Home » Uncategorized (Page 87)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Etelcalcetide, AMG 416, KAI-4169, velcalcetide
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2

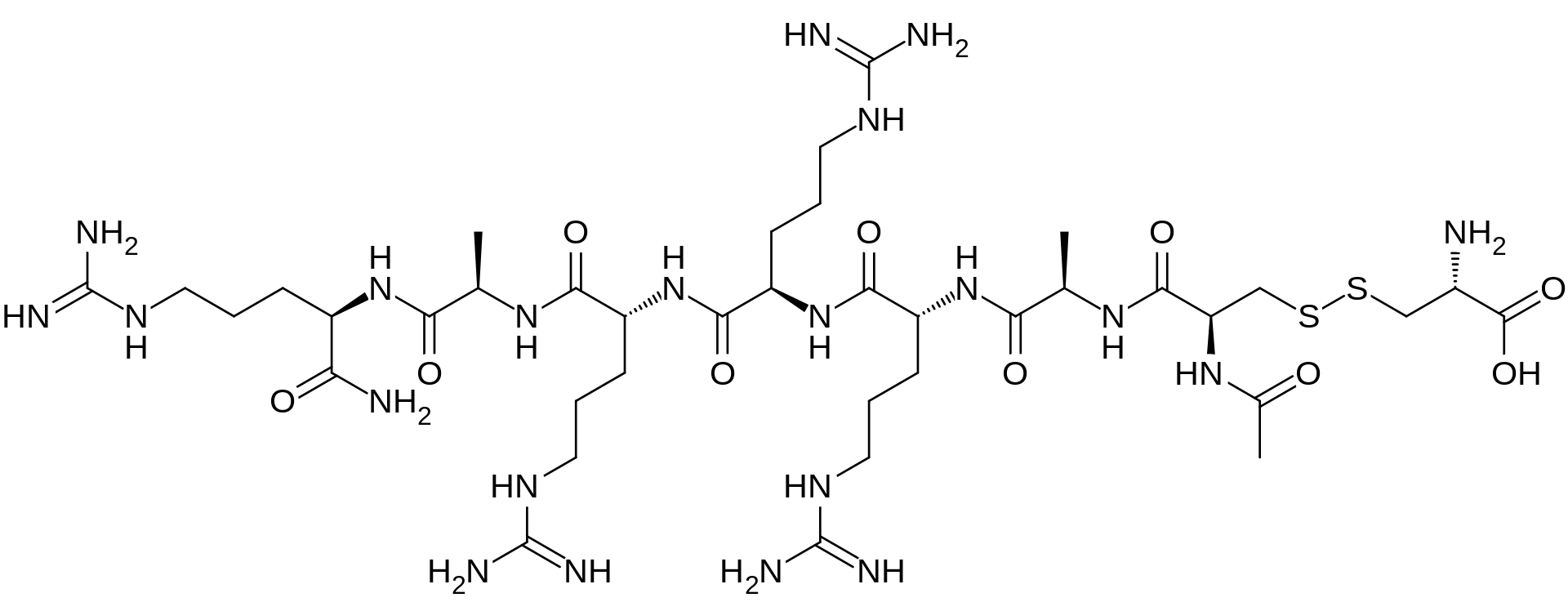
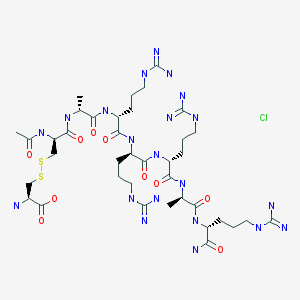
AMG 416 IS (Ac-D-Cys(L-Cys-OH)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2)
Etelcalcetide (AMG 416, KAI-4169, velcalcetide)
The main chain has 7 amino acids, all in the D-configuration. The side-chain cysteine residue is in the L-configuration. The molecular formula of AMG 416 (free base) is C38H73N21O10S2, and has a calculated average molecular mass of 1048.3 Da.
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine, hydrochloride (1:?)
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
http://www.amgenpipeline.com/pipeline/
WO 2011/014707. , the compound may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The main chain has 7 amino acids, all in the D-configuration and the side-chain cysteine residue is in the L-configuration. The amino terminal is acetylated and the carboxyl-terminal is amidated. This compound (“AMG-416”) has utility for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis patients. A liquid formulation comprising AMG-416 may be administered to a subject intravenously. The hydrochloride salt of AMG-416 may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
Therapeutic peptides pose a number of challenges with respect to their formulation. Peptides in general, and particularly those that contain a disulfide bond, typically have only moderate or poor stability in aqueous solution. Peptides are prone to amide bond hydrolysis at both high and low pH.
Disulfide bonds can be unstable even under quite mild conditions (close to neutral pH). In addition, disulfide containing peptides that are not cyclic are particularly prone to dimer formation. Accordingly, therapeutic peptides are often provided in lyophilized form, as a dry powder or cake, for later reconstitution.
A lyophilized formulation of a therapeutic peptide has the advantage of providing stability for long periods of time, but is less convenient to use as it requires the addition of one or more diluents and there is the potential risk for errors due to the use of an improper type or amount of diluent, as well as risk of contamination. In addition, the lyophilization process is time consuming and costly.
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
Generic Name:Etelcalcetide
Synonym:KAI-4169
CAS Number:1262780-97-1
N-acetyl-D-cysteinyl-S-(L-cysteine disulfide)-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide
Mechanism of Action:Activates calcium sensing receptor on parathyroid glands reducing PTH synthesis and secretion
Indication: secondary hyperparathyroidism associated with chronic kidney disease
Development Stage: Phase III
Developer:KAI Pharmaceuticals/Amgen Inc.
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
HYDROCHLORIDE
Generic Name:Etelcalcetide Hydrochloride
AMG 416, KAI-4169, previously also known as velcalcetide hydrochloride
CAS :1334237-71-6
Chemical Name:N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
Mechanism of Action:Activates calcium sensing receptor on parathyroid glands reducing PTH synthesis and secretion
Indication: secondary hyperparathyroidism associated with chronic kidney disease
Development Stage: Phase III
Developer:KAI Pharmaceuticals/Amgen Inc.
Method for preparing etelcalcetide and its salts, particularly hydrochloride. See WO2014210489, for a prior filing claiming stable liquid formulation of etelcalcetide. Amgen, following its acquisition of KAI Pharmaceuticals, and Japanese licensee Ono Pharmaceuticals are developing etelcalcetide, a long-acting iv isozyme-selective peptide-based protein kinase C epsilon inhibitor and agonist of the calcium-sensing receptor, for treating secondary hyperparathyroidism (SHPT) in patients with end-stage renal disease receiving dialysis.
In August 2015, an NDA was submitted seeking approval of the drug for SHPT in patients with chronic kidney disease (CKD) on hemodialysis (HD) in the US.
In September 2015, Amgen filed an MAA under the centralized procedure in the EU for the approval of etelcalcetide for treating SHPT in patients with CKD on HD therapy.
KAI is also investigating a transdermal patch formulation of the drug for treating primary HPT.
- 25 Aug 2015 Preregistration for Secondary hyperparathyroidism in USA (IV)
- 29 May 2015 Pooled analysis efficacy and adverse events data from two phase III trials in secondary hyperparathyroidism released by Amgen
- 21 Apr 2015 Amgen plans to submit Biological License Application to USFDA and Marketing Authorisation Application to EMA for Secondary hyperparathyroidism
PATENT
WO2011014707
http://www.google.com/patents/WO2011014707A2?cl=en
PATENT
WO 2015154031
The hydrochloride salt of AMG 416 has the chemical structure:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
(SEQ ID NO:l)
The main chain has 7 amino acids, all in the D-configuration. The side-chain cysteine residue is in the L-configuration. The molecular formula of AMG 416 (free base) is C38H73N21O10S2, and has a calculated average molecular mass of 1048.3 Da.
AMG 416 and a method for its preparation are described in International Pat. Publication No. WO 2011/014707, which is incorporated herein by reference for any purpose. As described in International Pat. Publication No. WO 2011/014707, AMG 416 may be assembled by solid-phase synthesis from the corresponding Fmoc-protected D-amino acids. After cleavage from the resin, the material may be treated with Boc-L-Cys(NPyS)-OH to form the disulfide bond. The Boc group may then be removed with trifluoroacetate (TFA) and the resulting product purified by reverse-phase high pressure liquid chromatography (HPLC) and isolated as the TFA salt form by lyophilization. The TFA salt can be converted to a pharmaceutically acceptable salt by carrying out a subsequent salt exchange procedure. Such procedures are well known in the art and include, e.g., an ion exchange technique, optionally followed by purification of the resultant product (for example by reverse phase liquid chromatography or reverse osmosis).
There is a need for an efficient method of producing AMG 416, or a pharmaceutically acceptable salt thereof (e.g., AMG 416 HC1), and particularly one appropriate for commercial scale manufacturing.
In a first aspect, provided is a method for preparing AMG 416, the method comprising: providing a resin-bound peptide having a structure selected from the group consisting of Fmoc-D-Cys(Trt)-D-Ala-D- Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:2) and Ac-D-Cys(Trt)-D-Ala-D- Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:3); cleaving the peptide from the solid support; and activating the side chain of the D-Cys residue of the cleaved peptide.
In a second aspect, provided is a method for preparing AMG 416, the method comprising: providing a peptide having a structure of Ac-D-Cys(SPy)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 (SEQ ID NO:4); and contacting the peptide with L-Cys to produce a conjugated product.
In yet a third aspect provided is a method for preparing AMG 416, the method comprising: providing a resin-bound peptide having a structure selected from the group consisting of Fmoc-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:2) and Ac-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-[Resin] (SEQ ID NO:3); cleaving the peptide from the solid support, i.e., to provide an unsupported peptide, and activating the side chain of the D-Cys residue of the unsupported peptide to generate an AMG 416 SPy intermediate (where SPy is 2-pyridinesulfenyl or S-Pyr), dissolving the AMG 416 SPy intermediate in an aqueous 0.1% TFA (trifluoroacetic acid solution), and purifying the AMG 416 SPy derivative by HPLC.
The term “AMG 416”, also known as etelcalcetide, formerly known as velcalcetide or KAI-4169, refers to a compound having the chemical name: N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-arginamide disulfide with L-cysteine, which has the following structural formula:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
Reference to AMG 416, or to any compound or AMG 416 fragment, intermediate, or precursor as described herein, is intended to encompass neutral, uncharged forms thereof, as well as pharmaceutically acceptable salts, hydrates and solvates thereof.
The terms “AMG 416 hydrochloride” and “AMG 416 HC1” are interchangeable and refer to a hydrochloride salt form of AMG 416 having the following structural formula:
H-L-Cys-OH
I
s— s
I
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · xHCl
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1 shows the chemical structure of AMG 416 (Ac-D-Cys(L-Cys-OH)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2) (SEQ ID NO: l).
FIG. 2 shows the chemical structure of Rink Amide AM resin and Ac-D-Cys(Trt)- D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-Resin (SEQ ID NO:3).
FIG. 3 shows a reaction scheme in which the SPy intermediate product (Ac-D-Cys(SPy)-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2) (SEQ ID NO:4) is formed from the peptidyl-resin (Ac-D-Cys(Trt)-D-Ala-D-Arg(Pbf)-D-Arg(Pbf)-D-Arg(Pbf)-D-Ala-D-Arg(Pbf)-NH-Resin) (SEQ ID NO:3).
FIG. 4 shows a reaction scheme in which a TFA salt of AMG 416 is formed from the SPy intermediate (AA1_7(SPy)).
FIG. 5 shows a reaction scheme in which the HC1 salt of AMG 416 is formed from the TFA salt of AMG 416.
FIG. 6 shows a reaction scheme in which Boc-D-Arg(Pbf)-OH is formed from Boc-D-Arg-OH.
FIG. 7 shows a reaction scheme in which D-Arg(Pbf)-OH is formed from Boc-D-Arg(Pbf)-OH.
EXAMPLE 5
Purification of the SPy Intermediate and Production of AMG 416 HC1
An alternative method for preparation of AMG 416 HC1 salt is described here. As described in Example 2 above, the SPy intermediate product was dried at 20°C under full vacuum after cleavage from the resin, precipitation and filtration. The precipitate was then dissolved in a 0.1% TFA aqueous solution and loaded onto a C-18 column for HPLC purification. The column was run at <60 bar and the solution temperature was 15-25 °C throughout. The eluents were 0.1% TFA in acetonitrile and 0.1% TFA in water. The fractions were stored at 5°C, they were sampled and then fractions were pooled. The combined pools from two runs were diluted and a concentration/purification run was performed using the same HPLC column to decrease the total volume and remove additional impurities. The fractions were stored at 5°C.
The fractions containing the AMG 416 SPy intermediate were subjected to azeotropic distillation to change the solvent from the 0.1% TFA to a 15% water in IPA solution, charging with IPA as needed. To the resultant AMG 416 SPy intermediate in IPA solution was then added L-Cysteine 1.15 eq and the reaction was allowed to proceed at room temperature for conjugation to occur and to form the AMG 416 TFA salt as described above in Example 4. The AMG 416 TFA solution was added to a solution of 12M aqueous HC1, 0.27 L/kg and IPA 49.4 L/kg over 3 hours via subsurface addition, resulting in direct precipitation of the AMG 416 4.5 HC1 salt. The batch was aged for 3 hours and sampled for analysis.
The material was filtered and slurry washed with 96 wt% IPA, 10 L/kg. The cake was then re-slurried for 4 hours in 10 L/kg of 96% wt% IPA. The material was filtered and further slurry washed with 96% IPA, 10 L/kg and then IPA 10 L/kg. The material was dried under full vacuum at 25°C. The dry cake was dissolved in water 8 L/kg and the batch was concentrated via distillation to remove residual IPA and achieve the desired concentration. The solution temperature was kept below 25 °C throughout the distillation.
PATENT
WO2014210489
SEE
EXAMPLE 1
Solubility of AMG 416 in Succinate Buffered Saline
In this study, the solubility of AMG 416 in succinate buffered-saline was investigated. AMG 416 HC1 (103 mg powder, 80 mg peptide) was dissolved in 200 iL of sodium succinate buffered saline (25 mM succinate, 0.9% saline, pH 4.5). After briefly vortexing, a clear solution was obtained with a nominal concentration of 400 mg/mL. Because expansion of the solution volume was not determined, the solubility of AMG 416 can be conservatively stated as at least 200 mg/mL. Although the maximal solubility was not determined in this experiment, AMG 416 is soluble in pH 4.5 succinate buffered saline to concentrations of at least 200 mg/mL.
REFERENCES
- “Amgen Submits New Drug Application For Novel Intravenous Calcimimetic Etelcalcetide (AMG 416)”
- “Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects
- “Evidence for Chronic Kidney Disease-Mineral and Bone Disorder Associated With Metabolic Pathway Changes”
KAI-4169, a novel calcium sensing receptor agonist, decreases serum iPTH, FGF-23 and improves serum bone markers in a phase 2 study in hemodialysis subjects with chronic kidney disease-mineral and bone disorder
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO054
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
| WO2011014707A2 | Jul 29, 2010 | Feb 3, 2011 | Kai Pharmaceuticals, Inc. | Therapeutic agents for reducing parathyroid hormone levels |
//////////////Etelcalcetide, AMG 416, KAI-4169, velcalcetide, peptide drugs
Monoclonal Antibody Therapy: What is in the name or clear description?
Leaders in Pharmaceutical Business Intelligence Group, LLC, Doing Business As LPBI Group, Newton, MA


