
On 9 November, São Paulo began building a facility to produce 100 million doses a year.[55] On 10 December, João Doria said Butantan aimed to fill and finish 1 million doses per day on its production line for a vaccination campaign starting 25 January. Doria said 11 Brazilian states have contacted Butantan seeking doses of CoronaVac.[56]
In Malaysia, Pharmaniaga will manufacture, fill, and finish CoronaVac. Pharmaniaga signed a deal to obtain bulk supply of the vaccine as well as technology transfer from Sinovac.[57]
In Egypt, the government was in “advanced stage” discussions with Sinovac to manufacture CoronaVac for local use and export to African countries.[58]
Market and deployment
As of March 21, 70 million doses of CoronaVac had been administered worldwide.[23]
| show Full authorizationshow Emergency authorization Eligible COVAX recipient (assessment in progress)[80] |
South America
.jpg)
São Paulo State Secretary of Health Jean Gorinchteyn (left) and Instituto Butantan chairman Dimas Covas (right) holding single-dose prefilled syringes of CoronaVac, part of the fourth shipment of Sinovac-manufactured vaccine to arrive in Brazil
In Brazil, São Paulo governor João Doria signed a $90 million contract with Sinovac in September to receive the initial 46 million doses of CoronaVac.[81] The price for CoronaVac was announced to be US$10.3 (about R$59).[82] In January, Brazil announced it would obtain 100 million total doses.[83] On 17 January, ANVISA approved emergency use of CoronaVac, with a 54-year-old nurse in São Paulo being the first to receive a vaccine outside of clinical trials in the country.[16] In early February, Brazil said it intends to buy an additional 30 million doses to be produced locally on top of the existing 100 million doses.[84]
In January, Bolivia authorized use of CoronaVac. Butantan Institute had opened negotiations with South American countries to sell the vaccine, which would be produced in São Paulo.[85]
In October, Chile signed an agreement to purchase 20 million doses of CoronaVac[86] which was approved for emergency use on 20 January.[87] By early March, the country had received 10 million doses of CoronaVac and had vaccinated 4.1 million people.[88]
In February, Colombia had purchased 5 million doses of CoronaVac and was in talks for an additional 5 million doses,[89] which had been approved for emergency use on February 5.[90]
In February, Ecuador signed a deal for 2 million doses of CoronaVac which had been approved for emergency use.[91] Chile donated 20,000 doses of CoronaVac to Ecuador on March 6.[92]
In March, Paraguay received a donation of 20,000 doses of CoronaVac from Chile.[92] Paraguay began vaccinations with CoronaVac on March 10.[93]
In January, Uruguay announced the purchased of 1.75 million doses of CoronaVac.[94] The first 192,000 doses arrived on 25 February and vaccinations started on 1 March.[18]
Europe
In March, Albania received 192,000 doses of a first batch of 1 million doses purchased through Turkey.[95]
In November, Turkey signed a contract to buy 50 million doses of CoronaVac.[96] Turkey approved emergency use on 13 January[97] and President Recep Tayyip Erdoğan received his first dose at Ankara City Hospital.[98] In February, Turkey signed a deal for another 50 million doses for a total of 100 million doses.[21] By March 10.7 million doses had been administered, and 852 of the 1.3 million people who had received both doses were later diagnosed with the disease. 53 were hospitalized, but none of those hospitalized were intubated or died.[99]
In December, Ukraine signed a contract to purchase 1.8 million doses of CoronaVac. One dose of CoronaVac would cost 504 hryvnias (around $18).[100] On March 9, Ukraine granted approval for use of CoronaVac.[101]
Asia
On 19 January, Azerbaijan launched its vaccination campaign with CoronaVac. Azerbaijan plans to receive 4 million doses of the vaccine and aims to vaccinate 40% of the population.[102]
In February, Cambodia approved Coronavac[103] for emergency use and later ordered 1.5 million doses to arrive on March 26.[104]
In late August, China approved CoronaVac for emergency use to vaccinate high-risk groups such as medical staff.[105] In early February, China approved CoronaVac for general use.[15]
In December, Hong Kong ordered 7.5 million doses of CoronaVac.[106] The vaccination campaign with CoronaVac began on 26 February.[107]
In August, Indonesia’s Foreign Minister Retno Marsudi said an agreement was signed with Sinovac for 50 million doses,[108] which later increased to 140 million doses.[109] Indonesia approved emergency use authorization on 11 January and[35] President Joko Widodo received the first shot of the vaccine, which would be free for all Indonesian citizens.[13] By March, Indonesia had received 53.5 million doses of CoronaVac.[110]
On 26 January, Malaysia ordered 12 million doses.[57] CoronaVac was approved for emergency use on 2 March.[111] Malaysian Science, Technology and Innovation Minister Khairy Jamaluddin received the first dose with CoronaVac on 18 March as part of the vaccination campaign.[112]
In January, the Philippine’s announced the country had secured 25 million doses.[113] The vaccine was approved on 22 February but not for all health workers as it had lower efficacy when used with health workers compared to healthy individuals aged 18-59. The first 600,000 doses of CoronaVac arrived on 28 February.[114]
Singapore has signed advance purchase agreements for CoronaVac.[115] In February, the first doses arrived in the country.[116]
In early January, Thailand’s Ministry of Public Health announced an order for 2 million doses of CoronaVac,[117] which was approved for emergency use on 22 February.[118] Thailand started its vaccination program on 27 February.[14] In March, Thailand was in talks to purchase an additional 5 million doses.[119]
North America
By March 8, Dominican Republic had vaccinated 400,000 people and had reserved delivery for 10 million additional doses of CoronaVac.[19]
In February, Mexico approved emergency use of CoronaVac.[120] The country has ordered 20 million doses,[121] of which the first 200,000 doses arrived on 20 February.[122] It is currently used as part of the national vaccination campaign.[20]
Africa
In March, Benin received 203,000 doses of CoronaVac with vaccinations to start with health workers and the medically vulnerable.[123]
In March, South Africa’s drug regulator began assessing CoronaVac for use in the country.[124] South African firm Numolux said it could supply 5 million doses once it secured regulatory clearances.[125]
In March, Tunisia’s Ministry of Health approved marketing authorization of CoronaVac in the country.[126]
In March, Zimbabwe approved CoronaVac for emergency use.[127]
Oceania
In March, Fiji said it would be receiving a donation of CoronaVac.[128]
Controversies
Politicization
CoronaVac has been championed by the governor of São Paulo, João Doria, who many believe will challenge Jair Bolsonaro for the presidency in 2022.[129] A political showdown began in October 2020, when Bolsonaro vetoed a deal between the Brazilian health ministry and the São Paulo government for the purchase of 46 million doses of the vaccine.[130] After Instituto Butantan announced CoronaVac’s efficacy rate, Bolsonaro mocked the vaccine’s effectiveness against COVID-19.[131] Critics against the politicization of vaccines have warned that failure to follow international testing and safety protocols risks undermining public trust and can increase people’s hesitancy to inoculation.[129] Doctors in São Paulo said they were struggling to convince patients that CoronaVac would be safe.[132]
In March 2021, the Paraná Pesquisas opinion polling institute found that the vaccines preferred by Brazilians are CoronaVac and the Oxford–AstraZeneca vaccine, chosen by 23.6% and 21.2% of Brazilians interviewed, respectively, against 11.3% of those who would prefer the Pfizer–BioNTech vaccine.[133]
Delays in releasing results
On 23 December 2020, researchers in Brazil said the vaccine was more than 50% effective, but withheld full results at Sinovac’s request, raising questions again about transparency as it was the third delay in releasing results from the trials.[134] São Paulo Health Secretary Jean Gorinchteyn later said the vaccine didn’t reach 90% efficacy. Turkey said its trial showed an estimated efficacy rate of 91.25%, though that was based on only 29 infected cases.[32] When São Paulo state officials announced the protection rate, they declined to provide a more detailed breakdown of the trial, such as information about age groups and side effects of the vaccine.[32] Scientists said the lack of transparency about the data ran the risk of damaging CoronaVac’s credibility, with Brazilians and others world-wide already reluctant to take it.[30] Nikolai Petrovsky, a professor at the College of Medicine and Public Health at Flinders University said, “There is enormous financial and prestige pressure for these trials to massively overstate their results.”[135]
References
- ^ Corum, Jonathan; Zimmer, Carl. “How the Sinovac Vaccine Works”. The New York Times. ISSN 0362-4331. Retrieved 1 March 2021.
- ^ Nidhi Parekh (22 July 2020). “CoronaVac: A COVID-19 Vaccine Made From Inactivated SARS-CoV-2 Virus”. Retrieved 25 July2020.
- ^ “New coronavirus vaccine trials start in Brazil”. AP News. 21 July 2020. Retrieved 7 October 2020.
- ^ “Chile initiates clinical study for COVID-19 vaccine”. Chile Reports. 4 August 2020. Retrieved 7 October 2020.
- ^ “248 volunteers have received Sinovac vaccine injections in Bandung”. Antara News. 30 August 2020. Retrieved 7 October2020.
- ^ “DOH eyes 5 hospitals for Sinovac vaccine Phase 3 clinical trial”. PTV News. 16 September 2020. Retrieved 7 October 2020.
- ^ “Turkey begins phase three trials of Chinese Covid-19 vaccine”. TRT World News. 1 September 2020. Retrieved 7 October 2020.
- ^ Zimmer, Carl; Corum, Jonathan; Wee, Sui-Lee. “Coronavirus Vaccine Tracker”. The New York Times. ISSN 0362-4331. Retrieved 12 February 2021.
- ^ “CoronaVac: Doses will come from China on nine flights and can…” AlKhaleej Today (in Arabic). 1 November 2020. Retrieved 12 February 2021.
- ^ “Sinovac: Brazil results show Chinese vaccine 50.4% effective”. BBC News. 13 January 2021. Retrieved 12 February 2021.
- ^ AGENCIES, DAILY SABAH WITH (25 December 2020). “Turkey set to receive ‘effective’ COVID-19 vaccine amid calls for inoculation”. Daily Sabah. Retrieved 12 February 2021.
- ^ hermesauto (11 January 2021). “Indonesia grants emergency use approval to Sinovac’s vaccine, local trials show 65% efficacy”. The Straits Times. Retrieved 12 February 2021.
- ^ Jump up to:a b TARIGAN, EDNA; MILKO, VICTORIA (13 January 2021). “Indonesia starts mass COVID vaccinations over vast territory”. Associated Press. Retrieved 15 January 2021.
- ^ Jump up to:a b “Thailand Kicks Off Covid-19 Vaccine Program With Sinovac Shots”. Bloomberg.com. Retrieved 28 February 2021.
- ^ Jump up to:a b “China approves Sinovac vaccines for general public use”. South China Morning Post. 6 February 2021. Retrieved 6 February2021.
- ^ Jump up to:a b Fonseca, Jamie McGeever, Pedro (17 January 2021). “Brazil clears emergency use of Sinovac, AstraZeneca vaccines, shots begin”. Reuters. Retrieved 17 January 2021.
- ^ Miranda, Natalia A. Ramos (28 January 2021). “Chile receives two million-dose first delivery of Sinovac COVID-19 vaccine”. Reuters. Retrieved 30 January 2021.
- ^ Jump up to:a b “BNamericas – Uruguay prepares to launch COVID-19 vaccinat…” BNamericas.com. Retrieved 1 March 2021.
- ^ Jump up to:a b “Anticovid vaccines run out as Dominican Republic awaits arrival of more doses”. Dominican Today. Retrieved 10 March2021.
- ^ Jump up to:a b “Venustiano Carranza next up for Covid vaccination in Mexico City”. Mexico News Daily. 15 March 2021. Retrieved 16 March2021.
- ^ Jump up to:a b “Turkey aims to vaccinate 60 percent of population: Minister – Turkey News”. Hürriyet Daily News. Retrieved 12 February 2021.
- ^ Jump up to:a b Liu, Roxanne (3 March 2021). “Sinovac eyes two billion doses in annual capacity of virus vaccine by June”. Reuters. Retrieved 3 March 2021.
- ^ Jump up to:a b Liu, Roxanne (21 March 2021). “China steps up COVID-19 vaccination, considers differentiated visa policies”. Reuters. Retrieved 21 March 2021.
- ^ Tan Y (16 December 2020). “Covid: What do we know about China’s coronavirus vaccines?”. BBC News. Retrieved 18 December 2020.
- ^ Zimmer C, Corum J, Wee SL (10 June 2020). “Coronavirus Vaccine Tracker”. The New York Times. ISSN 0362-4331. Retrieved 27 December 2020.
- ^ “CoronaVac: Doses will come from China on nine flights and can…” AlKhaleej Today (in Arabic). 1 November 2020. Archivedfrom the original on 16 December 2020. Retrieved 1 November2020.
- ^ Staff (7 September 2020). “China’s Sinovac coronavirus vaccine candidate appears safe, slightly weaker in elderly”. Reuters. Archived from the original on 7 October 2020. Retrieved 6 October 2020.
- ^ WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection (2020). “A minimal common outcome measure set for COVID-19 clinical research”. The Lancet Infectious Diseases. 20 (8): e192–e197. doi:10.1016/S1473-3099(20)30483-7. PMC 7292605. PMID 32539990.
- ^ Mariz, Fabiana; Caires, Luiza (7 January 2021). “Eficaz em prevenir doença grave e morte por covid, Coronavac deve ter impacto em frear pandemia”. Jornal da USP (in Portuguese). Retrieved 7 January 2021.
- ^ Jump up to:a b c Pearson, Samantha; Magalhaes, Luciana (12 January 2021). “Chinese Covid-19 Vaccine Is Far Less Effective Than Initially Touted in Brazil”. The Wall Street Journal. Retrieved 12 January2021.
- ^ Gielow, Igor; Lopes Batista, Everton; Bottallo, Ana (12 January 2021). “Coronavac tem eficácia geral de 50,38% no estudo feito pelo Butantan”. Folha de S. Paulo (in Portuguese). Retrieved 12 January 2021.
- ^ Jump up to:a b c d “Sinovac’s Covid Shot Proves 78% Effective in Brazil Trial”. Bloomberg L.P. 7 January 2021. Retrieved 7 January 2021.
- ^ Kucukgocmen, Ali (3 March 2021). “Turkish study revises down Sinovac COVID-19 vaccine efficacy to 83.5%”. Reuters. Retrieved 7 March 2021.
- ^ “China’s Sinovac vaccine efficacy 83.5 percent: Turkish researchers – Turkey News”. Hürriyet Daily News. Retrieved 7 March 2021.
- ^ Jump up to:a b hermesauto (11 January 2021). “Indonesia grants emergency use approval to Sinovac’s vaccine, local trials show 65% efficacy”. The Straits Times. Retrieved 11 January 2021.
- ^ “Why did the efficacy of China’s top vaccine drop from 78% to 50%?”. Fortune. Retrieved 14 January 2021.
- ^ “Coronavac tem eficácia de 78% contra a Covid-19 em estudo no Brasil”. Folha de S.Paulo (in Portuguese). 7 January 2021. Retrieved 7 January 2021.
- ^ Jump up to:a b “Estudos mostram eficácia da CoronaVac contra três variantes do vírus”. Agência Brasil (in Portuguese). 10 March 2021. Retrieved 18 March 2021.
- ^ Zhang Y, Zeng G, Pan H, Li C, Hu Y, Chu K, et al. (November 2020). “Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18–59 years: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial”. The Lancet. Infectious Diseases. 21 (2): 181–192. doi:10.1016/S1473-3099(20)30843-4. PMC 7832443. PMID 33217362. S2CID 227099817. Archived from the original on 16 December 2020. Retrieved 18 November 2020.
- ^ Clinical trial number NCT04551547 for “A Randomized, Double-Blinded, Placebo-Controlled, Phase I/II Clinical Trial, to Evaluate the Safety and Immunogenicity of the SARS-CoV-2 Inactivated Vaccine (Vero Cell) in Healthy Population Aged 3–17 Years” at ClinicalTrials.gov
- ^ Wu, Zhiwei; Hu, Yaling; Xu, Miao; Chen, Zhen; Yang, Wanqi; Jiang, Zhiwei; Li, Minjie; Jin, Hui; Cui, Guoliang; Chen, Panpan; Wang, Lei (3 February 2021). “Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine (CoronaVac) in healthy adults aged 60 years and older: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial”. The Lancet Infectious Diseases. 0. doi:10.1016/S1473-3099(20)30987-7. ISSN 1473-3099. PMC 7906628. PMID 33548194.
- ^ Savarese M (21 July 2020). “New coronavirus vaccine trials start in Brazil”. Associated Press. Archived from the original on 13 August 2020. Retrieved 15 August 2020.
- ^ Palacios R, Patiño EG, de Oliveira Piorelli R, Conde MT, Batista AP, Zeng G, et al. (October 2020). “Double-Blind, Randomized, Placebo-Controlled Phase III Clinical Trial to Evaluate the Efficacy and Safety of treating Healthcare Professionals with the Adsorbed COVID-19 (Inactivated) Vaccine Manufactured by Sinovac – PROFISCOV: A structured summary of a study protocol for a randomised controlled trial”. Trials. 21 (1): 853. doi:10.1186/s13063-020-04775-4. PMC 7558252. PMID 33059771. Archived from the original on 16 December 2020. Retrieved 28 October 2020.
- ^ “World’s vaccine testing ground deems Chinese COVID candidate ‘the safest, most promising'”. Fortune. Archived from the original on 31 October 2020. Retrieved 9 November 2020.
- ^ “Doria says it guarantees purchase of 100 million doses of CoronaVac…” AlKhaleej Today (in Arabic). 29 October 2020. Archived from the original on 1 November 2020. Retrieved 30 October 2020.
- ^ “Brazil Clears Sinovac Trial to Resume Two Days After Halting It”. Bloomberg L.P. 11 November 2020. Archived from the original on 11 November 2020. Retrieved 11 November 2020.
- ^ “Brazil’s health regulator says China’s Sinovac can resume Covid-19 vaccine trial after suspension”. CNBC. 12 November 2020. Archived from the original on 13 November 2020. Retrieved 17 November 2020.
- ^ “Chile initiates clinical study for COVID-19 vaccine”. Government of Chile. 4 August 2020. Archived from the original on 11 October 2020. Retrieved 28 August 2020.
- ^ Health Institutes of Turkey (8 October 2020). “Randomized, Double-Blind, Placebo-Controlled Phase III Clinical Trial For Evaluation of Efficacy and Safety of SARS-CoV-2 Vaccine (Vero Cell), Inactivated”. ClinicalTrials. Archived from the original on 20 October 2020. Retrieved 21 October 2020.
- ^ “Chinese COVID-19 vaccine to be free, 1st doses to be delivered soon: Turkey’s health minister”. Daily Sabah. 23 November 2020. Archived from the original on 23 November 2020. Retrieved 23 November 2020.
- ^ “248 volunteers have received Sinovac vaccine injections in Bandung”. Antara News. Archived from the original on 30 September 2020. Retrieved 22 September 2020.
- ^ antaranews.com. “Phase 3 Sinovac clinical trial running smoothly: research team”. Antara News. Archived from the original on 3 November 2020. Retrieved 3 November 2020.
- ^ “Virus vaccine waiting on Saudi ‘green light'”. Arab News. 31 October 2020. Archived from the original on 16 December 2020. Retrieved 1 November 2020.
- ^ hermesauto (12 October 2020). “Indonesia aims to start administering coronavirus vaccines in early November”. The Straits Times. Archived from the original on 13 October 2020. Retrieved 12 October 2020.
- ^ “Sao Paulo starts building production plant for China’s Sinovac vaccine – governor”. Financial Post. Archived from the original on 29 November 2020. Retrieved 9 November 2020.
- ^ Mano A, Simões (10 December 2020). “Chinese vaccine draws demand across Latin America, say Brazilian officials”. Reuters. Archived from the original on 10 December 2020. Retrieved 10 December 2020.
- ^ Jump up to:a b Choong, Jerry (26 January 2021). “Health Ministry: Malaysia secures 18.4 million doses of Russian, Chinese Covid-19 vaccines”. The Malay Mail. Retrieved 26 January 2021.
- ^ Mourad, Mahmoud (22 March 2021). “Egypt aims for deal to produce Sinovac COVID-19 vaccines”. Reuters. Retrieved 22 March 2021.
- ^ Jump up to:a b c Liu R (6 February 2021). “China approves Sinovac Biotech COVID-19 vaccine for general public use”. Reuters. Retrieved 7 February 2021.
- ^ Sipalan, Joseph; Donovan, Kirsten (3 March 2021). “Malaysia approves Sinovac, AstraZeneca COVID-19 vaccines for use”. Reuters. Retrieved 7 March 2021.
- ^ Aliyev J. “Azerbaijan kicks off COVID-19 vaccination”. Anadolu Agency. Retrieved 7 February 2021.
- ^ “Bolívia autoriza uso de vacinas Sputnik V e CoronaVac contra covid-19”. noticias.uol.com.br (in Portuguese). Retrieved 6 January 2021.
- ^ McGeever J, Fonseca P (17 January 2021). “Brazil clears emergency use of Sinovac, AstraZeneca vaccines, shots begin”. Reuters. Retrieved 17 January 2021.
- ^ Chanritheara, Torn. “Cambodia Approves AstraZeneca and Sinovac Vaccines for COVID-19 Emergency Use”. Cambodianess. Retrieved 12 February 2021.
- ^ “Chile aprueba el uso de emergencia de la vacuna china de Sinovac contra covid-19”. France 24. 20 January 2021. Retrieved 30 January 2021.
- ^ Aliyev J. “Colombia approves emergency use of CoronaVac vaccine”. Anadolu Agency. Retrieved 7 February 2021.
- ^ “Anticovid vaccines run out as Dominican Republic awaits arrival of more doses”. DominicanToday. Retrieved 10 March 2021.
- ^ “Ecuador signs agreement with Sinovac for 2 million COVID-19 vaccine: minister”. National Post. Retrieved 26 February 2021.
- ^ “Use of Sinovac vaccine authorised”. Government of Hong Kong. 18 February 2021. Retrieved 19 February 2021.
- ^ Soeriaatmadja W (11 January 2021). “Indonesia grants emergency use approval to Sinovac’s vaccine, local trials show 65% efficacy”. The Straits Times. Retrieved 11 January 2021.
- ^ “BPOM Grants Emergency Use Authorization for Sinovac Vaccine”. Tempo. 11 January 2021. Retrieved 11 January 2021.
- ^ Barrera, Adriana (11 February 2021). “Mexico approves China’s CanSino and Sinovac COVID-19 vaccines”. Reuters. Retrieved 11 February 2021.
- ^ “CoronaVac, vacuna de alta eficacia”. Ministerio de Salud Publica Y Bienestar Social.
- ^ “Philippines approves Sinovac’s COVID-19 vaccine for emergency use”. Reuters. 22 February 2021.
- ^ Thepgumpanat, Panarat (22 February 2021). “Thailand allows emergency use of Sinovac’s COVID-19 vaccine”. Reuters. Retrieved 23 February 2021.
- ^ “Tunisia approva vaccino cinese Sinovac” (in Italian). Agenzia Nazionale Stampa Associata (in Italian). 5 March 2021. Retrieved 7 March 2021.
- ^ “Turkey to begin COVID-19 vaccine jabs by this weekend”. Anadolu. 11 January 2021. Retrieved 11 January 2021.
- ^ Zinets, Natalia (9 March 2021). “Ukraine approves China’s Sinovac COVID-19 vaccine”. Reuters. Retrieved 10 March 2021.
- ^ “Covid-19: Zimbabwe authorises Sputnik V, Sinovac vaccines for emergency use”. news24.com. 9 March 2021.
- ^ “Regulation and Prequalification”. World Health Organization. Retrieved 12 March 2021.
- ^ Simoes E (30 September 2020). “Brazil’s Sao Paulo signs agreement with Sinovac for COVID vaccine doses”. Reuters. Archived from the original on 1 October 2020. Retrieved 1 October 2020.
- ^ Fonseca I (30 October 2020). “CoronaVac May Be Four Times More Costly Than Flu Vaccine”. The Rio Times. Archived from the original on 3 November 2020. Retrieved 30 October 2020.
- ^ “Em meio a críticas por atrasos, Pazuello diz que Brasil está preparado para iniciar vacinação em janeiro”. Folha de S.Paulo(in Portuguese). 6 January 2021. Retrieved 7 January 2021.
- ^ Rochabrun, Marcelo. “Brazil health ministry says plans to order 30 million more Coronavac doses | The Chronicle Herald”. http://www.thechronicleherald.ca. Retrieved 26 February 2021.
- ^ “Bolívia autoriza uso de vacinas Sputnik V e CoronaVac contra covid-19”. noticias.uol.com.br (in Portuguese). Retrieved 7 January 2021.
- ^ “Government meets with Sinovac for first COVID-19 vaccine clinical trial in Chile”. Government of Chile. 13 October 2020. Archived from the original on 17 October 2020. Retrieved 8 November 2020.
- ^ Presse, AFP-Agence France. “Chile Approves Chinese Coronavirus Vaccine”. barrons.com. Retrieved 21 January 2021.
- ^ “Fifth shipment with over two million Sinovac vaccines arrives to Chile”. Chile Reports. Retrieved 12 March 2021.
- ^ “Colombia extends health state of emergency, seeks more Sinovac vaccines”. Reuters. Retrieved 26 February 2021.
- ^ MENAFN. “Colombia declares emergency use of Sinovac vaccines”. menafn.com. Retrieved 4 February 2021.
- ^ “Ecuador signs agreement with Sinovac for 2 million COVID-19 vaccine: minister”. nationalpost. Retrieved 26 February 2021.
- ^ Jump up to:a b Valencia, Alexandra (7 March 2021). “Chile donates 40,000 doses of Sinovac vaccine to Ecuador and Paraguay”. Reuters. Retrieved 7 March 2021.
- ^ “CoronaVac, vacuna de alta eficacia”. Ministerio de Salud Publica Y Bienestar Social.
- ^ “Uruguay will receive first batches of Pfizer and Sinovac vaccines late February or early March: US$ 120 million investment”. MercoPress. Retrieved 24 January 2021.
- ^ “Albania gets 192,000 doses of Chinese Sinovac vaccine”. CNA. Retrieved 25 March 2021.
- ^ “Turkey signs 50 million dose COVID-19 vaccine deal, health minister says”. Reuters. 25 November 2020. Archived from the original on 1 December 2020. Retrieved 27 November 2020.
- ^ “Turkey grants emergency authorization to Sinovac’s CoronaVac: Anadolu”. Reuters. 13 January 2021. Retrieved 15 January 2021.
- ^ “Turkish president gets COVID-19 vaccine”. Anadolu Agency. 14 January 2021. Retrieved 20 January 2021.
- ^ SABAH, DAILY (12 March 2021). “Few virus infections reported among vaccinated people in Turkey”. Daily Sabah. Retrieved 12 March 2021.
- ^ “Ukraine signs up for China’s Sinovac vaccine, with doses expected soon”. Reuters. 30 December 2020. Retrieved 30 December 2020.
- ^ Zinets, Natalia (9 March 2021). “Ukraine approves China’s Sinovac COVID-19 vaccine”. Reuters. Retrieved 9 March 2021.
- ^ Aliyev, Jeyhun (19 January 2021). “Azerbaijan kicks off COVID-19 vaccination”. Anadolu Agency.
- ^ “Cambodian PM okays two more Covid-19 vaccines – Sinovac and AstraZeneca – for emergency use | The Star”. http://www.thestar.com.my. Retrieved 19 March 2021.
- ^ “Have no fear about shortage of vaccines, 1.5 million doses of Sinovac arriving on March 26”. Khmer Times. 19 March 2021. Retrieved 19 March 2021.
- ^ “Sinovac’s coronavirus vaccine candidate approved for emergency use in China – source”. Reuters. 29 August 2020. Archived from the original on 31 August 2020. Retrieved 30 August 2020.
- ^ “Government announces latest development of COVID-19 vaccine procurement” Archived 11 December 2020 at the Wayback Machine (Hong Kong Government Press Releases, 12 December 2020)
- ^ “Hong Kong kicks off COVID-19 vaccinations with Sinovac jab”. AP NEWS. 26 February 2021. Retrieved 7 March 2021.
- ^ “Indonesia books 50 million coronavirus vaccine doses from Sinovac”. Reuters. 21 August 2020. Archived from the original on 29 August 2020. Retrieved 21 August 2020.
- ^ “Sinovac vaccine has no critical side effects, BPOM says”. The Jakarta Post. Retrieved 21 December 2020.
- ^ Arkyasa, Mahinda (25 March 2021). “16 Million Sinovac Vaccines Material Arrives in Indonesia”. Tempo. Retrieved 25 March 2021.
- ^ “Malaysia’s NPRA Approves AstraZeneca, Sinovac Covid-19 Vaccines”. CodeBlue. 2 March 2021. Retrieved 2 March 2021.
- ^ Babulal, Veena (18 March 2021). “KJ gets first dose of Sinovac vaccine [NSTTV] | New Straits Times”. NST Online. Retrieved 19 March 2021.
- ^ “Duque says deal sealed for 25M doses of Sinovac COVID-19 vaccine”. GMA News Online. Retrieved 10 January 2021.
- ^ “Philippines receives COVID-19 vaccine after delays”. AP NEWS. 28 February 2021. Retrieved 28 February 2021.
- ^ Chen F (24 December 2020). “Brazil joins ranks of Chinese vaccine backers”. Asia Times Online. Retrieved 30 December2020.
- ^ “Singapore receives China’s Sinovac vaccine ahead of approval”. The Star. 25 February 2021. Retrieved 26 February2021.
- ^ “Thailand to get 2 million shots of China’s Sinovac”. Bangkok Post. Bangkok Post Public Company. Retrieved 4 January 2021.
- ^ “Thailand gives emergency use authorisation for Sinovac’s COVID-19 vaccine – official”. Reuters. 22 February 2021. Retrieved 23 February 2021.
- ^ Limited, Bangkok Post Public Company. “Thailand in talks to buy another 5m Sinovac shots”. Bangkok Post. Retrieved 20 March2021.
- ^ “Mexico approves China’s CanSino and Sinovac COVID-19 vaccines”. Reuters. 11 February 2021. Retrieved 11 February2021.
- ^ Jorgic, Drazen (10 March 2021). “Mexico leans on China after Biden rules out vaccines sharing in short term”. Reuters. Retrieved 10 March 2021.
- ^ Exteriores, Secretaría de Relaciones. “The Mexican Government receives 200,000 Sinovac COVID-19 vaccines”. gob.mx (in Spanish). Retrieved 7 March 2021.
- ^ “Lutte contre la Covid-19 : 203.000 doses de vaccins s dont 100.000 offertes par la Chine au Bénin”. Concentrées d’informations sur le Bénin et le monde à votre service depuis 2009(in French). 23 March 2021. Retrieved 25 March 2021.
- ^ Winning, Alexander. “South Africa’s drugs regulator to start assessing Sinovac COVID-19 vaccine”. U.S. Retrieved 12 March2021.
- ^ Nijini, Felix (18 March 2021). “Sinovac May Supply South Africa With 5 Million Vaccines: Report – BNN Bloomberg”. BNN. Retrieved 19 March 2021.
- ^ “Covid: Tunisia approva vaccino cinese Sinovac”. Agenzia Nazionale Stampa Associata (in Italian). 5 March 2021. Retrieved 7 March 2021.
- ^ Dzirutwe, MacDonald (10 March 2021). “Zimbabwe authorises Sputnik V, Sinovac coronavirus vaccines for emergency use”. Reuters. Retrieved 13 March 2021.
- ^ “China to donate Sinovac Vaccine to Fiji”. Fiji Broadcasting Corporation. Retrieved 17 March 2021.
- ^ Jump up to:a b Phillips, Tom (10 November 2020). “Jair Bolsonaro claims ‘victory’ after suspension of Chinese vaccine trial”. The Guardian. Retrieved 18 January 2021.
- ^ Baptista, Eduardo (11 December 2020). “China-made coronavirus vaccine at heart of political showdown in Brazil”. South China Morning Post. Retrieved 18 January 2021.
- ^ Carvalho, Daniel (14 January 2021). “‘Is 50% Good?’, Asks Bolsonaro, Mocking Coronavac’s Effectiveness”. Folha de S.Paulo. Retrieved 18 January 2021.
- ^ Pearson, Samantha; Magalhaes, Luciana (10 November 2020). “Brazil’s Medical Experts Worry Politics Is Hampering Covid-19 Vaccine Progress”. The Wall Street Journal. Retrieved 18 January 2021.
- ^ “Covid: 70% dos brasileiros não fazem questão de escolher vacina” [Covid: 70% of Brazilians do not make a point of choosing vaccine]. R7.com (in Portuguese). 3 March 2021. Retrieved 9 March2021.
- ^ Fonseca P. “Brazil institute says CoronaVac efficacy above 50%, but delays full results”. Reuters. Retrieved 25 December 2020.
- ^ Hong, Jinshan (12 January 2021). “How Effective Is China’s Sinovac Vaccine? Data Confuse Experts”. Bloomberg News. Retrieved 12 January 2021.
External links
- Clinical Research Protocol for CoronaVac Phase III Trials in Brazil
- Clinical Research Protocol for CoronaVac Phase III Trials in Chile
- “How the Sinovac Covid-19 Vaccine Works”. The New York Times.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Inactivated |
| Clinical data | |
| Routes of administration | Intramuscular injection |
| ATC code | None |
| Legal status | |
| Legal status | Emergency authorization for use in China, Indonesia, Brazil and Turkey |
| Identifiers | |
| DrugBank | DB15806 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
Sinovac Biotech Ltd. (Chinese: 北京科兴生物制品有限公司, Nasdaq: SVA) is a Chinese biopharmaceutical company that focuses on the research, development, manufacture and commercialization of vaccines that protect against human infectious diseases. The company is based in Haidian District, Beijing.[2] The company is listed on the NASDAQ but the exchange halted Sinovac’s trading in February 2019 due to a proxy fight.[3][4]
Vaccines
Sinovac’s commercialized vaccines include Healive (hepatitis A), Bilive (combined hepatitis A and B), Anflu (influenza), Panflu (H5N1) and PANFLU.1 (H1N1). Sinovac is currently developing a Universal Pandemic Influenza vaccine and a Japanese encephalitis vaccine.[5][better source needed]
Sinovac is also developing vaccines for enterovirus 71 and human rabies. Its wholly owned subsidiary, Tangshan Yian, is conducting field trials for independently developed inactivated animal rabies vaccines.[citation needed]
COVID-19 vaccine development
Main article: CoronaVac
CoronaVac is an inactivated virus COVID-19 vaccine developed by Sinovac.[6] It has been in Phase III clinical trials in Brazil,[7] Chile,[8] Indonesia,[9] Malaysia,[10] Philippines,[11] and Turkey.[12]
It relies on traditional technology similar to BBIBP-CorV and BBV152, other inactivated-virus COVID-19 vaccines in Phase III trials.[13] CoronaVac does not need to be frozen, and both the vaccine and raw material for formulating the new doses could be transported and refrigerated at 2–8 °C (36–46 °F), temperatures at which flu vaccines are kept.[14]
Brazil announced results on January 13, 2021 showing 50.4% effective at preventing symptomatic infections, 78% effective in preventing mild cases needing treatment, and 100% effective in preventing severe cases.[15] Final Phase III results from Turkey announced on 3 March 2021 showed an efficacy of 83.5%.[16] Interim results in Indonesia were announced on 11 January 2021 with an efficacy of 65.3%.[17]
CoronaVac is being used in vaccination campaigns by certain countries in Asia,[18][19][20] South America,[21][22] and Europe.[23] In March, a Sinovac spokesman told Reuters production capacity for CoronaVac could reach 2 billion doses a year by June 2021.[24] As of 27 February 36 million doses had been administered in total.[25]
See also
References
- ^ “China’s Vaccine Front-Runner Aims to Beat Covid the Old-Fashioned Way”. Bloomberg. 24 August 2020.
- ^ “Home (English)”. Sinovac. Retrieved 2021-03-06.
Add: No. 39 Shangdi Xi Road, Haidian District, Beijing, P.R.C. 100085
– Chinese address: “地址:中国· 北京 海淀区上地西路39号北大生物城(100085)” - ^ Dou, Eva (December 4, 2020). “As China nears a coronavirus vaccine, bribery cloud hangs over drugmaker Sinovac”. The Washington Post. ISSN 0190-8286. Archived from the original on December 4, 2020. Retrieved 2020-12-06.
- ^ Levine, Matt (May 22, 2020). “A Vaccine With a Poison Pill”. Bloomberg News. Archived from the original on June 21, 2020. Retrieved December 6, 2020.
- ^ Google Finance, url=https://www.google.com/finance?q=Sinovac
- ^ Nidhi Parekh (22 July 2020). “CoronaVac: A COVID-19 Vaccine Made From Inactivated SARS-CoV-2 Virus”. Retrieved 25 July2020.
- ^ “New coronavirus vaccine trials start in Brazil”. AP News. 21 July 2020. Retrieved 2020-10-07.
- ^ “Chile initiates clinical study for COVID-19 vaccine”. Chile Reports. 4 August 2020. Retrieved 2020-10-07.
- ^ “248 volunteers have received Sinovac vaccine injections in Bandung”. Antara News. 30 August 2020. Retrieved 2020-10-07.
- ^ “Malaysia Receives China’s Sinovac Vaccine For Regulatory Testing”. Bloomberg.com. 2021-02-27. Retrieved 2021-03-02.
- ^ “DOH eyes 5 hospitals for Sinovac vaccine Phase 3 clinical trial”. PTV News. 16 September 2020. Retrieved 2020-10-07.
- ^ “Turkey begins phase three trials of Chinese Covid-19 vaccine”. TRT World News. 1 September 2020. Retrieved 2020-10-07.
- ^ Zimmer, Carl; Corum, Jonathan; Wee, Sui-Lee. “Coronavirus Vaccine Tracker”. The New York Times. ISSN 0362-4331. Retrieved 2021-02-12.
- ^ “CoronaVac: Doses will come from China on nine flights and can…” AlKhaleej Today (in Arabic). 2020-11-01. Retrieved 2021-02-12.
- ^ “Sinovac: Brazil results show Chinese vaccine 50.4% effective”. BBC News. 2021-01-13. Retrieved 2021-02-12.
- ^ AGENCIES, DAILY SABAH WITH (25 December 2020). “Turkey set to receive ‘effective’ COVID-19 vaccine amid calls for inoculation”. Daily Sabah. Retrieved 12 February 2021.
- ^ hermesauto (11 January 2021). “Indonesia grants emergency use approval to Sinovac’s vaccine, local trials show 65% efficacy”. The Straits Times. Retrieved 12 February 2021.
- ^ TARIGAN, EDNA; MILKO, VICTORIA (13 January 2021). “Indonesia starts mass COVID vaccinations over vast territory”. Associated Press. Retrieved 15 January 2021.
- ^ Aliyev, Jeyhun (19 January 2021). “Azerbaijan kicks off COVID-19 vaccination”. Anadolu Agency.
- ^ “China approves Sinovac vaccines for general public use”. South China Morning Post. 6 February 2021. Retrieved 6 February2021.
- ^ Fonseca, Jamie McGeever, Pedro (17 January 2021). “Brazil clears emergency use of Sinovac, AstraZeneca vaccines, shots begin”. Reuters. Retrieved 17 January 2021.
- ^ Miranda, Natalia A. Ramos (28 January 2021). “Chile receives two million-dose first delivery of Sinovac COVID-19 vaccine”. Reuters. Retrieved 30 January 2021.
- ^ “Turkey aims to vaccinate 60 percent of population: Minister – Turkey News”. Hürriyet Daily News. Retrieved 12 February 2021.
- ^ Liu, Roxanne (2021-03-03). “Sinovac eyes two billion doses in annual capacity of virus vaccine by June”. Reuters. Retrieved 2021-03-03.
- ^ “Malaysia receives first batch of Sinovac Covid-19 vaccine today”. Bernama. 27 February 2021. Retrieved 27 February 2021– via The Malay Mail.
External links
- Official website
- Business data for Sinovac Biotech:
| Type | Public |
|---|---|
| Traded as | Nasdaq: SVA (American Depository Receipts) |
| Founded | 1999; 22 years ago |
| Founder | Yin Weidong[1] |
| Headquarters | Beijing,China |
| Website | http://www.sinovac.com/ |
| Sinovac Biotech | |
|---|---|
| Simplified Chinese | 北京科兴生物制品有限公司 |
| Traditional Chinese | 北京科興生物製品有限公司 |
| hideTranscriptionsStandard MandarinHanyu PinyinBěijīng Kē Xìng Shēngwù Zhìpǐn Yǒuxiàn Gōngsī |
/////////Sinovac COVID-19 vaccine, CoronaVac, corona virus, covid 19, vaccine, china, Sinovac Biotech, PiCoVacc
#Sinovac COVID-19 vaccine, #CoronaVac, #corona virus, #covid 19, #vaccine, #china, #Sinovac Biotech, #PiCoVacc
Sputnik V, Gam-COVID-Vac, Gamaleya
Sputnik V
Gam-COVID-Vac
Gamaleya
- Gam-COVID-Vac Lyo

