
Home » Uncategorized (Page 97)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
NEW DRUG APPROVALS STATS TODAY
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
FLAGS AND HITS
DR ANTHONY MELVIN CRASTO

ORGANIC SPECTROSCOPY
New comprehensive GMP Inspection Database available
DRUG REGULATORY AFFAIRS INTERNATIONAL
Just recently a so called “inspection tracker” was launched by Health Canada. Now, the agency offers an additional database which contains 3,821 inspections (per March 2015) which have been performed since 2012 – many of them outside Canada, e.g. in Europe or Asia. The information is available in an online database. The use of the database is very easy and search results are excellent.
By using the database even inspections in progress can be displayed. This is a service no other agency can provide. The database also offers further information about past inspections at the same production site. No further search is necessary because the information about past inspections will be displayed in the search result for a given production site. The rating of the inspection is also provided, and in case of GMP non compliance a detailed and very structured information about the findings is provided. The quality…
View original post 157 more words
The “Industry Coalition” gives practical advice for the control of elemental impurities in active substances and excipients
DRUG REGULATORY AFFAIRS INTERNATIONAL
.jpg)
The requirements of the “Guideline for Elemental Impurities ICH Q3D” published in December of last year mean a considerable expense for the affected pharmaceutical companies and drug manufacturers in terms of laboratory and personnel upgrading (see also our news about “ICH Q3D – Elemental Impurities” of 07 January 2015). In addition, the deadlines for the implementation of this guideline are quite tight. (June 2016 for newly approved drugs and December 2017 for already approved drugs, see our news “CHMP adopts ICH Q3D Guideline as “Scientific Guideline” of 21 January 2015).
In the March issue of “Pharmaceutical Technology Europe”, an article of the “Industry Coalition” has been published with the title “Implementation of ICH Q3D Elemental Impurities Guideline: Challenges and Opportunities“, which is intended to support the efffected companies with a number of pragmatic pieces of advice in the implementation of these requirements.
View original post 275 more words
What is SMU-B?

cas 1253286-89-3
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 5-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B
or is it
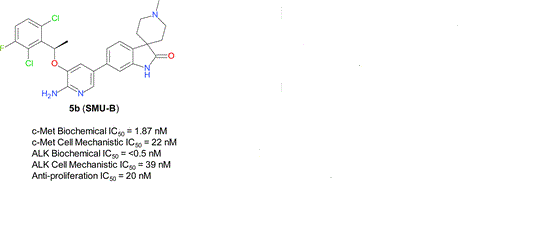
1253286-90-6
Spiro[3H-indole-3,4′-piperidin]-2(1H)-one, 6-[6-amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridinyl]-1′-methyl-
SMU-B

A series of novel aminopyridyl/pyrazinyl-substituted spiro[indoline-3,4′-piperidine]-2-ones were designed, synthesized, and tested in various in vitro/in vivo pharmacological and antitumor assays. 6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B) was identified as a potent, highly selective, well-tolerated, and orally efficacious c-Met/ALK dual inhibitor, which showed pharmacodynamics effect by inhibiting c-Met phosphorylation in vivo and significant tumor growth inhibitions (>50%) in GTL-16 human gastric carcinoma xenograft models.
see..http://pubs.acs.org/doi/abs/10.1021/ml400203d
cas 1253286-90-6
6-[6-Amino-5-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-3-pyridyl]-1′-methylspiro[indoline-3,4′-piperidine]-2-one (compound 5b or SMU-B)
SEE

1_4,3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-nitro-approved P set

obtained in Step 1-3 (IS) -I- (2,6- dichloro-3-fluorophenyl) ethanol (2. 09g, IOmmol) was dissolved in dry THF (80 ml). Then, at room temperature under a nitrogen atmosphere, a solution of 3-hydroxy-2-nitro-pyridine (1.54g, llmmol) and triphenylphosphine (3. 409g, 13mmol), and so is completely dissolved, cooled to 0 ° C, was added Diisopropyl azodicarboxylate (DIAD, 2.63g, 13mmol), After the addition, the mixture was stirred at 0 ° C for 16 hours, the solvent was removed by rotary evaporation and the oily residue was purified by silica gel column chromatography (petroleum ether / ethyl acetate : 4/1) to give the desired product as a white solid (3. 046g, yield: 92%) o 1H-NMR (CDClySOOMHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), 6 . 10 (q, J = 6. 4Hz, 1H), 7. 09 (dd, J = 7. 6,8. 8Hz, 1H), 7. 21 (dd, J = 8. 4, I. 2Hz, 1H ), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H),
7. 37 (dd, J = 4. 8,8. OHz, 1H), 8. 04 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 330 94 [M + H, 35C1,35Cl], 332. 92 [M + H, 35Cl, 37Cl].
1_5,3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere based grant given P

to take steps 1-4 to get the 3 – [(lR) _l- (2,6_ dichloro-3-fluorophenyl) ethoxy] -2_ nitro than Li Jie (2. 649g, 8mmol) was dissolved in ethanol (15mL) was added iron powder (3. 575g, 64mmol) were mixed under nitrogen with vigorous stirring at 90 ° C oil bath, was added via syringe 0.8mL IM HCl (aq), after 10 minutes, was added 0. 8mL IMHCl (aq). Stirring was continued for 30 minutes, TLC showed the reaction. Cooled to room temperature, filtered through Celite, the filter residue washed with ethanol (3X IOmL). The combined organic phase was removed by rotary evaporation of the solvent gave the desired product as a light brown solid (2. 41g, yield: 100%) o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H ), 5. 03 (s, br, 2H), 6. 01 (q, J = 6. 8Hz, 1H), 6. 47 (dd, J = 4. 8,7. 6Hz, 1H), 6. 70 (d, J = 8. OHz, 1H), 7. 05 (t, J = 8. 8Hz, 1H), 7. 28 (dd, J = 4. 0,8. 0Hz, 1H), 7. 57 ( d, J = 5.2Hz, lH). Mass spectrum m / z:. 301 00 [M + H, 35Cl, 35Cl], 302. 77 [M + H, 35Cl, 37Cl].
l-6,5_ desert _3_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -2_ atmosphere base than Li Jie

The steps 1-5 obtained 3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl atmosphere than Li Jie (1.506g, 5mmol) dissolved in acetonitrile (20mL) in. Then, at 0 ° C and the degree of stirring in added portionwise N- bromosuccinimide (0.908g, 5. Lmmol), After the addition, stirring was continued for 30 minutes. The solvent was removed by rotary evaporation, the crude product was obtained as a white solid was the desired product (1.045g, yield: 55%) was purified by column chromatography on silica gel. 1H-NMR (⑶Cl3,500MHz): 8 (ppm) I. 81 (d, J = 6. 8Hz, 3H), 4 85 (s, br, 2H), 6 98 (q, J = 6. 8Hz.. , 1H), 6. 82 (d, J =
2. 0Hz, 1H), 7. 08 (t, J = 8. 4Hz, 1H), 7. 31 (dd, J = 4. 8,8. 8Hz, 1H), 7. 65 (d, J = 2 . OHz, 1H). Mass spectrum m / z:… 378 84 [M + H, 35Cl, 35Cl, 79Br], 380 82 [M + H, 35Cl, 35Cl, 81Br or 35Cl, 37Cl, 79Br], 382 80 [M + H, 35Cl , 37Cl, 81Bror 37Cl, 37Cl, 79Br].
Step 2, I ‘- methyl-5- (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one

2-1,5- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] _2_ one

[0300] 5-bromo – indol-2-one (I. 272g, 6mmol) was suspended THF (15mL) at, and cooled to -78 ° C, added dropwise with stirring IM NaN (SiMe3) THF solution of 2 (30mL, 30mmol). After the addition was stirred at _78 ° C 30 min, then 2-chloro -N- (2- chloro-ethyl) -N- methyl-ethylamine hydrochloride solid (I. 155g, 6mmol). After the addition stirring was continued for 30 minutes, then warmed to room temperature and stirred for two days. TLC showed the reaction was completed, to the pink suspension was carefully added aqueous 4M hydrochloric acid (IOmL), and then adjusted with concentrated aqueous ammonia to pH ^ 9, and extracted with DCM (3 X 80mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (I. 38g, yield: 78%) was purified. 1H-NMR (CD3ODjOOMHz):. 8 (ppm) I. 86-1 92 (m, 2H), I 94-1 98 (m, 2H), 2 44 (s, 3H), 2 62-…. 2. 68 (m, 2H), 2. 86-2. 91 (m, 2H), 6. 76 (d, J = 7. 6Hz, 1H), 7. 33 (dd, J = I. 2,7 . 6Hz, 1H), 7. 44 (d, J = I. 6Hz, 1H), 7. 81 (s, br, 1H). Mass spectrum m / z:. 294 99 [M + H, 79Br], 296 82 [M + H, 81Br]..
2-2, V – methyl-5- (4,4,5,5-tetramethyl–1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

Under nitrogen, obtained in Step 2-1 to 5-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] _2_ one (147. 6mg, 0. 5mmol) , the United pinacols drop acid unitary purpose (140mg, 0. 55mmol) and acetic acid Bell (147mg, I. 5mmol) in DMSO (0. 2ml) was added in PdCl2 (dppf) • CH2Cl2 (20. 4mg, 0. 025mmol ), to the resulting solution was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 16 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (2mL), extracted with DCM (3X5mL). The organic phases were combined, dried (Na2SO4), and concentrated to give the desired product (170mg, yield: 100%) o MS m / z:. 342 07 [M + H], 343. 08 [M + H, 100%], 344. 11 [M + H].
Step 3,5_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ batch P fixed base] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2_ one
The steps 1-6 5_ desert obtained _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] -2-yl batch atmosphere pyridine (75. 8mg , 0. 2mmol), I’- step 2_2 obtained methyl 5- (4,4,5,5-tetramethyl-l, 3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4′-piperidin] -2-one (82mg, 0. 24mmol) and potassium carbonate (82. 9mg, 0. 6mmol) was dissolved in DME / water mixture solution (4 / 1,2. Oml ). Then, under nitrogen, was added Pd (PPh3) 4 (II. 6mg, 0. Olmmol), to the resulting mixture was bubbled with nitrogen for 2 minutes, and then stirred at 80 ° C of 18 hours. LC-MS showed completion of the reaction, after cooling to room temperature, water (5mL), extracted (3 X IOmL) with DCM. The organic phases were combined, dried (Na2SO4), and concentrated to give the crude product was purified by silica gel column chromatography (7M NH3 in methanol solution / DCM: 5/95) to give the desired product (88. 6mg, yield: 86%) was purified. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I 93-2 02 (m, 4H), 2 44 (s, 3H),…
2. 66-2. 72 (m, 2H), 2. 89-2. 93 (m, 2H), 4. 87 (s, br, 2H), 6. ll (q, J = 6. 4Hz, 1H ), 6. 88 (d, J =
8. OHz, 1H), 6. 94 (d, J = I. 2Hz, 1H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 19 (dd, J = I. 2,8 . OHz, 1H),
7. 31 (m, 1H), 7. 36 (s, 1H), 7. 66 (s, br, 1H), 7. 80 (d, J = 2. OHz, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
Example 2: 6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3_ than Li Jie base] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one

Step I, I ‘- methyl-6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indoline Spray – 3,4 ‘- piperidin] -2_ one
1-1,6- bromo -I ‘- methyl-spiro [indoline-3,4’ – piperidin] -2_ one

As described in Example I steps 2-1 of the method from the commercially available 6-bromo – indol-2-one was prepared, Yield: 82%. Analysis of the data obtained the desired product are = 1H-Nmr (Cd3OdJOOmHz): 8 (ppm) 1.90-1.98 (m, 4H),
2. 44 (s, 3H), 2. 64-2. 68 (m, 2H), 2. 86-2. 92 (m, 2H), 7. 05 (d, J = 2. 0Hz, 1H), 7. 16-7. 21 (m, 2H), 7. 91 (s, br, 1H). Mass spectrum m / z: 295 00 [M + H, 79Br], 296 78 [M + H, 81Br]… [0312] 1-2, 1 ‘- methyl-6- (4,4,5,5-tetramethyl-_1,3,2_ dioxolane Borane _2_ yl) spiro [indoline – 3,4 ‘- piperidin] -2_ one

In the step 1-1 of the obtained 6-bromo -I ‘- methyl-spiro [indoline-_3,4’ – piperidin] -2_ ketone and commercially available linking pinacol boronic ester material, the method of Example I was prepared in accordance with steps 2-2, Yield: 95%. Analysis of the data obtained of the target product are as follows: Mass spectrum m / z:. 342 06 [M + H], 343 04 [M + H, 100%], 344. 12 [M + H]..
Step 2,6_ [6_ atmosphere base _5_ [(IR) -I- (2,6_ two gas -3- gas phenyl) ethoxy] -3 ratio Li Jie base] -I ‘- methyl-spiro [indoline-3,4 ‘- piperidin] -2_ one
Example I steps 1-6 to obtain 5-bromo -3 – [(IR) -I- (2,6- dichloro-3-fluorophenyl) ethoxy] -2-amino- pyridine, I obtained in Example 1-2 of the present embodiment in step ‘- methyl-6- (4,4,5,5-tetramethyl-l, 3,2-dioxolane-2-yl borane) spiro [indoline-_3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 82%. 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 86 (d, J = 6. 4Hz, 3H), I. 91-1 95 (m, 2H), I 97-2 03 (m, 2H… ), 2. 45 (s, 3H), 2. 65-2. 72 (m, 2H), 2. 89-2. 95 (m, 2H), 5. 12 (s, hr, 2H),
6. 12 (q, J = 6. 4Hz, 1H), 6. 94-7. 00 (m, 3H), 7. 06 (t, J = 8. 4Hz, 1H), 7. 31 (m, 1H ), 7. 35 (d, J = 7. 2Hz, 1H), 7. 90 (d, J = 2. 0Hz, 1H), 9. 28 (s, br, 1H). Mass spectrum m / z:.. 515 05 [M + H, 35Cl, 35Cl], 517 03 [M + H, 35Cl, 37Cl].
5- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’–methyl-spiro [indole: 3 [0317] Example morpholine-3,4 ‘- piperidin] -2-one
H2N N


Step I, 5_ desert _3_ (2,6_ two gas -3- integrity oxy) _2_ atmosphere based grant given P
1-1,2,6_ two gas acid gas _3_
Cl OF
Sodium hydroxide (13g, 325mmol) in water (IlOmL) was cooled to _5 ° C was added dropwise under vigorous stirring of liquid bromine (12. 5g, 78. 2mmol), added after the addition of pre-cooled to 10 ° C dioxane (75mL). The above mixture under vigorous stirring was added dropwise a pre-cooled to 5 ° C of I- (2,6- dichloro-3-fluorophenyl) ethanone (5g, 21. 2mmol) in dioxane (330mL) and water (90mL) was added. After the addition, at room temperature for 2 hours Lan Xiang, Xiang Lan then 90 C for 30 minutes. TLC was not shown with the S starting material disappeared, and was acidified with concentrated hydrochloric acid to PH~9. The resulting mixture was rotary evaporated to dryness, added water (20mL), and extracted with diethyl ether (2X80mL), the organic phases were combined, dried (Na2SO4), and concentrated to give an oily product solidified after cooling to a transparent, slightly yellow solid (3. 4g, Yield: 67%). 1H-Nmr (Cdci3AOOmHz):. 8 (ppm) 7. 21 (. Dd, J = 8. 0,8 8Hz, 1H), 7 35 (. Dd, J = 4. 4,9 2Hz, 1H), 9 . 79 (s, br, 1H). Mass spectrum m / z (ES “:. 207 11 [M_H, 35Clj35Cl], 209 10 [MH, 35Cl, 37Cl]..
1-2,2,6–dichloro-3-fluoro-benzyl alcohol
^ Coh
F
[0325] To be filled with 2,6-dichloro-3-fluoro benzoic acid (3g, 14. 35mmol) added dropwise to the flask IM BH3. THF (43mL, 43mmol), added after the mixture was stirred under reflux for 24 hours. TLC showed the reaction was complete, methanol (50mL) to destroy excess borane, and the solvent was distilled off under reduced pressure and the resulting trimethyl borate, the process is repeated twice more to give a viscous product 2. I g, yield: 75% . 1H-Nmr (Cdci3JOOmHz): 8 (ppm) 2. 09 (t, J = 6. 4Hz, 1H), 4. 97 (d, J = 6. 4Hz, 2H), 7 09 (t, J = 8. . 8Hz, 1H), 7. 32 (dd, J = 4. 8,9. 1Hz, 1H). Mass spectrum m / z (ES-):.. 193 08 [M_H, 35Cl, 35Cl], 195 12 [MH, 35Cl, 37Cl].
1-3,3_ (2,6-gas _3_ integrity oxy) _2_ nitro grant given P