Monoclonal Antibody Therapy: What is in the name or clear description?
Curator: Demet Sag, PhD, CRA, GCP
What is in the name?
Nomenclature is important part of the scientific community so we can stay on the same page in all kinds of communications for clarity. Therefore, a defined nomenclature scheme for assigning generic, or nonproprietary, names to monoclonal antibody drugs is used by the World Health Organization’s International Nonproprietary Names (INN) and the United States Adopted Names (USAN). In general, word stems are used to identify classes of drugs, in most cases placed at the end of the word.
Knowing what Antibody relies on understanding of immune response system so that one can modify the cells, choose correct biomarkers from the primary pathways (like Notch, WNT etc), know signaling from outside to inside (like GPCRs, MAPKs, nuclear transcription receptors), personalized gene make up (genomics) and key gene regulation mechanisms. Thus…
View original post 2,927 more words
Daprodustat, ダプロデュスタット

Daprodustat, GSK1278863
ダプロデュスタット
CAS 960539-70-2
GSK1278863; GSK 1278863; GSK-1278863; Daprodustat
C19H27N3O6
Exact Mass: 393.18999
(1,3-dicyclohexyl-2,4,6-trioxohexahydropyrimidine-5-carbonyl)glycine
N-[(l,3-dicyclohexyl-6-hydroxy-2,4-dioxo-l,2,3,4- tetrahydro-5-pyrimidinyl)carbonyl]glycine
2-(1,3-dicyclohexyl-2,4,6-triohexahydropyrimidine-5-carboxamide acetic acid
Mechanism of Action: HIF-prolyl hydroxylase inhibitor
Indication: anemia, diabetic wounds, and reduction of ischemic complications
Development Stage: Phase II
Developer:GlaxoSmithKline
UNII:JVR38ZM64B
ダプロデュスタット
Daprodustat

C19H27N3O6 : 393.43
[960539-70-2]
Daprodustat , also known as GSK1278863, is a novel HIF-prolyl hydroxylase inhibitor. Hypoxia inducible factor (HIF) stabilization by HIF-prolyl hydroxylase (PHD) inhibitors may improve ischemic conditions such as peripheral artery disease (PAD). Short-term treatment with a novel HIF-prolyl hydroxylase inhibitor (GSK1278863) failed to improve measures of performance in subjects with claudication-limited peripheral artery disease
- Originator GlaxoSmithKline
- Class Antianaemics; Pyrimidines; Small molecules
- Mechanism of ActionErythropoiesis stimulants; Prolyl hydroxylase inhibitors
- Phase II Anaemia; Perioperative ischaemia
- Phase I Diabetic foot ulcer; Tendon injuries
- DiscontinuedPeripheral arterial disorders
Most Recent Events
- 27 Jul 2015No recent reports of development identified – Phase-II for Anaemia in India and New Zealand (PO)
- 27 Jul 2015Daprodustat is still in phase II trials for Anaemia in the USA, Australia, Canada, Czech Republic, Denmark, France, Germany, Hungary, Japan, Poland, Russia, Spain, South Korea, and United Kingdom
- 01 Jun 2015GlaxoSmithKline completes a phase I trial in Tendon injuries (In volunteers) in USA (PO) (NCT02231190)
| WHO ATC code: | B03 (Antianemic Preparations)C (Cardiovascular System)
C01 (Cardiac Therapy) D03 (Preparations for Treatment of Wounds and Ulcers) M09A-X (Other drugs for disorders of the musculo-skeletal system) |
| EPhMRA code: | B3 (Anti-Anaemic Preparations)C1 (Cardiac Therapy)
C6A (Other Cardiovascular Products) D3A (Wound Healing Agents) M5X (All Other Musculoskeletal Products) |
Daprodustat (INN) (GSK1278863) is a drug which acts as a HIF prolyl-hydroxylase inhibitor and thereby increases endogenous production of erythropoietin, which stimulates production of hemoglobin and red blood cells. It is in Phase III clinical trials for the treatment of anemia secondary to chronic kidney disease.[1][2] Due to its potential applications in athletic doping, it has also been incorporated into screens for performance-enhancing drugs.[3]
SYN 1

SYN 2

PATENT
WO 2007150011
https://www.google.com.ar/patents/WO2007150011A2
Illustrated Methods of preparation
Scheme 1

a) 1. NaH, THF, rt 2. R1NCO, 60 0C; b) 1. NaH, THF or dioxane, rt 2. R4NCX, heat; c) H2NCH2CO2H, DBU, EtOH, 1600C, microwave.
Scheme 2

a) R1NH2, CH2Cl2 or R1NH2-HCl, base, CH2Cl2; b) CH2(C(O)Cl)2, CH2Cl2, reflux or CH2(CO2Et)2, NaOEt, MeO(CH2)2OH, reflux or 1. EtO2CCH2COCl, CHCl3, 70 0C 2.
DBU, CHCl3, 70 0C; c) 1. YCNCH2CO2Et,, EtPr’2N, CHCl3 or CH2Cl2 2. aq NaOH, EtOH, rt. Scheme 3 (for R1 = R4)
a) CDI,

DMF, 70 0C or , EtOAc, rt
Scheme 4

a) OCNCH2CO2Et, EtPr’2N, CHCl3 or CH2Cl2; b) 1. R1HaI, Na/K2CO3, DMF or DMA, 100 0C or R1HaI, pol-BEMP, DMF, 120 0C, microwave 2. aq NaOH, MeOH or EtOH, rt.
Scheme 5

a) 1. CH2(CO2H)2, THF, O 0C – rt 2. EtOH, reflux; b) 1. OCNCH2CO2Et, EtPr’2N, CH2Cl2 2. aq NaOH, EtOH, rt.
Scheme 6

