
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

ETRIPAMIL



ETRIPAMIL
CAS 1593673-23-4
AS ACETATE 512.64 CAS 2891832-59-8
HCL SALT 2560549-35-9
WeightAverage: 452.595
Monoisotopic: 452.267507647
Chemical FormulaC27H36N2O4
12/12/2025, FDA 2025, APPROVALS 2025
Benzoic acid, 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]methylamino]ethyl]-, methyl ester
methyl 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]-methylamino]ethyl]benzoate
- Methyl 3-[2-[[(4S)-4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl]methylamino]ethyl]benzoate
- (-)-MSP 2017
- MSP 2017
- OriginatorMilestone Pharmaceuticals
- DeveloperCorxel Pharmaceuticals; Milestone Pharmaceuticals
- ClassAmines; Antiarrhythmics; Benzoates; Esters; Ischaemic heart disorder therapies; Small molecules
- Mechanism of ActionCalcium channel antagonists
- PreregistrationParoxysmal supraventricular tachycardia
- Phase IIAtrial fibrillation
- Phase IUnspecified
- No development reportedAngina pectoris
- 14 May 2025Milestone Pharmaceuticals has patent protection for etripamil in the USA
- 28 Mar 2025Milestone pharmaceuticals plans to request a Type A meeting with USFDA to discuss the issues raised in the complete response letter
- 28 Mar 2025USFDA has issued a Complete Response Letter (CRL) regarding New Drug Application (NDA) for Etripamil for Paroxysmal supraventricular tachycardia
Etripamil has been used in trials studying the treatment of Paroxysmal Supraventricular Tachycardia (PSVT).
Etripamil (MSP-2017) is a short-acting, L-type calcium-channel antagonist. Etripamil inhibits calcium influx through slow calcium channels, thereby slowing AV node conduction and prolonging the AV node refractory period. Etripamil increases heart rate and decreases systolic blood pressure. Etripamil can be used in the study of paroxysmal supraventricular tachycardia (PSVT).
To treat episodes of paroxysmal supraventricular tachycardia
SCHEME
SIDE CHAIN

MAIN

SYN
US20180110752/ U.S. Patent No. 10,117,848,
EXAMPLES
Example 1: Synthesis methyl 3-(2-((4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl)(methyl)amino)ethyl)benzoate
Part I: Synthesis of 5-Bromo-2-(3,4-dimethoxyphenyl)-2-isopropylpentanenitrile
Part II: Synthesis of methyl 3-(2-(methylamino)ethyl)benzoate
Part III: Reaction of Compound II with Compound III Produced Compound I
| Analysis of the product by mass spectrometry revealed a peak with a mass-to-charge ratio (m/z) of 453, corresponding to the M+H molecular ion of compound I. |
Example 2: Concentrated Solution of Acetate Salt of Compound I
| A concentrated aqueous solution of the acetate salt of compound I is formed according to the following protocol: |
| This protocol readily can be adapted to provide a concentrated solution of the methanesulfonate salt of compound I. |
PRED BY CHIRAL SEPERATION
US20230065401
WO2016165014
EP4119137 chiral sepn done
[0034] In one embodiment the present invention is a kit for treating a cardiac arrhythmia (e.g., PSVT or atrial fibrillation), angina, or a migraine in a subject in need thereof wherein the kit comprises a nasal delivery system comprising two doses of a therapeutically effective amount of compound I having a structure according to the formula:
and instructions for nasally administering to the subject (i) a first dose, and, optionally, (ii) a second dose of an aqueous composition comprising a pharmaceutically acceptable acetate or methanesulfonate salt of compound I, or a racemate or enantiomer thereof, wherein the acetate or methanesulfonate salt of compound I, or the racemate or enantiomer thereof, is dissolved in the aqueous composition at a concentration of 350 mg/mL± 50 mg/mL, and wherein the second dose of the compound is to be administered between 5 minutes and 60 minutes after the first dose.
Cross ref U.S. Patent No. 10,117,848,
[0336]
- 1. A method of treating a cardiac arrhythmia in a subject in need thereof with a therapeutically effective amount of compound I having a structure according to the formula:
the method comprising nasally administering to the subject (i) a first dose, and (ii) a second dose of an aqueous composition comprising a pharmaceutically acceptable acetate or methanesulfonate salt of compound I, or a racemate or enantiomer thereof, wherein the acetate or methanesulfonate salt of compound I, or the racemate or enantiomer thereof, is dissolved in the aqueous composition at a concentration of 350 mg/mL ± 50 mg/mL, and wherein the second dose of the compound is administered between 5 minutes and 25 minutes after the first dose.
PATENT
Journal of the American College of Cardiology (2018), 72(5), 489-497
American Heart Journal (2022), 253, 20-29
Expert Opinion on Investigational Drugs (2020), 29(1), 1-4
EP4119137 WO2016165014
EP-2170050-B1
US-9737503-B2
US-4968717-A
EP-0231003-A2
- [1]. Stambler BS, et al. Etripamil Nasal Spray for Rapid Conversion of Supraventricular Tachycardia to Sinus Rhythm. J Am Coll Cardiol. 2018 Jul 31;72(5):489-497. [Content Brief][2]. Milestone Pharmaceuticals Announces USAN Approval of Generic Name “Etripamil” for its Phase 2 Clinical Development Product for the Treatment of Paroxysmal Supraventricular Tachycardia.[3]. Ascah A, et al. Cardiovascular and Pharmacokinetic Profiles of Intravenous Etripamil in Conscious Telemetered Cynomolgus Monkeys. Int J Toxicol. 2025 Apr 1:10915818251327963. [Content Brief][4]. Pion J, et al. Preclinical Safety Evaluation of Etripamil Nasal Spray in Cynomolgus Macaques (Macaca fascicularis) to Assess for Safety in Patients With Paroxysmal Supraventricular Tachycardia. Int J Toxicol. 2024 Sep-Oct;43(5):503-510. [Content Brief]
//////////ETRIPAMIL, (-)-MSP 2017, MSP 2017, FDA 2025, APPROVALS 2025
Ervogastat


Ervogastat
CAS 2186700-33-2
Non-alcoholic Steatohepatitis (NASH) with Liver Fibrosis (FAST TRACK – U.S.)
- 2-[5-[(3-Ethoxy-2-pyridinyl)oxy]-3-pyridinyl]-N-[(3S)-tetrahydro-3-furanyl]-5-pyrimidinecarboxamide
- (S)-2-(5-((3-Ethoxypyridin-2-yl]oxy]pyridin-3-yl)-N-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
- PF 06865571
- BSOIY5AKQW
407.4 g/mol, C21H21N5O4
2-[5-(3-ethoxypyridin-2-yl)oxypyridin-3-yl]-N-[(3S)-oxolan-3-yl]pyrimidine-5-carboxamide
- OriginatorPfizer
- ClassAmides; Ethers; Furans; Hepatoprotectants; Pyridines; Pyrimidines; Small molecules
- Mechanism of ActionDiacylglycerol O-acyltransferase inhibitors
Phase IINon-alcoholic fatty liver disease; Non-alcoholic steatohepatitis
- 08 Jan 2025Chemical structure information added.
- 21 Feb 2024Pfizer completes a phase II trial in Non-alcoholic steatohepatitis (Combination therapy) in Slovakia, Japan, Bulgaria, Canada, China, Hong Kong, India, Poland, Puerto Rico, South Korea, Taiwan (PO) (NCT04321031) (EudraCT2019-004775-39)
- 21 Feb 2024Pfizer completes a phase II trial in Non-alcoholic steatohepatitis (Monotherapy) in Slovakia, Japan, Bulgaria, Canada, China, Hong Kong, India, Poland, Puerto Rico, South Korea, Taiwan (PO) (NCT04321031) (EudraCT2019-004775-39)
Ervogastat is an experimental small-molecule drug and selective diacylglycerol O-acyltransferase 2 inhibitor developed by Pfizer for non-alcoholic steatohepatitis.[1] Its development was previously halted by the company but resumed in 2022.[2]
Scheme
SIDE CHAIN

