

| Patent | Submitted | Granted |
|---|---|---|
| Compound useful for the treatment of degenerative and inflammatory diseases [US8088764] | 2010-12-30 | 2012-01-03 |
| NOVEL COMPOUNDS USEFUL FOR THE TREATMENT OF DEGENERATIVE AND INFLAMMATORY DISEASES [US2011190260] | 2011-08-04 |
Home » Uncategorized (Page 91)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New Parathyroid Disease Drug Etelcalcetide Seeks FDA Approval
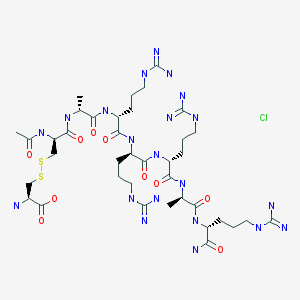
Etelcalcetide, AMG 416
AMG-416; Etelcalcetide hydrochloride; KAI-4169; KAI-4169-HCl; ONO-5163; Telcalcetide; Velcalcetide; Velcalcetide hydrochloride
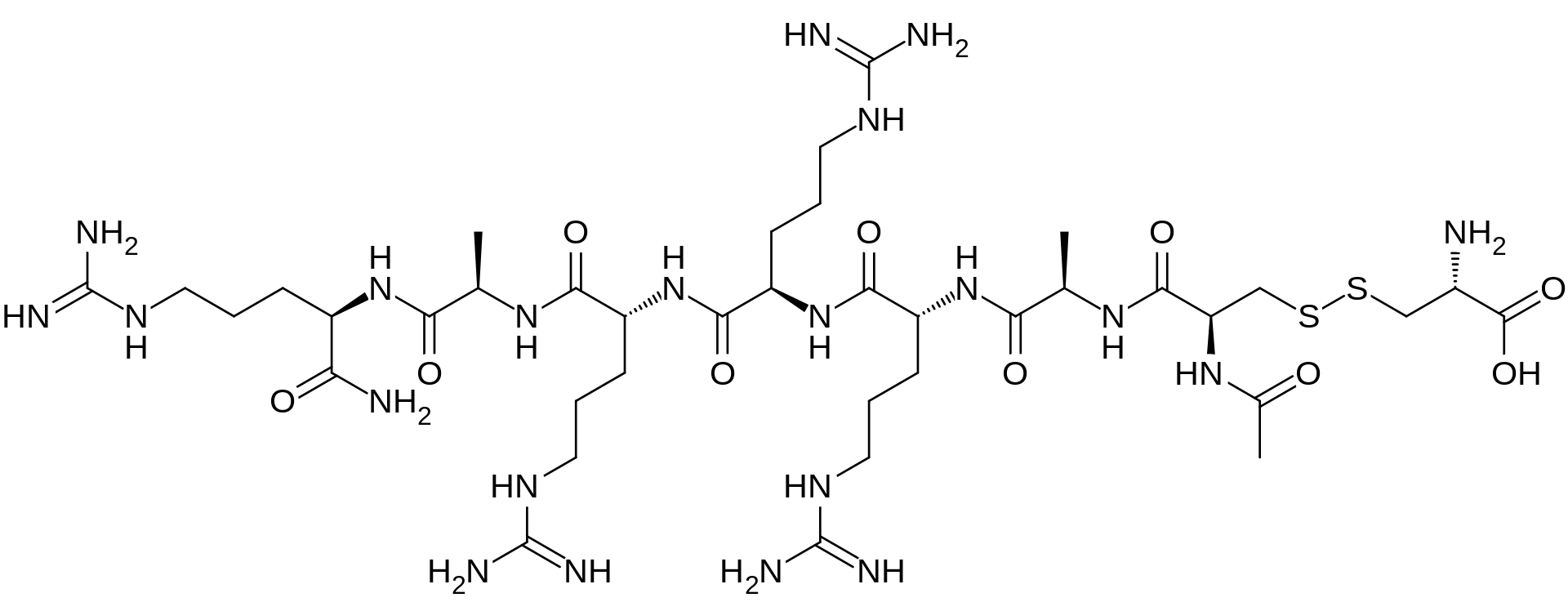
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine,
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine
Secondary hyperparathyroidism
- Originator KAI Pharmaceuticals…Kai Pharmaceuticals, Inc.
- Developer Amgen; KAI Pharmaceuticals; Ono Pharmaceutical
- ClassDisulfides; Peptides
- Mechanism of ActionCalcium-sensing receptor agonists
New Parathyroid Disease Drug Seeks FDA Approval
Amgen is seeking FDA approval for etelcalcetide (AMG 461), the first calcimimetic agent administered intravenously after dialysis to treat secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD).
SHPT is a common and serious condition that is often progressive among CKD patients. It usually manifests as high amounts of parathyroid hormone (PTH) associated with abnormal calcium and phosphorus levels in the body.
– See more at: http://www.pharmacytimes.com/product-news/new-parathyroid-disease-drug-seeks-fda-approval
Etelcalcetide is a D-amino peptide calcimimetic undergoing clinical evaluation for the treatment of secondary hyperparathyroidismfor patients with chronic kidney disease (CKD) on hemodialysis. Etelcalcetide is administered intravenously at the end of each dialysis session.[1][2] It exerts a pharmacological effect by binding to and activating the calcium-sensing receptor (CaSR) in theparathyroid gland, resulting in parathyroid hormone (PTH) reduction and suppression.[1] Elevated PTH is often observe in patients with CKD.[3]
On August 25, 2015 Amgen Inc. announced its submission of a New Drug Application to the Food and Drug Administration for etelcalcetide.[1]
| CAS Registry Number | 1262780-97-1 |
|---|---|
| Synonyms | Velcalcetide |
| Chemical data | |
| Formula | C38H73N21O10S2 |
| Molecular mass | 1,048.26 g·mol−1 |
Etelcalcetide hydrochloride
RN: 1334237-71-6
UNII: 72PT5993DU
The term “AMG 416” refers to the compound having the chemical name: JV-acetyl-D- cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-arginamide disulfide with L- cysteine, which may be represented as:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The terms “AMG 416 hydrochloride” or “AMG 416 HQ” are interchangeable and refer to the compound having the chemical name: N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl- D-arginyl-D-alanyl-D-arginamide disulfide with L-cysteine hydrochloride, which may be represented as:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · xHCl
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine, hydrochloride (1:?)
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
Amgen today announced the submission of a New Drug Application (NDA) with the United States Food and Drug Administration (FDA) for etelcalcetide (formerly AMG 416) for the treatment of secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD) on hemodialysis. If approved, etelcalcetide will be the first calcimimetic agent that can be administered intravenously at the end of the dialysis session.
“Secondary hyperparathyroidism is a serious, progressive disease that can lead to significant clinical consequences and is also associated with a high pill burden for patients,” said Sean E. Harper, M.D., executive vice president of Research and Development at Amgen. “We look forward to working with regulatory authorities during the review process to bring this important treatment to market, helping to fill an unmet need for the many patients impacted by this disease.”
Etelcalcetide is a novel calcimimetic agent that suppresses the secretion of parathyroid hormone and is in clinical development for the treatment of SHPT in patients with CKD on hemodialysis. Etelcalcetide is administered intravenously three times per week at the end of each dialysis session. It acts by binding to and activating the calcium-sensing receptor on the parathyroid gland, thereby causing decreases in parathyroid hormone (PTH). Sustained elevations in PTH are known to be associated with significant clinical consequences for patients with CKD.
The submission includes data from three Phase 3 studies, all of which met the primary endpoints, including two pooled placebo-controlled trials in more than 1,000 patients and a head-to-head study evaluating etelcalcetide compared with cinacalcet.
About Secondary Hyperparathyroidism
SHPT is a common and serious condition that is often progressive among patients with CKD, and it affects many of the approximately two million people throughout the world who are receiving dialysis, including 450,000 people in the U.S. The disorder develops early in the course of CKD and usually manifests as increased levels of PTH as a result of increased production from the parathyroid glands (four small glands in the neck). Patients with end stage renal disease who require maintenance dialysis often have substantial elevations of PTH that are commonly associated with abnormal calcium and phosphorus levels and an increased risk of significant clinical consequences.
About Etelcalcetide (AMG 416)
Etelcalcetide is a novel calcimimetic agent in clinical development for the treatment of SHPT in CKD patients on hemodialysis that is administered intravenously at the end of the dialysis session. Etelcalcetide binds to and activates the calcium-sensing receptor on the parathyroid gland, thereby decreasing PTH levels.
About Sensipar® (cinacalcet)
Sensipar® (cinacalcet) is the first oral calcimimetic agent approved by the FDA for the treatment of SHPT in adult patients with CKD on dialysis. Sensipar is not indicated for use in adult patients with CKD who are not on dialysis because of an increased risk of hypocalcemia. The therapy is also approved in the U.S. for treatment of hypercalcemia in adult patients with parathyroid carcinoma and hypercalcemia in adult patients with primary HPT for whom parathyroidectomy would be indicated on the basis of serum calcium levels, but who are unable to undergo parathyroidectomy. Sensipar binds to the calcium-sensing receptor, resulting in a drop in PTH levels by inhibiting PTH synthesis and secretion. In addition, the reductions in PTH lower serum calcium and phosphorus levels.
…………………
WO 2011014707
http://www.google.com/patents/WO2011014707A2?cl=en
……………………..
WO 2014210489
http://www.google.com/patents/WO2014210489A1?cl=en
A variety of compounds having activity for lowering parathyroid hormone levels have been described. See International Publication No. WO 2011/014707. In one embodiment, the compound may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The main chain has 7 amino acids, all in the D-configuration and the side-chain cysteine residue is in the L-configuration. The amino terminal is acetylated and the carboxyl-terminal is amidated. This compound (“AMG-416”) has utility for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis patients. A liquid formulation comprising AMG-416 may be administered to a subject intravenously. The hydrochloride salt of AMG-416 may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
Therapeutic peptides pose a number of challenges with respect to their formulation. Peptides in general, and particularly those that contain a disulfide bond, typically have only moderate or poor stability in aqueous solution. Peptides are prone to amide bond hydrolysis at both high and low pH. Disulfide bonds can be unstable even under quite mild conditions (close to neutral pH). In addition, disulfide containing peptides that are not cyclic are particularly prone to dimer formation. Accordingly, therapeutic peptides are often provided in lyophilized form, as a dry powder or cake, for later reconstitution. A lyophilized formulation of a therapeutic peptide has the advantage of providing stability for long periods of time, but is less convenient to use as it requires the addition of one or more diluents and there is the potential risk for errors due to the use of an improper type or amount of diluent, as well as risk of contamination. In addition, the lyophilization process is time consuming and costly.
Accordingly, there is a need for an aqueous liquid formulation comprising a peptide agonist of the calcium sensing receptor, such as AMG 416. It would be desirable for the liquid formulation to remain stable over a relevant period of time under suitable storage conditions and to be suitable for administration by intravenous or other parenteral routes.
…………………………………
Milestones
- 25 Aug 2015Preregistration for Secondary hyperparathyroidism in USA (IV)
- 29 May 2015Pooled analysis efficacy and adverse events data from two phase III trials in secondary hyperparathyroidism released by Amgen
- 21 Apr 2015Amgen plans to submit Biological License Application to USFDA and Marketing Authorisation Application to EMA for Secondary hyperparathyroidism
References
- “Amgen Submits New Drug Application For Novel Intravenous Calcimimetic Etelcalcetide (AMG 416)”
- “Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects
- “Evidence for Chronic Kidney Disease-Mineral and Bone Disorder Associated With Metabolic Pathway Changes”
KAI-4169, a novel calcium sensing receptor agonist, decreases serum iPTH, FGF-23 and improves serum bone markers in a phase 2 study in hemodialysis subjects with chronic kidney disease-mineral and bone disorder
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO054
KAI-4169, a novel peptide agonist of the calcium sensing receptor, attenuates PTH and soft tissue calcification and restores parathyroid gland VDR levels in uremic rats
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
Long term safety and efficacy of velcalcetide (AMG 416), a calcium-sensing receptor (CaSR) agonist, for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis (HD) patients
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
Preclinical PK and PD relationship for KAI-4169, a novel calcimimetic
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
KAI-4169, a novel calcimimetic for the treatment of secondary hyperparathyroidism
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
Characterization of KAI-4169, a novel peptide for the treatment of chronic kidney disease – Mineral and bone disorder, in a phase I study in healthy males
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
| WO2011014707A2 | Jul 29, 2010 | Feb 3, 2011 | Kai Pharmaceuticals, Inc. | Therapeutic agents for reducing parathyroid hormone levels |
////Etelcalcetide, Parathyroid Disease, Amgen Inc, AMG 416, KAI-4169; KAI-4169-HCl, ONO-5163, Telcalcetide, Velcalcetide, Velcalcetide hydrochloride
FDA approves Repatha to treat certain patients with high cholesterol
08/27/2015 05:10 PM EDT
The U.S. Food and Drug Administration today approved Repatha (evolocumab) injection for some patients who are unable to get their low-density lipoprotein (LDL) cholesterol under control with current treatment options.
August 27, 2015
Release
The U.S. Food and Drug Administration today approved Repatha (evolocumab) injection for some patients who are unable to get their low-density lipoprotein (LDL) cholesterol under control with current treatment options.
Repatha, the second drug approved in a new class of drugs known as PCSK9 inhibitors, is approved for use in addition to diet and maximally-tolerated statin therapy in adult patients with heterozygous familial hypercholesterolemia (HeFH), homozygous familial hypercholesterolemia (HoFH), or clinical atherosclerotic cardiovascular disease, such as heart attacks or strokes, who require additional lowering of LDL cholesterol.
Familial hypercholesterolemia (encompassing both HeFH and HoFH) is an inherited condition that causes high levels of LDL cholesterol. A high level of LDL cholesterol in the blood is linked to cardiovascular or heart disease. Heart disease is the number one cause of death for Americans, both men and women. According to the Centers for Disease Control and Prevention, about 610,000 people die of heart disease in the United States every year– that equals one in every four deaths.
“Repatha provides another treatment option in this new class of drugs for patients with familial hypercholesterolemia or with known cardiovascular disease who have not been able to lower their LDL cholesterol enough with statins,” said John Jenkins, M.D., director of the Office of New Drugs, Center for Drug Evaluation and Research. “Cardiovascular disease is a serious threat to the health of Americans, and the FDA is committed to facilitating the development and approval of effective and safe drugs to address this important public health problem.”
Repatha is an antibody that targets a specific protein, called PCSK9. PCSK9 reduces the number of receptors on the liver that remove LDL cholesterol from the blood. By blocking PCSK9’s ability to work, more receptors are available to get rid of LDL cholesterol from the blood and, as a result, lower LDL cholesterol levels.
The efficacy and safety of Repatha were evaluated in one 52-week placebo-controlled trial and eight 12-week placebo-controlled trials in participants with primary hyperlipidemia, including two that specifically enrolled participants with HeFH and one that enrolled participants with HoFH. In one of the 12-week studies, 329 participants with HeFH, who required additional lowering of LDL cholesterol despite statins with or without other lipid-lowering therapies, were randomized to receive Repatha or placebo for 12 weeks. Participants taking Repatha had an average reduction in LDL cholesterol of approximately 60 percent, compared to placebo.
The most common side effects of Repatha include nasopharyngitis, upper respiratory tract infection, flu, back pain, and reactions such as redness, pain, or bruising where the injection is given. Allergic reactions, such as rash and hives, have been reported with the use of Repatha. Patients should stop using Repatha and get medical help if they experience symptoms of a serious allergic reaction.
Multiple clinical trials have demonstrated that statins lower the risk of having a heart attack or stroke. A trial evaluating the effect of adding Repatha to statins for reducing cardiovascular risk is ongoing.
Repatha is marketed by Amgen Inc., of Thousand Oaks, Calif.
Mahendra Chemicals gets FDA Warning Letter with Focus on “Data Integrity”
DRUG REGULATORY AFFAIRS INTERNATIONAL

A Warning Letter issued by the US Food & Drug Administration (FDA) to an Indian API manufacturer on 13 July 2015 shows again a clear focus on the missing integrity of data. Specifically, the following issues are addressed:
1. Activities were not recorded at the time they were carried out and original data were deleted:
Entries in the manufacturing protocols were made only days after the relevant activities had been conducted. Further, batches were released before all results were available.
In particular the use of “rough notes” was criticised as these original data were completely destroyed after transfer in the batch records.
2. Due to unauthorised access to data systems, data could be modified or deleted:
Specifically HPLC, GC, and Karl Fischer Titrators were concerned. For instance, for the GC instrument multiple copies of raw data were found in the waste. And there was no password regulation for the data systems…
View original post 100 more words
Non compliance at Parabolic drugs
DRUG REGULATORY AFFAIRS INTERNATIONAL

Statement “non compliance GMP”. Officina Farmaceutica: Parabolic Drugs Limited – INDIA (30/07/2015)
Following the inspection, conducted by the inspectorate Italian, under the program of inspections of the EDQM, at the Indian site in question, the same was not “in compliance” with the GMP.
It calls on companies to verify, with urgency, if the medicines containing the following active substances / intermediate production Dicloxacillin SODIUM, amoxicillin trihydrate, PIVAMPICILLIN, Flucloxacillin SODIUM, SODIUM cloxacillin, AMPICILLIN trihydrate, AMPICILLIN ANHYDROUS, Bacampicillin HYDROCHLORIDE authorized for the Italian market and / or products for export, showing this as a possible supplier of active / intermediate Officina Farmaceutica: PARABOLIC DRUGS LIMITED, PDL-2 – Plot No. 45, Industrial Area, Phase II, Panchkula District of Haryana, 134113 , INDIA .
The communication must be sent only by all companies Holders of marketing authorizations or Officine pharmaceutical manufacturers of medicines containing these materials pharmacologically active / production intermediates produced at…
View original post 194 more words
AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR

 Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
Dolutegravir (I) is chemically known as (4/?,12aS)-N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2//-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide. Dolutegravir is a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor (INSTI) indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection. Dolutegravir is being marketed under the trade name Tivicay®. US 8,129,385 disclosed Dolutegravir or its pharmaceutically acceptable salts thereof. US ‘385 also discloses a process for the preparation of Dolutegravir (I). The process involves the condensation of 5-benzyloxy-4-hydroxy-6-hydroxymethyl nicotinic acid (II) with 2,4-difluorobenzylamine (III) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-4-hydroxy-6-hydroxymethyl nicotinic acid amide (IV), which is further under goes oxidation using manganese dioxide (Mn02) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-6-formyl-4-hydroxy-nicotinic acid amide (V). This amide compound (V) is reacted with sodium chlorite (NaClCh) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4- hydroxy-pyridine-2-carboxylic acid (VI), which is further treated with methanol (MeOH) to produce 3-benzyloxy-5-(2,4-difluorobenzyl)-4-hydroxy-pyridine-2-carboxylic acid methyl ester (VII).

