
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

PILOCARPINE
PILOCARPINE
- Molecular FormulaC11H16N2O2
- Average mass208.257 Da
2(3H)-Furanone, 3-ethyldihydro-4-[(1-methyl-1H-imidazol-5-yl)methyl]-, (3S-cis)-
202-128-4[EINECS]92-13-7 CAS
54-71-7[RN]
(+)-pilocarpine
(3S,4R)-3-Ethyl-4-[(1-methyl-1H-imidazol-5-yl)methyl]dihydro-2(3H)-furanone
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Pilocarpine hydrochloride | 0WW6D218XJ | 54-71-7 | RNAICSBVACLLGM-GNAZCLTHSA-N |
| Pilocarpine nitrate | M20T465H6J | 148-72-1 | PRZXEPJJHQYOGF-GNAZCLTHSA-N |
PilocarpineCAS Registry Number: 92-13-7
CAS Name: (3S-cis)-3-Ethyldihydro-4-[(1-methyl-1H-imidazol-5-yl)methyl]-2(3H)-furanone
Trademarks: Ocusert Pilo (Cusi)
Molecular Formula: C11H16N2O2, Molecular Weight: 208.26
Percent Composition: C 63.44%, H 7.74%, N 13.45%, O 15.36%
Literature References: Cholinergic principle from Pilocarpus jaborandi Holmes, Rutaceae. Isoln: Petit, Polanovski, Bull. Soc. Chim. [3] 17, 557, 702 (1897). Structure: Jowett, J. Chem. Soc.77, 473, 851 (1900); 83, 438 (1903). Stereoisomeric with isopilocarpine: Polonovski, Polonovski, Bull. Soc. Chim. [4] 31, 1314 (1922). Has the cis configuration; isopilocarpine is trans: Zav’yalov, Dokl. Akad. Nauk SSSR82, 257 (1952). Absolute configuration: Hill, Barcza, Tetrahedron22, 2889 (1966). Synthesis: Preobrashenski et al.,Ber.66, 1187 (1933); Samokhvalov, Med. Prom. SSSR11, no. 2, 10 (1957); DeGraw, Tetrahedron28, 967 (1972); Link, Bernauer, Helv. Chim. Acta55, 1053 (1972). Stereoselective synthesis: A. Noordam et al.,Rec. Trav. Chim.98, 467 (1979). Review: Langenbeck, Angew. Chem.60, 297 (1948); van Rossum et al.,Experientia16, 373 (1960). Toxicity studies: Beccari, Boll. Chim. Farm.106, 8 (1967). Comprehensive description: A. A. Al-Badr, H. Y. Aboul-Enein, Anal. Profiles Drug Subs.12, 385-432 (1983). Clinical trial in Sjögren’s syndrome: F. B. Vivino et al., Arch. Intern. Med.159, 174 (1999); in radiation-induced xerostomia: J.-C. Horiot et al.,Radiother. Oncol.55, 233 (2000).
Properties: Oil or crystals, mp 34°. bp5 260° (partial conversion to isopilocarpine). [a]D18 +106° (c = 2). pK1 (20°) 7.15; pK2 (20°) 12.57. Sol in water, alcohol, chloroform; sparingly sol in ether, benzene. Almost insol in petr ether.
Melting point: mp 34°
Boiling point: bp5 260° (partial conversion to isopilocarpine)
pKa: pK1 (20°) 7.15; pK2 (20°) 12.57
Optical Rotation: [a]D18 +106° (c = 2)
Derivative Type: Hydrochloride
CAS Registry Number: 54-71-7
Trademarks: Akarpine (Akorn); Almocarpine (Ayerst); Isopto Carpine (Alcon); Pilogel (Alcon); Pilopine HS (Alcon); Pilostat (Bausch & Lomb); Salagen (MGI)
Molecular Formula: C11H16N2O2.HCl, Molecular Weight: 244.72
Percent Composition: C 53.99%, H 7.00%, N 11.45%, O 13.08%, Cl 14.49%
Properties: Hygroscopic crystals from alcohol, mp 204-205°. [a]D18 +91° (c = 2). Freely sol in water, alcohol. Practically insol in ether, chloroform. Keep well closed and protected from light.
Melting point: mp 204-205°
Optical Rotation: [a]D18 +91° (c = 2)
Derivative Type: Nitrate
CAS Registry Number: 148-72-1
Trademarks: Chibro Pilocarpine (Chibret); Licarpin (Allergan); Pilo (Novopharma); Pilofrin (Allergan); Pilagan (Allergan)
Molecular Formula: C11H16N2O2.HNO3, Molecular Weight: 271.27
Percent Composition: C 48.70%, H 6.32%, N 15.49%, O 29.49%
Properties: mp 173.5-174.0° (dec). Poisonous! [a]D +77 to +83° (c = 10). One gram dissolves in 4 ml water, 75 ml alcohol. Insol in chloroform, ether. Incompat. Silver nitrate, mercury bichloride, iodides, gold salts, tannin, calomel, KMnO4, alkalies.
Melting point: mp 173.5-174.0° (dec)
Optical Rotation: [a]D +77 to +83° (c = 10)
Derivative Type: Isopilocarpine
Additional Names: b-Pilocarpine
Properties: Hygroscopic oily liquid or prisms. bp10 261°. [a]D18 +50° (c = 2). pK1 (18°) 7.17. Miscible with water and alcohol; very sol in chloroform; less sol in benzene, ether. Almost insol in petr ether.
Boiling point: bp10 261°
pKa: pK1 (18°) 7.17
Optical Rotation: [a]D18 +50° (c = 2)
Derivative Type: Isopilocarpine hydrochloride hemihydrate
Molecular Formula: C11H16N2O2.HCl.½H2O, Molecular Weight: 253.73
Percent Composition: C 52.07%, H 7.15%, N 11.04%, O 15.76%, Cl 13.97%
Properties: Scales from alcohol + ether, mp 127°; when anhydr, mp 161°. [a]D18 +39° (c = 5). Sol in 0.27 part water; 2.1 parts alcohol.
Melting point: mp 127°; mp 161°
Optical Rotation: [a]D18 +39° (c = 5)
Derivative Type: Isopilocarpine nitrate
Molecular Formula: C11H16N2O2.HNO3, Molecular Weight: 271.27Percent Composition: C 48.70%, H 6.32%, N 15.49%, O 29.49%
Properties: Prisms from water, scales from alcohol, mp 159°. [a]D18 +39° (c = 2). Sol in 8.4 parts water, in 350 parts abs alcohol.
Melting point: mp 159°
Optical Rotation: [a]D18 +39° (c = 2)
Therap-Cat: Antiglaucoma agent; miotic; sialogogue.
Therap-Cat-Vet: Parasympathomimetic; miotic; gastric secretory stimulant.
Keywords: Antiglaucoma; Miotic; Sialagogue.
Pilocarpine is a muscarinic cholinergic agonist used on the eye to treat elevated intraocular pressure, various types of glaucoma, and to induce miosis. Also available orally to treat symptoms of dry mouth associated with Sjogren’s syndrome and radiotherapy.
Pilocarpine is a medication used to reduce pressure inside the eye and treat dry mouth.[1][3] As eye drops it is used to manage angle closure glaucoma until surgery can be performed, ocular hypertension, primary open angle glaucoma, and to bring about constriction of the pupil following its dilation.[1][4][5] However, due to its side effects it is no longer typically used in the long term management.[6] Onset of effects with the drops is typically within an hour and lasts for up to a day.[1] By mouth it is used for dry mouth as a result of Sjögren syndrome or radiation therapy.[7]
Common side effects of the eye drops include irritation of the eye, increased tearing, headache, and blurry vision.[1] Other side effects include allergic reactions and retinal detachment.[1] Use is generally not recommended during pregnancy.[8] Pilocarpine is in the miotics family of medication.[9] It works by activating cholinergic receptors of the muscarinic type which cause the trabecular meshwork to open and the aqueous humor to drain from the eye.[1]
Pilocarpine was isolated in 1874 by Hardy and Gerrard and has been used to treat glaucoma for more than 100 years.[10][11][12] It is on the World Health Organization’s List of Essential Medicines.[13] It was originally made from the South American plant Pilocarpus.[10]

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////////////////////////////
Pilocarpine hydrochloride, KSS-694, MGI-647, Pilobuc, Pilocar, Isopto carpine, Spersacarpin, Pilo, Isopto-pilocarpine, Pilocarpina lux, Pilogel, PilaSite(sustained release), Salagen, Pilopine HS
SYN
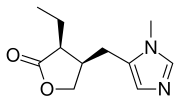
The alkylation of pilosine (I) with ethyl chloride (II) by means of LDA in THF gives trans-pilocarpine (III), which is isomerized with LDA in THF, yielding a mixture of cis- and trans-pilocarpine (IV). Finally, this mixture is resolved by crystallization with di-p-toluoyl tartaric acid.
SYN
Journal of Organic Chemistry, 58(1), 62-4; 1993
https://pubs.acs.org/doi/abs/10.1021/jo00053a016
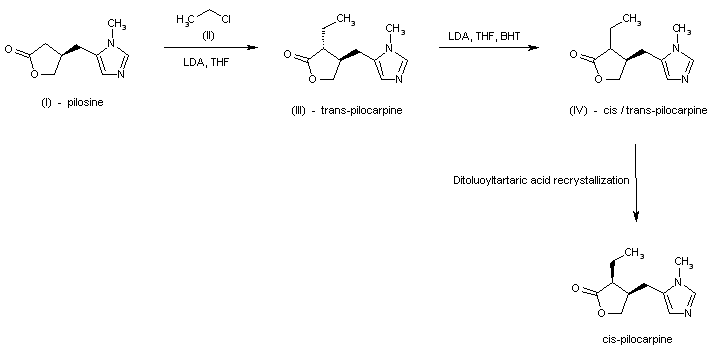
SYN
Tetrahedron, 65(39), 8283-8296; 2009
SYN
Science of Synthesis, 20b, 987-1046; 2006
SYN
https://linkinghub.elsevier.com/retrieve/pii/S0040402008014002
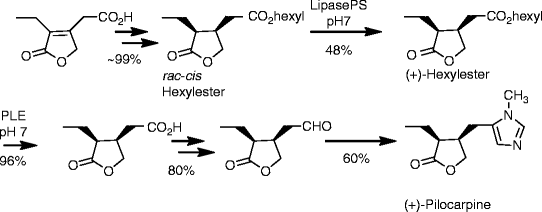

SYN
https://www.mdpi.com/1420-3049/26/12/3676/htm
Schmidt, Theresa et alFrom Molecules, 26(12), 3676; 2021
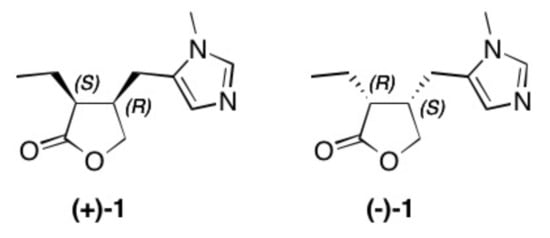
Figure 1. Structure of natural occurring pilocarpine (+)-1 and its enantiomer (–)-1.
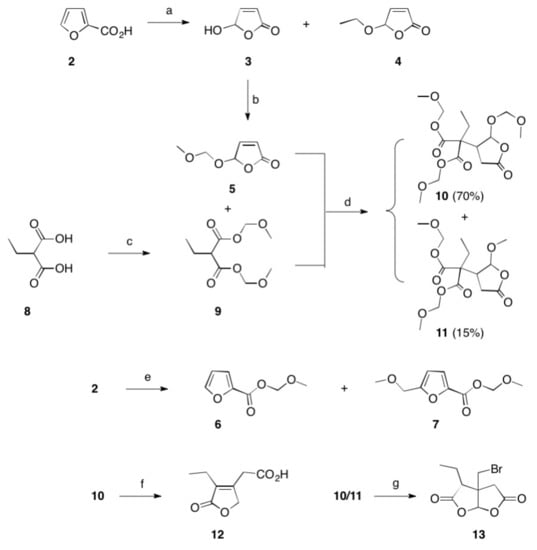
Scheme 1. Reactions and conditions: (a) hν, Bengal rosa, 8 h, 20 °C, 76% (of 3) and 5% (of 4); (b) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 98%; (c) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 99%; (d) THF, Na, 25 °C, 15 h, 72%; (e) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 77% (of 6) and 19% (of 7); (f) HBr, reflux, 2 d, 83%; (g) HBr, reflux, 4 d, 4%.
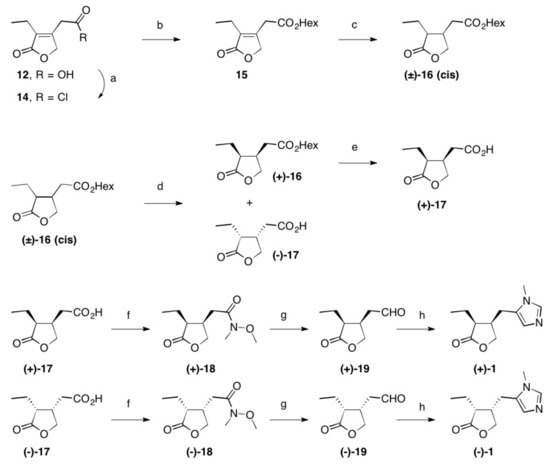
Scheme 2. Reactions and conditions: (a) SOCl2, reflux, 3 h, quant.; (b) Hex-OH, reflux, 16 h, 98%; (c) Rh/Al2O3, H2 (1 at), THF, 5 d, quant.; (d) Lipase PS, pH = 7.0, 2 d, 22 °C, 48% (of (±)-16) and 42% (of (–)-17); (e) PLE, pH = 7.0, 22 °C, 2 d, 96%; (f) N-methylmorpholine, iBu-chloroformate, N,O-dimethylhydroxylamine hydrochloride, 23 °C, 1 d, 84% (of (+)-18) and 85% of (–)-18); (g) LiAlH4, Et2O, 23 °C, 30 min, 95% (of (+)-19) and 95% of (–)-19; (h) CH3NH2, TosMic, DCM, benzene, NEt3, 7 d, 23 °C, 59% (of (+1)-1 and 60% of (–)-1; Hex stands for n-hexyl.
(+)-Pilocarpine [(+)-1]
Following the procedure given for the synthesis of its enantiomer, (+)-1 (1.92 g, 59%) was obtained as a colorless oil; Rf = 0.60 (SiO2, DCM/MeOH/aq NH4OH (25%), 95:4:1); [α]D = +115.7° (c 0.6, CHCl3), ee > 99% (by HPLC, Chiralcel OC, n-hexane/ethanol, 3:7, 0.3 mL/min, UV-detection λ = 215 nm; tR = (+)-1 47.1 min, tR = (–)-1 = 52.32 min); IR (film), 1H-NMR, 13C-NMR and MS (ESI, MeOH) were identical to the enantiomer (vide supra); analysis calcd. for C11H16N2O2 (208.26): C 63.44, H 7.74, N 13.45; found: C 63.31, H 7.98, N 13.32
PAPERBy Fuerstner, AloisFrom e-EROS Encyclopedia of Reagents for Organic Synthesis, 1-7; 2001
| Clinical data | |
|---|---|
| Trade names | Isopto Carpine, Salagen, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a608039 |
| Pregnancy category | AU: B3 |
| Routes of administration | Topical eye drops, by mouth |
| Drug class | Miotic (cholinergic)[1] |
| ATC code | N07AX01 (WHO) S01EB01 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Elimination half-life | 0.76 hours (5 mg), 1.35 hours (10 mg)[2] |
| Excretion | urine |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 92-13-7 54-71-7 (hydrochloride) |
| PubChem CID | 5910 |
| IUPHAR/BPS | 305 |
| DrugBank | DB01085 |
| ChemSpider | 5699 |
| UNII | 01MI4Q9DI3 |
| KEGG | D00525 |
| ChEBI | CHEBI:8207 |
| ChEMBL | ChEMBL550 |
| CompTox Dashboard (EPA) | DTXSID1021162 |
| ECHA InfoCard | 100.001.936 |
| Chemical and physical data | |
| Formula | C11H16N2O2 |
| Molar mass | 208.261 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
Medical uses
Pilocarpine stimulates the secretion of large amounts of saliva and sweat.[14] It is used to prevent or treat dry mouth, particularly in Sjögren syndrome, but also as a side effect of radiation therapy for head and neck cancer.[15]
It may be used to help differentiate Adie syndrome from other causes of unequal pupil size.[16][17][clarification needed]
It may be used to treat a form of dry eye called aqueous deficient dry eye (ADDE)[18]
Surgery
Pilocarpine is sometimes used immediately before certain types of corneal grafts and cataract surgery.[19][20] In ophthalmology, pilocarpine is also used to reduce symptomatic glare at night from lights when the patient has undergone implantation of phakic intraocular lenses; the use of pilocarpine would reduce the size of the pupils, partially relieving these symptoms.[dubious – discuss] The most common concentration for this use is pilocarpine 1%.[citation needed] Pilocarpine is shown to be just as effective as apraclonidine in preventing intraocular pressure spikes after laser trabeculoplasty.[21]
Presbyopia
In 2021, the US Food and Drug Administration approved pilocarpine hydrochloride as an eyedrop treatment for presbyopia, age-related difficulty with near-in vision. Marketed as vuity, the effect lasts for 7 to 10 hours.[22]
Other
Pilocarpine is used to stimulate sweat glands in a sweat test to measure the concentration of chloride and sodium that is excreted in sweat. It is used to diagnose cystic fibrosis.[23]
Adverse effects
Use of pilocarpine may result in a range of adverse effects, most of them related to its non-selective action as a muscarinic receptor agonist. Pilocarpine has been known to cause excessive salivation, sweating, bronchial mucus secretion, bronchospasm, bradycardia, vasodilation, and diarrhea. Eye drops can result in brow ache and chronic use in miosis.
Pharmacology
Pilocarpine is a drug that acts as a muscarinic receptor agonist. It acts on a subtype of muscarinic receptor (M3) found on the iris sphincter muscle, causing the muscle to contract – resulting in pupil constriction (miosis). Pilocarpine also acts on the ciliary muscle and causes it to contract. When the ciliary muscle contracts, it opens the trabecular meshwork through increased tension on the scleral spur. This action facilitates the rate that aqueous humor leaves the eye to decrease intraocular pressure. Paradoxically, when pilocarpine induces this ciliary muscle contraction (known as an accommodative spasm) it causes the eye’s lens to thicken and move forward within the eye. This movement causes the iris (which is located immediately in front of the lens) to also move forward, narrowing the Anterior chamber angle. Narrowing of the anterior chamber angle increases the risk of increased intraocular pressure.[24]
Society and culture
Preparation
Plants in the genus Pilocarpus are the only known sources of pilocarpine, and commercial production is derived entirely from the leaves of Pilocarpus microphyllus (Maranham Jaborandi). This genus grows only in South America, and Pilocarpus microphyllus is native to several states in northern Brazil.[25]
Pilocarpine is extracted from the powdered leaf material in a multi-step process. First the material is treated with ethanol acidified with hydrochloric acid, and the solvents removed under reduced pressure. The resultant aqueous residue is neutralized with ammonia and put aside until the resin has completely settled. It is then filtered and concentrated by sugar solution to a small volume, made alkaline with ammonia, and finally extracted with chloroform. The solvent is removed under reduced pressure.[verification needed]
Cost
Pilocarpine is one of the lowest cost medications for glaucoma.[26]
Trade names
Pilocarpine is available under several trade names such as: Diocarpine (Dioptic), Isopto Carpine (Alcon), Miocarpine (CIBA Vision), Ocusert Pilo-20 and -40 (Alza), Pilopine HS (Alcon), Salagen (MGI Pharma), Scheinpharm Pilocarpine (Schein Pharmaceutical), Timpilo (Merck Frosst) and Vuity (Abbvie).
Research
Pilocarpine is used to induce chronic epilepsy in rodents, commonly rats, as a means to study the disorder’s physiology and to examine different treatments.[27][28] Smaller doses may be used to induce salivation in order to collect samples of saliva, for instance, to obtain information about IgA antibodies.
Veterinary
Pilocarpine is given in moderate doses (about 2 mg) to induce emesis in cats that have ingested foreign plants, foods, or drugs. One feline trial determined it was effective, even though the usual choice of emetic is xylazine.
References
- ^ Jump up to:a b c d e f g “Pilocarpine”. The American Society of Health-System Pharmacists. Archived from the original on 28 December 2016. Retrieved 8 December 2016.
- ^ Gornitsky M, Shenouda G, Sultanem K, Katz H, Hier M, Black M, Velly AM (July 2004). “Double-blind randomized, placebo-controlled study of pilocarpine to salvage salivary gland function during radiotherapy of patients with head and neck cancer”. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 98 (1): 45–52. doi:10.1016/j.tripleo.2004.04.009. PMID 15243470.
- ^ Tarascon Pocket Pharmacopoeia 2019 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. 2018. p. 224. ISBN 9781284167542.
- ^ World Health Organization (2009). Stuart MC, Kouimtzi M, Hill SR (eds.). WHO Model Formulary 2008. World Health Organization. p. 439. hdl:10665/44053. ISBN 9789241547659.
- ^ “Glaucoma and ocular hypertension. NICE guideline 81”. National Institute for Health and Care Excellence. November 2017. Retrieved 19 September 2019.
Ocular hypertension… alternative options include carbonic anhydrase inhibitors such as brinzolamide or dorzolamide, a topical sympathomimetic such as apraclonidine or brimonidine tartrate, or a topical miotic such as pilocarpine, given either as monotherapy or as combination therapy.
- ^ Lusthaus J, Goldberg I (March 2019). “Current management of glaucoma” (PDF). The Medical Journal of Australia. 210 (4): 180–187. doi:10.5694/mja2.50020. PMID 30767238. S2CID 73438590.
Pilocarpine is no longer routinely used for long term IOP control due to a poor side effect profile
- ^ Hamilton R (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 415. ISBN 9781284057560.
- ^ “Pilocarpine ophthalmic Use During Pregnancy | Drugs.com”. http://www.drugs.com. Archived from the original on 28 December 2016. Retrieved 28 December 2016.
- ^ British national formulary : BNF 69 (69 ed.). British Medical Association. 2015. p. 769. ISBN 9780857111562.
- ^ Jump up to:a b Sneader W (2005). Drug Discovery: A History. John Wiley & Sons. p. 98. ISBN 978-0-471-89979-2. Archived from the original on 2016-12-29.
- ^ Rosin A (1991). “[Pilocarpine. A miotic of choice in the treatment of glaucoma has passed 110 years of use]”. Oftalmologia (in Romanian). 35 (1): 53–5. PMID 1811739.
- ^ Holmstedt, B; Wassén, SH; Schultes, RE (January 1979). “Jaborandi: an interdisciplinary appraisal”. Journal of Ethnopharmacology. 1 (1): 3–21. doi:10.1016/0378-8741(79)90014-x. PMID 397371.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “Pilocarpine”. MedLinePlus. U.S. National Library of Medicine. Archived from the original on 2010-03-06.
- ^ Yang, WF; Liao, GQ; Hakim, SG; Ouyang, DQ; Ringash, J; Su, YX (1 March 2016). “Is Pilocarpine Effective in Preventing Radiation-Induced Xerostomia? A Systematic Review and Meta-analysis”. International Journal of Radiation Oncology, Biology, Physics. 94 (3): 503–11. doi:10.1016/j.ijrobp.2015.11.012. hdl:10722/229069. PMID 26867879.
- ^ Kanski JJ, Bowling B (2015-03-24). Kanski’s Clinical Ophthalmology E-Book: A Systematic Approach. Elsevier Health Sciences. p. 812. ISBN 9780702055744.
- ^ Bartlett JD, James SD (October 2013). “Drug Affect the Autonomous Nervous System”. Clinical Ocular Pharmacology. Elsevier. p. 118. ISBN 9781483193915.
- ^ Mannis, Mark J; Holland, Edward J (September 2016). “Chapter 33: Dry Eye”. Cornea E-Book. Elsevier Health Sciences. p. 388. ISBN 978-0-323-35758-6. OCLC 960165358.
- ^ Parker, Jack (2017). Descemet Membrane Endothelial Keratoplasty (DMEK): A Review (PDF) (Thesis). Leiden University.
- ^ Ahmed E, E A (2010). Comprehensive Manual of Ophthalmology. JP Medical Ltd. p. 345. ISBN 9789350251751.
- ^ Zhang L, Weizer JS, Musch DC (February 2017). “Perioperative medications for preventing temporarily increased intraocular pressure after laser trabeculoplasty”. The Cochrane Database of Systematic Reviews. 2 (2): CD010746. doi:10.1002/14651858.CD010746.pub2. PMC 5477062. PMID 28231380.
- ^ Bankhead, Charles (2021-11-01). “First Eye Drop Treatment for Presbyopia Wins FDA Approval”. http://www.medpagetoday.com. Retrieved 2021-11-02.
- ^ Prasad RK (2017-07-11). Chemistry and Synthesis of Medicinal Agents: (Expanding Knowledge of Drug Chemistry). BookRix. ISBN 9783743821415.
- ^ Shaarawy TM, Sherwood MB, Hitchings RA, Crowston JG (September 2014). “Lsser Peripheral Iridoplasty”. Glaucoma E-Book. Elsevier Health Sciences. p. 718. ISBN 9780702055416.
- ^ De Abreu IN, Sawaya AC, Eberlin MN, Mazzafera P (November–December 2005). “Production of Pilocarpine in Callus of Jaborandi (Pilocarpus microphyllus Stapf)”. In Vitro Cellular & Developmental Biology – Plant. Society for In Vitro Biology. 41 (6): 806–811. doi:10.1079/IVP2005711. JSTOR 4293939. S2CID 26058596.
- ^ Schwab, Larry (2007). Eye Care in Developing Nations. CRC Press. p. 110. ISBN 9781840765229.
- ^ Károly N (2018). Immunohistochemical investigations of the neuronal changes induced by chronic recurrent seizures in a pilocarpine rodent model of temporal lobe epilepsy (Thesis). University of Szeged. doi:10.14232/phd.9734.
- ^ Morimoto K, Fahnestock M, Racine RJ (May 2004). “Kindling and status epilepticus models of epilepsy: rewiring the brain”. Progress in Neurobiology. 73 (1): 1–60. doi:10.1016/j.pneurobio.2004.03.009. PMID 15193778. S2CID 36849482.
External links
- “Pilocarpine”. Drug Information Portal. U.S. National Library of Medicine.
CLIP
Firms Team Up To Sustain Natural Pilocarpine
Sustainable harvest is key to a new pharmaceutical chemicals venture
https://cen.acs.org/articles/93/i11/Firms-Team-Sustain-Natural-Pilocarpine.html
Last summer, Andrew Badrot bought a portfolio of plant-sourced pharmaceutical chemicals from Boehringer Ingelheim and acquired BI’s distribution rights for pilocarpine, a plant-derived glaucoma treatment.
For BI, the transactions were small ones. The German drugmaker had been exiting its private-label active pharmaceutical ingredients (API) business, scaling back to produce only the chemicals it uses to manufacture its own drugs.
But for Badrot the deals were potentially big. He leads the company that bought the businesses—Centroflora CMS, a joint venture between the Brazilian botanicals firm Centroflora and CMS Pharma, Badrot’s custom chemicals consultancy. Together, Centroflora and Centroflora CMS are committed to nurturing the natural source of pilocarpine, an alkaloid used medicinally for more than 100 years, and to expanding into other APIs neglected by larger firms.
Pilocarpine’s source, Pilocarpus microphyllus, better known as jaborandi, had been harvested vigorously in the wild by Merck KGaA, which in 1975 built a factory in Parnaíba in northern Brazil to extract pilocarpine. By the mid-1980s, however, jaborandi had been overharvested, and the government declared it a protected species. Merck began obtaining the leaves from a plantation in the northern Brazilian state of Maranhão.
Demand for the drug as a glaucoma treatment began to decline, and Merck eventually closed the plant. When the market for the drug revived with new indications as a dry-mouth remedy, the company saw an opportunity to sell the site and did so in 2002.
The buyer was Centroflora, which was founded in 1957 in São Paulo. The firm was interested in adding pilocarpine to its botanical extracts business, according to its chief executive, Peter Andersen, a native of Brazil whose coffee-trader father bought into Centroflora in 1983. Along with the purchase, Centroflora signed a deal for BI to distribute the drug.
The company wanted to revitalize natural harvesting of jaborandi and began working with the Brazilian government to promulgate sustainable practices in the field. Centroflora also worked closely with a German government agency, Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ), which promotes sustainable harvesting internationally and had been working in the north of Brazil for decades.
Centroflora’s distribution agreement with BI arose through connections at GIZ, according to Andersen. BI also had been Merck’s biggest customer for pilocarpine.
But ecological sustainability was only half of the problem, Andersen says. Centroflora also found itself dealing with middlemen who would collect the jaborandi from poor family farms in remote areas and pay them next to nothing. Establishing a direct supply channel was not easy.

“I can spend a few days telling you about that process,” he says. “Stories of difficult relationships and difficult moments. But in some cases we managed to hire some of the middlemen to work for us on a salary basis. They made less money, but they had a job.”
Today, farmers in Brazil are paid at least twice what they were paid by intermediaries, Andersen says.
Key to the process was a program Centroflora launched in 2004 called Partnerships for a Better World to train and certify growers, establish community associations to support growers, and maintain sustainable harvesting practices.
Centroflora is the leading supplier of pilocarpine. Its only competitor, Sourcetech, with a plant near São Paulo, accesses jaborandi from the plantation that supplied Merck, now owned by U.S.-based Quercegen.
Pilocarpine accounts for only about 5% of Centroflora’s $95 million in annual sales. The company produces a long list of botanical extracts, including nutritional supplements and herbal medicines such as acai, acerola, coffee powder, and powdered fruit.The company manufactures at four facilities in Brazil, including the former Merck plant, which is dedicated to pilocarpine. But Andersen sees the partnership with CMS as a route to increase phytochemical API manufacturing at that site.
“The facility has the capacity to produce 12 metric tons per year of alkaloids,” Andersen says. It currently makes less than three metric tons. “So there is a lot of space to produce more, and the idea is that we can do some of the APIs we got from Boehringer Ingelheim.”
Those include atropine, digoxin, homatropine, and dihydroergotamine mesylate. Centroflora CMS also obtained distribution rights to BI’s scopolamine N-butyl bromide. All are derived from botanicals harvested on farms around the world.
Badrot was vice president of strategy for Lonza’s exclusive synthesis division before starting CMS in 2010 to consult on manufacturing and mergers and acquisitions in the custom chemicals business. “But for me, the dream was to return to manufacturing APIs,” he says.
The phytochemicals portfolio, including some of the oldest APIs made by BI, for which CMS has done consulting work, seemed like an ideal reentry to manufacturing, according to Badrot. “They are niche products that maybe fly a bit under the radar,” he says. “They seemed to fit us well because we can give them some attention.”
Centroflora CMS’s first order of business, he says, is to establish manufacturing for the BI products, which BI will continue to make until then. Badrot says Centroflora is well suited to manufacture at least the digoxin and atropine, but decisions have not been finalized. The partners will likely use contract manufacturers for some of the products. And Badrot says Centroflora CMS seeks to replicate the kind of deal it has with BI.
“We are looking for other companies with APIs that represent 0–1% of sales, products that lack focus,” he says. “We would take them over.”
Badrot and Andersen say they are also interested in sharing the Partnerships for a Better World program with other companies involved in harvesting natural products. And Centroflora looks for other ways to support its supply chain. Last month, it was approved as a trading member of the Union for Ethical BioTrade, a nonprofit that promotes sustainable development and biodiversity. As a member, Centroflora commits to sustainable sourcing practices and will be required to undergo periodic audits.
Last year, Centroflora received government recognition for its efforts on both the environmental and social fronts. The National Confederation of Industry in Brazil named Centroflora’s jaborandi harvesting program one of the country’s 10 most sustainable business practices. And Banco do Brasil, the national bank, recognized the firm for its work to improve conditions for farmers in the northern forest region of the country.
As the joint venture starts to work with its new portfolio of phytochemicals, both Andersen and Badrot look back at the jaborandi success as the road forward, a template for fostering a plant-based API business that may inspire other companies.
For Andersen, Partnerships for a Better World is an essential foundation of trust for the ecological and socially responsible harvesting of botanicals in Brazil. “There were a lot of problems along the way,” he says. “But we are at peace with it today.”
////////////////PILOCARPINE, Pilocarpine hydrochloride, KSS-694, MGI-647, Pilobuc, Pilocar, Isopto carpine, Spersacarpin, Pilo, Isopto-pilocarpine, Pilocarpina lux, Pilogel, PilaSite(sustained release), Salagen, Pilopine HS
CC[C@H]1[C@@H](CC2=CN=CN2C)COC1=O