Gam-COVID-Vac was created by Gamaleya Research Institute of Epidemiology and MIcrobiology in Russia. The vaccine candidate is a heterologous COVID-19 vaccine containing two components, recombinant adenovirus type 26 (rAd26) vector and recombinant adenovirus type 5 (rAd5) vector which both carry the SARS-CoV-2 spike glycoprotein. The vaccine is offered in both a frozen (Gam-COVID-Vac) and freeze-dried formulation (lyophilizate; Gam-COVID-Vac Lyo). Phase 1/2 human trials with 76 participants evaluated the safety, tolerability, and immunogenicity of both frozen (Gam-COVID-Vac;NCT04436471) and freeze-dried (Gam-COVID-Vac Lyo;NCT04437875) vaccine candidates in June 2020, and were completed in early August 2020. Preliminary results suggested that all participants developed antibodies to the SARS-CoV-2 glycoproteins with a good safety profile in both trials.
Sputnik V (Russian: Спутник V, literally Traveler V) is a COVID-19 vaccine developed by the Gamaleya Research Institute of Epidemiology and Microbiology. Registered on 11 August 2020 by the Russian Ministry of Health as Gam-COVID-Vac (Russian: Гам-КОВИД-Вак, romanized: Gam-KOVID-Vak),[2][3] Sputnik V is an adenovirus viral vector vaccine. The “V” in the name is the letter V, not the Roman numeral for five.[4]
Gam-COVID-Vac was initially approved for distribution in Russia on the preliminary results of Phase I–II studies eventually published on 4 September 2020.[5] The quick approval in early August of Gam-COVID-Vac was met with criticism in mass media and precipitated discussions in the scientific community whether this decision was justified in the absence of robust scientific research confirming the safety and efficacy of the vaccine.[2][3][6][7][8] On 2 February 2021, an interim analysis from the trial was published in The Lancet, indicating 91.6% efficacy without unusual side effects.[9]
Emergency mass-distribution of the vaccine began in December 2020 in multiple countries including Russia, Argentina, Belarus, Hungary, Serbia and the United Arab Emirates. As of February 2021, over a billion doses of the vaccine were ordered for immediate distribution globally.[10]

NEW DRUG APPROVALS
ONE TIME
$10.00
Technology
President Putin‘s meeting with government members, on 11 August 2020 via videoconference, at which he announced a conditionally registered vaccine against COVID-19.[2][3] Medical worker in Moscow with the vaccineSee also: COVID-19 vaccine
Gam-COVID-Vac is a viral two-vector vaccine based on two human adenoviruses – a common cold virus – containing the gene that encodes the full-length spike protein (S) of SARS-CoV-2 to stimulate an immune response.[5][11][12] The Gam-COVID-Vac vaccine was developed by a cellular microbiologists team of the government-backed Gamaleya Research Institute of Epidemiology and Microbiology. The group was led by MD and RAS associate member Denis Logunov, who also worked on vaccines for the Ebolavirus and the MERS-coronavirus.[13]
The recombinant adenovirus types 26 and 5 are both used as vectors in the vaccine. They were biotechnology-derived and contain the SARS-CoV-2 S protein cDNA. Both of them are administered into the deltoid muscle: the Ad26-based vaccine is used on the first day and the Ad5 vaccine is used on the 21st day to boost immune response.[11][14][15]
The vaccine can be formulated as frozen (storage temperature must be −18 °C or 0 °F or lower) and freeze-dried (“Gam-COVID-Vac-Lyo”, storage temperature is 2–8 °C or 36–46 °F) dosage forms.[16] The first formulation was developed for large-scale use, it is cheaper and easier to manufacture. The production of a lyophilized formulation takes much more time and resources, although it is more convenient for storage and transportation. Gam-COVID-Vac-Lyo was developed especially for vaccine delivery to hard-to-reach regions of Russia.[17] The head of the Gamaleya Research Institute Alexander Ginzburg estimates that it will take 9–12 months to vaccinate the vast majority of the Russian population, assuming in-country resources are adequate.[18][19] A single-dose version is also being developed to speed up vaccination outside Russia. It will offer less protection than the two-dose versions, but it is still expected to reach an efficacy of 85%.[20][21]