MAIN

https://doi.org/10.1021/acs.jmedchem.2c01200

SYN
WO2023026180
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023026180&_cid=P11-MB5XF4-40032-1
Preparation of Intermediates and Examples
Preparation of Intermediate 1 and Example 1 (Forms 1 and 2) were described in WO2018/033832 and are reproduced below.
Intermediate 1 : 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid
Step 1 : 3-Ethoxypyridine
Cesium carbonate (12 mol, 1.5 equiv) and ethyl iodide (9.7 mol, 1.2 equiv) were added to a solution of 3-hydroxypyrdine (8.10 mol, 1.0 equiv) in acetone (12 L) at 15 °C. The reaction mixture was stirred at room temperature for 24 hours. The reaction mixture was filtered and the organic layer was concentrated to give crude product. Ethyl acetate (20 L) was added and washed with water (3×5 L). The organic layer was dried over sodium sulfate, filtered and concentrated to give 3-ethoxypyridine (620 g, 62%) as an oil. 1H NMR (400 MHz, CDCh) 5 1.44 (t, 3H), 4.07 (q, 2H), 7.15-7.23 (m, 2H), 8.20 (dd, 1 H), 8.30 (d, 1 H).
Step 2: 3-Ethoxypyridine-1 -oxide
m-Chloroperoxybenzoic acid (6.5 mol, 1.3 equiv) was added to a solution of 3-ethoxypyridine (5.0 mol, 1.0 equiv) in dichloromethane (12 L) at 10 °C. The reaction mixture was stirred at room temperature for 24 hours. Sodium thiosulfate (4 kg, in 5 L of water) was added. The reaction mixture was stirred at 15 °C for 2 hours. Another portion of sodium thiosulfate (1.5 kg, in 5 L of water) was added. The reaction mixture was stirred at 15 °C for 1 hour. The mixture was extracted with dichloromethane (16×10 L). The combined organic layers were concentrated to give crude product. The crude product was purified by silica gel column chromatography (dichloromethane:methanol; 100:1-10:1) to give the title compound (680 g, 97%) as brown oil. This was further purified by trituration with petroleum ether (4 L) at room temperature for 24 hours to give 3-ethoxypyridine-1 -oxide (580 g, 83%) as yellow solid. 1H NMR (400 MHz, CDCh) 5 1.41 (t, 3H), 4.02 (q, 2H), 6.84 (dd, 1 H), 7.12 (dd, 1 H), 7.85 (d, 1 H), 7.91-7.95 (m, 1 H).
Step 3: 2-((5-Bromopyridin-3-yl)oxy)-3-ethoxypyridine
This reaction was carried out in five parallel batches.
Diisopropylethylamine (2.69 mol, 3.7 equiv) and bromotripyrrolidinophosphonium hexafluorophosphate (0.93 mol, 1.3 equiv) were added to a stirred solution of 3-ethoxypyridine-1-oxide (0.72 mol, 1.0 equiv) and 3-bromo-5-hydroxypyridine (0.72 mol, 1.0 equiv) in tetrahydrofuran (2500 mL) at room temperature. The reaction mixture was stirred at room temperature for 2 days then the separate batches were combined to a single batch. The resulting suspension was concentrated to dryness and dissolved in dichloromethane (25 L). The organic layer was washed with 1 N sodium hydroxide (15 L), water (3×20 L), and brine (20 L). The organic layer was dried over sodium sulfate, filtered and concentrated to give an oil. The crude oil was purified by silica gel column chromatography (petroleum ether : ethyl acetate; 10:1-1 :1) to give crude product as brown solid. This solid was triturated with methyl tert-butyl ether: petroleum ether (1 :10; 11 L) to afford 2-((5-bromopyridin-3-yl)oxy)-3-ethoxypyridine (730 g, 69%) as off yellow solid. 1H NMR (400 MHz, CDCh) 5 1.49 (t, 3H), 4.16 (q, 2H), 7.04 (dd, 1 H), 7.25 (dd, 1 H), 7.68-7.73 (m, 2H), 8.44 (d, 1 H), 8.49 (d, 1 H). MS (ES+) 297.1 (M+H).
Step 4: Ethyl 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate
A solution of 2-((5-bromopyridin-3-yl)oxy)-3-ethoxypyridine (300 mmol, 1.0 equiv) in tetrahydrofuran (1.3 L) was degassed with nitrogen for 30 minutes. Turbo Grignard
(390 mmol, 1.3 equiv, 1.3 M in tetrahydrofuran) was added at room temperature at a rate to maintain the internal temperature below 30 °C. The reaction mixture was allowed to cool to room temperature and stirred for 3 hours. The reaction was cooled to 10 °C and zinc chloride (390 mmol, 1.3 equiv, 1.9 M in 2-methyltetrahydrofuran) was added at a rate to maintain the temperature below 15 °C. The resulting suspension was warmed to room temperature until all the precipitate was dissolved and then cooled back to 10 °C. Ethyl 2-chloropyrimidine-5-carboxylate (360 mmol, 1.2 equiv) and dichloro[bis(2-(diphenylphosphino)phenyl)ether]palladium(ll) (6.00 mmol, 0.02 equiv) were added as solids. The resulting suspension was degassed with nitrogen for 30 minutes then heated to 50 °C for 16 hours. The reaction was worked up under aqueous conditions then treated sequentially with ethylenediaminetetraacetic acid disodium salt, thiosilica, and charcoal to remove metal impurities. The crude compound was recrystallized from methanol (450 mL) to yield ethyl 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate (77 g, 70%) as a pale, yellow solid. 1H NMR (400 MHz, CDCI3) 5 1.44 (t, 3H), 1.50 (t, 3H), 4.19 (q, 2H), 4.46 (q, 2H), 7.00-7.04 (m, 1 H), 7.25 (s, 1 H), 7.71 (d, 1 H), 8.59 (s, 1 H), 8.66 (d, 1 H), 9.32 (s, 2H), 9.55 (s, 1 H).
Step 5: 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid
Sodium hydroxide (307 mmol, 1.5 equiv, 4M aqueous) and methanol (50 mL) were added to a suspension of 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylate (205 mmol, 1.0 equiv) in tetrahydrofuran (300 mL). The resulting solution was stirred at room temperature for 3 hours. The reaction mixture was diluted with water (400 mL) and extracted with 2:1 diethyl ether: heptanes (2x 300 mL). The aqueous layer was acidified to pH of 4 with 4M hydrochloric acid. The resulting suspension was stirred at room temperature for 1 hour. The solid was filtered, washed with water, and dried to yield 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid (69 g, 100%) as a pale, yellow solid. 1H NMR (400 MHz, DMSO-de) 51.37 (t, 3H), 4.18 (q, 2H), 7.19 (dd, 1 H), 7.58 (dd, 1 H), 7.70 (dd, 1 H), 8.35-8.40 (m, 1 H), 8.66 (d, 1 H), 9.33 (s, 2H), 9.41 (d, 1 H), 13.9 (br. s, 1 H).
Example 1 : (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/V-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
Preparation of Form 1 of (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/- (tetrahydrofuran-3-yl)pyrimidine-5-carboxamide
Oxalyl chloride (13.8 mL, 160 mmol, 1.2 equiv) and dimethylformamide (0.510 mL, 6.65 mmol, 0.05 equiv) were added to a suspension of 2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)pyrimidine-5-carboxylic acid (45.0 g, 133 mmol, 1.0 equiv) in dichloromethane (500 mL). The suspension was stirred for 2 hours when a solution was achieved. The reaction mixture was concentrated to yield crude acid chloride as a red solid. A solution of (S)-tetrahydrofuran-3-amine (12.2 g, 140 mmol, 1.05 equiv) and diisopropylethylamine (51.0 mL, 293 mmol, 2.2 equiv) in tetrahydrofuran (100 mL) was added dropwise to a solution of the crude acid chloride in dichloromethane (200 mL) at 0 °C. The reaction was allowed to warm to room temperature and stirred for 16 hours. Water (1.0 L) and ethyl acetate (600 mL) were added and the organic layer was separated, washed with saturated sodium bicarbonate, dried over magnesium sulfate, and filtered. The filtrate was treated with activated charcoal (20 g) was stirred at 65 °C for 20 minutes. The suspension was filtered warm and filtrate was concentrated to a pale, yellow solid which was recrystallized from methanol in ethyl acetate (1 :4, 1 L) to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (43.5 g, 81%) as a colorless solid. The title compound was combined with previous batches (108.7 g, 266.8 mmol) prepared in the same manner and slurried with ethyl acetate (1 .0 L) at 80 °C for 4 hours. The suspension was allowed to cool to room temperature and stirred for 4 days. The solid was filtered, washed with ethyl acetate (3×200 mL) and dried under high vacuum at 50 °C for 24 hours to yield (S)-2-(5-((3-ethoxypyridin-2-yl)oxy)pyridin-3-yl)-/\/-(tetrahydrofuran-3-yl)pyrimidine-5-carboxamide (100.5 g, 92%) as a colorless solid. 1H NMR (300 MHz, DMSO-de) 5 1.38 (t, 3H), 1.89-1.98 (m, 1 H), 2.15-2.26 (m, 1 H), 3.65 (dd, 1 H), 3.70-3.78 (m, 1 H), 3.85-3.92 (m, 2H), 4.18 (q, 2H), 4.46-4.55 (m, 1 H), 7.18 (dd, 1 H), 7.58 (dd, 1 H), 7.69 (dd, 1 H), 8.37 (dd, 1 H), 8.64 (d, 1 H), 8.95 (d, 1 H), 9.28 (s, 2H), 9.39 (d, 1 H). MS (ES+) 408.4 (M+H). Melting point 177.5 °C. Elemental analysis for C21H21N5O4: calculated C, 61.91 ; H, 5.20; N, 17.19; found C, 61.86; H, 5.18; N, 17.30.
PATENT
WO2020234726
WO2020044266
WO2018033832
WO2021171164
WO2016036636 EG 1
References
- ^ Futatsugi, Kentaro; Cabral, Shawn; Kung, Daniel W.; Huard, Kim; Lee, Esther; Boehm, Markus; Bauman, Jonathan; Clark, Ronald W.; Coffey, Steven B.; Crowley, Collin; Dechert-Schmitt, Anne-Marie; Dowling, Matthew S.; Dullea, Robert; Gosset, James R.; Kalgutkar, Amit S.; Kou, Kou; Li, Qifang; Lian, Yajing; Loria, Paula M.; Londregan, Allyn T.; Niosi, Mark; Orozco, Christine; Pettersen, John C.; Pfefferkorn, Jeffrey A.; Polivkova, Jana; Ross, Trenton T.; Sharma, Raman; Stock, Ingrid A.; Tesz, Gregory; Wisniewska, Hanna; Goodwin, Bryan; Price, David A. (24 November 2022). “Discovery of Ervogastat (PF-06865571): A Potent and Selective Inhibitor of Diacylglycerol Acyltransferase 2 for the Treatment of Non-alcoholic Steatohepatitis”. Journal of Medicinal Chemistry. 65 (22): 15000–15013. doi:10.1021/acs.jmedchem.2c01200. PMID 36322383. S2CID 253257260.
- ^ “With the right partner, Pfizer gains fast-track tag for previously shelved NASH drug”. Retrieved 20 November 2023.
| Clinical data | |
|---|---|
| Other names | PF-06865571 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2186700-33-2 |
| PubChem CID | 134262752 |
| ChemSpider | 114929473 |
| UNII | BSOIY5AKQW |
| ChEMBL | ChEMBL4760665 |
| Chemical and physical data | |
| Formula | C21H21N5O4 |
| Molar mass | 407.430 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
////////////Ervogastat, PF 06865571, fast track, BSOIY5AKQW, PFIZER, PHASE 2,
Flotufolastat F 18



Flotufolastat F 18
- Flotufolastat F-18 Gallium
- POSLUMA
- CAS 2305081-64-3
- 18FrhPSMA-7.3
- 18F-rhPSMA-7.3
- 1537.3 g/mol
- C63H96FGaN12O25Si
gallium;2-[7-[(1S)-1-carboxy-4-[[(2R)-1-[[(1R)-1-carboxy-5-[[4-[[(4R)-4-carboxy-4-[[(4S)-4-carboxy-4-[[(1S)-1,3-dicarboxypropyl]carbamoylamino]butanoyl]amino]butyl]amino]-4-oxobutanoyl]amino]pentyl]amino]-3-[[4-[ditert-butyl(fluoranyl)silyl]benzoyl]amino]-1-oxopropan-2-yl]amino]-4-oxobutyl]-4,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate

WeightAverage: 1470.63
Monoisotopic: 1469.662295938
Chemical FormulaC63H99FN12O25Si
2639294-14-5 CAS
FDA 2023, Posluma, 5/25/2023, To use with positron emission tomography imaging in certain patients with prostate cancer
Drug Trials Snapshot
Flotufolastat (18F), sold under the brand name Posluma, is a radioactive diagnostic agent for use with positron emission tomography (PET) imaging for prostate cancer.[1] The active ingredient is flotufolastat (18F).[1]
Flotufolastat (18F) was approved for medical use in the United States in May 2023.[1][2]
SYNTHESIS
Bejot, R., et al. (2022). Methods of preparation of 18F labelled silyl-fluoride compounds (WO 2023047138 A1). World Intellectual Property Organization. https://patents.google.com/patent/WO2023047138A1/en?oq=WO2023047138A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023047138&_cid=P12-MB54HA-36492-1
EXAMPLES
rhPMSA-7, rhPMSA-10 & 2C013
Synthesis protocols for the 19F compounds 19F-rhPSMA-7.1 , 19F-rhPSMA-7.2, 19F-rhPSMA-7.3, 19F-rhPSMA-7.4, 19F-rhPSMA-10.1 , 19F-rhPSMA-10.2 and 19F-2C013 (shown below) are provided in WO2019/020831 , W02020/157177, WO2020/157184 and EP21157154.2.
rhPMSA-7.1
rhPMSA-7.2
rhPMSA-7.4
2C013
18F-Fluorination of rhPSMA-7.3
Aqueous 18F’ was passed through a quaternary methyl ammonium carbonate anion exchange cartridge (Sep-Pak Accell Plus QMA Carbonate), which was preconditioned with 5 mL of water. 18F’ was eluted with a 15 mg/mL cryptand 222 and 2.0 mg/mL potassium carbonate solution in acetonitrile/water (9/1 v/v). The resulting [18F]fluoride, cryptand and potassium carbonate solution was then azeotropically dried by heating at approx. 100 °C. Before radiolabelling, a 160 mM solution of acetic acid in DMSO was used to dissolve 0.27 pmol of rhPSMA-7.3. The resulting rhPSMA-7.3 solution was added to azeotropically-dried [18F]fluoride and the reaction mixture was incubated for 5 minutes at room temperature. For purification, a solid-phase extraction cartridge containing a hydrophobic resin (Sep-Pak Plus Short tC18 cartridge), preconditioned with 5 mL EtOH, followed by 10 mL of H2O was used. The labelling mixture was diluted with 5 mL citrate buffer (pH 5) and passed through the cartridge followed by 24 mL of citrate buffer. The 18F-rhPSMA-7.3 was eluted with 3 mL of a 1 :1 mixture (v/v) of EtOH in water.
Previously the process made use of oxalic acid and the impact of oxalic acid content, with on-cartridge drying of alkaline [18F]fluoride/K222, on radionuclide incorporation with rhPSMA-7.3 and similar silicon-fluorine acceptors (Kostikov, A. P. et al. Bioconjugate Chem. 2012, 23, 106-114) was evaluated. Maximum 18F-radiolabelling was reached when using approx.
30 pmol oxalic acid for 90 pmol of potassium hydroxide (acid-base molar ratio -0.6:1) (Wurzer, A. et al. EJNMMI radiopharm. chem. 6, 4 (2021)).
Although used in a limited quantity, oxalic acid may be toxic. Hence further development was conducted to replace oxalic acid with acetic acid, a common excipient for parenteral administration. Therefore, oxalic acid (dicarboxylic acid, 30 pmol) was replaced with 2 molar equivalents of acetic acid (monocarboxylic acid 60 pmol) and was shown to yield 18F-rhPSMA- 7.3 successfully using the Scintomics GRP synthesis module.
Implementation of the process with azeotropic drying of [18F]fluoride requires inverse addition, i.e., addition of acidified precursor solution to alkaline [18F]fluoride/K222 instead of addition of alkaline [18F]fluoride/K222 to the acidic precursor solution. As shown in Figure 1 , a higher amount of acid was required to prevent isomerisation of 18F-rhPSMA-7.3 or 19F-rhPSMA-7.3 in the presence of carbonate to related Compound A shown below. A decrease of radiolabelling conversion was also observed with increasing acid content. The optimised acetic acid amount for each process is provided in Table 1.
Compound A
Table 1 : Nominal acetic acid amounts for 18F-radiolabelling
BRIEF DESCRIPTION OF THE FIGURES
Figure 1 : Impact of acetic acid content on isomerisation (formation of Related Compound A) and yield.
Medical uses
Flotufolastat (18F) is indicated for positron emission tomography of prostate-specific membrane antigen positive lesions in men with prostate cancer.[1][3]
References
- ^ Jump up to:a b c d e “Posluma- flotufolastat f-18 injection”. DailyMed. 2 June 2023. Retrieved 25 June 2023.
- ^ “U.S. FDA Approves Blue Earth Diagnostics’ Posluma (Flotufolastat F 18) Injection, First Radiohybrid PSMA-targeted PET Imaging Agent for Prostate Cancer” (Press release). Blue Earth Therapeutics. 30 May 2023. Retrieved 25 June 2023 – via Business Wire.
- ^ Heo YA (September 2023). “Flotufolastat F 18: Diagnostic First Approval”. Molecular Diagnosis & Therapy. 27 (5): 631–636. doi:10.1007/s40291-023-00665-y. PMID 37439946. S2CID 259843992.
External links
- Clinical trial number NCT04186819 for “Imaging Study to Investigate the Safety and Diagnostic Performance of rhPSMA 7.3 (18F) in Newly Diagnosed Prostate Cancer (LIGHTHOUSE)” at ClinicalTrials.gov
- Clinical trial number NCT04186845 for “Imaging Study to Investigate Safety and Diagnostic Performance of rhPSMA 7.3 (18F) PET Ligand in Suspected Prostate Cancer Recurrence (SPOTLIGHT)” at ClinicalTrials.gov
| Flotufolastat F-18 gallium | |
| Clinical data | |
|---|---|
| Trade names | Posluma |
| Other names | 18F-rhPSMA-7.3, flotufolastat F18 (USAN US) |
| License data | US DailyMed: Flotufolastat f-18 |
| Routes of administration | Intravenous |
| ATC code | V09IX18 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| CAS Number | 2639294-14-5 |
| PubChem CID | 166177191 |
| DrugBank | DB17851 |
| UNII | 811W19E3OL |
| KEGG | D12606 |
| Chemical and physical data | |
| Formula | C63H9918FN12O25Si |
| Molar mass | 1537.3 g·mol−1 |
| 3D model (JSmol) | Interactive imageInteractive image |
| showSMILES | |
| showInChI | |
/////////Flotufolastat F 18, Posluma, FDA 2023, APPROVALS 2023, Flotufolastat F-18 Gallium, 18FrhPSMA-7.3, 18F-rhPSMA-7.3
ELUBIOL


ELUBIOL
Dichlorophenyl imidazoldioxolan
CAS 67914-69-6
- Elubiol
- 67914-69-6
- OristaR DCI
- Dichlorophenyl imidazoldioxolan
- (+/-)-Dichlorophenyl imidazoldioxolan
AMY 925
C27H30Cl2N4O5, 561.5 g/mol
ethyl 4-[4-[[(2R,4S)-2-(2,4-dichlorophenyl)-2-(imidazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazine-1-carboxylate
Elubiol (Dichlorophenyl imidazoldioxolan) has moderate sebum-inhibiting activity and can be used in the treatment of oily skin or dandruff.
SCHEME

PATENT
DE2804096
https://patentscope.wipo.int/search/en/detail.jsf?docId=DE102084041&_cid=P20-MB323Q-91006-1
PATENT
US4358449
https://patentscope.wipo.int/search/en/detail.jsf?docId=US37288536&_cid=P20-MB3265-92366-1
PATENT
CN102070620
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84648943&_cid=P20-MB329Q-94515-1
| Example 1 |
| According to the method of the present invention, bacteriostatic ester (±) cis-4-[4-[[2-(2,4-dichlorophenyl)-2-(1-H-imidazolemethyl)-1,3-dioxolane-4-yl]methoxy]phenyl]-1-piperazinecarboxylic acid ethyl ester is prepared, comprising the following steps: |
| 1. Condensation reaction |
| In a dry 500ml three-necked flask, add 473g of dimethyl sulfoxide, 130g of active lipid, 50g of N-(4-hydroxyphenyl)piperazine, and 21g of potassium hydroxide. Control the temperature at 30℃ and keep the reaction for 24 hours. After the reaction, add 520g of purified water. After the addition is completed, cool to 5℃, stir and keep warm for 2h, and filter to obtain the antibacterial ester condensate. The condensation yield is about 85%. |
| 2. Esterification reaction |
| In a three-necked flask, 322g of dichloromethane, 50g of antibacterial ester condensate, and 52g of potassium carbonate were added, and then 11.9g of ethyl chloroformate was slowly added. After the addition was completed, the temperature was controlled at 25°C and the reaction was kept warm for 4 hours. After the reaction was completed, 108g of purified water was slowly added. After the addition was completed, stirring was continued for 2h. The organic layer was washed three times with purified water until the pH reached 7. After washing, dichloromethane was evaporated under reduced pressure. After evaporation, 60ml of methyl isobutyl ketone was added and the temperature was kept at 0-5°C for 2-4h. The antibacterial ester was obtained by suction filtration, and the esterification yield was about 80%. |
| Heat the antibacterial ester and dissolve it in 8 times the amount of acetone, add 0.5 times the amount of activated carbon, reflux and keep warm for 0.5 hours, cool down to no reflux, filter and remove the activated carbon, concentrate the filtrate to 5 times the weight of the antibacterial ester, add water and cool down to 0-5°C after concentration, keep warm for 1-3 hours under stirring, and filter to obtain an off-white crystalline powder. After analysis, the antibacterial ester content is greater than 97%. |
PATENT
CN101665490
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN83857361&_cid=P20-MB32BE-95479-1