NEW DRUG APPROVALS
ONE TIME
$10.00
loteprednol etabonate
loteprednol etabonate
- Molecular FormulaC24H31ClO7
- Average mass466.952 Da
cas 82034-46-6
chloromethyl (8S,9S,10R,11S,13S,14S,17R)-17-ethoxycarbonyloxy-11-hydroxy-10,13-dimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthrene-17-carboxylate(11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxo-androsta-1,4-diene-17-carboxylic acid chloromethyl ester
(11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxoandrosta-1,4-diene-17-carboxylic Acid Chloromethyl Ester
(8S,9S,10R,11S,13S,14S,17R)-17-[(éthoxycarbonyl)oxy]-11-hydroxy-10,13-diméthyl-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodécahydro-3H-cyclopenta[a]phénanthrène-17-carboxylate de chlorométhyle
129260-79-3[RN]
17a-Ethoxycarbonyloxy-D’-cortienic Acid Chloromethyl Ester
82034-46-6[RN]
Androsta-1,4-diene-17-carboxylic acid, 17-((ethoxycarbonyl)oxy)-11-hydroxy-3-oxo-, chloromethyl ester, (11β,17α)-
Androsta-1,4-diene-17-carboxylic acid, 17-[(ethoxycarbonyl)oxy]-11-hydroxy-3-oxo-, chloromethyl ester, (11β,17α)-
Loteprednol Etabonate
CAS Registry Number: 82034-46-6
CAS Name: (11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxoandrosta-1,4-diene-17-carboxylic acid chloromethyl ester
Additional Names: chloromethyl 17a-ethoxycarbonyloxy-11b-hydroxyandrosta-1,4-diene-3-one-17b-carboxylate; 17a-ethoxycarbonyloxy-D¢-cortienic acid chloromethyl ester
Manufacturers’ Codes: CDDD-5604; HGP-1; P-5604
Trademarks: Alrex (Bausch & Lomb); Lotemax (Bausch & Lomb)
Molecular Formula: C24H31ClO7, Molecular Weight: 466.95
Percent Composition: C 61.73%, H 6.69%, Cl 7.59%, O 23.98%
Literature References: Ophthalmic corticosteroid. Prepn: N. S. Bodor, BE889563 (1981 to Otsuka); idem,US4996335 (1991). Physicochemical properties: M. Alberth et al.,J. Biopharm. Sci.2, 115 (1991). HPLC determn in plasma and urine: G. Hochhaus et al.,J. Pharm. Sci.81, 1210 (1992). NMR structural studies: S. Rachwal et al.,Steroids61, 524 (1996); idem et al., ibid. 63, 193 (1998). Metabolism and transdermal permeability: N. Bodor et al.,Pharm. Res.9, 1275 (1992). Evaluation of effect on intraocular pressure: J. D. Bartlett et al.,J. Ocul. Pharmacol.9, 157 (1993). Clinical trial in keratoconjunctivitis sicca: S. C. Pflugfelder et al.,Am. J. Ophthalmol.138, 444 (2004). Review of ophthalmic clinical studies: J. F. Howes, Pharmazie55, 178-183 (2000).
Properties: Crystals from THF + hexane, mp 220.5-223.5°. Soly at 25° (mg/ml): 0.0005 in water; 0.037 in 50% propylene glycol + water. Lipophilicity (log K): 3.04.
Melting point: mp 220.5-223.5°
Therap-Cat: Anti-inflammatory (topical).
Keywords: Glucocorticoid.
Research Code:HGP-1; CDDD-5604; P-5604Trade Name:Lotemax® / Alrex®MOA:CorticosteroidIndication:Acne rosacea; Superficial punctate keratitis; Postoperative inflammation and pain following ocular surgery; Iritis; Herpes zoster keratitis; Allergic conjunctivitis; CyclitisCompany:Bausch & Lomb (Originator)Sales:ATC Code:S01BA14
Loteprednol etabonate was approved by the U.S. Food and Drug Administration (FDA) on Mar 9, 1998. It was developed and marketed as Lotemax® by Bausch & Lomb.
Loteprednol etabonate is a corticosteroid used in ophthalmology. It is indicated for the treatment of steroid responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea and anterior segment of the globe such as allergic conjunctivitis, acne rosacea, superficial punctate keratitis, herpes zoster keratitis, iritis, cyclitis, selected infective conjunctivitides.
Lotemax® is available as drops for ophthalmic use, containing 0.5% of Loteprednol etabonate. The recommended dose is one to two drops into the conjunctival sac of the affected eyes four times daily.
Loteprednol (as the ester loteprednol etabonate) is a corticosteroid used to treat inflammations of the eye. It is marketed by Bausch and Lomb as Lotemax[1] and Loterex.
It was patented in 1980 and approved for medical use in 1998.[2]
Loteprednol Etabonate is the etabonate salt form of loteprednol, an ophthalmic analog of the corticosteroid prednisolone with anti-inflammatory activity. Loteprednol etabonate exerts its effect by interacting with specific intracellular receptors and subsequently binds to DNA to modify gene expression. This results in an induction of the synthesis of certain anti-inflammatory proteins while inhibiting the synthesis of certain inflammatory mediators. Loteprednol etabonate specifically induces phospholipase A2 inhibitory proteins (collectively called lipocortins), which inhibit the release of arachidonic acid, thereby inhibiting the biosynthesis of potent mediators of inflammation, such as prostaglandins and leukotrienes.
Loteprednol etabonate is an etabonate ester, an 11beta-hydroxy steroid, a steroid ester, an organochlorine compound, a steroid acid ester and a 3-oxo-Delta(1),Delta(4)-steroid. It has a role as an anti-inflammatory drug. It derives from a loteprednol.
Loteprednol Etabonate (LE) is a topical corticoid anti-inflammatory. It is used in ophthalmic solution for the treatment of steroid responsive inflammatory conditions of the eye such as allergic conjunctivitis, uveitis, acne rosacea, superficial punctate keratitis, herpes zoster keratitis, iritis, cyclitis, and selected infective conjunctivitides. As a nasal spray, it can be used for the treatment and management of seasonal allergic rhinitis. Most prescription LE products, however, tend to be indicated for the treatment of post-operative inflammation and pain following ocular surgery. A number of such new formulations that have been approved include Kala Pharmaceutical’s Inveltys – the first twice-daily (BID) ocular corticosteroid approved for this indication, designed specifically to enhance patient compliance and simplified dosing compared to all other similar ocular steroids that are dosed four times daily. Moreover, LE was purposefully engineered to be a ‘soft drug’, one that is designed to be active locally at the site of administration and then rapidly metabolized to inactive components after eliciting its actions at the desired location, thereby subsequently minimizing the chance for adverse effects.
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-09-28 | New dosage form | Lotemax | Postoperative inflammation and pain following ocular surgery | Gel | 0.5% | Bausch & Lomb | |
| 2011-04-15 | New dosage form | Lotemax | Postoperative inflammation and pain following ocular surgery | Ointment | 0.5% | Bausch & Lomb | |
| 1998-03-09 | First approval | Lotemax | Allergic conjunctivitis,Acne rosacea,Superficial punctate keratitis,Herpes zoster keratitis,Iritis,Cyclitis | Suspension/ Drops | 0.5% | Bausch & Lomb |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-11-26 | Marketing approval | 露达舒/Lotemax | Allergic conjunctivitis,Acne rosacea,Superficial punctate keratitis,Herpes zoster keratitis,Iritis,Cyclitis,Postoperative inflammation and pain following ocular surgery | Suspension | 滴眼剂,0.5%(2.5ml:12.5mg,5ml:25mg) | Bausch & Lomb | |
| 2011-11-05 | Marketing approval | 露达舒/Lotemax | Allergic conjunctivitis,Acne rosacea,Superficial punctate keratitis,Herpes zoster keratitis,Iritis,Cyclitis,Postoperative inflammation and pain following ocular surgery | Suspension | 滴眼剂,0.5%(2.5ml:12.5mg,5ml:25mg); 滴眼剂,0.5%(10ml:50mg,15ml:75mg) | Bausch & Lomb |
Reference:1. US4710495A / US4996335A.Route 2
Reference:1. CN103183714A.
SYN
doi:10.1016/0960-0760(91)90120-T doi: 10.1016/j.steroids.2011.01.006

| Clinical data | |
|---|---|
| Trade names | Lotemax |
| Other names | 11β,17α,Dihydroxy-21-oxa-21-chloromethylpregna-1,4-diene-3,20-dione 17α-ethylcarbonate |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Routes of administration | Eye drops |
| Drug class | Corticosteroid; glucocorticoid |
| ATC code | S01BA14 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | None |
| Protein binding | 95% |
| Metabolism | Ester hydrolysis |
| Metabolites | Δ1-cortienic acid and its etabonate |
| Onset of action | ≤2 hrs (allergic conjunctivitis) |
| Elimination half-life | 2.8 hrs |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 82034-46-6 |
| PubChem CID | 444025 |
| IUPHAR/BPS | 7085 |
| DrugBank | DB14596 |
| ChemSpider | 392049 |
| UNII | YEH1EZ96K6 |
| KEGG | D01689 |
| ChEBI | CHEBI:31784 |
| ChEMBL | ChEMBL1200865 |
| CompTox Dashboard (EPA) | DTXSID2046468 |
| ECHA InfoCard | 100.167.120 |
| Chemical and physical data | |
| Formula | C24H31ClO7 |
| Molar mass | 466.96 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 220.5 to 223.5 °C (428.9 to 434.3 °F) |
| Solubility in water | 0.0005 mg/mL (20 °C) |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
////////////////////////////////////////
Medical uses
Applications for this drug include the reduction of inflammation after eye surgery,[1] seasonal allergic conjunctivitis, uveitis,[3] as well as chronic forms of keratitis (e.g. adenoviral and Thygeson’s keratitis), vernal keratoconjunctivitis, pingueculitis, and episcleritis.[citation needed]
Contraindications
As corticosteroids are immunosuppressive, loteprednol is contraindicated in patients with viral, fungal or mycobacterial infections of the eye.[1][3][4]
Adverse effects
The most common adverse effects in patients being treated with the gel formulation are anterior chamber inflammation (in 5% of people), eye pain (2%), and foreign body sensation (2%).[5]
Interactions
Because long term use (more than 10 days) can cause increased intraocular pressure, loteprednol may interfere with the treatment of glaucoma. Following ocular administration, the drug is very slowly absorbed into the blood, therefore the blood level is limited to an extremely small concentration, and interactions with drugs taken by mouth or through any route other than topical ophthalmic are very unlikely.[1]
Pharmacology
Mechanism of action
Main article: Glucocorticoid § Mechanism of action
Pharmacokinetics
Neither loteprednol etabonate nor its inactive metabolites Δ1–cortienic acid and Δ1-cortienic acid etabonate are detectable in the bloodstream, even after oral administration. A study with patients receiving loteprednol eye drops over 42 days showed no adrenal suppression, which would be a sign of the drug reaching the bloodstream to a clinically relevant extent.[1]
Steroid receptor affinity was 4.3 times that of dexamethasone in animal studies.[1]
Retrometabolic drug design
Loteprednol etabonate was developed using retrometabolic drug design. It is a so-called soft drug, meaning its structure was designed so that it is predictably metabolised to inactive substances. These metabolites, Δ1-cortienic acid and its etabonate, are derivatives of cortienic acid, itself an inactive metabolite of hydrocortisone.[1][4][6]
- Cortisol, a naturally occurring corticosteroid, known as hydrocortisone when used as a drug
- Δ1-Cortienic acid, inactive metabolite of loteprednol
- Cortienic acid, inactive metabolite of hydrocortisone
Chemistry
Loteprednol etabonate is an ester of loteprednol with etabonate (ethyl carbonate). The pure chemical compound has a melting point between 220.5 °C (428.9 °F) and 223.5 °C (434.3 °F). Its solubility in water is 1:2,000,000,[4] therefore it is formulated for ophthalmic use as either an ointment, a gel, or a suspension.[7]
Loteprednol is a corticosteroid. The ketone side chain of classical corticosteroids such as hydrocortisone is replaced by a cleavable ester, which accounts for the rapid inactivation.[8] (This is not the same as the etabonate ester.)


Loteprednol etabonate
Chemical synthesis
References
- ^ Jump up to:a b c d e f g Haberfeld H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 488. ISBN 9783527607495.
- ^ Jump up to:a b Loteprednol Professional Drug Facts.
- ^ Jump up to:a b c Dinnendahl V, Fricke U (2008). Arzneistoff-Profile (in German). 6 (22 ed.). Eschborn, Germany: Govi Pharmazeutischer Verlag. ISBN 978-3-7741-9846-3.
- ^ “Highlights of Prescribing Information: Lotemax” (PDF). 2012.
- ^ Bodor N, Buchwald P (2002). “Design and development of a soft corticosteroid, loteprednol etabonate”. In Schleimer RP, O’Byrne PM, Szefler SJ, Brattsand R (eds.). Inhaled Steroids in Asthma. Optimizing Effects in the Airways. Lung Biology in Health and Disease. 163. Marcel Dekker, New York. pp. 541–564.
- ^ “Loteprednol (Professional Patient Advice)”. Retrieved October 4, 2018.
- ^ Pavesio CE, Decory HH (April 2008). “Treatment of ocular inflammatory conditions with loteprednol etabonate”. The British Journal of Ophthalmology. 92 (4): 455–9. doi:10.1136/bjo.2007.132621. PMID 18245274. S2CID 25873047.
- ^ Druzgala P, Hochhaus G, Bodor N (February 1991). “Soft drugs–10. Blanching activity and receptor binding affinity of a new type of glucocorticoid: loteprednol etabonate”. The Journal of Steroid Biochemistry and Molecular Biology. 38 (2): 149–54. doi:10.1016/0960-0760(91)90120-T. PMID 2004037. S2CID 27107845.
Further reading
- Stewart R, Horwitz B, Howes J, Novack GD, Hart K (November 1998). “Double-masked, placebo-controlled evaluation of loteprednol etabonate 0.5% for postoperative inflammation. Loteprednol Etabonate Post-operative Inflammation Study Group 1”. Journal of Cataract and Refractive Surgery. 24 (11): 1480–9. doi:10.1016/s0886-3350(98)80170-3. PMID 9818338. S2CID 24423725.
////////////loteprednol etabonate
CCOC(=O)OC1(CCC2C1(CC(C3C2CCC4=CC(=O)C=CC34C)O)C)C(=O)OCCl

NEW DRUG APPROVALS
ONE TIME
$10.00
Regdanvimab

| (Heavy chain) QITLKESGPT LVKPTQTLTL TCSFSGFSLS TSGVGVGWIR QPPGKALEWL ALIDWDDNKY HTTSLKTRLT ISKDTSKNQV VLTMTNMDPV DTATYYCARI PGFLRYRNRY YYYGMDVWGQ GTTVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL YSLSSVVTVP SSSLGTQTYI CNVNHKPSNT KVDKRVEPKS CDKTHTCPPC PAPELLGGPS VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVKFNWYV DGVEVHNAKT KPREEQYNST YRVVSVLTVL HQDWLNGKEY KCKVSNKALP APIEKTISKA KGQPREPQVY TLPPSRDELT KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPVLD SDGSFFLYSK LTVDKSRWQQ GNVFSCSVMH EALHNHYTQK SLSLSPGK (Light chain) ELVLTQPPSV SAAPGQKVTI SCSGSSSNIG NNYVSWYQQL PGTAPKLLIY DNNKRPSGIP DRFSGSKSGT SATLGITGLQ TGDEADYYCG TWDSSLSAGV FGGGTELTVL GQPKAAPSVT LFPPSSEELQ ANKATLVCLI SDFYPGAVTV AWKADGSPVK AGVETTKPSK QSNNKYAASS YLSLTPEQWK SHRSYSCQVT HEGSTVEKTV APTECS (Disulfide bridge: H22-H97, H155-H211, H231-L215, H237-H’237, H240-H’240, H272-H332, H378-H436, H’22-H’97, H’155-H’211, H’231-L’215, H’272-H’332, H’378-H’436, L22-L89, L138-L197, L’22-L’89, L’138-L’197) |
>Regdanvimab light chain: ELVLTQPPSVSAAPGQKVTISCSGSSSNIGNNYVSWYQQLPGTAPKLLIYDNNKRPSGIP DRFSGSKSGTSATLGITGLQTGDEADYYCGTWDSSLSAGVFGGGTELTVLGQPKAAPSVT LFPPSSEELQANKATLVCLISDFYPGAVTVAWKADGSPVKAGVETTKPSKQSNNKYAASS YLSLTPEQWKSHRSYSCQVTHEGSTVEKTVAPTECS
>Regdanvimab heavy chain: QITLKESGPTLVKPTQTLTLTCSFSGFSLSTSGVGVGWIRQPPGKALEWLALIDWDDNKY HTTSLKTRLTISKDTSKNQVVLTMTNMDPVDTATYYCARIPGFLRYRNRYYYYGMDVWGQ GTTVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHT FPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPC PAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKT KPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVY TLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSK LTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Regdanvimab
レグダンビマブ;
EMA APPROVED, 2021/11/12, Regkirona
Treatment of adults with coronavirus disease 2019 (COVID-19)
MONOCLONAL ANTIBODY, ANTI VIRAL, PEPTIDE
CAS: 2444308-95-4, CT-P59
Regdanvimab, sold under the brand name Regkirona, is a human monoclonal antibody used for the treatment of COVID-19.[1] The antibody is directed against the spike protein of SARS-CoV-2. It is developed by Celltrion.[2][3] The medicine is given by infusion (drip) into a vein.[1][4]
The most common side effects include infusion-related reactions, including allergic reactions and anaphylaxis.[1]
Regdanvimab was approved for medical use in the European Union in November 2021.[1]
Regdanvimab is a monoclonal antibody targeted against the SARS-CoV-2 spike protein used to treat patients with COVID-19 who are at risk of progressing to severe COVID-19.
Regdanvimab (CT-P59) is a recombinant human IgG1 monoclonal antibody directed at the receptor binding domain (RBD) of the SARS-CoV-2 spike protein.4 It blocks the interaction between viral spike proteins and angiotensin-converting enzyme 2 (ACE2) that allows for viral entry into the cell, thereby inhibiting the virus’ ability to replicate. Trials investigating the use of regdanvimab as a therapeutic candidate for the treatment of COVID-19 began in mid-2020.1,3 It received its first full approval in South Korea in September 2021,3 followed by the EU in November 2021.5
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Synthesis Reference
Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5.
Celltrion’s Monoclonal Antibody Treatment regdanvimab, Approved by the European Commission for the Treatment of COVID-19
- The European Commission (EC) granted marketing authorisation for Celltrion’s regdanvimab following positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) last week (11/11/2021)
- Celltrion continues to discuss supply agreements with regulatory agencies and contractors in more than 30 countries in Europe, Asia and LATAM to accelerate global access to regdanvimab
- The use of regdanvimab across the Republic of Korea is rapidly increasing to address the ongoing outbreaks
November 14, 2021 08:04 PM Eastern Standard Time
INCHEON, South Korea–(BUSINESS WIRE)–Celltrion Group announced today that the European Commission (EC) has approved Regkirona (regdanvimab, CT-P59), one of the first monoclonal antibody treatments granted marketing authorisation from the European Medicines Agency (EMA). The EC granted marketing authorisation for adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19. The decision from the EC follows a positive opinion by the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on November 11th, 2021.1
“Today’s achievement, coupled with CHMP positive opinion for regdanvimab, underscores our ongoing commitment to addressing the world’s greatest health challenges,” said Dr. HoUng Kim, Ph.D., Head of Medical and Marketing Division at Celltrion Healthcare. “Typically, the recommendations from the CHMP are passed on to the EC for rapid legally binding decisions within a month or two, however, given the unprecedented times, we have received the EC approval within a day. As part of our global efforts to accelerate access, we have been communicating with the governments and contractors in 30 countries in Europe, Asia and LATAM. We will continue working with all key stakeholders to ensure COVID-19 patients around the world have access to safe and effective treatments.”
Monoclonal antibodies are proteins designed to attach to a specific target, in this case the spike protein of SARS-CoV-2, which works to block the path the virus uses to enter human cells. The EC approval is based on the global Phase III clinical trial involving more than 1,315 people to evaluate the efficacy and safety of regdanvimab in 13 countries including the U.S., Spain, and Romania. Data showed regdanvimab significantly reduced the risk of COVID-19 related hospitalisation or death by 72% for patients at high-risk of progressing to severe COVID-19.
Emergency use authorisations are currently in place in Indonesia and Brazil, and the monoclonal antibody treatment is fully approved in the Republic of Korea. In the U.S., regdanvimab has not yet been approved by the Food and Drug Administration (FDA), but the company is in discussion with the FDA to submit applications for an Emergency Use Authorisation (EUA).
As of November 12th, 2021, more than 22,587 people have been treated with regdanvimab in 129 hospitals in the Republic of Korea.
Notes to Editors:
About Celltrion Healthcare
Celltrion Healthcare is committed to delivering innovative and affordable medications to promote patients’ access to advanced therapies. Its products are manufactured at state-of-the-art mammalian cell culture facilities, designed and built to comply with the US FDA cGMP and the EU GMP guidelines. Celltrion Healthcare endeavours to offer high-quality cost-effective solutions through an extensive global network that spans more than 110 different countries. For more information please visit: https://www.celltrionhealthcare.com/en-us.
About regdanvimab (CT-P59)
CT-P59 was identified as a potential treatment for COVID-19 through screening of antibody candidates and selecting those that showed the highest potency in neutralising the SARS-CoV-2 virus. In vitro and in vivo pre- clinical studies showed that CT-P59 strongly binds to SARS-CoV-2 RBD and significantly neutralise the wild type and mutant variants of concern. In in vivo models, CT-P59 effectively reduced the viral load of SARS-CoV-2 and inflammation in lung. Results from the global Phase I and Phase II/III clinical trials of CT-P59 demonstrated a promising safety, tolerability, antiviral effect and efficacy profile in patients with mild-to-moderate symptoms of COVID-19.2 Celltrion also has recently commenced the development of a neutralising antibody cocktail with CT-P59 against new emerging variants of SARS-CoV-2.
Medical uses
In the European Union, regdanvimab is indicated for the treatment of adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19.[1]
Society and culture
Names
Regdanvimab is the proposed international nonproprietary name (pINN).[5]
Legal status
In March 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on regdanvimab.[6][7] In October 2021, the EMA started evaluating an application for marketing authorization for the monoclonal antibody regdanvimab (Regkirona) to treat adults with COVID-19 who do not require supplemental oxygen therapy and who are at increased risk of progressing to severe COVID 19.[8] The applicant is Celltrion Healthcare Hungary Kft.[8] The European Medicines Agency (EMA) concluded that regdanvimab can be used for the treatment of confirmed COVID-19 in adults who do not require supplemental oxygen therapy and who are at high risk of progressing to severe COVID-19.[4]
In November 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) recommended granting a marketing authorization in the European Union for regdanvimab (Regkirona) for the treatment of COVID-19.[9][10] The company that applied for authorization of Regkirona is Celltrion Healthcare Hungary Kft.[10] Regdanvimab was approved for medical use in the European Union in November 2021.[1]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | Regkirona |
| Other names | CT-P59 |
| License data | EU EMA: by INN |
| Routes of administration | Intravenous infusion |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 2444308-95-4 |
| DrugBank | DB16405 |
| UNII | I0BGE6P6I6 |
| KEGG | D12241 |
- Tuccori M, Ferraro S, Convertino I, Cappello E, Valdiserra G, Blandizzi C, Maggi F, Focosi D: Anti-SARS-CoV-2 neutralizing monoclonal antibodies: clinical pipeline. MAbs. 2020 Jan-Dec;12(1):1854149. doi: 10.1080/19420862.2020.1854149. [Article]
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, Jeong JH, Kim M, Kim JI, Kim P, Bae JS, Shim EY, Lee MS, Kim MS, Noh H, Park GS, Park JS, Son D, An Y, Lee JN, Kwon KS, Lee JY, Lee H, Yang JS, Kim KC, Kim SS, Woo HM, Kim JW, Park MS, Yu KM, Kim SM, Kim EH, Park SJ, Jeong ST, Yu CH, Song Y, Gu SH, Oh H, Koo BS, Hong JJ, Ryu CM, Park WB, Oh MD, Choi YK, Lee SY: A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat Commun. 2021 Jan 12;12(1):288. doi: 10.1038/s41467-020-20602-5. [Article]
- Syed YY: Regdanvimab: First Approval. Drugs. 2021 Nov 1. pii: 10.1007/s40265-021-01626-7. doi: 10.1007/s40265-021-01626-7. [Article]
- EMA Summary of Product Characteristics: Regkirona (regdanvimab) concentrate for solution for intravenous infusion [Link]
- EMA COVID-19 News: EMA recommends authorisation of two monoclonal antibody medicines [Link]
- EMA CHMP Assessment Report: Celltrion use of regdanvimab for the treatment of COVID-19 [Link]
- Protein Data Bank: Crystal Structure of COVID-19 virus spike receptor-binding domain complexed with a neutralizing antibody CT-P59 [Link]
References
- ^ Jump up to:a b c d e f g “Regkirona EPAR”. European Medicines Agency. Retrieved 12 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Celltrion Develops Tailored Neutralising Antibody Cocktail Treatment with CT-P59 to Tackle COVID-19 Variant Spread Using Its Antibody Development Platform” (Press release). Celltrion. 11 February 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ “Celltrion Group announces positive top-line efficacy and safety data from global Phase II/III clinical trial of COVID-19 treatment candidate CT-P59” (Press release). Celltrion. 13 January 2021. Retrieved 4 March 2021 – via Business Wire.
- ^ Jump up to:a b “EMA issues advice on use of regdanvimab for treating COVID-19”. European Medicines Agency. 26 March 2021. Retrieved 15 October 2021.
- ^ World Health Organization (2020). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 124 – COVID-19 (special edition)” (PDF). WHO Drug Information. 34 (3): 660–1.
- ^ “EMA starts rolling review of Celltrion antibody regdanvimab for COVID-19” (Press release). European Medicines Agency (EMA). 24 February 2021. Retrieved 4 March 2021.
- ^ “EMA review of regdanvimab for COVID-19 to support national decisions on early use” (Press release). European Medicines Agency (EMA). 2 March 2021. Retrieved 4 March 2021.
- ^ Jump up to:a b “EMA receives application for marketing authorisation Regkirona (regdanvimab) treating patients with COVID-19”. European Medicines Agency. 4 October 2021. Retrieved 15 October 2021.
- ^ “Regkirona: Pending EC decision”. European Medicines Agency. 11 November 2021. Retrieved 11 November 2021.
- ^ Jump up to:a b “COVID-19: EMA recommends authorisation of two monoclonal antibody medicines”. European Medicines Agency (EMA) (Press release). 11 November 2021. Retrieved 11 November 2021.
Further reading
- Kim C, Ryu DK, Lee J, Kim YI, Seo JM, Kim YG, et al. (January 2021). “A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein”. Nature Communications. 12 (1): 288. doi:10.1038/s41467-020-20602-5. PMC 7803729. PMID 33436577.
External links
- “Regdanvimab”. Drug Information Portal. U.S. National Library of Medicine.
///////////Regdanvimab, Regkirona, MONOCLONAL ANTIBODY, ANTI VIRAL, EU 2021, APPROVALS 2021, EMA 2021, COVID 19, CORONAVIRUS, PEPTIDE, レグダンビマブ , CT-P59, CT P59

NEW DRUG APPROVALS
ONE TIME
$10.00
Diroximel fumarate

Diroximel fumarate
ジロキシメルフマル酸エステル;
| Formula |
C11H13NO6
|
|---|---|
| CAS | 1577222-14-0 |
| Mol weight |
255.224
|
2021/11/15 EMA APPROVED, VUMERITY
Treatment of multiple sclerosis
Diroximel fumarate, sold under the brand name Vumerity, is a medication used for the treatment of relapsing forms of multiple sclerosis (MS).[1][3][4]
Diroximel fumarate was approved for medical use in the United States in October 2019,[5] and in the European Union in November 2021.[2]
History
This drug was formulated by Alkermes in collaboration with Biogen.[6]
Society and culture
Legal status
Diroximel fumarate was approved for medical use in the United States in October 2019.[5]
On 16 September 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Vumerity, intended for the treatment of adults with relapsing remitting multiple sclerosis.[7] The applicant for this medicinal product is Biogen Netherlands B.V.[7] It was approved for medical use in the European Union in November 2021.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
US 8669281
https://patents.google.com/patent/US8669281B1/en
PATENT
WO 2014152494
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014152494
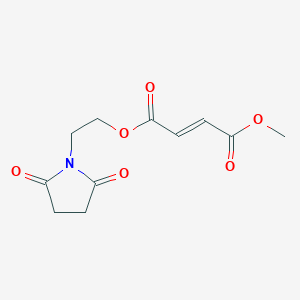
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (14)
2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate 14 was synthesized following general procedure 1 (1.03 g, 35 %).
1H NMR (400 MHz, DMSO): δ 6.81 (2H, dd, J = 15.8 Hz); 4.36 (2H, t, J = 5.3 Hz); 3.84 (2H, t, J = 5.1 Hz); 3.80 (3H, s); 2.73 (4H, s). [M+H]+ = 256.07.
General Procedure 1
To a mixture of monomethyl fumarate (MMF) (1.0 equivalent) and HBTU (1.5 equivalents) in DMF (25 ml per g of MMF) was added Hünigs base (2.0 equivalents). The dark brown solution was stirred for 10 minutes, where turned into a brown suspension, before addition of the alcohol (1.0 – 1.5 equivalents). The reaction was stirred for 18 hours at room temperature. Water was added and the product extracted into ethyl acetate three times. The combined organic layers were washed with water three times, dried with magnesium sulphate, filtered and concentrated in vacuo at 45 ºC to give the crude product. The crude product was purified by silica chromatography and in some cases further purified by trituration with diethyl ether to give the clean desired ester product. All alcohols were either commercially available or made following known literature procedures.
As an alternative to HBTU (N,N,N’,N’-Tetramethyl-O-(1H-benzotriazol-1 -yl)uronium hexafluorophosphate), any one of the following coupling reagents can be used: EDCI/HOBt (N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride/hydroxybenzotriazole hydrate); COMU ((1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate); TBTU (O-(benzotriazol-1 -yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TATU (O-(7-azabenzotriazole-1-yl)-1,1 ,3,3-tetramethyluronium tetrafluoroborate); Oxyma (ethyl (hydroxyimino)cyanoacetate); PyBOP ((benzotriazol-1 -yloxy)tripyrrolidinophosphonium hexafluorophosphate); HOTT (5-(1-oxido-2-pyridyl)-N,N,N’,N’-tetramethylthiuronium hexafluorophosphate); FDPP (pentafluorophenyl diphenylphosphinate); T3P (propylphosphonic anhydride); DMTMM (4-(4,6-dimethoxy-1,3,5-triazin-2-y1)-4-methylmorpholinium tetrafluoroborate); PyOxim ([ethyl
cyano(hydroxyimino)acetato-O2]tri-1-pyrrolidinylphosphonium hexafluorophosphate); TSTU (N,N,N’,N’-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate); TDBTU (O-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-yl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TPTU (O-(2-oxo-1(2H)pyridyl)-N,N,N’,N’-tetramethyluronium tetrafluoroborate); TOTU (O-[(ethoxycarbonyl)cyanomethylenamino]-N,N,N’,N’-tetramethyluronium tetrafluoroborate); IIDQ (isobutyl 1,2-dihydro-2-isobutoxy- 1-quinolinecarboxylate); or PyCIU
(chlorodipyrrolidinocarbenium hexafluorophosphate),
As an alternative to Hünig’s base (diisopropylethylamine), any one of the following amine bases can be used: triethylamine; tributylamine; triphenylamine; pyridine; lutidine (2,6-dimethylpyridine); collidine (2,4,6-trimethylpyridine); imidazole; DMAP (4-(dimethylamino)pyridine); DABCO (1 ,4-diazabicyclo[2.2.2]octane); DBU (1 ,8-
diazabicyclo[5.4.0]undec-7-ene); DBN (1,5-diazabicyclo[4.3.0]non-5-ene); or proton sponge® (N,N,N’,N’-tetramethyl-1 ,8-naphthalenediamine).