Clinical research
Phase I–II
A phase I safety trial began on 18 June.[2] On 4 September, data on 76 participants in a phase I–II trial were published, indicating preliminary evidence of safety and an immune response.[5] The results were challenged by international vaccine scientists as being incomplete, suspicious, and unreliable when identical data were reported for many of the trial participants,[22] but the authors responded that there was a small sample size of nine, and the measured results of titration could only take discrete values (800, 1600, 3200, 6400). Coupled with the observation that values tended to reach a plateau after three to four weeks, they contend that it is not unlikely that several participants would show identical results for days 21 to 28.[23]
Phase III
Sputnik V, efficacy for different conditions. The error bars indicate the confidence interval containing the efficacy with 95% probability
In early November 2020, Israel Hadassah Medical Center director-general Prof. Zeev Rotstein stated that Hadassah’s branch in Moscow’s Skolkovo Innovation Center was collaborating on a phase III clinical trial.[24]
The ongoing phase III study is a randomised, double-blind, placebo-controlled, multi-centre clinical trial involving 40,000 volunteers in Moscow, and is scheduled to run until May 2021.[25] In 2020–2021, phase III clinical studies were also being conducted in Belarus,[26] UAE,[27] India[28] and Venezuela.[29]
On 2 February 2021, an interim analysis from the Moscow trial was published in The Lancet, indicating 91.6% efficacy (95% CI 85.6–95.2) after the second vaccination, without unusual side effects.[30] The trial started on 7 September 2020 using the frozen liquid form of the vaccine, and data was analysed up to the second database lock on 24 November 2020. The over-60-years-old group in the trial (oldest participant was 87) had essentially the same efficacy (91.8%) as for all ages. The lowest age participants were 18 years old.[9][31]
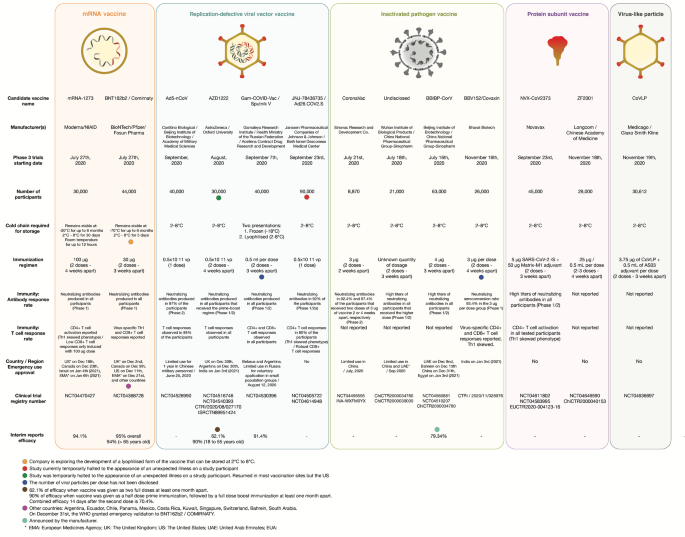
Sputnik–AstraZeneca COVID-19 vaccine trials
On 21 December 2020 the Russian Direct Investment Fund (RDIF), the Gamaleya National Center, AstraZeneca and R-Pharm have signed an agreement aimed at the development and implementation of a clinical research program to assess the immunogenicity and safety of the combined use of one of the components of the Sputnik V vaccine developed by the Gamaleya Center, and one of the components of the AZD1222 vaccine, developed by AstraZeneca and the University of Oxford.[32] The study program will last 6 months in several countries, and it is planned to involve 100 volunteers in each study program. On 9 February 2021, the Ministry of Health of the Republic of Azerbaijan allowed clinical studies in the country for the combined use of the Sputnik V vaccine and the vaccine developed by AstraZeneca, stating that the trials would begin before the end of February 2021.[33][34]
Composition
The Gam-COVID-Vac is a two-vector vaccine.[1] The active component for both vectors is a modified (recombinant) replication-defective adenovirus of a different serotype (Serotype 26 for the first vaccination and serotype 5 for the second vaccination), which has been modified to include the protein S-expressing gene of SARS-CoV-2.[1]
The other ingredients (excipients) are the same, both quantitatively and qualitatively, in the two components.[35]
- Tris(hydroxymethyl)aminomethane
- Sodium chloride
- Sucrose
- Magnesium chloride hexahydrate
- Disodium EDTA dihydrate (buffer)
- Polysorbate 80
- Ethanol 95%
- Water
As per the official datasheet, no further components or ingredients, including other adjuvants, should be included in the vaccine.[1]
History
In May 2020, the Gamaleya Research Institute of Epidemiology and Microbiology announced that it had developed the vaccine without serious side effects. By August 2020, phases I and II of two clinical trials (involving 38 patients each) were completed. Only one of them used the formulation which later obtained marketing authorization under limited conditions.[36][37] This vaccine was given the trade name “Sputnik V”, after the world’s first artificial satellite.[3][7][38]
During preclinical and clinical trials, 38 participants who received one or two doses of the Gam-COVID-Vac vaccine had produced antibodies against SARS-CoV-2’s spike protein, including potent neutralizing antibodies that inactivate viral particles.[2] On 11 August 2020, the Russian minister of Health Mikhail Murashko announced at a government briefing with the participation of President Vladimir Putin regulatory approval of the vaccine for widespread use. The state registration of the vaccine was carried out “conditionally” with post-marketing measures according to the decree of the Government of the Russian Federation. The registration certificate for the vaccine stated that it could not be used widely in Russia until 1 January 2021, and before that, it may be provided to “a small number of citizens from vulnerable groups”, such as medical staff and the elderly, according to a Ministry of Health spokesperson.[3] The license under register number No. ЛП-006395 (LP-006395) was issued on 11 August by the Russian Ministry of Health. Although the announcement was made even before the vaccine candidate had been entered into Phase III trials, the practice of marketing authorization “on conditions” also exists in other countries.[39][40] On 26 August, certificate No. ЛП-006423 (LP-006423) was issued for the lyophilized formulation “Gam-COVID-Vac-Lyo”.[2][3][7][41][5]
The commercial release of the Gam-COVID-Vac was first scheduled for September 2020. In October, Mikhail Murashko said that the Gam-COVID-Vac will be free for all Russian citizens after the launching of mass production.[42][43] Later on, Russian Ministry of Health registered maximum ex-factory price equal to 1,942 rubles for two components and included them into The National List of Essential medicines.[44] There were also suggestions to include the vaccine in the National Immunisation Calendar of Russia.[44]
According to Russian media, the mass production of the Gam-COVID-Vac was launched by 15 August. By that moment, the Russian Federation has already received applications from 20 countries for the supply of 1 billion doses of vaccine. Three facilities were able to produce about a million doses per month at each with a potential doubling of capacity by winter. By the end of 2020, Gamaleya Research Institute’s production, according to an interview with the organization’s spokesperson, was planned to produce 3–5 million doses.[45][46]
On 9 March 2021, an agreement was signed by the RDIF sovereign wealth fund and Swiss-based pharmaceutical company Adienne to produce the vaccine in Italy. Kirill Dmitriev, RDIF’s head, told Russian state TV his fund had also struck deals with production facilities in Spain, France and Germany for local manufacturing of the vaccine.[47]
Scientific assessment
Balram Bhargava, director of the Indian Council of Medical Research, said that Russia had managed to fast-track a COVID-19 vaccine candidate through its early phases.[48]
On 11 August 2020, a World Health Organization (WHO) spokesperson said, “… prequalification of any vaccine includes the rigorous review and assessment of all required safety and efficacy data”.[8]
- A WHO assistant director said, “You cannot use a vaccine or drugs or medicines without following through all of these stages, having complied with all of these stages”.[49]
- Francois Balloux, a geneticist at University College London, called the Russian government’s approval of Gam-COVID-Vac a “reckless and foolish decision”.[2] Professor Paul Offit, the director of the Vaccine Education Center at Children’s Hospital of Philadelphia, characterized the announcement was a “political stunt”, and stated that the untested vaccine could be very harmful.[8]
Stephen Griffin, Associate Professor in the School of Medicine, University of Leeds, said “that we can be cautiously optimistic that SARS-CoV2 vaccines targeting the spike protein are effective.” Moreover, as the Sputnik antigen is delivered via a different modality, namely using a disabled Adenovirus rather than formulated RNA, this provides flexibility in terms of perhaps one or other method providing better responses in certain age-groups, ethnicities, etc., plus the storage of this vaccine ought to be more straightforward.[50][failed verification][51]
Stephen Evans, professor of pharmacoepidemiology at the London School of Hygiene and Tropical Medicine, said “the data [is] compatible with the vaccine being reasonably effective … These results are consistent with what we see with other vaccines, because the really big message for global health scientists is that this disease [COVID-19] is able to be addressed by vaccines.”[50]
Julian Tang, clinical virologist at the University of Leicester, said: “Despite the earlier misgivings about the way this Russian Sputnik V vaccine was rolled out more widely – ahead of sufficient Phase 3 trial data – this approach has been justified to some extent now.”[52]
Ian Jones, a professor of virology at the University of Reading, and Polly Roy, professor and Chair of Virology at The London School of Hygiene and Tropical Medicine, commenting on phase III results published in the Lancet in February 2021, said “The development of the Sputnik V vaccine has been criticised for unseemly haste, corner cutting, and an absence of transparency. But the outcome reported here is clear and the scientific principle of vaccination is demonstrated, which means another vaccine can now join the fight to reduce the incidence of COVID-19.”[53]
Hildegund C. J. Ertl, a vaccine scientist at the Wistar Institute, called the phase-III results published on 2 February 2021 “great”: “Good safety profile, more than 90% efficacy across all age groups, 100% efficacy against severe disease or death, can be stored in the fridge and low cost. What more would we want?”[54]
According to preliminary review by experts,[who?] the lyophilized formulation of Gam-COVID-Vac is similar to the smallpox vaccine, circumventing the need for continuous “colder chain” or cold-chain storage – as required for the Pfizer–BioNTech and Moderna vaccines respectively – and allowing transportation to remote locations with reduced risk of vaccine spoilage.[55][56]
On 6 March 2021, Director of the U.S. National Institute of Allergy and Infectious Diseases (NIAID), Anthony Fauci, said that the data from Sputnik V “looked pretty good” to him.[57]
Distribution, vaccination and public perception
Early perception
An opinion poll of Canadians conducted by Léger in August 2020 found that a majority (68%) would not take the Russian vaccine if offered a free dose, compared to 14% who said they would take it. When Americans were asked the same question, 59% would not take the Russian vaccine if offered a free dose, compared to 24% who said they would take it.[58][59]
- At that time, British and American officials stated that the Gam-COVID-Vac vaccine would likely be rejected due to concerns that the normally rigorous process of vaccine clinical testing was not followed.[60] One public health expert said the quick approval of Gam-COVID-Vac by the Russian government was “cutting corners”, and may harm public confidence if the vaccine proves to be unsafe or ineffective.[7] “There is a huge risk that confidence in vaccines would be damaged by a vaccine that received approval and was then shown to be harmful”, said immunologist Peter Openshaw.[7]
As for early September 2020, according to public opinion polls, only half of the Russian population would take the vaccine voluntarily.[61]
In Russia
Vaccination of military personnel and civilian specialists of the Northern Fleet with the second component of the drug “Gam-COVID-Vac” (“Sputnik V”).
In the beginning of December 2020, Russian authorities announced the start of a large-scale free of charge vaccination with Gam-COVID-Vac for Russian citizens: the “immunization” program was launched on 5 December 2020 (with 70 Moscow-based medical centers providing vaccinations).[62]
Doctors and other medical workers, teachers, and social workers were given priority due to their highest risk of exposure to the disease.[63] The age for those receiving shots was initially capped at 60, later this restriction was lifted.[64]
Potential recipients were notified via text messaging, which says “You are working at an educational institution and have top-priority for the COVID-19 vaccine, free of charge”. Patients are asked a few general health questions before getting the vaccine. Program’s leaflet is handed to the patient, which warns of possible side effects, suggesting those are most likely to be mild and last a couple of days at most.[65][66][67] People with certain underlying health conditions, pregnant women, and those who have had a respiratory illness for the past two weeks are barred from vaccination.[63] Vaccine vial is removed from medical centre’s freezer about 15 minutes before use.
In early December 2020, the Minister of Health, Mikhail Murashko, said that Russia had already vaccinated more than 100,000 high-risk people.[68] Forty thousand of those are volunteers in Sputnik V’s Phase 3 trials, another 60,000 medics and doctors have also taken the vaccine.[69] The head of the Russian Direct Investment Fund, Kirill Dmitriev, said in an interview with the BBC that Russian medics expect to give about 2 million people coronavirus vaccinations in December.[70]
Up to the beginning of December 2020, Generium (which is supervised by Pharmstandard) and Binnopharm (which is supervised by AFK Sistema) companies produced Gam-COVID-Vac on a large scale.
On 10 December, Deputy Prime Minister Tatyana Golikova announced that approximately 6.9 million doses of the Sputnik V vaccine will enter civilian circulation in Russia before the end of February 2021.[71] Moscow Mayor Sergei Sobyanin announced that the newly-opened Moscow-based “R-Pharm” will become a leading manufacturer of Russia’s Sputnik V coronavirus vaccine. Working at full capacity, the factory will produce up to 10 million doses a month.[72]
Outside of Russia
In dark green are the countries that ordered (Russian or licensed domestic production; China also plans to produce Sputnik V on its territory.) or approved Sputnik V vaccine against COVID-19 (w/disputed Crimea). In light green are the countries that have shown interest in obtaining the vaccine.
According to the Russian Direct Investment Fund, they had received orders for more than 1.2 billion doses of the vaccine as of December 2020. Over 50 countries had made requests for doses, with supplies for the global market being produced by partners in India, Brazil, China, South Korea, Hungary, and other countries.[73][74] In August 2020, according to the Russian authorities, there were at least 20 countries that wanted to obtain the vaccine.[75]
While free in Russia, the cost per dose would be less than US$10 (or less than US$20 for the two doses needed to vaccinate one person) on international markets, which makes it much more affordable compared to mRNA vaccines from other manufacturers. Kirill Dmitriev, head of the fund, told reporters that over 1 billion doses of the vaccine are expected to be produced in 2021 outside of Russia.[76][77]
The Israeli Hadassah Medical Center has signed a commercial memorandum of understanding to obtain 1.5–3 million doses.[78]
- According to The New York Times’ sources,[79] to secure the release of an Israeli civilian held in Syria, Israel agreed to finance a supply of Russian-made Covid-19 vaccines for Damascus.
Argentina had agreed to buy 25 million doses of Russia’s Covid-19 vaccine.[80] The vaccine was registered and approved in Argentina in late December 2020.[81] The Brazilian state of Bahia has also signed an agreement to conduct Phase III clinical trials of the Sputnik V vaccine and plans to buy 50 million doses to market in northeastern Brazil.[82]
On 21 January 2021, the Argentine president Alberto Fernández became the first Latin American leader to be inoculated against the disease via the then recently approved Sputnik V.[83][84]
Due to the delay in shipping of doses from Italy and the European Union, San Marino imported doses of the Sputnik V vaccine (not approved by the E.M.A.) and started a mass vaccination on 28 February of its healthcare workers.[85]
EMA’s human medicines committee (CHMP) has started a rolling review of Sputnik V (Gam-COVID-Vac), a COVID-19 vaccine developed by Russia’s Gamaleya National Centre of Epidemiology and Microbiology. [86] Asked about the prospect of Austria taking the same step (as some other European countries chose to do), EMA management board chair Christa Wirthumer-Hoche told Austria’s ORF broadcaster: “It’s somewhat comparable to Russian roulette. I would strongly advise against a national emergency authorisation,” she said, pointing to the fact that there was not yet sufficient safety data about those who had already been given the vaccine. “We could have Sputnik V on the market in future, when we’ve examined the necessary data,” she said, adding that the vaccine needed to match up to European criteria on quality control and efficacy.[87]
Although vaccination rates in Russia are below those of other developed nations (as of March 2021),[88] Russia is pursuing deals to supply its vaccine abroad.[89]
Emergency use authorization
| show Full authorizationshow Emergency authorizationshow Ordered doses Eligible COVAX recipient (assessment in progress)[143] EMA review in progress[144] |
As of December 2020, Belarus and Argentina granted emergency use authorization for the vector-based vaccine.[145] On 21 January 2021, Hungary became the first European Union country to register the shot for emergency use, as well as the United Arab Emirates in the Gulf region.[146][147][148][149][150]
On 19 January 2021, the Russian authorities applied for the registration of Sputnik V in the European Union, according to the RDIF.[151] On 10 February, the European Medicines Agency (EMA) said that they had “not received an application for a rolling review or a marketing authorisation for the vaccine”. The developers have only expressed their interest that the vaccine be considered for a rolling review, but EMA’s Human Medicines Committee (CHMP) and the COVID-19 EMA pandemic Task Force (COVID-ETF) need to give their agreement first before developers can submit their application for initiation of the rolling review process.[152] On 4 March 2021, the Committee for Medicinal Products for Human Use (CHMP) of the EMA started a rolling review of Sputnik V.[153] The EU applicant is R-Pharm Germany GmbH.[153]
Emergency use has also been authorized in Algeria, Bolivia, Serbia, the Palestinian territories,[154] and Mexico.[155]
On 25 January 2021, Iran approved the vaccine, with Foreign Minister Mohammad Javad Zarif saying the country hopes to begin purchases and start joint production of the shot “in the near future”, after Supreme Leader Ayatollah Ali Khamenei banned the government from importing vaccines from the United States and United Kingdom.[156][157]
On 1 March 2021, Slovakia bought two million Sputnik V vaccines. Slovakia received the first batch of 200,000 vaccines, and expects to receive another 800,000 doses in March and April. Another 1 million vaccines are set to arrive in May and June.[158] The Czech Republic is also considering buying Sputnik V.[159]
On 18 March 2021, German regional leaders including State Premiers and the major of Berlin called for the swift approval of the Russian vaccine by the European Medicines Agency to counteract the acute shortages of effective vaccines in Europe. German medical experts have recommended its approval also, and consider the Sputnik Vaccine “clever” and “highly safe”.[160]
On 19 March 2021, the Philippine Food and Drug Administration granted emergency use authorization for Sputnik V, the fourth COVID-19 vaccine to be given authorization. The Philippine government is planning to buy 20 million doses of the vaccine.[161][162]
As of March 23, 2021, 56 countries have granted Sputnik V emergency use authorization.[163]
Production
As of March 2021, RDIF has licensed production in India, China, South Korea and Brazil. In the EU, RDIF has signed production agreements, subject to European Medicines Agency approval, with companies in Germany, Spain and France, and is in negotiations with a Swiss/Italian company. By the end of March 2021 RDIF anticipates 33 million doses will have been manufactured in Russia, less than 5% of which will have been exported.[164]
An agreement for the production of over 100 million doses of vaccine in India has been made with Dr. Reddy’s Laboratories, who on 11 January 2021 submitted mid-stage trial data to the Indian regulator and recommended moving onto late-stage trials.[154] The RDIF announced plans to sell 100 million doses to India, 35 million to Uzbekistan, and 32 million to Mexico, as well as 25 million each to Nepal and Egypt.[165]
In March 2021, the Italian-Russian Chamber of Commerce announced that Italy would be the first EU country to manufacture the two-dose COVID-19 vaccine under license. From July to the end of 2021, the Italian-Swiss pharmaceutical company Adienne in Caponago will manufacture 10 million doses. The announcement came in a time of acute vaccine shortages in Europe while the Sputnik V vaccine was still under review by the European Medicines Agency. Russian authorities said they would be able to provide a total of 50 million doses to European countries beginning in June 2021.[166]
The Sputnik V doses to be manufactured in South Korea are not for domestic use. The vaccine is to be exported to Russia, Algeria, Argentina, Hungary, Iran and the United Arab Emirates.[167]
References
- ^ Jump up to:a b c d “Sputnik V”. Russian drug reference. Medum.ru.
- ^ Jump up to:a b c d e f g Callaway E (August 2020). “Russia’s fast-track coronavirus vaccine draws outrage over safety”. Nature. 584(7821): 334–335. doi:10.1038/d41586-020-02386-2. PMID 32782400.
- ^ Jump up to:a b c d e f Cohen J (11 August 2020). “Russia’s approval of a COVID-19 vaccine is less than meets the press release”. Science. Retrieved 13 August 2020.
- ^ How Sputnik V works, Gamaleya Research Institute of Epidemiology and Microbiology, 11 January 2021, retrieved 18 March 2021
- ^ Jump up to:a b c d Logunov DY, Dolzhikova IV, Zubkova OV, Tukhvatullin AI, Shcheblyakov DV, Dzharullaeva AS, et al. (September 2020). “Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: two open, non-randomised phase 1/2 studies from Russia”. Lancet. 396 (10255): 887–897. doi:10.1016/S0140-6736(20)31866-3. PMC 7471804. PMID 32896291.
- ^ Mahase E (August 2020). “Covid-19: Russia approves vaccine without large scale testing or published results”. BMJ. 370: m3205. doi:10.1136/bmj.m3205. PMID 32816758.
- ^ Jump up to:a b c d e Burki TK (November 2020). “The Russian vaccine for COVID-19”. The Lancet. Respiratory Medicine. 8 (11): e85–e86. doi:10.1016/S2213-2600(20)30402-1. PMC 7837053. PMID 32896274.
- ^ Jump up to:a b c Berkeley Jr L (11 August 2020). “Scientists worry whether Russia’s Sputnik V’ coronavirus vaccine is safe and effective”. CNBC. Retrieved 11 August 2020.
- ^ Jump up to:a b Logunov DY, Dolzhikova IV, Shcheblyakov DV, Tukhvatulin AI, Zubkova OV, Dzharullaeva AS, et al. (2 February 2021). “Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia”. The Lancet. 397 (10275): 671–681. doi:10.1016/s0140-6736(21)00234-8. PMC 7852454. PMID 33545094.
- ^ Kramer, Andrew. “Russia is offering to export hundreds of millions of vaccine doses, but can it deliver?”. nytimes.com. Retrieved 20 February 2021.
- ^ Jump up to:a b “An Open Study of the Safety, Tolerability and Immunogenicity of the Drug ‘Gam-COVID-Vac’ Vaccine Against COVID-19”. ClinicalTrials.gov. 22 June 2020.
- ^ “Coronavirus Vaccine Trials Advance in Race for Covid-19 Protection”. Bloomberg. Retrieved 10 August 2020.
- ^ “Russia’s RDIF & Prominent Government Lab Progress COVID-19 Vaccine: Production Facility Readied in the Moscow Region”. trialsitenews.com. 10 June 2020. Retrieved 11 August 2020.
- ^ Sokolov A (12 December 2020). “Сколько хотят заработать на прививках от коронавируса”. Vedomosti. Archived from the original on 12 August 2020. Retrieved 20 December 2020.
- ^ “Нормативная документация ЛП-006395-110820” (PDF) (in Russian). Russian Ministry of Health. 2020. Retrieved 21 September 2020.
- ^ Rinat, Sagdiev; Ivanova, Polina (17 November 2020). “Russia focuses on freeze-dried vaccine doses as transport fix”. Reuters. Moscow. Retrieved 16 March 2021.
- ^ Logunov DY, Dolzhikova IV, Zubkova OV, Tukhvatullin AI, Shcheblyakov DV, Dzharullaeva AS, et al. (September 2020). “Safety and immunogenicity of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine in two formulations: two open, non-randomised phase 1/2 studies from Russia”. Lancet. 396 (10255): 887–897. doi:10.1016/S0140-6736(20)31866-3. PMC 7471804. PMID 32896291.
- ^ “Центр Гамалеи назвал сроки вакцинации большей части населения России”. RBK. 4 September 2020. Retrieved 6 September 2020.
- ^ Sagdiev R, Ivanova P, Nikolskaya P, Swift R, Smout A (17 November 2020). Mason J, Macfie N (eds.). “Russia focuses on freeze-dried vaccine doses as transport fix”. Reuters. Moscow. Retrieved 20 November 2020.
- ^ “Russia to try out ‘Sputnik-Light’ COVID vaccine to make it go further”. Reuters. Moscow. 11 January 2021. Retrieved 20 March2021.
- ^ “Study to Evaluate Efficacy, Immunogenicity and Safety of the Sputnik-Light (SPUTNIK-LIGHT)”. ClinicalTrials.gov. National Institutes of Health. 19 February 2021. Retrieved 20 March 2021.
- ^ Ellyatt H (10 September 2020). “Scientists question ‘strange’ data in Russian coronavirus vaccine trial after ‘unlikely’ patterns”. CNBC. Retrieved 10 September 2020.
- ^ Logunov DY, Dolzhikova IV, Tukhvatullin AI, Shcheblyakov DV (October 2020). “Safety and efficacy of the Russian COVID-19 vaccine: more information needed – Authors’ reply”. Lancet. 396(10256): e54–e55. doi:10.1016/S0140-6736(20)31970-X. PMC 7503057. PMID 32971043. S2CID 221805026.
- ^ “Hadassah bringing 1.5 million doses of Russian COVID-19 vaccine to Israel”. The Jerusalem Post | JPost.com. Retrieved 19 November 2020.
- ^ “Clinical Trial of Efficacy, Safety, and Immunogenicity of Gam-COVID-Vac Vaccine Against COVID-19”. National Library of Medicine. Retrieved 28 September 2020.
- ^ “Clinical Trial of Efficacy, Safety, and Immunogenicity of Gam-COVID-Vac Vaccine Against COVID-19 in Belarus”. clinicaltrials.gov. Retrieved 14 January 2021.
- ^ “UAE volunteers receive Russian Covid-19 vaccine”. Khaleej Times. 10 January 2021.
- ^ Bharadwaj S (15 January 2021). “Dr Reddy’s gets DCGI nod for Covid-19 vaccine Sputnik V Phase III trials”. The Times Of India.
- ^ Clinical trial number NCT04642339 for “Clinical Trial of the Immunogenicity, Safety, and Efficacy of the Gam-COVID-Vac Vaccine Against COVID-19 in Venezuela” at ClinicalTrials.gov
- ^ Logunov, Denis (2 February 2021). “Sputnik V COVID-19 vaccine candidate appears safe and effective”. The Lancet. doi:10.1016/S0140-6736(21)00234-8.
- ^ The Guardian: Sputnik V vaccine has 91.6% efficacy against symptomatic Covid, Russian trial suggests
- ^ “RDIF, The Gamaleya National Center, AstraZeneca and R-Pharm sign an agreement to cooperate on COVID-19 vaccine development”. The Russian Direct Investment Fund. 21 December 2020.
- ^ “Azerbaijan allowed for the first in the world to study a combination of “Sputnik V” vaccine and COVID-19 vaccine developed by “AstraZeneca” Company”. MoH of Azerbaijan. 9 February 2021.
- ^ “Study in Adults to Determine the Safety and Immunogenicity of AZD1222, a Non-replicating ChAdOx1 Vector Vaccine, Given in Combination With rAd26-S, Recombinant Adenovirus Type 26 Component of Gam-COVID-Vac Vaccine, for the Prevention of COVID-19”. ClinicalTrials.gov. U.S. National Library of Medicine. 14 January 2021. NCT04686773. Retrieved 9 February 2021.
- ^ “ИНСТРУКЦИЯ ПО МЕДИЦИНСКОМУ ПРИМЕНЕНИЮ ЛЕКАРСТВЕННОГО ПРЕПАРАТА Гам-КОВИД-Вак, Комбинированная векторная вакцина для профилактики коронавирусной инфекции, вызываемой вирусом SARS-CoV-2” (PDF). МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ.
- ^ “Russia plans to start producing coronavirus vaccine in September”. Daily Sabah. 13 June 2020. Retrieved 10 August2020.
- ^ Ermakov A (11 August 2020). “Вакцина от COVID-19 – в словах чиновников и документах разработчика” [Vaccine for COVID-19 – in the words of officials and developer documents] (in Russian). Fontanka.ru. Retrieved 21 September 2020.
- ^ Tétrault-Farber G, Soldatkin V (11 August 2020). “Russia becomes first country to approve a COVID-19 vaccine, says Putin”. Reuters. Moscow. Retrieved 11 August 2020.
- ^ “About the vaccine to prevent the novel coronavirus infection COVID-19, “Gam-COVID-Vac”, developed by the National Research Centre for Epidemiology and Microbiology named after academician N. F. Gamalei of the Ministry of Health of the Russian Federation”. Federal Service for Surveillance in Healthcare. 12 August 2020. Retrieved 18 August 2020.
- ^ “Ministry of Health of the Russian Federation has issued a registration certification № ЛП-006395 dated 11 August 2020 for a vaccine to prevent the novel coronavirus infection COVID-19”. Federal Service for Surveillance in Healthcare. 11 August 2020. Retrieved 21 September 2020.
- ^ “Coronavirus: Putin says vaccine has been approved for use”. BBC. 11 August 2020. Retrieved 11 August 2020.
- ^ “Минздрав пообещал россиянам бесплатную вакцинацию от COVID-19” [Ministry of Health promised Russians free vaccination against COVID-19]. RBC.ru (in Russian). Retrieved 23 October 2020.
- ^ “В будущем вакцинация от COVID-19 будет проходить бесплатно” [In the future, vaccination against COVID-19 will be free of charge]. Russian Newspaper (in Russian). Retrieved 23 October 2020.
- ^ Jump up to:a b “Минздрав зарегистрировал предельную отпускную цену на вакцину “Спутник V”” [The Ministry of Health registered the maximum selling price for the Sputnik V vaccine] (in Russian). RIA. 5 December 2020. Retrieved 21 September 2020.
- ^ “Голикова назвала сроки выпуска первых партий вакцин НИЦ Гамалеи и “Вектора”” [Golikova announced the timing of the release of the first batches of vaccines of the Research Center of Gamaleya and “Vector”] (in Russian). Interfax. 29 July 2020.
- ^ “Запущено производство российской антикоронавирусной вакцины” [Production of Russian anti-coronavirus vaccine launched] (in Russian). Vesti. 15 August 2020. Retrieved 21 September 2020.
- ^ Osborn A, Tétrault-Farber G (9 March 2021). “Russia’s Sputnik V COVID-19 vaccine could be produced in western Europe for first time after reported deals”. The Globe and Mail. Retrieved 9 March 2021.
- ^ “Russia has successfully fast tracked Covid-19 vaccine development, says India”. Mint. 14 July 2020. Retrieved 11 August 2020.
- ^ Nebehay S (11 August 2020). Shields M (ed.). “WHO says discussing new COVID-19 vaccine with Russia”. Reuters. Geneva. Retrieved 11 August 2020.
- ^ Jump up to:a b Brown C (28 November 2020). “Russia says its COVID vaccine is 95% effective. So why is there still Western resistance to it?”. http://www.cbc.ca. Retrieved 3 December 2020.
- ^ Reuters Staff (2020-11-11). “Instant view-Russia says its Sputnik V COVID-19 vaccine is 92% effective”
- ^ Zamira Rahim. “Russia’s Sputnik V vaccine is 91.6% effective against symptomatic Covid-19, interim trial results suggest”. CNN. Retrieved 9 February 2021.
- ^ Jones, Ian; Roy, Polly (2 February 2021). “Sputnik V COVID-19 vaccine candidate appears safe and effective”. The Lancet. 397(10275): 642–643. doi:10.1016/S0140-6736(21)00191-4. ISSN 0140-6736. PMC 7906719. PMID 33545098.
- ^ Taylor, Adam; Johnson, Carolyn Y. “Russian vaccine Sputnik V more than 90% effective in interim trial”. Washington Post. ISSN 0190-8286. Retrieved 9 February 2021.
- ^ Balakrishnan VS (1 October 2020). “The arrival of Sputnik V”. The Lancet Infectious Diseases. 20 (10): 1128. doi:10.1016/S1473-3099(20)30709-X. PMC 7511201. PMID 32979327.
- ^ Irfan U (11 December 2020). “Why staying cold is so important to a Covid-19 vaccine. The Moderna and Pfizer vaccines need to be stored at low temperatures. Are global health systems prepared?”. Vox. Retrieved 27 December 2020.
- ^ “US’ top infectious disease official commends Russia’s Sputnik”. United News of India. 6 March 2021.
- ^ Cyr E (24 August 2020). “Leger’s Weekly Survey” (PDF). leger360.com. Archived from the original (PDF) on 5 September 2020. Retrieved 5 September 2020.
- ^ Leger’s Weekly Survey on Russian vaccine; saved copy on 5 38.com
- ^ Mullin J, Malnick E (1 August 2020). “Britain unlikely to use Russia’s ‘untrustworthy’ Covid vaccine”. The Telegraph. Archived from the original on 19 August 2020. Retrieved 6 September 2020.
- ^ Ullah Z, Chernova A (6 September 2020). “Putin’s vaccine meets opposition from frontline workers in Russia”. CNN. Retrieved 6 September 2020.
- ^ “Covid: Russia begins vaccinations in Moscow”. BBC. 5 December 2020. Retrieved 11 December 2020.
- ^ Jump up to:a b Soldatkin V, Oziel C (5 December 2020). Char P (ed.). “Moscow rolls out Sputnik V COVID-19 vaccine to most exposed groups”. Reuters.
- ^ “Russia approves Sputnik V COVID-19 vaccine for people over 60: media”. Reuters. 26 December 2020.
- ^ “Covid: Russia begins vaccinations in Moscow”. BBC News. 5 December 2020.
- ^ “Moscow delivers Russia’s Sputnik V coronavirus vaccine to clinics”. The Guardian. Reuters. 5 December 2020.
- ^ “Coronavirus: Russia rolls out COVID vaccination in Moscow”. Deutsche Welle. 5 December 2020.
- ^ Marrow A, Ostroukh A (2 December 2020). “Putin orders Russia to begin a large-scale voluntary COVID-19 vaccination program next week”. The Globe and Mail. Retrieved 3 December 2020.
- ^ “COVID-19: Moscow opens Sputnik V clinics – but 100,000 have already had it”. Sky News.
- ^ “Russia to vaccinate two million against COVID-19 in Dec – RDIF head to BBC”. Reuters. 4 December 2020. Retrieved 21 September 2020.
- ^ “About 6.9 mln doses of Sputnik V vaccine to enter circulation in Russia by end of February”. TASS. 10 December 2020. Retrieved 21 September 2020.
- ^ “Coronavirus in Russia: The Latest News”. The Moscow Times. 22 December 2020. Retrieved 21 September 2020.
- ^ Rodgers J. “Facing Record COVID-19 Case Rise, Russia Rolls Out Sputnik V Vaccine”. Forbes.
- ^ Arkhipov I, Kravchenko S (2 December 2020). “Putin Orders Start of Mass Covid-19 Shots Hours After U.K. News”. Bloomberg News.
- ^ Meyer H, Arkhipov I. “Russia Defends First Covid-19 Vaccine as Safe Amid Skepticism”. Bloomberg News. Retrieved 12 August2020.
- ^ Litvinova D (24 November 2020). “Russian virus vaccine to cost less than $10 per dose abroad”. Associated Press.
- ^ Osborn A, Nikolskaya P (24 November 2020). “Russia’s Sputnik COVID-19 vaccine to cost less than $20 per person internationally”. The Globe and Mail. Retrieved 28 November2020.
- ^ Jaffe-Hoffman M (12 November 2020). “Israel to receive Russia’s 92% effective COVID vaccine”. The Jerusalem Post. Retrieved 19 November 2020.
- ^ Kingsley, Patrick; Bergman, Ronen; Kramer, Andrew E. (21 February 2021). “Israel Secretly Agrees to Fund Vaccines for Syria as Part of Prisoner Swap”. The New York Times. ISSN 0362-4331. Retrieved 3 March 2021.
- ^ “Argentina agrees to buy 25 million doses of Russia’s Covid-19 vaccine”. http://www.batimes.com.ar. 30 November 2020.
- ^ “Argentina Approves Russian Vaccine With Plane Waiting in Moscow”. Bloomberg.com. 23 December 2020.
- ^ Boadle A (24 October 2020). Wallis D (ed.). “Second Brazilian company to produce Russia’s Sputnik V COVID-19 vaccine”. Reuters.
- ^ “Argentina’s president sits for Russian Covid jab”. France 24. 21 January 2021.
- ^ Centenera M (21 January 2021). “Alberto Fernández, primer presidente de América Latina en vacunarse contra la covid-19 (in Spanish)”. EL PAÍS (in Spanish).
- ^ Camparsi, Maria Letizia. “Vaccino Sputnik, a San Marino 400 dosi al giorno dal 1 marzo: “Sicurezza? Confortati dagli studi. Per ora lo diamo solo ai nostri cittadini” (in Italian). Il Fatto Quotidiano. Retrieved 1 March 2021.
- ^ “EMA starts rolling review of the Sputnik V COVID-19 vaccine”.
- ^ “EU medical official warns of Sputnik jab ‘Russian roulette'”.
- ^ “Coronavirus (COVID-19) Vaccinations – Statistics and Research”. Our World in Data. Retrieved 3 March 2021.
- ^ “Putin Battles to Sell Russia’s Vaccine in New Rift With West”. Bloomberg.com. 31 December 2020. Retrieved 3 March 2021.
- ^ Jump up to:a b c “Angola, Congo Republic and Djibouti approve Russia’s Sputnik V vaccine”. Reuters. 3 March 2021.
- ^ “Hungarian drug regulator approves Sputnik V vaccine: website”. The Moscow Times. 7 February 2021.
- ^ Jump up to:a b c “Sputnik V vaccine registered in Bosnia and Herzegovina’s Republika Srpska”. TASS. 5 February 2021. Retrieved 8 February2021.
- ^ “Sputnik V registered in Kyrgyzstan”. Gamaleya Center (Press release). 23 February 2021.
- ^ “Syria authorizes use of Sputnik-V”. Roya. 22 February 2021.
- ^ “Turkmenistan is the first in Central Asia to have registered “Sputnik V” vaccine”. Orient. 18 January 2021.
- ^ “Uzbekistan Certifies Russia’s Sputnik Vaccine For Mass Use”. Agence France-Presse (Barron’s). 17 February 2021.
- ^ “Covid19: National Pharmaceuticals Agency registers Sputnik V vaccine”. Algeria Press service. 10 January 2021.
- ^ “Argentina has registered the Sputnik V vaccine based on Russian clinical trial data” (Press release). Gamaleya Center. Retrieved 1 January 2021.
- ^ “Armenia approves Russia’s Sputnik V coronavirus vaccine -Russia’s RDIF”. Reuters. 1 February 2021. Retrieved 1 February2021.
- ^ “Bahrain authorises Sputnik V COVID-19 vaccine for emergency use – Bahrain TV”. Reuters. 10 February 2021. Retrieved 19 February 2021.
- ^ “Belarus registers Sputnik V vaccine, in first outside Russia – RDIF”. Reuters. 21 December 2020. Retrieved 22 December2020.
- ^ “Ministerio de Salud de Bolivia – Bolivia y Rusia firman contrato para adquirir 5,2 millones de dosis de la vacuna Sputnik-V contra la COVID-19”. minsalud.gob.bo. Retrieved 1 January 2021.
- ^ “COVID-19: Egypt authorises Sputnik V, AstraZeneca virus jabs”. Gulf News. Retrieved 24 February 2021.
- ^ “Sputnik V authorised in Gabon” (Press release). Gamaleya Center. Retrieved 17 February 2021.
- ^ “Ghana approves Russia’s Sputnik V vaccine for emergency use – RDIF”. Reuters. 20 February 2021.
- ^ “Guatemala to receive Russia’s Sputnik vaccine in coming weeks”. Reuters. 24 February 2021.
- ^ “Guinea Begins Administering Russia’s Sputnik V Covid-19 Vaccine”. Africa news. 31 December 2020.
- ^ “Russia’s Sputnik V vaccine expands its reach in Latin America”. CNN. 3 March 2021.
- ^ “Honduras approves use of Sputnik V vaccine against COVID-19”. Xinhua News Agency. 25 February 2021.
- ^ “Iran approves Russian coronavirus vaccine Sputnik V”. Reuters. 26 January 2021.
- ^ “Sputnik V authorized in Iraq” (Press release). PharmiWeb.com. 4 March 2021.
- ^ “Jordan approves Russia’s Sputnik V vaccine for use against COVID-19” (Press release). Reuters. 10 March 2021.
- ^ “Kazakhstan begins mass vaccination by Russian Sputnik V”. 1 February 2021. Retrieved 19 February 2021.
- ^ “Morocco, Kenya approve Russian coronavirus vaccine for use – RDIF”. 10 March 2021. Retrieved 12 March 2021.
- ^ “Laos declares Covid-19 vaccinations safe, more to be inoculated next week | The Star”. The Star. Malaysia. Retrieved 19 February2021.
- ^ “Lebanon authorises emergency use of Russia’s Sputnik V vaccine”. Reuters. 5 February 2021.
- ^ “Mexico, Germany warm to Russia’s Sputnik V virus vaccine”. The Jakarta Post. 3 February 2021.
- ^ “Mongolia Approves Russia’s Sputnik V Coronavirus Vaccine – RDIF”. Urdu Point. 9 February 2021.
- ^ “Montenegro and St. Vincent approve Russia’s Sputnik V vaccine – RDIF”. Reuters. 12 February 2021.
- ^ “Morocco orders one million doses of Russia’s Sputnik V vaccine”. Yabiladi. 11 March 2021.
- ^ “Myanmar registers Russia’s Sputnik V COVID-19 vaccine”. TASS. Retrieved 19 February 2021.
- ^ “Namibia becomes the 50th country to authorize Sputnik V”(Press release). Moscow: Gamaleya Research Institute of Epidemiology and Microbiology. 11 March 2021. Retrieved 15 March 2021.
- ^ “Nicaragua approves Russian COVID-19 vaccine”. wsoctv. 3 February 2021.
- ^ “NRussia’s Sputnik V COVID 19 vaccine registered in North Macedonia”. TASS. 7 March 2021.
- ^ “Govt okays Russian vaccine for ’emergency use'”. Dawn. 24 January 2021.
- ^ “Palestine has become the first country in the Middle East to register Sputnik V vaccine”. RFID. 11 January 2021.
- ^ “Paraguay approves Russia’s Sputnik V vaccine: RDIF”. Reuters. 15 January 2021. Retrieved 15 January 2021.
- ^ “Russia’s Sputnik V approved for emergency use in PH”. CNN Philippines. 19 March 2021. Retrieved 19 March 2021.
- ^ Burki TK (November 2020). “The Russian vaccine for COVID-19”. The Lancet. Respiratory Medicine. 8 (11): e85–e86. doi:10.1016/S2213-2600(20)30402-1. PMC 7837053. PMID 32896274.
- ^ “Public Health (Emergency Authorisation of COVID-19 Vaccine) Rules, 2021” (PDF). Government of Saint Vincent and the Grenadines. 11 February 2021. Retrieved 12 February 2021.
- ^ “San Marino buys the Sputnik vaccine: “First doses already in the next few days””. Unioneonline. 20 February 2021.
- ^ “Agencija odobrila uvoz ruske vakcine Sputnjik V u Srbiju”. N1(in Serbian). 31 December 2020.
- ^ “Sputnik V approved for use in Slovakia”. rdif.ru. Retrieved 1 March 2021.
- ^ “Sri Lanka approves Russia’s Sputnik V vaccine”. The Hindu. 4 March 2021.
- ^ “Sputnik V vaccine authorized in Tunisia” (Press release). Gamaleya Center. Retrieved 30 January 2021.
- ^ “UAE approves Russia’s Sputnik vaccine for emergency use”. Khaleej Times. 21 January 2021. Retrieved 21 January 2021.
- ^ “Venezuela firma contrato para la adquisición de la vacuna rusa Sputnik V” (in Spanish). Reuters. 29 December 2020.
- ^ “Vietnam approves US, Russia Covid-19 vaccines for emergency use”. VnExpress. Retrieved 26 February 2021.
- ^ “Covid-19: Zimbabwe authorises Sputnik V, Sinovac vaccines for emergency use”. news24.com. 9 March 2021.
- ^ McCluskey, Mitchell; Pozzebon, Stefano; Arias, Tatiana; Lister, Tim (3 March 2021). “Russia’s Sputnik V vaccine expands its reach in Latin America”. CNN. Retrieved 15 March 2021.
- ^ “COVID vaccine: Italy to be first EU country to make RussiaN Sputnik V jab”. Euronews. Agence France-Presse. 9 March 2021. Retrieved 15 March 2021.
- ^ “RDIF inks contract with Malaysia to supply Sputnik V vaccine”. TASS. 26 January 2021. Retrieved 21 March 2021.
- ^ “Regulation and Prequalification”. World Health Organization. Retrieved 12 March 2021.
- ^ “EMA starts rolling review of the Sputnik V COVID-19 vaccine”. European Medicines Agency. 4 March 2021. Retrieved 12 March2021.
- ^ “Belarus registers Sputnik V vaccine, in first outside Russia – RDIF”. Reuters. 21 December 2020. Retrieved 22 December2020.
- ^ Turak N (21 January 2021). “Russia’s Sputnik vaccine gets its first approval in the EU, greenlight from UAE amid ongoing trials”. CNBC.
- ^ “Coronavirus: Hungary first in EU to approve Russian vaccine”. BBC News. 21 January 2021.
- ^ Walker S (21 January 2021). “Hungary breaks ranks with EU to license Russian vaccine”. The Guardian.
- ^ “Hungary Becomes First in EU to Approve Russian Covid Vaccine”. Bloomberg.com. 21 January 2021.
- ^ “COVID: Hungary fast-tracks Russian vaccine with EU approval in the works | DW | 21.01.2021”. DW.COM.
- ^ “Russia files for Sputnik vaccine registration in EU”. Euractiv.com. 20 January 2021.
- ^ “Clarification on Sputnik V vaccine in the EU approval process”(Press release). European Medicines Agency (EMA). 10 February 2021.
- ^ Jump up to:a b “EMA starts rolling review of the Sputnik V COVID-19 vaccine” (Press release). European Medicines Agency (EMA). 4 March 2021. Retrieved 4 March 2021.
- ^ Jump up to:a b Ahmed A, Kumar AM (11 January 2021). “Russia’s Sputnik V vaccine found safe in India mid-stage trial – Dr.Reddy’s”. Reuters. Retrieved 26 January 2021.
- ^ “Da la Cofepris autorización para que la vacuna Sputnik V se aplique en México”. Diario de Yucatán (in Spanish). 2 February 2021.
- ^ “Iran approves Russia’s Sputnik V COVID-19 vaccine”. Al Jazeera.
- ^ Reuters Staff (26 January 2021). “Iran approves Russian coronavirus vaccine Sputnik V”. Reuters.
- ^ “Sputnik V vaccines landed in Slovakia”. The Slovak Spectator. 1 March 2021. Retrieved 2 March 2021.
- ^ “Czech Republic turns to Russian vaccine amid soaring COVID cases”. Al Jazeera. 28 February 2021. Retrieved 1 March 2021.
- ^ “German leaders urge quick EU approval of Russia’s Sputnik V jab” thelocal.de. Retrieved 20 March 2021.
- ^ “Philippines grants emergency authorization for Russia’s Sputnik V vaccine”. ABS-CBN News. 19 March 2021. Retrieved 19 March2021.
- ^ “Russia’s Sputnik V approved for emergency use in PH”. CNN Philippines. 19 March 2021. Retrieved 19 March 2021.
- ^ “SPUTNIK V APPROVED IN VIETNAM”. sputnikvaccine.com. 23 March 2021. Retrieved 23 March 2021.
- ^ Foy, Henry; Seddon, Max; Sciorilli, Silvia Borrelli (10 March 2021). “Russia seeks to make Sputnik V in Italy as overseas demand surges”. Financial Times. Retrieved 10 March 2021.
- ^ “More Countries Line Up for Russia’s Sputnik V Coronavirus Vaccine”. The Moscow Times. 13 November 2020.
- ^ COVID vaccine: Italy to be first EU country to make Russian Sputnik V jab Euronews. Retrieved 11 March 2021.
- ^ Shim, Elizabeth (25 February 2021). “South Korean consortium to make 500 million doses of Sputnik V vaccine”. UPI. Retrieved 1 March 2021.
External links
| Scholia has a profile for Gam-COVID-Vac (Q98270627). |
| Russian Ministry of Health image of Gam-COVID-Vac vials | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Viral vector |
| Clinical data | |
| Trade names | Sputnik V[1]Спутник V |
| Other names | Gam-COVID-VacГам-КОВИД-Вак |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | Registered in Russia on 11 August 2020 AE, AG, DZ, BO, BY, HU, IR, PS, RS: EUA only |
| Identifiers | |
| DrugBank | DB15848 |
////////SARS-CoV-2, corona virus, covid 19, Gam-COVID-Vac Lyo, Sputnik V, Gam-COVID-Vac, Gamaleya, russia
#SARS-CoV-2, #corona virus, #covid 19, #Gam-COVID-Vac Lyo, #Sputnik V, #Gam-COVID-Vac, #Gamaleya, #russia, #vaccine
Johnson & Johnson COVID-19 vaccine, JNJ 78436735