| Synthesis of 4-(4-hydroxyphenyl)piperazine: |
| Example 1 |
| In a 1000ml reaction bottle, under nitrogen protection, add 500g of water, 178.5g of dichloroethylamine hydrochloride and 109g of p-hydroxyaniline, heat to 100°C, add 160g of 50% sodium hydroxide solution (80g of sodium hydroxide dissolved in 80g of water), reflux for 10 hours. Then cool to 35°C, add 400g of methanol, adjust the pH value to 8 with ammonia water, filter, and dry the filter cake in vacuum at 40°C to obtain 128g of 4-(4-hydroxyphenyl)piperazine (HPLC content greater than 98%), with a yield of 71.9%. |
| Example 2 |
| In a 1000ml reaction bottle, under nitrogen protection, add 500g of water, 178.5g of dichloroethylamine hydrochloride and 218g of p-hydroxyaniline, heat to 70°C, add 112g of 50% potassium hydroxide solution (56g of potassium hydroxide dissolved in 56g of water), and react for 5 hours. Then cool to 35°C, add 400g of methanol, adjust the pH value to 8 with ammonia water, filter, and dry the filter cake in vacuum at 40°C to obtain 112g of 4-(4-hydroxyphenyl)piperazine (HPLC content greater than 98%), with a yield of 62.9%. |
| Example 3 |
| In a 1000ml reaction bottle, under nitrogen protection, add 500g of water, 312g of dichloroethylamine hydrobromide and 150g of p-hydroxyaniline, stir at room temperature (25°C), add 200g of 50% potassium bicarbonate solution (100g of potassium bicarbonate dissolved in 100g of water), react for 1 hour, then cool to 35°C, add 400g of methanol, adjust the pH value to 8 with ammonia water, filter, and dry the filter cake in vacuum at 40°C to obtain 87g of 4-(4-hydroxyphenyl)piperazine (HPLC content greater than 98%), with a yield of 48.8%. |
| Example 4 |
| In a 1000ml reaction bottle, under nitrogen protection, add 500g of water, 452g of dichloroethylamine hydroiodide and 327g of p-hydroxyaniline, heat to 100°C, add 480g of 50% sodium hydroxide solution (240g of sodium hydroxide dissolved in 240g of water), reflux for 10 hours. Then cool to 35°C, add 600g of methanol, adjust the pH value to 8 with ammonia water, filter, and dry the filter cake in vacuum at 40°C to obtain 154g of 4-(4-hydroxyphenyl)piperazine (HPLC content greater than 98%), with a yield of 86.5%. |
| Synthesis of Ethyl [4-(4-Hydroxyphenyl)]-1-piperazinecarboxylate |
| Example 5 |
| In a 2000 ml reaction bottle, add 178 g of 4-(4-hydroxyphenyl)piperazine, 150 g of sodium bicarbonate and 500 g of acetone, cool to -20°C with ice brine, add 110 g of ethyl chloroformate dropwise, and keep the temperature in the bottle not higher than zero degrees. After the addition is complete, heat to room temperature and react for 5 hours; |
| Add 700g of water, stir for 1 hour and filter. Add the filter cake obtained by filtration to a 1000ml reaction bottle, add 300g of 75% ethanol solution by volume, heat to dissolve, cool to zero degrees with ice brine, filter, and dry the filter cake in vacuum at 40°C to obtain 146g of [4-(4-hydroxyphenyl)]-1-piperazinecarboxylic acid ethyl ester (HPLC content greater than 99%), 58.4%. |
| Example 6 |
| In a 2000ml reaction bottle, add 178g of 4-(4-hydroxyphenyl)piperazine, 180g of sodium carbonate and 500g of acetone, cool to -10°C with ice brine, add 165g of ethyl chloroformate dropwise, and keep the temperature in the bottle not higher than zero degrees. After the addition is complete, heat to 50 degrees and react for 1 hour; |
| Add 700g of water, stir for 1 hour and filter. Add the filter cake obtained by filtration to a 1000ml reaction bottle, add 300g of 75% ethanol solution by volume, heat to dissolve, cool to zero degrees with ice brine, filter, and dry the filter cake in vacuum at 40°C to obtain 156g of [4-(4-hydroxyphenyl)]-1-piperazinecarboxylic acid ethyl ester (HPLC content greater than 99%), 62.4%. |
| Example 7 |
| In a 2000ml reaction bottle, add 178g of 4-(4-hydroxyphenyl)piperazine, 400g of potassium bicarbonate and 1000g of acetone, cool to 0°C with ice brine, add 440g of ethyl chloroformate dropwise, and keep the temperature in the bottle not higher than zero degrees. After the addition is completed, react at about 0°C for 10 hours; |
| Add 1000g of water, stir for 1 hour and filter. Add the filter cake obtained by filtration to a 1000ml reaction bottle, add 500g of 75% ethanol solution by volume, heat to dissolve, cool to zero degrees with ice brine, filter, and dry the filter cake in vacuum at 40°C to obtain 216g of [4-(4-hydroxyphenyl)]-1-piperazinecarboxylic acid ethyl ester (HPLC content greater than 99%), 86.4%. |
| Example 8 |
| In a 2000ml reaction bottle, add 178g of 4-(4-hydroxyphenyl)piperazine, 140g of triethylamine, and 500g of acetone; cool to -10°C with ice brine, add 110g of ethyl chloroformate dropwise, and keep the temperature in the bottle not higher than zero degrees. After the addition is complete, react at -10°C for 10 hours; |
| Add 700g of water, stir for 1 hour and filter. Add the filter cake obtained by filtration to a 1000ml reaction bottle, add 300g of 75% ethanol solution by volume, heat to dissolve, cool to zero degrees with ice brine, filter, and dry the filter cake in vacuum at 40°C to obtain 126g of [4-(4-hydroxyphenyl)]-1-piperazinecarboxylic acid ethyl ester (HPLC content greater than 99%), 50.4%. |
| Synthesis of Ketoconazole Derivatives: |
| Example 9 |
| In a 1000ml reaction bottle, add 45g of cis-[2-(2,4-dichlorophenyl)-2(1H-imidazol-1-yl-methyl)-1,3-dioxopentyl]-4-methyl-p-toluenesulfonate, 25g of ethyl [4-(4-hydroxyphenyl)]-1-piperazinecarboxylate, 5.6g of potassium hydroxide and 180g of dimethyl sulfoxide; react at 25°C for 20 hours. After the reaction, add 450g of ice water to the reaction bottle to reduce the temperature in the reaction bottle to 10°C, and filter; wash the filter cake with water until it is neutral and dry; obtain 42g of crude ketoconazole derivative (HPLC content is 94%). |
| In a 1000ml reaction bottle, add 42g of crude ketoconazole derivative and 350g of ethyl acetate, heat to dissolve, add 0.5g of activated carbon, reflux for half an hour, filter, wash the filter cake with hot ethyl acetate, combine the ethyl acetate, and concentrate to 230g; cool naturally to room temperature, then continue to cool to 0°C with ice water, and keep warm for 1 hour, filter, and vacuum dry to obtain 39g of white powder (HPLC content greater than 99%), with a yield of 73.6%. |
| Example 10 |
| In a 1000 ml reaction bottle, add 45 g of cis-[2-(2,4-dichlorophenyl)-2(1H-imidazol-1-yl-methyl)-1,3-dioxolane]-4-methyl-p-toluenesulfonate, 50 g of ethyl [4-(4-hydroxyphenyl)]-1-piperazinecarboxylate, 11.2 g of sodium hydroxide and 200 g of dioxane; react at 50° C. for 10 hours. After the reaction, add 450 g of ice water to the reaction bottle to reduce the temperature in the reaction bottle to 10° C. and filter; wash the filter cake with water until it is neutral and dry; obtain 41 g of crude ketoconazole derivative (HPLC content is 94%). |
| In a 1000ml reaction bottle, add 41g of crude ketoconazole derivative and 340g of ethyl acetate, heat to dissolve, add 0.5g of activated carbon, reflux for half an hour; filter, wash the filter cake with hot ethyl acetate, combine ethyl acetate, and concentrate to 230g; cool naturally to room temperature, then continue to cool to 0°C with ice water, and keep warm for 1 hour, filter, and vacuum dry to obtain 37g of white powder (HPLC content greater than 99%), with a yield of 69.8%. |
| Embodiment 11 |
| In a 1000ml reaction bottle, add 45g of cis-[2-(2,4-dichlorophenyl)-2(1H-imidazol-1-yl-methyl)-1,3-dioxopentyl]-4-methyl-p-toluenesulfonate, 100g of ethyl [4-(4-hydroxyphenyl)]-1-piperazinecarboxylate, 22.4g of sodium methoxide and 300g of tetrahydrofuran; react at 0°C for 50 hours. After the reaction, add 500g of ice water to the reaction bottle to reduce the temperature in the reaction bottle to 10°C, filter; wash the filter cake with water until neutral and dry; obtain 49g of crude ketoconazole derivative (HPLC content is 94%). |
| In a 1000ml reaction bottle, add 49g of crude ketoconazole derivative and 350g of ethyl acetate, heat to dissolve, add 0.5g of activated carbon, reflux for half an hour; filter, wash the filter cake with hot ethyl acetate, combine ethyl acetate, and concentrate to 250g; cool naturally to room temperature, then continue to cool to 0°C with ice water, and keep warm for 1 hour, filter, and vacuum dry to obtain 43.9g of white powder (HPLC content greater than 99%), with a yield of 82.8%. |
| Example 12 |
| In a 1000ml reaction bottle, add 45g of cis-[2-(2,4-dichlorophenyl)-2(1H-imidazol-1-yl-methyl)-1,3-dioxopentyl]-4-methyl-p-toluenesulfonate, 42g of ethyl [4-(4-hydroxyphenyl)]-1-piperazinecarboxylate, 15g of sodium ethoxide and 300g of N,N-dimethylformamide; react at 10°C for 30 hours. After the reaction, add 500g of ice water to the reaction bottle to reduce the temperature in the reaction bottle to 10°C, and filter; wash the filter cake with water until it is neutral and dry; obtain 51.2g of crude ketoconazole derivative (HPLC content is 94%). |
| In a 1000ml reaction bottle, add 51.2g of crude ketoconazole derivative and 400g of ethyl acetate, heat to dissolve, add 0.5g of activated carbon, reflux for half an hour; filter, wash the filter cake with hot ethyl acetate, combine ethyl acetate, and concentrate to 250g; cool naturally to room temperature, then continue to cool to 0°C with ice water, and keep warm for 1 hour, filter, and vacuum dry to obtain 44.8g of white powder (HPLC content greater than 99%), with a yield of 84.5%. |
REF
[1]. Pierard GE, et al. Modulation of sebum excretion from the follicular reservoir by a dichlorophenyl-imidazoldioxolan. Int J Cosmet Sci. 1996 Oct;18(5):219-27. [Content Brief]
////////////ELUBIOL, AMY 925, Dichlorophenyl imidazoldioxolan, OristaR DCI
Elfucose



Elfucose
Cas 87-96-7
Chemical Formula: C6H12O5
Exact Mass: 164.07
Molecular Weight: 164.157
L-fucopyranose (6-deoxy-L-galactopyranose)
(3S,4R,5S,6S)-6-methyloxane-2,3,4,5-tetrol
- 6-Deoxy-L-galactose (ACI)
- Fucose, L- (8CI)
- (-)-Fucose
- 46: PN: US20220380460 SEQID: 47 claimed sequence
- 6-Desoxygalactose
- L-(-)-Fucose
- L-Fucose
- L-Galactomethylose
- L-Galactopyranose, 6-deoxy-
- CERC 803
- Elfucose
- Fucose
- NSC 1219
- congenital glycosylation disorders
- 6-Deoxy-L-galactopyranose
- L-galactomethylose
- 87-96-7
- Fucose, L-
- 6-deoxy-galactose
Fucose is under investigation in clinical trial NCT03354533 (Study of ORL-1F (L-fucose) in Patients With Leukocyte Adhesion Deficiency Type II).
L-fucopyranose is the pyranose form of L-fucose. It has a role as an Escherichia coli metabolite and a mouse metabolite. It is a L-fucose and a fucopyranose.
SCHEME