a) 1. Phthalimide, DIAD, PPh3, THF 2. (NH2)2, EtOH, reflux.
Scheme 7

a) Ac2O, AcOH, 130 0C.
Example 18
N-T(1 ,3-Dicvclohexyl-6-hydroxy-2,4-dioxo- 1 ,2,3,4-tetrahvdro-5-pyrimidinyl)carbonyl1grycine Method 1
18.1a) h3-Dicvclohexyl-2A6(lH,3H,5H)-pyrimidinetrione. Dicyclohexylurea (3.0 g, 13.39 mmoles) was stirred in chloroform (80 mL) and treated with a solution of malonyl dichloride (1.3 mL, 13.39 mmoles) in chloroform (20 mL), added dropwise under argon. The mixture was heated at 500C for 4 hours, wasahed with 1 molar hydrochloric acid and evaporated onto silica gel. Flash chromatography (10-30% ethyl acetate in hexane) to give the title compound (2.13 g, 55%). 1Η NMR (400 MHz, OMSO-d6) δ ppm 4.46 (tt, J=12.13, 3.54 Hz, 2 H), 3.69 (s, 2 H), 2.15 (qd, J=12.46, 3.28 Hz, 4 H), 1.77 (d, J=13.14 Hz, 4 H), 1.59 (t, J=12.76 Hz, 6 H), 1.26 (q, J=12.97 Hz, 4 H), 1.04 – 1.16 (m, 2 H)
18.1b) N-r(1.3-Dicvclohexyl-6-hvdroxy-2.4-dioxo-1.2.3.4-tetrahvdro-5- pyrimidinvDcarbonyll glycine. Ethyl isocyanatoacetate (802 uL, 7.15 mmoles) was added to a mixture of l,3-dicyclohexyl-2,4,6(lH,3H,5H)-pyrimidinetrione (2.1 g, 7.15 mmoles) and diisopropylethylamine (2.47 mL, 14.3 mmoles) in dichloromethane (100 mL) and stirred overnight. The reaction mixture was washed with 1 molar hydrochloric acid (x2) and evaporated. The residue was dissolved in ethanol (10 mL) and treated with 1.0 molar sodium hydroxide (5 mL). The mixture was stirred for 72 hours, acidified and extracted into ethyl acetate. Some ester remained, therefore the solution was evaporated and ther residue was dissolved in 1 molar soldium hydroxide solution with warming and strred for 2 hours. The mixture was acidified with IM HCl and extracted with ethyl acetate (x2). The combined extracts were washed with 1 molar hydrochloric acid , dried and evaporated to a solid which was slurried in a mixture of diethyl ether and hexane, collected, washed with the same solvent mixture and dried to give the title compound (1.86 g, 66%). IH NMR (400 MHz, DMSO-^6) δ ppm 13.07 (br. s., 1 H), 10.19 (t, J=5.31 Hz, 1 H), 4.63 (t, J=10.99 Hz, 2 H), 4.12 (d, J=5.56 Hz, 2 H), 2.27 (q, J=I 1.71 Hz, 4 H), 1.79 (d, J=12.88 Hz, 4 H), 1.50 – 1.69 (m, 6 H), 1.28 (q, J=12.97 Hz, 4 H), 1.12 (q, J=12.72 Hz, 2 H)
Method 2
18.2a) 1.3-Dicvclohexyl-2.4.6πH.3H.5H)-pyrimidinetrione. A solution of N5N- dicyclohexylcarbodiimide (254 g; 1.23 mol.) in anhydrous TΗF (700 mL) was added dropwise to a cold (0 0C) solution of malonic acid (64.1 g; 0.616 mol.) in anhydrous TΗF (300 mL) over a period of- 30 minutes. The mixture was stirred and allowed to warm to room temperature over 2 h. (After 1 h, the mixture became very thick with precipitate so further anhydrous TΗF (500 mL) was added to facilitate agitation.). The mixture was filtered and the filtrate evaporated to afford a yellow solid which was immediately slurried in ethanol (1 L) and heated to reflux temperature. The mixture was then allowed to cool to room temperature then filtered and the solid washed with cold ethanol (250 mL) to afford the title compound (129.4 g; 72%) as a colorless solid. 1Η NMR (400 MHz, DMSO-(Z6) δ ppm 1.03 – 1.18 (m, 2 H) 1.18 – 1.34 (m, 4 H) 1.59 (t, J=13.14 Hz, 6 H) 1.76 (d, J=12.88 Hz, 4 H) 2.04 – 2.24 (m, 4 H) 3.69 (s, 2 H) 4.35 – 4.54 (m, 2 H).
18.2b) Ethyl N-[(l .3-dicvclohexyl-6-hvdroxy-2.4-dioxo- 1.2.3.4-tetrahydro-5- pyrimidinyPcarbonyll glycinate. A solution of l,3-dicyclohexyl-2,4,6(lH,3H,5H)-pyrimidinetrione (120.0 g; 0.41 mol.) and diisopropylethylamine (105.8 g; 0.82 mol.) in dichloromethane (1 L) was stirred and treated dropwise with a solution of ethyl isocyanatoacetate (53.0 g; 0.41 mol.) in dichloromethane (500 mL) and the mixture was then stirred at room temperature overnight. The mixture was then treated dropwise with 6M aq. hydrochloric acid (500 mL) and the separated organic layer was dried and evaporated. The resulting solid was slurried in hexanes (500 mL) and heated to reflux temperature. The mixture was then allowed to cool and filtered to afford ethyl N- [(1 ,3-dicyclohexyl-6-hydroxy-2,4-dioxo- 1 ,2,3,4-tetrahydro-5-pyrimidinyl)carbonyl]glycinate (159.1 g; 92%) as a cream powder. IH NMR (400 MHz, CHLOROFORM-,/) δ ppm 1.24 (s, 2 H) 1.37 (s, 7 H) 1.52 – 1.76 (m, 6 H) 1.78 – 1.94 (m, 4 H) 2.25 – 2.48 (m, 4 H) 4.17 (d, J=5.81 Hz, 2 H) 4.28 (q, J=7.24 Hz, 2 H) 4.74 (s, 2 H) 10.37 (t, J=4.67 Hz, 1 H). 18.2c)
N-rπ^-Dicyclohexyl-ό-hydroxy^^-dioxo-l^J^-tetralivdro-S- pyrimidinyDcarbonyll glycine. A stirred suspension of ethyl Ν-[(l,3-dicyclohexyl-6-hydroxy-2,4- dioxo-l,2,3,4-tetrahydro-5-pyrimidinyl)carbonyl]glycinate (159.0 g; 0.377 mol.) in ethanol (1.5 L) was treated dropwise with 6M aq. Sodium hydroxide (250 mL) and stirred at room temperature for 3 h. The solution was then acidified by the dropwise addition of 6M aq. hydrochloric acid (300 mL), diluted with water (IL) and then filtered. The crude solid was slurried in water (2 L) then stirred vigorously and heated at 35 0C for 1 h and filtered and dried. The solid material (~ 138 g) was then crystallized from glacial acetic acid (1.5 L) (with hot filtration to remove a small amount of insoluble material). The solid, which crystallized upon cooling, was collected and washed with cold glacial acetic acid (3 x 100 mL) to afford N-[(l,3-dicyclohexyl-6-hydroxy-2,4-dioxo-l,2,3,4- tetrahydro-5-pyrimidinyl)carbonyl]glycine (116.2 g; 78%) as a colorless solid.
IH NMR (400 MHz, DMSO-(Z6) δ ppm 1.11 (d, J=12.88 Hz, 2 H) 1.27 (q, J=12.80 Hz, 4 H) 1.62 (s, 6 H) 1.70 – 1.90 (m, J=12.88 Hz, 4 H) 2.11 – 2.44 (m, 4 H) 4.11 (d, J=5.81 Hz, 2 H) 4.45 – 4.77 (m, 2 H) 10.19 (t, J=5.81 Hz, 1 H) 13.08 (s, 1 H).
References
- Jump up^ Schmid H, Jelkmann W. Investigational therapies for renal disease-induced anemia. Expert Opin Investig Drugs. 2016 Aug;25(8):901-16. . doi:10.1080/13543784.2016.1182981. PMID 27122198. Missing or empty
|title=(help) - Jump up^ Ariazi JL, Duffy KJ, Adams DF, Fitch DM, Luo L, Pappalardi M, Biju M, DiFilippo EH, Shaw T, Wiggall K, Erickson-Miller C. Discovery and Preclinical Characterization of GSK1278863 (Daprodustat), a Small Molecule Hypoxia Inducible Factor-Prolyl Hydroxylase Inhibitor for Anemia. J Pharmacol Exp Ther. 2017 Dec;363(3):336-347. . doi:10.1124/jpet.117.242503. PMID 28928122. Missing or empty
|title=(help) - Jump up^ Thevis M, Milosovich S, Licea-Perez H, Knecht D, Cavalier T, Schänzer W. Mass spectrometric characterization of a prolyl hydroxylase inhibitor GSK1278863, its bishydroxylated metabolite, and its implementation into routine doping controls. Drug Test Anal. 2016 Aug;8(8):858-63. . doi:10.1002/dta.1870. PMID 26361079. Missing or empty
|title=(help)
 |
|
| Clinical data | |
|---|---|
| Synonyms | GSK1278863 |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| Chemical and physical data | |
| Formula | C19H27N3O6 |
| Molar mass | 393.44 g/mol |
| 3D model (JSmol) | |
//////////////Daprodustat, GSK1278863, ダプロデュスタット , HIF-prolyl hydroxylase inhibitor, anemia, diabetic wounds, reduction of ischemic complications, Phase II, GlaxoSmithKline
- Daprodustat
- 960539-70-2
- GSK1278863
- UNII-JVR38ZM64B
- GSK-1278863
- JVR38ZM64B
- N-((1,3-Dicyclohexylhexahydro-2,4,6-trioxopyrimidin-5-yl)carbonyl)glycine
- Daprodustat [USAN:INN]
- GSK 1278863
- D0F6JC
- Daprodustat(GSK1278863)
- Daprodustat; GSK1278863
- Daprodustat (JAN/USAN/INN)
- GTPL8455
- Daprodustat (GSK1278863)
- CHEMBL3544988
- BCP16766
- EX-A1121
- KS-00000M8Z
- s8171
C1CCC(CC1)N2C(=O)C(C(=O)N(C2=O)C3CCCCC3)C(=O)NCC(=O)O
Sparsentan, PS433540, RE-021

Sparsentan (PS433540, RE-021)
- C32H40N4O5S
- Average mass592.749
FDA APPROVED 2023/2/17, Filspari
4′-((2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl)-N-(4,5-dimethylisoxazol-3-yl)-2′-(ethoxymethyl)-[1,1′-biphenyl]-2-sulfonamide
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide
Sparsentan
PS433540; RE-021, formerly known as DARA
CAS :254740-64-2
4-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(4,5- dimethylisoxazol-3-yl)-2-(ethoxymethyl)biphenyl-2-sulfonamide
Mechanism of Action:acting as both an Endothelin Receptor Antagonist (ERA) and Angiotensin Receptor Blocker (ARB).
Indication: Focal Segmental Glomerulosclerosis (FSGS).Focal Segmental Glomerulosclerosis (FSGS) is a rare and severe nephropathy which affects approximately 50,000 patients in the United States. Most cases of FSGS are pediatric.
Development Stage: Phase II
Developer:Retrophin, Inc
- OriginatorBristol-Myers Squibb
- DeveloperRetrophin
- ClassAntihypertensives; Isoxazoles; Small molecules; Spiro compounds; Sulfonamides
- Mechanism of ActionAngiotensin type 1 receptor antagonists; Endothelin A receptor antagonists
- Orphan Drug Status Yes – Focal segmental glomerulosclerosis
-
- 09 Jan 2015 Sparsentan receives Orphan Drug status for Focal segmental glomerulosclerosis in USA
- 31 Dec 2013 Phase-II/III clinical trials in Focal segmental glomerulosclerosis in USA (PO)
- 07 May 2012I nvestigation in Focal segmental glomerulosclerosis in USA (PO)
Sparsentan is an investigational therapeutic agent which acts as both a selective endothelin receptor antagonist and an angiotensin receptor blocker. Retrophin is conducting the Phase 2 DUET trial of Sparsentan for the treatment of FSGS, a rare and severe nephropathy that is a leading cause of end-stage renal disease. There are currently no therapies approved for the treatment of FSGS in the United States. Ligand licensed worldwide rights of Sparsentan (RE-021) to Retrophin in 2012 .The Food and Drug Administration (FDA) has granted orphan drug designation for Retrophins sparsentan for the treatment of focal segmental glomerulosclerosis (FSGS) in January 2015.
In 2006, the drug candidate was licensed to Pharmacopeia by Bristol-Myers Squibb for worldwide development and commercialization. In 2012, a license was obtained by Retrophin from Ligand. In 2015, Orphan Drug Designation was assigned by the FDA for the treatment of focal segmental glomerulosclerosis.
Sparsentan, also known as RE-021, BMS346567, PS433540 and DARA-a, is a Dual angiotensin II and endothelin A receptor antagonist. Retrophin intends to develop RE-021 for orphan indications of severe kidney diseases including Focal Segmental Glomerulosclerosis (FSGS) as well as conduct proof-of-concept studies in resistant hypertension and diabetic nephropathy. RE-021, with its unique dual blockade of angiotensin and endothelin receptors, is expected to provide meaningful clinical benefits in mitigating proteinuria in indications where there are no approved therapies
Sparsentan, sold under the brand name Filspari, is a medication used for the treatment of primary immunoglobulin A nephropathy.[1] Sparsentan is an endothelin and angiotensin II receptor antagonist.[1][4] It is taken by mouth.[1]
The most common side effects include swelling of the extremities, low blood pressure, dizziness, high blood potassium, anemia, injury to the kidney, and increased liver enzymes in the blood.[5]
It was approved for medical use in the United States in February 2023.[5][6][7] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[8]
PATENT
WO 2000001389
https://www.google.co.in/patents/WO2000001389A1?cl=en


