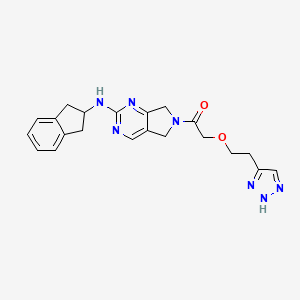
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Join me on Facebook
FACEBOOK
...................................................................Join me on twitter
..................................................................Join me on google plus
Googleplus
MYSELF
DR ANTHONY MELVIN CRASTO Ph.D ( ICT, Mumbai)
, INDIA
36Yrs Exp. in the feld of Organic Chemistry,Working for AFRICURE PHARMA as ADVISOR earlier with GLENMARK PHARMA at Navi Mumbai, INDIA. Serving chemists around the world. Helping them with websites on Chemistry.Million hits on google,
NO ADVERTISEMENTS , ACADEMIC , NON COMMERCIAL SITE, world acclamation from industry, academia, drug authorities for websites, blogs and educational contribution, ........amcrasto@gmail.com..........+91 9323115463, Skype amcrasto64
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
The GDUFA (Generic Drug User Fee Amendments) is a legislative package which came into force in 2012 and entitles the US-American FDA to collect fees from generic drug manufacturers, who strive for a marketing authorisation for the American market. An annual fee has to be paid after the successful registration.
The core of the document is the obligation to “Self-Identify” for those companies that have to submit essential site-related information to the FDA. The details of this self-identification are set in a Guidance for Industry entitled “Self-Identification of Generic Drug Facilities, Sites, and Organizations” published on 22 September 2016 by the FDA in the finalised form.
The Guidance describes the following elements:
1. Which types of generic facilities, sites, and organizations are required to self-identify?
2. What information is requested?
3. What technical standards are to be used for electronically submitting the requested information?
4. What is the penalty for failing to self-identify?
Hereinafter, you will find a short summary of these four topics:
1. Companies that manufacture finished generic medicinal products for human use or the APIs for them, or both are required to self-identify as well as companies that package the finished generic drug into the primary container and label it. Besides, sites that – pursuant to a contract with the applicant (generic drug manufacturer) – repack/redistribute the finished drug from a primary container into a different primary container are also required to submit a self-identification as well as sites that perform bioequivalence/bioavailability studies. Last but not least, the obligation to self-identify also concerns sites that are listed in the application dossier as contract laboratories for the sampling and performing of analytical testing.
2. Essential data are: the D-U-N-S number (a unique nine-digit sequence specific for each site / each distinct physical location of an entity), the “Facility Establishment Identifier, FEI” (an identifier used by the FDA for the planning and tracking of inspections) and general information with regard to the facility (company owner, type of business operation, contact data, information about the manufacture of non generic drugs).
4. Companies that fail to self-identify do not have to expect an explicit penalty. However, such a failure leads to two drawbacks: first, the likelihood of a site inspection by the FDA prior to approval is higher. The second drawback which is much more serious is that all the APIs or finished drugs from a manufacturer who hasn’t self-identified are deemed misbranded. For the FDA, such products are not allowed for importation in the USA.
To the satisfaction of the FDA, the regulations set in the GDUFA and the provisions laid down in the new Guidance represent a major contribution to an enhanced transparency in particular of complex supply chains.
//////////GDUFA, FDA, new Guidance, Self-Identification, Generic Drug Manufacturers
Class Antivirals; Aza compounds; Cyclic ethers; Cyclopentanes; Cyclopropanes; Quinoxalines; Small molecules
Mechanism of Action Hepatitis C virus NS3 protein inhibitors
Phase II Hepatitis C
Most Recent Events
18 Apr 2016 Pooled efficacy and adverse event data from the phase II SURVEYOR-I and SURVEYOR-2 trials for Hepatitis C presented at The International Liver Congress™ 2016 (ILC-2016)
15 Apr 2016 Updated efficacy data from a phase II MAGELLAN 1 study were reported by Enanta Pharmaceuticals
15 Apr 2016 Updated safety and efficacy data from a phase II MAGELLAN 1 study were presented at the International Liver Congress™ (ILC-2016)
AbbVie’s investigational, pan-genotypic regimen of glecaprevir (ABT-493) / pibrentasvir (ABT-530) (G/P) has received breakthrough therapy designation from the US Food and Drug Administration (FDA) to treat chronic hepatitis C virus (HCV).
HCV is the principal cause of non-A, non-B hepatitis and is an increasingly severe public health problem both in the developed and developing world. It is estimated that the virus infects over 200 million people worldwide, surpassing the number of individuals infected with the human immunodeficiency virus (HIV) by nearly five fold. HCV infected patients, due to the high percentage of individuals inflicted with chronic infections, are at an elevated risk of developing cirrhosis of the liver, subsequent hepatocellular carcinoma and terminal liver disease. HCV is the most prevalent cause of hepatocellular cancer and cause of patients requiring liver transplantations in the western world.
There are considerable barriers to the development of anti-HCV therapeutics, which include, but are not limited to, the persistence of the virus, the genetic diversity of the virus during replication in the host, the high incident rate of the virus developing drug-resistant mutants, and the lack of reproducible infectious culture systems and small-animal models for HCV replication and pathogenesis. In a majority of cases, given the mild course of the infection and the complex biology of the liver, careful consideration must be given to antiviral drugs, which are likely to have significant side effects.
Only two approved therapies for HCV infection are currently available. The original treatment regimen generally involves a 3-12 month course of intravenous interferon-α (IFN-α), while a new approved second-generation treatment involves co-treatment with IFN-α and the general antiviral nucleoside mimics like ribavirin. Both of these treatments suffer from interferon related side effects as well as low efficacy against HCV infections. There exists a need for the development of effective antiviral agents for treatment of HCV infection due to the poor tolerability and disappointing efficacy of existing therapies.
In a patient population where the majority of individuals are chronically infected and asymptomatic and the prognoses are unknown, an effective drug would desirably possess significantly fewer side effects than the currently available treatments. The hepatitis C non-structural protein-3 (NS3) is a proteolytic enzyme required for processing of the viral polyprotein and consequently viral replication. Despite the huge number of viral variants associated with HCV infection, the active site of the NS3 protease remains highly conserved thus making its inhibition an attractive mode of intervention. Recent success in the treatment of HIV with protease inhibitors supports the concept that the inhibition of NS3 is a key target in the battle against HCV.
HCV is a flaviridae type RNA virus. The HCV genome is enveloped and contains a single strand RNA molecule composed of circa 9600 base pairs. It encodes a polypeptide comprised of approximately 3010 amino acids.
The HCV polyprotein is processed by viral and host peptidase into 10 discreet peptides which serve a variety of functions. There are three structural proteins, C, E1 and E2. The P7 protein is of unknown function and is comprised of a highly variable sequence. There are six non-structural proteins. NS2 is a zinc-dependent metalloproteinase that functions in conjunction with a portion of the NS3 protein. NS3 incorporates two catalytic functions (separate from its association with NS2): a serine protease at the N-terminal end, which requires NS4A as a cofactor, and an ATP-ase-dependent helicase function at the carboxyl terminus. NS4A is a tightly associated but non-covalent cofactor of the serine protease.
The NS3/4A protease is responsible for cleaving four sites on the viral polyprotein. The NS3-NS4A cleavage is autocatalytic, occurring in cis. The remaining three hydrolyses, NS4A-NS4B, NS4B-NS5A and NS5A-NS5B all occur in trans. NS3 is a serine protease which is structurally classified as a chymotrypsin-like protease. While the NS serine protease possesses proteolytic activity by itself, the HCV protease enzyme is not an efficient enzyme in terms of catalyzing polyprotein cleavage. It has been shown that a central hydrophobic region of the NS4A protein is required for this enhancement. The complex formation of the NS3 protein with NS4A seems necessary to the processing events, enhancing the proteolytic efficacy at all of the sites.
A general strategy for the development of antiviral agents is to inactivate virally encoded enzymes, including NS3, that are essential for the replication of the virus. Current efforts directed toward the discovery of NS3 protease inhibitors were reviewed by S. Tan, A. Pause, Y. Shi, N. Sonenberg, Hepatitis C Therapeutics: Current Status and Emerging Strategies, Nature Rev. Drug Discov. 1, 867-881 (2002).
The acid 1-6a (21 mg, 0.0356 mmol) was dissolved in DCM (1.5 ml), and to this solution was added sulfonamide 1-7e (13.0 mg, 0.0463 mmol), HATU (17.6 mg, 0.0462 mmol) and DIPEA (12.4 ul, 0.0712 mmol). The mixture was stirred for 3 h, and then diluted with DCM. The organic layer was washed with 1 N HCl, water, brine, dried and concentrated in vacuo. The residue was purified by HPLC to afford the title compound. MS-ESI m/z 839.41 (M+H)+.
US FDA grants breakthrough therapy designation to AbbVie’s G/P to treat HCV
4 October 2016
AbbVie’s investigational, pan-genotypic regimen of glecaprevir (ABT-493) / pibrentasvir (ABT-530) (G/P) has received breakthrough therapy designation from the US Food and Drug Administration (FDA) to treat chronic hepatitis C virus (HCV).
The HCV is a bloodborne virus commonly transmitted through injecting drug use due to the sharing of injection equipment, reuse or inadequate sterilisation of medical equipment, and the transfusion of unscreened blood and blood products.
The designation facilitates the use of AbbVie’s G/P to treat chronic HCV patients who failed previous therapy with direct-acting antivirals (DAAs) in genotype 1 (GT1), including therapy with an NS5A inhibitor and / or protease inhibitor.
AbbVie research and development executive vice-president Michael Severino said: “AbbVie is committed to advancing HCV care and addressing areas of continued unmet need for people living with chronic HCV.
“AbbVie is committed to advancing HCV care and addressing areas of continued unmet need for people living with chronic HCV.”
“The FDA’s breakthrough therapy designation is an important step in our effort to bring our pan-genotypic regimen to market, which we are also investigating as an eight-week path to virologic cure for the majority of patients.”
AbbVie said that G/P is currently in Phase III trials evaluating the safety and efficacy of the regimen across all major HCV genotypes (genotypes 1-6).
Figures released by the World Health Organisation revealed that an estimated 700,000 people die each year from hepatitis C-related liver diseases.
There is currently no vaccine for hepatitis C, although research in this area is underway at present.
Vadadustat, also known as AKB-6548 and PG-1016548, is a potent Hypoxia-inducible factor-proline dioxygenase inhibitor. AKB-6548 works by inhibiting hypoxia inducible factor-prolyl hydroxylase (HIP-PH), leading to stabilization and increased levels of HIFα. In turn HIFα improves production of hemoglobin and red blood cells (RBCs), while maintaining normal levels of erythropoietin (EPO) in patients. We believe this differentiated mechanism of action has the potential to be safer than that of injectable recombinant erythropoietin stimulating agents (rESAs), avoiding supra-physiological levels of EPO and saturation of EPO receptors for prolonged periods of time.
Akebia Therapeutics, under license from Procter & Gamble, and sublicensee Mitsubishi Tanabe Pharma are developing vadadustat, an orally active small-molecule hypoxia-inducible factor prolyl hydroxylase (HIF-PH) inhibitor that stabilize HIF2-α, as a once-daily formulation, for treating anemia. Also the company is investigating AKB-6899, an oral HIF-PH inhibitor, for treating cancer and ocular diseases. In March 2016, the IND application was opened. Aerpio Therapeutics, a spinoff of Akebia, is investigating AKB-4924, a HIF2-α stabilizer, which inhibits HIF prolyl hydroxylase-2, for treating inflammatory bowel disease and wound healing
Hypoxia-inducible factor (HIF) is a transcription factor that is a key regulator of responses to hypoxia. In response to hypoxic conditions, i.e., reduced oxygen levels in the cellular environment, HIF upregulates transcription of several target genes, including those encoding erythropoietin. HIF is a heteroduplex comprising an alpha and beta subunit. While the beta subunit is normally present in excess and is not dependent on oxygen tension, the HIF-alpha subunit is only detectable in cells under hypoxic conditions. In this regard, the accumulation of HIF-alpha is regulated primarily by hydroxylation at two proline residues by a family of prolyl hydroxylases known as HIF prolyl hydroxylases, wherein hydroxylation of one or both of the proline residues leads to the rapid degradation of HIF-alpha. Accordingly, inhibition of HIF prolyl hydroxylase results in stabilization and accumulation of HIF-alpha {i.e., the degradation of HIF-alpha is reduced), thereby leading to an increase in the amount of HIF-alpha available for formation of the HIF heterodimer and upregulation of target genes, such as the Erythropoietin gene. Conversely, activation of HIF prolyl hydroxylase results in destabilization of HIF-alpha {i.e., the degradation of HIF-alpha is increased), thereby leading to a decrease in the amount of HIF-alpha available for formation of the HIF heterodimer and downregulation of target genes, such as VEGF.
The family of hypoxia inducible factors includes HIF- 1 -alpha, HIF-2-alpha, and HIF-3 -alpha.
A new class of prolyl hydroxylase inhibitors and their use to treat or prevent diseases ameliorated by modulation of hypoxia-inducible factor (HIF) prolyl hydroxylase are described in U.S. Patent No. 7,811,595, which is incorporated herein by reference in its entirety. The synthesis of such prolyl hydroxylase inhibitors is described in U.S. Patent Publication No.2012/0309977, which is incorporated herein by reference in its entirety. Such compounds inhibit HIF prolyl hydroxylase, thereby stabilizing HIF-alpha. As a consequence of stabilizing HIF-alpha, endogenous erythropoietin (EPO) production is increased. As with all drugs, proper doses and dosing regimens for treating patients having diseases such as anemia are essential for achieving a desired or optimal therapeutic effect without adverse effects or unwanted side-effects. Indeed, many active compounds fail in clinical trials because an effective and safe dosing regimen cannot be found.
Vadadustat (also known as AKB-6548) in anemia secondary to chronic kidney disease (CKD)
We are developing our lead product candidate, vadadustat, to be the potential best-in-class hypoxia inducible factor–prolyl hydroxylase inhibitor for the treatment of anemia secondary to CKD.
HIF inhibitor Vadadustat (Code AKB-6548) The chemical name N- [5- (3- chlorophenyl) -3-hydroxypyridine-2-carbonyl] glycine,
Vadadustat is a treatment for anemia associated with chronic kidney disease oral HIF inhibitor, is an American biopharmaceutical company Akebia Therapeutics invention in the research of new drugs, has completed Phase II pivotal clinical trial treatment studies, successfully met the researchers set given the level of hemoglobin in vivo target and good security, a significant effect, and phase III clinical trials.
U.S. Patent Publication US20120309977 synthetic route for preparing a Vadadustat: A 3-chlorophenyl boronic acid and 3,5_-dichloro-2-cyanopyridine as starting materials, by-catalyzed coupling methoxy substituted, cyano hydrolysis and condensation and ester hydrolysis reaction Vadadustat, process route is as follows:
Since the entire synthetic route 12 steps long, complicated operation, high cost.U.S. Patent No. 1 2 ^ ¥ disclosed 20070299086 & (^ (Scheme 3 1118 seven seven to 3,5-dichloro-2-cyanopyridine starting material, first-dichloro substituted with benzyloxy, then cyano hydrolysis, condensation, hydrogenation and deprotection trifluorosulfonyl, to give N- [5- trifluoromethanesulfonyloxy-3-hydroxypyridine-2-carbonyl) glycine methyl ester, 3-chlorophenyl and then boronic acid catalyzed coupling reactions, the final ester hydrolysis reaction Vadadustat, process route is as follows:
The synthesis steps long, intermediate products and final products contain more impurities and byproducts, thus purified requires the use of large amounts of solvents, complicated operation, low yield, and because the hydrogenation reaction is a security risk on the production, not conducive to the promotion of industrial production, it is necessary to explore a short process, simple operation, low cost synthetic method whereby industrial production Vadadus tat fit.