The methyl ester compound (VII) is reacted with 3-bromopropene to produce l-allyl-3-benzyloxy-5-(2,4-difluorobenzyl)-4-oxo-l,4-dihydro-pyridine-2- carboxylic acid methyl ester (VIII), which is further reacted with potassium osmate dihydrate (K2OSO4.2H2O) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4-oxo-l-(2-oxo-ethyl)-l,4-dihydropyridine-2-carboxylic acid methyl ester (IX). The compound (IX) is reacted with (R)-3-amino-l-butanol (X) to produce benzyloxy Dolutegravir (XI), which is deprotected by treating with TFA to produce Dolutegravir (I). The process is as shown in scheme-I below:
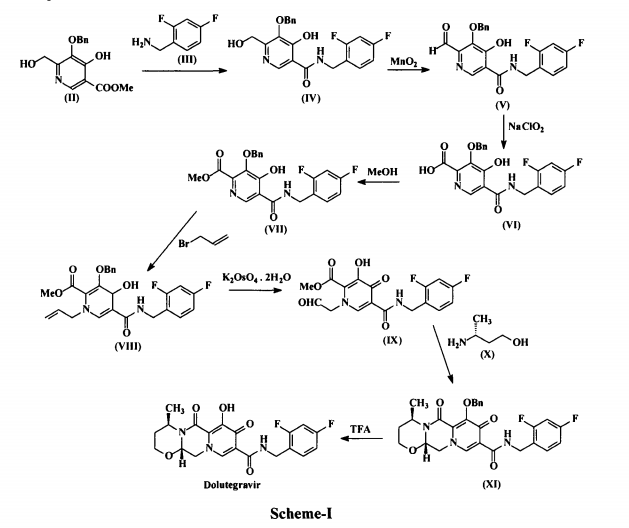
The major disadvantage with the above prior-art process is that it involves large no of steps and tedious work-up procedures to isolate the required product. This results a longer period of time cycle is required to produce Dolutegravir (I), which in turn renders the process more costly and less eco friendly. Further the above processes are low yielding and with less purity. US 8,217,034 discloses variant process for the preparation of Dolutegravir.
This process involves the reaction of methyl l-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-l,4-dihydro-2-pyridine carboxylate (XII) with (R)-3-amino-l-butanol (X) to produce (4R, 12o5)-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2//-pyrido[ 1 \2′,4,5] pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIII), which is further undergoes bromination using NBS to produce (4R,12aS)-9-bromo-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIV). The bromo Compound (XIV) is condensed with 2,4-difluorobenzylamine (III) in the presence of Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) to produce benzyloxy Dolutegravir (XI), which is hydrogenated in the presence of Pd/C to produce Dolutegravir (I). The process is as shown in Scheme-II below:

The major disadvantage with the above prior art process of preparing Dolutegravir is the use of expensive reagent tetrakis(triphenylphosphine)palladium (Pd(PPh3)4> in coupling step. Use of this reagent on industrial scale is not preferred, which makes the process more expensive. WO 2011/119566 discloses another variant process for the preparation of Dolutegravir.
This process involves the reaction of l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l,4-dihydropyridine-3-carboxylic acid (XV) with acetic acid in presence of methane sulfonic acid to produce 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), which is further condensed with (R)-3-amino-l-butanol (X) to produce (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[ 1 ‘,2’:4,5]pyrazino[2,1 -b] [ 1,3]-oxazine-9-carboxylic acid (XVII). This acid Compound XVII is acylated with 2,4-difluorobenzylamine (III) in the presence of carbonyldiimidazole (CDI) to produce methoxy Dolutegravir (XVIII), which is demethylated in the presence of lithium bromide (LiBr) to produce Dolutegravir (I).
The process is as shown in Scheme-3 below:
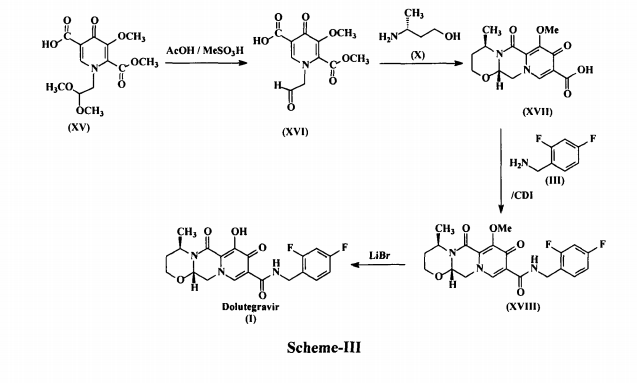
The major disadvantage of the above prior art process of preparing Dolutegravir is the use of expensive and highly moisture sensitive reagent, 1,1-carbonyldiimidazole (CDI), during acylation. Use of this reagent on industrial scale is not preferred due to anhydrous conditions required in the process. However, there is always a need for alternative preparative routes, which for example, involve fewer steps, use reagents that are less expensive and/or easier to handle, consume smaller amounts of reagents, provide a higher yield of product, have smaller and/or more eco-friendly waste products, and/or provide a product of higher purity. Hence, there is a need to develop cost effective and commercially viable process for the preparation of Dolutegravir of formula (I). The present invention is related to a process for the preparation of pure Dolutegravir of formula (I), wherein optically active acid addition salt of (R)-3-amino-l-butanol (X) is directly condensed with 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI) instead of condensing with free base of (R)-3-amino-1-butanol (X). The present invention is also related to a process for the preparation of pure Dolutegravir of formula (I), wherein, inexpensive and easily handling condensing reagents in the condensation of (4R, 12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[l’,2′:4,5]pyrazino [2,l-b][l,3]oxazine-9-carboxylic acid (XVII) with 2,4-difluorobenzylamine (III).


AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR
| APPLICATION NUMBER | 1361/CHE/2013 |
| APPLICANT NAME | AUROBINDO PHARMA LTD |
| DATE OF FILING | 27/03/2013 |
| PUBLICATION DATE (U/S 11A) | 16/01/2015 |
In another embodiment, 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4- dihydropyridine-3-carboxylic acid (XVI) used in the present invention is prepared by reacting 4-methoxyacetoacetate (XIX) with N,N-dimethyl-l,l- bis(methyloxy)methanamine (DMF-DMA) (XX) to produce methyl-2- (dimethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-(dimethylamino)-2 [(methyloxy)acetyl]-2-propenoate) (XXI), which is reacted with aminoacetaldehyde dimethyl acetal (XXII) to produce methyl-2-(2,2-dimethoxyethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-{[2,2-bis(methyloxy)ethyl]amino}-2-[(methyloxy) acetyl]-2-propenoate) (XXIII).
The compound (XXIII) is contacted with dimethyl ethanedioate in presence of alkali metal alkoxide to produce dimethyl-1-(2,2-dimethoxyethyl)-3-methoxy-4-oxo-l ,4-dihydropyridine-2,5-dicarboxylate (XXIV), which is selectively hydrolyzed with a base to produce l-[2,2-bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l ,4-dihydro-3-pyridinecarboxylic acid (XV). The compound (XV) is treated with a catalytic amount of a strong protic acid in the presence of acetic acid in an organic solvent to produce a reaction mixture containing 5- methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), The process is as shown in Scheme-IV below:

The following examples illustrate the nature of the invention and are provided for illustrative purposes only and should not be construed to limit the scope of the invention.
Example-1:
EXAMPLES: Example-1: Process for the preparation of Dolutegravir
Step-i: Preparation of (/?)-3-amino-l-butanol tartarate salt: D-(+) Tartaric acid (12.7 g, 0.085 mol) was added in to a solution of (i?,5)-3-amino-l-butnaol (7.5 g, 0.084 mol) in methanol (100 ml) at 40 °C. The reaction mixture was stirred for about 1 hour at 35-40 °C and the reaction mass was cooled to 0-5°C and maintained for 30-40 minutes. The obtained solid was filtered and washed with chilled methanol (10 ml) at 0-5 °C. The solid was dried to get (i?)-3-amino-l-butanol tartarate salt (8.0 g, 40%).
Step-ii: Preparation of (4rt,12a£)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[l’,2′;4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxylic acid (XVII): l-[2,2-Bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l,4-dihydro-3-pyridinecarboxylic acid (XV) (lOOg; 0.3175 moles) was suspended in acetonitrile (800 ml) and heated to 80-82°C. A mixture of acetic acid (95.25 g), methanesulfonic acid (9.14 g; 0.09525 moles) and acetonitrile (200 ml) were added to the slurry at 80-82°C. The reaction mass was continued at 80-82°C to complete the reaction. After completion of the reaction, anhydrous sodium acetate (65 g) and (/?)-3-amino-l-butanol tartrate salt (79.68g; 0.3334 moles) were added at 20-25°C and stirred at 60-65°C to complete the reaction. The reaction mass was concentrated and acidified with IN aqueous hydrochloric acid (750 ml) and extracted with methylene chloride (1500 ml) at ice cold temperature. The organic layer was separated, concentrated, treated with hot methanol (350 ml) for 2 h, filtered, washed with methanol and dried to yield (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxylic acid (XVII) (72 g; HPLC purity: 99.07%).
Step-iii: Process for the preparation of Dolutegravir (I). Method A: Triethylamine (3.61 g; 0.0357 moles) was added to the suspension of (4R,12aS)-7- methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 – b][l,3]oxazine-9-carboxylic acid (XVII) (10 g; 0.0325 moles) in methylene chloride (50 ml), and cooled to 10-15°C. Pivaloyl chloride (4.3 g; 0.0357 moles) was added to the reaction mass, and stirred at 10-15°C for 1 h. Thereafter, 2,4-difiuorobenzylamine (5.58 g; 0.0389 moles) was added at 10-15°C and then warmed to 20-25°C to complete the reaction. After completion of the reaction, IN aqueous hydrochloric acid (20 ml) was added, organic layer was separated, washed with 5% w/w aqueous sodium bicarbonate solution (10 ml) followed by 15% w/w aqueous sodium chloride solution (10 ml) and concentrated. To the concentrated mass, acetonitrile (100 ml) and Lithium bromide (5.08 g; 0.0584 moles) were added and heated to 65-70°C for 3 h to complete the reaction. After completion of the reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml), concentrated to about 50 ml and DM water was added to crystallize the product at 20-25°C. The slurry was stirred for 2 h, filtered, washed with DM water and dried to yield (4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxamide (I) (11.5 g, HPLC purity: 99.63%).
Method B: Isobutyl chloroformate (4.65 gm, 0.03404 moles) in methylene chloride (10 ml) was added to the solution of N-methylmorpholine (3.45 gm, 0.03410 moles) and (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino-[2,1 -b][l,3]oxazine-9-carboxy!ic acid (XVII) (10.0 gm, 0.03245 moles) in methylene chloride (60 ml) at -10 to 0°C in about 1 h. 2,4-Difloro benzyl amine (4.88 gm, 0.03409 moles) in methylene chloride (10 ml) was added to the cold reaction mass, and stirred at 20-30°C for completion of reaction. After completion of reaction, the reaction mass was washed with 5%w/w aqueous sodium bicarbonate solution (20 ml), IN hydrochloric acid (20 ml), DM water (20 ml) and concentrated. Acetonitrile (120 ml) and lithium bromide (4.8 gm, 0.05516 moles) were added to the concentrated mass, and stirred at 70-80°C for 3 h to complete the reaction. After completion of reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml) and concentrated to about 50 ml. DM Water (100 ml) was added to the concentrated reaction mass and stirred for 2 h at 25-30°C to crystallize the product. The product was filtered, washed with DM Water (50 ml) and dried to yield Dolutegravir (I) (10.7 gm, HPLC purity: 99.60%).
Example-2: Process for the preparation of Dolutegravir (I) (4R, 12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide (XVIII) (2 g, 0.0046 moles) was suspended in isopropyl alcohol (20 ml) and lithium bromide (0.8 g, 0.00924 moles) was added and stirred at 70-80°C for 15 h to complete the reaction. After completion of reaction the reaction mass was acidified with 5N aqueous hydrochloric acid (5 ml) and concentrated. DM Water (20 ml) was added to the concentrated mass and stirred at 25-30°C to crystallize the product. The product was filtered, washed with DM Water and dried to yield Dolutegravir (I) (1.5 g, HPLC purity: 97.93%).


/////
FDA warns Mylan about cGMP violations at its Indian facilities
DRUG REGULATORY AFFAIRS INTERNATIONAL

The US FDA has warned Mylan about manufacturing concerns at three of its plants in India.
In a warning letter to the generic drug manufacturer, the FDA said it had found ‘significant violations of current good manufacturing practice’ during inspections at the plants in August and September last year and in February this year.
The inspections relate to Mylan’s Agila Specialty Formulation Facility (SFF), Sterile Product Division (SPD), and Onco Therapies Limited (OTL) sites in Bangalore.

