
Home » Uncategorized (Page 9)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Amezalpat
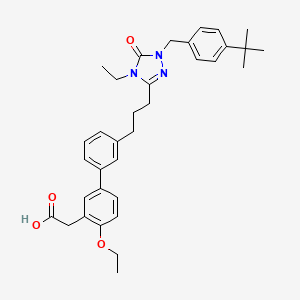
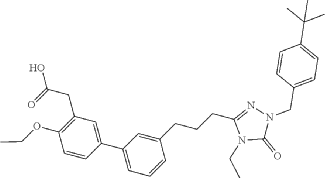
Amezalpat
CAS 1616372-41-8
MF C34H41N3O4 MW555.7 g/mol
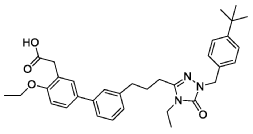
- [1,1′-Biphenyl]-3-acetic acid, 3′-[3-[1-[[4-(1,1-dimethylethyl)phenyl]methyl]-4-ethyl-4,5-dihydro-5-oxo-1H-1,2,4-triazol-3-yl]propyl]-4-ethoxy-
- 2-(3′-(3-(1-(4-(tert-Butyl)benzyl)-4-ethyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)-4-ethoxy-[1,1′-biphenyl]-3-yl)acetic acid
- 3′-(3-(1-((4-(1,1-DIMETHYLETHYL)PHENYL)METHYL)-4-ETHYL-4,5-DIHYDRO-5-OXO-1H-1,2,4-TRIAZOL-3-YL)PROPYL)-4-ETHOXY(1,1′-BIPHENYL)-3-ACETIC ACID
2-[5-[3-[3-[1-[(4-tert-butylphenyl)methyl]-4-ethyl-5-oxo-1,2,4-triazol-3-yl]propyl]phenyl]-2-ethoxyphenyl]acetic acid
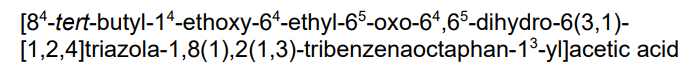
peroxisome proliferator-activated receptor alpha (PPARα) antagonist, antineoplastic, TPST 1120, FDA Fast Track, Orphan Drug, 1EQ4LQN9N3
Amezalpat (formerly TPST-1120) is an investigational, oral, small-molecule inhibitor targeting peroxisome proliferator-activated receptor alpha (PPAR being developed by Tempest Therapeutics. It works by directly targeting tumor cells and reducing immune suppression in the tumor microenvironment. In combination with atezolizumab and bevacizumab, it has shown improved survival in hepatocellular carcinoma (HCC) patients, receiving FDA Fast Track and Orphan Drug designations.
Key Details on Amezalpat
- Indication: Primarily being studied for unresectable or metastatic hepatocellular carcinoma (liver cancer).
- Mechanism: A selective, competitive antagonist of PPAR
, which plays a role in fatty acid metabolism in cancer cells.
- Clinical Efficacy: A phase 1b/2 study indicated that adding amezalpat to standard-of-care (atezolizumab + bevacizumab) improved median overall survival to 21 months compared to 15 months for the control, according to Tempest Therapeutics.
- Trial Status: A pivotal Phase 3 study (NCT06680258) to evaluate this combination as a first-line treatment is planned for 2025.
- Other Potential Uses: Preclinical data suggests potential activity in other advanced solid tumors, including renal cell carcinoma.
Disclaimer: Amezalpat is an investigational agent and is not yet approved by the FDA for widespread clinical use.
Amezalpat is an orally bioavailable, small molecule, selective and competitive antagonist of peroxisome proliferator activated receptor alpha (PPARa), with potential immunomodulating and antineoplastic activities. Upon oral administration, amezalpat targets, binds to and blocks the activity of PPARa, thereby blocking transcription of PPARa target genes leading to an intracellular metabolism shift from fatty acid oxidation (FAO) to glycolysis in FAO-dependent tumors and reducing the production of fatty acids in the tumor microenvironment (TME). As fatty acids are essential for tumor cell growth in FAO-dependent tumor cells and are needed for the metabolism of suppressive immune cells in the TME, including regulatory T-cells (Tregs), reducing the amount of fatty acids leads to a direct killing of FAO-dependent tumor cells. It also skews macrophages from the immune suppressive M2 phenotype to an effector M1 phenotype and facilitates the cytotoxicity of immune effector cells, thereby stimulating an anti-tumor immune response and further killing tumor cells. Amezalpat also restores the natural inhibitor of angiogenesis thrombospondin-1 (TSP-1) and stimulator of interferon genes (STING) in the TME. PPARa, a ligand-activated nuclear transcription factor and metabolic checkpoint, regulates the expression of FAO genes and lipid metabolism. It plays a key role in immunosuppression in the TME. FAO is a metabolic pathway essential to tumor growth, survival and immunosuppression.
SYN
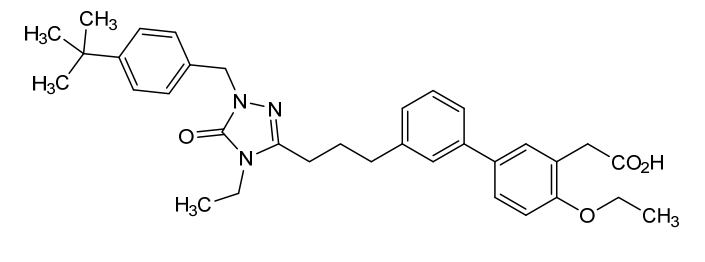
Example 6: 2-(3′-(3-(1-(4-(tert-Butyl)benzyl)-4-ethyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)-4-ethoxy-[1,1′-biphenyl]-3-yl)acetic acid
SYN
SYN
WO2014099503 TRIAZOLONE COMPOUNDS AND USES THEREOF
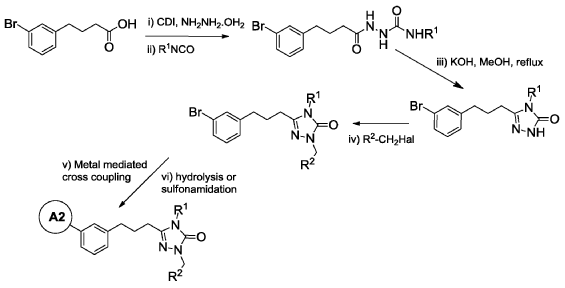
Example 6: 2-(3′-(3-(1-(4-(tert-Butyl)benzyl)-4-ethyl-5-oxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)propyl)-4-ethoxy-[1, 1′-biphenyl]-3-yl)acetic acid
Pat
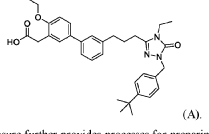
WO2025235527 CRYSTALLINE FORMS OF A PPAR ALPHA ANTAGONIST
2-(3′-(3-(l-(4-(tertbutyl)benzyl)-4-ethyl-5-oxo-4,5-dihydro-lH-l,2,4-triazol-3-yl)propyl)-4-ethoxy-[1,T-biphenyl]-3-yl)acetic acid, depicted below as Compound A
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

{kind=link}
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Triazolone compounds and uses thereofPublication Number: US-2017239223-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: WO-2014099503-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: US-10568871-B2Priority Date: 2012-12-20Grant Date: 2020-02-25
- Triazolone compounds and uses thereofPublication Number: US-2024041837-A1Priority Date: 2012-12-20
- Compound or pharmaceutically acceptable salt thereof, pharmaceutical composition and uses thereofPublication Number: BR-112015013350-B1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: US-2015344446-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: US-11666557-B2Priority Date: 2012-12-20Grant Date: 2023-06-06
- Triazolone compounds and uses thereofPublication Number: US-2020138790-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: US-9676754-B2Priority Date: 2012-12-20Grant Date: 2017-06-13
- Triazolone compounds and uses thereofPublication Number: CA-2894281-CPriority Date: 2012-12-20Grant Date: 2021-04-20
- Triazolone compounds and uses thereofPublication Number: WO-2024102620-A2Priority Date: 2022-11-09
- Triazolone compounds and uses thereofPublication Number: AU-2013363398-B2Priority Date: 2012-12-20Grant Date: 2017-06-01
- Triazolone compounds and uses thereofPublication Number: EP-2935228-B9Priority Date: 2012-12-20Grant Date: 2017-12-06
- Triazolone compounds and uses thereofPublication Number: CA-2894281-A1Priority Date: 2012-12-20
- Triazolone compounds and uses thereofPublication Number: EP-2935228-B1Priority Date: 2012-12-20Grant Date: 2017-08-02
/////////////amezalpat, ANAX LAB, antineoplastic, TPST 1120, FDA Fast Track, Orphan Drug, 1EQ4LQN9N3
Alnodesertib
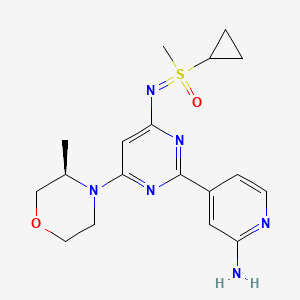
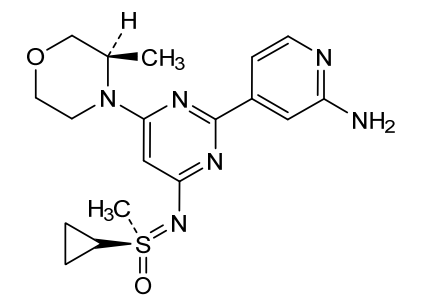
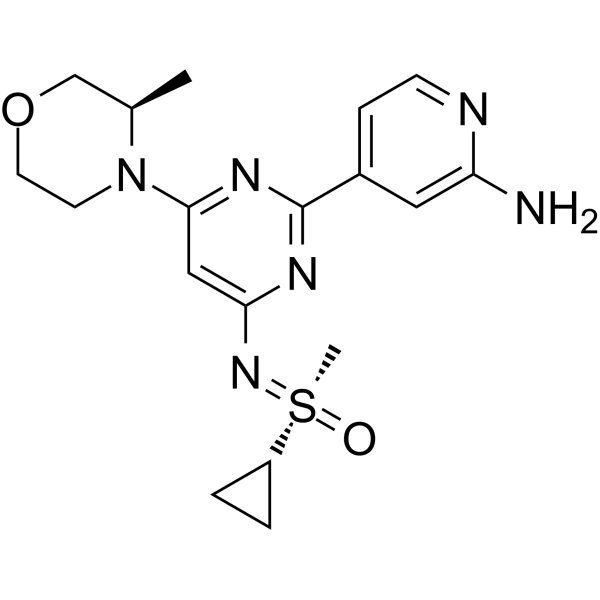
Alnodesertib
CAS 2267316-76-5
MF C18H24N6O2S MW388.49
4-[4-[(cyclopropyl-methyl-oxo-λ6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine
4-[4-[(cyclopropyl-methyl-oxo-lambda6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine
(S)-({2-(2-aminopyridin-4-yl)-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}imino)(cyclopropyl)(methyl)-λ6
-sulfanone
serine/threonine kinase inhibitor, antineoplastic, ART 0380, EX-A9085
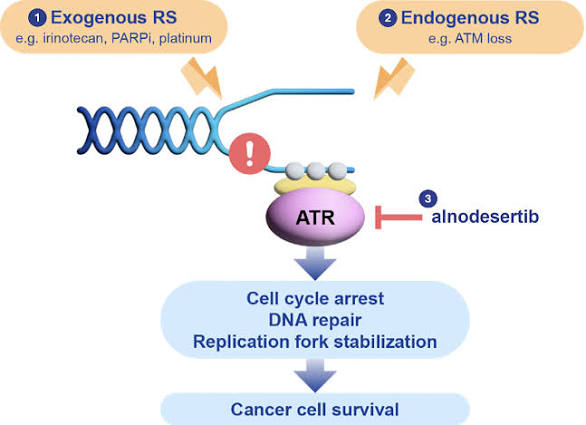
Alnodesertib (formerly known as ART0380) is an investigational, orally administered drug designed to treat various types of cancer. It is a selective inhibitor of ATR (Ataxia-Telangiectasia and Rad3-related protein), a key kinase involved in DNA repair and cell cycle progression.
Mechanism of Action
Alnodesertib works by disrupting the DNA Damage Response (DDR):
- Targets ATR Kinase: It selectively inhibits ATR, which cancer cells rely on to fix DNA damage caused by rapid replication.
- Blocks Signaling: By blocking the phosphorylation of CHK1, it prevents the activation of DNA damage checkpoints.
- Induces Apoptosis: Inhibiting these repair pathways prevents cancer cells from surviving replication stress, ultimately leading to cell death (apoptosis).

Clinical Status and Indications
As of early 2026, alnodesertib is undergoing several clinical trials:
- Metastatic Colorectal Cancer (mCRC): The FDA granted Fast Track designation in September 2025 for alnodesertib in combination with irinotecan for adult patients with ATM-negative mCRC in the third-line setting.
- Ovarian Cancer: In March 2026, Artios Pharma reported that a Phase 2a study reached its primary endpoint, showing that adding a low dose of alnodesertib to gemcitabine improved progression-free survival in patients with platinum-resistant high-grade serous ovarian carcinoma (HGSOC).
- Other Solid Tumours: It is being evaluated in the ongoing STELLA Phase 1/2a study for its potential across multiple solid tumour types characterized by high replication stress.
Key Facts
| Feature | Details |
|---|---|
| Developer | Artios Pharma Limited |
| Drug Class | ATR Kinase Inhibitor |
| Administration | Oral |
| Current Phase | Phase 2 clinical trials |
| FDA Status | Fast Track Designation (for ATM-negative mCRC) |
SYN
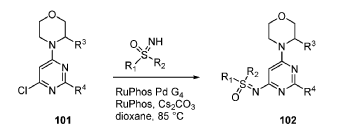
EXAMPLES 39a and 39b
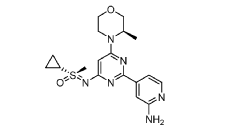
(R)-((2-(2-aminopyridin-4-yl)-6-((R)-3-methylmorpholino)pyrimidin-4- yl)imino)(cyclopropyl)(methyl)-λ6-sulfanone
and
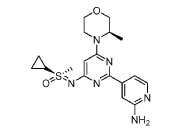
(S)-((2-(2-aminopyridin-4-yl)-6-((R)-3-methylmorpholino)pyrimidin-4- yl)imino)(cyclopropyl)(methyl)-λ6-sulfanone
[0648] Synthesis is similar to that described for Example 24. The mixture of diastereomers (26.8 mg, 0.069 mmol) was separated by Chiral SFC (Mobile phase: n-hexane (0.1% DEA):EtOH(0.1% DEA) = 70:30; Flow rate: 80 g / min; 20 min; Column temperature: 35 °C; Back pressure: 100 bar; Column: Gilson-281, AY 20 x 250mm, 10 μm) to afford the two diastereomers of unknown absolute stereochemistry at the sulfur atom, title compounds 39a (6.6mg, 25% yield, >99% ee) as a white solid and 39b (7.1mg, 27% yield, >99% ee) as a white solid.
[0649] 39a ((R)-cyclopropyl(methyl)-λ6-sulfanone or (S)-cyclopropyl(methyl)-λ6-sulfanone): 1H NMR (500 MHz, CD3OD) δ 8.03 – 7.91 (m, 1H), 7.53 (s, 1H), 7.49 (dd, J =5.5, 1.4 Hz, 1H), 5.97 (s, 1H), 4.48 (d, J = 4.6 Hz, 1H), 4.11 (d, J = 12.0 Hz, 1H), 4.02 (dt, J = 11.3, 3.6 Hz, 1H), 3.82 (d, J = 11.4 Hz, 1H), 3.75 (dt, J = 11.5, 3.0 Hz, 1H), 3.65 – 3.56 (m, 4H), 3.25 (tdJ, = 12.8, 3.8 Hz, 1H), 3.01 (td, J = 7.9, 4.0 Hz, 1H), 1.42 (dd, J = 10.2, 5.4 Hz, 1H), 1.31 (dt, J = 11.1, 6.2 Hz, 4H), 1.20 (dt,J = 11.3, 5.7 Hz, 2H); MS (ES+) C18H24N6O2S requires: 388, found: 389 [M+H]+; Rt = 11.35 min.
[0650] 39b ((R)-cyclopropyl(methyl)-λ6-sulfanone or (S)-cyclopropyl(methyl)-λ6-sulfanone): 1H NMR (500 MHz, CD3OD) δ 7.97 (d, J = 5.4 Hz, 1H), 7.53 (s, 1H), 7.49 (dt, J = 5.5, 1.3 Hz, 1H), 5.97 (s, 1H), 4.50 (s, 1H), 4.08 (d, J = 12.7 Hz, 1H), 4.02 (dd, J = 11.4, 3.7 Hz, 1H), 3.82 (d, J = 11.3 Hz, 1H), 3.75 (dt, J = 11.4, 3.0 Hz, 1H), 3.66 – 3.55 (m, 4H), 3.25 (tdJ, = 12.9, 3.9 Hz, 1H), 3.05 – 2.97 (m, 1H), 1.41 (dt, J = 10.6, 5.2 Hz, 1H), 1.31 (dd, J = 11.8, 5.8 Hz, 4H), 1.20 (dt, J = 11.1, 5.6 Hz, 2H); MS (ES+) C18H24N6O2S requires: 388, found: 389 [M+H]+; Rt = 15.22 min.