PATENT
WO2016150629
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016150629&_cid=P10-MB1MYE-34318-1
Examples
The invention will now be illustrated in more detail by the following non-limiting examples.
Example 1: Production of L-fucose by biocatalytic oxidation of L-fucitol with galactose oxidase in the presence of peroxidase and catalase
A solution of L-fucitol (6.0 mL aqueous solution containing 600 mg L-fucitol, CAS 13074-06-1, Santa Cruz Biotechnology) was added to a round-bottom three-neck bottle (50 mL), followed by the addition of 1.2 mL K2HPO4 / KH2PO4 ( 1000 mM , pH=7.0) and 0.095 mL catalase (from bovine liver, SIGMA, 21,300 U/mg, 34 mg/mL), 0.120 mL peroxidase (from horseradish, 173 U/mg solid, SIGMA ) and 2.218 mL galactose oxidase (38.4 mg/mL, 2,708 U/mL). The resulting solution was purged with O 2 at room temperature until all L-fucitol was converted to L-fucose. The reaction was monitored by HLPC. The final product was isolated and analyzed by 1 H and 13C NMR. The results are summarized in Table 1.
[Table 1]
Reaction time [h] Conversion [%]
0
3,5 54,0
24 95,8
29 96,0
PATENT
WO2010022244
WO2007021879
////////////Elfucose, 6-Deoxy-L-galactose, Fucose, L- , (-)-Fucose, 6-Desoxygalactose, L-(-)-Fucose, L-Fucose, L-Galactomethylose, L-Galactopyranose, 6-deoxy-, CERC 803, Elfucose, Fucose, NSC 1219, congenital glycosylation disorders, 6-Deoxy-L-galactopyranose, L-galactomethylose, 87-96-7, Fucose, L-, 6-deoxy-galactose
Durlobactam
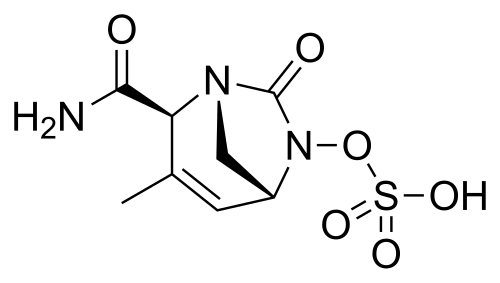
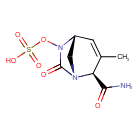
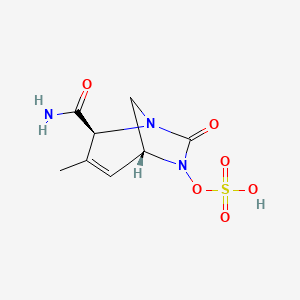
Durlobactam
CAS 1467829-71-5
WeightAverage: 277.25
Monoisotopic: 277.03685626
Chemical FormulaC8H11N3O6S
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Durlobactam sodium | F78MDZ9CW9 | 1467157-21-6 | WHHNOICWPZIYKI-IBTYICNHSA-M |
FDA 5/23/2023, Xacduro, To treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex
Press Release
Drug Trials Snapshots
(2S,5R)-2-CARBAMOYL-3-METHYL-7-OXO-1,6-DIAZABICYCLO(3.2.1)OCT-3-EN-6-YL SULFATE
SULFURIC ACID, MONO((2S,5R)-2-(AMINOCARBONYL)-3-METHYL-7-OXO-1,6-DIAZABICYCLO(3.2.1)OCT-3-EN-6-YL) ESTER
ETX 2514, ETX-2514, ETX2514, WHO 10824
Durlobactam is a non-beta-lactam, beta-lactamase inhibitor used to treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia.
Durlobactam is a beta-lactamase inhibitor used in combination with sulbactam to treat susceptible strains of bacteria in the genus Acinetobacter[1] It is an analog of avibactam.
The combination therapy sulbactam/durlobactam was approved for medical use in the United States in May 2023.[1]
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9309245 | No | 2016-04-12 | 2033-04-02 |  |
| US9623014 | No | 2017-04-18 | 2033-04-02 | |
| US9968593 | No | 2018-05-15 | 2035-11-17 | |
| US10376499 | No | 2019-08-13 | 2035-11-17 | |
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Durlobactam (1) is a copackaged antibiotic combination being developed by Entasis Therapeutics for the treatment of infections caused by Acinetobacter baumannii-calcoaceticus. 13,14
Entasis Therapeutics obtained the worldwide development rights for durlobactam (1) from AstraZeneca in 2015.13 The drug combination was approved by the USFDA in 2023 for use in patients 18 years of age and older as an intravenous infusion.13 Acinetobacter baumannii is a critical bacterial pathogen that has
become highly resistant to various β-lactam antibiotics for Gram-negative infections, including penicillin.15,16 The inventors targeted β-lactam resistance via coadministration of a β-lactamase inhibitor to restore the activity of β-lactam antibiotics. Sulbactam is a β-lactam antibiotic that inhibits penicillin binding proteins (PBP 1 and 3) essential for cell wall synthesis. Durlobactam is a β-lactamase inhibitor that protects sulbactam from degradation by Ambler class A, C, and D serine β-lactamases produced byAcinetobacter baumannii-calcoaceticus. Durlobactam binds covalently with these β-lactamases by
carbamoylating the active site serines, thus safeguarding sulbactam from enzymatic degradation.17,18 The covalent bond between durlobactam and the active site serine isreversible due to the ability of sulfated amine of durlobactam to recyclize back into urea. This allows durlobactam to exchange from one
enzyme molecule to another via a mechanism known as acylation exchange
(13) Keam, S. J. Sulbactam/Durlobactam: first approval. Drugs 2023, 83, 1245−1252.
(14) El-Ghali, A.; Kunz Coyne, A. J.;Caniff, K.; Bleick,C.; Rybak, M. J.Sulbactam-durlobactam: a novel β-lactam-β-lactamase inhibitor combination targeting carbapenem-resistant Acinetobacter baumannii
infections. Pharmacotherapy 2023, 43, 502−513.
(15) O’Donnell, J.; Tanudra, A.; Chen, A.; Miller, A. A.; McLeod, S.M.; Tommasi, R. In vitro pharmacokinetics/pharmacodynamics of the β-lactamase inhibitor, durlobactam, in combination with sulbactam against Acinetobacter baumannii-calcoaceticus complex. Antimicrob.Agents Chemother. 2024, 68, e00312-23.
(16) Arya, R.; Goldner, B. S.; Shorr, A. F. Novel agents in development for multidrug-resistant Gram-negative infections: potential new options facing multiple challenges. Curr. Opin. Infect. Dis. 2022, 35, 589−594.
(17) Shapiro, A. B.; Moussa, S. H.; McLeod, S. M.; Durand-Réville, T.; Miller, A. A. Durlobactam, a new diazabicyclooctane β-lactamase inhibitor for the treatment of Acinetobacter infections in combination
with Sulbactam. Front. Microbiol. 2021, 12, No. 709974.
(18) Iyer, R.; Moussa, S. H.; Durand-Reville, T. F.; Tommasi, R.; Miller, A. Acinetobacter baumannii OmpA is a selective antibiotic permeant porin. ACS Infect. Dis. 2018, 4, 373−381.
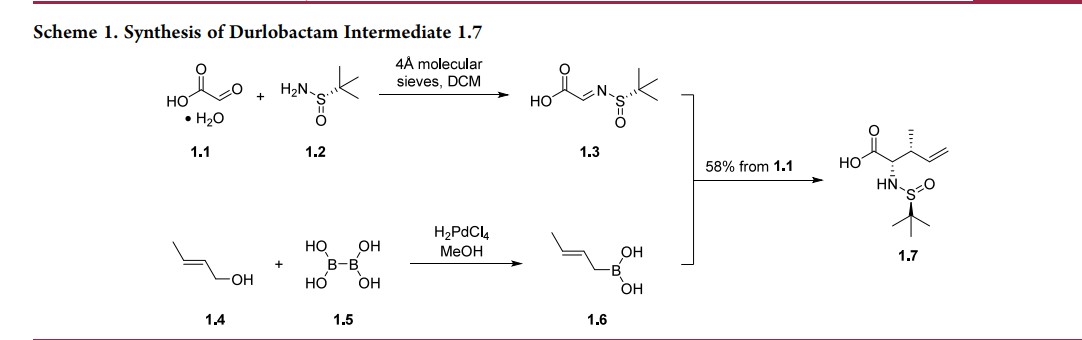
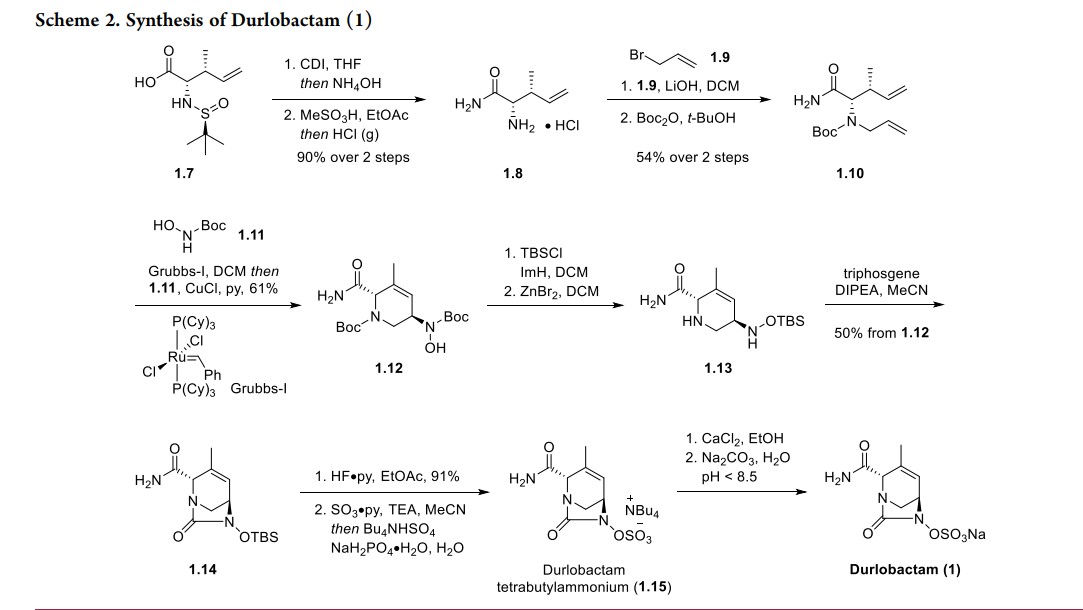
The route below was chosen as it was demonstrated on a multikilogram scale (Scheme 1), although some reagents (e.g.,triphosgene) are not typical for large-scale manufacturing.20,22 The synthesis commenced with the condensation of glyoxylic acid monohydrate (1.1) with (S)-tert-butylsulfinamide (1.2) to generate a solution of 2-(tert-butylsulfinylimino)acetic acid 1.3. In parallel, commercially available trans-crotyl alcohol (1.4) was treated with diboronic acid (1.5) in the presence of a palladium catalyst to produce a solution of crotylboronic acid 1.6. These two solutions were mixed to afford chiral α-amino acid 1.7 in
58% overall yield. Diastereo- and enantioselectivity were not reported for the transformation.
Conversion of 1.7 into durlobactam sodium (1) is described in Scheme 2. First, the carboxylic acid 1.7 was converted to an amide and the sulfinamide was removed to afford amino amide 1.8 as an HCl salt. The primary amine in 1.8 was subsequently alkylated with allyl bromide (1.9) and the resulting allyl amine
was protected with Boc anhydride to provide olefin metathesis precursor 1.10. Bisolefin 1.10 was then subjected to Grubbs first-generation catalyst (Grubbs-I) to generate a tetrahydropyridine precursor, which participated in a one-pot nitroso-ene reaction with N-Boc hydroxylamine (1.11) to produce allyl
hydroxylamine 1.12 in 61% overall yield. This key transformation efficiently installed the amine stereocenter required for formation of the bridged urea. Next, the hydroxyl moiety in 1.12 was protected as the TBS ether and the two Boc groups were removed with ZnBr2 to unveil bis-amine 1.13. The intramolecular urea formation was accomplished by the treatment with triphosgene to generate diazabicyclooctene 1.14 in 50% yield over 3 steps. The TBS ether was then removed, and the hydroxyl urea intermediate was treated with sulfur trioxide-pyridine complex and tetrabutylammonium
hydrogen sulfate to afford durlobactam tetrabutylammonium salt 1.15. Finally, tetrabutylammonium durlobactam 1.15 was converted to a calcium salt and subsequently to the targeted sodium salt providing 1. The authors mentioned that the salt formations were required to improve the purity of the final API
(>99%), however, the yields of these steps were not reported.22
(19) McGuire, H.; Bist, S.; Bifulco, N.; Zhao, L.; Wu, Y.; Huynh, H.; Xiong, H.; Comita-Prevoir, J.; Dussault, D.; Geng, B.; et al. Preparation of oxodiazabicyclooctenyl hydrogen sulfate derivatives for use as betalactamase inhibitors. WO 2013150296, 2013.