Johnson & Johnson COVID-19 vaccine, JNJ 78436735
- Ad26.COV2.S
- JNJ-78436735
- Ad26COVS1
- VAC31518
- UNII: JT2NS6183B
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Covid-19 Vaccine Janssen | Injection, suspension | 0.95 Inf. U | Intramuscular | Janssen Cilag International Nv | 2021-03-17 | Not applicable |  | |
| Janssen COVID-19 Vaccine | Injection, suspension | 50000000000 {VP}/0.5mL | Intramuscular | Janssen Products, LP | 2021-01-04 | Not applicable |  |
| NAME | INGREDIENTS | DOSAGE | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Janssen COVID-19 Vaccine | Ad26.COV2.S (50000000000 {VP}/0.5mL) | Injection, suspension | Intramuscular | Janssen Products, LP | 2021-01-04 | Not applicable | |
| FORM | ROUTE | STRENGTH |
|---|---|---|
| Injection, suspension | Intramuscular | 0.95 Inf. U |
| Injection, suspension | Intramuscular | 50000000000 {VP}/0.5mL |
The Johnson & Johnson COVID-19 vaccine is a human adenovirus viral vector COVID-19 vaccine[12] developed by Janssen Vaccines in Leiden in The Netherlands,[13] and its Belgian parent company Janssen Pharmaceuticals,[14] subsidiary of American company Johnson & Johnson (J&J).[15][16]
The vaccine is based on a human adenovirus that has been modified to contain the gene for making the spike protein of the SARS-CoV-2 virus that causes COVID-19.[3] The vaccine requires only one dose and does not need to be stored frozen.[17]
The vaccine started clinical trials in June 2020, with Phase III trials involving around 43,000 people.[18] On 29 January 2021, Janssen announced that the vaccine was 66% effective in a one-dose regimen in preventing symptomatic COVID-19, with an 85% efficacy in preventing severe COVID-19.[19][20][21] The most common side effects were pain at the injection site, headache, fatigue, muscle aches and nausea.[22] Most of these side effects were mild to moderate in severity and lasted one or two days.
The vaccine has been granted an Emergency Use Authorization by the US Food and Drug Administration[23] and a conditional marketing authorisation by the European Medicines Agency.[11][24][25]
Ad26.COV2.S is a lead recombinant vaccine candidate that contains an adenovirus serotype 26 (Ad26) vector expressing a stabilized SARS-CoV-2 spike protein. The vaccine was created in collaboration with Johnson and Johnson (J&J), Janssen Pharmaceutical, and the Beth Israel Deaconess Medical Center. This vaccine lead candidate uses Janssen’s AdVac® and PER.C6® technologies. A preclinical study in hamsters infected with SARS-COV-2 infection1 showed a single immunization with the vaccine elicited neutralizing responses and protected against SARS-CoV-2 induced pneumonia and mortality, providing protection against the disease progression. Follow up preclinical studies in rhesus monkeys2 showed that the Ad26 vaccine produced a robust response and provided near perfect protection in nasal swabs and bronchoalveolar lavage following SARS-COV-2 challenge. As of June 2020, a Phase 1/2 clinical trial in adult humans was announced to evaluate the safety, immunogenicity, and efficacy of the ad26.COV.S vaccine in 1045 healthy adults between the ages of 18-55 (NCT04436276).
NEW DRUG APPROVALS
one time
$10.00
Description
The Johnson & Johnson COVID-19 vaccine consists of a replication-incompetent recombinant adenovirus type 26 (Ad26) vector expressing the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike (S) protein in a stabilized conformation.[26][4] The stabilized version of the spike protein – that includes two mutations in which the regular amino acids are replaced with prolines – was developed by researchers at the National Institute of Allergy and Infectious Diseases‘ Vaccine Research Center and the University of Texas at Austin.[27][28][29] The vaccine also contains the following inactive ingredients: citric acid monohydrate, trisodium citrate dihydrate, ethanol (alcohol), 2-hydroxypropyl-β-cyclodextrin (HBCD) (hydroxypropyl betadex), polysorbate 80, sodium chloride, sodium hydroxide, and hydrochloric acid.[26][1]
Characteristics
The Johnson & Johnson COVID-19 vaccine can remain viable for months in a standard refrigerator.[30][31][32] Unlike the Pfizer–BioNTech COVID-19 vaccine and the Moderna COVID-19 vaccine, the Johnson & Johnson COVID-19 vaccine is administered as a single dose instead of two separate doses and it is not shipped frozen.[33][17]
The storage and handling information in the Fact Sheet supersedes the storage and handling information on the carton and vial labels.[17] The vaccine should not be stored frozen.[17] Unpunctured vials may be stored between 9 to 25 °C (48 to 77 °F) for up to twelve hours.[26][17]
Development
During the COVID-19 pandemic, Johnson & Johnson committed over US$1 billion toward the development of a not-for-profit COVID-19 vaccine in partnership with the Biomedical Advanced Research and Development Authority (BARDA) Office of the Assistant Secretary for Preparedness and Response (ASPR) at the U.S. Department of Health and Human Services (HHS).[34][35] Johnson & Johnson stated that its vaccine project would be “at a not-for-profit level” as the company viewed it as “the fastest and the best way to find all the collaborations in the world to make this happen”.[36]

