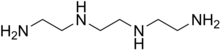
TRIENTINE
- Molecular Formula C6H18N4
- Average mass 146.234 Da
112-24-3 CAS
曲恩汀, KD-034, MK-0681, MK-681, TECZA, TETA, TJA-250
1,2-Ethanediamine, N1,N2-bis(2-aminoethyl)-
1,8-diamino-3,6-diazaoctane
TRIENTINE HYDROCHLORIDE
- Molecular Formula C6H19ClN4
- Average mass 182.695 Da
38260-01-4 CAS
Launched – 1986 VALEANT, WILSONS DISEASE


塩酸トリエンチン
Trientine Hydrochloride

C6H18N4 2HCl : 219.16
2HCl : 219.16
[38260-01-4]
UPDATE CDSCO INDIA Trientine 08.06.2021 APPROVED
Trientine Tetrahydrochloride bulk and
Trientine Tetrahydrochloride capsules 333 mg
(Each capsule contains Trientine
tetrahydrochloride 333mg equivalent to
Trientine 167mg base)
For the treatment of Wilson’s disease
(hepatolenticular degeneration) in patients
intolerant to Penicillamine. It should be
used when continued treatment with
Penicillamine is no longer possible because
of intolerable or life endangering side
effects.
Aton Pharma, a subsidiary of Valeant Pharmaceuticals, has developed and launched Syprine, a capsule formulation of trientine hydrochloride, for treating Wilson disease.

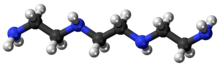
Triethylenetetramine, abbreviated TETA and trien and also called trientine (INN), is an organic compound with the formula [CH2NHCH2CH2NH2]2. This oily liquid is colorless but, like many amines, assumes a yellowish color due to impurities resulting from air-oxidation. It is soluble in polar solvents. The branched isomer tris(2-aminoethyl)amine and piperazine derivatives may also be present in commercial samples of TETA.[1]
Trientine hydrochloride is a metal antagonist that was first launched by Merck, Sharp & Dohme in the U.S. in 1986 under the brand name Syprine for the oral treatment of Wilson’s disease.
Orphan drug designation has also been assigned in the U.S. for the treatment of patients with Wilson’s disease who are intolerant or inadequately responsive to penicillamine and in the E.U. by Univar for the treatment of Wilson’s disease

By condensation of ethylenediamine (I) with 1,2-dichloroethane (II)
Trientine hydrochloride is N,N’-bis (2-aminoethyl)-1,2-ethanediamine dihydrochloride. It is a white to pale yellow crystalline hygroscopic powder. It is freely soluble in water, soluble in methanol, slightly soluble in ethanol, and insoluble in chloroform and ether.
The empirical formula is C6H18N4·2HCI with a molecular weight of 219.2. The structural formula is:
NH2(CH2)2NH(CH2)2NH(CH2)2NH2•2HCI
Trientine hydrochloride is a chelating compound for removal of excess copper from the body. SYPRINE (Trientine Hydrochloride) is available as 250 mg capsules for oral administration. Capsules SYPRINE contain gelatin, iron oxides, stearic acid, and titanium dioxide as inactive ingredients.

Production
TETA is prepared by heating ethylenediamine or ethanolamine/ammonia mixtures over an oxide catalyst. This process gives a variety of amines, which are separated by distillation and sublimation.[2]
Uses
The reactivity and uses of TETA are similar to those for the related polyamines ethylenediamine and diethylenetriamine. It was primarily used as a crosslinker (“hardener”) in epoxy curing.[2]
The hydrochloride salt of TETA, referred to as trientine hydrochloride, is a chelating agent that is used to bind and remove copper in the body to treat Wilson’s disease, particularly in those who are intolerant to penicillamine. Some recommend trientine as first-line treatment, but experience with penicillamine is more extensive.[3]
Coordination chemistry
TETA is a tetradentate ligand in coordination chemistry, where it is referred to as trien.[4] Octahedral complexes of the type M(trien)Cl3 can adopt several diastereomeric structures, most of which are chiral.[5]
Trientine, chemically known as triethylenetetramine or N,N’-bis(2-aminoethyl)-l,2-ethanediamine belongs to the class of polyethylene polyamines. Trientine dihydrochloride is a chelating agent which is used to bind and remove copper in the body in the treatment of Wilson’s disease.