Example 1
A) Preparation of N- (3,5_-dichloro-2-carbonyl) glycine methyl ester:
3,5-dichloro-2-pyridinecarboxylic acid (19.2g, 0.10mol) and N, N’_ carbonyldiimidazole (24.3g, 0.15mol) was dissolved in N, N- dimethylformamide (100 mL ), was added glycine methyl ester hydrochloride (15.18,0.12111〇1), 11 was added dropwise diisopropylethylamine (51.7g, 0.40mol), the reaction mixture was stirred 35 ° C for 8 hours, TLC determined the completion of reaction gussets The reaction solution was concentrated by rotary evaporation to dryness, dilute hydrochloric acid was adjusted to neutral by adding ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, and recrystallized from methanol to give N- (3,5- dichloro-pyridin-2 – carbonyl) glycine methyl ester, an off-white solid (21.6g), a yield of 82.0%, this reaction step is as follows:
1234567 B) Preparation of N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester: 2
1 (3,5-dichloro-2-carbonyl) glycine methyl ester (20 (^, 〇1 76111111), 3-chlorophenyl boronic acid (13.18, 3 83.7mmol), [l, l’- bis (diphenylphosphino) ferrocene] dichloropalladium (2.8g, 3.8mmol), potassium carbonate (14.2g, 4 0. lmo 1) and N, N- dimethylformamide (75mL) was added The reaction flask, the reaction mixture was heated to 60 ° C for 20 hours the reaction was stirred for 5:00, point TLC plates to determine completion of the reaction, the reaction solution was cooled to room temperature, was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, washed with brine, sulfuric acid 6 magnesium dried and concentrated by rotary evaporation to dryness, a mixed solvent of ethyl acetate and n-hexane was recrystallized to give N- [5- (3- chlorophenyl) -3-7-chloro-2-carbonyl] glycine methyl ester, white solid (19.7g), yield 76.4%, this reaction step is as follows:
C) Preparation of N_ [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine:
N- [5- (3- chlorophenyl) -3-chloropyridine-2-carbonyl] glycine methyl ester (19 (^, 56111 111〇1) and sodium methoxide (7.6g, 0.14mol) was dissolved in methanol (150 mL), the reaction mixture was heated to 65 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution was cooled to room temperature, water (300mL) was stirred for 3h, cooled to 0 ° C, stirred for 2h, precipitated solid was filtered, the filter cake was dried to give N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine, off-white solid (17.4 g of), a yield of 96.5%, of the reaction steps are as follows:
D) Preparation Vadadustat:
N- [5- (3- chlorophenyl) -3-methoxy-pyridine-2-carbonyl] glycine (16.68,51.7111111〇1) and 48% hydrobromic acid solution (52mL, 0.46mol) added to the reaction bottle, the reaction mixture was heated to 100 ° C, the reaction was stirred at reflux for 24 hours, TLC determined gussets completion of the reaction the reaction solution cooled square ~ 5 ° C, was slowly added 50% sodium hydroxide solution was adjusted to pH 2 at 0 -5 ° C under crystallization 3h, the filter cake washed with ethyl acetate and n-hexane mixed solvent of recrystallization, in finished Vadadustat, off-white solid (15.6g), a yield of 98.0%, this reaction step is as follow
Lanthier et al. (U.S. Patent Application 2012/0309977) described a procedure for synthesizing a compound of Formula (II) starting from 3-chloroboronic acid and 3,5-dichloropicolinonitrile, as shown in the scheme below:
which has an X-ray powder diffraction pattern as shown in FIG. 1. In certain embodiments, Form A of Compound (I) has an X-ray powder diffraction pattern comprising one, two, three, four, or five peaks at approximately 18.1 , 20.3, 22.9, 24.0, and 26.3 °2Θ; and wherein the crystalline Compound (I) is substantially free of any other crystalline form of Compound (I).
Compound (I) as prepared according to e.g., U.S. 7,811,595 and/or U.S. Patent Application No. 13/488,554 and then subjecting the resulting Compound (I)
(I),
to a procedure comprising
a) preparing a solution of Compound (I) in 2-methyltetrahydrofuran;
b) adding n-heptane;
c) heating the suspension {e.g., to about 40-50 °C);
d) cooling the suspension {e.g., to about 0-10 °C); and
c) isolating the crystals.
SYNTHESIS
US 2015361043
Synthesis of vadadustat and its intermediates is described. The process involves Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile with (3-chlorophenyl)boronic acid, selective chloride displacement, simultaneous hydrolysis of nitrile and methyl ether, activation with CDI, condensation with methyl glycinate hydrochloride and finally ester hydrolysis. The process is simple and provides high product yield with high quality. Vadadustat is expected to be useful for the treatment of renal failure anemia (1). Suzuki coupling of 3,5-dichloropyridine-2-carbonitrile (I) with (3-chlorophenyl)boronic acid (II) in the presence of PdCl2(dppf) and K2CO3 in DMF yields 3-chloro-5-(3-chlorophenyl)pyridine-2-carbonitrile (III), which upon selective chloride displacement with NaOMe in refluxing MeOH affords methyl ether (IV). Hydrolysis of nitrile and methyl ether in intermediate (IV) with HBr or HCl at 100 °C furnishes 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (V). After activation of carboxylic acid (V) with CDI or pivaloyl chloride and DIEA in DMSO, condensation with methyl glycinate hydrochloride (VI) in the presence of DIEA provides vadadustat methyl ester (VII). Finally, hydrolysis of ester (VII) with NaOH in H2O/THF produces the target vadadustat (1).
FIG. 1 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitors.
FIG. 2 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor ester prodrugs.
FIG. 3 depicts an outline of one embodiment for preparing the disclosed prolyl hydroxylase inhibitor amide prodrugs.
Example 1 describes a non-limiting example of the disclosed process for the preparation of a prolyl hydroxylase ester pro-drug
EXAMPLE 1Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): To a 100 mL round bottom flask adapted for magnetic stirring and equipped with a nitrogen inlet was charged (3-chlorophenyl)boronic acid (5 g, 32 mmol), 3,5-dichloro-2-cyanopyridine (5.8 g, 34 mmol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (0.1 g, 0.13 mmol), dimethylformamide (50 mL) and water (5 mL). The reaction solution was agitated and heated to 45° C. and held at that temperature for 18 hours after which the reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to room temperature and the contents partitioned between ethyl acetate (250 mL) and saturated aqueous NaCl (100 mL). The organic phase was isolated and washed a second time with saturated aqueous NaCl (100 mL). The organic phase was dried for 4 hours over MgSO4, the MgSO4 removed by filtration and the solvent removed under reduced pressure. The residue that remained was then slurried in methanol (50 mL) at room temperature for 20 hours. The resulting solid was collected by filtration and washed with cold methanol (50 mL) then hexanes (60 mL) and dried to afford 5.8 g (73% yield) of an admixture containing a 96:4 ratio of the desired regioisomer. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H)
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): To a 500 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (10 g, 40 mmol), sodium methoxide (13.8 mL, 60 mmol) and methanol (200 mL). With stirring, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to room temperature and combined with water (500 mL). A solid began to form. The mixture was cooled to 0° C. to 5° C. and stirred for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 9.4 g (96% yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a reflux condenser was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (1 g, 4 mmol) and a 48% aqueous solution of HBr (10 mL). While being stirred, the reaction solution was heated to reflux for 20 hours. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction contents was then cooled to 0° C. to 5° C. with stirring and the pH was adjusted to approximately 2 by the slow addition of 50% aqueous NaOH. Stirring was then continued at 0° C. to 5° C. for 3 hours. The resulting solid was collected by filtration and washed with water, then hexane. The resulting cake was dried in vacuo at 40° C. to afford 1.03 g (quantitative yield) of the desired product as an off-white solid. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): To a 50 mL round bottom flask adapted for magnetic stirring and fitted with a nitrogen inlet tube was charged 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (1 gm, 4 mmol), N,N′-carbonyldiimidazole (CDI) (0.97 g, 6 mmol) and dimethyl sulfoxide (5 mL). The reaction mixture was stirred at 45° C. for about 1 hour then cooled to room temperature. Glycine methyl ester hydrochloride (1.15 g, 12 mmol) is added followed by the dropwise addition of diisopropylethylamine (3.2 mL, 19 mmol). The mixture was then stirred for 2.5 hours at room temperature after which water (70 mL) was added. The contents of the reaction flask was cooled to 0° C. to 5° C. and 1N HCl was added until the solution pH is approximately 2. The solution was extracted with dichloromethane (100 mL) and the organic layer was dried over MgSO4 for 16 hours. Silica gel (3 g) is added and the solution slurried for 2 hours after which the solids are removed by filtration. The filtrate is concentrated to dryness under reduced pressure and the resulting residue was slurried in methanol (10 mL) for two hours. The resulting solid was collected by filtration and washed with cold methanol (20 mL) then hexane and the resulting cake is dried to afford 0.85 g of the desired product as an off-white solid. The filtrate was treated to afford 0.026 g of the desired product as a second crop. The combined crops afford 0.88 g (68% yield) of the desired product. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use
EXAMPLE 2Methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4)
Preparation of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine (1): A 20 L reactor equipped with a mechanical stirrer, dip tube, thermometer and nitrogen inlet was charged with (3-chlorophenyl)boronic acid (550 g, 3.52 mol), 3,5-dichloro-2-cyanopyridine (639 g, 3.69 mol), K2CO3 (5.5 g, 40 mmol), [1,1′-bis(diphenyphosphino)ferrocene]dichloro-palladium(II) [PdCl2(dppf)] (11.5 g, 140 mmol), and dimethylformamide (3894 g, 4.125 L). The reaction solution was agitated and purged with nitrogen through the dip-tube for 30 minutes. Degassed water (413 g) was then charged to the reaction mixture while maintaining a temperature of less than 50° C. 25 hours. The reaction was determined to be complete due to the disappearance of 3,5-dichloro-2-cyanopyridine as measured by TLC analysis using ethyl acetate/methanol (4:1) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction solution was then cooled to 5° C. and charged with heptane (940 g, 1.375 L) and agitated for 30 minutes. Water (5.5 L) was charged and the mixture was further agitated for 1 hour as the temperature was allowed to rise to 15° C. The solid product was isolated by filtration and washed with water (5.5 L) followed by heptane (18881 g, 2750 ML). The resulting cake was air dried under vacuum for 18 hours and then triturated with a mixture of 2-propanol (6908 g, 8800 mL0 and heptane (1 g, 2200 mL0 at 50° C. for 4 hours, cooled to ambient temperature and then agitated at ambient temperature for 1 hour. The product was then isolated by filtration and washed with cold 2-propanol (3450 g, 4395 mL) followed by heptane (3010 g, 4400 mL). The resulting solid was dried under high vacuum at 40° C. for 64 hours to afford 565.9 g (65% yield) of the desired product as a beige solid. Purity by HPLC was 98.3. 1H NMR (DMSO-d6) δ 9.12 (d, 1H), 8.70 (d, 1H), 8.03 (t, 1H) 7.88 (m, 1H), and 7.58 (m, 2H).
Preparation of 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine (2): A 20 L reactor equipped with a mechanical stirred, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine, 1, (558 g, 2.24 mol) and sodium methoxide (25% solution in methanol, 726.0 g, 3.36 mol). With agitation, the reaction solution was heated to reflux for 24 hours, resulting in a beige-colored suspension. The reaction was determined to be complete due to the disappearance of 5-(3-chlorophenyl)-3-chloro-2-cyanopyridine as measured by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components. The reaction mixture was cooled to 5° C. and then charged with water (5580 mL). The resulting slurry was agitated for 3 hours at 5° C. The solid product was isolated by filtration and washed with water (5580 mL) until the filtrate had a pH of 7. The filter cake was air dried under vacuum for 16 hours. The filter cake was then charged back to the reactor and triturated in MeOH (2210 g, 2794 mL) for 1 hour at ambient temperature. The solid was collected by filtration and washed with MeOH (882 g, 1116 mL, 5° C.) followed by heptane (205 mL, 300 mL), and dried under high vacuum at 45° C. for 72 hours to afford 448 g (82% yield) of the desired product as an off-white solid. Purity by HPLC was 97.9%. 1H NMR (DMSO-d6) δ 8.68 (d, 1H), 8.05 (d, 1H), 8.01 (s, 1H) 7.86 (m, 1H), 7.59 (s, 1H), 7.57 (s, 1H) and 4.09 (s, 3H).
Preparation of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid (3): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer, nitrogen inlet and 25% aqueous NaOH trap was charged 5-(3-chlorophenyl)-3-methoxy-2-cyanopyridine, 2, (440.6 g, 1.8 mol) and 37% aqueous solution of HCl (5302 g). While being agitated, the reaction solution was heated to 102° C. for 24 hours. Additional 37% aqueous HCl (2653 g) was added followed by agitation for 18 hours at 104° C. The reaction contents was then cooled to 5° C., charged with water (4410 g) and then agitated at 0° C. for 16 hours. The resulting precipitated product was isolated by filtration and washed with water until the filtrate had a pH of 6 (about 8,000 L of water). The filter cake was pulled dry under reduced pressure for 2 hours. The cake was then transferred back into the reactor and triturated in THF (1958 g, 2201 mL) at ambient temperature for 2 hours. The solid product was then isolated by filtration and washed with THF (778 g, 875 mL) and dried under reduced pressure at 5° C. for 48 hours to afford 385 g (89% yield) of the desired product as an off-white solid. HPLC purity was 96.2%. 1H NMR (DMSO-d6) δ 8.52 (d, 1H), 7.99 (d, 1H), 7.95 (s, 1H) 7.81 (t, 1H), 7.57 (s, 1H), and 7.55 (s, 1H).
Preparation of methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetate (4): A 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged with 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (380 g, 1.52 mol) and diisopropylethylamine (DIPEA) (295 g, 2.28 mol). With agitation, the solution was cooled to 3° C. and charged with trimethylacetyl chloride (275.7 g, 2.29 mol) while maintaining a temperature of less than 11° C., The mixture was then agitated at ambient temperature for 2 hours. The mixture was then cooled to 10° C. and charged with a slurry of glycine methyl ester HCl (573.3 g, 4. 57 mol) and THF (1689 g, 1900 mL), then charged with DIPEA (590.2 g, 4.57 mol) and agitated at ambient temperature for 16 hours. The mixture was then charged with EtOH (1500 g, 1900 mL) and concentrated under reduced pressure to a reaction volume of about 5.8 L. The EtOH addition and concentration was repeated twice more. Water (3800 g) was then added and the mixture was agitated for 16 hours at ambient temperature. The resulting solid product was isolated by filtration and washed with a mixture of EtOH (300 g, 380 mL) and water (380 g), followed by water (3800 g), dried under reduced pressure for 18 hours at 50° C. to afforded 443 g (91% yield) of the desired product as an off-white solid. Purity by HPLC was 98.9%. 1H NMR (DMSO-d6) δ 12.3 (s, 1H), 9.52 (t, 1H), 8.56 (d, 1H), 7.93 (s, 1H), 7.80 (q, 2H), 7.55 (t, 2H), 4.12 (d, 2H), and 3.69 (s, 3H).
Scheme II herein below outlines and Example 2 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase inhibitor from an ester prodrug.
EXAMPLE 3{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3 -chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 50 mL flask is charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (0.45 g, 1.4 mmol), tetrahydrofuran (4.5 mL) and 1 M NaOH (4.5 mL, 4.5 mmol). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was adjusted to pH 1 with concentrated HCl and the solution was heated at 35° C. under vacuum until all of the tetrahydrofuran had been removed. A slurry forms as the solution is concentrated. With efficient stirring the pH is adjusted to ˜2 with the slow addition of 1 M NaOH. The solid which forms was collected by filtration, washed with water, followed by hexane, then dried under vacuum to afford 0.38 g (88% yield) of the desired product as a white solid. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
The formulator can readily scale up the above disclosed synthesis. Disclosed herein below is a synthesis wherein the disclosed process is scaled up for commercial use.