[0651] Alternatively, Example 39a can also be prepared from Int. CC, Isomer lb.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- HETEROCYCLIC INHIBITORS OF KINASE ATRPublication Number: WO-2019014618-A1Priority Date: 2017-07-13
- Heterocyclic inhibitors of ATR kinasePublication Number: US-11434233-B2Priority Date: 2017-07-13Grant Date: 2022-09-06
- Heterocyclic inhibitors of ATR kinasePublication Number: US-10800769-B2Priority Date: 2017-07-13Grant Date: 2020-10-13
- Heterocyclic inhibitors of ATR kinasePublication Number: US-10392376-B2Priority Date: 2017-07-13Grant Date: 2019-08-27
- Heterocyclic inhibitors of atr kinasePublication Number: US-2019016713-A1Priority Date: 2017-07-13
- Heterocyclic inhibitors of atr kinasePublication Number: US-2020102296-A1Priority Date: 2017-07-13
- Heterocyclic inhibitors of atr kinasePublication Number: US-2021047311-A1Priority Date: 2017-07-13
///////alnodesertib, ANAX LAB, serine/threonine kinase inhibitor, antineoplastic, ART 0380, EX-A9085
Alixorexton
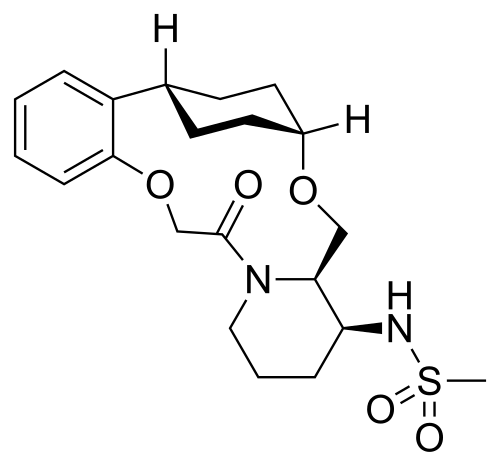
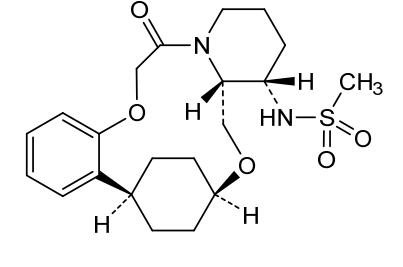
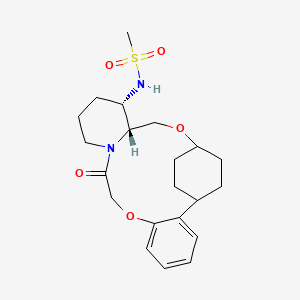
Alixorexton
CAS 2648347-56-0
MF C21H30N2O5S MW 422.5 g/mol
N-((21S,24S,52R,53S)-6-oxo-3,8-dioxa-5(2,1)-piperidina-1(1,2)-benzena-2(1,4)-cyclohexanacyclooctaphane-53-yl)methanesulfonamide
N-[(15S,16R)-10-oxo-8,18-dioxa-11-azatetracyclo[17.2.2.02,7.011,16]tricosa-2,4,6-trien-15-yl]methanesulfonamide
- Methanesulfonamide, N-[(4S,4aR,7alpha,10alpha)-1,2,3,4,4a,5,7,8,9,10,16,17-dodecahydro-17-oxo-7,10-ethanopyrido[1,2-d][1,7,4]benzodioxaazacyclotridecin-4-yl]-
- N-[(4S,4aR,7alpha,10alpha)-1,2,3,4,4a,5,7,8,9,10,16,17-Dodecahydro-17-oxo-7,10-ethanopyrido[1,2-d][1,7,4]benzodioxaazacyclotridecin-4-yl]methanesulfonamide
- N-[7,10-cis-(4S,4aR)-17-oxo-1,2,3,4,4a,5,7,8,9,10,16,17-dodecahydro-7,10-ethanopyrido[1,2-d][1,7,4]benzodioxaazacyclotridecin-4-yl]methanesulfonamide
N-[7,10-cis-(4S,4aR)-17-oxo-1,2,3,4,4a,5,7,8,9,10,16,17-dodecahydro-7,10-ethanopyrido[1,2-
d][1,7,4]benzodioxaazacyclotridecin-4-yl]methanesulfonamide
orexin-2 receptor agonist, ALKS 2680, Breakthrough Therapy designation, 3BRW4ARU66, TAK 994, firazorexton
Alixorexton (formerly ALKS 2680) is an investigational, once-daily, oral, selective orexin 2 receptor (OX2R) agonist being developed by Alkermes for treating narcolepsy and idiopathic hypersomnia. It has shown significant efficacy in Phase 2 trials (Vibrance-1, -2, -3) for improving wakefulness and received FDA Breakthrough Therapy designation in January 2026 for narcolepsy type 1.
- OriginatorAlkermes
- ClassSleep disorder therapies
- Mechanism of ActionOrexin receptor type 2 agonists
- Phase IIIdiopathic hypersomnia; Narcolepsy
- 09 Mar 2026Alkermes plans the phase III Brilliance NT1 trial for Narcolepsy in an undisclosed location (PO), in March 2026 (NCT07455383)
- 06 Jan 2026Alixorexton receives Breakthrough Therapy status for Narcolepsy in USA
- 12 Nov 2025Efficacy and adverse event data from the phase II VIBRANCE-2 trial in Narcolepsy released by Alkermes
Key Details About Alixorexton (ALKS 2680):
- Mechanism of Action: As an OX2R agonist, it targets the orexin system to treat the underlying cause of excessive daytime sleepiness by activating wake-promoting neurons.
- Target Indications: The drug is developed for Narcolepsy Type 1 (NT1), Narcolepsy Type 2 (NT2), and Idiopathic Hypersomnia (IH).
- Clinical Trial Results:
- Vibrance-1 (NT1): Demonstrated significant improvements in wakefulness, reduced cataplexy rates, and improved alertness in phase 2 trials.
- Vibrance-2 (NT2): Showed positive results, meeting primary endpoints in improving sleep latency and reducing daytime sleepiness.
- Safety: Generally well-tolerated, with most adverse events reported as mild to moderate.
- Development Status: Following positive Phase 2 data, Alkermes is preparing for phase 3 development, with plans to advance the program in early 2026.
Alixorexton is distinct as a potential once-daily treatment in a novel, rapidly advancing therapeutic category for sleep disorders
Alixorexton (INNTooltip International Nonproprietary Name; developmental code name ALKS-2680) is an orexin receptor agonist which is under development for the treatment of sleep disorders, including narcolepsy and idiopathic hypersomnia.[1][2][3] Alixorexton is being developed by Alkermes[4] As of July 2025, it is in phase 2 clinical trials with planned advancement to a phase 3 study for the treatment of narcolepsy type 1.[4]
Alixorexton, also known as TAK-994 or firazorexton, is a potent, orally bioavailable, brain-penetrant agonist that selectively targets the orexin 2 receptor (OX2R), with additional activity at the orexin 1 receptor (OX1R). Its mechanism of action involves mimicking the endogenous orexin neuropeptides, thereby promoting wakefulness. Preclinical studies have demonstrated that Alixorexton effectively suppresses fragmentation of wakefulness and reduces cataplexy-like episodes in mouse models of narcolepsy, indicating its potential as a therapeutic agent for sleep disorders.
- A Long-Term Study of ALKS 2680 in Subjects With Narcolepsy and Idiopathic HypersomniaCTID: NCT06767683Phase: Phase 2/Phase 3Status: RecruitingDate: 2026-03-04
- A Study to Evaluate the Safety and Effectiveness of ALKS 2680 in Subjects With Narcolepsy Type 2CTID: NCT06555783Phase: Phase 2Status: CompletedDate: 2026-02-27
- A Study to Evaluate the Safety and Effectiveness of ALKS 2680 in Subjects With Idiopathic HypersomniaCTID: NCT06843590Phase: Phase 2Status: RecruitingDate: 2026-02-12
- A Study to Evaluate the Safety and Effectiveness of ALKS 2680 in Subjects With Narcolepsy Type 1 (ALKS 2680-201)CTID: NCT06358950Phase: Phase 2Status: CompletedDate: 2025-10-14
SYN
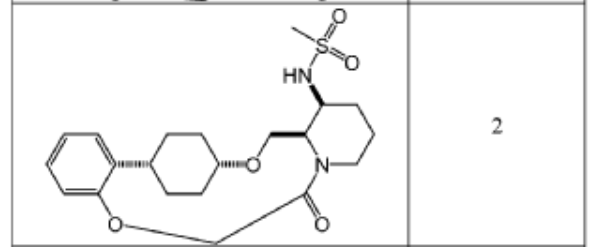
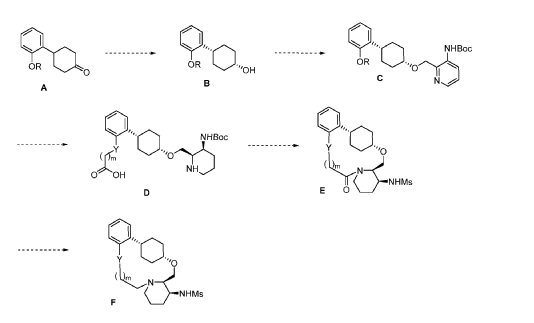
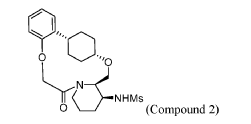
To (21S,24S,52R, 535)-53-amino-3,8-dioxa-5(2,1)-piperidina-1(1,2)-benzena-2(1,4)-cyclohexanacyclooctaphan-6-one 32.1 g (900 mg, 2.613 mmol, 1 equiv.) and DIPEA (1.69 g, 13.064 mmol, 5 equiv.) in DCM (148 mL) was added MsCl (900 mg, 7.858 mmol, 3 equiv.) dropwise at room temperature under nitrogen atmosphere. The resulting solution was stirred for 2 hr at room temperature. The reaction was then quenched by the addition of 50 mL of water. The resulting solution was extracted with 3 x 200 mL of dichloromethane, dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product was purified by Prep-HPLC to afford racemic crude product (800 mg, 72.46%) as a solid. The crude product was purified by prep-SFC to afford N-((21S,24S,52R,53S)-6-oxo-3,8-dioxa-5(2, 1)-piperidina-1(1, 2)-benzena-2(1, 4)-cyclohexanacyclooctaphane-53-yl)methanesulfonamide (270.6 mg, 27.1%) as a solid and its enantiomer, N-((21R,24R,52,53R)-6-oxo-3,8-dioxa-5(2, 1)-piperidina-1(1, 2)-benzena-2(1, 4)-cyclohexanacyclooctaphane-53-yl)methanesulfonamide (361.4mg, 36.1%) as a solid. LCMS (ESI): m/z calculated for C21H30N2O5S [M+H]+ = 423.19, found [M+H]+ = 423.15; 1H NMR (400 MHz, CDCl3): δ 7.19 (td, J = 7.7, 1.8 Hz,
1H), 7.11 (dd, J = 7.5, 1.7 Hz, 1H), 6.96 – 6.86 (m, 1H), 6.77 (dd, J = 8.0, 1.1 Hz, 1H), 5.24 (dd, J = 9.8, 4.9 Hz, 1H), 5.14 (d, J = 10.5 Hz, 1H), 4.34 (dd, J = 14.1, 9.4 Hz, 2H), 3.84 (t, J = 9.2 Hz, 1H), 3.73 (d, J = 14.9 Hz, 3H), 3.60 – 3.46 (m, 2H), 3.09 (s, 3H), 2.77 – 2.49 (m, 2H), 2.33 – 2.16 (m, 1H), 2.06 (d, J = 12.8 Hz, 2H), 2.00 – 1.82 (m, 2H), 1.68 (d, J = 11.5 Hz, 2H), 1.10 – 1.45 (m, 4H).
SYN
CREDIT MEDKOO
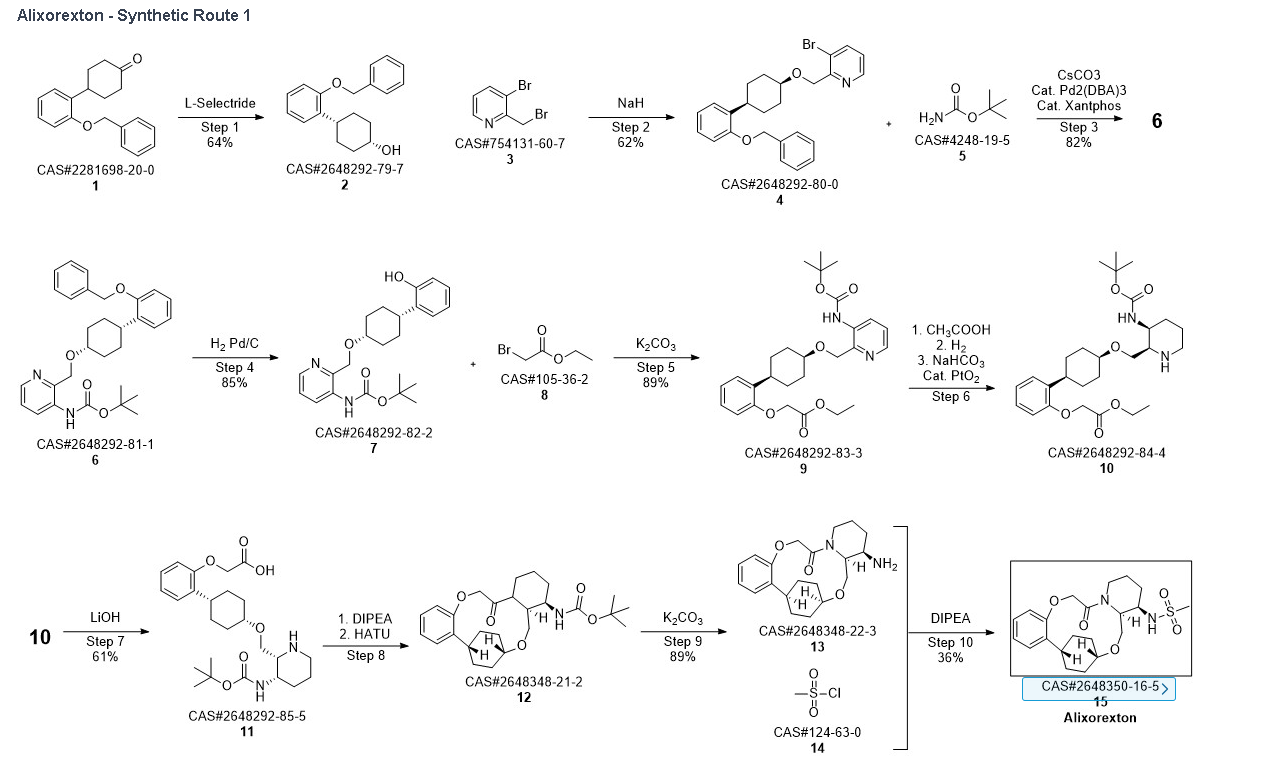
Synthetic Reference
Preparation of substituted macrocyclic derivatives for use in the treatment of narcolepsy or cataplexy
Assignee: Alkermes, Inc.
Inventors: Pennington, Lewis D.; et al
United States
Patent#US20210155636 A1
Example 1.1
Into a 5-L 4-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed 4-[2-(benzyloxy)phenyl]cyclohexan-1-one (210 g, 749 mmol, 1.00 equiv.) in tetrahydrofuran (2.1 L). This was followed by the addition of L-selectride (1 mol/L in THF) (1123 mL, 5257 mmol, 1.50 equiv.) dropwise with stirring at 0 degrees C. The resulting solution was stirred for 4 hr at room temperature. The reaction was then quenched by the addition of water/ice. The resulting solution was extracted with ethyl acetate, and the organic phase was washed with brine. The mixture was dried over anhydrous sodium sulfate, filtered, concentrated under vacuum. The residue was purified by silica gel column chromatography with ethyl acetate/petroleum ether (1:100-1:5) to give 137 g (64%) of (1s,4s)-4-[2-(benzyloxy)phenyl]cyclohexan-1-ol as a solid. 1H NMR (400 MHz, CDCl 3): δ 7.45-7.26 (6H, m), 7.16 (1H, dd), 6.98-6.90 (2H, m), 5.09 (2H, s), 4.13 (1H, s), 3.12-3.02 (1H, m), 1.93-1.82 (4H, m), 1.73-1.41 (4H, m).
Into a 2-L 4-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed NaH (60% wt, 26.9 g, 2.00 equiv.) in tetrahydrofuran (200 mL). This was followed by the addition of a solution of (1s, 4s)-4-[2-(benzyloxy)phenyl]cyclohexan-1-ol (95 g, 336 mmol, 1.00 equiv.) in THF (200 mL) dropwise with stirring at 50-55 degrees C. After stirring for 2 hr, to this was added a solution of 3-bromo-2-(bromomethyl)pyridine (143.5 g, 571 mmol, 1.70 equiv.) in THF (550 mL) dropwise with stirring at 50-55 degrees C. The resulting solution was stirred for 14 hr at 50-55 degrees C. The reaction mixture was cooled. The reaction was then quenched by the addition of water. The resulting solution was extracted with ethyl acetate and the organic layers combined and dried over anhydrous sodium sulfate. The solids were filtered out. The resulting mixture was concentrated under vacuum. The residue was purified by silica gel column chromatography with ethyl acetate/petroleum ether (1:100-1:2) to give 94 g (62%) of 3-bromo-2-([[(1s,4s)-4-[2-(benzyloxy) phenyl]cyclohexyl]oxy]methyl)pyridine as a solid. 1H NMR (400 MHz, CDCl 3): δ 8.57 (1H, d), 7.90 (1H, dd), 7.48-7.26 (6H, m), 7.18-7.14 (2H, m), 6.98-6.91 (2H, m), 5.12 (2H, s), 4.77 (2H, s), 3.86 (1H, s), 3.17-3.10 (1H, m), 2.20-2.15 (2H, m), 1.98-1.88 (2H, m), 1.69-1.57 (4H, m).
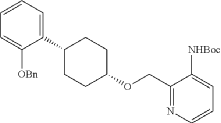
Into a 2-L 4-necked round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed Xantphos (10.7 g, 18 mmol, 0.10 equiv.), Cs 2CO 3 (84 g, 258 mmol, 1.39 equiv.), 3-bromo-2-([[(1s,4s)-4-[2-(benzyloxy) phenyl]cyclohexyl]oxy]methyl)pyridine (84 g, 185 mmol, 1.00 equiv.), Pd 2(dba) 3 (8.5 g, 9 mmol, 0.05 equiv.) and tert-butyl carbamate (26 g, 222 mmol, 1.20 equiv.) in dioxane (840 mL). The resulting solution was stirred for 5 hr at 100 degrees C. The solids were filtered out. The filtrate was concentrated under vacuum. The residue was purified by silica gel column chromatography with ethyl acetate/petroleum ether (1:100-1:4) to provide 74 g (82%) of tert-butyl N-[2-([[(1s,4s)-4-[2-(benzyloxy)phenyl]cyclohexyl]oxy]methyl)pyridin-3-yl]carbamate as a solid.
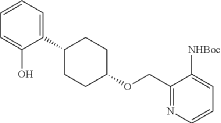
Into a 2-L 3-necked round-bottom flask, was placed tert-butyl N-[2-([[(1s,4s)-4-[2-(benzyloxy)phenyl]cyclohexyl]oxy]methyl)pyridin-3-yl]carbamate (74 g, 151 mmol, 1.00 equiv.) and Pd/C (7.4 g, 10% wt) in ethyl alcohol (740 mL), then, hydrogen gas was through in. The resulting solution was stirred for 14 hr at room temperature. The solids were filtered out. The filtrate was concentrated under vacuum. The residue was purified by silica gel column chromatography with ethyl acetate/petroleum ether (1:100-1:2) to provide 51.36 g (85%) of tert-butyl N-[2-([[(1s,4s)-4-(2-hydroxyphenyl)cyclohexyl]oxy]methyl)pyridin-3-yl]carbamate as a solid. LCMS (ESI): m/z [M+H] +=399.1; 1H NMR (300 MHz, CDCl 3): δ 8.65 (1H, s), 8.47 (1H, d), 8.19 (1H, q), 7.26-7.21 (1H, m), 7.09-7.03 (1H, m), 6.92-6.86 (1H, m), 6.75 (1H, q), 5.77 (1H, s), 4.84 (1H, s), 3.80 (1H, s), 2.94-2.93 (1H, m), 2.15-2.06 (2H, m), 1.88-1.47 (7H, m), 1.45 (9H, s), 1.26 (1H, d).
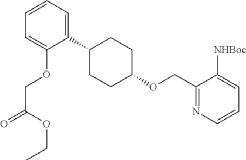
Into a 250-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed tert-butyl N-[2-([[(1s,4s)-4-(2-hydroxyphenyl)cyclohexyl]oxy]methyl)pyridin-3-yl]carbamate (8 g, 20.075 mmol, 1 equiv.), K 2CO 3 (13.97 g, 100.35 mmol, 5 equiv.), acetone (120 mL) and ethyl bromoacetate (5.03 g, 30.119 mmol, 1.5 equiv.). The resulting solution was stirred for 24 hr at 50 degrees C. The solids were filtered out. The filtrate was concentrated under vacuum. The residue was purified by silica gel column chromatography with ethyl acetate/petroleum ether (1:2) to provide ethyl 2-[2-[(1s,4s)-4-([3-[(tert-butoxycarbonyl)amino]pyridin-2-yl]methoxy)cyclohexyl]phenoxy]acetate (8.7 g, 89.43%) as a yellow oil. LCMS (ESI): m/z [M+H] +=485.