Trientine dihydrochloride (1)
Trientine dihydrochloride formulation, developed by Aton with the proprietary name SYPRINE, was approved by USFDA on November 8, 1985 for the treatment of patients with Wilson’s disease, who are intolerant to penicillamine. Trientine dihydrochloride, due to its activity on copper homeostasis, is being studied for various potential applications in the treatment of internal organs damage in diabetics, Alzheimer’s disease and cancer.
Various synthetic methods for preparation of triethylenetetramine (TETA) and the corresponding dihydrochloride salt have been disclosed in the prior art.
U.S. 4,806,517 discloses the synthesis of triethylenetetramine from ethylenediamine and monoethanolamine using Titania supported phosphorous catalyst while U.S. 4,550,209 and U.S. 5,225,599 disclose catalytic condensation of ethylenediamine and ethylene glycol for the synthesis of linear triethylenetetramine using catalysts like zirconium trimethylene diphosphonate, or metatungstate composites of titanium dioxide and zirconium dioxide.
U.S. 4,503,253 discloses the preparation of triethylenetetramine by reaction of an alkanolamine compound with ammonia and an alkyleneamine having two primary amino groups in the presence of a catalyst, such as supported phosphoric acid wherein the support is comprised of silica, alumina or carbon.
The methods described above for preparation of triethylenetetramine require high temperatures and pressure. Further, due to the various possible side reactions and consequent associated impurities, it is difficult to control the purity of the desired amine.
CN 102924289 discloses a process for trientine dihydrochloride comprising reduction of Ν,Ν’-dibenzyl-,N,N’-bis[2-(l,3-dioxo-2H-isoindolyl)ethyl]ethanediamine using hydrazine hydrate to give N,N’-dibenzyl-,N,N’-bis(2-aminoethyl)ethanediamine, which, upon condensation with benzyl chloroformate gave N,N’-dibenzyl-,N,N’-bis[2-(Cbz-amino)ethyl]ethanediamine, and further reductive deprotection to give the desired compound.
CS 197,093 discloses a process comprising reaction of triethylenetetramine with concentrated hydrochloric acid to obtain the crystalline tetrahydrochlonde salt. Further reaction of the salt with sodium ethoxide in solvent ethanol, filtration of the solid sodium chloride which is generated in the process, followed by slow cooling and crystallization of the filtrate provided the dihydrochloride salt. Optionally, aqueous solution of the tetrahydrochloride salt was passed through a column of an anion exchanger and the eluate containing free base was treated with a calculated amount of the tetrahydrochloride, evaporated, and the residue was crystallized from aqueous ethanol to yield the dihydrochloride salt.
The process is quite circuitous and cumbersome, requiring use of strong bases, filtration of sodium chloride and results in yields as low as 60%.
US 8,394,992 discloses a method for preparation of triethylenetetramine dihydrochloride wherein tertiary butoxycarbonyl (boc) protected triethylenetetramine is first converted to its tetrahydrochloride salt using large excess of hydrochloric acid in solvent isopropanol, followed by treatment of the resulting tetrahydrochloride salt with a strong base like sodium alkoxide to produce the amine free base (TETA) and sodium chloride salt in anhydrous conditions. The free amine is extracted with tertiary butyl methyl ether (TBME), followed by removal of sodium chloride salt and finally the amine free base TETA is treated with hydrochloric acid in solvent ethanol to give trientine hydrochloride salt.
PATENT
WO-2017046695

EXAMPLES
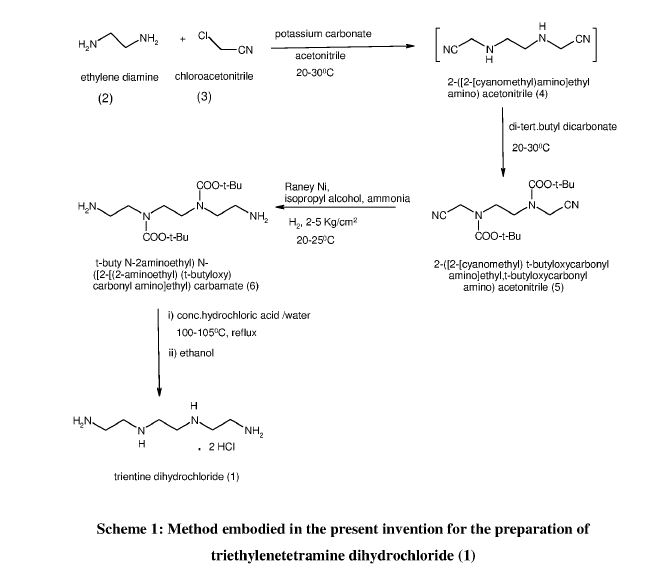
Example 1: Preparation of 2-([2-[cyanomethyl]-t-butyloxycarbonylamino]ethyl- 1-butyloxy carbonylamino)acetonitrile (5)
Potassium carbonate (481.9 g) was added to a stirred mixture of ethylenediamine (100.0 g) in acetonitrile (800 ml) and cooled to around 10°C. Chloroacetonitrile (263.8 g) was gradually added at same temperature and stirred at 25-30°C, till completion of the reaction, as monitored by HPLC. The mixture was cooled to 5-15°C and Boc-anhydride (762. lg) was added to it, followed by stirring at the same temperature. The temperature was raised to 25-30°C and the mass was stirred till completion of the reaction, as monitored by HPLC.
