
Home » Uncategorized (Page 96)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ECA and PQG publish Chapter 6 of the interpretation of the ECA and PQG publish Chapter 6 of the interpretation of the EU GDP Guideline
DRUG REGULATORY AFFAIRS INTERNATIONAL

The ECA Foundation and the Pharmaceutical Quality Group (PQG) have been working on the interpretation of different chapters of the EU GDP Guideline. Now the group has finalized the work on chapter 6 – Complaints, Returns, Suspected Falsified Medicinal Products & Medicinal Product Recalls. Read more about the GDP Guidance Chapter 6.
The ECA Foundation and the Pharmaceutical Quality Group (PQG) have been working on the interpretation of different chapters of the EU GDP Guideline. The interpretation of five chapters have been published already. The following 5 Guidance chapters on the EU GDP Guideline are available:
Chapter 1: Quality Management
Chapter 9: Transportation (also contains a template for a Technical Agreement)
Chapter 7: Outsourced Activities
Chapter 2: Personnel
Chapter 5: Operations
Now the group has finalized the work on chapter 6 – Complaints, Returns, Suspected Falsified Medicinal Products & Medicinal Product Recalls. Chapter 6 of the EU GDP…
View original post 124 more words
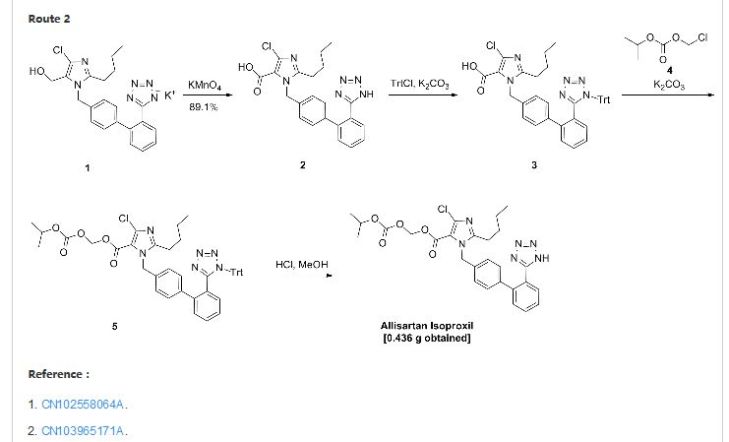
Allisartan isoproxil

Allisartan isoproxil
CAS: 947331-05-7
553.01, C27 H29 Cl N6 O5
An angiotensin II receptor antagonist used to treat mild to moderate essential hypertension.
Approved china, cfda July 1 2012
![]()
Shanghai Allist Pharmaceutical, Inc.
Allist Shanghai Pharmaceutical Co., Ltd.

2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carboxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester,
2-Butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)biphenyl-4-ylmethyl]-1H-imidazole-5-carboxylic acid isopropoxycarbonyloxymethyl ester
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester
Allisartan is an orally-available angiotensin AT1 antagonist in phase II clinical trials at Shanghai Allist Pharmaceutical for the treatment of mild to moderate essential hypertension.
Shanghai Allist Pharmaceutical PHASE 2 for Hypertension

CN200710094021.4 and CN201110289695.6 disclose the preparation of Alicante medoxomil, the inventor repeated, the proceeds of crystal and Chinese patent CN200710094131.0 consistent disclosed.

Allisartan isoproxil
Angiotensin II AT-1 receptor antagonist
Essential hypertension
Amorphous form of allisartan isoproxil is claimed in WO 2015062498. Useful for treating hypertension. Shenzhen Salubris Pharmaceuticals, in collaboration with Allist, has developed and launched allisartan isoproxil. In October 2012, Shenzhen Salubris signed a strategic cooperation framework agreement with Allist Pharmaceutical for the production and marketing of allisartan isoproxil. Family members of the product case of allisartanWO2007095789, expire in the EU and in the US in 2026. For a prior filing see WO2009049495 (assigned to Allist Pharmaceuticals), claiming the crystalline form of allisartan and its method of preparation.
The compound of formula (I) is an Ang II receptor antagonist. Its chemical name is 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carb-oxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester. Chinese Patent CN101024643A describes the structure, and its use as antihypertensive drugs.

As regards to the solid physical properties of the compound of formula (I), the patent document of CN101024643A discloses that it is a white solid, and its melting point is 134.5-136° C. However, CN101024643A dose not disclose the crystalline structure of the compound of formula (I).

 CHINA
CHINA
NEW PATENT
WO-2015062498
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015062498
……………………..
PATENT
http://www.google.com/patents/CN103965171A?cl=en
Hypertension is a major disease threat to human health, looking for efficiency, low toxicity anti-hypertensive drugs can help relieve social pressures and family responsibilities, with good social and economic benefits.
Angiotensin II (Ang II) is the renin – angiotensin – aldosterone system (RAAS) main vasoconstrictor hormone, which plays an important role in the pathobiology of many chronic diseases, particularly its the role of blood pressure regulation is particularly prominent, and therefore Ang II receptor is believed to be a good target for the development of anti-hypertensive drugs.
EP0253310 discloses a series of imidazole derivatives, DuPont declared and obtained by the study of losartan potassium-listed in 1994, was the first non-peptide Ang II receptor antagonist anti-hypertensive drugs. Thereafter, he listed a series of losartan antihypertensive drugs: candesartan cilexetil, valsartan, irbesartan, telmisartan and olmesartan medoxomil, etc. (EP0253310, W02005049587, GB2419592, EP1719766, US5196444) .
The losartan potassium in the body, the active metabolite EXP3174 has a stronger antihypertensive effect than losartan potassium, but EXP3174 polar molecular structure, is difficult to form passive absorption by diffusion through the cell membrane. US5298915 discloses five carboxyl ester group transformation EXP3174 is a series of derivatives, focusing on the compound HN-65021, and discloses hypotensive test results HN-65021 administered by the oral route, its hypotensive activity with chlorine Similar losartan potassium (BritishJouurnal ofClinical Pharmacology, 40,1995,591).
CN200680000397.8 _5_ discloses a class of imidazole carboxylic acid derivatives, namely Alicante medoxomil compound 8 has a good blood pressure lowering effect, the structure of formula I, the preparation method disclosed in this patent document follows the route A, losartan potassium by oxidation, the protecting group into an ester, deprotected to give a compound of formula I, the route step oxidation process of hydroxyl to carboxyl groups, will be reduced to very fine granular potassium permanganate, manganese dioxide, filtration This manganese mud time-consuming, inefficient, polluting; the second step conversion was about 70%, and post-processing cumbersome; byproducts and produced the first two steps more. This makes the high cost of the entire route, not suitable for the production of amplification.

CN200710094021.4 discloses another method for preparing the compounds of formula I, the following route B, the starting material by nucleophilic substitution, oxidation, an ester, a tetrazole ring to obtain a compound of formula I, the first step of the method nucleophilic substitution easy to generate an imidazole ring -3 para isomer impurities difficult to remove; the last step into the ring to use sodium azide, operating dangerous.

CN201210020174.5 disclosed a series of anti-hypertensive compound and preparation method, the following line C, the temperature control in the first step of its preparation O ~ 5 ° C, a mixed solution of acetone and water, with a 5% aqueous solution of sodium hypochlorite oxidation, yield 70%, the second step use of potassium permanganate, manganese dioxide will produce the same, and a yield of only 40%, the first two steps total yield of 28%, is very low, and the post-treatment methods are by column separation, the first two steps are used are organic and inorganic mixed solvent is not conducive to recovery, not suitable for scale-up.






Example 8 2-Butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil crude)

To a 20L reactor 9800ml of methanol, stirring was started, the rotational speed is added at 200r / min 1225.3g solid compound of formula II, and heated to reflux. The reaction 8-10h evacuation HPLC detection, the formula II compound residue <1.0% seen as a response endpoint. After reaching the end of the reaction the heating was stopped, continued stirring speed of 180r / min. About 3_4h fell 20_25 ° C, colorless transparent crystalline solid precipitated. The reaction mixture was cooled to continue to 15-20 ° C, to maintain 15-20 ° C with stirring 3h, the reaction mixture was filtered to give a pale yellow clear filtrate. The filtrate was concentrated under reduced pressure to move 20L flask, vacuum degree of 0.075MPa, 40_45 ° C methanol distilled off under until no distillate. 800ml of absolute ethanol was added, a vacuum degree of 0.075MPa, 40-45 ° C under distillation until no distillate.
900ml of absolute ethanol was added, heated to reflux. N-heptane was added slowly 1100ml, reflux 15min, to -10 ° c / h speed cooled to 15 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration lh, was Allie medoxomil crude (800.lg, yield 93.8%).Purification was used directly in the next step without drying.
Example 9 2-butyl-4-chloro-_1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole-5-carboxylic acid, 1 – [(isopropylamino oxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

850ml of absolute ethanol was added to the 3L reaction vessel was charged with crude Alicante medoxomil (800.lg, 1.45mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1300ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration, the purified Alicante medoxomil (780.9g, 97.6% yield).
Example 10 2-butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

950ml of absolute ethanol was added to the 5L reaction vessel was charged with crude Alicante medoxomil (549.9g, 1.72mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1200ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = cake was washed with a mixture of 1/3, and dried under reduced pressure after filtration to obtain a purified Alicante medoxomil (540.0g, 98.2% yield).
Example 122-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester (compound 8)

To a 100 ml of one-necked flask, 0.523 g of material, 0.124 g of potassium carbonate, 5 ml of N,N-dimethylacetamide were added in turn. The solution was stirred at room temperature for 20 minutes. Then 0.562 g of 1-chloromethyl isopropyl carbonate was added and the mixture was reacted at 45-50° C. for 16 hours. After the reaction was completed, the mixture solution was filtered, and 30 ml of water was added into the filtrate. The resulting mixture was extracted with 30 ml of ethyl acetate twice. The organic phase was dried and concentrated to give 1.724 g of oil, which was directly used in the next reaction without purification.
10 ml of dioxane and 5 ml of 4 mol/L HCl were added, and the resulting mixture was reacted at room temperature for 16 hours. The reaction was stopped and the solution was adjusted to pH 6-7 using aqueous sodium bicarbonate solution. The solution went turbid, and was extracted with ethyl acetate. The organic phase was washed with saturated brine, dried, concentrated to give 0.436 g of 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester.
In addition, the following reaction condition can be used to deprotect the protecting group. To 1.7 g of oily product, 5 ml absolute methanol was added and the mixture was heated slowly to reflux and stirred for 8 hours. When the insoluble solid disappeared totally, the mixture was discontinued to heating and cooled to 5° C. The white solid precipitated, and was separated by filtration, and the filter cake was washed with a small quantity of methanol. The combined filtrate was concentrated to dryness to give 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester with the yield of 70%.
1H-NMR (CDCl3) δ H (ppm): 0.89 (t, 3H, J=14.6), 1.24 (d, 6H, J=6.3), 0.37 (m, 2H, J=22.1), 1.69 (m, 2H, J=30.5), 2.64 (t, 2H, J=15.5), 4.81 (m, 1H, J=12.4), 5.54 (s, 2H), 5.86 (s, 2H), 6.95-7.64 (8H), 8.08 (d, 1H, J=7.42)
ESI(+) m/z: 552.7
Mp: 134.5-136° C.




| WO2005011646A2 * | 20 Jul 2004 | 10 Feb 2005 | Nicoletta Almirante | Nitrooxy derivatives of losartan, valsatan, candesartan, telmisartan, eprosartan and olmesartan as angiotensin-ii receptor blockers for the treatment of cardiovascular diseases |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8455526 * | 6 Jun 2008 | 4 Jun 2013 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| US20100168193 * | 6 Jun 2008 | 1 Jul 2010 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| USRE44873 | 31 Jul 2006 | 29 Apr 2014 | Salubris Asset Management Co., Ltd. | Imidazole-5-carboxylic acid derivatives, the preparation method therefor and the uses thereof |
| CN101024643A | 20 Feb 2006 | 29 Aug 2007 | 上海艾力斯医药科技有限公司 | Imidazo-5-carboxylic-acid derivatives, its preparing method and use |
| US5298519 * | 24 Sep 1992 | 29 Mar 1994 | Chemish Pharmazeutische Forschungsgesellschaft M.B.H. | Acylals of imidazole-5-carboxylic acid derivatives, and their use as angiotensin (II) inhibitors |
……………….
update……………..
Example 1
Weigh 25g 2- butyl-4-chloro-1- [2 ‘- (1-trityl–1H- tetrazol-5-yl) -1,1’-biphenyl – methyl] – imidazole 5-carboxylic acid, 1 – [(isopropoxy) – carbonyloxy] -, methyl ester, was added to a 500ml three-necked flask, methanol was added 200ml, refluxed for 9h, methanol was distilled off under reduced pressure to give crude Alicante medoxomil .
To the residue (i.e., medoxomil crude Alicante) were added 33ml of isopropanol and 66ml of n-heptane, heated to 76 ℃ stirred for 2h. After cooling to 60 ℃ stirring for 1h, and then the system was slowly cooled to 0 ℃, stirring was continued for 3h. Filtered, the filter cake was washed with n-heptane. At 40 ℃ 8 hours and dried in vacuo to give 15.3g Alicante medoxomil (purity 99.3%) as a XRD spectrum as shown in Figure, the main peak of the diffraction peaks as shown in the following table, the DSC spectrum shown in figure II . Compared with the published crystal, the crystal obtained by the absence of significant electrostatic phenomena.
Shanghai , CHINA