EXAMPLE 4{[5-(3-Chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5)
Preparation of {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}acetic acid (5): To a 20 L reactor equipped with a mechanical stirrer, condenser, thermometer and nitrogen inlet was charged methyl {[5-(3-chlorophenyl)-3-hydroxypyridin-2-yl]amino}-acetate, 4, (440 g, 1.42 mol), tetrahydrofuran (3912 g, 4400 mL) and 1 M NaOH (4400 mL). The mixture was stirred for 2 hours at room temperature after which it was determined by TLC analysis using hexane/ethyl acetate (6:3) as the mobile phase and UV 435 nm to visualize the reaction components that the reaction was complete. The reaction solution was acidified to a pH of 2 with slow addition of 2M HCl (2359 g). The resulting mixture was concentrated under reduced pressure to a volume of about 7.5 L. Ware (2210 g) was added and the solution cooled to ambient temperature and agitated for 18 hours. The solid product was isolated by filtration and washed with water (6 L). the crude product was transferred back into the reactor and triturated with 2215 g o deionized water at 70° C. for 16 hours. The mixture was cooled to ambient temperature, The solid product was isolated by filtration and washed with water (500 mL) and dried under reduced pressure at 70° C. for 20 hours to afford 368 g (87% yield) of the desired product as an off-white solid. Purity by HPLC was 99.3%. 1H NMR (DMSO-d6) δ 12.84 (s, 1H), 12.39 (s, 1H), 9.39 (t, 1H), 8.56 (d, 1H), 7.94 (s, 1H), 7.81 (m, 2H), 7.55 (q, 2H), and 4.02 (d, 2H).
Scheme III herein below outlines and Example 3 describes a non-limiting example of the disclosed process for preparing a prolyl hydroxylase amide prodrug.
EXAMPLE 55-(3-Chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide
Preparation of 5-(3-chlorophenyl)-N-(2-amino-2-oxoethyl)-3-hydroxylpyridin-2-yl amide (6): To a solution of 5-(3-chlorophenyl)-3-hydroxypyridine-2-carboxylic acid, 3, (749 mg, 3 mmol) in DMF (20 mL) at room temperature under N2 is added 1-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide (EDCI) (0.925 g, 5.97 mmol) and 1-hydroxybenzo-triazole (HOBt) (0.806 g, 5.97 mmol). The resulting solution is stirred for 15 minutes then 2-aminoacetamide hydrochloride (0.66 g, 5.97 mmol) and diisopropylethylamine (1.56 ml, 8.96 mmol) are added. The reaction is monitored by TLC and when the reaction is complete the reaction mixture is concentrated under reduced pressure and H2O added. The product can be isolated by normal work-up: The following data have been reported for compound (6). 1H NMR (250 MHz, DMSO-d6) δ ppm 12.46 (1H, s), 9.17 (1H, t, J=5.9 Hz), 8.55 (1H, d, J=2.0 Hz), 7.93 (1H, d, J=0.9 Hz), 7.75-7.84 (2H, m), 7.49-7.60 (3H, m), 7.18 (1H, s), 3.91 (2H, d, J=5.9 Hz). HPLC-MS: m/z 306 [M+H]+.
Scheme IV herein below depicts a non-limiting example the hydrolysis of an amide pro-drug to a prolyl hydroxylase inhibitor after removal of a R10 protecting group
Beuck S, Schänzer W, Thevis M. Hypoxia-inducible factor stabilizers and other small-molecule erythropoiesis-stimulating agents in current and preventive doping analysis. Drug Test Anal. 2012 Nov;4(11):830-45. doi: 10.1002/dta.390. Epub 2012 Feb 24. Review. PubMed PMID: 22362605.
Solid forms of 2-(5-(3-fluorophenyl)-3-hydroxypicolinamido)acetic acid, compositions, and uses thereof
clip
Oct 6, 2015
Akebia Reaches Agreement with FDA and EMA on Vadadustat Global Phase 3 Program
Plans to Initiate Phase 3 PRO2TECT™ Clinical Program by Year-End
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Akebia Therapeutics, Inc. (NASDAQ: AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced the successful completion of the End-of-Phase 2 Meeting process with the United States Food and Drug Administration (FDA) and the Scientific Advice Process with the European Medicines Agency (EMA) for its lead product, vadadustat (formerly AKB-6548), for patients with anemia related to non-dialysis dependent chronic kidney disease (NDD-CKD). The company has reached agreement with both the FDA and EMA regarding key elements of the Phase 3 program, known as the PRO2TECT™ program, and expects to launch the program later this year.
The PRO2TECT™program includes two separate studies and will collectively enroll approximately 3,100 NDD-CKD patients across 500 sites globally. The correction study will address anemia patients not currently being treated with recombinant erythropoiesis stimulating agents (rESAs). The conversion study includes patients currently receiving rESA who will be converted to either vadadustat or the active control with the goal of maintaining their baseline hemoglobin levels. Both studies will include a 1:1 randomization and an open label, active-control, non-inferiority design. Primary endpoints include an efficacy assessment of the hemoglobin response and an assessment of cardiovascular safety measured by major adverse cardiovascular events.
“Akebia’s Phase 3 program is designed to provide the medical community and regulators with a clear understanding of vadadustat’s potential benefit and safety advantages over rESAs, the current standard of care worldwide and, with a positive outcome, to establish vadadustat as the best-in-class treatment option for patients with renal anemia,” stated John P. Butler, President and Chief Executive Officer of Akebia. “We are pleased that the regulators are in agreement regarding the importance of an active-control trial as this design is the most clinically relevant and commercially valuable, and will allow us the quickest path to full enrollment. We are now moving rapidly to launch these studies and advance our goal of bringing forward new treatment options for patients suffering from renal anemia.”
“This Phase 3 program builds on the positive data from our Phase 2 program in NDD-CKD patients which demonstrated that once-daily vadadustat can control and maintain hemoglobin levels in a clinically relevant range while minimizing fluctuations in hemoglobin levels that are associated with increased cardiovascular safety risks,” stated Brad Maroni, M.D., Chief Medical Officer at Akebia. “These two Phase 3 event-driven studies are designed to establish the safety and efficacy of vadadustat in the setting of contemporary clinical practice patterns, and support regulatory approvals globally.”
In addition, Akebia discussed with the FDA and EMA a parallel Phase 3 program, known as the INNO2VATE™ program, for vadadustat in patients with anemia related to chronic kidney disease who are undergoing dialysis (DD-CKD). Akebia expects to formalize its Phase 3 program in DD-CKD patients after presenting the results from its recently completed Phase 2 study to both regulatory agencies.
About Vadadustat (Formerly AKB-6548)
Vadadustat is an oral therapy currently in development for the treatment of anemia related to chronic kidney disease (CKD). Vadadustat is designed to stabilize HIF, a transcription factor that regulates the expression of genes involved with red blood cell (RBC) production in response to changes in oxygen levels, by inhibiting the hypoxia-inducible factor prolyl hydroxylase (HIF-PH) enzyme. Vadadustat exploits the same mechanism of action used by the body to naturally adapt to lower oxygen availability associated with a moderate increase in altitude. At higher altitudes, the body responds to lower oxygen availability with increased production of HIF, which coordinates the interdependent processes of iron mobilization and erythropoietin (EPO) production to increase RBC production and, ultimately, improve oxygen delivery.
As a HIF stabilizer with best-in-class potential, vadadustat raises hemoglobin levels predictably and sustainably, with a dosing regimen that allows for a gradual and controlled titration. Vadadustat has been shown to improve iron mobilization, potentially eliminating the need for intravenous iron administration and reducing the overall need for iron supplementation.
About Anemia Related to CKD
Approximately 30 million people in the United States have CKD, with an estimated 1.8 million of these patients suffering from anemia. Anemia results from the body’s inability to coordinate RBC production in response to lower oxygen levels due to the progressive loss of kidney function, which occurs in patients with CKD. Left untreated, anemia significantly accelerates patients’ overall deterioration of health with increased morbidity and mortality. Renal anemia is currently treated with injectable rESAs, which are associated with inconsistent hemoglobin responses and well-documented safety risks.
About Akebia Therapeutics
Akebia Therapeutics, Inc. is a biopharmaceutical company headquartered in Cambridge, Massachusetts, focused on delivering innovative therapies to patients with kidney disease through HIF biology. The company has completed Phase 2 development of its lead product candidate, vadadustat, an oral therapy for the treatment of anemia related to CKD in both non-dialysis and dialysis patients.
clip
Akebia Announces Positive Top-Line Results from its Phase 2 Study of Vadadustat in Dialysis Patients with Anemia Related to Chronic Kidney Disease
-Treatment with Vadadustat Successfully Maintained Mean Hemoglobin Levels Following Conversion from rESA Therapy-
-Vadadustat Demonstrated a Favorable Safety Profile with Once Daily and Three Times per Week Dosing-
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Akebia Therapeutics, Inc. (NASDAQ:AKBA), a biopharmaceutical company focused on delivering innovative therapies to patients with kidney disease through the biology of hypoxia inducible factor (HIF), today announced positive top-line results from its Phase 2 study of vadadustat (formerly AKB-6548) in dialysis patients with anemia related to chronic kidney disease (CKD). The study achieved its primary objective, indicating that vadadustat maintained stable hemoglobin (HGB) levels throughout the 16-week treatment period following conversion from recombinant erythropoiesis-stimulating agent (rESA) therapy. Vadadustat demonstrated a favorable safety profile with no drug-related serious adverse events and no deaths. The results highlight the potential of vadadustat, dosed either once daily or three times per week, to safely and predictably manage and sustain HGB levels in CKD patients undergoing dialysis.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy”
The open-label, multi-center, 94 patient study was designed to evaluate the ability of vadadustat to maintain hemoglobin levels in patients undergoing hemodialysis who were previously being treated with rESAs. Patients were assigned to one of three dose cohorts: once daily vadadustat at a starting dose of 300mg, once daily vadadustat at a starting dose of 450mg, or vadadustat three times per week in conjunction with the patient’s hemodialysis schedule at a starting dose of 450mg. The study achieved its primary endpoints of maintaining stable hemoglobin levels over 16 weeks of treatment in all three cohorts of patients converting from rESAs to vadadustat.
Vadadustat was well tolerated among patients in all three dose cohorts. Treatment-emergent adverse events (TEAEs) with vadadustat were balanced across the cohorts. Serious adverse events (SAEs) were reported in 13 subjects (13.8%), well within the expected range for this patient population. There were no drug-related SAEs and no deaths reported in the study.
“This study was a clear success, demonstrating the potential of vadadustat to effectively and safely treat anemia in dialysis patients switching from injectable rESA therapy,” said Brad Maroni, M.D., Chief Medical Officer at Akebia. “We are impressed with the consistency in hemoglobin levels across the duration of the study, which highlights the ability of vadadustat to control and maintain hemoglobin levels in this patient population. Furthermore, the results indicate that daily and three times per week dosing regimens are both viable options for patients on dialysis.”
John P. Butler, President and Chief Executive Officer of Akebia, stated, “These results further confirm vadadustat as a potential best-in-class anemia treatment for CKD patients, and reinforce our confidence in this product candidate as we advance toward our Phase 3 program. Adding these results to the 12 other clinical studies we have completed, we are confident in the potential for vadadustat to treat anemia in a broad array of patients with CKD. We are pleased to have successfully completed this stage of our drug development and look forward to initiating Phase 3 studies.”
Complete efficacy and safety data from this Phase 2 study will be presented at an upcoming medical meeting.
About the Phase 2 Study Design of Vadadustat in Dialysis Patients with Anemia Related to CKD
The Phase 2 multi-center, open-label study evaluated 94 patients over 16 weeks of treatment, at 20 dialysis centers in the United States, including an assessment of HGB response to the starting dose of vadadustat during the first 8 weeks, followed by an assessment of HGB response to algorithm-guided dose adjustments of vadadustat during the subsequent 8 weeks of treatment. The study enrolled three cohorts, each consisting of approximately 30 CKD patients with anemia undergoing dialysis who were switched from injectable rESA therapy to vadadustat. Patients in the first two cohorts received once daily doses of vadadustat, while patients in the third cohort received vadadustat three times per week in conjunction with their hemodialysis schedule.
Jump up^Gupta N, Wish JB. Hypoxia-Inducible Factor Prolyl Hydroxylase Inhibitors: A Potential New Treatment for Anemia in Patients With CKD. Am J Kidney Dis. 2017 Jun;69(6):815-826. . doi:10.1053/j.ajkd.2016.12.011. PMID28242135. Missing or empty |title= (help)
Jump up^Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM. Clinical Trial of Vadadustat in Patients with Anemia Secondary to Stage 3 or 4 Chronic Kidney Disease. Am J Nephrol. 2017;45(5):380-388. . doi:10.1159/000464476. PMID28343225. Missing or empty |title= (help)
Vadadustat (Vafseo). Vadadustat (28) is a hypoxia inducible factor prolyl hydroxylase (HIF-PH) inhibitor developed by Akebia Therapeutics. It was approved by the European Commission in April 2023, and recently also by the USFDA, for the treatment of symptomatic anemia associated with chronic kidney disease in adults receiving chronic maintenance dialysis. Vadadustat acts by inhibiting HIFPH, 214 which results in increases of endogenous erythropoietin production, red blood cell synthesis, and iron mobilization. 215 While a number of syntheses of vadadustat (28) have been published in previous patents 216−228 and a journal article, 229 Akebia Therapeutics has published two patents regarding the large-scale preparation of vadadustat (Scheme 52). 218,226 The key intermediate nitrile 28.3 could be accessed in two steps: the neat SNAr reaction between commercially available 2,3,5trichloropyridine (28.1) and 4-DMAP to generate pyridinium salt 28.2, followed by a second SNAr reaction of 28.2 with NaCN. The Suzuki coupling between 28.3 and 3-chlorophenyl boronic acid (28.4) gave the biaryl 28.5, and the subsequent SNAr reaction of 28.5 with NaOMe replaced the 3-chloro substitution on the pyridine ring with a methoxy group, generating intermediate 28.6. Global acidic hydrolysis of both methyl ether and nitrile group in 28.6 gave the 3 hydroxypicolinic acid 28.7. Treatment of 28.7 with DIPEA and excess pivaloyl chloride (PivCl) resulted in the formation of mixed anhydride 28.8 with concomitant acylation of the 3 hydroxy group. Without isolation of 28.8, glycine methyl ester hydrochloride (28.9) was then charged with additional DIPEA to generate the corresponding amide 28.10. The residual amount (∼0.5%) of 28.7 in 28.10 was hard to remove, but this impurity could be effectively rejected with an extra amount of DIPEA during workup and solvent switch. Finally, the Opivaloyl group and methyl ester were both removed via basic hydrolysis, giving vadadustat (28) in about 90% yield from 28.7.
REF
(215) Pergola, P. E.; Spinowitz, B. S.; Hartman, C. S.; Maroni, B. J.; Haase, V. H. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016, 90, 1115−1122. (216) Lanthier, C. M.; Gorin, B.; Oudenes, J.; Dixon, C. E.; Lu, A. Q.; Copp, J. D.; Janusz, J. M. Preparation of [(3-hydroxypyridine-2carbonyl)amino]alkanoic acids, esters and amides as prolyl hydroxylase inhibitors. US 20120309977, 2012. (217) Li, X.; Chen, J. Process for the preparation of vadadustat. CN105837502, 2016. (218) Gorin, B. I.; Lanthier, C. M.; Luong, A. B. C.; Copp, J. D.; Gonzalez, J. Process for preparing 2-[[5-(3-chlorophenyl)-3-hydroxypyridine-2-carbonyl]amino]acetic acid. WO 2019217550, 2019. (219) Kou, J.; Li, Y.; Xiao, Q.; Lin, B.; Sun, J.; Wang, Z.; Luo, Z.;Huang, F. Preparation method of vadadustat. CN 110903238, 2020. (220) Machida, K.; Yasukouchi, H.; Nishiyama, A. Method for producing vadadustat intermediate. WO 2020217733, 2020. (221) Xiao, Q.; Lin, B.; Kou, J.; Sun, J.; Qiu, X.; Wang, Z.; Luo, Z.;Huang, F. Preparation of vadadustat intermediate. CN 111848505,2020
(222) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Li, Y.; Sun, J.; Jin, L.; Luo, Z.; Huang, F. Preparation of vadadustat and intermediate thereof. CN 111205222, 2020. (223) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Luo, Z.; Huang, F.; Li, Y. Preparation of vadadustat and intermediate thereof. CN 111423367, 2020. (224) Xiao, Q.; Qiu, X.; Lin, B.; Kou, J.; Li, Y.; Sun, J.; Wang, Z.; Luo, Z.; Huang, F. Preparation of vadadustat. CN 111320577, 2020. (225) Xiao, Q.; Lin, B.; Wang, Z.; Kou, J.; Qiu, X.; Cai, X.; Li, Y.; Luo, Z.; Huang, F. Method for preparing vadadustat and intermediate thereof. WO 2021179540, 2021. (226) Jurkauskas, V.; Jung, Y. C.; Kwon, T.; Kannan, A.; Gondi, V. B. Manufacturing process for 3,5-dichloropicolinonitrile for synthesis of vadadustat. WO 2022006427, 2022. (227) Chen, Z.; Zheng, Y.; Zhang, L.; Yu, C.; Liu, L.; He, B. Preparation of a pyridine compound used for the preparation of vadadustat. CN 117843565, 2024. (228) Patel, K. R.; Thakrar, V. H.; Mehta, T. B.; Wagh, A. G.; Patel, J. A.; Patil, R. R.; Solanki, Y. U.; Ladumor, C. B. A process for the preparation of Vadadustat or salts thereof. WO 2024079708, 2024. (229) Lin, B. Y.; Kou, J. P.; Wu, S. M.; Cai, X. R.; Xiao, Q. B.; Li, Y. L.; Hu, J.; Li, J. B.; Wang, Z. Q. Development of a robust and scalable process for the large-scale preparation of Vadadustat. Org. Process Res. Dev. 2021, 25, 960−968.