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
PATENT
WO 2016124960
PATENT
WO 2017108960
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017108960
Example 3b: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester
Procedure A:
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (3 g; 20.96 mmol) and maleic acid anhydride (2.26 g; 23.1 mmol) in toluene (10 mL) were heated to 60°C under stirring for 29 hours. The temperature was raised to 80°C and heated for another 19 hours. Acetyl chloride (0.3 mL; 4.2 mmol) was added and heating (80°C) was continued for 24 hours. The reaction mixture was cooled to RT. The biphasic system was separated, the upper layer was discarded. The lower layer (viscous oil) crystallized. The crystallized compound was suspended in acetone (50 mL) and stirred for 15 minutes before being filtrated off. The product was dried at 50°C for 5 hours and 8 mbar to yield the 1st crop (1.65 g). The mother liquor was evaporated and the obtained oil/solid was suspended in acetone (5 mL) and stirred overnight at RT. The product was filtrated off and dried at 50°C for 5 hours and 8 mbar to yield the 2nd crop (1.41 g). The mother liquor was evaporated and the obtained oil/solid was suspended in a mixture of diethylether/acetone (5 mL/1 mL) and stirred overnight at RT. The product was filtrated off and dried at 8mbar/50°C for 3 hours (3rd crop, 0.37 g).<a name=”
Yield: 3.43 g (68% of theory)
Purity: 1st crop 96.8 area%; 2nd crop 96.0 area-%; 3rd crop 85.4 area-% (HPLC/UV, method A, λ=200nm; tr: 3.8 min.)
1H NMR (400 MHz, DMSO-d6) δ ppm: 2.61 (s, 4 H) 3.66 (t, J=5.47 Hz, 2 H) 4.23 (t, J=5.47 Hz, 2 H) 6.51 – 6.72 (m, 2 H) 6.60 (s, 1 H) 6.63 (s, 1 H) 13.21 (br s, 1 H)
Procedure B:
Reaction performed in a reactor (Mettler Toledo, Optimax):
Distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (20 g; 0.14 mol) and maleic acid anhydride (15 g; 0.15 mol) in toluene (70 mL) were heated to 80°C under stirring (150 rpm) for 29 hours. Acetyl chloride (2 mL; 0.03 mol) was added and heating (80°C) was continued overnight. Stirring speed was raised to 200 rpm) after 15.5 hours (at 80°C) (product precipitated upon raising stirring speed. The reaction mixture was cooled to 20°C within 1 hour, directly after highering stirring speed. The reaction mixture was stirred for 4 hours, before being filtrated off. The filtrated precipitate was washed with toluene (30 mL) and then with heptane (70 mL), the product was dried at 60°C and 18 mbar. The crude product (26.26 g) with -90% purity was suspended in a mixture of acetone (30 mL)/heptane (30 mL) and stirred at RT for 2 days. The product was filtrated off, washed with heptane (30 mL) and dried at 50°C and 7 mbar.
Yield: 24.12 g (72% of theory)
Purity: 97.4 area-% at 200 nm
Procedure C:
a) Ethylene carbonate (8.89 g; 0.1 mol), succinimide (10 g; 0.1 mol) and sodium carbonate (0.53 g, 5 mmol) were heated to 100°C, the temperature was hold overnight. The product was cooled down yielding a brownish solid (13.73 g) which was grinded in a mortar.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g, 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (33 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (0.5 mL; 7 mmol) was added and heating<a name=”
(80°C) was continued overnight. Heating was stopped and after stirring for another 2 hours the product was filtered off. The product was dried for 2 hours at 60°C and 8 mbar, yielding 15.82 g of crude product.
purity: 63 area-% at 200nm; 80 area-% at 220 nm
Procedure D
a) Ethylene carbonate (44.43 g; 0.5 mol), succinimide (50 g; 0.5 mol) and sodium carbonate (2.67 g; 25 mmol) were heated to 100°C. The reaction mixture was stirred at 100°C for overnight. The mixture was cooled to RT, yielding 72.4 g of the raw product.
40 g of the raw product were suspended in ethylacetate (40 mL) and heated to reflux for 30 minutes. The turbid mixture was cooled to RT and left stirring O/N. The product was filtrated off and dried under vacuum at RT to yield 29.19 g.
b) 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (10 g; 69.9 mmol) from sequence a) and maleic acid anhydride (6.85 g; 69.9 mmol) in toluene (30 mL) were heated to 80°C under stirring. Acetyl chloride (0.5 mL; 7 mmol) was added after 19 hours and heating (80°C) was continued overnight. Heating was stopped and stirring was continued for 2 days. The product was filtrated off and dried at 23 mbar and 60°C.
purity: 82 area% at 200 nm; 91 area-% at 220 nm
Procedure E:
a) Succinimide (500 g; 5.0 mol), ethylene carbonate (444.34 g; 5.0 mol) and sodium carbonate (26.74 g; 0.25 mol) were mixed and slowly heated to 130°C under stirring for 7 hours. The product was distilled via vacuum distillation to yield the product as colourless substance (628.14 g; 87% of theory)
b) The distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (150 g; 1.05 mol) from sequence a) and maleic acid anhydride (102.76 g; 1.05 mol) in toluene (350 mL) were heated to 80°C under stirring for 23 hours. Acetyl chloride (7 mL; 0.01 mol) was added and heating (80°C) was continued. After 6 hours, the reaction mixture was cooled to 20°C within 30 minutes. The product was filtrated off and washed with toluene (200 mL), yielding 221.8 g of a white crystalline product (crude product).
purity: 91 area% at 200 nm; 92 area-% at 220 nm<a name=”
Procedure F:
a) Ethylene carbonate (9.78 g; 0.11 mol), succinimide (10 g; 0.10 mol) and triethylamine (0.7 mL; 5mmol) were heated to 98°C. The reaction mixture was stirred at this temperature overnight. The mixture was cooled to RT, yielding a colourless liquid, which crystallizes upon standing at RT to a colorless solid (14.89 g).
b) The crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione from sequence a) (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (25 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene and dried at 50°C and 8 mbar for 3 hours. Yield: 6.52 g (77%)purity: 93 area% at 200 nm; 94 area-% at 220 nm
Procedure F’
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in a reactor, succinimide (173.07 g, 1.747 mol) and Et3N (12.2 mL, 87.350 mmol) were added and the reaction mixture was warmed up to 90°C and stirred for 24h. Reaction mixture was cooled to 50°C, 500 mL of acetone was added, followed by addition of maleic anhydride (164.19 g, 1.674 mol) and Et3N (10.15 mL, 72.772 mmol). Reaction mixture was stirred at 50-55°C for 4h, cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid.
Yield: 274 g (65%)
Purity: 97.23 area % at 200 nm
Procedure F”
(Z)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (250 g, 1.036 mol) was suspended in acetone (500 mL) in 1-L reactor, acetyl chloride (5.53 mL, 77.736 mmol) was added drop wise at 20-25°C and reaction mixture was warmed up to 50-55°C and stirred for 20h. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone <a name=”(2×50 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II).
Yield: 231.3 g (92.5%)
Purity: 99.47 area % at 200 nm
Summary:
Procedure B and E, using distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, showed purities of -90-91 area-% of the crude product, ongoing crystallization of the target compound could improve the purity to -97% also shown in procedure A. Distillation of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione needs harsh conditions (Ex. 3a; procedure A). Using the crude 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, produced with Na2CO3 lead to low product purities of 63 area-% (procedure C).
Crystallization of 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (procedure D) lead to product purities comparable to procedure A, B and E with distilled 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione, but crystallization is compounded by a significant product loss of – 25%.
The raw 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione could be used without any disadvantageous impact on product quality by substituting Na2CO3 with triethylamine as shown in procedure F with a purity of 93 area-%.
Procedure G
Two experiments were performed in parallel:
Each with 1 g (7 mmol) 1-(2-hydroxy-ethyl)-pyrrolidine-2,5-dione and 0.75 g (7.7 mmol) maleic acid anhydride in 6 mL acetonitrile in screw capped vials. To one of the reaction mixtures was given 0.1 mL triethylamine. Both mixtures were stirred at RT. Samples were taken and investigated by NMR (in DMSO).
product formation after 1 hour (quantified by NMR):
mixture without triethylamine: 0%
mixture with triethylamine: 55%<a name=”
product formation after 2 hours:
mixture without triethylamine: 0%
mixture with triethylamine: 71 %
Procedure H (isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for -24 hours. The reaction was cooled to RT, first a biphasic layer was observed, then the product solidified (sticking to glass wall and stirrer). The product was filtrated off after 2.5 hours of stirring, washed with toluene (50 mL) and dried under vacuum. The dried product was milled and suspended again in toluene (60 mL) at RT, after 30 minutes the product was filtrated off and dried under atmospheric conditions to yield 7.24 g of the cis intermediate (86% of theory). The intermediate product was suspended in toluene (30 mL) and heated to 80°C, acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for 5 hours. The reaction mixture was cooled to RT and stirred for 2 hours. The product was filtrated off, washed with toluene (30 mL) and dried at 50°C and 8 mbar O/N.
purity: 95.6 area-% at 200nm; (0.2% of Impurity I)
Procedure H (without isolation of cis intermediate):
1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione (5 g; 35 mmol) and maleic acid anhydride (3.43 g; 35 mmol) in toluene (30 mL) were heated to 80°C under stirring for 24 hours. Acetyl chloride (0.25 mL; 3.5 mmol) was added and heating (80°C) was continued for ~4 hours. The reaction mixture was cooled to RT. The product was filtrated off washed with toluene (30 mL) and dried at 50°C and 8 mbar for 3 hours.
purity: 93.2 area-% at 200nm; (1.3% of Impurity I)
Procedure I (scale-up without cis isolation)
Maleic acid (959.09 g; 9.8 mol) was added to a reactor under stirring, which was already loaded with toluene (7 L), then 1-(2-Hydroxyethyl)-pyrrolidine-2,5-dione<a name=”
(1400 g; 9.8 mol) was added. Then the mixture was heated to 76°C within ~1 h (up to ~50°C the mixture is a suspension with the tendency of conglomeration of solids, very difficult consistency) at 50°C a turbid solution resulted. Stirring was continued at 80°C for 2 days. Acetyl chloride (138 mL; 1.96 mol) was added under enhanced stirring at 80°C. After -5-10 minutes a crystalline precipitate was formed, which transformed into a pasty/syrupy solid, sticking to reactor walls (difficult handling). Heating was continued overnight (reaction completed after 5 hours as IPC showed). Mixture is still an emulsion, seeding was added and the product precipitated. Stirring at 80°C was continued for ~2 hours then the mixture was cooled to RT. The solid was filtrated off and dried at 50°C and 12 mbar overnight to yield 1818.74 g of the product.
purity: 96.34 area-% at 218 nm; (1.5% of Impurity I)
Procedure J:
2L flask (reaction volume ~1 L): Succinimide (460 g; 4.6 mol), ethylene carbonate (450 g; 5.1 mol) and triethylamine (32 mL; 0.23 mol) were heated to 85°C under stirring overnight. Temperature was raised to 95°-97C and heating was continued O/N. The mixture was cooled to 50°C. Acetonitrile (1600 mL) was charged into a 10 L reactor. To the reaction mixture was added acetonitrile (1000 mL) at 50°C and the solution was transferred to the reactor (reactor T ~22°C), triethylamine (35 mL) was added, then maleic acid anhydride (500.81 g; 5.1 mol). The mixture was heated to 55°C for 5.5 hours. A part of the solvent was distilled off (~1200 mL). Then toluene (1200 mL) was added. The mixture was heated to 90°C. The mixture was cooled to 50°C. At 60°C (clear solution), seeding was added ~300 mg, after -3 minutes a suspension resulted. The mixture was further cooled down to 20°C within 10 hours and kept on stirring O/N. The white crystalline product was filtrated off, washed with toluene (1000 mL) and dried at 55°C and 9 mbar for 2 h to yield 908.99 g (81% yield).
905 g of the isolated, crystallized product was suspended in acetonitrile (2.9 L). Acetyl chloride (23 mL) was added and the mixture was heated to 80°C (clear, colorless solution) for 4 hours. Toluene (1000 mL) was added and the mixture was cooled to RT within 2 hours (linear). The mixture was further cooled to 0°C within 60 minutes. The <a name=”product was filtrated off and washed with toluene (1000 mL). The product was dried overnight at 9 mbar and 50°C.
Yield: 742.06 g (66%)
purity: 99.9 area% at 200 nm
Summary:
Isolation of the cis intermediate leads to a significantly lower content of impurities, in particular of Impurity I. Toluene as solvent leads to disadvantageous conditions regarding consistency of the reaction mixture (procedure H). The use of acetonitrile or acetone (procedure I/F) leads to improved reaction conditions and product quality.
Example 3c
Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) from ethylene carbonate and succinimide (without isolation of
intermediates)
Procedure
Ethylene carbonate (161.50 g, 1.834 mol) was melted at 50°C in an 1-L reactor, succinimide (173.07 g, 1.747 mol) and Et3N (24.4 mL, 0.175 mol) were added and the reaction mixture was warmed up to 90-92°C and stirred for 24h. Distillation column<a name=”
was set up on the reactor and the remaining Et3N was distilled off. Reaction mixture was cooled to 40-45°C, 500 mL of acetone was added, followed by addition of maleic anhydride (184 g, 1.878 mol) and Et3N (10.96 mL, 78.615 mmol). Reaction was stirred at 40°C for 6h (precipitation occurred after 3h), cooled to 20-25°C and acetyl chloride (20.86 mL, 0.293 mol) was added drop wise. Reaction mixture was then warmed up to 50-55°C and stirred for 20h. Orange solution crystallized upon seeding. Reaction mixture was cooled to 0°C and stirred for 3h. Resulting white suspension was filtered off and solid was washed with cold acetone (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford (E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid. Yield: 352.8 g (83.7%)
Purity: 99.69 area % at 200 nm
Example 4: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester
Procedure A:
The starting material (E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (5 g; 20 mmol) was suspended in dichloromethane (60 mL) and cooled to 0°C, triethylamine (3.16 mL; 22.8 mmol) was added, resulting a clear solution. To this solution methylchloroformate (3.3 mL; 20.7 mmol) was carefully added within 30 minutes via syringe (reaction very exothermic). After 15 min of stirring at 0°C, DMAP (0.25 g; 2.1 mmol) was added into the reaction mixture at 0°C, stirring was continued for 3 hours at 0°C. The reaction mixture was poured into water (200 mL) and additional dichloromethane (100 mL) was added. The organic layer was separated and the aqueous layer was extracted once again with dichloromethane (50 mL). The combined organic layers were washed with brine (50 mL). The solvent was evaporated at 52°C. To the brown oil, which solidified, was added acetone (20 mL) and the mixture was stirred overnight. The product was filtrated off (white solid, part I) (2.73 g) and to the mother <a name=”liquor silica was added, the mixture was evaporated. Acetone (50 mL) was added and silica was filtrated off. The solvent was evaporated and diethylether (30 mL) was added to the solid, the mixture was stirred for ~1 hour. The product was filtrated off (part II) (1.6 g).
Overall yield: 4.33 g (82%)
Purity: part I 100 area-% at 200 nm; part II 97.96 area-% at 200 nm
Procedure A’
(E)-4-(2-(2,5-dioxopyrrolidin-1-yl)ethoxy)-4-oxobut-2-enoic acid (Formula II) (200 g, 0.829 mol) was suspended in acetone (2000 mL) in 3-L reactor at 20-25°C and cooled to 0°C. Et3N (150.31 mL, 1.078 mol) was added drop wise at 0-5°C. Into resulting solution, methyl chloroformate (83.27 mL, 1.072 mol) was added drop wise at 0-5°C. Reaction mixture was warmed up to 45°C and stirred for 2h. Upon completion, reaction mixture was cooled to 20-25°C and water (600 mL) was added drop wise with maintaining the temperature at 20-25°C resulting with off white to yellowish solution. pH was adjusted to 7 with 1M HCl. One more volume of water was added and pH corrected if needed. Part of acetone from the reaction mixture (5 volumes or 1000 mL) was distilled off under diminished pressure and reactor walls were washed with 1 more volume of water (200 mL), thus resulting in a solution of acetone/water mixture 1:1 (total 10 volumes). Reaction mixture was gradually cooled to 0°C and stirred for 20h. Resulting white suspension was filtered off and solid was washed with cold water (2×200 mL) and dried for 6h at 50°C and 30 mbar to afford crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (Formula I).
Yield: 183.7 g (86.8%)
Purity: 100.00 area % at 200 nm
Crude 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate (170 g) was suspended in acetone (850 mL) at 20-25°C and warmed up to 50°C resulting with colorless solution. Water (850 mL) was added in portions at 50°C and solution was cooled gradually. Crystallization started at 32°C. Reaction mixture was stirred at crystallization temperature for 30 minutes and cooled further to 0°C, stirred at 0°C for 2h and resulting <a name=”white suspension was filtered off and solid was washed with cold water (2×170 mL) and dried for 6h at 50°C and 30 mbar to afford crystalline 2-(2,5-dioxopyrrolidin-1-yl)ethyl methyl fumarate.
Yield: 152.5 g (89.7%)
Purity: 100.00 area % at 200 nm
Procedure B:
The starting material (5 g, 20 mmol) was suspended in toluene (25 mL). Acetyl chloride (0.29 mL) and methanol (2.5 mL) were added, the reaction mixture was heated to 55°C and stirred for 3 hours. The reaction mixture was poured into water (100 mL) and extracted with ethylacetate (100 mL). The organic layer was separated and dried over sodium sulfate. The solvent was evaporated (crude product 4.7 g, main impurities dimetylfumarate (13%) and fumaric acid (1%) (HPLC at 200 nm)).
Yield: 4.7 g (88%)
Purity: 82.1 area-% at 200 nm
Procedure C:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form A; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (without isolation of (Z)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (30 g; 0.12 mol) was suspended in dichloromethane (DCM, 160 mL) and cooled to 0°C, triethylamine (TEA, 19 mL; 0.14 mol) was added, resulting a clear solution. To this solution methyl chloroformate (19.74 mL; 0.12 mol) was added carefully within 30 minutes via syringe. Stirring was continued for ~2 hours. Water (200 mL) was added to the reaction mixture and stirring was continued for 5-10 minutes. The organic layer was separated and the aqueous layer was washed with another portion of DCM (100 mL). The combined organic layers were dried over sodium sulfate, before being evaporated. To the crude product was added acetone (50 mL) and the mixture was stirred for 3 hours before being filtered off. The product was washed with heptane (50 mL) and dried at 50°C and 21 mbar for 1 h.<a name=”
Yield: 20.52 g (65%)
Purity: 98.7 area-% at 220 nm; (0.3% of Impurity I)
XRPD diffraction peaks: 7.1, 11.6, 13.5, 13.7, 16.3, 16.7, 18.0, 18.4, 21.1, 22.1, 23.1, 23.9, 24.4, 25.5, 27.0, 27.5, 28.0, 28.6, 30.8, 31.2, 31.9, 32.3, 33.7, 34.2, 34.4, 34.9, 35.1, 35.7, 36.0, 36.8, 38.3, 40.1, 40.5, 41.7, 42.4, 43.0, 43.4, 45.0, 45.3, 46.2, 46.4, 47.0, 48.6, 49.4, 49.9, 52.0 + 0.2 degrees two theta.
The Form A according to Procedure C showed a habitus as depicted in Figure 7a
Procedure D:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A The starting material (10 g; 41.5 mmol) was suspended in toluene (70 mL) at 23°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added. Methyl chloroformate (6.58 mL; 41.5 mmol) was slowly added within -30 minutes. After stirring for 2 hours water (40 mL) was added and shortly after acetone (110 mL), stirring was continued for ~2 minutes. The organic layer was separated and washed with brine (15 mL). After drying over sodium sulfate, the solvent was evaporated, yielding a slightly grey solid as crude product (9.42 g). The raw product was suspended in acetone (20 mL) and heptane (20 mL). The mixture was heated to reflux for 15 minutes resulting in a clear solution with just a small amount of solid. The mixture was cooled to RT and stirred overnight (precipitation started at 45°C, cooling: flask left in cooling oil bath ~lh to RT). The resulting product showed polymorphic form A.
Yield: 7.83 g (74%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure D showed a habitus as depicted in Figure 7b<a name=”
Procedure E:
The starting material (1 g; 4.15 mmol) was suspended in dichloromethane (50 mL) at RT. Methyl chloroformate (0.64 mL; 8.3 mmol) was added and stirring was continued overnight, in process control by HPLC showed no conversion.
Procedure F:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form A
The starting material (7 g; 0.03 mol) and Na2CO3 were suspended in ethylacetate (50 mL). To the suspension was added methyl chloroformate (3.37 mL; 0.04 mol) in one portion. The reaction mixture was heated to 70°C. The temperature was kept for 15.5 h. The reaction mixture was cooled to 20°C and ethyl acetate (70 mL) was added to the white suspension. The solids were filtrated off and the ethyl acetate layer was washed with water (40 mL), dried over Na2S04 and evaporated to yield 6.4 g of the white crystalline crude product.
The crude product was suspended in a mixture of ethylacetate (10 mL) and heptane (10 mL). The suspension was heated to reflux for 30 minutes, then cooled to 23°C and stirred overnight. The product was filtrated off and dried at 8 mbar and 50°C overnight.
Yield: 5.62 g (75%)
Purity: 99.4 area-% at 200 nm
The form A according to Procedure E showed a habitus as depicted in Figure 7c
Procedure G:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form B; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B
(A) 9 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methylester was heated to 115°C. The melted compound was stirred for -20 minutes and then dropped into a precooled mortar (0°C).<a name=”
Purity of form B: 98.8 area-% at 200nm
XRPD-pattern: (Figure 4)
(B) 3.00 g of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester (form A) was suspended in 150 mL of dibutyl ether. Suspension was heated to 120° C while mixing. Solution was left at 25°C for 2 days. Crystallized material was filtered and dried at 23 °C at 12 mbar.
XRPD-pattern: (Figure 4′)
A measure of the relative volume change of a solid as a response to pressure change is called compressibility. An API should exhibit good compressibility which is dependent on the polymorphic state.
Experimental data:
The compressibility of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester form B and form A was assessed using a die and a flat-faced punch fitted on a TA-XT2 Texture analyser (Stable Micro Systems Ltd., Godalming, UK). 200 mg of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester sample is compressed in a steel mould (with the rate of displacement 0.03 mm/s). Cyclic procedure (similar to tapping) was performed: compressing, then retracting, relaxation for 15 s and then repeated compressive steps (altogether 10 steps). Each step exerts 0.2 MPa pressure on to the sample. Sample density is calculated by dividing the weight by the sample volume for each cycle. Maximum density is reached within 10 steps. Measurements were performed in duplicates for each sample, results are expressed as an average of duplicate measurements.
Results:
<a name=”
Form B of (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester exhibits a higher density at compression, indicating superior compressibility compared to form A.
Procedure H:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form C; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form C
(E)-But-2-enedioic acid mono-[2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl] ester (10 g; 41.5 mmol) was suspended in dichloromethane (DCM; 100 mL) and cooled to 0°C, triethylamine (TEA; 6.3 mL; 45.6 mmol) was added, resulting a clear solution. To the reaction mixture was added methyl chloroformate (6.58 mL; 41.5 mmol) within 30 minutes via a syringe pump. After 15 min of stirring at 0°C, DMAP (0.51 g; 4 mmol) was added into the reaction mixture at 0°C. The resulting solution was stirred at 0°C for 2.5 hours, then the cold suspension was poured into water (70 mL), the reactor was washed with further DCM (20 mL), which was added also to the DCM/water mixture. The organic layer was separated and washed with HCl (32% aq) (5 mL) in water (60 mL), then with water (50 mL) and finally with brine (50 mL). To the obtained deep red to brown solution was added silica (40-63 um) and the mixture was stirred for 5 minutes, before being filtered off to yield a colorless solution, which was evaporated to yield a colorless oil (crude product). The obtained oil was dissolved in a mixture of ethyl acetate/heptane (1/4) (20 mL). The mixture was stirred for 2 days before being filtered off. The product was dried under vacuum.
Yield: 2.87 g (26%)
Purity: 90.9 area-% at 200 nm
XRPD diffraction peaks: 11.2, 11.8, 13.0, 13.6, 13.6, 16.8, 18.1, 19.6, 20.6, 21.2, 21.5, 22.3, 23.2, 23.7, 24.3, 24.4, 25.2, 25.6, 26.5, 27.6, 28.4, 29.1, 30.3, 31.1, 32.0, 33.1, 33.8, 36.1, 36.7, 37.5, 38.4, 38.9, 41.6, 42.5, 43.2, 44.8, 46.5, 48.7, 49.6, 49.9 + 0.2 degrees two theta.<a name=”
Procedure I:
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester in polymorphic form D; short: (E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form D
(E)-But-2-enedioic acid 2-(2,5-dioxo-pyrrolidin-1-yl)-ethyl ester methyl ester Form B (1 g) was suspended in acetonitrile (3 mL). The suspension was stirred for 7 days in a closed screw cap vials followed by slow evaporation of the solvent under ambient conditions within 3 days.
Purity of form D: 96.3 area-% at 200 nm
XRPD diffraction peaks: 6.9, 11.7, 13.6, 13.9, 16.4, 16.9, 18.2, 20.9, 21.3, 22.3, 23.3, 24.0, 24.6, 25.7, 27.5, 27.7, 31.0, 31.3, 32.1, 32.4, 33.9, 35.3, 35.7, 38.4, 41.9, 42.7, 43.1, 43.6, 44.4, 46.5, 48.9 + 0.2 degrees two theta.
Procedure J:
The starting material (obtained via isolation of (Z)-But-2-enedioic acid mono-[2-(2, 5-dioxo-pyrrolidin-1-yl)-ethyl] ester) (400 g; 1.7 mol) and Na2CO3 (264 g; 2.5 mol) were suspended in ethylacetate (2.7 L). To the suspension was added methyl chloroformate (193 mL; 2.5 mol) at 20°C. The reaction mixture was heated to 45°C within 90 minutes (linear heated). The mixture was kept on stirring for 5.5 hours. Ethylacetate (4 L) was added to the white suspension (at 45 °C). The suspension was stirred for 15 minutes before being filtrated off (45 °C suspension). The reactor was rinsed with another portion of ethylacetate (1 L). The filtrated solids were discarded. To the ethylacetate solution was added a mixture of HClaq (32%) (50 mL) and water (1 L) and the mixture was vigorously stirred for 10 minutes (at 35°C). Then the ethylacetate layer was separated (at ~35°C). The ethylacetate layer was transferred back to the reactor and stirred over sodium sulfate for 30 minutes, sodium sulfate was filtrated off and the ethylacetate layer was reduced to 900 mL. The suspension was transferred into a 3 L flask, equipped with a KPG stirrer and reflux condenser. The mixture was heated to reflux (stirring speed 160 rpm), the suspension was stirred until a clear solution was obtained (-30 minutes). Then heptane (550 mL) was added dropwise within 30 minutes<a name=”
under reflux conditions. Then the mixture (still solution) was slowly cooled to RT. The mixture was stirred O/N. The product was filtrated off and the filter cake was rinsed with heptane (500 mL) to yield the crystalline product (362.56 g; 86%).
purity: 99.8 area% at 218 nm (no Impurity I).
Alternative Procedure: Synthesis of (E)-But-2-enedioic acid 2-(2,5-dioxo- pyrrolidin-1-yl) -ethyl ester methyl ester
Procedure A
Monomethylfumarate (20 g, 0.15 mmol) was suspended in dry dichloromethane (400 mL) at RT, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (32.42 g, 0.17 mol), N-(2-hydroxyethyl)succinimide (21.57 g, 0.15 mol) and dimethylaminopyridine (0.94 g, 7.7 mmol) were added. The solution was stirred O/N at RT. The formed yellow solution was diluted with dichloromethane (300 mL) and washed twice with water (2×500 mL). The organic layer was dried over sodium sulfate and concentrated under reduced pressure. To the crude product was added methyl tert. butyl ether (850 mL) and the reaction mixture was refluxed for 2.5 hours, cooled to RT, then filtrated and heated to reflux again for ~2 hours. After cooling to RT, the mixture was stored at ~5°C for 4 days. The white precipitate was filtrated off and washed with isopropylacetate (25 mL). The crystalline product was dried at 50°C and 7 mbar.
Yield: 10.8 g (28%)
Procedure B
Monomethylfumarate (1.5 g; 11.5 mmol) was suspended in dry DCM (30 mL) at 0°C. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid hydrochloride (2.47 g; 12.8 mmol), N- (2-hydroxyethyl)succinimide (1.62 g; 11.3 mmol) and DMAP (0.07 g; 0.6 mmol) were added. The solution was stirred overnight at RT. The formed yellow solution was diluted with DCM (50 mL) and washed with water twice (2×35 mL). The organic layer<a name=”
was dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-heptane:ethyl acetate 1:1->1:2). The final product showed polymorphic form A. The form A according to alternative Procedure B showed a prismatic habitus as depicted in Figure 7d
Yield: 2.3 g (78%)
Purity: 99.5 area-% at 200 nm
Example 5: Kinetic investigations
Monomethyl maleate was prepared in analogy to WO 2014/197860. Samples of 13.2 grams of monomethyl maleate in 50 mL of toluene and 0.1 equivalents of the isomerization catalyst were reacted at 80°C. Samples were taken after the given times and analyzed by HPLC at 200 nm. The absorbance ratio of monomethyl fumarate (3.8 min.) to monomethyl maleate (2.8 min.) was taken as conversion parameter. The results are shown in Figure 1. As it can be seen from Figure 1 the conversion of monomethyl maleate to monomethyl fumarate in the presence of is TMS (trimethylsilylchloride) is advantageously enhanced compared to the one in the presence of AcCl (acetyl chloride).
Example 6: Yield determination
Six samples of 13.2 g (0.1 mol) monomethyl maleate were diluted with toluene (50 mL) and 0.1 eq of the isomerization catalyst (trimethylsilylchloride or acetyl chloride) were added, three samples with trimethylsilylchloride and three samples with acetyl chloride. The resulting reaction mixtures were heated to the temperatures of 45 °C, 51°C and 80°C. After 22 hours the reaction mixtures were cooled to room temperature, the product was filtrated off and dried at 50°C/8-16 mbar overnight. The results are shown in Figure 2 As it can be seen from Figure 2 the isolated yields of the conversion of monomethyl maleate to monomethyl fumarate in the presence of TMS (trimethylsilylchloride) is at any
PATENT
CN 110698442
PATENT
WO 2021053476
PATENT
IN 201921037120
PATENT
WO 2021074842
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021074842
The drug compound having the adopted name “Diroximel fumarate” has chemical name: 2-(2,5-Dioxopyrrolidin-l-yl)ethyl methyl fumarate as below.
Diroximel fumarate is an investigational, novel oral fumarate with a distinct chemical structure and developed by Alkermes pic, for the treatment of relapsing-remitting multiple sclerosis (RRMS) and is currently under review by U.S. Food and Drug Administration. Biogen, under an exclusive license from Alkermes, intends to market Diroximel fumarate under the brand name VUMERITY™.
US 8669281 B1 first disclosed Diroximel fumarate, its preparation, composition and use thereof for treating multiple sclerosis. US 10080733 B2 further discloses the crystalline solid form of Diroximel fumarate having an X-ray powder diffraction pattern comprising 2Q peaks at 11.6, 21.0, 24.3, 27.4, and 27.9 ±0.2 2Q.
WO 2017/108960 A1 also discloses various alternative synthetic approaches to make Diroximel fumarate and crystalline solid forms thereof, designated as Polymorphic forms A to D.
Hence, there remains a need for alternate solid forms of Diroximel fumarate and preparative processes thereof, exhibiting desired bioavailability and stability. Hence, it is desirable to provide a viable solid form of Diroximel fumarate. The known processes for the preparation of Diroximel fumarate are not viable at industrial scale due to the use of expensive reagents and catalyst such as coupling agents disclosed in US 8669281 Bl, with very low yields. Hence, there remains a need for the improved process to make Diroximel fumarate.
In another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of monomethyl fumarate with (2,5-dioxopyrrolidin-l-yl)ethanol in the presence of an acid halide.
n another aspect, the present application provides a process for the preparation of Diroximel fumarate, comprising the step of esterification of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid with methylation agent selected from the group consisting of 2,2-dimethoxypropane, trimethyl orthoformate and dimethyl carbonate.
Diroximel Fumarate
Example-7: Preparation of l-(2-hydroxyethyl)pyrrolidine-2,5-dione
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 0 °C; methyl /ert-butyl ether (300 mL) was added and the resulting mixture was stirred for 30 minutes at the same temperature. The solid was filtered and dried under vacuum for 5 minutes. The solid was combined with ethyl acetate (100 mL) at 0 °C and stirred at the same temperature for 30 minutes. The solid was filtered and dried in rotatory vacuum dryer at 40 °C for 30 minutes to obtain 142.5 g of the title compound as off-white solid with HPLC purity of 99.6%.
Example-8: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (4.0 g) and dichloromethane (40 mL) at 5 °C, Oxalyl chloride (5.85 g) was added slowly in 10 minutes, then a drop of DMF was added at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (5.06 g) and dichloromethane (35 mL), diisopropylethylamine (DIPEA) (9.93 g) was added and cooled the reaction mixture to 5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane was slowly added to this later mixture at 5 °C for 20 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with saturated ammonium chloride solution and the organic layer was separated. Organic layer was washed with 10% citric acid solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (15 mL) at 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with chilled acetone (3 mL) and then with cyclohexane (4 mL). The wet solid was dried at 40 °C under vacuum to obtain 3.3 g of the title compound with HPLC purity of 99.95 %
Example-9: Preparation of Diroximel fumarate
To a mixture of (E)-4-methoxy-4-oxobut-2-enoic acid (100.0 g) and dichloromethane (1000 mL) at 5 °C, Oxalyl chloride (117 g) was added slowly in 15 minutes, then catalytic DMF (1 mL) was added slowly at the same temperature and allowed the reaction mixture to warm up to 27 °C. After complete evolution of the gas, solvent was evaporated from the reaction mixture. To a mixture of l-(2-hydroxyethyl)pyrrolidine-2,5-dione (110 g) and dichloromethane (900 mL), diisopropylethylamine (DIPEA) (139 g) was added and cooled the reaction mixture to -5 °C. The former mixture of (E)-4-methoxy-4-oxobut-2-enoic acid chloride in dichloromethane (100 mL) was slowly added to this later mixture at – 5 °C for 60 minutes and stirred at the same temperature for 1 hour. The reaction mixture was quenched with water and the organic layer was separated. Organic layer was washed with 10% citric acid solution, 10% NaHC03 solution and then with brine solution. The solvent from the separated organic layer was evaporated completely at 30 °C and the resultant solid was combined with acetone (400 mL) at 27 °C. The reaction mixture was heated to 45 °C and stirred at the same temperature for 1 hour. The mixture was cooled to 27 °C and stirred for 8 hours at the same temperature. The solid was filtered and the cake was washed with methanol (200 mL). The wet solid was dried at 45 °C under vacuum to obtain 120 g of the title compound with HPLC purity of 99.97 %
Example-10: Preparation of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid
A mixture of succinimide (100 g), ethylene carbonate (70.6 mL) and triethylamine (14 mL) was heated to 90 °C and stirred at the same temperature for 24 hours. The reaction mixture was cooled to 50 °C and triethylamine was removed by evaporation under vacuum. The reaction mixture was heated to 90 °C to distill out the traces of triethylamine under vacuum. The reaction mixture was cooled to 40 °C. Acetone (300 mL), maleic anhydride (106.2 g) and triethylamine (6.31 mL) were added. The resulting mixture was stirred at 40 °C for 6 hours. The mixture was cooled to 20 °C and acetyl chloride (12 mL) was added slowly over a period of 30 minutes. The mixture was slowly heated to 50 °C and stirred for 20 hours at the same temperature followed 2 hours at 0 °C. The solid was filtered and washed with cold acetone (2 x 120 mL).The wet solid was dried at 40 °C for 2 hours to obtain 194.2 g of the title compound as white solid with HPLC purity of 99.55%
Example-11: Preparation of Diroximel fumarate
Diroximel Fumarate
To a mixture of (E)-4-(2-(2,5-dioxopyrrolidin-l-yl)ethoxy)-4-oxobut-2-enoic acid (5 g) and 2,2-dimethoxypropane (50 mL) at 29 °C, concentrated hydrochloric acid (1 mL) and water (5 mL) were added and stirred at the same temperature for 17 hours at the same temperature. The pH of the reaction mixture was adjusted to 7 with a saturated aqueous solution of NaHCCh and the solvent was evaporated completely at 40 °C. To the resultant solid, water (50 mL) was added at 29 °C and stirred for 15 minutes. The solid was filtered and dried under vacuum at 29 °C for 5 hours. The resultant solid was combined with methyl /er/-butyl ether (50 mL) at 29 °C and stirred for 20 hours at the same temperature. The solid obtained was filtered and washed with diethyl ether (20 mL). The wet solid was dried under vacuum for 3 hours at 29 °C to obtain 3.3 g of the title compound as white solid with HPLC purity of 98.11%
Example-12: Preparation of Amorphous solid dispersion of Diroximel fumarate with Copovidone
Diroximel fumarate (100 mg) and Copovidone (500 mg) were dissolved in acetone (30 mL) at 30 °C. The clear solution was filtered to make it particle free and the solvent was evaporated in a rotavapor at 45 °C under reduced pressure to obtain the title amorphous solid dispersion. The solid dispersion (100 mg) obtained was combined with Syloid (500 mg) and ground for 20 minutes to obtain the admixture of title compound. XRPD: Amorphous.
Example-13: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and methyl tert. butyl ether (30 mL) was added to the clear solution. A suspension of crystalline Diroximel fumarate seed (0.25 g) in methyl tert. butyl ether (10 m) was added at 40 °C and stirred the mixture at the same temperature for 30 minutes. Methyl tert. butyl ether (280 mL) was added slowly for 2 hours at 41 °C. The mixture was cooled to 25 °C in 3
hours and then to 0 °C in 1 hour. The mixture was stirred at 0 °C for 1 hour and the solid was filtered. The wet solid was washed with methyl tert. butyl ether (40 mL) and dried at 42 °C for 6 hours to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.776 pm, Dv (50) 31.292 pm & Dv (90) 133.437 pm
Example-14: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 43 °C and DM water (100 mL) was added to the clear solution. A crystalline Diroximel fumarate seed (0.20 g) was added at 42 °C and stirred the mixture at the same temperature for 10 minutes. DM water (100 mL) was added slowly at 41 °C. The mixture was cooled to 28 °C in 1 hour. The mixture was stirred at 28 °C for 2 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 8.59 pm, Dv (50) 61.08 pm & Dv (90) 187.07 pm
Example-15: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in acetone (80 mL) at 45 °C and DM water (400 mL) was added to the clear solution. The mixture was cooled to 30 °C in 1 hour and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.22 pm, Dv (50) 45.5 pm &
Dv (90) 136.7 pm
Example-16: Crystallization of Diroximel fumarate
Diroximel fumarate (20 g) was dissolved in Isopropyl acetate (360 mL) at 55 °C and cooled to 28 °C. A crystalline Diroximel fumarate seed (0.25 g) was added at 28 °C and cool to 5 °C. The mixture was stirred for 1 hour at the same temperature and the solid was filtered to obtain the title compound.
PXRD: Crystalline; Malvern particle size: Dv (10) 7.3 pm, Dv (50) 43.18 pm & Dv (90) 133.56 pm
PATENT
IN 201941042131
References
- ^ Jump up to:a b “Vumerity- diroximel fumarate capsule”. DailyMed. Retrieved 1 February 2021.
- ^ Jump up to:a b c “Vumerity EPAR”. European Medicines Agency. 14 September 2021. Retrieved 24 November 2021.
- ^ Wang Y, Bhargava P (July 2020). “Diroximel fumarate to treat multiple sclerosis”. Drugs of Today. 56 (7): 431–437. doi:10.1358/dot.2020.56.7.3151521. PMID 32648853. S2CID 220471534.
- ^ Kourakis S, Timpani CA, de Haan JB, Gueven N, Fischer D, Rybalka E (October 2020). “Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility?”. Pharmaceuticals (Basel, Switzerland). 13 (10): 306. doi:10.3390/ph13100306. PMC 7602023. PMID 33066228.
- ^ Jump up to:a b “Drug Approval Package: Vumerity”. U.S. Food and Drug Administration (FDA). 21 April 2020. Retrieved 1 February 2021.
- ^ “Diroximel fumarate”.
- ^ Jump up to:a b “Vumerity: Pending EC decision”. European Medicines Agency. 15 September 2021. Retrieved 17 September 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Diroximel fumarate”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Vumerity |
| Other names | ALKS-8700 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620002 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code |
|
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C11H13NO6 |
| Molar mass | 255.226 g·mol−1 |
| 3D model (JSmol) | |
/////////Diroximel fumarate, EU 2021, EMA 2021, APPROVALS 2021, VUMERITY, ジロキシメルフマル酸エステル , K0N0Z40J3W, RDC-5108, дироксимела фумарат , ديروكسيميل فومارات , 富马地罗昔美 ,

NEW DRUG APPROVALS
ONE TIME
$10.00
AVIPTADIL


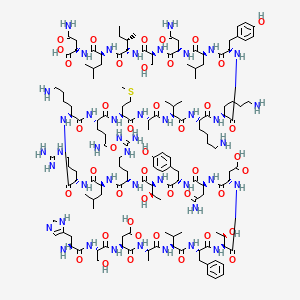
AVIPTADIL
- Molecular FormulaC147H237N43O43S
37221-79-7[RN]
6J2WVD66KR
L-Asparagine, L-histidyl-L-seryl-L-α-aspartyl-L-alanyl-L-valyl-L-phenylalanyl-L-threonyl-L-α-aspartyl-L-asparaginyl-L-tyrosyl-L-threonyl-L-arginyl-L-leucyl-L-arginyl-L-lysyl-L-glutaminyl-L-met hionyl-L-alanyl-L-valyl-L-lysyl-L-lysyl-L-tyrosyl-L-leucyl-L-asparaginyl-L-seryl-L-isoleucyl-L-leucyl-
Vasoactive intestinal octacosapeptide
Invicorp (aviptadil + phentolamine)
(2S)-4-amino-2-[[(2S)-2-[[(2S,3S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-5-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]propanoyl]amino]-3-methylbutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-carboxypropanoyl]amino]-4-oxobutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxybutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-4-methylpentanoyl]amino]-5-carbamimidamidopentanoyl]amino]hexanoyl]amino]-5-oxopentanoyl]amino]-4-methylsulfanylbutanoyl]amino]propanoyl]amino]-3-methylbutanoyl]amino]hexanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-3-hydroxypropanoyl]amino]-3-methylpentanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoic acid