Inside of an Emergent BioSolutions facility where, in collaboration with Johnson & Johnson, vaccines are produced.
Janssen Vaccines, in partnership with Beth Israel Deaconess Medical Center (BIDMC), is responsible for developing the vaccine candidate, based on the same technology used to make its Ebola vaccine.[16][37][38]
Clinical trials
Phase I-II
In June 2020, Johnson & Johnson and the National Institute of Allergy and Infectious Diseases (NIAID) confirmed its intention to start a clinical trials of the Ad26.COV2.S vaccine in September 2020, with the possibility of Phase I/IIa human clinical trials starting at an accelerated pace in the second half of July.[39][40][41]
A Phase I/IIa clinical trial started with the recruitment of the first subject on 15 July 2020, and enrolled study participants in Belgium and the US.[42] Interim results from the Phase I/IIa trial established the safety, reactogenicity, and immunogenicity of Ad26.COV2.S.[43][44]
Phase III
A Phase III clinical trial called ENSEMBLE started enrollment in September 2020, and completed enrollment on 17 December 2020. It was designed as a randomized, double-blind, placebo-controlled clinical trial designed to evaluate the safety and efficacy of a single-dose vaccine versus placebo in adults aged 18 years and older. Study participants received a single intramuscular injection of Ad26.COV2.S at a dose level of 5×1010 virus particles on day one.[45] The trial was paused on 12 October 2020, because a volunteer became ill,[46] but the company said it found no evidence that the vaccine had caused the illness and announced on 23 October 2020, that it would resume the trial.[47][48] On 29 January 2021, Janssen announced safety and efficacy data from an interim analysis of ENSEMBLE trial data, which demonstrated the vaccine was 66% effective at preventing the combined endpoints of moderate and severe COVID-19 at 28 days post-vaccination among all volunteers. The interim analysis was based on 468 cases of symptomatic COVID-19 among 43,783 adult volunteers in Argentina, Brazil, Chile, Colombia, Mexico, Peru, South Africa, and the United States. No deaths related to COVID-19 were reported in the vaccine group, while five deaths in the placebo group were related to COVID-19.[49] During the trial, no anaphylaxis was observed in participants.[49]
A second Phase III clinical trial called ENSEMBLE 2 started enrollment on 12 November 2020. ENSEMBLE 2 differs from ENSEMBLE in that its study participants will receive two intramuscular (IM) injections of Ad26.COV2.S, one on day 1 and the next on day 57.[50]
Manufacturing
In April 2020, Johnson & Johnson entered a partnership with Catalent who will provide large-scale manufacturing of the Johnson & Johnson vaccine at Catalent’s Bloomington, Indiana facility.[51] In July 2020, the partnership was expanded to include Catalent’s Anagni, Italy facility.[52]
In July 2020, Johnson & Johnson pledged to deliver up to 300 million doses of its vaccine to the U.S., with 100 million upfront and an option for 200 million more. The deal, worth more than $1 billion, will be funded by the Biomedical Advanced Research and Development Authority (BARDA) and the U.S. Defense Department.[53][54] The deal was confirmed on 5 August.[55]
In September 2020, Grand River Aseptic Manufacturing agreed with Johnson & Johnson to support the manufacture of the vaccine, including technology transfer and fill and finish manufacture, at its Grand Rapids, Michigan facility.[56]
In December 2020, Johnson & Johnson and Reig Jofre, a Spanish pharmaceutical company, entered into an agreement to manufacture the vaccine at Reig Jofre’s Barcelona facility.[57] If the European Medicines Agency (EMA) grants approval to the vaccine by March 2021, a European Union regulator said that Johnson & Johnson could start supplying vaccines to EU states starting on April 2021.[58][59]
In August 2020, Johnson & Johnson signed a contract with the U.S. federal government for US$1 billion, agreeing to deliver 100 million doses of the vaccine to the U.S. following the U.S. Food and Drug Administration (FDA) grant of approval or emergency use authorization (EUA) for the vaccine.[54] Under its agreement with the U.S. government, Johnson & Johnson was targeted to produce 12 million doses by the end of February 2021, more than 60 million doses by the end of April 2021, and more than 100 million doses by the end of June 2021. However, in January 2021, Johnson & Johnson acknowledged manufacturing delays would likely prevent it from meeting its contract of 12 million doses delivered to the U.S. by the end of February.[60] In late February 2021 congressional testimony by a company executive, however, Johnson & Johnson indicated that the company could deliver 20 million doses to the U.S. government by the end of March, and 100 million doses in the first half of 2021.[61]
In February 2021, Sanofi and Johnson & Johnson struck a deal for Sanofi to provide support and infrastructure at Sanofi’s Marcy-l’Étoile, France facility to manufacture approximately 12 million doses of the Johnson & Johnson vaccine per month once authorized.[62]
In March 2021, Merck & Co and Johnson & Johnson struck a deal for Merck to manufacture the Johnson & Johnson vaccine at two facilities in the United States to help expand the manufacturing capacity of the vaccine using provisions of the Defense Production Act.[63]
Regulatory approval process
Europe
Beginning on 1 December 2020, clinical trial of the vaccine candidate has been undergoing a “rolling review” process by the Committee for Medicinal Products for Human Use of the European Medicines Agency (EMA), a step to expedite EMA consideration of an expected conditional Marketing Authorisation Application.[58][78] On 16 February 2021, Janssen applied to the EMA for conditional marketing authorization of the vaccine.[3][79] The Committee for Medicinal Products for Human Use (CHMP) approved the COVID-19 Vaccine Janssen on 11 March.[11][25] Shipments of the vaccine are scheduled to start in the second half of April, with a commitment to deliver at least 200 million doses to the EU in 2021.[80]
United States
On 4 February 2021, Janssen Biotech applied to the U.S. Food and Drug Administration (FDA) for an EUA, and the FDA announced that its Vaccines and Related Biological Products Advisory Committee (VRBPAC) would meet on 26 February to consider the application.[30][33][81][82] Johnson & Johnson announced that it planned to ship the vaccine immediately following authorization.[49] On 24 February, ahead of the VRBPAC meeting, briefing documents from Janssen and the FDA were issued; the FDA document recommends granting the EUA, concluding that the results of the clinical trials and safety data are consistent with FDA EUA guidance for COVID-19 vaccines.[83][84][26][85] At the 26 February meeting, VRBPAC voted unanimously (22–0) to recommend that a EUA for the vaccine be issued.[86] The FDA granted the EUA for the vaccine the following day.[9][10][87] On 28 February, the CDC Advisory Committee on Immunization Practices (ACIP) recommended the use of the vaccine for those aged 18 and older.[88][23]
Elsewhere
On 11 February 2021, Saint Vincent and the Grenadines issued an EUA for the Johnson & Johnson vaccine, as well as the Moderna vaccine, the Pfizer–BioNTech vaccine, the Sputnik V vaccine, and the Oxford–AstraZeneca vaccine.[89]
In December 2020, Johnson & Johnson entered into an agreement in principle with Gavi, the Vaccine Alliance to support the COVAX Facility. On 19 February 2021, Johnson & Johnson submitted its formal request and data package to the World Health Organization for an Emergency Use Listing (EUL); an EUL is a requirement for participation in COVAX. Johnson & Johnson anticipates providing up to 500 million doses through 2022 for COVAX.[90][31][91]
On 25 February 2021, Bahrain authorized the vaccine for emergency use.[92][93]
On 26 February 2021, the South Korean Ministry of Food and Drug Safety began a review of Johnson & Johnson’s application for approval of its vaccine.[94]
In late November 2020, Johnson & Johnson submitted a rolling review application to Health Canada for approval of its vaccine.[95] The Canadian government has placed an order with Johnson & Johnson for 10 million doses, with an option to purchase up to 28 million additional doses; on 5 March, the vaccine became the fourth to receive Health Canada approval.[96]
In February 2021, the vaccine received emergency authorization in South Africa.[97][98][99]
Deployment and impact
Given the Johnson & Johnson vaccine is a single dose and has a lower cost, it is expected that it will play an important role in low and middle-income countries.[100] With lower costs and lower requirements of storage and distribution in comparison to the COVID-19 vaccines by Pfizer and Moderna, the Johnson & Johnson vaccine will be more easily transported, stored, and administered.[101] South African health minister Zweli Mkhize announced on 9 February 2021 that the country would sell or swap its one million doses of AstraZeneca vaccine.[102] Once it did so, South Africa began vaccination using the Johnson & Johnson vaccine on 17 February 2021,[99] marking the vaccine’s first use outside of a clinical trial.[103]
Ethical concerns
The United States Conference of Catholic Bishops has expressed ethical concerns about the vaccine due to the use of tissue from aborted fetuses in the 1980s.[104]
See also
Notes
- ^ US authorization also includes the three sovereign nations in the Compact of Free Association: Palau, the Marshall Islands, and Micronesia.[75][76]
References
- ^ Jump up to:a b c “Janssen COVID-19 Vaccine- ad26.cov2.s injection, suspension”. DailyMed. Retrieved 27 February 2021.
- ^ “Janssen COVID-19 Emergency Use Authorization (EUA) Official Website”. Janssen. 28 February 2021. Retrieved 28 February2021.
- ^ Jump up to:a b c “EMA receives application for conditional marketing authorisation of COVID-19 Vaccine Janssen” (Press release). European Medicines Agency (EMA). 16 February 2021. Retrieved 16 February 2021.
- ^ Jump up to:a b c d e “A Randomized, Double-blind, Placebo-controlled Phase 3 Study to Assess the Efficacy and Safety of Ad26.COV2.S for the Prevention of SARS-CoV-2-mediated COVID-19 in Adults Aged 18 Years and Older ENSEMBLE Protocol VAC31518COV3001; Phase 3” (PDF). Janssen Vaccines & Prevention.
- ^ Jump up to:a b c d “A Randomized, Double-blind, Placebo-controlled Phase 3 Study to Assess the Efficacy and Safety of Ad26.COV2.S for the Prevention of SARS-CoV-2-mediated COVID-19 in Adults Aged 18 Years and Older ENSEMBLE 2 Protocol VAC31518COV3009; Phase 3” (PDF). Janssen Vaccines & Prevention.
- ^ Jump up to:a b “Johnson & Johnson Initiates Pivotal Global Phase 3 Clinical Trial of Janssen’s COVID-19 Vaccine Candidate”. Johnson & Johnson (Press release). Retrieved 23 September 2020.
- ^ “Regulatory Decision Summary – Janssen COVID-19 Vaccine – Health Canada”. Health Canada. 5 March 2021. Retrieved 5 March 2021.
- ^ “Janssen COVID-19 Vaccine monograph” (PDF). Janssen. 5 March 2021.
- ^ Jump up to:a b c “FDA Issues Emergency Use Authorization for Third COVID-19 Vaccine”. U.S. Food and Drug Administration (FDA) (Press release). 27 February 2021. Retrieved 27 February 2021.
- ^ Jump up to:a b c https://www.fda.gov/media/146303/download
- ^ Jump up to:a b c “COVID-19 Vaccine Janssen EPAR”. European Medicines Agency (EMA). 5 March 2021. Retrieved 16 March 2021.
- ^ “A Study of Ad26.COV2.S for the Prevention of SARS-CoV-2-Mediated COVID-19 in Adult Participants (ENSEMBLE)”. ClinicalTrials.gov. Retrieved 30 January 2021.
- ^ “Leiden developed Covid-19 vaccine submitted to EMA for approval”. 16 February 2021.
- ^ “Clinical trial COVID-19 vaccine candidate underway”. Janssen Belgium. Retrieved 13 March 2021.
- ^ “EMA recommends Johnson & Johnson Covid vaccine for approval; Developed in Leiden”. NL Times.
- ^ Jump up to:a b Saltzman J (12 March 2020). “Beth Israel is working with Johnson & Johnson on a coronavirus vaccine”. The Boston Globe.
- ^ Jump up to:a b c d e “Fact Sheet for Vaccine Providers & full EUA PI”(PDF). Janssen. Retrieved 28 February 2021.
- ^ Commissioner, Office of the (2 March 2021). “FDA Issues Emergency Use Authorization for Third COVID-19 Vaccine”. FDA. Retrieved 7 March 2021.
- ^ Salzman S (29 January 2021). “Johnson & Johnson single-shot vaccine 85% effective against severe COVID-19 disease”. ABC News.
- ^ Gallagher J (29 January 2021). “Covid vaccine: Single dose Covid vaccine 66% effective”. BBC News. Retrieved 29 January 2021.
- ^ Sohn R (29 January 2021). “J&J’s Covid vaccine is 66% effective, a weapon but not a knockout punch”. Stat. Retrieved 29 January2021.
- ^ Commissioner, Office of the (2 March 2021). “FDA Issues Emergency Use Authorization for Third COVID-19 Vaccine”. FDA. Retrieved 7 March 2021.
- ^ Jump up to:a b “Media Statement from CDC Director Rochelle P. Walensky, MD, MPH, on Signing the Advisory Committee on Immunization Practices’ Recommendation to Use Janssen’s COVID-19 Vaccine in People 18 and Older”. Centers for Disease Control and Prevention (CDC) (Press release). 28 February 2021. Retrieved 1 March 2021.
- ^ “EMA recommends COVID-19 Vaccine Janssen for authorisation in the EU” (Press release). European Medicines Agency (EMA). 11 March 2021. Retrieved 11 March 2021.
- ^ Jump up to:a b “COVID-19 Vaccine Janssen”. Union Register of medicinal products. Retrieved 16 March 2021.
- ^ Jump up to:a b c d FDA Briefing Document Janssen Ad26.COV2.S Vaccine for the Prevention of COVID-19 (PDF) (Report). U.S. Food and Drug Administration (FDA). Lay summary.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “The tiny tweak behind COVID-19 vaccines”. Chemical & Engineering News. 29 September 2020. Retrieved 1 March 2021.
- ^ Kramer, Jillian (31 December 2020). “They spent 12 years solving a puzzle. It yielded the first COVID-19 vaccines”. National Geographic.
- ^ Mercado NB, Zahn R, Wegmann F, Loos C, Chandrashekar A, Yu J, et al. (October 2020). “Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques”. Nature. 586 (7830): 583–88. Bibcode:2020Natur.586..583M. doi:10.1038/s41586-020-2607-z. PMC 7581548. PMID 32731257. S2CID 220893461.
- ^ Jump up to:a b Johnson CY, McGinley L (4 February 2021). “Johnson & Johnson seeks emergency FDA authorization for single-shot coronavirus vaccine”. The Washington Post.
- ^ Jump up to:a b Weintraub K. “One-dose J&J COVID-19 vaccine meets criteria as safe and effective, FDA report finds”. USA Today. Retrieved 25 February 2021.
- ^ Mole B (29 January 2021). “COVID variants throw J&J vaccine a curveball, lowering efficacy to 66%”. Ars Technica. Retrieved 26 February 2021.
- ^ Jump up to:a b Chander V (4 February 2021). “J&J files COVID-19 vaccine application with U.S. FDA”. Reuters. Retrieved 4 February 2021.
- ^ Vecchione A (13 March 2020). “J&J collaborates to accelerate COVID-19 vaccine development”. NJBIZ. Retrieved 22 April2020.
- ^ “Prisma Health collaborates with Ethicon Inc. to make, distribute VESper Ventilator Expansion Splitter Device”. WSPA 7News. 6 April 2020. Retrieved 22 April 2020.
- ^ “Coronavirus: Johnson & Johnson vows to make ‘not-for-profit’ vaccine”. Sky News. Retrieved 22 April 2020.
- ^ “A Beth Israel researcher helped create a COVID-19 vaccine that awaits approval. It could be a ‘game changer'”. The Boston Globe. 16 January 2021. Retrieved 28 February 2021.
- ^ “FDA grants third COVID-19 vaccine, developed in part at BIDMC, emergency use authorization”. Beth Israel Deaconess Medical Center (BIDMC). 27 February 2021. Retrieved 28 February 2021.
- ^ Coleman J (10 June 2020). “Final testing stage for potential coronavirus vaccine set to begin in July”. TheHill. Retrieved 11 June 2020.
- ^ “Moderna, AstraZeneca and J&J coronavirus shots rev up for NIH tests beginning in July: WSJ”. FiercePharma. Retrieved 11 June2020.
- ^ “Johnson & Johnson to start human testing of COVID-19 vaccine next week”. FiercePharma. Retrieved 20 July 2020.
- ^ “A Study of Ad26.COV2.S in Adults (COVID-19)”. ClinicalTrials.gov. Retrieved 19 February 2021.
- ^ Sadoff J, Le Gars M, Shukarev G, Heerwegh D, Truyers C, de Groot AM, et al. (January 2021). “Interim Results of a Phase 1-2a Trial of Ad26.COV2.S Covid-19 Vaccine”. New England Journal of Medicine. doi:10.1056/NEJMoa2034201. PMC 7821985. PMID 33440088.
- ^ “Johnson & Johnson COVID-19 Vaccine Candidate Interim Phase 1/2a Data Published in New England Journal of Medicine”. Johnson & Johnson (Press release). Retrieved 16 January 2021.
- ^ “Fourth large-scale COVID-19 vaccine trial begins in the United States”. National Institutes of Health. Retrieved 30 January 2021.
- ^ Hughes V, Thomas K, Zimmer C, Wu KJ (12 October 2020). “Johnson & Johnson halts coronavirus vaccine trial because of sick volunteer”. The New York Times. ISSN 0362-4331. Retrieved 12 October 2020.
- ^ “Johnson & Johnson Prepares to Resume Phase 3 ENSEMBLE Trial of its Janssen COVID-19 Vaccine Candidate in the U.S.”Johnson & Johnson (Press release). 23 October 2020. Retrieved 28 October 2020.
- ^ Edwards E, Miller SG (23 October 2020). “AstraZeneca, Johnson & Johnson resume late-stage Covid-19 vaccine trials”. NBC News. Retrieved 28 October 2020.
- ^ Jump up to:a b c “Johnson & Johnson Announces Single-Shot Janssen COVID-19 Vaccine Candidate Met Primary Endpoints in Interim Analysis of its Phase 3 ENSEMBLE Trial”. Johnson & Johnson(Press release). Retrieved 1 February 2021.
- ^ “A Study of Ad26.COV2.S for the Prevention of SARS-CoV-2-mediated COVID-19 in Adults (ENSEMBLE 2)”. ClinicalTrials.gov. Retrieved 30 January 2021.
- ^ Vecchione A (29 April 2020). “Catalent to lead US manufacturing for J&J’s lead COVID-19 vaccine candidate”. NJBIZ. Retrieved 13 November 2020.
- ^ “J&J expands COVID-19 vaccine pact with Catalent for finishing work at Italian facility”. FiercePharma. Retrieved 13 November2020.
- ^ “HHS, DOD Collaborate With Johnson & Johnson to Produce Millions of COVID-19 Investigational Vaccine Doses”. HHS.gov(Press release). 5 August 2020. Retrieved 6 August 2020.
- ^ Jump up to:a b “Johnson & Johnson Announces Agreement with U.S. Government for 100 Million Doses of Investigational COVID-19 Vaccine”. Johnson & Johnson (Press release). Retrieved 6 August 2020.
- ^ “US to Pay Johnson and Johnson $1 Billion for COVID-19 Vaccine”. Voice of America. Retrieved 5 August 2020.
- ^ “Ramping Up COVID-19 Vaccine Fill and Finish Capacity”. Contract Pharma. 3 November 2020.
- ^ Allen JF (15 December 2020). “Spain’s Reig Jofre to manufacture J&J’s COVID-19 vaccine, shares soar”. Reuters.
- ^ Jump up to:a b Guarascio F (13 January 2021). “J&J COVID-19 vaccine could be available in Europe in April: source”. Reuters.
- ^ “EMA expected to approve Johnson & Johnson vaccine by March – CEO of Janssen Italy to paper”. Reuters. 10 February 2021. Retrieved 13 February 2021.
- ^ Zimmer C, LaFraniere S, Weiland N (13 January 2021). “Johnson & Johnson Expects Vaccine Results Soon but Lags in Production”. The New York Times.
- ^ Sarah Owermohle, Johnson & Johnson says it can provide 20M vaccine doses by late March, Politico (22 February 2021).
- ^ France’s Sanofi to help Johnson & Johnson manufacture COVID-19 vaccine, Reuters (22 February 2021).
- ^ “Biden Administration Announces Historic Manufacturing Collaboration Between Merck and Johnson & Johnson to Expand Production of COVID-19 Vaccines”. HHS (Press release). 2 March 2021. Retrieved 4 March 2021.
- ^ “European Commission authorises fourth safe and effective vaccine against COVID-19”. European Commission (Press release). 11 March 2021.
- ^ Jump up to:a b “EU-Kommissionen har i dag udstedt en betinget markedsføringstilladelse til Johnson & Johnsons COVID-19- vaccine. Tilladelsen gælder i Danmark”. Lægemiddelstyrelsen (in Danish). Retrieved 12 March 2021.
- ^ “The Icelandic Medicines Agency have issued a conditional marketing authorisation”. covid.is. Retrieved 12 March 2021.
- ^ “The European Commission has now approved the Johnson & Johnson vaccine. This means that the vaccine is approved for use in the EU and Norway”. vg. Retrieved 12 March 2021.
- ^ “Informació en relació amb la vacunació contra la COVID-19”(PDF). Govern d’Andorra. Retrieved 14 March 2021.
- ^ “Bahrain first to approve Johnson & Johnson COVID-19 vaccine for emergency use”. Reuters. 25 February 2021. Retrieved 25 February 2021.
- ^ “Bahrain becomes 1st nation to grant J&J shot emergency use”. ABC News. 25 February 2021. Retrieved 25 February 2021.
- ^ “Johnson & Johnson COVID-19 vaccine becomes 4th to receive Health Canada approval | CBC News”. Canadian Broadcasting Corporation. Retrieved 5 March 2021.
- ^ https://www.jnj.com/johnson-johnson-single-shot-covid-19-vaccine-granted-conditional-marketing-authorization-by-european-commission
- ^ “Public Health (Emergency Authorisation of COVID-19 Vaccine) Rules, 2021” (PDF). Government of Saint Vincent and the Grenadines. 11 February 2021. Retrieved 12 February 2021.
- ^ “Coronavirus: South Africa rolls out vaccination programme”. BBC News. 17 February 2021. Retrieved 19 February 2021.
- ^ “Interior Applauds Inclusion of Insular Areas through Operation Warp Speed to Receive COVID-19 Vaccines” (Press release). United States Department of the Interior (DOI). 12 December 2020. Retrieved 13 January 2021. This article incorporates text from this source, which is in the public domain.
- ^ Dorman B (6 January 2021). “Asia Minute: Palau Administers Vaccines to Keep Country Free of COVID”. Hawaii Public Radio. Retrieved 13 January 2021.
- ^ “WHO approves J&J’s COVID-19 vaccine for emergency listing”. Channel News Asia. 13 March 2021. Retrieved 13 March2021.
- ^ “Johnson & Johnson Announces Initiation of Rolling Submission for its Single-dose Janssen COVID-19 Vaccine Candidate with the European Medicines Agency” (Press release). Johnson & Johnson. 1 December 2020.
- ^ Johnson & Johnson Announces Submission of European Conditional Marketing Authorisation Application to the EMA for its Investigational Janssen COVID-19 Vaccine Candidate
- ^ M, Muvija; Aripaka, Pushkala (11 March 2021). “Europe clears J&J’s single-shot COVID-19 vaccine as roll-out falters”. Reuters. Retrieved 16 March 2021.
- ^ “FDA Announces Advisory Committee Meeting to Discuss Janssen Biotech Inc.’s COVID-19 Vaccine Candidate” (Press release). U.S. Food and Drug Administration. 4 February 2021. Retrieved 4 February 2021.
- ^ “VRBPAC February 26, 2021 Meeting Announcement”. U.S. Food and Drug Administration. Retrieved 19 February 2021.
- ^ Janssen Biotech, Inc. COVID-19 Vaccine Ad26.COV2.S VRBPAC Briefing Document (PDF) (Report). Janssen Biotech.
- ^ Janssen Biotech, Inc. COVID-19 Vaccine Ad26.COV2.S VRBPAC Briefing Document Addendum (PDF) (Report). Janssen Biotech.
- ^ Christensen J (24 February 2021). “FDA says Johnson & Johnson Covid-19 vaccine meets requirements for emergency use authorization”. CNN.
- ^ Lovelace Jr B (26 February 2021). “FDA panel unanimously recommends third Covid vaccine as J&J wins key vote in path to emergency use”.
- ^ McGinley L, Johnson CY (27 February 2021). “FDA authorizes Johnson & Johnson’s single-shot coronavirus vaccine, adding to the nation’s arsenal against the pandemic”. The Washington Post.
- ^ Feuer W (28 February 2021). “CDC panel recommends use of J&J’s single-shot Covid vaccine, clearing way for distribution”. CNBC. Retrieved 28 February 2021.
- ^ “Public Health (Emergency Authorisation of COVID-19 Vaccine) Rules, 2021” (PDF). Government of Saint Vincent and the Grenadines. 11 February 2021. Retrieved 12 February 2021.
- ^ Johnson & Johnson Announces Submission to World Health Organization for Emergency Use Listing of Investigational Single-Shot Janssen COVID-19 Vaccine Candidate, Johnson & Johnson (19 February 2021).
- ^ Heeb G. “Johnson & Johnson Applies For Emergency Use Vaccine Approval At W.H.O.” Forbes. Retrieved 25 February2021.
- ^ “Bahrain first to approve Johnson & Johnson COVID-19 vaccine for emergency use”. Reuters. 25 February 2021. Retrieved 25 February 2021.
- ^ “Bahrain becomes 1st nation to grant J&J shot emergency use”. ABC News. 25 February 2021. Retrieved 25 February 2021.
- ^ South Korea launches review of Johnson & Johnson’s COVID-19 vaccine, Reuters (26 February 2021).
- ^ Terry Haig, Novavax submits its vaccine for Health Canada approval, Radio Canada International (1 February 2021).
- ^ “Johnson & Johnson COVID-19 vaccine becomes 4th to receive Health Canada approval”. CBC.
- ^ “SA is the first country to roll out Johnson & Johnson vaccine – what you need to know about the jab”. BusinessInsider. 17 February 2021. Retrieved 4 March 2021.
- ^ Browdie, Brian (20 February 2021). “South Africa to be first to use Johnson Johnson Covid-19 vaccine”. Quartz. Retrieved 4 March2021.
- ^ Jump up to:a b Steinhauser G (17 February 2021). “South Africa Rolls Out J&J Covid-19 Vaccine to Healthcare Workers”. The Wall Street Journal.
- ^ Grady D (29 January 2021). “Which Covid Vaccine Should You Get? Experts Cite the Effect Against Severe Disease”. The New York Times. Retrieved 9 February 2021.
- ^ Brueck H. “Moderna vaccine creator calls Johnson & Johnson’s competing shot a ‘darn good’ tool to fight the pandemic”. Business Insider. Retrieved 9 February 2021.
- ^ Winning A, Roelf W (9 February 2021). “South Africa may sell AstraZeneca shots as it switches to J&J vaccine to fight variant”. Yahoo!. Reuters. Retrieved 11 February 2021.
- ^ “Johnson & Johnson applies to WHO for emergency use listing of COVID-19 vaccine”. Reuters. 19 February 2021. Retrieved 19 March 2021.
- ^ “Some US bishops discourage Catholics from getting Johnson & Johnson vaccine if others are available”. CNN. 3 March 2021. Retrieved 20 March 2021.
External links
| Scholia has a profile for Ad26.COV2.S (Q98655215). |
- “How the Johnson & Johnson Covid-19 Vaccine Works”. The New York Times.
- “Janssen COVID-19 Vaccine (Johnson & Johnson)”. Centers for Disease Control and Prevention (CDC).
| A vial of Janssen COVID-19 Vaccine | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Viral vector |
| Clinical data | |
| Trade names | Janssen COVID-19 Vaccine,[1][2] COVID-19 Vaccine Janssen[3] |
| Other names | Ad26.COV2.S[4][5][6]JNJ-78436735[4][5][6]Ad26COVS1[4][5]VAC31518[4][5] |
| License data | US DailyMed: Janssen_COVID-19_Vaccine |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | CA: Schedule D; Authorized by interim order [7][8]US: Unapproved (Emergency Use Authorization)[9][1][10]EU: Conditional marketing authorization granted [11] |
| Identifiers | |
| DrugBank | DB15857 |
| UNII | JT2NS6183B |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
////////////////Johnson & Johnson, COVID-19 vaccine, JNJ 78436735, Ad26.COV2.S, JNJ-78436735, Ad26COVS1, VAC31518, vaccine, corona virus, covid 19
#Johnson & Johnson, #COVID-19 vaccine, #JNJ 78436735, #Ad26.COV2.S, #JNJ-78436735, #Ad26COVS1, VAC31518, #vaccine, #corona virus, #covid 19
NOVAWAX, NVX-CoV2373,
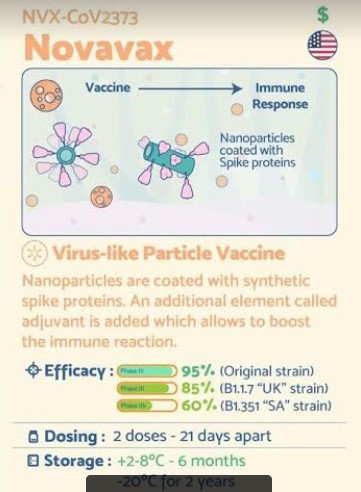
NOVAWAX
SARS-CoV-2 rS Nanoparticle Vaccine
MCDC OTA agreement number W15QKN-16-9-1002
Novavax COVID-19 vaccine, Coronavirus disease 19 infection
SARS-CoV-2 rS, TAK 019
Novavax, Inc. is an American vaccine development company headquartered in Gaithersburg, Maryland, with additional facilities in Rockville, Maryland and Uppsala, Sweden. As of 2020, it had an ongoing Phase III clinical trial in older adults for its candidate vaccine for seasonal influenza, NanoFlu and a candidate vaccine (NVX-CoV2373) for prevention of COVID-19.
NVX-CoV2373 is a SARS-CoV-2 rS vaccine candidate and was shown to have high immunogenicity in studies. The vaccine is created from the genetic sequence of COVID-19 and the antigen derived from the virus spike protein is generated using recombinant nanoparticle technology. The vaccine was developed and tested by Novavax. As of May 2020, the company is pursuing a Phase 1 clinical trial (NCT04368988) to test the vaccine.
History
Novavax was founded in 1987. It focused principally on experimental vaccine development, but did not achieve a successful launch up to 2021.[4]
In June 2013, Novavax acquired the Matrix-M adjuvant platform with the purchase of Swedish company Isconova AB and renamed its new subsidiary Novavax AB.[5]
In 2015, the company received an $89 million grant from the Bill & Melinda Gates Foundation to support the development of a vaccine against human respiratory syncytial virus for infants via maternal immunization.[6][7][8][9]
In March 2015 the company completed a Phase I trial for its Ebola vaccine candidate,[10] as well as a phase II study in adults for its RSV vaccine, which would become ResVax.[11] The ResVax trial was encouraging as it showed significant efficacy against RSV infection.[11]
2016 saw the company’s first phase III trial, the 12,000 adult Resolve trial,[11] for its respiratory syncytial virus vaccine, which would come to be known as ResVax, fail in September.[3] This triggered an eighty-five percent dive in the company’s stock price.[3] Phase II adult trial results also released in 2016 showed a stimulation of antigencity, but failure in efficacy.[11] Evaluation of these results suggested that an alternative dosing strategy might lead to success, leading to plans to run new phase II trials.[3] The company’s difficulties in 2016 led to a three part strategy for 2017: cost reduction through restructuring and the termination of 30% of their workforce; pouring more effort into getting ResVax to market; and beginning clinical trials on a Zika virus vaccine.[3]
Alongside the adult studies of ResVax, the vaccine was also in 2016 being tested against infant RSV infection through the route of maternal immunization.[11]
In 2019, late-stage clinical testing of ResVax, failed for a second time, which resulted in a major downturn in investor confidence and a seventy percent reduction in capital value for the firm.[12][13] As a secondary result, the company was forced to conduct a reverse stock split in order to maintain Nasdaq minimum qualification, meaning it was in risk of being delisted.[13]
The company positions NanoFlu for the unmet need for a more effective vaccine against influenza, particularly in the elderly who often experience serious and sometimes life-threatening complications. In January 2020, it was granted fast track status by the U.S. Food and Drug Administration (FDA) for NanoFlu.
External sponsorships
In 2018, Novavax received a US$89 million research grant from the Bill and Melinda Gates Foundation for development of vaccines for maternal immunization.[14]
In May 2020, Novavax received US$384 million from the Coalition for Epidemic Preparedness Innovations to fund early-stage evaluation in healthy adults of the company’s COVID-19 vaccine candidate NVX-CoV2373 and to develop resources in preparation for large-scale manufacturing, if the vaccine proves successful.[15] CEPI had already invested $4 million in March.[15]
Drugs in development
ResVax is a nanoparticle-based treatment using a recombinant F lipoprotein or saponin, “extracted from the Quillaja saponaria [or?] Molina bark together with cholesterol and phospholipid.”[16] It is aimed at stimulating resistance to respiratory syncytial virus infection, targeting both adult and infant populations.[11]
In January 2020, Novavax was given Fast Track status by the FDA to expedite the review process for NanoFlu, a candidate influenze vaccine undergoing a Phase III clinical trial scheduled for completion by mid-2020.[17]
COVID-19 vaccine candidate
See also: NVX-CoV2373 and COVID-19 vaccine
In January 2020, Novavax announced development of a vaccine candidate, named NVX-CoV2373, to establish immunity to SARS-CoV-2.[18] NVX-CoV2373 is a protein subunit vaccine that contains the spike protein of the SARS-CoV-2 virus.[19] Novavax’s work is in competition for vaccine development among dozens of other companies.
In January 2021, the company released phase 3 trials showing that it has 89% efficacy against Covid-19, and also provides strong immunity against new variants.[20] It has applied for emergency use in the US and UK but will be distributed in the UK first.Novavax COVID-19 Vaccine Demonstrates 89.3% Efficacy in UK Phase 3 TrialJan 28, 2021 at 4:05 PM ESTDownload PDF
First to Demonstrate Clinical Efficacy Against COVID-19 and Both UK and South Africa Variants
- Strong efficacy in Phase 3 UK trial with over 50% of cases attributable to the now-predominant UK variant and the remainder attributable to COVID-19 virus
- Clinical efficacy demonstrated in Phase 2b South Africa trial with over 90% of sequenced cases attributable to prevalent South Africa escape variant
- Company to host investor conference call today at 4:30pm ET
GAITHERSBURG, Md., Jan. 28, 2021 (GLOBE NEWSWIRE) — Novavax, Inc. (Nasdaq: NVAX), a biotechnology company developing next-generation vaccines for serious infectious diseases, today announced that NVX-CoV2373, its protein-based COVID-19 vaccine candidate, met the primary endpoint, with a vaccine efficacy of 89.3%, in its Phase 3 clinical trial conducted in the United Kingdom (UK). The study assessed efficacy during a period with high transmission and with a new UK variant strain of the virus emerging and circulating widely. It was conducted in partnership with the UK Government’s Vaccines Taskforce. Novavax also announced successful results of its Phase 2b study conducted in South Africa.
“With today’s results from our UK Phase 3 and South Africa Phase 2b clinical trials, we have now reported data on our COVID-19 vaccine from Phase 1, 2 and 3 trials involving over 20,000 participants. In addition, our PREVENT-19 US and Mexico clinical trial has randomized over 16,000 participants toward our enrollment goal of 30,000. NVX-CoV2373 is the first vaccine to demonstrate not only high clinical efficacy against COVID-19 but also significant clinical efficacy against both the rapidly emerging UK and South Africa variants,” said Stanley C. Erck, President and Chief Executive Officer, Novavax. “NVX-CoV2373 has the potential to play an important role in solving this global public health crisis. We look forward to continuing to work with our partners, collaborators, investigators and regulators around the world to make the vaccine available as quickly as possible.”
NVX-CoV2373 contains a full-length, prefusion spike protein made using Novavax’ recombinant nanoparticle technology and the company’s proprietary saponin-based Matrix-M™ adjuvant. The purified protein is encoded by the genetic sequence of the SARS-CoV-2 spike (S) protein and is produced in insect cells. It can neither cause COVID-19 nor can it replicate, is stable at 2°C to 8°C (refrigerated) and is shipped in a ready-to-use liquid formulation that permits distribution using existing vaccine supply chain channels.
UK Phase 3 Results: 89.3% Efficacy
The study enrolled more than 15,000 participants between 18-84 years of age, including 27% over the age of 65. The primary endpoint of the UK Phase 3 clinical trial is based on the first occurrence of PCR-confirmed symptomatic (mild, moderate or severe) COVID-19 with onset at least 7 days after the second study vaccination in serologically negative (to SARS-CoV-2) adult participants at baseline.
The first interim analysis is based on 62 cases, of which 56 cases of COVID-19 were observed in the placebo group versus 6 cases observed in the NVX-CoV2373 group, resulting in a point estimate of vaccine efficacy of 89.3% (95% CI: 75.2 – 95.4). Of the 62 cases, 61 were mild or moderate, and 1 was severe (in placebo group).
Preliminary analysis indicates that the UK variant strain that was increasingly prevalent was detected in over 50% of the PCR-confirmed symptomatic cases (32 UK variant, 24 non-variant, 6 unknown). Based on PCR performed on strains from 56 of the 62 cases, efficacy by strain was calculated to be 95.6% against the original COVID-19 strain and 85.6% against the UK variant strain [post hoc].
The interim analysis included a preliminary review of the safety database, which showed that severe, serious, and medically attended adverse events occurred at low levels and were balanced between vaccine and placebo groups.
“These are spectacular results, and we are very pleased to have helped Novavax with the development of this vaccine. The efficacy shown against the emerging variants is also extremely encouraging. This is an incredible achievement that will ensure we can protect individuals in the UK and the rest of the world from this virus,” said Clive Dix, Chair, UK Vaccine Taskforce.
Novavax expects to share further details of the UK trial results as additional data become available. Additional analysis on both trials is ongoing and will be shared via prepublication servers as well as submitted to a peer-reviewed journal for publication. The company initiated a rolling submission to the United Kingdom’s regulatory agency, the MHRA, in mid-January.
South Africa Results: Approximately 90% of COVID-19 cases attributed to South Africa escape variant
In the South Africa Phase 2b clinical trial, 60% efficacy (95% CI: 19.9 – 80.1) for the prevention of mild, moderate and severe COVID-19 disease was observed in the 94% of the study population that was HIV-negative. Twenty-nine cases were observed in the placebo group and 15 in the vaccine group. One severe case occurred in the placebo group and all other cases were mild or moderate. The clinical trial also achieved its primary efficacy endpoint in the overall trial population, including HIV-positive and HIV-negative subjects (efficacy of 49.4%; 95% CI: 6.1 – 72.8).
This study enrolled over 4,400 patients beginning in August 2020, with COVID-19 cases counted from September through mid-January. During this time, the triple mutant variant, which contains three critical mutations in the receptor binding domain (RBD) and multiple mutations outside the RBD, was widely circulating in South Africa. Preliminary sequencing data is available for 27 of 44 COVID-19 events; of these, 92.6% (25 out of 27 cases) were the South Africa escape variant.
Importantly in this trial, approximately 1/3 of the patients enrolled (but not included in the primary analyses described above) were seropositive, demonstrating prior COVID-19 infection at baseline. Based on temporal epidemiology data in the region, the pre-trial infections are thought to have been caused by the original COVID-19 strain (i.e., non-variant), while the subsequent infections during the study were largely variant virus. These data suggest that prior infection with COVID-19 may not completely protect against subsequent infection by the South Africa escape variant, however, vaccination with NVX-CoV2373 provided significant protection.
“The 60% reduced risk against COVID-19 illness in vaccinated individuals in South Africans underscores the value of this vaccine to prevent illness from the highly worrisome variant currently circulating in South Africa, and which is spreading globally. This is the first COVID-19 vaccine for which we now have objective evidence that it protects against the variant dominating in South Africa,” says Professor Shabir Maddi, Executive Director of the Vaccines and Infectious Diseases Analytics Research Unit (VIDA) at Wits, and principal investigator in the Novavax COVID-19 vaccine trial in South Africa. “I am encouraged to see that Novavax plans to immediately begin clinical development on a vaccine specifically targeted to the variant, which together with the current vaccine is likely to form the cornerstone of the fight against COVID-19.”
Novavax initiated development of new constructs against the emerging strains in early January and expects to select ideal candidates for a booster and/or combination bivalent vaccine for the new strains in the coming days. The company plans to initiate clinical testing of these new vaccines in the second quarter of this year.
“A primary benefit of our adjuvanted platform is that it uses a very small amount of antigen, enabling the rapid creation and large-scale production of combination vaccine candidates that could potentially address multiple circulating strains of COVID-19,” said Gregory M. Glenn, M.D., President of Research and Development, Novavax. “Combined with the safety profile that has been observed in our studies to-date with our COVID-19 vaccine, as well as prior studies in influenza, we are optimistic about our ability to rapidly adapt to evolving conditions.”
The Coalition for Epidemic Preparedness Innovations (CEPI) funded the manufacturing of doses of NVX-CoV2373 for this Phase 2b clinical trial, which was supported in part by a $15 million grant from the Bill & Melinda Gates Foundation.
Significant progress on PREVENT-19 Clinical Trial in US and Mexico
To date, PREVENT-19 has randomized over 16,000 participants and expects to complete our targeted enrollment of 30,000 patients in the first half of February. PREVENT-19 is being conducted with support from the U.S. government partnership formerly known as Operation Warp Speed, which includes the Department of Defense, the Biomedical Advanced Research and Development Authority (BARDA), part of the U.S. Department of Health and Human Services (HHS) Office of the Assistant Secretary for Preparedness and Response, and the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health (NIH) at HHS. BARDA is also providing up to $1.75 billion under a Department of Defense agreement.
PREVENT-19 (the PRE-fusion protein subunit Vaccine Efficacy Novavax Trial | COVID-19) is a Phase 3, randomized, placebo-controlled, observer-blinded study in the US and Mexico to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373 with Matrix-M in up to 30,000 subjects 18 years of age and older compared with placebo. The trial design has been harmonized to align with other Phase 3 trials conducted under the auspices of Operation Warp Speed, including the use of a single external independent Data and Safety Monitoring Board to evaluate safety and conduct an unblinded review when predetermined interim analysis events are reached.
The trial’s primary endpoint is the prevention of PCR-confirmed, symptomatic COVID-19. The key secondary endpoint is the prevention of PCR-confirmed, symptomatic moderate or severe COVID-19. Both endpoints will be assessed at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2.
Conference Call
Novavax will host a conference call today at 4:30pm ET. The dial-in numbers for the conference call are (877) 212-6076 (Domestic) or (707) 287-9331 (International), passcode 7470222. A replay of the conference call will be available starting at 7:30 p.m. ET on January 28, 2021 until 7:30 p.m. ET on February 4, 2021. To access the replay by telephone, dial (855) 859-2056 (Domestic) or (404) 537-3406 (International) and use passcode 7470222.
A webcast of the conference call can also be accessed on the Novavax website at novavax.com/events. A replay of the webcast will be available on the Novavax website until April 28, 2021.
About NVX-CoV2373
NVX-CoV2373 is a protein-based vaccine candidate engineered from the genetic sequence of SARS-CoV-2, the virus that causes COVID-19 disease. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike (S) protein and is adjuvanted with Novavax’ patented saponin-based Matrix-M™ to enhance the immune response and stimulate high levels of neutralizing antibodies. NVX-CoV2373 contains purified protein antigen and can neither replicate, nor can it cause COVID-19. Over 37,000 participants have participated to date across four different clinical studies in five countries. NVX-CoV2373 is currently being evaluated in two pivotal Phase 3 trials: a trial in the U.K that completed enrollment in November and the PREVENT-19 trial in the U.S. and Mexico that began in December.
About Matrix-M™
Novavax’ patented saponin-based Matrix-M™ adjuvant has demonstrated a potent and well-tolerated effect by stimulating the entry of antigen presenting cells into the injection site and enhancing antigen presentation in local lymph nodes, boosting immune response.
About Novavax
Novavax, Inc. (Nasdaq: NVAX) is a biotechnology company that promotes improved health globally through the discovery, development and commercialization of innovative vaccines to prevent serious infectious diseases. The company’s proprietary recombinant technology platform combines the power and speed of genetic engineering to efficiently produce highly immunogenic nanoparticles designed to address urgent global health needs. Novavax is conducting late-stage clinical trials for NVX-CoV2373, its vaccine candidate against SARS-CoV-2, the virus that causes COVID-19. NanoFlu™, its quadrivalent influenza nanoparticle vaccine, met all primary objectives in its pivotal Phase 3 clinical trial in older adults and will be advanced for regulatory submission. Both vaccine candidates incorporate Novavax’ proprietary saponin-based Matrix-M™ adjuvant to enhance the immune response and stimulate high levels of neutralizing antibodies.
For more information, visit www.novavax.com and connect with us on Twitter and LinkedIn.
Candidate: NVX-CoV2373
Category: VAX
Type: Stable, prefusion protein made using Novavax’ proprietary nanoparticle technology, and incorporating its proprietary saponin-based Matrix-M™ adjuvant.
2021 Status: Novavax on March 11 announced final efficacy of 96.4% against mild, moderate and severe disease caused by the original COVID-19 strain in a pivotal Phase III trial in the U.K. of NVX–CoV2373. The study enrolled more than 15,000 participants between 18-84 years of age, including 27% over the age of 65.
The company also announced the complete analysis of its Phase IIb trial in South Africa, showing the vaccine had an efficacy of 55.4% among a cohort of HIV-negative trial participants, and an overall efficacy of 48.6% against predominantly variant strains of SARS-CoV-2 among 147 PCR-positive cases (51 cases in the vaccine group and 96 in the placebo group). Across both trials, NVX-CoV2373 demonstrated 100% protection against severe disease, including all hospitalization and death.
Philippines officials said March 10 that they secured 30 million doses of NVX-CoV2373 through an agreement with the Serum Institute of India, the second vaccine deal signed by the national government, according to Agence France-Presse. The first was with AstraZeneca for 2.6 million doses of its vaccine, developed with Oxford University.
The Novavax vaccine will be available from the third quarter, at a price that has yet to be finalized. The government hopes to secure 148 million doses this year from seven companies—enough for around 70% of its population.
In announcing fourth quarter and full-year 2020 results on March 1, Novavax said it could file for an emergency use authorization with the FDA in the second quarter of 2021. Novavax hopes it can use data from its Phase III U.K. clinical trial in its FDA submission, and expects the FDA to examine data in May, a month after they are reviewed by regulators in the U.K., President and CEO Stanley C. Erck said on CNBC. Should the FDA insist on waiting for U.S. data, the agency may push the review timeline by one or two months, he added.
The company also said that NVX-CoV2373 showed 95.6% efficacy against the original strain of COVID-19 and 85.6% against the UK variant strain, and re-stated an earlier finding that its vaccine met the Phase III trial’s primary endpoint met with an efficacy rate of 89.3%.
Novavax said February 26 that it signed an exclusive license agreement with Takeda Pharmaceutical for Takeda to develop, manufacture, and commercialize NVX-CoV2373 in Japan.
Novavax agreed to transfer the technology for manufacturing of the vaccine antigen and will supply its Matrix-M™ adjuvant to Takeda. Takeda anticipated the capacity to manufacture over 250 million doses of the COVID-19 vaccine per year. Takeda agreed in return to pay Novavax undisclosed payments tied to achieving development and commercial milestones, plus a portion of proceeds from the vaccine.
Takeda also disclosed that it dosed the first participants in a Phase II clinical trial to test the immunogenicity and safety of Novavax’ vaccine candidate in Japanese participants.
Novavax on February 18 announced a memorandum of understanding with Gavi, the Vaccine Alliance (Gavi), to provide 1.1 billion cumulative doses of NVX-CoV2373 for the COVAX Facility. Gavi leads the design and implementation of the COVAX Facility, created to supply vaccines globally, and has committed to working with Novavax to finalize an advance purchase agreement for vaccine supply and global distribution allocation via the COVAX Facility and its partners.
The doses will be manufactured and distributed globally by Novavax and Serum Institute of India (SII), the latter under an existing agreement between Gavi and SII.
Novavax and SK Bioscience said February 15 that they expanded their collaboration and license agreement, with SK finalizing an agreement to supply 40 million doses of NVX-CoV2373 to the government of South Korea beginning in 2021, for an undisclosed price. SK also obtained a license to manufacture and commercialize NVX-CoV2373 for sale to South Korea, as a result of which SK said it will add significant production capacity.
The agreement also calls on Novavax to facilitate technology transfer related to the manufacturing of its protein antigen, its Matrix M adjuvant, and support to SK Bioscience as needed to secure regulatory approval.
Rolling review begins—On February 4, Novavax announced it had begun a rolling review process for authorization of NVX-CoV2373 with several regulatory agencies worldwide, including the FDA, the European Medicines Agency, the U.K. Medicines and Healthcare products Regulatory Agency (MHRA), and Health Canada. The reviews will continue while the company completes its pivotal Phase III trials in the U.S. and U.K., and through initial authorization for emergency use granted under country-specific regulations, and through initial authorization for emergency use.
A day earlier, Novavax executed a binding Heads of Terms agreement with the government of Switzerland to supply 6 million doses of NVX-CoV2373, to the country. Novavax and Switzerland plan to negotiate a final agreement, with initial delivery of vaccine doses slated to ship following successful clinical development and regulatory review.
On January 28, Novavax electrified investors by announcing that its COVID-19 vaccine NVX-CoV2373 showed efficacy of 89.3% in the company’s first analysis of data from a Phase III trial in the U.K., where a variant strain (B.1.1.7) accounted for about half of all positive cases.
However, NVX-CoV2373 achieved only 60% efficacy in a Phase IIb trial in South Africa, where that country’s escape variant of the virus (B.1.351, also known as 20H/501Y.V2) was seen in 90% of cases, Novavax said.
Novavax said January 7 it executed an Advance Purchase Agreement with the Commonwealth of Australia for 51 million doses of NVX-CoV2373 for an undisclosed price, with an option to purchase an additional 10 million doses—finalizing an agreement in principle announced in November 2020. Novavax said it will work with Australia’s Therapeutics Goods Administration (TGA), to obtain approvals upon showing efficacy in clinical studies. The company aims to deliver initial doses by mid-2021.
2020 Status: Phase III trial launched—Novavax said December 28 that it launched the pivotal Phase III PREVENT-19 trial (NCT04611802) in the U.S. and Mexico to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373. The randomized, placebo-controlled, observer-blinded study will assess the efficacy, safety and immunogenicity of NVX-CoV2373 in up to 30,000 participants 18 years of age and older compared with placebo. The trial’s primary endpoint is the prevention of PCR-confirmed, symptomatic COVID-19. The key secondary endpoint is the prevention of PCR-confirmed, symptomatic moderate or severe COVID-19. Both endpoints will be assessed at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2.
Two thirds of the participants will be assigned to randomly receive two intramuscular injections of the vaccine, administered 21 days apart, while one third of the trial participants will receive placebo. Trial sites were selected in locations where transmission rates are currently high, to accelerate the accumulation of positive cases that could show efficacy. Participants will be followed for 24 months following the second injection
PREVENT-19 is being conducted with support from federal agencies involved in Operation Warp Speed, the Trump administration’s effort to promote development and distribution of COVID-19 vaccines and drugs. Those agencies include the Department of Defense (DoD), the NIH’s National Institute of Allergy and Infectious Diseases (NIAID), and the Biomedical Advanced Research and Development Authority (BARDA)—which has committed up to $1.6 billion to Novavax under a DoD agreement (identifier MCDC OTA agreement number W15QKN-16-9-1002).
Novavax is also conducting a pivotal Phase III study in the United Kingdom, a Phase IIb safety and efficacy study in South Africa, and an ongoing Phase I/II trial in the U.S. and Australia. Data from these trials are expected as soon as early first quarter 2021, though timing will depend on transmission rates in the regions, the company said.
Novavax said November 9 that the FDA granted its Fast Track designation for NVX-CoV2373. By the end of November, the company expected to finish enrollment in its Phase III U.K. trial, with interim data in that study expected as soon as early first quarter 2021.
Five days earlier, Novavax signed a non-binding Heads of Terms document with the Australian government to supply 40 million doses of NVX-CoV2373 to Australia starting as early as the first half of 2021, subject to the successful completion of Phase III clinical development and approval of the vaccine by Australia’s Therapeutic Goods Administration (TGA). The vaccine regimen is expected to require two doses per individual, administered 21 days apart.
Australia joins the U.S., the U.K., and Canada in signing direct supply agreements with Novavax. The company is supplying doses in Japan, South Korea, and India through partnerships. Australian clinical researchers led the global Phase I clinical trial in August, which involved 131 Australians across two trial sites (Melbourne and Brisbane). Also, approximately 690 Australians have participated in the Phase II arm of the clinical trial, which has been conducted across up to 40 sites in Australia and the U.S.
Novavax joined officials in its headquarters city of Gaithersburg, MD, on November 2 to announce expansion plans. The company plans to take 122,000 square feet of space at 700 Quince Orchard Road, and has committed to adding at least 400 local jobs, nearly doubling its current workforce of 450 worldwide. Most of the new jobs are expected to be added b March 2021.
Maryland’s Department of Commerce—which has prioritized assistance to life sciences companies—approved a $2 million conditional loan tied to job creation and capital investment. The state has also approved a $200,000 Partnership for Workforce Quality training grant, and the company is eligible for several tax credits, including the Job Creation Tax Credit and More Jobs for Marylanders.
Additionally, Montgomery County has approved a $500,000 grant tied to job creation and capital investment, while the City of Gaithersburg said it will approve a grant of up to $50,000 from its Economic Development Opportunity Fund. The city accelerated its planning approval process to accommodate Novavax’ timeline, given the company’s role in fighting COVID-19 and resulting assistance from Operation Warp Speed, the Trump administration’s effort to accelerate development of COVID-19 vaccines.
On October 27, Novavax said that it had enrolled 5,500 volunteers in the Phase III U.K. trial, which has been expanded from 10,000 to 15,000 volunteers. The increased enrollment “is likely to facilitate assessment of safety and efficacy in a shorter time period,” according to the company.
The trial, which is being conducted with the U.K. Government’s Vaccines Taskforce, was launched in September and is expected to be fully enrolled by the end of November, with interim data expected by early first quarter 2021, depending on the overall COVID-19 attack rate. Novavax has posted the protocol for the Phase III U.K. trial online. The protocol calls for unblinding of data once 152 participants have achieved mild, moderate or severe endpoints. Two interim analyses are planned upon occurrence of 66 and 110 endpoints.
Novavax also said it expects to launch a second Phase III trial designed to enroll up to 30,000 participants in the U.S. and Mexico by the end of November—a study funded through the U.S. government’s Operation Warp Speed program. The patient population will reflect proportional representation of diverse populations most vulnerable to COVID-19, across race/ethnicity, age, and co-morbidities.
The company cited progress toward large-scale manufacturing while acknowledging delays from original timeframe estimates. Novavax said it will use its contract manufacturing site at FUJIFILM Diosynth Biotechnologies’ Morrisville, NC facility to produce material for the U.S. trial.
On September 25, Novavax entered into a non-exclusive agreement with Endo International subsidiary Par Sterile Products to provide fill-finish manufacturing services at its plant in Rochester, MI, for NVX-CoV2373. Under the agreement, whose value was not disclosed, the Rochester facility has begun production of NVX-CoV2373 final drug product, with initial batches to be used in Novavax’ Phase III clinical trial in the U.S. Par Sterile will also fill-finish NVX-CoV2373 vaccine intended for commercial distribution in the U.S.
A day earlier, Novavax launched the U.K. trial. The randomized, placebo-controlled, observer-blinded study to evaluate the efficacy, safety and immunogenicity of NVX-CoV2373 with Matrix-M in up to 10,000 subjects 18-84 years of age, with and without “relevant” comorbidities, over the following four to six weeks, Novavax said. Half the participants will receive two intramuscular injections of vaccine comprising 5 µg of protein antigen with 50 µg Matrix‑M adjuvant, 21 days apart, while half of the trial participants will receive placebo. At least 25% of the study population will be over age 65.
The trial’s first primary endpoint is first occurrence of PCR-confirmed symptomatic COVID-19 with onset at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2. The second primary endpoint is first occurrence of PCR-confirmed symptomatic moderate or severe COVID-19 with onset at least seven days after the second study vaccination in volunteers who have not been previously infected with SARS-CoV-2
“The data from this trial is expected to support regulatory submissions for licensure in the UK, EU and other countries,” stated Gregory M. Glenn, M.D., President, Research and Development at Novavax.
Maryland Gov. Larry Hogan joined state Secretary of Commerce Kelly M. Schulz and local officials in marking the launch of Phase III studies with a tour of the company’s facilities in Gaithersburg: “The coronavirus vaccine candidate that’s been developed by Novavax is one of the most promising in the country, if not the world.”
On August 31, Novavax reached an agreement in principle with the government of Canada to supply up to 76 million doses of NVX-CoV2373. The value was not disclosed. Novavax and Canada did say that they expect to finalize an advance purchase agreement under which Novavax will agree to supply doses of NVX-CoV2373 to Canada beginning as early as the second quarter of 2021.
The purchase arrangement will be subject to licensure of the NVX-CoV2373 by Health Canada, Novavax said. The vaccine is in multiple Phase II clinical trials: On August 24, Novavax said the first volunteers had been enrolled in the Phase II portion of its ongoing Phase I/II clinical trial (NCT04368988), designed to evaluate the immunogenicity and safety of two doses of of NVX-CoV2373 (5 and 25 µg) with and without 50 µg of Matrix‑M™ adjuvant in up to 1,500 volunteers ages 18-84.
The randomized, placebo-controlled, observer-blinded study is designed to assess two dose sizes (5 and 25 µg) of NVX-CoV2373, each with 50 µg of Matrix‑M. Unlike the Phase I portion, the Phase II portion will include older adults 60-84 years of age as approximately half of the trial’s population. Secondary objectives include preliminary evaluation of efficacy. The trial will be conducted at up to 40 sites in the U.S. and Australia, Novovax said.
NVX-CoV2373 is in a pair of Phase II trials launched in August—including a Phase IIb study in South Africa to assess efficacy, and a Phase II safety and immunogenicity study in the U.S. and Australia.
On August 14, the U.K. government agreed to purchase 60 million doses of NVX-CoV2373 from the company, and support its planned Phase III clinical trial in the U.K., through an agreement whose value was not disclosed. The doses are set to be manufactured as early as the first quarter of 2021.
The trial will be designed to evaluate the ability of NVX-CoV2373 to protect against symptomatic COVID-19 disease as well as evaluate antibody and T-cell responses. The randomized, double-blind, placebo-controlled efficacy study will enroll approximately 9,000 adults 18-85 years of age in the U.K., and is expected to start in the third quarter.
Novavax also said it will expand its collaboration with FUJIFILM Diosynth Biotechnologies (FDB), which will manufacture the antigen component of NVX-CoV2373 from its Billingham, Stockton-on-Tees site in the U.K., as well as at U.S. sites in Morrisville, NC, and College Station, TX. FDB’s U.K. sitevis expected to produce up to 180 million doses annually.
On August 13, Novavax said it signed a development and supply agreement for the antigen component of NVX-CoV2373 with Seoul-based SK bioscience, a vaccine business subsidiary of SK Group. The agreement calls for supply to global markets that include the COVAX Facility, co-led by Gavi, the Coalition for Epidemic Preparedness Innovations (CEPI) and the World Health Organization.
Novavax and SK signed a letter of intent with South Korea’s Ministry of Health and Welfare to work toward broad and equitable access to NVX-CoV2373 worldwide, as well as to make the vaccine available in South Korea. SK bioscience agreed to manufacture the vaccine antigen component for use in the final drug product globally during the pandemic, at its vaccine facility in Andong L-house, South Korea, beginning in August. The value of the agreement was not disclosed.
On August 7, Novavax licensed its COVID-19 vaccine technology to Takeda Pharmaceutical through a partnership by which Takeda will develop, manufacture, and commercialize NVX‑CoV2373 in Japan, using Matrix-M adjuvant to be supplied by Novavax. Takeda will also be responsible for regulatory submission to Japan’s Ministry of Health, Labour and Welfare (MHLW).
MHLW agreed to provide funding to Takeda—the amount was not disclosed in the companies’ announcement—for technology transfer, establishment of infrastructure, and scale-up of manufacturing. Takeda said it anticipated the capacity to manufacture over 250 million doses of NVX‑CoV2373 per year.
Five days earlier, Serum Institute of India agreed to license rights from Novavax to NVX‑CoV2373 for development and commercialization in India as well as low- and middle-income countries (LMIC), through an agreement whose value was not disclosed. Novavax retains rights to NVX-CoV2373 elsewhere in the world.
Novavax and Serum Institute of India agreed to partner on clinical development, co-formulation, filling and finishing and commercialization of NVX-CoV2373. Serum Institute will oversee regulatory submissions and marketing authorizations in regions covered by the collaboration. Novavax agreed to provide both vaccine antigen and Matrix‑M adjuvant, while the partners said they were in talks to have the Serum Institute manufacture vaccine antigen in India. Novavax and Seerum Institute plan to split the revenue from the sale of product, net of agreed costs.
A day earlier, Novavax announced positive results from the Phase I portion of its Phase I/II clinical trial (NCT04368988), designed to evaluate two doses of NVX-CoV2373 (5 and 25 µg) with and without Matrix‑M™ adjuvant in 131 healthy adults ages 18-59. NVX-CoV2373, adjuvanted with Matrix-M, elicited robust antibody responses numerically superior to human convalescent sera, according to data submitted for peer-review to a scientific journal.
All participants developed anti-spike IgG antibodies after a single dose of vaccine, Novavax said, many also developing wild-type virus neutralizing antibody responses. After the second dose, all participants developed wild-type virus neutralizing antibody responses. Both anti-spike IgG and viral neutralization responses compared favorably to responses from patients with clinically significant COVID‑19 disease, the company said—adding that IgG antibody response was highly correlated with neutralization titers, showing that a significant proportion of antibodies were functional.
For both dosages of NVX‑CoV2373 with adjuvant, the 5 µg dose performed “comparably” with the 25 µg dose, Novavax said. NVX‑CoV2373 also induced antigen-specific polyfunctional CD4+ T cell responses with a strong bias toward the Th1 phenotype (IFN-g, IL-2, and TNF-a).
Based on an interim analysis of Phase I safety and immunogenicity data, the trial was expanded to Phase II clinical trials in multiple countries, including the U.S. The trial—which began in Australia in May—is being funded by up-to $388 million in funding from the Coalition for Epidemic Preparedness Innovations (CEPI). If the Phase I/II trial is successful, CEPI said, it anticipates supporting further clinical development that would advance NVX-CoV2373 through to licensure.
On July 23, Novavax joined FDB to announce that FDB will manufacture bulk drug substance for NVX-CoV2373, under an agreement whose value was not disclosed. FDB’s site in Morrisville, NC has begun production of the first batch of NVX-CoV2373. Batches produced at FDB’s Morrisville site will be used in Novavax’s planned pivotal Phase III clinical trial, designed to assess NVX-CoV2373 in up to 30,000 participants, and set to start this fall.
The Phase III trial is among R&D efforts to be funded through the $1.6 billion awarded in July to Novavax through President Donald Trump’s “Operation Warp Speed” program toward late-stage clinical trials and large-scale manufacturing to produce 100 million doses of its COVID-19 vaccine by year’s end. Novavax said the funding will enable it to complete late-stage clinical studies aimed at evaluating the safety and efficacy of NVX-CoV2373.
In June, Novavax said biotech investor and executive David Mott was joining its board as an independent director, after recently acquiring nearly 65,000 shares of the company’s common stock. Also, Novavax was awarded a $60 million contract by the U.S. Department of Defense (DoD) for the manufacturing of NVX‑CoV2373. Through the Defense Health Program, the Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense Enabling Biotechnologies (JPEO-CBRND-EB) agreed to support production of several vaccine components to be manufactured in the U.S. Novavax plans to deliver this year for DoD 10 million doses of NVX‑CoV2373 that could be used in Phase II/III trials, or under an Emergency Use Authorization (EUA) if approved by the FDA.
Also in June, AGC Biologics said it will partner with Novavax on large-scale GMP production of Matrix-M– significantly increasing Novavax’ capacity to deliver doses in 2020 and 2021—through an agreement whose value was not disclosed. And Novavax joined The PolyPeptide Group to announce large-scale GMP production by the global CDMO of two unspecified key intermediate components used in the production of Matrix-M.
In May, Novavax acquired Praha Vaccines from the India-based Cyrus Poonawalla Group for $167 million cash, in a deal designed to ramp up Novavax’s manufacturing capacity for NVX-CoV2373. Praha Vaccines’ assets include a 150,000-square foot vaccine and biologics manufacturing facility and other support buildings in Bohumil, Czech Republic. Novavax said the Bohumil facility is expected to deliver an annual capacity of over 1 billion doses of antigen starting in 2021 for the COVID-19 vaccine.
The Bohumil facility is completing renovations that include the addition of Biosafety Level-3 (BSL-3) capabilities. The site’s approximately 150 employees with “significant experience” in vaccine manufacturing and support have joined Novavax, the company said.
On May 11, Novavax joined CEPI in announcing up to $384 million in additional funding for the company toward clinical development and large-scale manufacturing of NVX-CoV2373. CEPI agreed to fund preclinical as well as Phase I and Phase II studies of NVX-CoV2373. The funding multiplied CEPI’s initial $4 million investment in the vaccine candidate, made two months earlier. Novavax’s total $388 million in CEPI funding accounted for 87% of the total $446 million awarded by the Coalition toward COVID-19 vaccine R&D as of that date.
Novavax identified its COVID-19 vaccine candidate in April. The company said NVX-CoV2373 was shown to be highly immunogenic in animal models measuring spike protein-specific antibodies, antibodies that block the binding of the spike protein to the receptor, and wild-type virus neutralizing antibodies. High levels of spike protein-specific antibodies with ACE-2 human receptor binding domain blocking activity and SARS-CoV-2 wild-type virus neutralizing antibodies were also seen after a single immunization.
In March, Emergent Biosolutions disclosed it retained an option to allocate manufacturing capacity for an expanded COVID-19 program under an agreement with Novavax to provide “molecule-to-market” contract development and manufacturing (CDMO) services to produce Novavax’s NanoFlu™, its recombinant quadrivalent seasonal influenza vaccine candidate.
Earlier in March, Emergent announced similar services to support clinical development of Novavax’s COVID-19 vaccine candidate, saying March 10 it agreed to produce the vaccine candidate and had initiated work, anticipating the vaccine candidate will be used in a Phase I study within the next four months. In February, Novavax said it had produced and was assessing multiple nanoparticle vaccine candidates in animal models prior to identifying an optimal candidate for human testing.
References
- ^ “Company Overview of Novavax, Inc”. Bloomberg.com. Archived from the original on 24 February 2017. Retrieved 2 June2019.
- ^ https://www.globenewswire.com/news-release/2021/03/01/2184674/0/en/Novavax-Reports-Fourth-Quarter-and-Full-Year-2020-Financial-Results-and-Operational-Highlights.html
- ^ Jump up to:a b c d e Bell, Jacob (November 14, 2016). “Novavax aims to rebound with restructuring, more trials”. BioPharma Dive. Washington, D.C.: Industry Dive. Archived from the original on 2017-03-29. Retrieved 2017-03-28.
- ^ Thomas, Katie; Twohey, Megan (2020-07-16). “How a Struggling Company Won $1.6 Billion to Make a Coronavirus Vaccine”. The New York Times. ISSN 0362-4331. Retrieved 2021-01-29.
- ^ Taylor, Nick Paul (3 June 2013). “Novavax makes $30M bid for adjuvant business”. FiercePharma. Archived from the original on 14 September 2016. Retrieved 9 September 2016.
- ^ “Gaithersburg Biotech Receives Grant Worth up to $89 million”. Bizjournals.com. Archived from the original on 2017-04-01. Retrieved 2017-03-28.
- ^ “With promising RSV data in hand, Novavax wins $89M Gates grant for PhIII | FierceBiotech”. Fiercebiotech.com. Archivedfrom the original on 2017-04-14. Retrieved 2017-03-28.
- ^ “Novavax RSV vaccine found safe for pregnant women, fetus”. Reuters. 2016-09-29. Archived from the original on 2016-10-07. Retrieved 2017-03-28.
- ^ Herper, Matthew. “Gates Foundation Backs New Shot To Prevent Babies From Dying Of Pneumonia”. Forbes. Archived from the original on 2016-09-21. Retrieved 2017-03-28.
- ^ “Novavax’s Ebola vaccine shows promise in early-stage trial”. Reuters. 2017-07-21. Archived from the original on 2016-10-02. Retrieved 2017-03-28.
- ^ Jump up to:a b c d e f Adams, Ben (September 16, 2016). “Novavax craters after Phase III RSV F vaccine failure; seeks path forward”. FierceBiotech. Questex. Archived from the original on 18 August 2020. Retrieved 25 Jan 2020.
- ^ Shtrubel, Marty (December 12, 2019). “3 Biotech Stocks That Offer the Highest Upside on Wall Street”. Biotech. Nasdaq. Archived from the original on 2020-01-26. Retrieved 25 Jan 2020.
- ^ Jump up to:a b Budwell, George (January 20, 2020). “3 Top Biotech Picks for 2020”. Markets. Nasdaq. Novavax: A catalyst awaits. Archivedfrom the original on 2020-01-25. Retrieved 25 Jan 2020.
- ^ Mark Terry (February 16, 2018). “Why Novavax Could be a Millionaire-Maker Stock”. BioSpace. Archived from the original on 22 November 2020. Retrieved 6 March 2020.
- ^ Jump up to:a b Eric Sagonowsky (2020-05-11). “Novavax scores $384M deal, CEPI’s largest ever, to fund coronavirus vaccine work”. FiercePharma. Archived from the original on 2020-05-16. Retrieved 2020-05-12.
- ^ “Novavax addresses urgent global public health needs with innovative technology”. novavax.com. Archived from the original on 10 September 2020. Retrieved 30 August 2020.
- ^ Sara Gilgore (January 15, 2020). “Novavax earns key FDA status for its flu vaccine. Wall Street took it well”. Washington Business Journal. Archived from the original on 10 November 2020. Retrieved 6 March 2020.
- ^ Sara Gilgore (February 26, 2020). “Novavax is working to advance a potential coronavirus vaccine. So are competitors”. Washington Business Journal. Archived from the original on March 16, 2020. Retrieved March 6, 2020.
- ^ Nidhi Parekh (July 24, 2020). “Novavax: A SARS-CoV-2 Protein Factory to Beat COVID-19”. Archived from the original on November 22, 2020. Retrieved July 24, 2020.
- ^ “Covid-19: Novavax vaccine shows 89% efficacy in UK trials”. BBC news. Retrieved 1 February 2021.
Further reading
- “Novavax, Inc. Common Stock (NVAX) News Headlines”. Market Activity. Nasdaq. Retrieved 25 Jan 2020. Continuously updated listing of Nasdaq publications related to Novavax, newest items first.
External links
- Official website
- Business data for Novavax, Inc.:
General References
| Type | Public |
|---|---|
| Traded as | Nasdaq: NVAX Russell 2000 Component |
| Industry | Biotechnology |
| Founded | 1987; 34 years ago [1] |
| Headquarters | Gaithersburg, Maryland,United States |
| Area served | Worldwide |
| Key people | Stanley Erck (CEO) |
| Products | Vaccines |
| Revenue | |
| Number of employees | 500+[3] |
| Website | www.novavax.com |
The Novavax COVID-19 vaccine, codenamed NVX-CoV2373, and also called SARS-CoV-2 rS (recombinant spike) protein nanoparticle with Matrix-M1 adjuvant, is a COVID-19 vaccine candidate developed by Novavax and Coalition for Epidemic Preparedness Innovations (CEPI). It requires two doses[1] and is stable at 2 to 8 °C (36 to 46 °F) (refrigerated).[2]
Description
NVX-CoV2373 has been described as both a protein subunit vaccine[3][4][5] and a virus-like particle vaccine,[6][7] though the producers call it a “recombinant nanoparticle vaccine”.[8]
The vaccine is produced by creating an engineered baculovirus containing a gene for a modified SARS-CoV-2 spike protein. The baculovirus then infects a culture of Sf9 moth cells, which create the spike protein and display it on their cell membranes. The spike proteins are then harvested and assembled onto a synthetic lipid nanoparticle about 50 nanometers across, each displaying up to 14 spike proteins.[3][4][8]
The formulation includes a saponin-based adjuvant.[3][4][8]
Development
In January 2020, Novavax announced development of a vaccine candidate, codenamed NVX-CoV2373, to establish immunity to SARS-CoV-2.[9] Novavax’s work is in competition for vaccine development among dozens of other companies.[10]
In March 2020, Novavax announced a collaboration with Emergent BioSolutions for preclinical and early-stage human research on the vaccine candidate.[11] Under the partnership, Emergent BioSolutions will manufacture the vaccine at large scale at their Baltimore facility.[12] Trials have also taken place in the United Kingdom, and subject to regulatory approval, at least 60 million doses will be manufactured by Fujifilm Diosynth Biotechnologies in Billingham for purchase by the UK government.[13][14] They also signed an agreement with Serum Institute of India for mass scale production for developing and low-income countries.[15] It has also been reported, that the vaccine will be manufactured in Spain.[16] The first human safety studies of the candidate, codenamed NVX-CoV2373, started in May 2020 in Australia.[17][18]
In July, the company announced it might receive $1.6 billion from Operation Warp Speed to expedite development of its coronavirus vaccine candidate by 2021—if clinical trials show the vaccine to be effective.[19][20] A spokesperson for Novavax stated that the $1.6 billion was coming from a “collaboration” between the Department of Health and Human Services and Department of Defense,[19][20] where Gen. Gustave F. Perna has been selected as COO for Warp Speed. In late September, Novavax entered the final stages of testing its coronavirus vaccine in the UK. Another large trial was announced to start by October in the US.[21]
In December 2020, Novavax started the PREVENT-19 (NCT04611802) Phase III trial in the US and Mexico.[22][full citation needed][23]
On 28 January 2021, Novavax reported that preliminary results from the United Kingdom trial showed that its vaccine candidate was more than 89% effective.[24][2] However, interim results from a trial in South Africa showed a lower effectiveness rate against the 501.V2 variant of the virus, at around 50-60%.[1][25]
On 12 March 2021, they announced their vaccine candidate was 96.4% effective in preventing the original strain of COVID-19 and 86% effective against the U.K variant. It proved 55% effective against the South African variant in people without HIV/AIDS. It was also 100% effective at preventing severe illness.[citation needed]
Deployment
On 2 February 2021, the Canadian Prime Minister Justin Trudeau announced that Canada has signed a tentative agreement for Novavax to produce millions of doses of its COVID-19 vaccine in Montreal, Canada, once it’s approved for use by Health Canada, making it the first COVID-19 vaccine to be produced domestically.[26]
References
- ^ Jump up to:a b Wadman M, Jon C (28 January 2021). “Novavax vaccine delivers 89% efficacy against COVID-19 in UK—but is less potent in South Africa”. Science. doi:10.1126/science.abg8101.
- ^ Jump up to:a b “New Covid vaccine shows 89% efficacy in UK trials”. BBC News. 28 January 2021. Retrieved 28 January 2021.
- ^ Jump up to:a b c Wadman M (November 2020). “The long shot”. Science. 370 (6517): 649–653. Bibcode:2020Sci…370..649W. doi:10.1126/science.370.6517.649. PMID 33154120.
- ^ Jump up to:a b c Wadman M (28 December 2020). “Novavax launches pivotal U.S. trial of dark horse COVID-19 vaccine after manufacturing delays”. Science. doi:10.1126/science.abg3441.
- ^ Parekh N (24 July 2020). “Novavax: A SARS-CoV-2 Protein Factory to Beat COVID-19”. Archived from the original on 22 November 2020. Retrieved 24 July 2020.
- ^ Chung YH, Beiss V, Fiering SN, Steinmetz NF (October 2020). “COVID-19 Vaccine Frontrunners and Their Nanotechnology Design”. ACS Nano. 14 (10): 12522–12537. doi:10.1021/acsnano.0c07197. PMC 7553041. PMID 33034449.
- ^ Medhi R, Srinoi P, Ngo N, Tran HV, Lee TR (25 September 2020). “Nanoparticle-Based Strategies to Combat COVID-19”. ACS Applied Nano Materials. 3 (9): 8557–8580. doi:10.1021/acsanm.0c01978. PMC 7482545.
- ^ Jump up to:a b c “Urgent global health needs addressed by Novavax”. Novavax. Retrieved 30 January 2021.
- ^ Gilgore S (26 February 2020). “Novavax is working to advance a potential coronavirus vaccine. So are competitors”. Washington Business Journal. Archived from the original on 16 March 2020. Retrieved 6 March 2020.
- ^ “COVID-19 vaccine tracker (click on ‘Vaccines’ tab)”. Milken Institute. 11 May 2020. Archived from the original on 6 June 2020. Retrieved 12 May 2020. Lay summary.
- ^ Gilgore S (10 March 2020). “Novavax’s coronavirus vaccine program is getting some help from Emergent BioSolutions”. Washington Business Journal. Archived from the original on 9 April 2020. Retrieved 10 March 2020.
- ^ McCartney R. “Maryland plays an outsized role in worldwide hunt for a coronavirus vaccine”. Washington Post. Archived from the original on 7 May 2020. Retrieved 8 May 2020.
- ^ Boseley S, Davis N (28 January 2021). “Novavax Covid vaccine shown to be nearly 90% effective in UK trial”. The Guardian. Retrieved 29 January 2021.
- ^ Brown M (14 August 2020). “60m doses of new covid-19 vaccine could be made in Billingham – and be ready for mid-2021”. TeesideLive. Reach. Retrieved 29 January 2021.
- ^ “Novavax signs COVID-19 vaccine supply deal with India’s Serum Institute”. Reuters. 5 August 2020.
- ^ “Spain, again chosen to produce the vaccine to combat COVID-19”. This is the Real Spain. 18 September 2020.
- ^ Sagonowsky E (11 May 2020). “Novavax scores $384M deal, CEPI’s largest ever, to fund coronavirus vaccine work”. FiercePharma. Archived from the original on 16 May 2020. Retrieved 12 May 2020.
- ^ “Novavax starts clinical trial of its coronavirus vaccine candidate”. CNBC. 25 May 2020. Archived from the original on 26 May 2020. Retrieved 26 May 2020.
- ^ Jump up to:a b Thomas K (7 July 2020). “U.S. Will Pay $1.6 Billion to Novavax for Coronavirus Vaccine”. The New York Times. Archived from the original on 7 July 2020. Retrieved 7 July 2020.
- ^ Jump up to:a b Steenhuysen J (7 July 2020). “U.S. government awards Novavax $1.6 billion for coronavirus vaccine”. Reuters. Archived from the original on 14 September 2020. Retrieved 15 September 2020.
- ^ Thomas K, Zimmer C (24 September 2020). “Novavax Enters Final Stage of Coronavirus Vaccine Trials”. The New York Times. ISSN 0362-4331. Archived from the original on 28 September 2020. Retrieved 28 September 2020.
- ^ Clinical trial number NCT04611802 for “A Study Looking at the Efficacy, Immune Response, and Safety of a COVID-19 Vaccine in Adults at Risk for SARS-CoV-2” at ClinicalTrials.gov
- ^ “Phase 3 trial of Novavax investigational COVID-19 vaccine opens”. National Institutes of Health (NIH). 28 December 2020. Retrieved 28 December 2020.
- ^ Lovelace B (28 January 2020). “Novavax says Covid vaccine is more than 89% effective”. CNBC.
- ^ Facher L, Joseph A (28 January 2021). “Novavax says its Covid-19 vaccine is 90% effective in late-stage trial”. Stat. Retrieved 29 January 2021.
- ^ “Canada signs deal to produce Novavax COVID-19 vaccine at Montreal plant”. CP24. 2 February 2021. Retrieved 2 February2021.
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Subunit |
| Clinical data | |
| Other names | NVX-CoV2373 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15810 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
////////////// Novavax, COVID-19, vaccine, CORONA VIRUS, NVX-CoV2373, SARS-CoV-2 rS, TAK 019
#Novavax, #COVID-19, #vaccine, #CORONA VIRUS, #NVX-CoV2373, #SARS-CoV-2 rS, #TAK 019
UPDATE
SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373
SARS-CoV-2 rS;
Novavax Covid-19 vaccine (TN);
Nuvaxovid (TN)
SARS-CoV-2 rS;
組換えコロナウイルス (SARS-CoV-2) ワクチン;
コロナウイルス(SARS-CoV-2)スパイク糖タンパク質抗原nvx-cov2373ワクチン;
SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373;
SARS-CoV-2 rS
APPROVED JAPAN Nuvaxovid, 2022/4/19
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////SARS-CoV-2 Spike glycoprotein vaccine antigen nvx-cov2373, JAPAN 2022, APPROVALS 2022, VACCINE, COVID 19. CORONA VACCINE, SARS-CoV-2
BBIBP-CorV, Sinopharm COVID-19 vaccine