Example 41
4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.4! non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l, l’-biphenyl! -2-sulfonamide

A. 4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl)-N-[(2-trimethylsilylethoxy)methyl]-2′- hydroxym ethyl [1, l’-biphenyl] -2-sulfonamide P14 (243 mg, 0.41 mmol) was used to alkylate 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene hydrochloride according to General Method 4. 41A (100 mg, 35% yield) was isolated as a slightly yellow oil after silica gel chromatography using 1:1 hexanes/ethyl acetate as eluant. B. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methvn -N-0.4- dimethyl-5-isoxazolyl)-2′-hydroxymethyl[l,l’-biphenyn-2- sulfonamide
Deprotection of 41A (100 mg, 0.14 mmol) according to General Method 8 (ethanol) gave the title compound as white solid in 46% yield following silica gel chromatography (96:4 methanol/chloroform eluant):
MS m/e 565 (ESI+ mode); HPLC retention time 3.21 min (Method A);
HPLC purity >98%.
Example 42
4′-[(2-Butyl-4-oxo-1.3-diazaspiro[4.41non-l-en-3-yl)methvn-N-(3,4- dimethyl-5-isoxazolyl)-2′-ethoxymethyl [ 1 , l’-biphenyll -2-sulfonamide

A. 4′- [(2-Butyl-4-oxo- 1 ,3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3 ,4- dimethyl-5-isoxazolyl)-N-[(2-methoxyethoxy)methyll-2′- hvdroxym ethyl [1 , l’-biphenyl] -2-sulfonamide
Triethylsilane (6 ml) and TFA (6 ml) were added to a solution of 5F (960 mg, 1.5 mmol) in 15 ml dichloromethane at RT. The mixture was stirred at RT for 2 h and was then concentrated. The residue was taken up in ethyl acetate and was washed successively with aqueous sodium bicarbonate, water, and brine. The organic layer was dried over sodium sulfate and concentrated. The residue was chromatographed on silica gel using 100:2 dichloromethane/methanol to afford 42A (740 mg, 77%) as a colorless gum. Rf=0.13, silica gel, 100:5 dichloromethane/methanol. B. 4′- [(2-Butyl-4-oxo- 1.3-diazaspiro [4.41 non- l-en-3-yl)methyll -N-(3.4- dimethyl-5-isoxazolyl)-N-r(2-methoxyethoxy)methyll-2′- ethoxymethyl[l.l’-biphenyll-2-sulfonamide A mixture of 42A (100 mg, 0.15 mmol), iodoethane (960 mg, 6.1 mmol) and silver (I) oxide (180 mg, 0.77 mmol) in 0.7 ml DMF was heated at 40 ° C for 16 h.. Additional iodoethane (190 mg, 1.2 mmol) and silver (I) oxide (71 mg, 0.31 mmol) were added and the reaction mixture was heated at 40 ° C for an additional 4 h. The mixture was diluted with 1:4 hexanes/ethylacetate and was then washed with water and brine. The organic layer was dried over sodium sulfate and was then concentrated. The residue was chromatographed on silica gel using 200:3 dichloromethane/methanol as eluant to afford 42B (51mg, 49%) as a colorless gum. Rf=0.35, silica gel, 100:5 dichloromethane/methanol.
C. 4,-[(2-Butyl-4-oxo-1.3-diazaspirof4.41non-l-en-3-yl)methyll-N-(3.4- dimethyl-5-isoxazolyl )-2′-ethoxym ethyl [ 1. l’-biphenyll -2-sulfonamide
42B (51 mg) was deprotected according to General Method 7 to afford the title compound in 80% yield following preparative reverse-phase HPLC purification: white solid; m.p. 74-80 ° C (amorphous); IH NMR (CDCL, )δ0.87(tr, J=7Hz, 3H), 0.99(tr, J=7Hz, 3H), 1.32(m, 2H), 1.59(m, 2H), 1.75-2.02(m, 11H), 2.16(s, 3H), 2.35(m, 2H), 3.38 (m, 2H), 4.23(m, 2H), 4.73(s, 2H), 7.11-7.85 (m, 7H); MS m/e 593 (ESI+ mode); HPLC retention time 18.22 min. (Method E); HPLC purity >97%.
PATENT
WO 2001044239
http://www.google.co.in/patents/WO2001044239A2?cl=en
……………………
Dual angiotensin II and endothelin A receptor antagonists: Synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides with improved potency and pharmacokinetics
J Med Chem 2005, 48(1): 171
 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPDThe ETA receptor antagonist (2) (N-(3,4-dimethyl-5-isoxazolyl)-4‘-(2-oxazolyl)-[1,1‘-biphenyl]-2-sulfonamide, BMS-193884) shares the same biphenyl core as a large number of AT1 receptor antagonists, including irbesartan (3). Thus, it was hypothesized that merging the structural elements of 2 with those of the biphenyl AT1 antagonists (e.g., irbesartan) would yield a compound with dual activity for both receptors. This strategy led to the design, synthesis, and discovery of (15) (4‘-[(2-butyl-4-oxo-1,3-diazaspiro[4.4]non-1-en-3-yl)methyl]-N-(3,4-dimethyl-5-isoxazolyl)-2‘-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-[1,1‘-biphenyl]-2-sulfonamide, BMS-248360) as a potent and orally active dual antagonist of both AT1 and ETAreceptors. Compound 15 represents a new approach to treating hypertension.

Scheme 2 a DIFFERENT COMPD
a (a) DIBAL, toluene; (b) NaBH4, MeOH; (c) (Ph)3P, CBr4, THF (51% from 9); (d) compound 7, NaH, DMF; (e) 1 N HCl; (f) compound 4, (Ph3P)4Pd, aqueous Na2CO3, EtOH/toluene; (g) 6 N aqueous HCl/EtOH (60% from 10); (h) 13, sodium triacetoxy borohydride, AcOH, (i) diisopropylcarbodiimide, CH2Cl2 (31% from 12).
PATENT
WO 2010135350
http://www.google.com/patents/WO2010135350A2?cl=en
Compound 1 :

Scheme IV

Scheme V

Formula IV 1
Scheme VII

Formula Vl

A solution of 2-(2,4-dimethylphenyl)benzenesulfonic acid (Compound 12) (0.5 g, 1.9 mmol) in 50 mL of anhydrous acetonitrile was prepared and transferred to a round-bottom flask. After flushing with nitrogen gas, N-bromosuccinimide (0.75 g, 4.2 mmol) was added followed by 50 mg (0.2 mmol) of benzoyl peroxide. The solution was heated at reflux for 3 hours. The solvent was removed in-vacuo and the resulting syrup purified by silica gel chromatography (1 :1 hexanes/EtOAc) to yield Compound 13 as a white solid. 1H NMR (500 MHz, CD3CN) 8.12 (d, J = 7.5 Hz, IH), 7.92 (t, J = 7.5 Hz, IH), 7.78 (d, J= 7.5 Hz, IH), 7.74-7.71 (m, 2H), 7.68-7.65 (m, 2H), 5.12 (s, 2H), 4.70 (s, 2H). Example 4 2-(4-Bromomethyl-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 14)

A solution of 20 mg (0.058 mmol) of (l-bromomethylbenzo[3,4- d])benzo[l,2-f]-2-oxa-l,l-dioxo-l-thiocycloheptane (Compound 13) in ethanol was stirred at elevated temperature until the starting material was consumed to give crude product (compound 14) that was used directly in the next step without isolation or purification.
Example 5
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonic acid (Compound 15)

To the above ethanol solution of crude 2-(4-bromomethyl-2- ethoxymethylphenyl)benzenesulfonic acid (Compound 14) described in Example 4 was added approximately 25 mL of anhydrous DMF. The ethanol was removed from the system under reduced pressure. Approximately 15 mg (0.065 mmol) of 2-butyl-l,3- diazaspiro[4.4]non-l-en-4-one (compound 7 in Scheme IV) was added followed by 300 μL of a IM solution of lithium bis-trimethylsilylamide in THF. The solution was allowed to stir at room temperature for 3 hours. The solvents were removed under reduced pressure and the remaining residue purified by preparative RP-HPLC employing a Cl 8 column and gradient elution (H2O:MeCN) affording the title compound as a white solid; [M+H]+ calcd for C27H34N2O5S 499.21, found, 499.31 ; 1H NMR (500 MHz, CD3CN) 8.04 (t, J= 5.5 Hz, IH), 7.44-7.10 (m, 2H), 7.28 (s, IH), 7.22 (d, J= 8.0 Hz, 2H), 7.08- 7.04 (m, 2H), 4.74 (br s, 2H), 4.32 (d, J= 13.0 Hz IH), 4.13 (d, J= 13.0 Hz IH), 3.40- 3.31 (m, 2H), 2.66 (t, J= 8 Hz, 2H), 2.18-2.13 (m, 5H), 1.96-1.90 (m, 2H obscured by solvent), 1.48 (m, 2H), 1.27 (s, J= 7 Hz, 2H), 1.16 (t, J= 7 Hz, 3H), 0.78 (t, J= 7.5 Hz, 3H).
Example 6
2-(4-((2-Butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3-yl)methyl>2- ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16)

To a solution of DMF (155 μL, 2 mmol, 2 equiv.) in dichloromethane (5 mL) at 0 0C was added dropwise oxalyl chloride (175 μL, 2 mmol, 2 equiv.) followed by a dichloromethane (5 mL) solution of 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l- en-3-yl)methyl)-2-ethoxymethylphenyl)benzenesulfonic acid (Compound 15) (0.50 g, 1.0 mmol). The resulting mixture was stirred at 0 0C for ~2 hours, diluted with additional dichloromethane (25 mL), washed with saturated sodium bicarbonate solution (10 mL), water (10 mL), and brine (10 mL), dried over sodium sulfate, and then concentrated to give crude sulfonyl chloride (compound 16) that was used without purification.
Example 7
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

[0062] To a solution of 5-amino-3,4-dimethylisoxazole (60 mg, 0.54 mmol) in THF at -60 °C was added dropwise potassium tert-butoxide (1 mL of 1 M solution) followed by a solution of crude 2-(4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non-l-en-3- yl)methyl)-2-ethoxymethylphenyl)benzenesulfonyl chloride (Compound 16) (0.28 g, 0.54 mmol) in THF (4 mL). The resulting mixture was stirred at about -60 °C for 1 hour, allowed to warm to room temperature overnight, and then quenched with IN HCl solution to about pH 4. Standard workup of extraction with ethyl acetate, washing with water, drying, and concentration provided the final compounds as a white solid. 1H NMR (400 MHz, CDCl3) 8.03 (dd, J = 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J = 7.5, 2H), 1.37 (sextet, J = 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
Example 8 l-Bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17)