Aviptadil Acetate
CAS#: 40077-57-4 (free base)
Chemical Formula: C155H253N43O51S
Exact Mass:
Molecular Weight: 3567.039
H-His-Ser-Asp-Ala-Val-Phe-Thr-Asp-Asn-Tyr-Thr-Arg-Leu-Arg-Lys-Gln-Met-Ala-Val-Lys-Lys-Tyr-Leu-Asn-Ser-Ile-Leu-Asn-NH2 tetraacetic acid.
Aviptadil had been in phase II clinical trials for the treatment of pulmonary arterial hypertension and idiopathic pulmonary fibrosis. But these researches were discontinued in 2011.
In 2006, Orphan Drug Designations were granted in the E.U. for the treatment of pulmonary arterial hypertension, and sarcoidosis and acute lung injury in 2006, and 2008, respectively.
The compound was co-developed by Lung Rx (subsidiary of United Therapeutics) and Mondobiotech.
Aviptadil (INN) is an injectable synthetic formulation of human vasoactive intestinal peptide (VIP).[1] VIP was discovered in 1970, and has been used to treat various inflammatory conditions, such as acute respiratory distress syndrome (ARDS), asthma and chronic obstructive pulmonary disease (COPD).
| Clinical data | |
|---|---|
| Trade names | RLF-100 / Zyesamiô |
| AHFS/Drugs.com | International Drug Names |
| ATC code | none |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 40077-57-4 |
| PubChem CID | 16132300 |
| ChemSpider | 17288959 |
| UNII | A67JUW790C |
| KEGG | D12127 |
| ChEMBL | ChEMBL2106041 |
| CompTox Dashboard (EPA) | DTXSID7048584 |
| Chemical and physical data | |
| Formula | C147H237N43O43S |
| Molar mass | 3326.83 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
Regulatory history
ARDS in COVID-19
Studies have found that aviptadil may be beneficial for severely ill patients with COVID-19 related ARDS.[2] ACTIV-3, a trial examining aviptadil acetate (Zyesami), is recruiting patients as of 2 July 2021.[3] A separate trial is examining inhaled aviptadil for patients with high risk for ARDS, is ongoing as of 21 May 2021.[4] A trial for intravenous aviptadil for the same indication concluded in February 2021.[5]
U.S.-Israeli NeuroRx Inc partnered with Relief Therapeutics to develop aviptadil in the United States. In June 2020, the U.S. Food and Drug Administration granted fast-track designation to aviptadil for treatment of respiratory distress in COVID-19.[6] In September 2020, NeuroRX submitted a request for an Emergency Use Authorization to the US FDA for its use in patients in intensive care.[7] May 2021: NRx Pharmaceuticals Announces Positive Results for ZYESAMI™ (Aviptadil-acetate) and Submits Emergency Use Authorization Application to USFDA to Treat Critical COVID-19 in Patients Suffering from Respiratory Failure.[8]
Jan, 2021: Zuventus healthcare Ltd seeks approval for Aviptadil from India’s drug controller for emergency use in COVID-19 treatment. Mumbai’s Zuventus healthcare Ltd. has got the nod to conduct Phase 3 clinical trials of Aviptadil injectable formulation. The SEC noted that Zuventus had presented revised Phase 3 clinical trial protocol before the committee, and after “detailed deliberation”, it recommended grant of permission of Phase 3 trials with the drug.[9] [10]
Aviptadil/phentolamine combination for Erectile Dysfunction (ED)
October 2000 UK (Invicorp): Aviptadil, an injectable formulation of vasoactive intestinal polypeptide (VIP) in combination with the adrenergic drug phentolamine is approved as an effective alternative therapy for erectile dysfunction (ED) patients. 1 dose intracavernosal injection contains 25 micrograms aviptadil and 2 mg of phentolamine mesilate for the treatment of erectile dysfunction. Aviptadil dose used for treatment of erectile dysfunction is far lesser as compared to dose used for the treatment of ARDS.[11][12]
Vasoactive intestinal peptide (VIP)
Vasoactive intestinal peptide (VIP) is a 28-residue amino acid peptide first characterized in 1970 that was initially isolated from porcine duodenum. A member of the secretin/glucagon hormone superfamily. VIP was initially discovered owing to its potent vasodilatory effects (as its name implies). VIP is widely distributed in the central and peripheral nervous system as well as in the digestive, respiratory, reproductive, and cardiovascular systems as a neurotransmitter and neuroendocrine releasing factor. These effects contribute to an extensive range of physiological and pathological processes related to development, growth, and the control of neuronal, epithelial, and endocrine cell function.[13]
VIP Receptors
VIP acts on two receptors – VPAC1 and VPAC2, which are class B of G-protein-coupled receptors (GPCRs).VPAC1 is mainly present in the lung and T-lymphocytes, whereas VPAC2 is mainly seen in the smooth muscle,mast cells and the basal parts of the lung mucosa.[14]
Expression of VIP
VIP is produced in the neurons in the central and peripheral nervous systems. VIP is mainly localized in the myenteric and submucosal neurons and nerve terminals in the GI tract. Endogenous VIP is released by numerous stimuli such as acetylcholine (ACh), ATP, serotonin (5-HT), substance P (SP), GLP-2 from at least two populations of VIP-positive nerves: cholinergic and non-cholinergic VIP-releasing nerves. In guinea pig small intestine, most VIP-positive nerves in the mucosa and submucosa are non-cholinergic secretomotor neurons and well colocalized with neuronal nitric oxide synthase (nNOS) in human colonic circular muscles. VIP is also expressed in immune cells, such as activated T cells and therefore present in lymphoid tissues including Peyer’s patches, the spleen, and lymph nodes, in addition to the VIP-ergic innervation in lymphoid tissues. Beside the neuronal source, VIP is also expressed and released from endocrine organs – Heart, Thyroid, Kidney and GI tracts.[15]
Localization of VIP
- VIP is highly localised in lungs (70%) and binds with alveolar type II (AT II) cells via VPAC1.[2] The biological (vasodilator) activity of vasoactive intestinal peptide (VIP) was discovered in the lungs before the peptide was isolated and chemical identity characterized from intestine. Although VIP levels are consideralbly high in the brain or gut:VIP is localized in key sites in the lung, has potent activities on its major functions, and appears to play an important role in pulmonary physiology and disease.[16]
- The principal localization of VIP-containing neurons in the tracheobronchial tree is in the smooth muscle layer, around submucosal mucous glands and in the walls of pulmonary and bronchial arteries. Immunoreactive VIP is also present in neuronal cell bodies forming microganglia that provide a source of intrinsic innervation of pulmonary structures.[16]
Vasoactive Intestinal Peptide (VIP) and SARS-CoV-2
VIP is highly localised in lungs and binds with alveolar type II (AT II) cells via VPAC1 receptor. AT II cells constitute only 5% of pulmonary epithelium. Angiotensin Converting Enzyme 2 (ACE 2) surface receptors arepresent in AT II cells. AT II cells produces surfactant and plays an important role in the maintenance of type 1epithelial cells. SARS-CoV-2 enters into AT II cells by binding to ACE 2 surface receptors with its spike protein. SARS CoV-2 attack mainly type II cells (not type I alveolar cells) and results in the death of alveolar type II (AT 11) cells which produces surfactant, resulting in[2]
- Profound defect in oxygenation
- Leading to hypoxia
Mechanism of action of Aviptadil
- Pulmonary alveolar type II Cells have a high concentration of ACE 2 receptors on their cell membrane
- Investigators have confirmed that the SARS-CoV family of viruses selectively attack pulmonary Alveolar Type II (ATII) cells because of their ACE2 receptors, in contrast to other pulmonary epithelial cells.
- SARS-CoV Viruses bind to ACE2 receptors in order to enter the cell. Viral replication and rupture liberates inflammatory cytokines and destroys surfactant production
- VIP binds uniquely to receptors on Alveolar Type II cells in the lung, the same cells that bind the SARS-CoV-2 virus via their ACE2 receptors
- VIP is heavily concentrated in the lung and binds specifically to VIP receptors on alveolar type II cells. VIP exerts a broad anti-cytokine effect on immune system cells
- VIP specifically upregulates surfactant production via upregulation of C-Fos protein and protects type II cells from cytokine
- Upregulating the production of surfactant, the loss of which is increasingly implicated in COVID-19 respiratory failure [17]
Aviptadil a synthetic form of VIP results in rapid clinical recovery in patients with SARS-CoV-2 infection.[2]
Effect of Aviptadil on Lungs in COVID-19
Preservation of Pulmonary Tissue
Preserving surfactant production in the lung and in protecting type 2 alveolar cells. Significantly delayed the onset of edematous lung injury, effective in preventing ischemia-reperfusion injury, Prevents NMDA-induced caspase-3 activation in the Lung.[18]
Inhibits alveolar epithelial cell Apoptosis
VIP is a proven inhibitor of activation-induced perforin, as well as of granzyme B and therefore actively contributes to the reduction of deleterious proinflammatory and cell death-inducing processes, particularly in the lungs. Aviptadil restores barrier function at the endothelial/alveolar interface and thereby protects the lung and other organs from failure.[18]
VIP Promotes synthesis of pulmonary surfactant
Studies have demonstrated that VIP binds on type II cells and increases the incorporation of methyl-choline into phosphatidylcholine – the major component of the pulmonary surfactants by enhancing the activity of the enzyme choline-phosphate cytidylyltransferase. VIP upregulates C-Fos protein expression in cultured type II alveolar cells, which is instrumental in promoting synthesis of pulmonary surfactant phospholipids (Li 2007) and induces surfactant protein A expression in ATII cells through activation of PKC/c-Fos pathway.[18]
VIP decreases Pulmonary Inflammation
Anti-cytokine effect- Inhibits IL-6,TNF-α production and inhibit NF-kB activation. Protects against HCl-induced pulmonary edema.[18]
Pharmacokinetic Properties
Half-life: Its plasma half-life of elimination is 1 to 2 minutes.[2] Metabolism/Distribution: After injection of 1 µg radioactively labelled Aviptadil as bolus to patients a very rapid tissue distribution was observed Within 30 min about 45% of the radioactivity was found in the lungs Over an observation period of 24 hrs only minimal activity was detected in the GI tract & almost no activity was found in the liver or spleen Radioactivity in the lungs decreased within four hours to 25% and within 24 hours to 10% Apparent volume of distribution: Aviptadil has a volume of distribution of 14 ml/kg.[2] Tissue Distribution:Aviptadil binds to its receptors in discrete locations within the gastrointestinal, respiratory, and genital tracts. Aviptadil is localized on respiratory epithelium, smooth muscles of the airways, blood vessels and alveolar walls. Elimination:After injection of radiolabelled Aviptadil radioactivity was almost completely eliminated by the kidneys, 35% within 4 hours, and 90% within 24 hours
Justification for Aviptadil use in the treatment of ARDS
COVID-19-related death is primarily caused by Acute Respiratory Distress Syndrome (ARDS). The trigger for ARDS is widely attributed to a cytokine storm in the lungs, in which the virus causes release of inflammatory cytokines. As a result, alveolae of the lungs fill with fluid and become impermeable to oxygen, even in the setting of mechanical ventilation. SARS-CoV-2 is known to cause respiratory failure, which is the hallmark of Acute COVID-19. Tragically, survival of patients with COVID-19 who progress to Acute Respiratory Distress is dismal. There is an urgent need for a treatment approach that goes right into the heart of the matter – the alveolar type 2 cells which are vulnerable entry points and hosts for the SARS-CoV-2 virus.[19]
Aviptadil-Evidence from Studies in ARDS
Phase III Study-Increased Recovery and Survival in Patients With COVID-19 Respiratory Failure Following Treatment with Aviptadil
A multicenter, randomized, placebo-controlled trial in 196 patients with PCR+ COVID-19 receiving intensive care at 10 U.S. hospitals – 6 tertiary care and 4 regional hospitals to determine whether intravenous aviptadil (synthetic VIP) is superior to placebo in achieving recovery from respiratory failure and survival at 60 days post treatment. Primary, prespecified endpoint was “alive and free from respiratory failure at day 60.” Across all patients and sites of care, patients treated with aviptadil were significantly more likely to be alive and free from respiratory failure at 60 days, compared to those treated with placebo (P=.02) and demonstrated improvement in survival alone (P<.001). Advantages in survival for aviptadil-treated patients were seen in both the subgroup classified as 2 on the National Institute of Allergy and Infectious Disease (NIAID) ordinal scale (58.6% vs. 0%; p=.001) and the NIAID=3 subgroup (83.1% vs. 62.8%; p=.03). Among patients who recovered successfully, those treated with Aviptadil had a median 10-day reduction in length of hospital stay compared to placebo patients (P=.025). Treatment with aviptadil demonstrates multi-dimensional efficacy in improving the likelihood of recovery from respiratory failure and survival to 60 days, and markedly reduced hospital stay in critically ill patients with respiratory failure caused by COVID-19.[20]
Case report: Rapid Clinical Recovery from Critical COVID-19 Pneumonia with Aviptadil
A 54 year old man with double lung transplant presented with headache, fever and productive cough. COVID-19 infection was confirmed by positive RT-PCR of nasopharyngeal swab. The patient required only supportive care for 3 days and was discharged home. Two weeks later he presented with worsening dyspnea, fever and severe hypoxemia requiring high flow O2 and ICU admission. Chest CT showed diffuse bilateral consolidations. He had markedly elevated inflammatory markers. He was treated with dexamethasone and tocilizumab without improvement. He was not a candidate for Remdesivir due to chronic kidney disease. Convalescent plasma was not available, Pro-BNP level was normal; echocardiogram showed preserved biventricular function. He received Aviptadil, a total of three doses, per an open label access under an emergency use approved by USFDA. Rapid improvement in oxygenation and radiologic findings were noticed. No adverse effects were recorded. Patient was transferred out of the ICU 24 hours following the third dose and discharged home on room air 15 days later. This case report of lung transplant recipient with critical COVID-19 pneumonia treated with Aviptadil demonstrates rapid clinical and radiologic improvement.This is consistent with that VIP protects ATII cells, ameliorating the inflammation and improving oxygenation in critical COVID-19 pneumonia.[21]
Posology and method of administration
Aviptadil intravenous infusion is administered by infusion pump in escalating doses for 3 successive days
- Day 1 : Aviptadil 0.166 mcg/kg/hr (equivalent to 1 vial of Aviptadil Injection)
- Day 2 : Aviptadil 0.332 mcg/kg/hr (equivalent to 2 vials of Aviptadil Injection)
- Day 3 : Aviptadil 0.498 mcg/kg/hr (equivalent to 3 vials of Aviptadil Injection)
Duration of infusion depends on the patient’s body weight
- Body weight < 60 kg – 14 hour infusions of Aviptadil at escalating doses on 3 successive days
- Body weight 60 – 90 kg – 12 hour infusions of Aviptadil at escalating doses on 3 successive days
- Body weight > 90 kg – 10 hour infusions of Aviptadil at escalating doses on 3 successive days
Undesirable Effects
Gastrointestinal Disorders – Diarrhea, Vascular disorders – Hypotension, cutaneous flushing, facial flushing & Infusion related reactions[20]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Keijzers GB (April 2001). “Aviptadil (Senatek)”. Current Opinion in Investigational Drugs. 2 (4): 545–9. PMID 11566015. Archived from the original on 2010-09-02. Retrieved 2020-04-01.
- ^ Jump up to:a b c d e f Raveendran, A; Al Dhuhli, K.; Harish Kumar, G. (2021). “Role of Aviptadil in COVID-19”. BMH Medical Journal. 8 (2): 77-83.
- ^ National Institute of Allergy and Infectious Diseases (NIAID) (2021-06-25). “A Multicenter, Adaptive, Randomized, Blinded Controlled Trial of the Safety and Efficacy of Investigational Therapeutics for Hospitalized Patients With COVID-19”. International Network for Strategic Initiatives in Global HIV Trials (INSIGHT), University of Copenhagen, Medical Research Council, Kirby Institute, Washington D.C. Veterans Affairs Medical Center, AIDS Clinical Trials Group.
- ^ Leuppi, Jörg (2021-05-20). “Inhaled Aviptadil for the Treatment of COVID-19 in Patients at High Risk for ARDS: A Randomized, Placebo Controlled, Multicenter Trial”. Clinicaltrials.gov.
- ^ NeuroRx, Inc. (2021-02-23). “ZYESAMI (Aviptadil) for the Treatment of Critical COVID-19 With Respiratory Failure”. Lavin Consulting, LLC.
- ^ “Critically ill COVID-19 patients make quick recovery with treatment RLF-100”. New York Post. 2 August 2020. Retrieved 3 August 2020.
- ^ NeuroRx. “NeuroRx submits request for Emergency Use Authorization for RLF-100™ (aviptadil) in the treatment of patients with Critical COVID-19 and Respiratory Failure who have exhausted approved therapy”. http://www.prnewswire.com. Retrieved 2020-09-24.
- ^ Pharmaceuticals, NRx. “NRx Pharmaceuticals Announces Positive Results for ZYESAMI™ (Aviptadil-acetate) and Submits Emergency Use Authorization Application to USFDA to Treat Critical COVID-19 in Patients Suffering from Respiratory Failure”. http://www.prnewswire.com.
- ^ Das, Sohini (2021-01-25). “Dr Reddy’s, Zuventus get nod to conduct Covid-19 trials on repurposed drugs”. Business Standard India.
- ^ SECmeeting, e COVID-19. “Recommendations of the SECmeeting to examine COVID-19 related proposals under accelerated approval process made in its 140thmeeting held on 18.01.2021 & 19.01.2021 at CDSCO, HQ New Delhi” (PDF). CDSCO. Retrieved 1 July 2021.
- ^ Keijzers, GB (April 2001). “Aviptadil (Senatek)”. Current Opinion in Investigational Drugs. 2 (4): 545–9. PMID 11566015.
- ^ Procivni, Aviptadil/phentolamine mesilate. “Scientific discussion” (PDF).
- ^ Iwasaki, M; Akiba, Y; Kaunitz, JD (2019). “Recent advances in vasoactive intestinal peptide physiology and pathophysiology: focus on the gastrointestinal system”. F1000Research. 8: 1629. doi:10.12688/f1000research.18039.1. PMC 6743256. PMID 31559013.
- ^ Mathioudakis, A; Chatzimavridou-Grigoriadou, V; Evangelopoulou, E; Mathioudakis, G (January 2013). “Vasoactive intestinal Peptide inhaled agonists: potential role in respiratory therapeutics”. Hippokratia. 17 (1): 12–6. PMC 3738270. PMID 23935337.
- ^ Iwasaki, M; Akiba, Y; Kaunitz, JD (2019). “Recent advances in vasoactive intestinal peptide physiology and pathophysiology: focus on the gastrointestinal system”. F1000Research. 8: 1629. doi:10.12688/f1000research.18039.1. PMC 6743256. PMID 31559013.
- ^ Jump up to:a b Said, Sami I. (June 1988). “Vasoactive Intestinal Peptide in the Lung”. Annals of the New York Academy of Sciences. 527 (1 Vasoactive In): 450–464. Bibcode:1988NYASA.527..450S. doi:10.1111/j.1749-6632.1988.tb26999.x. PMID 2898912. S2CID 26804295.
- ^ Javitt, Jonathan C (2020-07-25). “Vasoactive Intestinal Peptide treats Respiratory Failure in COVID-19 by rescuing the Alveolar Type II cell”. doi:10.22541/au.159569209.99474501. S2CID 221509046.
- ^ Jump up to:a b c d Javitt, Jonathan C (2020-05-13). “Perspective: The Potential Role of Vasoactive Intestinal Peptide in treating COVID-19”. doi:10.22541/au.158940764.42332418. S2CID 219771946.
- ^ “Relief Therapeutics and NeuroRx Announce Final Manufacturing Validation of RLF-100 for Phase 2b/3 Clinical Trial in Patients with COVID-19 Associated Acute Respiratory Distress Syndrome”. GlobeNewswire News Room. 2020-05-14.
- ^ Jump up to:a b Youssef, Jihad G.; Lee, Richard; Javitt, Jonathan; Lavin, Philip; Lenhardt, Rainer; Park, David J; Perez Fernandez, Javier; Morganroth, Melvin; Jayaweera, Dushyantha (2021). “Increased Recovery and Survival in Patients With COVID-19 Respiratory Failure Following Treatment with Aviptadil: Report #1 of the ZYESAMI COVID-19 Research Group”. SSRN 3830051.
- ^ Beshay, S.; Youssef, J.G.; Zahiruddin, F.; Al-Saadi, M.; Yau, S.; Goodarzi, A.; Huang, H.; Javitt, J. (April 2021). “Rapid Clinical Recovery from Critical COVID-19 Pneumonia with Vasoactive Intestinal Peptide Treatment”. The Journal of Heart and Lung Transplantation. 40 (4): S501. doi:10.1016/j.healun.2021.01.2036. PMC 7979412. S2CID 232282732.
//////////AVIPTADIL, RLF 100, DK 1000
CCC(C)C(C(=O)NC(CC(C)C)C(=O)NC(CC(=O)N)C(=O)O)NC(=O)C(CO)NC(=O)C(CC(=O)N)NC(=O)C(CC(C)C)NC(=O)C(CC1=CC=C(C=C1)O)NC(=O)C(CCCCN)NC(=O)C(CCCCN)NC(=O)C(C(C)C)NC(=O)C(C)NC(=O)C(CCSC)NC(=O)C(CCC(=O)N)NC(=O)C(CCCCN)NC(=O)C(CCCNC(=N)N)NC(=O)C(CC(C)C)NC(=O)C(CCCNC(=N)N)NC(=O)C(C(C)O)NC(=O)C(CC2=CC=C(C=C2)O)NC(=O)C(CC(=O)N)NC(=O)C(CC(=O)O)NC(=O)C(C(C)O)NC(=O)C(CC3=CC=CC=C3)NC(=O)C(C(C)C)NC(=O)C(C)NC(=O)C(CC(=O)O)NC(=O)C(CO)NC(=O)C(CC4=CN=CN4)N

NEW DRUG APPROVALS
ONE TIME
$10.00
Marbofloxacin

Marbofloxacin
- Molecular FormulaC17H19FN4O4
- Average mass362.356 Da
115550-35-1[RN]
2,3-Dihydro-9-fluoro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido[3,2,1-ij][4,1,2]benzoxadiazine-6-carboxylic Acid
6807
7H-1,3,4-Oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid, 9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-
8X09WU898T
марбофлоксацин
ماربوفلوكساسين
马波沙星
Marbofloxacin is a carboxylic acid derivative third generation fluoroquinolone antibiotic. It is used in veterinary medicine under the trade names Marbocyl, Forcyl, Marbo vet and Zeniquin. A formulation of marbofloxacin combined with clotrimazole and dexamethasone is available under the name Aurizon (CAS number 115550-35-1).
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
CN 107383058,
https://patents.google.com/patent/CN107383058B/enMarbofloxacin (Marbofloxacin) is fluoroquinolone antibacterial agent for animals, the entitled fluoro- 3- methyl-1 0- of 9- of chemistry (4- methylpiperazine-1-yl) -7- oxo -2,3- dihydro -7H- pyridine [3,2,1-ij] [4,1,2] benzo oxadiazines -6- carboxylic acid, It is developed by Roche Holding Ag, and is further developed by French Vetoquinol (method national strength and prestige are grand) company earliest, in nineteen ninety-five in Europe Listing.Marbofloxacin is after Enrofloxacin (Enrofloxacin), Danofloxacin (Danofloxacin), sarafloxacin (Sarafloxacin) etc. another third generation carbostyril family antibacterial drugs after, the drug have extensive antibacterial activity simultaneously With very good dynamic characteristic, sterilizing power is strong, absorbs fastly, widely distributed in vivo, with other antimicrobials without crossing drug resistant Property, easy to use, adverse reaction is small.Pharmacokinetic is studies have shown that Marbofloxacin removes long half time in animal body, biology Availability, almost without residual in the blood of animal, excrement and tissue, is well suited for clinically to antibiosis for animals close to 100% The requirement of element, structural formula are as follows:
Structure is complicated for Marbofloxacin, not only contains methyl piperazine substituent group, but also aromatic moieties contain pyridine benzo evil two Piperazine skeleton has had many documents and patent report at present and has reviewed its synthetic method, such as patent US4801584, ZL94190968.9, EP2010/067828, CN101619068, CN102060860, CN102617595, document J.Org. Chem., 1992,57 (2), 744-766, ” chemical reagent ” 2007,29 (11), 701-703., ” Chinese Journal of Pharmaceuticals ” 2002,33 (1), 1358-1363 etc..Patent US4801584 reports fluoro- via the fluoro- 4,8- dihydroquinoline -3- carboxylic acid, ethyl ester of 6,7- bis- preparation 6,7- bis- The method of 8- hydroxyl -1- (methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline -3- carboxylic acid, ethyl ester, this method are related to using valuableness And commercialization is not easy amination reagent O- (2, the 4- dinitrophenyl) oxyammonia largely purchased in 1 upper amino, by multistep reaction After complete the preparation of fluoro- 8- hydroxyl -1- (the methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline -3- carboxylic acid, ethyl ester of 6,7- bis-, passed through after It crosses and paraformaldehyde, N methyl piperazine reacts the preparation for realizing Marbofloxacin.Correlated response formula is as follows:
The patent literature reports such as patent ZL94190968.9 are that raw material prepares Ma Bosha from 2,3,4,5 tetra fluoro benzoic acid The synthetic route of star, this method are not only related to the multisteps hazardous reactions such as carboxylic acyloxy chlorination, Grignard Reagent preparation reaction, synthesize road Wire length, and 3- (the N- methyl formyl hydrazono-) ethyl acrylate for being difficult to prepare is used, and yield is low, be not suitable for industrially putting Mass production, correlated response formula are as follows:
Patent CN101619068 is condensed using 2,3,4,5- phenyl tetrafluoride carbamoylalkyl esters and inferior amine salt, obtained N- bis- Methyl substituted enamine derivates react the enamine for preparing the substitution of N- methyl-N- acyl group under organic acid catalysis with N- methylhydrazide Derivative, then 6,7,8- tri- fluoro- 1- (methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline-are completed in cyclization and hydrolysis under alkaline condition The preparation of 3- carboxylic acid realizes Ma Bosha finally by with N methyl piperazine, dimethyl formal (or diethyl formal) reaction The preparation of star.The technique uses the dimethyl suflfate and the height hazardous reaction reagent such as sodium hydride or alkalide of severe toxicity, because And it is subject to certain restrictions in commercial process.Correlated response formula is as follows:
In conclusion there are various deficiencies, such as chemistry examinations in the synthetic route of existing synthesis Marbofloxacin The defects of agent is expensive, reaction route is too long, using the chemical reagent for being unfavorable for industrialized production, the present inventor are real after study It tests, invents a kind of new method for preparing Marbofloxacin.The preparation of embodiment 1:1,1,1- tri- chloro- 4- (4- methylpiperazine-1-yl) butyl- 3- alkene -2- ketone(E) -1,1,1- tri- chloro-4-methoxy butyl- 3- alkene -2- ketone (Formulas I, R=Me) (10.18g, 50mmol), 1- methyl The mixture of piperazine (6.0g, 60mmol) and mesitylene (50mL) is heated to reflux temperature and stirs 6 hours, and system is natural Be cooled to room temperature, remove organic solvent under high vacuum reduced pressure, residue (14.2g, crude product do not purify) without further purification, directly It connects for reacting in next step.Embodiment 2:(6,8- bis- fluoro- 7- (4- methylpiperazine-1-yl) -4- oxo -3- (2,2,2- trichloroacetyl) quinoline Quinoline -1 (4H)-yl) urethanes (Formula VII) preparationUnder nitrogen protection, the product (14.2g is not purified, is directly used) of embodiment 1 is dissolved in toluene (120mL), then body Triethylamine (72mL, 514mmol) is added in system, system is heated to reflux temperature.Under reflux temperature, slowly dripped into reaction system Add toluene (60mL) solution of 2,3,4,5- phenyl tetrafluoride formyl chloride (16g, 75.3mmol).Rear system reflux is added dropwise 30min, then system slow cooling is to 60 DEG C, heat filtering.Filtrate is transferred in 500ml reaction flask, and carbazic acid second is then added Ester (Formula V, R2=Et) (6.25g, 60mmol).System is reacted 12 hours at a temperature of 60-65 DEG C after addition.To reaction H is slowly added in system2O (150mL) quenching reaction, system are naturally cooling to room temperature.Filtering, obtains solid, and solid uses heptan Alkane/ethyl acetate system mashing processing, obtains solid (Formula VII, R2=Et) (21.2g).Embodiment 3:1- amino -6- fluoro- 8- hydroxyl -7- (4- methylpiperazine-1-yl) -4- oxo -1,4- dihydroquinoline -3- The preparation of carboxylic acid (Formula VIII)2 obtained solid of embodiment (21.2g) is placed in 200ml reaction flask, ethyl alcohol (50mL) is added into reaction system With water (50mL), system is heated to flowing back.The aqueous solution (30mL) of KOH (7.0g) is slowly added under counterflow condition to system, is dripped System maintains the reflux for state response 96 hours after adding.System is naturally cooling to room temperature, and H is added in system2O (100mL) and CH2Cl2(50ml) stands after stirring and separates organic phase, and water phase reuses CH2Cl2It is extracted twice (2 × 50mL).Water phase uses salt Sour regulation system is to acid (pH=3-4), and then water phase reuses CH2Cl2It is extracted twice (2 × 100mL), merges organic phase, subtract Pressure-off obtains solid (Formula VIII) (12.4g) after removing organic solvent.The preparation of embodiment 4:1,1,1- tri- chloro- 4- (4- methylpiperazine-1-yl) butyl- 3- alkene -2- ketoneSequentially added in reaction flask the chloro- 4- ethyoxyl butyl- 3- alkene -2- ketone (Formulas I, R=Et) of (E) -1,1,1- three (14.1g, 65mmol) and 1- methyl piperazine (7.0g, 70mmol).Then system is heated to 130-155 DEG C and is stirred to react 5 hours.System is cold But to room temperature, the complete raw material of a little unreacted of high vacuum removed under reduced pressure, residue (16.8g, crude product do not purify) is without pure Change, is directly used in and reacts in next step.Embodiment 5:(6,8- bis- fluoro- 7- (4- methylpiperazine-1-yl) -4- oxo -3- (2,2,2- trichloroacetyl) quinoline Quinoline -1 (4H)-yl) t-butyl carbamate (Formula VII, R2=tBu) preparationUnder nitrogen protection, the product (16.0g is not purified, is directly used) of embodiment 4 is dissolved in toluene (125mL), then N is added in system, N- diisopropylethylamine (104.5mL, 600mmol), system is heated to reflux temperature.Under reflux temperature, to Toluene (70mL) solution of 2,3,4,5- phenyl tetrafluoride formyl chloride (18.8g, 88mmol) is slowly added dropwise in reaction system.It is added dropwise Starting material Formula II is tracked to HPLC within system reflux 1 hour afterwards to disappear.Then system slow cooling is to 60 DEG C or so, hot mistake Filter.Filtrate is transferred in 500mL reaction flask, and tert-butyl carbazate (Formula V, R is then added2=tBu)(9.3g,70mmol).It is added After system reacted 48 hours at a temperature of 60-65 DEG C.H is slowly added into reaction system2O (150mL) quenching reaction, body System is naturally cooling to room temperature.Filtering obtains solid, and solid is handled using heptane/ethyl acetate system mashing, obtains solid (formula VII,R2=tBu) (19.3g) is directly used in next step without further purification.Embodiment 6:1- amino -6- fluoro- 8- hydroxyl -7- (4- methylpiperazine-1-yl) -4- oxo -1,4- dihydroquinoline -3- The preparation of carboxylic acid (Formula VIII)By 5 obtained solid of embodiment (19.0g) as in 200mL reaction flask, methanol (55mL) is added into reaction system With water (55mL), system is heated to flowing back.The aqueous solution (30mL) of CsOH (13.5g) is slowly added under counterflow condition to system, Rear system is added dropwise and maintains the reflux for state response 96 hours.System is naturally cooling to room temperature, and H is added in system2O (100mL) and CH2Cl2(50mL) stands after stirring and separates organic phase, and water phase reuses CH2Cl2It is extracted twice (2 × 50mL).Water phase uses salt Sour regulation system is to acid (pH=3-4), and then water phase reuses CH2Cl2It is extracted twice (2 × 100mL), merges organic phase, subtract Pressure-off obtains solid (Formula VIII) (8.8g) after removing organic solvent.Embodiment 7: the preparation of Marbofloxacin1- amino-6- fluoro- 8- hydroxyl-7- (4- methylpiperazine-1-yl) oxo-1-4- is sequentially added in 100mL reaction flask, 4- dihydroquinoline -3- carboxylic acid (Formula VIII, 6.0g), 85% formic acid (30mL) and 36.5% formalin (6.0mL). System is carefully slowly heated to 75 DEG C or so reactions 1 hour after addition.Then system is cooled to 10 DEG C hereinafter, being carefully added into 25% ammonium hydroxide (25mL), stir 0.5 hour.Then activated carbon (1g) is added into system, mistake after 1 hour is sufficiently stirred Filter, filtrate methylene chloride extract 2 times (2 × 100mL).Merge organic phase, anhydrous sodium sulfate dries, filters, organic phase high vacuum Removed under reduced pressure solvent obtains Marbofloxacin crude product (5.4g).H is added in the crude product2In O (50mL), first acid for adjusting pH value is slowly added dropwise To 3.2 (pH meter detections), 4 hours are stood, filtering, filtrate added drop-wise sodium bicarbonate aqueous solution adjusting pH value to 6.2 (pH meter detections), A large amount of solids are precipitated, and ice salt bath cooling system stirs 1 hour to 0 DEG C or so, filtering, obtain Marbofloxacin after product drying (4.72g)。

Patent
Publication numberPriority datePublication dateAssigneeTitleUS4801584A *1986-09-121989-01-31Hoffmann-La Roche Inc.Pyrido(3,2,1-IJ)-1,3,4 benzoxadiazine derivativesCN1116849A *1993-01-231996-02-14辉瑞大药厂Process for the manufacture of a tricyclic compoundCN102060860A *2011-01-072011-05-18安徽美诺华药物化学有限公司Preparation method of MarbofloxacinCN102617595A *2012-03-232012-08-01江西华士药业有限公司Preparation method of fluoroquinolone antibacterial medicament marbofloxacinCN102712598A *2009-11-192012-10-03新梅斯托克尔卡·托瓦纳·兹德拉维尔公司A process for a preparation of marbofloxacin and intermediate thereof
CN110283186A *2019-07-192019-09-27海门慧聚药业有限公司A kind of crystal form of Marbofloxacin and preparation method thereof
PATENT
CN 107522718
PATENT
CN 102617595,
PATENT
Indian Pat. Appl., 2009CH00164,
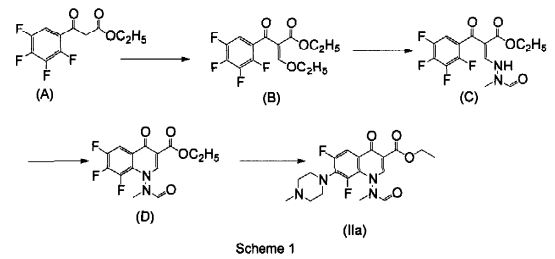