Following the procedure of steps 1-4 of Example I, was prepared from 2,6-dichloro-3-fluoro-benzyl alcohol and 3-hydroxy-2-nitropyridine prepared in yield (in this example embodiment steps 1_2) : 90%. 1H-Nmr (Cdci3AOOmHz): 8 (ppm) 5. 45 (s, 2H), 7 20, 7 37 (dd, J = 4. 8. (Dd, J = 8. 0,9 2Hz, 1H.). , 9. 2Hz, 1H), 7. 59 (dd, J = 4. 4,8. 4Hz, 1H),
7. 74 (dd, J = L 2,8. 4Hz, 1H), 8. 17 (dd, J = L 6,4. 4Hz, 1H). Mass spectrum m / z:. 316 89 [M + H, 35Cl, 35Cl], 318. 89 [M + H, 35Cl, 37Cl].
1_4,3_ (2,6-gas _3_ integrity oxy) _2_ atmosphere based grant given P

The method according to Example I step 1_5 from 3- (2,6-gas -3- integrity oxy) _2_ nitro Jie ratio 唳 preparation (in this case, steps 1-3), that Yield: 95% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 65 (s, br, 2H), 5 31 (s, 2H), 6 66 (dd, J = 5. 2,8.. . 0Hz, 1H), 7. 14 (dd, J = I. 2,8. 0Hz, 1H), 7. 18 (dd, J =
8. 4,9. 2Hz, 1H), 7. 37 (dd, J = 4. 8,8. 8Hz, 1H), 7. 73 (dd, J = I. 6,5. 6Hz, 1H). Mass spectrum m / z:. 286 95 [M + H, 35Cl, 35Cl], 288 85 [M + H, 35Cl, 37Cl]..
1-5,5_ desert -3- (2,6-gas -3_ integrity oxy) ~ 2 ~ atmosphere based grant given P

Following the procedure of Example I step 1_6 embodiment, starting from 3- (2,6-gas _3_ integrity yloxy) _2_ atmosphere group given the preparation of the batch P (in the example of the present embodiment in step 1-4), Yield: 60% o 1H-Nmr (Cdci3JOOmHz):. 8 (ppm) 4 68 (s, br, 2H), 5 28 (s, 2H), 7 21 (dd, J = 8. 0,8.. . 8Hz, lH), 7. 24 (dd, J = 2. OHz, 1H), 7. 39 (dd, J = 4. 8,
9. 2Hz, 1H), 7. 78 (d, J = 2. OHz, 1H). Mass spectrum m / z:. 364 83 [M + H, 35Cl, 36Cl, 79Br], 366 77 [M + H], 368 69 [M + H]…
Step 2,5_ [6_ atmosphere base _5_ [(2,6_ two gas -3- gas) methoxy] -3_ than Li Jie base] -I-methyl-spiro [indoline _ 3,4 ‘- piperidin] -2-one
The present embodiment 5_ desert steps 1_5 obtained _3_ (2,6_ two gas _3_ integrity yloxy) pyridine ~ 2 ~ atmosphere, Examples 2-2 obtained in step I I ‘- methyl-5- (4,4,5,5-tetramethyl-borane _1,3,2- dioxolane-2-yl) spiro [indoline-_3,4’ – piperidine ] -2-one, prepared as in Example I Step 3. Yield: 85 V0o 1H-Nmr (Cdci3JOOmHz):.. 8 (ppm) I. 92-2 02 (m, 4H), 2. 43 (s, 3H), 2. 65-2 71 (m, 2H) , 2. 90-2. 91 (m, 2H), 4. 92 (s, br, 2H), 5. 52 (s, 2H), 6. 89 (d, J = 8. 4Hz, 1H), 6 . 90 (d, J = L 2Hz, 1H), 7. 06 (t, J = 8. OHz, 1H), 7. 21 (dd, J = L 2,8. OHz, 1H), 7. 31 ( m, 1H),
7. 37 (s, 1H), 7. 79 (s, br, 1H), 7. 80 (d, J = 2.0Hz, lH). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
6- [6-amino-5 – [(2,6-dichloro-3-fluorophenyl) methoxy] _3_ pyridinyl] -I’- methyl-spiro [indole: 4 [0337] Example morpholine _3,4 ‘- piperidin] -2-one



H2N N
Following the procedure in Example I step of Example 3, the procedure of Example 3 to give 5-bromo-1-5 _3_ (2,6-dichloro-3-fluoro-benzyloxy) -2-amino-pyridine and Step 2 in Example I to give the embodiment 1-2 ‘- methyl-6- (4,4,5,5-tetramethyl-1,3,2-dioxolane Borane 2-yl) spiro [ indoline-3,4 ‘- piperidine] _2_ ester -one, yield:. 78 V0o 1H-Nmr (Cdci3JOOmHz): 8 (ppm) I. 96-2 00 (m, 2H), 2. 01 -2. 12 (m, 2H), 2. 46 (s, 3H), 2. 66-2. 73 (m, 2H), 2. 90-2. 96 (m, 2H), 5. 30 (s , hr, 2H), 6. 94-7. 01 (m, 3H), 7. 07 (t, J =
8. 4Hz, 1H), 7. 30 (m, 1H), 7. 34 (d, J = 7. 2Hz, 1H), 7. 89 (d, J = 2. OHz, 1H), 8. 56 ( s, br, 1H). MS m / z:. 501 06 [M + H, 35Cl, 35Cl], 503 04 [M + H, 35Cl, 37Cl]..
Example 5: 5_ [5_ atmosphere base -6- [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one
J0A = o
. | J: too
[0342] Step 1,5_ desert _2_ atmosphere base _3_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Jie than exposure
Cl 6, / ISL / Br
xy
H2N N
In at 0 ° C, NaH (80mg of NaH in mineral oil, 2mmol) force the mouth (1R) -1_ (2,6- dichloro-3-fluorophenyl) ethanol (418mg, 2mmol. See example Example I Step 1_3) in anhydrous THF (6mL) and stirred for half an hour, a solution of 2-amino-3,5-dibromo-pyrazine (506mg, 2mmol) in THF (6mL) was added. The resulting mixture was warmed to room temperature, heated under reflux for 20 hours. TLC showed the reaction was substantially complete. After cooling to room temperature, water was added (IOmL), the mixture was extracted three times with ethyl acetate (3x20mL), the organic phases were combined, dried, concentrated, and the residue to give 594mg product was purified by column chromatography (l-3Me0H inhexanes), yield: 78%. 1H-NMR (O) Cl3, 500MHz):. 8 (ppm) I. 83 (d, J = 7. 2Hz, 3H), 5. 12 (s, br, 2H), 6 73 (q, J = 6 . 8Hz, 1H), 7. 05 (t, J = 8. OHz, 1H), 7. 28 (dd, J = 4. 8,
8. 8Hz, 1H), 7. 58 (s, 1H). Mass spectrum m / z:. 379 83 [M + H, 35Cl, 35Cl, 79Br], 381. 81 [M + H, 35Cl, 35Cl, 81Br], 383 79 [M + H, 35Cl, 37Cl, 81Br]..
Step 2,5_ [5_ atmosphere base _6_ [(IR) -I- (2,6_ two gas _3_ gas phenyl) ethoxy] Batch-2-yl] -I ‘- A group spiro [indoline-3,4 ‘- piperidin] -2-one
5_ bromide present embodiment obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, implemented I’- methyl step 2-2 obtained in Example I-5 (4,4,5,5-tetramethyl -I, 3,2- dioxolane boron
2-yl) spiro [indoline-3,4 ‘- piperidin] -2-one, prepared as in Example I Step 3. Yield: 54%. 1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 85-1 88 (m, 2H), I 97-2 04 (m, 2H…. ), 2. 46 (s, 3H), 2. 76-2. 82 (m, 2H), 2. 97-3. 02 (m, 2H), 6. 74 (q, J = 6. 4Hz, 1H ), 6. 85 (d, J = 8. OHz, 1H), 7. 15 (t, J = 8. 4Hz, 1H), 7. 41 (dd, J = 4. 8,9. 2Hz, lH) , 7. 54 (dd, J = I. 6,
8. OHz, 1H), 7. 69 (d, J = I. 8Hz, 1H), 7. 81 (dt, J = 2. 0,8. 0Hz, 1H), 7. 87 (s, 1H). Mass spectrum m / z:. 515 92 [M + H, 35Cl, 35Cl], 517. 90 [M + H, 35Cl, 37Cl].
Example 6: 6- [5-amino -6 – [(lR) -l_ (2,6- dichloro _3_ fluorophenyl) ethoxy] pyrazin-2-yl] -I ‘ – methyl-spiro [indoline-3,4 ‘- piperidin] -2-one

The embodiment of Example 5, 5_ bromo obtained in step I _2_ amino _3_ [(IR) -I- (2,6_ dichloro _3_ fluorophenyl) ethoxy] pyrazine, Example I’- methyl-2 obtained in steps 1-2 6- (4,4,5,5-tetramethyl–I, 3,2- dioxolane boron-2-yl) spiro [indole morpholine -3,4’_ piperidin] -2-one, prepared as in Example I Step 3. Yield: 67% 0
1H-NMR (CD3ODjOOMHz): 8 (ppm) I. 85 (d, J = 6. 8Hz, 3H), I 88-1 96 (m, 4H), 2 48 (s… , 3H), 2. 76-2. 82 (m, 2H), 2. 98-3. 05 (m, 2H), 6. 75 (q, J = 6. 4Hz, 1H), 7. 16 (t , J = 8. 8Hz, 1H), 7. 31 (d, J = 2. OHz, 1H), 7. 36-7. 43 (m, 3H), 7. 88 (s, 1H).
Mass spectrum m / z:. 515 99 [M + H, 35Clj35Cl], 517 90 [M + H, 35Cl, 37Cl]..
SEE
Bioorganic & Medicinal Chemistry Letters (2014), 24(16), 3673-3682.
School of Pharmaceutical Sciences, Southern Medical University,



 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com





 Stockholm, Sweden
Stockholm, Sweden Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
Despite the cold weather, public came and enjoyed different activities. The famous chef, Paul Svensson who works in one of the fanciest and most famous …
1.5 lakh views on Top Organic Spectroscopy blog in the world
ORGANIC SPECTROSCOPY INTERNATIONAL
1.5 lakh views on this blog………http://orgspectroscopyint.blogspot.in/
Techniques for Organic Compounds ie NMR, MASS, IR, UV Etc. Starters,
Learners, advanced, all alike, contains content which is basic or advanced,
by Dr Anthony Melvin Crasto, Worlddrugtracker,
email me ……….. amcrasto@gmail.com, call +91 9323115463

1.5 lakh views on this blog………http://orgspectroscopyint.blogspot.in/
FRANCE
.jpg)
Republican march, Place de la République, Paris.



The St. Bartholomew’s Day massacre (1572) was the climax of theFrench Wars of Religion, which were brought to an end by the Edict of Nantes (1598).

One of the Lascaux paintings: a horse – Dordogne, approximately 18,000 BC

With Clovis‘ conversion to Catholicism in 498, theFrankish monarchy, electiveand secular until then, became hereditary and ofdivine right.

Frankish expansion from 481 to 843/870.

The Storming of the Bastille on 14 July 1789 was the starting event of theFrench Revolution.

Louis XIV, the “sun king” was the absolute monarch of France and made France the leading European power.

Napoleon, Emperor of the French, and his Grande Armée built a vast Empire across Europe. He helped spread the French revolutionary ideals and his legal reforms had a major influence worldwide.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
VENLAFAXINE PART 1/3


 SEE
SEEpart 1………http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine.html / https://newdrugapprovals.org/2015/04/09/venlafaxine-part-12/
part 2……..https://newdrugapprovals.org/2015/04/09/venlafaxine-22/
PART 3…..http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine-part-33.html
SEE ALSO
http://www.google.com/patents/WO2008059525A2?cl=en
WILL BE UPDATED………..
Chemistry

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (RS)-1-[2-dimethylamino-1-(4-methoxyphenyl)-ethyl]cyclohexanol | |
| Clinical data | |
| Trade names | Effexor XR, Effexor, Trevilor |
| AHFS/Drugs.com | monograph |
| Licence data | US Daily Med:link |
|
|
|
|
| Oral | |
| Pharmacokinetic data | |
| Bioavailability | 42±15%[1] |
| Protein binding | 27±2% (parent compound), 30±12% (active metabolite,desvenlafaxine)[2] |
| Metabolism | Hepatic (~50% of the parent compound is metabolised on first pass through the liver)[1][2] |
| Half-life | 5±2 h (parent compound for immediate release preparations), 15±6 h (parent compound for extended release preparations), 11±2 h (active metabolite)[1][2] |
| Excretion | Renal (87%; 5% as unchanged drug; 29% asdesvenlafaxine and 53% as other metabolites)[1][2] |
| Identifiers | |
| 93413-69-5 |
|
| N06AX16 | |
| PubChem | CID 5656 |
| DrugBank | DB00285 |
| ChemSpider | 5454 |
| UNII | GRZ5RCB1QG |
| ChEBI | CHEBI:9943 |
| ChEMBL | CHEMBL637 |
| Chemical data | |
| Formula | C17H27NO2 |
| 277.402 g/mol | |

HSQC

1H NMR PREDICT OF HCL

13C NMR PREDICT OF HCL


BASE


External links[Drug information
- U.S. Food and Drug Administration information on Effexor
- Efexor patient information leaflet (UK)
- Effexor XR prescribing information for healthcare professionals (pdf) (USA only) Archived from the original on 17 September 2006.
- Detailed Patient/Parent Information on Effexor
- List of international brand names for Venlafaxine
- U.S. National Library of Medicine: Drug Information Portal -Venlafaxine
Diagnostic tools
Patient experiences
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
VENLAFAXINE PART 2/3

part 1………http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine.html / https://newdrugapprovals.org/2015/04/09/venlafaxine-part-12/
part 2……..https://newdrugapprovals.org/2015/04/09/venlafaxine-22/
PART 3…..http://orgspectroscopyint.blogspot.in/2015/04/venlafaxine-part-33.html
http://www.google.com/patents/WO2008059525A2?cl=en
WILL BE UPDATED………..
………………….

……………………….
PAPER
DOI: 10.1039/C4RA00840E
http://pubs.rsc.org/en/content/articlelanding/2014/ra/c4ra00840e#!divAbstract
A protecting group free asymmetric total synthesis of (−)-venlafaxine is reported. The strategy employs Sharpless epoxidation and regio-selective epoxide ring opening by an in situgenerated Gilman reagent as key steps. This paper reports a 53% overall yield in 6 steps for total synthesis of (−)-venlafaxine.