To a solution of ethyl 4-bromo-3-ethoxymethylbenzoate (9.4 g, 33 mmol) in toluene (56 mL) at about -10 0C was added 51 g of a 20% diisobutylaluminum hydride solution in toluene (ca. 70 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy 5% NaOH solution to keep the temperature below about 10 °C. Organic phase of the resulting mixture was separated and the aqueous phase was extracted with toluene. The combined organic phase was concentrated in vacuo to a final volume of ~60 mL toluene solution of l-bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) that was used in next step without purification.
Example 9 l-Bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18)

To a solution of 1 -bromo-2-ethoxymethyl-4-hydroxymethylbenzene (Compound 17) (8.4 g, 33 mmol) in toluene (60 mL) prepared in Example 8 at about -10 °C was added methanesulfonyl chloride (7.9 g, 68 mmol). The reaction was stirred at the same temperature for about 30 minutes until the reduction was completed, and then quenched with icy water to keep the temperature at about 0 °C. The organic layer was separated and washed again with icy water to provide a crude product solution of 1 – bromo-2-ethoxymethyl-4-methanesulfonyloxymethylbenzene (Compound 18) that was used without purification.
Example 10
1 -Bromo-4-((2-butyl-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 -yl)methy l)-2- ethoxymethylbenzene bisoxalic acid salt (Compound 19)

To the crude solution of 1 -bromo-2-ethoxymethyl-4- methanesulfonyloxymethylbenzene (Compound 18) (1 1 g, 33 mmol) in toluene (80 mL) prepared in Example 9 was added a 75% solution of methyltributylammonium chloride in water (0.47 mL). The resulting mixture was added to a solution of 2-butyl-4-oxo-l,3- diazaspiro[4.4]non-l-ene (compound 7 in Scheme VI) (7.5 g, 32 mmol) in dichloromethane (33 mL) pretreated with a 10 M NaOH solution (23 mL). The reaction mixture was stirred at room temperature for 2 hours until compound 18 was not longer detectable by HPLC analysis and then was quenched with water (40 mL). After stirring about 10 minutes, the organic layer was separated and aqueous layer was extracted with toluene. The combined organic phase was washed with water and concentrated to a small volume. Filtration through a silica gel pad using ethyl acetate as solvent followed by concentration yielded 1 -bromo-4-((2-buty 1-4-oxo- 1 ,3 -diazaspiro [4.4]non- 1 -en-3 – yl)methyl)-2-ethoxymethylbenzene as a crude oil product.
The crude oil was dissolved in ethyl acetate (22 mL) and warmed to around 50 °C. Anhydrous oxalic acid (4.6 g) was added to the warm solution at once and the resulting mixture was stirred until a solution was obtained. The mixture was cooled gradually and the bisoxalic acid salt (compound 19) was crystallized. Filtration and drying provided pure product (compound 19) in 50-60% yield from ethyl 4-bromo-3- ethoxymethylbenzoate in 3 steps. 1H NMR (400 MHz, CDCl3) 12.32 (bs, 4H), 7.58 (d, J = 7.8, IH), 7.36 (s, IH), 7.12 (d, J= 7.8, IH), 4.90 (s, 2H), 4.56 (s, 2H), 3.68 (q, J= 7.5, 2H), 2.87-2.77 (m, 2H), 2.40-1.95 (m, 8H), 1.62-1.53 (m, 2H), 1.38-1.28 (m, 4H), and 1.82 (t, J= 7.5, 3H).
Example 11
N-(3,4-Dimethyl-5-isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en- 3yl)methyl-2-ethoxymethylphenyl)phenylsulfonamide (Compound 1)

To a suspension of l-bromo-4-((2-butyl-4-oxo-l,3-diazaspiro[4.4]non- l-en-3-yl)methyl)-2-ethoxymethylbenzene bisoxalic acid salt (Compound 19) (5.0 g, 8.3 mmol) in toluene (20 niL) under nitrogen was added water (30 mL) and pH was adjusted to 8-9 by addition of a 2 M NaOH solution at room temperature. The organic phase was separated and mixed with 2-(N-(3,4-dimethyl-5-isoxazolyl)-N- methoxymethylamino)sulfonylphenylboronic acid pinacol ester (Scheme VII, Formula IX, where R8is methoxymethyl and M = boronic acid pinacol ester) (3.6 g, 8.5 mmol), bis(dibenzylideneacetone)palladium(0) (Pd(dba)2) (0.12 g), and a standard phosphine ligand. After a 2 M sodium carbonate solution was added, the reaction mixture was warmed to 70 0C and stirred until the reaction was complete by HPLC analysis. The reaction was cooled to room temperature and quenched with water, and then separated in phases. The organic phase was treated with activated carbon, filtered through a pad of silica gel, and was concentrated to afford a crude mixture.
The crude reaction mixture was dissolved in ethanol (40 mL) after palladium catalyst was removed and was treated with 6 M HCl solution (ca. 40 mL). The mixture was warmed to 75-80 °C and stirred for about 2 hours until the reaction was completed by HPLC analysis. After the mixture was cooled to room temperature, the pH of the mixture was adjusted to 8 by addition of 10 M NaOH solution. The mixture was stirred for 2 more hours and the pH was adjusted to 6 by adding 2 M HCl and the crystal seeds. Filtration of the crystalline solid followed by drying provided N-(3,4-dimethyl-5- isoxazolyl)-2-(4-(2-butyl-4-oxo-l,3-diazospiro[4.4]non-l-en-3yl)methyl-2- ethoxymethylphenyl)phenylsulfonamide (Compound 1) as a white solid.1H NMR (400 MHz, CDCIa) 8.03 (dd, J= 8.0 and 1.2, IH), 7.60 (td, J = 7.5 and 1.5, IH), 7.50 (td, J = 7.7 and 1.5, IH), 7.36 (s, IH), 7.28 (d, J= 2.1, 1 H), 7.25 (dd, J = 7.5 and 1.2, IH), 7.09 (dd, J= 7.9 and 1.6, IH), 6.61 (bs, IH), 4.77 (AB quartet, J= 15.5 and 8.1, 2H), 4.18 (AB quartet, J= 12.0 and 35, 2H), 3.45-3.32 (m, 2H), 2.39 (t, J= 7.5, 2H), 2.26 (s, 3H), 2.02- 1.84 (m, 8H), 1.82 (s, 3H), 1.63 (quint, J= 7.5, 2H), 1.37 (sextet, J= 7.3, 2H), 1.07 (t, J = 7.0, 3H), and 0.90 (t J= 7.3, 3H).
| US20040002493 * | Aug 20, 2001 | Jan 1, 2004 | Kousuke Tani | Benzoic acid derivatives and pharmaceutical agents comprising the same as active ingredient |
| US20070054806 * | Sep 6, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Novel sulfonamide-comprising solid formulations |
| US20070054807 * | Sep 8, 2006 | Mar 8, 2007 | Bayer Cropscience Gmbh | Storage-stable formulations of sulfonamides |
.
 |
|
| Clinical data | |
|---|---|
| Trade names | Filspari |
| Other names | RE-021, PS433540 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a623018 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| UNII | |
| KEGG | |
| ChEBI | |
| ECHA InfoCard | 100.275.317 |
| Chemical and physical data | |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
References
- ^ Jump up to:a b c d e f “Filspari- sparsentan tablet, film coated”. DailyMed. 17 February 2023. Retrieved 6 March 2023.
- ^ Jump up to:a b c d “Filspari EPAR”. European Medicines Agency (EMA). 22 February 2024. Retrieved 24 February 2024. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Filspari Product information”. Union Register of medicinal products. 23 April 2024. Retrieved 7 September 2024.
- ^ Chiu AW, Bredenkamp N (September 2023). “Sparsentan: A First-in-Class Dual Endothelin and Angiotensin II Receptor Antagonist”. The Annals of Pharmacotherapy. 58 (6): 645–656. doi:10.1177/10600280231198925. PMID 37706310. S2CID 261743204.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Drug Trials Snapshots: Filspari”. U.S. Food and Drug Administration (FDA). 17 February 2023. Retrieved 7 September 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Travere Therapeutics Announces FDA Accelerated Approval of Filspari (sparsentan), the First and Only Non-immunosuppressive Therapy for the Reduction of Proteinuria in IgA Nephropathy” (Press release). Travere Therapeutics. 17 February 2023. Retrieved 17 February 2023 – via GlobeNewswire.
- ^ Syed YY (April 2023). “Sparsentan: First Approval”. Drugs. 83 (6): 563–568. doi:10.1007/s40265-023-01864-x. PMC 10232600. PMID 37022667.
- ^ New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ “PHARMACOPEIA LAUNCHES STUDY OF DARA COMPOUND | FDAnews”. http://www.fdanews.com.
- ^ “Ligand Licenses DARA Program to Retrophin”. investor.ligand.com. 21 February 2012.
- ^ https://www.fiercebiotech.com/biotech/retrophin-sheds-shkreli-connection-new-name-travere-therapeutics.
{{cite news}}: Missing or empty|title=(help) - ^ “Ongoing Non-malignant Hematological, Neurological, and Other Disorder Indications Accelerated Approvals”. U.S. Food and Drug Administration (FDA). 21 August 2024. Retrieved 7 September 2024.
- ^ “Travere Therapeutics Announces Full FDA Approval of Filspari (sparsentan), the Only Non-Immunosuppressive Treatment that Significantly Slows Kidney Function Decline in IgA Nephropathy” (Press release). Travere Therapeutics. 5 September 2024. Retrieved 7 September 2024 – via GlobeNewswire.
- ^ “Despite trial scare, Travere’s Filspari gains full FDA nod in kidney disease showdown with Novartis”. fiercepharma.com.
External links
- Clinical trial number NCT03762850 for “A Study of the Effect and Safety of Sparsentan in the Treatment of Patients With IgA Nephropathy (PROTECT)” at ClinicalTrials.gov
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Sparsentan (Filspari). Sparsentan (27), marketed by Travere Therapeutics, is an oral, dual endothelin angiotensin receptor antagonist that received accelerated USFDA approval in February 2023 for reducing proteinuria in adults with primary immunoglobulin A (IgA) nephropathy who are at risk of rapid
disease progression.205206,207 Also known as Berger’s disease, IgAnephropathy is an immune-complex mediated disease characterized by deposits of IgA in the kidneys, resulting in inflammation and damage which can eventually lead to kidney failure. Typical treatment of IgA nephropathy has focused
on supportive care to slow kidney decline, for example, lowering blood pressure, reducing proteinuria, and minimizing lifestyle risk factors; immunosuppressive therapy has also been utilized, though it is controversial and carries risks.208 Sparsentan is the first nonimmunosuppressive treatment for IgA nephropathy and has received first-in-class and orphan drug designations. Accelerated approval was based on reduction of proteinuria (which is a risk factor for disease progression) during interim
analysis in phase III clinical trials. 209 endothelin type A (ETASparsentan blocks ) and angiotensin II type 1 receptors(AT1), interrupting the signaling pathway that contributes to disease progression. 210
The structure of the drug combines 211,212 elements that target both of these receptor types.
213 Thesynthesis of sparsentan (27), as shown in Scheme 50 and Scheme 51, was disclosed by Retrophin Pharmaceuticals (now Travere Therapeutics). Its telescoped sequences and isolation of intermediates as salts suggest that this route may be suitable for large-scale manufacturing.
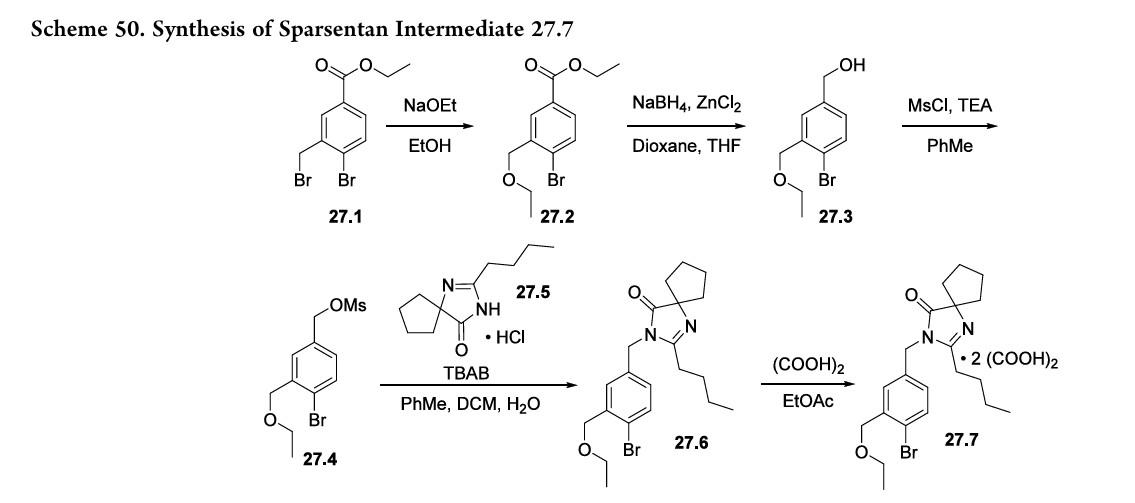
The synthesis of the spirocyclic imidazolinone intermediate 27.7 is shown in Scheme 50.
Displacement of the benzylic bromide in 27.1 with sodium ethoxide produced ether 27.2. Reduction of the ester with sodium borohydride and zinc chloride yielded alcohol 27.3 which was then converted to mesylate 27.4. Reaction with spirocyclic imidazolinone 27.5 under phase transfer conditions
yielded 27.6 whichwasisolatedasthebisoxalatesalt (27.7).The sequence from 27.1 to 27.7 is telescoped, and no yields were given in the patent.
The construction of the biphenyl framework is shown in Scheme 51. Treatment of aryl bromide 27.8 with n-BuLi and triisopropyl borate followed by reaction with pinacol yielded boronic ester 27.9. Intermediates 27.7 and 27.9 were coupled via a Suzuki reaction to form the biphenyl which was isolated as
the camphorsulfonate salt (27.10). The synthesis was finished with deprotection of the methoxymethyl group under acidic conditions followed by recrystallization from isopropanol and heptane to yield sparsentan (27).
(206) Donadio, J. V.; Grande, J. P. IgA nephropathy. N. Engl. J. Med.2002, 347, 738−748.
(207) Fabiano, R. C. G.; Pinheiro, S. V. B.; Simões e Silva, A. C.Immunoglobulin A nephropathy: a pathophysiology view. Inflammation Res. 2016, 65, 757−770.
(208) Floege, J.; Rauen, T.; Tang, S. C. W. Current treatment of IgAnephropathy. Springer Semin. Immunopathol. 2021, 43, 717−728.
(209) Rovin, B.H.; Barratt, J.; Heerspink, H. J. L.; Alpers, C. E.; Bieler,S.; Chae, D.-W.; Diva, U. A.; Floege, J.; Gesualdo, L.; Inrig, J. K.; et al.Efficacy and safety of sparsentan versus irbesartan in patients with IgA
nephropathy (PROTECT): 2-year results from a randomised, active controlled, phase 3 trial. Lancet 2023, 402, 2077−2090.
(210) Komers, R.; Plotkin, H. Dual inhibition of renin-angiotensin aldosterone system and endothelin-1 in treatment of chronic kidney disease. Am. J. Physiol.: Regul., Integr. Comp. Physiol. 2016, 310, R877−
R884.
(211) Murugesan, N.; Tellew, J. E.; Gu, Z.; Kunst, B. L.; Fadnis, L.;Cornelius, L. A.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; et al. Discovery of N-isoxazolyl biphenylsulfonamides as potent dual
angiotensin II and endothelin A receptor antagonists. J. Med. Chem.2002, 45, 3829−3835.
(212) Murugesan, N.; Gu, Z.; Fadnis, L.; Tellew, J. E.; Baska, R. A. F.; Yang, Y.; Beyer, S. M.; Monshizadegan, H.; Dickinson, K. E.; Valentine,M.T.; et al. Dual angiotensin II and endothelin A receptor antagonists:
synthesis of 2′-substituted N-3-isoxazolyl biphenylsulfonamides withimproved potencyandpharmacokinetics. J. Med. Chem. 2005, 48, 171−179.
(213) Komers, R.; Shih, A. Biphenyl sulfonamide compounds for the treatment of kidney diseases or disorders. WO 2018071784, 2018.