(20) Basarab, G. S.; Moss, B.; Comita-Prevoir, J.; Durand-Reville, T. F.; Gauthier, L.; O’Donnell, J.; Romero, J.; Tommasi, R.; Verheijen, J.C.; Wu, F.; et al. Preparation of substituted 2-(1,6-diazabicyclo[3.2.1]-
oct-3-en-6-yloxy)acetates as beta-lactamase inhibitors. WO2018053215, 2018.
(21) Durand-Reville, T. F.;Comita-Prevoir, J.; Zhang, J.; Wu, X.; MayDracka, T. L.; Romero, J. A. C.; Wu, F.; Chen, A.; Shapiro, A. B.; Carter, N. M.; et al. Discovery of an orally available diazabicyclooctane
inhibitor (ETX0282) of class A, C, and D serine β-lactamases. J. Med.Chem. 2020, 63, 12511−12525.
(22) Durand-Reville, T. F.; Wu, F.; Liao, X.; Wang, X.; Zhang, S.Preparation of Durlobactam crystalline forms. WO 2023206580, 2023
syn
https://www.mdpi.com/1424-8247/15/3/384
Synthesis of Durlobactam
Chemically, durlobactam is [(2S,5R)-2-carbamoyl-3-methyl-7-oxo-1,6-diazabicyclo [3.2.1] oct-3-en-6-yl] hydrogen sulfate which can be prepared from the key intermediate hydroxyurea 6-hydroxy-3-methyl-7-oxo-1,6-diaza-bicyclo [3.2.1] oct-3-ene-2-carboxylic acid amide I, which is the structural isomer of III prepared to synthetize ETX-1317 [101]. Then, according to Scheme 15, compound 1 obtained in the synthesis of III (Scheme 14) was reacted with penta-1,3-diene in place of isoprene, and, by an aza-Diels−Alder reaction, compound 2 was obtained.
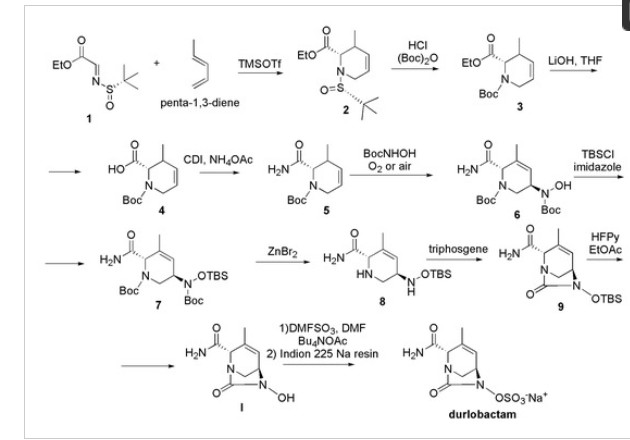
Scheme 15. Synthesis of durlobactam (C8H11N3O6S, MW = 277.36, IUPAC name, [(2S,5R)-2-carbamoyl-3-methyl-7-oxo-1,6-diazabicyclo [3.2.1] oct-3-en-6-yl] hydrogen sulphate.
Compound 2 underwent deprotection of the tert-butyl sulfinyl group to afford 3, subsequently Boc protected, to give compound 4. The saponification of the ester followed by amide coupling using ammonium acetate afforded compound 5. The reaction of alkene 5 with N-Boc-hydroxylamine in the presence of oxygen or air gave the desired compound 6 in a single step. Compound 6 was then protected with TBS group, using TBSCl to afford 7, which was Boc deprotected using zinc bromide obtaining compound 8. Cyclization of the diamine 8 with tri-phosgene provided the corresponding cyclic urea 9, which was TBS deprotected with HFPy to give the key intermediate I. This compound was then immediately sulfated with the DMF:SO3 complex to obtain the sulfate, which was isolated as its tetrabutylammonium salt 10 by reacting with tetrabutylammonium acetate. The tetrabutylammonium salt was converted to durlobactam in the form of sodium salt by passing 10 through a column filled with Indion 225 sodium resin.
REF
https://sioc-journal.cn/Jwk_yjhx/EN/abstract/abstract350784.shtml
References
- ^ Jump up to:a b “FDA Approves New Treatment for Pneumonia Caused by Certain Difficult-to-Treat Bacteria”. U.S. Food and Drug Administration (Press release). 24 May 2023. Retrieved 24 May 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
Further reading
- Shapiro AB, Moussa SH, McLeod SM, Durand-Réville T, Miller AA (2021). “Durlobactam, a New Diazabicyclooctane β-Lactamase Inhibitor for the Treatment of Acinetobacter Infections in Combination With Sulbactam”. Frontiers in Microbiology. 12: 709974. doi:10.3389/fmicb.2021.709974. PMC 8328114. PMID 34349751.
- Papp-Wallace KM, McLeod SM, Miller AA (May 2023). “Durlobactam, a Broad-Spectrum Serine β-lactamase Inhibitor, Restores Sulbactam Activity Against Acinetobacter Species”. Clinical Infectious Diseases. 76 (Supplement_2): S194 – S201. doi:10.1093/cid/ciad095. PMC 10150275. PMID 37125470.
| Clinical data | |
|---|---|
| Other names | ETX2514 |
| Routes of administration | Intravenous |
| Drug class | Antibacterial, beta-lactamase inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only co-packaged with sulbactam |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1467829-71-5 |
| PubChem CID | 89851852 |
| DrugBank | DB16704DBSALT003190 |
| ChemSpider | 5761778471060725 |
| UNII | PSA33KO9WAF78MDZ9CW9 |
| KEGG | D11591D11592 |
| ChEMBL | ChEMBL4298137ChEMBL4297378 |
| Chemical and physical data | |
| Formula | C8H11N3O6S |
| Molar mass | 277.25 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
/////////Durlobactam, Xacduro, FDA 2023, APPROVED 2023, ETX 2514, ETX-2514, ETX2514, WHO 10824
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
Durlobactam, developed by Entasis Therapeutics, is a novel β-lactamase inhibitor designed to combat multidrug-resistant (MDR) Acinetobacter baumannii infections [83]. It is co-formulated with sulbactam, a
β-lactam antibiotic, and marketed under the brand name XACDURO. In 2024, the NMPA approved XACDURO for the treatment of hospital-acquired bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP) caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex in adults [84]. Durlobactam inhibits a broad spectrum of β-lactamases, including class A, C, and D enzymes, which are commonly produced by A. baumannii. By protecting sulbactam from enzymatic degradation, it restores sulbactam’s antibacterial activity against these resistant pathogens. The
clinical efficacy of sulbactam-durlobactam was demonstrated in the PhaseIII ATTACK trial (NCT03894046), a randomized, active-controlled study comparing sulbactam-durlobactam to colistin in patients with infections caused by carbapenem-resistant A. baumannii [85]. In this trial, the primary efficacy endpoint was achieved. It demonstrated non-inferiority in terms of 28-day all-cause mortality. The mortality rate in the sulbactam – durlobactam group was 19.0 %, while that in the colistin group reached 32.3 %. Moreover, the incidence of nephrotoxicity was remarkably lower in the sulbactam-durlobactam
group. From the perspective of toxicity, sulbactam-durlobactam was typically well-tolerated by the subjects. The most common adverse reactions included liver function test abnormalities, diarrhea, and hypokalemia. Notably, the incidence of nephrotoxicity was lower compared to colistin, highlighting a more favorable safety profile. The approval of XACDURO provides a targeted therapeutic option for managing severe infections caused by MDR A. baumannii, addressing a critical need in the
treatment of these challenging pathogens [86–88].
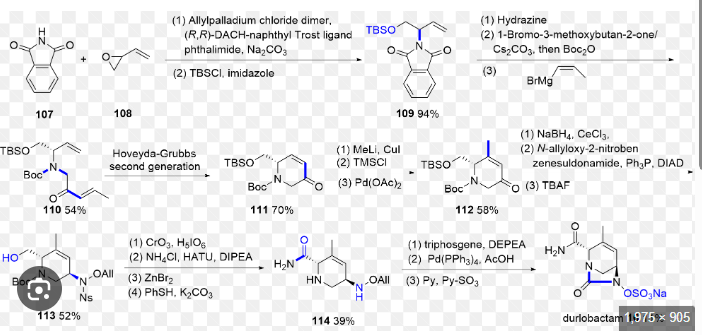
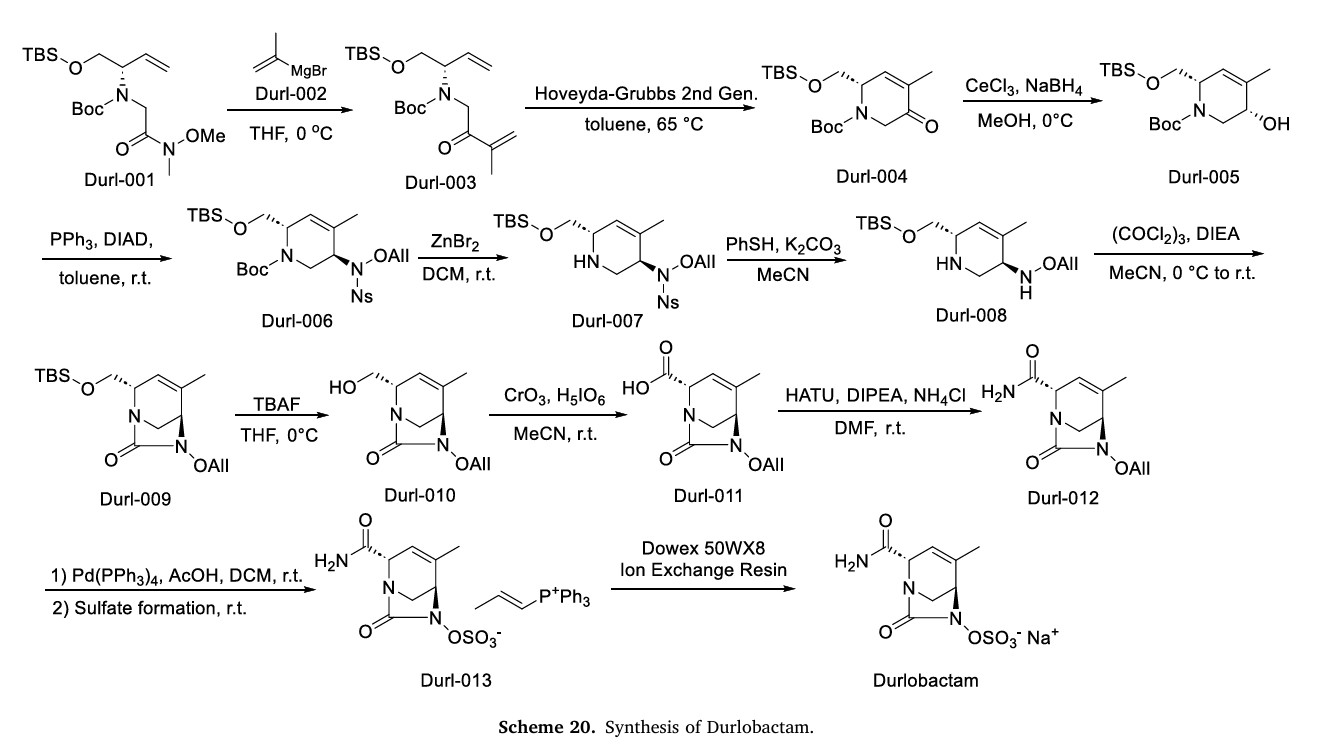
The synthetic route of Durlobactam, shown in Scheme 20, commences with a Grignard substitution between Durl-001 and Durl-002, affording Durl-003 [89]. This intermediate undergoes Diels-Alder
cyclization to form Durl-004, followed by reduction to Durl-005. Mitsunobu reaction of Durl-005 generates Durl-006, which is subjected to sequential deprotections yielding Durl-007 and subsequently Durl-008. Amidation of Durl-008 produces Durl-009, followed by TBAF-mediated deprotection to afford Durl-010. Oxidation of Durl-010Ngives carboxylic acid Durl-011, which undergoes amidation to form
Durl-012. Palladium-catalyzed coupling of Durl-012 produces Durl-013, with final ion exchange affording Durlobactam.
83-89
[83] A.B. Shapiro, S.H. Moussa, S.M. McLeod, T. Durand-R´ eville, A.A. Miller,
Durlobactam, a new diazabicyclooctane β-Lactamase inhibitor for the treatment of
acinetobacter infections in combination with sulbactam, Front. Microbiol. 12
(2021) 709974.
[84] G. Granata, F. Taglietti, F. Schiavone, N. Petrosillo, Durlobactam in the treatment
of multidrug-resistant Acinetobacter baumannii infections: a systematic review, J. Clin. Med. 11 (2022) 3258.
[85] K.M. Papp-Wallace, S.M. McLeod, A.A. Miller, Durlobactam, a broad-spectrum
serine β-lactamase inhibitor, restores sulbactam activity against acinetobacter
species, Clin. Infect. Dis. 76 (2023) S194–s201.
[86] Sulbactam and Durlobactam, Drugs and Lactation Database (Lactmed®), National
Institute of Child Health and Human Development, Bethesda (MD), 2006.
[87] S.J. Keam, Sulbactam/durlobactam: first approval, Drugs 83 (2023) 1245–1252.
[88] Y. Fu, T.E. Asempa, J.L. Kuti, Unraveling sulbactam-durlobactam: insights into its
role in combating infections caused by Acinetobacter baumannii, Expert Rev. Anti
Infect. Ther. 23 (2024) 1–12.
[89] H. McGuire, S. Bist, N. Bifulco, L. Zhao, Y. Wu, H. Huynh, H. Xiong, J. Comita-
Prevoir, D. Dussault, B. Geng, B. Chen, T. Durand-Reville, S. Guler, Preparation of
Oxodiazabicyclooctenyl Hydrogen Sulfate Derivatives for Use as beta-lactamase
Inhibitors, 2013 US9623014B2.
Edelinontrine