The reaction mass was filtered and the filtrate was concentrated. Toluene was added to the residue, and the mixture was heated to around 70°C followed by cooling and filtration to give 2-([2-[cyanomethyl)-t-butyloxycarbonylamino]ethyl-t-butyloxycarbonylamino) acetonitrile (5).
Yield: 506.8 g
% Yield: 89.9 %
Example 2: Preparation of t-butyl( N-2-aminoethyl)N-([2-[(2-aminoethyl)t-butyloxy)carbonylamino] ethyl) carbamate (6)
Raney nickel (120.0 g) in isopropanol (100 ml) was charged into an autoclave, followed by a mixture of Compound 5 (200 g) in isopropanol (400 ml). Cooled ammonia solution prepared by purging ammonia gas in 1400 ml isopropanol, equivalent to 125 g ammonia was gradually charged to the autoclave and the reaction was carried out around 15-25°C under hydrogen pressure of 2-5 Kg/cm2.
After completion of the reaction, as monitored by HPLC, the mass was filtered, concentrated, and methyl tertiary butyl ether was added to the residue. The mixture was heated to around 50°C, followed by cooling of the mass, stirring, optional seeding with compound 6 and filtration to give tertiary butyl-(N-2-aminoethyl)N-([2-[(2-aminoethyl)-(tert-butyloxy) carbonylamino] ethyl) carbamate.
Yield: 174 g
%Yield: 85 %
Example 3: Preparation of triethylenetetramine dihydrochloride (1)
Concentrated hydrochloric acid (121.5 g) was gradually added to a stirred mixture of tertiary-butyl-N-(2-aminoethyl)-N-2-[(2-aminoethyl)-(tert-butoxy) carbonyl] amino] ethyl} carbamate (Compound 6, 200.0 g) and water (1400 ml) at 20-30°C. The reaction mixture was heated in the temperature range of 100-105°C till completion of the reaction, as monitored by HPLC, with optionally distilling out water, if so required.