FDA approves Raplixa to help control bleeding during surgery

April 30, 2015
Release
The U.S. Food and Drug Administration today approved Raplixa (fibrin sealant [human]), the first spray-dried fibrin sealant approved by the agency. It is used to help control bleeding during surgery.
Raplixa is a biological product approved for use in adults to help control bleeding from small blood vessels when standard surgical techniques, such as suture, ligature or cautery, are ineffective or impractical. When applied to a bleeding site, Raplixa is dissolved in the blood and a reaction starts between the fibrinogen and thrombin proteins. This results in the formation of blood clots to help stop the bleeding.
Raplixa contains fibrinogen and thrombin, two proteins found in human plasma, the liquid portion of blood. The two protein components are individually purified using a manufacturing process that includes virus inactivation and removal steps to help reduce the risk for the transmission of blood-borne viruses. The fibrin sealant components are then spray-dried, blended and packaged in a vial. Raplixa can be applied directly from the original product vial or by spraying with a delivery device onto a bleeding site. It is approved for use in conjunction with an absorbable gelatin sponge.
“This approval provides surgeons an additional option to help control bleeding during surgery when needed,” said Karen Midthun, M.D., director of the FDA’s Center for Biologics Evaluation and Research. “The spray-drying process used to manufacture Raplixa produces dried powders that can be combined into a single vial. This eliminates the need to combine the fibrinogen and thrombin before use and allows the product to be stored at room temperature.”
In support of approval, the FDA reviewed data from a clinical study involving 719 participants, over 11 months, undergoing different types of surgical procedures. The study demonstrated Raplixa’s effectiveness by comparing the reduction in the time needed for bleeding to stop when using this fibrin sealant and the time needed for bleeding to stop when using an absorbable sponge alone.
The most commonly reported adverse reactions were surgical pain, nausea, constipation, fever and decreased blood pressure.
Raplixa is manufactured by ProFibrix BV, a wholly owned subsidiary of The Medicines Company, based in Parsippany, New Jersey.
Will WFI from membrane-based technologies now become an alternative for Europe?
DRUG REGULATORY AFFAIRS INTERNATIONAL

In an EDQM paper published in March 2015 the topic production of WFI by means of membrane-based technologies is discussed again and not excluded any more. Read more about WFI from membrane-based technologies.
In an EDQM paper published in Pharmeuropa in March 2015 the topic production of WFI (water for injections) by means of membrane technologies (reverse osmosis coupled with other suitable techniques) is discussed again and not excluded any more. So far distillation is the only permitted procedure for the production of WFI in Europe. It was already pointed out in the paper on the revision of Annex 1 published in February that alternative procedures for the manufacture of WFI might become possible.
The first part of the new document describes the history of the long lasting discussion of the question whether other procedures than distillation should be allowed for the production of WFI. In the end this led to…
View original post 266 more words
Doxylamine succinate

Doxylamine succinate
CAS NO. 562-10-7,
Sperber et al. Journal of the American Chemical Society, 1949 , vol. 71, p. 887,889
see
Application of Toluene in the Synthesis of Doxylamine Succinate KC. Chaluvaraju1*, MD. Karvekar2 and AR. Ramesha3 1Department of Pharmaceutical Chemistry, Govt. College of Pharmacy, Bengaluru, Karnataka, India. 2Department of Pharmaceutical Chemistry, Krupanidhi College of Pharmacy, Bengaluru, Karnataka, India. 3R&D, R L Fine Chemicals, Bengaluru, Karnataka, India.
ABSTRACT In the present study an efficient method for the synthesis of Doxylamine succinate in the presence of toluene is described. The yield and purity of the product prepared by this method has been found to be better in comparison to reported method. The structure of the synthesized compound was characterised by its melting point and spectral data’s (IR, I HNMR, 13CNMR and Mass spectra). The data obtained are in good agreement with the literature found for Doxylamine succinate.
m.p-102-103°C.
1HNMR (CDC13) δ ppm: 8.5 (d, J = 2.4 Hz ,1H; Het-H) ,7.6-7.0 (m,8H; Ar-H+ Het-H), 3.5-3.3 (t, J = 6.6 Hz, 2H;-OCH2), 2.6-2.5 (t, J = 3.0 Hz, 2H; – CH2), 2.3-2.2 (s, 6H, -N(CH3)2).2.0-1.9 (s, 3H, -CH3).
I3CNMR (CDC13) δ ppm: 148.17, 145.55, 136.17, 127.84, 126.62, 126.21, 121.50, 120.77, 81.81, 61.11, 59.39, 45.91, 23.76.
MS (EI) m/z: 271 (M+ ), 257, 226, 182.
nmr…………http://file.selleckchem.com/downloads/nmr/S424001-Doxylamine-succinate-HNMR-Selleck.pdf

nmr predict of succinate


nmr predict of free base
CAS NO. 469-21-6, N,N-dimethyl-2-(1-phenyl-1-pyridin-2-ylethoxy)ethanamine H-NMR spectral analysis


http://www.google.com/patents/CN102108059B?cl=en
Doxylamine succinate following structural formula:

CAS Number: 562-10-7
Formula = C21H28N2O5
Molecular weight: 388.46
III SUMMARY OF THE INVENTION
The present invention aims to provide a class of antihistamines ethanol as doxylamine succinate, the technical problem to be solved is the selection of a new simple synthetic methods.
The synthesis of doxylamine succinate process route is:

The synthesis of 2-acetyl-pyridine as starting materials, including synthetic and doxylamine salt-forming reaction and the separation and purification process of each unit, wherein the first synthetic doxylamine by The reaction of 2-acetyl pyridine Grignard reagent with bromobenzene and magnesium to produce 2-pyridyl generated methylcarbinol, then 2-pyridyl-methyl-phenyl methanol with sodium amide and sequentially generates 2-dimethylamino ethyl chloride reaction Doxylamine, most 后多西拉敏 a salt with succinic acid to give the title product doxylamine succinate.
the synthesis of doxylamine
150ml three-necked flask of xylene 40ml, weighed 2. 34g (0. 06mol) was added sodium amide three-neck flask, weighed 10g (0.05mol) 2- pyridyl methylcarbinol dissolved in 20ml of xylene was slowly added dropwise, followed by stirring.After the addition was complete, the oil bath was heated 150 ° C, maintained under reflux of xylene, the reaction was refluxed for 5 hours. Color from pale yellow reaction solution gradually turned dark brown, solid gradually dissolved.
The dried mixture of 2-dimethylamino ethyl chloride was added 20ml of xylene dropping funnel was slowly added dropwise to the three-necked flask. After the addition was complete, maintaining at reflux for 20 hours. TLC monitoring of the reaction process, the reactants and products change (V petroleum ether: V ethyl acetate = 5: 1).
After stopping the reaction, the oil bath was removed, and the reaction solution was cooled to room temperature, with ice-bath, was slowly added dropwise to the reaction solution 50ml of ice water, stirred for half an hour. The reaction solution was separated, the organic phase was retained and the aqueous phase was extracted with xylene (3 * 40ml), the combined organic
Phase. Drying, filtration, rotary evaporation to remove xylene.
The obtained crude product was subjected to silica gel mixed with the sample, the liquid sample with the silica mass ratio of 1: 2, dissolved in ethyl acetate, and stirred for half an hour, the solvent was removed by rotary evaporation. The mixed sample was subjected to column chromatography on silica gel, eluting with a mixed solvent (V petroleum ether: V ethyl acetate = 2: 1) petroleum ether and ethyl acetate eluent until the 2-pyridyl-methyl-phenyl The complete collection of components of methanol to stop the elution. The eluent was collected and the solvent was removed by rotary evaporation, after recycling the recovered 2-pyridyl-methyl-phenyl methanol and dried in vacuo.
The chromatography column of silica gel and the eluent was poured into the remaining single-necked flask, and the crude product was added mass of diethylamine, stirred for half an hour, filtration, and the solvent was removed by rotary evaporation and the liquid diethyl amine, to give doxylamine 7. 3g, 54% yield. Gas content was 99%. (Column chamber temperature 250 ° C, detection temperature 300 ° C, vaporization temperature of 300 ° C).
1HNMr (CDCI3), δ: 8 · 51 (1Η, m), 7 · 60-7 61 (2Η, m), 7 · 40 (2Η, m), 7 · 27 (2Η, m),. 7. 18 (1Η, m), 7. 09 (1H, m), 3. 41 (2H, m), 2. 59 (2H, m), 2. 27 (6H, s), 1. 98 (3H , s).
3, doxylamine succinate synthesis of
Doxylamine 1. 35g (0. 005mol) and succinic 0. 59g (0. 005mol) was added IOml single-necked flask, adding acetone 7ml, heating and stirring until dissolved, stirring was continued for half an hour, the heating was stopped. Cooled to room temperature and then placed in the refrigerator freezer -20 ° C for 24 hours. Filtration, the solid was placed in a vacuum desiccator the residual solvent was distilled off, and dried for 6 hours. The crude product was dissolved by heating continued recrystallized from acetone (Ig doxylamine succinate: 2.5mL acetone). Steps above, doxylamine succinate, and recrystallized to give 1.6g, 82% yield. Mp 101-103 ° C.
] 1HNMr (CDCI3), δ: 8 · 54 (1Η, m), 7 · 69 (1Η, m), 7 · 51 (1Η, m), 7 · 32 (2Η, m), 7 · 30 ( 2Η, m), 7. 23 (1Η, m), 7. 16 (1H, m), 3. 63 (2H, m), 3. 18 (2H, m), 2. 80 (6H, s), 2. 54 (4H, s), 1. 99 (3H,S) O
| CN1447694A | Jun 21, 2001 | Oct 8, 2003 | 达切斯内公司 | Rapid onset formulation |
| Reference | ||
|---|---|---|
| 1 | Bachman, G. Bryant等.Heterogeneous bimolecular reduction. II. Direct acylation of pyridine and its homologs and analogs.《Journal of Organic Chemistry》.1957,第22卷1302-1308. | |
| 2 | CHARLESH . TILFORD等.Histamine Antagonists. Basically Substituted Pyridine Derivatives.《Journal of the American Chemical Society 》.1948,第70卷4001-4009. | |
……………….


 Morning street in Bhaktapur, a UNESCO World Heritage Site on the east corner of the Kathmandu Valley, Bagmati, Nepal. Bhaktapur is an ancient Newar town, .
Morning street in Bhaktapur, a UNESCO World Heritage Site on the east corner of the Kathmandu Valley, Bagmati, Nepal. Bhaktapur is an ancient Newar town, .


 .
.

India’s Wockhardt to recall some drugs made in India after U.S. FDA concerns
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
Indian generic drugmaker Wockhardt Ltd said on Tuesday it would recall some drugs manufactured at its two plants in India before the U.S. Food and Drug Administration (FDA) banned those sites due to quality concerns.
The FDA banned U.S. exports from Wockhardt’s Waluj and Chikalthana plants in central India in 2013, citing manufacturing quality lapses.
see
Fosamprenavir

Fosamprenavir
BASE

| Systematic (IUPAC) name | |
|---|---|
| {[(2R,3S)-1-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-3-({[(3S)-oxolan-3-yloxy]carbonyl}amino)-4-phenylbutan-2-yl]oxy}phosphonic acid | |
| Clinical data | |
| Trade names | Lexiva |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a604012 |
|
|
|
|
| Oral | |
| Pharmacokinetic data | |
| Bioavailability | Unknown |
| Protein binding | 90% |
| Metabolism | Hydrolysed to amprenavirand phosphate in GI tractepithelium |
| Half-life | 7.7 hours |
| Excretion | Fecal (as metabolites of amprenavir) |
| Identifiers | |
| 226700-81-8 |
|
| J05AE07 | |
| PubChem | CID 131536 |
| DrugBank | DB01319 |
| ChemSpider | 116245 |
| UNII | WOU1621EEG |
| ChEMBL | CHEMBL1664 |
| NIAID ChemDB | 082186 |
| Chemical data | |
| Formula | C25H36N3O9PS |
| 585.608 g/mol 623.700 g/mol (calciumsalt) |
|

Fosamprenavir (marketed by ViiV Healthcare as the calcium salt), under the trade names Lexiva (U.S.) and Telzir (Europe) is apro-drug of the protease inhibitor and antiretroviral drug amprenavir. The FDA approved it October 20, 2003, while the EMEA approved it on July 12, 2004. The human body metabolizes fosamprenavir in order to form amprenavir, which is the active ingredient. That metabolization increases the duration that amprenavir is available, making fosamprenavir a slow-release version of amprenavir and thus reducing the number of pills required versus standard amprenavir.
A head-to-head study with lopinavir[1] showed the two drugs to have comparable potency, but patients on fosamprenavir tended to have a higher serum cholesterol. Fosamprenavir’s main advantage over lopinavir is that it is cheaper.
PATENT
http://www.google.com/patents/WO2012032389A2
Fosamprenavir calcium has HIV aspartyl protease inhibitory activity and is particularly well suited for inhibiting HIV-1 and HIV-2 viruses; it is chemically known as calcium (3S) tetrahydro-3-furanyl(l S,2R)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]- l-benzyl-2- (phosphonooxy)propyl carbamate and represented by formula la.
(la)
There are very few references available in the literature for preparation of fosamprenavir and its intermediates. Patent US 5 585 397 provides process for preparation of fosamprenavir intermediate (IV), as depicted in scheme 1 , wherein it is purified using silica gel chromatography, however it does not provide any purity data. Purification by column chromatography is not suitable on commercial scale, since it is time consuming, requires large volume of solvents and is very much laborious.