.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Generalised anxiety disorder; Major depressive disorder
Most Recent Events
10 May 2016Discontinued – Phase-I for Generalised anxiety disorder in USA, Japan (PO)
10 May 2016Discontinued – Phase-I for Major depressive disorder in USA, Japan (PO)
30 Jul 2015Tedatioxetine is still in phase I trials for Major depressive disorders and Generalised anxiety disorder in USA and Japan
Tedatioxetine (Lu AA24530) is an antidepressant that was discovered by scientists at Lundbeck; in 2007 Lundbeck and Takedaentered into a partnership that included tedatioxetine but was focused on another, more advanced Lundbeck drug candidate,vortioxetine.[1]
Tedatioxetine chemical name 4- (2- (4-methylphenyl group)) phenylpiperidine by Lundbeck developed for the treatment of severe depression, it is a monoamine reuptake inhibitor, a monoamine reuptake transporter inhibitors, 5-HT3 antagonists and 5-HT2c receptor antagonist. For the treatment of major depressive disorder and generalized anxiety, II clinical study in. Tedatioxetine has the following structure:
According to the literature, the current synthesis routes are the following:
WO 2003/029232 discloses Tedatioxetine first preparation method, as shown in the following Scheme,
The method of low yield, the product is not easy purification by column chromatography requires; more important is the preparation of the compound N-Boc- piperidin-4-ol of the need to use butyl lithium, and reaction was carried out at lower temperatures, not conducive to industrial production.
WO 2009109541 provides a, as shown in the above-described method for improved routes following synthetic route,
Bn- replaced with Boc-, dehydroxylation switch to TFA and Et 3 of SiH, yield improved despite increased. But there are many shortcomings.Deficiencies mainly reflected in the following aspects: the compound used in the expensive starting 2-bromo benzene iodine source and a catalyst of palladium and a bidentate phosphine ligand 3, an increase of production cost; preparation of compound needed 4:00 butyl lithium reagent to the more dangerous, the need at a low temperature reaction. This will bring in the production of a big security risk, is not conducive to the operation; when dehydroxylation
Preparation of 2- (4-methyl-phenyl mercapto) phenylpiperidine hydrobromide, to use a lot of trifluoroacetate (15eq), post-processing is too much trouble and the environment have a greater pollution.
Given 4- [2- (4-methylphenyl) phenyl] piperidine and salts thereof possess excellent pharmacological properties, and deficiencies of the prior processes, is necessary to develop a suitable industrial production, easy to operate and environmentally friendly preparation process.
2- (4-methyl-phenylthio) benzaldehyde prepared as in Example 1
Direction of Na 2 CO. 3 stirred mixture (11g, 105mmol) and 30mlDMF added 4-methyl-thiophenol (12.4g, 100mmol), stirred for 20 minutes. To the mixture was slowly added 2-bromobenzaldehyde (18.4g, 100mmol); a pending completion of the addition, under nitrogen, was heated to 100 deg.] C for 6 hours. After completion of the reaction, the reaction solution was cooled to room temperature, 100ml of water was added and stirred for 30 minutes. Filtered, washed with water (30ml) and dried in vacuo to give the filter cake was washed with 20.5g pale green solid; After n-hexane to give 18.5g pale yellow solid was recrystallized from 2- (4-phenylthio) benzaldehyde (mp: 52- 54 ℃), 81% yield. 2- (4-methyl-phenylthio) benzaldehyde Example 2 Preparation of
To the K 2 CO. 3 stirred mixture (15g, 110mmol) and 30mlDMA added 4-methyl-thiophenol (12.6g, 102mmol), stirred for 20 minutes. To the mixture was slowly added 2-chlorobenzaldehyde (14g, 100mmol); a pending completion of the addition, under nitrogen, the reaction was heated to 100 deg.] C for 7 hours. After completion of the reaction, the reaction solution was cooled to room temperature, 100ml of water was added and stirred for 30 minutes. Filtered, washed with water (30ml) and dried in vacuo to give the filter cake was washed with 19.7g pale green solid; After n-hexane to give 17g as a pale yellow solid was recrystallized from 2- (4-phenylthio) benzaldehyde (melting point: 51-53 ℃), a yield of 77.5%
2- (4-methyl-phenylthio) benzaldehyde Example 3 Preparation of
Ask NaOH (4.2g, 105mmol) and stirred 50ml 1,4-dioxane was added 4-methyl-thiophenol (12.4g, 100mmol), stirred for 30 minutes. To the mixture was slowly added 2-iodo-benzaldehyde (23.1g, 100mmol); a pending completion of the addition, under nitrogen, was heated under reflux for 5 hours.After completion of the reaction, the reaction solution was cooled to room temperature, 50ml of water was added, extraction separated; the organic phase was washed with 50ml of ethyl acetate, and the combined organic phases were washed with 20% aqueous ammonium chloride solution and saturated brine, dried over anhydrous magnesium sulfate, filtration and concentration gave 21g viscous liquid, and cooled to solidify; after n-hexane to give 18.1g pale yellow solid was recrystallized from 2- (4-phenylthio) benzaldehyde (m.p.: 53-54 ℃), close rate of 79%.
Example 4 Preparation of 3- [2 (4-methyl) phenyl] pentanedioic acid
1) Preparation of ethyl-2-cyano-3- (2- (4-methyl) phenyl) acrylate
2- (4-methylphenyl thio) benzaldehyde (4g, 17.5mmol), ethyl cyanoacetate (2.4g 21mmol) and toluene (30ml) was added a mixture of glacial acetic acid (5ml) and piperidine (0.3 ml of) stirred for 10 minutes; heated to reflux, and isolating the resulting water trap. Completion of the reaction, cooled to room temperature; the reaction was washed with 30ml water and 30ml saturated sodium bicarbonate solution, dried over anhydrous magnesium sulfate; filtered, and concentrated to give 5.0g yellow liquid (solidifies on cooling), yield 86%. It was used directly in the next reaction without purification.
2) Preparation of Diethyl 2,4-diethyl-3- (2- (4-methyl) phenyl) glutarate
Sodium methoxide (1.9g, 35mmol) and dry THF (30ml) was stirred and cooled to mix 0-5 ℃, was added dropwise diethyl malonate (4.6g, 35mmol), stirred for 15 minutes at room temperature dropwise Bi; dropwise obtained above in step 2-cyano-3- (2- (4-methyl) phenyl) acrylate (5g, 15.4mmol) and dry tetrahydrofuran (40ml) solution; BI dropwise, at room temperature stirred for 13 hours. Completion of the reaction, the reaction mixture was added 150ml20% aqueous ammonium chloride solution, followed by extraction separated; the aqueous phase was extracted with ethyl acetate, the combined organic phase was dried over anhydrous magnesium sulfate; filtered, and concentrated to give 5.4 g of a viscous liquid, yield 78%. It was used directly in the next reaction without purification.
Was added 6N hydrochloric acid (70ml), was heated at reflux for 3 days the material obtained in the above step (5.4 g of); completion of the reaction, slowly cooled to room temperature, added 50ml of ethyl acetate, stirred for 30 minutes to precipitate a solid from the solution, filtered and washed with 20ml washed with ethyl acetate, and dried in vacuo at 50 ℃ 10 hours to give 2.7g of white solid 3- [2 (4-methylphenyl) phenyl] glutaric acid (melting point: 191-195 ℃), in 58% yield.
Example 5 Preparation of 3- [2- (4-phenylthio) phenyl] pentanedioic acid
To ethyl acetoacetate (13g, 100mmol) and piperidine (1.7g, 10mmol) was added a mixture of 2- (4-methyl-phenylthio) benzaldehyde (11.5g, 50mmol), room temperature for 1 day to give a yellow viscous semi-solid, 2.7g of sodium methoxide was added. after stirring for 1 hour cure, stand for 2 days.To the above mixture was added ethanol (180ml) and 40% aqueous sodium hydroxide (140ml) was stirred and heated to reflux for 4-5 hours the reaction. Completion of the reaction the heating was stopped, and after cooling to room temperature, the solvent was distilled off under reduced pressure; the residue after distillation under cooling in an ice water bath, and treated dropwise with concentrated hydrochloric acid (150ml) adjusted to pH 1-2. 300ml ethyl acetate was added, the aqueous phase was extracted with 300ml of ethyl acetate, and the combined organic phases were washed with 300ml water; the organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated to 500ml of the solvent. The residue was cooled to room temperature, stirred for 2 hours. The title compound was isolated by filtration through with ethyl acetate (20ml) and was washed and dried at 50 deg.] C in vacuo overnight to give 21.5g of white solid 3- [2 (4-methylphenyl) phenyl] glutaric acid (melting point: 194-196 ℃) yield 65%.
Example 6 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine-2,6-dione
Mixing the compound 3- [2 (4-methyl) phenyl] glutaric acid (10g, 30mmol) and urea (5.4g, 90mmol) prepared in Step stirred and heated to 146 deg.] C for 4 hours ; after completion of the reaction was monitored by TLC, cooled to 80 deg.] C, was slowly added 70ml of water and 70ml of ethanol was stirred for 30 minutes; cooled to room temperature and stirred for 1 hour. The title compound was filtered absolute ethanol (170ml) and recrystallized from 50 deg.] C overnight and dried in vacuo to give 8.0g white solid 4- [2- (4-methylphenyl) phenyl] piperidine-2,6-di -one (mp: 164-166 ℃), yield 86%
7 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine-2,6-dione Example
In four of 250ml equipped with a condenser reaction flask was added 3- [2 (4-methyl) phenyl] glutaric acid (10g, 30mmol) and urea (14.4g, 240mmol) and the mixture was stirred and heated to 146 deg.] C for 4 hours; TLC monitoring completion of the reaction, cooled to 100 deg.] C, was slowly added 70ml of water and 70ml of ethanol was stirred for 30 minutes; cooled to room temperature and stirred for 1 hour. The title compound was filtered absolute ethanol (170ml) and recrystallized from 50 deg.] C overnight and dried in vacuo to give 7.8g white solid 4- [2- (4-methylphenyl) phenyl] piperidine-2,6-di -one (mp: 165-166 ℃), yield 84%.
8 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine-2,6-dione Example
In four of 250ml equipped with a condenser reaction flask was added 3- [2 (4-methyl) phenyl] pentanedioic acid (5g, 15mmol) and urea (1.8g, 30mmol) and the mixture was stirred and heated to 143 deg.] C for 4 hours; cool to 100 deg.] C, was slowly added 35ml of water and 35ml of ethanol was stirred for 30 minutes; cooled to room temperature and stirred for 1 hour. The title compound was filtered absolute ethanol (70ml) and recrystallized from 50 deg.] C overnight and dried in vacuo to give an off-white solid 2.9g of 4- [2- (4-methylphenyl) phenyl] piperidine-2,6-dione (Melting point: 163-166 ℃), a yield of 63%.
9 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine-2,6-dione Example
The compound prepared in the step of 3- [2 (4-methyl) phenyl] glutaric acid (10g, 30mmol) and urea (3.6g, 60mmol) were mixed and stirred and heated to 146 deg.] C for 4 hours ; after completion of the reaction was monitored by TLC, cooled to 80 deg.] C, was slowly added 70ml of water and 70ml of ethanol was stirred for 30 minutes; cooled to room temperature and stirred for 1 hour. The title compound was filtered, absolute ethanol (45 ml of) and recrystallized from 50 deg.] C overnight and dried in vacuo to give 8.0g white solid 4- [2- (4-methylphenyl) phenyl] piperidine-2,6 dione (melting point: 164-166 ℃), yield 86%.
10 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine-2,6-dione Example
A step of preparing the compound 3- [2 (4-methylphenyl) phenyl] glutaric acid (19.8g, 60mmol) and urea (21.6g, 360mmol) were mixed and stirred and heated to 144 deg.] C for 4 hours; after completion of the reaction was monitored by TLC, cooled to 100 deg.] C, slowly added water 140ml 140ml ethanol and stirred for 30 min; cooled to room temperature and stirred for 1 hour. The title compound was filtered, absolute ethanol (350ml) and recrystallized from 50 deg.] C overnight and dried in vacuo to give a white solid 16.5g of 4- [2- (4-methylphenyl) phenyl] piperidine-2,6 dione (melting point: 164-166 ℃), yield 88%.
Example 11 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine
Tetrahydro lithium aluminum (5.1g, 39mmol) with 140ml of tetrahydrofuran were mixed and stirred ice bath cooled to 8 ℃, under nitrogen, was added dropwise 4- (2-mercapto-methylphenyl) piperidine-2,6-phenyl one (7g) in tetrahydrofuran (140ml) solution, so that the temperature does not exceed 20 ℃; dropping was completed, the reaction at room temperature for 5 hours. The reaction solution was cooled in an ice-water bath, was slowly added dropwise 30ml of water, stirred for 20 minutes. The reaction mixture was added sodium sulfate (20g), stirred for 30 minutes. Filtered and the filtrate was concentrated to give a colorless liquid (4.5g), cooled to solidify to a white solid of 4- [2- (4-methylphenyl) phenyl] piperidine.
Example 12 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine
The reaction flask was added 100ml four 1mol / l borane tetrahydrofuran solution (40ml, 40mmol), cooled to ice bath 5 ℃; under nitrogen was added dropwise 4- (2-mercapto-methylphenyl) piperidine-2-phenyl , 6-dione (3.1g) in tetrahydrofuran (40ml) solution, so that the temperature does not exceed 10 ℃; dropping was completed, the reaction at room temperature for 20 hours. The reaction solution was cooled to 0 deg.] C, and slowly added dropwise 1mol / l HCl (30mL), dropwise finished warming at reflux for 5 hours; of THF was removed and concentrated, 30ml of ethyl acetate and washed with an aqueous solution, a saturated aqueous sodium bicarbonate was added to adjust the pH> 10 , followed by addition of 50ml of ethyl acetate, the organic phase was dried, filtered and concentrated to give 1.8g of a colorless liquid, and cooled to solidify to a white solid of 4- [2- (4-methyl) phenyl] piperidine.
Example 13 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine
The a 2 mol / L the BH 3 .CH 3 the SCH 3 (20 mL) and diethylene glycol dimethyl ether 20ml were mixed and stirred ice bath cooled to 10 ℃, solution of 4- (2-mercapto-methyl-phenyl) phenylpiperidine pyridine 2,6-dione (3.1g) in diethylene glycol dimethyl ether (60ml) solution, so that the temperature does not exceed 20 ℃; dropping was completed, the reaction at room temperature 0.5 hours, then slowly heated to 120 deg.] C for 10 hours. The reaction solution was cooled to 0 deg.] C, and slowly added dropwise 30ml of methanol, a dropping was completed, the mixture was stirred overnight at room temperature; was added 4mol / l HCl / EA (10ml ), was heated to 100 deg.] C for 4 hours; the resulting residue was distilled under reduced pressure was dissolved in 30ml water, saturated aqueous sodium bicarbonate was added to adjust the pH> 10, followed by addition of 50ml of ethyl acetate, the organic phase was dried, filtered and concentrated to give a pale red liquid; after column chromatography (hexane – acetic acid – ethanol 10 : 1.5: 0.5) to give a white solid (0.9g) 4- [2- (4- methylphenylsulfanyl) phenyl] piperidine after purification.