Some of the violations cited were failure to establish and follow appropriate written procedures designed to prevent microbiological contamination of drug products, such as the use of gloves with tears and pinholes, as well as deficiencies in environmental monitoring and poor monitoring of staff……..http://www.manufacturingchemist.com/news/article_page/FDA_warns_Mylan_about_cGMP_violations_at_its_Indian_facilities/111318/cn48579?dm_i=8EU,3MBVR,9ETTTY,D0ENC,1

Recently the Food and Drug Administration (FDA) began ramping up inspections of offshore manufacturing facilities and the results are shocking. Although cGMP violations have been found worldwide, experts are…
View original post 508 more words
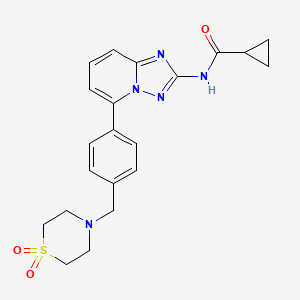
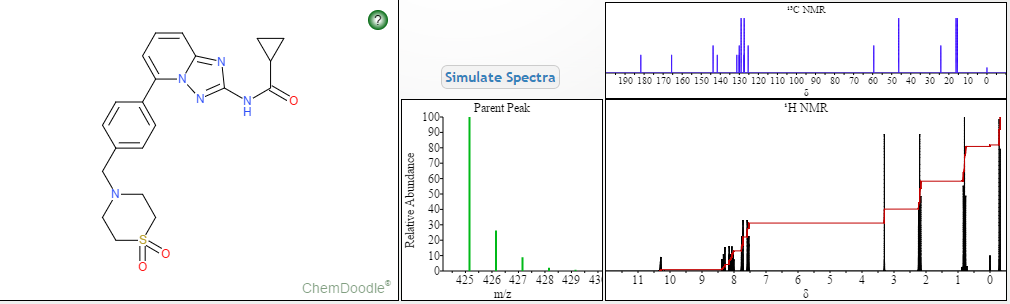
Filgotinib

Filgotinib
EU APPROVED 2020/9/24, JYSELECA
JAPAN APPROVED2020/9/25
- C21H23N5O3S
- MW425.504
- Elemental Analysis: C, 59.28; H, 5.45; N, 16.46; O, 11.28; S, 7.54
1206161-97-8
Cyclopropanecarboxamide, N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]-
G146034
GLPG0634
N-(5-(4-((1,1-dioxidothiomorpholino)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide
Galapagos Nv INNOVATOR
PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulcerative colitis
Filgotinib is an orally available inhibitor of JAK1/JAK2 and TYK2 in phase III clinical development at Galapagos and Gilead for the treatment of rheumatoid arthritis, moderate or severe Crohn’s disease and ulcerative colitis
IL-6 antagonist; Jak1 tyrosine kinase inhibitor; Tyk2 tyrosine kinase inhibitor; Jak3 tyrosine kinase inhibitor; Jak2 tyrosine kinase inhibitor
Autoimmune disease; Cancer; Colitis; Crohns disease; Inflammatory disease; Neoplasm; Rheumatoid arthritis; Transplant rejection
In 2017, orphan drug designation was assigned to the compound in the U.S. for the treatment of pediatric Crohn’s disease and pediatric ulcerative colitis.
GlaxoSmithKline had been developing filgotinib preclinically for the treatment of rheumatoid arthritis pursuant to a license; however, in 2010, the compound was re-acquired by Galapagos. In 2012, the product was licensed to Abbott for development and marketing. In January 2013, Abbott spun-off its research-based pharmaceutical business into a newly-formed company AbbVie. The license agreement between Galapagos and Abbott was terminated in September 2015, Galapagos regaining all rights to the product. The same year, Galapagos and Gilead entered into a global partnership and Gilead obtained the global rights of codevelopment and commercialization for the treatment of inflammatory diseases
Filgotinib (GLPG0634), by the Belgian biotech company Galápagos NV, is a drug which is currently under investigation for the treatment of rheumatoid arthritis and Crohn’s disease.
Filgotinib (GLPG0634) is an orally-available, selective inhibitor of JAK1 (Janus kinase 1) for the treatment of rheumatoid arthritis and potentially other inflammatory diseases. Filgotinib (GLPG0634) dose-dependently inhibited Th1 and Th2 differentiation and to a lesser extent the differentiation of Th17 cells in vitro. GLPG0634 was well exposed in rodents upon oral dosing, and exposure levels correlated with repression of Mx2 expression in leukocytes. The JAK1 selective inhibitor GLPG0634 (Filgotinib) is a promising novel therapeutic with potential for oral treatment of rheumatoid arthritis and possibly other immune-inflammatory diseases. Filgotinib (GLPG0634) is currently in a Phase 2 study in Crohn’s disease.

Mechanism of action
Filgotinib is a Janus kinase inhibitor with selectivity for subtype JAK1 of this enzyme. It is considered a promising agent as it inhibits JAK1 selectively. Less selective JAK inhibitors (e.g. tofacitinib) are already being marketed. They show long-term efficacy in the treatment of various inflammatory diseases. However, their lack of selectivity leads to dose-limiting side effects.[1] It is thought that inhibition of all JAK isoenzymes is beneficial in rheumatoid arthritis. However, pan-JAK inhibition might also lead to unwanted side effects that might not outweigh its benefits. This is the rationale for the development of newer and more selective inhibitors like filgotinib.
The signal transmission of large numbers of proinflammatory cytokines is dependent on JAK1. Inhibition of JAK2 may also contribute to the efficacy against RA. Nonetheless it is thought that JAK2 inhibition might lead to anemia and thrombopenia by interference witherythropoietin and thrombopoietin and granulocyte-macrophage colony-stimulating factor. Therefore one might prefer to choose a more selective JAK1 inhibitor as a primary therapeutic option. Filgotinib exerts a 30-fold selectivity for JAK1 compared to JAK2.[2] It is however still to be seen to what extent JAK2 inhibition should be avoided.
Novel crystalline forms of filgotinib salts, particularly hydrochloride salt, useful for treating JAK-mediated diseases eg inflammatory diseases, autoimmune diseases, proliferative diseases, allergy and transplant rejection. Galapagos and licensee AbbVie are developing filgotinib, a selective JAK-1 inhibitor, for treating rheumatoid arthritis (RA) and Crohn’s disease (CD). In August 2015, the drug was reported to be in phase 2 clinical development for treating RA and CD. The drug is also being investigated for the treatment of colitis and was discovered as part of the company’s arthritis alliance with GSK; however in August 2010 Galapagos reacquired the full rights. See WO2013189771, claiming use of filgotinib analog for treating inflammatory diseases. Also see WO2010010190 (co-assigned with GSK and Abbott) and WO2010149769 (assigned to Galapagos) claiming filgotinib, generically and specifically, respectively.
Clinical trials and approval
The efficacy of filgotinib is currently studied in a phase2b program (DARWIN trial 1, 2) with involvement of 886 rheumatoid arthritis patients and 180 Crohn’s disease patients.
Phase 1 study
It was shown in phase 1 studies that the pharmacokinetics of filgotinib metabolism is independent of hepatic CYP450 enzymatic degradation. The drug metabolism is however mediated by carboxylesterases. There is no interference reported with the metabolism of methotrexate nor with any of the investigated transport proteins.[3]
Phase 2 study: Proof of concept (2011)
In november 2011 Galápagos released the results of their phase 2 study (identification: NCT01384422, Eudract: 2010-022953-40) in which 36 patients were treated who showed a suboptimal clinical response to methotrexate treatment. Three groups of twelve patients were treated either with 200 mg filgotinib in a single dose, 200 mg divided in two doses or placebo. The primary end-point was the ACR20 score, which monitors improvements in the symptomatology of the patient. After the scheduled 4 weeks of treatment, 83% of the respondents showed an improved ACR20-score. Half of the treated patients showed a complete (or near complete) remission of the disease. There were no reports ofanemia nor changes in lipidemia. The company stated in their press release that filgotinib is the first selective JAK1 inhibitor that shows clinical efficacy. As a result of this study, the company stated that “GLPG0634 shows one of the highest initial response rates ever reported for rheumatoid arthritis treatments”.[4]
DARWIN 1 trial
The DARWIN 1 trial is a 24 week double blind placebo-controlled trial with 599 rheumatoid arthritis patients enrolled. All participants have moderate to severe RA and showed an insufficient response to standard methotrexate treatment. The trial compares three dosages of filgotinib as a once or twice per day regimen. During the trial all participants remain on their methotrexate treatment. According to the company, the results of this trial are expected in July 2015.[5]
DARWIN 2 trial
The DARWIN 2 trial is a double blind placebo-controlled trial with 280 rheumatoid arthritis patients enrolled who show an insufficient response to standard methotrexate treatment. This trial, in contrast to the previous DARWIN 1 trial, methotrexate is discontinued. Therefore, this trial investigates filgotinib as a monotherapy.[6] The recruitment of DARWIN trial 2b ended in november 2014.[7] Preliminary results are expected in the second quarter of 2015 and a full completion of the study is expected in the third quarter of 2015.
DARWIN 3 trial
Patients who complete DARWIN 1 and 2 will be eligible for DARWIN 3.
COSY PREDICT
Time line
- june 2011: results of first phase 2 trial
- november 2014: initiation of DARWIN 1 and 2 trials
- april 2015: expected date of DARWIN 1 trial results
- june 2015: expected date of DARWIN 2 trial results
NMR FROM NET….ABMOLE, DMSOD6
NMR MEDKOO DMSOD6

CHEMIETEK
1H NMR PREDICT


13C NMR PREDICT

……………………
MORE PREDICTS
1H NMR PREDICT
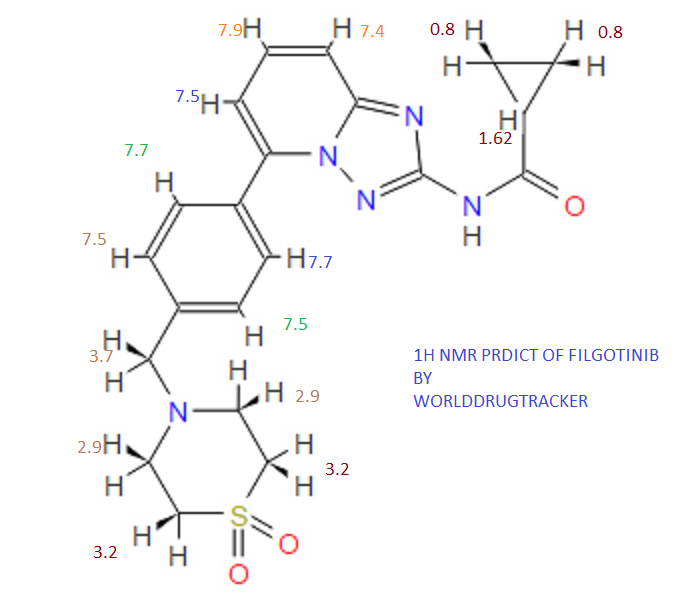
13C NMR PREDICT
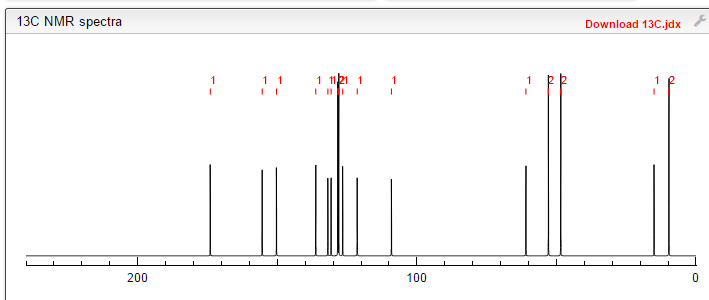
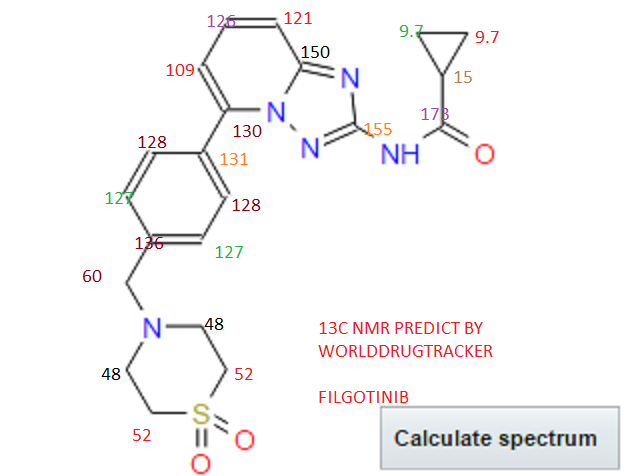
PRODUCT PATENT
http://www.google.com/patents/WO2010149769A1?cl=en
| Applicants: | GALAPAGOS NV [BE/BE]; Generaal De Wittelaan L11/A3 B-2800 Mechelen (BE) (For All Designated States Except US). MENET, Christel Jeanne Marie [FR/BE]; (BE) (For US Only). SMITS, Koen Kurt [BE/BE]; (BE) (For US Only) |
| Inventors: | MENET, Christel Jeanne Marie; (BE). SMITS, Koen Kurt; (BE) |
| International Filing Date: | 25.06.2010 |
ESTIMATED EXP 2030
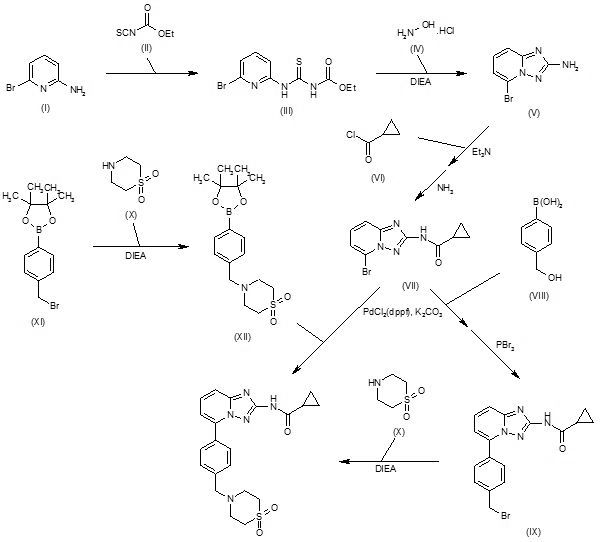
Condensation of 2-amino-6-bromopyridine (I) with ethoxycarbonyl isothiocyanate (II) in CH2Cl2 gives 1-(6-bromopyridin-2-yl)-3-carboethoxythiourea (III), which upon cyclization with hydroxylamine hydrochloride (IV) in the presence of DIEA in EtOH/MeOH yields 2-amino-5-bromo[1,2,4]triazolo[1,5-a]pyridine (V). N-Acylation of amine (V) with cyclopropanecarbonyl chloride (VI) using Et3N in acetonitrile, and subsequent treatment with methanolic ammonia furnishes the carboxamide (VII) (1-3), which upon Suzuki coupling with 4-(hydroxymethyl)phenylboronic acid (VIII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C, followed by bromination with PBr3 in CHCl3 affords intermediate (IX). Condensation of benzyl bromide derivative (IX) with thiomorpholine-1,1-dioxide (X) using DIEA in CH2Cl2/MeOH yields filgotinib (1,2). Alternatively, condensation of (4-bromomethylphenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (XI) with thiomorpholine 1,1-dioxide (X) in the presence of DIEA in CH2Cl2/MeOH gives intermediate (XII), which undergoes Suzuki coupling with aryl bromide (VII) in the presence of PdCl2(dppf) and K2CO3 in dioxane/H2O at 90 °C to afford the target filgotinib
The present invention is based on the discovery that the compound of the invention is able to act as an inhibitor of JAK and that it is useful for the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6. In a specific aspect the compound is an inhibitor of JAKl and JAK2. The present invention also provides methods for the production of this compound, a pharmaceutical composition comprising this compound and methods for treating inflammatory conditions, autoimmune diseases, proliferative diseases, transplantation rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 by administering the compound of the invention.
Accordingly, in a first aspect of the invention, a compound of the invention is provided having a formula (I):

[0017] The compound of the invention is a novel inhibitor of JAK that appears to exhibit a dramatically improved in vivo potency as compared to structurally similar compounds. In a particular embodiment the compound of the invention is an inhibitor of JAKl and JAK2. In particular it appears to exhibit this increase in potency at lower in vivo exposure levels compared to structurally similar compounds. The use of a compound with these improvements is expected to result in a lower dosage requirement (and therefore an improved dosing schedule).
General Synthetic Method Scheme 1

1. RCOCI, Et3N 2. NH3 / MeOH CH3CN, 20 0C 2O 0C

wherein Ar represents phenyl-Ll-heterocycloalkyl, where Ll is a bond, -CH2– or -CO- and the heterocycloalkyl group is optionally substituted.
General
1.1.1 l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

(2)
[00117] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5 0C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20 0C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
7.7.2 5-Bromo-[l, 2, 4]triazolo[l, 5-a]pyridin-2-ylamine (3)

[00118] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH
(1 :1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 0C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition Of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-t/β) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (IH, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 :1, M+H+, 100%).
7.7.3 General procedure for mono-acylation to afford intermediate (4):

[00119] To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN
(150 mL) at 5 0C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2x50mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A
Preparation of compounds of the invention via Suzuki coupling (5):
[00120] An appropriate boronic acid (2eq.) is added to a solution of bromo intermediate (4) in
1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters
XBridge Prep Cl 8 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using
MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

Bl. 4 4-[2-(Cyclopropanecarbonyl-amino)-[ 1 , 2, 4]triazolo[l, 5-a] pyridin-5-yl] -benzoyl chloride

[00121] 2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)- [l,2,4]triazolo[l,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide formation (General Method)

[00122] An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 00C. The acid chloride (Bl, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method C

Wherein R3a or R3b together with the nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reductive alkylation (general method)
[00123] An appropriate amine (2 eq.), cyclopropanecarboxylic acid (for example cyclopropanecarboxylic acid [5-(4-formyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridine-2-yl]-amide) prepared by method A (1 eq.) and Ti(OPr)4 are mixed and stirred at room temperature for 3 hrs. The mixture is diluted in ethanol and Na(CN)BH3 (leq.) is added. The resulting solution is stirred at room temperature for 16 hrs. The mixture is diluted in water and filtered. The filtrate is washed with ethanol. The combined solvent phases are evaporated under vacuum. The final compound is isolated by preparative HPLC.
Method D 
wherein R1 and R2 together with the Nitrogen atom to which they are attached, may form a heterocycloalkyl.
Reaction ofalkylation