//////////////Sparsentan, PS433540, RE-021, Bristol-Myers Squibb, ORPHAN DRUG, Retrophin, FDA 2023, APPROVALS 2023
O=S(C1=CC=CC=C1C2=CC=C(CN3C(CCCC)=NC4(CCCC4)C3=O)C=C2COCC)(NC5=NOC(C)=C5C)=O,
Synthesis of a fluorinated Ezetimibe analogue
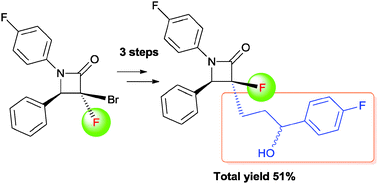
DOI: 10.1039/C5NJ01969A, Paper
E-mail: aando@pharm.setsunan.ac.jp
A facile and efficient synthesis of a fluorinated Ezetimibe analogue was achieved by radical allylation, Wacker oxidation, and nucleophilic arylation of [small alpha]-bromo-[small alpha]-fluoro-[small beta]-lactam
The synthesis of an α-fluoro-β-lactam-containing Ezetimibe analogue was accomplished starting from α-bromo-α-fluoro-β-lactam which was readily prepared from ethyl dibromofluoroacetate. A facile and efficient method for the introduction of the C3 alkyl side chain was realized via radical allylation. The diastereoselective allylation of α-bromo-α-fluoro-β-lactam was successfully applied to construct the relative configuration of the β-lactam nucleus between C3 and C4. Further modification of the allyl side chain gave the 3′-(4-fluorophenyl)-3′-hydroxypropyl group through Wacker oxidation and nucleophilic arylation.
Acceptability of Draft Labeling to Support ANDA Approval Guidance for Industry

Acceptability of Draft Labeling to Support ANDA Approval Guidance for Industry
INTRODUCTION This guidance provides recommendations and information related to the submission of proposed labeling with abbreviated new drug applications (ANDAs) under section 505(j)(2)(A)(v) of the Federal Food, Drug, and Cosmetic Act (the Act) and FDA’s implementing regulations (21 CFR 314.94(a)(8)). This guidance is intended to assist applicants submitting ANDAs under section 505(j) of the Act to the Office of Generic Drugs (OGD) in the Center for Drug Evaluation and Research (CDER). It explains FDA’s interpretation of the regulatory provision related to the submission of copies of applicants’ proposed labeling in ANDAs and clarifies that OGD will accept draft labeling and does not require the submission of final printed labeling (FPL) in order to approve an ANDA. FDA is implementing this guidance without prior public comment because the Agency has determined that prior public participation is not feasible or appropriate (see 21 CFR 10.115(g)(2) and (g)(3)). FDA made this determination because this guidance presents a less burdensome policy that is consistent with the public health. In general, FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
DISCUSSION OGD is issuing this guidance to provide regulated industry and other interested persons with our current thinking on the requirement that ANDA applicants submit copies of proposed labeling in their applications. Specifically, OGD is clarifying whether submission of FPL as opposed to draft labeling is required in order for OGD to approve an ANDA…………http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm465628.pdf
//////
EZETIMIBE POSTER
The synthesis of ezetimibe with high stereochemical purity
Krzysztof Bańkowski , Katarzyna Sidoryk , Katarzyna Filip , Joanna Zagrodzka
Pharmaceutical Research Institute (IF), Rydygiera 8, Warszawa 01-793, Poland
Ezetimibe, (3R,4S)-1-(4-fluorophenyl)-3-((3S)-3-(4-fluorophenyl)- 3-hydroxypropyl)-4-(4-hydroxyphenyl)-2-azetidinone, is an anti-hyperlipidemic drug which is used to lower cholesterol level. It acts by decreasing cholesterol absorption in the intestine.
The three chiral centers in the ezetimibe molecule give rise to eight stereoisomers and the synthesis of stereochemical pure ezetimibe is a significant challenge. The synthesis of ezetymibe is described in many patents and patent applications, however the problem of stereochemical purity of the final product and its intermediates is almost completely omitted.
The synthesis of ezetimibe was realized by a procedure shown below, according to Schering Co. patents No US 6,207,822, EP 1137634:

We have investigated the sterochemical course of all steps of this process and found that for the preparation of optical pure ezetimibe the providing of pure (S,R,S,S) – EZ-6 is cru-cial. This diastereomer (product of anti-condensation of EZ-4 + EZ-5) is usually contaminated with (S,R,R,S) – EZ-6 isomer (syn-condensation), and also with (R,R,S,S) – EZ-6 isomer derived from small amount of (R,S)-alcohol EZ-4 which is usually occurring in required (S,S)-alcohol. The presence of (R,R,S,S) – EZ-6 diastereomer leads to (R,R,S) -“iso-ezetimibe” which is very difficult to remove from ezetimibe.
The synthesis of ezetimibe was optimized, all chemical and sterochemical impurities were isolated and/or synthesized and characterized by NMR, MS and HPLC techniques. The method for the purification of desired key intermediate (S,R,S,S)-6 was elaborated. These al-lowed us to develop the large scale efficient synthesis of pharmaceutical pure Ezetimibe (HPLC > 99,5 %, (R,R,S)-isomer < 0,1 %, single unknown impurity < 0,1 %, total impurities < 0,6 % ).
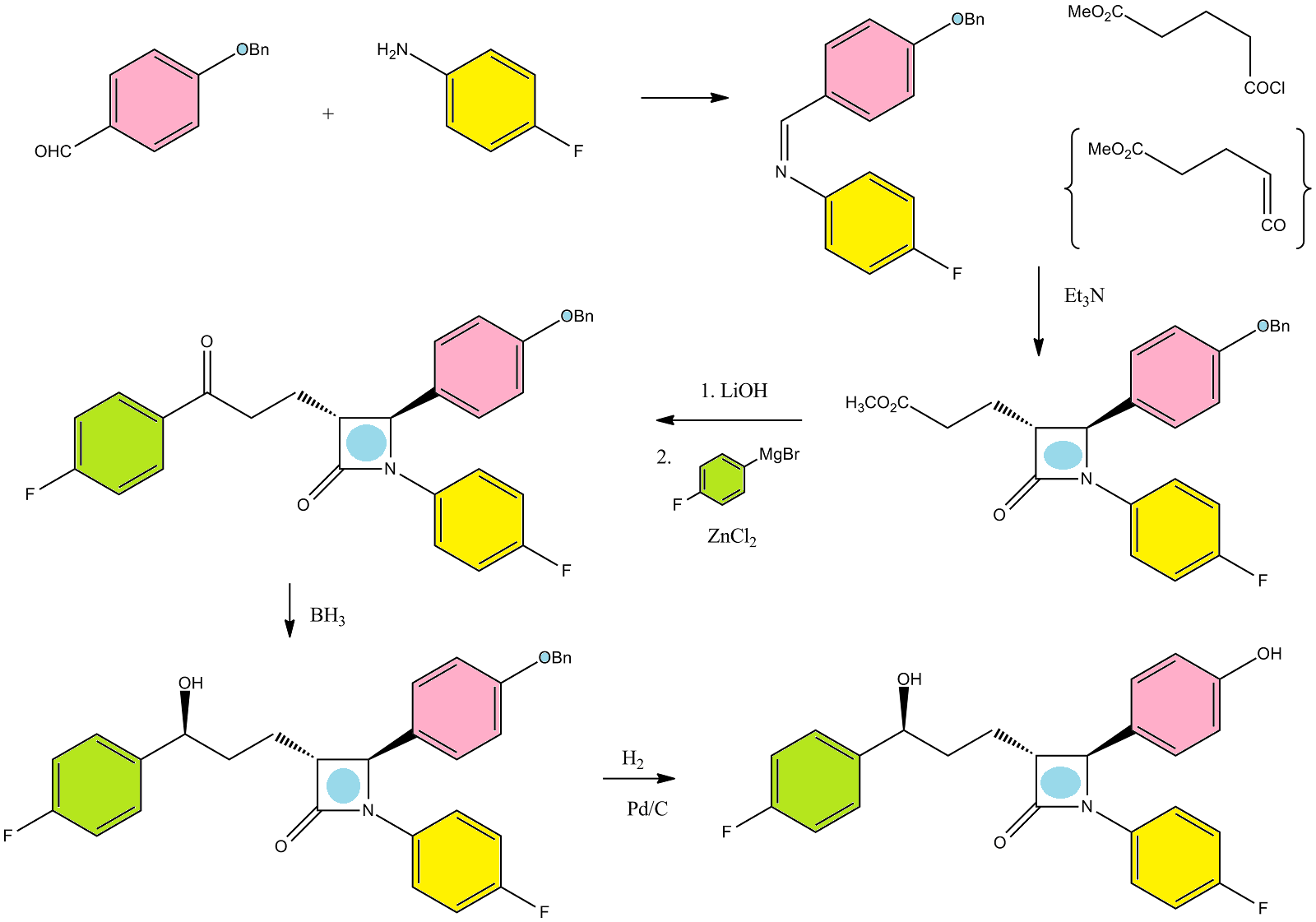
Ezetimibe has the chemical name 1-(4-fluorophenyl)-3(R)-[3-(4-fluorophenyl)-3(S)-hydroxypropyl]-4(S)-(4-hydroxyphenyl)-2-azetidinone (hereinafter referred to by its adopted name “ezetimibe”) and is structurally represented by Formula I.

Ezetimibe is in a class of lipid lowering compounds that selectively inhibit the intestinal absorption of cholesterol and related phytosterols. It is commercially available in products sold using the trademark ZETIA as a tablet for oral administration containing 10 mg of ezetimibe, and in combination products with simvastatin using the trademark VYTORIN.
U.S. Pat. No. 6,096,883 discloses generically and specifically ezetimibe and its related compounds along with their pharmaceutical compositions
The preparation of ezetimibe ezetimibe first disclosed in U.S. Patent US 5767115.

Hydrogen Debenzylation get ezetimibe, the method disclosed in this patent require the use of several key intermediates purified by column chromatography, increasing the difficulty and cost of industrial production.
US patent US5767115 to improve the synthesis process have also been reported. For example: W02006137080 US5767115 on the basis of synthesis of intermediate compound 3 were improved optimization, using pivaloyl chloride and the formation of a mixed anhydride intermediate compound 2, and then with the chiral auxiliary (S) -4- phenyl-2- oxazolidinone reaction intermediate compound 3; US Patent US6133001 discloses a microbial catalytic asymmetric reduction of carbonyl to give chiral hydroxy, instead US5767115 Synthesis of (R) -CBS catalyst;
W02008089984 reported the use of a rhodium catalyst [(S, S) -N- (piperidyl-N-sulfonyl) -I, 2-diphenyl ethylenediamine] (η 6-mesitylene) Ruthenium right of intermediate compound 9Said reduction.
W02008032338 reports by reacting the intermediate compound 8 with a salt of an aliphatic amine which was purified manner, although effectively improve the purity, but adds steps, and the yield was significantly reduced.
In addition to the synthetic route based on open Pu Xi US5767115, US Patent US6207822, US Patent US5856473, US patent US5886171, W02005066120, W02005113496, W02006050634, W02007017705 also disclose the ezetimibe different preparation methods.
Patent W02007072088 discloses another synthetic route for preparing ezetimibe ezetimibe, which is a small step synthesis reaction, the specific synthetic route is as follows:

Another US: 5739321; US: 1 5886171 reported the route: the (4S) – hydroxytetrahydrofuran _2_ one and N- (4- fluorophenyl) -4-benzyloxy-benzylidene methylamine as starting Preparation of raw ezetimibe, the reaction scheme is as follows:

…………

Since the first report since the synthesis method, there are already several ezetimibe ezetimibe synthetic route reports, such as document US 5856473, US 5739321, EP 1137634, EP 720599, WO 1995/08532 EP 0720599, provides ezetimibe ezetimibe synthetic route.
Example 9 Preparation of Compound 8 embodiment.
Hydrogenation bottle was added 7a (2.14 g, 4.3 mmol), methanol (30 mL), was added Pd / C (50 mg :), transferred into the autoclave, and replaced with hydrogen three times, filled with hydrogen 5 atm, room temperature and stirred for 6 hours, venting of hydrogen, filtered through Celite, with a small amount of methanol (10 mL), dried and concentrated, the residue was mixed solvent of methyl t-butyl ether, and recrystallized from n-hexane to give compound 8, 78% yield

8, 1H-NMR (300 MHz, DMSO 6 ) [delta] = 9.50 (s, 1H), 7.41-7.07 (m, 10H), 6.79 (d, J = 8.6 Hz, 2H), 5.27-5.25 (m, 1H) , 4.78-4.71 (m, 1H), 4.47-4.44 (m, 1H), 3.07-3.08 (m, 1H), 1.85-1.75 (m, 4H) ppm. 10 Compound la (P = Bn benzyl)
Example 3 (Preparation -2a of.
The reaction is as follows: Under an argon atmosphere, [Pd (C 3 H 5 ) Cl] 2 (54.8 mg, 0.15 mmol) and (&& 5 Lc (193 mg, 0.25 mmol) were added to a Schlenk tube, was added anhydrous CH 2 C1 2 C50 mL), stirred at room temperature for 10 minutes, the substrate was added successively lb (4.12 g, 10 mmol), K 2 C0 3(1.0 M solution, 30 mL, 30 mmol) and p-fluoroaniline (3.33 g, 30 mmol ). After stirring at room temperature for three hours, liquid separation, the aqueous phase was extracted with dichloromethane (3 x 50 mL), The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated

(I?) – 2a, 85% yield, 93% ee.
Example 4 Compound Example (Preparation -2b of.
The reaction is as follows: Under an argon atmosphere, [Pd (C 3 H 5 ) Cl] 2 (54.8 mg, 0.15 mmol) and (&& 5) -La (165 mg, 0.25 mmol) were added to a Schlenk tube, was added anhydrous CH 2 C1 2 C50 mL), stirred at room temperature for 10 minutes, the substrate was added successively lb (3.78 g, 10 mmol), K 2 C0 3(1.0 M solution, 30 mL, 30 mmol) and p-fluoroaniline (3.33 g, 30 mmol). After stirring at room temperature for three hours, liquid separation, the aqueous phase was extracted with dichloromethane (3 x 50 mL), The combined organic phase was dried over anhydrous sodium sulfate, filtered and concentrated, purified by column chromatography to give asymmetric amination the product of (i?) – 2b. The reaction formula is as follows:

(R) -2b, colorless liquid, yield 86%, [a] D 2Q = -89.1 (c 1.00, CHC1 3 ), EE 95% [determined by high-performance liquid chromatography, chiral AD-H column, n hexane / isopropanol = 95: 5, 1.0 mL / min, 254 nm; t R (minor) = 4.15 min; t R . (Major) = 4.60 min] 1H NMR (300 MHz, CDCl 3 ) [delta] = 7.20 (d, J = 8.4 Hz, 2H), 6.89-6.78 (m, 4H), 6.51-6.47 (m, 2H), 6.34 (s, 1H), 5.88 (s, 1H), 5.26 (s, 1H), 4.19-4.08 (m, 2H), 4.00 (s, br, 1H), 1.20 (t, J = 7.2 Hz, 3H), 0.97 (s, 9H), 0.18 (s, 6H) ppm; 13 C NMR (100 MHz, CDCl 3 ) [delta] = 166.1, 155.8 (d, J (F , C) = 234.3 Hz), 155.1 (s), 143.0 (d, J (F , C) = 1.9 Hz), 140.4 (s), 133.1 (s), 128.5 (s), 125.2 (s), 120.0 (s), 115.4 (d, J (F , C) = 22.3 Hz), 114.1 (d, J (F , C) = 7.4 Hz), 60.6 , 58.9, 25.5, 18.0, 13.9, -4.5 ppm; 19 F-NMR (376 MHz, CDCl 3 ) [delta] -127.5 ppm.
Preparation Example 5 Compound 4a embodiment.
(I?) – 2a (3.44 g, 8.48 mmol) and nucleophiles 3a (2.82 g, 12.7 mmol) was added in an eggplant-shaped flask, tetrahydrofuran (100 mL), DBU (4.25 g, 16.96 mmol was stirred at room temperature for 12 hours, thin layer chromatography until starting material disappeared by TLC the reaction mixture was concentrated and purified by column chromatography, to obtain compound 4a, 82% yield (Note: Allyl allyl).
ESI-MS m / z: 628.4 [M + H + ]; HRMS (ESI) m / z:. calcd for C 37 H 36 N0 6 F 2 +1 : 628.2505, Found:
+ ]. After the reaction system may also not treated directly in the next step. The reaction formula is as follows:

: Example 6 Preparation of Compound 5a.
To the reaction system of Example 5 is continued morpholine (4.43 g, 50.88 mmol) and Pd (PPh 3 ) 4 , and stirring was continued at room temperature for 6 hours, concentrated purified by chromatography (98 mg, 0.0848 mmol) after column .
The total yield from the compound 2a to 5a rate of 71%. Compound 5a is composed of a pair of non-enantiomer at a ratio of 2 or 3: 1. No need to separate the non-enantiomer, can be used directly in the next step.
ESI-MS m / z: 544.2 [M + H +]; HRMS (ESI) m / z:. Calcd for C33H31NO4 F 2 Na +1 : 566.2113, Found: 566.2113 [M + Na + ].
Preparation Example 7 Compound 6a embodiment.
Compound 5a (3.5 g, 6.4 mmol) and anhydrous tetrahydrofuran (50 mL) was added an eggplant-shaped flask, and cooled to -20 ° C under slowly added dropwise amino lithium hexamethyldisilazide (LiHMDS) (1.0 M THF, 14 ml, 14 mmol). The reaction system was stirred at this temperature continued for 40 minutes, 5 mL of water was added to quench the reaction, and extracted with dichloromethane (3 x 100 mL), the organic phase was dried over anhydrous sodium sulfate