Edelinontrine
CRD740, PF04447943, cas 1082744-20-4
| Molecular Weight | 395.46 |
|---|---|
| Formula | C20H25N7O2 |
6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(oxan-4-yl)-5H-pyrazolo[3,4-d]pyrimidin-4-one
- 7N969W8Y4O
- 6-((3S,4S)-4-Methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl)-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo(3,4-d)pyrimidin-4-one
- 6-[(3S,4S)-4-METHYL-1-(PYRIMIDIN-2-YLMETHYL)PYRROLIDIN-3-YL]-1-(OXAN-4-YL)-5H-PYRAZOLO[3,4-D]PYRIMIDIN-4-ONE
- 6-((3S,4S)-4-Methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl)-1-(tetrahydro-2H-pyran-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one
Edelinontrine (PF-04447943) is a potent inhibitor of human recombinant PDE9A (IC50=12 nM) with >78-fold selectivity, respectively, over other PDE family members (IC50>1000 nM).

PF-04447943 is a potent, selective brain penetrant PDE9 inhibitor (Ki of 2.8, 4.5 and 18 nM) for human, rhesus and rat recombinant PDE9 respectively and high selectivity for PDE9 versus PDEs1-8 and 10-11. PF-04447943 was being developed by Pfizer for the treatment of cognitive disorders. PF-04447943 attenuates a scopolamine-induced deficit in a novel rodent attention task. PF-04447943 enhances synaptic plasticity and cognitive function in rodents. PF-04447943 has completed Phase II clinical trials in subjects with mild to moderate AD in 2013 but this research was discontinued. Pfizer completes a phase I trial in Sickle cell anaemia.
SCHEME
SUDECHAIN

MAIN

Patent
WO2023114995 PFIZER
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023114995&_cid=P20-MB07JE-44537-1
PAPER
Journal of Medicinal Chemistry (2012), 55(21), 9045-9054
PATENT
WO2008139293
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008139293&_cid=P20-MB07LY-46583-1
EXAMPLE 111
6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyiτolidin-3-yl]-1-αetrahydro-2H- pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one

///////Edelinontrine, CRD740, PF04447943, CRD 740, PF 04447943, PHASE 1
Sulbactam



Sulbactam
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Sulbactam benzathine | 49MU89FVBV | 83031-43-0 | YSEPFTSCLHUBNH-HFKSPEPWSA-N |
| Sulbactam sodium | DKQ4T82YE6 | 69388-84-7 | NKZMPZCWBSWAOX-IBTYICNHSA-M |
WeightAverage: 233.242
Monoisotopic: 233.035793157
Chemical FormulaC8H11NO5S
(2S,5R)-3,3-dimethyl-4,4,7-trioxo-4λ6-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid
Betamaze, Penicillanic Acid Sulfone, Sulbactamum, CP 45899, CP-45899, CP45899
FDA 2023, Xacduro, 5/23/2023, To treat hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia caused by susceptible isolates of Acinetobacter baumannii-calcoaceticus complex
Press Release
Drug Trials Snapshots
Sulbactam is a β-lactamase inhibitor. This drug is given in combination with β-lactam antibiotics to inhibit β-lactamase, an enzyme produced by bacteria that destroys the antibiotics.[1]
It was patented in 1977 and approved for medical use in 1986.[2]
Sulbactam is a beta (β)-lactamase inhibitor and a derivative of the basic penicillin nucleus. When given in combination with β-lactam antibiotics, sulbactam produces a synergistic effect as it blocks the enzyme responsible for drug resistance by hydrolyzing β-lactams.
PATENT
| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US9309245 | No | 2016-04-12 | 2033-04-02 | |
| US9623014 | No | 2017-04-18 | 2033-04-02 | |
| US9968593 | No | 2018-05-15 | 2035-11-17 | |
| US10376499 | No | 2019-08-13 | 2035-11-17 | |
doi:10.1016/S0040-4039(00)89275-8