BBIBP-CorV, Sinopharm COVID-19 vaccine
- Inactivated novel coronavirus (2019-CoV) vaccine (Vero cells)
- Purified inactivated SARS-CoV-2 Vaccine
ref Lancet Infectious Diseases (2021), 21(1), 39-51.
BBIBP-CorV, also known as the Sinopharm COVID-19 vaccine,[1] is one of two inactivated virus COVID-19 vaccines developed by Sinopharm. In late December 2020, it was in Phase III trials in Argentina, Bahrain, Egypt, Morocco, Pakistan, Peru, and the United Arab Emirates (UAE) with over 60,000 participants.[2]
On December 9, the UAE announced interim results from Phase III trials showing BBIBP-CorV had a 86% efficacy against COVID-19 infection.[3] In late December, Sinopharm announced that its internal analysis indicated a 79% efficacy.[4] While mRNA vaccines like the Pfizer–BioNTech COVID-19 vaccine and mRNA-1273 showed higher efficacy of +90%, those present distribution challenges for some nations as they require deep-freeze facilities and trucks. BIBP-CorV could be transported and stored at normal refrigerated temperatures.[5]
BBIBP-CorV shares similar technology with CoronaVac and BBV152, other inactivated virus vaccines for COVID-19 being developed in Phase III trials.[6][7]
BBIBP-CorV is being used in vaccination campaigns by certain countries in Asia,[8][9][10] Africa,[11][12][13] South America,[14][15] and Europe.[16][17][18] Sinopharm expects to produce one billion doses of BBIBP-CorV in 2021.[19] By February 21, Sinopharm said more than 43 million doses of the vaccine had been administered in total.[20]
BBIBP-CorV vaccine contains a SARS-CoV-2 strain inactivated inside Vero Cells. Investigation shows this vaccine induces neutralizing antibodies in several mammalian species while also showing protective efficacy with SARS-CoV-2 challenge in rhesus macaques2. As of August 2020, this vaccine is being tested for prophylaxis against COVID-19 in human clinical trials.