Scheme 1: Process for preparation of fosamprenavir intermediate (IV) as given in US 5
585 397 Another patent US 6 281 367, provides process for preparation of fosamprenavir intermediate (IV) as depicted in scheme 2, but it does not provide any method for purification of compound (IV).

P= amine protecting
group deprotection

Scheme 2: Process for preparation of fosamprenavir intermediate (VI) as given in US 6
281 367
The patent US 6 514 953 provides process for preparation of fosamprenvair calcium (la) utilizing compound (IV), as depicted in Scheme 3, however it does not provide purity of fosamprenavir calcium (la) or the intermediates thereof.

Aq. soln. of Ca(OAc)2
monohydrate

(la) crude (la)
Scheme 3: Process for preparation of fosamprenavir Calcium (la) as given in US 6 514
953 Another patent, US 6 436 989, which is product patent for fosamprenavir salts, provide process for preparation of fosamprenavir sodium salt (VII) from compound (IV) as depicted in Scheme 4:

(VIA)
(V)
3 eq. NaHC03
resin column,
lyophilize

Scheme 4: Process for preparation of fosamprenavir sodium (VII) as given in US 6 436989. US 6 436 989 provides compound (V) and (VIA) with an HPLC purity of 90% and 92% respectively, however purity of fosamprenavir sodium salt (VII) is not mentioned. This patent provides fosmaprenavir salt intermediates with very low HPLC purity. The prior art literature describes synthesis of fosamprenavir calcium and its intermediates and like any synthetic compound, fosmaprenavir calcium can contain number of impurities from various source like starting material, reaction by-products, degradation, isomeric impurities etc. The prior art documents for fosamprenavir calcium does not provide any information for the impurities that may have been formed from the various synthetic processes provided therein.
Fosamprenavir calcium i.e. calcium (3S) tetrahydro-3-furanyl(lS,2R)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]- 1 -benzyl-2-(phosphonooxy)propyl carbamate (la), is a chiral substrate containing three asymmetrical carbon centre resulting into eight stereoisomers.
Different isomers of a chiral drug molecule bind differently to target receptors, one isomer of a drug may have a desired beneficial effect while the other may cause serious and undesired side effects or sometimes even beneficial but entirely different effects, hence in the drug molecules the effective isomer is preferred in pure form, free of other undesired isomers, thus fosamprenavir calcium free of its other stereoisomer would always be preferred.
The methods described above for preparation of fosamprenavir does not describe suitable methods to minimize formation of R-isomer impurity (lb)

(lb)
One of the approach to minimize R-isomer impurity (lb) is to use highly pure intermediate (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate (Ila), in the synthesis of fosamprenavir. US 5 585 397 provides process for preparation of N-succinimidlyl-(S)-3-tetrahydrofuryl carbonate (Ila), however it does not provide any method for purification neither does it provide any purity data for the same. The PCT application WO 94/18192 provides process for preparation (S)-3-tetrahydrofuranyl- N-succinimidyl carbonate (Ila) as depicted in scheme 5. The application discloses recrystallization of compound (Ila) from EtOAc/hexane. At our hands, crystallization of compound (Ila) from ethyl acetate/hexane provided compound (Ila) containing the intermediate R-isomer impurity compound (lib) upto 0.37% area percentage of HPLC, which is not suitable for its use in the synthesis of fosamprenavir substantially free of R-isomer impurity (lb).

(VIII) (IX) (II)
a= S-isomer a= S-isomer
b= R-isomer b= R-isomer
Scheme 5: process for preparation of (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate
Commercially available (S)-3-tetrahydrofuranol (Villa) contains upto 5% area percentage of HPLC of (R)-3-tetrahydrofuranyl (Vlllb), which on reaction with N,N-disuccinimidyl carbonate (IX) results in (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate (Ila) containing upto 2.5% area percentage of HPLC of the R-isomer impurity, (R)-3-tetrahydrofuranyl-N- succinimidyl carbonate (lib). This impure (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate (Ila) when converted to fosamprenavir calcium (la) by series of reaction, results into fosamprenavir calcium containing upto 2.0 % area percentage of HPLC of (3R) tetrahydro-3- furanyl(l S,2R)-3-[[(4-aminophenyl) sulfonyl] (isobutyl) amino]- 1 -benzy 1-2- (phosphonooxy)propyl carbamate (lb), which is undesired isomer of fosamprenavir calcium. Impurities of any form are undesirable in the active pharmaceutical product since it may have adverse effect on the patient to be treated.
The purity of API produced is clearly a necessary condition for commercialization. The impurities produced in the manufacturing process must be limited to very small amount and are preferred to be substantially absent. The ICH Q7A guidance for API manufacturers requires that process impurities must be maintained below set limits utilizing various parameters. In the United States the Food and Drug Administration guidelines, would mostly limit the amount of impurities present in the API, similarly in other countries the impurity levels would be defined in their respective pharmacopeias.
The process for preparation of fosamprenavir calcium (la) of present invention is as depicted in scheme 5.

crude fosamprenavir calcium (la)
Example 2: Preparation of pure fosamprenavir calcium (I).
Mixture of 100 g (0.23 mol) (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4- nitrobenzene sulphonamide (III), 65 g (0.28 mol) (S)-3-tetrahydrofuranyl-N-succinimidyl carbonate (Ila) (of Example 1) and 24 g (0.23) triethylamine in 800 ml dichloromethane was stirred at ambient temperature for 4 hours, extracted with 10% sodium bicarbonate solution. The organic layer was separated, washed with water and concentrated. To the concentrated mass was added 1000 ml methanol and heated to 60-65°, cooled to 25°C and solid was filtered, washed with methanol and dried. Mixture of 100 g (0.186 mol) (3S)-tetrahydro-3-furyl N-[(l S,2R)-l-benzyl-2-hydroxy-3-(N- isobutyl-4-nitrobenzene sulphonamido) propyl] carbamate (IV) and 200 ml pyridine was cooled to 0-10°C and 70.0 g (0.456 mol) of POCl3 was added and stirred at ambient temperature for 4 hours, 400 ml methyl isobutyl ketone was added, cooled and 1 : 1 cone. HC1- water was added. Mixture was heated to 50°C for 1 hour, cooled to 25-30°C. Organic layer was separated, washed with water and partially concentrated; 500 ml water and 31.5 g sodium bicarbonate was added and stirred. The organic layer was separated and 100 ml ethylacetate, 400 ml methanol and 5.0 g Pd/C was added. The reaction mass was stirred under hydrogen pressure for 4 hours at 30°C. The mixture was filtered, catalyst washed with methanol. The filtrate was heated to 50°C and 33.0 g (0.186 mol) calcium acetate monohydrate in 100 ml water was added and stirred for 30 minutes. Cooled to 30°C and stirred. Solid was filtered, washed with 1 : 1 mixture of methanol-water and dried to obtain crude fosamprenavir calcium. 65 g (0.104 mol) crude fosamprenavir calcium and 1 170 ml denatured ethanol was heated to 70-72°C, charcaolized. Water (138 ml) was added and mixture stirred for 30 minutes. Cooled to ambient temperature and stirred. Solid filtered, washed with 1 : 1 ethanol-water and dried. Methanol (315 ml) was added to the solid, stirred and filtered. The filtrate was concentrated under vacuum to obtain solid, which was dried to obtain 37.5 g pure fosamprenavir calcium. HPLC purity: fosamprenavir calcium (la): 99.85%; R-isomer impurity (lb): 0.05%; all other individual impurities less than 0.1%.


Fosamprenavir sodium, GW-433908A, 908, VX-175(free acid)
………………………………….
PAPER
DOI: 10.1039/B404071F
http://pubs.rsc.org/en/content/articlelanding/2004/ob/b404071f#!divAbstract
Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors(Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.


…………………
EP 0659181; EP 0885887; JP 1996501299; US 5585397; WO 9405639

The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding the dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) (prepared by condensation of tetrahydrofuran-3(S)-ol (VIII) with phosgene and N-hydroxysuccinimide (IX)) and DIEA in acetonitrile to provide the corresponding carbamate (X). The deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide (XIII). Finally, the nitro group of (XIII) is reduced with H2 over Pd/C in ethyl acetate to afford the target
………………………….

The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) — obtained by reaction of tetrahydrofuran-3(S)-ol (VIII) first with phosgene and then with N-hydroxysuccinimide (IX) — and DIEA in acetonitrile to provide the corresponding carbamate (X). Deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide intermediate (XIII).
……………………………

Esterification of the OH group of compound (XIII) with PO3H3 by means of DCC in hot pyridine gives the corresponding phosphite (XVII), which is oxidized with bis(trimethylsilyl)peroxide in bis(trimethylsilyl)azane to yield the expected phosphate (XVIII). Reduction of the nitro group of (XVIII) with H2 over Pd/C in ethyl acetate affords fosamprenavir (XIX). Finally, fosamprenavir (XIX) is treated with aqueous NaHCO3 or with calcium acetate in water to provide the corresponding salts. Alternatively, the phosphate (XIX) can be obtained directly by reaction of intermediate (XIII) with POCl3 in pyridine, followed by hydrolysis with 2N HCl.
………………………………………..
HIV protease inhibitor; water soluble prodrug of amprenavir, q.v. Prepn: R. D. Tung et al., WO 9933815;eidem, US 6559137 (1999, 2003 both to Vertex).
Prepn of crystalline calcium salt: I. G. Armitage et al., WO 0004033 (2000 to Glaxo); eidem, US 6514953 (2003 to SKB).
Clinical pharmacokinetics: C. Falcoz et al., J. Clin. Pharmacol. 42, 887 (2002).
Review of pharmacology and clinical experience in HIV: T. M. Chapman et al., Drugs 64, 2101-2124 (2004); C. Arvieux, O. Tribut,ibid. 65, 633-659 (2005).
References
- Eron J Jr, Yeni P, Gathe J Jr et al. (2006). “The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial”. Lancet 368 (9534): 476–82.doi:10.1016/S0140-6736(06)69155-1. PMID 16890834.
| WO1994005639A1 * | Sep 7, 1993 | Mar 17, 1994 | Vertex Pharma | Sulfonamide inhibitors of hiv-aspartyl protease |
| WO1994018192A1 | Feb 7, 1994 | Aug 18, 1994 | Merck & Co Inc | Piperazine derivatives as hiv protease inhibitors |
| INKO02772010A | Title not available | |||
| US5585397 | Sep 7, 1993 | Dec 17, 1996 | Vertex Pharmaceuticals, Incorporated | Viricides |
| US6281367 | Mar 18, 1999 | Aug 28, 2001 | Glaxo Wellcome Inc. | Process for the synthesis of HIV protease inhibitors |
| US6436989 | Dec 24, 1997 | Aug 20, 2002 | Vertex Pharmaceuticals, Incorporated | Prodrugs of aspartyl protease inhibitors |
| US6514953 | Jul 15, 1999 | Feb 4, 2003 | Smithkline Beecham Corporation | Calcium (3S) tetrahydro-3-furanyl(1S,2R)-3-[[(4-aminophenyl)sulfonyl](isobutyl)amino]-1-benzyl-2-(phosphonooxy)propylcarbamate |
| Reference | ||
|---|---|---|
| 1 | * | EKHATO I VICTOR ET AL: “Isotope labeled ‘HEA/HEE’ moiety in the synthesis of labeled HIV-protease inhibitors. Part II“, JOURNAL OF LABELLED COMPOUNDS AND RADIOPHARMACEUTICALS, JOHN WILEY, CHICHESTER, GB, vol. 48, no. 3, 1 January 2005 (2005-01-01), pages 179-193, XP009112607, ISSN: 0362-4803 |
| 2 | * | MOON KIM B ET AL: “SYNTHESIS OF A CHIRAL AZIRIDINE DERIVATIVE AS A VERSATILE INTERMEDIATE FOR HIV PROTEASE INHIBITORS“, ORGANIC LETTERS, AMERICAN CHEMICAL SOCIETY, US, vol. 3, no. 15, 1 January 2001 (2001-01-01), pages 2349-2351, XP001179485, ISSN: 1523-7060, DOI: 10.1021/OL016147S |
| 3 | * | SORBERA, L. A. ET AL.: “FOSAMPRENAVIR“, DRUGS OF THE FUTURE, PROUS SCIENCE, ES, vol. 26, no. 3, 1 March 2001 (2001-03-01), pages 224-231, XP009001334, ISSN: 0377-8282, DOI: 10.1358/DOF.2001.026.03.615590 |
![]()

