
FLAGS AND HITS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO FACEBOOK
...................................................................Join me on twitter
FACEBOOK
...................................................................Join me on twitter
 ..................................................................Join me on google plus
..................................................................Join me on google plus
 Googleplus
GoogleplusMYSELF

Pfizer’s PF 04937319 glucokinase activators for the treatment of Type 2 diabetes

PF 04937319
N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide
MW 432.43
CLINICAL TRIALS
A trial to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of single doses of PF-04937319 in subjects with type 2 diabetes mellitus (NCT01044537)
SYNTHESIS

Glucokinase is a key regulator of glucose homeostasis and small molecule activators of this enzyme represent a promising opportunity for the treatment of Type 2 diabetes. Several glucokinase activators have advanced to clinical studies and demonstrated promising efficacy; however, many of these early candidates also revealed hypoglycemia as a key risk. In an effort to mitigate this hypoglycemia risk while maintaining the promising efficacy of this mechanism, we have investigated a series of substituted 2-methylbenzofurans as “partial activators” of the glucokinase enzyme leading to the identification of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as an early development candidate.

Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type Il diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM. As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity. Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type 2 diabetes and the metabolic syndrome” Nature 414; 821 -827, (2001 )): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents. Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication No’s. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.
Designing glucokinase activators with reduced hypoglycemia risk: discovery of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide as a clinical candidate for the treatment of type 2 diabetes mellitus
E-mail: jeffrey.a.pfefferkorn@pfizer.com
Tel: +860 686 3421
DOI: 10.1039/C1MD00116G
http://pubs.rsc.org/en/content/articlelanding/2011/md/c1md00116g/unauth#!divAbstract
http://www.rsc.org/suppdata/md/c1/c1md00116g/c1md00116g.pdf
N,N-Dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)carbamoyl)-benzofuran-4- yloxy)pyrimidine-2-carboxamide (28). To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethoxyethane (315 mL) in a 3-neck flask equipped with overhead stirring and a condenser at 0 o C was added Me2AlCl (1 M solution in hexanes) (715 mL). The mixture was warmed to room temperature and stirred for 1.5 h. In a separate flask, 26 (52.6 g, 142.5 mmol) was dissolved in dimethoxyethane (210 mL). This mixture was then added to the amine mixture. A gum precipitated and upon scratching the flask it dissipated into a solid. The reaction was refluxed for 3.5 h. Aq. Rochelle’s salt (5 L) and 2-MeTHF (2 L) was added to the mixture and this was allowed to stir with overhead stirring for 14 h, after which time, a yellow solid precipitated. The solid was collected by filtration, washing with 2-MeTHF. The resulting solid was dried in a vacuum oven overnight to afford the desired material (50.0g) in 81% yield.
1 H NMR (400MHz, CDCl3) δ 9.54 (d, J = 1.56 Hz, 1H), 8.50 (s, 2H), 8.37 (s, 1H), 8.14 (d, J = 0.78 Hz, 1H), 7.88 – 7.92 (m, 1H), 7.52 (d, J = 1.37 Hz, 1H), 6.28 (t, J = 0.98 Hz, 1H), 3.14 (s, 3H), 2.98 (s, 3H), 2.55 (s, 3H), 2.49 (d, J = 1.17 Hz, 3H);
MS(ES+ ): m/z 433.4 (M+1), MS(ES- ): m/z 431.3 (M-1).
PAPER

http://pubs.rsc.org/en/content/articlelanding/2013/md/c2md20317k#!divAbstract
PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(16), 4571-4578
http://www.sciencedirect.com/science/article/pii/S0960894X13007452

Figure 1.
Glucokinase activators 1 and 2.
PATENT
WO 2010103437
https://www.google.co.in/patents/WO2010103437A1?cl=en
Scheme I outlines the general procedures one could use to provide compounds of the present invention having Formula (I).


Preparations of Starting Materials and Key Intermediates
Preparation of Intermediate (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but- 3-enoic acid (I- 1a):

(Ma) To a vigorously stirred solution of 5-methyl-2-furaldehyde (264 ml_, 2650 mmol) and diethyl succinate (840 ml_, 5050 mmol) in ethanol (1.820 L) at room temperature was added sodium ethoxide (0.93 L of a 21 weight % solution in ethanol) in one portion. The reaction mixture was then heated at reflux for 13 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was partitioned between ethyl acetate (1 L) and hydrochloric acid (1 L of a 2M aqueous solution). After separation, the aqueous layer was extracted with ethyl acetate (2 x 1 L). The combined organic extracts were then extracted with sodium hydrogen carbonate (2 x 1 L of a saturated aqueous solution). These aqueous extracts were combined and adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give desired (E)-3-(ethoxycarbonyl)-4-(5-methylfuran-2-yl)but-3-enoic acid (J1 Ia: 34.34 g, 5%). The original organic extract was extracted with sodium hydroxide (2 L of a 2M aqueous solution). This aqueous extract was adjusted to pH 2 with hydrochloric acid (2M aqueous solution) then extracted with ethyl acetate (2 x 1 L). These organic extracts were combined and concentrated in vacuo to give additional desired materials (395.2 gram, 63%) as red liquid. 1H NMR (CDCI3, 300 MHz) δ ppm 7.48 (s, 1 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.24 (q, 2H), 3.87 (s, 2H), 2.32 (s, 3H), 1.31 (t, 3H).
Preparation of Intermediate ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (1-1 b):

(M b) To a vigorously stirred solution of (E)-3-(ethoxycarbonyl)-4-(5- methylfuran-2-yl)but-3-enoic acid (1-1 a: 326.6 g, 1 .371 mol) in acetic anhydride (1 .77 L, 18.72 mol) at room temperature was added sodium acetate (193 g, 2350 mmol) in one portion. The reaction mixture was then heated at reflux for 2.5 hours. After cooling to room temperature, the mixture was concentrated in vacuo (all batches were combined at this point). The resulting residue was suspended in dichloromethane (1 .5 L) and filtered, washing the solids with dichloromethane (3 x 500 ml_). The combined filtrate and washings were then washed with sodium hydrogencarbonate (2 x 1 L of a saturated aqueous solution) and brine (2 L), then concentrated in vacuo to give desired ethyl 4-acetoxy-2-methylbenzofuran-6-carboxylate (H b: 549.03 g, quantitative). 1H NMR (CDCI3, 300 MHz) δ ppm 8.00-7.99 (m, 1 H), 7.64 (d, 1 H), 6.32-6.32 (m, 1 H), 4.38 (q, 2H), 2.47 (d, 3H), 2.37 (s, 3H), 1 .39 (t, 3H).
Preparation of Intermediate ethyl 4-hydroxy-2-methylbenzofuran-6- carboxylate (1- 1 c):

(He) To a stirred solution of ethyl 4-acetoxy-2-methylbenzofuran-6- carboxylate (Hb: 549.03 g, 1 .37 mol) in ethanol (4.00 L) at room temperature was added potassium carbonate (266 g, 1 .92 mol) in one portion. The reaction mixture was then heated at 600C for 3 hours. Potassium carbonate (100 g, 0.720 mol) was then added in one portion and the reaction mixture was heated at 600C for a further 3 hours. After cooling to room temperature the mixture was diluted with dichloromethane (2 L) and the suspension filtered, washing the solids with dichloromethane (2 x 1 L) (all batches were combined at this point). The combined filtrate and washings were then washed with citric acid (2.5 L of a 1 M aqueous solution), then concentrated in vacuo and the resulting residue purified by dry flash chromatography (hexane then 2:1 hexane:ethyl acetate). All fractions containing the desired product were combined and concentrated in vacuo. The resulting residue, which solidified on standing, was slurried with cold toluene and filtered. The solids were then stirred with hot toluene and decolourising charcoal for 1 hour, followed by filtration of the hot mixture through a pad of celite. The filtrate was allowed to cool and the resulting precipitate isolated by filtration to give desired ethyl 4-hydroxy-2- methylbenzofuran-6-carboxylate (1-1 c: 360 g, 90%) as orange powder.
1H NMR (CDCI3, 300 MHz) δ ppm 7.73-7.73 (m, 1 H), 7.45 (d, 1 H), 6.51 -6.50 (m, 1 H), 5.85 (s, 1 H), 4.39 (q, 2H), 2.48 (d, 3H), 1.40 (t, 3H). LCMS (liquid chromatography mass spectrometry): m/z 221.06 (96.39 % purity).
Preparation of SM-25-bromo-N,N-dimethylpyrimidine-2-carboxamide (SM-
£1:

(SM-2) Oxalyl chloride (47.4g, 369mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (5Og, 250mmol) in dichloromethane (821 ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture. The acid dissolved after 30 minutess. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 100%). The 5-Bromo-pyrimidine-2-carbonyl chloride (55g, 250mmol) was dissolved in tetrahydrofuran (828ml) and dimethyl-amine (2M solution in tetrahydrofuran) (373ml, 745mmol) was added portionwise at room temperature. The reaction was stirred at room temperature under nitrogen for 16 hours, after which time, LCMS indicated completion. The mixture was diluted with ethyl acetate (500ml) and washed with H2O (500ml). The water layer was further extracted with CH2CI2 (5x500ml), all organics combined, and dried over magnesium sulfate. The filtrate was concentrated in vacuo and then suspended in methyl-/-butylether (650ml). The solution was then heated to reflux. The hot solution was allowed to cool overnight to afford pink crystals. The crystals were filtered and washed with cold methyl-t-butylether (100ml) the solid was dried in a vacuum oven at 550C for 12 hourrs to afford the title compound 5-bromo-N,N-dimethylpyhmidine-2-carboxamide (SM-2: 44g, 77%) as a pink solid.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 2.94 (s, 3 H) 3.13 (s, 3 H) 8.85 (s, 2 H) m/z (M+1 ) = 232.
Preparation of Intermediate Ethyl 4-(2-(dimethylcarbamoyl)Dyrimidin-5- yloxy)-2-methylbenzofuran-6-carboxylate (l-2a):

A mixture of Cs2CO3 (62.1 g, 191 mmol), 5-bromo-N,N- dimethylpyrimidine-2-carboxamide (SM-2: 24g, 104mmol) and ethyl 4- hydroxy-2-methylbenzofuran-6-carboxylate (1-1 c: 2Og, 91 mmol); 1 ,10- phenanthroline (1.64g, 9.07mmol) and copper iodide (864mg, 4.54mmol) in dimethylformamide (200ml) was purged with N2 gas and then heated to 90°C using a mechanical stirrer. The heterogeneous reaction mixture was stirred at this temperature for 18 hours. HPLC indicated near completion. The reaction mixture was cooled to 350C and diluted with ethyl acetate (300ml). The mixture was filtered to remove any cesium carbonate. The filtrate was then partitioned between water (500ml) and ethyl acetate (500ml); however, no separation was observed. Concentrated HCL (20ml) was added to the mixture. When the aqueous phase was about pH1 , the phases separated. The organics were separated and the aqueous layer reextracted with ethyl acetate (2x500ml). All organics were combined and back extracted with water (200ml) and brine (500ml). The organics were separated and treated with activated charcoal (10g) and magnesium sulfate. The mixture was allowed to stir for 10 minutes and then filtered through a plug of celite to afford a crude yellow solution. The filter cake was washed with ethyl acetate (100 ml_). The organics were concentrated in vacuo to afford a crude solid this was dried under high vacuum for 4 days. The dry crude solid was triturated using methanol (80 ml_). The solids were dispersed into a fine light orange crystalline powder with a red liquor. The solids were isolated by filtration and rinsed with methanol (20 ml_). The solid was dried in the vacuum oven at 550C for 12 hours to afford ethyl 4-(2- (dimethylcarbamoyl)pyrimidin-5-yloxy)-2-methylbenzofuran-6-carboxylate (J1 2a) as a yellow solid (18.2g, 54%)
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.41 (t, J=7.12 Hz, 3 H) 2.50 (d, J=0.98 Hz, 3 H) 3.00 (s, 3 H) 3.17 (s, 3 H) 4.41 (d, J=7.22 Hz, 2 H) 6.29 (s, 1 H) 7.62 (d, J=1.17 Hz, 1 H) 8.06 (s, 1 H) 8.50 (s, 2 H). m/z (M+1 ) = 370.5
Preparation of Starting material 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3):

(SM-3) Oxalyl chloride (1 .45g, 1 1 .1 mmol) was added to a suspension of 5-
Bromo-pyrimidine-2-carboxylic acid (1 .5g, 7.4mmol) in dichloromethane (50ml) at room temperature followed by 1 -2 drop of dimethylformamide. The reaction mixture was stirred under nitrogen for 2 hours LCMS in methanol indicated the presence of the methyl ester and some acid. Dimethylformamide (0.2ml) was added to the reaction mixture and all of the acid dissolved after 30 minutes. LCMS showed corresponding methyl ester and no starting material peak was observed. The solvent was removed and dried in vacuo to afford the crude 5-Bromo-pyrimidine-2-carbonyl chloride (1 -6g). 5-Bromo-pyrinnidine-2-carbonyl chloride (1600mg, 7.225mnnol) was dissolved in dichloromethane (25ml) and triethylamine (4.03ml, 28.9mmol) was added followed by ethyl-methyl-amine (0.68 mL, 7.92 mmol). The reaction was stirred at room temperature under nitrogen for 16 ours, after which time, LCMS indicated completion. The mixture was diluted with dichloromethane (50ml) and washed with water (50ml) followed by 10% citric acid (50ml) and brine (50ml). The organic layer was separated and dried over MgSO4, the residue was filtered and the solvent was removed in vacuo to afford the title compound 5-bromo-N-ethyl-N-methylpyrimidine-2- carboxamide (SM-3): (1.4g, 79.4%) as a brown oil.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.08 – 1.31 (m, 3 H) 2.99 (d, J=79.05 Hz, 3 H) 3.19 (q, J=7.22 Hz, 1 H) 3.59 (q, J=7.22 Hz, 1 H) 8.84 (d, J=3.12 Hz, 2 H)
Example 2
Preparation of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2- yl)carbamoyl)-benzofuran-4-yloxy)Dyrimidine-2-carboxamide (2):

(2)
To a solution of the 5-methyl-2-aminopyrazine (38.9 g, 356 mmol) in dimethylether (315 ml_) in a 3-neck flask equipped with overhead stirring and a condensor at O0C was added Me2AICI (1 M solution in hexanes) (715 ml_). The mixture was warmed at room temperature and stirred for 1.5 hours. In a separate flask, ethyl 4-(2-(dimethylcarbamoyl)pyrimidin-5-yloxy)-2- methylbenzofuran-6-carboxylate (l-2a: 52.6g, 142.5mmol) was dissolved in dimethylether (210 ml_). This mixture was then added to the complexed amine. A gum precipitated upon scratching the flask and dissipated into a solid. The resultant reaction was refluxed for 3.5 hours HPLC indicated 93% complete. Five liters of Rochelles salt made up in water and 2 liters of 2- methyltetrahydrofuran was added to the mixture. The reaction mixture was then poured into the biphasic system. The mixture was allowed to stir with overhead stirring for 14 hours, after which time, a yellow solid precipitated. The solid was collected through filteration. The solid retained was washed with 2-methyltetrahydrofuran. The resultant solid was dried in vacuo oven overnight to afford the title compound N,N-dimethyl-5-(2-methyl-6-((5- methylpyrazin-2-yl)carbamoyl)benzofuran-4-yloxy)pyhmidine-2-carboxamide (2): (49.98g, 81 %)
1H NMR (400 MHz, CHLOROFORM-d) d ppm 2.49 (d, J=1 .17 Hz, 3H) 2.55 (s, 3H) 2.98 (s, 3 H) 3.14 (s, 3 H) 6.28 (t, J=0.98 Hz, 1 H) 7.52 (d, J=1 .37 Hz, 1 H) 7.88 – 7.92 (m, 1 H) 8.14 (d, J=0.78 Hz, 1 H) 8.37 (s, 1 H) 8.50 (s, 2 H) 9.54 (d, J=1 .56 Hz, 1 H).
m/z (M+1 ) = 433.4, m/z (M-1 )= 431 .5
REFERENCES
Beebe, D.A.; Ross, T.T.; Rolph, T.P.; Pfefferkorn, J.A.; Esler, W.P.
The glucokinase activator PF-04937319 improves glycemic control in combination with exercise without causing hypoglycemia in diabetic rats
74th Annu Meet Sci Sess Am Diabetes Assoc (ADA) (June 13-17, San Francisco) 2014, Abst 1113-P
Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A.
Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes
Diabetes Obes Metab 2015, 17(8): 751
Study to compare single dose of three modified release formulations of PF-04937319 with immediate release material-sparing-tablet (IR MST) formulation previously studied in adults with type 2 diabetes mellitus (NCT02206607)
OTHERS

///////////Pfizer , PF 04937319, glucokinase activators, Type 2 diabetes
TARANABANT