…………………
http://www.google.com/patents/EP2181982B1?cl=en
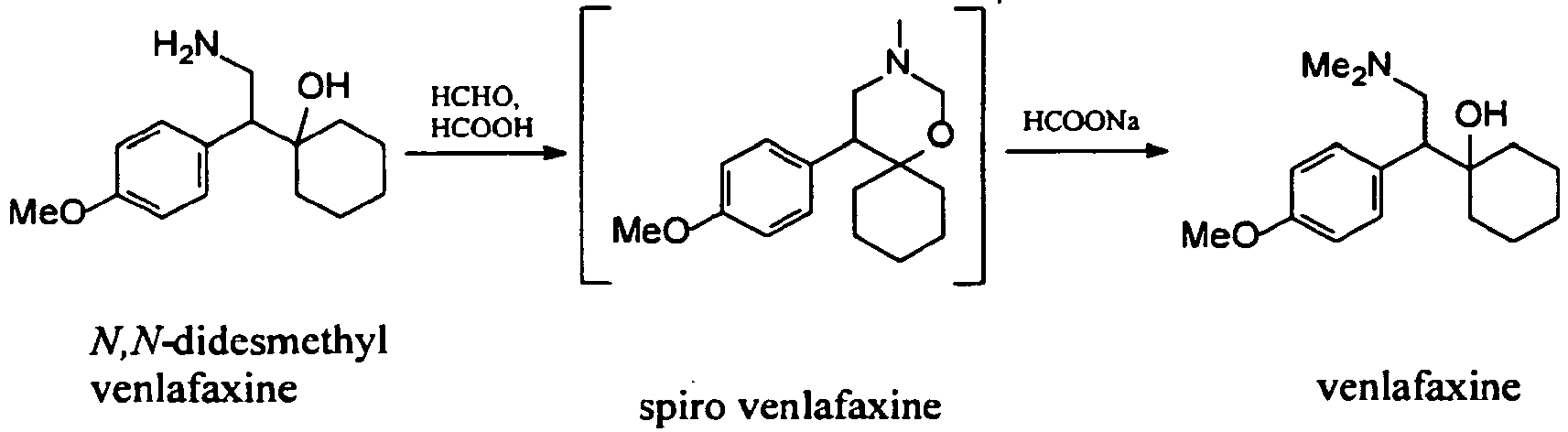
Examples:Example 1 – Preparation of venlafaxine from N,N-didesmethyl venlafaxine hydrochloride
-
A 50 % aqueous NaOH solution (4 ml, 74 mmol) was added to a stirred solution of N,N-didesmethyl venlafaxine hydrochloride (5.72 g, 20 mmol) in water (16 ml) at room temperature. Formic acid (98 %, 11.5 ml, 305 mmol) and 37 % aqueous solution of formaldehyde (8.4 ml, 113 mmol) were added to this mixture. The mixture was stirred under reflux temperature and the conversion was completed in 5 h (HPLC: 98.67 area %). Then the solution was cooled to room temperature and adjusted with 50 % aequous NaOH to pH 12. The mixture was extracted twice with 66 ml of isopropyl acetate. The collected organic phases were washed three times with water (66 ml). The isolated solution of venlafaxine base was very pure (HPLC: 98.9 area%).
Example 2 – Preparation of venlafaxine hydrochloride form I from the solution of venlafaxine base in isopropyl acetate
-
To the solution of venlafaxine base in isopropyl acetate from example 1 (66 ml, 10 mmol) 5 ml of 2 M aqueous HCl were added. The mixture was heated and water was removed by azeotropic distillation using a Dean-Starck trap. When all water was removed from the mixture, the product began slowly to crystallize. The obtained suspension was heated under reflux temperature for 1.5 h, then cooled and filtered. 2.75 g (88 % from N,N-didesmethyl venlafaxine hydrochloride) of pure venlafaxine hydrochloride form I (HPLC: 99.65 area %) were obtained.
Example 3 (exemplary) – Preparation of venlafaxine hydrochloride form I from the solution of venlafaxine base in isopropyl acetate
-
The solution of venlafaxine in isopropyl acetate from example 1 (66 ml, 10 mmol) was concentrated to ½ of the volume. Then 10 to 50 mg of venlafaxine hydrochloride form I was added to the solution. Subsequently, a 2.5 M solution of HCl in ethanol (4.0 ml) was slowly added within 30 min. After the whole amount of acid was added, the obtained suspension was stirred for another 2 h. Then the mixture was filtered and the product was washed with isopropyl acetate and dried. We obtained 2.69 g (86 % from N,N-didesmethyl venlafaxine hydrochloride) of pure venlafaxine hydrochloride form I (HPLC: 99.65 area %).
…………………..
PATENT
http://www.google.com/patents/WO2006035457A1?cl=en
Venlafaxine is known by the chemical name 1-[2-dimethylamino-1-(4 methoxyphenyl ethyl Cyclohexanol hydrochloride and structure of formula (V).

(V)
Venlafaxine is a useful pharmaceutical agent as an antidepressant. Venlafaxine, the intermediates in the manufacture of Venlafaxine, the process of preparing said Venlafaxine and their intermediates are well known from US Patents 4,535,186, US Patent No. 6,350,912, and CN 1225356.
Further International Publication No. WO03/050074 discloses the manufacture of Venlafaxine Hydrochloride and crystalline polymorphs Form I, Form II, Form III and optically pure (R) and (S) enantiomers exhibiting different crystalline structures of Venlafaxine hydrochloride. The preparation of all the forms of Venlafaxine and their inter-conversion are also described in said WO03/0500074 publication. U.S. Patents 4535186, 4761501 disclose a process for manufacture of 1-[2- amino-i-(p-methoxyphenyl) ethyljcyclohexanol (free base of formula IV), an intermediate produced during the preparation of Venlafaxine in two stages by reacting p-methoxyphenyl acetonitrile with cyclohexanone in the presence of n- butyl lithium (Stage 1) to form 1-[cyano(p-methoxyphenyl) methyl] Cyclohexanol of formula III

(111)
This process is commonly used for the preparation of formula III. The US Patent 4,535,186 produces a yield of about 30% based on p-methoxyphenyl acetonitrile.
WO/03/050074 suggests an alternate way of preparing compound of formula III without using butyl lithium i.e. by reacting p-methoxyphenyl acetonitrile with cyclohexanone in the presence of alkali metal hydroxide in a mixture of toluene and hexane. The publication WO/03/050074 also suggests a material yield of 74% based upon p-methoxyphenyl acetonitrile and purity.
The drop wise addition of butyl lithium to p-methoxyphenyl acetonitrile is hazardous and hence it requires skill and safety measures to be taken by the person skilled in the art for handling butyl lithium over the addition period to avoid any accidents during the preparation process.
The second stage i.e. conversion of compound of formula III to formula IV described in US patent US 4,535,186 is by hydrogenating compound of formula III using Rhodium on alumina. The catalyst Rhodium is recycled by filtering and washing the catalyst with ethanol and the combined filtrate evaporated and dried under vacuum yielding free base as an oil. However, the cost of Rhodium catalyst is very high and hence the catalyst has to be recovered.
WO/02/500017 suggests the use of a Nickel or cobalt catalyst for the hydrogenation, which is highly economical when compared with the Rhodium catalyst as suggested by US Patent No. 4,535,186. The International Publication WO/02/500017 teaches that the hydrogenation reaction of Stage Il may be carried out in the presence of an organic solvent preferably an alcohol. The international publication also suggests the pretreatment of the catalyst with ethanol.
The US Patent 4,535,186 describes the third stage in the process of preparing Venlafaxine i.e. conversion of compound of formula IV (free base) to compound V i.e. Venlafaxine by methylating the compound of formula IV (free base) with a mixture of formaldehyde and formic acid in water.

(IV)
US Patent Publication No. 2005/0033088 describes a process for preparing phenylethylamine derivative, an intermediate of Venlafaxine hydrochloride; said process comprising steps of reduction of compound of formula III with palladium on charcoal in an organic acid selected from formic acid, acetic acid or propionic acid, preferably acetic acid in an autoclave at a pressure of 5 to 25 kg/cm2 preferably 10 to 15 kg/cm2 at a temperature in the range of 30 to 75°C, preferably at 50 to 55°C till the hydrogenation substantially complete, filtering the palladium catalyst and evaporating the filtrate. Extracting the filtrate with halogenated hydrocarbon solvent and purifying the same. The process also describes the preparation of Venlafaxine hydrochloride without isolation of freebase.
The route of synthesis for Venlafaxine (formula V) and intermediate of Venlafaxine (formula IV) is depicted in the following scheme:
Step -I

(I) (II) (III) Step-ll

…………..(III) ……………………(IV)
Step-ll I

(IV) (V)
Accordingly, the present invention relates to an improved process for the preparation of compound of formula IV

(IV)
comprising the step of hydrogenating a compound of formula

(Hi)
in the presence of toluene, water, and a catalyst wherein the said process yields 66% formula (IV) with 99% HPLC purity.
The compound of formula IV is further methylated using formaldehyde and formic acid mixture to form Venlafaxine (formula V) followed by the treatment with HCL gas dissolved in Isopropanol.

(IV) (V)
According to the preferred embodiment of the present invention, nickel catalyst, preferably Raney nickel catalyst is used. The catalyst is washed in water to remove the alkali. No pretreatment of the nickel catalyst is required.

(HI) (IV)
According to another embodiment of the present invention, compound of formula IV is prepared by hydrogenating compound of formula III in the presence of Raney Nickel and water. According to another embodiment of the present invention, compound of formula III is prepared by charging p-methoxyphenyl acetonitrile into butyl lithium at -70 to -75°C and tetrahydrofuran; cooling the reaction mixture to about -50°C to -750C; adding cyclohexanone at a temperature below -500C quenching with ice and saturated ammonium chloride solution below 0°C; and stirring and filtering the product of formula (III) wherein the said process yields 89% of compound of formula III with 99.8% purity. The reaction scheme is depicted as follows:

…………(I) …………(H)………………………………. (III)
Example 1 :
Preparation of 1-[cyano-(4-methoxyphenyl) methyl Icvclohexanol.
In a 2 ltr 4 necked round bottom flask equipped with a overhead stirrer, thermometer and dropping funnel, 100 ml dry THF followed by 210 ml Butylithium (1.6 M solution in Hexane) was charged. The reaction mixture was cooled to – 700C. Added gradually a solution of 50 gm p-methoxyphenyl acetonitrile dissolved in 50 ml dry THF at -70 to -75°C. After 30 min, added solution of 33.1 gm Cyclohexanone in 50 ml THF. After the addition, maintained at -65 to -700C and monitored by TLC. After 4 hrs, reaction mixture was gradually added over mixture of ice and 150 ml saturated ammonium chloride solution below 0°C and adjusted pH to 7 with dilute Hydrochloric acid. Stirred for 1 hr and filtered the product. Washed the product with 200 ml hexane and dried to obtain 74.3 gm. (The yield based on p-methoxyphenyl acetonitrile 89%, Melting range 123- 125°C, HPLC purity of 99.8%).
Example 2:
Preparation of 1-[2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate
In an autoclave are charged 100 gm 1-[cyano(4- methoxyphenyl)ethyl]cyclohexanol, 100 ml toluene and 400 ml water at RT. Stirred and cooled to 10°C. Charged 20 gm Raney Nickel (which was prewashed with water to make it free of Alkali) and 100 ml liquor ammonia (20%). Then pressurized the autoclave with hydrogen to 4 – 5 kg pressure and maintained for 120 minutes below 120C. Then the reaction temperature slowly raised to bout 500C along with the increase in the hydrogen pressure to 7 to 8 kg. Maintained between 45 – 50°C for 8 hr. After the completion of the reaction, cooled the reaction to RT, released the hydrogen pressure and charged 400 ml toluene. Filtered the catalyst and washed bed with 100 ml toluene. Separated the organic layer from the filtrate. The organic layer was washed with 10% Sodium chloride solution. To the organic layer was added 40 ml methanol followed by 10 ml acetic acid. Stirred for 15 minutes and then again charged 10 ml acetic acid. Then heated to 75-8O0C and maintained for 15 minutes. Cooled to 0 – 50C. Filtered the product. Washed the product with 100 ml ethyl acetate and dried: 83.5 gm (Yield 66%, Melting range 164-166°C, HPLC purity 99%).
Example 3:
Preparation of H2-dimethylamino-1-(4-methoxyphenyl) ethvH Cyclohexanol Hydrochloride
To a stirred solution of 100 gm of 1-[2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate in 300 ml water was added 117 gm of formic acid (88%) and 91 gm of formaldehyde (40% solution). The solution was heated to 98°C and maintained for 20 hrs. Reaction mixture was cooled to about 100C and added 500 ml ethyl acetate. The pH was adjusted to about 7 with sodium hydroxide solution and further to 10 – 10.5 with ammonium hydroxide solution. Layers were separated. Aqueous layer was extracted with ethyl acetate. Combined organic layers were washed with water. Combined organic extract was stirred with activated carbon (5 gm) and filtered. Filtrate was concentrated in vacuum to completely remove ethyl acetate. Residue was dissolved in isopropanol (300 ml) and acidified at 300C (pH 1-1.5) with the solution of HCI in isopropanol. Temperature was then raised to 600C and maintained for 60 to 90 min. The reaction mass was cooled under agitation to 10°C and maintained under agitation at 10°C for 60 min. Product was isolated by filtration. Finally it was washed with isopropanol and dried at 60°C.
Dry wt. : 85 gm (84% yield, HPLC 99.9% purity with all individual impurities below 0.1% concentration). This material exhibited following characteristic x-ray powder diffraction pattern with characteristic peaks expressed in d-values (A) at.
(The abbreviations in brackets mean : (vs) = Relative intensity above 80%; (s) = 30% – 80%; (m) = 15% – 30%; ![]() = 8% to 15% and (vw) = below 8%.) 2.23 (VW), 2.29(VW), 2.32 (VW)1 2.35(VW), 2.38(VW), 2.43(VW), 2.46(VW), -2.48(VW)12.55(M), 2.64(W), 2.69(W), 2.73(VW), 2.8(W), 2.83(W), 2.88(W), 2.93(VW), 3.09(VW), 3.12(M), 3.26(VM), 3.31(VM), 3.38(W), 3.45(VW), 3.5(VW), 3.55(M), 3.69(VW), 3.87(VW), 3.99(VW), 4.07(M), 4.18(S), 4.35(VS), 4.48(VW), 4.68(M), 5.1 (VW), 5.27(W), 5.42(VW), 5.55(VW), 5.63(M), 5.68(M), 5.76(VW), 6.5(S), 6.95(VS), 8.65(VW), 10.56(M), 13.06(M).
= 8% to 15% and (vw) = below 8%.) 2.23 (VW), 2.29(VW), 2.32 (VW)1 2.35(VW), 2.38(VW), 2.43(VW), 2.46(VW), -2.48(VW)12.55(M), 2.64(W), 2.69(W), 2.73(VW), 2.8(W), 2.83(W), 2.88(W), 2.93(VW), 3.09(VW), 3.12(M), 3.26(VM), 3.31(VM), 3.38(W), 3.45(VW), 3.5(VW), 3.55(M), 3.69(VW), 3.87(VW), 3.99(VW), 4.07(M), 4.18(S), 4.35(VS), 4.48(VW), 4.68(M), 5.1 (VW), 5.27(W), 5.42(VW), 5.55(VW), 5.63(M), 5.68(M), 5.76(VW), 6.5(S), 6.95(VS), 8.65(VW), 10.56(M), 13.06(M).
Example 4 :
Preparation of 1-f2-amino-(4-methoxyphenyl) ethyl] Cyclohexanol acetate (IV)
In an autoclave charged 150 gm 1-[cyano(4-methoxyphenyl)ethyl]cyclohexanol, and 675 ml water at RT. Stirred and cooled to 1O0C. Charged 30 gm Raney Nickel (prewashed with water to make it free of Alkali) and 150 ml liquor Ammonia (20%). Then pressurized the autoclave with hydrogen to 4 – 5 kg pressure and maintaind for 120 minutes below 12°C. After completion of 120 minutes slowly raised the temperature to about 500C along with the increase in the hydrogen pressure to 7 to 8 kg. Maintained between 45 – 5O0C for about 20 hrs. Monitored reaction by TLC to ensure disappearance of starting material. After the completion of the reaction cooled the reaction to RT, released the hydrogen pressure and filtered through celite bed. Washed bed with 300 ml toluene. To the filtrate added 300 ml toluene. Shaken well and separated the organic layer. The organic layer was washed with 5% Sodium chloride solution. To the organic layer was added 45 ml methanol and 15 ml acetic acid. Stirred for 15 minutes and then again charged 15 ml acetic acid. Then heated to 75-8O0C and maintained for 15 mins. Cooled to 0 – 5°C. Filtered the product. Washed the product with 100 ml ethyl acetate and dried: 104 gm (Yield 53%, Melting range 152-153°C, HPLC purity 90%).
……………….
PATENT
http://www.google.com/patents/WO2008059525A2?cl=en
Venlafaxine acts by inhibiting re-uptake of norepinephrine and serotonin. It has been reported that its (-) enantiomer is a more potent inhibitor of norepinephrine synaptosomal uptake while its (+) enantiomer is more selective in inhibiting serotonin uptake (J. Med. Chem. 1990, 33(10), 2899-2905) In humans, venlafaxine is transformed by a metabolic pathway into two minor metabolites, N-desmethylvenlafaxine of formula II, N,O-di- desmethylvenlafaxine of formula IV and one major metabolite, O-desmethylvenlafaxine of formula III.

formula I formula II

formula III formula IV
In the literature there are several processes reported for the synthesis of venlafaxine of formula I and venlafaxine hydrochloride of formula Ia.

formula Ia
The synthesis of venlafaxine from 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile (hereinafter called as cyano-intermediate and represented by formula V) involving two step synthesis is known in the prior art.

formula V
US 4,535,186 discloses the preparation of venlafaxine of formula I by the reaction of p- methoxyphenylacetonitrile with cyclohexanone at -78 0C in the presence of n-butyllithium as a base which yields 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile of formula V. Reduction of the cyano-intermediate under hydrogen pressure with rhodium on alumina catalyst gives l-[2-amino-l-(4-methoxyphenyl)ethyl]cyclohexanol. N-Methylation of the amino compound is accomplished employing formaldehyde and formic acid (Eschweiler- Clarke reaction) to give venlafaxine of formula I.
The reaction is as shown in the Scheme- 1.