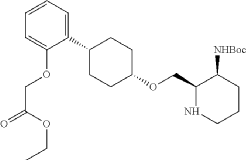
To a stirred mixture of ethyl 2-[2-[(1s,4s)-4-([3-[(tert-butoxycarbonyl)amino]pyridin-2-yl]methoxy)cyclohexyl]phenoxy]acetate (7.89 g, 16.268 mmol, 1 equiv.) in MeOH (142 mL) and AcOH (15.8 mL) were added PtO 2 (1.85 g, 8.142 mmol, 0.50 equiv.) at room temperature under hydrogen atmosphere. The resulting mixture was stirred for 2 hr at room temperature under hydrogen atmosphere. The resulting mixture was filtered, the filter cake was concentrated under reduced pressure. The reaction was quenched with sat. NaHCO 3 (aq.) at 0 degrees C. The resulting mixture was extracted with CH 2Cl 2 (3×500 mL). The combined organic layers were washed with brine (3×200 mL), dried over anhydrous Na 2SO 4. After filtration, the filtrate was concentrated under reduced pressure to afford diastereomeric cis and trans mixture (7 g, 88.7%) as a solid. The crude product was purified by Prep-TLC (DCM/MeOH=20:1) to afford cis-racemic mixture of ethyl 2-(2-((1S,4s)-4-((3-((tert-butoxycarbonyl)amino)piperidin-2-yl)methoxy)cyclohexyl)phenoxy)acetate (4.1 g) and trans-racemic mixture (1.7 g). LCMS (ESI): m/z [M+H] +=491.
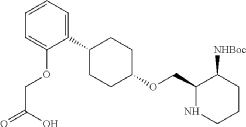
Into a 500 mL round-bottom flask purged and maintained with an atmosphere of nitrogen, was placed cis-racemic mixture of ethyl 2-(2-((1S,4s)-4-((3-((tert-butoxycarbonyl)amino)piperidin-2-yl)methoxy)cyclohexyl)phenoxy)acetate (4.1 g, 8.356 mmol, 1 equiv.), MeOH (30 mL), THF (60 mL), H 2O (30 mL) and lithium hydroxide (83 mg, 3.465 mmol, 5 equiv.). The reaction was stirred for 2 hr at room temperature. The reaction was concentrated and the residue was purified by reverse phase flash with the following conditions, then freezing-drying to afford 2-(2-((1s,4s)-4-((3-((tert-butoxycarbonyl)amino)piperidin-2-yl)methoxy)cyclohexyl)phenoxy)acetic acid (2.35 g, 60.8%) as a solid. LCMS (ESI): m/z [M+H] +=463
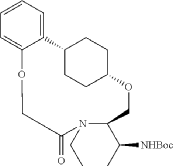
Into a 2000-mL round-bottom flask was added 2-(2-((1s,4s)-4-((3-((tert-butoxycarbonyl)amino)piperidin-2-yl)methoxy)cyclohexyl)phenoxy)acetic acid (100 mg, 0.216 mmol, 1 equiv.), MeCN (36 mL), DMF (9 mL), HATU (124 mg, 0.326 mmol, 1.51 equiv.) and DIPEA (56 mg, 0.436 mmol, 2.02 equiv.) under nitrogen atmosphere. The resulting solution was stirred for 3 hr at room temperature. LCMS showed full conversation. The resulting mixture was concentrated. The crude product tert-butyl ((2 1S,2 4S,5 2R,5 3S)-6-oxo-3,8-dioxa-5(2,1)-piperidina-1(1,2)-benzena-2(1,4)-cyclohexanacyclooctaphane-5 3-yl)carbamate was used directly for the next step without purification. LCMS (ESI): m/z [M+H] +=445.
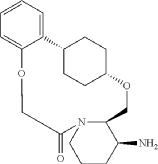
Into a 500-mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed crude mixture tert-butyl ((2 1S,2 4S,5 2R,5 3S)-6-oxo-3,8-dioxa-5(2,1)-piperidina-1(1,2)-benzena-2(1,4)-cyclohexanacyclooctaphane-5 3-yl)carbamate (2 g, 4.499 mmol, 1 equiv.), DCM (120 mL), TFA (40 mL). The resulting solution was stirred for 1 hr at 25 degrees C. LCMS showed full conversation. The resulting mixture was concentrated under vacuum. The crude product was purified by Prep-HPLC to afford (2 1,S,2 4S,5 2R,5 3S)-5 3-amino-3,8-dioxa-5(2,1)-piperidina-1(1,2)-benzena-2(1,4)-cyclohexanacyclooctaphan-6-one 32.1 g (800 mg, 51.6%) as a solid. LCMS (ESI): m/z [M+H] +=345.
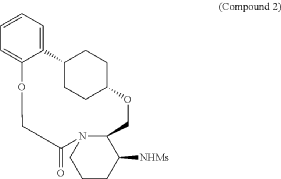
To (2 1S,2 4S,5 2R,5 3S)-5 3-amino-3,8-dioxa-5(2,1)-piperidina-1(1,2)-benzena-2(1,4)-cyclohexanacyclooctaphan-6-one 32.1 g (900 mg, 2.613 mmol, 1 equiv.) and DIPEA (1.69 g, 13.064 mmol, 5 equiv.) in DCM (148 mL) was added MSCl (900 mg, 7.858 mmol, 3 equiv.) dropwise at room temperature under nitrogen atmosphere. The resulting solution was stirred for 2 hr at room temperature. The reaction was then quenched by the addition of 50 mL of water. The resulting solution was extracted with 3×200 mL of dichloromethane, dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product was purified by Prep-HPLC to afford racemic crude product (800 mg, 72.46%) as a solid. The crude product was purified by prep-SFC to afford N-((2 1S,2 4S,5 2R,5 3S)-6-oxo-3,8-dioxa-5(2,1)-piperidina-1(1,2)-benzena-2(1,4)-cyclohexanacyclooctaphane-5 3-yl)methanesulfonamide (270.6 mg, 27.1%) as a solid and its enantiomer, N-((2 1R,2 4R,5 2S,5 3R)-6-oxo-3,8-dioxa-5(2,1)-piperidina-1(1,2)-benzena-2(1,4)-cyclohexanacyclooctaphane-5 3-yl)methanesulfonamide (361.4 mg, 36.1%) as a solid. LCMS (ESI): m/z calculated for C 21H 30N 2O 5S [M+H] +=423.19, found [M+H] +=423.15; 1H NMR (400 MHz, CDCl 3): δ 7.19 (td, J=7.7, 1.8 Hz, 1H), 7.11 (dd, J=7.5, 1.7 Hz, 1H), 6.96-6.86 (m, 1H), 6.77 (dd, J=8.0, 1.1 Hz, 1H), 5.24 (dd, J=9.8, 4.9 Hz, 1H), 5.14 (d, J=10.5 Hz, 1H), 4.34 (dd, J=14.1, 9.4 Hz, 2H), 3.84 (t, J=9.2 Hz, 1H), 3.73 (d, J=14.9 Hz, 3H), 3.60-3.46 (m, 2H), 3.09 (s, 3H), 2.77-2.49 (m, 2H), 2.33-2.16 (m, 1H), 2.06 (d, J=12.8 Hz, 2H), 2.00-1.82 (m, 2H), 1.68 (d, J=11.5 Hz, 2H), 1.10-1.45 (m, 4H).
REF
CHEMICAL.AI
Published Synthetic Route to ALKS 2680 (Alkermes)
The total synthesis of ALKS 2680 was originally reported in US2021/0155636 A1, beginning with a stereoselective reduction of ketone 4a to afford alcohol 5a (Scheme 1). The patented route employed L-selectride for facial selectivity.
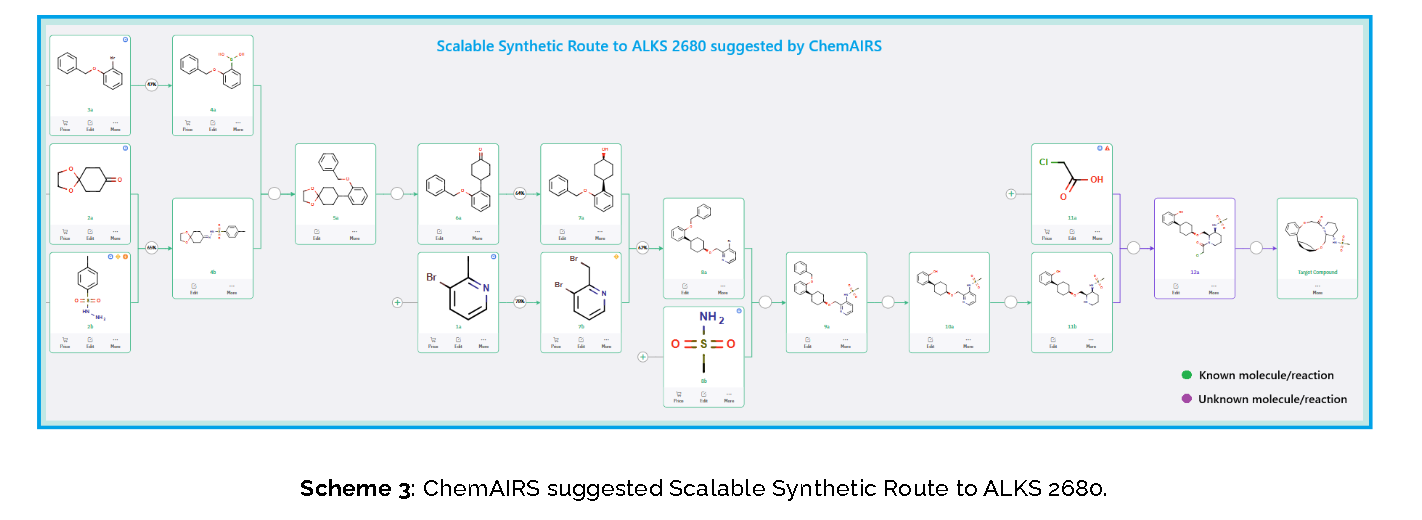
To optimize this step, ChemAIRS proposed an alternative asymmetric hydrogenation utilizing an Ir/f-AmphBINOL catalyst system. This enantioselective methodology has demonstrated excellent control over both enantio- and diastereoselectivity, delivering chiral alcohols in up to 99% ee and a cis/trans ratio of 99:1. [DOI: 10.1021/acs.orglett.3c03550]
Another key transformation in the Alkermes route is the macrocyclization of intermediate 11a using HATU/DIPEA in a mixed MeCN/DMF solvent system to deliver macrocycle 12a.
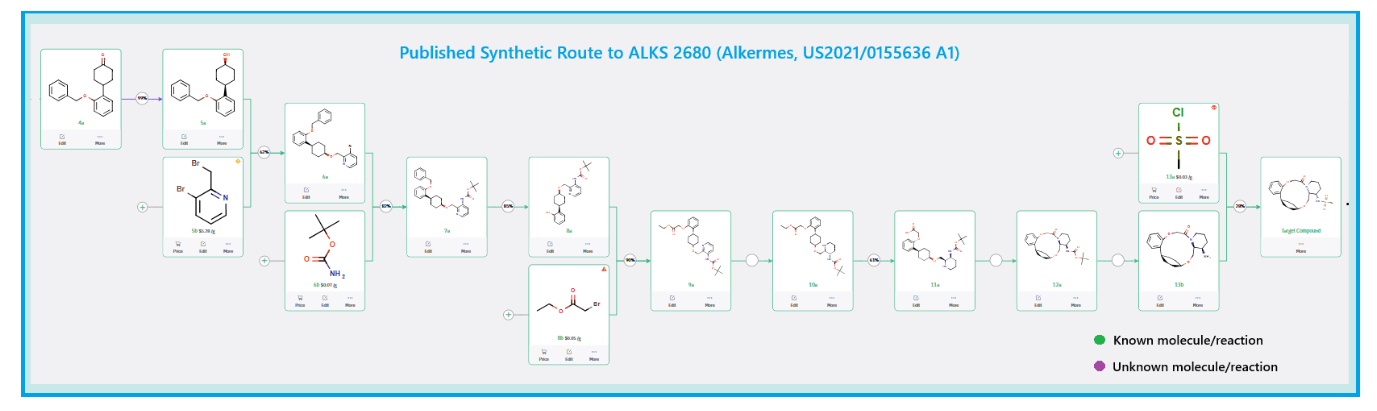
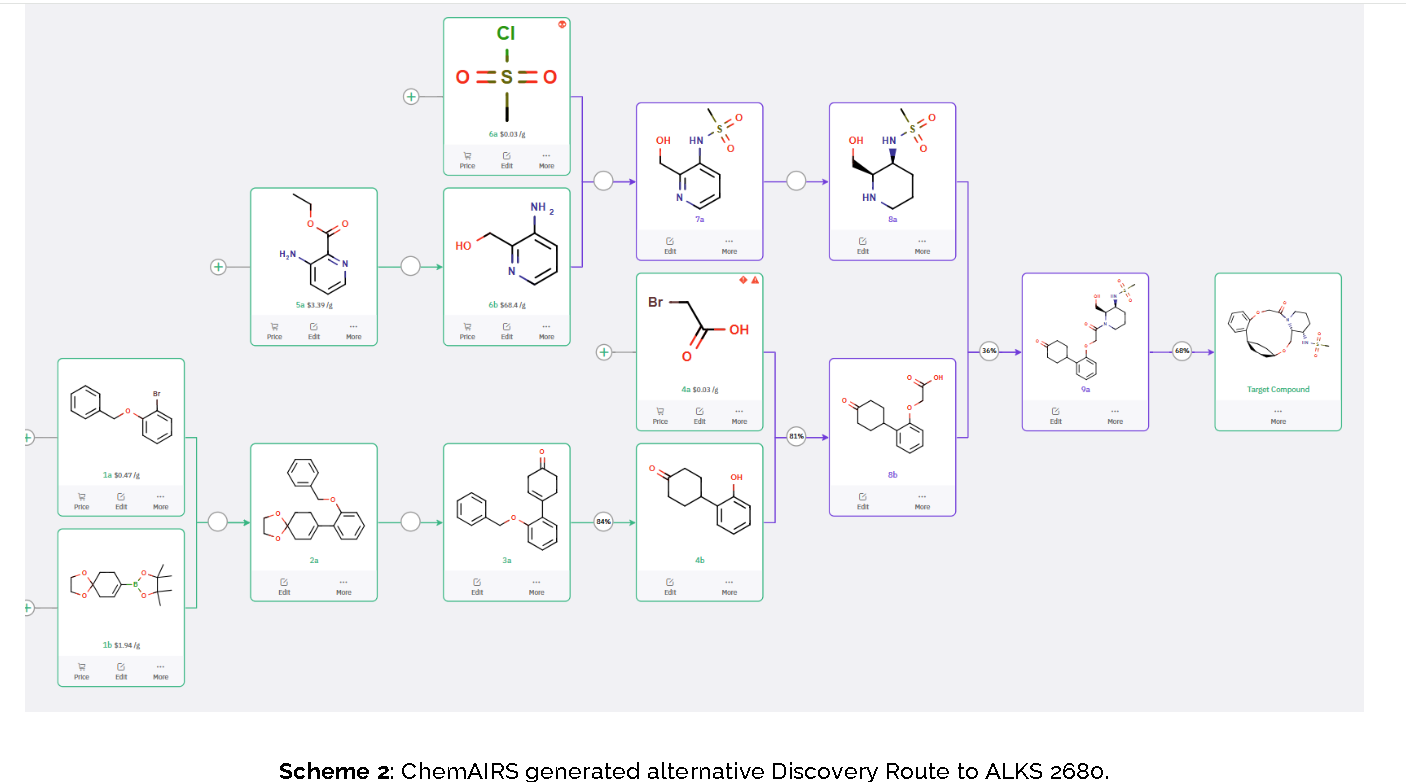
The conversion of (pyridin-3-yl)methanesulfonamide 7a to (piperidin-3-yl)methanesulfonamide 8a was facilitated by Rh/C-catalyzed hydrogenation in EtOH/AcOH, followed by kinetic resolution using (+)-mandelic acid for stereochemical enrichment (WO2017/135306).
The macrocycle was constructed via intramolecular reductive etherification using Et₃SiH/TMSOTf in DCM (doi.org/10.1002/ejoc.201300135), providing the final scaffold in a late-stage cyclization strategy conducive to structural diversification.
PAT
- Substituted macrocyclic compounds and related methods of treatmentPublication Number: US-11542276-B2Priority Date: 2019-11-25Grant Date: 2023-01-03
- Substituted Macrocyclic Compounds and Related Methods of TreatmentPublication Number: US-2021155636-A1Priority Date: 2019-11-25
- Substituted macrocyclic compounds and related methods of treatmentPublication Number: US-2023339969-A1Priority Date: 2019-11-25
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- “Alixorexton – Alkermes”. AdisInsight. Springer Nature Switzerland AG. Retrieved 2025-07-30.
- Morse AM (May 2025). “Enhancing the Management of Hypersomnia: Examining the Role of the Orexin System”. Seminars in Neurology. 45 (3): 410–419. doi:10.1055/a-2589-3825. PMID 40239951.
- Prakash BA, Shah I, Ni G, Vasudevan S, Jagannath A, Foster RG (May 2025). “Dreaming of Better Treatments: Advances in Drug Development for Sleep Medicine and Chronotherapy”. Journal of Sleep Research. 34 (5) e70087. doi:10.1111/jsr.70087. PMID 40346938.
- Chen E (2025-07-21). “Alkermes drug helped narcolepsy patients stay awake in Phase 2 trial”. STAT. Retrieved 2025-07-30.
| Clinical data | |
|---|---|
| Other names | ALKS-2680; ALKS2680 |
| Routes of administration | Oral |
| Drug class | Orexin OX2 receptor agonist |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2648347-56-0 |
| PubChem CID | 156417714 |
| IUPHAR/BPS | 13879 |
| UNII | 3BRW4ARU66 |
| Chemical and physical data | |
| Formula | C21H30N2O5S |
| Molar mass | 422.54 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
/////////alixorexton, anax lab, orexin-2 receptor agonist, ALKS 2680, Breakthrough Therapy designation, 3BRW4ARU66, TAK 994, firazorexton, CHEMICAL AI
Zeprumetostat
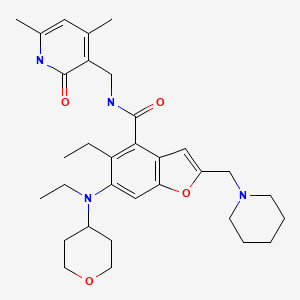
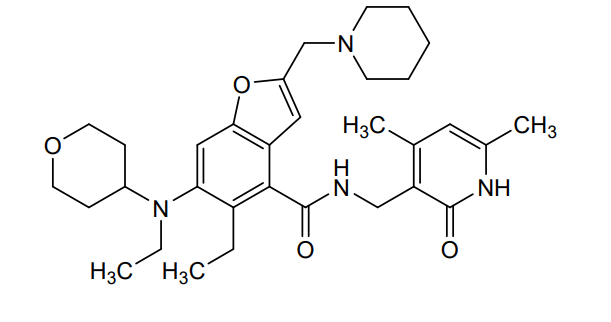
Zeprumetostat
CAS 2098545-98-1
MF C32H44N4O4 MW 548.7 g/mol
CHINA 2025, APPROVALS 2025
- 4-Benzofurancarboxamide, N-[(1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl]-5-ethyl-6-[ethyl(tetrahydro-2H-pyran-4-yl)amino]-2-(1-piperidinylmethyl)-
- N-[(1,2-Dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl]-5-ethyl-6-[ethyl(tetrahydro-2H-pyran-4-yl)amino]-2-(1-piperidinylmethyl)-4-benzofurancarboxamide
N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-5-ethyl-6-[ethyl(oxan-4-yl)amino]-2-[(piperidin-1-yl)methyl]-1-benzofuran-4-carboxamide
enhancer of zeste homolog 2 (EZH2) inhibitor, antineoplastic, Airijing® (China), EZH2-IN-15, SHR 2554
The chemical structure for zeprumetostat was obtained from WHO proposed INN list 131 (August 2024). The INN record describes the compound as an enhancer of zeste homolog 2 (EZH2) inhibitor and antineoplastic. The chemical structure is claimed in patent WO2017084494A1 [3]. Based on Hengrui’s declared development pipeline, we predicted at that time that zeprumetostat was likely the INN for their EZH2 inhibitor clinical lead SHR2554.