Taranabant ( MK-0364)
701977-09-5
N-[3-(4-Chlorophenyl)-2(S)-(3-cyanophenyl)-1(S)-methylpropyl]-2-methyl-2-[5-(trifluoromethyl)pyridin-2-yloxy]propionamide
N-[(2S,3S)-4-(4-chlorophenyl)-3-(3-cyanophenyl)butan-2-yl]-2-methyl-2-[5-(trifluoromethyl)pyridin-2-yl]oxypropanamide
Taranabant (codenamed MK-0364) is a cannabinoid receptor type 1 inverse agonist being investigated as a potential treatment forobesity due to its anorectic effects.[1][2] It was discovered by Merck & Co.
In October 2008, Merck has stopped its phase III clinical trials with the drugs due to high level of central side effects, mainlydepression and anxiety.[3][4][5][6]
The compound had also been in clinical evaluation in chronic cigarette smokers as an aid for smoking cessation.
Paper
 .
.
http://pubs.rsc.org/en/content/articlelanding/2013/cs/c2cs35410a#!divAbstract
PATENT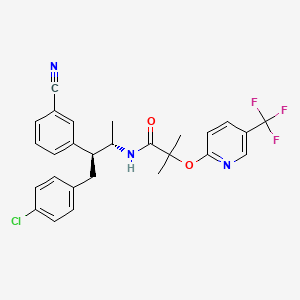
WO 2003077847
http://www.google.co.in/patents/WO2003077847A2?cl=en
PAPERS
Convenient total synthesis of taranabant (MK-0364), a novel cannabinoid-1 receptor inverse agonist as an anti-obesity agent
Tetrahedron 2007, 63(52): 12845
Wallace, D.J.; Campos, K.R.; Shultz, S.; Klapars, A.; et al.
New efficient asymmetric synthesis of taranabant, a CB1R inverse agonist for the treatment of obesity
Org Process Res Dev 2009, 13(1): 84
Lin, L.S.; Lanza, T.J. Jr.; Jewell, J.P.; Liu, P.; Shah, S.K.; Qi, H.; Tong, X.; Wang, J.; Xu, S.S.; Fong, T.M.; Shen, C.P.; Lao, J.; Xiao, J.C.; Shearman, L.P.; Stribling, D.S.; Rosko, K.; Strack, A.; Marsh, D.J.; Feng, Y.; Kumar, S.; Samuel, K.; Yin, W.; Ploeg, L.H.; Goulet, M.T.; Hagmann, W.K.
Discovery of N-[(1S,2S)-3-(4-Chlorophenyl)-2- (3-cyanophenyl)-1-methylpropyl]-2-methyl-2- [[5-(trifluoromethyl)pyridin-2-yl]oxy]propanamide (MK-0364), a novel, acyclic cannabinoid-1 receptor inverse agonist for the treatment of obesity
J Med Chem 2006, 49(26): 7584
Cole, P.; Serradell, N.; Rosa, E.; Bolos, J. Taranabant Drugs Fut 2008, 33(3): 206
PAPER
Chen, C.-Y.; Frey, L.F.; Shultz, S.; et al. Catalytic, enantioselective synthesis of taranabant, a novel, acyclic cannabinoid-1 receptor inverse agonist for the treatment of obesity
Org Process Res Dev 2007, 11(3): 616
http://pubs.acs.org/doi/abs/10.1021/op700026n

Chiral amide 1 (MK-0364, taranabant) is a potent, selective, and orally bioavailable cannabinoid-1 receptor (CB-1R) inverse agonist indicated for the treatment of obesity. An asymmetric synthesis featuring a dynamic kinetic resolution via hydrogenation for the preparation of the bromo alcohol 5 is disclosed. Conversion of the alcohol intermediate to the chiral amide 1 is accomplished in good overall yield.
N-[(1S,2S)-3-(4-Chlorophenyl)-2-(3-cyanophenyl)-1-methylpropyl]-2-methyl-2-{[5-(trifluoromethyl)pyridin-2-yl]oxy}propanamide (1, MK-0364). hemisolvate (approximately 94 wt %, 94% isolated yield from amine salt).
1H NMR (CDCl3): δ 8.35 (s, 1H), 7.83 (dd, J = 2.38, 8.70 Hz, 1H), 7.45 (d, J = 7.57 Hz, 1H), 7.31 (t, J = 7.99 Hz, 1H), 7.24 (m, 2H), 7.07 (d, J = 8.34 Hz, 2H), 6.88 (d, J = 8.63 Hz, 1H), 6.72 (d, J = 8.33 Hz, 2H), 5.88 (d, J = 8.95 Hz, 1H), 4.34 (m, 1H), 3.13 (dd, J = 3.04, 12.72 Hz, 1H), 2.82 (m, 2H), 1.76 (s, 3H), 1.72 (s, 3H), 0.87 (d, J = 6.72 Hz, 3H).
13C NMR (CDCl3): δ 173.4, 163.9, 144.5 (q, J = 4.30 Hz), 142.4, 137.5, 136.3 (q, J = 3.02 Hz), 133.0, 132.2, 132.0, 130.7, 130.0, 129.3, 128.5, 123.7 (q, J = 271.45 Hz), 121.1 (q, J = 33.32 Hz), 118.6, 112.7, 112.6, 82.1, 53.6, 48.6, 38.2, 25.4, 25.1, 18.4.
Anal. Calcd for C27H25ClF3N3O2: C 62.85; H 4.88; N 8.14. Found: C 62.95; H 4.74; N 8.00.
References
- Armstrong HE, Galka A, Lin LS, Lanza TJ Jr, Jewell JP, Shah SK, et al. “Substituted acyclic sulfonamides as human cannabinoid-1 receptor inverse agonists.” Bioorganic & Medicinal Chemistry Letters. 2007 Apr 15;17(8):2184-7. PMID 17293109. doi:10.1016/j.bmcl.2007.01.087
- Fong TM, Guan XM, Marsh DJ, Shen CP, Stribling DS, Rosko KM, et al. “Antiobesity efficacy of a novel cannabinoid-1 receptor inverse agonist, N-[(1S,2S)-3-(4-chlorophenyl)-2-(3-cyanophenyl)-1-methylpropyl]-2-methyl-2-[[5-(trifluoromethyl)pyridin-2-yl]oxy]propanamide (MK-0364), in rodents.” Journal of Pharmacology and Experimental Therapeutics. 2007 Jun;321(3):1013-22. PMID 17327489.doi:10.1124/jpet.106.118737
- “Press release by Merck”. Retrieved October 2008.
- Aronne LJ, Tonstad S, Moreno M, Gantz I, Erondu N, Suryawanshi S, Molony C, Sieberts S, Nayee J, Meehan AG, Shapiro D, Heymsfield SB, Kaufman KD, Amatruda JM (May 2010). “A clinical trial assessing the safety and efficacy of taranabant, a CB1R inverse agonist, in obese and overweight patients: a high-dose study”. International Journal of Obesity (2005) 34 (5): 919–35. doi:10.1038/ijo.2010.21.PMID 20157323.
- Kipnes MS, Hollander P, Fujioka K, Gantz I, Seck T, Erondu N, Shentu Y, Lu K, Suryawanshi S, Chou M, Johnson-Levonas AO, Heymsfield SB, Shapiro D, Kaufman KD, Amatruda JM (June 2010). “A one-year study to assess the safety and efficacy of the CB1R inverse agonist taranabant in overweight and obese patients with type 2 diabetes”. Diabetes, Obesity & Metabolism 12 (6): 517–31. doi:10.1111/j.1463-1326.2009.01188.x. PMID 20518807.
- Proietto J, Rissanen A, Harp JB, Erondu N, Yu Q, Suryawanshi S, Jones ME, Johnson-Levonas AO, Heymsfield SB, Kaufman KD, Amatruda JM (August 2010). “A clinical trial assessing the safety and efficacy of the CB1R inverse agonist taranabant in obese and overweight patients: low-dose study”. International Journal of Obesity (2005) 34 (8): 1243–54. doi:10.1038/ijo.2010.38. PMID 20212496.
| Radiolabeled cannabinoid-1 receptor modulators [US7754188] | 2006-06-01 | 2010-07-13 |
| Combination therapy for the treatment of obesity [US2006270650] | 2006-11-30 | |
| CERTAIN CRYSTALLINE DIPHENYLAZETIDINONE HYDRATES, PHARMACEUTICAL COMPOSITIONS THEREOF AND METHODS FOR THEIR USE [US8003636] | 2009-08-13 | 2011-08-23 |
| NOVEL DIPHENYLAZETIDINONE SUBSTITUTED BY PIPERAZINE-1-SULFONIC ACID AND HAVING IMPROVED PHARMACOLOGICAL PROPERTIES [US2009264402] | 2009-10-22 | |
| Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, process for preparing them, medicaments comprising these compounds, and their use [US7759366] | 2009-08-27 | 2010-07-20 |
| COMPOUNDS WITH A COMBINATION OF CANNABINOID CB1 ANTAGONISM AND SEROTONIN REUPTAKE INHIBITION [US8138174] | 2008-09-04 | 2012-03-20 |
| Substituted imidazoline-2,4-diones, process for preparation thereof, medicaments comprising these compounds and use thereof [US2011112097] | 2011-05-12 | |
| Novel phenyl-substituted imidazolidines, process for preparation thereof, medicaments comprising said compounds and use thereof [US2011178134] | 2011-07-21 | |
| HETEROCYCLIC COMPOUNDS, PROCESSES FOR THEIR PREPARATION, MEDICAMENTS COMPRISING THESE COMPOUNDS, AND THE USE THEREOF [US2011183998] | 2011-07-28 | |
| Cyclic pyridyl-N-[1,3,4]-thiadiazol-2-yl-benzene sulfonamides, processes for their preparation and their use as pharmaceuticals [US2011224263] | 2011-09-15 |
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[(2S,3S)-4-(4-chlorophenyl)-3-(3-cyanophenyl)-2-butanyl]-2-methyl-2-{[5-(trifluoromethyl)-2-pyridinyl]oxy}propanamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Identifiers | |
| CAS Number | 701977-09-5 |
| ATC code | A08AX |
| PubChem | CID: 11226090 |
| UNII | X9U622S114 |
| Chemical data | |
| Formula | C27H25ClF3N3O2 |
| Molecular mass | 515.95 g/mol |
///////////CC(C(CC1=CC=C(C=C1)Cl)C2=CC=CC(=C2)C#N)NC(=O)C(C)(C)OC3=NC=C(C=C3)C(F)(F)F
C[C@@H]([C@@H](CC1=CC=C(C=C1)Cl)C2=CC=CC(=C2)C#N)NC(=O)C(C)(C)OC3=NC=C(C=C3)C(F)(F)F
Revised USP Chapter on Visual Inspection published
DRUG REGULATORY AFFAIRS INTERNATIONAL

After the long-awaited Chapter <1790> on visual inspection of injections was first published in the PF 41(1) as a draft the USP has now submitted a revised draft in the PF41 (6). Read more about the revised draft of the USP Chapter <1790>.
After the long-awaited Chapter <1790> Visual Inspection of Injections was first published in the Pharmacopeial Forum 41(1) as a draft the USP has now submitted a revised draft in the PF41 (6). Through its number >1000, the monograph <1790> is not binding but rather offers an explanation to Chapter <790> Visible Particulates in Injections.
With regard to the content, several comments recommended by the industry have been included. The new Chapter 9 represents the biggest change and describes the evaluation of marketed products where anomalies had been observed regarding particles. The test procedure for it is described in Chapter <790>. Yet, this topic was missing…
View original post 149 more words
New Antibacterial oxazolidinones in pipeline by Wockhardt

WCK ?
(5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
(5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
MF C19 H25 F2 N3 O5, MW 413.42
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-hydroxy-4-(methoxymethyl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
CAS 957796-51-9
Antibacterial oxazolidinones
Wockhardt Ltd, Innovator
THIS MAY BE WCK 4086?????….WATCHOUT THIS POST FOR UPDATION
PATENTS
WO 2015173664, US8217058, WO 2012059823, IN 2011MU03726

Oxazolidinone represent a novel chemical class of synthetic antimicrobial agents. Linezolid represents the first member of this class to be used clinically. Oxazolidinones display activity against important Gram-positive human and veterinary pathogens including Methicillin-Resistant Staphylococcus aureus (MRSA), Vancomycin Resistant Enterococci (VRE) and β-lactam Resistant Streptococcus pneumoniae (PRSP). The oxazolidinones also show activity against Gram-negative aerobic bacteria, Gram-positive and Gram-negative anaerobes. (Diekema D J et al., Lancet 2001 ; 358: 1975-82).
Various oxazolidinones and their methods of preparation are disclosed in the literature. International Publication No. WO 1995/25106 discloses substituted piperidino phenyloxazolidinones and International Publication No. WO 1996/13502 discloses phenyloxazolidinones having a multisubstituted azetidinyl or pyrrolidinyl moiety. US Patent Publication No. 2004/0063954, International Publication Nos. WO 2004/007489 and WO 2004/007488 disclose piperidinyl phenyl oxazolidinones for antimicrobial use.
Pyrrolidinyl/piperidinyl phenyl oxazohdinone antibacterial agents are also described in Kim H Y et al., Bioorg. & Med. Chem. Lett., (2003), 13:2227-2230. International Publication No. WO 1996/35691 discloses spirocyclic and bicyclic diazinyl and carbazinyl oxazolidinone derivatives. Diazepeno phenyloxazolidinone derivatives are disclosed in the International Publication No. WO 1999/24428. International Publication No. WO 2002/06278 discloses substituted aminopiperidino phenyloxazolidinone derivatives.
Various other methods of preparation of oxazolidinones are reported in US Patent No. 7087784, US Patent No. 6740754, US Patent No. 4948801 , US Patent No. 3654298, US Patent No. 5837870, Canadian Patent No. 681830, J. Med. Chem., 32, 1673 (1989), Tetrahedron, 45, 1323 (1989), J. Med. Chem., 33, 2569 (1990), Tetrahedron Letters, 37, 7937-40 (1996) and Organic Process Research and Development, 11 , 739-741(2007).
Indian Patent Application No. 2534/MUM/2007 discloses a process for the preparation of substituted piperidino phenyloxazolidinones. International Publication No. WO2012/059823 further discloses the process for the preparation of phosphoric acid mono-(L-{4-[(5)-5-(acetylaminomethyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}4-methoxymethyl piperidine-4-yl)ester.
US Patent No. 8217058 discloses (5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide as an antibacterial agent and its process for preparation.
PATENT
WO2015173664

In some embodiments, there is provided a process for preparation of a compound of Formula (I) as shown in Scheme 1

(I I) (I N)
Scheme 1

Example 1
Preparation of (55)-iV-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide (I)
To a stirred solution of lithium teri-butoxide (59.1 g, 0.74 mol) in tetrahydrofuran (500 ml) was added a solution of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-carbamic acid benzyl ester (II) (100 g, 0.25 mol) in 500 ml of tetrahydrofuran slowly at room temperature. The resulting mixture was stirred for 3 hours at room temperature (formation of lumps observed). The reaction mixture was cooled to temperature of 10°C to 15°C and acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) (95.2 g, 0.49 mol) was added in one lot, after 5 minutes methanol (2.36 g, 0.075 mol) was added in one portion. The resulting mixture was stirred further at temperature of 10°C to 15°C. After 5 hours the reaction mixture was allowed to warm to room temperature and stirring continued further for 16 hours. An aqueous solution of saturated ammonium chloride (100 ml) was added to the reaction mixture, the resulting mixture was stirred well and the solvent evaporated under reduced pressure (35°C, 150 mm Hg). The residual mixture was diluted with water (1 L stirred well and filtered under suction, the residual solid was washed with additional fresh water (100 ml). The residual mass was suspended in acetone (500 ml), stirred well and the mixture diluted with hexane (1 L), slowly. The mixture was stirred further for 1 hour and filtered under suction. The residual solid was washed with a 2:1 mixture of acetone and water (100 ml). The residual solid was dried at 45°C, for 3.5 hour at 4 mm Hg, to obtain the 78 g of (55)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l -yl)-phenyl]-2-oxo-oxazolidin-5-ylmethylj -acetamide (I) as white solid, in 77% yield.
Analysis:
Mass: 414 (M+l ); for Molecular Weight: 413 and Molecular Formula: ![]()
Melting Point: 178-179°C;
1H NMR (400 MHz, DMSO): δ 8.18-8.21 (m, 1H), 7.19-7.25 (d, 2H), 4.07-4.71 (m, 1H), 4.32 (s, 1H), 4.02-4.07 (t, 1H), 3.64-3.68 (t, 1H), 3.14 (s, 2H), 2.81-2.83 (d, 2H), 1.81 (s, 3H), 1.63-1.69 (t, 2H), 1.42-1.45 (d, 2H);
Purity as determined by HPLC: 97.65%.
Example 2
Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III)
Step-I: Preparation of l-amino-3-chloro-propan-2-ol hydrochloride (VI)
Benzaldehyde (118.67 g, 1.03 mol) was dissolved in ethanol (297 ml) under stirring and the solution was cooled to 18-19°C. To this solution aqueous ammonia solution (25%) (101.58 ml) was added slowly, followed by slow addition of S-epichlorohydrin (100 g, 1 mol). The resulting mixture was warmed to 40°C and stirred for 7 hours. The mixture was allowed to cool to room temperature and stirred further. After 16 hours, the reaction mixture was concentrated to 50% volume under reduced pressure. Toluene (228 ml) was added to the reaction mixture followed by addition of aqueous hydrochloric acid (162 ml of concentrated hydrochloric acid diluted with 152 ml of water). The mixture thus obtained for 3 hours at 45°C, the resulting mixture was allowed to cool to room temperature and the toluene layer separated. The toluene layer was further extracted with water (56 ml). The combined aqueous layer was diluted with ethanol (56 ml) and the mixture evaporated under reduced pressure. This process was repeated again. To the final concentrate was added ethanol (180 ml), stirred for 10 minutes and the mixture cooled to -28°C to -30°C and maintained at this temperature for 2 hours. The separated solid was filtered under suction and the residue washed with cold (-30°C) ethanol (50 ml). The residue was dried at 45°C, under reduced pressure (4 mm Hg) for 3 hours, to obtain 96 g of l-amino-3-chloro-propan-2-ol hydrochloride (VI) as white solid in 61% yield.
Analysis:
Mass: 110 (M+l) as free base; for Molecular Weight: 145.5 and Molecular Formula: ![]()
1H NMR (400 MHz, D20): δ 4.02-4.08 (m, 1H), 3.51-3.61 (m, 2H), 3.12-3.16 (dd, 1H), 2.93 -2.99 (dd, 1H).
Step-II: Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III).
A stirred solution of dichloromethane (220.8 ml) containing the step-I salt (96 g, 0.66 mol) was cooled to 18-20°C. Acetic anhydride (154.78 g, 1.5175 mol) was added slowly (slight exothermic). Pyridine (67.76 g, 0.8577 mol) was added slowly (exothermic) while maintaining the temperature at 18-20°C. The resulting mixture was heated to 40°C for 5 hours. The reaction mixture was allowed to cool to room temperature and stirring continued for further 16 hours. The reaction mass was cooled to 3-6°C and diluted with 170 ml of fresh water. To this was added an aqueous solution of potassium carbonate (191.2 g of K2CO3 in 382 ml water). The reaction mixture was further diluted with additional dichloromethane (170 ml) and water (425 ml). The reaction mass was stirred well and the dichloromethane layer separated. The aqueous layer was further extracted with 2×170 ml dichloromethane. The combined dichloromethane layer was washed with aqueous sodium chloride solution (13.6 g of sodium chloride in 493 ml water). The solvent was evaporated till a volume of 170 ml and the residual layer was diluted with toluene (340 ml), stirred well and the solvent was evaporated completely at 40°C under reduced pressure (4 mm Hg). To the residue ethyl acetate (170 ml) and hexane (187 ml) were added and the mixture stirred for 30 minute. The separated solid was filtered under suction and the residue washed with 50 ml of a 1 :1 mixture of ethyl acetate and hexane. The solid obtained was dried under reduced pressure (4 mm Hg) at 45°C for 3.5 hours, to obtain 96 g of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) as a white solid, in 75% yield.
Analysis:
Mass: 194 (M+l); for Molecular Weight: 193 and Molecular Formula: C7Hi2ClN03; 1H NMR (400 MHz, CDC13): 5 5.69 (s, 1H), 5.0-5.1 (m, 1H), 3.4-3.7 (m, 4H), 2.1 (s, 3H), 1.9 (s, 3H).
PATENT
http://www.google.st/patents/WO2007132314A2?cl=en