[00124] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and Et3N (2 eq) (or AgCO3) are dissolved in DCM/MeOH (4:1 v:v) under N2 and an amine (2 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSθ4. Organic layers are filtered and evaporated. The final compound is isolated by flash chromatography.
Suzuki coupling

[00125] The obtained boronic acid (2eq.) is added to a solution of cyclopropanecarboxylic acid
(5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide (4) in 1 ,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140 0C for 30 min (This reaction can also be carried out by traditional heating in an oil bath at 900C for 16h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5μm ODB 19mm ID x 100mm L (Part No.186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Synthesis of the compound of the invention and comparative examples
Compound l(the compound of the invention)
Step 1:

[00126] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (leq) and DIPEA
(2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgS O4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
Step 2: Suzuki coupling

[00127] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide
(l.leq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00128] Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 500C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative route to Compound l(the compound of the invention):
Step 1:

[00129] 4-(Hydroxymethyl)phenylboronic acid (l.leq.) was added to a s o luti o n o f cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 900C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSθ4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

[00130] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)- [l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSθ4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

[00131] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin- 2-yl]-amide (leq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO^ Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
PATENT
WO 2010010190
WO 2013173506
WO 2013189771
WO 2015117980
WO 2015117981
POLYMORPH
CN 105061420
https://encrypted.google.com/patents/CN105061420A?cl=en
JAK inhibitor N-(5-(4-(1,1-dioxothiomorpholinyl)methyl)phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl)cyclopropanecarboxamide, and methods for preparing the four crystal forms, wherein the four crystal forms respectively are a crystal form H1, a crystal form H2, a crystal form H3 and a crystal form H4,
POLYMORPH
E CRYSTAL
CN 105111206
D CRYSTAL
CN 105111207
H CRYSYAL
CN 105198876
G
CN 105198877
F CN 105198878
C CN 105198880
POLYMORPH
WO 2016105453
POLYMORPH
Preparation of solid forms of filgotinib free base
WO 2017012773
The present invention relates to cryst. filgotinib free base (I), a method of its prepn. and a pharmaceutical compn. comprising the same. Thus, reacting the compd. II with thiomorpholine dioxide afforded 92% I (purity of 98.6%) which was then converted into its hydrochloride salt. Pharmaceutical compn. comprising compd. I was disclosed.
POLYMORPH
CN 105669669
The present invention provides a crystal form A, B, D, G and M of N-[5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl][1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide hydrochloride.
PAPER
Future Medicinal Chemistry (2015), 7(2), 203-235. | Language: English, Database: CAPLUSA review. The discovery of the JAK-STAT pathway was a landmark in cell biol. The identification of these pathways has changed the landscape of treatment of rheumatoid arthritis and other autoimmune diseases. The two first (unselective) JAK inhibitors have recently been approved by the US FDA for the treatment of myelofibrosis and rheumatoid arthritis and many other JAK inhibitors are currently in clin. development or at the discovery stage. Research groups have demonstrated the different roles of JAK member and the therapeutic potential of targeting them selectively. ………..
https://www.future-science.com/doi/10.4155/fmc.14.149
PAPER
Journal of Pharmaceutical Sciences (Philadelphia, PA, United States) (2018), 107(6), 1624-1632.
PATENT
US2010/331319 A1, ; Page/Page column 13-14
http://www.google.com/patents/US20100331319
Synthetic Preparation of the Compound of the Invention and Comparative Examples
The compound of the invention and the comparative examples can be produced according to the following scheme.

wherein Ar represents phenyl-L1-heterocycloalkyl, where L1 is a bond, —CH2— or —CO— and the heterocycloalkyl group is optionally substituted.
General 1.1.1 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2)

To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5° C. is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture is then allowed to warm to room temp. (20° C.) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea may be used as such for the next step without any purification. 1H (400 MHz, CDCl3) δ 12.03 (1H, br s, NH), 8.81 (1H, d, J=7.8 Hz, H-3), 8.15 (1H, br s, NH), 7.60 (1H, t, J=8.0 Hz, H-4), 7.32 (1H, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.2 5-Bromo-[1,2,4]triazolo[1,5-a]pyridin-2-ylamine (3)

To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1:1, 900 mL) is added N,N-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20° C.) for 1 h. 1-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3 h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H2O (250 mL) and filtration. The combined solids are washed successively with H2O (250 mL), EtOH/MeOH (1:1, 250 mL) and Et2O (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. 1H (400 MHz, DMSO-d6) δ 7.43-7.34 (2H, m, 2×aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1:1, M+H+, 100%).
1.1.3 General Procedure for Mono-Acylation to Afford Intermediate (4)

To a solution of the 2-amino-triazolopyridine (3) (7.10 g, 33.3 mmol) in dry CH3CN (150 mL) at 5° C. is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material (3) is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp. (for 1-16 h) to hydrolyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et2O (50 mL). The solids are collected by filtration, washed with H2O (2×50 mL), acetone (50 mL) and Et2O (50 mL), then dried in vacuo to give the required bromo intermediate (4).
Method A Preparation of Compounds of the Invention Via Suzuki Coupling (5):
An appropriate boronic acid (2 eq.) is added to a solution of bromo intermediate (4) in 1,4-dioxane/water (5:1). K2CO3 (2 eq.) and PdCl2dppf (5%) are added to the solution. The resulting mixture is then heated in a microwave at 140° C. for 30 min (this reaction can also be carried out by traditional heating in an oil bath at 90° C. for 16 h under N2). Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhyd. MgSO4 and evaporated in vacuo. The final compound is obtained after purification by flash chromatography or preparative HPLC. HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
Method B

B1. 4 4-[2-(Cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoyl chloride

2 Drops of DMF are added to a solution of 4-[2-(cyclopropanecarbonyl-amino)-[1,2,4]triazolo[1,5-a]pyridin-5-yl]-benzoic acid (1 eq) obtained by Method A using 4-carboxyphenylboronic acid in DCM under N2 atmosphere. Then oxalyl chloride (2 eq) is added dropwise to this resulting solution (gas release). The mixture is stirred at room temperature for 2 hours. After completion of the reaction by LCMS, the solvent is removed. The crude acid chloride is used without further purification in next step.
B2. Amide Formation (General Method)

An appropriate amine (1.1 eq) and Et3N (5 eq) are dissolved in DCM under N2 atmosphere and cooled at 0° C. The acid chloride (B1, 1 eq) dissolved in DCM is added dropwise to this solution. The reaction is stirred at room temperature for 16 h. After this time, reaction is complete. The compound is extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers are filtered and evaporated. The final compound is isolated by preparative HPLC. Preparative HPLC: Waters XBridge Prep C18 5 μm ODB 19 mm ID×100 mm L (Part No. 186002978). All the methods are using MeCN/H2O gradients. H2O contains either 0.1% TFA or 0.1% NH3.
…
Synthesis of the Compound of the Invention and Comparative Examples Compound 1 (the Compound of the Invention) Step 1:

2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[1,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (2 eq) was added portionwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
STEP 2: Suzuki coupling

4-[4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-1,1-dioxide (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
Alternatively, after completion of the reaction, a palladium scavenger such as 1,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cooled down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HCl is added, and after stirring at RT, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at RT, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H2O, treated with a palladium scavenger (e.g. SMOPEX 234) at 50° C., the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the title compound as a free base.
Alternative Route to Compound 1 (the Compound of the Invention): Step 1:

4-(Hydroxymethyl)phenylboronic acid (1.1 eq.) was added to a solution of cyclopropanecarboxylic acid (5-bromo-[1,2,4]triazolo[1,5-a]pyridin-2-yl)-amide in 1,4-dioxane/water (4:1). K2CO3 (2 eq.) and PdCl2dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90° C. for 16 h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhyd. MgSO4 and evaporated in vacuo. The resulting mixture was used without further purification.
Step 2:

To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1.0 eq) in chloroform was slowly added phosphorus tribromide (1.0 equiv.). The reaction mixture was stirred at room temperature for 20 hours, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer was dried over anhyd. MgSO4, filtered and concentrated to dryness. The resulting white residue was triturated in dichloromethane/diethyl ether 2:1 to afford the expected product as a white solid.
Step 3:

Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[1,2,4]triazolo[1,5-a]pyridin-2-yl]-amide (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1-dioxide (1.1 eq) was added dropwise. The resulting solution was stirred at room temperature for 16 h. After this time, the reaction was complete. The solvent was evaporated. The compound was dissolved in DCM, washed with water and dried over anhyd. MgSO4. Organic layers were filtered and evaporated. The final compound was isolated by column chromatography using EtOAc to afford the desired product.
…………………….
PATENT
Novel salts and pharmaceutical compositions thereof for the treatment of inflammatory disorders
Also claims a method for preparing filgotinib hydrochloride trihydrate. The present filing forms a pair with this week’s filing, WO2015117980, claiming a tablet composition comprising filgotinib hydrochloride.
The compound cyclopropanecarboxylic acid {5-[4-(l,l-dioxo-thiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5-a]pyridin-2-yl -amide (Compound 1), which has the chemical structure:

is disclosed in our earlier application WO 2010/149769 (Menet C. J., 2010) as being an inhibitor of JAK and as being useful in the treatment of inflammatory conditions, autoimmune diseases, proliferative diseases, allergy, transplant rejection, diseases involving impairment of cartilage turnover, congenital cartilage malformations, and/or diseases associated with hypersecretion of IL6 or interferons. Hereafter this compound is named Compound 1. The data presented in WO 2010/149769 demonstrate that despite similar in vitro activities, Compound 1 has unexpectedly high in vivo potency compared with structurally similar compounds.
Example 1. Preparation of Compound 1
1.1. Route 1
1.1.1. 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide

[00205] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) is added portionwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated without further purification.
1.1.2. Cyclopropanecarboxylic acid (5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-yl)-amide

1.1.2.1. Step i): l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea
[00206] To a solution of 2-amino-6-bromopyridine (1) (253.8 g, 1.467 mol) in DCM (2.5 L) cooled to 5°C is added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction
mixture is then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gives a solid which may be collected by filtration, thoroughly washed with petrol (3 x 600 niL) and air-dried to afford the desired product. The thiourea may be used as such for the next step without any purification. lH (400 MHz, CDC13) δ 12.03 (1H, br s), 8.81 (1H, d), 8.15 (1H, br s), 7.60 (1H, t), 7.32 (1H, dd), 4.31 (2H, q), 1.35 (3H, t).
1.1.2.2. Step ii): 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine
[00207] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) is added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture is stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) is then added and the mixture slowly heated to reflux (Note: bleach scrubber is required to quench H2S evolved). After 3h at reflux, the mixture is allowed to cool and filtered to collect the precipitated solid. Further product is collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids are washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound may be used as such for the next step without any purification. lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2.3. Step Hi): Cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide
[00208] To a solution of the 2-amino-triazolopyridine obtained in the previous step (7.10 g, 33.3 mmol) in dry MeCN (150 mL) at 5°C is added Et3N (11.6 mL, 83.3 mmol) followed by cyclopropanecarbonyl chloride (83.3 mmol). The reaction mixture is then allowed to warm to ambient temperature and stirred until all starting material is consumed. If required, further Et3N (4.64 mL, 33.3 mmol) and cyclopropanecarbonyl chloride (33.3 mmol) is added to ensure complete reaction. Following solvent evaporation in vacuo the resultant residue is treated with 7 N methanolic ammonia solution (50 mL) and stirred at ambient temp, (for 1-16 h) to hydro lyse any bis-acylated product. Product isolation is made by removal of volatiles in vacuo followed by trituration with Et20 (50 mL). The solids are collected by filtration, washed with H20 (2x50mL), acetone (50 mL) and Et20 (50 mL), then dried in vacuo to give the desired compound.
1.1.3. Compound 1

[00209] 4-[4-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)-benzyl] hiomoφholine , l -dioxide (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo.
[00210] The final compound is obtained after purification by flash chromatography.
[00211] Alternatively, after completion of the reaction, a palladium scavenger such as 1 ,2-bis(diphenylphosphino)ethane, is added, the reaction mixture is allowed to cool down and a filtration is performed. The filter cake is reslurried in a suitable solvent (e.g. acetone), the solid is separated by filtration, washed with more acetone, and dried. The resulting solid is resuspended in water, aqueous HC1 is added, and after stirring at room temperature, the resulting solution is filtered on celite (Celpure P300). Aqueous NaOH is then added to the filtrate, and the resulting suspension is stirred at room temperature, the solid is separated by filtration, washed with water and dried by suction. Finally the cake is re-solubilised in a mixture of THF/H20, treated with a palladium scavenger (e.g. SMOPEX 234) at 50°C, the suspension is filtered, the organic solvents are removed by evaporation, and the resulting slurry is washed with water and methanol, dried and sieved, to obtain the desired compound as a free base.
1.2. Route 2
1.2.1. Step 1: cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2, 4]triazolo[l, 5- a] pyridin-2-yl] -amide

[00212] 4-(Hydroxymethyl)phenylboronic acid (l . l eq.) is added to a solution of cyclopropanecarboxylic acid (5-bromo-[l ,2,4]triazolo[l ,5-a]pyridin-2-yl)-amide in 1 ,4-dioxane/water
(4:1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) are added to the solution. The resulting mixture is then heated in an oil bath at 90°C for 16h under N2. Water is added and the solution is extracted with ethyl acetate. The organic layers are dried over anhydrous MgS04 and evaporated in vacuo. The resulting mixture is used without further purification.
1.2.2. Step 2: Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5- a Jpyridin-2-ylJ -amide

[00213] To a solution of cyclopropanecarboxylic acid [5-(4-hydroxymethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl] -amide (1.0 eq) in chloroform is slowly added phosphorus tribromide (1.0 eq.). The reaction mixture is stirred at room temperature for 20 h, quenched with ice and water (20 mL) and extracted with dichloromethane. The organic layer is dried over anhydrous MgSO i, filtered and concentrated to dryness. The resulting white residue is triturated in dichloromethane/diethyl ether 2:1 to afford the desired product.
1.2.3. Step 3:

[00214] Cyclopropanecarboxylic acid [5-(4-bromomethyl-phenyl)-[l,2,4]triazolo[l,5-a]pyridin-2-yl]-amide (l eq) and DIPEA (2 eq) are dissolved in DCM/MeOH (5: 1 v:v) under N2 and thiomorpho line 1,1-dioxide (1.1 eq) is added dropwise. The resulting solution is stirred at room temperature for 16h. After this time, the reaction is complete. The solvent is evaporated. The compound is dissolved in DCM, washed with water and dried over anhydrous MgSO i. Organic layers are filtered and evaporated. The final compound is isolated by column chromatography using EtOAc to afford the desired product.
…………………
PATENT
http://www.google.co.in/patents/WO2013189771A1?cl=en
Example 1. Synthesis of the compounds
1.1. Route 1
1.1.1. Synthesis of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (Intermediate 3)

led to 5 °C was added ethoxycarbonyl isothiocyanate (173.0 mL, 1.467 mol) dropwise over 15 min. The reaction mixture was then allowed to warm to room temp. (20 °C) and stirred for 16 h. Evaporation in vacuo gave a solid which was collected by filtration, thoroughly washed with petrol (3×600 mL) and air-dried to afford (2). The thiourea was used as such in the next step without any purification.
[00157] lH (400 MHz, CDC13) δ 12.03 (IH, br s, NH), 8.81 (IH, d, J 7.8 Hz, H-3), 8.15 (IH, br s, NH), 7.60 (IH, t, J 8.0 Hz, H-4), 7.32 (IH, dd, J 7.7 and 0.6 Hz, H-5), 4.31 (2H, q, J 7.1 Hz, CH2), 1.35 (3H, t, J 7.1 Hz, CH3).
1.1.1.2. 5-Bromo-f 1,2, 4]triazolo[ 1 ,5-a] pyridin-2-ylamine (3)
[00158] To a suspension of hydroxylamine hydrochloride (101.8 g, 1.465 mol) in EtOH/MeOH (1 : 1, 900 mL) was added NN-diisopropylethylamine (145.3 mL, 0.879 mol) and the mixture was stirred at room temp. (20 °C) for 1 h. l-(6-Bromo-pyridin-2-yl)-3-carboethoxy-thiourea (2) (89.0 g, 0.293 mol) was then added and the mixture slowly heated to reflux (Note: bleach scrubber was required to quench H2S evolved). After 3 h at reflux, the mixture was allowed to cool and filtered to collect the precipitated solid. Further product was collected by evaporation in vacuo of the filtrate, addition of H20 (250 mL) and filtration. The combined solids were washed successively with H20 (250 mL), EtOH/MeOH (1 : 1, 250 mL) and Et20 (250 mL) then dried in vacuo to afford the triazolopyridine derivative (3) as a solid. The compound was used as such in the next step without any purification.
[00159] lH (400 MHz, DMSO-i¼) δ 7.43-7.34 (2H, m, 2 x aromatic-H), 7.24 (1H, dd, J 6.8 and 1.8 Hz, aromatic-H), 6.30 (2H, br, NH2); m/z 213/215 (1 : 1, M+H+, 100%).
1.1.2. Synthesis of 4-[ 4-(4, 4, 5, 5-Tetramethyl-f 1, 3,2] ‘ dioxaborolan-2-yl) -benzyl] ‘- thiomor holine- 1, 1 -dioxide (Intermediate 4)