Zeprumetostat is an orally available selective inhibitor of the histone lysine methyltransferase (HMT) enhancer of zeste homolog 2 (EZH2), with potential antineoplastic activity. Upon oral administration, zeprumetostat selectively targets, binds to and inhibits the activity of EZH2. Inhibition of EZH2 specifically prevents the methylation of histone H3 on lysine 27 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways and results in decreased proliferation of EZH2-expressing cancer cells. EZH2, an HMT class enzyme and the catalytic subunit of the polycomb repressive complex 2 (PRC2), is overexpressed or mutated in a variety of cancer cells and plays a key role in tumor cell proliferation; its expression is correlated with tumor initiation, progression, stem cell self-renewal, migration and angiogenesis.
Zeprumetostat is a small molecule drug. The usage of the INN stem ‘-metostat’ in the name indicates that Zeprumetostat is a histone N-methyltransferase inhibitor. Zeprumetostat has a monoisotopic molecular weight of 548.34 Da.
- Zeprumetostat, Azacitidine Combined With Lipo-MIT in R/R PTCLCTID: NCT07372352Phase: Phase 2Status: RecruitingDate: 2026-01-28
- EZH2 Inhibitor Zeprumetostat in Combination Therapy for Patients With Relapsed or Refractory Mature T-cell and NK-cell LymphomasCTID: NCT07339527Phase: Phase 1/Phase 2Status: Not yet recruitingDate: 2026-01-14
PAT
WO2017084494
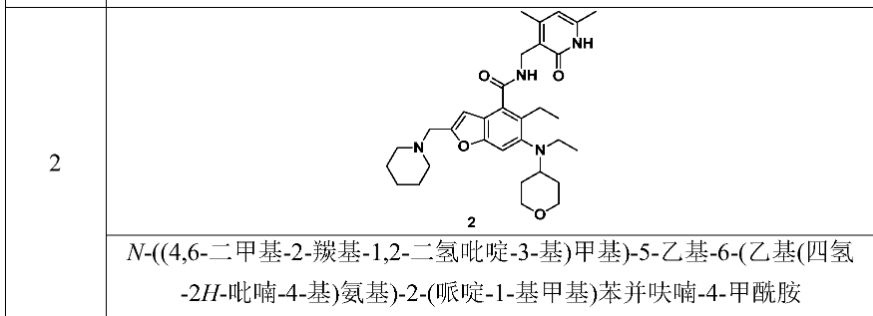
[0234]N-((4,6-dimethyl-2-carbonyl-1,2-dihydropyridin-3-yl)methyl)-5-ethyl-6-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-(piperidin-1-ylmethyl)benzofuran-4-carboxamide
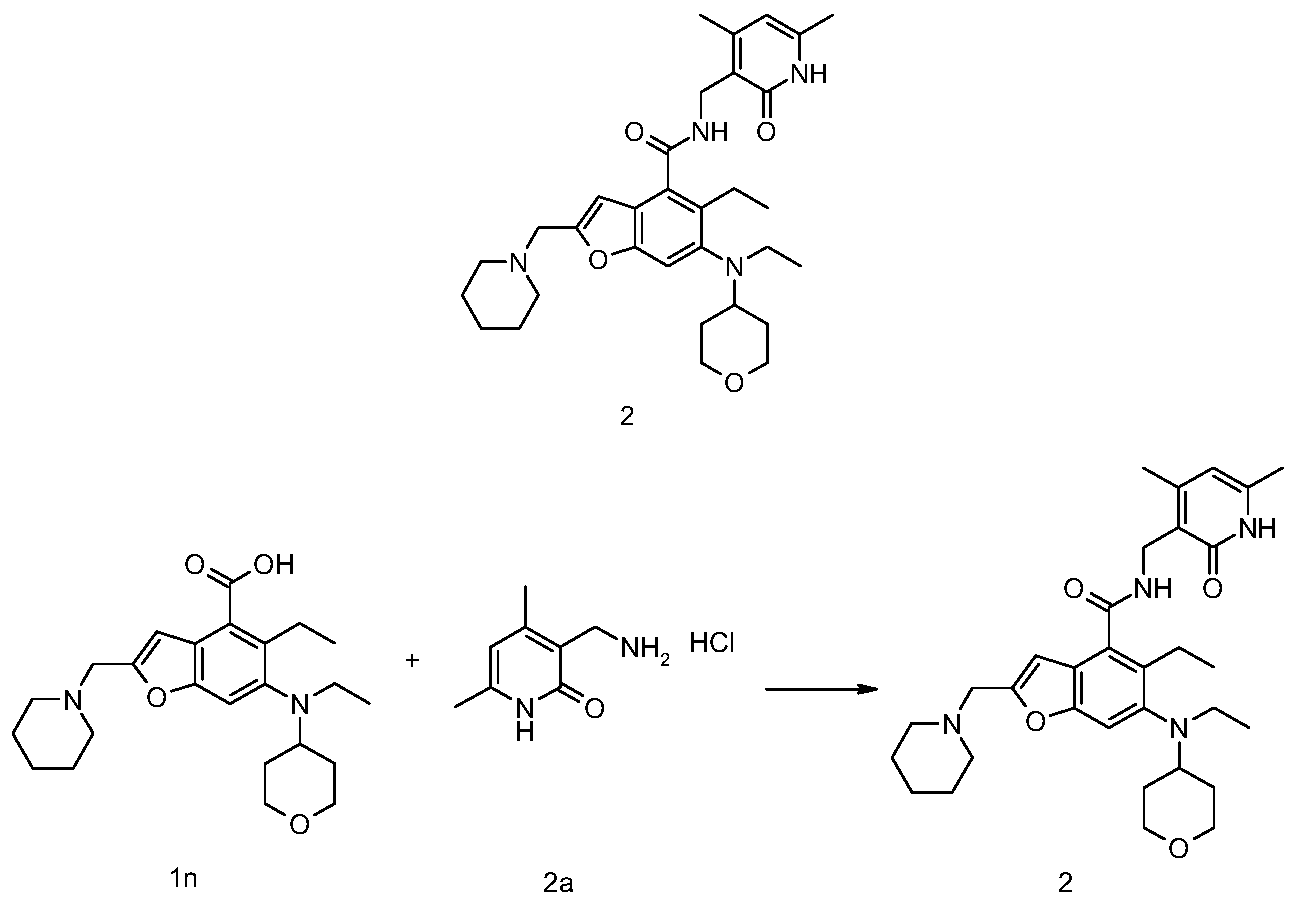
5-Ethyl-6-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-(piperidin-1-ylmethyl)benzofuran-4-carboxylic acid 1n (1.0 g, 2.4 mmol) was dissolved in 30 mL of N,N-dimethylformamide, and 1-ethyl-3-(3-dimethylpropylamine)carbodiimide (696 mg, 3.6 mmol), 1-hydroxybenzotriazole (490 mg, 3.6 mmol), and N,N-diisopropylethylamine (1.56 g, 12.1 mmol) were added. The mixture was stirred for 1 hour, and then 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one hydrochloride 2a (593 mg, 3.0 mmol, prepared by the method disclosed in patent application “WO2014097041”) was added. The mixture was stirred at room temperature for 12 hours. After the reaction was complete, excess water was added, and the mixture was extracted with a mixed solvent of dichloromethane and methanol (V:V = 8:1). The organic phases were combined, washed with water and saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using eluent system A to give the title product N-((4,6-dimethyl-2-carbonyl-1,2-dihydropyridin-3-yl)methyl)-5-ethyl-6-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-(piperidin-1-ylmethyl)benzofuran-4-carboxamide 2 (750 mg, white solid), yield: 57%.
[0238]
1H NMR(400MHz,DMSO-d 6):δ11.48(s,1H),8.15(t,1H),7.39(s,1H),6.46(s,1H),5.86(s,1H),4.32(d,2H),3.83(d,2H),3.54(s,2H),3.21(t,2H),3.01-3.07(m,2H),2.92-2.97(m,1H),2.77-2.82(m,2H),2.39(brs,4H),2.23(s,3H),2.11(s,3H),1.64-1.67(brd,2H),1.47-1.55(m,6H),1.36-1.37(brd,2H),1.02(t,3H),0.82(t,3H).
PAT
WO2019091450]
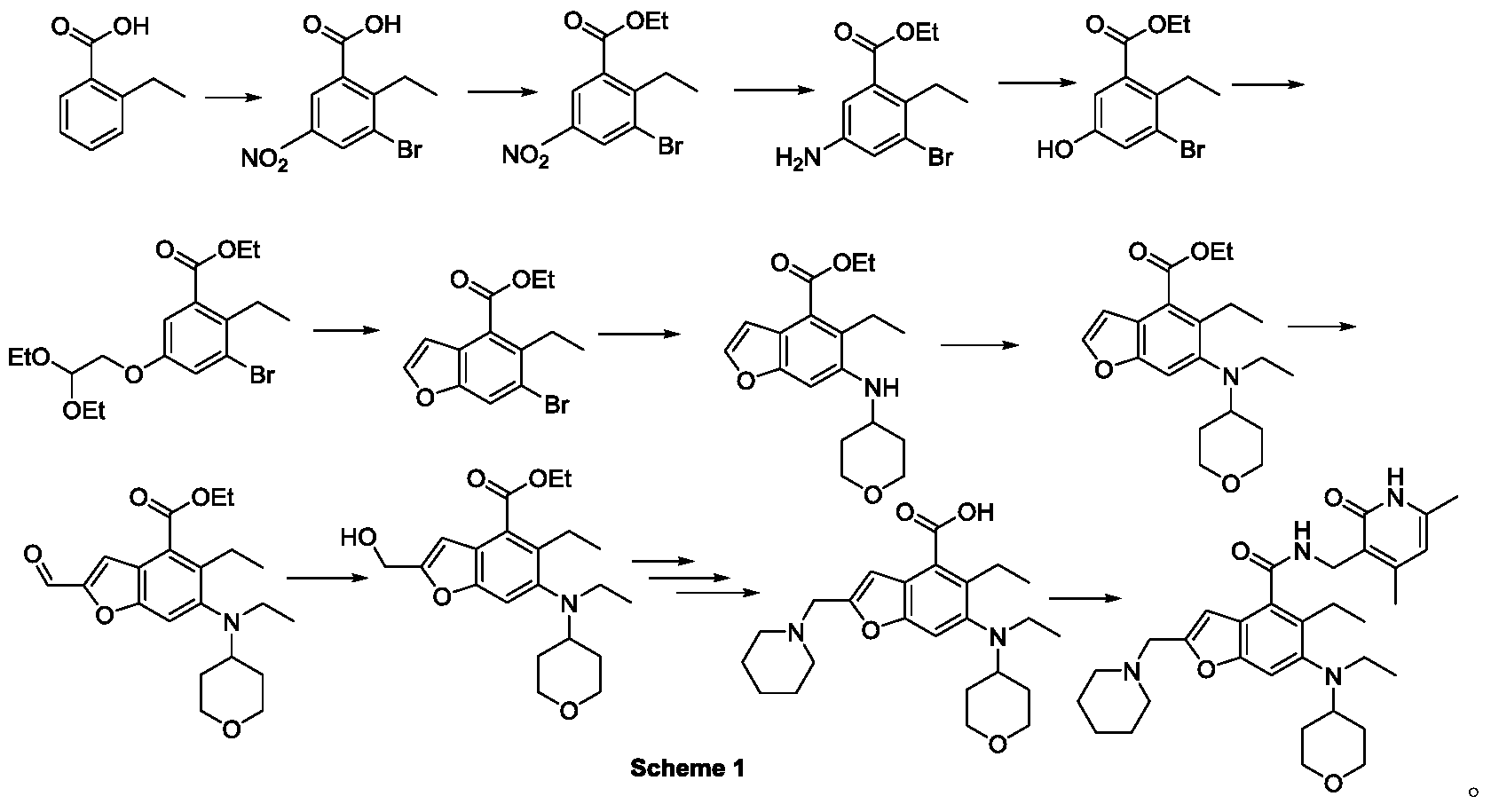
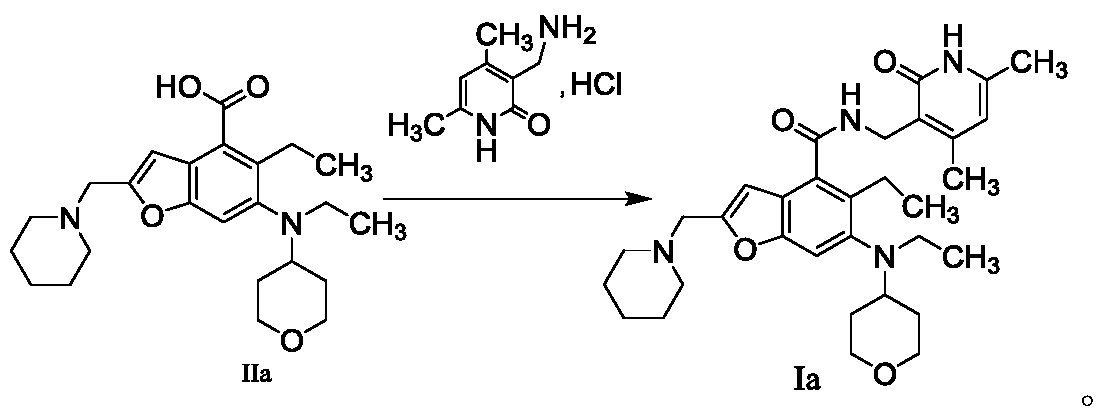
The method for preparing compounds from Formula IIa to Formula Ia provided by this invention can be specifically referred to in the methods for preparing amides disclosed in PCT applications WO2017084494A, WO2012142513, WO2013039988, WO2015-141616, and WO2011140325.