Vertex Pharmaceuticals’ Boston Campus, United States of America

Olopatadine

| Systematic (IUPAC) name | |
|---|---|
| {(11Z)-11-[3-(dimethylamino)propylidene]-6,11- dihydrodibenzo[b,e]oxepin-2-yl}acetic acid (Z)-11-(3-dimethylaminopropylidene)-6,11-dihydrodibenz[b,e] oxepin-2- acetic acid |
|
| Clinical data | |
| Trade names | Patanol and others |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602025 |
|
|
| Ophthalmic, intranasal, oral | |
| Pharmacokinetic data | |
| Half-life | 3 hours |
| Identifiers | |
| 113806-05-6 |
|
| S01GX09 R01AC08 | |
| PubChem | CID 5281071 |
| DrugBank | DB00768 |
| ChemSpider | 4444528 |
| UNII | D27V6190PM |
| KEGG | D08293 |
| ChEMBL | CHEMBL1189432 |
| Chemical data | |
| Formula | C21H23NO3 |
| 337.412 g/mol | |
Olopatadine hydrochloride is an antihistamine (as well as anticholinergic and mast cell stabilizer), sold as a prescription eye drop(0.2% solution, Pataday (or Patanol S in some countries), manufactured by Alcon). It is used to treat itching associated with allergicconjunctivitis (eye allergies). Olopatadine hydrochloride 0.1% is sold as Patanol (or Opatanol in some countries). A decongestantnasal spray formulation is sold as Patanase, which was approved by the FDA on April 15, 2008.[1] It is also available as an oral tablet in Japan under the tradename Allelock, manufactured by Kyowa Hakko Kogyo.[2]
It should not be used to treat irritation caused by contact lenses. The usual dose for Patanol is 1 drop in each affected eye 2 times per day, with 6 to 8 hours between doses.
There is potential for Olopatadine as a treatment modality for steroid rebound (red skin syndrome.) [3]
Olopatadine was developed by Kyowa Hakko Kogyo.[4]
Synthesis

Olopatadine free base is specifically described in U.S. Patent No. 5,116,863. This U.S. patent does not provide any example describing the preparation of olopatadine hydrochloride.
It is believed that the preparation of olopatadine hydrochloride was first disclosed in J. Med. Chem. 1992, 35, 2074-2084.
Olopatadine free base can be prepared according to the processes described in U.S. Patent Nos. 4,871,865 and 5,116,863, and olopatadine hydrochloride can be prepared according to the process described in J. Med. Chem. 1992, 35, 2074-2084, as shown in Scheme 1 below:

Scheme 2 below:


Grignard r**ctlon

OlopDtadins hydrochloride
Olopatadine and its pharmaceutically acceptable salts are disclosed in EP 0214779, U.S. Patent No. 4,871,865, EP 0235796 and U.S. Patent No. 5,116,863. There are two general routes for the preparation of olopatadine which are described in EP 0214779: One involves a Wittig reaction and the other involves a Grignard reaction followed by a dehydration step. A detailed description of the syntheses of olopatadine and its salts is also disclosed in Ohshima, E., et al., J. Med Chem. 1992, 35, 2074-2084. EP 0235796 describes a preparation of olopatadine derivatives starting from 1 l-oxo-6,11- dihydroxydibenz[b,e]oxepin-2-acetic acid, as well as the following three different synthetic routes for the preparation of corresponding dimethylaminopropyliden-dibenz[b,e]oxepin derivatives, as shown in schemes 1-3 below:
Scheme 1:

HaIMgCH2CH2CH2NMe2

Scheme 2:

R1OH or
R2CI

R1 = R2 = alkyl group R1 = H, R2 = trityl group
HaIMgCH2CH2CH2NMe2



Scheme 3:Ph3P Hal’ sHal

R3 = COOH, etc.
The syntheses of several corresponding tricyclic derivatives are disclosed in the same manner in EP 0214779, in which the Grignard addition (analogous to Scheme 1) and the Wittig reaction (analogous to Scheme 3) are described as key reactions.
The synthetic routes shown above in Schemes 2 and 3 for the preparation of olopatadine are also described in Ohshima, E., et al., J Med. Chem. 1992, 35, 2074-2084 (schemes 4 and 5 below). In contrast to the above-identified patents, this publication describes the separation of the Z/E diastereomers (scheme 5). Scheme 4:

65% Ph3CCI

81% CIMgCH2CH2CH2NMe2

A significant disadvantage of the synthetic route depicted in Scheme 4 is the diastereoselectivity of the dehydration step, which gives up to 90% of the undesired E-isomer. The last step (oxidation) is not described in this publication.Scheme 5 below depicts a prior art method disclosed in Ohshima, E., et al., supra.
Scheme 5:

Each of the prior art methods for synthesis of olopatadine have significant cost and feasibility disadvantages. Specifically with the respect to the method set forth in Scheme 5, the disadvantages include: (1) the need for excess reagents, e.g. 4.9 equivalents Wittig reagent and 7.6 equivalents of BuLi as the base for the Wittig reaction, which can be expensive;
(2) the need to use Wittig reagent in its hydrobromide salt form, so that additional amounts of the expensive and dangerous butyllithium reagent are necessary for the “neutralization” of the salt (i.e., excess butyllithium is required because of the neutralization);
(3) because 7.6 equivalents of the butlylithium are used (compared to 9.8 equivalents of the (Olo-IM4) Wittig reagent), the Wittig reagent is not converted completely to the reactive ylide form, and thus more than 2 equivalents of the Wittig reagent are wasted;
(4) the need for an additional esterifϊcation reaction after the Wittig reaction (presumably to facilitate isolation of the product from the reaction mixture) and the purification of the resulting oil by chromatography;
(5) the need to saponify the ester and to desalinate the reaction product (a diastereomeric mixture) with ion exchange resin, prior to separating the diastereomers;
(6) the need, after the separation of the diastereomers, and liberation of the desired diastereomer from its corresponding pTsOH salt, to desalinate the product (olopatadine) again with ion exchange resin;
(7) the formation of olopatadine hydrochloride from olopatadine is carried out using 8 N HCl in 2-propanol, which may esterify olopatadine and give rise to additional impurities and/or loss of olopatadine; and
(8) the overall yield of the olopatadine, including the separation of the diastereomers, is only approximately 24%, and the volume yield is less than 1%.
As noted above, the known methods for preparing olopatadine in a Wittig reaction use the intermediate compounds 6,11-dihydro-l l-oxo-dibenz[b,e]oxepin-2-acetic acid and 3- dimethylaminopropyltriphenylphosphonium bromide hydrobromide. Preparation of these chemical intermediates by prior art syntheses present a number of drawbacks that add to the cost and complexity of synthesizing olopatadine.
One known method for preparation of the compound 6,11-dihydro-l 1-oxo- dibenz[b,e]oxepin-2-acetic acid is depicted in Scheme 6, below. See also, U.S. Patent No. 4,585,788; German patent publications DE 2716230, DE 2435613, DE 2442060, DE 2600768; Aultz, D.E., et al., J Med. Chem. (1977), 20(1), 66-70; and Aultz, D.E., et al., J Med. Chem. (1977), 20(11), 1499-1501. Scheme 6:
COOE

In addition, U.S. Patent No. 4,417,063 describes another method for the preparation of 6,11-dihydro-l l-oxo-dibenz[b,e]oxepin-2-acetic acid, which is shown in Scheme 7. Scheme 7:

Ueno, K., et al., J Med. Chem. (1976), 19(7), 941, describes yet another prior art method for preparing 6,11-dihydro-l l-oxo-dibenz[b,e]oxepin-2-acetic acid, which is shown below in Scheme 8. Scheme 8:

acidFurther, as depicted in Scheme 9, below, U.S. Patent Nos. 4,118,401; 4,175,209; and 4, 160,781 disclose another method for the synthesis of 6, 11 -dihydro- 11 -oxo-dibenz[b,e]oxepin-2- acetic acid.
Scheme 9:
AICI3


6,11 -dihydro-11 -oxo-dibenz- [b,e]oxepin-2-acetic acid
JP 07002733 also describes the preparation of 6,11 -dihydro- 1 l-oxo-dibenz[b,e]oxepin-2- acetic acid, as follows in Scheme 10, below.
Scheme 10:

acidSpecific methods and reagents for performing the intramolecular Friedel-Crafts reaction for cyclizing 4-(2-carboxybenzyloxy)-phenylacetic acid to form 6,11 -dihydro-11-oxo- dibenz[b,e]oxepin-2-acetic acid are described in (1) EP 0068370 and DE 3125374 (cyclizations were carried out at reflux with acetyl chloride or acetic anhydride in the presence of phosphoric acid, in toluene, xylene or acetic anhydride as solvent); (2) EP 0069810 and US 4282365 (cyclizations were carried out at 70-80° C with trifluoroacetic anhydride in a pressure bottle); and (3) EP 0235796; US 5,116,863 (cyclizations were carried out with trifluoroacetic anhydride in the presence of BF3 »OEt2 and in methylene chloride as solvent).
Turning to the Wittig reagent for use in preparing olopatadine, 3- dimethylaminopropyltriphenylphosphonium bromide-hydrobromide and methods for its preparation are described in U.S. Patent Nos. 3,354,155; 3,509,175; 5,116,863, and EP 0235796, and depicted in Scheme 11 below. Scheme 11:

Corey, E. J., et al, Tetrahedron Letters, Vol. 26, No. 47, 5747-5748, 1985 describes a synthetic method for the preparation of 3-dimethylaminopropyltriphenylphosphonium bromide (free base), which is shown below in Scheme 12. Scheme 12:

The prior art methods for preparing olopatadine and the chemical intermediates 6,11- dihydro-ll-oxo-dibenz[b,e]oxepin-2-acetic acid, and 3- dimethylaminopropyltriphenylphosphonium bromide-hydrobromide (and its corresponding free base) are not desirable for synthesis of olopatadine on a commercial scale. For example, due to high reaction temperatures and the absence of solvents, the synthesis described in Ueno, K., et al., J. Med. Chem. (1976), 19(7), 941 and in U.S. Patent No. 4,282,365 for preparation of the intermediate 4-(2-carboxybenzyloxy)phenylacetic acid is undesirable for a commercial scale process, although the synthesis described in JP 07002733, and set forth in Scheme 13 below, is carried out in an acceptable solvent. Scheme 13:

OIO-1M1
The processes described in the literature for the intramolecular Friedel-Crafts acylation used to prepare 6,11-dihydro-l l-oxo-dibenz[b,e]oxepin-2-acetic acid are undesirable for commercial scale synthesis because they generally require either drastic conditions in the high boiling solvents (e.g. sulfolane) or they require a two step synthesis with the corresponding acid chlorides as intermediate. Furthermore the procedures for synthesizing 6,11-dihydro-l 1-oxo- dibenz[b,e]oxepin-2-acetic acid as set forth in European patent documents EP 0069810 and EP 0235796 use excess trifluoroacetic anhydride (see Scheme 14), and are carried out without solvent in a pressure bottle at 70-80° C (EP 0069810) or at room temperature in methylene chloride using catalytic amounts of BF3^Et2O (EP 0235796). Scheme 14:

According to the teachings in EP 0235795, a suspension of 3- bromopropyltriphenylphosphonium bromide (Olo-IM4) in ethanol was reacted with 13.5 equivalents of an aqueous dimethylamine solution (50%) to provide dimethylaminopropyltriphenylphosphonium bromide HBr. After this reaction, the solvent was distilled off and the residue was recrystallized (yield: 59%).
U.S. Patent No. 3,354,155 describes a reaction of 3-bromopropyltriphenylphosponium bromide with 4.5 equivalents dimethylamine. The solution was concentrated and the residue was suspended in ethanol, evaporated and taken up in ethanol again. Gaseous hydrogen bromide was passed into the solution until the mixture was acidic. After filtration, the solution was concentrated, whereupon the product crystallized (yield of crude product: 85%). The crude product was recrystallized from ethanol. A significant disadvantage of the prior art processes for making 3- dimethylaminopropyltriphenylphosphonium bromide hydrobromide involves the need for time consuming steps to remove excess dimethylamine, because such excess dimethylamine prevents crystallization of the reaction product. Thus, to obtain crystallization, the prior art processes require, for example, repeated evaporation of the reaction mixture (until dryness), which is undesirable for a commercial scale synthesis of olopatadine.
Corey, EJ., et al., Tetrahedron Letters, Vol. 26, No. 47, 5747-5748 (1985) describes the preparation of 3-dimethylaminopropyltriphenylphosphonium bromide (free base) from its corresponding hydrobromide salt. But the preparation of the free base, which uses an extraction step with methylene chloride as the solvent, is undesirable for commercial production because of the poor solubility of the free base in many of the organic solvents that are desirable for commercial production of chemical products, and because of the high solubility of the free base in water, causing low volume yields and loss of material. Furthermore according to this publication, the work up procedure gave an oil, which crystallized only after repeated evaporation in toluene.

-
Olopatadine and pharmaceutically acceptable salts thereof are described in patents EP 214779 , US 4871865 , EP 235796 andUS 5116863 . Patent EP 214779 describes two general processes for the production of Olopatadine, one of them involving a Wittig reaction and the other a Grignard reaction followed by a dehydration step.
-
Patent US 5116863 describes the production of Olopatadine hydrochloride by several different processes, two of which include a Grignard reaction for introducing the side chain in position 11 and a third process (called “Process C” in said patent) in which said side chain is introduced in position 11 by means of a Wittig reaction. In a specific embodiment (Example 9), the Wittig reaction is performed on the 6,11-dihydro-11-oxodibenz[b,e]oxepin-2-acetic acid (3) substrate, also known as Isoxepac, which is reacted with (3-dimethylaminopropyl)-triphenylphosphonium bromide hydrobromide, in the presence of n-butyl lithium giving rise to a Z/E mixture of Olopatadine together with salts of phosphorus which, after purifying by means of transforming it into the methyl ester of Olopatadine (2) and subsequent hydrolysis, provides Olopatadine hydrochloride (1), as shown in reaction scheme 1.