Reduction


Scheme-1
Another prior art reference, Zhou Jinpei et.al, J. China Pharm. University, 1999, 30(4), 249- 50) discloses the preparation of venlafaxine starting from anisole. Anisole is acylated to the chloroacetyl derivative, which is then animated using N,N-dirnethylamine. The carbonyl group of this compound is reduced to the alcohol using KBH4 and is converted to the bromo- derivative using PBr3 which in turn when reacted with Mg and cyclohexanone undergoes a Grignard reaction to provide venlafaxine of formula I.
The reaction is as shown in the Scheme-2.

Scheme-2
US 2005033088 discloses a two step process for venlafaxine starting from the cyano- intermediate. The cyano-intermediate is reduced in the presence of palladium on charcoal in acetic acid at a hydrogen pressure of 5-25 kg/cm2 at a temperature in the range of 30-75 0C. The product of step 1 is N-methylated using formic acid, formaldehyde solution at a temperature of 90-98 0C for 19 hrs to yield venlafaxine, which is then converted to its hydrochloride salt.
WO2006035457 also discloses a process of making venlafaxine and its intermediates. The process comprises the step of hydrogenating the cyano-intermediate in the presence of toluene, water, and Raney nickel where in the said process yields 66% of an intermediate with 99% HPLC purity. This reaction is carried out at 10-12 0C and at 4-5 kg/cm2 of hydrogen pressure for 2 hrs and further at 50 0C at 7-8 kg/cm2 for 7-8 hrs. This intermediate is N-methylated using formaldehyde and formic acid mixture to form venlafaxine, which is treated with IPA/HC1 to get venlafaxine hydrochloride.
US 6,350,912 discloses a one pot process for the preparation of venlafaxine in 15-28 % yield from the cyano-intermediate. In the said patent venlafaxine has been prepared by the reduction of cyano-intermediate in the presence of Raney nickel and without isolation of intermediate l-[2-amino-l-(4-methoxyphenyl) ethyl] cyclohexanol
CN 1850781 discloses a process for the preparation of venlafaxine by following steps: (1) carrying out condensation of 4-methoxyphenylacetonitrile and cyclohexanone in presence of base to obtain 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile, (2) reacting 2-(l- hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile with cuprous chloride and dimethylamine to obtain 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)-N,N- dimethylacetimidamide, and (3) reacting 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)- N,N-dimethylacetimidamide with KBH4 to obtain 1 – [2-dimethylamino)- 1 -(4-methoxyphenyl) ethyl)cyclohexanol (venlafaxine).
The reaction is as shown in the Scheme-3.

Scheme-3
The processes disclosed in the prior art have many disadvantages. Most of the prior art processes employ formaldehyde as a reactant for N-methylation step which is known to be a carcinogen. Acute exposure of the same is highly irritating to the eyes, nose and throat. Ingestion of formaldehyde is fatal and long term exposure causes respiratory problems and skin irritation.
Another disadvantage is the formation of an impurity (represented by formula VI), which is formed during N-methylation step using formaldehyde as a reagent.

formula VI
As may be appreciated, all the above well-known processes share the same strategy of synthesis, consisting of two steps for the synthesis of venlafaxine from cyano-intermediate or are prepared in one pot with poor yield.
Yet another drawback of the processes disclosed in the prior art is the use of expensive catalysts like rhodium on alumina and use OfBF3 etherate which is highly corrosive.
The prior art references disclose the synthesis of alkoxyphenylethyldimethylamine from alkoxyphenylacetonitrile using excess dimethylamine and palladium catalyst in methanol solution which is firstly reported by Kindler and Hensse. (1. Kindler and Hesse; Arch. Pharm., 1933, 271, 439. 2. Johannes S. Buck, Richard Baltzly and Walter S. Ide; J. Am. Chem. Soc. 1938, 60(8), 1789-1792; 3. Albert J. Schuster and Eugene R. Wagner; J. Labelled Compounds and Radiopharmaceuticals 1992, XXXIII(3), 213-217).
The reaction is as shown in the Scheme-5.

Scheme-5
According to an aspect of the invention there is provided a novel single step process for the synthesis of venlafaxine of formula I and N-desmethylvenlafaxine of formula II from 2-(l- hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile of formula V comprising reaction of 2- (l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile with an alkylamine and/or its salt in a solvent in the presence of a transition metal catalyst, under hydrogen atmosphere.

formula I formula II formula V DETAILED DESCRIPTION OF THE INVENTION
The present invention describes a single step process for venlafaxine and its analog starting from the cyano-intermediate. The present invention circumvents the difficulties encountered in the prior art and is an economically viable process.
The present invention particularly relates to a single step synthesis of venlafaxine of formula I, and N-desmethylvenlafaxine of formula II from the cyano-intermediate of formula V.
The reaction of the present invention is as shown in Scheme-4:

formula V formula L R = CH3 (Venlafaxine) formula II. R = H (N-desmethylvenlafaxine)

formula Ia. R = CH3, X = Cl formula Ha. R = H, X = Cl
Example-1:
General procedure for synthesis of venlafaxine and N-desmethylvenlafaxine To the, stirred solution of 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile (1.0 equiv.) in methanol (10-20 volumes), alkylamine (3-5 equiv.) was added and the mixture was stirred for 5-10 minutes to get a clear solution. Palladium catalyst (10-50 wt %) was added under nitrogen atmosphere to the above reaction mixture. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 5 to 40 hrs. The progress of the reaction was monitored by TLC and HPLC. After completion of the reaction, the catalyst was filtered through celite and washed with methanol. The combined filtrate was concentrated to dryness under reduced pressure and the residue was poured in water. The aqueous layer was basifϊed with 10% aq. NaOH to pH 8-10 and extracted with ethyl acetate (3 times). The combined ethyl acetate layers were washed with brine and dried over sodium sulphate. Ethyl acetate was evaporated under vacuum to obtain the title compound.
Example-2: Venlafaxine
To the stirred solution of 2-(l-hydroxycyclohexyl)-2-(4-methoxyphenyl)acetonitrile (20.0 g, 1 equiv.) in methanol (460.0 ml), dimethylamine hydrochloride (26.6 g, 4 equiv.) was added and the mixture was stirred for 5-10 minutes at room temperature to obtain a clear solution. 5% Palladium on alumina (4.0 g, 20 wt %) was added under nitrogen to the above clear solution. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 20 hrs. After completion of the reaction product was isolated by the procedure as described in Example- 1 above to obtain light yellow viscous liquid (18.Og) which was directly converted to its hydrochloride salt using IPA/HC1. HPLC purity of the crude reaction mixture (18.Og) = 81.64%.
Venlafaxine-free base:
1H NMR in CDCl3 (300 MHz): δ 7.05 (d, 2H), 6.81 (d, 2H), 3.79 (s, 3H), 3.27 (t, IH), 2.94 , (dd, IH), 2.31 (s, 6H), 2.28 (dd, IH), 1.71-0.94 (m, 10H). 13C NMR in CDCl3: δ 158.16,’ 132.65, 130.01, 113.20, 74.14, 61.14, 55.05, 51.52, 45.37, 37.99, 31.07, 25.91, 21.52, 21.23.
IR (KBr): 3152, 2980, 2941, 2895, 1728, 1607, 1512, 1462, 1439, 1358, 1279, 1204, 1186, 1177, 1146, 1103, 1040, 1011, 968, 851 cm“1. HPLC Purity: 99.37 % (area %). GC-MS: 178 (M+H+).
Venlafaxine hydrochloride salt:
1H NMR in D2O (300 MHz): δ 7.23 (d, 2H), 6.91 (d, 2H), 3.71 (s, 3H), 3.54 (t, IH), 3.48 (dd, IH), 2.97 (dd, IH), 2.69 (s, 6H), 1.41-1.0 (m, 10H).
13C NMR in D2O: δ 158.52, 130.86, 128.16, 114.25, 73.23, 58.26, 55.25, 50.38, 44.87, 41.41, 35.07, 33.34, 24.76, 21.08, 20.86.
IR (KBr): 3321, 2941, 2928, 2675, 2644, 2623, 2611, 2587, 2521, 2482, 2359, 2330, 1512, 1441, 1242, 1179, 1038, 829 cm“1. HPLC Purity: 99.64 % (area %). GC-MS: 178 (M+H+).
Example-3: N-Desmethyl venlafaxine To the stirred solution of 2-(l -hydroxy cyclohexyl)-2-(4-methoxyphenyl)acetonitrile (20.0 g, 1 equiv.) in methanol (350.0 ml), monomethylamine hydrochloride (27.7 g, 4 equiv.) was added and the mixture was stirred for 5-10 minutes at room temperature to obtain a clear solution. 5% Palladium on alumina (4.0 g, 20 wt %) was added under nitrogen to the above clear solution. The reaction mixture was purged with hydrogen gas (three times) and allowed to stir under hydrogen (1-2 atmospheric pressure.) at room temperature for 24 hrs. After completion of the reaction product was isolated by the procedure as described in Example- 1 above to obtain light yellow viscous liquid (18.5g) which was directly converted to its hydrochloride salt using IPA/HCl.
HPLC conversion to N-desmethylvenlafaxine (in the crude reaction mixture = 18.5g): 35.46%. N-Desmethyl venlafaxine-free base:
1H NMR in CDCl3 (300 MHz): δ 7.23 (d, 2H), £91 (d, 2H), 3.71 (s, 3H), 3.54 (t, IH), 3.48 (dd, IH), 2.97 (dd, IH), 2.69 (s, 3H), 1.41-1.0 (m, 10H)
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003050074A1 * | Mar 19, 2002 | Jun 19, 2003 | Cadila Healthcare Ltd | Manufacture of venlafaxine hydrochloride and crystalline polymorphs thereof |
| US4535186 * | Oct 26, 1983 | Aug 13, 1985 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008059525A2 * | Oct 1, 2007 | May 22, 2008 | Calyx Chemicals And Pharmaceut | An improved process for the preparation of venlafaxine and its analogs |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006035457A1 * | Sep 16, 2005 | Apr 6, 2006 | Amoli Organics Ltd | A process for the manufacture of venlafaxine and intermediates thereof |
| US4535186 * | Oct 26, 1983 | Aug 13, 1985 | American Home Products Corporation | 2-Phenyl-2-(1-hydroxycycloalkyl or 1-hydroxycycloalk-2-enyl)ethylamine derivatives |
| US6350912 * | Feb 28, 2001 | Feb 26, 2002 | Council Of Scientific And Industrial Research | One pot process for the preparation of 1-[2-dimethylamino-(4-methoxyphenyl)-ethyl]cyclohexanol |
| US20050033088 * | Jun 7, 2004 | Feb 10, 2005 | Dr. Reddy’s Laboratories Limited | Catalytic hydrogenation of phenylacetonitrile using palladium on carbon supports |
| Reference | ||
|---|---|---|
| 1 | * | CHAVAN S P ET AL: “An efficient and green protocol for the preparation of cycloalkanols: a practical synthesis of venlafaxine” TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 45, no. 39, 20 September 2004 (2004-09-20), pages 7291-7295, XP004558985 ISSN: 0040-4039 |
| 2 | * | J.S. BUCK ET AL: “beta-Phenylethylamine Derivatives. Tertiary and quaternary salts” J.AM.CHEM.SOC., 1938, pages 1789-1792, XP002478651 cited in the application |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010100520A1 * | Mar 4, 2009 | Sep 10, 2010 | Hikal Limited | A process for preparation of phenethylamine derivative |
| WO2011124190A2 | Apr 6, 2011 | Oct 13, 2011 | Zentiva, K.S. | Method of producing 4-(2-(substituted)-1-(1-hydroxycyclohexyl)ethyl)phenols by o- demethylation of their methylethers by means of inodorous aromatic thiols |



Chernivtsi central square

KIEV

Independence Square (Maidan Nezalezhnosty)

DONETSK
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMRDRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
: The views expressed are my personal and in no-way suggest the views
of the professional body or the company that I represent.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
OLANZEPINE VISITED PART 3/3

PART 1…..https://newdrugapprovals.org/2015/04/08/olanzepine/
PART 2….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-22/
PART 3…….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-33/
review……http://cosmos.ucdavis.edu/archives/2011/cluster8/JAIN_VINEET.pdf
WATCHOUT………..
…………….
DOI: 10.1039/C3OB27424A
http://pubs.rsc.org/en/Content/ArticleLanding/2013/OB/c3ob27424a#!divAbstract
A new strategy for converting antipsychotic drug olanzapine into PDE4 inhibitors is describedvia the design and Pd/C mediated synthesis of novel N-indolylmethyl olanzapine derivatives. One compound showed good inhibition (IC50 1.1 μM) and >10 fold selectivity towards PDE4B over D that was supported by docking studies. This compound also showed significant inhibition of TNF-α and no major toxicities in cell lines and a zebrafish embryo model except the teratogenic effects to be re-assessed in rodents.

…………..
http://www.google.com/patents/US7329747
Olanzapine or 2-methyl-4-[4-methyl-1-piperazinyl]-10H-thieno[2,3b][1,5]-benzodiazepine is a pharmaceutically active compound that can be represented by the formula (1).

It was disclosed in EP 454436 and corresponding U.S. Pat. No. 5,229,382 as a useful antipsychotic agent. Olanzapine acts as a serotonin (5-HT2) and dopamine (D1/D2) receptor antagonist with anticholinergic activity. In commercially available final forms, the active substance is marketed as a free base, which is a white to yellow crystalline solid that is insoluble in water.
One synthetic route for making olanzapine starts from “des-methylpiperazine olanzapine precursor” of formula (3), which reacts with piperazine to form a “des-methyl olanzapine precursor” of formula (2) (see Jun-Da Cen, Chinese Journal of Pharmaceuticals 2001, 32(9),391-393). The compound (2) can be methylated to form olanzapine (see U.S. Pat. No. 4,115,568 for such suggestion). The methylation reaction can be carried out using formaldehyde under conditions of Eschweiler-Clarke reaction (see Jun-Da Cen) or by classical methylation agents such as methyl iodide (see WO 04-000847).