A vaccination certificate of BBIBP-CorV (Beijing Institute of Biological Products, Sinopharm).
Clinical research
Main article: COVID-19 vaccine
Phases I and II
In April 2020, China approved clinical trials for a candidate COVID-19 vaccine developed by Sinopharm‘s Beijing Institute of Biological Products[21] and the Wuhan Institute of Biological Products.[22] Both vaccines are chemically-inactivated whole virus vaccines for COVID-19.
On October 15, the Beijing Institute of Biological Products published results of its Phase I (192 adults) and Phase II (448 adults) clinical studies for the BBIBP-CorV vaccine, showing BBIBP-CorV to be safe and well-tolerated at all tested doses in two age groups. Antibodies were elicited against SARS-CoV-2 in all vaccine recipients on day 42. These trials included individuals older than 60.[21]
On August 13, the Wuhan Institute of Biological Products published interim results of its Phase I (96 adults) and Phase II (224 adults) clinical studies. The report noted the inactivated COVID-19 vaccine had a low rate of adverse reactions and demonstrated immunogenicity, but longer-term assessment of safety and efficacy would require Phase III trials.[22]
BIBP-CorV may have characteristics favorable for vaccinating people in the developing world. While mRNA vaccines, such as the Pfizer–BioNTech COVID-19 vaccine and Moderna COVID-19 vaccine showed higher efficacy of +90%, mRNA vaccines present distribution challenges for some nations, as some may require deep-freeze facilities and trucks. By contrast, BIBP-CorV can be transported and stored at normal refrigeration temperatures.[23] While Pfizer and Moderna are among developers relying on novel mRNA technology, manufacturers have decades of experience with the inactivated virus technology Sinopharm is using.[23]
Phase III
Africa and Asia
On July 16, Sinopharm began conducting a Phase III vaccine trial of 31,000 volunteers in the UAE in collaboration with G42 Healthcare, an Abu Dhabi-based company.[24] By August, all volunteers had received their first dose and were to receive the second dose within the next few weeks.[25] On December 9, UAE’s Ministry of Health and Prevention announced the official registration of BBICP-CorV, after an interim analysis of the Phase III trial showed BBIBP-CorV to have a 86% efficacy against COVID-19 infection.[26] The vaccine had a 99% sero-conversion rate of neutralizing antibodies and 100% effectiveness in preventing moderate and severe cases of the disease.[27]
On September 2, Sinopharm began a Phase III trial in Casablanca and Rabat on 600 people.[28][29] In September, Egypt opened registration for a Phase III trial to last one year and enroll 6,000 people.[30]
In August 2020, Sinopharm began a Phase III clinical trial in Bahrain on 6,000 citizens and resident volunteers.[31][32] In a November update, 7,700 people had volunteered in the trials.[33] Also in late August, Sinopharm began a Phase III clinical trial in Jordan on 500 volunteers at Prince Hamzah Hospital.[34][35]
In Pakistan, Sinopharm began working with the University of Karachi on a trial with 3,000 volunteers.[36]
South America
On September 10, Sinopharm began a Phase III trial in Peru with the long-term goal of vaccinating a total of 6,000 people between the ages of 18 and 75.[37] In October, the trials were expanded to include an additional 6,000 volunteers.[38] On January 26, a volunteer in the placebo group of the vaccine trials had died.[39]
On September 16, Argentina began a Phase III trial with 3,000 volunteers.[40]
Manufacturing
Sinopharm’s Chariman Yang Xioyun has said the company could produce one billion doses in 2021.[19]
In October, Dubai’s G42 Healthcare reached manufacturing agreements to provide UAE and other regional states with BBIBP-CorV, with the UAE producing 75 to 100 million doses in 2021.[41]
In December, Egypt announced an agreement between Sinopharm and Egyptian Holding Company for Biological Products & Vaccines (VACSERA) for the vaccine to be manufactured locally,[42] which would also be exported to other African countries.[43]
In December, AP reported Morocco plans to produce BBIBP-CorV locally.[44]
In March, Serbia announced plans to produce 24 million doses of BBIBP-CorV annually starting in October. The production volume would be sufficient to meet the needs of Serbia and all of its neighbors, deputy prime minister Branislav Nedimović noted.[45]
In March, Belarus was looking to produce BBIBP-CorV locally.[18]
Marketing and Distribution
| show Full authorizationshow Emergency authorizationshow Received donated doses Eligible COVAX recipient (assessment in progress)[86] |
On February 21, 2021 Sinopharm said more than 43 million doses of BBIBP-CorV had been administered so far, including more than 34 million administered in China and the rest internationally.[20]
Asia
In February, Afghanistan was pledged 400,000 doses of BBIBP-CorV by China.[82]
In November 3, 2020 Bahrain granted emergency use authorization of BBIBP-CorV for frontline workers.[33] In December, Bahrain approved Sinopharm’s vaccine, citing data from Phase III clinical trials that showed an 86% efficacy rate.[87]
In February, Brunei received the first batch of Sinopharm vaccines donated by China.[84]
In January, Cambodia said China would provide a million doses.[88] Cambodia granted emergency use authorization on February 4[89] and started the vaccination campaign on February 10 with the first 600,000 doses.[90]
In China, Sinopharm obtained an EUA in July.[91] In October, it began offering the vaccine for free to students going abroad for higher studies.[92] On December 30, China‘s National Medical Products Administration approved BBIBP-CorV for general use.[93][8] In February, Macau received the first 100,000 doses of 400,000 doses.[94]
In October, Indonesia reached an agreement with Sinopharm to deliver 15 million dual-dose vaccines in 2020.[95]
In February, Iran approved emergency use of BBIBP-CorV,[96] and received the first batch of 250,000 doses on February 28.[97]
In January, Iraq approved BBIBP-CorV for emergency use[98] and has signed agreements for 2 million doses. The first doses arrived on March 2.[99]
In January, Jordan approved BBIBP-CorV for emergency use[100] and started its vaccination campaign on January 13.[101]
In March, Kyrgyzstan received a donation of 150,000 doses of the vaccine.[102]
In January, Laos began vaccinating medical workers at hospitals in Vientiane [103] and received another 300,000 doses in early February.[104]
In March, Lebanon received a donation of 50,000 doses at its request,[105] for which it granted emergency use authorization on March 2.[106]
In March, Maldives granted emergency approval for use. At the time of approval, the country had received 18,000 doses and was awaiting 200,000 additional doses.[107]
In February, Mongolia received a donation of 300,000 doses.[108] On March 10, Governor of Ulaanbaatar D. Sumiyabazar and Deputy Prime Minister S. Amarsaikhan received the first doses of BBIBP-CorV.[109]
In February, Nepal approved the vaccine for emergency use, allowing a donation of 500,000 doses to enter the country.[110]
In December, Pakistan‘s purchased 1.2 million doses,[111] which was approved for emergency use on January 18,[112] and began a vaccination campaign on February 2.[10]
In March, Palestine said it would receive 100,000 doses donated by China.[113]
In March 19, Sri Lanka approved the vaccine for emergency use, allowing a donation of 600,000 doses by China to enter the country.[114]
On 14 September 2020, the United Arab Emirates approved the vaccine for front-line workers following successful interim Phase III trials.[24] In December, the country registered BBIBP-CorV after it reviewed the results of the interim analysis.[26] In March, a small number of people who have reduced immunity against diseases, have chronic illnesses, or belong to high-risk groups have been given a 3rd booster shot.[115]
Africa
In February, Algeria received a donation of 200,000 doses.[83]
Egypt plans to buy 40 million doses of Sinpharm’s vaccine[116] which was approved for regulatory use on January 3.[116] President Abdel Fattah el-Sisi announced a vaccination campaign starting 24 January.[11]
In February, Equatorial Guinea received a donation of 100,000 doses which arrived on February 10. The country began vaccinations on February 15.[56]
In March, Gabon received a donation of 100,000 doses which was the second vaccine approved for use in the country.[117]
Morocco placed orders for 41 million vaccine doses from Sinopharm and 25 million from AstraZeneca, for a total of 66 million doses.[118] Morocco granted emergency use approval on January 23,[119] and the first 500,000 doses arrived on January 27.[12]
In February, Mozambique received a donation of 200,000 doses[120] and planned to start vaccinations on March 8.[121]
In March, Namibia received a donation of 100,000 doses and announced the start of vaccinations in the Khomas and Erongo regions.[122]
In March, Niger received a donation of 400,000 doses with vaccinations to begin on March 27.[123]
In February, Senegal received 200,000 doses in Dakar[124] and began vaccinating health workers on February 22.[125]
In February, Sierra Leone received a donation of 200,000 doses.[126] It was approved for emergency use and vaccinations began on March 15.[127]
In January, Seychelles said it would begin administering vaccinations on January 10 with 50,000 doses it had received as a gift from the UAE.[128]
In March, Republic of the Congo received 100,000 doses with vaccinations prioritizing the medically vulnerable and those over 50.[129]
In February, Zimbabwe purchased 600,000 doses on top of 200,000 doses donated by China,[130] and started vaccinations on February 18.[13] Zimbabwe later purchased an additional 1.2 million doses.[131]
North America
In February, the Dominican Republic ordered 768,000 doses of BBIBP-CorV.[132]
In March, Dominica received 20,000 doses of BBIBP-CorV which it began using in its vaccination campaign on March 4.[133]
In March, Mexico announced it would order 12 million doses of BBIBP-CorV pending approval by its health regulator.[134]
South America
In February, Argentina authorized emergency use of BBIBP-CorV[135] ahead of the arrival of 904,000 doses on February 26.[136]
In February, Bolivia purchased 400,000 doses on top of 100,000 doses donated by China,[137] and started its vaccination campaign on February 26.[15]
In March, Guyana received a donation of 20,000 doses of BBIBP-CorV.[138] Vaccinations were to start on March 7.[139]
In January, Peru purchased 38 million doses of BBIBP-CorV.[140] Peru granted emergency approval for BBIBP-CorV on January 27[141] and started vaccinations on February 9 with the first 300,000 doses.[14]
In March, Venezuela granted approval for BBIBP-CorV to be used in the country.[142] The first 500,000 doses arrived on March 2.[143]
Europe
In February, Belarus received a donation of 100,000 doses[144] and began using the vaccine on March 15.[18]
In January, Hungary became first EU member to approve BBIBP-CorV, signing a deal for 5 million doses.[145] The first 550,000 doses arrived in Budapest on February 16[146] and vaccinations started on February 24.[17] Prime Minister Viktor Orbán was vaccinated with BBIBP-CorV on February 28.[147]
In March, Moldova received 2,000 doses donated by the UAE[148] which will be used to vaccinate doctors at the State University of Mediecne and Pharmacy starting on March 22.[149]
In March 3, Montenegro received a donation of 30,000 doses of BBIBP-CorV.[85]
In February, North Macedonia signed an agreement for 200,000 doses of BBIBP-CorV, with which they hoped to launch their vaccination program later that month.[150]
In January, Serbia received one million doses, making it the first country in Europe to receive BBIBP-CorV.[151] On January 19, Serbia approved the vaccine and Health Minister Zlatibor Lončar became the first person to receive a shot.[16]
Controversies
Lack of public data
Unlike Moderna‘s MRNA-1273, Oxford–AstraZeneca‘s AZD1222, and Johnson & Johnson‘s Ad26.COV2.S, there is little public information about the Chinese vaccine’s safety or efficacy.[152] The UAE said it had reviewed Sinopharm’s interim data analysis which showed the vaccine was 100% effective to prevent moderate and severe instances of COVID-19, but did not say whether it had independently analyzed the case data in its review. It was unclear how Sinopharm drew conclusions, since the UAE announcement of the approval for BBIBP-CorV noticeably lacked details such as the number of COVID-19 cases in the placebo or active group or the volunteers ages.[153]
As of December 30, 2020, no detailed efficacy data of the vaccine has been released to the public. A Sinopharm executive said detailed data would be released later and published in scientific journals in China and internationally.[8]
Sinopharm president Wu Yonglin said the trial results exceeded the WHO’s requirements, but a director at a large pharmaceutical company in Shanghai expressed skepticism over the trials and the expectation that drug regulators in Bahrain and the UAE would not hold the same standard as the U.S. Food and Drug Administration.[154]
Unauthorized use in Asia
On December 30, Philippine Defense Secretary Delfin Lorenzana said in an interview that at least one minister and president Rodrigo Duterte‘s bodyguards were provided BBIBP-CorV which were “smuggled” but that he felt what happened was “justified”. Brigadier General Jesus Durante, head of the Presidential Security Guard (PSG), said he felt compelled and “took the risk” to have some of his men vaccinated because they provide close-in security to Duterte, who at 75 is highly vulnerable to COVID-19.[155] Ingming Aberia, an author at The Manila Times commented that FDA director-general Enrique Domingo had reason to believe Sinopharm may cause harm to the consuming public given that no COVID-19 vaccine license was issued, but out of “self-preservation”, he would not initiate charges against PSG.[156]
On January 1, Mainichi Shimbun reported that 18 wealthy people, including several owners of leading Japanese companies, have been vaccinated with Sinopharm vaccines since November 2020. The vaccines were brought in by a Chinese consultant close to a senior member of the Chinese Communist Party.[157] The Chinese embassy in Japan later expressed its dissatisfaction at the unverified claims by Japanese news media.[158]
References
- ^ https://www.nytimes.com/interactive/2020/health/sinopharm-covid-19-vaccine.html
- ^ Reuters Staff (2020-11-19). “China Sinopharm’s coronavirus vaccine taken by about a million people in emergency use”. Reuters. Retrieved 2020-12-09.
- ^ “UAE: Ministry of Health announces 86 per cent vaccine efficacy”. gulfnews.com. Retrieved 2020-12-13.
- ^ Wee, Sui-Lee; Qin, Amy (2020-12-30). “China Approves Covid-19 Vaccine as It Moves to Inoculate Millions”. The New York Times. ISSN 0362-4331. Retrieved 2021-02-12.
- ^ “China State-Backed Covid Vaccine Has 86% Efficacy, UAE Says”. Bloomberg.com. 2020-12-09. Retrieved 2020-12-09.
- ^ Cohen J (December 2020). “China’s vaccine gambit”. Science. 370 (6522): 1263–1267. Bibcode:2020Sci…370.1263C. doi:10.1126/science.370.6522.1263. PMID 33303601.
- ^ Tan Y (16 December 2020). “Covid: What do we know about China’s coronavirus vaccines?”. BBC News. Retrieved 18 December 2020.
- ^ Jump up to:a b c Liu R (2020-12-31). “China gives its first COVID-19 vaccine approval to Sinopharm”. Reuters. Retrieved 2020-12-31.
- ^ Turak, Natasha (2021-01-18). “The UAE is on track to have half its population vaccinated by the end of March”. CNBC. Retrieved 2021-01-21.
- ^ Jump up to:a b Dawn.com (2021-02-02). “PM Imran kicks off Pakistan’s Covid-19 vaccination drive”. DAWN.COM. Retrieved 2021-02-03.
- ^ Jump up to:a b Reuters Staff (2021-01-24). “Sisi says Egypt to begin COVID-19 vaccinations on Sunday”. Reuters. Retrieved 2021-01-24.
- ^ Jump up to:a b Dumpis, Toms (2021-01-27). “Morocco Receives Half a Million Doses of Chinese Sinopharm Vaccine”. Morocco World News. Retrieved 2021-01-28.
- ^ Jump up to:a b “Zimbabwe starts administering China’s Sinopharm vaccines”. thestar.com. 2021-02-18. Retrieved 2021-02-20.
- ^ Jump up to:a b Aquino, Marco (2021-02-10). “‘The best shield’: Peru launches inoculation drive with Sinopharm vaccine”. Reuters. Retrieved 2021-02-10.
- ^ Jump up to:a b “Bolivia begins inoculation with Sinopharm jabs | The Star”. http://www.thestar.com.my. Retrieved 2021-02-28.
- ^ Jump up to:a b “Serbia Becomes First European Nation To Use China’s Sinopharm Vaccine”. RadioFreeEurope/RadioLiberty. Retrieved 2021-01-21.
- ^ Jump up to:a b “Hungary first EU nation to use China’s Sinopharm vaccine against COVID”. euronews. 2021-02-24. Retrieved 2021-02-26.
- ^ Jump up to:a b c d “Belarus begins COVID-19 vaccinations with Chinese shots”. eng.belta.by. 2021-03-15. Retrieved 2021-03-16.
- ^ Jump up to:a b “Which companies will likely produce the most COVID-19 vaccine in 2021?”. Pharmaceutical Processing World. 2021-02-05. Retrieved 2021-02-28.
- ^ Jump up to:a b hermesauto (2021-02-22). “More than 43 million doses of China’s Sinopharm Covid-19 vaccines used globally”. The Straits Times. Retrieved 2021-02-22.
- ^ Jump up to:a b Xia S, Zhang Y, Wang Y, Wang H, Yang Y, Gao GF, et al. (October 2020). “Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: a randomised, double-blind, placebo-controlled, phase 1/2 trial”. The Lancet. Infectious Diseases. 21 (1): 39–51. doi:10.1016/s1473-3099(20)30831-8. PMC 7561304. PMID 33069281.
- ^ Jump up to:a b Xia S, Duan K, Zhang Y, Zhao D, Zhang H, Xie Z, et al. (September 2020). “Effect of an Inactivated Vaccine Against SARS-CoV-2 on Safety and Immunogenicity Outcomes: Interim Analysis of 2 Randomized Clinical Trials”. JAMA. 324 (10): 951–960. doi:10.1001/jama.2020.15543. PMC 7426884. PMID 32789505.
- ^ Jump up to:a b “China State-Backed Covid Vaccine Has 86% Efficacy, UAE Says”. Bloomberg.com. 2020-12-09. Retrieved 2020-12-09.
- ^ Jump up to:a b Maxwell C. “Coronavirus: UAE authorises emergency use of vaccine for frontline workers”. The National. Retrieved 14 September 2020.
- ^ “Coronavirus: 15,000 register as volunteers for Covid-19 vaccine trial in UAE”. The National. 13 August 2020. Retrieved 15 August2020.
- ^ Jump up to:a b Reuters Staff (2020-12-09). “UAE says Sinopharm vaccine has 86% efficacy against COVID-19”. Reuters. Retrieved 2020-12-09.
- ^ “UAE: Ministry of Health announces 86 per cent vaccine efficacy”. gulfnews.com. Retrieved 2020-12-09.
- ^ “Morocco orders R-Pharm Covid-19 vaccine | The North Africa Post”. northafricapost.com. Retrieved 2020-10-07.
- ^ “Chinese Clinical Trial Register (ChiCTR) – The world health organization international clinical trials registered organization registered platform”. http://www.chictr.org.cn. Retrieved 2020-10-21.
- ^ “Egypt to start receiving volunteers for COVID-19 vaccine trials”. Egypt Independent. 2020-09-12. Retrieved 2020-09-21.
- ^ “Bahrain starts Phase III trial of Sinopharm’s Covid-19 vaccine”. Clinical Trials Arena. 24 August 2020.
- ^ Manama TD. “Vaccine trial continues | THE DAILY TRIBUNE | KINGDOM OF BAHRAIN”. DT News. Retrieved 2020-10-22.
- ^ Jump up to:a b Barrington L (3 November 2020). “Bahrain allows Sinopharm COVID-19 vaccine candidate use in frontline workers”. Reuters. Retrieved 3 November 2020.
- ^ Liu R (5 September 2020). “China’s CNBG, Sinovac find more countries to test coronavirus vaccines”. Reuters. Retrieved 6 September 2020.
- ^ “Jordan starts phase 3 trial of China’s COVID-19 vaccine”. Jordan Times. 2020-08-30. Retrieved 2020-10-22.
- ^ “Coronavirus vaccine should be available in Pakistan ‘within 6-8 weeks'”. http://www.geo.tv. Retrieved 2020-11-14.
- ^ “Third Phase of Human Trials for Coronavirus Vaccine Underway in Peru | Voice of America – English”. http://www.voanews.com. Retrieved 2020-09-11.
- ^ “6,000 additional volunteers required for trials of Sinopharm’s COVID-19 vaccine” (in Spanish). Andina. Retrieved 17 October2020.
- ^ Aquino, Marco (2021-01-27). “Peru volunteer in Sinopharm vaccine trial dies of COVID-19 pneumonia, university says”. Reuters. Retrieved 2021-01-27.
- ^ “Clinical Trial to Evaluate the Efficacy, Immunogenicity and Safety of the Inactivated SARS-CoV-2 Vaccine (COVID-19) – Full Text View – ClinicalTrials.gov”. clinicaltrials.gov. Retrieved 2020-09-28.
- ^ Greaves J (2020-10-08). “UAE company nears end of Chinese Covid-19 vaccine trial”. Reuters. Retrieved 2020-10-10.
- ^ “Chinese COVID-19 vaccine effective: Egypt’s MoH”. EgyptToday. 2020-12-13. Retrieved 2020-12-15.
- ^ “Health Min: a new production line to produce Sinopharm’s Chinese vaccine in Egypt and will be exported to African countries”. EgyptToday. 2020-12-12. Retrieved 2020-12-31.
- ^ “Morocco acquires 65 million vaccine doses from China, UK”. ABC News. Retrieved 2020-12-26.
- ^ “Serbia to produce 24 mln doses of China’s Sinopharm vaccine annually – deputy PM”. seenews.com. Retrieved 2021-03-17.
- ^ “Bahrain approves China’s Sinopharm coronavirus vaccine”. Arabian Business Industries. 13 December 2020.
- ^ Kuo L. “China approves Sinopharm coronavirus vaccine, the country’s first for general use”. The Washington Post.
- ^ “President Ramkalawan and First Lady receives second dose SinoPharm Vaccine”. statehouse.gov.sc. Retrieved 5 February2021.
- ^ Wee S (9 December 2020). “Chinese Covid-19 Vaccine Gets Key Push, but Doubts Swirl”. The New York Times. Retrieved 12 December 2020.
- ^ “Coronavirus: UAE authorises emergency use of vaccine for frontline workers”. The National. Retrieved 24 November 2020.
- ^ Biannchi, Walter (21 February 2021). “Argentina approves Sinopharm COVID-19 vaccine for emergency use”. Reuters. Retrieved 23 February 2021.
- ^ “Bolivia begins inoculation with Sinopharm jabs | The Star”. The Star. Malaysia. Retrieved 28 February 2021.
- ^ Rinith T (4 February 2021). “Health Ministry grants Emergency Use Authorization to China’s Sinopharm vaccine”. Khmer Times. Retrieved 4 February 2021.
- ^ “Dominica: Melissa Skerrit receives the Sinopharm COVID-19 vaccine”. WIC News. 4 March 2021. Retrieved 7 March 2021.
- ^ “Egypt licenses China’s Sinopharm COVID-19 vaccine for emergency use: health minister – Xinhua | English.news.cn”. Xinhua News Agency.
- ^ Jump up to:a b “Equatorial Guinea President receives 1st dose of Chinese COVID-19 vaccine”. dailynewsegypt.com. Retrieved 2021-03-10.
- ^ “Gabon receives 100,000 doses of Sinopharm’s vaccine from China”. Gabon 24. 2021-03-12. Retrieved 2021-03-13.
- ^ “Sinopharm vaccine rollout starts this weekend”. Stabroek News. 6 March 2021. Retrieved 7 March 2021.
- ^ “Hungary signs deal for Chinese Sinopharm’s COVID-19 vaccine, first in EU”. National Post.
- ^ “Iran Launches Phase Two of Mass Inoculation Campaign”. Financial Tribune. 22 February 2021. Retrieved 23 February 2021.
- ^ “Iraq approves Sinopharm, AstraZeneca vaccines”. Big News Network.com. Retrieved 30 January 2021.
- ^ “First batch of Chinese Sinopharm vaccine arrives in Jordan”. Roya News. Retrieved 9 January 2021.
- ^ “Laos declares Covid-19 vaccinations safe, more to be inoculated next week | The Star”. The Star. Malaysia. Retrieved 19 February2021.
- ^ Crean, Rosabel. “China donates 50,000 doses of Sinopharm vaccine to Lebanon | News , Lebanon News | THE DAILY STAR”. Daily Star. Retrieved 2 March 2021.
- ^ “Macau receives first batch of COVID-19 vaccines”. IAG. 7 February 2021. Retrieved 24 February 2021.
- ^ “MFDA approves Pfizer, Sinopharm Covid-19 vaccines for emergency use”. raajje.mv. Retrieved 2021-03-15.
- ^ Cristina (2021-03-19). “O mie de studenți și medici-rezidenți din cadrul USMF vor fi imunizați anti-COVID cu vaccinul BBIBP-CorV, produs de către Sinopharm Beijing Institute of Biological Products”. Ziarul de Gardă (in Romanian). Retrieved 2021-03-19.
- ^ “Deputy PM and City Governor get the first dose of Sinopharm vaccine”. MONTSAME News Agency. Retrieved 2021-03-15.
- ^ “Covid-19: Morocco authorizes use of the Sinopharm vaccine”. en.yabiladi.com.
- ^ Reuters Staff (6 March 2021). “Mozambique expects to vaccinate 16 million against coronavirus by 2022”. Reuters. Retrieved 7 March 2021.
- ^ Namibian, The. “Khomas, Erongo first to get vaccinated”. The Namibian. Retrieved 2021-03-17.
- ^ Poudel, Arjun. “China’s Shinopharm vaccine gets emergency use authorisation in Nepal”. Kathmandu Post. Retrieved 19 February2021.
- ^ “Covid-19 : Le Niger réceptionne 400.000 doses de vaccin SINOPHARM, un don de la Chine | Agence Nigérienne de Presse”. http://www.anp.ne. Retrieved 2021-03-21.
- ^ Shahzad A (19 January 2021). “Pakistan approves Chinese Sinopharm COVID-19 vaccine for emergency use”. Reuters.
- ^ “Peru grants ‘exceptional’ approval for Sinopharm COVID-19 vaccine – government sources”. Reuters. 27 January 2021.
- ^ Asala, Kizzi. “Senegal Kicks Off COVID-19 Vaccination Campaign with China’s Sinopharm”. Africanews. Retrieved 23 February2021.
- ^ “Serbia Becomes First European Nation To Use China’s Sinopharm Vaccine”. RadioFreeEurope/RadioLiberty.
- ^ Thomas, Abdul Rashid (2021-03-15). “Sierra Leone’s President Bio leads the way in taking COVID-19 Vaccine”. SIERRA LEONE TELEGRAPH. Retrieved 2021-03-15.
- ^ “NMRA approves sinopharm vaccine for emergency use”. Colombo Gazette. 2021-03-19. Retrieved 2021-03-20.
- ^ Sequera, Vivian (1 March 2021). “Venezuela approves use of China’s Sinopharm coronavirus vaccine”. Reuters. Retrieved 2 March 2021.
- ^ Mutsaka, Farai (18 February 2021). “Zimbabwe starts administering China’s Sinopharm vaccines”. The Star. Retrieved 20 February 2021.
- ^ Jump up to:a b “China ‘to provide 400,000 COVID vaccine doses’ to Afghanistan”. http://www.aljazeera.com. Retrieved 2021-03-01.
- ^ Jump up to:a b Presse, AFP-Agence France. “Algeria Receives 200,000 Coronavirus Jabs From China”. http://www.barrons.com. Retrieved 2021-02-26.
- ^ Jump up to:a b “Vaccine donation from China arrives | The Star”. http://www.thestar.com.my. Retrieved 2021-02-12.
- ^ Jump up to:a b Radomir, Ralev. “Montenegro receives 30,000 doses of China’s COVID-19 vaccine”.
- ^ “Regulation and Prequalification”. World Health Organization. Retrieved 18 March 2021.
- ^ “Bahrain approves Chinese COVID-19 vaccine for use”. ABC News. Retrieved 2020-12-13.
- ^ Reuters Staff (2021-01-15). “Cambodia says China donates 1 million doses of COVID-19 vaccines”. Reuters. Retrieved 2021-01-16.
- ^ “Health Ministry grants Emergency Use Authorization to China’s Sinopharm vaccine”. Khmer Times. 2021-02-04. Retrieved 2021-02-04.
- ^ “Lt Gen Manet first to be inoculated today with the Sinopharm vaccine”. Khmer Times. 2021-02-09. Retrieved 2021-02-10.
- ^ “Sinovac’s coronavirus vaccine candidate approved for emergency use in China – source”. Reuters. 2020-08-29. Retrieved 2020-08-30.
- ^ Vivek V (15 October 2020). “China’s Sinopharm offers experimental COVID-19 vaccines to students: WSJ”. Reuters. Retrieved 15 October 2020.
- ^ “China gives conditional approval to coronavirus vaccine made by Sinopharm”. Global News. Retrieved 2020-12-31.
- ^ “First Sinopharm Covid-19 vaccines to arrive today”. Macau Business. 2021-02-06. Retrieved 2021-02-07.
- ^ Taufiqurrahman M. “Indonesia can be manufacturing hub for COVID-19 vaccine, says Chinese foreign minister”. Jakarta Post. Retrieved 13 October 2020.
- ^ “Iran Launches Phase Two of Mass Inoculation Campaign”. Financial Tribune. 2021-02-22. Retrieved 2021-02-23.
- ^ “Iran Gets Chinese Vaccine for Coronavirus – Society/Culture news”. Tasnim News Agency. Retrieved 2021-02-28.
- ^ Jangiz, Khazan. “Iraq approves the emergency use of two more COVID-19 vaccines”. http://www.rudaw.net. Retrieved 2021-01-21.
- ^ “Iraq receives first Covid vaccines, gift from China”. France 24. 3 March 2021.
- ^ “Jordan approves China’s Sinopharm Covid vaccine”. France 24. 2021-01-09. Retrieved 2021-03-07.
- ^ Omari, Raed. “Jordan begins COVID-19 vaccination drive as physician, 87, gets first jab”. Arab News.
- ^ KHARIZOV, Ruslan (2021-03-19). “150,000 doses of Sinopharm coronavirus vaccine delivered to Kyrgyzstan”. 24.kg. Retrieved 2021-03-20.
- ^ Thanabouasy, Phayboune (2021-01-27). “Laos Begins Vaccinations for Over 600 Medical Workers”. Laotian Times. Retrieved 2021-01-27.
- ^ Limited, Bangkok Post Public Company. “Laos receives 300,000 vaccine doses from China”. Bangkok Post. Retrieved 2021-02-10.
- ^ “China donates 50,000 doses of Sinopharm vaccine to Lebanon | News , Lebanon News | THE DAILY STAR”. http://www.dailystar.com.lb. Retrieved 2021-03-02.
- ^ March 2021, Naharnet Newsdesk 01; 20:39. “Lebanon Authorizes Use of Chinese Vaccine Sinopharm”. Naharnet. Retrieved 2021-03-07.
- ^ “MFDA approves Pfizer, Sinopharm Covid-19 vaccines for emergency use”. raajje.mv. Retrieved 2021-03-15.
- ^ “Mongolia receives COVID-19 vaccine donation from China – The Manila Times”. http://www.manilatimes.net. Retrieved 2021-02-28.
- ^ “Deputy PM and City Governor get the first dose of Sinopharm vaccine”. MONTSAME News Agency. Retrieved 2021-03-15.
- ^ “China’s Shinopharm vaccine gets emergency use authorisation in Nepal”. kathmandupost.com. Retrieved 2021-02-19.
- ^ Peshimam GN (2020-12-31). “Pakistan to purchase 1.2 million COVID-19 vaccine doses from China’s Sinopharm”. Reuters. Retrieved 2020-12-31.
- ^ Shahzad, Asif (2021-01-19). “Pakistan approves Chinese Sinopharm COVID-19 vaccine for emergency use”. Reuters. Retrieved 2021-01-21.
- ^ “Palestine to receive 100,000 doses of Sinopharm Covid-19 vaccine”. WAFA Agency. Retrieved 2021-03-12.
- ^ “NMRA approves sinopharm vaccine for emergency use”. Colombo Gazette. 2021-03-19. Retrieved 2021-03-20.
- ^ Sircar, Nandini. “UAE Covid vaccine: Third dose to help those with weak immunity”. Khaleej Times. Retrieved 2021-03-19.
- ^ Jump up to:a b “Egypt approves Chinese COVID vaccine, roll-out likely this month”. http://www.aljazeera.com. Retrieved 2021-01-03.
- ^ “Gabon receives 100,000 doses of Sinopharm’s vaccine from China”. Gabon 24. 2021-03-12. Retrieved 2021-03-13.
- ^ Eljechtimi, Ahmed (2021-01-26). “Morocco prepares to launch COVID-19 vaccination programme”. Reuters. Retrieved 2021-01-27.
- ^ “Moroccan health ministry grants emergency approval to Sinopharm’s Covid-19 vaccine”. wam. Retrieved 2021-01-27.
- ^ “China, Africa and the Vaccine Donations”. Modern Ghana. Retrieved 2021-03-05.
- ^ Mucari, Manuel (2021-03-06). “Mozambique expects to vaccinate 16 million against coronavirus by 2022”. Reuters. Retrieved 2021-03-07.
- ^ Namibian, The. “Khomas, Erongo first to get vaccinated”. The Namibian. Retrieved 2021-03-17.
- ^ “Covid-19 : Le Niger réceptionne 400.000 doses de vaccin SINOPHARM, un don de la Chine | Agence Nigérienne de Presse”. http://www.anp.ne. Retrieved 2021-03-21.
- ^ Staff, Reuters (2021-02-18). “Senegal takes delivery of China’s Sinopharm vaccine”. Reuters. Retrieved 2021-02-19.
- ^ AfricaNews (2021-02-23). “Senegal begins covid-19 vaccination with doses from China’s Sinopharm”. Africanews. Retrieved 2021-02-23.
- ^ AFP. “Sierra Leone to receive 200,000 virus vaccine doses”. ewn.co.za. Retrieved 2021-02-26.
- ^ Thomas, Abdul Rashid (2021-03-15). “Sierra Leone’s President Bio leads the way in taking COVID-19 Vaccine”. SIERRA LEONE TELEGRAPH. Retrieved 2021-03-15.
- ^ “Seychelles to start vaccinations with Chinese-made Sinopharm”. AP NEWS. 2021-01-08. Retrieved 2021-01-08.
- ^ “Covid-19: le Congo-Brazzaville reçoit des milliers de doses du vaccin chinois Sinopharm”. RFI (in French). 2021-03-10. Retrieved 2021-03-12.
- ^ Banya, Nelson (2021-02-11). “Zimbabwe purchases 600,000 Sinopharm COVID-19 vaccinations -information minister”. Reuters. Retrieved 2021-02-11.
- ^ Staff, Reuters (2021-02-24). “Zimbabwe to buy 1.2 million more COVID-19 vaccine doses from China”. Reuters. Retrieved 2021-02-26.
- ^ Lopez, Ezequiel Abiu (2021-02-16). “Dominican Republic launches COVID-19 vaccination campaign”. Reuters. Retrieved 2021-02-28.
- ^ “Dominica: Melissa Skerrit receives the Sinopharm COVID-19 vaccine”. WIC News. 2021-03-04. Retrieved 2021-03-05.
- ^ Jorgic, Drazen (2021-03-10). “Mexico leans on China after Biden rules out vaccines sharing in short term”. Reuters. Retrieved 2021-03-10.
- ^ Biannchi, Walter (2021-02-21). “Argentina approves Sinopharm COVID-19 vaccine for emergency use”. Reuters. Retrieved 2021-02-22.
- ^ “Buenos Aires Times | Shipment of 900,000 Sinopharm vaccine doses arrives in Argentina”. http://www.batimes.com.ar. Retrieved 2021-02-26.
- ^ Ramos, Danny (2021-02-11). “Bolivia signs deal with China´s Sinopharm for coronavirus vaccine”. Reuters. Retrieved 2021-02-11.
- ^ “China-donated Sinopharm vaccine received”. Guyana Chronicle. 2021-03-03. Retrieved 2021-03-03.
- ^ “Sinopharm vaccine rollout starts this weekend”. Stabroek News. 2021-03-06. Retrieved 2021-03-07.
- ^ Reuters Staff (2021-01-06). “Peru inks deals with Sinopharm, AstraZeneca for coronavirus vaccines -president”. Reuters. Retrieved 2021-01-07.
- ^ Aquino, Marco (2021-01-27). “Peru grants ‘exceptional’ approval for Sinopharm COVID-19 vaccine – government sources”. Reuters. Retrieved 2021-01-28.
- ^ Sequera, Vivian (2021-03-01). “Venezuela approves use of China’s Sinopharm coronavirus vaccine”. Reuters. Retrieved 2021-03-02.
- ^ Sequera, Vivian (2021-03-02). “Venezuela receives donated coronavirus vaccine from China”. Reuters. Retrieved 2021-03-02.
- ^ “China sends 100,000 coronavirus vaccines to Belarus”. eng.belta.by. 2021-02-19. Retrieved 2021-02-19.
- ^ “Hungary signs deal for Chinese Sinopharm’s COVID-19 vaccine, first in EU”. nationalpost. Retrieved 2021-01-29.
- ^ Staff, Reuters (2021-02-16). “First 550,000 doses of Chinese Sinopharm’s vaccine arrive in Hungary”. Reuters. Retrieved 2021-02-18.
- ^ “Hungary’s PM Viktor Orbán vaccinated against COVID with Chinese Sinopharm vaccine”. euronews. 2021-02-28.
- ^ “Emiratele Arabe Unite au donat Republicii Moldova un lot de vaccin împotriva COVID-19”. TV8 (in Romanian). 13 March 2021.
- ^ Cristina (2021-03-19). “O mie de studenți și medici-rezidenți din cadrul USMF vor fi imunizați anti-COVID cu vaccinul BBIBP-CorV, produs de către Sinopharm Beijing Institute of Biological Products”. Ziarul de Gardă (in Romanian). Retrieved 2021-03-19.
- ^ “Vaccine delay in North Macedonia stirs political tension”. ABC News. Retrieved 2021-02-12.
- ^ Reuters Staff (2021-01-16). “Serbia receives million doses of China’s Sinopharm COVID-19 vaccine”. Reuters. Retrieved 2021-01-16.
- ^ “Abu Dhabi starts COVID-19 vaccinations”. Arab News. 2020-12-14. Retrieved 2020-12-17.
- ^ Wee SL (9 December 2020). “Chinese Covid-19 Vaccine Gets Key Push, but Doubts Swirl”. The New York Times. Retrieved 21 December 2020.
- ^ Yu, Sun (December 31, 2020). “China approves first domestic Covid-19 vaccine for general use”. Financial Times. Retrieved January 12, 2021.
- ^ Dancel R. “Philippine officials under fire from critics, health authorities for unsanctioned Covid-19 vaccinations”. The Straits Times.
- ^ Aberia, Ingming (6 January 2021). “Did Sinopharm forget that Duque exists?”. The Manila Times. Retrieved 9 January 2021.
- ^ “水面下で出回る中国ワクチン 富裕層から永田町へ? 狙われる日本市場”. Mainichi Daily News (in Japanese). 2020-12-31. Retrieved 2021-01-02.
- ^ Elmer, Keegan (January 3, 2021). “Beijing responds to claims Japanese were given unapproved Sinopharm jabs”. South China Morning Post. Retrieved January 9, 2021.
External links
- “How the Sinopharm Covid-19 Vaccine Works”. The New York Times.
| A vial of the BBIBP-CorV COVID‑19 vaccine | |
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | Inactivated |
| Clinical data | |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | Authorization for use in Bahrain, China, Egypt, Iraq, Pakistan, Serbia, United Arab Emirates, Iran (emergency use) |
| Identifiers | |
| CAS Number | 2503126-65-4 |
| DrugBank | DB15807 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 Portal |
| vte |
How the Sinopharm Vaccine Works
By Jonathan Corum and Carl ZimmerUpdated March 22, 2021Leer en español

In early 2020, the Beijing Institute of Biological Products created an inactivated coronavirus vaccine called BBIBP-CorV. Clinical trials run by the state-owned company Sinopharm showed that it had an efficacy rate of 79 percent. China approved the vaccine and soon began exporting it to other countries.
A Vaccine Made From Coronaviruses
BBIBP-CorV works by teaching the immune system to make antibodies against the SARS-CoV-2 coronavirus. The antibodies attach to viral proteins, such as the so-called spike proteins that stud its surface.

Spikes
Spike
protein
gene
CORONAVIRUS
To create BBIBP-CorV, the Beijing Institute researchers obtained three variants of the coronavirus from patients in Chinese hospitals. They picked one of the variants because it was able to multiply quickly in monkey kidney cells grown in bioreactor tanks.
Killing the Virus
Once the researchers produced large stocks of the coronaviruses, they doused them with a chemical called beta-propiolactone. The compound disabled the coronaviruses by bonding to their genes. The inactivated coronaviruses could no longer replicate. But their proteins, including spike, remained intact.

Beta-
propiolactone
INACTIVATED
CORONAVIRUS
Inactivated
genes
The researchers then drew off the inactivated viruses and mixed them with a tiny amount of an aluminum-based compound called an adjuvant. Adjuvants stimulate the immune system to boost its response to a vaccine.
Inactivated viruses have been used for over a century. Jonas Salk used them to create his polio vaccine in the 1950s, and they’re the bases for vaccines against other diseases including rabies and hepatitis A.
Prompting an Immune Response
Because the coronaviruses in BBIBP-CorV are dead, they can be injected into the arm without causing Covid-19. Once inside the body, some of the inactivated viruses are swallowed up by a type of immune cell called an antigen-presenting cell.