SYN
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
On May 23, 2023, the FDA granted approval to Xacduro for the treatment of Baumannii-sensitive strains causing hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia in
patients aged 18 years or older [4]. Xacduro consists of Sulbactam and Durlobactam. Sulbactam, a medication with a similar structure to Penicillin, has the ability to eliminate Acinetobacter baumannii. On the other hand, Durlobactam shields Sulbactam from being broken down by enzymes that may be produced by Acinetobacter baumannii [5].
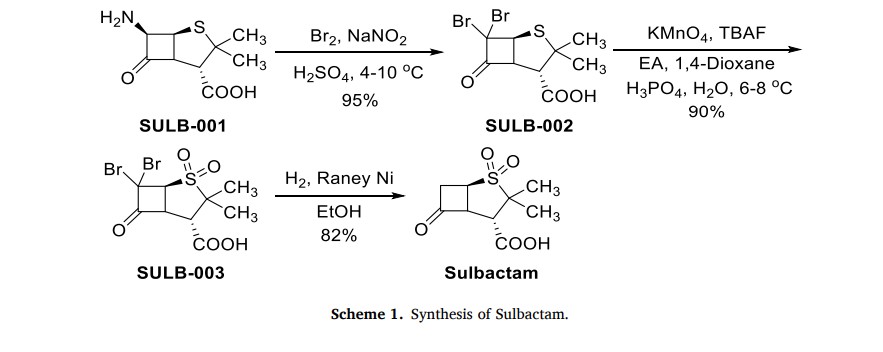
The production process of Sulbactam started with 6-aminopenicilanic acid (6-APA) (SULB-001) as the starting material (Scheme 1) [6]. It underwent bromination reaction with sodium nitrite and bromine
in the presence of sulfuric acid. Then, SULB-002 was oxidized by potassium permanganate to obtain sulfone SULB-003. Finally, the sulfonewas catalytically hydrogenated and dehalogenated in the presence of Raney nickel to get Sulbactam
[5] A. El-Ghali, A.J. Kunz Coyne, K. Caniff, C. Bleick, M.J. Rybak, Sulbactamdurlobactam: a novel β-lactam-β-lactamase inhibitor combination targeting
carbapenem-resistant Acinetobacter baumannii infections, Pharmacotherapy 43
(2023) 502–513.
[6] Z.M. Song, W. Liu, J. Yang, Y. Sun, Improvement on the synthetic process of
sulbactam, Chin. J
PATENT
https://patents.google.com/patent/CN101967155A/en
Embodiment 1
In the four-hole boiling flask of 2000ML, add 600ML methylene dichloride and 180ML2.5N sulfuric acid, stirring is cooled to below 0 ℃, add 28ML bromine and 25g Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 40g 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 100ML dichloromethane extraction 3 times, merge organic phase, with 100ML saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
To go up 6, the 6-dibromo penicillanic acid changes in the 2000ML beaker mutually. and add 250ML distilled water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 80ML deionized water, merge water, water changes in the 2000ML four-hole boiling flask, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (44g KMn04+10.8ML H3P04+700MLH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 500ML ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add the color fade of 27.5% hydrogen peroxide (about 45g) to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 250ML ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 100ML saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam;
To go up 6,6-dibromo sulbactam acid organic phase changes in the 2000ML four-hole boiling flask, adds 350ML water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 25ML methyl alcohol, add the 26g zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 25ML ethyl acetate and 25ML water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 150ML ethyl acetate extraction 4 times, washs to redness with the 50ML-100ML5% potassium permanganate solution earlier at organic layer and does not take off, again with 150ML saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 2g activated carbon decolorizing, the 15g anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 32g, the product yield is 74%, the product colour pure white was placed 30 days the color no change under the room temperature.
Embodiment 2
In the reactor of 2000L, add 600L methylene dichloride and 180L2.5N sulfuric acid, stirring is cooled to below 0 ℃, add 28L bromine and 25Kg Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 40Kg 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 100L dichloromethane extraction 3 times, merge organic phase, with 100L saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
Will on obtain 6, the 6-dibromo penicillanic acid changes in the 2000L reactor mutually. add the 250L tap water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 80L deionized water, merge water, water changes in the 2000L reactor, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (44Kg KMn04+10.8L H3P04+700LH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 500L ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add 28% hydrogen peroxide (about 44Kg) color fade to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 250L ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 100L saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam;
To go up organic phase and contain 6, the acid of 6-dibromo sulbactam changes in the 2000L reactor, adds 350L water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 25L methyl alcohol, add the 26Kg zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 25L ethyl acetate and 25L water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 150L ethyl acetate extraction 4 times, washs to redness with the 30-50L10% potassium permanganate solution earlier at organic layer and does not take off, again with 150L saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 2Kg activated carbon decolorizing, the 15Kg anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 31.5Kg, the product yield is 72.8%, the product colour pure white was placed 30 days the color no change under the room temperature.
Embodiment 3
In the four-hole boiling flask of 1000ML, add 300ML methylene dichloride and 90ML2.5N Hydrogen bromide, stirring is cooled to below 0 ℃, add 14ML bromine and 12.5g Sodium Nitrite, 0 ± 0.2 ℃, gradation adds 20g 6-APA, and controlled temperature is lower than 5 ℃, stirring reaction 1h, be cooled to then below 0 ℃, 20% aqueous solution of sodium bisulfite of dropping below 0 ℃ leaves standstill phase-splitting to the color fade of bromine, water 50ML dichloromethane extraction 3 times, merge organic phase, with 50ML saturated sodium-chloride water solution washing 2 times, obtain 6, the 6-dibromo penicillanic acid;
To go up 6, the 6-dibromo penicillanic acid changes in the 1000ML beaker mutually. and add 125ML distilled water and stir, be cooled to below 5 ℃, drip 4NNaHCO 3The aqueous solution leaves standstill phase-splitting to pH=7, organic phase extracts 3 times with the 40ML deionized water, merge water, water changes in the 1000ML four-hole boiling flask, stirring is cooled to 0 ℃, and beginning dropping oxidizing agent (22g KMn04+5.4ML H3P04+300MLH20 stirring and dissolving) dripped in 30 minutes, controlled temperature is lower than 10 ℃ in the dropping process, keep 0~5 ℃ then, stirring reaction 1h adds the 250ML ethyl acetate, drip 6N sulfuric acid to pH=1.25, be cooled to 0 ℃, slowly add 25% hydrogen peroxide (about 29g) color fade to KMn04, during continue to keep pH=1.25 with 6N sulfuric acid, controlled temperature is lower than 10 ℃, reaction 10mi n filters, and adds sodium-chlor in the filtrate to no longer dissolving, leave standstill the branch phase of anhydrating, water 125ML ethyl acetate extraction 4 times merge organic phase, and wash 2 times with the 50ML saturated sodium-chloride water solution, organic phase contains 6, the acid of 6-dibromo sulbactam.
To go up organic phase and contain 6, the acid of 6-dibromo sulbactam changes in the 1000ML four-hole boiling flask, adds 175ML water, is cooled to below 5 ℃, uses 4N NaHCO 3The aqueous solution is transferred pH to 5.0, and add 12.5ML methyl alcohol, add the 13g zinc powder in batches, and drip 6N sulfuric acid maintenance pH:4.5~5.5, after adding zinc powder, keep stirring reaction 4h below 5 ℃, keep pH=4.5~5.5 with 6N sulfuric acid simultaneously, filter, with 12.5ML ethyl acetate and 12.5ML water washing, merging filtrate is transferred pH to 2.0 with 6N sulfuric acid, add sodium-chlor to water insoluble till, leave standstill the branch phase of anhydrating, water merges organic phase with 75ML ethyl acetate extraction 4 times, washs to redness with the 15ML-35ML7% potassium permanganate solution earlier at organic layer and does not take off, again with 75ML saturated sodium-chloride water solution washing 2 times, layering, organic layer add the 1g activated carbon decolorizing, the 7.5g anhydrous magnesium sulfate drying, suction filtration, be evaporated to feed liquid and be creamy white, cool to 0 ℃ after centrifuging, after the oven dry product Sulbactam (sulbactam acid) 15.9g, the product yield is 73.5%, the product colour pure white was placed 30 days the color no change under the room temperature.
PATENT
https://patents.google.com/patent/US4420426A/en
Medical uses
The combination ampicillin/sulbactam (Unasyn) is available in the United States.[3]
The combination cefoperazone/sulbactam (Sulperazon) is available in many countries but not in the United States.[4]
The co-packaged combination sulbactam/durlobactam was approved for medical use in the United States in May 2023.[5]
Mechanism
Sulbactam is primarily used as a suicide inhibitor of β-lactamase, shielding more potent beta-lactams such as ampicillin.[6] Sulbactam itself contains a beta-lactam ring, and has weak antibacterial activity by inhibiting penicillin binding proteins (PBP) 1 and 3, but not 2.[7]
References
- ^ Totir MA, Helfand MS, Carey MP, Sheri A, Buynak JD, Bonomo RA, Carey PR (August 2007). “Sulbactam forms only minimal amounts of irreversible acrylate-enzyme with SHV-1 beta-lactamase”. Biochemistry. 46 (31): 8980–8987. doi:10.1021/bi7006146. PMC 2596720. PMID 17630699.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 492. ISBN 9783527607495.
- ^ “Unasyn- ampicillin sodium and sulbactam sodium injection, powder, for solution”. DailyMed. U.S. National Library of Medicine. 29 March 2023. Retrieved 25 May 2023.
- ^ “Sulperazon”. drugs.com.
- ^ “FDA Approves New Treatment for Pneumonia Caused by Certain Difficult-to-Treat Bacteria”. U.S. Food and Drug Administration (Press release). 24 May 2023. Retrieved 24 May 2023.
- ^ Crass RL, Pai MP (February 2019). “Pharmacokinetics and Pharmacodynamics of β-Lactamase Inhibitors”. Pharmacotherapy. 39 (2): 182–195. doi:10.1002/phar.2210. PMID 30589457. S2CID 58567725.
- ^ Penwell WF, Shapiro AB, Giacobbe RA, Gu RF, Gao N, Thresher J, et al. (March 2015). “Molecular mechanisms of sulbactam antibacterial activity and resistance determinants in Acinetobacter baumannii”. Antimicrobial Agents and Chemotherapy. 59 (3): 1680–1689. doi:10.1128/AAC.04808-14. PMC 4325763. PMID 25561334.
Further reading
Singh GS (January 2004). “Beta-lactams in the new millennium. Part-II: cephems, oxacephems, penams and sulbactam”. Mini Reviews in Medicinal Chemistry. 4 (1): 93–109. doi:10.2174/1389557043487547. PMID 14754446.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a693021 |
| Routes of administration | Intravenous, intramuscular |
| ATC code | J01CG01 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | 29% |
| Elimination half-life | 0.65–1.20 hrs |
| Excretion | Mainly kidneys (41–66% within 8 hrs) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 68373-14-8 |
| PubChem CID | 130313 |
| ChemSpider | 115306 |
| UNII | S4TF6I2330 |
| KEGG | D08533 |
| ChEBI | CHEBI:9321 |
| ChEMBL | ChEMBL403 |
| CompTox Dashboard (EPA) | DTXSID1023605 |
| ECHA InfoCard | 100.063.506 |
| Chemical and physical data | |
| Formula | C8H11NO5S |
| Molar mass | 233.24 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 148 to 151 °C (298 to 304 °F) |
| showSMILES | |
| showInChI | |
//////////Sulbactam, Xacduro, FDA 2023, APPROVALS 2023, Betamaze, Penicillanic Acid Sulfone, Sulbactamum, CP 45899, CP-45899, CP45899
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}