This synthetic pathway has the disadvantage that the reaction with piperazine may lead to formation of dimeric impurities and that the methylation with formaldehyde or other methylation agent may lead to side products, e.g. products of multiple methylation. All these contaminants are difficult to remove from the product. Also, methylation agents are, in general, toxic and mutagenic compounds.
An alternative of the above process was suggested in WO 04/000847 and comprises converting the compound (2) into a “formyl-olanzapine precursor” of formula (4) by a reaction with a methyl formate, and converting the compound (4) into olanzapine by a reduction with a metal borohydride.

In comparison with the preceding procedure, the alternate procedure is one step longer and suffers from the same problems in the step of making compound (2). Furthermore, the reported purity of the actually obtained olanzapine product is only 88%, which is not sufficient for pharmaceutical applications.
The present invention relates to the formation, purification and/or use of an N-formyl olanzapine. Accordingly, a first aspect of the present invention relates to a process, which comprises reacting a des-piperazine olanzapine of formula (3) or a salt thereof

with an N-formyl piperazine of formula (5)

to form an N-formyl olanzapine of formula (4) or a salt thereof

The reaction can be carried out in an inert solvent, generally a dipolar aprotic solvent, and is typically accomplished by heating. The N-formyl olanzapine can be converted to olanzapine.
Another aspect of the invention relates to a process for making an olanzapine salt, which comprises: reducing an N-formyl olanzapine of formula (4) or a salt thereof

with a reducing agent in a solvent to form olanzapine or a salt thereof dissolved in said solvent; reacting said dissolved olanzapine or a salt thereof with an acid to form an acid addition salt of olanzapine; and precipitating said olanzapine acid addition salt from said solution. Precipitating the salt of olanzapine can avoid the formation of technical grade olanzapine. That is, the olanzapine salt can be obtained in a purified state and then converted to olanzapine base, if desired, in high purity.
A further aspect of the present invention relates to purifying the N-formyl olanzapine, which process which comprises:
(1) dissolving and/or slurrying an N-formyl olanzapine of formula (4)

or a salt thereof in a solvent selected from the group consisting of an aliphatic alcohol, an aromatic hydrocarbon, and mixtures thereof, at a temperature of at least 35° C. to form a crystallization treatment medium;
(2) cooling said crystallization treatment medium; and
(3) isolating solid N-formyl olanzapine of formula (4) having improved purity. The steps (1)-(3) can be repeated if necessary until the desired purity is reached. Generally, such a process can achieve purity of greater than 95% and preferably greater than 98%.
An overall synthetic scheme for making olanzapine, which combines various aspects of the present invention, is set forth below:

Example 1
N-Formyl Olanzapine (4)
In a 1000 ml flask, a mixture of 12.0 g of “des-methylpiperazine olanzapine precursor” (compound of formula (2)) hydrochloride and 40 ml of N-formyl piperazine in a mixture of 60 ml of dimethylsulfoxide and 60 ml of toluene was heated at reflux under a nitrogen atmosphere overnight. Progress was monitored by HPLC. After cooling to 40° C., 160 ml of water was added. The resulting mixture was cooled and stirred at 0° C. The solid material was isolated by filtration and washed with 2×40 ml of water. Wet solid was dried overnight at ambient conditions and subsequently at 40° C. under vacuum.
Isolated yield: 12.19 gram, Purity (HPLC): 91.6%
Example 2
Crystallization of the Compound (4)
8.0 g of crude N-formyl olanzapine precursor (compound (4)) of a purity of about 89% (HPLC) was suspended in 50 ml of methanol and heated at 60° C. for 3 hours. The hot suspension was allowed to cool to room temperature and was subsequently cooled to 5° C. under stirring. The solid material was isolated by filtration, washed with 5 ml of cold methanol and 10 ml of cold diethyl ether and dried overnight at 40° C. under vacuum.
Yield: 3.97 g, purity 96.7% (HPLC)
……………….
PATENT
http://www.google.com/patents/WO2004056833A1?cl=en
…………………………….
http://www.google.com/patents/WO2002018390A1?cl=en
Olanzapine is represented by the following structure.

Olanzapine
Olanzapine is useful for treating psychotic patients and mild anxiety states. Preparation of Olanzapine and its acid salts, having pharmaceutical properties particularly in the treatment of disorders ofthe central nervous system has been discussed in U.S. Patent No. 5,229,382.
U.S. Patent No. 5,229,382 does not refer to any specific polymorphic crystalline form of Olanzapine. European patent specification No. 733635A1 claims Form-2 of Olanzapine. The process under this patent describes preparation of Form-2 from ethyl acetate. This patent also designated the product obtained according to the process described in U.S. Patent No. 5,229,382 as Form-1. Furthermore, EP 733635 Al discloses the d values for Form-1 and Form-2 from their X-ray Diffractograms. The values are: d value d value
Form-1 Form-2 9.94 10.26
8.55 8.57 8.24 7.47 6.88 7.12 6.37 6.14 6.24 6.07 5.58 5.48 5.30 5.21 4.98 5.12 4.83 4.98 4.72 4.76 4.62 4.71 4.53 4.47 4.46 4.33 4.29 4.22 4.23 4.14 4.08 3.98 3.82 3.72 3.74 3.56 3.69 3.53 3.58 3.38 3.50 3.25 3.33 3.12 3.28 3.08
3.21 3.06
3.11 3.01
3.05 2.87 2.94 2.81
2.81 ‘ 2.72
2.75 2.64
2.65 2.60
2.63 2.59
It is noteworthy to mention that EP 0 831 098 A2 discloses the preparation of a series of dihydrates of olanzapine namely Dihydrate B, Dihydrate D and Dihydrate E.
The d values from the X-ray Diffractograms for these forms are listed in EP 0 831
098 A2. We conducted experiments to obtain Olanzapine Form I by recrystallization of olanzapine from acetonitrile using the process described in Example 1, sub example 4 of U.S. Patent No. 5,229,382. The process is described herein for reference: A mixture of 4-amino-2-methyl-10H-thieno-[ 2,3-b] [l,5]benzodiazepine HCl (100 g),
N-methyl piperizine (350ml), DMSO (465 ml) and toluene (465 ml) was heated to reflux. The reaction mass was maintained at reflux for 19 hours and then cooled to
50°C and water was added. The reaction mass was cooled to 0-10°C and stirred at the same temperature for 6 hours. The crude Olanzapine separated was filtered and dried in oven to a constant weight (76.5 g). The crude compound was added to acetonitrile (750 ml) at boiling temperature. The mixture was boiled for further 5 minutes. The mixture was filtered to remove the undissolved solid. The filtrate was treated with carbon and filtered. The filtrate was distilled to a minimum volume, cooled to 0-5 °C and maintained at the same temperature for 1.0 hour and filtered.
The compound was dried to a constant weight in an oven (51.6g).
The polymorphic form obtained from these experiments was characterized for its X-ray Powder Diffraction on Rigaku D / Max 2200. As clearly observed, the d values for this product (Fig. 1) matched with those of Olanzapine Form-2 claimed in EP 733635A1. It is therefore inferred that the recrystallization of Olanzapine in acetonitrile produces Form-2 and not Form-1.
Accordingly, the present invention provides a novel method for preparation of hydrates of olanzapine, which are different from those reported in the literature. These hydrates are named Olanzapine monohydrate-I and Olanzapine dihydrate-I for convenience.
Accordingly, the present invention also provides a novel method for preparation of Olanzapine Form-1 by recrystallization of olanzapine or its hydrates in dichloromethane . The present invention also provides a novel method for converting Olanzapine
Form-2 to Olanzapine Form-1
PREPARATION OF OLANZAPINE MONOHYDRATE-1
EXAMPLE 1 A mixture of 4-amino-2-methyl-10H-thieno-[2,3-b][l,5]benzodiazepine hydrochloride (20 Kg), N-methyl piperazine (42 lit), dimethyl sulfoxide (40 lit) and toluene (95 lit) was heated to reflux. The reaction mass was maintained at reflux for
17 hours and 15 minutes and then cooled to 40-50°C. Water (95 lit) was added slowly at40-50°C. The reaction mass was cooled to -0.6 to 1.2°C and stirred at the same temperature for six hours. The Olanzapine crude that separated was filtered and washed with water (10 lit). The product was dried at 30.5 to 31.8°C for 10 hrs and 50 minutes. Yield: 20 Kg. A 20 gm sample from the above material after prolonged heating for an additional 72 hours gave the product with a moisture content of 5.22%.
PREPARATION OF OLANZAPINE DIHYDRATE-I EXAMPLE 2
A mixture of 4-amino-2-methyl-l OH-thieno- [2,3-b] [ 1 ,5]benzodiazepine hydrochloride (200 g), N-methyl piperazine (420 ml), dimethyl sulfoxide (200 ml) and toluene (940 ml) was heated to reflux. The reaction mass was maintained at reflux for 12 hours and then cooled to 40°C. Water (940 ml) was added slowly at 40-44°C. The reaction mass was cooled to 0-5°C and stirred at the same temperature for five hours. The Olanzapine crude that separated was filtered and washed with water (100 ml). The solid obtained was dried atmospherically (25-35°C) for 24 hours (Yield :
241 g).
PREPARATION OF FORM-1
EXAMPLE 3 Crude 2-methyl-4-(4-methyl- 1 -piperazinyl)- 1 OH-thieno- [2,3 -b] [ 1 ,5] benzodiazepine (35.0 g) was suspended in dichloromethane (160.0 ml). The suspension was heated to reflux to obtain a clear solution. The resultant solution was then treated with carbon (3.5 g) followed by filtration. Upon completion of this step the filtrate was cooled to 0 to 5°C and stirred at the same temperature for one hour. The separated solid was filtered and washed with chilled dichloromethane (10.0ml). The product obtained on drying in oven at 65 to 70°C to a constant weight gave Form-1 of Olanzapine (Yield 22.0 g).
CONVERSION OF FORM-2 TO FORM-1 EXAMPLE 4 The stirred suspension of pure form-2 of 2-methyl-4-(4-methyl-l-piperazinyl)-
10H-thieno-[2,3-b][l,5]benzodiazepine (20.0 g) in dichloromethane (90.0 ml) was heated to reflux to obtain a clear solution. The clear solution was filtered and the filtrate was then cooled to 3 to 5°C and stirred at same temperature for one hour. The crystalline solid separated was filtered and washed with dichloromethane (4.0 ml). Subsequent drying at 60 to 70°C to a constant weight yielded Olanzapine Form-1. (Yield: 12.7 g).
PREPARATION OF FORM-1 FROM MONOHYDRATE-I OF OLANZAPINE
EXAMPLE 5 Monohydrate-I of 2-methyl-4-(4-methyl-l-piperazinyl)-10H-thieno-[2,3- b][l,5] benzo- diazepine (25.0 g) prepared as per Example- 1 was suspended in dichloromethane (325.0 ml). The suspension was heated to reflux to obtain a clear solution. The resultant solution was then treated with carbon (2.5 g) followed by filtration. Upon completion of this step the filtrate was distilled to a minimum volume and then cooled to 2 to 4°C and stirred at the same temperature for 90 minutes. The product separated was filtered arid washed with chilled dichloromethane (10 ml). The product obtained on drying in oven at 60 to 70°C to a constant weight gave Form-1 of Olanzapine (Yield 16.5 g)
PREPARATION OF FORM-1 FROM DIHYDRATE-I OF OLANZAPINE
EXAMPLE 6 Dihydrate-I of 2-methyl-4-(4-methyl-l-piperazinyl)-10H-thieno-[2,3-b][l,5] benzodiazepine (40.0 g) prepared as per Example-2 was suspended in dichloromethane (520.0 ml). The suspension was heated to reflux to obtain a clear solution. The resultant solution was then treated with carbon (4.0 g) followed by filtration. Upon completion of this step the filtrate was distilled to a minimum volume and the left over reaction mass was cooled to 0 to 2°C and stirred at the same temperature for one hour. The separated solid was filtered and washed with dichloromethane (10.0ml). The product obtained on drying in oven at 65 to 70°C to a constant weight renders Form-1 of Olanzapine (Yield 26.0 g).
The aforementioned crystalline forms in examples 1 to 6 have been examined for their structural and analytical data viz., Powder X-Ray Diffraction, Differential Scanning Calorimetry, and Infrared Absorption Spectroscopy. The results obtained are discussed and the respective drawings attached (Fig. 2 -19).
………………………………..
http://www.google.com/patents/EP2292624A1?cl=en
-
Olanzapine is a pharmaceutical active substance from the group of antipsychotics, applicable for the treatment of different mental diseases and conditions, such as, for example, disorders of the central nervous system, schizophrenia, hallucination, acute mania, depression, and the like.
-
Chemically, it belongs to the group of the benzodiazepines and is 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine (formula 1).

-
Olanzapine and analogues thereof are encompassed for the first time within a general formula in the patent GB 1,533,235 and specifically described in EP 454 436 B1 . The patents disclose two different one-step processes for olanzapine preparation. The first described process is a reaction of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride with N-methylpiperazine in an organic solvent, such as anisole, toluene, dimethyl formamide or dimethyl sulfoxide, preferably at a temperature from 100 to 150°C to yield olanzapine (Scheme 1).

-
The second process disclosed in EP 454 436 B1 is the reaction of N-methylpiperazine with methyl-2-(2-aminoanilino)-5-methylthiophene-3-carboxylate in the presence of titanium tetrachloride (Scheme 2).

-
The same patent also mentions the formation of acid addition salts of olanzapine and their potential use as intermediates in olanzapine purification process and for a pharmaceutical use.
-
As disclosed in EP 454 436 B1 and US equivalent US 5,229,382 , olanzapine obtained according to the first synthesis (Scheme 1) is purified by recrystallization from acetonitrile, whereas olanzapine prepared according to the second route (Scheme 2) is further purified by column chromatography on Florisil® and recrystallization from acetonitrile. This purification procedure lacks on industrial applicability.
-
Some other synthetic approaches for the preparation of olanzapine describe two steps for creating 4-methylpiperazinyl side chain (Scheme 3). Firstly, 4-amino-2-methyl-10H-thieno [2,3-b][1,5] benzodiazepine hydrochloride reacts with piperazine to yield 2-methyl-4-(1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine (i.e. N-desmethylolanzapine; Bioorganic & Medicinal Che-mistry Letters, Vol. 7, No. 1, pp. 25-30, 1997), then the methyl group is introduced either by reductive N-methylation (using formaldehyde and metal boron hydride -WO 04/000847) or by nucleophylic substitution reaction with methyl iodide (WO 05/090359). The two-step approach reduces the dark colour appearance but it does not solve the problem of purification from similar by-products.

-
It is well known to a skilled person that most chemical reactions are not completely finished, may be reversible or are driven simultaneously with some other parallel reactions. Starting materials or side reaction products are usually found as impurities in the isolated main product which should therefore be further purified. The simplest way of purification includes various recrystallization and precipitation procedures which are usually less effective if the impurities have physico-chemical properties very similar to the main product.
-
In the case where olanzapine is prepared according to the one step processes disclosed in EP 454 436 B1 , the starting material, 4-amino-2-methyl-10H-thieno[2,3-b][1,5]-benzodiazepine, is found as an impurity in the final product olanzapine.
-
In the case of preparation of olanzapine via the two-step process, as disclosed also in the patent application WO 04/000847 , the presence of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]-benzodiazepine hydrochloride is not critical but various other similar compounds could be found as impurities, such as 4-(4-formylpiperazinyl)-2-methyl-10H-thieno[2,3-b][1,5]-benzo diazepine and N-desmethylolanzapine. In the case of preparation of olanzapine by a two-step process with methyl iodide, an overmethylated N,N-dimethylpiperazinium analogue can be formed.
-
A further undesired impurity which accompanies olanzapine is obtained when olanzapine compound is dissolved in methylene chloride. It is so called olanzapine-CM, being (E)-1-(chloromethyl)-1-methyl-4-(2-methyl-10H-benzo[b]thieno[2,3-e][1,4]diazepin-4-yl)piperazin-1-ium chloride). Olanzapine-CM is formed by alkylation of olanzapine with methylene chloride in methylene chloride solution, for example during evaporation of methylene chloride before the crystallization (Scheme 4).