[00160] 2-(4-Bromomethyl-phenyl)-4,4,5,5-tetramethyl-[l,3,2]dioxaborolane (1 eq) and DIPEA (2 eq) were dissolved in DCM/MeOH (5:1 v:v) under N2 and thiomorpholine 1,1 -dioxide (2 eq) was added portion wise. The resulting solution was stirred at room temperature for 16h. After this time, the reaction was complete. The solvent was evaporated. The compound was extracted with EtOAc and water, washed with brine and dried over anhydrous MgSO i. Organic layers were filtered and evaporated. The final compound was isolated without further purification.
1.1.3. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- a ridin-2-ylamine (Formula I)

[00161] 4-[4-(4,4,5,5-Tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzyl]-thiomorpholine-l,l-dioxide (l .leq.) was added to a solution of 5-bromo-[l,2,4]triazolo[l,5-a]pyrid in-2-ylamine (4: 1). K2CO3 (2 eq.) and PdC^dppf (0.03 eq.) were added to the solution. The resulting mixture was then heated in an oil bath at 90°C for 16h under N2. Water was added and the solution was extracted with ethyl acetate. The organic layers were dried over anhydrous MgSC>4 and evaporated in vacuo. The final compound was obtained after purification by flash chromatography.
[00162] lH (400 MHz, CDC13) δ 7.94-7.92 (d, 2H), 7.52-7.48 (m, 3H), 7.37-7.34 (m, 1H), 7.02-7.00 (m, 1H), 6.00 (d, 2H), 3.76 (d, 2H), 3.15-3.13 (m, 4H), 2.93-2.91 (m, 4H).
[00163] m/z 358.2 (M+H+, 100%). 1.2. Route 2
1.2.1. Cyclopropanecarboxylic acid {5-[4-(l, l-dioxo-thiomorpholin-4-ylmethyl)-phenylJ- [l,2,4]triazolo[l,5-a]pyridin-2-yl}-amide (Formula II)
[00164] The compound according to Formula II may be synthesized according to the procedure described in WO 2010/149769.
1.2.2. Synthesis of 5-[4-(l, l-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[l,2,4]triazolo[l,5- aJpyridin-2-ylamine (Formula I)
[00165] The compound according to Formula I can also be produced by hydrolysis of the compound accor ing to Formula II:

[00166] Hydrochloric acid 30% aq (12.06 kg; 3.9 rel. volumes) was added to a slurry of the compound according to Formula II (3.45 kg; 1.0 equiv.) in demineralized water (10.0 kg; 3.0 rel. volumes). Subsequently, a line rinse was performed with demineralized water (3.4 kg; 1.0 rel. volumes). The reaction mixture was heated to 80±5°C for 14.5 h. After completion of the reaction (conversion > 99%>), the reaction mixture was cooled to 20±5°C. The reaction mixture was diluted with demineralized water (6.8 kg; 2.0 rel. volumes) and sodium hydroxide 33%> aq (9.52 kg; 3.7 rel volumes) was dosed at such a rate that the temperature of the reactor contents remained below 35°C. An additional amount of sodium hydroxide 33%> aq (2.55 kg; 1.0 rel. volumes) was needed to get the pH > 10. The product was filtered off, washed twice with demineralized water (1.5 rel. volumes) and dried under vacuum for 1 h, thus yielding the crude compound according to Formula I.
[00167] The crude compound according to Formula I (5.70 kg) was re-slurried in demineralized water (23.0 kg; 8.5 rel. volumes). Hydrochloric acid 30%> aq (1.65 kg; 0.7 rel. volumes) and demineralized water (4.3 kg; 1.6 rel. volumes) were added and the reaction mixture was stirred at 20±5°C for 45 min. As the compound according to Formula I was not dissolved completely, the reaction mixture was stirred at 45±5°C for 1 h. The reaction mixture was filtered and the residue was washed with demineralized water (2.0 kg 0.75 rel. volumes). Sodium hydroxide 33%> aq (1.12 kg; 0.6 rel volumes) was added to the filtrate. An additional amount of sodium hydroxide 33%> aq (1.01 kg) was needed to get the pH > 10. The resulting reaction mixture was stirred at 20±5°C for about 3 h. The product was filtered off, washed twice with demineralized water (4.1 kg; 1.5 rel. volumes), and twice with methyl tert-butyl ether (MTBE; 3.0 kg; 1.5 rel. volumes) and dried under vacuum for 15.5 h on the filter. The product was further dried in a vacuum oven at 40±5°C for 202 h, thus affording the desired compound according to Formula I.
Update
Scheme 1: General S nthesis of Compounds of Formula I or A

Formula A
Scheme 7.

(16) (17) (18)
(18a): R3a=R3b=R2a=R (18b): R3a=R3b=D; R2a 18c): R3a=R3b=H; R2a

References
- Namour, Florence; Diderichsen, Paul Matthias; Cox, Eugène; Vayssière, Béatrice; Van der Aa, Annegret; Tasset, Chantal; Van’t Klooster, Gerben (2015-02-14). “Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection”. Clin Pharmacokinet. Epub ahead of print.doi:10.1007/s40262-015-0240-z.
- Van Rompaey, L; Galien, R; Van der Aar, E; Clement-Lacroix, P; Van der Aar, E; Nelles, L; Smets, B; Lepescheux, L; Cristophe, T; Conrath, K; Vandeghinste, N; Vayssiere, B; De Vos, S; Fletcher, S; Brys, R; Van’t Klooster, G; Feyen, J; Menet, C (2013-10-01). “Preclinical characterization of GLPG0634, a selective inhibitor of JAK1 for the treatment of inflammatory diseases”. J Immunol. 191(7). doi:10.4049/jimmunol.1201348.
- http://acrabstracts.org/abstracts/phase-1-and-phase-2-data-confirm-that-glpg0634-a-selective-jak1-inhibitor-has-a-low-potential-for-drug-drug-interactions/
- “Galapagos’ GLPG0634 shows excellent efficacy and safety in rheumatoid arthritis Phase II study” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos reports that the last patient in DARWIN 1 has completed 12 weeks of treatment” (PDF) (Press release). Retrieved 2015-02-26.
- “Galapagos completes recruitment for Darwin 1 study with GLPG0634 (filgotinib) in RA”. EuroInvestor. Retrieved 2015-02-26.
- NASDAQ OMX Corporate Solutions. “Galapagos completes recruitment for Darwin 2 monotherapy study with GLPG0634 (filgotinib) in RA”. Yahoo Finance. Retrieved 2015-02-26.
| US8551980 | Nov 17, 2010 | Oct 8, 2013 | Bayer Intellectual Property Gmbh | Substituted triazolopyridines |
| US8796457 | Jun 25, 2010 | Aug 5, 2014 | Galapagos Nv | Compound useful for the treatment of degenerative and inflammatory diseases |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-yl]cyclopropanecarboxamide
|
|
| Clinical data | |
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Biological half-life | 6 hours[1] |
| Identifiers | |
| CAS Registry Number | 1206161-97-8 |
| ATC code | L01XE18 |
| IUPHAR/BPS | 7913 |
| ChemSpider | 28189566 |
| UNII | 3XVL385Q0M |
| ChEMBL | CHEMBL3301607 |
| Chemical data | |
| Formula | C21H23N5O3S |
| Molecular mass | 425.50402 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter

 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
/////////Galapagos, GLPG0634, Filgotinib, PHASE 2, orphan drug designation, PHASE 3, Crohn’s disease, Rheumatoid arthritis, Ulceraticolitis
ve SMILES code: O=C(C1CC1)NC2=NN3C(C4=CC=C(CN5CCS(CC5)(=O)=O)C=C4)=CC=CC3=N2
Sacubitril

Sacubitril, AHU 377
NEPRILYSIN INHIBITOR
FOR HEART FAILURE
CAS 149709-62-6
CAS SODIUM SALT 149690-05-1
(2R,4S)-5-(biphenyl-4-yl)-4-((3-carboxypropionyl)amino)-2-methylpentanoic acid ethyl ester
5-(Biphenyl-4-yl)-4(S)-(3-carboxypropionamido)-2(R)-methylbutyric acid ethyl ester
N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methyl butanoic acid ethyl ester
[1,1′-Biphenyl]-4-pentanoic acid, γ-[(3-carboxy-1-oxopropyl)amino]-α-methyl-, α-ethyl ester, (αR,γS)-
- [1,1′-Biphenyl]-4-pentanoic acid, γ-[(3-carboxy-1-oxopropyl)amino]-α-methyl-, ethyl ester, [S-(R*,S*)]-
- (2R,4S)-4-[(3-Carboxy-1-oxopropyl)amino]-4-[(p-phenylphenyl)methyl]-2-methylbutanoic acid ethyl ester
- (2R,4S)-5-(Biphenyl-4-yl)-4-[(3-carboxypropionyl)amino]-2-methylpentanoic acid ethyl ester
-
Formula C24H29NO5 MW 411.49 g/mol
AHU377; AHU-377; Sacubitril; 149709-62-6; UNII-17ERJ0MKGI; Alpha-ethyl (alphaR,gammaS)-gamma-<(3-carboxy-1-oxopropyl)amino>-alpha-methyl<1,1′-biphenyl>-4-pentanoate
Sacubitril sodium
149690-05-1
Sacubitril is an antihypertensive drug used in combination with valsartan. The combination drug, valsartan/sacubitril, known during trials as LCZ696 and marketed under the brand name, Entresto, is a treatment for heart failure.[1] It was approved under the FDA’spriority review process for use in heart failure on July 7, 2015.


Mechanism of action
Sacubitril is a prodrug that is activated to LBQ657 by de-ethylation via esterases.[2] LBQ657 inhibits the enzyme neprilysin,[3] which is responsible for the degradation of atrial and brain natriuretic peptide, two blood pressure lowering peptides that work mainly by reducing blood volume.[4]


SYNTHESIS
CLICK ON IMAGE FOR CLEAR VIEW
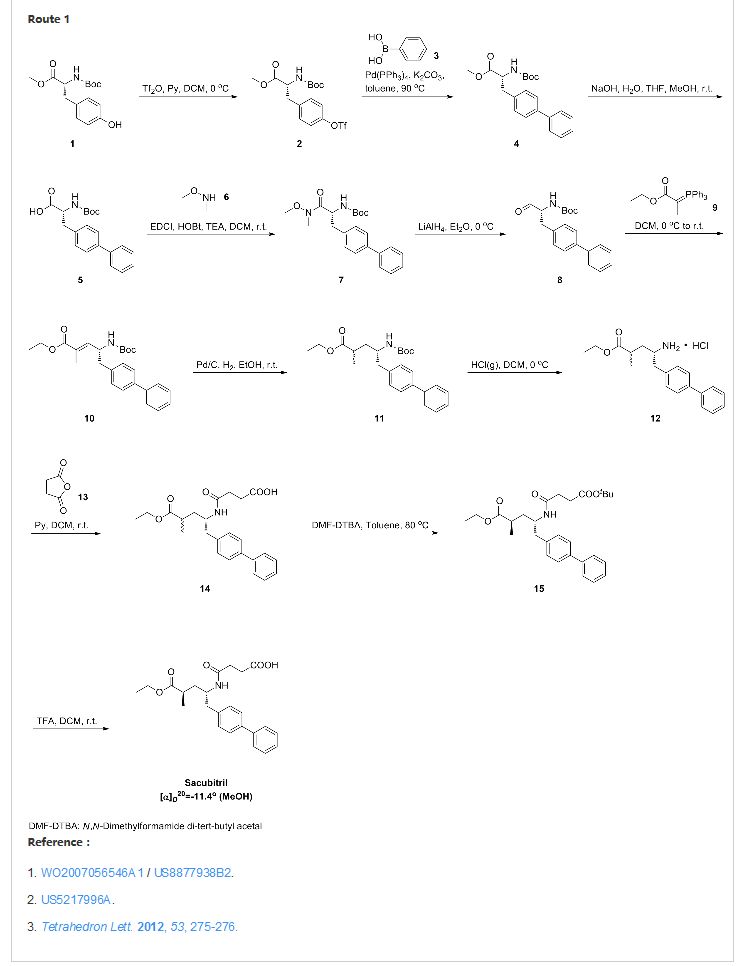
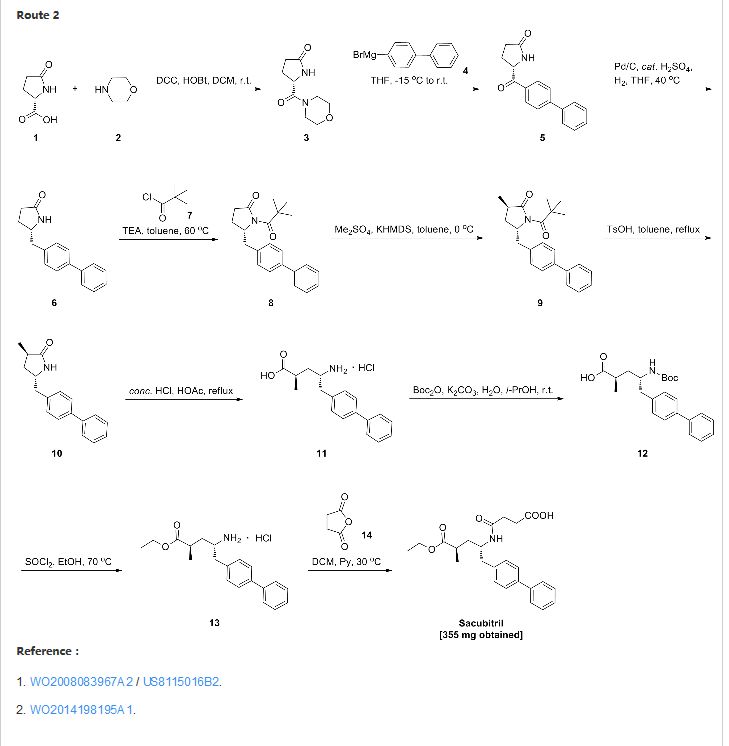
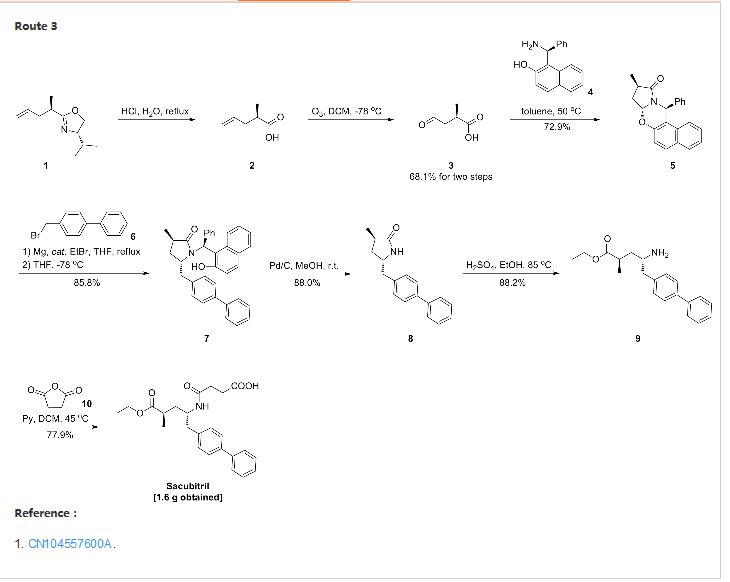
SYNTHESIS

WO-2008031567
http://www.google.com/patents/WO2008031567A1?cl=en
the following steps:


and optionally the following additional steps:


Example 1 :
(E)-(R)-5-biphenyl-4-yl-4-fert-butoxycarbonylamino-2-methylpent-2-enoic acid