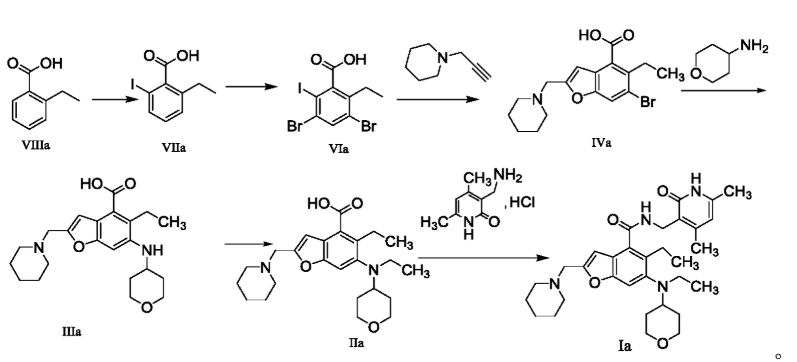
In a 25 mL three-necked flask, starter IIa (50 mg, 0.12 mmol), 1-ethyl-3-(3-dimethylpropylamine)carbodiimide (34.5 mg, 0.18 mmol), 1-hydroxybenzotriazole (23.67 mg, 0.18 mmol), and N,N-diisopropylethylamine (77.89 mg, 0.6 mmol) were mixed and dissolved in 3 mL of N,N-dimethylformamide and stirred until homogeneous. Then, starter 3-(aminomethyl)-4,6-dimethylpyridine-2(1H)-one hydrochloride (24.9 mg, 0.13 mmol) was added and the mixture was stirred at room temperature until the thin-layer chromatography showed that starter IIa had disappeared. The reaction was then terminated. Excess water was added to the reaction solution, and the mixture was extracted with a mixed solvent of dichloromethane and methanol. The organic phases were combined, washed with water, washed with saturated sodium chloride solution, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was purified by silica gel column chromatography with a dichloromethane-methanol eluent system to give 30.1 mg of white solid, yield 47.0%. [0151]m/z[M+H]
1H NMR(400MHz,DMSO-d6)ppm 11.51(s,1H)8.17(t,1H)7.39(s,1H)6.47(s,1H)5.86(s,1H)4.32(d,2H)3.83(d,2H)3.53(s,2H)3.21(t,2H)3.04(d,2H)2.94(br.s .,1H)2.79(d,2H)2.38(br.s.,4H)2.23(s,3H)2.08-2.14(m,3H)1.65(d,2H)1.44-1.56(m,6H)1.36(d,2H)1.02(t,3H)0.81(t,3H).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Benzofuran derivative, preparation method thereof and use thereof in medicinePublication Number: US-11059811-B2Priority Date: 2015-11-19Grant Date: 2021-07-13
- Derived from benzofuran, method of preparing it and using it in medicinePublication Number: ES-2760510-T3Priority Date: 2015-11-19Grant Date: 2020-05-14
- Benzofuran derivative, a method for production thereof and use thereof in medicinePublication Number: RU-2727198-C2Priority Date: 2015-11-19Grant Date: 2020-07-21
- Benzofuran derivative, preparation method thereof and use thereof in medicinePublication Number: US-2020354349-A1Priority Date: 2015-11-19
- BENZOFURAN DERIVATIVE, ITS USES AND ITS PREPARATION PROCESS, AND PHARMACEUTICAL COMPOSITIONPublication Number: BR-112018007876-B1Priority Date: 2015-11-19
- Benzofuran derivative, preparation method thereof and use thereof in medicinePublication Number: US-2018327394-A1Priority Date: 2015-11-19
- Benzofuran derivative, preparation method thereof and use thereof in medicinePublication Number: EP-3378859-B1Priority Date: 2015-11-19Grant Date: 2019-10-30
- Benzofuran derivative, preparation method thereof and use thereof in medicinePublication Number: US-10759787-B2Priority Date: 2015-11-19Grant Date: 2020-09-01
- Benzofuran derivative, preparation method thereof and use thereof in medicinePublication Number: EP-3378859-A1Priority Date: 2015-11-19
- Crystal of benzofuran derivative free base and preparation methodPublication Number: US-11155537-B2Priority Date: 2017-05-18Grant Date: 2021-10-26
- Use of ezh2 inhibitor combined with btk inhibitor in preparing drug for treating tumorPublication Number: US-2021030736-A1Priority Date: 2017-05-18
- Use of EZH2 inhibitor combined with BTK inhibitor in preparing drug for treating tumorPublication Number: US-11065239-B2Priority Date: 2017-05-18Grant Date: 2021-07-20
- Crystal of benzofuran derivative free base and preparation methodPublication Number: US-2021130333-A1Priority Date: 2017-05-18
- Determination and preparation method of benzofuran derivative free basePublication Number: KR-102612379-B1Priority Date: 2017-05-18Grant Date: 2023-12-12
//////////zeprumetostat, ANAX LAB, CHINA 2025, APPROVALS 2025, antineoplastic, Airijing® (China), EZH2-IN-15, SHR 2554
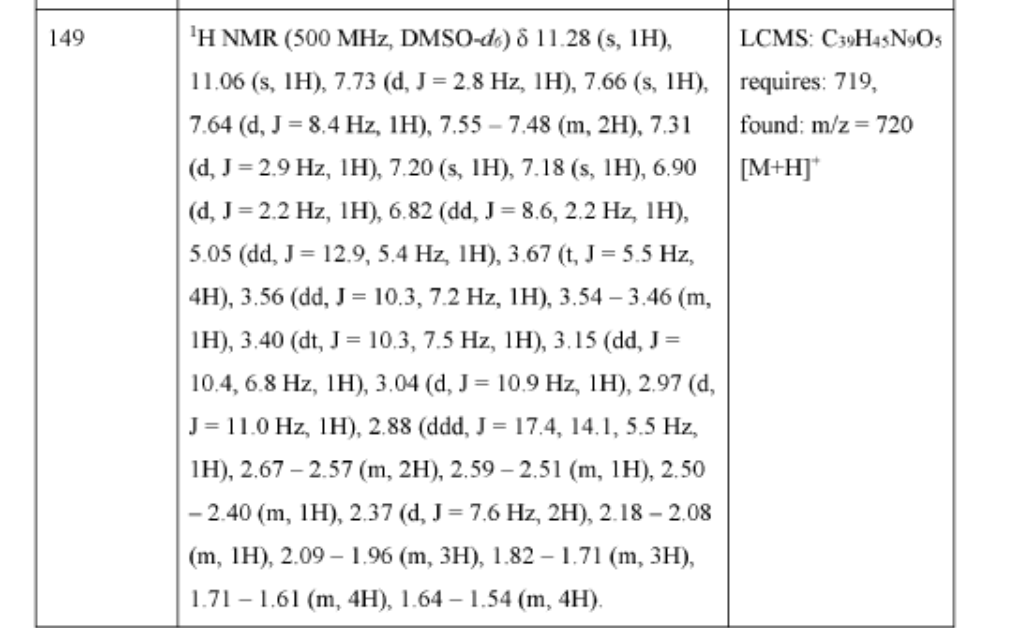
Zelebrudomide
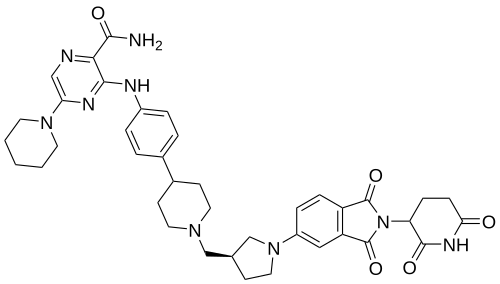
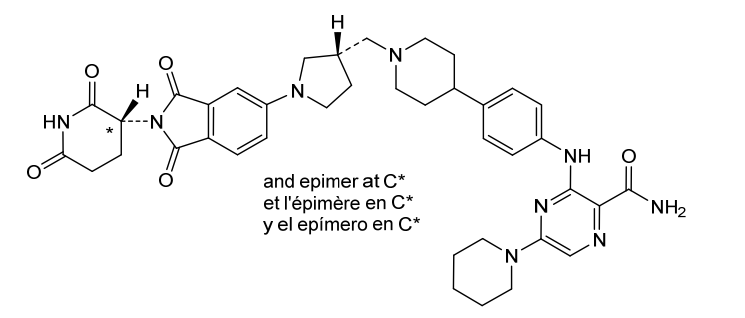
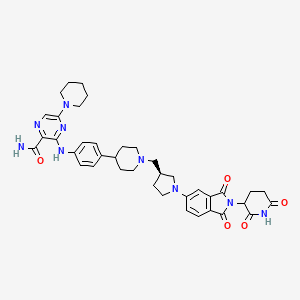
Zelebrudomide
CAS 2416131-46-7
MF C39H45N9O5 MW 719.8 g/mol
3-[4-[1-[[(3S)-1-[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-5-yl]pyrrolidin-3-yl]methyl]piperidin-4-yl]anilino]-5-piperidin-1-ylpyrazine-2-carboxamide

protein degrader, antineoplastic, NX 2127, LSC67HA8DE, NX-2127, BTK Degrader NX-2127
Zelebrudomide (NX-2127) is an investigational new drug that is being evaluated by Nurix Therapeutics for the treatment of relapsed or refractory B-cell malignancies such as chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL), and Waldenström macroglobulinemia (WM). It is an orally bioavailable proteolysis targeting chimera (PROTAC) designed to degrade Bruton’s tyrosine kinase (BTK) along with the immunomodulatory proteins Ikaros (IKZF1) and Aiolos (IKZF3).[1]
- OriginatorNurix
- ClassAntineoplastics; Small molecules
- Mechanism of ActionAgammaglobulinaemia tyrosine kinase degraders; IKZF1 protein degraders; IKZF3 protein degraders
Phase IChronic lymphocytic leukaemia; Diffuse large B cell lymphoma; Follicular lymphoma; Lymphoma; Mantle-cell lymphoma; Marginal zone B-cell lymphoma; Waldenstrom’s macroglobulinaemia
- 09 Dec 2024Pharmacodynamics data from a preclinical studies in Chronic lymphocytic leukaemia released by Nurix Therapeutics
- 11 Jul 2024NX 2127 is still in phase I development in Chronic-lymphocytic-leukaemia (Late-stage disease, Second-line therapy or greater) in USA (PO) (NCT04830137)
- 11 Jul 2024NX 2127 is still in phase I development in Diffuse large B cell lymphoma(Late-stage disease, Second-line therapy or greater) in USA (PO) (NCT04830137)
Zelebrudomide, (S)- is the S-enantiomer of zelebrudomide, an orally bioavailable chimeric targeting molecule (CTM) and targeted degrader of Bruton’s tyrosine kinase (BTK), with potential immunomodulatory drug (IMiD) and antineoplastic activities. Zelebrudomide is comprised of a cereblon (CRBN)-binding moiety conjugated, via a linker, to a BTK-binding moiety. Upon administration, zelebrudomide targets and binds to BTK with its BTK-targeting moiety. Upon binding, the CRBN-binding moiety recruits CRBN, a component of the CRL4-CRBN E3 ubiquitin ligase complex. This catalyzes ubiquitination and proteasome-mediated degradation of BTK, and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways. This leads to an inhibition of the growth of malignant B-cells that overexpress BTK. In addition, zelebrudomide catalyzes the degradation of CRBN neosubstrates Aiolos (IKZF3) and Ikaros (IKZF1), two transcription factors regulating T-cell function. This modulates the activity of the immune system and increases the activation of T-lymphocytes, thereby increasing T-cell-mediated anti-tumor effects. BTK, a member of the src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival. CRBN, the substrate recognition component of the CRL4-CRBN E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins. Compared to BTK inhibitors, zelebrudomide may overcome tumor resistance associated with BTK inhibitor-induced resistance mutations.
A Study of NX-2127 in Adults With Relapsed/Refractory B-cell Malignancies
CTID: NCT04830137
Phase: Phase 1
Status: Recruiting
Date: 2025-03-13
REF
- Discovery and Preclinical Pharmacology of NX-2127, an Orally Bioavailable Degrader of Bruton’s Tyrosine Kinase with Immunomodulatory Activity for the Treatment of Patients with B Cell MalignanciesPublication Name: Journal of Medicinal ChemistryPublication Date: 2024-02-01PMID: 38300987DOI: 10.1021/acs.jmedchem.3c01007
- The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacologyPublication Name: ImmunologyPublication Date: 2020-03-02PMCID: PMC7160657PMID: 32020584DOI: 10.1111/imm.13175
- Effect of osmotic pressure, ionic strength and dibutyryl cyclic adenosine monophosphate on the adhesion of hen erythrocytesPublication Name: Blut Zeitschrift für die gesamte BlutforschungPublication Date: 1976-07PMID: 10025DOI: 10.1007/bf01005212
SYN
compound 28 Journal of Medicinal ChemistryPublication Date: 2024-02-01PMID: 38300987DOI: 10.1021/acs.jmedchem.3c01007
SYN
WO2021219070A1
PAT
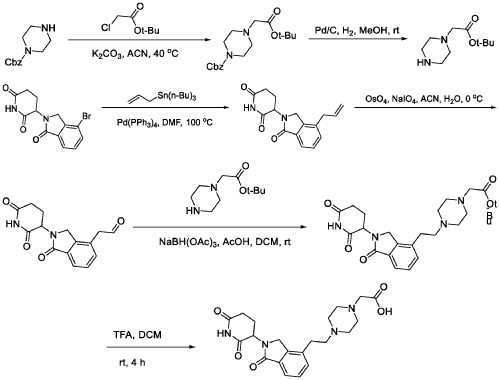
PAT
- Bifunctional compounds for degrading btk via ubiquitin proteosome pathwayPublication Number: US-2023227471-A1Priority Date: 2019-12-04
- Bifunctional compounds for degrading btk via ubiquitin proteosome pathwayPublication Number: WO-2021113557-A1Priority Date: 2019-12-04
- Bifunctional compounds for degrading btk via ubiquitin proteosome pathwayPublication Number: US-2023029378-A1Priority Date: 2018-10-15
- Bifunctional compounds for degrading btk via ubiquitin proteosome pathwayPublication Number: WO-2020081450-A1Priority Date: 2018-10-15
- Bifunctional compounds for degrading BTK via ubiquitin proteosome pathwayPublication Number: US-11479556-B1Priority Date: 2018-10-15Grant Date: 2022-10-25
- Btk reducing molecules for treatment of cancers and immune system disordersPublication Number: WO-2023235691-A1Priority Date: 2022-05-31
- Piperidinylpyrazine-carboxamide compounds for treating and preventing cancer and for degrading btkPublication Number: US-2023149416-A1Priority Date: 2021-10-26
- Piperidinylpyrazine-carboxamide compounds for treating and preventing cancer and for degrading btkPublication Number: WO-2023076303-A1Priority Date: 2021-10-26
- Bifunctional compounds for degrading BTK via ubiquitin proteosome pathwayPublication Number: US-11820781-B2Priority Date: 2019-12-04Grant Date: 2023-11-21
- Bifunctional compounds for degrading btk via ubiquitin proteosome pathwayPublication Number: US-2021198280-A1Priority Date: 2019-12-04
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
References
- Salvaris RT, Brennan J, Lewis KL (February 2025). “BTK Is the Target That Keeps on Giving: A Review of BTK-Degrader Drug Development, Clinical Data, and Future Directions in CLL”. Cancers. 17 (3): 557. doi:10.3390/cancers17030557. PMC 11817010. PMID 39941922.
| Clinical data | |
|---|---|
| Other names | NX-2127 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2416131-46-7 |
| PubChem CID | 146559796 |
| ChemSpider | 128922006 |
| UNII | LSC67HA8DE |
| Chemical and physical data | |
| Formula | C39H45N9O5 |
| Molar mass | 719.847 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
//////////zelebrudomide, anax lab, protein degrader, antineoplastic, NX 2127, LSC67HA8DE, NX-2127, BTK Degrader NX-2127
Zanvipixant
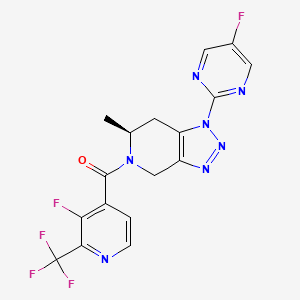
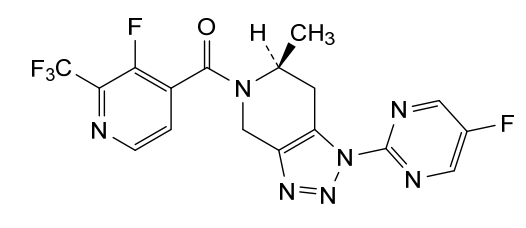
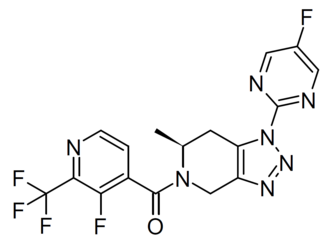
Zanvipixant
CAS 2166558-11-6
MF C17H12F5N7O MW 425.3 g/mol
[(6S)-1-(5-fluoropyrimidin-2-yl)-6-methyl-6,7-dihydro-4H-triazolo[4,5-c]pyridin-5-yl]-[3-fluoro-2-(trifluoromethyl)-4-pyridinyl]methanone
[(6S)-1-(5-fluoropyrimidin-2-yl)-6-methyl-1,4,6,7-tetrahydro-5H-[1,2,3]triazolo[4,5-c]pyridin-5-yl][3-fluoro-2-
(trifluoromethyl)pyridin-4-yl]methanone
purinoreceptor (P2X) antagonist, JNJ-55308942, JNJ 55308942, B7YN3CQ7S7,
JNJ-55308942 is under investigation in clinical trial NCT05328297 (A Study of JNJ-55308942 in the Treatment of Bipolar Depression).
JNJ-55308942 is an investigational drug that works as a P2X7 antagonist with a downstream effect of reducing interleukin-1β release.[1][2][3] It is developed by Janssen Pharmaceuticals for bipolar depression.[4]
Zanvipixant (JNJ-55308942) is an investigational small-molecule, brain-penetrant, and potent antagonist of the P2X7 receptor developed by Janssen (J&J). It is primarily studied for its potential to treat neuroinflammation-related conditions, including mood disorders and depression, by inhibiting P2X7-mediated IL-1β release.
Key Details for Zanvipixant:
- Target: P2X7 Receptor (P2X7R).
- Mechanism: Brain-penetrant antagonist reducing neuroinflammation, specifically inhibiting microglial IL-1β release.
- Chemical Properties: Structure typically includes a triazolopyridine moiety.
- Development Status: Evaluated as a potential CNS treatment for conditions related to neuroinflammation, with research highlighting its pharmacological profile, including good CNS partitioning.
- Alternative Name: JNJ-55308942.
Research indicates that the P2X7 receptor is activated by high levels of extracellular ATP during stress, leading to inflammation and behavioral deficits associated with depression. Zanvipixant has been studied in this context as a potential therapeutic intervention.
- A Study of JNJ-55308942 in the Treatment of Bipolar DepressionCTID: NCT05328297Phase: Phase 2Status: CompletedDate: 2025-07-11
- A Study in Healthy Participants to Evaluate the Effects of Multiple Doses of JNJ-55308942 on Cytochrome P450 Substrate Activity and on the Pharmacokinetics of Levonorgestrel/Ethinyl EstradiolCTID: NCT03547024Phase: Phase 1Status: CompletedDate: 2025-04-27
- A Positron Emission Tomography (PET) Study to Investigate P2X7 Receptor Occupancy by JNJ-55308942 Using [18F]-JNJ-64413739CTID: NCT03437590Phase: Phase 1Status: CompletedDate: 2025-04-27
- A Study to Investigate the Safety, Tolerability, and Pharmacokinetics of JNJ-55308942 in Healthy Male and Female ParticipantsCTID: NCT03151486Phase: Phase 1Status: CompletedDate: 2025-04-27
- A Randomized, Stratified, Double-blind, Placebo-Controlled Study to Investigate the Efficacy, Safety and Tolerability of JNJ-55308942 in Bipolar Depression
- EudraCT: 2021-004790-31
- Phase: Phase 2
- Status: Ongoing, Completed
- Date: 2022-05-31
SYN
Publication Name: Journal of Medicinal Chemistry
Publication Date: 2017-12-20
PMID: 29211470
DOI: 10.1021/acs.jmedchem.7b01279
SYN
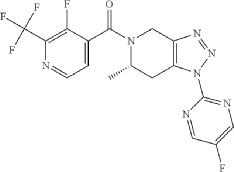
Example 228
(S*)-(3-fluoro-2-(trifluoromethyl)pyridin-4-yl)(1-(5-fluoropyrimidin-2-yl)-6-methyl-6,7-dihydro-1H-[1,2,3]triazolo[4,5-c]pyridin-5(4H)-yl)methanone
MS (ESI) mass calcd C 17H 12F 5N 7O, 425.1 m/z. found, 426.1 [M+H] +.
PAT
Example 228 (S)-(3 -fluoro-2-(trifluoromethyl)pyridin-4-yl)( 1 -(5 -fluoropyrimidin-2-yl)-6-methyl-6J-dihvdro-lH-ri,2,31triazolor4,5-c1pyridin-5(4H)-yl)methanone
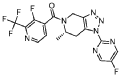
he title compound was prepared as described in Example 65, substituting (S)-l-(5-fluoropyrimidin-2-yl)-6-methyl-4,5,6,7-tetrahydro-lH-[l,2,3]triazolo[4,5-c]pyridine-hydrochloride salt (prepared as Step 2 intermediate of method II synthesis) in Example 220 for l-pyrimidin-2-yl-4,5,6,7-tetrahydro-lH-[l,2,3]triazolo[4,5-c]pyridine, 3-fluoro-2-(trifluoromethyl)isonicotinic acid for 2-chloro-3-(trifluoromethyl)benzoic acid and Hunig’s base (3.0 equiv) for Et3N. The powder x-ray diffraction pattern for this compound is shown in Figure 2. Alternatively, the title compound was synthesized using the following procedure:
Step 1 : (S)-l-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)-2-methylpiperidin-4-one
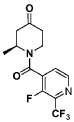
(S)-l-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)-2-methylpiperidin-4-one.: (S)-2-methylpiperidin-4-one, TFA salt (108 g, 478 mmol, 1.0 equiv.) was suspended in DCM (1.4 L). Et3 (265 mL, 1.9 mol, 4.0 equiv.) and 3-fluoro-2-(trifluoromethyl)isonicotinoyl
chloride (1 19 g, 526 mmol, 1.1 equiv.) were added sequentially. The reaction solution was stirred at room temperature for 1 h. The precipitated solid was filtered and washed with EtOAc. The filtrate solution was concentrated and the residue was re-dissolved in EtOAc (500 mL). The organic layer was washed with saturated aHC03 aqueous solution, water and brine, dried over Na2S04, and concentrated. The crude product was triturated from EtOAc/hexanes to afford (S)-l-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)-2-methylpiperidin-4-one (103 g, 368 mmol, 77%), which was used without further purification. MS = 305.1 (positive mode)
Step 2: (S)-(3-fluoro-2-(trifluoromethyl)pyridin-4-yl)(l-(5-fluoropyrimidin-2-yl)-6-methyl-6,7-dihvdro-lH-ri ,2,31triazolor4,5-c1pyridin-5(4H)-yl)methanone and (S)-(3-fluoro-2-(trifluoromethyl)pyridin-4-yl)(l-(5-fluoropyrimidin-2-yl)-4-methyl-6J-dihydro-lH
[ 1.2.3 Itriazolo [4.5 -c]pyridin-5 (4H)-yl)methanone
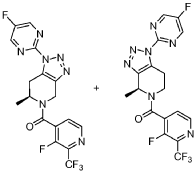
In a 5 L, three-neck, round-bottom flask equipped with a mechanic stirring, Dean-Stark trap, reflux condenser and an internal thermometer, to the solution of (S)-l-(3-fluoro-2-(trifluoromethyl)isonicotinoyl)-2-methylpiperidin-4-one, prepared in Step 1 above (100 g, 328 mmol, 1.0 equiv.) in toluene (1.5 L), -toluenesulphonic acid (0.62 g, 3.29 mmol, 0.01 equiv.), pyrrolidine (33 mL, 394 mmol, 1.2 equiv.) and 2-azido-5-fluoropyrimidine (59.4 g, 427 mmol, 1.3 equiv.) were added sequentially. The reaction mixture was heated to reflux temperature for 4 hours and then cooled to room temperature. aHC03 (55.2 g, 657 mmol, 2.0 equiv.) and mCPBA (162 g, 657 mmol, 2.0 equiv.) solution in EtOAC (-250 mL) were added sequentially. After stirring at room temperature for 2 hours, water and EtOAc were added. The organic layer was washed sequentially with 1M a2S03, saturated aHC03 aqueous solution, and brine, dried over Na2S04, and concentrated. The crude product was purified via column chromatography to afford a mixture of (S)-(3-fluoro-2-
(trifluoromethyl)pyridin-4-yl)(l-(5-flu^
[l,2,3]triazolo[4,5-c]pyridin-5(4H)-yl)methanone and (S)-(3-fluoro-2- (trifluoromethyl)pyridin-4-yl)(l-(5-fl^^
[l,2,3]triazolo[4,5-c]pyridin-5(4H)-yl)methanone in a 10: 1 ratio (97 grams).