-
According to the Regulatory Toxicology and Pharmacology 44, pp. 198-211, 2006, classification of potential genotoxic impurities, olanzapine-CM is a potential genotoxic substance due to its R-CH2-Cl structural element, which is known to be involved in reactions with DNA.
-
For all impurities that have a thienobenzodiazepine ring system as a part of the molecule skeleton and because it represents a great part of the molecule, said ring system is crucial for the similarity of physico-chemical properties of said impurities compared to olanzapine.
-
Different salts and crystal forms of an active pharmaceutical ingredient are an important tool for modulating pharmacokinetic properties but can be also a tool for purification.
-
WO 04/089313 discloses olanzapine acid salts, solvates and co-crystals and their use as active pharmaceutical ingredients in formulations. The preparation of fumaric, maleic and malonic acid addition salts of olanzapine is disclosed in WO 04/089313 . Olanzapine acid addition salts disclosed in this application exhibit specific aqueous solubility from 50 µg/ml to 100 mg/ml.
-
WO 05/090359 discloses a method for the purification of olanzapine which has on one hand been prepared by the one-step-process according to Scheme 1, and on the other hand by the process according to Scheme 3, by preparing an addition salt of at least one carboxylic salt, purification of said salt and transfer into purified olanzapine.
-
Neutral olanzapine can be isolated in various crystal forms, hydrates and other solvates. However crystal forms I and II are the most often mentioned as an active pharmaceutical ingredient. Both forms were first disclosed in EP 733 635 alleging that form II is thermodynamically more stable than form I which had been prepared already by the basic patent procedures ( EP 454 436 B1 ).
-
Crystal Growth & Design, Vol. 3, No. 6, pp. 897-907, 2003 discloses anhydrates and hydrates of olanzapine.
-
Olanzapine form I is thermodynamically less stable but it can possess specific kinetic properties which can be applied in designing a final dosage form. Many procedures are known how to prepare it but a person skilled in the art can soon find that the use of methylene chloride is unavoidable to develop a repeatable process. Because this solvent is used in the final step of preparation of olanzapine form I, previous purification methods cannot prevent the presence of the impurity olanzapine-CM in the final product.
-
WO 03/101997 A1 discloses a process for the preparation of a pure olanzapine form I by an addition of methyl-piperazine to the hydrochloride salt of the corresponding benzodiazepine derivative. The purification of olanzapine is conducted by recrystallization. Olanzapine-CM is not disclosed as a disturbing side product which can be removed by recrystallization.
-
WO 02/18390 discloses a process for the preparation of hydrates of olanzapine and the conversion thereof into crystalline forms of olanzapine by recrystallization from methylene dichloride. It is not disclosed that essentially pure olanzapine can be obtained by the removal of olanzapine-CM.
-
WO 04/056833 A1 discloses a process for the preparation of essentially pure olanzapine by removing olanzapine-CM, which is obtained from a solution of olanzapine in methylene chloride, by treating this solution with SiO2, followed by the removal of SiO2. According to the examples, olanzapine is obtained having a purity of 99.92 %, whereas olanzapine-CM is present in an amount of 0.05 %, corresponding to 500 ppm. But SiO2 is acidic, so it adsorbs well also the basic olanzapine what leads to considerable losses of the mother compound.
-
Olanzapin-CM has to be removed in order to obtain essentially pure olanzapine with a high purity which can be further used in pharmaceutical applications. It has been found that olanzapine cannot be efficiently separated from its highly related impurities, in particular from olanzapine-CM, using repeated crystallizations of crude olanzapine. It would therefore be desirable to develop a purification process, in order to provide pharmaceutically acceptable pure and discoloured olanzapine, in particular to provide pharmaceutically acceptable pure olanzapine, being essentially free of the olanzapine-CM impurity. A further object of the present invention is to provide a process for the purification of olanzapine which can also and preferably be conducted in a large scale synthesis.





Example 1 – Synthesis of olanzapine oxalic salt
-
[0093]A solution of 12.0 g of N-desmethylolanzapine in 90 mL of THF and 36 mL of dimethylacetamide (DMAc) is cooled to approx. -20 °C. At -20 °C, 8.19 g of diisopropylamine is added to the solution and afterwards, 8.14 g of methyl iodide is added over 30 min. After stirring the reaction mixture for 65 minutes at -20 °C, 6.4 mL of concentrated hydrochloric acid in 50 mL of water and a solution of 6.36 g of thiourea in 50 mL of water is added and the reaction mixture is stirred for 15 minutes at 20 °C. Then THF is evaporated off and 120 mL of methylene chloride is added and the pH is adjusted to 8.6 with a 40 % water solution of NaOH. After the separation of the phases, the water phase is washed twice with 60 and 30 mL of methylene chloride. The organic phases are combined and 380 mg of acetic anhydride is added and the mixture is stirred for 5 minutes. Then 6.20 g of oxalic acid in 24 mL of methanol is added within 15 minutes. The resulting suspension is stirred for about 1 hour at approx. 20 °C and afterwards 1 hour at approx. 0 °C. The product is isolated by filtration, washed with 100 mL of methylene chloride and dried for 15 hours at 25 °C in vacuo. Yield: 15 g (93 %).
Example 2 – Formation of olanzapine form I
-
[0094]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. After the solution is concentrated in vacuo to the volume of 15 mL, the solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of -20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 380 ppm IR Form I XRD Form I
Example 3 – Formation of olanzapine form I (scale up)
-
[0095]24 kg of olanzapine oxalate is dissolved in 240 L of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 2.4 kg of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 L of water. The filtrate and wash water are combined and after the addition of 300 L of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 15 minutes, the layers are separated and the water phase is extracted twice with 50 L of methylene chloride. The organic layers are combined and washed twice with 100 L of water. After the solution is concentrated in vacuo to the volume of 50 L, the solution is immediately cooled to -15 °C. The resulting suspension is stirred for 30 minutes, then olanzapine is isolated by filtration. The wet cake is washed with 10 L of methylene chloride of the temperature of -20 °C. The product is dried for 10 hours at 100 °C in vacuo.
HPLC-Purity: 99.7 % olanzapine-CM 1000 ppm IR Form I XRD Form I
Example 4 – Formation of olanzapine form I
- (
Al2O3 fluidized bed adsorption
-
[0096]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. After 0.2 g of Al2O3 is added and the methylene chloride suspension is stirred for 5 minutes, Al2O3 is filtered off and the methylene chloride solution is concentrated to the volume of 15 mL. The solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of -20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 214 ppm IR Form I XRD Form I
- )
Example 5 – Formation of olanzapine form I (Al2O3 fluidized bed adsorption
-
[0097]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. Afterwards, the methylene chloride solution is concentrated to 30 mL and 0.2 g of Al2O3is added. After stirring for 5 minutes, Al2O3 is filtered off and the methylene chloride solution is concentrated to the volume of 15 mL. The solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of -20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % Olanzapine-CM 321 ppm IR Form I XRD Form I
- )
Example 6 – Formation of olanzapine form I (Al2O3 fluidized bed adsorption
-
[0098]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. Then the methylene chloride solution is concentrated to 30 mL and 1 g of Al2O3 is added. After stirring for 5 minutes, Al2O3 is filtered off and the methylene chloride solution is concentrated to the volume of 15 mL. The solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of a temperature of -20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 138 ppm IR Form I XRD Form I
- )
Example 7 – Formation of olanzapine form I (Al2O3 short column adsorption)
-
[0099]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. Then the methylene chloride solution is concentrated to 30 mL and filtered through 20 g of Al2O3 (h = 2 cm). After the methylene chloride solution is concentrated to the volume of 15 mL, the solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of-20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 162 ppm IR Form I XRD Form I
Example 8 – Formation of olanzapine form I (Al2O3 short column adsorption)
-
[0100]5 g of olanzapine oxalate is dissolved in 50 mL of water and the pH of the solution is adjusted to 2.0 by the addition of 6 N HCl. To the resulting clear solution of olanzapine oxalate, 0.5 g of charcoal is added. After stirring for 5 minutes, the charcoal is filtered off and the cake is washed with 10 mL of water. The filtrate and wash water are combined and after the addition of 60 mL of methylene chloride, the pH of the combined mixture is adjusted to 9.0 by the addition of a 40 % water solution of NaOH. After stirring for 5 minutes, the layers are separated and the water phase is extracted twice with 10 mL of methylene chloride. The organic layers are combined and washed twice with 20 mL of water. Afterwards, the methylene chloride solution is concentrated to 30 mL and filtered through 20 g of Al2O3 (h = 5 cm). After the methylene chloride solution is concentrated to the volume of 15 mL, the solution is immediately cooled on an ice/salt bath. The resulting suspension is stirred for 15 minutes, and then olanzapine is isolated by filtration. The wet cake is washed with 3 mL of methylene chloride of the temperature of-20 °C. The product is dried for four hours at 100 °C in vacuo.
HPLC-Purity: 99.9 % olanzapine-CM 73 ppm IR Form I XRD Form I Table 1. Comparison and overview of beneficial effect of Al2O3 treatment
Ex. Scale Treatment Mass (Al2O3) [g] Al2O3 pad height [cm] CM assay [ppm] 2 5 g none / / 380 3 24 kg none / / 1000 4 5 g fluidized bed adsorption 0.2 / 214 5 5 g fluidized bed adsorption (concentrated to 30 mL) 0.2 / 321 6 5 g fluidized bed adsorption (concentrated to 30 mL) 1.0 / 138 7 5 g short column adsorption (concentrated to 30 mL) 20 2 162 8 5 g short column adsorption (concentrated to 30 mL) 20 5 73 Table 2. As comparative data, olanzapine obtained by processes according to the prior art comprises the following amounts of olanzapine-CM:
No. prior art amount of olanzapine-CM 1 WO 2005/090359 , lab. scale 300 – 400 ppm 2 WO 2005/090359 , industrial scale 800 – 2000 ppm 3 WO 2004/056833 <0.15%
………………………………
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| EP0454436B1 | Apr 24, 1991 | Sep 13, 1995 | Lilly Industries Limited | Pharmaceutical compounds |
| EP0733635A1 | Mar 22, 1996 | Sep 25, 1996 | Eli Lilly And Company | Crystal forms of a thieno(2,3-B)(1,5) benzodiazepine derivative and process for their preparation |
| GB1533235A | Title not available | |||
| US5229382 | May 22, 1992 | Jul 20, 1993 | Lilly Industries Limited | 2-methyl-thieno-benzodiazepine |
| WO2002018390A1 | Mar 7, 2001 | Mar 7, 2002 | Ramesh Chakka | Process for preparation of hydrates of olanzapine and their conversion into crystalline forms of olanzapine |
| WO2003055438A2 * | Dec 23, 2002 | Jul 10, 2003 | Vijay Chhangamal Chhabada | Crystalline form i of 2-methyl-4-(4-methyl-1-piperazinyl) 10h thieno [2,3-b][1,5]benzodiazepine |
| WO2003101997A1 | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004000847A1 | Jun 10, 2003 | Dec 31, 2003 | Adamed Sp Zoo | A process for the preparation of olanzapine and an intermediate therefor |
| WO2004056833A1 | Dec 15, 2003 | Jul 8, 2004 | Adamed Sp Zoo | A process for the preparation of a pharmaceutically pure polymorphic form i of olanzapine |
| WO2004089313A2 | Mar 31, 2004 | Oct 21, 2004 | Magali Bourghol Hickey | Novel olanzapine forms and related methods of treatment |
| WO2005090359A2 | Mar 17, 2005 | Sep 29, 2005 | Lek Pharmaceuticals | Synthesis of 2-methyl-4-(4-methyl-1-piperazinyl)-10h-thieno[2, 3-b][1,5]benzodiazepine and salts thereof |
| WO2008139228A2 * | May 14, 2008 | Nov 20, 2008 | Generics Uk Ltd | Process for the purification of olanzapine |
| Reference | ||
|---|---|---|
| 1 | BIOORGANIC & MEDICINAL CHE-MISTRY LETTERS vol. 7, no. 1, 1997, pages 25 – 30 | |
| 2 | BIOORGANIC & MEDICINAL CHEMISTRY LETTERS vol. 7, no. 1, 1997, pages 25 – 30 | |
| 3 | CRYSTAL GROWTH & DESIGN vol. 3, no. 6, 2003, pages 897 – 907 | |
| 4 | REGULATORY TOXICOLOGY AND PHARMACOLOGY vol. 44, 2006, pages 198 – 211 | |
| WO1998011893A1 * | Sep 18, 1997 | Mar 26, 1998 | Lilly Co Eli | Olanzapine dihydrate d |
| EP0733635A1 * | Mar 22, 1996 | Sep 25, 1996 | Eli Lilly And Company | Crystal forms of a thieno(2,3-B)(1,5) benzodiazepine derivative and process for their preparation |
| EP0831098A2 * | Sep 22, 1997 | Mar 25, 1998 | Eli Lilly And Company | Intermediates and process for preparing olanzapine |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2003037903A1 * | Oct 29, 2002 | May 8, 2003 | Cord Janet I | Olanzapine dihydrate-ii a process for its preparation and use thereof |
| WO2003090730A1 * | Apr 25, 2002 | Nov 6, 2003 | Generics Uk Ltd | Novel crystalline forms of celecoxib and other compounds |
| WO2003091260A1 * | Apr 22, 2003 | Nov 6, 2003 | Ramesh Chakka | Novel crystalline polymorph form-vi of olanzapine and a process for preparation thereof |
| WO2003097650A1 * | May 16, 2003 | Nov 27, 2003 | Urszula Fraczek | Methods for preparation of olanzapine polymorphic form i |
| WO2003101997A1 | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004006933A2 | Jul 14, 2003 | Jan 22, 2004 | Krka D D Novo Mesto | Crystal forms of olanzapine and processes for their preparation |
| WO2004056833A1 * | Dec 15, 2003 | Jul 8, 2004 | Adamed Sp Zoo | A process for the preparation of a pharmaceutically pure polymorphic form i of olanzapine |
| WO2004058773A1 * | Dec 24, 2003 | Jul 15, 2004 | Judith Aronhime | Novel crystal forms of olanzapine, methods for their preparation and method for the preparation of known olanzapine crystal forms |
| WO2005070937A1 * | Jan 26, 2005 | Aug 4, 2005 | Rolf Keltjens | A process for making olanzapine in a polymorph form i |
| WO2005080401A1 * | Jul 16, 2004 | Sep 1, 2005 | Shriprakash Dhar Dwivedi | Process for the preparation of olanzapine form 1 useful as antipsychotic drug |
| WO2005107375A2 * | Apr 4, 2005 | Nov 17, 2005 | Jyothi Basu Abbineni | Process for the preparation of olanzapine form-i |
| WO2006006185A1 * | Jul 13, 2005 | Jan 19, 2006 | Saravanan Govindaraju | Improved process for making form i of olanzapine. |
| WO2006025065A1 * | Aug 31, 2004 | Mar 9, 2006 | Lee Pharma Private Ltd | A process for the preparation of anhydrous olanzopine hydrochloride of form-1 |
| WO2008091169A2 | Jan 22, 2008 | Jul 31, 2008 | Tomasz Kozluk Nobilus Ent | Process for preparation of substantially pure polymorphic form i of olanzapine |
| WO2008139228A2 * | May 14, 2008 | Nov 20, 2008 | Generics Uk Ltd | Process for the purification of olanzapine |
| WO2011009831A1 | Jul 19, 2010 | Jan 27, 2011 | Lek Pharmaceuticals D.D. | Process for the purification of olanzapine |
| EP1863775A2 * | Mar 20, 2006 | Dec 12, 2007 | Dr. Reddy’s Laboratories Ltd. | Process for preparing crystalline form i of olanzapine |
| EP1997822A1 * | Nov 10, 2006 | Dec 3, 2008 | EGIS Gyógyszergyár Nyilvánosan Müködõ Részvénytársaság | Process for the preparation of 2-methyl-4-(4-methylpiperazin-1-yl)-10h-thieno-[2,3-b] [1,5] benzodiazepine dihydrochloride trihydrate, 2-methyl-4-(4-methyl-piperazin-1-yl) -10h-thieno[2,3-b] [1,5] benzodiazepine dihydrochloride trihydrate, pharmaceutical compositions comprising it and its use |
| EP2264016A2 | Jul 14, 2004 | Dec 22, 2010 | Jubilant Organosys Limited | A process for producing pure form form of 2-Methyl-4-(4-Methyl-1-Piperazinyl)-10h-thieno[2,3-B][1,5] benzodiazepine |
| EP2292624A1 | Jul 20, 2009 | Mar 9, 2011 | LEK Pharmaceuticals d.d. | Process for the purification of olanzapine |
| US7297789 | May 30, 2003 | Nov 20, 2007 | Sandoz, Inc. | From 4-amino-2-methyl-10H-thieno(2,3-b)(1,5) benzodiazpine HCl and 1-methylpiperazine in an aprotic high boiling solvent; purifying in an acid medium; basifying to pH of 7.5-9; and extracting with a low boiling organic solvent |
| US7323459 | Dec 24, 2003 | Jan 29, 2008 | Teva Pharmaceutical Industries Ltd. | Dissolving olanzapine in a solution of acetic acid and water; filtering the solution; stirring the solution; precipitating the crystalline form by adding a base; and isolating the crystals |
| US7329747 | Jan 27, 2005 | Feb 12, 2008 | Synthon Ip Inc. | Synthesis of olanzapine and intermediates thereof |
| US7425627 | Dec 22, 2004 | Sep 16, 2008 | Teva Pharmaceutical Industries Ltd. | Methods of synthesizing olanzapine |
| US7459449 | Jan 27, 2005 | Dec 2, 2008 | Rolf Keltjens | Olanzapine malonate, olanzapine glycolate, and olanzapine benzoate; antipsychotic agents; water soluble; can form tablets by direct compression; may be mixed with dibasic calcium phosphate and/or hydroxypropyl cellulose |
| US7538213 | May 16, 2003 | May 26, 2009 | Institut Farmaceutyczny | Condensation of molar excess of N-methylpiperazine with 4-amine-2-methyl-10H-thieno [2,3-b][1,5]benzodiazepine hydrochloride in dimethylsulfoxide; recovery and purification with methylene chloride |
| US7745429 | Jul 14, 2003 | Jun 29, 2010 | Krka, D.D. Novo Mesto | Crystal forms of olanzapine and processes for their preparation |
| US7759484 | Jul 7, 2005 | Jul 20, 2010 | Inke, S.A. | Mixed solvate of olanzapine, method for preparing it and method for preparing form I of olanzapine therefrom |
| US7829700 | Sep 5, 2005 | Nov 9, 2010 | Shasun Chemicals And Drugs Limited | Process for preparation of a pharmaceutically pure polymorphic form I of Olanzapine |
| US7834176 | Jan 26, 2006 | Nov 16, 2010 | Sandoz Ag | Polymorph E of Olanzapine and preparation of anhydrous non-solvated crystalline polymorphic Form I of 2-methyl-4(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5] benzodiazepine (Olanzapine Form I) from the polymorphic Olanzapine Form E |
| WO2002018390A1 * | Mar 7, 2001 | Mar 7, 2002 | Ramesh Chakka | Process for preparation of hydrates of olanzapine and their conversion into crystalline forms of olanzapine |
| WO2003097650A1 * | May 16, 2003 | Nov 27, 2003 | Urszula Fraczek | Methods for preparation of olanzapine polymorphic form i |
| WO2003101997A1 * | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004006933A2 * | Jul 14, 2003 | Jan 22, 2004 | Krka D D Novo Mesto | Crystal forms of olanzapine and processes for their preparation |
| EP0733367A1 * | Mar 22, 1996 | Sep 25, 1996 | Eli Lilly And Company | Oral olanzapine formulation |
| US5229382 * | May 22, 1992 | Jul 20, 1993 | Lilly Industries Limited | 2-methyl-thieno-benzodiazepine |
| US5703232 * | Jan 16, 1996 | Dec 30, 1997 | Eli Lilly And Company | Process and solvate of 2-methyl-thieno-benzodiazepine |
| US5736541 * | Jul 25, 1996 | Apr 7, 1998 | Eli Lilly And Company | Slurrying impure material in anhydrous ethyl acetate, then crystallization yields pure stable drug for treatment of anxiety, psychological disorders and gastrointestinal disorders |
| US6348458 * | Mar 31, 2000 | Feb 19, 2002 | U & I Pharmaceuticals Ltd. | Polymorphic forms of olanzapine |
| WO2006025065A1 * | Aug 31, 2004 | Mar 9, 2006 | Lee Pharma Private Ltd | A process for the preparation of anhydrous olanzopine hydrochloride of form-1 |
| WO2007052167A2 | Oct 27, 2006 | May 10, 2007 | Actavis Group Ptc Ehf | Stable composition for a pharmaceutical formulation containing olanzapine |
| WO2007144901A1 * | Jun 12, 2007 | Dec 21, 2007 | Jubilant Organosys Ltd | Process for stabilization of olanzapine polymorphic form i |
| WO2008091169A2 | Jan 22, 2008 | Jul 31, 2008 | Tomasz Kozluk Nobilus Ent | Process for preparation of substantially pure polymorphic form i of olanzapine |
| WO2011009831A1 | Jul 19, 2010 | Jan 27, 2011 | Lek Pharmaceuticals D.D. | Process for the purification of olanzapine |
| EP2292624A1 | Jul 20, 2009 | Mar 9, 2011 | LEK Pharmaceuticals d.d. | Process for the purification of olanzapine |
| US7323459 | Dec 24, 2003 | Jan 29, 2008 | Teva Pharmaceutical Industries Ltd. | Dissolving olanzapine in a solution of acetic acid and water; filtering the solution; stirring the solution; precipitating the crystalline form by adding a base; and isolating the crystals |
| US7829700 | Sep 5, 2005 | Nov 9, 2010 | Shasun Chemicals And Drugs Limited | Process for preparation of a pharmaceutically pure polymorphic form I of Olanzapine |
COCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE amcrasto@gmail.com
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1996038151A1 | May 30, 1995 | Dec 5, 1996 | Lilly Co Eli | Method for treating cognitive dysfunction |
| WO1999016312A1 * | Sep 25, 1998 | Apr 8, 1999 | Lilly Co Eli | Method for treating sexual dysfunction |
| WO2001047933A1 | Dec 22, 2000 | Jul 5, 2001 | Cipla Ltd | New polymorphic forms of olanzapine |
| WO2002018390A1 | Mar 7, 2001 | Mar 7, 2002 | Ramesh Chakka | Process for preparation of hydrates of olanzapine and their conversion into crystalline forms of olanzapine |
| WO2003097650A1 | May 16, 2003 | Nov 27, 2003 | Urszula Fraczek | Methods for preparation of olanzapine polymorphic form i |
| WO2003101997A1 * | May 30, 2003 | Dec 11, 2003 | Geneva Pharmaceuticals Inc | Process of preparation of olanzapine form i |
| WO2004056833A1 | Dec 15, 2003 | Jul 8, 2004 | Adamed Sp Zoo | A process for the preparation of a pharmaceutically pure polymorphic form i of olanzapine |
| WO2005107375A2 * | Apr 4, 2005 | Nov 17, 2005 | Jyothi Basu Abbineni | Process for the preparation of olanzapine form-i |
| WO2006006180A1 * | Jul 14, 2004 | Jan 19, 2006 | Akshat Bhatnagar | A PROCESS FOR PRODUCING PURE FORM OF 2-METHYL-4-(4-METHYL-1-PIPERAZINYL)-10H-THIENO[2,3-b][1,5]BENZODIAZEPINE |
| WO2006010620A2 | Jul 28, 2005 | Feb 2, 2006 | Krka Tovarna Zdravil D D Novo | Olanzapine salts and their conversion to olanzapine free base |
| EP0454436A1 | Apr 24, 1991 | Oct 30, 1991 | Lilly Industries Limited | Pharmaceutical compounds |
| EP0454436B1 | Apr 24, 1991 | Sep 13, 1995 | Lilly Industries Limited | Pharmaceutical compounds |
| EP0733635B1 | Mar 22, 1996 | Aug 16, 2001 | Eli Lilly And Company | Crystal forms of a thieno(2,3-B)(1,5) benzodiazepine derivative and process for their preparation |
| US5637584 | Mar 24, 1995 | Jun 10, 1997 | Eli Lilly And Company | Solvate of olanzapine |
Data Integrity – Again Import Alert issued for Indian company IPCA
DRUG REGULATORY AFFAIRS INTERNATIONAL