-
In the process shown in reaction scheme 1, the Wittig reagent [(Ph)3P+(CH2)3N(Me)2Br–HBr] is used in excess of up to 5 equivalents per equivalent of Isoxepac (3), a dangerous reagent (n-butyl lithium) is used; the process is very long and includes a number of extractions, changes of pH, in addition to esterification and subsequent saponification, the process therefore having very low yields and being rather expensive. The Z/E isomer ratio obtained in said process is not described.
-
Ohshima E., et al., in J. Med. Chem., 1992, 35:2074-2084 (designated inventors in US 5116863 ) describe several methods for synthesizing Olopatadine hydrochloride and other compounds of similar structure by means of Grignard reactions in some cases, and by means of Wittig reactions in other cases, for introducing the side chain (3-dimethylaminopropylidene). Following the synthetic scheme shown in reaction scheme 1, they start from type (3) compounds with free carboxylic acid and use (i) as base, n-butyl lithium, in a ratio relative to the type (3) compound of 7.5 equivalents of base/equivalent of type (3) compound and (ii) as Wittig reagent, (3-dimethylaminopropyl)-triphenylphosphonium bromide hydrobromide, in a ratio relative to the type (3) compound of 4.9 equivalents of the Wittig reagent/equivalent of type (3) compound. Once the Wittig reaction is carried out, in order to be able to better isolate the products, the acid is subsequently esterified; thus, and after purification by means of column chromatography, the obtained Z/E isomer ratio is 2:1. In said article, the authors (page 2077) acknowledge that when they try to perform this same Wittig reaction starting from a type (3) compound having an ester group instead of a carboxylic acid, the reaction does not occur and the starting material is recovered without reacting. This process has several drawbacks since it needs large amounts both of the Wittig reagent and of the base, n-butyl lithium (dangerous reagent, as already mentioned), it needs esterification, column purification, saponification and purification again, whereby the global process is not efficient.
-
Application WO 2006/010459 describes obtaining Olopatadine hydrochloride by means of a process in which a Wittig reaction is also performed but, this time, on an open substrate with final cyclization to form oxepin by means of Pd catalyst as can be seen in reaction scheme 2.



[R is an acid protecting group, especially C-C4alkyl]
-
The process shown in reaction scheme 2 has several drawbacks: high number of synthesis steps, the use of palladium catalysts which increase the cost of the process, the obtained Z/E isomer ratio is only 2.5:1 in favor of the Z isomer, and, finally, the need of using ionic exchange resins and chromatography columns, together with the use of dangerous reagents such as lithium aluminium hydride, n-butyl lithium or Jones reagent, make the process unfeasible on an industrial scale.
-
Application US2007/0232814 describes obtaining Olopatadine hydrochloride by means of a process which includes a Wittig reaction between Isoxepac (3) and the corresponding Wittig reagent [(3-dimethylaminopropyl)-triphenylphosphonium halides or salts thereof], using as base sodium hydride (NaH), whereby obtaining Olopatadine base which, after subsequent formation of an addition salt (essential for the production and isolation of the product of interest) and purification, yields Olopatadine hydrochloride (1), as shown in reaction scheme 3.

-
In the process shown in scheme 3, the amounts of Wittig reagent and of base used are very high since when the Wittig reagent is used in the form of salt 2.7 equivalents and 8.1 equivalents of base (NaH) are used, whereas if the free Wittig reagent is used 2.7 equivalents and 4.0 equivalents of base (NaH) are used. In these conditions, the reaction is very long (it can last more than one day) and the obtained Z/E isomer ratio is only 2.3:1, which results in a relatively low final yield and makes subsequent purification necessary. This process is, in addition, slow and tedious, therefore it is not very attractive from the industrial point of view.

EXAMPLE 4(Z)-11-(3-Dimethylaminopropylidene)-6,11-dihydrodibenz[b,e] oxepin-2-acetic acidPart A: (Z)-11-(3-dimethylaminopropylidene)-6,11-dihdrodibenz[b,e] oxepin-2-acetic acid ethyl ester
-
21.49 g (0.050 moles) of (3-dimethylaminopropyl)-triphenylphosphine bromide were suspended in 80 ml of tetrahydrofuran (THF) in a reaction flask under a N2 stream. 1.86 g (0.046 moles) of 60% NaH were carefully added, maintaining the obtained suspension at 20-25°C. Then, 10 ml of dimethylacetamide were slowly added to the previous suspension. The resulting mixture was heated at 35-40°C for 1 hour. At the end of this time period, 10 g (0.031 moles) of 6,11-dihydro-11-oxodibenz[b,e]oxepin-2-ethyl acetate dissolved in 30 ml of THF were added dropwise to the previous solution. The reaction mixture obtained was maintained at 35-40°C for 2 hours. After this time period, the reaction mixture was left to cool to a temperature lower than 10°C, then adding 150 ml of water on the reaction mixture. The solvent was eliminated by means of distillation under reduced pressure until obtaining an aqueous residue on which 100 ml of toluene were added. Subsequently, the organic and aqueous phases were decanted and separated. The organic phase was washed with concentrated HCl (2×50 ml). Then, the organic and aqueous phases were decanted and separated. The obtained aqueous phases were pooled and 100 ml of toluene and 2×10 ml of a solution of 20% Na2CO3 were added to them. The organic and aqueous phases were decanted and separated and the organic phase was concentrated under reduced pressure until obtaining a residue which was used without purifying in Part B.
-
The obtained product can be identified, after being purified by means of silica gel column chromatography. The compound of the title is eluted with a dichloromethane/methanol/ammonia (95/5/1) mixture, the spectroscopic properties of which compound are:
- 1H-NMR (CDCl3, 400 MHz), δ: 1.24 (t, 3H), 2.80 (s, 6H), 2.89 (m, 2H), 3.20 (m, 2H), 3.51 (s, 2H), 4.11 (m, 2H), 5.15 (bs, 2H), 5.63 (t, 1H), 6.82 (d, 1H), 7.04 (m, 2H), 7.25 (m, 4H) ppm.
- 13C-NMR (CDCl3, 400 MHz), δ: 14.41; 25.03; 40.12; 43.14; 57.33; 61.16; 70.93; 120.34; 123.95: 125.44; 126.34; 126.63; 127.72; 128.27; 129.33; 130.85; 131.64; 133.66; 143.74; 144.12; 154.96; 163.34; 172.27 ppm.
- MS, M++1: 366.06.
Part B: (Z)-11-(3-dimethylaminopropylidene)-6,11-dihydrodibenz[b,e] oxepin-2- acetic acid
-
The compound (Z)-11-(3-dimethylaminopropylidene)-6,11-dihydrodibenz[b,e]oxepin-2-acetic acid ethyl ester (residue obtained in Part A) was dissolved in 100 ml of acetone in a reaction flask. 3.4 ml (0.040 moles) of HCl were added to this solution. The reaction was heated under reflux for 10 hours, in which time the reaction passed from being a solution to being a suspension. After this time, the reaction was cooled until reaching 20-25°C. The solid was filtered, washed and the resulting product was dried in an oven with air circulation at 50-55°C, obtaining 5.2 g (0.015 moles, 50%) of a white solid identified as (Z)-11-(3-dimethylaminopropylidene)-6,11-dihydrodibenz[b,e] oxepin-2-acetic acid, isolated as hydrochloride, the spectroscopic properties of which are the following:
- 1H-NMR (DMSO, 400MHz), δ: 2.69 (s, 6H); 2.77 (m, 2H); 3.24 (m, 2H): 3.56 (s, 2H); 5.15 (bs, 2H); 5.62 (t, 1H); 6.76 (d, 1H); 7.06 (m, 2H); 7.30 (m, 4H) ppm.
- 13C-NMR (DMSO, 400MHz), δ: 25.12; 40.13; 42.44(2); 56.02; 70.26; 119.95; 123.43; 126.62; 127.64; 128.03; 128.47(2); 129.85; 131.34; 132.57; 134.12; 141.63; 145.25; 154.52; 173.67 ppm.
- MS, M’+1: 338.17
References
- Drugs.com, Alcon’s Patanase Nasal Spray Approved by FDA for Treatment of Nasal Allergy Symptoms
- Kyowa Hakko Kogyo Co., Ltd. (2007). “ALLELOCK Tablets 2.5 & ALLELOCK Tablets 5 (English)” (PDF). Retrieved2008-08-10.
- Tamura T, Matsubara M, Hasegawa K, Ohmori K, Karasawa A. (2005). “Olopatadine hydrochloride suppresses the rebound phenomenon after discontinuation of treatment with a topical steroid in mice with chronic contact hypersensitivity.”.
- Kyowa Hakko Kogyo Co., Ltd. (2002). “Company History”.Company Information. Kyowa Hakko Kogyo Co., Ltd. Retrieved16 September 2010.
- Ueno, K.; Kubo, S.; Tagawa, H.; Yoshioka, T.; Tsukada, W.; Tsubokawa, M.; Kojima, H.; Kasahara, A. (1976). “6,11-Dihydro-11-oxodibenz[b,e]oxepinacetic acids with potent antiinflammatory activity”. Journal of Medicinal Chemistry 19 (7): 941.doi:10.1021/jm00229a017.
Patent No. U.S. 8,877,947
AZILSARTAN SPECTRAL VISIT