Step 3 : (S)-(3-fluoro-2-(trifluoromethyl)pyridin-4-yl)(l -(5-fluoropyrimidin-2-yl)-6-methyl-6.7-dihvdro-lH-ri.2.31triazolor4.5-c1pyridin-5(4H)-yl)methanone
HPLC purification of the mixture from Step 2 was performed via achiral SFC (Stationary phase: Chiralcel OD-H 5μιη 250x30mm), (Mobile phase: 75% C02, 25% MeOH) to afford pure (S)-(3-fluoro-2-(trifluoromethyl)pyridin-4-yl)(l-(5-fluoropyrimidin-2-yl)-6-methyl-6,7-dihydro-lH-[l,2,3]triazolo[4,5-c]pyridin-5(4H)-yl)methanone (73 gram, 172 mmol, 52%). MS (ESI) mass calcd Ci7H12F5 70, 425.1 m/z found, 426.1 [M+H]+. XH NMR (500 MHz, CDC13) δ 8.84 – 8.69 (m, 2H), 8.68 – 8.57 (m, 1H), 7.71 – 7.49 (m, 1H), 5.90 -5.68 (d, J = 16.4 Hz, 0.5H), 5.66 – 5.54 (m, 0.5H), 4.76 – 4.58 (d, J = 15.8 Hz, 0.5H), 4.58 – 4.48 (m, 0.5H), 4.45 – 4.33 (m, 0.5H), 4.20 – 4.09 (m, 0.5H), 3.56 – 3.12 (m, 2H), 1.48 -1.18 (m, 3H).
2-chloro-3-(trifluoromethyl)benzoic acid and Hunig’s base (3.0 equiv) for Et3N. MS (ESI) mass calcd Ci7H12F5 70, 425.1 m/z found, 426.1 [M+H]+.
PAT
- P2X7 modulatorsPublication Number: US-9464084-B2Priority Date: 2013-03-14Grant Date: 2016-10-11
- P2X7 MODULATORSPublication Number: PT-2970267-TPriority Date: 2013-03-14
- P2X7 modulatorsPublication Number: US-11225478-B2Priority Date: 2013-03-14Grant Date: 2022-01-18
- P2X7 modulatorPublication Number: JP-6293861-B2Priority Date: 2013-03-14Grant Date: 2018-03-14
- P2X7 modulatorsPublication Number: AU-2019203186-A1Priority Date: 2013-03-14
- P2X7 modulatorsPublication Number: TW-201446762-APriority Date: 2013-03-14
- P2X7 regulatorsPublication Number: CN-110003200-APriority Date: 2013-03-14
- P2X7 modulatorsPublication Number: US-9066946-B2Priority Date: 2013-03-14Grant Date: 2015-06-30
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
“CTG Labs – NCBI”. clinicaltrials.gov. Retrieved 27 November 2023.
Bhattacharya, Anindya; Lord, Brian; Grigoleit, Jan-Sebastian; He, Yingbo; Fraser, Ian; Campbell, Shannon N.; Taylor, Natalie; Aluisio, Leah; O’Connor, Jason C.; Papp, Mariusz; Chrovian, Christa; Carruthers, Nicholas; Lovenberg, Timothy W.; Letavic, Michael A. (December 2018). “Neuropsychopharmacology of JNJ-55308942: evaluation of a clinical candidate targeting P2X7 ion channels in animal models of neuroinflammation and anhedonia”. Neuropsychopharmacology. 43 (13): 2586–2596. doi:10.1038/s41386-018-0141-6. ISSN 0893-133X. PMC 6224414. PMID 30026598.
Bhattacharya, Anindya; Ceusters, Marc (January 2020). “Targeting neuroinflammation with brain penetrant P2X7 antagonists as novel therapeutics for neuropsychiatric disorders”. Neuropsychopharmacology. 45 (1): 234–235. doi:10.1038/s41386-019-0502-9. ISSN 0893-133X. PMC 6879571. PMID 31477815.
Kolb, Hartmuth C.; Barret, Olivier; Bhattacharya, Anindya; Chen, Gang; Constantinescu, Cristian; Huang, Chaofeng; Letavic, Michael; Tamagnan, Gilles; Xia, Chunfang A.; Zhang, Wei; Szardenings, Anna Katrin (August 2019). “Preclinical Evaluation and Nonhuman Primate Receptor Occupancy Study of 18 F-JNJ-64413739, a PET Radioligand for P2X7 Receptors”. Journal of Nuclear Medicine. 60 (8): 1154–1159. doi:10.2967/jnumed.118.212696. ISSN 0161-5505. PMID 30733317. S2CID 73454130.
| Clinical data | |
|---|---|
| Trade names | Zanvipixant |
| Legal status | |
| Legal status | Investigational New Drug |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2166558-11-6 |
| PubChem CID | 90408860 |
| DrugBank | DB19110 |
| ChemSpider | 76771276 |
| UNII | B7YN3CQ7S7 |
| ChEMBL | ChEMBL3914857 |
| Chemical and physical data | |
| Formula | C17H12F5N7O |
| Molar mass | 425.323 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
////////zanvipixant, ANAX LAB, purinoreceptor (P2X) antagonist, JNJ-55308942, JNJ 55308942, B7YN3CQ7S7,
Vormatrigine
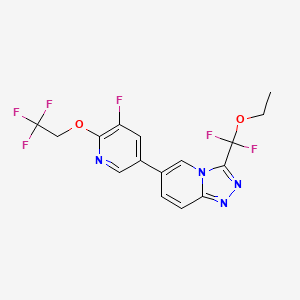
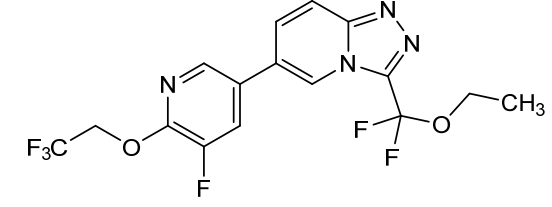
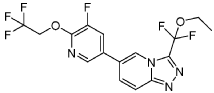
Vormatrigine
CAS 2392951-18-5
MF C16H12F6N4O2 MW406.28 g/mol
3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyridine
3-[ethoxydi(fluoro)methyl]-6-[5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl][1,2,4]triazolo[4,3-a]pyridine
sodium channel blocker, PRAX-628, PRAX 628, QU3C48T4NV,
Vormatrigine is a small molecule drug. The usage of the INN stem ‘-trigine’ in the name indicates that Vormatrigine is a sodium channel blocker, signal transduction modulator. Vormatrigine has a monoisotopic molecular weight of 406.09 Da.
Vormatrigine (formerly PRAX-628) is an oral, small-molecule, voltage-gated sodium channel inhibitor being developed by Praxis Precision Medicines for focal onset and generalized epilepsy. Phase II RADIANT trial data indicated that 22% of participants achieved complete seizure freedom, with a 56.3% median reduction in seizure frequency. IGMPI +2
Key Aspects of Vormatrigine:
- Mechanism: Targets the hyperexcitable state of NaV channels, acting as a functional state modulator.
- Efficacy: In the Phase II RADIANT trial (NCT06908356) for focal epilepsy, the drug showed significant seizure reduction (56.3% median reduction), with 60% of participants achieving a $\ge$50% reduction in seizures.
- Dosing: Developed for once-daily, oral administration without the need for complex titration.
- Safety Profile: Vormatrigine was generally well-tolerated in clinical studies, with mostly mild, transient adverse events reported, as noted in the Phase 1 PRAX-628-102 study.
- Future Development: Praxis is moving forward with Phase III trials (POWER2) following the successful Phase II results, with plans to potentially redefine treatment for patients with treatment-resistant focal epilepsy.
- Drug-Drug Interactions: Preliminary data suggests a favorable profile with minimal interaction risks, supporting its potential use in polytherapy.
Neurology Live +4
The drug is expected to be a major competitor in the epilepsy market if approved, with potential for high sales due to its efficacy profile compared to existing treatments. IGMPI
SYN
WO2019232209A1
- Assignee: Praxis Precision Medicines
- Title: Pyridin-3-yl substituted triazolopyrazines / triazolopyridines
- Year: 2019
- Covers:
- Vormatrigine structure (explicitly or as a close analog)
- General synthetic routes
- Multiple heterocycle construction strategies
This is the core patent used by all CRO/CDMO reverse synthesis work.Example ~178
Patent Landscape
The primary patent coverage for Vormatrigine and its related analogs is held by Praxis Precision Medicines, Inc. The chemical structure is a 1,2,4-triazolo[4,3-a]pyridine derivative.
- Primary Compound Patent:WO 2020/033839 (and its US equivalent US 11,447,489).
- Title: Preparation of pyridin-3-yl substituted triazolopyrazines, triazolopyridazines, and triazolopyridines as ion channel modulators.
- Scope: This patent covers the specific structure of Vormatrigine (Example 167 in some filings) and its use as a voltage-gated sodium channel (VGSC) inhibitor.
- Other Relevant Filings: * WO 2022/173918: Focuses on specific crystalline forms (polymorphs) and manufacturing improvements.
- CN 116444437 / CN 111087324: While these specific numbers often relate to broader triazolopyridine research (like Darolutamide, which you’ve researched previously), Praxis holds several Chinese counterparts for the PRAX-628 series.
SYN
SYN
Example 3: 3-[ethoxy(difluoro)methyl]-6-[5-fluoro-6-(2,2,2-trifluoroethoxy)-3-pyridyl]-[1,2,4]triazolo[4,3-a]pyridine
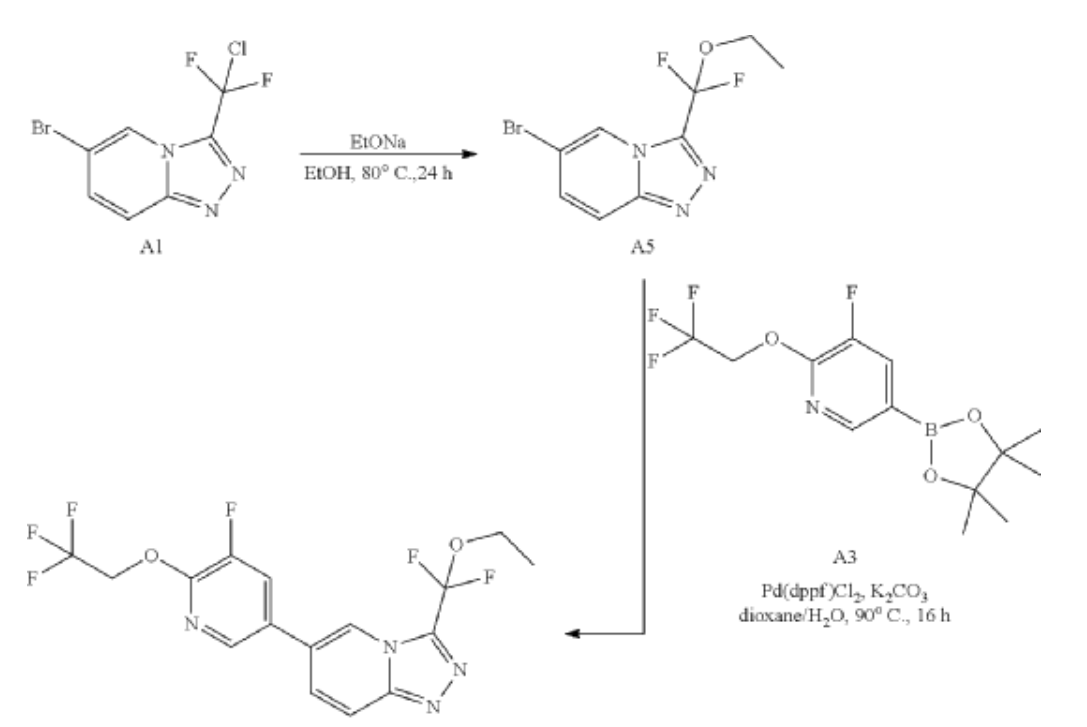
| Synthesis of A5: A mixture of 6-bromo-3-[chloro(difluoro)methyl]-[1,2,4]triazolo[4,3-a]pyridine (300 mg, 1.06 mmol) and EtONa (361.37 mg, 5.31 mmol) in Ethanol (10 mL) was stirred at 80° C. for 24 hours. After cooling to room temperature, the reaction was quenched with sat.NH 4Cl (10 mL), and the mixture was extracted with EtOAc (20 mL×2). The combined organic phase was washed with brine (10 mL), dried over Na 2SO 4, filtered and concentrated to give the crude product. The crude product was purified by flash chromatography on silica gel (EtOAc in PE=10% to 40%) to give the product (70 mg, 0.17 mmol) as a solid. LCMS R t=1.97 min in 4 min chromatography, MS ESI calcd. C 9H 9BrF 2N 3O [M+H+2] + 294.0, found 293.8. |
SYN
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
REF
PAT
- Ion channel modulatorsPublication Number: TW-202012403-APriority Date: 2018-05-30
- 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyridine as an ion channel modulatorPublication Number: US-11731978-B2Priority Date: 2018-05-30Grant Date: 2023-08-22
- Ion channel modulatorsPublication Number: EP-3801535-A1Priority Date: 2018-05-30
- Ion channel modulatorsPublication Number: EP-4487916-A2Priority Date: 2018-05-30
- Ion channel modulatorsPublication Number: US-2022024930-A1Priority Date: 2018-05-30
- 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyrazine as an ion channel modulatorPublication Number: US-11014931-B2Priority Date: 2018-05-30Grant Date: 2021-05-25
- Ion channel modulatorPublication Number: KR-20210024500-APriority Date: 2018-05-30
- Ion channel modulatorsPublication Number: US-2021163488-A1Priority Date: 2018-05-30
- Ion channel modulatorsPublication Number: US-2022220118-A1Priority Date: 2018-05-30
- 3-(ethoxydifluoromethyl)-6-(5-fluoro-6-(2,2,2-trifluoroethoxy)pyridin-3-yl)-[1,2,4]triazolo[4,3-a]pyridine as an ion channel modulatorPublication Number: US-11731976-B2Priority Date: 2018-05-30Grant Date: 2023-08-22
- Treatment of neurological disordersPublication Number: WO-2023211859-A1Priority Date: 2022-04-26
- Methods for the treatment of neurological disordersPublication Number: WO-2023211856-A1Priority Date: 2022-04-26
- Ion channel modulatorsPublication Number: US-11866439-B2Priority Date: 2018-05-30Grant Date: 2024-01-09
- Ion channel modulatorsPublication Number: US-2024132501-A1Priority Date: 2018-05-30
- Ion channel modulatorsPublication Number: US-2021087197-A1Priority Date: 2018-05-30
////////////vormatrigine, ANAX LAB, sodium channel blocker, PRAX-628, PRAX 628, QU3C48T4NV,
Veonetinib
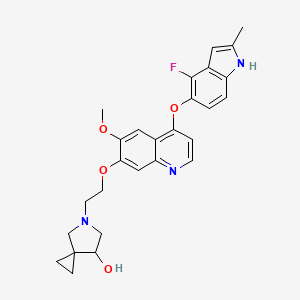
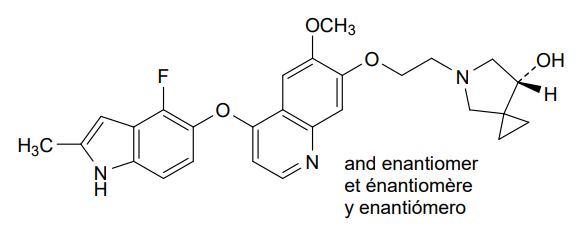
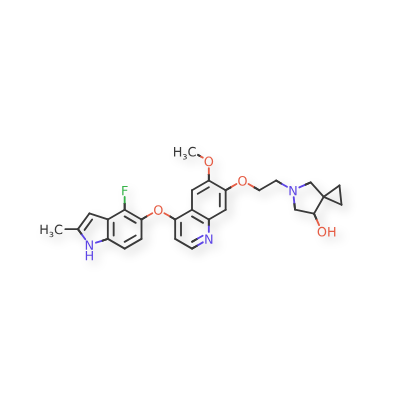
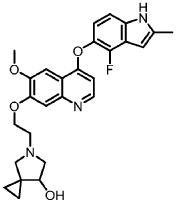
Veonetinib
👉CAS 1210828-09-3
MF C27H28FN3O4 MW 477.5 g/mol
5-[2-[4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxyquinolin-7-yl]oxyethyl]-5-azaspiro[2.4]heptan-7-ol
5-AZASPIRO(2.4)HEPTAN-7-OL, 5-(2-((4-((4-FLUORO-2-METHYL-1H-INDOL-5-YL)OXY)-6-METHOXY-7-QUINOLINYL)OXY)ETHYL)-
5-(2-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)ethyl)-5-azaspiro[2.4]-heptan-7-ol
(7RS)-5-[2-({4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxyquinolin7-yl}oxy)ethyl]-5-azaspiro[2.4]heptan-7-ol
tyrosine kinase inhibitor, antineoplastic, U7PA8S6XGJ
👉SYN
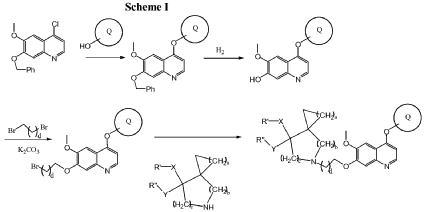
Example 3
5-(2-(4-(4-fluoro-2-methyl-lH-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)ethyl)-5-azaspiro[2.4]-heptan-7-ol
The above product from Example 2 (75 mg) was dissolved into MeOH (8 ml) and stirred at RT.