The reaction mass was concentrated and ethanol (600 ml) was added to the residue, followed by heating till a clear solution was obtained. The reaction mixture was gradually cooled with stirring, filtered and dried to provide triethylenetetramine dihydrochloride (1).
Yield: 88.9 g, (70 %)
Purity : > 99%
Patent
https://www.google.com/patents/US8394992
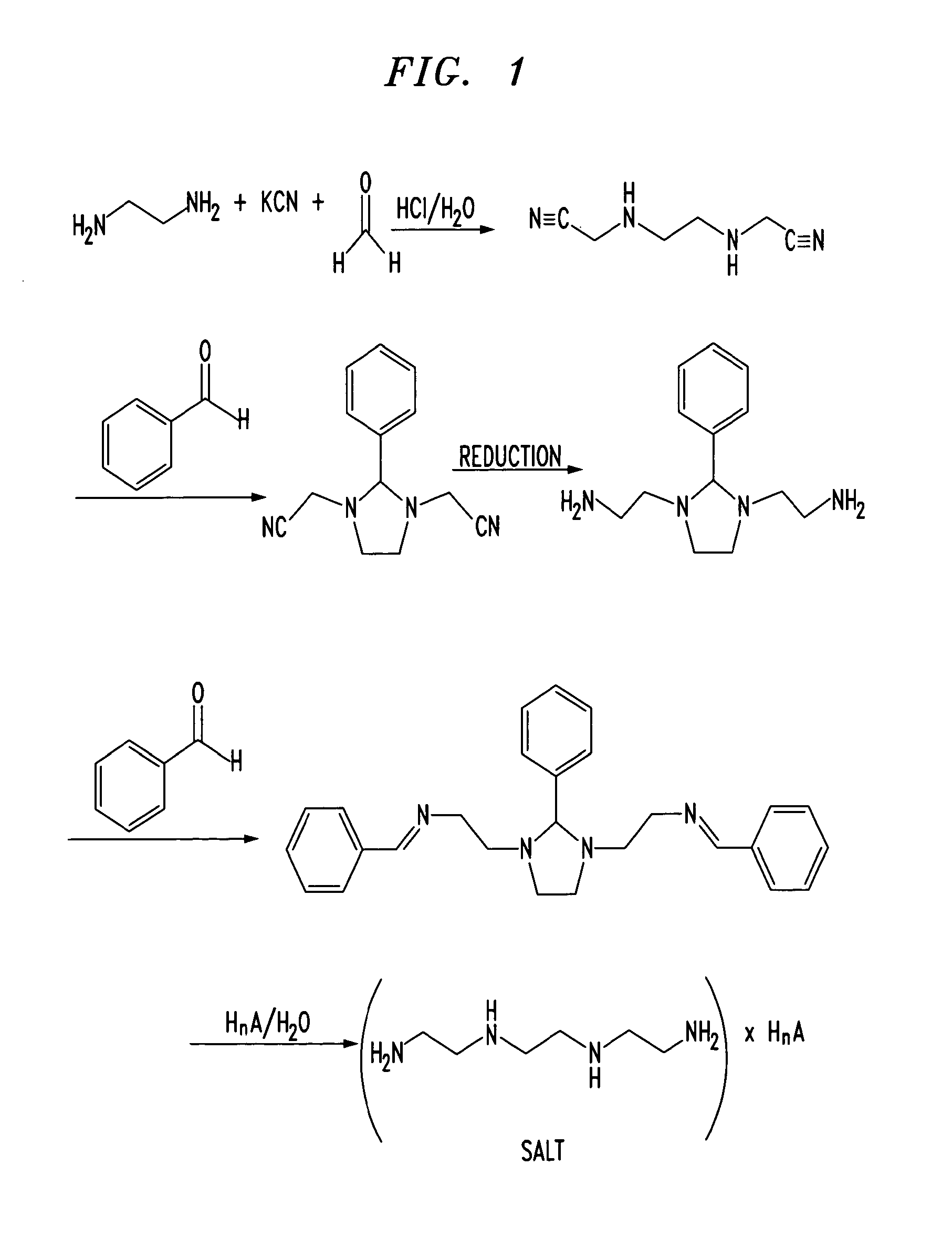
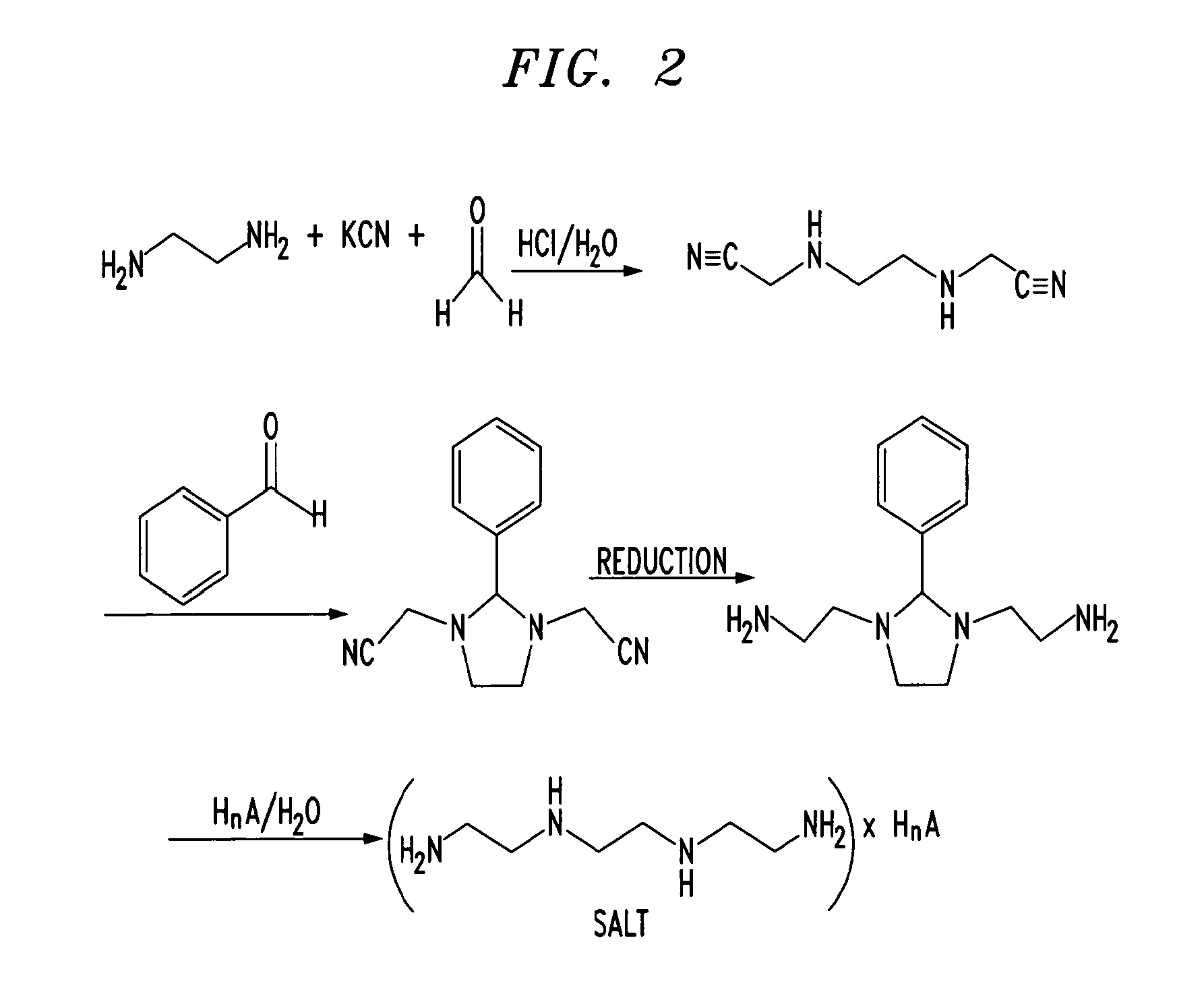
Trientine was said to be used in the synthesis of benzylidene-(2-{3-[2-(benzylidene-amino)-ethyl]-2-phenyl-imidazolidin-1-yl}-ethyl)-amine in French Patent No. FR2810035 to Guilard et al. Cetinkaya, E., et al., “Synthesis and characterization of unusual tetraminoalkenes,” J. Chem. Soc. 5:561-7 (1992), is said to be directed to synthesis of benzylidene-(2-{3-[2-(benzylidene-amino)-ethyl]-2-phenyl-imidazolidin-1-yl}-ethyl)-amine from trientine, as is Araki T., et al., “Site-selective derivatization of oligoethyleneimines using five-membered-ring protection method,” Macromol., 21:1995-2001 (1988). Triethylenetetramine may reportedly also be used in the synthesis of N-methylated triethylenetetramine, as reported in U.S. Pat. No. 2,390,766, to Zellhoefer et al.