Example 2: Preparation of ethyl 6,8-difluoro-1-(N~methylfomnamido)-7-(4-methyl-1- piperazinyl)-4-oxo-4H-quinoline-3-carboxylate hydrochloride of Formula (Ilia)
STR IIIA
Water (400 ml) and the compound of Formula (IIa) (200 g) are charged into a round bottom flask at 28°C and concentrated HCI (124 ml) is added slowly at a temperature below 40°C, and the mass is heated to 95-1OO0C. 300 ml of water and ethanol are distilled under vacuum at 1004C. The mass is cooled to 25-30°C. Acetone (400 ml) Is added and the mass is cooled to 0-5°C. The mass is maintained at 0-58C for 30-60 minutes and the product is filtered. The product is washed with pre-chilled acetone (200 ml) and dried under vacuum at 70-75°C for 12-15 hours to obtain the title compound. Yield: 181.0 g (95%). Example 3: Preparation of marbofloxacin from the compound of Formula (Ilia) Ethylene glycol (100 ml) and potassium hydroxide (17.3 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (Ilia) (10 g) is added and the mass is heated to 120-130’C, and then maintained for 24 hours. The mass is cooled to 30°C and water (15 ml) is added. Hydrochloric acid (36%, 18 ml) is slowly added below 404C.rformic acid (6 ml) is slowly added below 40°C and the mass is stirred for 20-30 minutes. Formaldehyde (5 ml) is added and the mass is then heated to 70-75°C and maintained for 1-2 hours. The mass is slowly cooled to 15-20°C and stirred for 30-60 minutes. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled demineralized water (5 ml). The material is suction dried for 2-3 hours. Methanol (50 ml), demineralized water (15 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes.
Ammonia solution (25%, 7.5 ml) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (5 ml) at 25- 35°C. The water and methanol are distilled at 60-70°C under vacuum until 20 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid is filtered at 0-5°C and the wet cake is washed with methanol (10 ml). The material is suction dried for 30-60 minutes and the product is dried at 60-70°C under vacuum for 18-20 hours. Yield: 6.51 g (70%). Example 4: preparation of marbofloxacin from a compound of Formula (Ilia) Ethylene glycol (150 ml) and potassium hydroxide (72.2 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (Ilia) (50 g) is added and the mass is heated to 115-1256C, and then is maintained for 10-12 hours at 115— 125°C. The mass is cooled to 25-35°C and water (150 ml) is added. Formic acid (98%, 100 m!) is slowly added below 45°C and the mass is stirred for 30-60 minutes. Formaldehyde (37-41%, 35 ml) is added to the mass, which is then heated to 70- 75°C and maintained for 1-2 hours. The mass is slowly cooled to 0-5°C and stirred for 1-2 hours. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled water (50 ml). The material is suction dried for 1 hour and washed with pre-chilled acetone (50 ml) and suction dried for 1 hour. Methanol (250 ml), water (100 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes. Ammonia solution (25%, 40 mi) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (50 ml) at 25-35°C. The filtrate is distilled at 60-70°C under vacuum until 75-100 ml remain. The mass is cooled to 10-15’C and maintained for 30-60 minutes. The solid free base is filtered at 10-15°C and the wet cake is washed with chilled methanol (50 ml). The material is suction dried for 30-^60 minutes and the product is dried at 60-70°C under vacuum for 10-12 hours. Yield: 33.0 g (70.8%). Example 5: Preparation of marbofloxacin from a compound of Formula (Ilia) Water (350 ml) and potassium hydroxide (86.6 g) are stirred for 10 minutes. A compound of Formula (Ilia) (50 g) is added and the mass is heated to 100-104°C. The mass is maintained for 105-110 hours at 100-1040C, then is copied to 25-35°C and water (65 ml) is added. Hydrochloric acid (36%, 125 ml) is slowly added below 40°C and the mass is stirred for 30 minutes. Formaldehyde (37%, 19 ml) is added and the mass is heated to 70-756C. The mass is maintained for 1-2 hours at 70-75 0C and then is slowly cooled to 0-5°C and maintained for 30-60 minutes. The obtained solid hydrochloride salt is filtered and the bed is washed with pre-chilled water (25 ml) at 0-5°C. The material is suction dried. Ethanol (250 ml), water (75 ml), ammonia solution (25%, 38 ml) and the wet cake are charged into a round-bottom flask and stirred for 1-2 hours at 25-35° C. The turbid solution is filtered and the bed is washed with ethanol (50 ml). The filtrate is distilled at 65-70°C under vacuum until 100 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid free base is filtered and the wet cake is washed with pre-chilled ethanol (50 ml). The product is dried under vacuum at 60-70°C for 15-^20 hours. Yield: 23.3 g (50%).
Example 6: Preparation of marbofloxacin from a compound of Formula (IIa) Ethylene glycol (60 ml) and potassium hydroxide (28.05 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (IId) (20 g) is added. The mass is heated to 120-135°C and maintained for 4-6 hours. The mass is cooled to 30°C and water (60 ml) is added. Formic acid (98-100%, 40 ml) is slowly added below 40°C and stirred for 20-30 minutes. Formaldehyde (37-41%, 12 ml) is added to the mass, which is heated to 70-75°C and maintained for 1-2 hours. The mass is slowly cooled to O-S6C and stirred for 30-60 minutes. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled water (20 ml). The material is suction dried for 2-3 hours. Methanol (100 ml), water (30 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes. Ammonia solution (25%, 20 ml) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (10 ml) at 25-35°C. The water and methanol are distilled at 60-70°C under vacuum until 40 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid free base is filtered at 0-5°C and the wet cake is washed with methanol (20 ml). The material is suction dried for 30-60 minutes and the product is dried at 60-70°C under vacuum for 18-20 hours. Yield: 12.6 g (71%)
Example 7: Purification of marbofloxacin To crude marbofloxacin (25 g) is added methanol (125 ml) and ammonia (18.75 ml). Half of the volume of the methanol and ammonia solution is removed by azeotropic distillation. The mass is slowly cooled and maintained for 1 hour. The product is filtered and washed with chilled methanol (25 ml). The product is suction dried for 30 minutes and dried under vacuum for 12 hours, to yield pure marbofloxacin of a purity 99.80%. XRD pattern, DSC thermogram, TGA1 and IR are substantially in accordance with Figs. 1, 2, 3, and 4, respectively. Yield: 22 g (88.0%),
PATENT
Indian Pat. Appl., 2009CH00163,
PATENT
WO 2011061292
PATENT
CN 102060860,
PATENT
CN 101619068,
PATENT
https://patents.google.com/patent/EP2332916A2/en
- Marbofloxacin is the common name for 9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyridol(3,2,1-ij)(4,2,1)benzoxadiazin-6-carboxylic acid, of the formula :
- [0003]
Marbofloxacin is a potent antibiotic of the fluoroquinolone group. - [0004]
EP 259804 describes marbofloxacin as well as a synthesis for the preparation thereof by a multistep process which is unpractical for a large scale manufacture, since it requires high temperatures and reagents not suitable for large-scale production, resulting in low over-all yields. The process for the preparation is disclosed in the reaction scheme 1. - [0005]
EP 680482 discloses an alternative approach for the preparation of marbofloxacin, wherein hydroxy group is introduced into molecule by means of reaction of intermediate with alkali metal hydroxide in aqueous media. The starting material used is 2,3,4,5-tetrafluorobenzoic acid. Disadvantages of this process are relatively high excess of alkali metal hydroxide and lengthy procedure. The process for the synthesis according to this patent is shown in the reaction scheme 2. - [0006]
Research Disclosure No. 291, 1988, pages 548-551 discloses an alternative route of synthesis also starting from 2,3,4,5-tetrafluorobenzoic acid. Later steps of the process are shown in the reaction scheme 3. - [0007]
IT 1313683 relates to a process for preparation of marbofloxacin by a process via benzyl ether. Ether was debenzylated in aqueous solution by hydrogenating over 5% Pd/BaSO4 and the obtained product is cyclized using HCOOH/HCOH. - [0008]
In view of the prior art there still exists a need for an improved method for preparation of marbofloxacin and intermediates thereof suitable for a large-scale production.
Examples
- [0068]
A high resolution HPLC method is used to determine an amount and purity compounds of formula I, II and IV. The tests are carried out in X-Bridge C18, 150 x 4.6mm, 3.5µm column. The mobile phase is gradient of A) 5mM NH4COOCH3 pH=7.0 B) acetonitrile. Gradient: 0’=10%B, 10’=20%B, 25′-30’=90%B, 32’=10%B. - [0069]
The chromatograph is equipped with a UV detector set at 250 nm and 315nm, the flow rate is 1.0 ml per minute at 30°C.
Example 1a) 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid and 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt
- [0070]
- [0071]
4.137g of Ethyl 6,8-difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (10.14mmol) was put into 40mL of 10% H2SO4 and stirred at 100°C for 7 hours. Reaction mixture was cooled and crystals were formed. Mixture was cooled to 4°C and filtered with suction. Filter cake was washed with a mixture of H2O/EtOH/THF (1/1/5) and dried. 3.260g of 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid as yellow crystals were obtained (91%). - [0072]
In case the sodium salt is desired the product obtained in previous step was put into 5mL of EtOH and 10mL of CH2Cl2 and 1.20g of NaOH dissolved in 2mL of water was added. Solution was stirred at room temperature. for 1h, dried with Na2SO4 and evaporated. 2.90g of pure title product was isolated (yellow powder, 7.71mmol, 76%).
b) 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
- [0073]
- [0074]
400mg of Ethyl 6,8-difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (0, 979mmol) was put into 2mL of 10% H2SO4 and stirred at 100°C for 2 hours. Reaction mixture was cooled and crystals were formed. To this mixture 1,7mL of 25% aq. NH3 was slowly added. At first very dense suspension was formed that dissolves with further addition of ammonia solution. At the end clear solution formed with pH of 9. Ammonium sulphate was precipitated by the addition of 10mL of EtOH , filtered off and washed with 5mL of H2O/EtOH (1/2). Mother liquor was dried on the rotary evaporator and 10 mL of EtOH/H2O mixture (7/3) was added to precipitate residual inorganic salt, which was again filtered off. Remaining yellow solution was dried on a rotary evaporator to obtain 321mg of yellow powder (0.912 mmol, 93%).
Example 26-fluoro-8-hydroxy-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
- [0075]
- [0076]
178 mg of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt (0.470mmol) was mixed with 360 mg of Me4NOH.5H2O (2.00 mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 0.10mL of HCOOH was added to neutralize hydroxide. 5mL of EtOH is added to precipitate the product, which was filtered with suction and filter cake was washed with 2mL of cold EtOH. 90mg of the product was obtained.
Example 39-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid formate salt
- [0077]
- [0078]
180 mg of 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt (0.481mmol) was mixed with 360 mg of Me4NOH.5H2O (2.00 mmol) and stirred at 100°C for 3 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 1 mL of HCOOH was added followed by addition of 0.4 mL of 37% aq. solution of HCHO and stirred at 70°C for additional hour. Reaction mixture was cooled to room temperature and 5mL of EtOH was added to precipitate the product, which was filtered with suction and filter cake was washed with 2mL of cold EtOH. 111 mg of grey powder was obtained.
Example 49-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid formate salt
- [0079]
- [0080]
1.14g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.00mmol) was mixed with 3.06g of Me4NOH.5H2O (16.96mmol) and stirred at 100°C for 5 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 1.44 mL of HCOOH (85% aq. sol) was added followed by addition of 0.5 mL of 37o aq. solution of HCHO and the flask was cooled on the water bath at 22°C. Another 1.44mL of 85% HCOOH was added and the reaction mixture was warmed to 70°C for 30min and after cooling 20mL of EtOH was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.23g of grayish powder was obtained (90%) .
Example 59-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0081]
- [0082]
1.145g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 2.72g of Me4NOH.5H2O (15.00mmol) and stirred at 100°C for 8 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved. After stirring at 70°C for 30min (precipitate was formed again after 5min) reaction flask was cooled to room temperature and 20mL of EtOH was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.165g of grayish powder was obtained (85%), with a purity of 97.11% (HPLC). - [0083]
Crude reaction product was mixed with 0.9mL of 25% NH3 aqueous solution and crystallized in a mixture of 26mL of EtOH and 14mL H2O. 0.673g of powder was obtained (61%) with a purity of 98.75% (HPLC).
Example 69-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0084]
- [0085]
1.140g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.00mmol) was mixed with 2.72g of Me4NOH.5H2O (15.01mmol) and stirred at 100°C for 8 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and 20mL of H2O was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.022g of greyish powder was obtained (75%). with a purity of 97.11% (HPLC). - [0086]
Crude reaction product was mixed with 0.9mL of 25% NH3 aqueous solution and crystallised in a mixture of 20mL of EtOH and 6mL CHCl3. 0.771g of yellow powder was obtained (71%) with a purity of 99.50% as determined by HPLC.
Example 79-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0087]
- [0088]
1.142 g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 3.26g of Me4NOH.5H2O (18.01mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure. After drying 1.147g of white powder was obtained (84%). - [0089]
Crude reaction product was mixed with 5mL of water and 2mL of 25% aqueous solution of NH3 and clear solution was obtained. To this solution, 7mL of EtOH was added and dried under reduced pressure. Product was crystallized in a mixture of 15mL of EtOH and 10mL CHCl3 to obtain 0.4321g of white powder (41%) with a purity of 98.63% as determined by HPLC
Example 89-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0090]
- [0091]
1.136 g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (2.98mmol) was mixed with 2.73g of Me4NOH.5H2O (15.00mmol) and stirred at 100°C for 7 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure. After drying 1.039g of grey powder was obtained (77%). - [0092]
Crude reaction product was neutralized with 2mL of 25% aqueous solution of NH3 and clear solution was diluted with 15mL of EtOH and 9mL of H2O. Solution was partially dried under reduced pressure until the formation of precipitate. At this point mixture was cooled in a refrigerator and precipitate was isolated by filtration under reduced pressure to obtain 0.675g of powder (65%) with a purity of 98.84% as determined by HPLC.
Example 99-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0093]
- [0094]
1.140g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 3.30g of Me4NOH.5H2O (18.20mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure to obtain 0.847g of solid, while mother liquid was diluted with EtOH and concentrated under reduced pressure until precipitate forms, which was filtered again to obtain additional 0.208g of solid. The yield of combined solid material is 1.055g, 77%. Crude reaction product (formate salt) was crystallized in H2O/EtOH (25mL/10mL) to obtain 0.722g (53%) of yellow powder. Formate salt was put in 20mL of EtOH/CH2Cl2 mixture (1/1) and 0.5mL of 25%aq. NH3 was added to obtain clear solution. Solution was dried with Na2SO4 and solvent evaporated under reduced pressure to obtain 0.580g of yellow powder (53%).
Example 109-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
- [0095]
- [0096]
100 mL reactor with a rotary stirrer was charged with 10,16g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (28,83mmol) and 26,50g of Me4NOH˙5H2O (146,25mmol) that was previously mixed together. Temperature of the heating jacket was set to 100°C and stirring to 100s-1, while water was allowed to evaporate out of the reactor during the reaction. Reaction was stirred at specified temperature for 5 hours and homogenous dark brown oil was obtained. Temperature of reactor was cooled to 20°C, 30mL of HCOOH was added and stirred well so that all oil is transformed into brown suspension. 4,5mL of 37% aq. HCHO was added drop-wise and heated at 70°C for 30min. Reaction mixture was cooled to 20°C and 20mL of water added to precipitate the product in the form of formate complex. Suspension was cooled to 0°C and filtered under reduced pressure and washed the filter cake with additional 10mL of cold water to obtain 8,38g of white powder. Mother liquor was partially evaporated under reduced pressure and when solid started to precipitate it was filtered again to obtain additional 0.80g of powder. 50mL of EtOH was added into the mother liquor to precipitate the product and after filtration at reduced pressure further 0.80g of white powder was obtained. Product was collected and 9,98g of white powder was suspended in a mixture of 50mL of EtOH and 50mL of CH2Cl2. Into the suspension 25% aq. NH3 was added to neutralize the formate complex and after addition of 12mL of NH3 all product was dissolved and small amount of solid material is formed. 5g of anhydrous Na2SO4 was added to dry the organic solution and it was filtered off and solvent evaporated under reduced pressure. 8.99g of slightly yellow powder was obtained in 86% yield.
Example 11Crystallization from ethanol/toluene/water 2:1:1
- [0097]
8.4g of crude marbofloxacin was suspended in a mixture of 83 ml of ethanol, 41ml of toluene and 41 ml of water and heated to reflux. From the clear yellow solution formed 83 ml of solvent mixture was distilled off, whereby the temperature rose from 74 to about 79°C, and a yellow precipitate was formed. The suspension was cooled to 20° – 25°C, stirred for 1 hour, filtered, and the filter cake was washed with 3 portions of 6 ml of ethanol to yield after drying in vacuum dryer the product in more than 95% yield.
Example 12Crystallization of marbofloxacin starting from marbofloxacin formate
- [0098]
26g of marbofloxacin formate was suspended in a mixture of 65ml of ethanol and 27ml of water. Under stirring a solution of 25% ammonia in ethanol (20ml 25%NH3/10ml EtOH) is slowly (about 30 minutes) added by drops until the substance is dissolved and pH value of 7-9 is reached. The reaction was stirred for about 15 minutes and filtered. The filtrate was evaporated at 110°C until about 60ml of the solvent was distilled off and marbofloxacin started to precipitate. After distillation the suspension was cooled and stirred for 0.5 to 1 hour at 0-5°C, filtered, to yield after drying at 40°C/50mbar for 3 to 5 hours the product in 100%yield.
Example 13Crystallization from ethanol
- [0099]
1g of marbofloxacin was dissolved under heating to reflux in 160ml of ethanol, after filtration, the solution is cooled and the crystallized product is recovered in more than 90% yield.
Example 146,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
- [0100]
- [0101]
10mmol of 6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester was put in the round bottomed flask. 20mL of 10% H2SO4 was added and stirred with the temperature of the sand bath of 100°C for the time periods specified in the following table. Reaction mixture was cooled down to 4°C, filtered and the cake washed with water and the conversion an yield were determined. - [0102]
The experiment was repeated but starting compound was mixed with 1.0mL of solvent (EtOH, AcOH or MeCN as specified in the following table) before adding the 10% H2SO4. - [0103]
The starting compound is insoluble in aqueous phase. By mixing the starting compound with a small amount of polar solvent (EtOH, MeCN, AcOH) a film is formed around the crystals which improves wetting of the crystals with the aqueous acid. Without addition of polar solvent prior to adding the aqueous acid solution wetting of the crystals is impaired and the reaction is slower.Exp.Reaction time (solvent)Conversion (yield)14.016h65%14.027h60%14.0324h100%14.0424h100% (94%)14.056h (0.1mL AcOH per mmol)91%14.066h (0.1mL EtOH per mmol)89%14.0721h (0.1mL MeCN per mmol)100% (97%)14.0821h (0.1mL MeCN per mmol)100% (96%)
Example 156,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
- [0104]
3.30g of 6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester (10.054 mmol) was put into the round-bottomed flask equipped with the magnetic stirrer. 1mL of MeCN was added and stirred for a minute. 20mL of 10% H2SO4 was added and stirred. The flask was put into the sand bath (T = 100°C) and stirred for 21h. Suspension was cooled down to 4°C and filtered under suction. Yellow powder was washed twice with cold water and dried. 2.646g of yellow powder was obtained (9.721 mmol, 96.7%) and identified by NMR spectroscopy to be title compound.
Example 166,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
- [0105]
6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester (6.868g, 20.92 mmol) was mixed with 1mL of EtOH (to decrease the hydrophobicity of the substrate). Next, 40mL of 10% aqueous H2SO4 solution was added and the mixture was stirred at the temperature of the bath of 100°C for 12h. A white suspension formed which was cooled to 0°C and filtered under reduced pressure. The white powder was washed with cold water and cold EtOH and dried. 5.135g of yellow powder was obtained and identified as title compound by 19F and 1H NMR spectroscopy. The yield of hydrolysis was 90%.
Example 176,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
- [0106]
- [0107]
6,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid (272mg, 1.0mmol, obtained as described in Example 16, and 400 mg of N-methylpiperazine (4.0mmol) were mixed with 1mL of EtOH and stirred under reflux temperature (jacket temperature Tj=100°C). After two hours of reaction clear solution formed, afterwards the product precipitated and a very dense suspension was formed. Reaction was stopped after three hours of stirring at Tj=100°C. A sample was put directly to the NMR analysis and only two signals were observed indicating reaction was quantitative. Crude reaction product was diluted with EtOH and neutralized by addition of aqueous solution of NH3 until pH of 8 was reached. Suspension was cooled to 0°C and product isolated by filtration under reduced pressure, washed further with 10mL of cool EtOH and dried. 138mg (39%) of product was obtained.
Example 186,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
- [0108]
6,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid (1.087g, 3.993mmol), 484mg of N-methylpiperazine (4.83mmol) and 484 mg of Et3N (4.78mmol) were mixed with 8mL of EtOH and stirred under reflux temperature (Tj=100°C). After 19h of reflux yellow solution and white precipitate are formed in the reaction flask. Solvent was evaporated under reduced pressure and put directly to the NMR analysis. Crude reaction product was mixed with 20mL of EtOH and suspension cooled in the refrigerator. The product (white precipitate) was isolated by filtration under reduced pressure, washed further with 10mL of cool EtOH and dried. 1.178g of white powder was obtained (3.375 mmol, 800).
Example 196,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
- [0109]
In accordance with examples 17 and 18 additional experiments were carried out using different reaction conditions for the conversion of 6,7,8-trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid into 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid. The experiments were performed according to the following general procedure: 1.0mm of starting compound was put in the round bottomed flask and N-methylpiperazine (NMP), base and solvent were added according to the following table. Reaction mixture was stirred at the corresponding temperature. Solvent was evaporated and crude reaction mixture analyzed directly by NMR (1H and 19F).
Example 206,8-Difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester
- [0111]
- [0112]
Substitution: 6,7,8-Trifluoro-1-(N-methylformamido)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester (1.0 mmol, 324mg) was mixed with 2 equivalents of N-methylpiperazine (220mg) and 400mg Et3N stirred for three hours at 100°C. Reaction mixture liquefied in 10 minutes and solidified again within 30 minutes of the reaction (that is the reason for higher amount of TEA). After 3 hours of stirring was reaction mixture cooled to room temperature and analyzed by NMR spectroscopy. - [0113]
Substitution: The above reaction was repeated but Et3N was replaced by 1 equivalent of DABCO. - [0114]
In both cases, substitution was quantitative and analysis of the crude reaction mixtures showed that there was some hydrolysis of the ethyl ester (EE) to the free carboxylic acid (CA) group resulting in a product mixture. The results are summarized in the following table. Ethyl ester is readily soluble in water.Exp.Reaction conditionsConversion (yield)20.012.5 NMP, 1 DABCO, 100°C, 3h100% (48% EE, 52% CA)20.022.5 NMP, 4 Et3N, 100°C, 3h100% (58% EE, 42% CA)
Example 216,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid (one-pot reaction)
- [0115]
- [0116]
6,7,8-Trifluoro-1-(N-methylformamido)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester (1.0 mmol, 324mg) was mixed with 2 equivalents of N-methylpiperazine (200mg) and stirred for one hour at 100°C. Reaction mixture liquefied in 10 minutes and solidified again within 30 minutes of reaction. After one hour of reaction the reaction mixture was cooled to room temperature and 10% aqueous H2SO4 (5mL) was added and stirred again at 100°C for two hours. Yellow solution was cooled to 0°C so that product precipitated. It was isolated by filtration under reduced pressure. Pure 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid in the form of sulfate salt was obtained (as determined by NMR) as slightly yellow powder (279mg, 58%).
Example 22Synthesis of 9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3, 7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid (Marbofloxacin, MBX)
- [0117]
13.5 g of 6,8-Difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride and ca. 63 g of tetramethylammonium hydroxide water solution 25 % were charged into a reactor and slowly heated to 100°C. When this temperature was reached, water was removed by distillation at reduced pressure (between 0.8 to 0.3 bar) in such a manner that ca. 25 to 32 ml of water were removed in 3 hours. The reaction mixture was stirred for another 3 hours and after completion of the conversion, the reaction mixture was cooled to 0 – 10 °C and ca. 40.5 ml of formic acid were slowly added with violent agitation. The temperature was maintained below 20°C, preferable between 0 – 10°C. Then ca. 6.1 ml of formaldehyde were slowly added. After addition the reaction mixture was heated to 70°C and maintained at this temperature for about 30 minutes. - [0118]
The reaction mixture was cooled to room temperature (20 – 30°C), ca. 27 ml of purified water were added and the mixture was stirred for 30 minutes. Then the reaction mixture was cooled to 0 – 5°C and stirred at this temperature for at least 2 hours. The product marbofloxacin formate (MBXBZ) was centrifuged and washed with 10 – 15 g of cooled (0 – 5°C) purified water. The product was spun dried and collected. - [0119]
Wet product MBXBZ was added to the mixture of 67 ml of ethanol, 67 ml of methylene chloride and 16.2 ml of ammonia solution (ca. 25 0). If phases did not separate, additional 63 ml of methylene chloride and 33 ml of purified water were added. The pH of the water phase was adjusted to be between 7 and 9.5, preferable between 7.5 and 8.5. The mixture was agitated for approximately 15 minutes to 1 hour and then the layers were separated and both phases were subjected to in process control (IPC) analysis. - [0120]
If IPC results showed that extraction was not complete, ca. 63 ml of methylene chloride were added to the water layer and the extraction was repeated until the IPC specification was met. - [0121]
The organic phases were combined and ca. 6.8 mg of sodium sulphate anhydrous and optionally 0.4 mg of activated charcoal were added. The mixture was mixed for at least 30 minutes and filtered, then organic solvent was distilled off to obtain crude marbofloxacin.
Purification of the crude Marbofloxacin
- [0122]
In an inert atmosphere 5 g of purified water, 12 g of ethanol 96 % and 4.3 g of toluene (ratio between the solvents was within the following ranges: ethanol : toluene : water : 1.8 – 2.8 : 1 : 1.1 – 1.2) were charged into a reactor and wet crude marbofloxacin (MBXCA) from the previous step was added under nitrogen. The mixture was slowly heated to reflux (70 – 80°C) until a clear solution was obtained. The solution was stirred for 0.5 hour under this temperature and then one half of the azeotrope solvent mixture (toluene : water : ethanol = 51 % : 6 % : 43 %) was evaporated. Then the remaining mixture was cooled slowly to 5°C (allowed interval is between 0 and 25 °C) with agitation (optionally 1 % mass of product of disodium-EDTA can be added). The mixture was mixed for 1 to 3 hours and the product was then isolated by centrifugation, washed with 13 g of ethanol, spun dry and collected. The product was dried at temperature 40 – 45°C, p < 100 mbar for 8 hour.
Example 23Purification of Marbofloxacin
- [0123]
Marbofloxacin was dissolved in 20 parts by weight of water by addition of acetic acid. Marbofloxacin was completely dissolved at pH of 5.3. Active charcoal was added and the mixture was stirred overnight. The mixture was then filtered using activated charcoal filter. The pH of the filtrate was adjusted to 7.2 by use of KOH, the obtained suspension was stirred for 1 hour at room temperature and then the precipitated product was recovered. Marbofloxacin with a purity of 99,9% (HPLC area) was obtained. - [0124]
HPLC analysis was performed on a pentafluorophenyl propyl (PFP) column (type Luna® PFP, 150 x 4.6mm, 3µm, Phenomenex, USA); detector: UV315 nm; flow rate: 0.8 ml/min; injection volume: 5 µl; mobile phase: A: 0.02M NaH2PO4xH2O+0,1% TEA, pH2.5; B: acetonitrile : methanol = 5:95 (v/v) ; gradient: 0’=10B, 25’=100B, 30’= 100B, 32’=10B. The HPLC chromatogram of marbofloxacin prior to purification is shown in Figure 1, the HPLC chromatogram after purification is shown in Figure 2. As evident from the chromatograms all products with retention time above 24min were successfully eliminated.
Mechanism of action
Its mechanism of action is not thoroughly understood, but it is believed to be similar to the other fluoroquinolones by impairing the bacterial DNA gyrase which results in rapid bactericidal activity.[1] The other proposed mechanisms include that it acts against nondividing bacteria and does not require protein and RNA synthesis, which block protein and RNA synthesis respectively.[2]
Activity
Marbofloxacin is a synthetic, broad spectrum bactericidal agent. The bactericidal activity of marbofloxacin is concentration dependent, with susceptible bacteria cell death occurring within 20–30 minutes of exposure. Like other fluoroquinolones, marbofloxacin has demonstrated a significant post-antibiotic effect for both gram– and + bacteria and is active in both stationary and growth phases of bacterial replication.[3]
It has good activity against many gram-negative bacilli and cocci, is effective against:
- Aeromonas
- Brucella
- Campylobacter
- Chlamydia trachomatis
- Enterobacter
- Escherichia coli
- Haemophilus
- Klebsiella spp
- Mycobacterium
- Mycoplasma
- Proteus
- Pseudomonas aeruginosa
- Salmonella
- Serratia
- Shigella
- Staphylococci (including penicillinase-producing and methicillin-resistant strains)
- Vibrio
- Yersinia
Application
Marbofloxacin can be used both orally and topically. It is particularly used for infections of the skin, respiratory system and mammary glands in dogs and cats, as well as with urinary tract infections. For dogs, a dose ranges from 2.75 – 5.5 mg/kg once a day. The duration of treatment is usually at least five days, longer if there is a concurrent fungal or yeast infection.[4] Maximum duration of treatment is 30 days.[3]
Contraindications and side effects
Marbofloxacin should usually be avoided in young animals because of potential cartilage abnormalities. In rare occasion, it can cause central nervous system (CNS) stimulation and should be used with caution in patients with seizure disorders.[3] Under certain conditions it can cause discomfort such as cramps, treatable with diazepam. Other adverse effects are usually limited to gastrointestinal tract (GI) distress (vomiting, anorexia, soft stools, diarrhoea) and decreased activity.[3]
References
- ^ Boothe, D.M. (2001) Antimicrobial drugs. In Small Animal ClinicalPharmacology and Therapeutics, pp. 150–173. W. B. Saunders Co., Philadelphia, PA.
- ^ Hunter RP, Koch DE, Coke RL, Carpenter JW, Isaza R. Identification and comparison of marbofloxacin metabolites from the plasma of ball pythons (Python regius) and blue and gold macaws (Ara ararauna). J Vet Pharmacol Ther. 2007 Jun;30(3):257-62.
- ^ Jump up to:a b c d Plumb DC (ed). Plumb’s Veterinary Handbook, 7th ed. Ames, IA: Wiley-Blackwell Publishing, 2011.
- ^ Rougier S, Borell D, Pheulpin S, Woehrlé F, Boisramé B (October 2005). “A comparative study of two antimicrobial/anti-inflammatory formulations in the treatment of canine otitis externa”. Veterinary Dermatology. 16 (5): 299–307. doi:10.1111/j.1365-3164.2005.00465.x. PMID 16238809. Archived from the original on 2013-01-05.
External links
| Clinical data | |
|---|---|
| Trade names | XeniQuin bolus & Injection (Opsonin Agrovet BD) |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | By mouth |
| ATCvet code | QJ01MA93 (WHO) |
| Legal status | |
| Legal status | Veterinary use only |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 115550-35-1 |
| ChemSpider | 54663 |
| UNII | 8X09WU898T |
| ChEMBL | ChEMBL478120 |
| CompTox Dashboard (EPA) | DTXSID4046600 |
| ECHA InfoCard | 100.168.181 |
| Chemical and physical data | |
| Formula | C17H19FN4O4 |
| Molar mass | 362.356 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////////Marbofloxacin, марбофлоксацин , ماربوفلوكساسين , 马波沙星 ,

NEW DRUG APPROVALS
ONE TIME
$10.00
STAUROSPORINE

STAUROSPORINE
(+)-Staurosporine
- Molecular FormulaC28H26N4O3
- Average mass466.531 Da
(2S,3R,4R,6R)-3-Methoxy-2-methyl-4-(methylamino)-29-oxa-1,7,17-triazaoctacyclo[12.12.2.12,6.07,28.08,13.015,19.020,27.021,26]nonacosa-8,10,12,14,19,21,23,25,27-nonaen-16-one
6,10-Epoxy-6H,16H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-16-one, 7,8,9,10,17,18-hexahydro-7-methoxy-6-methyl-8-(methylamino)-, (6S,7R,8R,10R)-
62996-74-1[RN]
AM-2282
Antibiotic 230
antibiotic am 2282
StaurosporineCAS Registry Number: 62996-74-1
CAS Name: (9S,10R,11R,13R)- 2,3,10,11,12,13-Hexahydro-10-methoxy-9-methyl-11-(methylamino)-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3¢,2¢,1¢-lm]pyrrolo[3,4-j][1,7]benzodiazonin-1-one
Manufacturers’ Codes: AM-2282; CGP-39360
Molecular Formula: C28H26N4O3, Molecular Weight: 466.53
Percent Composition: C 72.09%, H 5.62%, N 12.01%, O 10.29%
Literature References: Protein kinase C inhibitor; alkaloid isolated from Streptomyces staurosporeus. Isoln: S. Omura et al., J. Antibiot.30, 275 (1977). Crystal and molecular structure: A. Furusaki et al., J. Chem. Soc. Chem. Commun.1978, 800; eidem,Bull. Chem. Soc. Jpn.55, 3681 (1982). Corrected stereochemistry: N. Funato et al., Tetrahedron Lett.35, 1251 (1994). Total synthesis: J. T. Link et al., J. Am. Chem. Soc.117, 552 (1995); idem et al., ibid.118, 2825 (1996). Biosynthetic studies: D. Meksuriyen, G. A. Cordell, J. Nat. Prod.51, 884, 893 (1988); S.-W. Yang et al., ibid.62 1551 (1999). HPLC determn in blood and pharmacokinetics in rats: L. R. Gurley et al., J. Chromatogr. B712, 211 (1998). Inhibition of protein kinase C: T. Tamaoki et al., Biochem. Biophys. Res. Commun.135, 397 (1986); of other protein kinases: U. T. Rüegg, G. M. Burgess, Trends Pharmacol. Sci.10, 218 (1989). Induction of apoptosis: E. Falcieri et al., Biochem. Biophys. Res. Commun.193, 19 (1993); R. Bertrand et al., Exp. Cell Res.211, 314 (1994); of tyrosine phosphorylation: D. Rasouly, P. Lazarovici, Eur. J. Pharmacol.269, 255 (1994).
Properties: Pale yellow needles from chloroform-methanol as the methanol solvate, mp 270° (dec) (Omura). Also reported as yellow crystals from methanol, mp 288-291° (Meksuriyen, Cordell). [a]D25 +35.0° (c = 1 in methanol); [a]D22 +56.1° (c = 0.14 in methanol). uv max (methanol): 241.0, 266.0, 292.5, 321.5, 335.0, 355.0, 372.5 nm (log e 4.25, 4.26, 4.53, 3.88, 3.96, 3.81, 3.85). Sol in DMSO, DMF. Slightly sol in chloroform, methanol.
Melting point: mp 270° (dec); mp 288-291° (Meksuriyen, Cordell)
Optical Rotation: [a]D25 +35.0° (c = 1 in methanol); [a]D22 +56.1° (c = 0.14 in methanol)
Absorption maximum: uv max (methanol): 241.0, 266.0, 292.5, 321.5, 335.0, 355.0, 372.5 nm (log e 4.25, 4.26, 4.53, 3.88, 3.96, 3.81, 3.85)
Derivative Type: Hydrochloride
Molecular Formula: C28H26N4O3.HCl, Molecular Weight: 502.99
Percent Composition: C 66.86%, H 5.41%, N 11.14%, O 9.54%, Cl 7.05%
Properties: LD50 in mice (mg/kg): 6.6 i.p. (Omura).
Toxicity data: LD50 in mice (mg/kg): 6.6 i.p. (Omura)
Use: Pharmacological tool to study signal transduction pathways, tyrosine phosphorylation and to induce apoptosis.
An indolocarbazole that is a potent protein kinase C inhibitor which enhances cAMP-mediated responses in human neuroblastoma cells. (Biochem Biophys Res Commun 1995;214(3):1114-20)
Staurosporine (antibiotic AM-2282 or STS) is a natural product originally isolated in 1977 from the bacterium Streptomyces staurosporeus.[1] It was the first of over 50 alkaloids to be isolated with this type of bis-indole chemical structure. The chemical structure of staurosporine was elucidated by X-ray analysis of a single crystal and the absolute stereochemical configuration by the same method in 1994.[2]
Staurosporine was discovered to have biological activities ranging from anti-fungal to anti-hypertensive.[3] The interest in these activities resulted in a large investigative effort in chemistry and biology and the discovery of the potential for anti-cancer treatment.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Synthesis Reference
Chikara Murakata, Toshimitsu Takiguchi, Shigeo Katsumata, Akira Mihara, Keiichi Takahashi, Hiromitsu Saito, Shiro Akinaga, Masami Okabe, Yutaka Saito, “Process for producing staurosporine derivatives.” U.S. Patent US5344926, issued December, 1990.
SYN
CN 113122591
WO 2021127275
CN 110642872
WO 2020200945
CN 107603922
WO2006002422
PAPER
Journal of Antibiotics, 51(7), 679-682; 1998
PAPER
Journal of the American Chemical Society, 117(1), 552-3; 1995

PATENT
WO2006002422
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2006002422
Preparation of Staurosporine Analogs
|00501 ] As will become apparent to a skilled artisan, many of the bridged epoxy diindolopyrrolo-hexahydrobenzodiazocines are commercially available as final compounds or modifiable intermediates. Staurosporine was originally isolated from the bacterium Streptomyces staurosporeus. (S.Omura et al. J.Antibiotics, 30, 275
1977).
[00502] Synthesis of 9, 12-epoxy staurosporine analogs:
NOTE: 2) 120°, 6) 120°,
Reactantsi 4, Reagents : 7, Catalysts. 2, Solvents 18,
Steps: 9, Stages: 11, Most stages in any one stept 2
[00503] Greater detail is provided in Tetrahedron Letters, 36(46), 8383-6,
1995.
[00504] Alternative synthesis of 9,12-epoxy staurosporine analogs:
NOTEt 1) stereoselective, 5) Raney nickel present,
Reactantsi 5, Reagentsi 6, Catalystsi 4, Solvents! 5,
Stepsi 7, stagest 9, Most stages in any one step: 2 [00505] Greater detail is provided in Organic Letters, 3(11), 1689-1692; 2001.
[00506]
[00507] Synthesis of 9, 13-epoxy staurosporine analogs:
NOTE: 1) STEREOSELECTIVE, 3) (92%/65%/95%/92%) , 4) 100% OVERALL (5.5:1,
ALPHA: BETA), 5) STEREOSELECTIVE, KEY STEP, 8) (97%/91%), 9) PHOTOCHEM.,
12) STEREOSELECTIVE KEY STEP, 14) (92%/81%/82%),
Reactants: 10, Reagents: 20, Catalysts: 5, Solvents: 9,
Steps: 17, Stages: 35, Most stages in any one step: 6
[00508] Whereas, a more thorough description of reagents, reaction conditions, and other pertinent syntheses are described Journal of the American Chemical Society, 117(1), 552-3; 1995. Additionally, syntheses on staurosporine and analogs thereof are described by S.J Danishefsky et al, J.Am.Chem.Soc, 118, 28251996 and J.L.Wood et al, J.Am.Chem.Soc, 118, 106561996.
Table 1 : Staurosporine Analogs
PATENT
https://patents.google.com/patent/WO2020200945A1/enEXAMPLES

.

A2 B2

Example 1Method for obtaining crude midostaurin B1 from crude staurosporine A1


A reactor was loaded with crude staurosporine A1 (1 mol) and DMF (7 L). The solution was cooled to 0°C and subsequently DIPEA (1.5 mol) was added. Benzoyl chloride (1.2 mol) was added while keeping the temperature within the range 0-5°C. After 30 minutes from the end of the addition, an aqueous 1 % ammonium chloride solution (15 L) was added while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the suspension was filtered and the panel was washed with plenty of water. The solid was dried for 6 hours at 40°C, obtaining crude midostaurin B1 with 95% yield. Example 2Method for obtaining purified midostaurin B2 from crude midostaurin B1 – reduction of 3-hydroxymidostaurin to midostaurin with triethylsilanei. TFA/TESii. NaHC03iii. Crystallizationin MeTHFiv. Crystallization
in EtOH/H20


B4 B2

A reactor was loaded with crude midostaurin B1 (1 mol) and DCM (10 L). The solution was cooled to 0°C and subsequently added with TES (1 mol) and TFA (0.50 L) in this order, while keeping the temperature within the range 0-5°C. At the end of the additions the solution was brought to 20°C. After 3 hours the solution was added with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (30 L) and two changes of solvent at atmospheric pressure were carried out. The solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C and subsequently transferred into another reactor. Ethanol (7 L) was added and the mixture was heated at 75°C up to complete dissolution. Water (30 L) was added with a concurrent cooling to 20°C. The resulting suspension was filtered and the panel was washed with plenty of water. The solid was dried for 12 hours at 80°C, obtaining purified midostaurin B2 with 85% yield. Example 3Method for obtaining purified staurosporine A2 from crude staurosporine A1 – reduction of 3-hydroxystaurosporine to staurosporine with triethylsilane

A reactor was loaded with crude staurosporine A1 (1 mol) and DCM (10 L). The solution was cooled to 0°C and subsequently added with TES (1 mol) and TFA (0.50 L) in this order, while keeping the temperature within the range 0-5°C. After 1 hour from the end of the additions, the solution was added with MeOH (10 L) and, subsequently, with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (50 L) and two changes of solvent at atmospheric pressure were carried out. The warm solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C, obtaining purified staurosporine A2 with 80% yield. Example 4Method for obtaining purified staurosporine A2 from crude staurosporine A1 – derivatization of 3-hydroxystaurosporine with trifluoroacetic acid and purification by crystallization


A reactor was loaded with crude staurosporine A1 (1 mol) and DCM (10 L). The mixture was cooled to 0°C and added with TFA (0.50 L), while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the solution was added with MeOH (10 L) and, subsequently, with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (50 L) and two changes of solvent at atmospheric pressure were carried out. The warm solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C, obtaining purified staurosporine A2 with 80% yield. Example 5Method for obtaining purified midostaurin B2 from purified staurosporine A2 i. BzCI/DIPEAii. NH4CI/H2Oiii. Crystallizationin MeTHFiv. Crystallization
in EtOH/H20


A2 B2

A reactor was loaded with purified staurosporine A2 (1 mol) and DMF (7 L). The solution was cooled to 0°C and subsequently DIPEA (1.5 mol) was added. Benzoyl chloride (1.2 mol) was added while keeping the temperature within the range 0-5°C. After 30 minutes from the end of the addition, an aqueous 1 % ammonium chloride solution (15 L) was added while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the suspension was filtered and the panel was washed with plenty of water. The solid was dried for 6 hours at 40°C and subsequently transferred into another reactor. 2-MeTHF (30 L) was added and the suspension was heated under reflux up to complete dissolution. The solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C and subsequently transferred into another reactor. Ethanol (7 L) was added and the mixture was heated at 75°C up to complete dissolution. Water (30 L) was added with a concurrent cooling to 20°C. The resulting suspension was filtered and the panel was washed with plenty of water. The solid was dried for 12 hours at 80°C, obtaining purified midostaurin B2 with 85% yield.
ClaimsHide Dependent
1) A process for the preparation of midostaurin with high purity, that is with a content of 3-hydroxymidostaurin impurities (III) and (IV) lower than 0.1%, comprising the treatment with strong organic or inorganic acids in a water-immiscible solvent and, optionally, also with reducing silanes.2) The process for the preparation of midostaurin according to claim 1 , comprising the treatment of crude midostaurin with a reducing silane in the presence of a strong organic or inorganic acid.3) The process for the preparation of midostaurin according to claim 1 , comprising the treatment of crude staurosporine with a strong organic or inorganic acid, optionally with the concomitant addition of a reducing silane.4) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the water-immiscible solvent is an aprotic polar water-immiscible solvent.5) The process for the preparation of midostaurin according to claim 4 wherein the water-immiscible solvent is dichloromethane, dichloroethane, methyl tetrahydrofuran or methylethylketone, preferably dichloromethane.6) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the strong acid is trifluoroacetic acid.7) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the reducing silane is triethylsilane.8) The process for the preparation of midostaturin according to anyone of the preceding claims, further comprising the benzoylation reaction of staurosporine to midostaurin characterized in that the benzoylation reaction is quenched with an aqueous solution having a slightly acid pH.9) The process for the preparation of midostaurin according to claim 8 wherein the aqueous solution having a slightly acid pH is an aqueous ammonium chloride solution.10) The process for the preparation of midostaurin according to anyone of the preceding claims, comprising the obtainment of purified midostaurin by crystallization from 2-MeTHF and its further isolation by:dissolving the crystallized midostaurin in a water-miscible polar solvent, adding waterisolating purified midostaurin as an amorphous solid obtained by filtering and drying, with a content of organic solvents < 50ppm. 11) The process for the preparation of purified midostaurin according to claim 10, wherein the polar s
Patent
Publication numberPriority datePublication dateAssigneeTitleJPS5247055B21973-12-041977-11-30US5093330A1987-06-151992-03-03Ciba-Geigy CorporationStaurosporine derivatives substituted at methylamino nitrogenEP0575955A11992-06-221993-12-29Kyowa Hakko Kogyo Co., Ltd.Process for producing staurosporine derivativesWO2006048296A12004-11-052006-05-11Novartis AgOrganic compoundsWO2011064355A12009-11-302011-06-03Novartis AgPolymorphous forms iii and iv of n-benzoyl staurosporineWO2018165071A12017-03-062018-09-13Teva Pharmaceutical Works Ltd.Solid state forms of midostaurin
Biological activities
The main biological activity of staurosporine is the inhibition of protein kinases through the prevention of ATP binding to the kinase. This is achieved through the stronger affinity of staurosporine to the ATP-binding site on the kinase. Staurosporine is a prototypical ATP-competitive kinase inhibitor in that it binds to many kinases with high affinity, though with little selectivity.[4] Structural analysis of kinase pockets demonstrated that main chain atoms which are conserved in their relative positions to staurosporine contributes to staurosporine promiscuity.[5] This lack of specificity has precluded its clinical use, but has made it a valuable research tool. In research, staurosporine is used to induce apoptosis. The mechanism of how it mediates this is not well understood. It has been found that one way in which staurosporine induces apoptosis is by activating caspase-3.[6] At lower concentration, depending on the cell type, staurosporine induces specific cell cycle effects arresting cells either in G1 or in G2 phase of the cell cycle.[7]
Chemistry family
Main article: Indolocarbazole
Staurosporine is an indolocarbazole. It belongs to the most frequently isolated group of indolocarbazoles: Indolo(2,3-a)carbazoles. Of these, Staurosporine falls within the most common subgroup, called Indolo(2,3-a)pyrrole(3,4-c)carbazoles. These fall into two classes – halogenated (chlorinated) and non-halogenated. Halogenated indolo(2,3-a)pyrrole(3,4-c)carbazoles have a fully oxidized C-7 carbon with only one indole nitrogen containing a β-glycosidic bond, while non-halogenated indolo(2,3-a)pyrrole(3,4-c)carbazoles have both indole nitrogens glycosylated, and a fully reduced C-7 carbon. Staurosporine is in the non-halogenated class.[8]
Staurosporine is the precursor of the novel protein kinase inhibitor midostaurin (PKC412).[9][10] Besides midostaurin, staurosporine is also used as a starting material in the commercial synthesis of K252c (also called staurosporine aglycone). In the natural biosynthetic pathway, K252c is a precursor of staurosporine.
pyrrole(3,4-c)carbazol.svg)
Structure of an Indolo[2,3-a]pyrrole[3,4-c]carbazol