14 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine hydrochloride Example
The step resulting 4- [2- (4-methylphenyl) phenyl] piperidine (4g, 14mmol) was added to absolute ethanol (30ml) and heated to 50 deg.] C to dissolve; 4mol slowly added dropwise / l hydrogen chloride – ethyl acetate solution (4ml), 40 minutes with the reaction temperature; cooled to 5-10 ℃ stirred for 2 hours, filtered through a cake when the ethanol (5ml) and washed with 44 ℃ overnight and dried in vacuo to give 3.2 g of white solid 4- [2- (4-methylphenyl) phenyl] piperidine hydrochloride (melting point: 222-225 ℃), 75% yield.
15 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine hydrochloride Example
4- [2- (4-methylphenyl) phenyl] piperidine (4g, 14mmol) was added to acetone (20ml) and heated to 50 deg.] C to dissolve; 37% was gradually added dropwise concentrated hydrochloric acid ( 1.5ml), 40 minutes with the reaction temperature; cooled with stirring to 5-10 ℃ 2 hours, filtered through a cake of acetone (5ml) and washed with 44 ℃ vacuum dried overnight to give 3.6g of white solid 4- [2- ( 4-methylphenyl) phenyl] piperidine hydrochloride (melting point: 224-227 ℃), in 80% yield.
Example 16 Preparation of 4- [2- (4-methylphenyl) phenyl] piperidine hydrochloride embodiment
Tetrahydro Lithium aluminum (19g, 500mmol) and 200ml of tetrahydrofuran were mixed and stirred at room temperature was added dropwise 4- (2-mercapto-methylphenyl) piperidine-2,6-dione phenyl (31.1g, 100mmol) and tetrahydrofuran ( 200ml) solution, the temperature does not exceed 35 ℃; dropping was completed, the reaction heated under reflux for 3 hours. The reaction solution was cooled in an ice-water bath, was slowly added dropwise 100ml of saturated aqueous sodium sulfate solution, stirred for 60 minutes. The reaction mixture was added ethyl acetate (200ml) and anhydrous magnesium sulphate (50g) was stirred for 60 minutes. Filtered and the filtrate was concentrated to give a colorless liquid. Was added to 80ml of acetone and heated to 40 ℃ dissolved, was added quickly 4mol / l hydrogen chloride – ethyl acetate solution (10ml), seeded, stirred for 20 minutes to precipitate a white solid. 40 ℃, slowly dropping the remaining hydrogen chloride – ethyl acetate solution (20ml). Drop Bi, 5-10 ℃ for 3 hours. The filtered cake in acetone (30ml) and washed with 44 ℃ when dried in vacuo overnight to give 20.8g of white solid 4- [2- (4-methylphenyl) phenyl] piperidine hydrochloride (melting point: 225-228 ℃), yield 66%.
4- [2- (4-METHYLPHENYLSULFANYD-PHENYL] PIPERIDINE WITH COMBINED SEROTONIN AND NOREPINEPHRINE REUPTAKE INHIBITION FOR THE TREATMENT OF ADHD, MELANCHOLIA, TREATMENT RESISTENT DEPRESSION OR RESIDUAL SYMPTOMS IN DEPRESSION
CRYSTALLINE FORMS OF 4- [2- (4-METHYLPHENYLSULFANYL) -PHENYL] PIPERIDINE WITH COMBINED SEROTONIN AND NOREPINEPHRINE REUPTAKE INHIBITION FOR THE TREATMENT OF NEUROPATHIC PAIN
CRYSTALLINE FORMS OF 4-[2-(4-METHYLPHENYLSULFANYL)-PHENYL] PIPERIDINE WITH COMBINED SEROTONIN AND NOREPINEPHRINE REUPTAKE INHIBITION FOR THE TREATMENT OF NEUROPATHIC PAIN
Clenbuterol, marketed as Dilaterol, Spiropent, Ventipulmin,[1] is a sympathomimeticamine used by sufferers of breathing disorders as a decongestant and bronchodilator. People with chronic breathing disorders such as asthma use this as a bronchodilator to make breathing easier. It is most commonly available as the hydrochloridesalt, clenbuterol hydrochloride.[2]
Clenbuterol is also prescribed for treatment of horses, but equine use is usually the liquid form.
Human use
Clenbuterol is approved for use in some countries, free or via prescription, as a bronchodilator for asthma patients.[3]
Legal status
Clenbuterol is not an ingredient of any therapeutic drug approved by the US Food and Drug Administration[3] and is now banned forIOC-tested athletes.[4] In the US, administration of clenbuterol to any animal that could be used as food for human consumption is banned by the FDA.[5][6]
Clenbuterol is a therapeutic drug for asthma and COPD, approved for human use in some countries in Europe (Bulgaria and Russia) and Asia (China).
Weight-loss drug
Although often used by bodybuilders during their “cutting” cycles,[citation needed] the drug has been more recently known to the mainstream, particularly through publicized stories of use by celebrities such as Victoria Beckham,[4]Britney Spears, and Lindsay Lohan, [7] for its off-label use as a weight-loss drug similar to usage of other sympathomimetic amines such as ephedrine, despite the lack of sufficient clinical testing either supporting or negating such use.
By bromination of 4-amino-3,5-dichloroacetophenone (I) with Br2 in CHCl3 to give 4-amino-3,5-dichloro-alpha-bromoacetophenone (II), m.p. 140-5 C, which is condensed with tert-butylamine (III) in CHCl3 to 4-amino-3,5-dichloro-alpha-tertbutylaminoacetophenone hydrochloride (IV), m.p. 252-7 C; this product is finally reduced with NaBH4 in methanol.
Synthesen von neuen Amino-Halogen-substituierten Phenyl-aminothanolen. Arzneim-Forsch Drug Res 1972, 22, 5, 861-869
CLIP
Synthesis and Characterization of Bromoclenbuterol
Ravi Kumar Kannasani*, Srinivasa Reddy Battula, Suresh Babu Sannithi, Sreenu Mula and Venkata Babu VV
R&D Division, RA Chem Pharma Limited, API, Hyderabad, Telangana, India
*Corresponding Author:
Ravi Kumar Kannasani
R&D Division, RA Chem Pharma Limited
API, Prasanth Nagar, Hyderabad, Telangana, India Tel: +919000443184 E-mail: kannasani.ravi@rachempharma.com
Citation: Kannasani RK, Battula SR, Sannithi SB, Mula S, Babu VVV (2016) Synthesis and Characterization of Bromoclenbuterol. Med Chem (Los Angeles) 6:546-549. doi:10.4172/2161-0444.1000397
Clenbuterol, it is most commonly available as the hydrochloride salt, clenbuterol hydrochloride. Clenbuterol, marketed as Dilaterol, Spiropent, Ventipulmin, and also generically as clenbuterol, is a sympathomimetic amine used for breathing disorders as a decongestant and bronchodilator. People with chronic breathing disorders such as asthma use this as a bronchodilator to make breathing easier. Clenbuterol is a β2 agonist with some structural and pharmacological similarities to epinephrine and salbutamol, but its effects are more potent and longerlasting as a stimulant and thermogenic drug. It causes an increase in aerobic capacity, central nervous system stimulation, blood pressure, and oxygen transportation. It increases the rate at which body fat is metabolized while increasing the body’s BMR. It is commonly used for smooth muscle-relaxant properties as a bronchodilator and tocolytic. Clenbuterol is also prescribed for treatment of horses, but equine use is usually the liquid form
Clenbuterol Hydrochloride was first synthesized at Thomae; a Boehringer Ingelheim research facility in Biberach, Germany, in 1967. The synthesis of Clenbuterol Hydrochloride was patented in the United States in 1970. After comprehensive clinical trials, Clenbuterol Hydrochloride was approved for the treatment of reversible airway obstruction in Germany in 1976 and later as a veterinary pharmaceutical for the treatment of bronchiolytic disorders in Germany in 1980. Boehringer Ingelheim markets Clenbuterol Hydrochloride as Spirospent for Human Pharmaceuticals and as Ventipulmin for Veterinary Pharmaceuticals. Clenbuterol Hydrochloride is not approved by the Federal Drug Administration for human use in the United States.
As per the available literature [4–7], clenbuterol hydrochloride was synthesized from 4-amino acetophenone (Scheme 1). Initially 4-amino acetophenone (1) was reacted with chlorine to afford 4-amino-3,5- dichloro acetopheneone (2) which was further reacted bromine to give 1-(4-amino-3,5-dichlorophenyl)-2-bromoethanone (3). The obtained bromo compound was reacted tertiary butyl amine to afford 2-(tertbutylamino)- 1-(4-amino-3,5-dichlorophenyl)ethanone (4), which was further reduced with sodium borohydride to give clenbuterol base (5) and converted in to hydrochloride salt by using alcoholic HCl to get clenbuterol hydrochloride (6).
In the synthesis of clenbuterol hydrochloride, first step was a double chlorination of 4-aminoacetophenone (1) through an electrophillic aromatic substitution reaction to yield 4-amino-3,5- dichloroacetophenone (2). Due to the ortho/para directing, amino group and the meta directing, electron withdrawing, acetyl group, chlorination of 4-aminoacetophenone occurs primarily at the 3 and 5 positions over the 2 and 6 positions. Therefore, under chlorination would produce only the mono chlorinated impurity, 4-amino-3- chloroacetophenone. Under these conditions, over chlorination does not result in the addition of chlorine to the 2 and 6 positions because the amino and acetyl groups do not direct that addition. Even though chlorides are ortho/para directing and direct to the 2 and 6 position, chlorides are also deactivating. After close observation on this chlorination reaction, it was noted that the formed mono chlorinated impurity (Scheme 2) (4-amino-3-chloro acetophenone) caused the formation of process related impurity (bromoclenbuterol) in clenbuerol synthesis.
GENENTECH, INC. [US/US]; 1 DNA Way South San Francisco, California 94080-4990 (US). CONSTELLATION PHARMACEUTICALS, INC. [US/US]; 215 First Street Suite 200 Cambridge, Massachusetts 02142 (US)
Chromatin is a complex combination of DNA and protein that makes up chromosomes. It is found inside the nuclei of eukaryotic cells and is divided between heterochromatin (condensed) and euchromatin (extended) forms. The major components of chromatin are DNA and proteins. Histones are the chief protein components of chromatin, acting as spools around which DNA winds. The functions of chromatin are to package DNA into a smaller volume to fit in the cell, to strengthen the DNA to allow mitosis and meiosis, and to serve as a mechanism to control expression and DNA replication. The chromatin structure is controlled by a series of post-translational modifications to histone proteins, notably histones H3 and H4, and most commonly within the “histone tails” which extend beyond the core nucleosome structure. Histone tails tend to be free for protein-protein interaction and are also the portion of the histone most prone to post-translational modification. These modifications include acetylation, methylation, phosphorylation, ubiquitinylation, and SUMOylation. These epigenetic marks are written and erased by specific enzymes that place the tags on specific residues within the histone tail, thereby forming an epigenetic code, which is then interpreted by the cell to allow gene specific regulation of chromatin structure and thereby transcription.
Of all classes of proteins, histones are amongst the most susceptible to post-translational modification. Histone modifications are dynamic, as they can be added or removed in response to specific stimuli, and these modifications direct both structural changes to chromatin and alterations in gene transcription. Distinct classes of enzymes, namely histone acetyltransferases (HATs) and histone deacetylases (HDACs), acetylate or de-acetylate specific histone lysine residues (Struhl K., Genes Dev., 1989, 12, 5, 599-606).
Bromodomains, which are approximately 1 10 amino acids long, are found in a large number of chromatin-associated proteins and have been identified in approximately 70 human proteins, often adjacent to other protein motifs (Jeanmougin F., et al., Trends Biochem. Sc , 1997, 22, 5, 151-153; and Tamkun J.W., et al., Cell, 1992, 7, 3, 561-572).
Interactions between bromodomains and modified histones may be an important mechanism underlying chromatin structural changes and gene regulation. Bromodomain-containing proteins have been implicated in disease processes including cancer, inflammation and viral replication. See, e.g., Prinjha et al,, Trends Pharm. Sci., 33(3):146-153 (2012) and Muller et al , Expert Rev. , 13 (29): 1 -20 (September 201 1 ).
Cell-type specificity and proper tissue functionality requires the tight control of distinct transcriptional programs that are intimately influenced by their environment.
Alterations to this transcriptional homeostasis are directly associated with numerous disease states, most notably cancer, immuno-inflammation, neurological disorders, and metabolic diseases. Bromodomains reside within key chromatin modifying complexes that serve to control distinctive disease-associated transcriptional pathways. This is highlighted by the observation that mutations in bromodomain-containing proteins are linked to cancer, as well as immune and neurologic dysfunction. Hence, the selective inhibition of bromodomains across a specific family, such as the selective inhibition of a bromodomain of CBP/EP300, creates varied opportunities as novel therapeutic agents in human dysfunction.
There is a need for treatments for cancer, immunological disorders, and other
To a solution of (^)-tetrahydrofuran-3-ol (25 g, 253.7 mmol) in DCM (250 mL) at 0 °C was added triethylamine (86 g, 851.2 mmol) and mesyl chloride (39 g, 340.48 mmol) dropwise. The mixture was stirred at room temperature for 12 h. The reaction was quenched with water (100 mL) and extracted with DCM (100 mL x 2). The combined organic layers were dried over anhydrous Na2S04, filtered and concentrated in vacuo to give the title compound (47 g, 99%) as a brown oil. Ή NMR (400 MHz, CDC13) δ 5.35 – 5.27 (m, 1H), 4.05 – 3.83 (m, 4H), 3.04 (s, 3 H), 2.28 – 2.20 (m, 2 H).
To a solution of tert-butyl 3-bromo-6,7-dihydro-lH-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate (Intermediate A, 24.8 g, 82 mmol) in DMF (200 mL) was added Cs2C03 (79 g, 246 mmol) and (/?)-tetrahydrofuran-3-yl methanesulfonate (17.4 g, 98 mmol). The mixture was heated to 80 °C for 12 h. After cooling the reaction to room temperature, the mixture was concentrated in vacuo. The crude residue was purified by silica gel chromatography
(petroleum ether / EtOAc = from 10 : 1 to 3 : 1) to give the title compound (Intermediate F, 50 g, 71 %) as a yellow oil. Ή NMR (400 MHz, DMSO-i ) δ 4.97 – 4.78 (m, 1H), 4.13 (s, 2H), 3.98 – 3.86 (m, 2H), 3.81 – 3.67 (m, 2H), 3.56 (t, J= 5.6 Hz, 2H), 2.68 (t, J= 5.6 Hz, 2H), 2.33 – 2.08 (m, 2H), 1.38 (s, 9H).
To a solution of (S)-tert-buty\ 3-bromo- 1 -(tetrahydrofuran-3-yl)-6,7-dihydro-lH-pyrazolo [4,3 -c]pyridine-5(4H)-carboxy late (29 g, 78 mmol) in DCM (300 mL) was added trifluroacetic acid (70 mL) dropwise. The mixture was stirred at room temperature for 2 h. The solvent was concentrated in vacuo and the crude residue was re -dissolved in DMF (100 mL). The mixture was cooled to 0 °C before triethylamine (30 g, 156 mmol) and acetic anhydride (8.7 g, 86 mmol) were added dropwise. The mixture was stirred at room temperature for an additional 2 h. The reaction was quenched with water (200 mL) at 0 °C and extracted with EtOAc (150 mL x 3). The combined organic layers were dried over anhydrous Na2S0 , filtered and concentrated in vacuo. The crude residue was purified by silica gel chromatography (DCM / MeOH = 30 : 1) to give the title compound (Intermediate G, 21.3 g, 87%) as a white solid. lH NMR (400 MHz, CDC13) δ 4.78 – 4.67 (m, 1H), 4.45 -4.29 (m, 2H), 4.15 – 4.06 (m, 2H), 3.96 – 3.92 (m, 2H), 3.88 – 3.70 (m, 2H), 2.71 – 2.67 (m, 2H), 2.38 – 2.34 (m, 2H), 2.16 (s, 3H).