(E)-(R)-5-Biphenyl-4-yl-4-tert-butoxycarbonylamino-2-methylpent-2-enoic acid ethyl ester (CAS# 149709-59-1) is hydrolysed using lithium hydroxide in ethanol to yield (£)-(f?)-5-biphenyl-4-yl-4-te/t-butoxycarbonylamino-2-methylpent-2-enoic acid as a white solid. δH (400 MHz; DMSO) 1.31 (9H1 s, (CH3J3), 1.59 (3H, s, 1- CH3), 2.68 (1H, dd, J 6.8, 13.2, 5-HA), 2.86 (1H, m, 5-HB), 4.44 (1H, m, 4-H), 6.51 (1H1 d, J 9.2, 3-H), 7.16 (1H, d, J 8.0, NH), 7.26 (2H, d, J 8.0, Ar-ortho- H(Ph)), 7.31 (1H, t, J 7.6, Ar-(Ph)-para-H), 7.40 (2H, t, J 8.0, Ar-(Ph)-metø-H), 7.54 (2H, d, J 8.0, Ar-mefa-H(Ph), 7.60 (2H, d, J 7.6, Ar-(Ph)-ort/vo-H), 12.26 (1H, s, CO2H); m/z (+ESI) 404 ([MNa]+, 17%), 382 ([MHf, 2), 326 (10), 264 (100), 167 (13).
Example 2:
(2/?,4S)-5-biphenyl-4-yl-4-fert-butoxycarbonylamino-2-methylpentanoic acid in crystalline form [2(i-a)]

2(i-a) To a suspension of (E)-(f?)-5-biphenyl-4-yl-4-te/t-butoxycarbonylamino-2- methylpent-2-enoic acid [2(ii-a)] (200 g, 524.3 mmol) in degassed ethanol (900 ml) at 40 °C a solution of diiodo(p-cymene)ruthenium(ll) dimer (0.052 g, 0.0524 mmol) and (αf?,αf?)-2,2>-bis(α-Λ/,Λ/-dimethylaminophenylmethyl)-(S,S)- 1 ,1′-bis[di(3,5-dimethyl-4-methoxyphenyl)phosphine]ferrocene (= Mandyphos SL-M004-1) (0.116 g, 0.110 mmol) is added in degassed ethanol (100 ml). The solution is degassed using vacuum and a pressure of 20 bar hydrogen applied. The mixture is stirred at 40 0C for 6 h. Vessel is then purged with nitrogen. Ethanol (700 ml) is removed by distillation, lsopropyl acetate (600 ml) is added. Solvent (600 ml) is removed by distillation, lsopropyl acetate (600 ml) is added. Solvent (600 ml) is removed by distillation, lsopropyl acetate (300 ml) is added and the solution is heated to reflux. Heptane fraction (1200 ml) is added and the mixture is cooled to room temperature. The solid is collected by filtration and washed with heptane fraction-isopropyl acetate 2 : 1 mixture (360 ml). The solid is dried overnight at 50 0C under 1-50 mbar vacuum to afford the title compound as a white/off-white solid [Ratio 2(i-a) : 2(i-b) 99 : 1, as determined by HPLC analysis]. Mpt 146-147 0C; δH (500 MHz; DMSO) 1.07 (3H1 d, J 7.0, 1-CH3), 1.34 (9H, s, (CH3)3), 1-38 (1H, m, 3-HA), 1.77 (1H, m, 3-HB), 2.43 (1H, m, 2-H), 2.70 (2H, d, J 7.0, 5-H)1 3.69 (1 H, m, 4-H), 6.74 (1 H, d, J 9.0, NH)1 7.27 (2H, d, J 8.0, Ar-ortA;o-H(Ph)), 7.36 (1H, t, J 7.0, Ar-(Ph)-para-H), 7.46 (2H, t, J 7.5, Ar-(Ph)- meta-H), 7.57 (2H, d, J 8.0, Ar-mefa-H(Ph), 7.64 (2H, d, J 7.5, Ar-(Ph)-orfΛo-H), 12.01 (1H, s, CO2H); δc (500 MHz, DMSO) 18.1 (1-CH3), 28.3 [(CH3)3], 35.9 (2- C), 37.9 (3-C), 40.7 (5-C), 50.0 (4-C), 77.4 [(C(CH3)3], 126.3, 126.5, 127.2, 128.9, 129.8 (Ar-CH), 137.7 (Ar-/pso-C(Ph)), 138.3 (Ar-para-C(Ph)), 140.1 (Ar- (Ph)-/pso-C), 155.2 (NCO), 177.2 (CO2H); mlz (+ESI) 406 ([MNa]+, 6%), 384 ([MH]+, 31 ), 328 (100), 284 (19); Found: [MH]+, 384.21691. C23H30NO4 requires MH 384.21693
PATENT
http://www.google.com/patents/EP0555175A1
Example 1….THE SODIUM SALT
To a solution of N-(3-carbo(t)butoxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methylbutanoic acid ethyl ester (0.80 g) in 15 ml of CH2CI2 at room temperature are added 3 ml of trifluoroacetic acid. The mixture is stirred overnight and concentrated. The residue is dissolved in tetrahydrofuran (THF), and 6.5 ml of 1 N NaOH is added. The mixture is concentrated and triturated with ether. The solid can be recrystallized from methylene chloride-hexane to give sodium N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2R-methyl butanoic acid ethyl ester melting at 159-160°C; [a]D20 = – 11.4° (methanol).
-
 SODIUM SALT
SODIUM SALT

The starting material is prepared as follows:
A solution of a-t-BOC-(R)-tyrosine methyl ester (5.9 g, 20 mmol) and pyridine (8 mL, 100 mmol) in methylene chloride (30 mL) is cooled to 0-5°C. Trifluoromethanesulfonic anhydride (4 mL, 23 mmol) is added at 0-5°C, and the resulting mixture is held for another 30 minutes. The reaction mixture is diluted with water (60 mL) and methylene chloride (100 mL), and washed sequentially with 0.5 N sodium hydroxide solution (1 x 50 mL), water (1 x 60 mL), 10% citric acid solution (2 x 75 mL) and water (1 x 60 mL). The organic phase is dried over MgS04 and concentrated to an oil. The oil is purified by column chromatography (silica gel, hexane/ethyl acetate, 2:1 to give methyl(R)-2-(t-butoxycarbonylamino)-3-[4-(trifluoromethylsulfonyloxy)phenyl]-propionate which crystallizes on standing; m.p. 46-48°C; [a]20 D-36.010 (c=1, CHCI3).
Nitrogen is passed through a suspension of (R)-2-(t-butoxycarbonylamino)-3-[4-(trifluoromethylsulfonyloxy)-phenyl]-propionate (1.75mmol), phenylboronic acid (3.5 mmol), anhydrous potassium carbonate (2.63 mmol) and toluene (17 mL) for 15 minutes. Tetrakis(triphenyiphosphine)paiiadium(0) is added, and the mixture is heated at 85-90° for 3 hours. The reaction mixture is cooled to 25°C, diluted with ethyl acetate (17 mL) and washed sequentially with saturated sodium bicarbonate (1 x 20 mL), water (1 x 20 mL), 10% citric acid (1 x 20 mL), water (1 x 20 mL) and saturated sodium chloride solution (1 x 20 mL). The organic phase is concentrated, and the residue is purified by column chromatography (silica gel, hexane/ethyl acetate 2:1) to yield methyl (R)-2-(t-butoxycarbonylamino)-3-(p-phenylphenyl)-propionate which can also be called N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine methyl ester.
To a solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine methyl ester (6.8 g) in 60 ml of THF and 20 ml of methanol are added 20 ml of aqueous 1 N sodium hydroxide solution. The mixture is stirred for 1 h at room temperature and then acidified with 21 ml of 1 N hydrochloric acid. The aqueous solution is extracted 3x with ethyl acetate. The combined organic extracts are dried (MgS04), filtered and concentrated to give N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine, m.p. 98-99°C; [a]2°D -18.59° (c=1, methanol).
To a solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine (4.8 g) in 70 ml of methylene chloride (CH2CI2) at 0°C with 1.65 g of N,O-dimethylhydroxylamine HCI, 1.7 g of triethylamine and 2.85 g of hydroxybenzotriazole are added 5.37 g of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride. The mixture is stirred 17 h at room temperature. The mixture is concentrated taken up in ethyl acetate (EtOAc) and washed with saturated sodium bicarbonate, 1N HCI and brine, then dried (MgS04), filtered and concentrated to give N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine N,O-dimethyl hydroxylamine amide.
To a 0°C solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine N,O-dimethyl hydroxylamine amide (5.2 g) in 250 ml of diethyl ether are added 0.64 g of lithium aluminum hydride. The reaction is stirred for 30 min. and quenched with aqueous potassium hydrogen sulfate. The mixture is stirred for additional 5 min., poured onto 1N HCI, extracted (3x) with EtOAc, dried (MgS04), filtered, and concentrated to give N-(R)-4-t-butoxycarbonyl-(p-phenylphenyl)-alanine carboxaldehyde as a colorless oil.
To a 0°C solution of N-(R)-t-butoxycarbonyl-(p-phenylphenyl)-alanine carboxaldehyde (4.4 g) in 200 ml of CH2CI2are added 10 g of carboethoxyethylidene phenyl phosphorane. The mixture is warmed to room temperature, stirred for 1 h, washed with brine, dried (MgS04), filtered and concentrated. The residue is chromatographed on silica gel eluting with (1:2) ether:hexane to give N-t-butoxycarbonyl-(4R)-(p-phenylphenylme- thyl)-4-amino-2-methyl-2-butenoic acid ethyl ester.
A solution of N-t-butoxycarbonyl-(4R)-(p-phenylphenylmethyl)-4-amino-2-methyl-2-butenoic acid ethyl ester (4.2 g) in 400 ml of ethanol is suspended with 2.0 g of 5% palladium on charcoal and then is hydrogenated at 50 psi for 6h. The catalyst is removed by filtration and the filtrate is concentrated to give N-t-butoxycarbonyl(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester as a 80:20 mixture of diastereomers.
To the N-t-butoxycarbonyl(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester (4.2 g) in 40 ml of CH2CI2 at 0°C is bubbled dry hydrogen chloride gas for 15 min. The mixture is stirred 2 h and concentrated to give (4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester hydrochloride as a 80:20 mixture of diastereomers.
To a room temperature solution of the above amine salt (3.12 g) in 15 ml of CH2CI2 and 15 ml of pyridine are added 13.5 g of succinic anhydride. The mixture is stirred for 17 h, concentrated, dissolved in ethyl acetate, washed with 1N HCI and brine, and dried (MgS04) to give N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenyl- methyl)-4-amino-2-methylbutanoic acid ethyl ester as a 80:20 mixture of diastereomers.
The above N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-2-methylbutanoic acid ethyl ester diastereomeric mixture (3.9 g) and N,N-dimethylformamide-di-t-butyl acetal (8.8 ml) are heated at 80°C in 40 ml of toluene for 2 h. The mixture is poured onto ice- 1N HCI, extracted with ether, chromatographed on silica gel eluting with (2:1) toluene:ethyl acetate to give N-(3-carbo(t)butoxy-1-oxopropyl)-(4S)-(p-phenylphe- nylmethyl)-4-amino-2R-methylbutanoic acid ethyl ester as the more polar material and the corresponding (S,S) diastereomer as the less polar material.
Example 2………THE ACID
To a solution of N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-(2R)-methylbutanoic acid ethyl ester (0.33 g) in 20 ml of (1:1) ethanol:tetrahydrofuran (THF) at room temperature are added 5 ml of 1 N sodium hydroxide solution (NaOH) and stirred for 17 h. The mixture is concentrated, dissolved in water and washed with ether. The aqueous layer is acidified with 1 N hydrochloric acid (HCI), extracted 3x with ethyl acetate (EtOAc), dried over magnesium sulfate (MgS04), filtered and concentrated. The residue is triturated with ether to yield N-(3-carboxy-1-oxopropyl)-(4S)-(p-phenylphenylmethyl)-4-amino-(2R)-methylbutanoic acid melting at 158-164°C, [α]D 20= -23.5° (methanol).
PATENT
CN 104557600
http://www.google.com/patents/CN104557600A?cl=zh

United States Patent US5217996 and international patent W02008031567, W02010136474 and W02012025501 reported a synthetic route follows to the chiral amino alcohols as raw materials, oxidized to the aldehyde, Victoria ladder tin reaction, chiral hydrogenation and amidation condensation reaction to obtain the objective product.

In addition, the international patent TO2008083967, TO2011088797, TO2012025502 and TO2014198195 reported that another type of preparation. The route through the 2-oxo-proline as raw material, carboxyl activating biphenyl substituted carbonyl reduction, chiral methylation, ring-opening reaction and amide condensation reaction to obtain the objective product.


Example Eight:
in the reaction flask was added (2R, 4S) -2- methyl-4-amino -5- (l, P- biphenyl-4-yl) – pentanoic acid ethyl ester (VII) (1.55g, 5mmol ), Jie of pyridine (1.2g, 15mmol) and dichloromethane burning 25mL, stirring to dissolve, butyric anhydride (1.0g, 10mmol), was heated to 4〇-45 ° C, the reaction was stirred for 6 hours. Fill Gaudin anhydride (0. 5g, 5mmol), the reaction was continued for 4 hours and the end of the reaction by TLC. Concentrated under reduced pressure, the residue was recrystallized from ethyl acetate and n-hexane to give an off-white solid sand sacubitril Kubica song (I) L 6g, a yield of 77.9%;
1H NMR (CDCl3) S 7.51 (d, 2H), 7.46 ( d, 2H), 7.36 (m, 2H), 7. 27 (m, 1H), 7. 17 (d, 2H), 5. 72 (d, 1H), 4. 19 (brs, 1H), 4. 06 (q, 2H), 2. 87-2. 72 (m, 2H), 2. 62-2. 54 (m, 2H), 2. 49 (brs, 1H), 2. 43-2. 33 ( m, 2H), I 88 (m 1H), I 54-1 43 (m, 1H), I. 18 (t, 3H), l 10 (d, 3H);…..
FAB-MSm / z : 412 [M + H] +.
PATENT
http://www.google.com/patents/WO2014198195A1?cl=en
Example 7
(2 Standby