Biosynthesis
The biosynthesis of staurosporine starts with the amino acid L-tryptophan in its zwitterionic form. Tryptophan is converted to an imine by enzyme StaO which is an L-amino acid oxidase (that may be FAD dependent). The imine is acted upon by StaD to form an uncharacterized intermediate proposed to be the dimerization product between 2 imine molecules. Chromopyrrolic acid is the molecule formed from this intermediate after the loss of VioE (used in the biosynthesis of violacein – a natural product formed from a branch point in this pathway that also diverges to form rebeccamycin. An aryl aryl coupling thought to be catalyzed by a cytochrome P450 enzyme to form an aromatic ring system occurs.[8]

This is followed by a nucleophilic attack between the indole nitrogens resulting in cyclization and then decarboxylation assisted by StaC exclusively forming staurosporine aglycone or K252c. Glucose is transformed to NTP-L-ristoamine by StaA/B/E/J/I/K which is then added on to the staurosporine aglycone at 1 indole N by StaG. The StaN enzyme reorients the sugar by attaching it to the 2nd indole nitrogen into an unfavored conformation to form intermediated O-demethyl-N-demethyl-staurosporine. Lastly, O-methylation of the 4’amine by StaMA and N-methylation of the 3′-hydroxy by StaMB leads to the formation of staurosporine.[8]
Research in clinical use
When encapsulated in liposome nanoparticle, staurosporine is shown to suppress tumors in vivo in a mouse model without the toxic side effects which have prohibited its use as an anti-cancer drug with high apoptotic activity. Researchers in UC San Diego Moores Cancer Center develop a platform technology of high drug-loading efficiency by manipulating the pH environment of the cells. When injected into the mouse glioblastoma model, staurosporine is found to accumulate primarily in the tumor via fluorescence confirmation, and the mice did not suffer weight loss compared to the control mice administered with the free compound, an indicator of reduced toxicity.[11][12]
References
- ^ Omura S, Iwai Y, Hirano A, Nakagawa A, Awaya J, Tsuchiya H, Takahashi Y, Masuma R (1977). “A new alkaloid AM-2282 of Streptomyces origin taxonomy, fermentation, isolation and preliminary characterization”. J. Antibiot. 30 (4): 275–282. doi:10.7164/antibiotics.30.275. PMID 863788.
- ^ Funato N, Takayanagi H, Konda Y, Toda Y, Harigaya Y, Omura S (1994). “Absolute configuration of staurosporine by X-ray analysis”. Tetrahedron Lett. 35 (8): 1251–1254. doi:10.1016/0040-4039(94)88036-0.
- ^ [1] Rüegg UT, Burgess GM. (1989) Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends in Pharmacological Science 10 (6): 218-220.
- ^ Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP (2008). “A quantitative analysis of kinase inhibitor selectivity”. Nat. Biotechnol. 26 (1): 127–132. doi:10.1038/nbt1358. PMID 18183025. S2CID 205273598.
- ^ Tanramluk D, Schreyer A, Pitt WR, Blundell TL (2009). “On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine”. Chemical Biology & Drug Design. 74 (1): 16–24. doi:10.1111/j.1747-0285.2009.00832.x. PMC 2737611. PMID 19519740.
- ^ Chae HJ, Kang JS, Byun JO, Han KS, Kim DU, Oh SM, Kim HM, Chae SW, Kim HR (2000). “Molecular mechanism of staurosporine-induced apoptosis in osteoblasts”. Pharmacological Research. 42 (4): 373–381. doi:10.1006/phrs.2000.0700. PMID 10987998.
- ^ Bruno S, Ardelt B, Skierski JS, Traganos F, Darzynkiewicz Z (1992). “Different effects of staurosporine, an inhibitor of protein kinases, on the cell cycle and chromatin structure of normal and leukemic lymphocytes”. Cancer Res. 52 (2): 470–473. PMID 1728418.
- ^ Jump up to:a b c Ryan KS (2008). “Structural studies of rebeccamycin, staurosporine, and violacein biosynthetic enzymes” (PDF). Ph.D. Thesis. Massachusetts Institute of Technology. Archived from the original (PDF) on 2012-03-14.
- ^ Midostaurin product page, Fermentek
- ^ Wang, Y; Yin, OQ; Graf, P; Kisicki, JC; Schran, H (2008). “Dose- and Time-Dependent Pharmacokinetics of Midostaurin in Patients With Diabetes Mellitus”. J Clin Pharmacol. 48 (6): 763–775. doi:10.1177/0091270008318006. PMID 18508951. S2CID 26657407.
- ^ News Release (21 October 2013). “Study Identifies Safe Delivery System for Tricky Yet Highly Potent Anti-Cancer Compounds”. UC San Diego Health System. Retrieved 27 October 2013.
- ^ Mukthavaram, Rajesh; Jiang, Pengei; Saklecha, Rohit; Simbery, Dmitri; Bharati, Ila; Nomura, Natsuko; Chao, Ying; Pastorino, Sandra (2013). “High-efficiency liposomal encapsulation of a tyrosine kinase inhibitor leads to improved in vivo toxicity and tumor response profile”. International Journal of Nanomedicine. 8 (1): 3991–4006. doi:10.2147/IJN.S51949. PMC 3808212. PMID 24174874.
| Clinical data | |
|---|---|
| ATC code | none |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 62996-74-1 |
| PubChem CID | 44259 |
| IUPHAR/BPS | 346 |
| DrugBank | DB02010 |
| ChemSpider | 40272 |
| UNII | H88EPA0A3N |
| ChEBI | CHEBI:15738 |
| ChEMBL | ChEMBL162 |
| PDB ligand | STU (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID30911019 DTXSID6041131, DTXSID30911019 |
| ECHA InfoCard | 100.109.946 |
| Chemical and physical data | |
| Formula | C28H26N4O3 |
| Molar mass | 466.541 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////STAUROSPORINE, AM-2282, CGP-39360
[H][C@]1(C[C@@]2([H])O[C@](C)(N3C4=CC=CC=C4C4=C5CNC(=O)C5=C5C6=CC=CC=C6N2C5=C34)[C@]1([H])OC)NC