To a solution of (R)-tetrahydrofuran-3-ol (25 g, 253.7 mmol) in DCM (250 mL) at 0° C. was added triethylamine (86 g, 851.2 mmol) and mesyl chloride (39 g, 340.48 mmol) dropwise. The mixture was stirred at room temperature for 12 h. The reaction was quenched with water (100 mL) and extracted with DCM (100 mL×2). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo to give the title compound (47 g, 99%) as a brown oil. 1H NMR (400 MHz, CDCl3) δ 5.35-5.27 (m, 1H), 4.05-3.83 (m, 4H), 3.04 (s, 3H), 2.28-2.20 (m, 2H).
To a solution of tert-butyl 3-bromo-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate (Intermediate A, 24.8 g, 82 mmol) in DMF (200 mL) was added Cs2CO3 (79 g, 246 mmol) and (R)-tetrahydrofuran-3-yl methanesulfonate (17.4 g, 98 mmol). The mixture was heated to 80° C. for 12 h. After cooling the reaction to room temperature, the mixture was concentrated in vacuo. The crude residue was purified by silica gel chromatography (petroleum ether/EtOAc=from 10:1 to 3:1) to give the title compound (Intermediate F, 50 g, 71%) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 4.97-4.78 (m, 1H), 4.13 (s, 2H), 3.98-3.86 (m, 2H), 3.81-3.67 (m, 2H), 3.56 (t, J=5.6 Hz, 2H), 2.68 (t, J=5.6 Hz, 2H), 2.33-2.08 (m, 2H), 1.38 (s, 9H).
To a solution of (S)-tert-butyl 3-bromo-1-(tetrahydrofuran-3-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate (29 g, 78 mmol) in DCM (300 mL) was added trifluroacetic acid (70 mL) dropwise. The mixture was stirred at room temperature for 2 h. The solvent was concentrated in vacuo and the crude residue was re-dissolved in DMF (100 mL). The mixture was cooled to 0° C. before triethylamine (30 g, 156 mmol) and acetic anhydride (8.7 g, 86 mmol) were added dropwise. The mixture was stirred at room temperature for an additional 2 h. The reaction was quenched with water (200 mL) at 0° C. and extracted with EtOAc (150 mL×3). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by silica gel chromatography (DCM/MeOH=30:1) to give the title compound (Intermediate G, 21.3 g, 87%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 4.78-4.67 (m, 1H), 4.45-4.29 (m, 2H), 4.15-4.06 (m, 2H), 3.96-3.92 (m, 2H), 3.88-3.70 (m, 2H), 2.71-2.67 (m, 2H), 2.38-2.34 (m, 2H), 2.16 (s, 3H).
The single bromodomain of the closely related transcriptional regulators CBP/EP300 is a target of much recent interest in cancer and immune system regulation. A co-crystal structure of a ligand-efficient screening hit and the CBP bromodomain guided initial design targeting the LPF shelf, ZA loop, and acetylated lysine binding regions. Structure–activity relationship studies allowed us to identify a more potent analogue. Optimization of permeability and microsomal stability and subsequent improvement of mouse hepatocyte stability afforded 59 (GNE-272, TR-FRET IC50 = 0.02 μM, BRET IC50 = 0.41 μM, BRD4(1) IC50 = 13 μM) that retained the best balance of cell potency, selectivity, and in vivo PK. Compound 59 showed a marked antiproliferative effect in hematologic cancer cell lines and modulates MYC expression in vivo that corresponds with antitumor activity in an AML tumor model.
Discovery of a Potent and Selective in Vivo Probe (GNE-272) for the Bromodomains of CBP/EP300
In a similar procedure to59, the title compound was prepared from (S)-tetrahydrofuran-3-yl
methanesulfonate and purified by Prep-TLC (DCM / MeOH = 15 : 1) to give the title
compound as a light yellow solid.
The crude residue was purified by silica gel chromatography (DCM / MeOH = 100:1) to give (S)-1-(3-((2-fluoro-4-(1- methyl-1H-pyrazol-4-yl)phenyl)amino)-1-(tetrahydrofuran-3-yl)-6,7-dihydro-1Hpyrazolo[4,3-c]pyridin-5(4H)-yl)ethanone as a light yellow solid.
(From left to right) Principal Investigator Associate Professor Gautam Sethi and NUS PhD candidate Ms Zhang Jingwen from the Department of Pharmacology at the NUS Yong Loo Lin School of Medicine led a research which found that a bioactive compound from the neem plant could significantly suppress development of prostate cancer.
Credit: National University of Singapore
Date:September 29, 2016Source:National University of SingaporeSummary:Oral administration of nimbolide, over 12 weeks shows reduction of prostate tumor size by up to 70 per cent and decrease in tumor metastasis by up to 50 per cent, report investigators.
Oral administration of nimbolide, over 12 weeks shows reduction of prostate tumor size by up to 70 per cent and decrease in tumor metastasis by up to 50 per cent
A team of international researchers led by Associate Professor Gautam Sethi from the Department of Pharmacology at the Yong Loo Lin School of Medicine at the National University of Singapore (NUS) has found that nimbolide, a bioactive terpenoid compound derived from Azadirachta indica or more commonly known as the neem plant, could reduce the size of prostate tumor by up to 70 per cent and suppress its spread or metastasis by half.
Prostate cancer is one of the most commonly diagnosed cancers worldwide. However, currently available therapies for metastatic prostate cancer are only marginally effective. Hence, there is a need for more novel treatment alternatives and options.
“Although the diverse anti-cancer effects of nimbolide have been reported in different cancer types, its potential effects on prostate cancer initiation and progression have not been demonstrated in scientific studies. In this research, we have demonstrated that nimbolide can inhibit tumor cell viability — a cellular process that directly affects the ability of a cell to proliferate, grow, divide, or repair damaged cell components — and induce programmed cell death in prostate cancer cells,” said Assoc Prof Sethi.
Nimbolide: promising effects on prostate cancer
Cell invasion and migration are key steps during tumor metastasis. The NUS-led study revealed that nimbolide can significantly suppress cell invasion and migration of prostate cancer cells, suggesting its ability to reduce tumor metastasis.
The researchers observed that upon the 12 weeks of administering nimbolide, the size of prostate cancer tumor was reduced by as much as 70 per cent and its metastasis decreased by about 50 per cent, without exhibiting any significant adverse effects.
“This is possible because a direct target of nimbolide in prostate cancer is glutathione reductase, an enzyme which is responsible for maintaining the antioxidant system that regulates the STAT3 gene in the body. The activation of the STAT3 gene has been reported to contribute to prostate tumor growth and metastasis,” explained Assoc Prof Sethi. “We have found that nimbolide can substantially inhibit STAT3 activation and thereby abrogating the growth and metastasis of prostate tumor,” he added.
The findings of the study were published in the April 2016 issue of the scientific journal Antioxidants & Redox Signaling. This work was carried out in collaboration with Professor Goh Boon Cher of Cancer Science Institute of Singapore at NUS, Professor Hui Kam Man of National Cancer Centre Singapore and Professor Ahn Kwang Seok of Kyung Hee University.
Neem — The medicinal plant
The neem plant belongs to the mahogany tree family that is originally native to India and the Indian sub-continent. It has been part of traditional Asian medicine for centuries and is typically used in Indian Ayurvedic medicine. Today, neem leaves and bark have been incorporated into many personal care products such as soaps, toothpaste, skincare and even dietary supplements.
Future Research
The team is looking to embark on a genome-wide screening or to perform a large-scale study of proteins to analyse the side-effects and determine other potential molecular targets of nimbolide. They are also keen to investigate the efficacy of combinatory regimen of nimbolide and approved drugs such as docetaxel and enzalutamide for future prostate cancer therapy.
Journal Reference:
Jingwen Zhang, Kwang Seok Ahn, Chulwon Kim, Muthu K. Shanmugam, Kodappully Sivaraman Siveen, Frank Arfuso, Ramar Perumal Samym, Amudha Deivasigamanim, Lina Hsiu Kim Lim, Lingzhi Wang, Boon Cher Goh, Alan Prem Kumar, Kam Man Hui, Gautam Sethi. Nimbolide-Induced Oxidative Stress Abrogates STAT3 Signaling Cascade and Inhibits Tumor Growth in Transgenic Adenocarcinoma of Mouse Prostate Model. Antioxidants & Redox Signaling, 2016; 24 (11): 575 DOI:10.1089/ars.2015.6418
Autotaxin is an enzyme reported to be the source of lysophosphatidic acid (LPA) which up-regulates pain-related proteins through one if its cognate receptors, LPAi. LPA is an intracellular lipid mediator which influences a multiplicity of biological and biochemical processes. Targeted inhibition of autotaxin-mediated LPA biosynthesis may provide a novel mechanism to prevent nerve injury-induced neuropathic pain.
Compounds that inhibit autotaxin are desired to offer a potential treatment option for patients in need of treatment for pain.
Pain associated with osteoarthritis (OA) is reported to be the primary symptom leading to lower extremity disability in OA patients. Over 20 million Americans have been diagnosed with OA, the most common of the arthropathies. The currently approved treatments for OA pain may be invasive, lose efficacy with long term use, and may not be appropriate for treating all patients. Additional treatment options for patients suffering from pain associated with OA are desired. Compounds that inhibit autotaxin represent another possible treatment option for patients with pain associated with OA.
U.S. Patent 7,524,852 (‘852) discloses substituted bicyclic pyrimidine derivatives as anti-inflammatory agents.
PCT/US2011/048477 discloses indole compounds as autotoxin inhibitors.
There is a need for novel compounds that provide autotaxin inhibition. The present invention provides novel compounds which are autotaxin inhibitors. The present invention provides certain novel compounds that inhibit the production of LPA.
Autotaxin inhibitor compounds are desired to provide treatments for autotaxin mediated conditions, such as pain and pain associated with OA.
PAPER
In an effort to develop a novel therapeutic agent aimed at addressing the unmet need of patients with osteoarthritis pain, we set out to develop an inhibitor for autotaxin with excellent potency and physical properties to allow for the clinical investigation of autotaxin-induced nociceptive and neuropathic pain. An initial hit identification campaign led to an aminopyrimidine series with an autotaxin IC50 of 500 nM. X-ray crystallography enabled the optimization to a lead compound that demonstrated favorable potency (IC50 = 2 nM), PK properties, and a robust PK/PD relationship.
Novel Autotaxin Inhibitors for the Treatment of Osteoarthritis Pain: Lead Optimization via Structure-Based Drug Design
Synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]-2-[2-(lH- l ,2,3-triazol-4-yl)ethoxy]ethanone.
Stir a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid
(20.22 g; 70.90 mmol), N-(2,3-dihydro- lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (27.99 g; 81.54 mmol) and triethylamine (98.83 mL; 709.03 mmol) in dimethylformamide (404.40 mL) at 0°C. Add a solution of 1-propanephosphonic acid cyclic anhydride (50% solution in DMF; 51.89 mL; 81.54 mmol) over 30 minutes, and stir the mixture at room temperature for 18 hours.
Concentrate the reaction mixture under reduced pressure to give a residue. Add water (200 mL) and extract the mixture with ethyl acetate (4 x 250 mL) and
dichloromethane (4 x 250 mL). Wash the combined organic layers with saturated aqueous sodium bicarbonate (2 x 100 mL) and brine (100 mL), then dry over anhydrous sodium sulfate. Filter the mixture and concentrate the solution under reduced pressure to give a red solid (25.70 g) that is slurried in ethyl acetate/methanol (9: 1 mixture; 200 mL) for 2 hours at room temperature. Filter the resulting solid and wash with cold ethyl acetate (50 mL) to give a solid (ca.18.2 g) that is re-slurried in ethyl acetate (200 mL) at reflux for 1 hour. On cooling to room temperature, stir the mixture for 1 hour and filter the resulting light pink solid.
Slurry the light pink solid in water/methanol (1 : 1 mixture; 200 mL) and heat the mixture at 50°C for 30 minutes. Add ammonium hydroxide solution (32% ; 50 mL) and continue to heat the mixture at 50°C for 30 minutes. Upon cooling to room temperature, add additional ammonium hydroxide solution (32% ; 50 mL) and continue stirring for 1 hour at room temperature. Filter the resulting light gray solid, dry and slurry again in ethyl acetate (200 mL) for 1 hour to afford a light gray solid that is filtered, washed with ethyl acetate (25 mL), and dried to give the title compound (12.42 g; 43%) as a gray solid. MS (m/z): 406 (M+l).
Synthesis of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid.
Pressurize 1 atmosphere of hydrogen (g) to a flask containing [2-(l-benzyl-lH- l,2,3-triazol-5-yl)ethoxy]acetic acid (10.1 g; 1.00 equiv; 38.66 mmoles) and palladium (II) chloride (3 g; 16.92 mmoles; 3.00 g) in isopropyl alcohol (300 mL) and water (60 mL). Maintain the flask under a hydrogen atmosphere for 3 h, then filter through Celite™ and concentrate. Add toluene (2×50 mL) and concentrate to afford the title compound (7.96 g, 100%). ]H NMR (d6-DMSO): 2.86 (t, / = 7 Hz, 2 H), 3.65 (t, / = 7 Hz, 2 H), 3.98 (s, 2 H), 7,77 (s, 1 H), 13.4 – 13.6 (br s, 2 H).
Example 1
Synthesis of l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin- 6(5H)-yl]-2-[2-(lH-l,2,3-triazol-4- l)ethoxy]ethanone.
Add N-indan-2-yl-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-2-amine (4.2 g, 15.8 mmol) to a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid (2.7 g, 15.8 mmol), 1-hydroxybenzotriazole (3.20 g, 23.7 mmol), and dimethylaminopropyl)-3- ethylcarbodiimide hydrochloride (5.44 g, 28.4 mmol) in dichloromethane (40 mL) at 25 °C. Add triethylamine (4.40 mL, 31.6 mmol) to the reaction mixture and stir for 16 h. Wash with water (2 x 50 mL) and concentrate the organic layer. Purify by silica gel column chromatography, eluting with ethyl acetate/methanol, to give the title compound (4.0 g, 60%) as a solid. MS (m/z): 420 (M + Η). Preparation 8
Synthesis of 2-chloro-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8-dihydropyrido[4,3- d]pyrimidin-6(5H)-yl]ethanone.
To N-indan-2-yl-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-2-amine (11.0 g, 41.3 mmol) and triethylamine (7.48 mL, 53.7 mmol) in dichloromethane (200 mL), add 2- chloroacetyl chloride (3.61 mL, 5.13 g, 45.4 mmol) dropwise over five minutes at 23 °C. Stir for 30 minutes and pour the reaction mixture into 1 : 1 50% saturated aqueous sodium bicarbonate: dichloromethane (75 mL). Separate the organic layer from the aqueous layer and further extract the aqueous layer with dichloromethane (2 x 25 mL). Combine the organic extracts and dry over anhydrous sodium sulfate, filter, and concentrate. Dissolve the residue in chloroform (10 mL) and purify via silica gel column chromatography (gradient elution: 25% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (9.75 g, 69%). ]H NMR (CDC13, * = minor amide rotamer) δ 2.77* (t, 2H), 2.84 (dd, 2H), 2.87 (t, 2H), 3.35 (dd, 2H), 3.76 (t, 2H), 3.85* (t, 2H), 4.12 (s, 2H), 4.52* (s, 2H), 4.57 (s, 2H), 4.72-4.82 (m, IH), 5.48-5.64 (m, IH), 7.12-7.21 (m, 4H), 8.03-8.10 (m, IH).
Preparation 9
Synthesis of 2-(but-3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-7,8- dihydropyrido[4,3-d]p rimidin-6(5H)-yl]ethanone.