(3) (4)
Scheme -1

(6) Formula π Scheme-2

Formula II Formula in

Formula I(a) Scheme-4
Example -11 : (5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
The example- 10 (54.86 g, 0.144 mol) was suspended in methanol (1100 ml) under stirring at RT. Sodium metal (4 g, 0.174 mol) was added in small lots in 2 min to the above suspension under stirring. The reaction mixture was warmed to 40-420C and was stirred at this temperature for about 40 hrs. After completion of the reaction (TLC), the solvent was evaporated under reduced pressure to obtain a thick slurry. The thick slurry thus obtained was gradually added to water (1100 ml) under stirring. After the complete addition, the pH of the aqueous suspension was adjusted to 7 by adding sufficient quantity of glacial acetic acid. The separated solid was filtered and the residue was washed with water. The obtained solid was further purified by column chromatography over silica gel to obtain the product as a white solid, 32.7 g, 55 % yield.
M.P.: 173-1740C;
MS : M+l= 414(MH+, 100%); for M.F.: Ci9H25F2N3O5
1H-NMR (400 MHz, CDCl3): δ 7.0-7.1 (m, 2H5Ar-H), 6.0 (t, IH, NH), 4.70-4.80 (m, IH), 4.00 (t,lH), 3.70-3.75 (m, 2H), 3.5-3.7 (m, IH), 3.43 (s, 3H, OCH3), 3.37-3.42 (m, 2H), 3.30 (s, 2H, -OCH2), 3.0-3.05 (m, 2H), 2.22(bs,lH ,-OH),2.04 (s, 3H, COCH3), 1.70-1.75 (m, 4H).
Patent
INDIAN 3049/MUM/2010
Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxy methyl-piperidin-4-yl) ester

Specific intermediate compounds of the invention include:
6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane;
1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol;
[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one;
(5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one; and
(5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide.
Examples
Preparation of Intermediate-1: 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one
Chloroform (9.3 L) was charged in a 20 L reaction assembly and 4-piperidone hydrochloride (1.17 Kg, 7.62 mol) was added under stirring followed by triethylamine (2.14 Kg, 2.95 L, 21.1 mol). After 30 minutes of stirring, 3,4,5-trifluoronitrobenzene (1.5 Kg, 8.47 mol) was added to the mixture in one lot and the contents were heated to 65-70ºC for 8 h. After completion of the reaction, chloroform was removed under vacuum to obtain a syrupy mass. At this stage, water (10 L) was added to the mass and the chloroform recovery was continued under vacuum below 65oC till the chloroform was removed completely. The slurry was cooled to RT and filtered. The solid product was washed with water (3 L) followed by hexanes (2 L). The product was dried in a vacuum oven below 70oC to obtain the product as a yellow solid, 1.88 Kg ; Yield 97%.
M.P.: 130-132oC; MS: 257(M+1); M.F.: C11H10F2N2O3.
Preparation of Intermediate 3: 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
Method A:
Preparation of Intermediate–2: (Stage-I): 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane
A solution of trimethylsulfoxonium iodide (1.504kg, 6.836mol) in acetonitrile (7L) was cooled to 0 to 5oC. , under argon atmosphere. Potassium tert-butoxide (0.736kg, 6.552 mol) was added in small lots over 0.5h. The resulting solution was stirred for 2h at the same temperature. To this solution was added 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one ( 1.4kg, 5.46mol) in small lots over a period of 1h, while maintaining the temp. between 5-10oC. The resulting mixture was stirred for 1h. The solvent was evaporated to a minimum amount possible, under reduced pressure while maintaining the temperature below 10oC. The residue was poured in water( 18L) and the pH adjusted to neutral with dilute acetic acid. The resulting slurry was stirred well and the separated solid filtered under suction. The solid was washed with fresh water till the filtrate was free of acetic acid. The solid was dried at 80oC, for 6h, under reduced pressure to obtain the product as pale yellow solid, 1.264kgs, yield 85%.
M.P.: 96-97oC; MS: M+1: 271; M.F.: C12H12F2N2O3,.
Preparation of Intermediate-3: (Stage-II): 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
To a solution of sodium methoxide (236g, 4.35mol) in methanol (3L), at RT, was added 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro [2.5]octane (964g, 3.57mol) in small portions and the reaction mixture was stirred for 26h at RT. Acetic acid (265g, 4.44mol) was added slowly to neutralize the pH of the solution. The resulting mixture was poured into chilled water(18L) and stirred for 1h. The separated solid was filtered under suction. The solid was washed with additional water till the filtrate was free of acetic acid. The solid was dried for 10hat RT under reduced pressure, to obtain the product as a pale yellow solid, 973g, yield, 90%
M.P.: 84-86oC; MS: 303 (M+1); M.F.: C13H16F2N2O4
Method B:
Dimethylsulfoxide (DMSO, 100 ml) and methanol (500 ml) were charged in a 1 L glass reaction assembly. Potassium hydroxide (59.2g, 0.898 mol) was charged in the assembly followed by trimethylsulfoxonium iodide (94.5 g, 0.43 mol) and the contents were stirred for 30 minutes and then cooled to 10oC-15oC. To the cooled contents was added 1-(2,6-difluoro-4-nitrophenyl)-piperidin-4-one (100 g, 0.39 mol) in small lots. After the addition, the temperature was allowed to raise to RT and the contents were further stirred for 24 h (ring opening of the epoxide intermediate viz. 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane takes place).
[Physical data of the intermediate: M.P.: 96-970C, MS: 271(M+1); M.F.: C12H12F2N2O3, .
After completion of the reaction the contents were poured slowly in ice-water (600g crushed ice in 600 ml water). The precipitated solid product was filtered and was washed with water:methanol, 2:1 (100 ml X 2). The wet product was used in the next step.
M.P.: 84-86oC; MS: 303 (M+1);.M.F.: C13H16F2N2O4,:
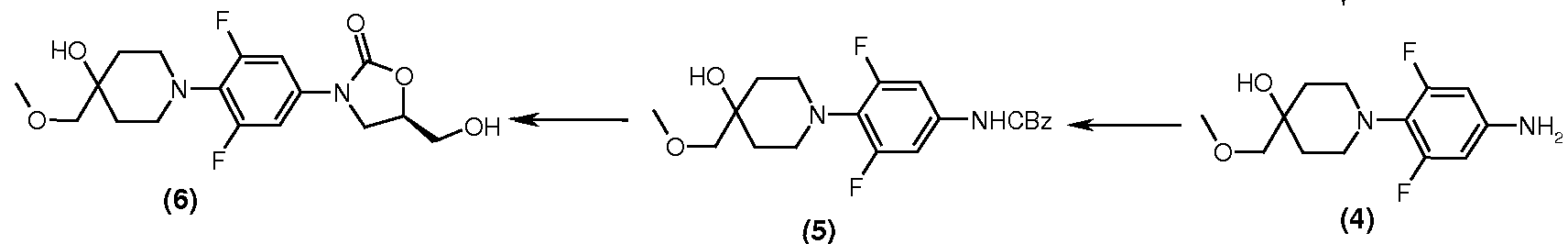
Preparation of Intermediate -5: [3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester
Method A: Preparation of Intermediate 4: ( Stage-I)
Water (1.19 L) and methanol (595 ml) were charged in a 3 L glass reaction assembly, followed by 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (85 g, 0.281 mol) and the contents were stirred. Sodium dithionite (288 g, 1.407 mol) was added in one lot and the reaction mixture was heated to 80oC for 8 h. After completion of the reaction (TLC), methanol was recovered under vacuum below 65oC. After the recovery, the aqueous residue was extracted with chloroform (400 ml X 3). The combined chloroform extract (containing the intermediate 1-(4-amino-2,6-difluoro-phenyl)-4-methoxymethyl-piperidin-4-ol) was dried over anhydrous Sodium sulfate and used in the next step (carbamate formation).
Preparation of Intermediate -5: (Stage-II):
The above chloroform extract was charged in a 3 L glass reaction assembly. Sodium bicarbonate (70 g, 0.843 mol) was added to the extract and the contents were cooled to 15oC-20oC. Benzylchloroformate solution (50% in toluene, 48 g, 96 ml, 0.281 mol) was added slowly to the above mixture under stirring. After completion of the addition, the reaction mixture was stirred at RT for 2 h. After completion of the reaction (TLC), the contents were filtered on a Buchner assembly and the solid cake was washed with chloroform (85 ml X 2). The combined filtrate was evaporated under vacuum below 50oC to obtain yellowish oily mass, which was poured slowly in hexanes (850 ml) under stirring to obtain a precipitate. The precipitated product was filtered and washed with hexanes (100 ml X 2). The product was dried in a vacuum oven below 65oC to obtain 60.2 g brownish product (Yield = 38% on the basis of step-I input).
M.P.: 138-140oC; MS: 407(M+1); M.F.: C21H24F2N2O4.:.
Method B: : Preparation of Intermediate 4: ( Stage-I): To a solution of 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (973g, 3.22 mol) in ethyl acetae (10L) was added 10% Pd-C, (250g, 50% wet) and the resulting miture was hydrogenated in a pressure at 30 PSI, 45-55oC, for 3h. The catakyst was filtered and the residue was washed with additional ethyl acetate( 200ml). The combined filtrates were used as such for the next reaction (carbamate formation)
Preparation of Intermediate -5: (Stage-II):
To the above filtrate was added sodium bicarbonate(406g, 4.83 mol) and the mixture warmed to 40-45oC. To this mixture was added a 50% solution of Benzyl chloroformate in toluene(1.373L, 4.025 mol), drop-wise, over a period of 1h. Stir the resulting mixture for 1h and filter the insoluble material. The residue was washed with 300ml of ethyl acetate. The filtrates were combined and the solvent evaporated under reduced pressure, below 55oC.. Cool the residue and dilute it with hexane(10L). The resulting slurry was stirred well and the separated solid was filtered under suction. The residue was washed with additional hexane ( 2L). The solid was dried for 10h at RT, to obtain the product as dark brown solid, 1200g, yield, 96%.
M.P.: 138-140oC; MS: 407( M+1); M.F.: C21H24F2N2O.
Preparation of Intermediate -6:
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one
To a mixture of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester (100g, 0.237 mol) in dry tetrahydrofuran (THF) (2 L) at 40ºC was added drop-wise n-BuLi in hexane (1.6M, 45.5 g, 455 ml, 0.711 mol) under nitrogen atmosphere. The contents were stirred for 1 h at 40ºC and R-(-)-glycidyl butyrate (68.25 g, 0.474 mol) was added gradually. After the addition of R-(-)-glycidyl butyrate, the reaction mixture was stirred for 5-6 h at 40oC till completion of the reaction (TLC). After completion of the reaction, a solution of sodium methoxide (2 g) in methanol (66 ml) was added to the contents followed by water (8 ml) and the contents were stirred for an additional 0.5 h. Water (1 L) was added to the solution and the contents were extracted with ethyl acetate (1 L). The aqueous layer was further extracted with ethyl acetate (3 X 500 ml). The combined organic layer was evaporated under vacuum to obtain a thick residue. tert-Butyl methyl ether (1 L) was added to the residue and the contents were stirred for about 1 h to obtain a solid product, which was filtered and washed with tert-butyl methyl ether (2 X 100 ml). The product was dried under vacuum below 60ºC to obtain the product as a 46.5 g dark brown compound, 46.5g ,yield 51%.
M.P.: 117-119oC; MS: 373(M+1); M.F.: C17H22F2N2O5..
Preparation of Intermediate -7: (5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester
To a mixture of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one (45 g, 0.121 mol) in dichloromethane (0.3 L), was added triethylamine (24.5 g, 34 ml, 0.242 mol) while stirring. Methanesulfonyl chloride (18 g, 12.2 ml, 0.157 mol) was added to the above solution over a period of 1 h at 10oC -20oC and the reaction mixture was stirred for additional 2 h at RT. After completion of the reaction (TLC), the contents were evaporated under vacuum at 40oC to obtain an oily residue. Water (450 ml) was added to the residue and the traces of dichloromethane were removed under vacuum. The solid product thus obtained was filtered, washed with water (2 X 50 ml) and dried under vacuum at 70oC to obtain 50.6 g brownish compound. Yield = 93%; M.P.:106-108oC; MS: 451(M+1); M.F.: C18H24F2N2O7S.
Preparation of Intermediate 8a: (5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one
Method A:
To a solution of (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl)oxazolidin-2-one (2g, 5.3 mmol),in tetrahydrofuran (20 mL), under argon , was added diphenylphosphoryl azide (1.63mL, 5.9 mmol). The solution was cooled to 0oC in an ice-bath. 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) (0.76mL, 4.9mmol) was added drop-wise over 15min..The reaction was stirred at same temperature for 1 hr, and then warmed to room temperature and stirred under for 16 hr. The reaction mixture was diluted with ethyl acetate (20 mL), and water (20mL). After separation of water layer, the organic layer was washed with water and 0.5M citric acid monohydrate (10 mL). The organic layer was dried over sodium sulfate and the solvent evaporated under reduced pressure.The residue was triturated with ether to obtain the product as a buff colored solid, 1.32g (62%).
M.P.: 106-108oC; M.S.- 398(M+1); M.F.- C17H21F2N5O4,
Method B:
To a solution of (5R)-methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester (20 g, 0.044 mol, wet) in N,N-dimethylformamide (30 ml), was added sodium azide (8.6 g, 0.133 mol) in a single lot. The reaction mixture was gradually heated and the temperature was maintained at 70ºC for 8 h. After completion of the reaction (TLC), the contents were cooled to 20-25ºC and poured slowly into chilled water (300 ml). The solid product thus obtained was filtered and washed with water (2 x 50 ml). The wet product was air dried to obtain 16.5g dark brown compound (being an azide, it was NOT exposed to heat during drying) Yield ~ 93%.
M.P.: 106-108oC; MS : 398(M+1); M.F.: C17H21F2N5O4;:
Preparation of Intermediate 8b: (5S)-N-2-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-phthalimide
Method A:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate(10g, 0.022 mol), Potassium phthalimide (12.2g, 0.066 mol) and DMF (50ml) was heated, with stirring, at 90oC for 4h. The resulting mixture was cooled to RT and poured over ice-water mixture. The separated solid was filtered, washed with water and dried under suction to obtain the product as a white solid, 9.46g, in 85% yield.
M.P.: 154-156 oC; MS: 502 (M+1); M.F. C25H25F2N3O6.
Method B:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 minute phthalimide (1.18g, 8 mmol)) was added and after a further stirring for 10 minute, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 8 hrs ice-cold water (4 ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 1.56g, yield 58%.
M.P.: 154-156 oC; MS : 502 (M+1); M.F. C25H25F2N3O6.
Preparation of Intermediate 10: (5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40oC to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HCl solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70oC to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50oC.. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume.
To the above ethyl acetate solution was added Triethyl amine (19.1g, 0.189 mol), and acetic anhydride (16.1g, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5oC, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate(100ml) and dried at 70oC under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.011mol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(1.11g, 0.011mol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate (1gm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 ºC. for 15hrs. Then a mixture of conc. HCl (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75ºC for 5hrs. The mixture was concentrated under reduced pressure at 60-75 ºC. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 ºC for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50ºC to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Preparation of Intermediate -11: (S)-N-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryloxy-piperi din-1yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
To a solution of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)-phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.2 mmol) and tetrazole (0.6 mmol) in dichloromethane (5 ml) was added dibenzyl N,N,diisopropylphosphoramidite (0.4 mmol) and the resulting mixture was stirred for 4h. The resulting solution was cooled to 0 oC and 0.6 ml of 0.5M m-chloroperbenzoic acid solution in dichloromethane was added. After 4h, the solvent was evaporated under residue pressure and the residue chromatographed on a column of silica gel to obtain the product as a off-white solid in 75% yield,
MS: 674 (M+1); M.F. C33H38F2N3O8P;
Example A: Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxymethyl-piperidin-4-yl) ester
To a suspension of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryl- oxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.15 mmol) and 20 % palladium hydroxide (20 mg) in 20 ml of a mixture of dichloromethane /aqueous methanol was stirred at room temperature for 6h. The catalyst was filtered and the residue evaporated under reduced pressure. The residue obtained was triturated with acetone to obtain a white solid as product in 70% yield.
MP. >140 °C; MS : 494(M+1) M.F.: C19H26F2N3O8P.
PATENT
http://www.google.co.in/patents/WO2012059823A1?cl=en


c) Converting intermediate of Formula (5) into intermediate of structure (6)



f) Converting intermediate of Formula (11) into compound of Formula (A) or Pharmaceutically acceptable salts thereof



ormu a-
Scheme-1
Preparation of Intermediate 10: (5S)- N-{ 3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl- piperidin- 1 -yl)-phenyl] -2-oxo-oxazolidin-5-ylmethyl } -acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l- yl)-phenyl] -oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40°C to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HC1 solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4- hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70°C to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-5- azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50°C. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume. To the above ethyl acetate solution was added Triethyl amine (19. lg, 0.189 mol), and acetic anhydride (16. lg, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5°C, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate( 100ml) and dried at 70°C under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine- lyl)phenyl]-2-oxo-oxazolidin-5-yl methyl }-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.01 lmol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(l.l lg, 0.01 lmol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5. Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)phenyl]- 2-oxo-oxazolidin-5-yl methyl }-methanesulfonate (lgm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 °C. for 15hrs. Then a mixture of cone. HC1 (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75°C for 5hrs. The mixture was concentrated under reduced pressure at 60-75 °C. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 °C for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50°C to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.1 lg, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3- (3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-l-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5.
PATENT
http://www.google.co.in/patents/WO2008038092A2?cl=en