NEW DRUG APPROVALS
ONE TIME
$10.00
Alpha lipoic acid
Alpha lipoic acid
(+)-Thioctic acid
- Molecular FormulaC8H14O2S2
- Average mass206.326 Da
5-[3-(1,2-Dithiolanyl)]pentanoic Acid
5-19-07-00237[Beilstein]
62-46-4[RN](+)-Thioctic acid, (+)-α-Lipoic acid, (3R)-1,2-Dithiolane-3-pentanoic acid
(R)-(+)-1,2-Dithiolane-3-pentanoic acid, (R)-(+)-lipoic acid, (R)-(+)-α-Lipoic acid
(R)-6,8-Dithiooctanoic acid, (R)-6,8-thioctic acid, (R)-α-Lipoic Acid, (R)-α-Lipoic Acid
1,2-Dithiolane-3-pentanoic acid, (3R)-
5-[(3R)-1,2-Dithiolan-3-yl]pentanoic acidd-Thioctic acid, (R)-(+)-alpha-Lipoic acid, (R)-(+)-Thioctic acid, Dexlipotam
Thioctic Acid
CAS Registry Number: 62-46-4
CAS Name: 1,2-Dithiolane-3-pentanoic acid
Additional Names: 1,2-dithiolane-3-valeric acid; 6,8-thioctic acid; a-lipoic acid; 5-(1,2-dithiolan-3-yl)valeric acid; 5-[3-(1,2-dithiolanyl)]pentanoic acid; d-[3-(1,2-dithiacyclopentyl)]pentanoic acid; protogen A; acetate replacing factor; pyruvate oxidation factor
Trademarks: Biletan (Gador); Thioctacid (Viatris); Thioctan (Katwijk); Tioctan (Fujisawa)
Molecular Formula: C8H14O2S2, Molecular Weight: 206.33
Percent Composition: C 46.57%, H 6.84%, O 15.51%, S 31.08%
Literature References: Growth factor for many bacteria and protozoa; prosthetic group, coenzyme, or substrate in plants, microorganisms, and animal tissues. Isoln of naturally occurring d-form: L. J. Reed et al.,Science114, 93 (1951); eidem,J. Am. Chem. Soc.75, 1267 (1953); Patterson et al.,ibid.76, 1823 (1954). Syntheses of dl-form: Bullock et al.,ibid.74, 1868, 3455 (1952); Hornberger et al.,ibid. 2382; Reed, US2980716 and US3049549 (1961, 1962 to Res. Corp.); Lewis, Raphael, J. Chem. Soc.1962, 4263; Ose et al.,US3223712 (1965 to Yamanouchi); J. Tsuji et al.,J. Org. Chem.43, 3606 (1978). Biosynthesis via linoleic acid: J. P. Carreau Methods Enzymol.62, 152-158 (1974). Enantioselective synthesis of d-form: P. C. Bulmanpage et al.,Chem. Commun.1986, 1408. Clinical study in treatment of Wilson’s disease: S. F. Gomes da Costa, Arzneim.-Forsch.20, 1210 (1970). Use in treatment of mushroom poisoning: R. Plotzker et al.,Am. J. Med. Sci.283, 79 (1982); J. P. Hanrahan, M. A. Gordon, J. Am. Med. Assoc.251, 1057 (1984). Reviews: Wagner, Folkers, Vitamins and Coenzymes (Interscience, New York, 1964) pp 244-263; Schmidt et al.,Angew. Chem. Int. Ed.4, 846 (1965); Schmidt et al.,Adv. Enzymol. Relat. Areas Mol. Biol.32, 423 (1969).
Derivative Type: Sodium salt
CAS Registry Number: 2319-84-8
Molecular Formula: C8H13NaO2S2, Molecular Weight: 228.31
Percent Composition: C 42.09%, H 5.74%, Na 10.07%, O 14.02%, S 28.09%
Properties: White powder, sol in water. pH of aq solns about 7.4.
Derivative Type:d-Form
CAS Registry Number: 1200-22-2
Properties: Crystals by vacuum sublimation (at 85-90° and 25 microns). mp 46-48° (microblock). [a]D23 +104° (c = 0.88 in benzene). uv max (methanol): 333 nm (e 150). pKa 5.4. Practically insol in water. Sol in fat solvents.Melting point: mp 46-48° (microblock)
pKa: pKa 5.4
Optical Rotation: [a]D23 +104° (c = 0.88 in benzene)
Absorption maximum: uv max (methanol): 333 nm (e 150)
Derivative Type:dl-Form
CAS Registry Number: 1077-28-7
Properties: Yellow needles from cyclohexane, mp 60-61°. bp 160-165°. uv spectrum: Calvin, Fed. Proc.13, 703 (1954). Practically insol in water. Sol in fat solvents. Forms a water-soluble sodium salt.
Melting point: mp 60-61°
Boiling point: bp 160-165°
Derivative Type:l-Form
CAS Registry Number: 1077-27-6
Properties: Crystals from cyclohexane, mp 45-47.5° (microblock). [a]D23 -113° (c = 1.88 in benzene). uv max (methanol): 330 nm (e 140).
Melting point: mp 45-47.5° (microblock)
Optical Rotation: [a]D23 -113° (c = 1.88 in benzene)
Absorption maximum: uv max (methanol): 330 nm (e 140)
Derivative Type: Ethylenediamine
Trademarks: Tioctidasi (ISI)
Therap-Cat: Treatment of liver disease; antidote to poisonous mushrooms (Amanita species).
Keywords: Hepatoprotectant.
Lipoic acid (LA), also known as α-lipoic acid, alpha-lipoic acid (ALA) and thioctic acid, is an organosulfur compound derived from caprylic acid (octanoic acid).[3] ALA is made in animals normally, and is essential for aerobic metabolism. It is also manufactured and is available as a dietary supplement in some countries where it is marketed as an antioxidant, and is available as a pharmaceutical drug in other countries.[3]
Physical and chemical properties
Lipoic acid (LA), also known as α-lipoic acid,[3][4] alpha-lipoic acid (ALA), and thioctic acid[5] is an organosulfur compound derived from octanoic acid.[3] LA contains two sulfur atoms (at C6 and C8) connected by a disulfide bond and is thus considered to be oxidized although either sulfur atom can exist in higher oxidation states.[3]
The carbon atom at C6 is chiral and the molecule exists as two enantiomers (R)-(+)-lipoic acid (RLA) and (S)-(-)-lipoic acid (SLA) and as a racemic mixture (R/S)-lipoic acid (R/S-LA).
LA appears physically as a yellow solid and structurally contains a terminal carboxylic acid and a terminal dithiolane ring.
For use in dietary supplement materials and compounding pharmacies, the USP has established an official monograph for R/S-LA.[6][7]
Biological function
“Lipoate” is the conjugate base of lipoic acid, and the most prevalent form of LA under physiological conditions.[3] Most endogenously produced RLA are not “free” because octanoic acid, the precursor to RLA, is bound to the enzyme complexes prior to enzymatic insertion of the sulfur atoms. As a cofactor, RLA is covalently attached by an amide bond to a terminal lysine residue of the enzyme’s lipoyl domains. One of the most studied roles of RLA is as a cofactor of the pyruvate dehydrogenase complex (PDC or PDHC), though it is a cofactor in other enzymatic systems as well (described below).[3]
Only the (R)-(+)-enantiomer (RLA) exists in nature and is essential for aerobic metabolism because RLA is an essential cofactor of many enzyme complexes.[3]
Biosynthesis and attachment
The precursor to lipoic acid, octanoic acid, is made via fatty acid biosynthesis in the form of octanoyl-acyl carrier protein.[3] In eukaryotes, a second fatty acid biosynthetic pathway in mitochondria is used for this purpose.[3] The octanoate is transferred as a thioester of acyl carrier protein from fatty acid biosynthesis to an amide of the lipoyl domain protein by an enzyme called an octanoyltransferase.[3] Two hydrogens of octanoate are replaced with sulfur groups via a radical SAM mechanism, by lipoyl synthase.[3] As a result, lipoic acid is synthesized attached to proteins and no free lipoic acid is produced. Lipoic acid can be removed whenever proteins are degraded and by action of the enzyme lipoamidase.[8] Free lipoate can be used by some organisms as an enzyme called lipoate protein ligase that attaches it covalently to the correct protein. The ligase activity of this enzyme requires ATP.[9]
Cellular transport
Along with sodium and the vitamins biotin (B7) and pantothenic acid (B5), lipoic acid enters cells through the SMVT (sodium-dependent multivitamin transporter). Each of the compounds transported by the SMVT is competitive with the others. For example research has shown that increasing intake of lipoic acid[10] or pantothenic acid[11] reduces the uptake of biotin and/or the activities of biotin-dependent enzymes.
Enzymatic activity
Lipoic acid is a cofactor for at least five enzyme systems.[3] Two of these are in the citric acid cycle through which many organisms turn nutrients into energy. Lipoylated enzymes have lipoic acid attached to them covalently. The lipoyl group transfers acyl groups in 2-oxoacid dehydrogenase complexes, and methylamine group in the glycine cleavage complex or glycine dehydrogenase.[3]
2-Oxoacid dehydrogenase transfer reactions occur by a similar mechanism in:
- the pyruvate dehydrogenase complex
- the α-ketoglutarate dehydrogenase or 2-oxoglutarate dehydrogenase complex
- the branched-chain oxoacid dehydrogenase (BCDH) complex
- the acetoin dehydrogenase complex.
The most-studied of these is the pyruvate dehydrogenase complex.[3] These complexes have three central subunits: E1-3, which are the decarboxylase, lipoyl transferase, and dihydrolipoamide dehydrogenase, respectively. These complexes have a central E2 core and the other subunits surround this core to form the complex. In the gap between these two subunits, the lipoyl domain ferries intermediates between the active sites.[3] The lipoyl domain itself is attached by a flexible linker to the E2 core and the number of lipoyl domains varies from one to three for a given organism. The number of domains has been experimentally varied and seems to have little effect on growth until over nine are added, although more than three decreased activity of the complex.[12]
Lipoic acid serves as co-factor to the acetoin dehydrogenase complex catalyzing the conversion of acetoin (3-hydroxy-2-butanone) to acetaldehyde and acetyl coenzyme A.[3]
The glycine cleavage system differs from the other complexes, and has a different nomenclature.[3] In this system, the H protein is a free lipoyl domain with additional helices, the L protein is a dihydrolipoamide dehydrogenase, the P protein is the decarboxylase, and the T protein transfers the methylamine from lipoate to tetrahydrofolate (THF) yielding methylene-THF and ammonia. Methylene-THF is then used by serine hydroxymethyltransferase to synthesize serine from glycine. This system is part of plant photorespiration.[13]
Biological sources and degradation
Lipoic acid is present in many foods in which it is bound to lysine in proteins,[3] but slightly more so in kidney, heart, liver, spinach, broccoli, and yeast extract.[14] Naturally occurring lipoic acid is always covalently bound and not readily available from dietary sources.[3] In addition, the amount of lipoic acid present in dietary sources is low. For instance, the purification of lipoic acid to determine its structure used an estimated 10 tons of liver residue, which yielded 30 mg of lipoic acid.[15] As a result, all lipoic acid available as a supplement is chemically synthesized.
Baseline levels (prior to supplementation) of RLA and R-DHLA have not been detected in human plasma.[16] RLA has been detected at 12.3−43.1 ng/mL following acid hydrolysis, which releases protein-bound lipoic acid. Enzymatic hydrolysis of protein bound lipoic acid released 1.4−11.6 ng/mL and <1-38.2 ng/mL using subtilisin and alcalase, respectively.[17][18][19]
Digestive proteolytic enzymes cleave the R-lipoyllysine residue from the mitochondrial enzyme complexes derived from food but are unable to cleave the lipoic acid-L–lysine amide bond.[20] Both synthetic lipoamide and (R)-lipoyl-L-lysine are rapidly cleaved by serum lipoamidases, which release free (R)-lipoic acid and either L-lysine or ammonia.[3] Little is known about the degradation and utilization of aliphatic sulfides such as lipoic acid, except for cysteine.[3]
Lipoic acid is metabolized in a variety of ways when given as a dietary supplement in mammals.[3][21] Degradation to tetranorlipoic acid, oxidation of one or both of the sulfur atoms to the sulfoxide, and S-methylation of the sulfide were observed. Conjugation of unmodified lipoic acid to glycine was detected especially in mice.[21] Degradation of lipoic acid is similar in humans, although it is not clear if the sulfur atoms become significantly oxidized.[3][22] Apparently mammals are not capable of utilizing lipoic acid as a sulfur source.
Chemical synthesis
-Lipoic_acid_molecular_structure.png)
(R)-Lipoic acid (RLA, top) and (S)-lipoic acid (SLA, down). A 1:1 mixture (racemate) of (R)- and (S)-lipoic acid is called (RS)-lipoic acid or (±)-lipoic acid (R/S-LA).
SLA did not exist prior to chemical synthesis in 1952.[23][24] SLA is produced in equal amounts with RLA during achiral manufacturing processes. The racemic form was more widely used clinically in Europe and Japan in the 1950s to 1960s despite the early recognition that the various forms of LA are not bioequivalent.[25] The first synthetic procedures appeared for RLA and SLA in the mid-1950s.[26][27][28][29] Advances in chiral chemistry led to more efficient technologies for manufacturing the single enantiomers by both classical resolution and asymmetric synthesis and the demand for RLA also grew at this time. In the 21st century, R/S-LA, RLA and SLA with high chemical and/or optical purities are available in industrial quantities. At the current time, most of the world supply of R/S-LA and RLA is manufactured in China and smaller amounts in Italy, Germany, and Japan. RLA is produced by modifications of a process first described by Georg Lang in a Ph.D. thesis and later patented by DeGussa.[30][31] Although RLA is favored nutritionally due to its “vitamin-like” role in metabolism, both RLA and R/S-LA are widely available as dietary supplements. Both stereospecific and non-stereospecific reactions are known to occur in vivo and contribute to the mechanisms of action, but evidence to date indicates RLA may be the eutomer (the nutritionally and therapeutically preferred form).[32][33]
Pharmacology
Pharmacokinetics
A 2007 human pharmacokinetic study of sodium RLA demonstrated the maximum concentration in plasma and bioavailability are significantly greater than the free acid form, and rivals plasma levels achieved by intravenous administration of the free acid form.[34] Additionally, high plasma levels comparable to those in animal models where Nrf2 was activated were achieved.[34]
The various forms of LA are not bioequivalent.[25][non-primary source needed] Very few studies compare individual enantiomers with racemic lipoic acid. It is unclear if twice as much racemic lipoic acid can replace RLA.[34]
The toxic dose of LA in cats is much lower than that in humans or dogs and produces hepatocellular toxicity.[35]
Pharmacodynamics
The mechanism and action of lipoic acid when supplied externally to an organism is controversial. Lipoic acid in a cell seems primarily to induce the oxidative stress response rather than directly scavenge free radicals. This effect is specific for RLA.[4] Despite the strongly reducing milieu, LA has been detected intracellularly in both oxidized and reduced forms.[36] LA is able to scavenge reactive oxygen and reactive nitrogen species in a biochemical assay due to long incubation times, but there is little evidence this occurs within a cell or that radical scavenging contributes to the primary mechanisms of action of LA.[4][37] The relatively good scavenging activity of LA toward hypochlorous acid (a bactericidal produced by neutrophils that may produce inflammation and tissue damage) is due to the strained conformation of the 5-membered dithiolane ring, which is lost upon reduction to DHLA. In cells, LA is reduced to dihydrolipoic acid, which is generally regarded as the more bioactive form of LA and the form responsible for most of the antioxidant effects and for lowering the redox activities of unbound iron and copper.[38] This theory has been challenged due to the high level of reactivity of the two free sulfhydryls, low intracellular concentrations of DHLA as well as the rapid methylation of one or both sulfhydryls, rapid side-chain oxidation to shorter metabolites and rapid efflux from the cell. Although both DHLA and LA have been found inside cells after administration, most intracellular DHLA probably exists as mixed disulfides with various cysteine residues from cytosolic and mitochondrial proteins.[32] Recent findings suggest therapeutic and anti-aging effects are due to modulation of signal transduction and gene transcription, which improve the antioxidant status of the cell. However, this likely occurs via pro-oxidant mechanisms, not by radical scavenging or reducing effects.[4][37][39]
All the disulfide forms of LA (R/S-LA, RLA and SLA) can be reduced to DHLA although both tissue specific and stereoselective (preference for one enantiomer over the other) reductions have been reported in model systems. At least two cytosolic enzymes, glutathione reductase (GR) and thioredoxin reductase (Trx1), and two mitochondrial enzymes, lipoamide dehydrogenase and thioredoxin reductase (Trx2), reduce LA. SLA is stereoselectively reduced by cytosolic GR whereas Trx1, Trx2 and lipoamide dehydrogenase stereoselectively reduce RLA. (R)-(+)-lipoic acid is enzymatically or chemically reduced to (R)-(-)-dihydrolipoic acid whereas (S)-(-)-lipoic acid is reduced to (S)-(+)-dihydrolipoic acid.[40][41][42][43][44][45][46] Dihydrolipoic acid (DHLA) can also form intracellularly and extracellularly via non-enzymatic, thiol-disulfide exchange reactions.[47]
RLA may function in vivo like a B-vitamin and at higher doses like plant-derived nutrients, such as curcumin, sulforaphane, resveratrol, and other nutritional substances that induce phase II detoxification enzymes, thus acting as cytoprotective agents.[39][48] This stress response indirectly improves the antioxidant capacity of the cell.[4]
The (S)-enantiomer of LA was shown to be toxic when administered to thiamine-deficient rats.[49][50]
Several studies have demonstrated that SLA either has lower activity than RLA or interferes with the specific effects of RLA by competitive inhibition.[51][52][53][54][55]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Uses
R/S-LA and RLA are widely available as over-the-counter nutritional supplements in the United States in the form of capsules, tablets, and aqueous liquids, and have been marketed as antioxidants.[3]
Although the body can synthesize LA, it can also be absorbed from the diet. Dietary supplementation in doses from 200–600 mg is likely to provide up to 1000 times the amount available from a regular diet. Gastrointestinal absorption is variable and decreases with the use of food. It is therefore recommended that dietary LA be taken 30–60 minutes before or at least 120 minutes after a meal. Maximum blood levels of LA are achieved 30–60 minutes after dietary supplementation, and it is thought to be largely metabolized in the liver.[56]
In Germany, LA is approved as a drug for the treatment of diabetic neuropathy since 1966 and is available as a non-prescription pharmaceutical.[57]
Clinical research
According to the American Cancer Society as of 2013, “there is no reliable scientific evidence at this time that lipoic acid prevents the development or spread of cancer”.[58] As of 2015, intravenously administered ALA is unapproved anywhere in the world except Germany for diabetic neuropathy, but has been proven reasonably safe and effective in four clinical trials; however another large trial over four years found no difference from placebo.[59] As of 2012, there was no good evidence alpha lipoic acid helps people with mitochondrial disorders.[60] A 2018 review recommended ALA as an anti-obesity supplement with low dosage (< 600 mg/day) for a short period of time (<10 weeks); however, it is too expensive to be practical as a complementary therapy for obesity.[61]
SYN
WO 0210151
DE 19709069; EP 0863125; US 6013833
A synthetic route based on the asymmetric reduction of oxo diesters has been reported. Meldrum’s acid (LII) was acylated by methyl adipoyl chloride (LI) in the presence of pyridine to produce the intermediate (LIII) which, upon alcoholysis with isobutanol, led to oxo diester (LIV). Enantioselective reduction of (LIV) by means of baker’s yeast furnished the (S)-hydroxy diester (LV). Alternatively, the analogous oxo diester (LVI) was prepared by acylation of methyl acetoacetate with methyl adipoyl chloride (LI), followed by deacetylation in the presence of ammonium hydroxide. Then, asymmetric chemical reduction of (LVI) by hydrogenation in the presence of the chiral catalyst Ru2Cl4[(S)-BINAP]2 provided the (S)-hydroxy diester (LVII). Regioselective reduction of either diester (LV) or (LVII) by means of NaBH4 in refluxing THF furnished dihydroxy ester (XLVIII). After conversion of (XLVIII) to the dimesylate (XLIX), displacement with potassium thioacetate afforded the bis(acetylthio) derivative (LVIII), which was further hydrolyzed with KOH to provide dihydrolipoic acid (LIX). In a related procedure, dihydrolipoic acid (LIX) was prepared by reaction of dimesylate (XLIX) with sodium disulfide, followed by reductive treatment with NaBH4 and NaOH. The title cyclic disulfide was then obtained by oxidation of the dithiol (LIX) using oxygen in the presence of FeCl3.
SYN
DE 10036516; WO 0210113
The key dihydroxy ester intermediate (XIII) was also obtained by asymmetric hydrogenation of hydroxy ketoester (XLIII) in the presence of (S)-BINAP-dichlororuthenium catalyst. The precursor hydroxy ketoester (XLIII) was prepared by two alternative procedures. In one method, the racemic dihydroxy ester (XLII) was selectively oxidized to (XLIII) by means of NaOCl. In another method, the unsaturated keto ester (XLIV) was epoxidized by means of sodium percarbonate, and the resultant epoxide (XLV) was then reduced to the hydroxy ketoester (XLIII) by catalytic hydrogenation over PtO2.
SYN
WO 0230919
Both enantiomers of racemic 8-chloro-6-hydroxyoctanoic acid (LX) were separated employing either (+)- or (-)-alpha-methylbenzylamine. Esterification of the (R)-(-)-enantiomer with HCl-MeOH provided the chloro hydroxy ester (LXI). Further chlorination of (LXI) with SOCl2 and pyridine proceeded with inversion of configuration at C-6 to furnish the (S)-dichloro derivative (LXII). The cyclic disulfide (L) was then prepared by treatment of chloride (LXII) with sulfur and sodium sulfide in boiling EtOH. Basic hydrolysis of the methyl ester group of (LXII) then afforded (R) alpha lipoic acid. The title compound was also obtained from the (S)-(+)-acid (LXIII). Reaction of hydroxy acid (LXIII) with methanesulfonyl chloride produced the chloro mesylate (LXIV), which was then cyclized to the target disulfide in the presence of sulfur and Na2S.
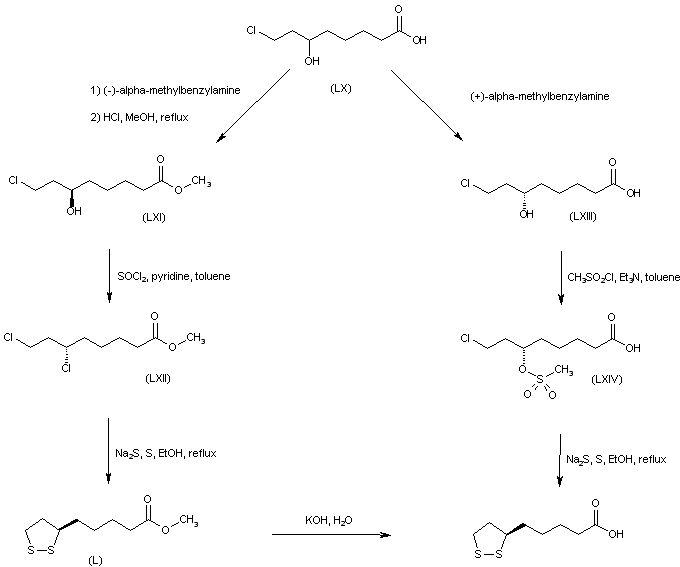
SYN
The reaction of the chiral dibenzoyloxy-dihydropyran (LXV) with H2SO4 and HgSO4 gives the unsaturated aldehyde (LXVI), which is condensed with the phosphorane (LXVII) to yield the hepatdienoic ester (LXVIII). The hydrogenation of (LXVIII) with H2 over Pd/C affords the heptanoic ester (LXIX), which is treated with Ts-Cl and pyridine to provide the tosyloxy derivative (LXX). The cyclization of (LXX) by means of K2CO3 gives the chiral epoxide (LXXI), which is condensed with vinylmagnesium bromide (LXXII) to yield 6(S)-hydroxy-8-nonenoic acid methyl ester (LXXIII). The oxidation of the terminal double bond of (LXXIII) with ozone affords the carbaldehyde (LXXIV), which is reduced with NaBH4 to provide 6(S),8-dihydroxyoctanoic acid methyl ester (XLVIII). The reaction of (XLVIII) with Ms-Cl and pyridine gives the dimesylate (XLIX), which is treated with Na2S2 to yield the lipoic acid methyl ester (L), which is hydrolyzed to the target acid with KOH in H2O.
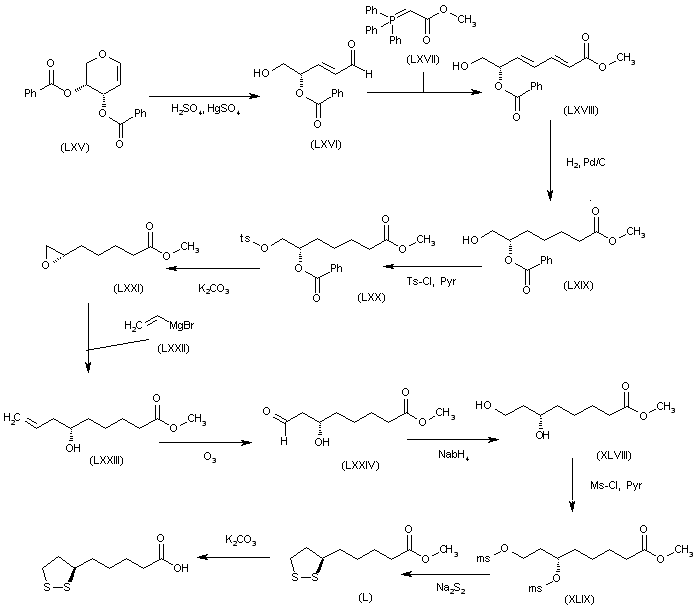
SYN
DE 3629116; EP 0261336
Alkylation of the lithio-dianion of propargyl alcohol (XIII) with 6-bromo-1-hexene (XIV), followed by in situ reduction of the resultant disubstituted acetylene with lithium metal gave the allylic alcohol (XV). Asymmetric Sharpless epoxidation of (XV) using tert-butyl hydroperoxide in the presence of L-(+)-diisopropyl tartrate afforded the (S,S)-epoxy alcohol (XVI). This was reduced to the chiral diol (XVII) employing Red-Al?in THF. After formation of the bis-mesylate (XVIII), oxidative cleavage of the terminal double bond by means of NaIO4 in the presence of ruthenium catalyst furnished the carboxylic acid (XIX). The mesylate groups were finally displaced by sodium disulfide to produce the desired cyclic disulfide compound.
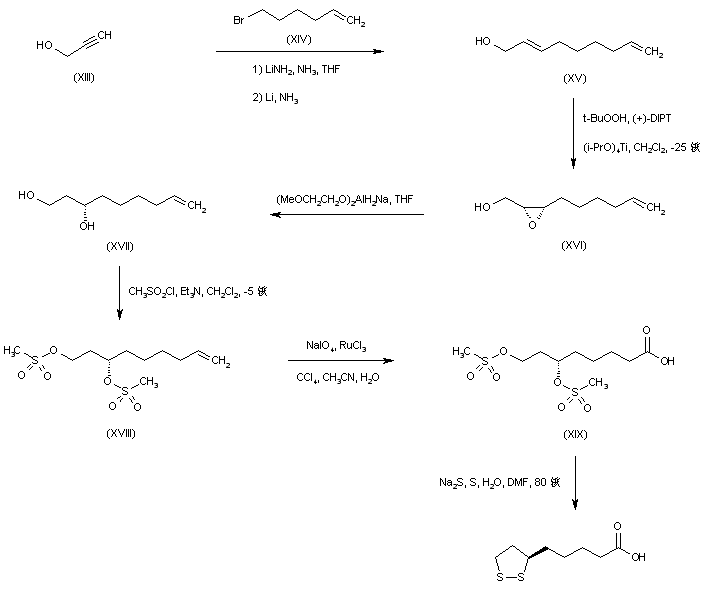
SYN
Both enantiomers of racemic 8-chloro-6-hydroxyoctanoic acid (LX) were separated employing either (+)- or (-)-alpha-methylbenzylamine. Esterification of the (R)-(-)-enantiomer with HCl-MeOH provided the chloro hydroxy ester (LXI). Further chlorination of (LXI) with SOCl2 and pyridine proceeded with inversion of configuration at C-6 to furnish the (S)-dichloro derivative (LXII). The cyclic disulfide (L) was then prepared by treatment of chloride (LXII) with sulfur and sodium sulfide in boiling EtOH. Basic hydrolysis of the methyl ester group of (LXII) then afforded (R) alpha lipoic acid. The title compound was also obtained from the (S)-(+)-acid (LXIII). Reaction of hydroxy acid (LXIII) with methanesulfonyl chloride produced the chloro mesylate (LXIV), which was then cyclized to the target disulfide in the presence of sulfur and Na2S.
| DE 19533881; EP 0763533; US 5731448 |
SYN
WO 9638437
A different strategy was based on the enantioselective oxidation of a cyclohexanone derivative by enzymic Baeyer-Villiger reaction. Keto ester (XXXVIII) was protected as the ethylene ketal (XXXIX) and subsequently reduced to alcohol (XL) using LiAlH4. Acetylation of alcohol (XL) to acetate (XLI), followed by acidic ketal hydrolysis afforded cyclohexanone (XLII) (9,10). The racemic ketone (XLII) was then subjected to oxidative cleavage by monooxigenase 2 obtained from Pseudomonas putida to furnish the (R)-lactone (XLIV) along with unreacted (S)-cyclohexanone (XLIII) (9-11). The use of cyclohexanone monooxigenase from Acinetobacter NCIMB 9871 has also been reported for this reaction (12). Methanolysis of lactone (XLIV) in the presence of NaOMe gave rise to the (R)-dihydroxy ester (XLV). Inversion of the configuration of (XLV) was accomplished by Mitsunobu coupling with p-nitrobenzoic acid (XLVI) to produce the (S)-p-nitrobenzoate ester (XLVII). Smooth hydrolysis of ester (XLVII) provided methyl (S)-6,8-dihydroxyoctanoate (XLVIII), which was processed through intermediates (XLIX) and (L), as for the isopropyl (X) (Scheme 29605101a) and ethyl (XXIX) (Scheme 29605103a) homologues, to afford the title compound.
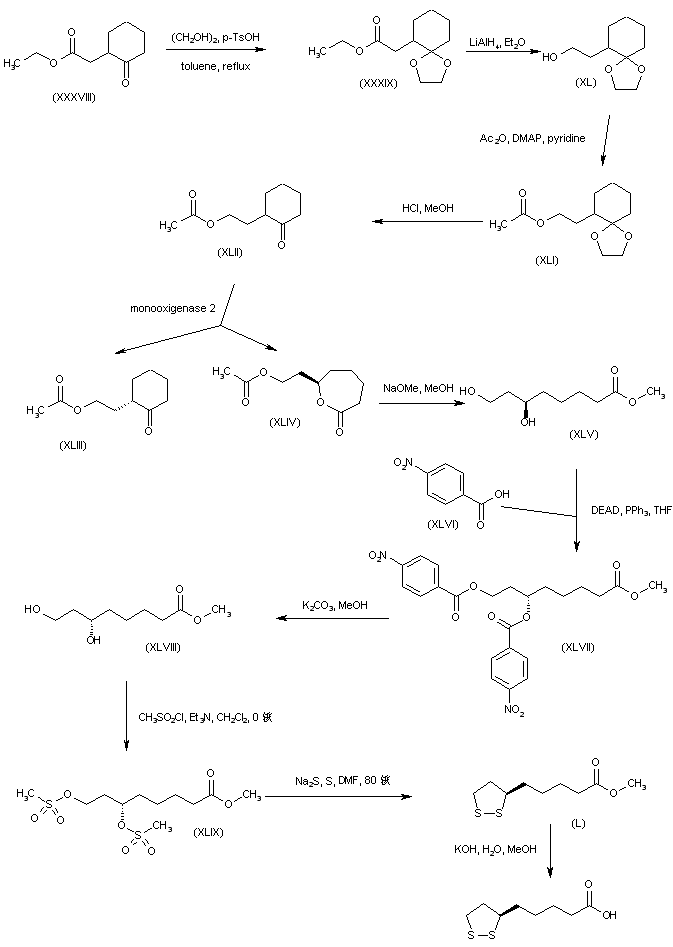
SYN
| Tetrahedron Lett 2001,42(29),4891 |
The olefinic diester (XXXVIII) was subjected to OsO4-catalyzed asymmetric dihydroxylation using hydroquinidine 1,4-phthalazinediyl diether [(DHQD)2-PHAL] as chiral ligand to afford diol (XXXIX). This was converted to the cyclic sulfate (XL) by treatment with SOCl2, followed by RuCl3-catalyzed NaIO4 oxidation of the intermediate sulfite. Regioselective reduction of sulfate (XL) at the alpha position with NaBH4 in DMA led to the (3S)-alcohol (XLI). Further selective reduction of the ethyl ester group of (XLI) was achieved by treatment with NaBH4-Et3N in MeOH-DMF, yielding the target intermediate dihydroxy ester (XIII).
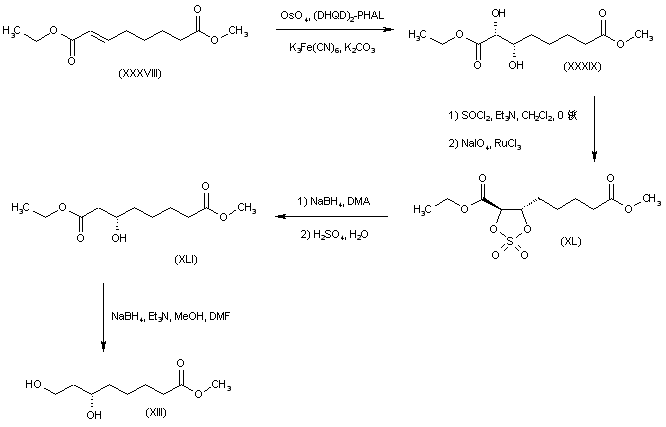
SYN
1,6-Hexanediol (I) was protected as the mono-tetrahydropyranyl ether (II), and the free hydroxyl group was subsequently oxidized to aldehyde (III) under Swern conditions. Reformatskii reaction of aldehyde (III) with the organozinc reagent generated from ethyl bromoacetate yielded the racemic hydroxy ester (IV). The requisite (S)-enantiomer (VI) was obtained via oxidation of (IV) to oxo ester (V) using pyridinium chlorochromate, and then asymmetric hydrogenation in the presence of (S)-(-)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl dichlororuthenium complex. Oxo ester (V) was also prepared by SnCl2-catalyzed insertion of ethyl diazoacetate into aldehyde (III). The chiral hydroxy ester (VI) was then reduced to diol (VII) by means of NaBH4-CuSO4. After conversion of (VII) to the corresponding dimesylate (VIII), removal of the tetrahydropyranyl protecting group under acidic conditions gave alcohol (IX). This was sequentially oxidized with PCC to aldehyde, and then with Ag2O to furnish the target dimesylate acid intermediate (X).
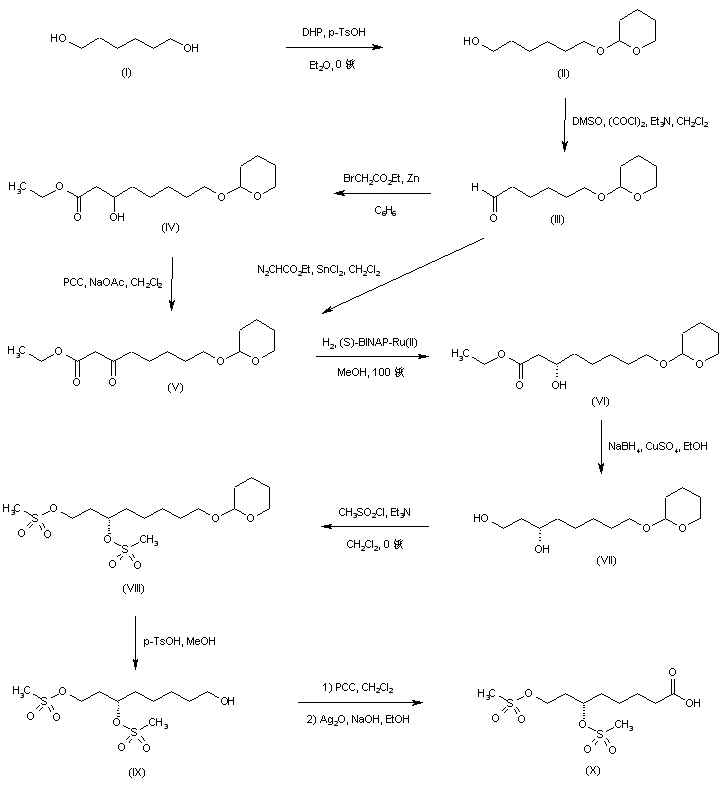
SYN
Tetrahedron Asymmetry 2000,11(4),879
The intermediate 6(S)-hydroxy-8-nonenoic acid methyl ester (III) has been obtained by enantioselective allylation of 6-oxohexanoic acid methyl ester (I) with allyltributylstannane (II) catalyzed by the chiral catalyst (R)-BINOL/Ti(O-iPr)4 in refluxing dichloromethane (other BINOL/metal catalysts have also been studied).
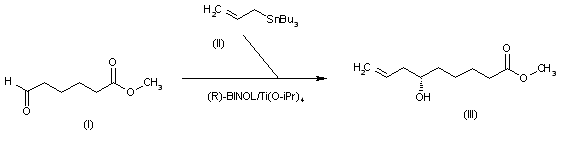
SYN
Tetrahedron Lett 1985,26(21),2535
Aldehyde (II), prepared by ozonolysis of cyclohexene (I), was ketalized with (S,S)-2,4-pentanediol (III) to afford dioxane (IV). Titanium chloride-mediated coupling of acetal (IV) with the ketene acetal (V) afforded diastereoselectively adduct (VI), which was subsequently hydrolyzed to carboxylic acid (VII) by means of trifluoroacetic acid. Removal of the pentanediol moiety to furnish the (R)-alcohol (IX) was accomplished via Jones oxidation of the secondary alcohol (VII) to ketone (VIII), followed by beta-elimination in the presence of piperidinium acetate. Reduction of the free carboxyl group by borane-tetrahydrofuran complex gave diol (X), which was further converted to dimesylate (XI). Disulfide displacement of the mesylate groups provided (+)-lipoic acid isopropyl ester (XII), which was finally hydrolyzed to the title acid using K2CO3 in MeOH/H2O.
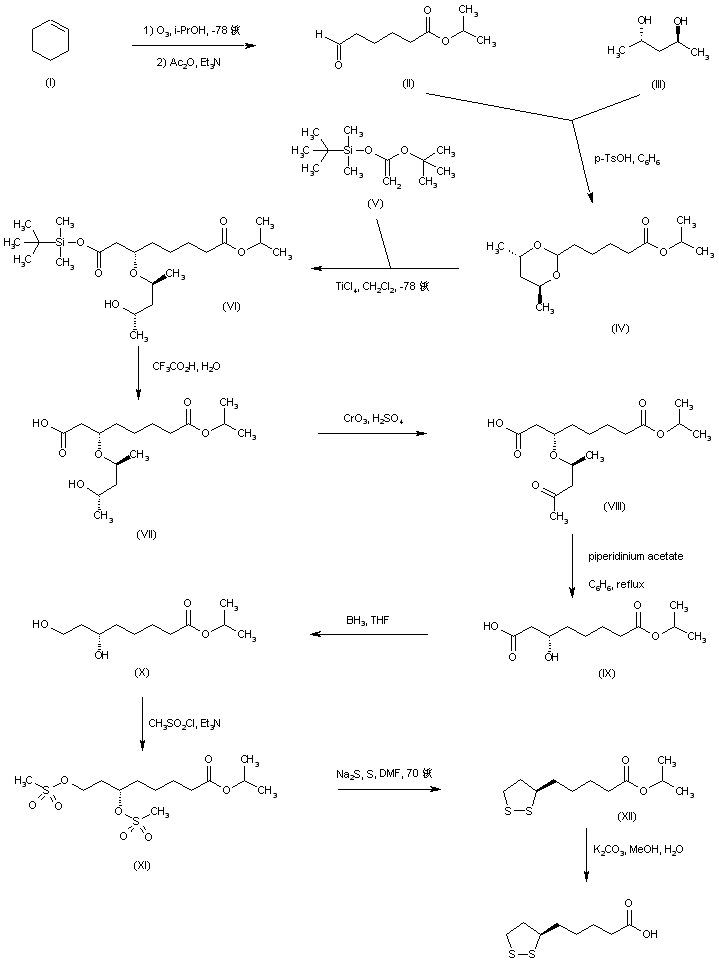
SYN
Tetrahedron Lett 1987,28(44),5313
A short synthetic strategy utilized the cyclic thioketal (XXXIII), derived from d-menthone (XXXII) and 1,3-propanedithiol, as the chiral template. Stereospecific oxidation of dithiane (XXXIII) employing NaIO4 produced sulfoxide (XXXIV). The carbanion generated from sulfoxide (XXXIV) was stereoselectively alkylated by 5-bromopentanoic acid (XXXV) in the presence of TMEDA to furnish the trans alkylated compound (XXXVI). Finally, acidic hydrolysis of (XXXVI) formed the intermediate mercapto sulfinic acid (XXXVII) which spontaneously cyclized to the desired dithiolane derivative.
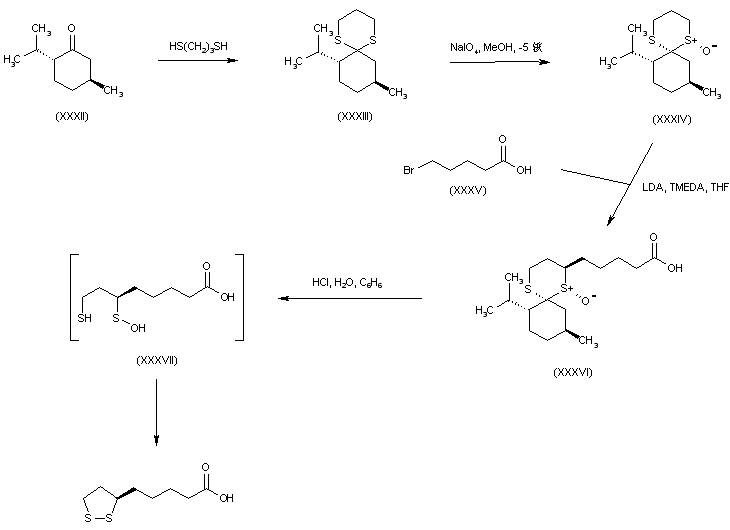
SYN
Tetrahedron Lett 1987,28(19),2183
Diisopropylidene mannitol (I) was first converted into the dibutyltin derivative (II), which was subsequently mono-benzylated to (III). Acetylation of (III) with acetic anhydride in pyridine gave (IV). After acidic hydrolysis of the isopropylidene ketals of (IV), the resultant tetraol (V) was converted into tetramesylate (VI). Reductive elimination in (VI) with Zn and NaI produced diene (VII). The acetate group of (VII) was then hydrolyzed to (VIII) using NaOMe. Intermediate (VIII) was reacted with triethyl orthoacetate in the presence of propionic acid to generate the allyl vinyl ether (IX), which underwent a Claisen rearrangement to the diene-ester (X). Selective hydroboration-oxidation of the terminal double bond of (X) yielded the primary alcohol (XI). Subsequent benzyl group hydrogenolysis in (XI) furnished the target intermediate diol (XII).
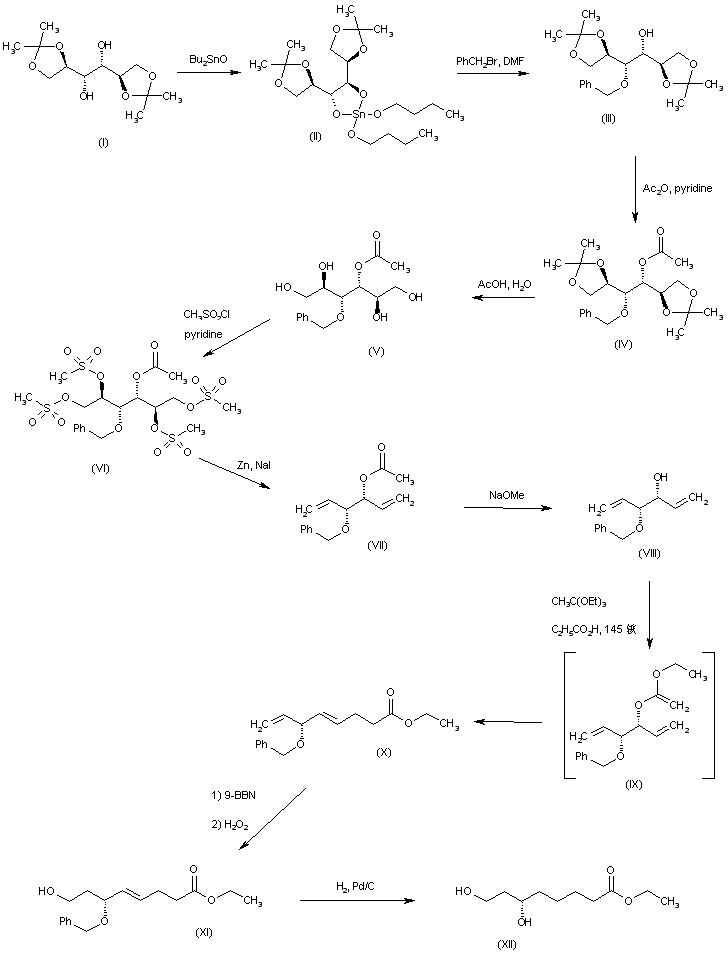
SYN
Esterification of diisopropylidene mannitol (I) with benzoyl chloride in pyridine afforded dibenzoate (II). Hydrolysis of the isopropylidene ketals of (II) with aqueous HOAc gave tetraol (III), which was further converted to tetramesylate (IV) on treatment with methanesulfonyl chloride and pyridine. Reductive elimination of the mesylate groups of (IV) using Zn dust and NaI yielded diene (V). The benzoate esters of (V) were then removed by treatment with sodium methoxide. The resultant divinylglycol (VI) was reacted with dibutyltin oxide to produce the tin derivative (VII), which was converted to the target intermediate, themono-benzyl ether (VIII), by treatment with benzyl bromide in hot DMF.
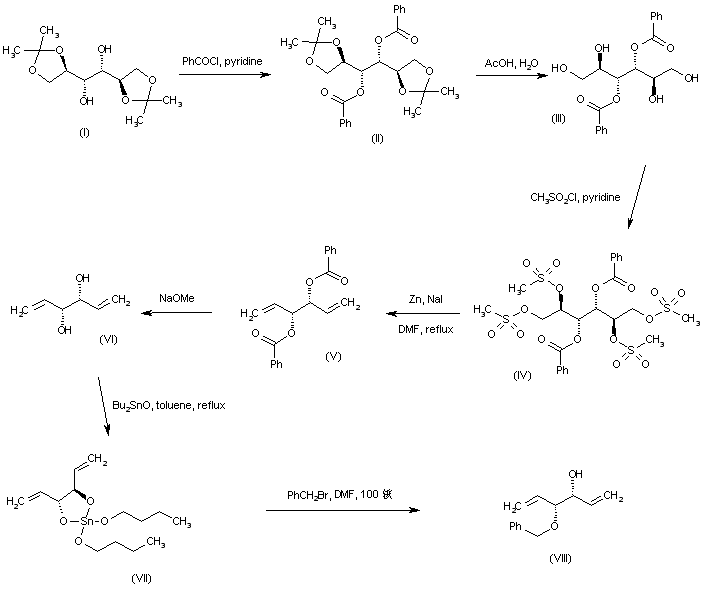
SYN
Tetrahedron Lett 1989,30(42),5705
Alkylation of the dianion of octyl acetoacetate (XIII) with 4-iodobutyronitrile (XIV) provided the cyano keto ester (XV). Enantiospecific reduction of (XV) utilizing baker’s yeast gave rise to the desired (S)-hydroxy ester (XVI) in high enantiomeric excess. Subsequent ester group reduction in (XVI) by means of LiBH4 provided diol (XVII). The target dihydroxy ester (XII) was then obtained by alcoholysis of nitrile (XVII) under acidic conditions.
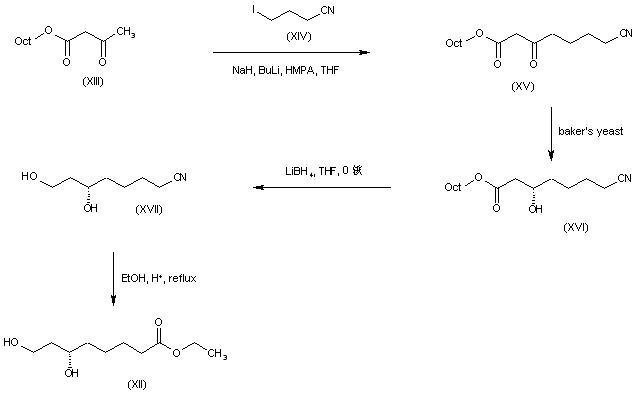
SYN
J Chem Soc Chem Commun 1995,(15),1563
A different strategy was based on the enantioselective oxidation of a cyclohexanone derivative by enzymic Baeyer-Villiger reaction. Keto ester (XXXVIII) was protected as the ethylene ketal (XXXIX) and subsequently reduced to alcohol (XL) using LiAlH4. Acetylation of alcohol (XL) to acetate (XLI), followed by acidic ketal hydrolysis afforded cyclohexanone (XLII) (9,10). The racemic ketone (XLII) was then subjected to oxidative cleavage by monooxigenase 2 obtained from Pseudomonas putida to furnish the (R)-lactone (XLIV) along with unreacted (S)-cyclohexanone (XLIII) (9-11). The use of cyclohexanone monooxigenase from Acinetobacter NCIMB 9871 has also been reported for this reaction (12). Methanolysis of lactone (XLIV) in the presence of NaOMe gave rise to the (R)-dihydroxy ester (XLV). Inversion of the configuration of (XLV) was accomplished by Mitsunobu coupling with p-nitrobenzoic acid (XLVI) to produce the (S)-p-nitrobenzoate ester (XLVII). Smooth hydrolysis of ester (XLVII) provided methyl (S)-6,8-dihydroxyoctanoate (XLVIII), which was processed through intermediates (XLIX) and (L), as for the isopropyl (X) (Scheme 29605101a) and ethyl (XXIX) (Scheme 29605103a) homologues, to afford the title compound.
SYN
Synthesis (Stuttgart) 1996,(5),594
Racemic tetrahydro-2-furylmethanol (I) was converted to tosylate (II), which was further displaced by KCN to yield nitrile (III). Basic hydrolysis of nitrile (III), followed by Fischer esterification of the resultant carboxylic acid (IV) provided ethyl ester (V). Enzymatic resolution of racemic ester (V) by means of the lipase from Candida cylindracea generated a mixture of the (R)-acid (VI) and the unreacted (S)-ester (VII), which were separated by column chromatography. The desired (S) ester (VII) was then reduced to alcohol (VIII) with LiAlH4 in cold Et2O. Regioselective opening of the cyclic ether (VIII) with iodotrimethylsilane in acetone furnished the acetonide of 6-iodo-1,3-hexanediol (IX). Alkylation of benzyl methyl malonate (X) with iodide (IX) provided malonate (XI). Hydrogenolysis of the benzyl ester group of (XI), followed by thermal decarboxylation led to ester (XII). The target dihydroxy ester precursor (XIII) was then obtained by acid-catalyzed hydrolysis of the acetonide function.
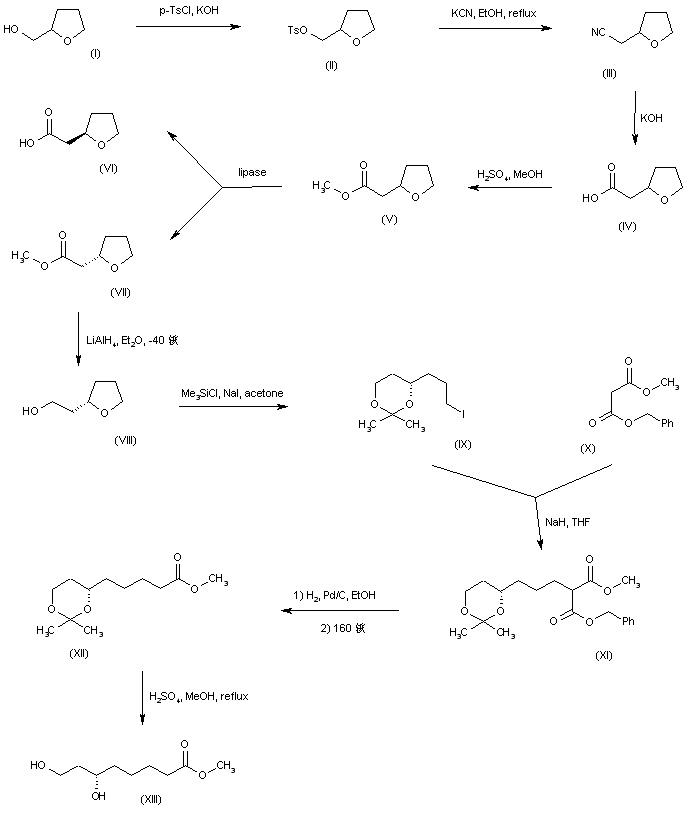
SYN
Synthesis (Stuttgart) 1996,(11),1289
Addition of vinylmagnesium bromide to 2-nitrocyclohexanone (XIV) afforded the nitro alcohol (XV). Ring cleavage of (XVI) in the presence of anhydrous CuSO4 absorbed on silica gel gave the nitro ketone (XVI). Nitro group hydrolysis in (XVI) by successive treatment with NaOMe and H2SO4 in MeOH furnished oxo ester (XVII) as the main product. This was enantiospecifically reduced with baker’s yeast to yield the (S)-alcohol (XVIII). Selective methyl ether cleavage with tetrabutylammonium iodide and BF3 provided the dihydroxy ester precursor (XIII).
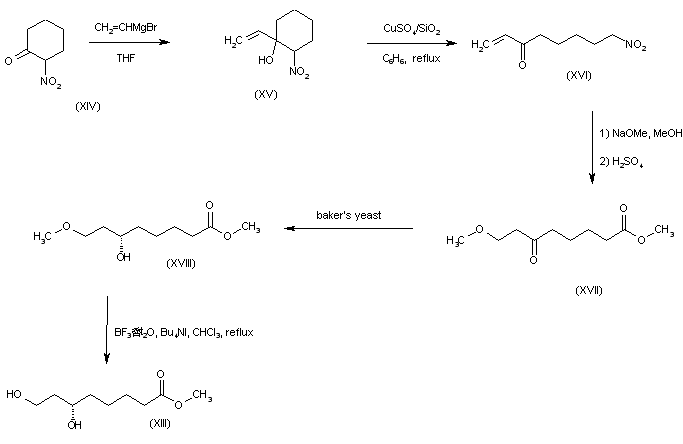
SYN
An alternative route to (+)-lipoic acid used ethyl 4,6-di-O-acetyl-2,3-dideoxy-alpha-D-erythro-hexopyranoside (XX), prepared from triacetyl-D-glucal, as the chiral starting point. Deacetylation of (XX) with sodium methoxide under Zemplen conditions gave diol (XXI) which, after conventional benzylation, led to the 4,6-di-O-benzyl derivative (XXII). Ring opening of the cyclic acetal (XXII) with propanediol in the presence of boron trifluoride afforded the dithiane derivative (XXIII). The free hydroxyl group of (XXIII) was converted into xanthate (XXIV) by reaction with NaH and CS2, followed by methyl iodide. Reductive cleavage of the xanthate group by means of Bu3SnH and AIBN provided (XXV). Hydrolysis of the thioacetal function with HgO and BF3 provided aldehyde (XXVI). Chain homologation was performed by Wittig reaction of aldehyde (XXVI) with phosphorane (XXVII) to afford the unsaturated ester (XXVIII). Simultaneous double bond hydrogenation and benzyl ether cleavage in the presence of Raney nickel led to dihydroxy ester (XXIX). This was converted to the corresponding dimesylate (XXX), which was further cyclized to disulfide (XXXI) using the in situ generated sodium disulfide as in the precedent Schemes. Finally, basic hydrolysis of the ethyl ester (XXXI) yielded the title carboxylic acid.
| Carbohydr Res 1986,148(1),51 |
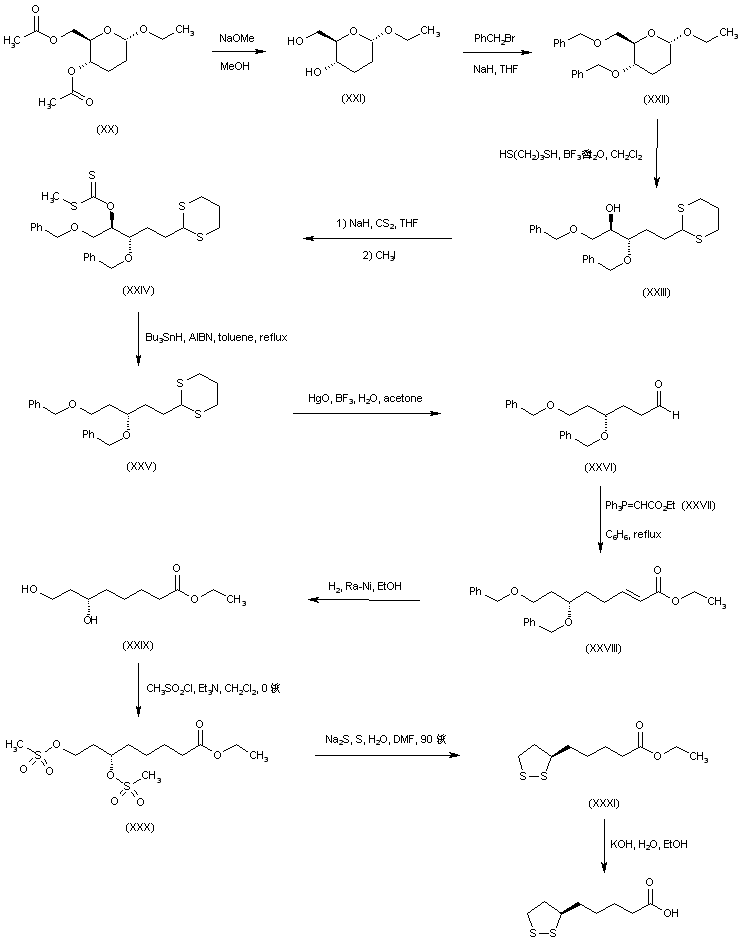
SYN
Diisopropylidene mannitol (I) was first converted into the dibutyltin derivative (II), which was subsequently mono-benzylated to (III). Acetylation of (III) with acetic anhydride in pyridine gave (IV). After acidic hydrolysis of the isopropylidene ketals of (IV), the resultant tetraol (V) was converted into tetramesylate (VI). Reductive elimination in (VI) with Zn and NaI produced diene (VII). The acetate group of (VII) was then hydrolyzed to (VIII) using NaOMe. Intermediate (VIII) was reacted with triethyl orthoacetate in the presence of propionic acid to generate the allyl vinyl ether (IX), which underwent a Claisen rearrangement to the diene-ester (X). Selective hydroboration-oxidation of the terminal double bond of (X) yielded the primary alcohol (XI). Subsequent benzyl group hydrogenolysis in (XI) furnished the target intermediate diol (XII).
| J Carbohydr Chem 1990,9(2-3),307 |
SYN
J Chem Soc Chem Commun 1986,(18),1408
SYN
https://www.sciencedirect.com/science/article/abs/pii/S1381117713003342