Synthesis of polyethylenepolyamines, including triethylenetetramines, from ethylenediamine and monoethanolamine using pelleted group IVb metal oxide-phosphate type catalysts was reported by Vanderpool et al. in U.S. Pat. No. 4,806,517. Synthesis of triethylenetetramine from ethylenediamine and ethanolamine was also proposed in U.S. Pat. No. 4,550,209, to Unvert et al. U.S. Pat. No. 5,225,599, to King et al. is said to be directed to the synthesis of linear triethylene tetramine by condensation of ethylenediamine and ethylene glycol in the presence of a catalyst. Joint production of triethylenetetramine and 1-(2-aminoethyl)-aminoethyl-piperazine was proposed by Borisenko et al. in U.S.S.R. Patent No. SU1541204. U.S. Pat. No. 4,766,247 and European Patent No. EP262562, both to Ford et al., reported the preparation of triethylenetetramine by reaction of an alkanolamine compound, an alkaline amine and optionally either a primary or secondary amine in the presence of a phosphorous containing catalyst, for example phosphoric acid on silica-alumina or Group IIIB metal acid phosphate, at a temperature from about 175° C. to 400° C. under pressure. These patents indicate that the synthetic method used therein was as set forth in U.S. Pat. No. 4,463,193, to Johnson. The Ford et al. ‘247 patent is also said to be directed to color reduction of polyamines by reaction at elevated temperature and pressure in the presence of a hydrogenation catalyst and a hydrogen atmosphere. European Patent No. EP450709 to King et al. is said to be directed to a process for the preparation of triethylenetetramine and N-(2-aminoethyl)ethanolamine by condensation of an alkylenamine and an alkylene glycol in the presence of a condensation catalyst and a catalyst promoter at a temperature in excess of 260° C.