IV

V
‘ Scheme-1 ‘
/////////
SEE FULL ZOLID SERIES…………http://drugsynthesisint.blogspot.in/p/zolid.html
Flow Chemistry India 2016, 21-22 January 2016, Mumbai, India

Flow Chemistry India 2016Date: Thursday, 21 January 2016 – Friday, 22 January 2016
|
SELECTBIO INDIA
http://selectbiosciences.com/conferences/index.aspx?conf=FCINDIA16&se=india
Register…………..http://selectbiosciences.com/conferences/registration.aspx?conf=FCINDIA16&se=india

venue
Hotel Ramada Powai and Convention Centre, Mumbai, India


Professor & Research Chair, Nelson Mandela Metropolitan University |

Professor, Synthetic Organic Chemistry , The University of Tokyo |

Deputy Director and Chair, National Chemical Laboratory |

Professor, Eindhoven University of Technology |

Scientific Director, University of Lyon |

Chairman, Flow Chemistry Society
Professor, University of Warsaw |






Maninderjit Singh Ahluwalia
Overview
SELECTBIO INDIA is delighted to welcome you all at the 4th International Conference Flow Chemistry India 2016 to be held in Mumbai on January 21-22, 2016 under the auspices of the Flow Chemistry Society. The society aims to unite and represent those who are actively working on this rapidly developing field. This meeting is dedicated to the integration of flow chemistry into everyday practice throughout the world by delivering the latest knowledge and making it available for the entire chemistry community.
Society members save 25% on the registration fee and non-members will receive their first year’s membership included in the fee.
Running alongside the conference will be an exhibition covering the latest technological advances in the area of flow chemistry.
Who Should Attend
• Scientists, Chemists, Chemical Engineers and Researchers working in Pharmaceutical and Fine Chemicals Research and Development including Drug Discovery, Medicinal Chemistry and Chemical Process Development
• Scientists, Chemists and Chemical Engineers working in Pharmaceutical and Fine Chemical Bulk Manufacturing Units
• Corporate Management, Scientists, Managers responsible for development of Pharmaceutical and Fine Chemicals R & D and Manufacturing activities
• Scientists, Chemists & Engineers belonging to the fields of Inorganic, Organic, Medicinal, Natural Products, Analytical, High-throughput and Process Chemistry in the Academic research as well as in Applied research and development in the area of Agrochemical, Petrochemical and Fragrance industry
• Scientists working in or interested in applications of Flow Chemistry in Material science, Green chemistry, Nanotechnology, Biotechnology, Theoretical Chemistry, Information technology and Flow synthesis instruments including Engineering & Automation
Conference Package – Includes Registration, 2 Nights Accommodation, Dinner & Airport Transfers (Valid up to January 5, 2016 only)
Call for Posters
You can also present your research on a poster while attending the meeting. Submit an abstract for consideration now!
Poster Submission Deadline: 30 November 2015
Agenda Topics
- Advances in Micro & Continuous Flow Reactors, Systems & Processes
- Applications in Pharmaceutical Industry & API Synthesis
- Engineering Aspects of Flow Chemistry
- Flow Reactor – Choosing the Right One
- Photochemistry & Multistep Synthesis in Flow
- Quality Issue and QbD in Flow Chemistry
- Scale up – From Micro to Commercial Scale
- Yield Improvement, Cost Cutting and Waste Reduction in Flow Chemistry
Sponsorship and Exhibition Opportunities
http://selectbiosciences.com/conferences/index.aspx?conf=FCINDIA16&se=india

Workshop Tutor
Charlotte Wiles
CEO CHEMTRIX
A Workshop on “Flow Chemistry Demonstrations (Lab & Plant Scale) for Chemical and Pharmaceutical Industry-” will be held one day prior to the training course i.e. on 20th January, 2016 from 10:00 am – 05:00 pm in Mumbai. This workshop is supported by Process Intensification will be jointly conducted by :
Dr. Dinesh Kudav (Mumbai University); Dr. Charlotte Wiles (Chemtrix BV-Neth); Mr. Wouter Stam (Flowid, NV-Neth); Mr. Manjinder Singh (CIPLA & VP-FCS-India Chapter); Dr. Viktor Gyollai, (AM Technology-UK); Dr. Prashant Kini (UPL Ltd.); Mr. Kumar Oza (Pi & TCPL); Mr. Madhav Sapre (Pi & Sharon Bio); et al .
This workshop is specially designed to demonstrate application/capabilities of Flow Chemistry running “live” reactions in Continuous Flow Reactors. The reactions likely to be demonstrated using Flow Chemistry includes :• Nitration • Organometallic reaction• Oxidation • Bi-phasic reaction• Nano-Particle preparation in Flow• Biocatalytic Reaction with enhanced enzyme life.
This workshop is free for the registered delegates of Flow Chemistry India 2016 Conference and Continuous Flow Reactors Training Course.
You can visit Mumbai city
Taj hotel, mumbai
Gateway of india
 Food in mumbai
Food in mumbai
 mumbai skyline
mumbai skyline

The Bandra-Worli Sea Link is a cable-stayed bridge that connects central Mumbai with its western suburbs

get in if you can
The Mumbai Suburban Railway system carries more than 6.99 million commuters on a daily basis. It has the highest passenger densities of any urban railway …

Chhatrapati shivaji in mumbai india
British-victoria terminus


VADA PAV


SELECTBIO CONFERENCES PICS










/////////
WCK Series by Wockhardt for treating the bacterial infection

Which WCK is it, WCK-4873 , WCK-4086, WCK ? for treating the bacterial infection.
(Not sure) will arrive at correct one….keep watching………..

CAS 1627163-98-7
- C14 H16 N2 O4 . Na,

- 1,6-Diazabicyclo[3.2.1]octane-2-carboxylic acid, 7-oxo-6-(phenylmethoxy)-, sodium salt (1:1), (1R,2S,5R)-
- SODIUM (2S, 5R)-6-(BENZYLOXY)-7-OXO-1,6-DIAZABICYCLO[3.2.1]OCTANE-2-CARBOXYLATE
sodium (2S,5R)-6-(benzyloxy)-7-oxo-1,6-diazabicyclo[3.2.1]octane-2-carboxylate…..WO2014135929

Patent
http://www.google.com/patents/WO2015136473A1?cl=en

EXAMPLES
The following examples illustrate the embodiments of the invention that are presently best known. However, it is to be understood that the following are only exemplary or illustrative of the application of the principles of the present invention. Numerous modifications and alternative compositions, methods, and systems may be devised by those skilled in the art without departing from the spirit and scope of the present invention. The appended claims are intended to cover such modifications and arrangements. Thus, while the present invention has been described above with particularity, the following examples provide further detail in connection with what are presently deemed to be the most practical and preferred embodiments of the invention.
Example 1
Synthesis of sodium (25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicvclor3.2.11octane-2- carboxylate
Step 1; Preparation of -Γl-Γ(feΓt-butyldimethylsilyl -oxymethyll-5-Γdimethyl(oxido -λ-4-sulfanylidenel-4-oxo-pentyll-carbamic acid tert-butyl ester (III):
To a suspension of trimethylsulfoxonium iodide (180.36 gm, 0.819 mol) in tetrahydrofuran (900 ml), sodium hydride (32.89 g, 0.819 mol, 60% in mineral oil) was charged in one portion at 30°C temperature. The reaction mixture was stirred for 15 minutes and then dropwise addition of dimethylsulphoxide (1.125 ml) was done over a period of 3 hours at room temperature to provide a white suspension. The white suspension was added to a pre-cooled a solution of 2-(feri-butyldimethylsilyl-oxymethyl)-5-oxo-pyrrolidine-l-carboxylic acid tert-buty\ ester (II) (225 g, 0.683 mol, prepared as per J. Org Chem.; 2011, 76, 5574 and WO2009067600) in tetrahydrofuran (675 ml) and triethylamine (123.48 ml, 0.887 mol) mixture at -13°C by maintaining the reaction mixture temperature below -10°C. The resulting suspension was stirred for additional 1 hour at -10°C. The reaction mixture was carefully quenched by addition of saturated aqueous ammonium chloride (1.0 L) at -10°C to 10°C. The reaction was extracted by adding ethyl acetate (1.5 L). The layers were separated and aqueous layer was re-extracted with ethyl acetate (500 ml x 3). The combined organic layer was washed successively with saturated aqueous sodium bicarbonate (1.0 L), water (2.0 L) followed by saturated aqueous sodium chloride solution (1.0 L). Organic layer was dried over sodium sulfate and evaporated under vacuum to provide 265 g of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-buty\ ester (III) as an yellow oily mass.
Analysis:
Mass: 422.3 (M+l); for Molecular weight: 421.68 and Molecular Formula: ![]()
1H NMR (CDC13): δ 4.77 (br d, 1H), 4.38 (br s, 1H), 3.58 (br s, 3H), 3.39 (s, 3H), 3.38 (s, 3H), 2.17-2.27 (m, 2H), 1.73-1.82 (m, 2H), 1.43 (s, 9H), 0.88 (s, 9H), 0.01 (s, 3H), 0.04 (s, 3H).
Step 2: Preparation of 5-r4-benzyloxyimino-l-(fert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyll-carbamic acid tert- butyl ester (IV):
To a suspension of 5-[l-[(ieri-butyldimethylsilyl)-oxymethyl]-5-[dimethyl(oxido)- -4-sulfanylidene]-4-oxo-pentyl]-carbamic acid tert-butyl ester (III) (440.0 g, 1.045 mol) in tetrahydrofuran (6.6 L), O-benzhydroxylamine hydrochloride (200.0 g, 1.254 mol) was charged. The reaction mixture was heated to 50°C for 2.5 hours. The reaction mixture was filtered through pad of celite and filtrate was concentrated to provide a residue. The residue was dissolved in ethyl acetate (5.0 L) and washed successively with saturated aqueous sodium bicarbonate (1.5 L), water (1.5 L) and saturated aqueous sodium chloride (1.5 L). Organic layer was dried over sodium sulfate. Solvent was evaporated under vacuum to yield 463.0 g of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) as an oily mass.
Analysis:
Mass: 486.1 (M+l); for Molecular weight: 485.4 and Molecular Formula: ![]()
1H NMR (CDCI3): δ 7.26-1 6 (m, 5H), 5.10 (s, 2H), 4.66 (br d, 1H), 3.58-4.27 (m, 2H), 3.56-3.58 (m, 3H), 2.40-2.57 (m, 2H), 1.68-1.89 (m, 2H), 1.44 (s, 9H), 0.89 (s, 9H), 0.02 (s, 3H), 0.04 (s, 3H).
Step 3: Preparation of 5-5-benzyloxyimino-2-(fert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V):
To a solution of 5-[4-benzyloxyimino-l-(tert-butyldimethylsilyl-oxymethyl)-5-chloro-pentyl]-carbamic acid tert-butyl ester (IV) (463.0 g 0.954 mol) in tetrahydrofuran (6.9 L), was charged potassium feri-butoxide (139.2 g, 1.241 mol) in portions over a period of 30 minutes by maintaining temperature -10°C. The resulting suspension was stirred for additional 1.5 hours at -10°C to -5°C. The reaction mixture was quenched by addition of saturated aqueous ammonium chloride (2.0 L) at -5°C to 10°C. The organic layer was separated and aqueous layer was extracted with ethyl acetate (1.0 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride solution (2.0 L). Organic layer was dried over sodium sulfate, and then evaporated under vacuum to yield 394.0 g of 5-5-benzyloxyimino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-butyl ester (V) as an yellow oily mass.
Analysis:
Mass: 449.4 (M+l) for Molecular weight: 448.68 and Molecular Formula: C24H4oN204Si;
1H NMR (CDC13): δ 7.25-1 3 (m, 5H), 5.04-5.14 (m, 2H), 4.35 (br s, 1H), 3.95 (br s, 1H), 3.63-3.74 (br d, 2H), 3.60-3.63 (m, 1H), 2.70-2.77 (m, 1H), 2.33-2.41 (m, 1H), 1.79-1.95 (m, 2H), 1.44 (s, 9H), 0.88 (s, 9H), 0.03 (s, 3H), 0.04 (s, 3H).
Step 4: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI):
To a solution of 5-5-benzyloxyimino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (V) (394.0 g, 0.879 mol) in dichloromethane (5.0 L) and glacial acetic acid (788 ml), was charged sodium cyanoborohydride (70.88 g, 1.14 mol) one portion. The resulting reaction mixture was stirred at temperature of about 25 °C to 30°C for 2 hours. The mixture was quenched with adding aqueous solution of sodium bicarbonate (1.3 kg) in water (5.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (2.0 L). The combined organic layer washed successively with water (2.0 L), saturated aqueous
sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to provide a residue. The residue was purified by silica gel column chromatography to yield 208 g of (25,5i?5)-5-benzyloxyamino-2-(ieri-butyldimethylsilyl-oxymethyl)-piperidine- 1 -carboxylic acid tert-buty\ ester (VI) as pale yellow liquid.
Analysis:
Mass: 451.4 (M+l); for Molecular weight: 450.70 and Molecular Formula: C24H42N204Si;
1H NMR (CDC13): δ 7..26-7.36 (m, 5H), 4.90-5.50 (br s, 1H), 4.70 (dd, 2H), 4.09-4.25 (m, 2H), 3.56-3.72 (m, 2H), 2.55-3.14 (m, 2H), 1.21-1.94 (m, 4H), 1.45 (s, 9H), 0.89 (s, 9H), 0.05 (s, 6H).
Step 5: Preparation of (25,5R5)-5-benzyloxyamino-2-(tert-butyldimethylsilyl-oxymethyl)-piperidine (VII):
To a solution of 5-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine-l-carboxylic acid tert-butyl ester (VI) (208 g, 0.462 mol) in dichloromethane (3.0 L), boron trifluoride diethyletherate complex (114.15 ml, 0.924 mol) was charged in one portion. The resulting reaction mixture was stirred at temperature of about 25°C to 35°C temperature for 2 hours. The reaction mixture was quenched with saturated aqueous sodium bicarbonate (2.0 L). The organic layer was separated and aqueous layer was extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed with saturated aqueous sodium chloride (1.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum to yield 159 g of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) as a yellowish syrup.
Analysis:
Mass: 351.3 (M+l); for Molecular weight: 350.58 and Molecular Formula: C19H34N202Si.
Step-6: Preparation of (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-r3.2.11octane (VIII):
Part 1; Preparation of (2S,5RS)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
To a solution of (25,5i?5)-5-benzyloxyamino-2-(feri-butyldimethylsilyl-oxymethyl)-piperidine (VII) (159.0 g, 0.454 mol) in a mixture of acetonitrile (2.38 L) and diisopropylethylamine (316.5 ml, 1.81 mol) was added triphosgene (59.27 gm, 0.199 mol) dissolved in acetonitrile (760 ml) at -15°C over 30 minutes under stirring. The resulting reaction mixture was stirred for additional 1 hour at -10°C. The reaction mixture was quenched by addition of saturated aqueous sodium bicarbonate (2.0 L) at -5°C to 10°C. Acetonitrile was evaporated from the reaction mixture under vacuum and to the left over aqueous phase, dichloromethane (2.5 L) was added. The organic layer was separated and aqueous layer extracted with dichloromethane (1.5 L x 2). The combined organic layer was washed successively with water (2.0 L), saturated aqueous sodium chloride (2.0 L) and dried over sodium sulfate. Solvent was evaporated under vacuum and the residue was passed through a silica gel bed to yield 83.0 g of diastereomeric mixture (25, 5i?5)-6-benzyloxy-2-(feri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio as a yellow liquid.
Part-2: Separation of diastereomers to prepare (25,5R)-6-benzyloxy-2-(fert-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane:
A mixture of diastereomers (2S,5Z?S)-6-benzyloxy-2-(teri-butyl-dimethylsilyl-oxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane in 50:50 ratio (47.0 gm, 0.125 mol), was dissolved in n-hexane (141 ml) and stirred at temperature of about 10°C to 15°C for 1 hour. Precipitated solid was filtered and washed with n-hexane (47 ml) to provide 12.0 g of diastereomerically pure (25,5i?)-6-benzyloxy-2-(tert-butyl-dimethylsilyl-oxymethyl)-7-oxo- 1,6-diaza-bicyclo-[3.2.1] octane (VIII) as a white crystalline material.
Analysis:
Mass: 377.3 (M+l); for Molecular weight: 376.58 and Molecular Formula: ![]()
1H NMR (CDCI3): δ Ί -Ί.ΑΑ (m, 5H), 4.95 (dd, 2H), 3.76-3.85 (ddd, 2H), 3.37-3.40 (m, 1H), 3.28-3.31 (m, 2H), 2.89 (brd, 1H), 1.90-2.02 (m, 2H), 1.62- 1.74 (m, 2H), 1.56 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
Diastereomeric purity as determined by HPLC: 99.85%
Step-7: Preparation of (25,5R)-6-benzyloxy-2-hvdroxymethyl)-7-oxo-l,6-diaza-bicvclo-r3.2.11octane (IX):
To a solution of (25,5i?)-6-benzyloxy-2-(ieri-butyl-dimethylsilyl-oxymethyl)-7-oxo- l,6-diaza-bicyclo-[3.2.1]octane (VIII) ( 12.0 g, 31.9 rnmol) in tetrahydrofuran (180 ml) was charged tetra 7? -butyl ammonium fluoride (38.0 ml, 38 mmol, 1 M in tetrahydrofuran) at room temperature. The reaction mixture was stirred for 2 hours. It was quenched with saturated aqueous ammonium chloride ( 100 ml). The organic layer was separated and aqueous layer extracted with dichloromethane (150 ml x 3). The combined organic layer was washed with saturated aqueous sodium chloride (150 ml), dried over sodium sulfate and evaporated under vacuum to yield 24.0 g of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l ,6-diaza-bicyclo-[3.2.1]octane (IX) as a yellow liquid. The compound of Formula (IX) was purified by silica gel (60-120 mesh) column chromatography using a mixture of ethyl acetate and hexane as an eluent.
Analysis:
Mass: 263.1 (M+l); for Molecular weight: 262.31 and Molecular Formula: C14H18N203
1H NMR (CDCb): δ 7.34-7.42 (m, 5H), 4.95 (dd, 2H), 3.67-3.73 (m, 1H), 3.53-3.60 (m, 2H), 3.32-3.34 (m, 1H), 2.88-3.01 (m, 2H), 2.09 (brs, 1H), 1.57-2.03 (m, 2H), 1.53- 1.57 (m, 1H), 1.37- 1.40 (m, 1H).
Step 8: Preparation of sodium salt of (25, 5R)-6-benzyloxy-7-oxo-l,6-diaza-bicvclor3.2.11-octane-2-carboxylic acid (I):
Step I:
Compound of Formula (IX) obtained in step 8 above was used without any further purification. To the clear solution of (25,5i?)-6-benzyloxy-2-hydroxymethyl)-7-oxo-l,6-diaza-bicyclo-[3.2.1]octane (IX) (24.0 g, 31.8 mmol) (quantities added based upon theoretical basis i.e 8.3 g ) in dichloromethane (160 ml), was added Dess-Martin reagent (24.1 g, 57.24 mmol) in portions over 15 minutes. The resulting suspension was stirred for 2 hours at 25°C. The reaction was quenched by adding a solution, prepared from saturated aqueous sodium hydrogen carbonate solution (160 ml) and 72.0 g of sodium thiosulfate. Diethyl ether (160 ml) was added to the reaction mixture and it was stirred for 5-10 minutes and filtered through celite. Biphasic layer from filtrate was separated. Organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (160 ml) followed by saturated aqueous sodium chloride solution (160 ml). Organic layer was dried over sodium sulfate and evaporated to dryness at 30°C to obtain 20.0 g of intermediate aldehyde, which was used immediately for the next reaction.
Step II:
To the crude intermediate aldehyde (20.0 g, 31.6 mmol) (quantities added based upon theoretical yield i.e. 8.2 g) obtained as above, was charged i-butyl alcohol (160 ml) and cyclohexene (10.8 ml, 110.6 mmol). The reaction mixture was cooled to temperature of about 10°C to 15°C. To this mixture was added clear solution prepared from sodium hypophosphate (14.8 g, 94.8 mmol) and sodium chlorite (5.7 g, 63.2 mmol) in water (82.0 ml) over a period of 30 minutes by maintaining temperature between 10°C to 15°C. The reaction mixture was further stirred for 1 hour and was quenched with saturated aqueous ammonium chloride solution. The reaction mixture was subjected to evaporation under vacuum at 40°C to remove i-butyl alcohol. Resulting mixture was extracted with dichloromethane (3 x 150 ml). Layers were separated. Combined organic layer was washed with aqueous brine solution, dried over sodium sulfate and evaporated to dryness under vacuum to obtain 16.0 g of crude residue. To this residue was added acetone (83 ml) to provide a clear solution and to it was added dropwise a solution of sodium 2-ethyl hexanoate (4.5 g) in acetone (24 ml). The reaction mixture was stirred for 15 hours at 25°C to 30°C to provide a suspension. To the suspension was added diethyl ether (215 ml) and stirred for 30 minutes. Resulting solid was filtered over suction, and wet cake was washed with cold acetone (83 ml) followed by diethyl ether (83 ml). The solid was dried under vacuum at 40°C to provide 3.6 g of off-white colored, non-hygroscopic sodium salt of (25, 5i?)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]-octane-2-carboxylic acid (I).
Analysis:
Mass: 275.2 as M-1 (for free acid) for Molecular Weight: 298 and Molecular Formula: ![]()
NMR (DMSO-d6): δ 7.43-7.32 (m, 5H), 4.88 (q, 2H), 3.48 (s, IH), 3.21 (d, IH), 2.73 (d, IH), 2.04-2.09 (m, IH), 1.77-1.74 (m, IH), 1.65-1.72 (m, IH), 1.55-1.59 (m, IH);
Purity as determined by HPLC: 97.47%;
[a]D25: -42.34° (c 0.5, water).


Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.

/////////
New molecules from Wochkardt to treat bacterial infections

WCK ?
( Not sure) Keep watching this post………..
TRANS-SULFURIC ACID MONO-{2-[5-(3-AZETIDINYLAMINO)-METHYL-[1,3,4]- OXADIAZOL-2-YL]-7-OXO-1,6-DIAZABICYCLO[3.2.1] OCT-6-YL} ESTER TRIFLUOROACETATE
trans-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[1,3,4]- oxadiazol-2-yl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl}ester trifluoroacetate
(25,5R)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[ l,3,4]-oxadiazol-2-yl)-7-oxo- l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester
2-(1 ,3,4-OXADIAZOL-2-YL)-7-OXO-1 ,6-DIAZABICYCLO[3.2.1 ]OCTANE DER
(25,5R)-Sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[i,3,41-oxadiazol-2-yl)-7-oxo-l,6-diaza- bicvclo[3.2.11 oct-6-yll ester
PCT International Patent Application No. PCT/US2013/034562.
Indian Patent Application No. 1635/MUM/2014
Molecular Weight: 488.3 and Molecular Formula:![]()

![]()
Scheme 1. Typically, compound of Formula (I) is prepared from sodium salt of 6-benzyloxy-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylic acid (III).
The sodium salt of 6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylic acid
(III) is reacted with 3-(ier^butoxycarbonyl-hydrazinocarbonylmethyl-amino)-azetidine-1-carbamic acid tert-buty\ ester (II) in presence of coupling agent at a temperature ranging from -15°C to 60°C for about 1 hour to about 24 hours to provide an intermediate compound of Formula (IV). Typical, non-limiting examples of coupling agent include EDC hydrochloride, dicyclohexylcarbodiimide, diisopropylcarbodiimide (DIC), (benzotriazol-l-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP), O-(benzotriazol- 1 -yl)-N,N,N’ ,Ν’ -tetramethyluroniumhexafluorophosphate (HBTU), O-(benzotriazol-l-yl)- Ν,Ν,Ν’,Ν’-tetramethyluroniumtetrafluoroborate (TBTU), 0-(7-azabenzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU), O-(6-ahlorobenzotriazol-l-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HCTU), 0-(3,4-dihydro-4-oxo-l,2,3-benzotriazine-3-yl)-N,N,N’,N’-tetramethyl uronium tetrafluoroborate(TDBTU), 3-(diethylphosphoryloxy)- 1 ,2,3-benzotriazin-4(3H)-one (DEPBT), carbonyldiimidazole (CDI), pivalyl chloride, HOBt and the like. In some embodiments, compound of Formula (II) is reacted with a compound of Formula (III) in presence of EDC hydrochloride and HOBt at a temperature of about 25°C to about 35°C for about 15 hours to provide an intermediate compound of Formula (IV). In some embodiments, a compound of Formula (II) is reacted with a compound of Formula (III) in presence of suitable solvent such as dimethylformamide, water or a mixture thereof.
The compound of Formula (IV) is cyclized to provide a compound of Formula (V). The cyclization of a compound of Formula (IV) is effected by treating with a reagent such as p-toluene sulfonyl chloride, p-nitrobenzene sulfonyl chloride, methane sulfonyl chloride or triphenylphosphine in a suitable solvent such as toluene, chloroform, dichloromethane, or N,N-dimethyl formamide at a temperature ranging from about -10° C to about 70°C for about 15 minutes to about 4 hours to provide 1,3,4-oxadiazole intermediate compound of Formula (V). In some embodiments, a compound of Formula
(IV) is cyclized in presence of triphenylphosphine, iodine and triethylamine, at a temperature of about -10°C to about 0°C for about 30 minutes to provide a compound of Formula (V). In some embodiments, compound of Formula (IV) is cyclized to a compound of Formula (V) in presence of dichloromethane as solvent.

Sulfonation

Scheme 1
Example 1
Synthesis of traras-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]- oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl]ester trifluoroacetate (I)
Step 1; Preparation of traras-{3-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane-2-carbonyl)-hydrazinocarbonyl]-2-oxo-ethyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (IV):
A solution of 3-(ier^butoxycarbonyl-hydrazinocarbonylmethyl-amino)-azetidine-1-carbamic acid tert-butyl ester (II) (2.8 g, 0.008 mol) in dimethylformamide (7 ml) was added to a stirred solution of sodium salt of 6-benzyloxy-7-bicyclo [3.2.1] octane-2-carboxylic acid (III) (2.43 g 0.008 mol) in water (41 ml). To this EDC.HCl (2.32 g, 0.012 mol) and HOBt (1.09 g, 0.008 mol) was added and stirred for 15 hours. Dichloro methane (50 ml) was added and layers were separated. Organic layer was dried over sodium sulfate and concentrated. The residue (6.1 gm) was purified by silica gel column chromatography using mixture of acetone and hexane as eluent to afford 3.4 g of ir ns-3-({2-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazino]-2-oxo-ethyl}-teri-butoxy carbonyl-amino)-azetidine-l -carboxylic acid tert-butyl ester (IV) in 70% yield.
Analysis:
Mass: 603.3 (M+l); for Molecular Weight: 602.6; Molecular Formula: ![]()
1H NMR (400 MHz, CDC13): δ 8.45. (bs, IH), 8.20 (bs, IH) 7.38-7.45 (m, 5H), 5.04 (d, IH), 4.91 (d, IH), 4.13 (m, 2H), 3.97-4.04 (m, 5H), 3.30 (s, IH), 3.07 (s, 2H), 2.91 (d, IH), 2.31 (m, IH), 2.20 (d, IH), 1.93-2.00 (m, 2H), 1.45 (s, 18H).
Step 2: Preparation of tr «s-{2-[5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-2-oxo-ethyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (V):
Triethyl amine (3.6 ml, 0.026 mol) was added to a cooled (0 °C) solution of iodine (1.62 gm, 0.0063 mol) and triphenylphosphine (1.67 g, 0.0063 mol) in dichloromethane (64 ml). After stirring for 15 minutes a solution of 3-({2-[N-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazino]-2-oxo-ethyl}-fert-butoxycarbonyl- amino)-azetidine-l-carboxylic acid tert-butyl ester (IV) (3.2 g, 0.0053 mol) in dichloromethane (16 ml) was added. Reaction mixture was stirred at -10°C to 0°C for another 30 minutes. Dichloromethane was concentrated and ethyl acetate (35 ml) was added; stirred and filtered to remove triphenylphosphine oxide. Filtrate was concentrated and purified by silica gel column chromatography using a mixture of methanol and chloroform as eluent to obtain 4.5 g of 3-{ [5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4] oxadiazol-2-yl-methyl]-tert-butoxycarbonyl-amino}-azetidine- 1 -carboxylic acid tert-buty\ ester (V).
Analysis:
Mass: 585.4 (M+l); for Molecular Weight: 584.6 and Molecular Formula: ![]()
1H NMR (400 MHz, CDC13): δ 7.64-7.68 (m, 6H), 7.52-7.56 (m, 3H) 7.42-7.48 (m, 7H), 7.36-7.38 (m, 2H), 5.07 (d, IH), 4.92 (d, 2H), 4.72 (s, IH), 4.68 (s, 2H), 4.15 (s, 2H), 4.01 (s, 2H), 3.36 (s, IH), 2.91 (d, IH), 2.79 (d, IH), 2.27-2.30 (m, 2H), 2.11-2.14 (m, IH), 1.97-1.99 (m, IH), 1.42 (s, 18H).
Step 3: Preparation of tr «s-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-tert-butoxycarbonyl-amino)-azetidine-l-carboxylic acid tert-butyl ester (VI):
Palladium on carbon (10%) was added to a stirred solution of 3-{ [5-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl-methyl]-feri-butoxy carbonyl-amino}-azetidine-l -carboxylic acid tert-butyl ester (V) (4.5 g) in methanol (45 ml). Resulting suspension was stirred under hydrogen gas pressure of about 50 psi for 15 hours at 25°C. The reaction mixture was filtered through celite bed and washed using additional methanol (5 ml). The filtrate was concentrated to obtain 3.5 g of ir ns-{2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-teri-butoxy carbonyl-amino)-azetidine-l -carboxylic acid tert-butyl ester (VI) in 92% yield.
Analysis:
Mass: 495.4 (M+l); for Molecualr Weight: 494.5 and Molecular Formula: ![]()
1H NMR (400 MHz, DMSO): δ 9.86 (s, 1H), 7.51-7.62 (m, 12H), 4.70 (s, 2H), 4.58 (d, 1H), 3.99 (d, 2H), 3.65 (s, 2H), 2.92 (d, 1H), 2.67 (d, 1H), 2.31 (s, 1H), 2.00-2.11 (m, 2H), 1.84 (m, 1H), 1.31 (s, 18H).
Step-4: Preparation of traras-tetrabutyl ammonium salt-methyl-{2-[5-(7-oxo-6-sulphooxy-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-tert-butoxycarbonyl-amino )-azetidine-l-carboxylic acid fert-butyl ester (VII):
Sulfur trioxide-pyridine complex (3.17 g, 0.019 mol) and triethyl amine (4.5 ml, 0.033 mol) was added to a stirred solution of ir ns- {2-[5-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]-oxadiazole-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-butyl ester (VI) (2.62 g, 0.0066 mol) in dichloromethane (20 ml). The reaction mixture was stirred for 2 hours. Aqueous solution of 0.5 N potassium dihydrogen phosphate (50 ml) followed by ethyl acetate (40 ml) was added, stirred for 10 minutes and aqueous layer was separated. Aqueous layer was again extracted with the mixture of dichloromethane (10 ml) and ethyl acetate (20 ml). Combined organic layers were concentrated. The residue was dissolved in water (50 ml), washed with diethyl ether (2 x 25 ml) to remove triphenylphosphine oxide (a side product carried from the step-2) and extracted with dichloromethane (2 x25 ml). Dichloromethane was dried over sodium sulfate and concentrated to give 2.7 g of residue (87%). This residue was again dissolved in dichloromethane (50 ml) followed by addition of triethylamine (5.70 ml, 0.042 mol). Tetrabutylammonium hydrogen sulphate (1.27 g, 0.0037 mol) was added and stirred for 2 hours. Water (30 ml) was added to the reaction mixture and layers were separated. Dichloromethane layer was dried on sodium sulfate and solvent was concentrated under vacuum. The residue (2.7 g) was purified by silica gel column chromatography using methanol and chloroform as eluent to get 2.1 g of irans-tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy- 1 ,6-diaza-
bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-buty\ ester (VII) in 48% yield.
Analysis:
Mass: 575.4 (M+l) as free sulfonic acid; for Molecular Weight: 816.6 and Molecular Formula: C22H34N6O10S. Ci6H36N;
1H NMR (400 MHz, CDC13): δ 4.63-4.69 (m, 5H), 4.40 (s, 2H), 4.16 (s, 2H), 4.02 (s, 2H), 3.28-3.32 (m, 12H), 3.23 (s, 1H), 2.84 (d, 1H), 2.24-2.32 (m, 2H), 2.02-2.04 (m, 1H), 1.63-1.71 (m, 12H), 1.46-1.56 (m, 12H), 1.44 (s, 18H), 0.99-1.02 (m, 18H).
Step 5: Preparation of traras-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo[3.2.1]oct-6-yl]ester trifluoroacetate (I)
irans-Tetrabutyl ammonium salt-methyl- {2-[5-(7-oxo-6-sulphooxy- 1 ,6-diaza-bicyclo[3.2.1]oct-2-yl)-[l,3,4]oxadiazol-2-yl]-methyl}-ieri-butoxycarbonyl-amino)-azetidine- 1 -carboxylic acid tert-butyl ester (VII) (2.1 g, 0.003 mol) was cooled to 0°C and to this was added trifluoro acetic acid cooled at 0°C in 15 minutes and the reaction mixture was stirred for 3 hours. The obtained reaction mixture was concentrated under high vacuum. Diethyl ether (20 ml) was added and solid precipitated was stirred and diethyl ether was decanted. This treatment was repeated twice. Solid separated was dried and dichloromethane (20 ml) was added and stirred; solid was allowed to settle and dichloromethane was decanted. Again this treatment was repeated twice and the solid was dried to get 1 g of irans-sulfuric acid mono-{2-[5-(3-azetidinylamino)-methyl-[l,3,4]-oxadiazol-2-yl]-7-oxo-l,6-diazabicyclo [3.2.1]oct-6-yl]ester trifluoroacetate (I) in 76% yield.
Analysis:
Mass: 375.2 (M+l) as free sulfonic acid; for Molecular Weight: 488.3 and Molecular Formula:![]()
CF3COOH;
1H NMR (400 MHz, DMSO): δ 4.64 (d, IH), 4.06 (s, 3H), 3.92 (s, 2H), 3.81-3.86 (m, IH), 3.73 (s, 2H), 2.94-2.97 (d, IH), 2.70 (d, IH), 2.16 -2.19 (m, IH), 1.88-2.14 (m, 2H), 1.86-1.88 (m, IH);
19F NMR (DMSO-d6): δ -74.41 (CF3COOH);
1 C NMR (DMSO-de as a TFA salt): δ 165.4, 165.1, 164.9, 159.2-158.2 (TFA-C), 57.7, 52.6 (2C), 52.3, 49.3, 46.1, 40.4, 20.1, 19.7.
PATENT
WO2015110963