To sodium hydride (60 wt% in mineral oil, 1.58 g, 39.6 mmol) in tetrahydrofuran (50 mL) at 23 °C, add 3-butyn-l-ol (7.93 g, 8.59 mL, 113.2 mmol) dropwise, then stir at 23 °C for 20 minutes. Add this solution to 2-chloro-l-[2-(2,3-dihydro-lH-inden-2- ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl]ethanone (9.70 g, 28.3 mmol) in tetrahydrofuran (150 mL) at 23 °C and stir for one hour. Pour the reaction mixture into 50% saturated aqueous sodium bicarbonate solution. Separate the organic layer and further extract the aqueous layer with ethyl ether (x 2) and ethyl acetate (x 2). Combine the organic extracts and wash with brine, then dry over anhydrous sodium sulfate, filter, and concentrate. Purify the resulting crude product by silica gel column chromatography (gradient elution: 20% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (8.16 g, 77%). MS (m/z): 377 (M + 1).
Example la
Alternative synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-7,8-dihydropyrido[4,3- d]pyrimidin-6(5H)-yl]-2-[2-(lH- l,2,3-triazol-4- l)ethoxy]ethanone.
Sparge a solution of 2-(but-3-yn- l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2- ylamino)-7,8-dihydropyrido[4,3-d]pyrimidin-6(5H)-yl]ethanone (8.15 g, 21.7 mmol) and L-ascorbic acid sodium salt (8.58 g, 43.3 mmol) in dimethylformamide (60 mL) and water (60 mL) with nitrogen for ten minutes, then evacuate and backfill with nitrogen three times. Add copper (II) sulfate pentahydrate (1.08 g, 4.33 mmol) and heat to 90 °C, then add azidotrimethylsilane (23.1 mL, 20.0 g, 173 mmol) dropwise and stir for one hour. Cool reaction mixture to 23 °C and pour into water (50 mL). Extract this mixture with ethyl acetate (4 x 50 mL). Combine the organic extracts and wash with saturated aqueous sodium chloride, dry over anhydrous sodium sulfate, filter, and concentrate.
Purify the resulting crude product by silica gel column chromatography (gradient elution: 0 to 10% methanol in ethyl acetate) to give the title compound (3.60 g, 40%). MS (m/z): 420 (M + 1). Preparation 10
Synthesis of tert-butyl-2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidine-6-carboxylate.
Charge 450 rriL (2.58 mol) of N-ethyl-N-isopropylpropan-2-amine into a 15 °C solution of tert-butyl 2-chloro-5,7-dihydro-6H-pyrrolo[3,4-d]pyrimidine-6-carboxylate (220 g, 860.37 mmol) and 2,3-dihydro-lH-inden-2-amine (137.7 g, 1.03 mol) in 1- methylpyrrolidin-2-one (3.6 L). Heat the resulting mixture to 80 °C for 16 h, then cool to 30 °C and transfer the resulting mixture into 5 L of water at 25 °C. Filter the resulting solid and rinse the filter cake with water (2 x 300 rriL). Reslurry the solid in ethyl acetate (350 iriL) for 45 min at 15 °C. Filter the slurry, rinsing with 15 °C ethyl acetate ( 2 x 250 rriL), and dry to give the title compound (226 g, 75%) as an off-white solid. ‘H NMR (d6-DMSO) 1.45 (s, 9 H), 2.87 (dd, /= 7.2, 15.8 Hz, 2 H), 3.24 (dd, /= 7.2, 15.8 Hz, 2 H), 4.36 (d, 10.4 Hz, 2 H), 4.44 (d, /= 12.8 Hz, 2 H), 4.60 (m, 1 H), 7.14 (m, 2 H), 7.20 (m, 2 H), 7.55 (d, /= 6.8 Hz, 1 H), 8.27 (d, /= 7.2 Hz, 1 H).
Preparation 11
Synthesis of N-(2,3-dihydro-lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4-d]pyrimidin-2- amine dihydrochloride hydrate.
Charge 670 rriL of 5 M hydrochloric acid (3.35 mol) to a solution of tert-butyl 2-
(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H pyrrolo[3,4-d]pyrimidine-6- carboxylate (226 g, 641.25 mmol) in tetrahydrofuran (2.0 L) at 17 °C, maintaining the internal temperature below 26 °C during the addition. Heat the resulting solution to 50 °C for 16 h, cool to 25 °C and dilute with 500 rriL of water and 500 mL of tert- butylmethylether. Separate the resulting layers and extract with tert-butylmethylether (3 x 1 L). Concentrate the water phase down to a reaction volume of ca. 200 mL, and filter the resulting slurry. Rinse the cake with tert-butylmethylether (2 x 200 mL) and dry to give the title product (177 g, 80%) as a light brown solid. MS (m/z): 253.2 (M-2HC1- H20+1).
Preparation 12
Syntheis of tert-butyl 2-but-3-ynox acetate.
Stir a mixture of but-3-yn-l-ol (6.00 g; 85.60 mmol), tetrabutylammonium sulfate (2.07 g; 8.54 mmol) and sodium hydroxide (40% wt/wt; 150 mL) in dichloromethane (150 mL) at 0°C. Add tert-butyl bromoacetate (19.34 mL; 128.40 mmol) dropwise and stir the mixture for 2.5 hours at room temperature. Dilute the reaction mixture with dichloromethane (200 mL) and water (100 mL), separate the layers, and further extract the aqueous layer with dichloromethane (2 x 100 mL). Wash the combined organic layers with brine (100 mL), dry over anhydrous sodium sulfate, and concentrate to afford the crude title compound as a brown oil (11.93 g). Purify the oil by silica gel column chromatography, eluting with hexane: ethyl acetate (0% to 10% mixtures) to give the title compound (11.35 g; 72%) as a colorless oil. ]H NMR (CDCI3) δ 1.48 (s, 9H), 2.00 (m, 1H), 2.52 (m, 2H), 3.67 (m, 2H), 4.01 (bs, 2H).
Preparation 13
Synthesis of tert-butyl 2-[2-(lH-triazol-5- l)ethoxy]acetate.
Stir tert-Butyl 2-but-3-ynoxyacetate (11.34 g; 61.55 mmol) and copper(I)iodide (584 mg; 3.07 mmol) in a mixture of dimethylformamide (56.70 mL) and methanol (11.34 mL) at 0°C. Add azido(trimethyl)silane (12.33 mL; 86.47 mmol) dropwise and heat the mixture at 90°C for 18 hours.
In a second batch, stir tert-butyl 2-but-3-ynoxyacetate (4.38 g; 23.77 mmol) and copper(I)iodide (226 mg; 1.19 mmol) in a mixture of dimethylformamide (22 mL) and methanol (6 mL) at 0°C. Add azido(trimethyl)silane (4.8 mL; 33.66 mmol) dropwise and the mixture heated at 90°C for 18 hours.
Upon cooling to room temperature, combine the crude products from both batches and concentrate the mixture to afford a greenish residue. Purify the crude product by filtration through a plug of silica eluting with dichloromethane: ethyl acetate (75% to 100% mixtures) to afford the title compound (14.15 g, 73%) as a colorless oil. MS (m/z): 228.15 (M+l).
Preparation 14
Synthesis of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid.
Stir a mixture of ieri-butyl 2-[2-(lH-triazol-5-yl)ethoxy]acetate (14.15 g; 62.26 mmol) and trifluoroacetic acid (70.75 mL, 935.69 mmol) in dichloromethane (70.75 mL) for 2 hours at room temperature. Concentrate the reaction mixture under reduced pressure to provide the title compound containing additional trifluoroacetic acid (20.22 g, >100%) as a brown solid. MS (m/z): 172.05 (M+l).
Example 2
Synthesis of l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]-2-[2-(lH- l ,2,3-triazol-4-yl)ethoxy]ethanone.
Stir a mixture of 2-[2-(lH-triazol-5-yl)ethoxy]acetic acid 2,2,2-trifluoroacetic acid
(20.22 g; 70.90 mmol), N-(2,3-dihydro- lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (27.99 g; 81.54 mmol) and triethylamine (98.83 mL; 709.03 mmol) in dimethylformamide (404.40 mL) at 0°C. Add a solution of 1-propanephosphonic acid cyclic anhydride (50% solution in DMF; 51.89 mL; 81.54 mmol) over 30 minutes, and stir the mixture at room temperature for 18 hours.
Concentrate the reaction mixture under reduced pressure to give a residue. Add water (200 mL) and extract the mixture with ethyl acetate (4 x 250 mL) and
dichloromethane (4 x 250 mL). Wash the combined organic layers with saturated aqueous sodium bicarbonate (2 x 100 mL) and brine (100 mL), then dry over anhydrous sodium sulfate. Filter the mixture and concentrate the solution under reduced pressure to give a red solid (25.70 g) that is slurried in ethyl acetate/methanol (9: 1 mixture; 200 mL) for 2 hours at room temperature. Filter the resulting solid and wash with cold ethyl acetate (50 mL) to give a solid (ca.18.2 g) that is re-slurried in ethyl acetate (200 mL) at reflux for 1 hour. On cooling to room temperature, stir the mixture for 1 hour and filter the resulting light pink solid.
Slurry the light pink solid in water/methanol (1 : 1 mixture; 200 mL) and heat the mixture at 50°C for 30 minutes. Add ammonium hydroxide solution (32% ; 50 mL) and continue to heat the mixture at 50°C for 30 minutes. Upon cooling to room temperature, add additional ammonium hydroxide solution (32% ; 50 mL) and continue stirring for 1 hour at room temperature. Filter the resulting light gray solid, dry and slurry again in ethyl acetate (200 mL) for 1 hour to afford a light gray solid that is filtered, washed with ethyl acetate (25 mL), and dried to give the title compound (12.42 g; 43%) as a gray solid. MS (m/z): 406 (M+l).
Preparation 15
Synthesis of 2-chloro- l-[2-(2,3-dihydro- lH-inden-2-ylamino)-5,7-dihydro-6H- pyrrolo[3,4-d]pyrimidin-6-yl]ethanone.
Stir a suspension of N-(2,3-dihydro-lH-inden-2-yl)-6,7-dihydro-5H-pyrrolo[3,4- d]pyrimidin-2-amine dihydrochloride hydrate (14.4 g, 41.9 mmol) and triethylamine (14.3 g, 19.7 mL, 141.4 mmol) in dichloromethane (200 mL) at 23 °C for 10 minutes, then cool to -30 °C. Add 2-chloroacetyl chloride (5.49 g, 3.86 mL, 48.6 mmol) over two minutes and warm to 23 °C over 10 minutes. Add methanol (5 mL) and remove the solvent in vacuo. Slurry the crude reaction mixture in methanol (30 mL), add 50 g silica gel and remove solvent in vacuo. Load the resulting residue onto a loading column and purify via silica gel column chromatography (gradient elution: 50% ethyl acetate in hexanes to ethyl acetate to 10% methanol in ethyl acetate) to give the title compound (11.5 g, 84%). MS (m/z): 329(M+1).
Preparation 16
Synthesis of 2-(but-3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro- 6H-pyrrolo[3,4-d]pyrimidin-6-yl]ethanone.
To sodium hydride (60 wt% in mineral oil, 2.06 g, 51.4 mmol) in tetrahydrofuran (86 mL) at 0 °C, add 3-butyn-l-ol (4.64 g, 5.03 mL, 64.3 mmol), then stir at 23 °C for 15 minutes. Add this solution to 2-chloro-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7- dihydro-6H-pyrrolo[3,4-d]pyrimidin-6-yl]ethanone (8.45 g, 25.7 mmol) in
tetrahydrofuran (86 mL) at 0 °C and stir for five minutes. Pour reaction mixture into 50% saturated aqueous sodium bicarbonate solution. Separate the organic layer and further extract the aqueous layer with ethyl ether and ethyl acetate (2 x 50 mL each). Combine the organic extracts and wash with brine, then dry over anhydrous sodium sulfate, filter, and concentrate. Combine the crude product with the crude product from a second reaction (run reaction under identical conditions and stoichiometry employing 2-chloro- 1- [2-(indan-2-ylamino)-5,7-dihydropyrrolo[3,4-d]pyrimidin-6-yl]ethanone (3.0 g, 9.1 mmol)) and purify by silica gel column chromatography (gradient elution: 25% ethyl acetate in hexanes to 100% ethyl acetate) to give the title compound (2.90 g, 23%). MS
(m/z): 363(M+1). Example 2a
Alternative synthesis of l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro- pyrrolo[3,4-d]pyrimidin-6-yl]-2-[2-(lH-l,2,3-triazol-4-yl)ethoxy]ethanone.