Russian Patent No. RU2186761, to Zagidullin, proposed synthesis of diethylenetriamine by reaction of dichloroethane with ethylenediamine. Ethylenediamine has previously been said to have been used in the synthesis of N-carboxylic acid esters as reported in U.S. Pat. No. 1,527,868, to Hartmann et al.
Japanese Patent No. 06065161 to Hara et al. is said to be directed to the synthesis of polyethylenepolyamines by reacting ethylenediamine with ethanolamine in the presence of silica-treated Nb205 supported on a carrier. Japanese Patent No. JP03047154 to Watanabe et al., is said to be directed to production of noncyclic polyethylenepolyamines by reaction of ammonia with monoethanolamine and ethylenediamine. Production of non-cyclic polyethylenepolyamines by reaction of ethylenediamine and monoethanolamine in the presence of hydrogen or a phosphorous-containing substance was said to be reported in Japanese Patent No. JP03048644. Regenerative preparation of linear polyethylenepolyamines using a phosphorous-bonded catalyst was proposed in European Patent No. EP115,138, to Larkin et al.
A process for preparation of alkyleneamines in the presence of a niobium catalyst was said to be provided in European Patent No. 256,516, to Tsutsumi et al. U.S. Pat. No. 4,584,405, to Vanderpool, reported the continuous synthesis of essentially noncyclic polyethylenepolyamines by reaction of monoethanolamine with ethylenediamine in the presence of an activated carbon catalyst under a pressure between about 500 to about 3000 psig., and at a temperature of between about 200° C. to about 400° C. Templeton, et al., reported on the preparation of linear polyethylenepolyamides asserted to result from reactions employing silica-alumina catalysts in European Patent No. EP150,558.
Production of triethylenetetramine dihydrochloride was said to have been reported in Kuhr et al., Czech Patent No. 197,093, via conversion of triethylenetetramine to crystalline tetrahydrochloride and subsequently to triethylenetetramine dihydrochloride. “A study of efficient preparation of triethylenetetramine dihydrochloride for the treatment of Wilson’s disease and hygroscopicity of its capsule,” Fujito, et al., Yakuzaigaku, 50:402-8 (1990), is also said to be directed to production of triethylenetetramine.
Preparation of triethylenetetramine salts used for the treatment of Wilson’s disease was said to be reported in “Treatment of Wilson’s Disease with Triethylene Tetramine Hydrochloride (Trientine),” Dubois, et al., J. Pediatric Gastro. & Nutrition, 10:77-81 (1990); “Preparation of Triethylenetetramine Dihydrochloride for the Treatment of Wilson’s Disease,” Dixon, et al., Lancet, 1(1775):853 (1972); “Determination of Triethylenetetramine in Plasma of Patients by High-Performance Liquid Chromatography,” Miyazaki, et al., Chem. Pharm. Bull., 38(4):1035-1038 (1990); “Preparation of and Clinical Experiences with Trien for the Treatment of Wilson’s Disease in Absolute Intolerance of D-penicillamine,” Harders, et al., Proc. Roy. Soc. Med., 70:10-12 (1977); “Tetramine cupruretic agents: A comparison in dogs,” Allen, et al., Am. J. Vet. Res., 48(1):28-30 (1987); and “Potentiometric and Spectroscopic Study of the Equilibria in the Aqueous Copper(II)-3,6-Diazaoctane-1,8-diamine System,” Laurie, et al., J.C.S. Dalton, 1882 (1976).
Preparation of Triethylenetetramine Salts by Reaction of Alcohol Solutions of Amines and acids was said to be reported in Polish Patent No. 105793, to Witek. Preparation of triethylenetetramine salts was also asserted in “Polycondensation of polyethylene polyamines with aliphatic dicarboxylic acids,” Witek, et al., Polimery, 20(3):118-119 (1975).
Baganz, H., and Peissker, H., Chem. Ber., 1957; 90:2944-2949; Haydock, D. B., and Mulholland, T. P. C., J. Chem. Soc., 1971; 2389-2395; and Rehse, K., et al., Arch. Pharm., 1994; 393-398, report on Strecker syntheses. Use of Boc and other protecting groups has been described. See, for example, Spicer, J. A. et al., Bioorganic & Medicinal Chemistry, 2002; 10: 19-29; Klenke, B. and Gilbert, I. H., J. Org. Chem., 2001; 66: 2480-2483.