Example-1
(25,5R)-Sulfuric acid mono-r2-(5-azetidin-3-ylmethyl-ri,3,41-oxadiazol-2-yl)-7-oxo-l,6-diaza- bicvclor3.2.11 oct-6-yll ester:

Step-1: Preparation of (25,5R)-2-{N’-[2-(5)-iV-tert-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazino carbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of sodium (2S, 5R)-7-oxo-6-benzyloxy-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (8.45 g, 28.3 mmol) (prepared according to the process disclosed in PCT/IB2013/059264) in water (100 ml) was added 3-(N-feri-butoxycarbonyl-azetidin-3-yl)-acetic acid hydrazide (5.9 g, 25.7 mmol), EDC hydrochloride (7.47 g, 38.6 mmol) and N-hydroxybenzotriazole (3.47 g, 25.7 mmol) at 25°C to 35°C under stirring. The reaction mixture was stirred for 18 hours. Precipitated solid was filtered under suction and washed with water (100 ml). It was dried to provide 10.01 g of (25,5R)-2-{N’-[2-(S)-N-fert-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazinocarbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo [3.2.1] octane in 80% yield.
Analysis:
Mass: 486.4 (M-l), for Molecular Formula of C24H33N5O6;
Purity as determined by HPLC: 89.90%.
Step-2: Preparation of (25,5R)-2-(5-(/V-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of (25,5i?)-2-{N’-[2-(5)-N-ieri-butoxycarbonyl-azetidin-2-yl-acetyl]-hydrazinocarbonyl}-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane (4 gm, 8.21 mmol) in chloroform (70 ml) was added p-toluenesulfonylchloride (2.34 gm, 12.3 mmol) followed by dnsopropylethylamine (4.4 ml, 24.6 mmol). The reaction mixture was heated under stirring at 75°C for 18 hours. The reaction mixture was concentrated under vacuum and the resulting mass was purified by using silica gel column chromatography, to provide (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane in 3.3 g quantity in 86% yield as a solid.
Analysis:
Mass: 470.4 (M+l), for Molecular Formula of C^HsiNsOs;
1H NMR: (CDCb): δ 7.36-7.44 (m, 5H), 5.08 (d, 1H), 4.93 (d, 1H), 4.68-4.71 (m, 1H), 4.10-4.15 (m, 2H), 3.68-3-72 (m, 2H), 3.37 (s, 1H), 3.13-3.15 (m, 2H), 2.90-3.11 (m, 2H), 2.77 (d, 1H), 2.25-2.31 (m, 2H), 2.10-2.19 (m, 1H), 1.87- 1.97 (m, 1H), 1.43 (s, 9H).
Step-3: Preparation of (25,5R)-2-(5-(iV-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To the solution of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-benzyloxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane ( 3.3 g, 7.0 rnmol) in methanol (35 ml) was subjected to catalytic hydrogenolysis using 10% palladium on charcoal (350 mg) under atmospheric hydrogen gas pressure at 25°C to 35°C for 2 hours. The reaction mixture was filtered through celite bed and was washed with methanol (30 ml). The filtrate was concentrated under vacuum below 35°C to provide 2.7 g of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-hydroxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane, which was used immediately for the next reaction.
Analysis:
Mass: 378.4 (M-l), for Molecular Formula of CnH^NsOs.
Step-4: Preparation of tetrabutylammonium salt of (2S,5R)-2-(5-(V-tert-butoxycarbonylazetidin-3-yl)-methyl-[l,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane:
To a solution of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)- 6- hydroxy-7-oxo- l,6-diaza-bicyclo[3.2.1] octane (2.7 gm, 7.12 mmol) in dichloromethane (50 ml) was added triethylamine (5 ml, 35 mmol) followed by sulfur trioxide pyridine complex (2.26 g 14.2 mmol) under stirring at 25°C to 35°C. The reaction mixture was stirred for 2 hours. To the reaction mixture was added aqueous 0.5 N potassium dihydrogen phosphate solution (100 ml). It was stirred for about 30 minutes and tetrabutyl ammonium hydrogen sulfate (2.17 gm 6.4 mmol) was added. It was stirred for 2 hours. Layers were separated and organic layer was concentrated under vacuum to provide a crude mass, which was purified by silica gel column chromatography to furnish 2.1 g of tetrabutylammonium salt of (25,5i?)-2-(5-(N-ieri-butoxycarbonylazetidin-3-yl)-methyl-[ 1 ,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo- 1 ,6-diaza-bicyclo[3.2.1] octane as solid in 43% yield.
Analysis:
Mass: 458.3 (M- l), as a free sulfonic acid, for Molecular Formula of C17H25N5O8S. N(C4H9)4; Purity as determined by HPLC: 94.87%.
Step-5: Preparation of (25,5R)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[l,3,4]-oxadiazol-2-yl)- 7- oxo-l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester:
To the solution of tetrabutylammonium salt of (25,5i?)-2-(5-(N-feri-butoxycarbonylazetidin-3-yl)-methyl-[ l,3,4]-oxadiazol-2-yl)-6-sulphooxy-7-oxo-l,6-diaza-bicyclo[3.2.1] octane (1.0 g, 2.2 mmol) in dichloromethane (5 ml) was charged trifluoroacetic acid (5 ml) with syringe at – 10°C under stirring. The reaction mixture was stirred for 1 hour. The mixture was evaporated under vacuum by maintaining temperature below 35 °C, to provide a residue, which was suspended in diethyl ether (25 ml) twice. The suspension was filtered and the solid was suspended further in dichloromethane (50 ml) and stirred for 30 minutes. The suspension was filtered and dried to afford the 310 mg of (25,5i?)-sulfuric acid mono-[2-(5-azetidin-3-ylmethyl-[ l,3,4]-oxadiazol-2-yl)-7-oxo- l,6-diaza-bicyclo[3.2.1] oct-6-yl] ester as a solid in 60% yield.
Analysis:
Mass: 358.2 (M-l), for Molecular Formula of C^HnNsOeS;
1H NMR (DMSO-d6): δ 8.50 (br s, IH), 8.62 br s, IH), 4.60 (d, IH), 4.05 (s, 3H), 3.82-3.84 (m, IH), 3.21-3.27 (m, 4H), 2.93-2.96 (m, IH), 2.75 (d, IH), 2.12-2.17 (m, IH), 1.96-2.05 (m, 2H), 1.82-1.88 (m, IH).
Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.
///////
Zidebactam, WCK 5107 in PHASE 1 FROM WOCKHARDT

Zidebactam, WCK 5107
![]()
Useful for treating bacterial infections
CAS 1436861-97-0, UNII: YPM97423DB, Wockhardt Biopharm
Molecular Formula, C13-H21-N5-O7-S
Molecular Weight, 391.4029
Disclosed in PCT International Patent Application No. PCT/IB2012/054290D
- 01 Aug 2015 Phase-I clinical trials in Bacterial infections (In volunteers, Combination therapy) in USA (IV) (NCT02532140)
trans- sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(2S, 5R)-sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(1R,2S,5R)-l,6-Diazabicyclo [3.2.1] octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-[2-[(3R)-3-piperidinylcarbonyl]hydrazide]
trans- sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(2S, 5R)-sulphuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7-oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl] ester
(lR,2S,5R)-l,6-Diazabicyclo [3.2.1] octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-[2-[(3R)-3 -piperidinylcarbonyl] hydrazide]
1,6-Diazabicyclo(3.2.1)octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-(2-((3R)-3-piperidinylcarbonyl)hydrazide), (1R,2S,5R)-

Zidebactam potassium
cas is 1706777-49-2
Zidebactam sodium ………..below

Cas 1706777-46-9
Sodium;[(2S,5R)-7-oxo-2-[[[(3R)-piperidine-3-carbonyl]amino]carbamoyl]-1,6-diazabicyclo[3.2.1]octan-6-yl] sulfate
UNII-NHY7N0Y9DG; NHY7N0Y9DG; Zidebactam sodium; Zidebactam sodium, (-)-; 1,6-Diazabicyclo(3.2.1)octane-2-carboxylic acid, 7-oxo-6-(sulfooxy)-, 2-(2-((3R)-3-piperidinylcarbonyl)hydrazide), sodium salt (1:1), (1R,2S,5R)-; 1706777-46-9;
| Molecular Formula: | C13H20N5NaO7S |
|---|---|
| Molecular Weight: | 413.381969 g/mol |
In September 2015, the drug was reported to be in phase I clinical trial.One of the family members US09132133, claims a combination of sulbactam and WCK-5107.
Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate.
Treatment of infections caused by resistant bacteria remains a key challenge for the clinician community. One example of such challenging pathogen is Acinetobacter baumannii (A. baumannii), which continues to be an increasingly important and demanding species in healthcare settings. The multidrug resistant nature of this pathogen and its unpredictable susceptibility patterns make empirical and therapeutic decisions more difficult. A. baumannii is associated with infections such as pneumonia, bacteremia, wound infections, urinary tract infections and meningitis.
Therefore, there is a need for development of newer ways to treat infections that are becoming resistant to known therapies and methods. Surprisingly, it has been found that a compositions comprising cefepime and certain nitrogen containing bicyclic compounds (disclosed in PCT/IB2012/054290) exhibit unexpectedly synergistic antibacterial activity, even against highly resistant bacterial strains.

PATENT
http://www.google.com/patents/WO2013030733A1?cl=en

Scheme-1

function with Boc group)
o ormua –
Scheme-2
Example-2 trans-sulfuric acid mono-r2-(N,-r(R)-piperidin-3-carbonyll-hvdrazinocarbonyl)-7-oxo-l,6- diaza-bicyclo Γ3.2.11 oct-6-νΠ ester
Step-1: Preparation of trans-3-[N’-(6-benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-1 of Example- 1 above, and by using trans-6- benzyloxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylic acid (25 gm, 0.084 mol), N,N- dimethyl formamide (625 ml), EDC hydrochloride (24 gm, 0.126 mol), HOBt (16.96 gm, 0.126 mol), (R)-N-tert-butoxycarbonyl-piperidin-3-carboxylic acid hydrazide (21.40 gm , 0.088 mol) to provide the title compound in 17.0 gm quantity, 41% yield as a white solid.
Analysis: MS (ES+) CzsHasNsOe = 502.1 (M+l);
I^NMR (CDCI3) = 8.40 (br s, IH), 7.34-7.44 (m, 5H), 5.05 (d, IH), 4.90 (d, IH), 4.00 (br d, IH), 3.82 (br s, IH), 3.30 (br s, IH), 3.16-3.21 (m, IH), 3.06 (br d, IH), 2.42 (br s, IH), 2.29-2.34 (m, IH), 1.18-2.02 (m, 4H), 1.60-1.75 (m, 4H), 1.45-1.55 (m, 2H),1.44 (s, 9H).
Step-2: Preparation of trans-3-[N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester:
By using the procedure described in Step-2 of Example- 1 above, and by using trans-3- [N ‘ -(6-benzyloxy-7-oxo- 1 ,6-diaza-bicyclo [3.2.1 ]octane-2-carbonyl)-hydrazinocarbonyl] -(R)- piperidin-l-carboxylic acid tert-butyl ester (16.5 gm , 0.033 mol), methanol (170 ml) and 10% palladium on carbon (3.5 gm) to provide the title compound in 13.5 gm quantity as a pale pink solid and it was used for the next reaction immediately.
Analysis: MS (ES+) CiglfeNsOe = 411.1 (M+l);
Step-3: Preparation of tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza- bicyclo [3.2.1] octane-2-carbonyl)-hydrazinocarbonyl] -(R)-piperidin- 1 -carboxylic acid tert- butyl ester:
By using the procedure described in Step-3 of Example- 1 above, and by using trans-3- [N’-(6-hydroxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2-carbonyl)-hydrazinocarbonyl]-(R)- piperidin-1 -carboxylic acid tert-butyl ester (13.5 gm , 0.033 mol), pyridine (70 ml) and pyridine sulfur trioxide complex (26.11 gm, 0.164 mol), 0.5 N aqueous potassium dihydrogen phosphate solution (400 ml) and tetrabutylammonium sulphate (9.74 gm, 0.033 mol) to provide the title compound in 25 gm quantity as a yellowish solid, in quantitative yield.
Analysis: MS (ES-)
as a salt = 490.0 (M-l) as a free sulfonic acid;
Step-4: trans-sulfuric acid mono-[2-(N’-[(R)-piperidin-3-carbonyl]-hydrazinocarbonyl)-7- oxo-l,6-diaza-bicyclo[3.2.1]oct-6-yl]ester:
By using the procedure described in Step-4 of Example- 1 above, and by using tetrabutylammonium salt of trans-3-[N’-(6-sulfooxy-7-oxo-l,6-diaza-bicyclo[3.2.1]octane-2- carbonyl)-hydrazinocarbonyl]-(R)-piperidin-l-carboxylic acid tert-butyl ester (24 gm , 0.032 mmol), dichloromethane (60 ml) and trifluoroacetic acid (60 ml) to provide the title compound in 10 gm quantity as a white solid, in 79% yield.
Analysis: MS (ES-)= C13H21N5O7S = 390.2 (M-l) as a free sulfonic acid;
HXNMR (DMSO-d6) = 9.97 (d, 2H), 8.32 (br s, 2H), 4.00 (br s, IH), 3.81 (d, IH), 3.10-3.22 (m, 3H), 2.97-3.02 (m, 2H), 2.86-2.91 (m, IH), 2.65-2.66 (m, IH), 1.97-2.03 (m, IH), 1.57-1.88 (m, 7H).
-32.6°, (c 0.5, water).
PATENT
http://www.google.com/patents/WO2015059643A1?cl=en

Both, cefepime and a compound of Formula (I) may be present in the composition in their free forms or in the form of their pharmaceutically acceptable derivatives (such as salts, pro-drugs, metabolites, esters, ethers, hydrates, polymorphs, solvates, complexes, or adducts).
Individual amounts of a compound of Formula (I) or a stereoisomer or a pharmaceutically acceptable derivative thereof, and cefepime or pharmaceutically acceptable derivative thereof in the composition may vary depending on clinical requirements. In some embodiments, a compound of Formula (I) or a stereoisomer or a pharmaceutically acceptable derivative thereof in the composition is present in an amount from about 0.01 gram to about 10 gram. In some other embodiments, cefepime or a pharmaceutically acceptable derivative thereof in the composition is present in an amount from about 0.01 gram to about 10 gram.
PATENT
http://www.google.com/patents/WO2015063653A1?cl=en
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015110885
Formula (I)

(a) hydrogenolysis of a compound of Formula (II) to obtain a compound of Formula (III);

convertin a compound of Formula (III) to a compound of Formula (IV);