References
- ^ “Lipoic Acid”. Pubmed. NCBI. Retrieved October 18, 2018.
- ^ Teichert, J; Hermann, R; Ruus, P; Preiss, R (November 2003). “Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers”. The Journal of Clinical Pharmacology. 43 (11): 1257–67. doi:10.1177/0091270003258654. PMID 14551180. S2CID 30589232.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y “Lipoic acid”. Micronutrient Information Center, Linus Pauling Institute, Oregon State University, Corvallis. 1 January 2019. Retrieved 5 November 2019.
- ^ Jump up to:a b c d e Shay, KP; Moreau, RF; Smith, EJ; Hagen, TM (June 2008). “Is alpha-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity”. IUBMB Life. 60 (6): 362–7. doi:10.1002/iub.40. PMID 18409172. S2CID 33008376.
- ^ Reljanovic, M; Reichel, G; Rett, K; Lobisch, M; et al. (September 1999). “Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (alpha-lipoic acid): A two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Alpha Lipoic Acid in Diabetic Neuropathy”. Free Radical Research. 31 (3): 171–9. doi:10.1080/10715769900300721. PMID 10499773.
- ^ USP32-NF27. p. 1042.
- ^ “Pharmacopeial Forum”. 34 (5): 1209.
- ^ Jiang, Y; Cronan, JE (2005). “Expression cloning and demonstration of Enterococcus faecalis lipoamidase (pyruvate dehydrogenase inactivase) as a Ser-Ser-Lys triad amidohydrolase”. Journal of Biological Chemistry. 280 (3): 2244–56. doi:10.1074/jbc.M408612200. PMID 15528186.
- ^ Cronan, JE; Zhao, X; Jiang, Y (2005). Poole, RK (ed.). Function, attachment and synthesis of lipoic acid in Escherichia coli. Advances in Microbial Physiology. 50. pp. 103–46. doi:10.1016/S0065-2911(05)50003-1. ISBN 9780120277506. PMID 16221579.
- ^ Zempleni, J.; Trusty, T. A.; Mock, D. M. (1997). “Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver”. The Journal of Nutrition. 127 (9): 1776–81. doi:10.1093/jn/127.9.1776. PMID 9278559.
- ^ Chirapu, S. R.; Rotter, C. J.; Miller, E. L.; Varma, M. V.; Dow, R. L.; Finn, M. G. (2013). “High specificity in response of the sodium-dependent multivitamin transporter to derivatives of pantothenic acid”. Current Topics in Medicinal Chemistry. 13 (7): 837–42. doi:10.2174/1568026611313070006. PMID 23578027.
- ^ Machado, RS; Clark, DP; Guest, JR (1992). “Construction and properties of pyruvate dehydrogenase complexes with up to nine lipoyl domains per lipoate acetyltransferase chain”. FEMS Microbiology Letters. 79 (1–3): 243–8. doi:10.1111/j.1574-6968.1992.tb14047.x. PMID 1478460.
- ^ Douce, R; Bourguignon, J; Neuburger, M; Rebeille, F (2001). “The glycine decarboxylase system: A fascinating complex”. Trends in Plant Science. 6 (4): 167–76. doi:10.1016/S1360-1385(01)01892-1. PMID 11286922.
- ^ Durrani, AI; Schwartz, H; Nagl, M; Sontag, G (October 2010). “Determination of free [alpha]-lipoic acid in foodstuffs by HPLC coupled with CEAD and ESI-MS”. Food Chemistry. 120 (4): 38329–36. doi:10.1016/j.foodchem.2009.11.045.
- ^ Reed, LJ (October 2001). “A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes”. Journal of Biological Chemistry. 276 (42): 38329–36. doi:10.1074/jbc.R100026200. PMID 11477096.
- ^ Hermann, R; Niebch, G; Borbe, HO; Fieger, H; et al. (1996). “Enantioselective pharmacokinetics and bioavailability of different racemic formulations in healthy volunteers”. European Journal of Pharmaceutical Sciences. 4 (3): 167–74. doi:10.1016/0928-0987(95)00045-3.
- ^ Teichert, J; Preiss, R (1997). High-performance Liquid Chromatography Methods for Determination of Lipoic and Dihydrolipoic Acid in Human Plasma. Methods in Enzymology. 279. pp. 159–66. doi:10.1016/S0076-6879(97)79019-0. ISBN 9780121821807. PMID 9211267.
- ^ Teichert, J; Preiss, R (October 1995). “Determination of lipoic acid in human plasma by high-performance liquid chromatography with electrochemical detection”. Journal of Chromatography B. 672 (2): 277–81. doi:10.1016/0378-4347(95)00225-8. PMID 8581134.
- ^ Teichert, J; Preiss, R (November 1992). “HPLC-methods for determination of lipoic acid and its reduced form in human plasma”. International Journal of Clinical Pharmacology, Therapy, and Toxicology. 30 (11): 511–2. PMID 1490813.
- ^ Biewenga, GP; Haenen, GR; Bast, A (September 1997). “The pharmacology of the antioxidant lipoic acid”. General Pharmacology. 29 (3): 315–31. doi:10.1016/S0306-3623(96)00474-0. PMID 9378235.
- ^ Jump up to:a b Schupke, H; Hempel, R; Peter, G; Hermann, R; et al. (June 2001). “New metabolic pathways of alpha-lipoic acid”. Drug Metabolism and Disposition. 29 (6): 855–62. PMID 11353754.
- ^ Teichert, J; Hermann, R; Ruus, P; Preiss, R (November 2003). “Plasma kinetics, metabolism, and urinary excretion of alpha-lipoic acid following oral administration in healthy volunteers”. Journal of Clinical Pharmacology. 43 (11): 1257–67. doi:10.1177/0091270003258654. PMID 14551180. S2CID 30589232.
- ^ Hornberger, CS; Heitmiller, RF; Gunsalus, IC; Schnakenberg, GHF; et al. (1953). “Synthesis of DL—lipoic acid”. Journal of the American Chemical Society. 75 (6): 1273–7. doi:10.1021/ja01102a003.
- ^ Hornberger, CS; Heitmiller, RF; Gunsalus, IC; Schnakenberg, GHF; et al. (1952). “Synthetic preparation of lipoic acid”. Journal of the American Chemical Society. 74 (9): 2382. doi:10.1021/ja01129a511.
- ^ Jump up to:a b Kleeman, A; Borbe, HO; Ulrich, H (1991). “Thioctic Acid-Lipoic Acid”. In Borbe, HO; Ulrich, H (eds.). Thioctsäure: Neue Biochemische, Pharmakologische und Klinische Erkenntnisse zur Thioctsäure [Thioctic Acid. New Biochemistry, Pharmacology and Findings from Clinical Practice with Thioctic Acid]. Symposium at Wiesbaden, DE, 16–18 February 1989. Frankfurt, DE: Verlag. pp. 11–26. ISBN 9783891191255.
- ^ Fontanella, L (1955). “Preparation of optical antipodes of alpha-lipoic acid”. Il Farmaco; Edizione Scientifica. 10 (12): 1043–5. PMID 13294188.
- ^ Walton, E; Wagner, AF; Bachelor, FW; Peterson, LH; et al. (1955). “Synthesis of (+)-lipoic acid and its optical antipode”. Journal of the American Chemical Society. 77 (19): 5144–9. doi:10.1021/ja01624a057.
- ^ Acker, DS; Wayne, WJ (1957). “Optically active and radioactive α-lipoic acids”. Journal of the American Chemical Society. 79 (24): 6483–6487. doi:10.1021/ja01581a033.
- ^ Deguchi, Y; Miura, K (June 1964). “Studies on the synthesis of thioctic acid and its related compounds. XIV. Synthesis of (+)-thioctamide”. Yakugaku Zasshi. 84 (6): 562–3. doi:10.1248/yakushi1947.84.6_562. PMID 14207116.
- ^ Lang, G (1992). In Vitro Metabolism of a-Lipoic Acid Especially Taking Enantioselective Bio-transformation into Account (Ph.D. thesis). Münster, DE: University of Münster.
- ^ US patent 5281722, Blaschke, G; Scheidmantel, U & Bethge, H et al., “Preparation and use of salts of the pure enantiomers of alpha-lipoic acid”, issued 1994-01-25, assigned to DeGussa.
- ^ Jump up to:a b Carlson, DA; Young, KL; Fischer, SJ; Ulrich, H. “Ch. 10: An Evaluation of the Stability and Pharmacokinetics of R-lipoic Acid and R-Dihydrolipoic Acid Dosage Forms in Plasma from Healthy Human Subjects”. Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. pp. 235–70. In Packer & Patel 2008.
- ^ Packer, L; Kraemer, K; Rimbach, G (October 2001). “Molecular aspects of lipoic acid in the prevention of diabetes complications”. Nutrition. 17 (10): 888–95. doi:10.1016/S0899-9007(01)00658-X. PMID 11684397.
- ^ Jump up to:a b c Carlson, DA; Smith, AR; Fischer, SJ; Young, KL; et al. (December 2007). “The plasma pharmacokinetics of R-(+)-lipoic acid administered as sodium R-(+)-lipoate to healthy human subjects” (PDF). Alternative Medicine Review. 12 (4): 343–51. PMID 18069903.
- ^ Hill, AS; Werner, JA; Rogers, QR; O’Neill, SL; et al. (April 2004). “Lipoic acid is 10 times more toxic in cats than reported in humans, dogs or rats”. Journal of Animal Physiology and Animal Nutrition. 88 (3–4): 150–6. doi:10.1111/j.1439-0396.2003.00472.x. PMID 15059240.
- ^ Packer, L; Witt, EH; Tritschler, HJ (August 1995). “Alpha-lipoic acid as a biological antioxidant”. Free Radical Biology and Medicine. 19 (2): 227–50. doi:10.1016/0891-5849(95)00017-R. PMID 7649494.
- ^ Jump up to:a b Shay, KP; Moreau, RF; Smith, EJ; Smith, AR; et al. (October 2009). “Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential”. Biochimica et Biophysica Acta (BBA) – General Subjects. 1790 (10): 1149–60. doi:10.1016/j.bbagen.2009.07.026. PMC 2756298. PMID 19664690.
- ^ Haenen, GRMM; Bast, A (1991). “Scavenging of hypochlorous acid by lipoic acid”. Biochemical Pharmacology. 42 (11): 2244–6. doi:10.1016/0006-2952(91)90363-A. PMID 1659823.
- ^ Jump up to:a b Shay, KP; Shenvi, S; Hagen, TM. “Ch. 14 Lipoic Acid as an Inducer of Phase II Detoxification Enzymes Through Activation of Nr-f2 Dependent Gene Expression”. Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. pp. 349–71. In Packer & Patel 2008.
- ^ Arnér, ES; Nordberg, J; Holmgren, A (August 1996). “Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase”. Biochemical and Biophysical Research Communications. 225 (1): 268–74. doi:10.1006/bbrc.1996.1165. PMID 8769129.
- ^ Biaglow, JE; Ayene, IS; Koch, CJ; Donahue, J; et al. (April 2003). “Radiation response of cells during altered protein thiol redox”. Radiation Research. 159 (4): 484–94. Bibcode:2003RadR..159..484B. doi:10.1667/0033-7587(2003)159[0484:RROCDA]2.0.CO;2. PMID 12643793.
- ^ Haramaki, N; Han, D; Handelman, GJ; Tritschler, HJ; et al. (1997). “Cytosolic and mitochondrial systems for NADH- and NADPH-dependent reduction of alpha-lipoic acid”. Free Radical Biology and Medicine. 22 (3): 535–42. doi:10.1016/S0891-5849(96)00400-5. PMID 8981046.
- ^ Constantinescu, A; Pick, U; Handelman, GJ; Haramaki, N; et al. (July 1995). “Reduction and transport of lipoic acid by human erythrocytes”. Biochemical Pharmacology. 50 (2): 253–61. doi:10.1016/0006-2952(95)00084-D. PMID 7632170.
- ^ May, JM; Qu, ZC; Nelson, DJ (June 2006). “Cellular disulfide-reducing capacity: An integrated measure of cell redox capacity”. Biochemical and Biophysical Research Communications. 344 (4): 1352–9. doi:10.1016/j.bbrc.2006.04.065. PMID 16650819.
- ^ Jones, W; Li, X; Qu, ZC; Perriott, L; et al. (July 2002). “Uptake, recycling, and antioxidant actions of alpha-lipoic acid in endothelial cells”. Free Radical Biology and Medicine. 33 (1): 83–93. doi:10.1016/S0891-5849(02)00862-6. PMID 12086686.
- ^ Schempp, H; Ulrich, H; Elstner, EF (1994). “Stereospecific reduction of R(+)-thioctic acid by porcine heart lipoamide dehydrogenase/diaphorase”. Zeitschrift für Naturforschung C. 49 (9–10): 691–2. doi:10.1515/znc-1994-9-1023. PMID 7945680.
- ^ Biewenga, GP; Haenen, GRMM; Bast, A (1997). “Ch. 1: An Overview of Lipoate Chemistry”. In Fuchs, J; Packer, L; Zimmer, G (eds.). Lipoic Acid In Health & Disease. CRC Press. pp. 1–32. ISBN 9780824700935.
- ^ Lii, CK; Liu, KL; Cheng, YP; Lin, AH; et al. (May 2010). “Sulforaphane and alpha-lipoic acid upregulate the expression of the pi class of glutathione S-transferase through c-jun and Nrf2 activation”. Journal of Nutrition. 140 (5): 885–92. doi:10.3945/jn.110.121418. PMID 20237067.
- ^ Gal, EM; Razevska, DE (August 1960). “Studies on the in vivo metabolism of lipoic acid. 1. The fate of DL-lipoic acid-S35 in normal and thiamine-deficient rats”. Archives of Biochemistry and Biophysics. 89 (2): 253–61. doi:10.1016/0003-9861(60)90051-5. PMID 13825981.
- ^ Gal, EM (July 1965). “Reversal of selective toxicity of (-)-alpha-lipoic acid by thiamine in thiamine-deficient rats”. Nature. 207 (996): 535. Bibcode:1965Natur.207..535G. doi:10.1038/207535a0. PMID 5328673. S2CID 4146866.
- ^ US patent 6271254, Ulrich, H; Weischer, CH & Engel, J et al., “Pharmaceutical compositions containing R-alpha-lipoic acid or S-alpha.-lipoic acid as active ingredient”, issued 2001-08-07, assigned to ASTA Pharma.
- ^ Kilic, F; Handelman, GJ; Serbinova, E; Packer, L; et al. (October 1995). “Modelling cortical cataractogenesis 17: In vitro effect of a-lipoic acid on glucose-induced lens membrane damage, a model of diabetic cataractogenesis”. Biochemistry and Molecular Biology International. 37 (2): 361–70. PMID 8673020.
- ^ Artwohl, M; Schmetterer, L; Rainer, G; et al. (September 2000). Modulation by antioxidants of endothelial apoptosis, proliferation, & associated gene/protein expression. 36th Annual Meeting of the European Association for the Study of Diabetes, 17–21 September 2000, Jerusalem, Israel. Diabetologia. 43 (Suppl 1) (published August 2000). Abs 274. PMID 11008622.
- ^ Streeper, RS; Henriksen, EJ; Jacob, S; Hokama, JY; et al. (July 1997). “Differential effects of lipoic acid stereoisomers on glucose metabolism in insulin-resistant skeletal muscle”. AJP: Endocrinology and Metabolism. 273 (1 Pt 1): E185–91. doi:10.1152/ajpendo.1997.273.1.E185. PMID 9252495.
- ^ Frölich, L; Götz, ME; Weinmüller, M; Youdim, MB; et al. (March 2004). “(r)-, but not (s)-alpha lipoic acid stimulates deficient brain pyruvate dehydrogenase complex in vascular dementia, but not in Alzheimer dementia”. Journal of Neural Transmission. 111 (3): 295–310. doi:10.1007/s00702-003-0043-5. PMID 14991456. S2CID 20214857.
- ^ McIlduff, Courtney E; Rutkove, Seward B (2011-01-01). “Critical appraisal of the use of alpha lipoic acid (thioctic acid) in the treatment of symptomatic diabetic polyneuropathy”. Therapeutics and Clinical Risk Management. 7: 377–385. doi:10.2147/TCRM.S11325. ISSN 1176-6336. PMC 3176171. PMID 21941444.
- ^ Ziegle, D.; Reljanovic, M; Mehnert, H; Gries, F. A. (1999). “α-Lipoic acid in the treatment of diabetic polyneuropathy in Germany”. Experimental and Clinical Endocrinology & Diabetes. 107 (7): 421–30. doi:10.1055/s-0029-1212132. PMID 10595592.
- ^ “Lipoic Acid”. American Cancer Society. November 2008. Retrieved 5 October 2013.
- ^ Javed, S; Petropoulos, IN; Alam, U; Malik, RA (January 2015). “Treatment of painful diabetic neuropathy”. Therapeutic Advances in Chronic Disease. 6 (1): 15–28. doi:10.1177/2040622314552071. PMC 4269610. PMID 25553239.
- ^ Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF (April 2012). “Treatment for mitochondrial disorders”. Cochrane Database Syst Rev (4): CD004426. doi:10.1002/14651858.CD004426.pub3. PMC 7201312. PMID 22513923.
- ^ Namazi, Nazli; Larijani, Bagher; Azadbakht, Leila (2018). “Alpha-lipoic acid supplement in obesity treatment: A systematic review and meta-analysis of clinical trials”. Clinical Nutrition. 37 (2): 419–428. doi:10.1016/j.clnu.2017.06.002. ISSN 0261-5614. PMID 28629898
| Names | |
|---|---|
| IUPAC name(R)-5-(1,2-Dithiolan-3-yl)pentanoic acid | |
| Other namesα-Lipoic acid; Alpha lipoic acid; Thioctic acid; 6,8-Dithiooctanoic acid | |
| Identifiers | |
| CAS Number | 1077-28-7 (racemate) 1200-22-2 (R) |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:30314 |
| ChEMBL | ChEMBL134342 |
| ChemSpider | 5886 |
| DrugBank | DB00166 |
| ECHA InfoCard | 100.012.793 |
| IUPHAR/BPS | 4822 |
| KEGG | C16241 |
| MeSH | Lipoic+acid |
| PubChem CID | 6112 |
| UNII | 73Y7P0K73Y (racemate) VLL71EBS9Z (R) |
| CompTox Dashboard (EPA) | DTXSID7025508 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C8H14O2S2 |
| Molar mass | 206.32 g·mol−1 |
| Appearance | Yellow needle-like crystals |
| Melting point | 60–62 °C (140–144 °F; 333–335 K) |
| Solubility in water | Very Slightly Soluble(0.24 g/L)[1] |
| Solubility in ethanol 50 mg/mL | Soluble |
| Pharmacology | |
| ATC code | A16AX01 (WHO) |
| Pharmacokinetics: | |
| Bioavailability | 30% (oral)[2] |
| Related compounds | |
| Related compounds | Lipoamide Asparagusic acid |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
//////////Alpha lipoic acid, d-Thioctic acid, (R)-(+)-alpha-Lipoic acid, (R)-(+)-Thioctic acid, Dexlipotam,

NEW DRUG APPROVALS
one time
$10.00
Maribavir

Maribavir
- Molecular FormulaC15H19Cl2N3O4
- Average mass376.235 Da
FDA APROVED 11/23/2021, Livtencity1263 W94, 1263W94
176161-24-3[RN]
1H-Benzimidazol-2-amine, 5,6-dichloro-N-(1-methylethyl)-1-β-L-ribofuranosyl-
UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94
Camvia, D04859, G1263, GW257406X
1263W94; BW-1263W94; GW-1263; GW-257406X; SHP-620; VP-41263
Company:GlaxoSmithKline (Originator) , Shire
MOA:UL97 kinase inhibitorIndication:CMV prophylaxis
To treat post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV
Press Release
Reference:1. WO9601833A1.
Syn
US 6204249
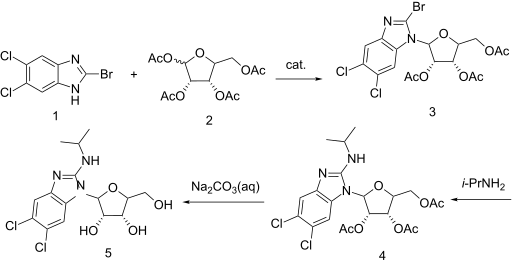

https://patents.google.com/patent/WO2001077083A1/enExample 7: 5,6-Dichloro-2-(isoproylamino)-1-(β-L-ribofuranosyl)-1 H-benzimidazolesoprylamino (10 mL) and 2-bromo-5,6-dichloro-1-(2,3,5-tri-0-acetyl-β-L- ribofuranosyl)-1 H-benzimidazole (1.0 g, 1.9 mmol) were combined with absolute ethanol (20 mL) and stirred at 75°C for 48 h. The reaction mixture was concentrated and purified on a silica gel column (2.5 vm x 16 cm, 230-400 mesh) with 1 :20 methanol: dichloromethane to give product contaminated with a small amount of higher Rf material. This was repurified on a chromatotron, fitted with a 2 mm silica gel rotor, with 1 :25 methanol.dichloromethane to give a white solid (0.43 g, 1.15 mmol, 60o/o); [a]20D=(-)22.4 (c=0.5 DMF); UVλ™* (E): pH 7.0:304 nm (95,00), 275 (1 ,800) 260 (8,300); 0.1 NaOH: 304 nm (9,900), 275 (19,00), 260 (8,100); MS (Cl): m/z (re/, intensity) 376 (100, M+1); ‘H NMR (DMSO-de) d 7.59 (s, 1 H, Ar-H), 7.35 (s, 1 H, Ar- H), 6.90 (d, 1 H, NH, J=7.8 Hz), 5.73 (d, 1 H, H-1′, J=6.5 Hz), 5.62 (t, 1 H, OH, J=4.2 Hz), 5.27-5.23 (m, 2H, OH), 4.27 (apparent dd, 1 H, J=13.4 Hz, J=7.6 Hz), 4.11 -3.99 (m, 2H), 3.97 (br. s, 1 H), 3.72-3.61 (m, 2H, H-5’), 1.18 (d, 6H, CH(CH3)2, J=6.6 Hz).Anal. Calcd. for

H2O: C, 45.70; H, 5.37; N, 10.66. Found: C, 45.75; H, 4.98; N, 10.50.
Maribavir was in phase II clinical trials for the treatment of cytomegalovirus (CMV) infection. It was granted orphan drug designation by the FDA for the indication.
The drug was originally developed by the University of Michigan and was licensed to GlaxoSmithKline. ViroPharma (now subsidiary of Shire) acquired worldwide rights to the drug from GlaxoSmithKline in 2003.
Maribavir, sold under the brand name Livtencity, is an antiviral medication that is used to treat post-transplant cytomegalovirus (CMV).[1][2]
The most common side effects include taste disturbance, nausea, diarrhea, vomiting and fatigue.[2]
Maribavir is a cytomegalovirus pUL97 kinase inhibitor that works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.[2]
Maribavir was approved for medical use in the United States in November 2021.[2][3]
Medical uses
Maribavir is indicated to treat people twelve years of age and older and weighing at least 35 kilograms (77 lb) with post-transplant cytomegalovirus infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for cytomegalovirus.[2]
Contraindications
Maribavir may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these medications is not recommended.[2]
History
Maribavir is licensed by ViroPharma from GlaxoSmithKline in 2003, for the prevention and treatment of human cytomegalovirus (HCMV) disease in hematopoietic stem cell/bone marrow transplant patients. The mechanism by which maribavir inhibits HCMV replication is by inhibition of an HCMV encoded protein kinase enzyme called UL97 or pUL97.[4] Maribavir showed promise in Phase II clinical trials and was granted fast track status, but failed to meet study goals in a Phase III trial.[5] However, the dosage used in the Phase III trial may have been too low to be efficacious.[6]
A Phase II study with maribavir demonstrated that prophylaxis with maribavir displayed strong antiviral activity, as measured by statistically significant reduction in the rate of reactivation of CMV in recipients of hematopoietic stem cell/bone marrow transplants.[7] In an intent-to-treat analysis of the first 100 days after the transplant, the number of subjects who required pre-emptive anti-CMV therapy was statistically significantly reduced with maribavir compared to placebo.
ViroPharma conducted a Phase III clinical study to evaluate the prophylactic use for the prevention of cytomegalovirus disease in recipients of allogeneic stem cell transplant patients. In February 2009, ViroPharma announced that the Phase III study failed to achieve its goal, showing no significant difference between maribavir and a placebo at reducing the rate at which CMV DNA levels were detected in patients.[8]
The safety and efficacy of maribavir were evaluated in a Phase III, multicenter, open-label, active-controlled trial that compared maribavir with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat cytomegalovirus: ganciclovir, valganciclovir, foscarnet, or cidofovir.[2] In the study, 352 transplant recipients with cytomegalovirus infections who did not respond (with or without resistance) to treatment randomly received maribavir or treatment assigned by a researcher for up to eight weeks.[2] The study compared the two groups’ plasma cytomegalovirus DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable.[2] Of the 235 participants who received maribavir, 56% had levels of cytomegalovirus DNA below what was measurable versus 24% of the 117 participants who received an investigator-assigned treatment.[2]
The U.S. Food and Drug Administration (FDA) granted the application for maribavir orphan drug, breakthrough therapy and priority review designations.[2][3][9][10] The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.[2][3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs
Approval is for Cytomegalovirus, a Type of Herpes Virus
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-common-type-post-transplant-infection-resistant-other-drugsFor Immediate Release:November 23, 2021
Today, the U.S. Food and Drug Administration approved Livtencity (maribavir) as the first drug for treating adults and pediatric patients (12 years of age and older and weighing at least 35 kilograms) with post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV. Livtencity works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.
“Transplant recipients are at a much greater risk for complications and death when faced with a cytomegalovirus infection,” said John Farley, M.D., M.P.H., director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research. “Cytomegalovirus infections that are resistant or do not respond to available drugs are of even greater concern. Today’s approval helps meet a significant unmet medical need by providing a treatment option for this patient population.”
CMV is a type of herpes virus that commonly causes infection in patients after a stem cell or organ transplant. CMV infection can lead to CMV disease and have a major negative impact on transplant recipients, including loss of the transplanted organ and death.
Livtencity’s safety and efficacy were evaluated in a Phase 3, multicenter, open-label, active-controlled trial that compared Livtencity with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat CMV: ganciclovir, valganciclovir, foscarnet or cidofovir. In the study, 352 transplant recipients with CMV infections who did not respond (with or without resistance) to treatment randomly received Livtencity or treatment assigned by a researcher for up to eight weeks.
The study compared the two groups’ plasma CMV DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable. Of the 235 patients who received Livtencity, 56% had levels of CMV DNA below what was measurable versus 24% of the 117 patients who received an investigator-assigned treatment.
The most common side effects of Livtencity include taste disturbance, nausea, diarrhea, vomiting and fatigue. Livtencity may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these drugs is not recommended. Virologic failure due to resistance can occur during and after treatment with Livtencity, therefore CMV DNA levels should be monitored and Livtencity resistance should be checked if the patient is not responding to treatment or relapses.
Livtencity received Breakthrough Therapy and Priority Review designations for this indication. Breakthrough Therapy designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s). Priority Review designation directs overall attention and resources to the evaluation of applications for drugs that, if approved, would be significant improvements in the safety or effectiveness of the treatment, diagnosis or prevention of serious conditions when compared to standard applications.
The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.
Related Information
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215596lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs”. U.S. Food and Drug Administration (FDA) (Press release). 23 November 2021. Retrieved 23 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c “Takeda’s Livtencity (maribavir) Approved by U.S. FDA as the First and Only Treatment for People Ages 12 and Older with Post-Transplant Cytomegalovirus (CMV), Refractory (With or Without Genotypic Resistance) to Conventional Antiviral Therapies”. Takeda (Press release). 23 November 2021. Retrieved 26 November 2021.
- ^ Biron KK, Harvey RJ, Chamberlain SC, Good SS, Smith AA, Davis MG, et al. (August 2002). “Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action”. Antimicrobial Agents and Chemotherapy. 46 (8): 2365–72. doi:10.1128/aac.46.8.2365-2372.2002. PMC 127361. PMID 12121906.
- ^ Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, et al. (April 2011). “Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial”. The Lancet. Infectious Diseases. 11 (4): 284–92. doi:10.1016/S1473-3099(11)70024-X. PMID 21414843.
- ^ Snydman DR (April 2011). “Why did maribavir fail in stem-cell transplants?”. The Lancet. Infectious Diseases. 11 (4): 255–7. doi:10.1016/S1473-3099(11)70033-0. PMID 21414844.
- ^ Phase 2 Data Shows Maribavir Markedly Reduced Rate Of Cytomegalovirus Infection And Disease In Bone Marrow Transplant Patients, Medical News Today, Jun 2, 2008
- ^ ViroPharma:Maribavir Phase III Study Missed Goal;Shares Plunge, CNN Money, February 09, 2009
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 February 2007. Retrieved 26 November 2021.
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 7 June 2011. Retrieved 26 November 2021.
External links
- “Maribavir”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02931539 for “Efficacy and Safety Study of Maribavir Treatment Compared to Investigator-assigned Treatment in Transplant Recipients With Cytomegalovirus (CMV) Infections That Are Refractory or Resistant to Treatment With Ganciclovir, Valganciclovir, Foscarnet, or Cidofovir” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Livtencity |
| Other names | 1263W94 |
| License data | USDailyMed: Maribavir |
| Routes of administration | By mouth |
| ATC code | J05AX10 (WHO) |
| Legal status | |
| Legal status | US:℞-only[1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 176161-24-3 |
| PubChemCID | 471161 |
| DrugBank | DB06234 |
| ChemSpider | 413807 |
| UNII | PTB4X93HE1 |
| ChEMBL | ChEMBL515408 |
| NIAID ChemDB | 070966 |
| CompTox Dashboard (EPA) | DTXSID60170091 |
| Chemical and physical data | |
| Formula | C15H19Cl2N3O4 |
| Molar mass | 376.23 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////Maribavir, APPROVALS 2021, FDA 2021, Livtencity, Takeda, Breakthrough Therapy, Priority Review , ORPHAN, UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94, Camvia, D04859, G1263, GW257406X, 1263W94, BW-1263W94, GW-1263, GW-257406X, SHP-620, VP-41263,

NEW DRUG APPROVALS
ONE TIME
$10.00
Pafolacianine

Pafolacianine
OTL-38
- Molecular FormulaC61H67N9O17S4
- Average mass1326.495 Da
FDA APPROVED NOV 2021
2-{(E)-2-[(3E)-2-(4-{2-[(4-{[(2-Amino-4-oxo-3,4-dihydro-6-pteridinyl)methyl]amino}benzoyl)amino]-2-carboxyethyl}phenoxy)-3-{(2E)-2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene ]ethylidene}-1-cyclohexen-1-yl]vinyl}-3,3-dimethyl-1-(4-sulfobutyl)-3H-indolium-5-sulfonate OTL-38Tyrosine, N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-O-[(6E)-6-[(2E)-2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[(E)-2-[3,3-dimethy l-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt
2-(2-(2-(4-((2S)-2-(4-(((2-amino-4-oxo-3,4-dihydropteridin-6-yl)methyl)amino)benzamido)-2-carboxyethyl)phenoxy)-3-(2-(3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene)ethylidene)cyclohex-1-en-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium inner salt,sodium salt (1:4)
- 3H-Indolium, 2-(2-(2-(4-((2S)-2-((4-(((2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl)amino)benzoyl)amino)-2-carboxyethyl)phenoxy)-3-(2-(1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene)ethylidene)-1-cyclohexen-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1 (4-sulfobutyl)-, inner salt,sodium salt (1:4)
1628423-76-6 [RN]
Pafolacianine sodium [USAN]
RN: 1628858-03-6
UNII: 4HUF3V875C
C61H68N9Na4O17S4+5
- Intraoperative Imaging and Detection of Folate Receptor Positive Malignant Lesions
Pafolacianine, sold under the brand name Cytalux, is an optical imaging agent.[1][2]
The most common side effects of pafolacianine include infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity.[2]
It was approved for medical use in the United States in November 2021.[2][3]
Pafolacianine is a fluorescent drug that targets folate receptor (FR).[1]
Medical uses
Pafolacianine is indicated as an adjunct for intraoperative identification of malignant lesions in people with ovarian cancer.[1][2]
History
The safety and effectiveness of pafolacianine was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.[2] Of the 134 women (ages 33 to 81 years) who received a dose of pafolacianine and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pafolacianine orphan drug, priority review, and fast track designations.[2][4] The FDA granted the approval of Cytalux to On Target Laboratories, LLC.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

WO 2014149073
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014149073
In another aspect of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0029] In a fourth embodiment of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0030]
[0032] wherein C is any carbon isotope. In this embodiment, the amino acid linker is selected from a group consisting of methyl 2-di-tert-butyl dicarbonate-amino-3-(4-phenyl)propanoate, 3-(4-hydroxyphenyl)-2-(di-tert-butyl-dicarbonate methylamino)propanoic acid, 2-amino-4-(4-hydroxyphenyl)butanoic acid, and Tert-butyl (2-di-tert-butyl dicarbonate- amino)-3-(4-hydroxyphenyl)propanoate . In a particular embodiment, the aqueous base is potassium hydroxide (KOH). The method of this embodiment may also further include purifying the compound by preparatory HPLC.
EXAMPLE 1 : General synthesis of Pte – L Tyrosine – S0456 (OTL-0038)
[0088] Scheme:
C33H37CIF3N
Reactants for Step I:
[0089] A 500 mL round bottom flask was charged with a stirring bar, pteroic acid
(12.0 g, 29.40 mmol, 1 equiv), (L)-Tyr(-OfBu)-OfBu- HCI (1 1 .63 g, 35.28 mmol, 1 .2
equiv) and HATU (13.45 g, 35.28 mmol, 1 .2 equiv) then DMF (147 mL) was added to give a brown suspension [suspension A]. DIPEA (20.48 mL, 1 17.62 mmol, 4.0 equiv) was added slowly to suspension A at 23 °C, over 5 minutes. The suspension turned in to a clear brown solution within 10 minutes of addition of DIPEA. The reaction was stirred at 23 °C for 2.5 h. Reaction was essentially complete in 30 minutes as judged by LC/MS but was stirred further for 2.5 h. The formation of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI (Figure 12) was confirmed by LC/MS showing m/z 409→m/z 684. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column . The reaction mixture was cannulated as a steady stream to a stirred solution of aq. HCI (2.0 L, 0.28 M) over the period of 30 minutes to give light yellow precipitate of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The precipitated Pte_N 10(TFA)_L_Tyr(- OfBu)-OfBu HCI was filtered using sintered funnel under aspirator vacuum, washed with water (8 * 300 mL) until the pH of the filtrate is between 3 and 4. The wet solid was allowed to dry under high vacuum for 12 hours on the sintered funnel. In a separate batch, where this wet solid (3) was dried under vacuum for 48 hours and then this solid was stored at -20 0 C for 48 h. However, this brief storage led to partial decomposition of 3. The wet cake (58 g) was transferred to a 500 mL round bottom flask and was submitted to the next step without further drying or purification.
Reactants for Step II:
The wet solid (58 g) was assumed to contain 29.40 mmol of the desired compound (3) (i. e. quantitative yield for the step I ).
[0090] A 500 mL round bottom flask was charged with a stirring bar, Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI as a wet cake (58 g, 29.40 mmol, 1 equiv). A solution of TFA:TIPS:H20 (95:2.5:2.5, 200 mL) was added at once to give a light brown suspension. The reaction content was stirred at 23°C for 1 .5 hours and was monitored by LC/MS. The suspension became clear dull brown solution after stirring for 5 minutes. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column. The formation of Pte_TFA_L_Tyr (Figure 12) was confirmed by showing m/z 684→m/z 572. Reaction time varies from 30 min to 1 .5 hours depending on the water content of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The reaction mixture was cannulated as a steady stream to a stirred MTBE (1 .8 L) at 23 °C or 100 °C to give light yellow precipitate of Pte_TFA_L_Tyr. The precipitated Pte_TFA_L_Tyr was filtered using sintered funnel under aspirator vacuum, washed with MTBE (6 * 300 mL) and dried under high vacuum for 8 hours to obtain Pte_TFA_L_Tyr (14.98 g, 83.98% over two steps) as a pale yellow solid. The MTBE washing was tested for absence of residual TFA utilizing wet pH paper (pH between 3-4). The yield of the reaction was between 80-85% in different batches. The deacylated side product was detected in 3.6% as judged by LC/MS. For the different batches this impurity was never more than 5%.
Reactants for Step III:
[0091] A 200 mL round bottom flask was charged with a stirring bar and Pte_TFA_L_Tyr (13.85 g, 22.78 mmol, 1 equiv), then water (95 mL) was added to give a yellow suspension [suspension B]. A freshly prepared solution of aqueous 3.75 M NaOH (26.12 mL, 97.96 mmol, 4.30 equiv), or an equivalent base at a corresponding temperature using dimethylsulfoxide (DMSO) as a solvent (as shown in Table 1 ), was added dropwise to suspension B at 23 °C, giving a clear dull yellow solution over 15 minutes [solution B]. The equivalence of NaOH varied from 3.3 to 5.0 depending on the source of 4 (solid or liquid phase synthesis) and the residual TFA. Trianion 5 (Figure 12) formation was confirmed by LC/MS showing m/z 572→m/z 476 while the solution pH was 9-10 utilizing wet pH paper. The pH of the reaction mixture was in the range of 9-10. This pH is crucial for the overall reaction completion. Notably, pH more than 10 leads to hydrolysis of S0456. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. The presence of hydrolysis by product can be visibly detected by the persistent opaque purple/blue to red/brown color.
TABLE 1 : Separate TFA deprotection via trianion formation; S0456
[0092] The precipitated OTL-0038 product could also be crashed out by adding the reaction solution steady dropwise to acetone, acetonitrile, isopropanol or ethyl acetate/acetone mixture. Acetone yields optimal results. However, viscous reactions could be slower due to partial insolubility and/or crashing out of S0456. In this reaction, the equivalence of the aqueous base is significant. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. This solution phase synthesis provides Pte_N10(TFA)_Tyr-OH »HCI salt and desires approximately 4.1 to approximately 4.8 equiv base as a source to hydrolyze the product. Particularly, precipitation of Pte_Tyr_S0456 was best achieved when 1 mL of reaction mixture is added dropwise to the stirred acetone (20 mL). Filtration of the precipitate and washing with acetone (3 x10 mL) gave the highest purity as judged from LC/MS chromatogram.
[0093] During experimentation of this solution-phase synthesis of Pte – L Tyrosine -S0456 (OTL-0038) at different stages, some optimized conditions were observed:
Mode of addition: Separate TFA deprotection via trianion formation; S0456 @ 23 °C; reflux.
Stability data of Pte – L Tyrosine – S0456 (OTL-0038):
Liquid analysis: At 40 °C the liquid lost 8.6% at 270 nm and 1 % at 774 nm. At room temperature the liquid lost about 1 .4% at 270 nm and .5% at 774 nm. At 5 °C the
270 nm seems stable and the 774 nm reasonably stable with a small degradation purity.
Source Purity Linker S0456 Base Solvent Duration % Conversion
4.3-4.6
Solution 0.95
95% 1 equiv equiv H20 15 min 100% phase equiv
K2C03
PATENT
US 20140271482
FDA approves pafolacianine for identifying malignant ovarian cancer lesions
On November 29, 2021, the Food and Drug Administration approved pafolacianine (Cytalux, On Target Laboratories, LLC), an optical imaging agent, for adult patients with ovarian cancer as an adjunct for interoperative identification of malignant lesions. Pafolacianine is a fluorescent drug that targets folate receptor which may be overexpressed in ovarian cancer. It is used with a Near-Infrared (NIR) fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
Efficacy was evaluated in a single arm, multicenter, open-label study (NCT03180307) of 178 women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer scheduled to undergo primary surgical cytoreduction, interval debulking, or recurrent ovarian cancer surgery. All patients received pafolacianine. One hundred and thirty-four patients received fluorescence imaging evaluation in addition to standard of care evaluation which includes pre-surgical imaging, intraoperative palpation and normal light evaluation of lesions. Among these patients, 36 (26.9%) had at least one evaluable ovarian cancer lesion detected with pafolacianine that was not observed by standard visual or tactile inspection. The patient-level false positive rate of pafolacianine with NIR fluorescent light with respect to the detection of ovarian cancer lesions confirmed by central pathology was 20.2% (95% CI 13.7%, 28.0%).
The most common adverse reactions (≥1%) occurring in patients were nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, pruritus, and hypersensitivity.
The recommended pafolacianine dose is 0.025 mg/kg administered intravenously over 60 minutes, 1 to 9 hours before surgery. The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of pafolacianine.
View full prescribing information for Cytalux.
This application was granted priority review, fast track designation, and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
USFDA approves new drug to help identify cancer lesions
This drug is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery.By The Health Master -December 2, 2021
The U.S. Food and Drug Administration (USFDA) has approved Cytalux (pafolacianine), an imaging drug intended to assist surgeons in identifying ovarian cancer lesions. The drug is designed to improve the ability to locate additional ovarian cancerous tissue that is normally difficult to detect during surgery.
Cytalux is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery. The drug is a diagnostic agent that is administered in the form of an intravenous injection prior to surgery.
Alex Gorovets, M.D., deputy director of the Office of Specialty Medicine in the FDA’s Center for Drug Evaluation and Research said, “The FDA’s approval of Cytalux can help enhance the ability of surgeons to identify deadly ovarian tumors that may otherwise go undetected.
By supplementing current methods of detecting ovarian cancer during surgery, Cytalux offers health care professionals an additional imaging approach for patients with ovarian cancer.”
The American Cancer Society estimates there will be more than 21,000 new cases of ovarian cancer and more than 13,000 deaths from this disease in 2021, making it the deadliest of all female reproductive system cancers.
Conventional treatment for ovarian cancer includes surgery to remove as many of the tumors as possible, chemotherapy to stop the growth of malignant cells or other targeted therapy to identify and attack specific cancer cells.
Ovarian cancer often causes the body to overproduce a specific protein in cell membranes called a folate receptor. Following administration via injection, Cytalux binds to these proteins and illuminates under fluorescent light, boosting surgeons’ ability to identify the cancerous tissue.
Currently, surgeons rely on preoperative imaging, visual inspection of tumors under normal light or examination by touch to identify cancer lesions. Cytalux is used with a Near-Infrared fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
The safety and effectiveness of Cytalux was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.
Of the 134 women (ages 33 to 81 years) who received a dose of Cytalux and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.
The most common side effects of Cytalux were infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity. Cytalux may cause fetal harm when administered to a pregnant woman.
The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of Cytalux. There is a risk of image interpretation errors with the use of Cytalux to detect ovarian cancer during surgery, including false negatives and false positives.
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214907s000lbl.pdf
- ^ Jump up to:a b c d e f g h i “FDA Approves New Imaging Drug to Help Identify Ovarian Cancer Lesions”. U.S. Food and Drug Administration (FDA) (Press release). 29 November 2021. Retrieved 30 November 2021. This article incorporates text from this source, which is in the public domain.
- ^ “On Target Laboratories Announces FDA Approval of Cytalux (pafolacianine) injection for Identification of Ovarian Cancer During Surgery”. On Target Laboratories. 29 November 2021. Retrieved 30 November 2021 – via PR Newswire.
- ^ “Pafolacianine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 23 December 2014. Retrieved 30 November 2021.
External links
- “Pafolacianine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Cytalux |
| Other names | OTL-0038 |
| License data | US DailyMed: Pafolacianine |
| Pregnancy category | Not recommended |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1628423-76-6 |
| PubChem CID | 135565623 |
| DrugBank | DB15413 |
| ChemSpider | 64880249 |
| UNII | F7BD3Z4X8L |
| ChEMBL | ChEMBL4297412 |
| Chemical and physical data | |
| Formula | C61H67N9O17S4 |
| Molar mass | 1326.49 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Pafolacianine, FDA 2021, APPROVALS 2021, Cytalux, OVARIAN CANCER, OTL 38,
[Na+].[Na+].[Na+].[Na+].CC1(C)\C(=C/C=C/2\CCCC(=C2Oc3ccc(C[C@H](NC(=O)c4ccc(NCc5cnc6N=C(N)NC(=O)c6n5)cc4)C(=O)O)cc3)\C=C\C7=[N](CCCCS(=O)(=O)O)c8ccc(cc8C7(C)C)S(=O)(=O)O)\N(CCCCS(=O)(=O)O)c9ccc(cc19)S(=O)(=O)O

NEW DRUG APPROVALS
ONE TIME
$10.00
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}