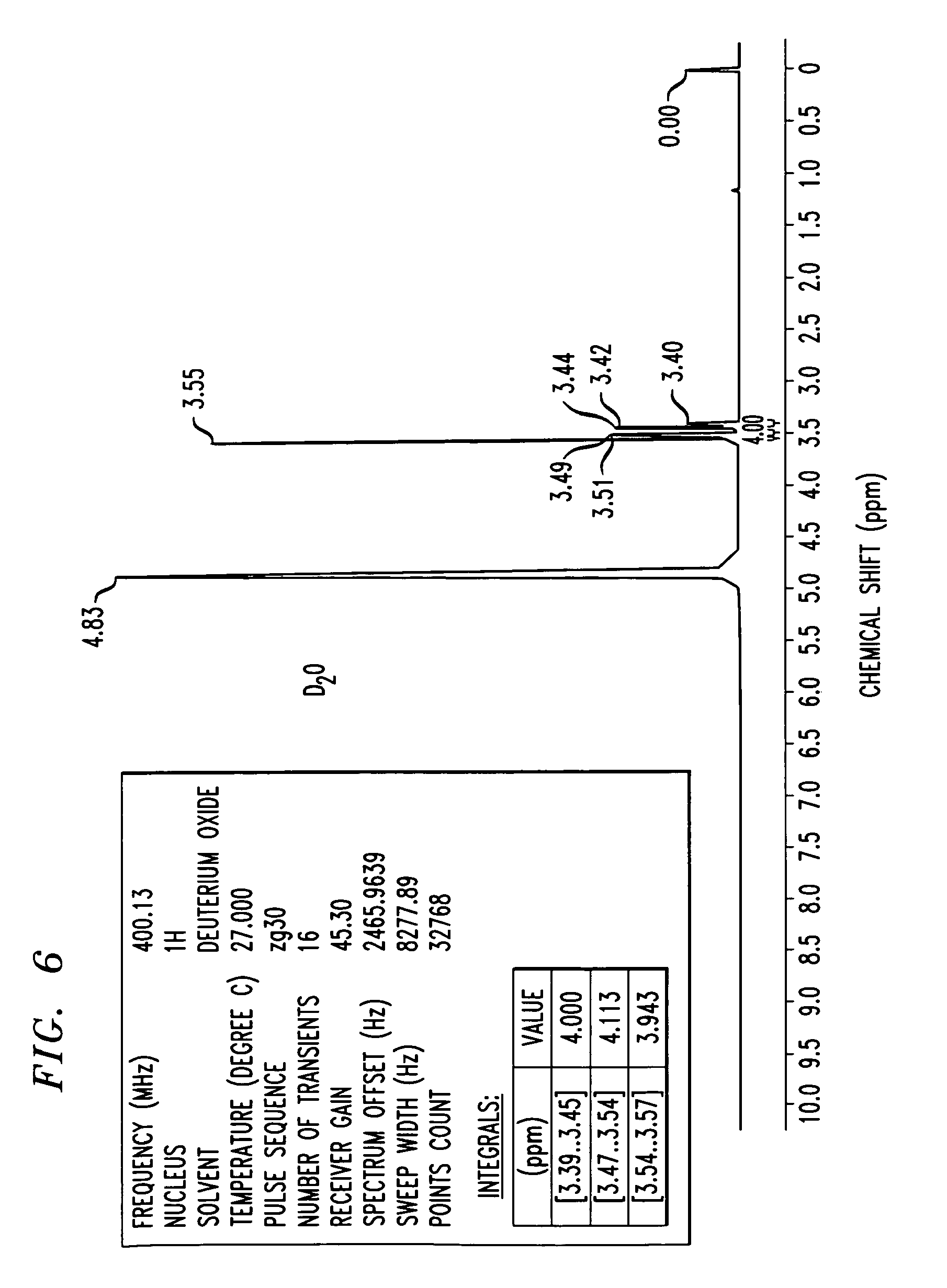
FIG. 6 shows an 1H-NMR spectrum of a triethylenetetramine hydrochloride salt in D2O, as synthesized in Example 3. NMR values include a frequency of 400.13 Mhz, a 1H nucleus, number of transients is 16, points count of 32768, pulse sequence of zg30, and sweep width of 8278.15 H

CLIP
Click to access JP17e_1.pdf
Method of purification: Dissolve Trientine Hydrochloride in water while warming, and recrystallize by addition of ethanol (99.5). Or dissolve Trientine Hydrochloride in water while warming, allow to stand after addition of activated charcoal in a cool and dark place for one night, and filter. To the filtrate add ethanol (99.5), allow to stand in a cool and dark place, and recrystallize. Dry the crystals under reduced pressure not exceeding 0.67 kPa at 409C until ethanol odor disappears.