Example 1
Synthesis of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (25, 5R)-6-hydroxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III):
(25, 5i?)-6-benzyloxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazino-carbonyl] -l,6-diazabicyclo[3.2.1]octane (II) (130 g, 0.259 mol) was dissolved in methanol (1040 ml) to obtain a clear solution. To this solution, was added 10% palladium on carbon (13 g, 0.26 mol). The suspension was stirred under 230-250 psi hydrogen atmosphere at temperature of about 30 °C for about 2 hour. The catalyst was filtered over celite bed and catalyst containing bed was washed with additional methanol (400 ml). The methanolic solution was re-filtered through fresh celite bed and washed with methanol (100 ml). The filtrate was concentrated under vacuum at temperature of about 30°C to obtain the off white solid as product. The so obtained solid was stirred with cyclohexane (750 ml). The solid was then filtered and washed with cyclohexane (320 ml) and dried under suction to obtain 107 g of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo [3.2.1]octane (III).
Analysis:
Mass: 412.4 (M+l); for Molecular Formula of C18H29N5O6 and Molecular Weight of 411.5; and
Purity as determined by HPLC: 98.02%.
Step-2: Preparation of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1] octane (IV):
A solution of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) (106 g, 0.26 mol) in dichloromethane was charged with triethyl amine (110 ml, 0.78 mol) under stirring. To this clear solution was added pyridine sulfur trioxide complex (82.5 g, 0.53 mol) under nitrogen atmosphere and stirred at temperature of about 30°C for about 2 hour. The reaction mixture was diluted with 0.5 N aqueous potassium dihydrogen phosphate solution (2100 ml) followed by ethyl acetate (2100 ml). The turbid solution was stirred for 15 minute and then the layers were separated. The aqueous layer was washed with dichloromethane (530 ml) and then with ethyl acetate (1060 ml). Tetrabutyl ammonium sulfate (79 g, 0.23 mol) was added to the separated aqueous layer and stirred for 12 hour. The extraction of the product was done using dichloromethane as solvent (1150 ml x 2). The organic layer was dried over sodium sulfate and then evaporated under vacuum at temperature below 40°C to furnish 108 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo
[3.2.1] octane (IV).
Analysis:
Mass: 490.3 (M-l) as free sulfonic acid; for Molecular Formula of Ci8H28N509S.N(C4H9)4 and Molecular weight of 733.0; and
Purity as determined by HPLC: 86.50 %.
Step-3: Preparation of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) (88 g, 0.12 mol) was dissolved in dichloromethane (225 ml). The reaction mass was cooled to about -10°C and to this trifluoroacetic acid (225 ml) was added slowly. The reaction mixture was stirred for 1 hour at temperature of about -10°C. The solvent was removed under high vacuum at about 30°C. The residue (280 g) was stirred with diethyl ether (1320 ml) for 1 hour. The precipitated solid was filtered and the cake was washed with fresh diethyl ether (440 ml). This process was repeated with fresh diethyl ether (1320 ml + 440 ml). The obtained white solid was dried at temperature of about 30°C and suspended in acetone (1320 ml). The pH of the suspension was adjusted to 6.5-7.0 using 10% solution of sodium 2-ethyl hexanoate in acetone. The resulting suspension was filtered under suction and the wet cake was washed with acetone (440 ml) to provide the crude solid. The solid was further dried under vacuum at 40°C to yield 40 g of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I).
Analysis:
Mass: 392.2 (M+l); for Molecular formula of C13H21N5O7S and Molecular Weight of 391.4;
Purity as determined by HPLC: 92.87%; and
Melting point as determined by DSC: 274°C.
Example 2
Synthesis of Pure (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (25, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III):
The procedure for the synthesis of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) is same as given in Step- 1 of Example 1.
Step-2: Preparation of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1] octane (IV):
A solution of (25, 5i?)-6-hydroxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (III) (106 g, 0.26 mol) in dichloromethane was charged with triethylamine (110 ml, 0.78 mol) under stirring to provide a clear solution. To this clear solution was added pyridine sulfur trioxide complex (82.5 g, 0.53 mol) under nitrogen atmosphere and stirred at temperature of about 30 °C for 2 hours. The reaction mixture was diluted with 0.5 N aqueous potassium dihydrogen phosphate solution (2100 ml) followed by ethyl acetate (2100 ml). The turbid solution was stirred for 15 minutes and then the layers were separated. The aqueous layer was washed with dichloromethane (530 ml) and then with ethyl acetate (1060 ml) respectively. Tetrabutyl ammonium sulfate (79 g, 0.23 mol) was added to the separated aqueous layer and stirred for 12 hours. The extraction of the product was done using dichloromethane as solvent (1150 ml x 2). Aliquot of the organic layer was dried over sodium sulfate for purity check. Considering the purity of the product as obtained above, silica gel (530 g) was added to the dichloromethane layer and stirred for 1 hour. This was filtered and again silica was taken in dichloromethane (3200 ml) and stirred for 45 minutes and filtered. Combined dichloromethane layer was filtered through the celite bed again and washed with additional 200 ml dichloromethane. The solvent was removed to obtain 88 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-!, 6-diaza-bicyclo[3.2.1]octane (IV) as white foam.
Analysis:
Mass: 490.3 (M-l) as a free sulfonic acid; for Molecular Formula of Ci8H28N509S.N(C4H9)4 and Molecular Weight of 733.0; and
Purity as determined by HPLC: 98.34%.
Step-3: Preparation of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
The above obtained tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) having purity of more than 98% (88 g, 0.12 mol) was dissolved in dichloromethane (225 ml). The reaction mass was cooled to temperature of about -10°C and to this trifluoroacetic acid (225 ml) was added slowly. The reaction mixture was stirred for 1 hour at about -10°C. The solvent was removed under high vacuum at temperature of about 30°C. The residue (280 g) was stirred with diethyl ether (1320 ml) for 1 hour. The precipitated solid was filtered and the cake was washed with fresh diethyl ether (440 ml). This process was repeated with fresh diethyl ether (1320 ml + 440 ml). The obtained white solid was dried at about 30°C and suspended in acetone (1320 ml). The pH of the suspension was adjusted to 6.5-7.0 using 10% solution of sodium 2-ethyl hexanoate in acetone. The resulting suspension was filtered under suction and the wet cake was washed with acetone (440 ml) to provide the crude solid. The solid was further dried under vacuum at 40°C to yield 40 g of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I).
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4; and
Purity as determined by HPLC: 98.7%.
Recovery of tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2-[((3R)-iV-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1] octane (IV):
The silica recovered from the Step-2 was stirred with dichloromethane containing 2%
methanol (2000 ml) for one hour. Silica was filtered, washed with additional same composition of solvents (500 ml). Combined dichloromethane was filtered through the celite bed and washed with same composition of solvents (200 ml), evaporated to afford 1 1 g of tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l , 6-diaza-bicyclo[3.2.1] octane (IV) as off white solid.
Repeating Step-3 with the above obtained tetrabutylammonium salt of (25, 5R)-6-sulfooxy-7-oxo-2- [((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1 , 6-diaza-bicyclo [3.2.1] octane (IV) produced additional 7 g of compound of Formula (I).
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of CnH^NsOvS and Molecular Weight of 391.4;
Purity as determined by HPLC: 98.7%; and
Assay as determined by HPLC: 104% against reference standard of compound of Formula (I).
Example 3
Preparation of amorphous form of (25, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1, 6-diaza-bicyclo[3.2. l]octane (I) :
Tetrabutylammonium salt of (25, 5i?)-6-sulfooxy-7-oxo-2-[((3i?)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l, 6-diaza-bicyclo[3.2.1]octane (IV) (60 g, 0.081 mol), obtained in Step-2 of Example-2 was dissolved in dichloromethane (150 ml, 2.5 volume) to obtain a clear solution. Reaction mass was cooled to about -10°C and to it trifluoroacetic acid (150 ml) was slowly added. The reaction mixture was stirred for 1 hour at about – 10°C. The solvent was removed under high vacuum at about 30°C. Diethyl ether (600 ml x 3) was added to the residue ( 184 g) and stirred for 15 minute every time. The solvent was decanted off and the residue was washed with acetonitrile (600 ml x 3). This process was also repeated with dichloromethane (600 ml x 3). The off white solid was
isolated and dried under high vacuum at about 35 °C for 3 hour to obtain 33 g of amorphous form of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I). The XRD is shown in Figure 1.
Analysis:
Mass: 392.2 (M+l); for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4;
HPLC purity: 92.26%; and
Melting point as determined by DSC: 210°C (loss of moisture below 100°C).
Example 4
Preparation of crystalline form of (25, 5R)-7-oxo-6-sulpho-oxy-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I):
The (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I) obtained as white solid (40 g) in Step-3 of Example 2 was dissolved in demineralised water (40 ml) to obtain a clear solution. To this isopropyl alcohol (280 ml) was added under stirring at room temperature. The obtained turbid solution became sticky initially then slowly started to convert into white solid, stirring continued for about 17 hours at temperature of about 30°C. The precipitated solid was filtered and washed with water: isopropyl alcohol mixture (20 ml: 140 ml). White solid was dried under high vacuum at temperature of about 45 °C for 5 hours to get 34 g of crystalline form of (25, 5i?)-7-oxo-6-sulphooxy-2-[((3i?)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1] octane (I).
Analysis:
Mass: 392.2 (M+l) for Molecular Formula of C13H21N5O7S and Molecular Weight of 391.4;
Purity as determined by HPLC: 98.7%;
Assay as determined by HPLC: 104% against reference standard of compound of Formula (I); and
Melting point as determined by DSC: 278°C (9% loss of moisture at 143-152°C).
X-ray powder diffraction pattern comprising a peak selected from the group consisting of 10.31 (± 0.2), 10.59 (± 0.2), 12.56 (± 0.2), 13.84 (± 0.2), 15.65 (± 0.2), 18.19 (± 0.2), 18.51(± 0.2), 20.38 (± 0.2), 20.65 (± 0.2), 24.30 (± 0.2), 24.85 (± 0.2) and 25.47 (± 0.2) degrees 2 theta.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014135931
Scheme 1.


Formula (I)
preparation of a compound of Formula (I), comprising:

Formula (I)
(a) reacting a compound of Formula (II) with a compound of Formula (III) to obtain a compound of Formula (IV);

Formula (II) Formula (III)

Formula (IV)
(b) hydrogenolysis of a compound of Formula (IV) to obtain a compound of Formula

X. Formula (V)
(c) sulfonating a compound of Formula (V) to obtain a compound of Formula (VI); and

Formula (VI)
(d) converting a compound of Formula (VI) into a compound of Formula (I).
Example -1
Preparation of (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (II):
Step-1: Preparation of (R)-Ethyl-N-Boc-piperidine-3-carboxylate (VIII)
To a solution of (R)-N-Boc-piperidine-3-carboxylic acid (1 kg. 4.36 mol) in N,N-dimethylacetamide (3 L) was charged potassium carbonate (0.664 kg, 4.80 mol) under mechanical stirring and the resulting suspension was stirred for 30 minutes at room temperature. To the reaction mass, ethyl iodide (0.75 kg, 4.80 mol) was charged via addition funnel and the reaction mass was stirred for 15 minutes at room temperature followed by at 50°C for 1 hour. The reaction was monitored using TLC (ethyl acetate: hexane 1:1). After the reaction was complete, the reaction mass was allowed to cool to room temperature and diluted with ethyl acetate (5 L). The suspension was filtered under suction and the wet cake was washed with ethyl acetate (5 L). The filtrate was stirred with 5% w/v sodium thio sulfate (15 L) and layers were separated. The aqueous layer was re-extracted with additional ethyl acetate (5 L). The combined organic layer was washed with water (5 L) and dried over sodium sulfate. The organic layer was evaporated under vacuum to provide semi-solid which solidifies upon standing as (R)-ethyl-N-Boc-piperidine-3-carboxylate in 1.1 kg quantity in 99.5% yield.
Analysis:
NMR: (CDC13): 4.63 (q, 2H), 3.90 (d, 1H), 2.87-2.95 (m, 2H), 2.73 (td, 1H), 2.32-2.39 (m, 1H), 1.66-2.01 (m, 2H), 1.52-1.68 (m, 2H), 1.39 (s, 9H), 1.19 (t, 3H).
Mass: (M+l): 258.1 for C13H23N04;
Step-2: Preparation of (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (II):
(R)-N-Boc-ethyl-piperidine-3-carboxylate (1.1 kg, 4.28 mol) was liquefied by warming and transferred to a round bottom flask (10 L), to this was charged hydrazine hydrate (0.470 kg, 9.41 mol) and stirring was started. The reaction mixture was stirred at about 120°C to 125°C for 5 hours. As the TLC showed (Chloroform: methanol 9:1) completion of reaction, the reaction mixture was cooled to room temperature and diluted with water (5.5 L) followed by dichloromethane (11 L) and was stirred for 20 minutes. The layers were separated and aqueous layer was extracted with additional dichloro methane (5.5 L). Combined organic layer was washed with water (2.75 L). The organic layer was dried over sodium sulfate and evaporated under vacuum to provide a thick gel which upon stirring and seeding in the presence of cyclohexane (5.5 L) provided white solid. The suspension was filtered and wet cake was washed with fresh cyclohexane (0.5 L). The cake was dried at 35°C under vacuum to provide (R)-N-Boc-piperidine-3-carboxylic acid hydrazide as a white solid in 0.90 kg quantity in 87% yield.
Analysis
NMR: (CDC13): 7.42 (br s, 1H), 3.92 (d, 1H), 3.88 (s, 2H), 3.54-3.65 (br s, 1H), 3.17 (br t, 1H), 2.98 (br s, 1H), 2.22-2.32 (br s, 1H), 1.82-1.90 (br m, 2H), 1.76 (s, 1H), 1.60-1.70 (m, 1H), 1.45 (s, 9H).
Mass (M+l): 244.1 for C11H21N303.
Specific rotation: [ ]25D = -53.5° (c 0.5, Methanol).
HPLC purity: 99%
Example 2
Preparation of (2S, 5R)-7-oxo-6-sulphooxy-2-[((3R)-piperidine-3-carbonyl)- hydrazinocarbonyl] -l,6-diaza-bicyclo[3.2.1]octane (I):
Step-1: Preparation of (2S, 5R)- 6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl] – 1 ,6-diaza-bicyclo [3.2.1 ] octane(IV) :
Sodium (2S, 5R)-7-oxo-6-benzyloxy-l,6-diaza-bicyclo[3.2.1]octane-2-carboxylate (III, 200 gm, 0.67 mol; prepared using a method disclosed in Indian Patent Application No 699/MUM/2013) was dissolved in water (2.8 L) to obtain a clear solution under stirring at room temperature. To the clear solution was added successively, (R)-N-Boc-piperidine-3-carboxylic acid hydrazide (171 gm, 0.70 mol), EDC hydrochloride (193 gm, 1.01 mol), and HOBt (90.6 gm, 0.67 mol) followed by water (0.56 L) under stirring at 35°C. The reaction mixture was stirred at 35°C for 20 hours. As maximum precipitation was reached, TLC (acetone: hexane 35:65) showed completion of reaction. The suspension was filtered under
suction and the wet cake was washed with additional water (2 L). The wet cake was suspended in warm water (10 L) and stirred for 5 hours. It was filtered under suction and dried under vacuum at 45°C to furnish (2S, 5R)-6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (IV) as a white powder in 270 gm quantity in 87% yield.
Analysis
NMR: (CDC13): 8.40 (br s, 1H), 7.34-7.44 (m, 5H), 5.05 (d, 1H), 4.90 (d, 1H), 4.00 (br d, 1H), 3.82 (br s, 1H), 3.30 (br s, 1H), 3.16-3.21 (m, 1H), 3.06 (br d, 1H), 2.42 (br s, 1H), 2.29-2.34 (m, 1H), 1.18-2.02 (m, 4H), 1.60-1.75 (m, 4H), 1.45-1.55 (m, 2H),1.44 (s, 9H).
Mass: (M+l) = 502.1 for C25H35N506
HPLC purity: 98.4%
Step-2: Preparation of (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2. l]octane (V):
(2S,5R)-6-benzyloxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino-carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (153 gm, 0.305 mol) was dissolved in methanol (1.23 L) to obtain a clear solution. To this solution, was added 10% Pd-C (15.3 gm, 50% wet) catalyst. The suspension was stirred for 3 hours under 100 psi hydrogen atmosphere at 35°C. As reaction showed completion on TLC (TLC system methanol: chloroform 10:90), the catalyst was filtered through celite under suction. The catalyst was washed with additional methanol (600 ml). The filtrate was evaporated under vacuum below 40°C to provide a crude residue. The residue was stirred with cyclohexane (1.23 L) for 1 hour. The solid was filtered at suction and the wet cake was washed with additional cyclohexane (0.25 L) to furnish (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2.1]octane (V) in 125 gm quantity as a solid in quantitative yield. The product being unstable was used immediately for the next reaction.
Analysis:
NMR: (CDC13): 9.0 (br s, 2H), 4.01 (br d, 2H), 3.80 (br s, 1H), 3.74 (br s, 1H), 3.48 (s, 1H), 3.13-3.26 (m, 3H), 2.96 (br s, 1H), 2.47 (br s, 1H), 2.28-2.32 ( br dd, 1H), 2.08 (br s, 1H), 1.90-2.0 (m, 3H),1.65-1.80 (m, 3H) 1.44 (s, 9H).
Mass: (M-l): 410.3 for C18H29N506
HPLC purity: 96.34%
Step-3: Preparation of Tetrabutyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazinocarbonyl]- 1 ,6-diaza-bicyclo[3.2.1 ] octane (VI) :
A solution of (2S, 5R)-6-hydroxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (113 gm, 0.274 mol), in dichloromethane (1.13 L) was charged with triethylamine (77 ml, 0.548 mol) under stirring to provide a clear solution. To the clear solution, was added pyridine sulfur trioxide complex (57 gm, 0.356 mol) under stirring at 35°C. The reaction mixture was stirred for 3 hours. The reaction mixture was worked up by adding 0.5 M aqueous potassium dihydrogen phosphate (1.13 L) followed by ethyl acetate (2.26 L) and the biphasic mixture was stirred for 15 minutes at 35°C. Layers were separated. Aqueous layer was re-extracted with dichloromethane ethyl acetate mixture (1:2 v/v, 2.26 L twice). Layers were separated. To the aqueous layer, was added solid tetrabutyl ammonium hydrogen sulfate (84 gm, 0.247 mol) and stirring was continued for 3 hours at room temperature. Dichloromethane (1.13 L) was added to the reaction mixture. Layers were separated. The aqueous layer was re-extracted with additional dichloromethane (0.565 L). Layers were separated. To the combined organic layer was added silica gel (226 gm) and the suspension was stirred for 1 hour. Suspension was filtered and silica gel was washed with dichloromethane (1 L). The combined filtrate was evaporated under vacuum to provide solid mass. To the solid mass was added cyclohexane (0.9 L) and stirred till complete solidification occurred (about 1 to 2 hours). The suspension was filtered under suction and the wet cake was dried under vacuum below 40°C to furnish tetrabutyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (VI) as a white solid in 122 gm quantity in 60% yield.
Analysis
NMR: (CDC13): 8.50 (br s, 2H), 4.32 (br s, 1H), 3.97 (d, 2H), 3.15-3.37 (m, 12H), 2.43 (br s, 1H), 2.33 (d, 1H), 2.10-2.2 (br m, 1H), 1.84-1.95 (m, 3H), 1.60-1.73 (m, 13H), 1.39-1.48 (m, 19H), 0.98 (t, 12H).
Mass: (M-l): 490.4 as a free sulfonic acid for C18H28N509S.N(C4H9)4;
HPLC purity: 96.3%
Step-4: Synthesis of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-piperidine-3-carbonyl)-hydrazinocarbonyl]-l,6-diaza-bicyclo[3.2. l]octane (I):
Tetra-butyl ammonium salt of (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-N-Boc-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (113 gm, 0.154 mol) was dissolved in dichloromethane (280 ml) and to the clear solution was slowly added trifluoroacetic acid (280 ml) between 0 to 5°C. The reaction mixture was stirred between 0 to 5°C for 1 hour. The solvent and excess trifluoroacetic acid was evaporated under vacuum below 40°C to approximately 1/3 of it’s original volume to provide pale yellow oily residue. The oily residue was stirred with diethyl ether (2.25 L) for 1 hour to provide a suspension. The precipitate was filtered under suction and transferred to a round bottom flask, to it was added diethyl ether (1.1 L) under stirring. The suspension was stirred for 30 minutes and filtered under suction to provide a solid. The solid was charged in a round bottom flask and to it was added acetone (1.130 L). The pH of suspension was adjusted to 4.5 to 5.5 by adding 10% solution of sodium-2-ethyl hexanoate in acetone carefully. The resulting suspension was filtered under suction and the wet cake was washed with acetone (550 ml) to provide a crude solid. The obtained solid was dried under vacuum below 40°C to furnish 65 gm of a crude mass. The crude mass was dissolved in water (65 ml) under stirring and to the clear solution was added isopropyl alcohol (455 ml). The suspension was stirred for 24 hours and filtered under suction. The wet cake was washed with isopropyl alcohol (225 ml) and dried under vacuum below 40°C to provide a crystalline (2S, 5R)-6-sulfooxy-7-oxo-2-[((3R)-piperidine-3-carbonyl)-hydrazino carbonyl]-l,6-diaza-bicyclo[3.2.1]octane (I) free from impurities in 48 gm quantity in 80% yield.
Analysis:
NMR: (DMSO-d6) = 9.97 (d, 2H), 8.32 (br s, 2H), 4.00 (br s, IH), 3.81 (d, IH), 3.10-3.22 (m, 3H), 2.97-3.02 (m, 2H), 2.86-2.91 (m, IH), 2.65-2.66 (m, IH), 1.97-2.03 (m, IH), 1.57-1.88 (m, 7H).
Mass: (M-l): 390.3 for C13H21N507S
HPLC purity: 95.78%
Specific rotation: [(X]25D: – 32.6° (c 0.5, water)
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 10.28 (+ 0.2), 10.57 (± 0.2), 12.53 (± 0.2), 13.82 (± 0.2), 15.62 (± 0.2), 18.16 (± 0.2), 18.49 (± 0.2), 20.35 (+ 0.2), 20.64 (± 0.2), 21.33 (+ 0.2), 22.99 (+ 0.2), 23.18 (+ 0.2), 24.27 (± 0.2), 24.81 (+ 0.2), 25.45 (± 0.2), 29.85 (+ 0.2), 30.45 (± 0.2), 32.39 (+ 0.2), 36.84 (± 0.2).
REFERENCES
Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of WCK-5107 Alone and in Combination With Cefepime (NCT02532140) https://clinicaltrials.gov/show/NCT02532140
ClinicalTrials.gov Web Site 2015, September 01, To evaluate the safety,tolerability and pharmacokinetics of single intravenous doses of WCK 5107 alone and in combination with cefepime in healthy adult human subjects.
| WO2013030733A1 * | Aug 24, 2012 | Mar 7, 2013 | Wockhardt Limited | 1,6- diazabicyclo [3,2,1] octan-7-one derivatives and their use in the treatment of bacterial infections |
| WO2014135931A1 * | Oct 12, 2013 | Sep 12, 2014 | Wockhardt Limited | A process for preparation of (2s, 5r)-7-oxo-6-sulphooxy-2-[((3r)-piperidine-3-carbonyl)-hydrazino carbonyl]-1,6-diaza-bicyclo [3.2.1]- octane |
| IB2012054290W | Title not available |
Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.
///////see………http://apisynthesisint.blogspot.in/2015/11/wck-5107-in-phase-1-from-wockhardt.html
SEE BACTAM SERIES…………..http://apisynthesisint.blogspot.in/p/bactam-series.html
C1C[C@H](CNC1)C(=O)NNC(=O)[C@@H]2CC[C@@H]3C[N@]2C(=O)N3OS(=O)(=O)O
or
O=C(NNC(=O)[C@@H]2CC[C@@H]1CN2C(=O)N1OS(=O)(=O)O)[C@@H]3CCCNC3
C1CC(CNC1)C(=O)NNC(=O)C2CCC3CN2C(=O)N3OS(=O)(=O)[O-].[Na+]
WCK 2349 in phase II trials by Wockhardt
 . CH3SO3H
. CH3SO3H
WCK 2349: A novel fluoroquinolone (FQ) prodrug-13 week oral (PO) safety profile in cynomolgus monkeys
47th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 17-20, Chicago) 2007, Abst F1-2133a
8-{4-[2(S)-Amino-propionyloxy] piperidine-l-yl}-9-fluoro-5 (S)-methyl-ό, 7-dihydro-l- oxo-lH, 5H-benzo[i,j]quinolizine-2-carboxylic acid of structural Formula I can be used to treat bacterial Gram-positive, Gram-negative and anaerobic infections; especially infections caused by resistant Gram-positive organism and Gram-negative organism, mycobacterial infections and emerging nosocomial pathogen infections.
Formula I
U.S. Patent Nos. 6,750,224 and 7,247,642 describes optically pure S-(-)-benzoquinolizine carboxylic acids, their derivatives, salts, pseudopolymorphs, polymorphs and hydrates thereof, their processes of preparation and their pharmaceutical compositions.
PATENT
WO 2007102061
http://www.google.co.in/patents/WO2007102061A2?cl=en