NaBH4 (75 mg) was added to the reaction and stirred at RT for 30 min. The reaction was evaporated and purified by column chromatography to give title compound (60 mg). Mass: (M + 1), 478
Patent Scope
- Covers:
- Quinoline–indole kinase inhibitors
- VEGFR / angiogenesis targets
- Broad Markush structures
Fragment A: Indole Phenol
4-fluoro-2-methyl-1H-indol-5-ol
Typical Preparation
- Fischer indole synthesis or substituted aniline cyclization
- Fluorination / directed substitution
- Hydroxyl introduction at C-5
Fragment B: Quinoline Electrophile
4-chloro-6-methoxy-7-(leaving group) quinoline
Typical Route
- Start from 6-methoxyaniline
- Skraup / Doebner–Miller → quinoline core
- Chlorination at C-4
- Functionalization at C-7 (OH or halide)
Fragment C: Chiral Spiro Amine
(R)-5-azaspiro[2.4]heptan-7-ol
- Usually from:
- Chiral pool OR
- Resolution of racemate
- Important: defines final stereochemistry
4. STEP-BY-STEP SYNTHESIS (PATENT-ALIGNED)
Step 1: Indole–Quinoline Ether Formation
Reaction: SNAr / Ullmann-type coupling
Indole phenol + 4-chloroquinoline → aryl ether
Conditions
- Base: K2CO3 / Cs2CO3
- Solvent: DMF / DMSO
- Temp: 80–120°C
Forms:
Indole–O–quinoline core
Step 2: Introduction of Linker (C-7 substitution)
If quinoline has OH:
Quinoline–OH + Br–CH2–CH2–X → O–CH2CH2–X
If halide:Direct alkylation
Conditions
- Base: NaH / K2CO3
- Solvent: DMF
- Temp: 50–90°C
Product:
Quinoline–O–CH2CH2–X
Step 3: Coupling with Spiro Amine
Quinoline–O–CH2CH2–X + spiro amine → final amine linkage
Reaction Type
- SN2 substitution
Conditions
- Base: DIPEA / Et3N
- Solvent: ACN / DMF
- Temp: 50–80°C
Step 4: Final Deprotection / Purification
- Remove protecting groups (if any)
- Chiral purity control
- Crystallization
Step 1: Preparation of Indole–Quinoline Ether
Starting materials:
- 4-fluoro-2-methyl-1H-indol-5-ol
→ 1.00 equiv (e.g., 5.0 g, ~30 mmol) - 4-chloro-6-methoxyquinoline
→ 1.10 equiv (~33 mmol)
Reagents:
- Potassium carbonate (K₂CO₃) → 2.0 equiv (~60 mmol)
- Solvent: DMF (50–60 mL)
Procedure:
- Charge indole phenol and K₂CO₃ in DMF under nitrogen.
- Add 4-chloroquinoline portionwise.
- Heat to 100–110°C.
- Stir for 8–12 h.
Workup:
- Cool to RT
- Pour into water (200 mL)
- Extract with EtOAc (3×)
- Wash with brine, dry (Na₂SO₄)
- Concentrate
Purification:
- Silica gel chromatography (EtOAc/hexane)
Yield: ~70–80%
Product: Indole–quinoline ether intermediate
Step 2: Installation of Ethylene Linker
Starting material: Step 1 product (~25 mmol)
Reagents:
- 1,2-dibromoethane → 1.5–2.0 equiv
- Base: K₂CO₃ → 2 equiv
- Solvent: DMF (40 mL)
Procedure:
- Dissolve intermediate in DMF
- Add K₂CO₃
- Add dibromoethane
- Heat to 80–90°C for 6–8 h
Workup:
- Pour into water
- Extract with EtOAc
- Dry and concentrate
Product: Quinoline–O–CH₂CH₂–Br
Yield: ~65–75%
Step 3: Coupling with Chiral Spiro Amine
Starting materials:
- Bromo intermediate → 1.0 equiv (~15–20 mmol)
- (R)-5-azaspiro[2.4]heptan-7-ol → 1.2 equiv
Reagents:
- DIPEA or Et₃N → 2 equiv
- Solvent: Acetonitrile or DMF (30–40 mL)
Procedure:
- Combine bromo intermediate and amine in solvent
- Add DIPEA
- Heat to 60–70°C
- Stir 12–16 h
Workup:
- Remove solvent
- Dissolve in EtOAc
- Wash with water + brine
- Dry and concentrate
Yield: ~70–85%
Step 4: Final Purification
Purification options:
- Silica chromatography OR
- Recrystallization (EtOAc/hexane or IPA)
Optional:
- Convert to pharmaceutically acceptable salt
Final Yield (overall): ~35–45%
PAT
EXAMPLE 1
4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-7-[2-(5,8-Dioxa-10-azadispiro[2.0.4.3]-undecane)ethoxy]quinoline
Preparation of 4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxy-7-benzyloxyquinoline
Method A:
Method B:
Preparation of Title Compound
Method C:
Method D:
EXAMPLE 2
5-(2-(4-(4-Fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)ethyl)-5-azaspiro[2.4]-heptan-7-one
EXAMPLE 3
5-(2-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)ethyl)-5-azaspiro[2.4]-heptan-7-ol
PAT
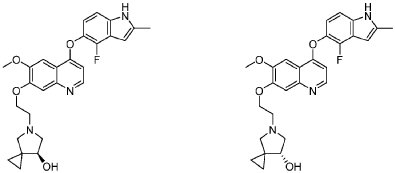
PAT
CN201710900497.6 CN201610649732.2 → leads to US10689361B2
PAT
- Compounds as kinase inhibitorsPublication Number: CA-2733250-CPriority Date: 2008-08-19Grant Date: 2016-06-21
- Compounds that act as kinase inhibitorsPublication Number: ES-2617678-T3Priority Date: 2008-08-19Grant Date: 2017-06-19
- Compounds as kinase inhibitorsPublication Number: WO-2010021918-A1Priority Date: 2008-08-19
- Compounds as kinase inhibitorsPublication Number: EP-2312950-A1Priority Date: 2008-08-19
- Compounds as kinase inhibitorsPublication Number: US-8211911-B2Priority Date: 2008-08-19Grant Date: 2012-07-03
- Compounds as kinase inhibitorsPublication Number: JP-2012500269-APriority Date: 2008-08-19
- Compounds as kinase inhibitorsPublication Number: KR-20110044749-APriority Date: 2008-08-19
- Compounds as kinase inhibitorsPublication Number: EP-2312950-B1Priority Date: 2008-08-19Grant Date: 2016-11-30
- Biological activities of 5-(2-(4-(4-fluoro-2-methyl-1h-indol-5-yloxy)-6-methoxyquinolin-7-yloxy)ethyl)-5-azaspiro[2.4]-heptan-7-ol crystalline, phosphoric acid salt and its enantiomersPublication Number: US-2023399313-A1Priority Date: 2022-06-10
- Btological activities of 5-(2-(4-(4-fluoro-2-methyl-1h-indol-5- yloxy)-6-methoxyquinolin-7-yloxy)ethyl)-5-azaspiro[2.4]-heptan- 7-ol crystalline, phosphoric acid salt and its enantiomersPublication Number: WO-2023239945-A1Priority Date: 2022-06-10
- Btological activities of 5-(2-(4-(4-fluoro-2-methyl-1h-indol-5- yloxy)-6-methoxyquinolin-7-yloxy)ethyl)-5-azaspiro[2.4]-heptan- 7-ol crystalline, phosphoric acid salt and its enantiomersPublication Number: EP-4536652-A1Priority Date: 2022-06-10
- Compounds as kinase inhibitorsPublication Number: CA-2733250-A1Priority Date: 2008-08-19
- Compounds As Kinase InhibitorsPublication Number: US-2010048599-A1Priority Date: 2008-08-19
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
REF
//////////veonetinib, ANAX LAB, tyrosine kinase inhibitor, antineoplastic, U7PA8S6XGJ
Ulacamten
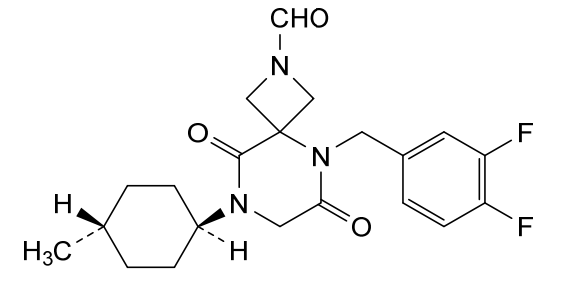
Ulacamten
CAS 2830607-59-3
MF C21H25F2N3O3 MW405.4 g/mol
5-[(3,4-difluorophenyl)methyl]-8-(4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde
5-[(3,4-difluorophenyl)methyl]-8-[(1r,4r)-4-methylcyclohexyl]-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde
cardiac myosin inhibitor, CK-586, CK-4021586, CK 586, CK 4021586, X325G97HZJ
Ulacamten (also known as CK-586 or CK-4021586) is an investigational drug developed by Cytokinetics that acts as a cardiac myosin inhibitor (CMI). It is currently being studied for the treatment of heart failure with preserved ejection fraction (HFpEF), a condition where the heart muscle is too stiff to fill properly
Key Characteristics and Development
- Mechanism of Action: Unlike earlier CMIs like mavacamten or aficamten, ulacamten is highly selective. It binds to the regulatory light chain (RLC) of myosin and only inhibits the “two-headed” form of cardiac myosin, potentially allowing for more precise control over heart muscle contraction.
- Clinical Status: As of March 2026, it is in Phase 2 clinical trials (specifically the AMBER-HFpEF study) to evaluate its safety, tolerability, and optimal dosage in patients with symptomatic HFpEF.
- Administration: It is designed as an orally active small molecule intended for once-daily dosing.
- Potential Benefits: Preclinical studies suggest it can reduce excessive myocardial contractility and improve left ventricular relaxation (diastolic function) without significantly compromising the heart’s overall pumping ability.
- AMBER-HFpEF: Assessment of CK-4021586 in a Multi-Center, Blinded Evaluation of Safety and Tolerability Results in HFpEFCTID: NCT06793371Phase: Phase 2Status: RecruitingDate: 2026-01-12
- A Single and Multiple Ascending Dose Study of CK-4021586 in Healthy Adult ParticipantsCTID: NCT05877053Phase: Phase 1Status: CompletedDate: 2025-05-01
SYN
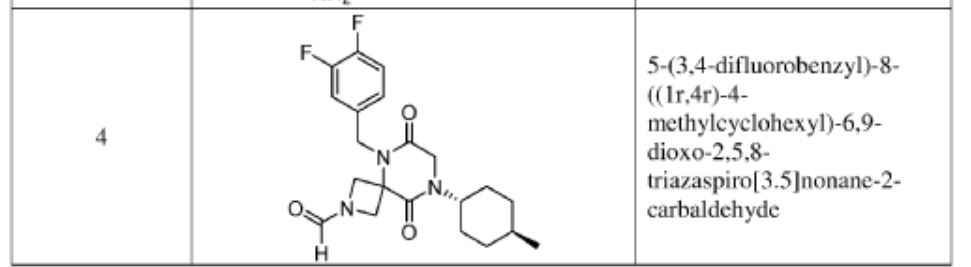
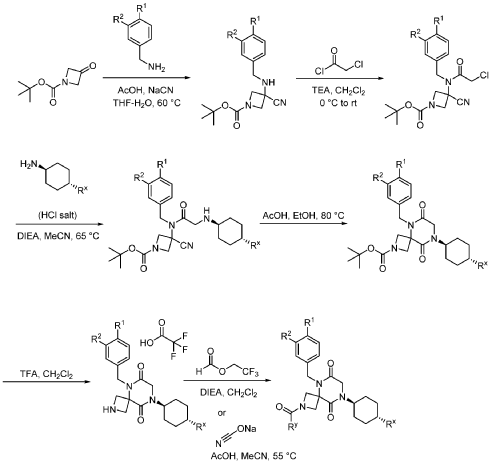
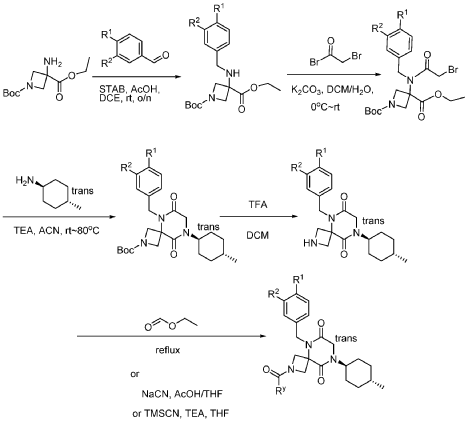
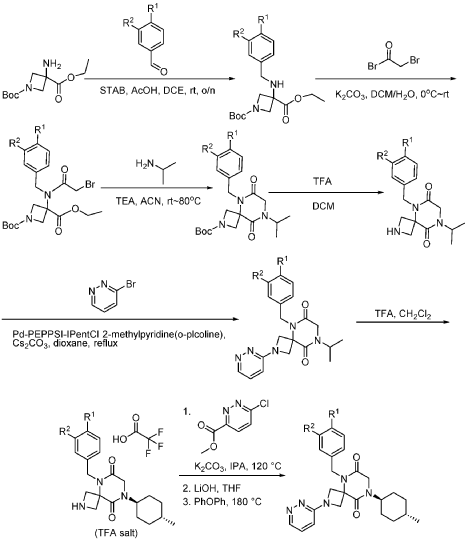
Example 1
Synthesis of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde
(Compound 4)
Step 1: Synthesis of 1-(tert-butyl) 3-ethyl 3-((3,4-difluorobenzyl)amino)azetidine-1,3-dicarboxylate:
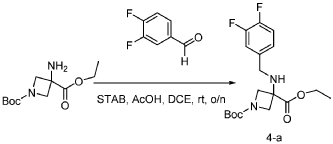
[0147] To a solution of 1-tert-butyl 3-ethyl 3-aminoazetidine-1,3-dicarboxylate (4.0 g, 16.4 mmol, 1.0 equiv) and 3,4-difluorobenzaldehyde (2.4 g, 19.6 mmol, 1.2 equiv) in DCE (40.0 mL) at 0 ˚C were added STAB (7.0 g, 32.8 mmol, 2.0 equiv) and AcOH (2.0 g, 32.8 mmol, 2.0 equiv). The resulting mixture was stirred at rt overnight, adjusted the pH to 8 with ammonium hydroxide, added water (50.0 mL) and extracted with DCM (50.0 mL) twice. The combined organic layers were washed with brine (50 mL) twice, dried over anhydrous Na 2 SO 4 , and concentrated under reduced pressure to afford 6.0 g of 1-tert-butyl 3-ethyl 3-(3,4-difluorobenzyl)amino)azetidine-1,3-dicarboxylate as a yellow oil. LRMS (ES) m/z 315 (M+H-56).
Step 2: Synthesis of 1-(tert-butyl) 3-ethyl 3-(2-bromo-N-(3,4-difluorobenzyl)acetamido)azetidine-1,3-dicarboxylate:
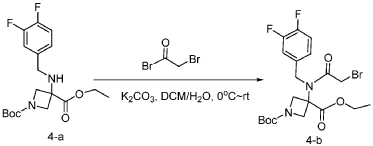
[0148] To a solution of 1-tert-butyl 3-ethyl 3-[[(3,4-difluorophenyl)methyl]amino]azetidine-1,3-dicarboxylate (6.0 g, 16.2 mmol, 1.0 equiv) in DCM (60.0 mL) at 0 ˚C were added a solution of K 2 CO 3 (3.4 g, 24.3 mmol, 1.50 equiv) in water (30 mL), and then bromoacetyl bromide (3.9 g, 19.4 mmol, 1.2 equiv) dropwise over a period of 10 min. The resulting mixture was stirred at rt overnight and extracted with DCM (50.0 mL) twice. The combined organic layers were washed with brine (100 mL) twice, dried over anhydrous Na 2 SO 4 , and concentrated under reduced pressure to afford 8.0 g of 1-(tert-butyl) 3-ethyl 3-(2-bromo-N-(3,4-difluorobenzyl)acetamido)azetidine-1,3-dicarboxylate as a yellow oil. LRMS (ES) m/z 435 (M+H-56).
Step 3: Synthesis of tert-butyl 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carboxylate:
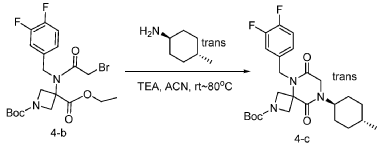
[0149] To a solution of 1-(tert-butyl) 3-ethyl 3-(2-bromo-N-(3,4-difluorobenzyl)acetamido)azetidine-1,3-dicarboxylate (8.0 g, 16.3 mmol, 1.0 equiv) in ACN (80 mL) were added TEA (4.9 g, 48.4 mmol, 3.0 equiv) and trans-(1r,4r)-4-methylcyclohexan-1-amine (2.8 g, 24.7 mmol, 1.5 equiv). The resulting mixture was stirred at rt for 1 h, gradually warmed to 80 ˚C, and stirred at 80 ˚C overnight. The mixture was cooled to rt, concentrated under reduced pressure, and triturated with a mixture of PE and EA (7/1; 80 mL) to afford 7 g (~80% purity) of tert-butyl 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carboxylate as an off-white solid. LRMS (ES) m/z 422 (M+H-56).
Step 4: Synthesis of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-2,5,8-triazaspiro[3.5]nonane-6,9-dione:
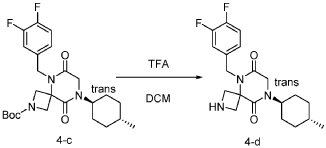
[0150] To a stirred solution of tert-butyl 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carboxylate (7.0 g, 14.7 mmol, 1.0 equiv) in DCM (70.0 mL) was added TFA (18.0 mL). The resulting mixture was stirred at rt for 3h, diluted with water (100.0 mL), adjusted the pH to 13-14 with aqueous NaOH solution (2 N), and extracted with DCM (100 mL) twice. The combined organic layers were washed with brine (100.0 mL) twice, dried over anhydrous Na 2 SO 4 , and concentrated under reduced pressure to afford 4.5 g (~80% purity) of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-2,5,8-triazaspiro[3.5]nonane-6,9-dione as a yellow semi-solid. LRMS (ES) m/z 378 (M+H).
Step 5: Synthesis of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde (Compound 4):
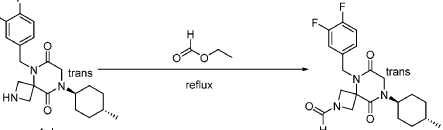
[0151] A solution of tert-butyl 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carboxylate (1.5 g, 4.0 mmol, 1.0 equiv) in ethyl formate (15.0 mL) was stirred at 80
o C overnight. The mixture was cooled to rt, concentrated under reduced pressure, and purified by C18 column chromatography, eluted with a mixture of water (0.05% NH
4 HCO
3 )/CH
3 CN (3:2) to afford 1.3 g (81%) of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde as an amorphous white solid. An experimental X-ray powder diffraction (XRPD) pattern of this amorphous white solid is shown in FIG. 1 LRMS (ES) m/z 406 (M+H);
1 H NMR (300 MHz, DMSO-d6) δ 7.96 (s, 1H), 7.47 – 7.29 (m, 2H), 7.10 (ddd, J = 9.4, 4.4, 2.0 Hz, 1H), 4.82 (s, 2H), 4.50 (d, J = 9.6 Hz, 1H), 4.15-4.28 (m, J = 3H), 4.01 (s, 2H), 3.96 (d, J = 10.8 Hz, 1H), 1.80 – 1.69 (m, 2H), 1.65 – 1.48 (m, 4H), 1.35 (d, J = 10.9 Hz, 1H), 1.13 – 0.93 (m, 2H), 0.88 (d, J = 6.5 Hz, 3H).
PAT
5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde, also called compound 1, having the structure shown below,
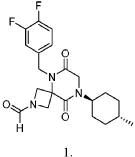
Example 1
Synthesis of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde (Compound 1)
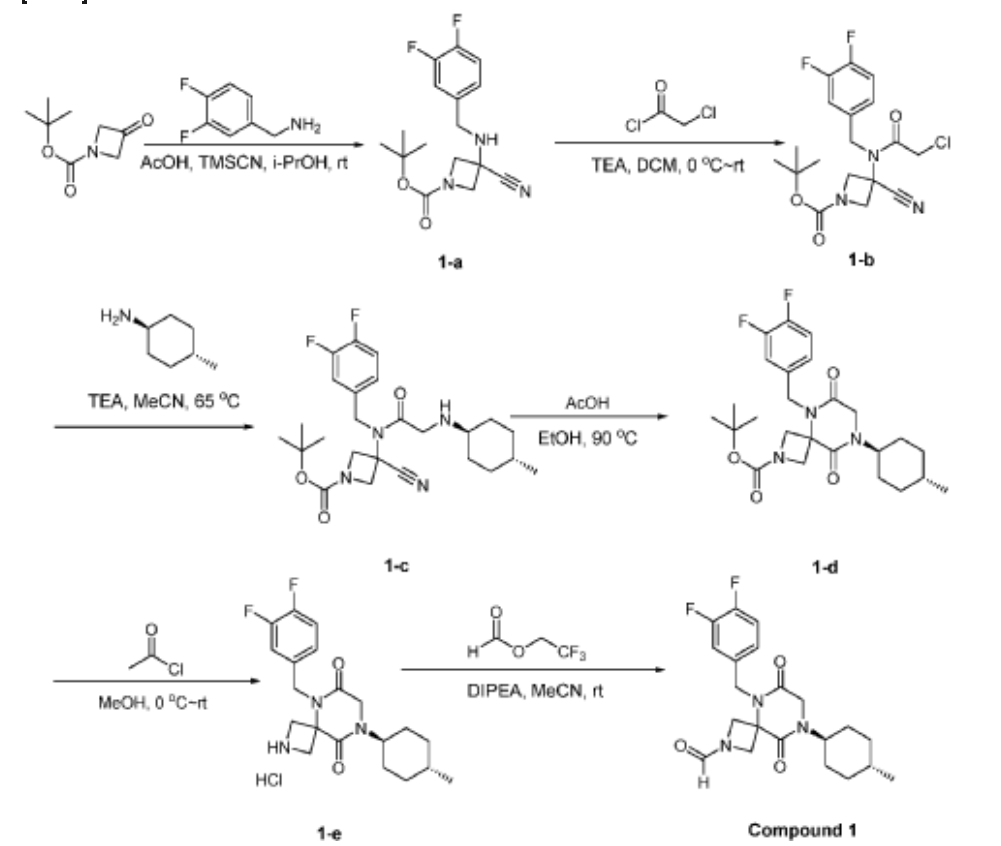
Step 6: Synthesis of 5-[(3,4-difluorophenyl)methyl]-6,9-dioxo-8-[(1r,4r)-4-methylcyclohexyl]-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde (Compound 1):
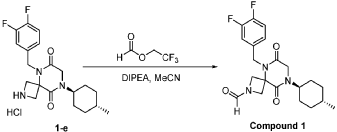
[0165] To a solution of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde (4.0 kg, 10.60 mol, 1 equiv) in MeCN (20 L) at r.t. were added 2,2,2-trifluoroethyl formate (1.63 kg, 12.72 mol, 1.2 equiv) and DIPEA (3.42 kg, 26.50 mol, 2.5 equiv) . The resulting mixture was stirred overnight at rt. The reaction was monitored by LCMS. The resulting mixture was concentrated under reduced pressure. The resulting mixture was diluted with EtOAc (10 L). The resulting mixture was quenched with NH 4Cl (6 L, sat.) and water (6 L), extracted with EtOAc (3×15 L). The combined organic layers were washed with NH 4Cl (aq.) (10 L) and brine (10 L), dried over anhydrous Na 2SO 4, concentrated under reduced pressure to give a crude brown oil, the crude oil was re-crystallized from cyclohexane and EtOAc (5:1, 4L, 80 °C to r.t.), filtered to afford 3 kg (1 st batch) 5-[(3,4-difluorophenyl)methyl]-6,9-dioxo-8-[(1r,4r)-4-methylcyclohexyl]-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde light yellow solid. The filtrate was concentrated under reduced pressure, re-crystallized with petroleum ether and EtOAc (10:1, 3 L, rt) to afford 800 g (2 nd batch) of light yellow solid. Two batches were combined, dried to afford 3.8 kg of Form I (m.p. at 133 °C) 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehyde light yellow solid. The overall yield of this step is 97%.
PAT
- Cardiac sarcomere inhibitorsPublication Number: US-12286437-B2Priority Date: 2021-03-04Grant Date: 2025-04-29
- Cardiac sarcomere inhibitorsPublication Number: US-11919909-B2Priority Date: 2021-03-04Grant Date: 2024-03-05
- Cardiac sarcomere inhibitorsPublication Number: US-2024309011-A1Priority Date: 2021-03-04
- Cardiac sarcomere inhibitorsPublication Number: WO-2022187501-A1Priority Date: 2021-03-04
- Cardiac sarcomere inhibitorsPublication Number: US-2022306642-A1Priority Date: 2021-03-04
- cardiac sarcomere inhibitorPublication Number: CN-117083275-APriority Date: 2021-03-04
- Cardiac sarcomere inhibitorsPublication Number: EP-4301760-A1Priority Date: 2021-03-04
- Crystalline forms of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehydePublication Number: US-2024132498-A1Priority Date: 2022-09-02
- Crystalline forms of 5- (3, 4-difluorobenzyl) -8- ((1 r,4 r) -4-methylcyclohexyl) -6, 9-dioxo-2, 5, 8-triazaspiro [3.5] nonane-2-carbaldehydePublication Number: CN-119998292-APriority Date: 2022-09-02
- Crystalline forms of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehydePublication Number: WO-2024050539-A1Priority Date: 2022-09-02
- Crystalline forms of 5-(3,4-difluorobenzyl)-8-((1r,4r)-4-methylcyclohexyl)-6,9-dioxo-2,5,8-triazaspiro[3.5]nonane-2-carbaldehydePublication Number: EP-4581034-A1Priority Date: 2022-09-02
- Cardiac sarcomere inhibitorsPublication Number: TW-202302594-APriority Date: 2021-03-04
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
REF
Publication Name: Scientific Reports
Publication Date: 2024-05-27
PMCID: PMC11130313
PMID: 38802475
DOI: 10.1038/s41598-024-62840-3
///////////////ulacamten, ANAX, cardiac myosin inhibitor, CK-586, CK-4021586, CK 586, CK 4021586, X325G97HZJ
Tigemocoxib
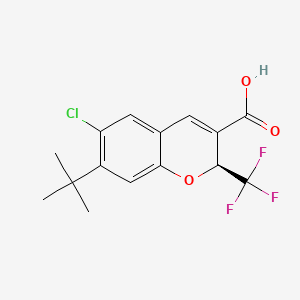
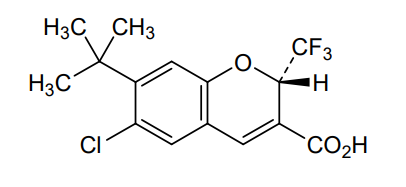
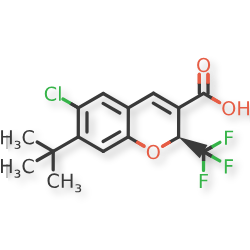
Tigemocoxib
MF C15H14ClF3O3 MW 334.72
2H-1-Benzopyran-3-carboxylic acid, 6-chloro-7-(1,1-dimethylethyl)-2-(trifluoromethyl)-, (2S)-
(2S)-7-tert-butyl-6-chloro-2-(trifluoromethyl)-2H-1-benzopyran-3-carboxylic acid
(2S)-7-tert-butyl-6-chloro-2-(trifluoromethyl)-2H-1-benzopyran-3-carboxylic acid
cyclo-oxygenase 2 (COX-2) inhibitor, anti-inflammatory, SC-75416, SC 75416, C8D42HX4WH
CAS 215122-74-0
Originator: Pfizer Inc.
Tigemocoxib is a small molecule drug. The usage of the INN stem ‘-coxib’ in the name indicates that Tigemocoxib is a selective cyclo-oxygenase inhibitor. Tigemocoxib has a monoisotopic molecular weight of 334.06 Da.
Tigemocoxib (also known as SC-75416) is a selective cyclo-oxygenase 2 (COX-2) inhibitor and nonsteroidal anti-inflammatory drug (NSAID) designed for reducing pain and inflammation. Initially developed as an orally bioavailable clinical lead for treating acute, postoperative, and chronic inflammatory pain, it acts by targeting the COX-2 enzyme
SYN
Bioorganic & Medicinal Chemistry LettersPublication Date: 2010-12-01PMID: 20709553DOI: 10.1016/j.bmcl.2010.07.054
PAT
WO2000037469
PAT
EXAMPLE 68
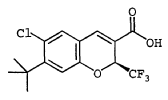
(S)-6-Chloro-7-(1,1-dimethylethyl)-2-(trifluoromethyl)-2H-1- benzopyran-3-carboxylic acid To a solution of 6-chloro-7-(1,1-dimethylethyl)-2-trifluoromethyl-2H-1-benzopyran-3-carboxylic acid (Example 8) (11.4 g, 34.1 mmol) and (S)-(-)-2-amino-3-phenyl-1-propanol (2.57 g, 17.00 mmol) was added n-heptane (200 mL) and the mixture set aside for 16 h. The resulting
suspension was filtered yielding a solid (3.8 g). This solid was recrystallized from 2-butanone (20 mL) and n-heptane (200 mL) yielding upon filtration a white solid (3.0 g).
This solid was dissolved in ethyl acetate (100 mL) and washed with 1 N hydrochloric acid (50 mL) and brine (2 × 50 mL), dried over MgSO4 and concentrated in vacuo yielding a white solid. This solid was recrystallized from n-heptane yielding the title compound of high optical purity as a crystalline solid (1.7 g, 30%): mp 175.4-176.9 °C. 1H NMR (acetone-d6/300 MHz) 7.86 (s, 1H), 7.52 (s, 1H), 7.12 (s,
1 H), 5.83 (q, 1H, J = 7.1 Hz), 1.48 (s, 9H). Anal. Calc’d for C15H14O3F3Cl : C, 53.83; H, 4.22; N, 0.0; Cl, 10.59. Found: C, 53.78; H, 4.20; N, 0.0; Cl, 10.65. This compound was determined to have an optical purity of greater than 90% ee. Chiral purity was determined as describe in Example 66.
EXAMPLE 8
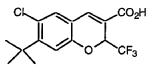
6-Chloro-7-(1,1-dimethylethyl)-2-trifluoromethyl- 2H-1-benzopyran-3-carboxylic acid
Step 1. Preparation of 4-tert-butylsalicylaldehyde.
A five liter three-neck round bottom flask equipped with overhead mechanical stirrer and condenser was charged with trifluoroacetic acid (2.4 L). A mixture of 3-tert-butylphenol (412 g, 2.8 mole) and HMTA (424 g, 3.0 mole) was added portion-wise causing an exotherm. With cooling, the temperature was maintained under 80 °C. The reaction was heated at 80 °C for one hour, then cooled, and water (2 L) added. After 0.5 hour additional water (4 L) was added and the mixture was extracted with ethyl acetate (6 L). The organic extract was washed with water and brine. The resulting organic phase was divided into 2 L volumes and each diluted with water (1 L), and solid NaHCO3 added until the mixture was neutralized. The organic phases were isolated and combined, dried over MgSO4, filtered and
concentrated in vacuo yielding an oil. This oil was
distilled at 95 °C (0.8 mm) yielding the desired
salicylaldehyde as an oil (272.9 g, 56 %) which was of sufficient purity to be used without further purification.
Step 2. Preparation of ethyl 7-(1,1-dimethylethyl)-2- (trifluoromethyl)-2H-1-benzopyran-3-carboxylate.
A one liter three-neck flask was charged with 4-tert-butylsalicylaldehyde (Step 1) (100.0 g, 0.56 mole),
dimethylformamide (110 mL), and potassium carbonate (79.9 g, 0.58 mole) causing the temperature of the mixture to rise to 40 °C. Ethyl 4,4,4-trifluorocrotonate (118.0 g, 0.70 mole) in dimethylformamide (110 mL) was added and the mixture heated to 60 °C at which time the reaction temperature rose to 70 °C. The reaction was cooled to 60 °C, maintained at 60
°C (with addedheating) for 8.5 hours and cooled to room temperature. Ethyl acetate (600 mL) and 3 N HCl (600 mL) were added, mixed, and the layers separated. The aqueous phase was extracted with ethyl acetate and the organic phases were combined. The combined organic phases were washed with brine-water (1:1), brine, dried over MgSO4, filtered and concentrated in vacuo, yielding a semi-solid. Hexane (600 mL) was added with mixing and the mixture was filtered. The filtrate was washed with brine, dried over
MgSO4, filtered and concentrated in vacuo yielding a solid. This solid was dissolved in hot ethanol (600 mL). Water (190 mL) was added which induced crystallization. Filtration of the mixture and drying of the product provided the desired ester as a crystalline solid (131.3 g, 71%): mp 91.0-94.9 °C. This material was of suitable purity to be used in subsequent steps without further purification.
Step 3. Preparation of ethyl 6-chloro-7-(1,1-dimethylethyl)-2-(trifluoromethyl)-2H-1-benzopyran-3-carboxylate.
A one liter three-neck flask equipped with mechanical stirrer and gas inlet tube was charged with the ester (Step 2) (100 g, 0.3 mole) and acetic acid (300 mL). While cooling (water bath) the reaction mixture, chlorine gas (37.6 g, 0.53 mole) was added which caused the temperature to rise to 48 °C. After stirring for two hours, the
reaction was cooled in an ice-water bath to 15 °C. Zinc powder (19.5 g, 0.3 mole) was added in one portion which caused the temperature to rise to 72 °C. After cooling to room temperature additional zinc powder (5.0 g, 0.08 mole) was added and the mixture was stirred for 0.5 hour longer. The crude mixture was filtered through diatomaceous earth and was concentrated in vacuo yielding an oil. The oil was dissolved in ethyl acetate (700 mL) washed with brine-water (1:1, 1 L) and brine (0.5 L). The resulting aqueous phase was extracted with ethyl acetate (700 mL). This ethyl acetate phase was washed with brine-water (1:1, 1 L) and brine (0.5 L). The combined organic phases were dried over
MgSO4, filtered and concentrated in vacuo yielding the title compound as a yellow oil (116 g, 106 %). This material, which contained some entrained ethyl acetate, was of suitable purity to be used in subsequent steps without further purification.
Step 4. Preparation of 6-chloro-7-(1,1-dimethylethyl)-2- (trifluoromethyl)-2H-1-benzopyran-3-carboxylic acid.
To a solution of the ester (Step 3) (116 g,
0.3 mole) in methanol (500 mL) and tetrahydrofuran (500 mL) in a one liter flask was added aqueous
sodium hydroxide (2.5 N, 240 mL, 0.6 mole). After
stirring overnight, the pH of the solution was
adjusted to 1 with concentrated hydrochloric acid
and the solution was extracted with ethyl acetate.
The ethyl acetate phase was dried over MgSO4,
filtered and concentrated in vacuo yielding a
solid. This solid was dissolved in hot ethanol
(500 mL). Water (500 mL) was added and upon
cooling to room temperature crystals formed which
were collected by vacuum filtration. The crystals
were washed with ethanol-water (3:7, 3 X 200 mL)
and dried providing the title acid as a
crystalline solid (91.6 g, 91 %) : mp 194.9-196.5
°C. 1H NMR (acetone-d6/300 MHz) 7.86 (s, 1H),
7.52 (s, 1H), 7.12 (s, 1H), 5.83 (q, 1H, J = 7.1
Hz), 1.48 (s, 9H). Anal. Calc’d for C15H14ClF3O3:
C, 53.83; H, 4.22; Cl, 10.59. Found: C, 53.92; H,
4.24; Cl, 10.50
REF
- The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacologyPublication Name: ImmunologyPublication Date: 2020-03-02PMCID: PMC7160657PMID: 32020584DOI: 10.1111/imm.13175
- Synthesis of Deuterated Benzopyran Derivatives as Selective COX-2 Inhibitors with Improved Pharmacokinetic PropertiesPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2014-08-06PMCID: PMC4190635PMID: 25313331DOI: 10.1021/ml500299q
- Leukotrienes, But Not Angiotensin II, Are Involved in the Renal Effects Elicited by the Prolonged Cyclooxygenase-2 Inhibition When Sodium Intake Is LowPublication Name: Journal of Cardiovascular PharmacologyPublication Date: 2013-04PMID: 23288201DOI: 10.1097/fjc.0b013e31828399ae
- The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-lifePublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2010-12-01PMID: 20709553DOI: 10.1016/j.bmcl.2010.07.054
- Evaluation of COX-1/COX-2 selectivity and potency of a new class of COX-2 inhibitorsPublication Name: European Journal of PharmacologyPublication Date: 2008-06-24PMID: 18457826DOI: 10.1016/j.ejphar.2008.03.057
- Modeling and Simulation to Support Dose Selection and Clinical Development of SC-75416, a Selective COX-2 Inhibitor for the Treatment of Acute and Chronic Pain
- Publication Name: Clinical pharmacology and therapeutics
- Publication Date: 2007-09-19
- PMID: 17882158
- DOI: 10.1038/sj.clpt.6100374
PAT
- Substituted benzopyran derivatives for the treatment of inflammationPublication Number: EP-0977748-A1Priority Date: 1997-04-21
- Endothermic reaction apparatusPublication Number: NO-308725-B1Priority Date: 1993-06-16
- History of Transformed Benzofiran, their preparation and pharmacological preparations for the treatment of inflammation containing themPublication Number: IL-132296-APriority Date: 1997-04-21
- Substituted benzopyran derivatives for the treatment of inflammationPublication Number: KR-100538258-B1Priority Date: 1997-04-21Grant Date: 2006-01-12
- Substituted benzopyran derivatives for the treatmentPublication Number: US-6806288-B1Priority Date: 1997-04-21Grant Date: 2004-10-19
- Benzopyran derivatives with substituents for the treatment of inflammationPublication Number: JP-4577534-B2Priority Date: 1997-04-21Grant Date: 2010-11-10
- Substituted benzopyran derivatives for the treatment of inflammationPublication Number: KR-20010020152-APriority Date: 1997-04-21
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////////tigemocoxib, ANAX, cyclo-oxygenase 2 (COX-2) inhibitor, anti-inflammatory, SC-75416, SC 75416, C8D42HX4WH