Add dimethylformamide (27 mL) and water (27 mL) to a flask containing 2-(but- 3-yn-l-yloxy)-l-[2-(2,3-dihydro-lH-inden-2-ylamino)-5,7-dihydro-6H-pyrrolo[3,4- d]pyrimidin-6-yl]ethanone (2.90 g, 8.00 mmol). Add copper (II) sulfate pentahydrate (400 mg, 1.60 mmol) and L-ascorbic acid sodium salt (3.17 g, 16.0 mmol). Evacuate flask and backfill with nitrogen (x 2), then add azidotrimethylsilane (7.37 g, 8.53 mL, 64.0 mmol) and heat the reaction to 90 °C for 70 minutes. Cool the reaction mixture to 23 °C and remove all solvent in vacuo. Suspend the residue in methanol/dichloromethane and then add silica gel and remove solvent in vacuo. Load this material onto a loading column and purify via silica gel column chromatography (gradient elution: 0-9% methanol in ethyl acetate) to give the title compound (980 mg, 30%). MS (m/z):
Balomenib CAS 2939850-17-4 MF C33H34F3N7O2 MW617.7 g/mol 4-methyl-1-[[(2S)-5-oxomorpholin-2-yl]methyl]-5-[[2-[6-(2,2,2-trifluoroethyl)quinazolin-4-yl]-2,7-diazaspiro[3.5]nonan-7-yl]methyl]indole-2-carbonitrile 4-methyl-1-{[(2S)-5-oxomorpholin-2-yl]methyl}-5-({2-[6-(2,2,2-trifluoroethyl)quinazolin-4-yl]-2,7-diazaspiro[3.5]nonan-7-yl}methyl)-1H-indole-2-carbonitrilemenin inhibitor, antineoplastic, ZE63-0302, 3BEG4BWN8E Balomenib (also known as ZE63-0302) is an oral, small-molecule menin inhibitor currently in clinical development for metabolic and…
Anvumetostat CAS 2790567-82-5 MF C22H19F3N4O3 MW444.4 g/mol (4-amino-1,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl)-[(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl]methanone (4-amino-1,3-dihydrofuro[3,4-c][1,7]naphthyridin-8-yl){(3S)-3-[4-(trifluoromethyl)phenyl]morpholin-4-yl}methanoneantineoplastic, AMG 193, QAT649EJ5E, PRMT5-IN-27, Anvumetostat (also known as AMG 193) is an orally available, small-molecule inhibitor of protein arginine methyltransferase…
Alnodesertib CAS 2267316-76-5 MF C18H24N6O2S MW388.49 4-[4-[(cyclopropyl-methyl-oxo-λ6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine 4-[4-[(cyclopropyl-methyl-oxo-lambda6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine (S)-({2-(2-aminopyridin-4-yl)-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}imino)(cyclopropyl)(methyl)-λ6-sulfanoneserine/threonine kinase inhibitor, antineoplastic, ART 0380, EX-A9085 Alnodesertib (formerly known as ART0380) is an investigational, orally administered drug designed to…
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO






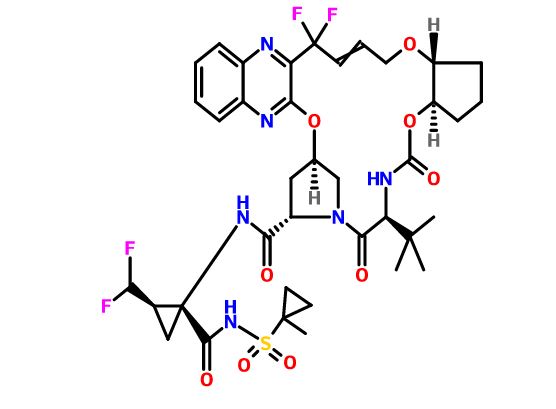


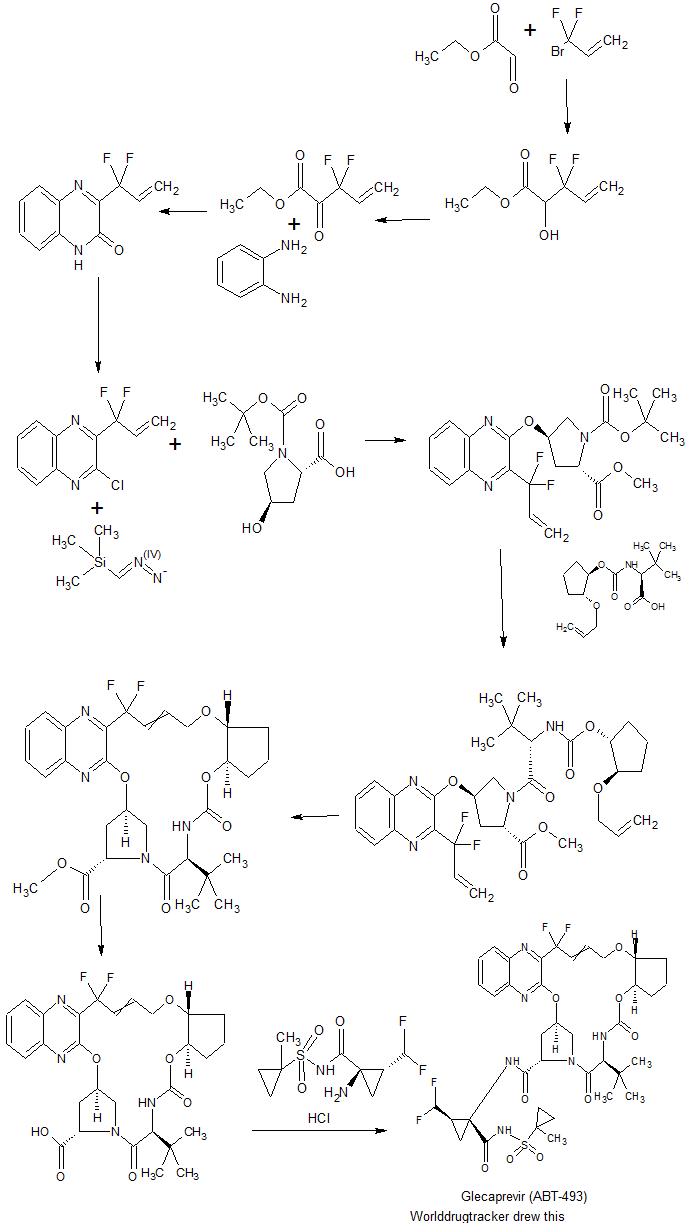



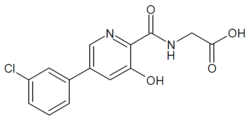
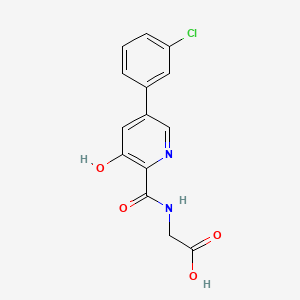



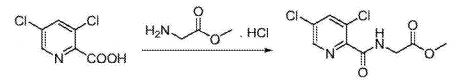
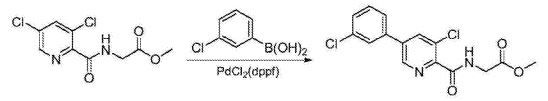
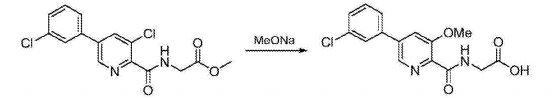
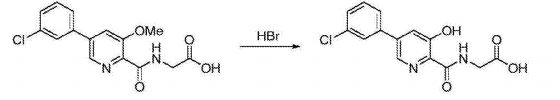


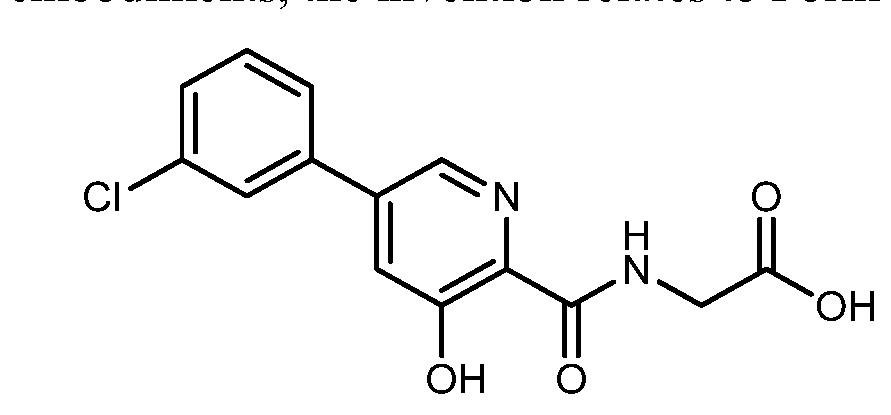
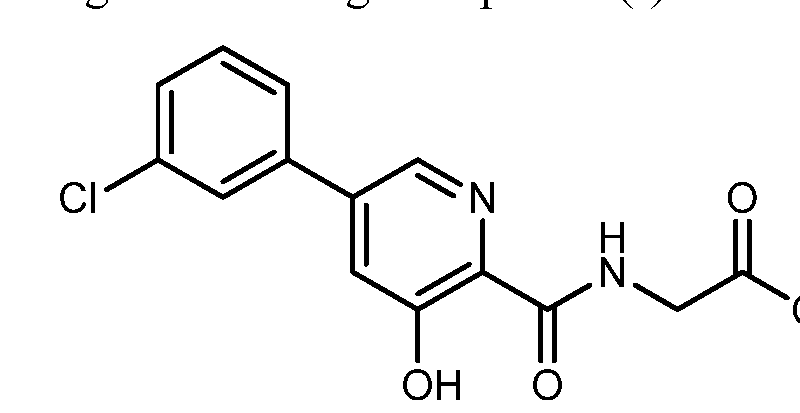







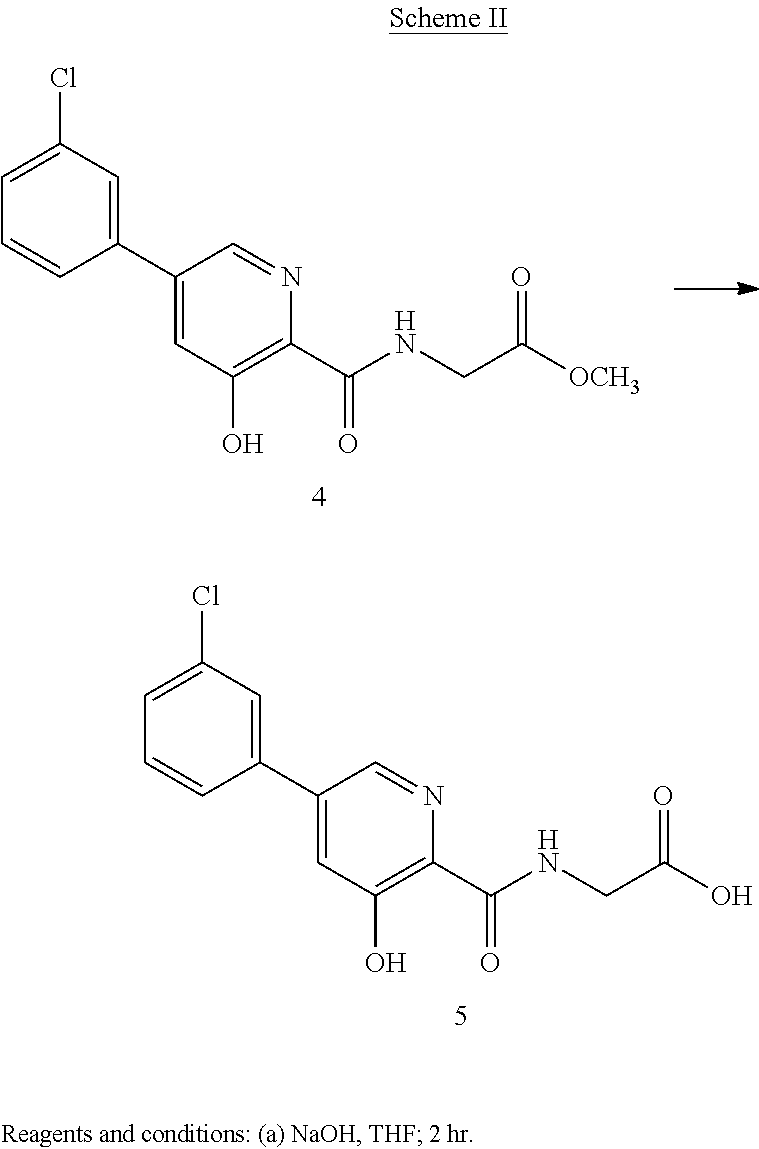
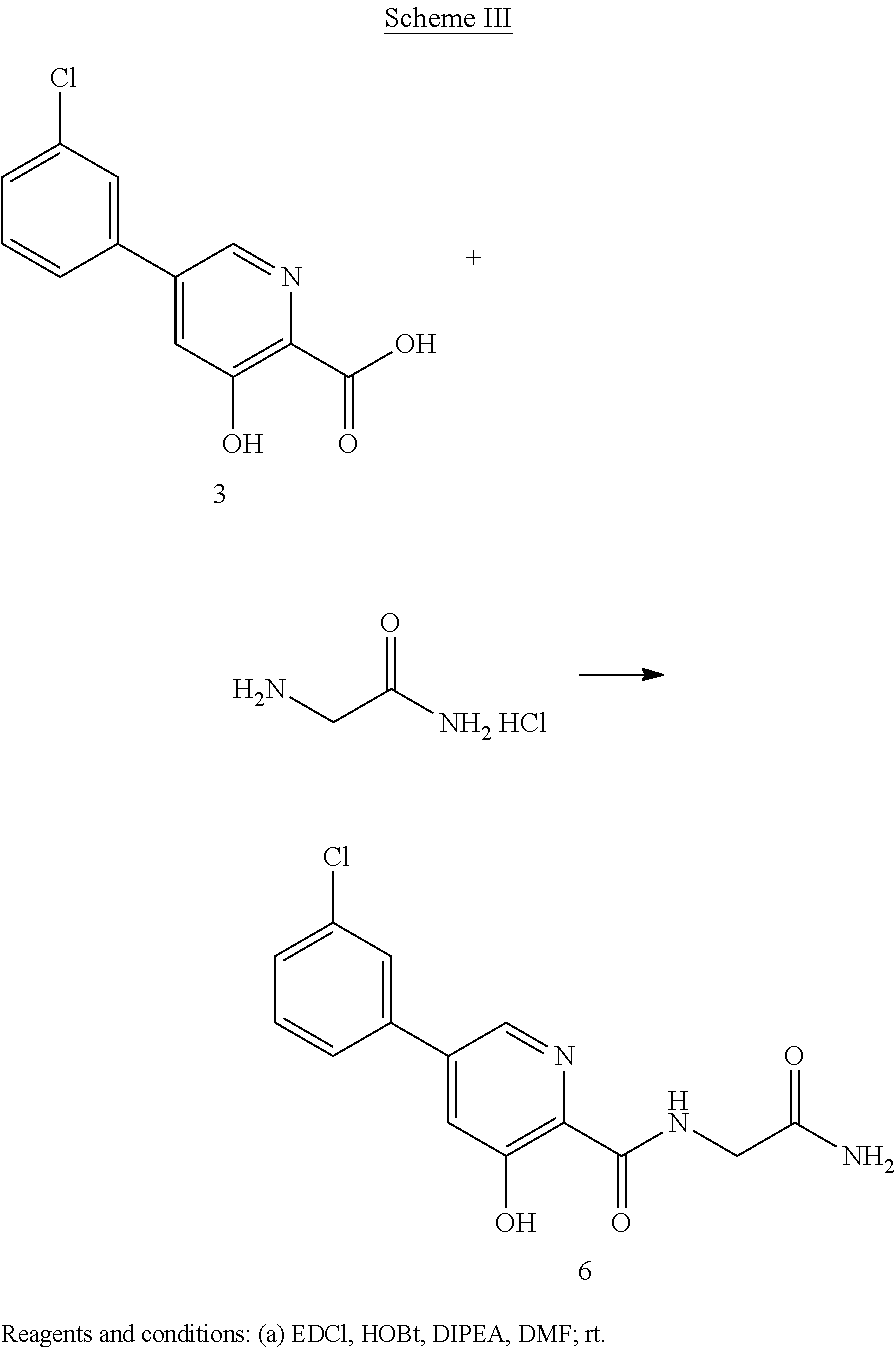




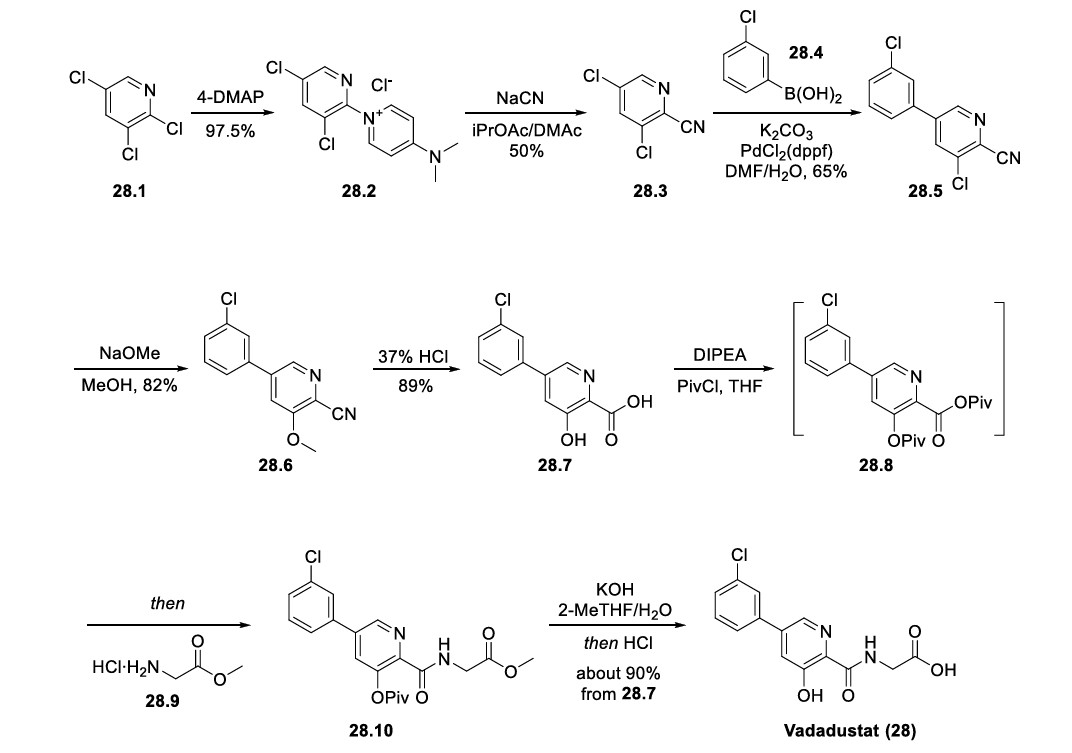











































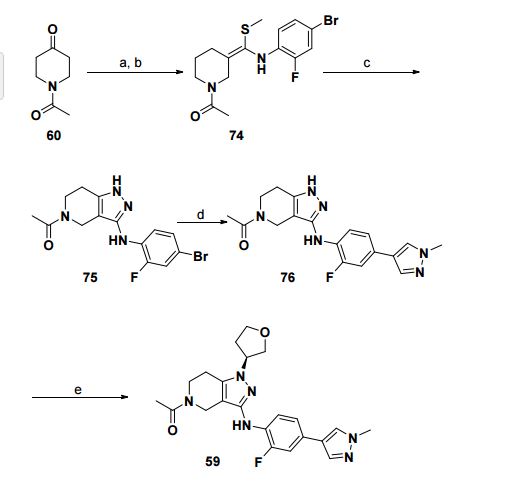

 The presentation will load below
The presentation will load below









 Dr Gautam Sethi
Dr Gautam Sethi