Scheme 1
Experimental:
(S)-9-Fluoro-6,7-dihydro-8-(4-hydroxypiperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[ij] quinolizine-2-carboxylic acid was prepared as per procedure described in Chem. Pharm. Bull. 1996, 44(4), 642-645.
Example-l
Preparation of (2’S,5S)-9-fluoro-6,7-dihydro-8-(4-(N-tert-butoxycarbonyI-L-aIaninyl- oxy)-piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid:
Method-1 : To a mixture of N-tert-butoxycarbonyl-L-alanine (473 g) in dichloromethane (2 L), dicyclohexylcarbodiimide (515 g) dissolved in dichloromethane (2 L) was charged at -10 to 0 0C to provide a turbid suspension. To the turbid suspension, 300 g of (S)-9-fluoro-6,7- dihydro-8-(4-hydroxy-piperidin- 1 -yl)-5-methyl- 1-oxo- lH,5H-benzo[i,j]quinolizine-2- carboxylic acid was added followed by 4-N,N-dimethylamino pyridine (58 g) and the reaction mixture was stirred at -10 to 5 °C temperature over a period of 2 h. Suspension was filtered and solid was washed with 500 ml of dichloromethane. The filtrate was washed with water. Filtrate was dried over anhydrous sodium sulfate. Dried organic layer was then concentrated to its half volume where upon solid was precipitated. The solid was filtered and washed with 300 ml of dichloromethane. Clear organic filtrate was concentrated to dryness to provided an oily mass. Oily mass was triturated with diethyl ether (4 L) to provide white solid. The solid was filtered under suction and washed with diethyl ether (1 L) to provide title compound in 415 g (94%) quantity.
Method-2: To a mixture of triethylamine (98.0 ml) and N-tert-butoxycarbonyl-L-alanine (110 g) in tetrahydrofuran (1050 ml) and N,N-dimethyl formamide (350 ml) mixture, was added 2,4,6-trichlorobenzoyl chloride (100 ml). The resultant mixture was stirred at a temperature -5 to 0 °C for 5 h. To the > reaction mixture 4-N,N-dimethylamino pyridine (24g) and (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l-yl)-5-methyl-l-oxo-lH,5H- benzo[i,j]quinolizine-2-carboxylic acid (70 g) was added. The reaction mixture was stirred for additional 7 h at -5 to 0 0C temperature. The suspension was filtered at room temperature and the filtrate was extracted with ethyl acetate after addition of water. The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 85 g quantity.
Method-3: To a solution N-tert-butoxycarbonyl-L-alanine (7.9 g) in tetrahydrofuran (75 ml) and N,N-dimethyl formamide (25 ml) mixture at -10 to 0°C was added methanesulfonyl chloride (2.42 ml) dropwise. To the above solution triethylamine (8.7 ml) was added dropwise over 5 min. the reaction was stirred for 1.5 h maintaining the temperature between at -10 to 0 0C. To the reaction mixture (S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-piperidin-l- yl)-5-methyl-l-oxo-lH,5H-benzo[ij]quinolizine-2-carboxylic acid (5.01 g) and 4-N5N- dimethylamino pyridine (1.70 g) was added. The reaction mixture was stirred for additional 1 h at -5 to 0 °C temperature. The suspension was filtered at room temperature and the filtrate was diluted with water (300 ml) and extracted with ethyl acetate (150 ml x 2). The evaporation of organic layer under reduced pressure provided a sticky solid, which upon triturating with diethyl ether provided a white solid in 6.38 g (86%) quantity.
Example-2
Preparation of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-L-alaninyl-oxy-piperidin-l-yl)-5-methyl- l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid methanesulfonic acid salt:
To a mixture of (2’S, 5S)-9-fluoro-6,7-dihydro-8-(4-N-tert-butoxycarbonyl-L-alaninyloxy- piperidin-l-yl)-5-methyl-l-oxo-lH,5H-benzo[i,j]quinolizine-2-carboxylic acid (415 g) in acetone (4.5 L) was charged methanesulfonic acid (66 ml). Reaction mixture was stirred at 65-67 °C temperature for overnight. The suspension was filtered at 40-45 0C. Solid was washed with acetone (1.5 L) followed by diethyl ether (1.5 L). Off white solid was dried under 40 to 45 mm vacuum at 55-60 °C temperature over the period of 3-4 h. Title compound was obtained as a free flowing off white material 383.0 g (93%).
For MF: C23H30FN3O8S, MS (ES+) m/z 432 (obtained as free base for MF: C22H26FN3O5);
M.P. 278.50 0C by DSC

PATENT

Patent
PATENT
The tablets may optionally be coated with film forming agents and/or pharmaceutically acceptable excipients. Particularly suitable for use are commercially available coating compositions comprising film-forming polymers marketed under various trade names, such as Opadry® and Eudragit®. The coating layers over the tablet may be applied as solution/dispersion of coating ingredients using conventional techniques known in the art.
The present invention is further illustrated by the following examples which are provided merely to be exemplary of the invention and do not limit the scope of the invention. Certain modifications and equivalents will be apparent to those skilled in the art and are intended to be included within the scope of the present invention.
Example 1 :
Table 1 provides the composition of batches of the present invention.
Table 1

Procedure: The compound of Formula I or pharmaceutically acceptable salts, esters or products thereof, lactose and croscannellose sodium were sifted and dry mixed in a rapid mixer granulator. The above mass was granulated by spraying aqueous solution of povidone. The granules were dried in a fluidized bed drier, sifted and oversize granules were milled in a Quadra mill. The resultant granules were mixed with talc, croscarmellose sodium, microcrystalline cellulose and sodium stearyl fumarate in a double cone blender. The lubricated granules were compressed into tablets using suitable tooling. Tablets were coated with aqueous dispersion of opadry.
Table 2 provides the dissolution data for the compound of formula I or pharmaceutically acceptable salts, esters or products thereof tablets prepared as per the formula given in Table 1. For determination of drug release rate, USP Type 2 Apparatus (rpm 50) was used wherein 0.1 N hydrochloric acid (900 ml) was used as a medium. Table 2: Dissolution data



NEW DELHI: Drug maker WockhardtBSE -1.83 % today said that two of its anti-infective drugs
have received Qualified Infectious Disease Product (QIDP) status from the US
health regulator.Two drugs – WCK 771 and WCK 2349 – have received QIDP
status, which allows fast-track review of the drug application by the US Food and Drug Administration (USFDA),
Wockhardt said in a statement.
http://economictimes.indiatimes.com/articleshow/41359481.cms?utm_source=contentofinterest&utm_medium=text&utm_campaign=cppst
RN: 306748-89-0
-
C19-H21-F-N2-O4.C6-H14-N4-O2
- MW: 534.5855
-
L-Arginine, mono((5S)-9-fluoro-6,7-dihydro-8-(4-hydroxy-1-piperidinyl)-5-methyl-1-oxo-1H,5H-benzo(ij)quinolizine-2-carboxylate)

J Med Chem 2005, 48(16): 5232
| WO1991012815A1 * | Feb 25, 1991 | Sep 5, 1991 | Squibb Bristol Myers Co | COMPOSITIONS AND METHODS FOR TREATING INFECTIONS CAUSED BY ORGANISMS SENSITIVE TO β-LACTAM ANTIBIOTICS |
| WO2000068229A2 * | May 8, 2000 | Nov 16, 2000 | S K Agarwal | (s)-benzoquinolizine carboxylic acids and their use as antibacterial agents |
| WO2001085095A2 * | May 3, 2001 | Nov 15, 2001 | Shiv Kumar Agarwal | Chiral fluoroquinolizinone arginine salt forms |
| WO2002009758A2 * | Jul 31, 2001 | Feb 7, 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| EP2062582A1 * | Aug 14, 2007 | May 27, 2009 | Tianjin Hemey Bio-Tech Co., Ltd. | The antibiotics composition comprising beta-lactam antibiotics and buffers |
| US4524073 * | Jul 20, 1983 | Jun 18, 1985 | Beecham Group P.1.C. | β-Lactam compounds |
| US6465428 * | Aug 25, 2000 | Oct 15, 2002 | Aventis Pharma S.A. | Pharmaceutical combinations based on dalfopristine and quinupristine, and on cefepime |
| US20040254381 * | Aug 15, 2003 | Dec 16, 2004 | Day Richard A. | Antibiotic compositions and methods of using the same |
| US20050148571 * | Nov 29, 2002 | Jul 7, 2005 | Nancy Niconovich | Method of treating bacterial infections using gemifloxacin or a salt thereof and a betha-Lactam antibiotic |
| US20090148512 * | Apr 17, 2008 | Jun 11, 2009 | Lannett Co Inc | Novel uses of chloramphenicol and analogous thereof |
| US20090232744 * | Feb 26, 2009 | Sep 17, 2009 | Pari Pharma Gmbh | Macrolide compositions having improved taste and stability |
| WO2002009758A2 * | 31 Jul 2001 | 7 Feb 2002 | Satish B Bhawsar | Inhibitors of cellular efflux pumps of microbes |
| US6750224 | 17 Aug 2000 | 15 Jun 2004 | Wockhardt Limited | Antibacterial optically pure benzoquinolizine carboxylic acids, processes, compositions and methods of treatment |
Mr Habil Khorakiwala, Chairman, Wockhardt Ltd.
///////////keywords USFDA, Qualified Infectious Disease Product status, Wockhardt, drugs, WCK 2349, QIDP
ORGANIC SPECTROSCOPY

Experimental Study on Holoptelia Integrifolia Planch. in Relation to Diabetes Mellitus Type 2

Experimental Study on Holoptelia Integrifolia in Relation to Diabetes Mellitus Type 2
http://www.ijpr.in/Data/Archives/2015/september/2407201501.pdf
see also
http://www.apjtb.com/zz/2012s2/130.pdf
Holoptelea integrifolia
|
Holoptelea integrifolia Planch. , Ann. Sci. Nat., Bot. III, 10: 259 1848. (Syn. Ulmus integrifolia Roxb.);
entire-leaved elm tree, jungle cork tree, south Indian elm tree • Bengali: নাটা করঞ্জা nata karanja • Gujarati: ચરલ charal, ચરેલ charel, કણઝો kanjho • Hindi: चिलबिल chilbil, कान्जू kanju, पपड़ी papri • Konkani: वांवळो vamvlo • Malayalam: ആവല് aaval • Marathi: ऐनसादडा ainasadada, वावळ or वावळा vavala • Nepalese: sano pangro • Oriya: dhauranjan • Sanskrit: चिरिविल्वः chirivilva • Tamil: ஆயா aya • Telugu: నాలి nali;
|
///////
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