Data Integrity – Again Import Alert issued for Indian company IPCA…http://www.gmp-compliance.org/enews_04789_Data-Integrity—Again-Import-Alert-issued-for-Indian-company-IPCA_9193,S-QSB_n.html
![]()
Data Integrity has become one of the most important GMP compliance issues in past two years. This has enormous consequences for the concerned companies but also for companies and authorities in EU and US. It was the US FDA that has first experienced huge data integrity problems in companies worldwide. Many sites in India have been found to violate GMP requirements by Data Integrity issues. Tests have been repeated and original data have been deleted. This is called “testing into compliance”. At the Webpage of the US FDA IPCA products are listed which are impacted by the Import Alert. Two facilities from IPCA have been found to be out of GMP compliance: One in Pithampur (Madhya Pradesh) and one in Piparia (Silvassa) (see also report by FiercePharma).
Products manufactured at those facilities might cause high risks to patients. The quality of the products…
View original post 207 more words
OLANZEPINE VISITED PART 1/3


PART 1…..https://newdrugapprovals.org/2015/04/08/olanzepine/
PART 2….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-22/
PART 3…….https://newdrugapprovals.org/2015/04/09/olanzepine-visited-part-33/

Olanzapine (sold under the brand names Zyprexa, Zypadhera and Lanzek or in combination with fluoxetine, Symbyax) is anatypical antipsychotic. It is approved by the U.S. Food and Drug Administration (FDA) for the treatment of schizophrenia and bipolar disorder.[4]
Olanzapine is structurally similar to clozapine and quetiapine, but is classified as a thienobenzodiazepine. The olanzapine formulations are manufactured and marketed by the pharmaceutical company Eli Lilly and Company; the drug went generic in 2011. Sales of Zyprexa in 2008 were $2.2B in the US, and $4.7B worldwide.[5]

Zyprexa (olanzapine) 10 mg tablets (AU)
Olanzapine has a higher affinity for 5-HT2A serotonin receptors than D2 dopamine receptors, which is a common property of all atypical antipsychotics, aside from the benzamide antipsychotics such as amisulpride. Olanzapine also had the highest affinity of any second-generation antipsychotic towards the P-glycoprotein in one in vitro study.[60] P-glycoprotein transports a number of drugs across a number of different biological membranes including the blood-brain barrier, which could mean that less brain exposure to olanzapine results from this interaction with the P-glycoprotein.[61]
Olanzapine is a potent antagonist of the muscarinic M3 receptor,[65] which may underlie its diabetogenic side effects.[64][66] Additionally, olanzapine also exhibits a relatively low affinity for serotonin 5-HT1, GABAA, beta-adrenergic receptors, and benzodiazepine binding sites.[67] [27]
Dosage forms
Olanzapine is marketed in a number of countries, with tablets ranging from 2.5 to 20 milligrams. Zyprexa (and generic olanzapine) is available as an orally-disintegrating “wafer” which rapidly dissolves in saliva. It is also available in 10 milligram vials for intramuscular injection.[4]
Research
Olanzapine has been investigated for use as an antiemetic, particularly for the control of chemotherapy-induced nausea and vomiting (CINV). A 2007 study demonstrated its successful potential for this use, achieving a complete response in the acute prevention of nausea and vomiting in 100% of patients treated with moderately and highly-emetogenic chemotherapy, when used in combination with palonosetron and dexamethasone.[85]
Olanzapine has been considered as part of an early psychosis approach for schizophrenia. The Prevention through Risk Identification, Management, and Education (PRIME) study, funded by the National Institute of Mental Health and Eli Lilly, tested the hypothesis that olanzapine might prevent the onset of psychosis in people at very high risk forschizophrenia. The study examined 60 patients with prodromalschizophrenia, who were at an estimated risk of 36–54% of developing schizophrenia within a year, and treated half with olanzapine and half with placebo.[86] In this study, patients receiving olanzapine did not have a significantly lower risk of progressing to psychosis. Olanzapine was effective for treating the prodromal symptoms, but was associated with significant weight gain.[87]
1H NMR PREDICT

13C NMR PREDICT


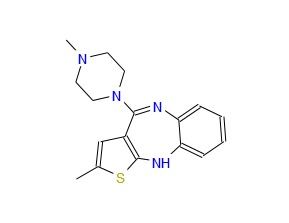
COSY

HMBC
…………………………………..WILL BE UPDATED
UV
IR
1H NMR
13C NMR
MASS
…………………………………….
INTERMEDIATE/S USED IN SYNTHESIS AND REFERENCE


LEK PHARMACEUTICALS D.D. Patent: WO2005/90359 A2, 2005 ; Location in patent: Page/Page column 21 ;


Apotex Pharmachem Inc. Patent: US2008/319189 A1, 2008 ; Location in patent: Page/Page column 2 ;

LEK PHARMACEUTICALS D.D. Patent: WO2005/90359 A2, 2005 ; Location in patent: Page/Page column 21 ;

Leyva-Perez, Antonio; Cabrero-Antonino, Jose R.; Corma, Avelino Tetrahedron, 2010 , vol. 66, # 41 p. 8203 – 8209
SEE
WATSON PHARMACEUTICALS, INC. Patent: WO2004/94390 A1, 2004 ; Location in patent: Page 15 ;

WO2006/6180 A1, ; Page/Page column 11 ;
 Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
Russian Journal of Bioorganic Chemistry, , vol. 31, # 4 p. 378 – 382
WO2006/6180 A1, ; Page/Page column 11 ;
 US5605897 A1, ;
US5605897 A1, ;
………………………………………
PATENT
http://www.google.com.na/patents/EP1730153B1?cl=en


Figure 2 shows the NMR spectrum of the solvate according to the invention. The peaks were assigned as follows (1H NMR; CDCl3, 300 MHz) :

| Chemical shift δ | Assignement |
| 1.20 (3H, d) | CH3 – isopropanol |
| 2.30 (3H, s) | 4′-CH3 |
| 2.34 (3H, s) | 2- CH3 |
| 2.20-2.40 (2H, br s) | H – water |
| 2.49 (4H, m) | 3′-CH2 |
| 3.52 (4H, m) | 2′-CH2 |
| 4.03 (0.5H, dq) | CH – isopropanol |
| 5.02 (H, broad s) | 10-NH |
| 6.29 (H, broad s) | 3-CH |
| 6.29-7.05 (4H, m) | 6, 7, 8, 9-H |
-
Olanzapine has shown to have high activity with regard to the central nervous system and is also useful for the treatment of schizophrenia, schizophreniform disorders, acute mania, mild anxiety states and psychosis.
-
Various polymorphic and pseudopolymorphic forms, such as solvates, of olanzapine have become known. Some of them are useful for conversion to other desirable forms.
-
The British patent GB 1 533 235 discloses antipsychotically effective thienobenzodiazepines by a generic formula which also covers olanzapine.
-
US patent 5,229,382 discloses olanzapine explicitly. The described process for its synthesis involves a crystallization from acetonitrile.
-
EP-B-733 635 claims crystalline form II olanzapine, and this polymorphic form is said to be more stable than the material obtained according to US 5,229,382 which is designated “form I olanzapine”. Both the form I and the form II of olanzapine are characterized by e. g. X-ray data. The preparation of the more stable form II of olanzapine is effected by dissolving technical grade olanzapine in ethyl acetate and crystallization from the resulting solution by any conventional process such as seeding, cooling, scratching the glass of the reaction vessel or other common techniques.
-
WO 02/18390 discloses the monohydrate form I and the dihydrate form I of olanzapine, a process for production thereof and a process for production of form I of olanzapine which comprises the steps of stirring olanzapine monohydrate form I or crude olanzapine or form II of olanzapine in methylene chloride at reflux, cooling, filtering and drying. It is also described that a repeating of the process described in US 5,229,382 Example 1, subexample 4 did not lead to formation of form I of olanzapine.
-
WO 03/101997 relates to processes for preparation of form I of olanzapine by regulation of the pH-value of the solution.
-
WO 03/055438 discloses the preparation of form I olanzapine by crystallization from ethanol and subsequent conversion of the obtained ethanol solvate.
-
US patent 5,637,584 discloses the (mono)methylene chloride solvate form of olanzapine and a method for its conversion to the polymorphic form I of olanzapine.
-
EP-B-733 634 discloses three specific solvates of olanzapine, namely the methanol, ethanol and 1-propanol solvates and a process for production of form II olanzapine by drying such a solvate.
-
WO 03/097650 describes two new, mixed solvate forms, the water/methylene chloride solvate and the water/DMSO solvate, methods for preparing them, and their transformation to polymorphic form I.
-
WO 2004/006933 discloses a process for the preparation of form I olanzapine, as well as various pseudopolymorphic forms, namely the isopropanol solvate, and the acetonitrile/methylene chloride/water and acetonitrile/water mixed solvates of olanzapine, and the polymorphic form A.