AzilsartanMedoxomil Potassium
(5-methyl-2-oxo-l,3-dioxol-4-yl)methyl 2- ethoxy- 1 – { [2′-(5-oxo-4,5-dihydro- 1 ,2,4-oxadiazol-3-yl)biphenyl-4yl]methyl} – 1 H- benzimidazole-7-carboxylate monopotassium salt
NMR http://file.selleckchem.com/downloads/nmr/S305702-Azilsartan-Medoxomil-NMR-Selleck.pdf
AzilsartanMedoxomil Potassium is chemically named as (5-Methyl-2-oxo-1, 3-dioxol-4yl) methyl 2-ethoxy-1-{[2- (5-oxo-4, 5-dihydro-1, 2, 4-oxadiazol-3-yl) biphenyl-4-yl]methyl}- 1H-benzimidazole-7-carboxylatemonopotassium salt. Azilsartanmedoxomil is the prodrug of 2-ethoxy-1-([2′-(5-oxo4,5 – dihydro-1,2,4-oxadiazol-3-yl)biphenyl-4- yl]methyl) 1H-benimidazole-7-carboxylic acid.It is a white crystalline powder insoluble in water, slightly soluble in solvents such as acetone, and acetonitrile, freely soluble in methanol, dimethylsulfoxide, and dimethylformamide, soluble in solvents such as acetic acid, and very slightly soluble in solvents tetrahydrofuran and 1-octanol.
The US Food and Drug Administration (FDA) has approved Edarbi tablet (AzilsartanMedoxomil Potassium) on February 25, 2011, to treat hypertension in adults. It is available in 80mg and 40 mg dosages, with the recommended dosage set at 80mg once in a day [1].
Angiotensin II hormone plays a vital role in activation of renin-angiotensinaldosterone systems well as in regulation of blood pressure, fluid-electrolyte balance, and also in pathophysiology of hypertension. Activation of type 1 angiotensin receptor which is a member of G protein coupled receptor efficiently controls the numerous effects of AII which are vasoconstriction, secretion of aldosterone and vasopressin and cellular proliferation. So blocking of AII receptor will also block receptor-1, and it will lead to termination of the whole course of action mentioned above;
so all blocker will be helpful in the management of cardiovascular and renal diseases as therapeutic agent.The active moiety of AMP is revealed by hydrolysis of the medoxomil ester and it converts into azilsartan which is an active angiotensin II receptor blocker and more effective in lowering blood pressure within 24 hours as compared to valsartan and olmesartan[2-5].There are several methods that are reported for preparation of azilsartan [6-15].
The presence of related substances in an active pharmaceutical ingredient (API) can have a significant impact on the quality and safety of the drug products. Therefore, it is necessary to study the impurity (related substance) profile of the API to be used in the manufacturing of the drug product. International Conference on Harmonization (ICH)guidelines recommends identifying and characterizing all related substancesthat are present at level less than 0.10% [16].
![2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylic acid](https://i0.wp.com/pic6.molbase.net/molpic/ad/02/198350.png)
AZILSARTAN
| 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylic acid | |
| CAS No.: | 147403-03-0 |
|---|---|
| Synonyms: |
|
| Formula: | C25H20N4O5 |
| Exact Mass: | 456.14300 |
1H NMR
![2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylic acid NMR spectra analysis, Chemical CAS NO. 147403-03-0 NMR spectral analysis, 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylic acid H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-26/000/198/350/147403-03-0-1h.png)
13 C NMR
![2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylic acid NMR spectra analysis, Chemical CAS NO. 147403-03-0 NMR spectral analysis, 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylic acid C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-26/000/198/350/147403-03-0-13c.png)
AZILSARTAN MEDOXIMIL
![(5-methyl-2-oxo-1,3-dioxol-4-yl)methyl 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate](https://i0.wp.com/pic2.molbase.net/molpic/70/54/1535556.png)
| (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate | |
| CAS No.: | 863031-21-4 |
|---|---|
| Synonyms: |
View More
|
| Formula: | C30H24N4O8 |
| Exact Mass: | 568.15900 |
![(5-methyl-2-oxo-1,3-dioxol-4-yl)methyl 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate NMR spectra analysis, Chemical CAS NO. 863031-21-4 NMR spectral analysis, (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-30/001/535/556/863031-21-4-1h.png)
![(5-methyl-2-oxo-1,3-dioxol-4-yl)methyl 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate NMR spectra analysis, Chemical CAS NO. 863031-21-4 NMR spectral analysis, (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl 2-ethoxy-3-[[4-[2-(5-oxo-2H-1,2,4-oxadiazol-3-yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-08-30/001/535/556/863031-21-4-13c.png)
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2012107814A1 * | Jan 24, 2012 | Aug 16, 2012 | Jubilant Life Sciences Limited | An improved process for the preparation of azilsartan medoxomil |
| US5243054 * | Jun 25, 1992 | Sep 7, 1993 | Takeda Chemical Industries, Ltd. | Compound which is angiotensin ii antagonist |
| US20050187269 * | Jan 7, 2005 | Aug 25, 2005 | Takeda Pharmaceutical Company Limited | Benzimidazole derivative and use thereof |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2013186792A2 * | Jun 6, 2013 | Dec 19, 2013 | Msn Laboratories Limited | Process for the preparation of (5-methyl-2-oxo-l,3-dioxoi-4-vl)methvl 2- ethoxv-l-{[2′-(5-oxo-4,5-dihvdro-l,2,4-oxadiazol-3-vl)biphenyi-4-vl]methyl}- lh-benzimidazole-7-carboxyiate and its salts |
REFERENCES [1] M. Gasparo, K. J. Catt, T. Inagami, J.W. Wright, T. Unger, Pharmacol. Rev.2000, 52, 415
[2] W.B. White, M.A. Weber, D. Sica, G.L. Bakris,A. Perez,C. Cao, S. Kupfer, Hypertension.2011, 57, 413.
[3] G.L.Bakris, D.Sica, M.Weber, W.B. White,A. Roberts,A. Perez, C. Cao, S. J. Kupfer, Clin. Hypertens.2011, 13, 81.
[4] D. Sica, W.B. White, M.A. Weber, G.L. Bakris, A. Perez,C. Cao,A. Handley, S. Kupfer, J. Clin. Hypertens. 2011, 13, 467.
[5] H. Rakugi, K. Enya, K. Sugiura, Y. Ikeda, Hypertens. Res.2012, 35, 552.
[6] Y. Kohara, E. Imamiya, K. Kubo, T. Wada, Y. Inada, T. Naka, Bioorg. Med. Chem. Lett.1995, 5, 1903.
[7] T. Naka,Y. Inada, U.S. Patent 5583141 (1996) [8] T. Naka, Y. Inada, Eur Pat. Appl. EP 0520423, (1992)
[9] Y. Kohara, K. Kubo,E. Imamiya, T. Wada, Y. Inada, T. Naka, J. Med. Chem.1996, 39, 5228.
[10] T. Kuroita, H. Sakamoto,M.Ojima, U.S. Pat. Appl. 0187269 (2005) [11] T. Kuroita, H. Sakamoto, M. Ojima, U.S. Pat. Appl. 7157584 (2007) [12] S. Radl, J. Cerny,J. Stach, Z. Gablíkova, Org. Process Res. Dev.2013, 17, 77
[13] A. Agarwal, D. Bansal, A.S. Choudhary, S.K. Dubey, H. Mishra, D. Vir, WO 2012107814 A8 (2012)
[14] S.D. Dwivedi, K.K. Singh, J.T. Gajera, US 20140113942 A1, (2014)
[15] D. Bansal,H. Mishra,S.K. Dubey, A.S. Choudhary, D. Vir, A. Agarwal, A. Daz, US 20130317230 A1 (2013)
[16] International Conference of Harmonization (ICH). Q3A(R) Related substance/Impurities in New Drug Substance, Feb. 2002.
Tazemetostat

Tazemetostat
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
HBR 1467052-75-0
タゼメトスタット臭化水素酸塩
Current developer: Epizyme, Inc., Cambridge, MA 02139.
EPZ-6438 (Tazemetostat)
CAS: 1403254-99-8
HBR
Chemical Formula: C34H44N4O4
Exact Mass: 572.33626
USFDA APPROVED 23/1/2020 AS HBR SALT, TAZVERIK, EPIZYME
N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[1,1′-biphenyl]-3-carboxamide
SIMLES: O=C(C1=CC(C2=CC=C(CN3CCOCC3)C=C2)=CC(N(CC)C4CCOCC4)=C1C)NCC5=C(C)C=C(C)NC5=O
(1,1′-Biphenyl)-3-carboxamide, N-((1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(4-morpholinylmethyl)-
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-(ethyl(oxan-4-yl)amino)-4-methyl-4′-((morpholin-4-yl)methyl)(1,1′-biphenyl)-3-carboxamide
UNII-Q40W93WPE1
Tazemetostat, sold under the brand name Tazverik, is a medication used for the treatment of adults and adolescents aged 16 years and older with metastatic (when cancer cells spread to other parts of the body) or locally advanced (when cancer has grown outside the organ it started in, but has not yet spread to distant parts of the body) epithelioid sarcoma not eligible for complete resection (surgically removing all of a tissue, structure, or organ).[1]
Tazemetostat is a cancer drug that acts as a potent selective EZH2 inhibitor.[2]
Tazemetostat blocks activity of the EZH2 methyltransferase, which may help keep the cancer cells from growing.[1] Most cases of epithelioid sarcoma begin in the soft tissue under the skin of an extremity, though it can start in other areas of the body.[1] Surgical removal is considered the main treatment when the cancer is localized to one area of the body.[1] Chemotherapy or radiation may also be given.[1] However, there is a high likelihood for local and regional spread of the disease even with treatment and approximately 50% of patients have metastatic disease at the time of diagnosis.[1] Metastatic disease is considered life-threatening to the patient.[1]
The most common side effects are pain, fatigue, nausea, decreased appetite, vomiting and constipation.[1] People taking tazemetostat are at increased risk of developing secondary malignancies including: T-cell lymphoblastic lymphoma (a type of blood cancer that affects the lymphatic system usually found in the lymph nodes), myelodysplastic syndrome (a disorder resulting from poorly formed or dysfunctional blood cells) and acute myeloid leukemia (a cancer of the blood and bone marrow).[1]
According to the NCI Drug Dictionary, “tazemetostat is an orally available, small molecule selective and S-adenosyl methionine (SAM) competitive inhibitor of histone methyl transferase EZH2, with potential antineoplastic activity. Upon oral administration, tazemetostat selectively inhibits the activity of both wild-type and mutated forms of EZH2. Inhibition of EZH2 specifically prevents the methylation of histone H3 lysine 27 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways and results in decreased tumor cell proliferation in EZH2 mutated cancer cells. EZH2, which belongs to the class of histone methyltransferases (HMTs), is overexpressed or mutated in a variety of cancer cells and plays a key role in tumor cell proliferation.”[3]
History
The U.S. Food and Drug Administration (FDA) approved tazemetostat in January 2020,[1] based on the results of a clinical trial (NCT02601950) enrolling 62 subjects with metastatic or locally advanced epithelioid sarcoma.[1][4] During the clinical trial, subjects received 800 milligrams (mg) of tazemetostat twice a day until the disease progressed or the subject reached an unacceptable level of toxicity.[1][4] Tumor response assessments were performed every eight weeks during the clinical trial.[1] The trial measured how many subjects experienced complete or partial shrinkage (by a certain amount) of their tumors during treatment (overall response rate).[1] The overall response rate was 15%, with 1.6% of subjects having a complete response and 13% having a partial response.[1] Of the nine subjects that had a response, six (67%) subjects had a response lasting six months or longer.[1]
The trial was conducted at 22 sites in France, United Kingdom, Taiwan, Italy, Canada, Belgium, and the United States.[4]
The FDA granted the application for tazemetostat accelerated approval and orphan drug designation.[1] The FDA granted the approval of Tazverik to Epizyme Inc.[1]
PATENT
PRODUCT PAT
US 8410088 EXP 21/1/2034
WO 2012142504
US 9090562 EXP 13/4/32
SEE Proceedings of the National Academy of Sciences of the United States of America (2013), 110(19), 7922-7927, S7922/1-S7922/5….http://www.pnas.org/content/110/19/7922.abstract
http://www.epizyme.com/wp-content/uploads/2014/11/Ribrag-ENA-FINAL.pdf
Tazemetostat, also known as EPZ-6438, is a potent, selective, and orally bioavailable small-molecule inhibitor of EZH2 enzymatic activity. EPZ-6438 induces apoptosis and differentiation specifically in SMARCB1-deleted MRT cells.
Treatment of xenograft-bearing mice with EPZ-6438 leads to dose-dependent regression of MRTs with correlative diminution of intratumoral trimethylation levels of lysine 27 on histone H3, and prevention of tumor regrowth after dosing cessation.
These data demonstrate the dependency of SMARCB1 mutant MRTs on EZH2 enzymatic activity and portend the utility of EZH2-targeted drugs for the treatment of these genetically defined cancers. EPZ-6438 is currently in clinical trials.
Epizyme, Inc., Eisai R&D Management Co.Ltd.
Epizyme is developing tazemetostat, a lead from several small molecule EZH2 inhibitors, for treating cancer (phase 1 clinical, as of April 2015). Japanese licensee Eisai was developing the program for the potential oral treatment of cancers, including non-Hodgkin’s lymphoma; however, in March 2015, Epizyme regained worldwide, ex-Japan, rights to the program.
It appeared that Eisai was planning to investigate the program in Japan .
WO-2015057859 From, Eisai Research Institute; Epizyme Inc, indicates Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
see WO2013155317, claiming novel hydrobromide salt of tazemetostat.
PREDICT

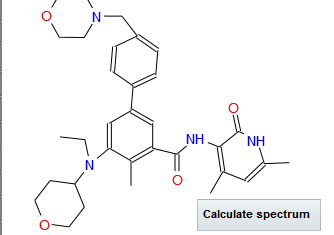
………………………………….
PATENT
WO 2012142504
http://www.google.com/patents/WO2012142504A1?cl=en

Example 44: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(moφholinomethyl)-[l , – biphenyl]-3-carboxamide

Compound 44
[Step 1 : Synthesis of 5-brom -2-methyl-3-nitrobenzoic acid

To stirred solution of 2-methyl-3-nitrobenzoic acid ( 100 g, 552 mmol) in cone. H2S04 (400 mL), 1 ,3-dibromo-5,5-dimethyl-2,4-imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid ( 140 g, 98%). The isolated compound was taken directly into the next step. Ή NMR (DMSO-4$, 400 MHz) δ 8.31 (s, 1 H), 8.17 (s, 1 H), 2.43 (s, 3H).
Step 2: Synthesis of methyl -bromo-2-methyl-3-nitrobenzoate

To a stirred solution of 5-bromo-2-methyl-3-nitrobenzoic acid (285 g, 1 105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate ( 1 L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).
Step 3: Synthesis of methyl 3-amino-5-bromo-2-methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-nitrobenzoate (290 g,
1058 mmol) in ethanol (1 .5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in-vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. Ή NMR (CDC13, 400 MHz) δ 7.37 (s, 1 H), 6.92 (s, 1 H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).
Step 4: Synthesis of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate

To a stirred solution of methyl 3-amino-5-bromo-2-methylbenzoate (15 g, 61 .5 mmol) and dihydro-2H-pyran-4(3)-one (9.2 g, 92 mmol) in dichloroethane (300 mL) was added acetic acid (22 g, 369 mmol) and the reaction mixture stirred at room temperature for 15 minutes, then the reaction mixture was cooled to 0°C and sodium triacetoxyborohydnde (39 g, 184 mmol) was added. The reaction mixture was stirred overnight at room temperature. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH of 7-8 was obtained. The organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a solid ( 14 g, 69%). ‘H NMR (DMSO-<fc, 400 MHz) δ 7.01 (s, 1 H), 6.98 (s, 1 H), 5.00 (d, 1 H, J=7.6 Hz), 3.84-3.87 (m, 2H), 3.79 (s, 31 1), 3.54-3.56 (mf 1 H), 3.43 (L 21 1, J 12 Hz), 2.14 (s. 31 1). 1 . 1 – 1 .84 (m: 211). 1 .47- 1 .55 (m, 2H).
Step 5: Synthesis of methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate

To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid ( 15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium
triacetoxyborohydnde (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). Ή NMR (DMSO-cfo 400 MHz) δ 7.62 (s, 1 H), 7.52 (s, 1 H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1 H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1 .37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).
Step 6: Synthesis of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3 -(ethyl (tetrahydro-2H-pyra -4-yl) amino)-2-methylbenzamide