INACTIVATED
CORONAVIRUS
Engulfing
the virus
ANTIGEN-
PRESENTING
CELL
Digesting
virus proteins
Presenting
virus protein
fragments
HELPER
T CELL
The antigen-presenting cell tears the coronavirus apart and displays some of its fragments on its surface. A so-called helper T cell may detect the fragment. If the fragment fits into one of its surface proteins, the T cell becomes activated and can help recruit other immune cells to respond to the vaccine.
Making Antibodies
Another type of immune cell, called a B cell, may also encounter the inactivated coronavirus. B cells have surface proteins in a huge variety of shapes, and a few might have the right shape to latch onto the coronavirus. When a B cell locks on, it can pull part or all of the virus inside and present coronavirus fragments on its surface.
A helper T cell activated against the coronavirus can latch onto the same fragment. When that happens, the B cell gets activated, too. It proliferates and pours out antibodies that have the same shape as their surface proteins.

ACTIVATED
HELPER
T CELL
INACTIVATED
CORONAVIRUS
Activating
the B cell
Matching
surface proteins
B CELL
SECRETED
ANTIBODIES
Stopping the Virus
Once vaccinated with BBIBP-CorV, the immune system can respond to an infection of live coronaviruses. B cells produce antibodies that stick to the invaders. Antibodies that target the spike protein can prevent the virus from entering cells. Other kinds of antibodies may block the virus by other means.

ANTIBODIES
LIVE
VIRUS
Remembering the Virus
Sinopharm’s clinical trials have demonstrated that BBIBP-CorV can protect people against Covid-19. But no one can yet say how long that protection lasts. It’s possible that the level of antibodies drops over the course of months. But the immune system also contains special cells called memory B cells that might retain information about the coronavirus for years or even decades.
Vaccine Timeline
January, 2020 Sinopharm begins developing an inactivated vaccine against the coronavirus.
June Researchers report the vaccine produces promising results in monkeys. A Phase 1/2 trial shows that the vaccine doesn’t cause any serious side effects and enables people to make antibodies against the coronavirus.

A Sinopharm production plant in Beijing.Zhang Yuwei/Xinhua, via Associated Press
July A Phase 3 trial begins in the United Arab Emirates.
August Phase 3 trials begin in Morocco and Peru.

Preparing a Sinopharm dose in Lima, Peru.Ernesto Benavides/Agence France-Presse
Sept. 14 The U.A.E. gives emergency approval for Sinopharm’s vaccine to use on health care workers. Government officials and others begin to receive it.
November The chairman of Sinopharm says almost a million people in China have received Sinopharm vaccines.
Nov. 3 The ruler of Dubai, Sheikh Mohammed bin Rashid al-Maktoum, announces he received the vaccine.

Sheikh Mohammed before receiving the vaccine.Agence France-Presse
Dec. 9 The U.A.E. gives full approval to BBIBP-CorV, announcing it has an efficacy rate of 86 percent. But the government did not release any details with their announcement, leaving it unclear how they had come to their conclusions.
Dec. 13 Bahrain also approves the vaccine.

Vials of the Sinopharm vaccine at a packaging plant.Zhang Yuwei/Xinhua, via Associated Press
Dec. 30 Sinopharm announces that the vaccine has an efficacy of 79.34 percent, leading the Chinese government to approve it. The company has yet to publish detailed results of their Phase 3 trial.
Jan. 3, 2021 Egypt authorizes the vaccine for emergency use.
Sources: National Center for Biotechnology Information; Science; The Lancet; Lynda Coughlan, University of Maryland School of Medicine; Jenna Guthmiller, University of Chicago.
Data
/////////////BBIBP-CorV, Sinopharm, COVID-19 vaccine, china, covid 19, corona virus, vaccine
#BBIBP-CorV, #Sinopharm, #COVID-19 vaccine, #china, #covid 19, #corona virus, #vaccine
Sitravatinib

Sitravatinib
1-N‘-[3-fluoro-4-[2-[5-[(2-methoxyethylamino)methyl]pyridin-2-yl]thieno[3,2-b]pyridin-7-yl]oxyphenyl]-1-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide
| シトラバチニブ; ситраватиниб , سيترافاتينيب , 司曲替尼 , |
| Formula | C33H29F2N5O4S |
|---|---|
| Cas | 1123837-84-2 |
| Mol weight | 629.6763 |
MGCD516
Antineoplastic, Receptor tyrosine kinase inhibitor
Sitravatinib (MGCD516) is an experimental drug for the treatment of cancer. It is a small molecule inhibitor of multiple tyrosine kinases.
Sitravatinib is being developed by Mirati Therapeutics.[1]
Ongoing phase II trials include a trial for liposcarcoma,[2] a combination trial for non-small cell lung cancer,[3] and a combination trial with nivolumab for renal cell carcinoma.[4]
Mirati Therapeutics and licensee BeiGene are developing sitravatinib, an oral multitargeted kinase inhibitor which inhibits Eph, Ret, c-Met and VEGF-1, -2 and -3, DDR, Trk, Axl kinases, CHR4q12, TYRO3 and Casitas B-lineage, in combination with immune checkpoint inhibitors, for treating advanced solid tumors.
In March 2021, sitravatinib was reported to be in phase 3 clinical development.

PDT PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009026717
WO2009026717 , in which sitravatinib was first disclosed, claiming heterocyclic compounds as multi kinase inhibitors.
Scheme 10
Example 52
N-(3-Fluoro-4-(2-(5-((2-methoxyethylamino)methyl)pyridin-2-yl)thieno[3,2-b]pyridin-7- yloxy)phenyl)-N-(4-fluorophenyl)cyclopropane- 1 , 1 -dicarboxamide
Step 1 : tert-Butyl (6-(7-(2-Fluoro-4-(1-(4-fluorophenylcarbamoyl)-cyclopropanecarboxamido)phenoxy)thieno [3 ,2-b]pyridin-2-yl)pyridin-3 -y l)methyl(2-methoxyethyl)carbamate (146)
To aniline 126 (0.58 g, 1.1 mmol) and DIPEA (0.58 mL, 0.43 g, 3.3 mmol) in dry DMF
(20 mL) was added 1-(4-fluorophenylcarbamoyl)cyclopropanecarbpxylic acid (0.35 g, 1.5 mmol) and HATU (0.72 g, 1.9 mmol) and the mixture was stirred at r.t. for 18 h. It was then partitioned between ethyl acetate and water, the organic phase was washed with water, IM NaOH, brine, dried (MgSO4), filtered, and concentrated. Silica gel chromatography (ethyl acetate) afforded title compound Ϊ46 (0.60 g, 74 % yield). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.40 (s, 1H), 10.01 (s, 1H), 8.52-8.49 (m, 2H), 8.33 (s, 1H), 8.27-8.24 (m, 1H), 7.92-7.88 (m, 1H), 7.78 (dd, J = 8.2, 2.1 Hz, 1H) 7.65-7.60 (m, 2H), 7.52-7.42 (m, 2H), 7.14 (t, J = 8.8 Hz, 2H), 6.65 (d, J = 5.1 Hz 1H), 4.47 (s, 2H), 3.42-3.30 (m, 4H), 3.22 (s, 3H), 1.46-1.30 (m, 13H). MS (m/z): 730.1 (M+H).
Step 2. N-(3-Fluoro-4-(2-(5-((2-methoxyethylamino)methyl)pyridin-2-yl)thieno[3,2-blpyridin-7-yloxy)phenyl)-N-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide (147)
To the compound 146 (0.59 g, 0.81 mmol) in dichloromethane (50 mL) was added TFA (3 mL). The solution was stirred for 18 h then concentrated. The residue was partitioned between dichloromethane and 1 M NaOH, and filtered to remove insolubles. The organic phase was collected, washed with IM NaOH, brine, dried (MgSO4), filtered, and concentrated to afford title compound 147 (0.35 g, 69 % yield).
1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.40 (s, 1H), 10.01 (s, 1H), 8.55 (d, J = 1.6 Hz, 1H), 8.51 (d, J = 5.3 Hz, 1H), 8.31 (s, 1H), 8.22 (d, J = 8.0 Hz, 1H), 7.92-7.87 (m, 2H), 7.65-7.61 (m, 2H), 7.52-7.43 (m, 2H), 7.17-7.12 (m, 2H), 6.64 (d, J = 5.5 Hz, 1H), 3.77 (s, 2H), 3.40 (t, J = 5.7 Hz, 2H), 3.23 (s, 3H), 2.64 (t, J = 5.7 Hz, 2H), 1.46 (br s, 4H). MS (m/z): 630.1 (M+H).
PATENT
WO 2009026720
https://patents.google.com/patent/WO2009026720A1
PATENT
WO-2021050580
Novel, stable crystalline polymorphic forms (form D) of sitravatinib , useful for treating a multi tyrosine kinase-associated cancer eg sarcoma, glioma, non-small cell lung, bladder, kidney, ovarian, gastric, breast or liver cancer.
International publication No. W02009/026717A disclosed compounds with the inhibition activities of multiple protein tyrosine kinases, for example, the inhibition activities of VEGF receptor kinase and HGF receptor kinase. In particular, disclosed N-(3-fluoro-4-((2-(5-(((2-methoxyethyl)amino)methyl)pyridin-2-yl)thieno[3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane- 1,1 -di carboxamide (Compound 1) is a multi-tyrosine kinase inhibitor with demonstrated potent inhibition of a closely related spectrum of tyrosine kinases, including RET, CBL, CHR4ql2, DDR and Trk, which are key regulators of signaling pathways that lead to cell growth, survival and tumor progression.
[003]
Compound 1
[004] Compound 1 shows tumor regression in multiple human xenograft tumor models in mice, and is presently in human clinical trials as a monotherapy as well as in combination for
treating a wide range of solid tumors. Compound 1 is presently in Phase 1 clinical trial for patients with advanced cancer, in Phase 2 studies for patients with advanced liposarcoma and non-small cell lung cancer (NSCLC).
[005] The small scale chemical synthesis of the amorphous Compound 1 had been disclosed in the Example 52 (compound 147) of W02009/026717A, however, in order to prepare the API of Compound 1 with high quality and in large quantity, crystalline forms of Compound 1 would be normally needed so the process impurities could be purged out by recrystallization.
Practically, it is difficult to predict with confidence which crystalline form of a particular compound will be stable, reproducible, and suitable for phamaceutical processing. It is even more difficult to predict whether or not a particular crystalline solid state form will be produced with the desired physical properties for pharmaceutical formulations.
[006] For all the foregoing reasons, there is a great need to produce crystalline forms of Compound 1 that provide manufacturing improvements of the pharmaceutical composition.
The present invention advantageously addresses one or more of these needs.
EXAMPLE 1
Preparation of N-(3-fluoro-4-((2-(5-(((2-methoxyethyl)amino)methyl)pyridin-2- yl)thieno[3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane-l,l- dicarboxamide (Compound 1)
[0085] This Example illustrates the preparation ofN-(3-fluoro-4-((2-(5-(((2-methoxyethyl)amino)methyl)pyridin-2-yl)thieno[3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4-fluorophenyl)cyclopropane- 1,1 -di carboxamide (Compound 1).
[0086] Step 1: N-(Y6-bromopyridin-3-vDmethvD-2-methoxyethan-l-amine (Compound 1A)
Compound 1A
[0087] To a stirred solution of 2-Methoxyethylamine (3.0 eq) in dichloromethane (DCM) (12 vol) was added Molecular sieves (0.3 w/w) and stirred for 2 hours at 25±5°C under nitrogen atmosphere. The reaction mass water content was monitored by Karl Fischer analysis until the water content limit reached 0.5 % w/w. Once the water content limit was reached, the reaction mass cooled to 5±5°C and 6-bromonicotinaldehyde (1.0 eq) was added lot wise over period of 30 minutes to the above reaction mass at 5±5°C. The reaction mass was stirred for 30±5 minutes at 5±5°C and acetic acid (1.05 eq) was added drop wise at 5±5°C. After completion of the addition, the mass was slowly warmed to 25±5°C and stirred for 8 h to afford Compound 1 A. The imine formation was monitored by HPLC.
[0088] Step 2: tert-butyl (Y6-brom opyri din-3 -vQmethvO(2-m ethoxy ethvDcarbamate (Compound
IB)
Compound 1B
[0089] Charged Compoud 1A (1.0 eq) in THF (5.0 vol) was added and the reaction mass was stirred for 30 minutes at 25±5°C under nitrogen atmosphere. The reaction mass was cooled to temperature of about 10±5°C. Di-tert- butyl dicarbonate (1.2 eq) was added to the reaction mass at 10±5°C under nitrogen atmosphere and the reaction mass temperature was raised to 25±5°C and the reaction mass for about 2 hours. The progress of the reaction was monitored by HPLC. After IPC completion, a prepared solution of Taurine (1.5 eq) in 2M aq NaOH (3.1 vol) was charged and stirred at 10±5°C for 16 h to 18 h. The reaction mass was further diluted with 1M aq.NaOH solution (3.7 vol) and the layers were separated. The aqueous layer was extracted with DCM (2 x 4.7vol) and the extract combined with the organic layer. The combined organic layers were washed with 1M aq.NaOH solution (3.94 vol), followed by water (2×4.4 vol), and dried over sodium sulfate (2.0 w/w) . The filtrate was concentrated under reduced pressure below 40° C until no distillate was observed. Tetrahydrofuran (THF) was sequentially added (1×4 vol and lx 6vol) and concentrated under reduced pressure below 40°C until no distillate was observed to obtained Compound IB as light yellow colored syrup liquid.
[0090] Step 3: tert-butyl 7-chlorothieno[3.2-b1pyridin-2-yl)pyridin-3-yl )methyl)(2-
methoxyethvDcarbamate (Compound 1C)
Compound 1C
[0091] To a stirred solution of 7-chlorothieno[3,2-b]pyridine (1.05 eq) in tetrahydrofuran (7 vol) was added n-butyl lithium (2.5 M in hexane) drop wise at -15±10°C and stirred for 90 minutes at same temperature under nitrogen atmosphere. Zinc chloride (1.05 eq) was added to the reaction mass at -15±10°C. The reaction mass was slowly warmed to 25±5°C and stirred for 45 minutes under nitrogen atmosphere to afford Compound 1C. The progress of the reaction was monitored by HPLC.
[0092] Step 4: tert-butyl (Y6-(7-(4-amino-2-fluorophenoxy)thieno[3.2-b1pyridin-2-v0pyridin-3-vDmethvD(2-methoxyethvDcarbamate (Compound ID)
Compound 1D
[0093] 3-fluoro-4-hydroxybenzenaminium chloride (1.2 eq) in DMSO (3.9 vol) at 25±5°C was charged under nitrogen atmosphere and the reaction mass was stirred until observance of a clear solution at 25±5°C. t-BuOK was added lot wise under nitrogen atmosphere at 25±10°C. The reaction mass temperature was raised to 45±5°C and maintained for 30 minutes under nitrogen atmosphere. Compound 1C was charged lot-wise under nitrogen atmosphere at 45±5°C and stirred for 10 minutes at 45± 5°C.The reaction mixture was heated to 100± 5°C and stirred for 2 hrs. The reaction mass is monitored by HPLC.
[0094] After reaction completion, the reaction mass was cooled to 10± 5°C and quenched with chilled water (20 vol) at 10±5°C. The mass temperature was raised to 25± 5°C and stirred for 7-8 h. The resulting Compound ID crude was collected by filtration and washed with 2 vol of water. Crude Compound ID material taken in water (10 vol) and stirred for up to 20 minutes at 25±5°C. The reaction mass was heated to 45±5°C and stirred for 2-3 h at 45±5°C, filtered and vacuum-dried.
[0095] Crude Compound ID was taken in MTBE (5 vol) at 25±5°C and stirred for about 20 minutes at 25±5°C. The reaction mass temperature was raised to 45±5°C, stirred for 3-4 h at 45±5°C and then cooled to 20±5°C. The reaction mass was stirred for about 20 minutes at 20±5°C, filtered, followed by bed wash with water (0. 5 vol) and vacuum-dried.
[0096] The crude material was dissolved in acetone (10 vol) at 25±5°C and stirred for about 2h at 25±5°C. The reaction mass was filtered through a celite bed and washed with acetone (2.5 vol). The filtrate was slowly diluted with water (15 vol) at 25±5°C. The reaction mass was stirred for 2-3 h at 25±5°C, filtered and bed washed with water (2 vol) & vacuum-dried to afford Compound ID as brown solid.
[0097] Step 5 : 1 -((4-((2-(5-(((tert-butoxycarbonv0(2-methoxy ethvOaminolmethvOpyri din-2 -yl )thieno[3.2-b]pyridin-7-yl )oxy)-3 -fluorophenyl icarbamoyl level opropane-1 -carboxylic acid (Compound IE)
Compound 1E
[0098] To a solution of Compound ID (1.0 eq.) in tetrahydrofuran (7 vol.), aqueous potassium carbonate (1.0 eq.) in water (8 vol.) was added. The solution was cooled to 5±5°C, and stirred for about 60 min. While stirring, separately triethylamine (2.0 eq.) was added to a solution of 1,1-cyclopropanedicarboxylic acid (2.0 eq.) in tetrahydrofuran (8 vol.), at 5±5°C, followed by thionyl chloride (2.0 eq.) and stirred for about 60 min. The acid chloride mass was slowly added to the Compound ID solution at 5±5°C. The temperature was raised to 25±5°C and stirred for 3.0 h. The reaction was monitored by HPLC analysis.
[0099] After reaction completion, the mass was diluted with ethyl acetate (5.8 vol.), water (5.1 vol.), 10% (w/w) aqueous hydrochloric acid solution (0.8 vol.) and 25% (w/w) aqueous sodium chloride solution (2 vol.). The aqueous layer was separated and extracted with ethyl acetate (2 x 5 vol.). The combined organic layers were washed with a 0.5M aqueous sodium bicarbonate solution (7.5 vol.). The organic layer was treated with Darco activated charcoal (0.5 w/w) and sodium sulfate (0.3 w/w) at 25±5°C for 1.0 h. The organic layer was filtered through celite and washed with tetrahydofuran (5.0 vol.). The filtrate was concentrated under vacuum below 50°C to about 3 vol and co-distilled with ethyl acetate (2 x 5 vol.) under vacuum below 50°C up to ~ 3.0 vol. The organic layer was cooled to 15±5°C, stirred for about 60 min., filtered, and the solid was washed with ethyl acetate (2.0 vol.). The material was dried under vacuum at 40±5°C until water content was less than 1% to afford Compound IE as brown solid.
[00100] Step 6: tert-butyl (Y6-(7-(2-fluoro-4-(T-(Y4-fluorophenvDcarbamovDcvclopropane-l-carboxamido)phenoxy)thieno[3.2-b]pyridin-2-v0pyri din-3 – (2-
methoxyethvDcarbamate (Compound IF)
[00101] Pyridine (1.1 eq.) was added to a suspension of Compound IE (1.0 eq.) in tetrahydrofuran (10 vol.) and cooled to 5±5°C. Thionyl chloride (2.0 eq.) was added and stirred for about 60 min. The resulting acid chloride formation was confirmed by HPLC analysis after quenching the sample in methanol. Separately, aqueous potassium carbonate (2.5 eq.) solution (7.0 vol. of water) was added to a solution of 4-fluoroaniline (3.5 eq.) in tetrahydrofuran (10 vol.), cooled to 5±5°C, and stirred for about 60 min. The temperature of the acid chloride mass at 5±5°C was raised to a temperature of about 25±5°C and stirred for 3 h. The reaction monitored by HPLC analysis.
[00102] After completion of the reaction, the solution was diluted with ethyl acetate (25 vol.), the organic layer was separated and washed with a 1M aqueous sodium hydroxide solution (7.5 vol.), a 1M aqueous hydrochloric acid solution (7.5 vol.), and a 25% (w/w) aqueous sodium chloride solution (7.5 vol.). The organic layer was dried and and filtered with sodium sulfate (1.0 w/w). The filtrate was concentrated ~ 3 vol under vacuum below 50°C and co-distilled with ethyl acetate (3 x 5 vol.) under vacuum below 50°C to ~ 3.0 vol. Ethyl acetate (5 vol.) and MTBE (10 vol.) were charged, heated up to 50±5°C and stirred for 30-60 min. The mixture was cooled to 15±5°C, stirred for about 30 min., filtered, and the solid was washed with ethyl acetate (2.0 vol.). MGB3 content was analyzed by HPLC analysis. The material was dried under vacuum at 40±5°C until the water content reached about 3.0% to afford Compound IF as brown solid.
[00103] Step 7 : N-(3-fluoro-4-((2-(5-(((2-methoxyethv0amino)methv0pyridin-2-yl )thieno[3.2-b]pyridin-7-yl )oxy)phenyl)-N-(4-fluorophenyl level opropane-1. 1 -dicarboxamide (Compound 1)
Compound 1
[0100] To a mixture of Compound IF in glacial acetic acid (3.5 vol.) concentrated hydrochloric acid (0.5 vol.) was added and stirred at 25±5°C for 1.0 h. The reaction was monitored by HPLC analysis.
[0101] After reaction completion, the mass was added to water (11 vol.) and stirred for 20±5°C for 30 min. The pH was adjusted to 3.0 ± 0.5 using 10% (w/w) aqueous sodium bicarbonate solution and stirred for 20±5°C for approximately 3.0 h.. The mass was filtered, washed with water (4 x 5.0 vol.) and the pH of filtrate was checked after every wash. The material was dried under vacuum at 50±5°C until water content was about 10%.
[0102] Crude Compound 1 was taken in ethyl acetate (30 vol.), heated to 70±10°C, stirred for 1.0 h., cooled to 25±5°C, filtered, and washed with ethyl acetate (2 vol.). The material was dries under vacuum at 45±5°C for 6.0 h.
[0103] Crude Compound 1 was taken in polish filtered tetrahydrofuran (30 vol.) and pre washed Amberlyst A-21 Ion exchange resin and stirred at 25±5°C until the solution became clear. After getting the clear solution, the resin was filtered and washed with polish filtered tetrahydrofuran (15 vol.). The filtrate was concentrated by -50% under vacuum below 50°C and co-distilled with polish filtered IPA (3 x 15.0 vol.) and concentrated up to -50% under vacuum below 50°C. Charged polish filtered IPA (15 vol.) was added and the solution concentrated under vacuum below 50°C to – 20 vol. The reaction mass was heated to 80±5°C, stirred for 60 min. and cooled to 25±5°C. The resultant reaction mass was stirred for about 20 hours at 25±5°C. The reaction mass was cooled to 0±5°C, stirred for 4-5 hours, filtered, and washed with polish filtered IPA (2 vol.). The material was dried under vacuum at 45±5°C, until the water content was about 2%, to obtain the desired product Compound 1. ¾-NMR (400 MHz, DMSO- d): 510.40 (s, 1H), 10.01 (s, 1H), 8.59 – 8.55 (m, 1H), 8.53 (d, J= 5.6 Hz, 1H), 8.32 (s, 1H), 8.23 (d, J= 8.0 Hz, 1H), 7.96 – 7.86 (m, 2H), 7.70 – 7.60 (m, 2H), 7.56 – 7.43 (m, 2H), 7.20 – 7.11 (m, 2H), 6.66 (d, J= 5.6 Hz, 1H), 3.78 (s, 2H), 3.41 (t, J= 5.6 Hz, 2H), 3.25 (s, 3H), 2.66 (t, J= 5.6 Hz, 2H), 1.48 (s, 4H)ppm. MS: M/e 630 (M+l)+.
EXAMPLE 2
Preparation of Crystalline Form D of N-(3-fluoro-4-((2-(5-(((2- methoxyethyl)amino)methyl)pyridin-2-yl)thieno[3,2-b]pyridin-7-yl)oxy)phenyl)-N-(4- fluorophenyl)cyclopropane-l, 1-dicarboxamide
EXAMPLE 2A: Preparation of Compound 1 Crystalline Form D
[0104] To a 50 L reactor, 7.15 Kg of Compound 1, 40 g of Form D as crystal seed and 21 L acetone (>99%) were added. The mixture was heated to reflux ( ~56 °C) for 1~2 h. The mixture was agitated with an internal temperature of 20±5 °C for at least 24 h. Then, the suspension was filtered and washed the filter cake with 7 L acetone. The wet cake was dried under vacuum at <45 °C, to obtain 5.33 kg of Compound 1 of desired Form D
[0105] X-Ray Powder Diffraction (XRPD)
The XRPD patterns were collected with a PAN alytical X’ Pert PRO MPD diffractometer using auincident beam of Cu radiation produced using au Optix long, fine-focus source. An elliptically graded multilayer mirror was used to focus Cu Ka X -rays through the specimens and onto the detector. Prior to the analysis, a silicon specimen (NIST SRM 640e) was analyzed to verify the observed position of the Si Ill peak is consistent with the NIST-certified position. A specimen of each sample was sandwiched between 3 -pm -thick films and analyzed in transmission geometly. A beam-stop, short autiscatter extension, and an autiscatter knife edge were used to minimize the background generated by air. Sober slits for the incident aud diffracted beauls were used to minimize broadening from axial divergence. The diffraction patterns were collected using a scanning position-sensitive detector (X’Celerator) located 240 mm from the specimens and Data Collector software v. 2.2b. Pattern Match v2.3.6 was used to create XRPD patterns.
[0106] The X-ray powder diffraction (XRPD) pattern was used to characterize the Compound 1 obtained, which showed that the Compound 1 was in Crystalline Form D of Compound 1 (Compound 1 Form D), see Figure 1A. The XRPD pattern yielded is substantially the same as that shown in Figure 3C.
[0107] Differential Scanning Calorimetry (DSC)
[0108] DSC was performed using a Mettler-Toledo DSC3+ differential scanning calorimeter. Temperature calibration was performed using octane, phenyl salicylate, indium, tin, and zinc. The TAWN sensitivity was 11.9. The samples were placed into aluminum DSC pans, covered with lids, and the weights were accurately recorded. A weighed aluminum pan configured as the sample pan was placed on the reference side of the cell. The pan lids were pierced prior to sample analyses. The method name on the thermograms is an abbreviation for the start and end temperature as well as the heating rate; e.g., -30-250-10 means “from ambient to 250°C, at 10°C/min.” The nitrogen flow rate was 50.0 mL/min. This instrument does not provide gas pressure value as required by USP because it is the same as atmospheric pressure.
[0109] A broad small endotherm with a peak maximum at approximately 57°C to 62°C (onset ~20°C to 22°C) followed by a sharp endotherm with a peak maximum at approximately 180°C (onset ~178°C) were observed. These events could be due to the loss of volatiles and a melt, respectively (see Figure IB).
[0110] In an alternative embodiment Form D was prepared as follows. Designated Material O was suspended in 600 pL of acetone. Initial dissolution was observed followed by re precipitation. The amount of suspended solids was not measured because the target of the experiment was to get a suspension with enough solids to slurry isolate and collect XRPD data. Based on the solubility of Form D in acetone a very rough estimate for the scale of the experiment is about 80-100mg. The suspension was stirred at ambient temperature for approximately 2 5 weeks after which the solids were isolated by centrifugation with filtration. XRPD data appeared to be consistent with Form D The sample was then dried in vacuum oven at ~40 °C for ~2 5 hours. The XRPD pattern of the final solids was consistent with Form D EXAMPLE 2B: Preparation of Compound 1 Form D
[0111] 427.0 mg of Compound 1 was dissolved in 5 mL of THF to obtain a clear brown solution. The resulting solution was filtered, and the filtrate evaporated under flow of nitrogen. A sticky solid was obtained, which was dried under vacuum in room temperature for ~5 min, still a sticky brown solid obtained. It was dissolved in 0.2 mL of EtOAc and sonicated to dissolve. The solution obtained was stirred at room temperature for 15 min and a solid precipitated. The resulting solid was added 0.4 mL of EtOAc and stirred in room temperature for 21 h 40 min to ontian a suspension. The solid was spparated from mother liquor by centrifugation, then the resulting solid was resuspended the in 0.6 mL of EtOAc and stirred in room temperature for 2 days. The solid was isolated by centrifugation, to obtain Compound 1 of desired Form D.
[0112] The X-ray powder diffraction (XRPD) pattern was used to characterize the Compound 1 obtained, which showed that the Compound 1 was in Crystalline Form D of Compound 1 (Compound 1 Form D).
EXAMPLE 2C: Preparation of Compound 1 Form D
[0113] Single crystal X-ray diffraction data of Compound 1 was collected at 180 K on a Rigaku XtaLAB PRO 007HF(Mo) diffractometer, with Mo Ka radiation (l = 0.71073 A). Data reduction and empirical absorption correction were performed using the CrysAlisPro program. The structure was solved by a dual-space algorithm using SHELXT program. All non-hydrogen atoms could be located directly from the difference Fourier maps. Framework hydrogen atoms were placed geometrically and constrained using the riding model to the parent atoms. Final structure refinement was done using the SHELXL program by minimizing the sum of squared deviations of F2 using a full-matrix technique.
Preparation of Compound 1 Form D ( a Single Crystal )
[0114] Compound 1 Form D was dissolved in a mixture of acetone/ ACN (1/2) with the concentration of Compound 1 at ~7 mg/mL. A block single crystal was obtained, which was a single crystal.
[0115] The XRPD pattern was used to characterize the single crystal of Compound 1 Form D obtained, see Figure 2A. The crystal structural data are summarized in Table IB. The refined single crystal structure were shown in Figure 2B. The single crystal structure of Compound 1 Form D is in the P-1 space group and the triclinic crystal system. The terminal long alkyl chain is found to have large ellipsoids, indicating high mobility with disordered atoms.
[0116] The theoretical XRPD calculated from the single crystal structure and experimental XRPD are essentially similar (Figure 2A). A few small peaks are absent or shift because of orientation preference, disorder and tested temperature (180 K for single crystal data and 293 K for experimental one).
[0117] Table IB. Crystal Data and Structure Refinement for Compound 1 Form D (a Single Crystal)
References
- ^ http://www.mirati.com/go/mgcd516/
- ^ “MGCD516 in Advanced Liposarcoma and Other Soft Tissue Sarcomas – Full Text View – ClinicalTrials.gov”.
- ^ “Phase 2 Study of Glesatinib, Sitravatinib or Mocetinostat in Combination With Nivolumab in Non-Small Cell Lung Cancer – Full Text View – ClinicalTrials.gov”.
- ^ “MGCD516 Combined With Nivolumab in Renal Cell Cancer (RCC) – Full Text View – ClinicalTrials.gov”.
| Identifiers | |
|---|---|
| showIUPAC name | |
| CAS Number | 1123837-84-2 |
| ChemSpider | 52083477 |
| UNII | CWG62Q1VTB |
| KEGG | D11140 |
| Chemical and physical data | |
| Formula | C33H29F2N5O4S |
| Molar mass | 629.68 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESCOCCNCc1ccc(nc1)c2cc3c(s2)c(ccn3)Oc4ccc(cc4F)NC(=O)C5(CC5)C(=O)Nc6ccc(cc6)F | |
| hideInChIInChI=1S/C33H29F2N5O4S/c1-43-15-14-36-18-20-2-8-25(38-19-20)29-17-26-30(45-29)28(10-13-37-26)44-27-9-7-23(16-24(27)35)40-32(42)33(11-12-33)31(41)39-22-5-3-21(34)4-6-22/h2-10,13,16-17,19,36H,11-12,14-15,18H2,1H3,(H,39,41)(H,40,42)Key:WLAVZAAODLTUSW-UHFFFAOYSA-N |
///////////// sitravatinib, phase 3, シトラバチニブ , MGCD516, MG-516, Sitravatinib (MGCD516), UNII-CWG62Q1VTB, CWG62Q1VTB, MGCD-516, ситраватиниб , سيترافاتينيب , 司曲替尼 , Antineoplastic, MGCD 516
#sitravatinib, #phase 3, #シトラバチニブ , #MGCD516, #MG-516#Sitravatinib (MGCD516), #UNII-#CWG62Q1VTB, #CWG62Q1VTB, #MGCD-516, ситраватиниб , سيترافاتينيب , 司曲替尼 , #Antineoplastic, #MGCD516
COCCNCC1=CN=C(C=C1)C2=CC3=NC=CC(=C3S2)OC4=C(C=C(C=C4)NC(=O)C5(CC5)C(=O)NC6=CC=C(C=C6)F)F
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.svg){kind=link}
{kind=link}
_%E2%80%93_(01).jpg){kind=link}
.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
_K_(cropped).jpeg){kind=link}
{kind=link}