Acetyl chloride (1 mL) 0 ° C was added ethanol (10 mL), and at room temperature for 0.5 hours, the compound (3R, 5S) -5- biphenyl-4-methyl-1- (2,2- methyl-propionyl) -3-methyl pyrrolidone (520 mg, 1.49 mmol), the reaction was refluxed for 3 days. After cooling to room temperature, and concentrated. The reaction mixture was dissolved in 8 mL of dichloromethane and pyridine 1: 1 mixed solution, and then butyryl anhydride (223 mg, 2.23 mmol). 30 ° C overnight. LC-MS detection, a small amount of starting material remaining, fill Gading anhydride (75 mg, 0.74 mmol), continue to reflect four hours. Concentrated and reverse phase column chromatography to give a white foam solid (2R, 4S) -5- biphenyl-4-yl-4- (3-carboxy – propionylamino) -2-methyl – acetic acid ester a (355 mg, 58%) and white solid (2R, 4S) -5- biphenyl-4-yl-4- (3-carboxy – propionylamino) -2-methyl – pentanoic acid b ( 13 mg, 2.3%).
a: 1H MR (400 MHz, CDCl 3 ) [delta] 7.51 (d, = 7.8 Hz, 2H), 7.46 (d, = 7.8 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.2 Hz, IH), 7.17 (d, J = 7.9 Hz, 2H), 5.72 (d, J = 8.1 Hz, IH), 4.19 (brs, IH), 4.06 (q, J = 7.0 Hz, 2H) , 2.87-2.72 (m, 2H), 2.62-2.54 (m, 2H), 2.49 (brs, IH), 2.43-2.33 (m, 2H), 1.88 (ddd, = 13.2, 9.5, 3.9 Hz, IH), 1.54-1.43 (m, IH), 1.18 (t, = 7.0 Hz, 3H), 1.10 (d, = 7.2 Hz, 3H).
LC-MS: t R = 3.43 min; [M + H] +: 412.0.
……………………
Paper
JOURNAL OF MEDICINAL CHEMISTRY, vol. 38, no. 10, 1995, pages 1689-1700,
http://pubs.acs.org/doi/pdf/10.1021/jm00010a014
NOTE———–DIACID
(aR,yS)-y-[ (3-Carbo-1-oxopropyl)aminol-a-methyl- [l,l’-biphenyllpentanoic Acid (21a).
To the sodium salt of 19a (0.73 g, 1.68 mM) in 20 mL of THF:EtOH was added 1 N NaOH (5.0 mL, 5.0 “01). The reaction mixture was stirred overnight and then washed with ether. The aqueous layer was acidified with 1 N HCI, re-extracted with EtOAc (3 x 10 mL), dried (MgSO& and evaporated to dryness. The solid was recrystallized from ethanol to yield 435 mg of 21a DIACID OF SACUBITRIL
melting at 165-167 “C:
[a] D25~ -28.73 (c = 10.1 in MeOH);
‘H NMR, DMSOD6
PPM 12.0, (s, 2H), 7.75 (d, 1H), 7.62 (d, 2H), 7.55 (d, 2H), 7.45 (t, 2H), 7.32 (t, lH), 7.25 (d, 2H), 4.92 (m, lH), 2.70 (d, 2H), 2.35 (t, 3H), 2.25 (m, 2H), 1.75 (m, lH), 1.32 (m, lH), 1.03 (d, 3H).
Anal. (C22H25N05) C,H,N
Note diacid is sacubitrilat (LBQ657)
NMR PREDICT
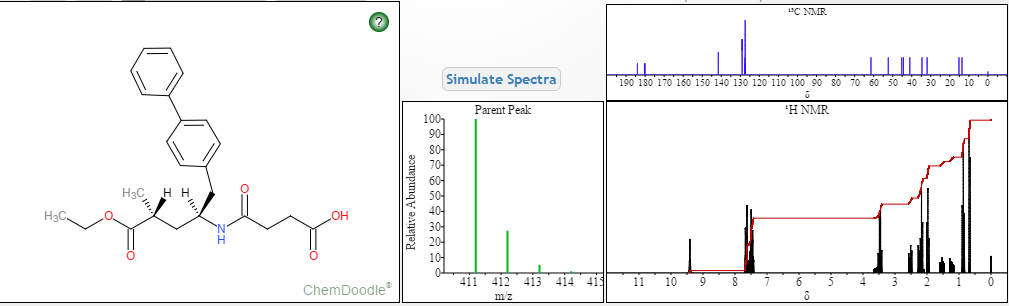
1H NMR PREDICT
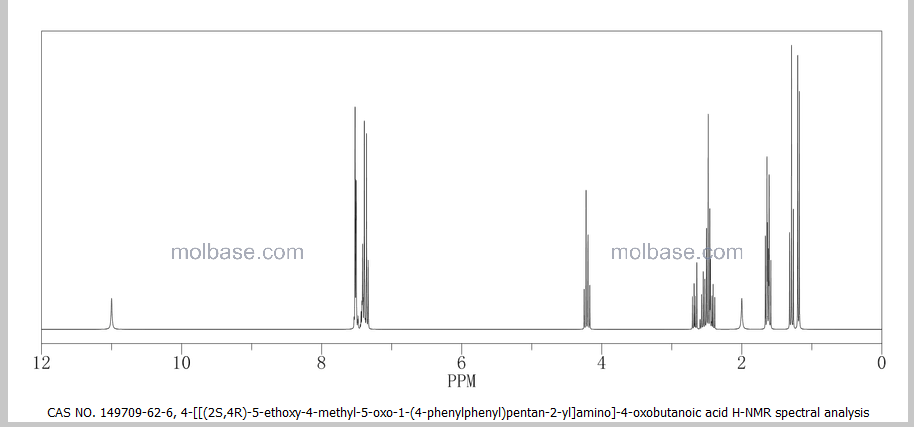
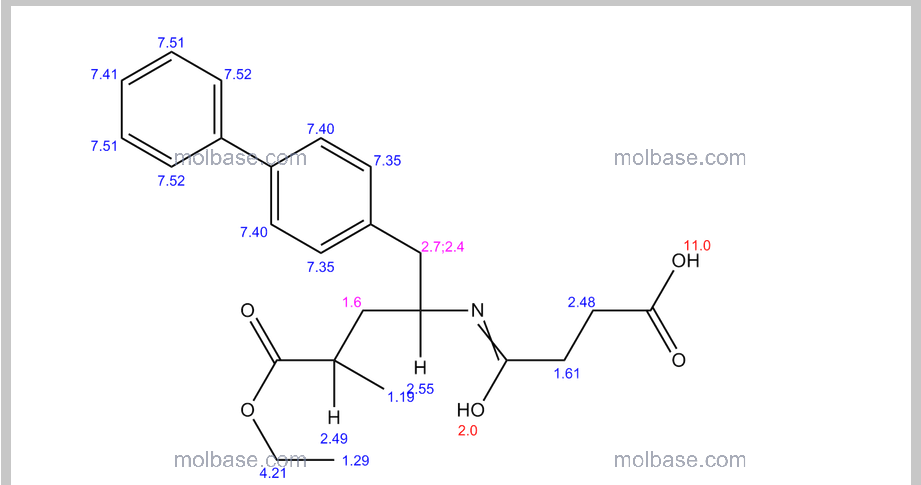
13C NMR PREDICT
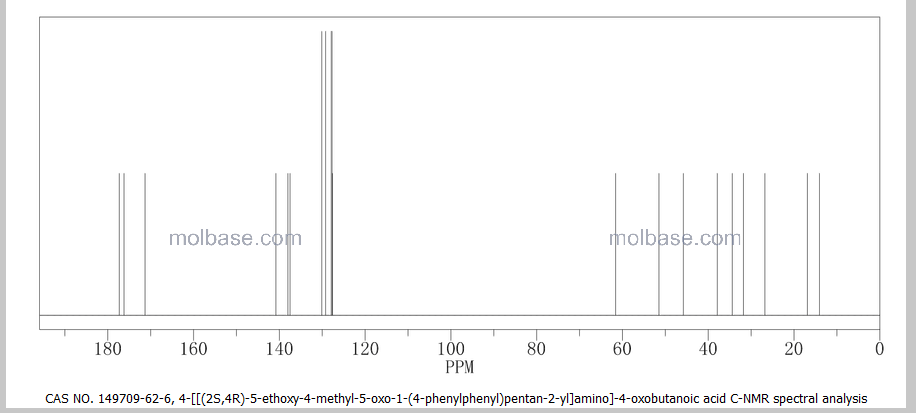
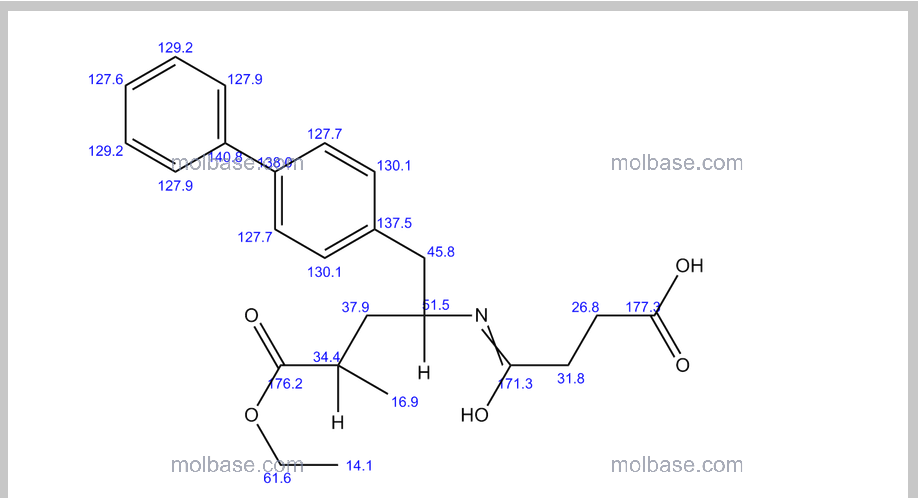
COSY PREDICT
…………….

NMR…..http://www.chemietek.com/Files/Line3/CHEMIETEK,%20AHU-377%20,%20Lot%2001,%20NMR-MeOD,%201.1.pdf
- Mol. Formula:C24H29NO5 ∙ C4H11NO3
- MW:532.6
- HPLC………http://www.chemietek.com/Files/Line2/CHEMIETEK,%20AHU-377%20,%20Lot%2001,%20HPLC.pdf
update………
PATENT
WO2016180275
Heart failure is a very high mortality syndrome, for patients with heart failure, so far no drug can significantly improve mortality and morbidity, and thus a new type of therapy is necessary. AHU-377 (CAS No. 149709-62-6) is an enkephalinase inhibitor, which is a prodrug ester groups can be lost through hydrolysis, converted to pharmaceutically active LBQ657, inhibit endorphin enzyme (NEP) the role of the main biological effects of NEP is to natriuretic peptides, bradykinin and other vasoactive peptide degradation failure. AHU-377 and angiotensin valsartan composition according to the molar ratio of 1 LCZ696. LCZ696 is an angiotensin receptor enkephalinase inhibitors, which can lower blood pressure, treat heart failure may become a new drug. Clinical data show, LCZ696 is more effective for the treatment of hypertension than valsartan alone.
Patents US 5,217,996 and US 5,354,892 reported the first synthesis of AHU-377, the synthetic route is as follows:
Reaction with unnatural D-tyrosine derivative as a substrate, more expensive, while the second step in the synthesis is necessary to use Pd-catalyzed Suzuki coupling reaction, whereby preparative route costs than the AHU-377 high.
Patent US 8,115,016 above routes also reported the departure from the pyroglutamate, through multi-step process for preparing a reaction AHU-377, which is more difficult methylation reaction, and the yield is not high. Patent US 8,580,974 also reported a carbonyl group of the a- introducing N, N- dimethyl enamine is converted to methyl, however, there are some problems in the route for constructing methyl chiral centers, are not suitable for scale-up synthesis route as follows:
About the latest AHU377 synthesis intermediates, Patent WO2014032627A1 reported using a Grignard reagent to react with epichlorohydrin, a quicker been important intermediates, synthetic route Compound AHU377 synthesized as follows:
However, the second step of the synthetic route use succinimide nitrogen atoms introduced by Mitsunobu reaction with hydrochloric acid hydrolysis to remove, then converted to Boc protected at the end of the synthesis process AHU377 Boc will have to take off protection, then any connection with succinic anhydride reaction product introduced into the structure of succinic acid portion, so that this method of atom economy and the economy of the steps are low.
Example 1
Synthesis of Compound 2
In inert atmosphere, a solution of three 500mL flask was added compound 1 (10g, 1eq), dissolved after 90mL THF, was added CuI (4.814g, 0.1eq), the system moves to the low temperature in the cooling bath to -20 ℃ when, biphenyl magnesium bromide dropwise addition, the internal temperature was controlled not higher than -10 ℃. Bi closed refrigeration drop, return to room temperature overnight. Completion of the reaction, the reaction solution was poured into saturated the NH 4 of Cl (10vol, 100 mL) was stirred at room temperature for 0.5h. Suction filtered, the filter cake was rinsed with a small amount of EA, and the filtrate was transferred to a separatory funnel carved, and the aqueous phase was extracted with EA (10vol × 2,100mL × 2) and the combined organic phases with saturated NaHC [theta] 3 , the NH 4 of Cl, each Brine 150mL (15vol) washed once, dried over anhydrous over MgSO 4 dried, suction filtered, and concentrated to give a white solid. Product obtained was purified by column 15.2g, yield 78%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.52 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.38-7.25 (m, 8H), 4.62-4.47 ( m, 2H), 4.09 (dd, J = 6.7,3.5Hz, 1H), 3.54 (dd, J = 9.5,3.5Hz, 1H), 3.43 (dd, J = 9.4 , 6.9Hz, 1H), 2.84 ( d, J = 6.6Hz, 2H), 2.38 (s, 1H).
Example 2
Synthesis of Compound 3
In an inert gas, at room temperature was added to the flask 500mL three Ph3P (18.54g, 2eq), 240mL DCM dissolution, butyryl diimide (of 6.44 g), compound 2 (15g), an ice-water bath cooling to 0 ℃ or so, was added dropwise DIAD (14mL) was complete, the reaction go to room temperature.Starting material the reaction was complete, the system was added to water (100 mL) quenched the reaction was stirred for 10min; liquid separation, the aqueous phase was extracted with DCM (100mL × 2), the combined organic phases with saturated Brine 100mL × 2), dried over anhydrous over MgSO 4 dried , filtration, spin dry to give a white solid; product was purified by column 15.4g, yield 82%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.56 (D, J = 7.4Hz, 2H), 7.49 (D, J = 8.0Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.37-7.30 (m, 3H), 7.27 ( d, J = 6.7Hz, 3H), 7.22 (d, J = 8.0Hz, 2H), 4.75 (s, 1H), 4.56 (d, J = 12.0Hz, 1H), 4.45 (d, J = 12.0Hz, 1H ), 4.06 (t, J = 9.6Hz, 1H), 3.70 (dd, J = 10.0,5.2Hz, 1H), 3.23 (dd, J = 13.8,10.3Hz, 1H) , 3.14-3.00 (m, 1H), 2.48 (d, J = 4.0Hz.4H).
Example 3
Synthesis of Compound 4
Protection of inert gas, at room temperature was added to the flask 1L three compound 3 (18.81g), 470mL EtOH was dissolved, was added Pd / C, replaced the H 2 three times, move heated on an oil bath at 60 ℃ reaction. Raw reaction was complete, the system was removed from the oil bath, the reaction solution was suction filtered through Celite and concentrated to give the crude product. It was purified by column pure 11.8g, a yield of 81.2%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.8Hz, 2H), 7.51 (D, J = 7.8Hz, 2H), 7.42 (T, J = 7.5Hz, 2H), 7.33 (T , J = 7.2Hz, 1H), 7.26 (d, J = 7.2Hz, 2H), 4.55 (d, J = 5.2Hz, 1H), 4.06-3.97 (m, 1H), 3.86 (dd, J = 12.0, 3.1Hz, 1H), 3.16 (dd , J = 8.1,2.9Hz, 2H), 2.58 (t, J = 7.0Hz, 4H), 1.26 (s, 2H).
Example 4
Synthesis of Compound 7
Protection of inert gas, at room temperature to a 25mL flask was added three Dess-Martin oxidant (767.7mg), 10mL DCM was dissolved, the system was cooled down to -10 deg.] C, was added 4 (500mg). Starting material the reaction was complete, to the system was added saturated NaHCO3 and Na2S2O3 each 5mL, quench the reaction stirred for 10min; aqueous phase was extracted with DCM (10mL × 3) and the combined organic phases with saturated NaHCO3, Brine 30mL each wash, dried over anhydrous MgSO4, filtration, spin dried to give the crude product used directly in the next reaction cast.
Example 5
Synthesis of Compound 8
Inert gas, at room temperature for three to 500mL flask 7 (497.5mg), 10mL DCM to dissolve an ice water bath to cool, added phosphorus ylide reagent (880.6mg), the system was removed from the ice water bath at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. Liquid separation, the aqueous phase was extracted with DCM (10mL × 2), organic phases were combined, washed with saturated Brine 20mL × 2, dried over anhydrous MgSO4, filtration, spin crude done. Product obtained was purified by column 563mg, 90% yield.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.60-7.53 (m, 2H), 7.51 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.33 (D, J = 7.3Hz, 1H), 7.23 (d , J = 8.1Hz, 2H), 7.13 (dd, J = 9.2,1.5Hz, 1H), 5.26 (td, J = 9.5,6.9Hz, 1H), 4.25-4.05 ( m, 2H), 3.40 (dd , J = 13.7,9.7Hz, 1H), 3.13 (dd, J = 13.8,6.7Hz, 1H), 2.53 (d, J = 2.2Hz, 4H), 1.85 (d, J = 1.4Hz, 3H), 1.30 ( t, J = 7.1Hz, 3H).
Example 6
Synthesis of Compound 9
Protection of inert gas, at room temperature to a 50mL flask was added three 8 (365mg, 1eq), 9mL of ethanol and stirred to dissolve, the system was replaced with hydrogen three times, was added Pd / C (25% w / w) at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. The reaction mixture was suction filtered through Celite and concentrated to give the crude product. Product was purified by column, yield 80.2%, purity 97.2%.
Example 7
Synthesis of Compound 10
Equipped with Compound 9 (100mg) acetic acid A reaction flask (9mL), hydrochloric acid (1mL). The reaction was heated oil bath at 80 deg.] C. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. After saturated NaHCO3 and extracted with EA and concentrated to give crude product. Product obtained was purified by column 90mg, yield 84%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.54 (m, 2H), 7.53-7.48 (m, 2H), 7.41 (dd, J = 10.5,4.9Hz, 2H), 7.31 (dd, J = 8.3 , 6.4Hz, 1H), 7.22 ( d, J = 8.2Hz, 2H), 5.93 (t, J = 9.7Hz, 1H), 4.34-4.00 (m, 3H), 2.91-2.71 (m, 2H), 2.68 -2.57 (m, 2H), 2.55 (ddd, J = 9.4,7.0,4.3Hz, 1H), 2.42 (dt, J = 13.3,6.8Hz, 2H), 1.97-1.74 (m, 1H), 1.64-1.46 (m, 1H), 1.23 ( td, J = 7.1,3.3Hz, 3H), 1.14 (dd, J = 7.1,3.9Hz, 3H)
Example 8
Synthesis of Compound 5
Example 8-1: The reaction flask was added compound 4 (1eq) was added water (2VOL), concentrated hydrochloric acid (2VOL), 110 ℃ reaction was heated in an oil bath overnight, complete conversion of starting material, the HPLC peak area 97%. 10% NaOH solution was added to adjust the pH to about 10, filtration products. Yield 85%.
Example 8-2: The reaction flask was added compound 4 (1eq) was added ethanol (5 vol), water (5 vol), potassium hydroxide (8 eq), was heated in an oil bath overnight at 110 ℃ reaction, complete conversion of the starting material, the HPLC peak area 99%. Water was added (5Vol), filtered to obtain the product. Yield 95%. Product was dissolved in toluene, was added ethanolic hydrochloric acid, the precipitated hydrochloride Compound 5.
NMR data for the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.31 (S, 3H), 7.70-7.61 (m, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.31 (m, 3H), 4.09 (the dq- , J = 42.6,7.1Hz, 1H), 3.62-3.51 (m, 1H), 3.50-3.41 (m, 1H), 3.11-3.00 (m, 1H), 2.95-2.84 (m, 1H), 1.30-1.10 (m, 1H).
EXAMPLE 9
Synthesis of Compound 6
To the reactor was added compound 5, was added absolute ethanol (3vol). Temperature of the outer set 30 ℃ heating, stirring was continued after the temperature reached 25 ℃ 20min. Was added 30% NaOH aqueous solution (1.1eq). External temperature 65 ℃ heating provided, after the internal temperature reached 60 deg.] C was slowly added (of Boc) 2 O (1.1 eq). Stirring 0.5h, reaction monitoring. After completion of the reaction, water was added slowly dropwise (8vol), turn off the heating and natural cooling. The system temperature was lowered to 25 deg.] C and continue stirring for 2h. Filter cake at 50 ℃ blast oven drying to obtain the product.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Synthesis of Intermediate Compound 6 to Compound 10, i.e., the AHU-377, a synthetic route in the background of the present invention, the cited patent application WO2014032627A1 loaded in detail, not in this repeat.
Example 10
Synthesis of Compound 2
Benzyl glycidyl ether preparation (50g) in THF (200mL) was added. Under inert gas protection, the biphenyl magnesium bromide (365mmol) was added to THF (1020mL) was added the reaction flask is placed in a low temperature bath -40 ℃ cooling. Cuprous iodide (O.leq) when the internal temperature dropped to -9 ℃. Continued to decrease the temperature of -23 ℃ dropwise addition of benzyl glycidyl ether in THF was added dropwise to control the internal temperature process of not higher than -15 deg.] C, 47 min when used, the addition was completed the cooling off the reaction was stirred overnight. The cooling system to -20 ℃ quenched with 1N HCl aqueous solution, <10 ℃ Go stirred 30min at room temperature. Liquid separation, the aqueous phase was extracted with THF, the combined THF phases. Respectively saturated ammonium chloride (250mL), saturated brine (250mL) washed. Rotary evaporation to remove THF, and water (200 mL) Continue rotary evaporation 1h, cool to precipitate a solid. Suction crude. Crude n-heptane was added 2Vol beating, suction filtration to obtain the product in a yield of 90 ~ 95%, HPLC peak area 94%. In another column purification was pure, columned yield 88.6%, HPLC 99.1%.
Example 11
Synthesis of Compound 3
Preparation Example 9, said compound taking the embodiment 2 (5g) added to the reaction flask, the reaction flask was added toluene (80mL), phthalimide (2.55 g of) and triphenylphosphine (5.35g of), the nitrogen was replaced protection. An ice-salt bath cooling to -5 deg.] C, was added dropwise DIAD (4.12g), dropwise addition was exothermic, the temperature was raised to 5 ℃. The reaction was continued 1h sampling HPLC test material substantially complete reaction. Join 12g silica spin column done to collect the product (including DIEA derivative).
Example 12
Synthesis of Compound 11
Compound 3 (3g) was added to the reaction flask embodiment taken in Preparation Example 10, was added ethanol (30 mL), with stirring. Was added hydrazine hydrate (2g) was heated in an oil bath reflux 1h, when supplemented with 20mL ethanol was stirred difficulties, the reaction was continued to 2.5h, HPLC showed the starting material the reaction was complete. Add EA / H2O 100mL each liquid separation, the EA phase was washed with water (100mL) and the combined organic phases were washed with water (100mL) and saturated brine (100mL) washed. Anhydrous magnesium sulfate and filtered spin column was done product 1.88g, yield 88%, HPLC 94%.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 7.64 (D, J = 7.2Hz, 2H), 7.57 (D, J = 8.1Hz, 2H), 7.45 (T, J = 7.6Hz, 2H), 7.39-7.32 ( m, 5H), 7.29 (d , J = 8.1Hz, 3H), 4.55-4.43 (m, 2H), 3.38-3.23 (m, 3H), 3.18-3.10 (m, 1H), 2.82-2.74 (m, 1H), 2.61-2.52 (m, 1H ).
Example 13
Synthesis of Compound 11
To the toluene solution of the compound 2 was added phthalimide (1.1 eq), triphenylphosphine (1.3 eq) with stirring. External bath set -10 ℃, to cool the system, the internal temperature dropped to 0 ~ 5 ℃, start dropping DIAD (1.3eq), control the internal temperature -5 ~ 5 ℃. Completion of the dropwise addition, the cooling bath was turned off outside the reaction was stirred at room temperature. The reaction was stirred for 1 to 4 hours. The reaction solution to give compound 3, administered directly in the next reaction. To the above reaction mixture was added hydrazine hydrate (6 eq), heated to 70 ~ 80 ℃, to complete the reaction, filtered hot, the filtrate. Aqueous sodium hydroxide solution (20vol 10%) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. Water was added (20vol) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. The toluene phase was added hydrochloric acid (20vol, 3N), stirred for 0.5h, to form a solid precipitate. Filtration and drying to obtain a product, i.e. compound 11, the hydrochloride salt, yield 60% in two steps.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.46 (S, 3H), 7.63 (dd, J = 16.4,7.7Hz, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.22 (m, 8H ), 4.56 (d, J = 12.1Hz, 1H), 4.48 (d, J = 12.1Hz, 1H), 3.58 (d, J = 7.9Hz, 2H), 3.47 (dd, J = 10.9,6.3Hz, 1H ), 3.11 (dd, J = 13.5,4.9Hz, 1H), 2.92 (dd, J = 13.4,9.1Hz, 1H).
Example 14
Synthesis of Compound 12
Weigh Compound 11 (1.38g) was added to the reaction flask. To the reaction flask plus DCM (14ml) and Et3N (462mg, 0.73ml). Weighed (of Boc) 2O (1.23 g of) was added to DCM (5ml) was dissolved. Room temperature (8 ℃), a solution (of Boc) 2 DCM solution O was added dropwise to the reaction, (2ml) rinsed with DCM. The reaction mixture was stirred at room temperature, detected by HPLC, the reaction ends 4h. Reaction mixture was washed (15ml) 3 times with Brine (15ml) The reaction solution was washed 1 times. Inorganic sulfate, concentrated and purified by column PE:EA = 15:1 give product 560mg, yield 30.8%, HPLC 99.92%.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.49 (D, J = 7.4Hz, 2H), 7.43 (T, J = 7.3Hz, 2H), 7.39-7.28 (m, 5H), 7.24 ( d, J = 9.0Hz, 3H), 5.00-4.80 (br, 1H), 4.51 (q, J = 11.8Hz, 2H), 4.08-3.85 (br, 1H), 3.43 ( d, J = 2.9Hz, 2H) , 3.02-2.77 (m, 2H), 1.42 (s, 9H).
Example 15
Synthesis of Compound 6
Weigh Compound 12 (250mg) and methanol (9ml) was added to the reaction flask. Added Pd / C (138mg, 1 / 4w / w, water content 55%). The H 2replaced 3 times, 50 ℃ stirred and heated. HPLC detection reaction, the reaction end 30h. Filtered off Pd / C, 40 ℃ concentrated under reduced pressure to remove methanol. PE:EA = 3:1 florisil column to give the product 196mg, 100% yield, 99.34% purity.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Method for preparing the AHU-377, characterized by comprising the steps of: (a) Compound (1) S- benzyl glycidyl ether and biphenyl Grignard reagent produced by the reaction of the compound (2) in an organic solvent; ( b) compound (2) with a succinimide or phthalimide Mitsunobu reaction occurs in an organic solvent to form a compound (3); (C) compound (3) in an organic solvent in the role of a catalyst under removal debenzylation protected form compound (4); (D) compound (4) with an oxidizing agent oxidation reaction occurs in an organic solvent to form a compound (7); (E) compound (7) with a phosphorus ylide reagent in an organic solvent to give the compound (8); (F.) compound (8) in an organic solvent in the selective catalytic hydrogenation of the compound (9); and (g) of the compound (9) in an organic solvent in the hydrolysis reaction of the amide compound occurs in the presence of an acid ( 10), i.e., AHU-377;
References
- John J.V. McMurray, Milton Packer, Akshay S. Desai, et al. for the PARADIGM-HF Investigators and Committees (August 30, 2014).“Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure”. N Eng J Med 371. doi:10.1056/NEJMoa1409077.
- Solomon, SD. “HFpEF in the Future: New Diagnostic Techniques and Treatments in the Pipeline”. Boston. p. 48. Retrieved 2012-01-26.
- Gu, J.; Noe, A.; Chandra, P.; Al-Fayoumi, S.; Ligueros-Saylan, M.; Sarangapani, R.; Maahs, S.; Ksander, G.; Rigel, D. F.; Jeng, A. Y.; Lin, T. H.; Zheng, W.; Dole, W. P. (2009). “Pharmacokinetics and Pharmacodynamics of LCZ696, a Novel Dual-Acting Angiotensin Receptor-Neprilysin Inhibitor (ARNi)”. The Journal of Clinical Pharmacology 50 (4): 401–414. doi:10.1177/0091270009343932.PMID 19934029.
- Schubert-Zsilavecz, M; Wurglics, M. “Neue Arzneimittel 2010/2011.” (in German)


| WO2004085378A1 * | Mar 15, 2004 | Oct 7, 2004 | Joseph D Armstrong Iii | Process for the preparation of chiral beta amino acid derivatives by asymmetric hydrogenation |
| WO2006057904A1 * | Nov 18, 2005 | Jun 1, 2006 | Merck & Co Inc | Stereoselective preparation of 4-aryl piperidine amides by asymmetric hydrogenation of a prochiral enamide and intermediates of this process |
| WO2006069617A1 * | Dec 5, 2005 | Jul 6, 2006 | Dsm Fine Chem Austria Gmbh | Process for transition metal-catalyzed asymmetric hydrogenation of acrylic acid derivatives, and a novel catalyst system for asymmetric transition metal catalysis |
| US5217996 * | Jan 22, 1992 | Jun 8, 1993 | Ciba-Geigy Corporation | Biaryl substituted 4-amino-butyric acid amides |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | KSANDER, GARY M. ET AL: “Dicarboxylic Acid Dipeptide Neutral Endopeptidase Inhibitors” JOURNAL OF MEDICINAL CHEMISTRY, vol. 38, no. 10, 1995, pages 1689-1700, XP002340280 cited in the application |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{[(2S,4R)-1-(4-Biphenylyl)-5-ethoxy-4-methyl-5-oxo-2-pentanyl]amino}-4-oxobutanoic acid
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 149709-62-6 |
| ATC code | None |
| PubChem | CID: 9811834 |
| ChemSpider | 7987587 |
| Synonyms | AHU-377; AHU377 |
| Chemical data | |
| Formula | C24H29NO5 |
| Molecular mass | 411.49 g/mol |
Relevant Clinical Literature
UK Guidance
Regulatory Literature
Other Literature
|
|
| Systematic (IUPAC) name | |
|---|---|
|
4-{[(2S,4R)-1-(4-Biphenylyl)-5-ethoxy-4-methyl-5-oxo-2-pentanyl]amino}-4-oxobutanoic acid
|
|
| Identifiers | |
| CAS Number | 149709-62-6 |
| ATC code | None |
| PubChem | CID: 9811834 |
| ChemSpider | 7987587 |
| Synonyms | AHU-377; AHU377 |
| Chemical data | |
| Formula | C24H29NO5 |
| Molecular mass | 411.49 g/mol |
Message
Comwinchem <comwinchem@foxmail.com>
Date: 1 September 2016 at 15:16
Subject: LCZ696 (SACUBITRIL+VALSARTAN)/ Changzhou Comwin Fine Chemicals Co,. Ltd
To: amcrasto <amcrasto@gmail.com>
Dear SirHow do you do! Sincerely hope my email will bring you more LCZ696 (SACUBITRIL+VALSARTAN) possibilities.I am Wang Zhuo of ComWin from China, and in charge of LCZ696 (SACUBITRIL+VALSARTAN) global marketing.
Date: 1 September 2016 at 15:16
Subject: LCZ696 (SACUBITRIL+VALSARTAN)/ Changzhou Comwin Fine Chemicals Co,. Ltd
To: amcrasto <amcrasto@gmail.com>
Dear SirHow do you do! Sincerely hope my email will bring you more LCZ696 (SACUBITRIL+VALSARTAN) possibilities.I am Wang Zhuo of ComWin from China, and in charge of LCZ696 (SACUBITRIL+VALSARTAN) global marketing.
LCZ696 (sacubitril/Valsartan) is a combination drug for use in heart failure developed by Novartis. It consists of valsartan and sacubitril, in a 1:1 mixture by molecule count. It was approved by US FDA in July 2015.
Sacubitril (Hemicalcium) is a neprilysin inhibitor, We have developed this project since 2nd half of 2014. At present, some intermediates are in commercial scale, and some are in pilot production.
We will put our most focus on this project from 2nd half of this year. Certainly we will file DMF for Sacubitril and make submission to regulartory market. We have sold Sacbutitril to some EU customers for evaluation purpose, such as Teva, Chemo. Also, we are doing the development of LCZ696 (the final API). It’s co-crystallized valsartan and sacubitril, in a one-to one molar ratio. One LCZ696 complex consists of six valsartan anions, six sacubitril anions, 18 sodium cations, and 15 molecules of water. Now we have the sample of LCZ696 in kilogram grade.
Best Regards
Wang Zhuo
Sales Executive
Changzhou ComWin Fine Chemicals Co.,Ltd.
24th Floor, Jiaye International Commercial Plaza
99 Yanling West Road, Changzhou
Jiangsu 213003 China
Tel: 0086 519 8663 2882
Fax: 0086 519 8661 3190
email: wang.zhuo@comwin-china.com
www.comwin-china.com


सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
////// antihypertensive drug, Heart Failure, Sacubitril, AHU 377
CCOC(=O)[C@H](C)C[C@@H](Cc1ccc(cc1)c2ccccc2)NC(=O)CCC(=O)O
CCOC(=O)[C@H](C)C[C@@H](CC1=CC=C(C=C1)C2=CC=CC=C2)NC(=O)CCC(=O)O
Updating of the HMPC-Guideline on the use of the CTD Format in the Registration of Traditional Herbal Medicinal Products
DRUG REGULATORY AFFAIRS INTERNATIONAL
 Updating of the HMPC-Guideline on the use of the CTD Format in the Registration of Traditional Herbal Medicinal Products
Updating of the HMPC-Guideline on the use of the CTD Format in the Registration of Traditional Herbal Medicinal Products
Compared to herbal medicinal products (HMPs) there is a simplified registration procedure for traditional herbal medicinal products (THMP).
EMA’s HMPC (Committee on Herbal Medicinal Products) published the draft of revision 2 on the use of the CTD format in the preparation of a registration application for traditional herbal medicinal products on 10 March 2015.
This guideline contains instructions on how to prepare a CTD for a registration application of traditional herbal medicinal products.
Now, there is a new annex 2 with a mock-up which shows by means of a concrete example where and to what extent information should be given on traditional herbal medicinal products in the dossier.
Appendix 1 is a best practice guide for module 3 on quality.
For further information please see the complete draft revision 2 of the…
View original post 33 more words
Indian and Chinese API Manufacturers in the Focus of European Authorities
DRUG REGULATORY AFFAIRS INTERNATIONAL
Indian and Chinese API Manufacturers in the Focus of European Authorities
The EudraGMP database was originally launched in April 2007 and is used to exchange information on compliance with the Good Manufacturing Practices (GMP) between the relevant regulatory authorities of the EU Member States – including Iceland, Liechtenstein and Norway. Since January 2011 the data of all national authorities can be accessed. Further, since April 2013 the database also contains information about GDP, why it is referred to as Eudra GMDP database now.
The database comprising the reports about deficiencies found in inspections by the European authorities – the “non-compliance reports” or, officially, “statement of non-compliance with GMP” – was extended by three reports last week: two of these reports related to Chinese firms, one report to a company in India. The inspections were conducted by inspectors of the Italian authority.
The inspection of the Indian site (antibiotic…
View original post 224 more words