Preparation of the water-isopropanol mixed solvate of olanzapine Example 1
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (120 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 200 ml were distilled off under vacuum. The residue was cooled to room temperature, isopropanol (180 ml) was added, and the solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After the crystallization was completed, the precipitate was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 25°C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C and after completion of the crystallization the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 22.84 g. Loss on drying (140°C): 13.6%. Water content (Karl Fischer): 5.12%.
Example 2
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (36 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 80 ml were distilled off under vacuum. The residue was cooled to room temperature, and isopropanol (180 ml) was added. The solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After the crystallization was completed, the precipitate was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 35 °C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C, upon finalization of the crystallization, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 21.98 g. Loss on drying (140°C): 13.2 %. Water content (Karl Fischer): 5.09%. Assay of isopropanol (GC): 8.55 %.
Example 3
-
A mixture of 4-amino-2-methyl-10H-thieno[2,3-b][1,5]benzodiazepine hydrochloride (26.6 g), 1-methylpiperazine (92 ml), dimethylsulfoxide (36 ml) and toluene (120 ml) was refluxed for 4 hours. The solution was cooled to 95°C and 120 ml were distilled off under vacuum. The residue was cooled to room temperature, and isopropanol (180 ml) was added. The solution was further cooled to 0°C and water (36 ml) was added to initialize crystallization. After completion of the crystallization, the precipitate.was filtered off and washed with isopropanol (20 ml). The wet product was suspended in isopropanol (200 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (3 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 35°C and water (6 ml) was added to start crystallization. The suspension was cooled to 0°C, upon completion of the crystallization, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 24.35 g. Loss on drying (140°C): 13.5%. Water content (Karl Fischer): 5.05%.
Example 4
-
Anhydrous olanzapine (10 g) was suspended in isopropanol (108 ml) and heated to reflux to obtain a clear solution. The solution was slowly cooled. Water (6 ml) was added at 57°C to start crystallization. The suspension was cooled to 0°C, upon finalization of the crystallization, the product was filtered off and washed with isopropanol (5 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 10.97 g. Loss on drying (140°C): 13.3%. Water content (Karl Fischer): 5.13%.
Example 5
-
60 g of olanzapine obtained from mother liquors was suspended in isopropanol (650 ml) and heated to reflux to obtain a clear solution. Ethylenediaminotetraacetic acid disodium salt (7.9 g) was added and the suspension was stirred for one hour. Undissolved material was removed by hot filtration. The clear solution was cooled to 25°C and water (16 ml) was added to start crystallization. The suspension was cooled to 0°C and, upon completion of the crystallization, the product was filtered off and washed with isopropanol (50 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 57.64 g. Loss on drying (140°C): 13.5%. Water content (Karl Fischer): 5.26%.
Example 6
-
The solution of 2,4-bis(4-methyl-1-piperazinyl)-3-propylidene-3H-[1,5]benzodiazepine (41.86 g, 0.11 mmol) (prepared according to WO 2004/065390 ), pyridinium p-toluenesulfonate (55.29 g, 0.22 mmol) and sulfur (11.99 g, 0.374 mmol) in benzonitrile (1100 mL) was stirred at 140°C for 11 h, cooled to 90°C and concentrated to an oily residue. The residue was diluted with dichloromethane and isopropanol (250 mL, 1 : 1). The precipitate was filtered off and washed with dichloromethane and isopropanol (20 ml, 1 : 1). The filtrate was extracted with HCl (250 ml, 2 M). The organic phase was further extracted with HCl (2 X 100 ml, 1 M). The combined aqueous phases were cooled in an ice bath and made alkaline by using 5 M NaOH. The obtained turbid solution was left in a refrigerator over night resulting in a suspension. This was separated by filtration and washed with isopropanol (2 X 25 ml). The wet material was suspended in isopropanol (215 ml) and heated to reflux to obtain a clear solution. The solution was hot filtered. Water (6.5 ml) was added to induce crystallization. The obtained’suspension was cooled to 0°C, and upon completion of crystallisation, the product was filtered off and washed with isopropanol (10 ml). The product was dried at room temperature under vacuum to a constant weight. Yield: 18.61 g. Loss on drying (140°C): 12.8 %. Water content (Karl Fischer): 5.29 %.
Example 7
-
The solution of 2,4-bis(4-methyl-1-piperazinyl)-3-propylidene-3H-[1,5]benzodiazepine (3.805 g, 10 mmol) (prepared according to WO 2004/065390 ), pyridinium p-toluenesulfonate (5.026 g, 20 mmol) and sulfur (1.122 g, 35 mmol) in benzonitrile (100 ml.) was stirred at 140°C for 8.5 h, cooled to 90°C and concentrated to an oily residue. The residue was diluted with isopropanol (50 ml) and dimethyl sulfoxide (5 ml). The precipitate was filtered off and washed with isopropanol (5 ml). Water (10 ml) and sodium hydroxide (1.00 g, 25 mmol) were added to the filtrate. The mixture was stirred at room temperature until the sodium hydroxide had dissolved. The turbid solution was left in a refrigerator over night resulting in a suspension. This was filtered off and washed with isopropanol (5 mL). The wet material was suspended in isopropanol (25 mL) and the suspension was heated to reflux. Then solids were hot filtered. Water (0.75 mL) was added to the filtrate to induce crystallization. The resulting suspension was cooled to 0°C, and upon completion of crystallisation, the product was filtered off and washed with isopropanol (1 mL). The product was then dried at room temperature under vacuum to a constant weight. Yield: 0.738 g.
………………………………..
PAPER
http://www.biomedcentral.com/1471-2210/12/8

Scheme 1
Synthesis of compounds 8a, 8b, and 8c. Reagents and conditions: (i) 1-fluoro-2-nitrobenzene, NaH, THF, rt, 20 h, 60%; (ii) SnCl2, EtOH, 80°C, 1 h, 80%; (iiia) N-methylhomopiperazine (5 equiv), no solvent, microwave heating, 80°C, 4 h,65%; (iiib) N-methylhomopiperazine (5 equiv), no solvent, microwave heating, 120°C, 3 h, 55%; (iiic) N-methylpiperazine (10 equiv), N-methylpiperazine hydrochloride (10 equiv), DMSO, 110–120°C, 20 h, 56%.
References
- Burton, Michael E.; Shaw, Leslie M.; Schentag, Jerome J.; Evans, William E. (May 1, 2005). Applied Pharmacokinetics & Pharmacodynamics: Principles of Therapeutic Drug Monitoring (4th ed.). Lippincott Williams & Wilkins. p. 815. ISBN 978-0-7817-4431-7.
- “PRODUCT INFORMATION OLANZAPINE SANDOZ® 2.5mg/5mg/7.5mg/10mg/15mg/20mg FILM-COATED TABLETS” (PDF). TGA eBusiness Services. Sandoz Pty Ltd. 8 June 2012. Retrieved 26 November 2013.
- “Zyprexa, Zyprexa Relprevv (olanzapine) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 26 November 2013.
- ^ Jump up to:a b c “Olanzapine Prescribing Information” (PDF). Eli Lilly and Company. 2009-03-19. Retrieved 2009-09-06.
- “Lilly 2008 Annual Report” (PDF). Lilly. 2009. Retrieved 2009-08-06.
- National Collaborating Centre for Mental Health (25 March 2009). “Schizophrenia: Full national clinical guideline on core interventions in primary and secondary care”. Retrieved 25 November 2009.
- Duggan L, Fenton M, Dardennes RM, El-Dosoky A, Indran S (2005). Duggan, Lorna, ed. “Olanzapine for schizophrenia”. Cochrane Database of Systematic Reviews (2): CD001359.doi:10.1002/14651858.CD001359.pub2. PMID 10796640.
- “Psychosis and schizophrenia in adults: treatment and management | Guidance and guidelines | NICE”. National Institute for Health and Care Excellence.
- Barnes TR (2011). “Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology”. J. Psychopharmacol. (Oxford) 25 (5): 567–620. doi:10.1177/0269881110391123.PMID 21292923.
- Hasan A, Falkai P, Wobrock T, Lieberman J, Glenthoj B, Gattaz WF, Thibaut F, Möller HJ (2013). “World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects”. World J. Biol. Psychiatry 14 (1): 2–44. doi:10.3109/15622975.2012.739708. PMID 23216388.
- Abou-Setta, AM; Mousavi, SS; Spooner, C; Schouten, JR; Pasichnyk, D; Armijo-Olivo, S; Beaith, A; Seida, JC; Dursun, S; Newton, AS; Hartling, L (August 2012).PMID 23035275. Missing or empty
|title=(help) - Zhang JP, Gallego JA, Robinson DG, Malhotra AK, Kane JM, Correll CU (July 2013).“Efficacy and safety of individual second-generation vs. first-generation antipsychotics in first-episode psychosis: a systematic review and meta-analysis”. Int. J. Neuropsychopharmacol. 16 (6): 1205–18. doi:10.1017/S1461145712001277.PMC 3594563. PMID 23199972.
- Citrome L (August 2012). “A systematic review of meta-analyses of the efficacy of oral atypical antipsychotics for the treatment of adult patients with schizophrenia”. Expert Opin Pharmacother 13 (11): 1545–73. doi:10.1517/14656566.2011.626769.PMID 21999805.
- Lepping P, Sambhi RS, Whittington R, Lane S, Poole R (May 2011). “Clinical relevance of findings in trials of antipsychotics: systematic review”. Br J Psychiatry 198 (5): 341–5.doi:10.1192/bjp.bp.109.075366. PMID 21525517.
- Désaméricq G, Schurhoff F, Meary A et al. (February 2014). “Long-term neurocognitive effects of antipsychotics in schizophrenia: a network meta-analysis”. Eur. J. Clin. Pharmacol. 70 (2): 127–34. doi:10.1007/s00228-013-1600-y. PMID 24145817.
- Leucht S, Cipriani A, Spineli L et al. (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”.Lancet 382 (9896): 951–62. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019.
- Osser DN, Roudsari MJ, Manschreck T (2013). “The psychopharmacology algorithm project at the Harvard South Shore Program: an update on schizophrenia”. Harv Rev Psychiatry 21 (1): 18–40. doi:10.1097/HRP.0b013e31827fd915. PMID 23656760.
- [+http://www.nice.org.uk/guidance/cg185/chapter/1-recommendations “Bipolar disorder: the assessment and management of bipolar disorder in adults, children and young people in primary and secondary care | 1-recommendations | Guidance and guidelines | NICE”].
- Yatham LN, Kennedy SH, O’Donovan C et al. (December 2006). “Canadian Network for Mood and Anxiety Treatments (CANMAT) guidelines for the management of patients with bipolar disorder: update 2007”. Bipolar Disord 8 (6): 721–39. doi:10.1111/j.1399-5618.2006.00432.x. PMID 17156158.
- Selle V, Schalkwijk S, Vázquez GH, Baldessarini RJ (March 2014). “Treatments for acute bipolar depression: meta-analyses of placebo-controlled, monotherapy trials of anticonvulsants, lithium and antipsychotics”. Pharmacopsychiatry 47 (2): 43–52.doi:10.1055/s-0033-1363258. PMID 24549862.
- Maglione M, Maher AR, Hu J et al. (2011). “Off-Label Use of Atypical Antipsychotics: An Update”. PMID 22132426.
- Review of olanzapine in the management of bipolar disorders Neuropsychiatr Dis Treat. 2007 October; 3(5): 579–587.
- Scott, Lisa (Winter 2006). “Genetic and Neurological Factors in Stuttering”. Stuttering Foundation of America.
- “Important Safety Information for Olanzapine”. Zyprexa package insert. Eli Lilly & Company. 2007. Archived from the original on 2007-11-23. Retrieved 2007-12-03.
Elderly patients with dementia-related psychosis treated with atypical antipsychotic drugs are at an increased risk of death compared to placebo. […] ZYPREXA (olanzapine) is not approved for the treatment of elderly patients with dementia-related psychosis.
- “Doctors ‘ignoring drugs warning'”. BBC News. 17 June 2008. Retrieved 2008-06-22.
- L-Z\In re Zyprexa\Documents Leak\Injunction Memo & Order\FINAL INJUNCTION MEMO 2.13.07.wpd
- EFF.org
- Press Releases: January, 2007 | Electronic Frontier Foundation
- Eli Lilly was Concerned by Zyprexa Side-Effects from 1998,[dead link] The Times (London), January 23, 2007
- Navari RM, Einhorn LH, Loehrer PJ, Passik SD, Vinson J, McClean J, Chowhan N, Hanna NH, Johnson CS (2007). “A phase II trial of olanzapine, dexamethasone, and palonosetron for the prevention of chemotherapy-induced nausea and vomiting: A Hoosier oncology group study”. Supportive Care in Cancer 15 (11): 1285–91. doi:10.1007/s00520-007-0248-5. PMID 17375339.
- McGlashan TH, Zipursky RB, Perkins D, Addington J, Miller TJ, Woods SW, Hawkins KA, Hoffman R, Lindborg S, Tohen M, Breier A (2003). “The PRIME North America randomized double-blind clinical trial of olanzapine versus placebo in patients at risk of being prodromally symptomatic for psychosis”. Schizophrenia Research 61 (1): 7–18.doi:10.1016/S0920-9964(02)00439-5. PMID 12648731.
- McGlashan TH, Zipursky RB, Perkins D, Addington J, Miller T, Woods SW, Hawkins KA, Hoffman RE, Preda A, Epstein I, Addington D, Lindborg S, Trzaskoma Q, Tohen M, Breier A (2006). “Randomized, Double-Blind Trial of Olanzapine Versus Placebo in Patients Prodromally Symptomatic for Psychosis”. American Journal of Psychiatry 163 (5): 790–9.
Literature References:
Serotonin (5-HT2) and dopamine (D1/D2) receptor antagonist with anticholinergic activity. Prepn: J. K. Chakrabarti et al., EP 454436; eidem, US 5229382 (1991, 1993 both to Lilly). PRODUCT PATENT
Comparative pharmacology: N. A. Moore et al., Curr. Opin. Invest. Drugs 2, 281 (1993). HPLC determn in human plasma: J. T. Catlow et al., J. Chromatogr. B 668, 85 (1995).
Clinical evaluation in schizophrenia: D. S. Baldwin, S. A. Montgomery, Int. Clin. Psychopharmacol. 10, 239 (1995); in mania of bipolar disorder: M. Tohen et al., Am. J. Psychiatry 156, 702 (1999).
Review of pharmacology and clinical experience: B. C. Lund, P. J. Perry, Expert Opin. Pharmacother. 1, 305-323 (2000).
External links
- Zyprexa.com – official Zyprexa brand website from Eli Lilly and Company
- Zyprexa package insert
- Berenson, Alex (December 17, 2006). “Lilly Settles With 18,000 Over Zyprexa”. New York Times.
COCK WILL TEACH YOU NMRCOCK SAYS MOM CAN TEACH YOU NMR
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE