
Home » Uncategorized (Page 90)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Pirarubicin Hydrochloride
 Pirarubicin Hydrochloride
Pirarubicin Hydrochloride 
(7S,9S)-7-((2R,4S,5S,6S)-4-amino-6-methyl-5-((R)-tetrahydro-2H-pyran-2-yloxy)-tetrahydro-2H-pyran-2-yloxy)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione HCl
(CAS 95343-20-7)
THP Hydrochloride
(7S,9S)-7-((2R,4S,5S,6S)-4-amino-6-methyl-5-((R)-tetrahydro-2H-pyran-2-yloxy)-tetrahydro-2H-pyran-2-yloxy)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione HCl
MF C32H38ClNO12
MW 664.1
BASE 72496-41-4
Pirarubicin
or Pinorubicin
or Therarubicin
or (8S,10S)-10-(((2R,4S,5S,6S)-4-Amino-6-methyl-5-(((R)-tetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H-pyran-2-yl)oxy)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione
or Pirarubicin
Pirarubicin Hcl is an analogue of the anthracycline anti-neoplastic doxorubicin, which is an inhibitor of Topo II.
Target: Topoisomerase
Pirarubicin is an anthracycline drug. An analogue of the anthracycline antineoplastic antibiotic doxorubicin. Pirarubicin intercalates into DNA and interacts with topoisomerase II, thereby inhibiting DNA replication and repair and RNA and protein synthesis. This agent is less cardiotoxic than doxorubicin and exhibits activity against some doxorubicin-resistant cell lines.

Pirarubicin (THP-adriamycin or THP-doxorubicin) was found during a search of new anthracycline antibiotics among 4′-O-substituted compounds having less toxicities than other anthracycline anticancer drugs in 1979 by Umezawa et al. In its preclinical studies, this compound possessed almost similar antitumor efficacies to doxorubicin, but was effective against doxorubicin-resistant P388 and other murine tumor cell lines. This compound was rapidly incorporated into tumor cells, inhibiting DNA polymerase alpha and subsequently DNA synthesis.
Inhibition of RNA synthesis was also noted. In the clinical studies, clinical responses were established against head and neck cancer, breast cancer, urogenital cancers, ovarian cancer, uterine cancer, acute leukemia, and malignant lymphoma, showing a wide antitumor spectrum clinically. Among the side effects, cardiac toxicity, alopecia and disturbance of the digestive organs were mild. From these results, THP-adriamycin seems to be a useful clinical drug for human solid tumors.
Pirarubicin (INN) is an anthracycline drug. An analogue of the anthracycline antineoplastic antibiotic doxorubicin. Pirarubicin intercalates into DNA and interacts with topoisomerase II, thereby inhibiting DNA replication and repair and RNA and protein synthesis. This agent is less cardiotoxic than doxorubicin and exhibits activity against some doxorubicin-resistant cell lines
 .
.
EP 0014853
https://www.google.com/patents/EP0014853B1?cl=en
")
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (3S)-3-glycoloyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl 3-amino-2,3,6-trideoxy-4-O-[(2R)-tetrahydro-2H-pyran-2-yl]-α-L–lyxo-hexopyranoside | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
|
|
| Identifiers | |
| 72496-41-4 |
|
| L01DB08 | |
| PubChem | CID 3033521 |
| ChemSpider | 2298189 |
| UNII | D58G680W0G |
| KEGG | D01885 |
| ChEMBL | CHEMBL1398373 |
| Synonyms | (9S)-7-[(2R,4S,5S,6S)-4-amino-6-methyl-5-[(2R)-oxan-2-yl]oxyoxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione |
| Chemical data | |
| Formula | C32H37NO12 |
| 627.63 g/mol | |
TAKE A TOUR
Bijapur, Karnataka, INDIA
-
Bijapur – Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/BijapurVijayapur city, formerly Bijapur, is the district headquarters of Bijapur District of Karnataka state. It is also the headquarters for Bijapur Taluka. Bijapur city is well …
.
 .
.







 GOLCONDA
GOLCONDA



Badami Cave Temple, near Bijapur





//////////
VX 787, PIMODIVIR, for Avian influenza

VRT-0928787
VX-787
vx 787
| Vertex Pharmaceuticals |
Janssen Pharmaceuticals, under license from Vertex Pharmaceuticals, is developing VX-787 and its back-up compound VX-353, an influenza A viral replication inhibitor, for treating influenza A virus infection, including pandemic and avian influenza strains. In May 2015, VX-787 was in phase II clinical trial.

Useful for treating influenza virus infection. For concurrent filing see WO2015073476 (claiming the polymorphic forms of VX-787) and WO2015073491 (claiming the composition comprising the hydrochloride salt of VX-787).
Polymorphic forms of hydrochloride (A,F and D) and tosylate salts (form A) of VX-787 are claimed. , useful for treating influenza virus infection. For concurrent filing see WO2015073481 (claiming the processes for the synthesis of VX-787 ) and WO2015073491 (claiming the composition comprising the hydrochloride salt of VX-787).
WO2010148197
http://www.google.com/patents/WO2010148197A1?cl=en
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
Compound 1070 was made in a similar fashion as described above for compounds 946 and 947.
………………….
WO 2013019828
http://www.google.com/patents/WO2013019828A1?cl=en
WO 2012083122
http://www.google.co.in/patents/WO2012083122A1?cl=en
Synthetic Scheme 1

(a) CHC13; (b) NaOMe, MeOH; (c) DPPA, Et3N, BnOH; (d) H2, Pd/C;
Synthetic Scheme 2

(a) Et3N, CH3CN; (b) cone. H2S04; (c) 9M H2S04; (d) Ag2C03, HOAc, DMSO, 100 °C; (e) X- phos, Pd2(dba)3, K3PO4, 2-methyl THF, H20, 120 °C (f) LiOH, THF, MeOH, 70 °C
Synthetic Scheme 3

(a) Et3N, THF; (b) chiral SFC separation; (c) 5-fluoro- l -(p-tolylsulfonyl)-3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-
………………………
See new patents
WO2015073491
……………………………..
Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2
J. Med. Chem., 2014, 57 (15), pp 6668–6678
DOI: 10.1021/jm5007275
http://pubs.acs.org/doi/abs/10.1021/jm5007275
Vertex Pharmaceuticals Inc

Vertex Licenses VX-787 to Janssen Pharmaceuticals for the Treatment of Influenza
Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced that it has entered into a licensing agreement with Janssen Pharmaceuticals, Inc. for the worldwide development and commercialization of VX-787, a novel medicine discovered by Vertex for the treatment of influenza. As part of the agreement, Vertex will receive an up-front payment of $30 million from Janssen and has the potential to receive additional development and commercial milestone payments as well as royalties on future product sales. Vertex completed a Phase 2a study of VX-787 in 2013 that showed statistically significant improvements in viral and clinical measurements of influenza infection. VX-787 is designed to directly inhibit replication of the influenza virus.
“With a deep history in developing new medicines for viral infections and diseases, Janssen is well-positioned to advance the global development of VX-787 for the treatment of influenza,” said Jeffrey Leiden, M.D., Ph.D., Chairman, President and Chief Executive Officer of Vertex. “This collaboration provides important support for the continued development of VX-787 in influenza and contributes to our financial strength to enable continued investment in our key development programs for cystic fibrosis and in research aimed at discovering new medicines.”
About the Collaboration
Under the terms of the collaboration, Janssen will have full global development and commercialization rights to VX-787. Vertex will receive a $30 million up-front payment from Janssen and could receive additional development and commercial milestone payments as well as royalties on future product sales. The collaboration, and the related $30 million up-front payment, is subject to the expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act.
About VX-787
VX-787 is an investigational medicine that is designed to directly inhibit replication of influenza A, including recent H1 (pandemic) and H5 (avian) influenza strains, based on in-vitro data. VX-787’s mechanism represents a new class of potential medicines for the treatment of influenza, distinct from neuraminidase inhibitors, the current standard of care for the treatment of influenza. VX-787 is intended to provide a rapid onset of action and an expanded treatment window.
In a Phase 2a influenza challenge study, statistically significant improvements in viral and clinical measurements of influenza infection were observed after treatment with VX-787. The study met its primary endpoint and showed a statistically significant decrease in the amount of virus in nasal secretions (viral shedding) over the seven-day study period. In addition, at the highest dosing regimen evaluated in the study, there was a statistically significant reduction in the severity and duration of influenza-like symptoms. In this study, VX-787 was generally well-tolerated, with no adverse events leading to discontinuation. Those who took part in the study volunteered to be experimentally exposed to an attenuated form of live H3N2 influenza A virus. H3N2 is a common type of influenza virus and was the most common type observed in the 2012/2013 influenza season in the United States.
VX-787 was discovered by Vertex scientists.
About Influenza
Often called “the flu,” seasonal influenza is caused by influenza viruses, which infect the respiratory tract.1 The flu can result in seasonal epidemics2 and can produce severe disease and high mortality in certain populations, such as the elderly.3 Each year, on average 5 to 20 percent of the U.S. population gets the flu4 resulting in more than 200,000 flu-related hospitalizations and 36,000 deaths.5 The overall national economic burden of influenza-attributable illness for adults is $83.3 billion.5 Direct medical costs for influenza in adults totaled $8.7 billion including $4.5 billion for adult hospitalizations resulting from influenza-attributable illness.5 The treatment of the flu consists of antiviral medications that have been shown in clinical studies to shorten the disease and reduce the severity of symptoms if taken within two days of infection.6 There is a significant need for new medicines targeting flu that provide a wider treatment window, greater efficacy and faster onset of action.
About Vertex
Vertex is a global biotechnology company that aims to discover, develop and commercialize innovative medicines so people with serious diseases can lead better lives. In addition to our clinical development programs focused on cystic fibrosis, Vertex has more than a dozen ongoing research programs aimed at other serious and life-threatening diseases.
Founded in 1989 in Cambridge, Mass., Vertex today has research and development sites and commercial offices in the United States, Europe, Canada and Australia. For four years in a row, Science magazine has named Vertex one of its Top Employers in the life sciences. For additional information and the latest updates from the company, please visit www.vrtx.com.
Vertex’s press releases are available at www.vrtx.com.
| WO2002024705A1 | 13 Sep 2001 | 28 Mar 2002 | Charles Jackson Barnett | Stereoselective process for preparing cyclohexyl amine derivatives |
| WO2003015798A1 | 13 Aug 2002 | 27 Feb 2003 | Toyama Chemical Co Ltd | Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 | 30 Mar 2005 | 13 Oct 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006069258A1 * | 20 Dec 2005 | 29 Jun 2006 | Amgen Inc | Substituted heterocyclic compounds and methods of use |
| WO2007084557A2 | 17 Jan 2007 | 26 Jul 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2008079346A1 | 21 Dec 2007 | 3 Jul 2008 | Vertex Pharma | 5-cyan0-4- (pyrrolo [2, 3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| WO2009073300A1 | 31 Oct 2008 | 11 Jun 2009 | Vertex Pharma | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 | 22 Jul 2009 | 28 Jan 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * | 17 Jun 2010 | 23 Dec 2010 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| WO2011008915A1 * | 15 Jul 2010 | 20 Jan 2011 | Abbott Laboratories | Pyrrolopyridine inhibitors of kinases |
| US20100038988 | 12 Aug 2008 | 18 Feb 2010 | Gannon Ramy | Stator and Method of Making the Same |
| WO2003015798A1 | Aug 13, 2002 | Feb 27, 2003 | Toyama Chemical Co Ltd | Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 | Mar 30, 2005 | Oct 13, 2005 | Vertex Pharma | Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 | Jan 17, 2007 | Jul 26, 2007 | Vertex Pharma | Azaindoles useful as inhibitors of janus kinases |
| WO2009073300A1 | Oct 31, 2008 | Jun 11, 2009 | Vertex Pharma | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 | Jul 22, 2009 | Jan 28, 2010 | Vertex Pharmaceuticals Incorporated | Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * | Jun 17, 2010 | Dec 23, 2010 | Vertex Pharmaceuticals Incorporated | Inhibitors of influenza viruses replication |
| US20100038988 | Aug 12, 2008 | Feb 18, 2010 | Gannon Ramy | Stator and Method of Making the Same |
……
.

Vertex Pharmaceuticals’ Boston Campus, United States of America
Lynette Hopkinson VP Commercial Regulatory Affairs, Global Regulatory Affairs Vertex Pharmaceuticals Incorporated, United States

swati Patel, a lead analyst, shared a toast with Mir Hussain, a systems engineer, at Vertex Pharmaceuticals during the Friday beer hour, which features beer and chips for employees.
On Fridays around 5 o’clock, after a hard week of work, Frank Holland likes to unwind with a beer. And he doesn’t have to leave work to get one.
Holland is a research scientist at Vertex Pharmaceuticals, which every Friday rings in “beer hour,” offering free adult beverages and munchies to its 1,300 Boston employees.
For Holland, the weekly ritual is a chance to escape the bubble of his chemistry lab and bump into colleagues from other departments — as well as Vertex’s top executives, who regularly attend. For those who prefer grapes to hops, there is also wine.
“Some of the other companies I worked at, you really had to go out of your way to meet people,” said Holland, 32. “At Vertex all you have to do is show up in the cafeteria on a Friday afternoon.”
Sure, free beer is common at hip tech offices; some even have their own bars. But Vertex, best known for its treatment for cystic fibrosis, was doing this way before it was cool. The beer-hour tradition goes back to the company’s founding days, in 1989. Back then, it was just two dozen people in a small office in Cambridge. Someone went to a corner store, bought a case of beer and some chips, and beer hour was born.

Virginia Carden Carnahan
Vice President, New Product Planning and Strategy, Vertex Pharmaceuticals

A scientist works in the lab at Boston-based Vertex Pharmaceuticals.
Vertex Pharmaceuticals Headquarters Lobby
…………
β-Sitosterol, 후박(厚朴)

β-Sitosterol
http://www.herbdb.co.kr/herb/dbsearch3/separation_view.asp?key=302
| C29H50O, 414.00 | |||
| White needles | |||
| m.p(℃) | 283-285 | ||
| IR(cm-¹) | νmax (KBr): 3400, 1680 | ||
| UV(nm) | λmax (MeOH): 216 | ||
| MS | EIMS m/z: 414 [M]+ | ||


β-Sitosterol (β-谷甾醇); CAS: 83-46-5

(300 MHz, CDCl3) δ: 5.36 (1H, d, J = 5.2 Hz, H-6), 3.53 (1H, m, H-3),1.01 (3H, s, CH3-19), 0.94 (3H, d, J = 6.5 Hz, CH3-21), 0.92 (3H, d, J = 6.5 Hz,CH3-26), 0.83 (3H, t, J = 6.6 Hz, CH3-29), 0.69 (3H, s, CH3-18)
13c nmr

(75 MHz, CDCl3) δ: 37.2 (C-1), 32.1 (C-2), 72.0 (C-3), 42.5 (C-4), 141.0 (C-5), 121.9 (C-6), 32.1 (C-7), 31.9 (C-8), 50.4 (C-9), 36.7 (C-10), 21.3 (C-11), 40.0 (C-12), 42.5 (C-13), 57.0 (C-14), 24.5 (C-15), 28.4 (C-16), 56.3 (C-17), 12.2 (C-18), 19.2 (C-19), 36.3 (C-20), 19.0 (C-21), 34.2 (C-22), 26.4 (C-23),46.1 (C-24), 29.4 (C-25), 19.6 (C-26), 20.0 (C-27), 23.3 (C-28),12.0 (C-29)

ZSTK 474

4-[4-[2-(difluoromethyl)benzimidazol-1-yl]-6-morpholin-4-yl-1,3,5-triazin-2-yl]morpholine
ZSTK474; 475110-96-4; 4,4′-(6-(2-(Difluoromethyl)-1H-benzo[d]imidazol-1-yl)-1,3,5-triazine-2,4-diyl)dimorpholine; ZSTK-474; ZSTK 474; TCMDC-137004;
2-(2-Difluoromethylbenzimidazol-1-yl)-4,6-bis(morpholino)-1,3,5-triazine
2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine
Zenyaku Kogyo (Innovator)
phase2………Treatment of Solid Tumors Therapy
ZSTK474 is a cell permeable and reversible P13K inhibitor with an IC₅₀ at 6nm. It was identified as part of a screening library, selected for its ability to block tumor cell growth. ZSTK474 has shown strong antitumor activities against human cancer xenographs when administered orally to mice without a significant toxic effect.
Phosphatidylinositol 3-kinase (PI3K) has been implicated in a variety of diseases including cancer. A number of PI3K inhibitors have recently been developed for use in cancer therapy. ZSTK474 is a highly promising antitumor agent targeting PI3K. We previously reported that ZSTK474 showed potent inhibition against four class I PI3K isoforms but not against 140 protein kinases.
However, whether ZSTK474 inhibits DNA-dependent protein kinase (DNA-PK), which is structurally similar to PI3K, remains unknown. To investigate the inhibition of DNA-PK, we developed a new DNA-PK assay method using Kinase-Glo. The inhibition activity of ZSTK474 against DNA-PK was determined, and shown to be far weaker compared with that observed against PI3K. The inhibition selectivity of ZSTK474 for PI3K over DNA-PK was significantly higher than other PI3K inhibitors, namely NVP-BEZ235, PI-103 and LY294002.
Other Names: ZSTK-474
Chemical Formula: C19H21F2N7O2
CAS Number: 475110-96-4
Molecular Weight: 417.41
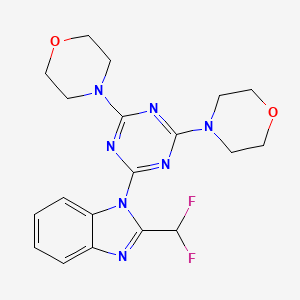
WO 2002088112
http://www.google.co.in/patents/EP1389617A1?cl=en
The condensation of 2,4-dichloro-6-(4-morpholinyl)-1,3,5-triazine

with 2-(difluoromethyl)-1H-benzimidazole by means of K2CO3 in DMF gives

2-chloro-4-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-6-(4-morpholinyl)-1,3,5-triazine ,

which is then condensed with morpholine by means of K2CO3 in DMF to afford the target trisubstituted triazine.
aReagents and conditions: (i) K2CO3, DMF, room temp; (ii) morpholine, DMF or THF, room temp; (iii) NaH or K2CO3, DMF or DMSO, 120 °C.

- 2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine(compound 19)
Melting point: 211-214°C
NMR(CDCl3) δ : 3.79(8H, t, J=4Hz), 3.88(8H, t, J=4Hz), 7.3-7.4(2H, m), 7.56(1H, t, J=53Hz), 7.88(1H, d, J=7Hz), 8.32(1H, d, J=7Hz)
MS m/z: 417(M+
……………………
1H NMR (CDCl3) δ 8.33 (dd, J = 7.3, 1.4 Hz, 1H), 7.89 (dd, J = 7.2, 1.5 Hz, 1H), 7.56 (t, JHF= 53.6 Hz, 1H), 7.46–7.37 (m, 2H), 3.91–3.86 (m, 8H), 3.81–3.76 (m, 8H).
TRIAZINE, PYRIMIDINE AND PYRIDINE ANALOGS AND THEIR USE AS THERAPEUTIC AGENTS AND DIAGNOSTIC PROBES [US2011275762]2011-11-10
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound and antitumor agent containing the same as active ingredient [US7071189] | 2004-06-17 | 2006-07-04 |
| Treatment of prostate cancer, melanoma or hepatic cancer [US2007244110] | 2007-10-18 | |
| Heterocyclic compound and antitumor agent containing the same as effective ingredient [US7307077] | 2006-11-02 | 2007-12-11 |
| IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US7750001] | 2008-05-15 | 2010-07-06 |
| PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLES AND THEIR USE IN CANCER THERAPY [US2011009405] | 2011-01-13 | |
| SUBSTITUTED PYRIMIDINES AND TRIAZINES AND THEIR USE IN CANCER THERAPY [US2011053907] | 2011-03-03 | |
| IMMUNOSUPPRESSIVE AGENT AND ANTI-TUMOR AGENT COMPRISING HETEROCYCLIC COMPOUND AS ACTIVE INGREDIENT [US2010267700] | 2010-10-21 | |
| AMORPHOUS BODY COMPOSED OF HETEROCYCLIC COMPOUND, SOLID DISPERSION AND PHARMACEUTICAL PREPARATION EACH COMPRISING THE SAME, AND PROCESS FOR PRODUCTION OF THE SAME [US8227463] | 2010-09-30 | 2012-07-24 |
| PYRAZOLO[1,5-a]PYRIDINES AND THEIR USE IN CANCER THERAPY [US2010226881] | 2010-09-09 | |
| PYRIMIDINYL AND 1,3,5-TRIAZINYL BENZIMIDAZOLE SULFONAMIDES AND THEIR USE IN CANCER THERAPY [US2010249099] | 2010-09-30 |
…………..
Zenyaku Kogyo






Lascufloxacin, KRP-AM1977, by Kyorin

Lascufloxacin
CAS 848416-07-9
Kyorin Pharmaceutical Co., Ltd., 杏林製薬株式会社
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-
7-((3S,4S)-3-((Cyclopropylamino)methyl)-4-fluoropyrrolidin-1-yl)-6-fluoro-1-(2-fluoroethyl)-8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
{(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
(KRP-AM1977X)
-
C21-H24-F3-N3-O4
- 439.4316
- SMILES……COc1c2c(cc(c1N3C[C@H](C(C3)CNC4CC4)F)F)c(=O)c(cn2CCF)C(=O)O
![]()
…………………………
Lascufloxacin hydrochloride
-
C21-H24-F3-N3-O4.Cl-H
- 475.8925
- CAS 1433857-09-0
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, hydrochloride (1:1)
……………….
Lascufloxacin mesylate
3-Quinolinecarboxylic acid, 7-((3S,4S)-3-((cyclopropylamino)methyl)-4-fluoro-1-pyrrolidinyl)-6-fluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, methanesulfonate (1:1)
-
C21-H24-F3-N3-O4.C-H4-O3-S
- 535.5372
- CAS 1433857-41-0
The other non-fluorinated quinolone under clinical development is KRP-AM1977, by Kyorin, which is in Phase I of clinical trials. The oral formulation of the compound (KRP-AM1977X) is being tested for treatment of respiratory infections and the I.V. formulation is under development for treatment of MRSA infections [1,2].
………………………………..
PATENT
WO 2013069297
http://www.google.co.in/patents/WO2013069297A1?cl=en
The present invention is represented by Formula (1) – {(3S, 4S) -3 – [(cyclopropylamino) methyl] -4-fluoro-1-yl} -6-fluoro-1- (2 – fluoroethyl) -8-methoxy-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (hereinafter, compound (1) crystals of a salt also referred to), and a method for their preparation.
Typically, the pharmaceutical, in addition to the therapeutic effects on diseases, such as safety and quality are required. Therefore, the compound is the active ingredient of drugs, a variety of conditions and that is excellent in storage stability in the (light, temperature, humidity etc. influence the compound) are determined. Also, if the medicament is a dosage form such as oral preparations and injections, it is preferred that higher solubility in active ingredients of the water contained.
Compound (1) is safe, not only exhibit a strong antimicrobial action, conventional hard Gram-positive bacteria antimicrobial agents shown efficacy, particularly MRSA, PRSP, to VRE such resistant strains, to exhibit strong antibacterial activity It is known (for example, Patent Document 1).
WO 2005/026147
Patent Document 1, as the physicochemical characteristics of the compound (1) only has been shown to be a light brown free crystals. Also, Patent Document 1, the solubility in water of Compound (1), stability, no disclosure whatsoever information including characteristics of the crystal.
The present invention aims to provide a technique capable of improving the solubility and storage stability in water of the compound (1).
(Reference Example 4)
Bis (acetato -O) – [6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylate -O 3, O 4] boron Under a nitrogen atmosphere, boric acid (catalyst preparation) 86.4 g (1.40mol) was added acetic anhydride 17.9 L (190mol), and was heated and stirred for 30 minutes at 70.0 ~ 77.7 ℃. It was then cooling the mixture to an internal temperature of 24.7 ℃ (hot water set temperature 23.0 ℃). Subsequently, it was added portionwise boric acid to 4 times to the mixture. Specifically, the addition of boric acid (1 time) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.7 ~ 27.4 ℃. The addition of boric acid (second) 842g of (13.6mol) to the mixture and stirred for 30 minutes at 24.3 ~ 26.3 ℃. In addition boric acid (third time) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 24.3 ~ 26.8 ℃. In addition boric acid (4 th) 842g the (13.6mol) to the mixture, and the mixture was stirred for 30 minutes at 25.1 ~ 28.3 ℃. The mixture was stirred for 30 minutes at 50.0 ~ 54.9 ℃, was with boric acid triacetate adjusted solution.
In the boric acid triacetate adjusted solution, 6,7-difluoro-1- (2-fluoro-ethyl) -8-methoxy-4-oxo-1,4-dihydro-3-carboxylic acid ethyl ester 4.60kg (14. In a reaction preparation solution are added 0mol), and stirred for 3 hours at 53.7 ~ 56.9 ℃. The reaction preparation was cooled to 30.0 ℃, and allowed to stand overnight at room temperature. The reaction preparation was allowed to dissolve with heating to precipitate up to 55.0 ℃, acetone 13.8L was added and the reaction solution (1).
Separately, under nitrogen atmosphere, it is mixed Tsunemizu 161L and aqueous ammonia (28%) 28.2L (464mol), and cooled the mixture to 1.6 ℃. To the mixture, it was added the reaction solution of the above (1), to obtain a crude crystal acquisition solution crowded washed with acetone 9.20L. After cooling the crude crystal acquisition solution to 15.0 ℃, it was stirred for 1 hour at 6.2 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with Tsunemizu 46.0L, to give 9.07kg of wet crude crystals. Set temperature 65.0 to about 16 hours and dried under reduced pressure at ℃, the crude crystals were obtained 5.89kg.
Under a nitrogen atmosphere, it is mixed acetone and 29.5L crude crystal, the resulting mixture was heated and dissolved (melting temperature 52.6 ℃). When heated, it was dropped until the crystallization of diisopropyl ether 58.9L in a mixture (dropping amount 10.0L; 52.8 → 48.7 ℃; crystallization temperature 49.0 ℃). After crystallization confirmation, stirred for 15 minutes the mixture at 49.0 ~ 50.1 ℃, it was dropped the rest of diisopropyl ether to the mixture (50.1 → 46.4 ℃), 46.7 ~ 51.7 It was stirred for 15 minutes mixture at ℃. After cooling the mixture to 15 ℃, it was stirred for 30 minutes at 8.1 ~ 15.0 ℃. And The precipitated crystals were filtered, washed with acetone and diisopropyl ether 5.89L 11.8L, to obtain 6.19kg of wet crystals. For about 20 hours drying under reduced pressure at warm water set temperature 65.0 ℃, bis (acetato -O) – [6,7-difluoro-1- (2-fluoroethyl) -8-methoxy-4-oxo-1,4- dihydro-3-carboxylate -O 3, O 4] was obtained 5.42kg boron (90.4% yield).
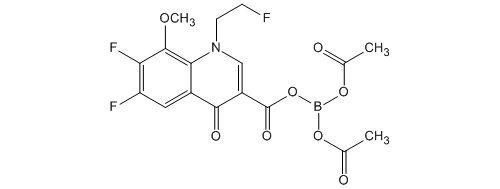
Melting point: 183 ~ 185 ℃ (dec).
Elemental analysis (%): calculated as C 17 H 15 BF 3 NO 8: C, 47.58; H, 3.52; N, 3.26.
Measured value: C, 47.91; H, 3.44; N, 3.04.
1 H-NMR (CDCl 3, 400 MHz) δ: 2.04 (6H, s), 4.22 (3H, d, J = 2.4Hz), 4.88 (2H, dt, J = 47.0 , 4.4Hz), 5.21 (2H, dt, J = 24.9,4.4Hz), 8.17 (1H, t, J = 8.8Hz), 9.11 (1H, s).
ESI MS (positive) m / z: 430 (M + H) +.
IR (KBr) cm -1: 3080,1703.
………………………………………….
WO 2005026147
http://www.google.com/patents/EP1666477A1?cl=en
KEY INTERMEDIATE

604798-54-1
3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-
| Chemical Name:3-Pyrrolidinemethanamine, N-cyclopropyl-4-fluoro-, (3R,4S)-CAS: 604798-54-1Molecular Formula: C8H15FN2Molecular Weight: 158.2165032 |
………………………….
KEY INTERMEDIATE
CAS 848498-67-9
-8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ
キシ]ボラン
| 化学物質名 | ビス(アセチルオキシ)[6,7-ジフルオロ-1-(2-フルオロエチル) -8-メトキシ-4-オキソ-1,4-ジヒドロキノリン-3-カルボニルオ キシ]ボラン |
|---|---|
| 構造別分類コード番号 | F60622212422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
|
| 安衛法官報通し番号 | 21534 |
| 安衛法官報公示整理番号 | 8-(1)-3764 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 848498-67-9 |
| 出典 | 厚生労働省 |
……………………………….
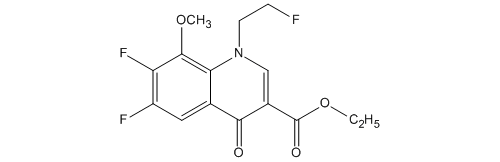
KEY INTERMEDIATE
3-Quinolinecarboxylic acid, 6,7-difluoro-1-(2-fluoroethyl)-1,4-dihydro-8-methoxy-4-oxo-, ethyl ester
114214-60-7
C15H14F3NO4
ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル
| 化学物質名 | 6,7-ジフルオロ-1-(2-フルオロエチル)-8-メトキシ-4-オキ ソ-1,4-ジヒドロキノリン-3-カルボン酸エチル |
|---|---|
| 構造別分類コード番号 | F60622322422 |
| 化学式、構造式
(マウス左クリックで拡大します。) |
 |
| 安衛法官報通し番号 | 21467 |
| 安衛法官報公示整理番号 | 8-(1)-3758 |
| 安衛法官報公示時期 | 平成24年9月27日 |
| 化審法官報公示整理番号 | - |
| CAS番号 | 114214-60-7 |
| 出典 | 厚生労働省 |
| WO2003076428A1 * | 8 Mar 2002 | 18 Sep 2003 | Toshifumi Akiba | Quinolonecarboxylic acid derivative |
| WO2005026147A1 | 8 Sep 2004 | 24 Mar 2005 | Yoshikazu Asahina | 7-(4-substituted 3- cyclopropylaminomethyl-1 pyrrolidinyl) quinolonecarboxylic acid derivative |
| WO2007082471A1 * | 18 Jan 2007 | 26 Jul 2007 | Guangzhou Baiyunshan Pharmaceu | Anti-infective compound, preparation method thereof and use thereof |
| CN1158846A * | 9 May 1995 | 10 Sep 1997 | 昆山市康壮达兽药厂 | Synthesis technology of norfluxacini hydrochloride |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014174846A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174847A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Solid pharmaceutical composition |
| WO2014174848A1 * | 24 Apr 2014 | 30 Oct 2014 | Kyorin Pharmaceutical Co., Ltd. | Tablet |
- Kyorin. Kyorin—Main R&D Activities-1 (4 February 2013 Release). Available online: http://www.kyorin-pharm.co.jp/en/business/pdf/main_rd_activities_20130204_en.pdf (accessed on 4 February 2013).
- Kyorin. Drug discovery, development, and lcm with medical professionals and patients in mind. Available online: http://www.kyorin-gr.co.jp/en/business/gensen/r_and_d.shtml (accessed on 11 April 2013).
-
……….
![]()




Ochyanomizu Sola City 16F,
Kanda Surugadai 4-6, Chiyoda-ku,
Tokyo 101-8311 Japan
TEL: 03-3525-4711
Access
One-minute walk from the Hijiribashi exit of Ochanomizu station on JR Chuo and Sobu lines
One-minute walk from the B2 exit of Shin-Ochanomizu station on Tokyo Metro Chiyoda line
Four-minutes walk from the No.1 exit of Ochanomizu station on Tokyo Metro Marunouchi line
Six-minutes walk from the B3 exit of Ogawamachi station on Toei Subway Shinjuku line



| Trade Name | KYORIN Pharmaceutical Co.,Ltd. |
|---|---|
| Business | Manufacture and sales of prescription medicines |
| Head Office | Ochyanomizu Sola City 16F, Kanda Surugadai 4-6, Chiyoda-ku, Tokyo 101-8311 Japan (Access Map) |
| Telephone | 03-3525-4711 |
| Foundation | 1923 |
| Establishment | 1940 |

 Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park –
Tochigi Wanpaku Park – Mibu-machi – Reviews of Tochigi Wanpaku Park – .
.

Motesanib (AMG-706)

Motesanib (AMG-706)
Amgen Inc.

Motesanib (AMG 706) is an experimental drug candidate originally developed by Amgen[1] but is now being investigated by theTakeda Pharmaceutical Company. It is an orally administered small molecule belonging to angiokinase inhibitor class which acts as an antagonist of VEGF receptors, platelet-derived growth factor receptors, and stem cell factor receptors.[2] It is used as thephosphatesalt motesanib diphosphate.
Motesanib, also known as AMG-706, is an orally administered multikinase inhibitor that selectively targets VEGF receptors, platelet-derived growth factor receptors, and Kit receptors.
Clinical trials
Motesanib was originally investigated for effectiveness against advanced nonsquamous non-small-cell lung cancer (NSCLC), withPhase II trials indicating an effectiveness comparable to bevacizumab when they were both used in combination withpaclitaxel/carboplatin.[3] However a later and more detailed Phase III trial failed to show any benefit for the treatment of NSCLC.[2][4]A second Phase III trial was started in 2012,[5] which focused on patients from Asian backgrounds (performed on the bases ofsubgroup analysis)[6] however this also failed to meet its primary endpoint.[7]
The drug has undergone a Phase II evaluation as first-line therapy for breast cancer[2] however this study found no evidence to support further investigation.[8] Phase II testing against persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas was also unsuccessful.[9]
There have also been 2 separate Phase II clinical trials for thyroid cancer which have both shown promising results.[10][11][12]
Developed at Amgen, the compound is also being evaluated as both monotherapy and in combination with other agents in the treatment of breast, colorectal, lung, thyroid and ovarian cancers. Clinical trials for the treatment of bladder cancer have been terminated.
The National Cancer Institute had been evaluating the potential of the drug in patients with low-grade neuroendocrine tumors; however, no recent development has been reported for this research. The FDA awarded fast track status to motesanib in 2004. In 2008, the compound was licensed to Takeda in Japan.

AMG-706 is synthesized as follows: 1-Acetyl-3,3-dimethyl-6-nitroindoline (I) is reduced by catalytic hydrogenation over Pd/C, giving the aminoindoline (II), which is then coupled with 2-chloronicotinoyl chloride (III) in the presence of DIEA to yield the corresponding nicotinamide (IV). Subsequent condensation of (IV) with neat 4-(aminomethyl)pyridine (V) at 120 °C affords the 2-aminonicotinamide derivative (VI). The N-acetyl group of (VI) is finally removed by acidic hydrolysis to furnish the title compound (1,2).
,………………………………………
US 2003125339
http://www.google.com/patents/US20030125339
………………………………………………….
US 2003225106
https://www.google.com/patents/US20030225106
EXAMPLE 133
[2295]

N-(3,3-Dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
Step A—Preparation of 1-acetyl-6-amino-3,3-dimethylindoline
1-Acetyl-3,3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL), the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc:CH2Cl2 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O—204.27.
Step B—Preparation of N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from 1-acetyl-6-amino-3,3-dimethylindoline (Step A) by the method described in Example 82.
Step C—Preparation of N-(3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide
The titled compound was prepared from N-(1-acetyl-3,3-dimethylindolin-6-yl){2-[(4-pyridylmethyl)amino](3-pyridyl)}carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N5O—373.45.
…………………….
http://www.google.com/patents/WO2012063085A3?cl=en
Example 133

N- (3, 3-Dimethy1indolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) }carboxamide Step A – Preparation of l-acetyl-6-amino-3 , 3- dimethylindoline l-Acetyl-3 , 3-dimethyl-6-nitroindoline (250 mg) was dissolved in MeOH (20 mL) , the mixture was bubbled with H2 for 10 min. 10% Pd/C (50 mg) was added and the mixture was stirred under H2 overnight. The mixture was filtered through Celite® and concentrated in vacuo. The crude material was purified by flash chromatography on silica gel with 1:1 EtOAc :CH2C12 to afford the title compound as a white crystalline material. MS: 205 (M+1). Calc’d. for C12H16N2O-204.27.
Step B – Preparation of N-(l-acetyl- 3 , 3-dimethylindolin-6- yl) (2-[ (4-pyridylmethyl) amino] (3-pyridyl) } carboxamide The titled compound was prepared from l-acetyl-6- amino-3 , 3-dimethylindoline (Step A) by the method described in Example 82.
Step C – Preparation of N- (3 , 3-dimethylindolin-6-yl) {2- [ (4- pyridylmethyl) amino] (3-pyridyl) }carboxamide
The titled compound was prepared from N-(l-acetyl- 3 , 3-dimethylindolin-6-yl) {2- [ (4-pyridylmethyl) amino] (3- pyridyl) } carboxamide (Step B) by the deacylation method described in Example 993. MS: 374 (M+1). Calc’d. for C22H23N50-373.45.
References
- Stafford, edited by Rongshi Li, Jeffrey A. (2009). “Chapter 5. Discovery of Motesanib”. Kinase inhibitor drugs. Hoboken, N.J.: Wiley. pp. 113–130. ISBN 978-0-470-27829-1.
- “Amgen and Takeda’s NSCLC Drug Fails in Phase III Study”. 30 Mar 2011.
- Blumenschein Jr, G. R.; Kabbinavar, F.; Menon, H.; Mok, T. S. K.; Stephenson, J.; Beck, J. T.; Lakshmaiah, K.; Reckamp, K.; Hei, Y.- J.; Kracht, K.; Sun, Y.- N.; Sikorski, R.; Schwartzberg, L. (14 February 2011). “A phase II, multicenter, open-label randomized study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced nonsquamous non-small-cell lung cancer”. Annals of Oncology 22 (9): 2057–2067. doi:10.1093/annonc/mdq731.
- Jump up^ Scagliotti, G. V.; Vynnychenko, I.; Park, K.; Ichinose, Y.; Kubota, K.; Blackhall, F.; Pirker, R.; Galiulin, R.; Ciuleanu, T.-E.; Sydorenko, O.; Dediu, M.; Papai-Szekely, Z.; Banaclocha, N. M.; McCoy, S.; Yao, B.; Hei, Y.-j.; Galimi, F.; Spigel, D. R. (2 July 2012). “International, Randomized, Placebo-Controlled, Double-Blind Phase III Study of Motesanib Plus Carboplatin/Paclitaxel in Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer: MONET1”. Journal of Clinical Oncology 30 (23): 2829–2836. doi:10.1200/JCO.2011.41.4987. PMID 22753922.
- “Takeda Initiates Phase 3 Trial of Motesanib in Japan and Additional Asian Countries”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J. H.; Shinkai, T.; Takeda, K.; Kim, S.- W.; Hsia, T.- C.; Li, R. K.; Tiangco, B. J.; Yau, S.; Lim, W.- T.; Yao, B.; Hei, Y.- J.; Park, K. (13 January 2014). “Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis”.Annals of Oncology 25 (2): 529–536. doi:10.1093/annonc/mdt552.
- Jump up^ “Takeda Announces Phase 3 MONET-A Study Evaluating Motesanib (AMG 706) in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer Does Not Meet Primary Endpoint”. Takeda Pharmaceutical Company Limited. Retrieved 19 February 2015.
- Martin, Miguel; Roche, Henri; Pinter, Tamas; Crown, John; Kennedy, M John; Provencher, Louise; Priou, Frank; Eiermann, Wolfgang; Adrover, Encarna; Lang, Istvan; Ramos, Manuel; Latreille, Jean; Jagiełło-Gruszfeld, Agnieszka; Pienkowski, Tadeusz; Alba, Emilio; Snyder, Raymond; Almel, Sachin; Rolski, Janusz; Munoz, Montserrat; Moroose, Rebecca; Hurvitz, Sara; Baños, Ana; Adewoye, Henry; Hei, Yong-Jiang; Lindsay, Mary-Ann; Rupin, Matthieu; Cabaribere, David; Lemmerick, Yasmin; Mackey, John R (April 2011). “Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study”. The Lancet Oncology 12 (4): 369–376. doi:10.1016/S1470-2045(11)70037-7. PMID 21429799.
- Schilder, R.J.; Sill, M.W.; Lankes, H.A.; Gold, M.A.; Mannel, R.S.; Modesitt, S.C.; Hanjani, P.; Bonebrake, A.J.; Sood, A.K.; Godwin, A.K.; Hu, W.; Alpaugh, R.K. (April 2013). “A phase II evaluation of motesanib (AMG 706) in the treatment of persistent or recurrent ovarian, fallopian tube and primary peritoneal carcinomas: A Gynecologic Oncology Group study”. Gynecologic Oncology 129 (1): 86–91. doi:10.1016/j.ygyno.2013.01.006. PMID 23321064.
- Motesanib Diphosphate Provides Anticancer Activity Among Patients with Progressive Thyroid Cancer, CancerConnect.com
- Jump up^ Schlumberger, M. J.; Elisei, R.; Bastholt, L.; Wirth, L. J.; Martins, R. G.; Locati, L. D.; Jarzab, B.; Pacini, F.; Daumerie, C.; Droz, J.-P.; Eschenberg, M. J.; Sun, Y.-N.; Juan, T.; Stepan, D. E.; Sherman, S. I. (29 June 2009). “Phase II Study of Safety and Efficacy of Motesanib in Patients With Progressive or Symptomatic, Advanced or Metastatic Medullary Thyroid Cancer”.Journal of Clinical Oncology 27 (23): 3794–3801. doi:10.1200/JCO.2008.18.7815. PMID 19564535.
- Sherman, Steven I.; Wirth, Lori J.; Droz, Jean-Pierre; Hofmann, Michael; Bastholt, Lars; Martins, Renato G.; Licitra, Lisa; Eschenberg, Michael J.; Sun, Yu-Nien; Juan, Todd; Stepan, Daniel E.; Schlumberger, Martin J. (3 July 2008). “Motesanib Diphosphate in Progressive Differentiated Thyroid Cancer”. New England Journal of Medicine 359 (1): 31–42.doi:10.1056/NEJMoa075853. PMID 18596272.
External links

Motesanib Diphosphate (AMG-706)
857876-30-3 diphosphate
453562-69-1 (free base)
N-(2,3-Dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide diphosphate
3-Pyridinecarboxamide, N-(2,3-dihydro-3,3-dimethyl-1H-indol-6-yl)-2-[(4-pyridinylmethyl)amino]-, phosphate (1:2)
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide diphosphate
| 569.4 | |
| Formula | C22H23N5O.2H3PO4 |
|---|
 |
|
| Names | |
|---|---|
| IUPAC name
N-(3,3-Dimethyl-2,3-dihydro-1H-indol-6-yl)-2-[(pyridin-4-ylmethyl)amino]pyridine-3-carboxamide
|
|
| Other names
AMG 706
|
|
| Identifiers | |
| 453562-69-1 |
|
| ChEMBL | ChEMBL572881 |
| ChemSpider | 9842625 |
| Jmol-3D images | Image |
| PubChem | 11667893 |
| Properties | |
| C22H23N5O | |
| Molar mass | 373.45 g·mol−1 |
Stats today
6.8 lakh views on this blog
…………………..
TAKEDA, JAPAN
![]()

TOKYO HO

Takeda Pharmaceutical CEO Yasuchika Hasegawa
 Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.
Takeda Pharmaceutical Co. President Christophe Weber is interviewed recently in Tokyo.

Christophe Weber (L), the new president of Takeda Pharmaceutical Co., and CEO Yasuchika Hasegawa pose

 Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
Dr. Paul Chapman of Takeda Pharmaceuticals colors in the eye…
 OSAKA
OSAKA

Dotonbori, Osaka, Japan
 OSAKA
OSAKA

AZD 3264 an IKK2 Inhibitor from Astra Zeneca

AZD 3264
MW 441.50
CAS 1609281-86-8
Inhibition of IkB-kinase IKK2 has been identified as one of the novel pathways to treat inflammatory conditions such as asthma, chronic pulmonary obstructive disorder (COPD) and rheumatoid arthritis
……………………..
PATENT
WO 2003010158
https://www.google.com/patents/WO2003010158A1?cl=en

The synthesis began with the aromatic nucleophilic substitution reaction of 2-fluorobromobenzene (2) with (S)-N-Boc-3-pyrrolidinol 3 to give the bromo intermediate 4, which was borylated via halogen metal exchange using n-hexLi in THF followed by treatment with triisopropyl borate and acidic work-up to give the boronic acid intermediate 5. Suzuki coupling of the boronic acid 5 with bromothiophene 6(2)afforded the intermediate 7. Intermediate 7 was subjected to regioselective bromination using bromine in acetic acid. This reaction was nonregioselective and yielded 17% of the required isomer 8. The bromo compound 8 was coupled with isoxazole boronate ester 9 by another Suzuki reaction to get the title compound. The overall yield of the synthesis was <6%.


………………………..
PAPER
http://pubs.acs.org/doi/full/10.1021/op500105n

An efficient and scalable synthesis of AZD3264 is described in which the differential reactivities of various halogen atoms have been employed. The process involves five linear chemical steps with three isolated stages starting from commercially available fragments.
Elemental impurities – A database to facilitate the risk assessment of active ingredients and excipients
DRUG REGULATORY AFFAIRS INTERNATIONAL

One of the main demands of the Guideline ICH Q3D is to carry out risk assessments on metallic impurities. A database with analytical data provides a valuable support. Learn more about the data sharing using the new elemental impurities database.
Released in December 2014, the ICH Q3D Guideline on Elemental Impurities contains extensive specifications for the control of a total of 24 elements (21 metals, 3 metalloids) that can be present as impurities in pharmaceutical products. Main sources can be
- Active ingredients
- Excipients (including water)
- Processing auxiliaries and catalysts
- Production equipment
- Container and closure systems
The Guideline ICH Q3D calls for a risk assessment with regard to the presence of metallic impurities in various dosage forms, taking into account the respective limit values. The main factors of influence are to be included (see fishbone diagram on p. 6 of the Guideline). The risks identified in a comprehensive analysis…
View original post 343 more words
ECA and PQG publish Chapter 6 of the interpretation of the ECA and PQG publish Chapter 6 of the interpretation of the EU GDP Guideline
DRUG REGULATORY AFFAIRS INTERNATIONAL

The ECA Foundation and the Pharmaceutical Quality Group (PQG) have been working on the interpretation of different chapters of the EU GDP Guideline. Now the group has finalized the work on chapter 6 – Complaints, Returns, Suspected Falsified Medicinal Products & Medicinal Product Recalls. Read more about the GDP Guidance Chapter 6.
The ECA Foundation and the Pharmaceutical Quality Group (PQG) have been working on the interpretation of different chapters of the EU GDP Guideline. The interpretation of five chapters have been published already. The following 5 Guidance chapters on the EU GDP Guideline are available:
Chapter 1: Quality Management
Chapter 9: Transportation (also contains a template for a Technical Agreement)
Chapter 7: Outsourced Activities
Chapter 2: Personnel
Chapter 5: Operations
Now the group has finalized the work on chapter 6 – Complaints, Returns, Suspected Falsified Medicinal Products & Medicinal Product Recalls. Chapter 6 of the EU GDP…
View original post 124 more words
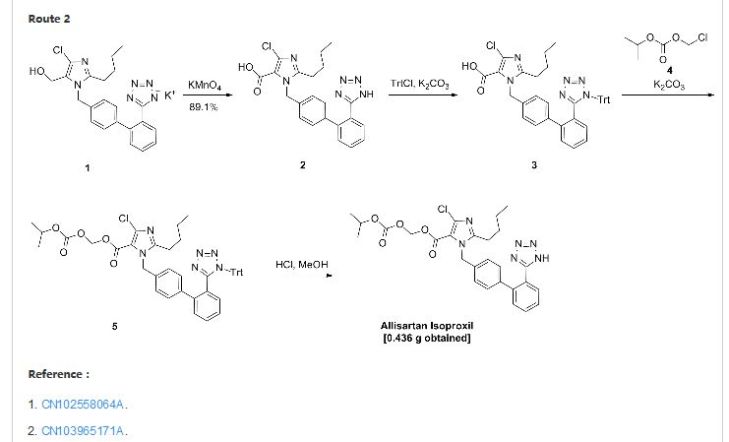
Allisartan isoproxil

Allisartan isoproxil
CAS: 947331-05-7
553.01, C27 H29 Cl N6 O5
An angiotensin II receptor antagonist used to treat mild to moderate essential hypertension.
Approved china, cfda July 1 2012
![]()
Shanghai Allist Pharmaceutical, Inc.
Allist Shanghai Pharmaceutical Co., Ltd.

2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carboxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester,
2-Butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)biphenyl-4-ylmethyl]-1H-imidazole-5-carboxylic acid isopropoxycarbonyloxymethyl ester
2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester
Allisartan is an orally-available angiotensin AT1 antagonist in phase II clinical trials at Shanghai Allist Pharmaceutical for the treatment of mild to moderate essential hypertension.
Shanghai Allist Pharmaceutical PHASE 2 for Hypertension

CN200710094021.4 and CN201110289695.6 disclose the preparation of Alicante medoxomil, the inventor repeated, the proceeds of crystal and Chinese patent CN200710094131.0 consistent disclosed.

Allisartan isoproxil
Angiotensin II AT-1 receptor antagonist
Essential hypertension
Amorphous form of allisartan isoproxil is claimed in WO 2015062498. Useful for treating hypertension. Shenzhen Salubris Pharmaceuticals, in collaboration with Allist, has developed and launched allisartan isoproxil. In October 2012, Shenzhen Salubris signed a strategic cooperation framework agreement with Allist Pharmaceutical for the production and marketing of allisartan isoproxil. Family members of the product case of allisartanWO2007095789, expire in the EU and in the US in 2026. For a prior filing see WO2009049495 (assigned to Allist Pharmaceuticals), claiming the crystalline form of allisartan and its method of preparation.
The compound of formula (I) is an Ang II receptor antagonist. Its chemical name is 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)-1,1′-biphenyl-methyl]-imidazole-5-carb-oxylic acid, 1-[(isopropoxy)-carbonyloxy] methyl ester. Chinese Patent CN101024643A describes the structure, and its use as antihypertensive drugs.

As regards to the solid physical properties of the compound of formula (I), the patent document of CN101024643A discloses that it is a white solid, and its melting point is 134.5-136° C. However, CN101024643A dose not disclose the crystalline structure of the compound of formula (I).

 CHINA
CHINA
NEW PATENT
WO-2015062498
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015062498
……………………..
PATENT
http://www.google.com/patents/CN103965171A?cl=en
Hypertension is a major disease threat to human health, looking for efficiency, low toxicity anti-hypertensive drugs can help relieve social pressures and family responsibilities, with good social and economic benefits.
Angiotensin II (Ang II) is the renin – angiotensin – aldosterone system (RAAS) main vasoconstrictor hormone, which plays an important role in the pathobiology of many chronic diseases, particularly its the role of blood pressure regulation is particularly prominent, and therefore Ang II receptor is believed to be a good target for the development of anti-hypertensive drugs.
EP0253310 discloses a series of imidazole derivatives, DuPont declared and obtained by the study of losartan potassium-listed in 1994, was the first non-peptide Ang II receptor antagonist anti-hypertensive drugs. Thereafter, he listed a series of losartan antihypertensive drugs: candesartan cilexetil, valsartan, irbesartan, telmisartan and olmesartan medoxomil, etc. (EP0253310, W02005049587, GB2419592, EP1719766, US5196444) .
The losartan potassium in the body, the active metabolite EXP3174 has a stronger antihypertensive effect than losartan potassium, but EXP3174 polar molecular structure, is difficult to form passive absorption by diffusion through the cell membrane. US5298915 discloses five carboxyl ester group transformation EXP3174 is a series of derivatives, focusing on the compound HN-65021, and discloses hypotensive test results HN-65021 administered by the oral route, its hypotensive activity with chlorine Similar losartan potassium (BritishJouurnal ofClinical Pharmacology, 40,1995,591).
CN200680000397.8 _5_ discloses a class of imidazole carboxylic acid derivatives, namely Alicante medoxomil compound 8 has a good blood pressure lowering effect, the structure of formula I, the preparation method disclosed in this patent document follows the route A, losartan potassium by oxidation, the protecting group into an ester, deprotected to give a compound of formula I, the route step oxidation process of hydroxyl to carboxyl groups, will be reduced to very fine granular potassium permanganate, manganese dioxide, filtration This manganese mud time-consuming, inefficient, polluting; the second step conversion was about 70%, and post-processing cumbersome; byproducts and produced the first two steps more. This makes the high cost of the entire route, not suitable for the production of amplification.

CN200710094021.4 discloses another method for preparing the compounds of formula I, the following route B, the starting material by nucleophilic substitution, oxidation, an ester, a tetrazole ring to obtain a compound of formula I, the first step of the method nucleophilic substitution easy to generate an imidazole ring -3 para isomer impurities difficult to remove; the last step into the ring to use sodium azide, operating dangerous.

CN201210020174.5 disclosed a series of anti-hypertensive compound and preparation method, the following line C, the temperature control in the first step of its preparation O ~ 5 ° C, a mixed solution of acetone and water, with a 5% aqueous solution of sodium hypochlorite oxidation, yield 70%, the second step use of potassium permanganate, manganese dioxide will produce the same, and a yield of only 40%, the first two steps total yield of 28%, is very low, and the post-treatment methods are by column separation, the first two steps are used are organic and inorganic mixed solvent is not conducive to recovery, not suitable for scale-up.






Example 8 2-Butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil crude)

To a 20L reactor 9800ml of methanol, stirring was started, the rotational speed is added at 200r / min 1225.3g solid compound of formula II, and heated to reflux. The reaction 8-10h evacuation HPLC detection, the formula II compound residue <1.0% seen as a response endpoint. After reaching the end of the reaction the heating was stopped, continued stirring speed of 180r / min. About 3_4h fell 20_25 ° C, colorless transparent crystalline solid precipitated. The reaction mixture was cooled to continue to 15-20 ° C, to maintain 15-20 ° C with stirring 3h, the reaction mixture was filtered to give a pale yellow clear filtrate. The filtrate was concentrated under reduced pressure to move 20L flask, vacuum degree of 0.075MPa, 40_45 ° C methanol distilled off under until no distillate. 800ml of absolute ethanol was added, a vacuum degree of 0.075MPa, 40-45 ° C under distillation until no distillate.
900ml of absolute ethanol was added, heated to reflux. N-heptane was added slowly 1100ml, reflux 15min, to -10 ° c / h speed cooled to 15 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration lh, was Allie medoxomil crude (800.lg, yield 93.8%).Purification was used directly in the next step without drying.
Example 9 2-butyl-4-chloro-_1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole-5-carboxylic acid, 1 – [(isopropylamino oxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

850ml of absolute ethanol was added to the 3L reaction vessel was charged with crude Alicante medoxomil (800.lg, 1.45mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1300ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = 1 mixture of filter cake was washed / 3, the back pressure dry vacuum filtration, the purified Alicante medoxomil (780.9g, 97.6% yield).
Example 10 2-butyl-4-chloro _1- [2 ‘- (1-tetrazol-5-yl biphenyl – methyl] imidazole
5-carboxylic acid, 1 – [(isopropoxy) carbonyl] -L-methoxy ester (Alicante medoxomil)

950ml of absolute ethanol was added to the 5L reaction vessel was charged with crude Alicante medoxomil (549.9g, 1.72mol), heated to reflux. After completely dissolved clear, slow addition of n-heptane 1200ml, reflux 15min, to -10 ° C / h speed cooled to 10 ± 2 ° C, keep stirring 3h. Filtered under reduced pressure, ethanol / n-heptane = cake was washed with a mixture of 1/3, and dried under reduced pressure after filtration to obtain a purified Alicante medoxomil (540.0g, 98.2% yield).
Example 122-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester (compound 8)

To a 100 ml of one-necked flask, 0.523 g of material, 0.124 g of potassium carbonate, 5 ml of N,N-dimethylacetamide were added in turn. The solution was stirred at room temperature for 20 minutes. Then 0.562 g of 1-chloromethyl isopropyl carbonate was added and the mixture was reacted at 45-50° C. for 16 hours. After the reaction was completed, the mixture solution was filtered, and 30 ml of water was added into the filtrate. The resulting mixture was extracted with 30 ml of ethyl acetate twice. The organic phase was dried and concentrated to give 1.724 g of oil, which was directly used in the next reaction without purification.
10 ml of dioxane and 5 ml of 4 mol/L HCl were added, and the resulting mixture was reacted at room temperature for 16 hours. The reaction was stopped and the solution was adjusted to pH 6-7 using aqueous sodium bicarbonate solution. The solution went turbid, and was extracted with ethyl acetate. The organic phase was washed with saturated brine, dried, concentrated to give 0.436 g of 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester.
In addition, the following reaction condition can be used to deprotect the protecting group. To 1.7 g of oily product, 5 ml absolute methanol was added and the mixture was heated slowly to reflux and stirred for 8 hours. When the insoluble solid disappeared totally, the mixture was discontinued to heating and cooled to 5° C. The white solid precipitated, and was separated by filtration, and the filter cake was washed with a small quantity of methanol. The combined filtrate was concentrated to dryness to give 2-butyl-4-chloro-1-[2′-(1H-tetrazol-5-yl)1,1′-biphenyl-methyl]imidazole-5-carboxylic acid, 1-[(isopropoxycarbonyl)oxy]methyl ester with the yield of 70%.
1H-NMR (CDCl3) δ H (ppm): 0.89 (t, 3H, J=14.6), 1.24 (d, 6H, J=6.3), 0.37 (m, 2H, J=22.1), 1.69 (m, 2H, J=30.5), 2.64 (t, 2H, J=15.5), 4.81 (m, 1H, J=12.4), 5.54 (s, 2H), 5.86 (s, 2H), 6.95-7.64 (8H), 8.08 (d, 1H, J=7.42)
ESI(+) m/z: 552.7
Mp: 134.5-136° C.




| WO2005011646A2 * | 20 Jul 2004 | 10 Feb 2005 | Nicoletta Almirante | Nitrooxy derivatives of losartan, valsatan, candesartan, telmisartan, eprosartan and olmesartan as angiotensin-ii receptor blockers for the treatment of cardiovascular diseases |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8455526 * | 6 Jun 2008 | 4 Jun 2013 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| US20100168193 * | 6 Jun 2008 | 1 Jul 2010 | Shanghai Allist Pharmaceuticals, Inc. | Therapeutic use of imidazole-5-carboxylic acid derivatives |
| USRE44873 | 31 Jul 2006 | 29 Apr 2014 | Salubris Asset Management Co., Ltd. | Imidazole-5-carboxylic acid derivatives, the preparation method therefor and the uses thereof |
| CN101024643A | 20 Feb 2006 | 29 Aug 2007 | 上海艾力斯医药科技有限公司 | Imidazo-5-carboxylic-acid derivatives, its preparing method and use |
| US5298519 * | 24 Sep 1992 | 29 Mar 1994 | Chemish Pharmazeutische Forschungsgesellschaft M.B.H. | Acylals of imidazole-5-carboxylic acid derivatives, and their use as angiotensin (II) inhibitors |
……………….
update……………..
Example 1
Weigh 25g 2- butyl-4-chloro-1- [2 ‘- (1-trityl–1H- tetrazol-5-yl) -1,1’-biphenyl – methyl] – imidazole 5-carboxylic acid, 1 – [(isopropoxy) – carbonyloxy] -, methyl ester, was added to a 500ml three-necked flask, methanol was added 200ml, refluxed for 9h, methanol was distilled off under reduced pressure to give crude Alicante medoxomil .
To the residue (i.e., medoxomil crude Alicante) were added 33ml of isopropanol and 66ml of n-heptane, heated to 76 ℃ stirred for 2h. After cooling to 60 ℃ stirring for 1h, and then the system was slowly cooled to 0 ℃, stirring was continued for 3h. Filtered, the filter cake was washed with n-heptane. At 40 ℃ 8 hours and dried in vacuo to give 15.3g Alicante medoxomil (purity 99.3%) as a XRD spectrum as shown in Figure, the main peak of the diffraction peaks as shown in the following table, the DSC spectrum shown in figure II . Compared with the published crystal, the crystal obtained by the absence of significant electrostatic phenomena.
Shanghai , CHINA