References
- “Ethyleneamines” (PDF). Huntsman. 2007.
- ^ Jump up to:a b Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. (2005). “Amines, Aliphatic”. Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a02_001.
- Jump up^ Roberts, E. A.; Schilsky, M. L. (2003). “A practice guideline on Wilson disease” (pdf). Hepatology. 37 (6): 1475–1492. doi:10.1053/jhep.2003.50252. PMID 12774027.
- Jump up^ von Zelewsky, A. (1995). Stereochemistry of Coordination Compounds. Chichester: John Wiley. ISBN 047195599X.
- Utsuno, S.; Sakai, Y.; Yoshikawa, Y.; Yamatera, H. (1985). “Three Isomers of the Trans-Diammine-[N,N′-bis(2-Aminoethyl)-1,2-Ethanediamine]-Cobalt(III) Complex Cation”. Inorganic Syntheses. 23: 79–82. doi:10.1002/9780470132548.ch16.
Triethylenetetramine
|
 |
 |
| Names |
Other names
N, N’-Bis(2-aminoethyl)ethane-1,2-diamine; TETA; Trien; Trientine ( INN); Syprine (brand name)
|
| Identifiers |
|
|
|
|
|
|
|
|
605448 |
| ChEBI |
|
| ChemSpider |
|
| ECHA InfoCard |
100.003.591 |
| EC Number |
203-950-6 |
|
|
27008 |
| KEGG |
|
| MeSH |
Trientine |
|
|
|
| RTECS number |
YE6650000 |
| UNII |
|
| UN number |
2259 |
|
|
|
|
| Properties |
|
|
C6H18N4 |
| Molar mass |
146.24 g·mol−1 |
| Appearance |
Colorless liquid |
| Odor |
Fishy, ammoniacal |
| Density |
982 mg mL−1 |
| Melting point |
−34.6 °C; −30.4 °F; 238.5 K |
| Boiling point |
266.6 °C; 511.8 °F; 539.7 K |
|
|
Miscible |
| log P |
1.985 |
| Vapor pressure |
<1 Pa (at 20 °C) |
|
|
1.496 |
| Thermochemistry |
|
|
376 J K−1 mol−1 (at 60 °C) |
| Pharmacology |
|
|
A16AX12 (WHO) |
| Hazards |
| GHS pictograms |
  |
| GHS signal word |
DANGER |
|
|
H312, H314, H317, H412 |
|
|
P273, P280, P305+351+338, P310 |
|
|
 C C |
| R-phrases |
R21, R34, R43, R52/53 |
| S-phrases |
(S1/2), S26, S36/37/39, S45 |
| Flash point |
129 °C (264 °F; 402 K) |
| Lethal dose or concentration (LD, LC): |
|
|
- 550 mg kg−1 (dermal, rabbit)
- 2.5 g kg−1 (oral, rat)
|
| Related compounds |
|
Related amines
|
|
|
Related compounds
|
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
///////////////TRIENTINE, 112-24-3, 曲恩汀 , KD-034 , MK-0681, MK-681, TECZA, TETA, TJA-250, Orphan drug
NCCNCCNCCN

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO

































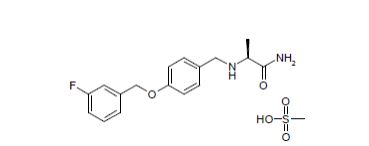
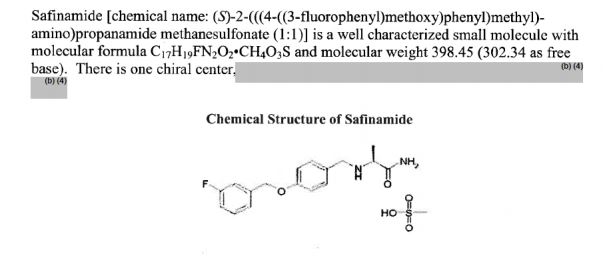











 …………….BASE
…………….BASE …………MESYLATE
…………MESYLATE










{kind=link}