To a stirred solution of 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2- methylbenzoate (14 g, 39.4 mmol) in ethanol ( 100 mL) was added aqueous NaOH (2.36 g, 59.2 mmol in 25mL water) and the resulting mixture was stirred at 60 °C for 1 h. Upon completion of the reaction as determined by TLC, the solvent was removed under reduced pressure and the residue obtained was acidified with IN HC1 until a pH 7 was obtained and then aqueous citric acid solution was added until a pH 5-6 was obtained. The aqueous layer was extracted with 10% MeOH in DCM (200mL X 3), the combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to give the respective acid (14 g, 100%).
The above acid (14 g, 40.9 mmol) was then dissolved in DMSO (70 mL) and 3- (amino methyl)-4, 6-dimethylpyridin-2( l H)-one ( 12.4 g, 81 .9 mmol) was added to it. The reaction mixture was stirred at room temperature for 15 minutes, then PYBOP (31.9 g, 61.4 mmol) was added and stirring was continued for overnight at room temperature. Upon completion of the reaction as determined by TLC, the reaction mixture was poured onto ice- cold water (700 mL), stirred for 30 minutes and the precipitated solid was collected by filtration, washed with water (500 mL) and air dried. The solid obtained was stirred with acetonitrile (75mL X 2), filtered and air dried. The solid obtained was again stirred with 5% MeOH in DCM ( l OOmL), filtered and dried completely under vacuum to afford the title compound as a solid ( 14 g, 74 %). Ή NMR (DMSO- 6, 400 MHz) δ 1 1.47 (s, 1 H), 8.23 (t, 1 H), 7.30 (s, 1 H), 7.08 (s, 1 H), 5.85 (s, 1 H), 4.23 (d, 2H, J=4.4 Hz), 3.81 (d, 2H, J=l 0.4 Hz), 3.20-3.26 (m, 2H), 3.00-3.07 (m, I H), 2.91 -2.96 (m, 2H), 2.18 (s, 3H), 2.14 (s, 3H), 2.10 (s, 3H), 1.58-1.60 (m, 2H), 1.45-1.50 (m, 2H), 0.78 (t, 3H, J=6.8 Hz).
Step 7: Synthesis of N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl) amino)-4-methyl-4′-(morpholinomethyl)-[l , l ‘-biphenyl]-3- carboxamide
 TITLE COMPD
TITLE COMPDTo a stirred solution of 5-bromo-N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzamide (14 g, 29.5 mmol) in dioxane/ water mixture (70 mL/ 14 mL) was added 4-(4-(4, 4, 5, 5-tetramethyl- l , 3, 2- dioxaborolan-2-yl) benzyl) morpholine (13.4 g, 44.2 mmol) followed by addition of Na2C03 (1 1 .2 g, 106.1 mmol). The solution was purged with argon for 15 minutes and then Pd (PPh3)4 (3.40 g, 2.94 mmol) was added and the solution was again purged with argon for a further 10 min. The reaction mixture was heated at 100°C for 4 h. After completion (monitored by TLC), the reaction mixture was diluted with water and extracted with 10% MeOH/DCM.
The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100- 200 mesh silica gel) eluting with methanol: DCM to the title compound as a solid (12 g, 71 %).
Analytical Data: LCMS: 573.35 (M + 1 )+; HPLC: 99.5% (@ 254 nm) (R,;3.999; Method: Column: YMC ODS-A 1 50 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol : 10 μΐ, Col. Temp.: 30 °C; Flow rate: 1 .4 mL/min.;
Gradient: 5% B to 95% B in 8 min, Hold for 1 .5 min, 9.51 -12 min 5% B);
Ή NMR (DMSO-i 6, 400 MHz) 5 1 1 .46 (s, I H), 8. 19 (t, 1 H), 7.57 (d, 2H, J=7.2 Hz), 7.36-7.39 (m, 3H), 7.21 (s, I H), 5.85 (s, I H), 4.28 (d, 2H, J=2.8 Hz), 3.82 (d, 2H, J=9.6 Hz), 3.57 (bs, 4H), 3.48 (s, 2H), 3.24 (t, 2H, J=10.8Hz), 3.07-3.09 (m, 2H), 3.01 (m, I H), 2.36 (m, 4H), 2.24 (s, 3H), 2.20 (s, 3H), 2.10 (s, 3H), 1 .64-1 .67 (m, 2H), 1 .51 – 1 .53 (m, 2H), 0.83 (t, 3H, J=6.4 Hz).
TRIHYDROCHLORIDE
Step 8: Synthesis of N-((4,6-dimethyl-2-oxo-l ,2-dihydropyridin-3-yl)methyl)-5- (ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[ 1 , 1 ‘-biphenyl]-3- carboxamide trihydrochloride

N-((4, 6-dimethyl-2-oxo-l , 2-dihydropyridin-3-yl) methyl)-5-(ethyl (tetrahydro- 21 l-pyran-4-yl) amino)-4-methyI-4′-(niorpholinornethyl)-[ 1 , 1 ‘-biphenyl]-3-carboxamide ( 12 g, 21.0 mmol) was dissolved in methanolic HC1 (200 mL) and stirred at room temperature for 3 h. After three hours of stirring, the reaction mixture was concentrated under reduced pressure. The solid obtained was stirred with ether ( l OOmL X 2) to afford the desired salt as a solid ( 1 1 g, 77 %).
Analytical Data of the tri-HCl salt: LCMS: 573.40 (M + 1 )+; HPLC: 99.1 % (@ 254 nm) (R,;3.961 ; Method: Column: YMC ODS-A 150 mm x 4.6 mm x 5 μ; Mobile Phase: A; 0.05% TFA in water/ B; 0.05% TFA in acetonitrile; Inj. Vol: 10 pL, Col. Temp.: 30 °C; Flow rate: 1.4 mL/min.; Gradient: 5% B to 95% B in 8 min, Hold for 1.5 min, 9.51 -12 min 5% B);
Ή NMR (D20 400 MHz) δ 7.92 (bs, I H,) 7.80 (s, I H), 7.77 (d, 2H, J=8 Hz), 7.63 (s, I H), 7.61 (s, I H), 6.30 (s, I H), 4.48 (s, 2H), 4.42 (s, 2H), 4.09-4.1 1 (m, 4H), 3.95-3.97 (m, 2H), 3.77 (t, 3H, J=10.4 Hz), 3.44-3.47 (m, 3H), 3.24-3.32 (m, 3H), 2.42 (s, 3H), 2.35 (s, 3H), 2.26 (s, 3H), 2.01 (m, 2H), 1 .76 (m, 2H), 1 .04 (t, 3H, J=6.8 Hz).
…………………………………………
PATENT
WO2013155317
http://www.google.com/patents/WO2013155317A1?cl=en
N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl] -3-carboxamide hydrobromide:

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl
(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3- carboxamide hydrobromide:

As used herein, “Compound I” refers to N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl (tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxamide. The hydrobromide of Compound I can be used to inhibit the histone methyltransferase activity of EZH2, either in a subject or in vitro. The hydrobromide of Compound I can also be used to treat cancer in a subject in need thereof.
Scheme 1

……………………………………..Compound I Compound I – HBr
HPLC
HPLC was conducted on an Agilent 1200 HPLC quaternary pump, low pressure mixing, with an in-line degasser. Analytical method conditions: 8 μΐ^ sample (20 mg of ER-581982-06 diluted with 50 mL of a methanol to provide approximately 0.4 mg/mL solution) was injected onto a Agilent Zorbax Eclipse XDB-C18 (4.6 x 150 mm, 3.5 um), Chromatography conditions: mobile phase A, water with 5mM ammonium formate; mobile phase B, 5 mM ammonium formate in 50/45/5 acetonitrile/methanol/water; flow rate, 1.5 ml/min.; gradient: isocratic at 10% B from 0 to 3 min; linear increase to 70% B from 3 to 7 min; isocratic at 70% B from 7 to 12 min; linear increase to 100% B from 12 to 15 min isocratic at 100% B from 15 to 20 min;
column temperature, 35 °C; detection, UV 230 nm. Approximate retention time of Compound I = 10.7 min.
Synthesis of Polymorph A

5-bromo-2-methyl-3-nitrobenzoic acid stirred solution of 2-methyl-3-nitrobenzoic acid (100 g, 552 mmol) in cone. H2S04 (400 mL), l,3-dibromo-5,5-dimethyl-2,4- imidazolidinedione (88 g, 308 mmol) was added in a portion wise manner at room temperature and the reaction mixture was then stirred at room temperature for 5 h. The reaction mixture was poured onto ice cold water, the precipitated solid was filtered off, washed with water and dried under vacuum to afford the desired compound as a solid (140 g, 98%). The isolated compound was taken directly into the next step. 1H NMR (DMSO-J6, 400 MHz) δ 8.31 (s, 1H), 8.17 (s, 1H), 2.43 (s, 3H).

Methyl 5-bromo-2-methyl-3-nitrobenzoate To a stirred solution of 5-bromo-2- methyl-3-nitrobenzoic acid (285 g, 1105 mmol) in DMF (2.8L) at room temperature was added sodium carbonate (468 g, 4415 mmol) followed by addition of methyl iodide (626.6 g, 4415 mmol). The resulting reaction mixture was heated at 60 °C for 8 h. After completion (monitored by TLC), the reaction mixture was filtered (to remove sodium carbonate) and washed with ethyl acetate (1L X 3). The combined filtrate was washed with water (3L X 5) and the aqueous phase was back extracted with ethyl acetate (1L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (290g, 97% yield). The isolated compound was taken directly into the next step. 1H NMR (CDC13, 400 MHz) δ 8.17 (s, 1H), 7.91 (s, 1H), 3.96 (s, 3H), 2.59 (s, 3H).

Methyl 3-amino-5-bromo-2-methylbenzoate (1) To a stirred solution of methyl 5- bromo-2-methyl-3-nitrobenzoate (290 g, 1058 mmol) in ethanol (1.5L) was added aqueous ammonium chloride (283 g, 5290 mmol dissolved in 1.5L water). The resulting mixture was stirred at 80°C to which iron powder (472 g, 8451 mmol) was added in a portion wise manner. The resulting reaction mixture was heated at 80 °C for 12 h. Upon completion as determined by TLC, the reaction mixture was hot filtered over celite® and the celite bed was washed with methanol (5L) followed by washing with 30% MeOH in DCM (5L). The combined filtrate was concentrated in- vacuo, the residue obtained was diluted with aqueous sodium bicarbonate solution (2L) and extracted with ethyl acetate (5L X 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the title compound as a solid (220 g, 85%). The compound was taken directly into the next step. 1H
NMR (CDCI3, 400 MHz) δ 7.37 (s, 1H), 6.92 (s, 1H), 3.94 (s, 3H), 3.80 (bs, 2H), 2.31 (s, 3H).

Methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (2) A reactor was charged with methyl 3-amino-5-bromo-2-methylbenzoate (455.8 g, 1.87 mol), 1,2- Dichloroethane (4.56 L), and acetic acid (535 ml, 9.34 mol). To the mixture were added dihydro-2H-pyran-4(3H)-one (280 g, 2.80 mol) and sodium triacetoxyborohydride (594 g, 2.80 mol) maintaining the internal temperature below 40 °C. The mixture was stirred at 25 °C for 2.5 h and then the reaction was quenched with a solution of sodium hydroxide (448 g, 11.20 mol) in water (5.61 L). After stirring for 20 minutes at ambient temperature, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (3.65 L). The organic layers were combined, washed with brine (1.5 L), and concentrated under vacuum.
The residue was treated with ethyl acetate (1.8 L) and heated to 65-70 °C. The mixture was stirred at 65-70 °C for 15 minutes to give a clear solution and then treated with n-heptane (7.3 L) maintaining the temperature between 60-70 °C. Once the heptane was completely added to the solution, the mixture was held at 65-70 °C for 15 minutes and then allowed to cool to 18- 22 °C over 3 h. The resulting suspension was stirred at 18-22 °C for 4 h, cooled to 0-5 °C over 1 h, and held at 0-5 °C for 2 h. The precipitate was filtered, washed twice with n-heptane (1.4 L), and dried under vacuum to give the title compound (540 g, 88%). The XRPD pattern of this compound is shown in Figure 17.

Methyl 5-bromo-3-(ethyl (tetrahydro-2H-pyran-4-yl) amino)-2-methylbenzoate (3)
To a stirred solution of methyl 5-bromo-2-methyl-3-((tetrahydro-2H-pyran-4-yl) amino) benzoate (14 g, 42.7 mmol) in dichloroethane (150 mL) was added acetaldehyde (3.75 g, 85.2 mmol) and acetic acid (15.3 g, 256 mmol). The resulting reaction mixture was stirred at room temperature for 15 minutes. The mixture was cooled to 0 °C and sodium triacetoxyborohydride (27 g, 128 mmol) was added. The reaction mixture was stirred at room temperature for 3 hours. Upon completion of the reaction as determined by TLC, aqueous sodium bicarbonate solution was added to the reaction mixture until a pH 7-8 was obtained, the organic phase was separated and the aqueous phase was extracted with ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude compound was purified by column chromatography (100-200 mesh silica gel) eluting with ethyl acetate: hexane to afford the desired compound as a viscous liquid (14 g, 93%). 1H NMR DMSO-d6, 400 MHz) δ 7.62 (s, 1H), 7.52 (s, 1H), 3.80 (bs, 5H), 3.31 (t, 2H), 2.97-3.05 (m, 2H), 2.87-2.96 (m, 1H), 2.38 (s, 3H), 1.52-1.61 (m, 2H), 1.37-1.50 (m, 2H), 0.87 (t, 3H, J=6.8 Hz).

Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-
[l,l’-biphenyl]-3-carboxylate (4): A mixture of methyl 5-bromo-3-(ethyl(tetrahydro-2H-pyran- 4-yl)amino)-2-methylbenzoate (580 g, 1.63 mol), 4-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)benzyl)morpholine (592 g, 1.95 mol), 1,4-dioxane (3.86 L), sodium carbonate (618 g, 5.83 mol), and water (771 ml) was degassed by bubbling nitrogen through the mixture at 20 °C for 20 minutes and treated with tetrakis(triphenylphosphine)palladium(0) (14.11 g, 12.21 mmol). The resulting mixture was degassed for an additional 20 minutes and then heated to 87-89 °C for 17 h. After cooling to 20 °C, the mixture was diluted with ethyl acetate (5.80 L) and a solution of (R)-2-Amino-3-mercaptopropionic acid (232 g) in water (2.320 L). After stirring for 1 h at 20 °C, the organic layer was separated and washed again with a solution of (R)-2-Amino-3- mercaptopropionic acid (232 g) in water (2.320 L). The aqueous layers were combined and extracted with ethyl acetate (5.80 L). The organic layers were combined, washed with a solution of sodium hydroxide (93 g) in water (2.32 L), and concentrated under vacuum at 35 °C to give the title compound as an orange oil (1.21 kg, 164% yield).

5-(Ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’- biphenyl]-3-carboxylic acid (5): Methyl 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylate (69.0 g, 152.5 mmol) (based on the theoretical yield from the previous step) was suspended in ethanol (380 mL) and treated with a solution of sodium hydroxide (24.84 g, 621.0 mmol) in water (207 mL). The mixture was stirred at 40°C for 18 h. After cooling to 0-5 °C, the mixture was neutralized to pH 6.5 with 1 N hydrochloric acid (580 mL) maintaining the temperature below 25 °C. Then, the mixture was extracted twice with a mixture of dichloromethane (690 mL) and methanol (69.0 mL). The organic layers were combined and concentrated under vacuum to give a crude product as a yellow solid (127g).
The crude product was dissolved in 2-methyltetrahydrofuran (656 mL) at 70 °C and then treated with IPA (828 mL). The mixture was allowed to cool to rt over 3-4 h and then stirred overnight at rt. The precipitate was filtered, washed twice with IPA (207 mL), and dried under vacuum to give the title compound as an off white solid (53.54 g, 80%). The XRPD pattern of this compound is shown in Figure 9.

N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)-5-(ethyl(tetrahydro-2H- pyran-4-yl)amino)-4-methyl-4′-(morpholinomethyl)-[l,l’-biphenyl]-3-carboxamide
(Compound I): A mixture of 5-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4-methyl-4′- (morpholinomethyl)-[l,l’-biphenyl]-3-carboxylic acid (540 g, 1.23 mol) and 3-(aminomethyl)- 4,6-dimethyl-dihydro-pyridin-2(lH)-one hydrochloride (279 g, 1.48 mol) was suspended in DMSO (2.70 L) and treated with triethylamine (223 ml, 1.60 mol). The mixture was stirred at 25 °C for 30 min and treated with EDC-HC1 (354 g, 1.85 mol) and HOBT hydrate (283 g, 1.85 mol). The reaction mixture was stirred at rt for 16 h. After addition of triethylamine (292 ml, 2.09 mol), the mixture was cooled to 15 °C, diluted with water (10.1 L) maintaining the temperature below 30 °C, and stirred at 19-25 °C for 4 h. The resulting precipitate was filtered, washed twice with water (2.70 L), and dried under vacuum to give a crude product (695 g, wt-wt analysis = 78%).
For the further purification of the product, recrystallization was conducted. A crude product (20.00 g, 34.92 mmol) was suspended in a mixture of ethanol (190 ml) and water (10.00 ml) and heated to 75°C until a clear solution was obtained. The solution was allowed to cool to rt overnight. The precipitate was filtered, washed twice with a mixture of ethanol (30.0 ml) and water (30.0 ml), and dried under vacuum at 35 °C to give the title compound as an off white solid (14.0 g, 70% recovery from the crude and 90% yield based on wt-wt assay).

4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′- (ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4-yl)methyl)morpholin- 4-ium bromide (Polymorph A): A crude N-((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)-5-(ethyl(tetrahydro-2H-pyran-4-yl)am
biphenyl]-3-carboxamide (595 g, 464 g based on wt-wt assay, 810.3 mmol) was suspended in ethanol (3.33 L). After heating to 70 °C, the mixture was treated with 48% aqueous HBr (97 ml, 850.8 mmol) and stirred at 70 °C for 30 min. The resulting orange-red solution was treated with ethyl acetate (3.33 L) maintaining the temperature above 60 °C. The mixture was slowly cooled to rt over 18 h. The mixture was cooled to 0 °C over 1 h and stirred at that temperature for 5.5 h. The resulting precipitate was filtered, washed twice with ethyl acetate (1.39 L), and dried under vacuum to give the title compound as an off white solid (515 g, 97% yield).
Recrystallization of Polymorph A: 4-((3′-(((4,6-dimethyl-2-oxo-l,2-dihydropyridin-3- yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′-methyl-[l,l’-biphenyl]-4- yl)methyl)morpholin-4-ium bromide (0.50 g, 0.77 mmol; 95.6% pure by HPLC) was suspended in ethanol (3.0 mL) and heated to 80 °C until a clear solution was obtained. To the solution was added MTBE (5.0 mL) slowly. The resulting solution was allowed to cool to 18-22 °C over 3 h and stirred at 18-22 °C for 15 h. The precipitate was filtered, washed twice with MTBE (2 mL) and dried under vacuum to give 0.45 g of the title compound (89% recovery, 96.6% pure by HPLC).
Compound I is protonated at the nitrogen of the morpholino substituent, providing a monohydrobromide of Compound I having the following structure:

This particular monohydrobromide can be referred to as “4-((3′-(((4,6-dimethyl-2-oxo- l,2-dihydropyridin-3-yl)methyl)carbamoyl)-5′-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-4′- methyl-[l, -biphenyl]-4-yl)methyl)morpholin-4-ium bromide.” Figure 11 depicts the X-ray crystal structure of this particular salt form.
…………………………………………………………..
see
WO-2015057859
Eisai Research Institute; Epizyme Inc
Novel crystalline polymorphic form C of tazemetostat, useful for treating an EZH2-mediated cancer, including non-Hodgkin’s lymphoma and breast cancer.
…………………
Synthesis
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| USA | Tazverik | Epizyme, 2020 |
Formulations
- oral tabs. and suspension
References
-
- Knutson, S. K. et al., Proc. Natl. Acad. Sci USA, (2013) 110(19), 7922-7927.
- WO 2012 142504 (Epizyme/Eisai Co; 18.10.2012; appl. 13.4.2012; USA-prior. 13.4.2011).
- WO 2013 155317 (Epizyme/Eisai Co; 17.10.2013; appl. 11.4.2013).
- WO 2015 057859 (Epizyme/Eisai Co; 23.4.2015; appl. 15.10.2014; USA-prior. 16.10.2013).
-
EZH2 inhibitors for treating lymphona:
- WO 2015 195848 (Epizyme; 23.12.2015; appl. 17.6.2015; USA-prior. 17.6.2014).
////////
PAPER
RSC Advances (2015), 5(33), 25967-25978
http://pubs.rsc.org/en/content/articlelanding/2015/ra/c5ra02365c#!divAbstract
DOI: 10.1039/C5RA02365C
The histone lysine methyltransferase EZH2 has been implicated as a key component in cancer aggressiveness, metastasis and poor prognosis. This study discovered a new class of hexahydroisoquinolin derivatives as EZH2 inhibitors. A structure–activity relationship study showed that the steric hindrance was important to the activity for EZH2. A preliminary optimization study led to the discovery of several potent compounds with low nanomolar to sub-nanomolar potency for EZH2. Biological evaluation indicated that SKLB1049 was a highly potent with improved solubility compared to EPZ6438, SAM-competitive, and cell-active EZH2 inhibitor that decreased global H3K27me3 in SU-DHL-6 and Pfeiffer lymphoma cells in a concentration- and time-dependent manner. Further study indicated that SKLB1049 caused cell arrest in G0/G1 phase. These compounds would be useful as chemical tools to further explore the biology of EZH2 and provided us with a start point to develop new EZH2 inhibitors.

|
In vitro protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
In vivo protocol: |
Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. |
|
References |
1: Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, Copeland RA, Pollock RM, Kuntz KW, Raimondi A, Keilhack H. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS One. 2014 Dec 10;9(12):e111840. doi: 10.1371/journal.pone.0111840. eCollection 2014. PubMed PMID: 25493630; PubMed Central PMCID: PMC4262195.
2: Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014 Apr;13(4):842-54. doi: 10.1158/1535-7163.MCT-13-0773. Epub 2014 Feb 21. PubMed PMID: 24563539
3. Inhibitors of human histone methyltransferase EZH2, and methods of use thereof for treating cancer. By Kuntz, Kevin W.; Knutson, Sarah K.; Wigle, Timothy James Nelson . From U.S. Pat. Appl. Publ. (2013), US 20130040906 A1 20130214.
4. Aryl-or heteroaryl-substituted benzamide compounds as anticancer agents and their preparation By Kuntz, Kevin Wayne; Chesworth, Richard; Duncan, Kenneth William; Keilhack, Heike; Warholic, Natalie; Klaus, Christine; Zheng, Wanjun; Seki, Masashi; Shirotori, Syuji; Kawano, Satoshi From PCT Int. Appl. (2012), WO 2012142504 A1 20121018.
5: Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013 May 7;110(19):7922-7. doi: 10.1073/pnas.1303800110. Epub 2013 Apr 25. PubMed PMID: 23620515; PubMed Central PMCID: PMC3651445.
| WO2013155317A1 * | Apr 11, 2013 | Oct 17, 2013 | Epizyme, Inc. | Salt form of a human hi stone methyltransf erase ezh2 inhibitor |
| WO2013155464A1 * | Apr 12, 2013 | Oct 17, 2013 | Epizyme, Inc. | Combination therapy for treating cancer |
| WO2014049488A1 * | Sep 16, 2013 | Apr 3, 2014 | Pfizer Inc. | Benzamide and heterobenzamide compounds |
| WO2014062732A1 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014062733A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2014172044A1 * | Mar 14, 2014 | Oct 23, 2014 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015004618A1 * | Jul 9, 2014 | Jan 15, 2015 | Glaxosmithkline Intellectual Property (No.2) Limited | Enhancer of zeste homolog 2 inhibitors |
| WO2015010049A1 * | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted benzene compounds |
| WO2015010078A2 | Jul 18, 2014 | Jan 22, 2015 | Epizyme, Inc. | Substituted 6,5-fused bicyclic heteroaryl compounds |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012142504A1 * | Apr 13, 2012 | Oct 18, 2012 | Eisai Co., Ltd. | Aryl-or heteroaryl-substituted benzene compounds |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014062720A2 * | Oct 15, 2013 | Apr 24, 2014 | Epizyme, Inc. | Methods of treating cancer |
| WO2011140324A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indoles |
| WO2011140325A1 * | May 5, 2011 | Nov 10, 2011 | Glaxosmithkline Llc | Indazoles |
| WO2012005805A1 * | May 5, 2011 | Jan 12, 2012 | Glaxosmithkline Llc | Azaindazoles |
| US4522811 | Jul 8, 1982 | Jun 11, 1985 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US5763263 | Jul 24, 1996 | Jun 9, 1998 | Dehlinger; Peter J. | Method and apparatus for producing position addressable combinatorial libraries |
| US7563589 | May 27, 2005 | Jul 21, 2009 | The University Of North Carolina At Chapel Hill | Including EED, EZH2 and SUZ12 wherein the reconstituted complex has histone methyltransferase (HMTase) activity for lysine 27 of histone H3 (H3-K27); cancer |
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “FDA approves first treatment option specifically for patients with epithelioid sarcoma, a rare soft tissue cancer”. U.S. Food and Drug Administration (FDA) (Press release). 23 January 2020. Retrieved 23 January 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Lue JK, Amengual JE (October 2018). “Emerging EZH2 Inhibitors and Their Application in Lymphoma”. Curr Hematol Malig Rep. 13 (5): 369–382. doi:10.1007/s11899-018-0466-6. PMID 30112706. S2CID 52010283.
- ^ “Tazemetostat”. NCI Drug Dictionary. National Cancer Institute.
- ^ Jump up to:a b c “Drug Trials Snapshots: Tazverik”. U.S. Food and Drug Administration (FDA). 23 January 2020. Retrieved 22 February 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Tazemetostat”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Tazverik |
| Other names | EPZ-6438 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620018 |
| License data |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C34H44N4O4 |
| Molar mass | 572.750 g·mol−1 |
| 3D model (JSmol) | |
Epizyme®, Inc.
400 Technology Square, 4th Floor
Cambridge, MA 02139
Phone: (617) 229-5872
Fax: (617) 349-0707
contact@Epizyme.com
![]()



 Jason Rhodes (left) has been appointed to president of Epizyme Inc.,
Jason Rhodes (left) has been appointed to president of Epizyme Inc.,

100 Technology Square