6a, 77% yield. [A] D 2Q = +1.9 (c 1.00, MeOH), 95% EE [by the high performance liquid chromatography, chiral OD-H column n is isopropanol = 70:30, 1.0 mL / min, 254 nm; t R (Major) = 19.60 min; t R . (minor) = 25.83 min] 1H NMR (400 MHz, CDCl 3 ) [delta] = 7.98-7.94 (m, 2H), 7.41-7.30 ( m, 5H), 7.25-7.23 (m, 4H), 7.09 (t, J = 8.8 Hz, 2H), 6.96-6.88 (m, 4H), 5.02 (s, 1H), 4.67 (d, J = 2.4 Hz , 1H), 3.31-3.23 (m, 1H), 3.17-3.08 (m, 2H), 2.42-2.20 (m, 2H) ppm; 13 C NMR (100 MHz, CDCl 3 ) [delta] = 197.2, 167.1, 165.6 ( d, J (F , C) = 253.9 Hz), 158.9, 158.8 (d, J (F , C) = 242.2 Hz), 136.5, 133.7 (d, J (F , C) = 2.7 Hz), 132.9 (d , J (F , C) = 2.8 Hz), 130.5 (d, J (F , C) = 9.4 Hz), 129.3, 128.5, 127.9, 127.3, 127.1, 118.2 (d, J (F , C) = 7.9 Hz ), 115.7 (d, J (F , C) = 8.4 Hz), 115.5 (d, J (F , c) = 8.3 Hz), 115.3, 69.9, 60.9, 59.6, 35.4, 23.0 ppm; 19 F NMR (376 MHz, CDCl 3 ) [delta] -104.8, -117.9 ppm.
Compound 6a is the same as reported in the literature specific rotation direction, the same NMR data reported in the literature. References:
(A) Wu, G; Wong, Y;. Chen, X .; Ding, ZJ Org Chem 1999, 64, 3714. (b) Sasikala, CHVA;. Padi, PR; Sunkara, V; Ramayya, P .; Dubey , PK; Uppala, VBR;… Praveen, C. Org Process Res Dev 2009, 13, 907. (c) Sova, M .; Mravljak, J .; Kovac, A .; Pecar, S .; Casar, Z .; Gobec, S .; Synthesis, 2010, 20, 3433.
Preparation Example 8 Compound 7a embodiment.
In dichloromethane (40 mL) and tetrahydrofuran (5 mL) were added to an eggplant-shaped flask, and cooled to 0 ° C, was added borane dimethyl sulfide complex (0.46 mL, 7.23 mmol) and ![]() – (+) – 2-methyl–CBS- oxazaborolidine (133 mg, 0.482 mmol). Compound 6 (; 2.4 § , 4.82 11 ^ 101) was dissolved in dichloromethane (2011 ^) in the join. Stirred at the same temperature for 5 hours. After completion of the reaction with methanol (10 mL) quenched the reaction was concentrated, added to 1 mol per liter of dilute hydrochloric acid, methylene-wan (X) was extracted, the organic phase was washed with saturated sodium chloride wash paint, concentrated, ethyl acetate – n-hexane to give the compound 7a, 90% yield,> 99%. Reaction
– (+) – 2-methyl–CBS- oxazaborolidine (133 mg, 0.482 mmol). Compound 6 (; 2.4 § , 4.82 11 ^ 101) was dissolved in dichloromethane (2011 ^) in the join. Stirred at the same temperature for 5 hours. After completion of the reaction with methanol (10 mL) quenched the reaction was concentrated, added to 1 mol per liter of dilute hydrochloric acid, methylene-wan (X) was extracted, the organic phase was washed with saturated sodium chloride wash paint, concentrated, ethyl acetate – n-hexane to give the compound 7a, 90% yield,> 99%. Reaction

6a 7a
7a, 1H-NMR (300MHz, CDCI3) δ = 7.47-7.21 (m, 11H), 7.07-6.92 (m, 6H), 5.05 (s, 2H), 4.75-4.72 (m, 1H), 4.58 (m, 1H), 3.17-3.09 (m, 1H), 2.04-1.85 (m, 4H) ppm.


1H NMR spectrum of C24H21F2NO3 in CDCL3 at 400 MHz.
Patent
http://www.google.com/patents/CN103086938A?cl=en
Another report line 2: (5S) – acetyl-5- (4-fluorophenyl) valeric acid as reaction intermediates for the preparation of ezetimibe, the synthesis process is as follows:






Seventh Embodiment
The 7 (20g, 0.04mol) was dissolved in methanol (25OmL) was added ammonium formate (25g, 0.4mol), 10% palladium / carbon (Ig) and formic acid (2mL, 0.04mol), stirred at room temperature 20min, filtered palladium / carbon, the filtrate was concentrated to dryness. The residue was dissolved in ethyl acetate, washed with saturated brine, and dried. The organic phase was concentrated to approximately 40mL, was slowly added thereto at room temperature, methyl tert-butyl ether, stirring lh, floc filtered and the filtrate was concentrated to dryness. The residue was dissolved in ethyl acetate, petroleum ether was added, stirred at room temperature 2h, filtered, and dried to give a white solid Ezetimibe 6.4g, yield 38.9%, [a] 2 ° D = _23.7. IH-NMR (DMS0-d6) δ: 9.51 (s, 1Η), 7.32-7.08 (m, 10Η), 6.75 (d, J = 8.4, 2Η), 5.27 (d, J = 4.5, 1Η), 4.80 ( d, J = 2.1, 1Η), 4.49 (m, 1Η), 3.08 (m, 1Η), 1.68-1.82 (m, 4Η).
PATENT
http://www.google.com/patents/CN102675177A?cl=en

ezetimibe ezetimibe synthesis and purification methods:
A Method: IOOml reactor was added to 60ml of ethanol, was added glacial acetic acid and 6g 2. 4g compound 10, followed by stirring for 20 minutes, O. 6g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen , under hydrogen atmosphere, 10 ° C at atmospheric pressure for 18 hours the reaction inches, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor was concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml ko alcohol, filtered, the mother liquor 56ml of purified water was slowly added dropwise, after the large amount of solid precipitated, suction filtered, the filter cake rinsed with an aqueous solution of an ice drained, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml ko alcohol, purified water was slowly added dropwise 160ml a large number of solid precipitation, filtration, alcohol use ko – after pumping out water rinse, 60 ° C and dried under vacuum to obtain the product 4. Og, yield: 81 · 4%.
B method: to IOOml reaction flask 60ml of methanol, acetic acid 2. 4g and 6g compound 10,
Stirred for 20 minutes, added I. 2g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen under a hydrogen atmosphere, the reaction for 18 hours at ambient temperature and pressure inch, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml isopropanol, filtered, and the mother liquor was slowly added dropwise 56ml of purified water, a large number of solid precipitation, filtration, filter cake washed with isopropanol – water rinse after pumping dried, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml isopropanol was slowly added dropwise 160ml of purified water, large amount of solid precipitated, suction filtered, washed with isopropanol – water rinse after draining, 60 ° C under vacuum drying products 3. 7g, yield: 75.3%.
C Method: To a IOOml 60ml of methanol was added to the reaction vessel, was added glacial acetic acid and 6g 2. 4g compound 10, followed by stirring for 20 minutes, O. 8g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen , under hydrogen atmosphere, 30 ° C at atmospheric pressure for 18 hours the reaction inches, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor was concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml ko alcohol, filtered, the mother liquor 56ml of purified water was slowly added dropwise, after the large amount of solid precipitated, suction filtered, the filter cake washed with methanol – water rinsing after drained, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml of methanol, 160ml of purified water was slowly added dropwise a large number of solid precipitation, filtration, washed with methanol – after pumping out water rinse, 60 ° C under vacuum drying products 3. 5g, Yield: 71.3%.
PATENT
http://www.google.co.in/patents/CN102531985A?cl=en
Scheme 1

PATENT
http://www.google.com/patents/US20070049748
Processes for preparation of ezetimibe and its intermediates have also been described in U.S. Pat. Nos. 6,207,822, 5,856,473, 5,739,321, and 5,886,171, International Application Publication No. WO 2006/050634, and in Journal of Medicinal Chemistry 1998, 41, 973-980, Journal of Organic Chemistry 1999, 64, 3714-3718, and Tetrahedron Letters, 44(4), 801-804.

EXAMPLE 1 DETERMINATION OF IMPURITIES IN EZETIMIBE
Determining the level of impurities in ezetimibe using HPLC. The HPLC analysis conditions are as described in Table 1.
| TABLE 1 | |||
| HPLC method for detecting the level of the impurities. | |||
| Column: | Zorbax SB-C18 150 × 4.6 mm, 3.5 μm | ||
| Flow: | 1.0 ml/minute | ||
| Column oven | Ambient | ||
| temperature: | |||
| Wave length: | 230 nm | ||
| Injection volume: | 10 μl | ||
| Run time: | 65 minutes | ||
| Elution: | Gradient | ||
| Diluent: | Acetonitrile | ||
| Gradient Program: | Time | % B | % A |
| (in minutes) | concentration. | concentration. | |
| 0.01 | 35 | 65 | |
| 10.0 | 35 | 65 | |
| 35.0 | 80 | 20 | |
| 55.0 | 80 | 20 | |
| 60.0 | 35 | 65 | |
| 65.0 | 35 | 65 | |
| Mobile phase A = Buffer:Acetonitrile is 80:20 (v/v) | |||
| Mobile phase B = Buffer:Acetonitrile is 20:80 (v/v) | |||
| Buffer: 2.76 g of sodium dihydrogen phosphate monohydrate was | |||
| dissolved in 1000 ml of water and the pH was adjusted to 5.0 with | |||
| dilute NaOH solution. | |||
| IMPURITY NAME | RRT | ||
| Benzyl ezetimibe impurity | 2.6 | ||
| Benzyl ezetimibe diol impurity | 2.2 | ||
| Lactam cleaved alcohol impurity | 1.8 | ||
| Ezetimibe diol impurity | 0.66 | ||
| Lactam cleaved acid impurity | 1.5 | ||
PATENT
http://www.google.com/patents/CN104230978A?cl=en

Example 4. Synthesis of ezetimibe
[0042] To a 500ml bottle of single oral Compound I (PG1 = PG2 = trimethylsilyl) (10g, 13.3mmol), BSA (lOml), TBAF (0. 2g, 0. 66mmol) and methyl tert-butyl ether 100ml. Stirred for 15 minutes at room temperature, the disappearance of the test compound 8, the reaction was terminated. The pre-mixed solution of isopropanol and 2N sulfuric acid was added to the above solution and stirred at room temperature for 1 hour. Crystallized from aqueous isopropanol final product, the product was filtered and washed with aqueous isopropanol, washed with water until the eluate pH is less than 5. Drying at 60 ° to give the final product 5g, yield: 91%, optical yield was 100%. ΐ NMR (400MHz, d6-D MS0): δ 1. 68 (m, 2H), 1 · 82 (m, 2H), 3. 07 (m, 1H), 4. 47 (d, 1H), 4. 79 (d, 1H), 5. 25 (d, 1H), 6. 75 (d, 2H), 7. 10 (m, 4H), 7. 21 (m, 4H), 7. 29 (m, 2H ), 9. 43 (s, 1H).
Patent
http://www.google.com/patents/CN102531985B?cl=en
CN2006 / 10150638 discloses another improved synthetic process, the intermediate acid chloride (IV) first converted to the Weinreb amide, and then reacted with a Grignard reagent to give the key intermediate I.
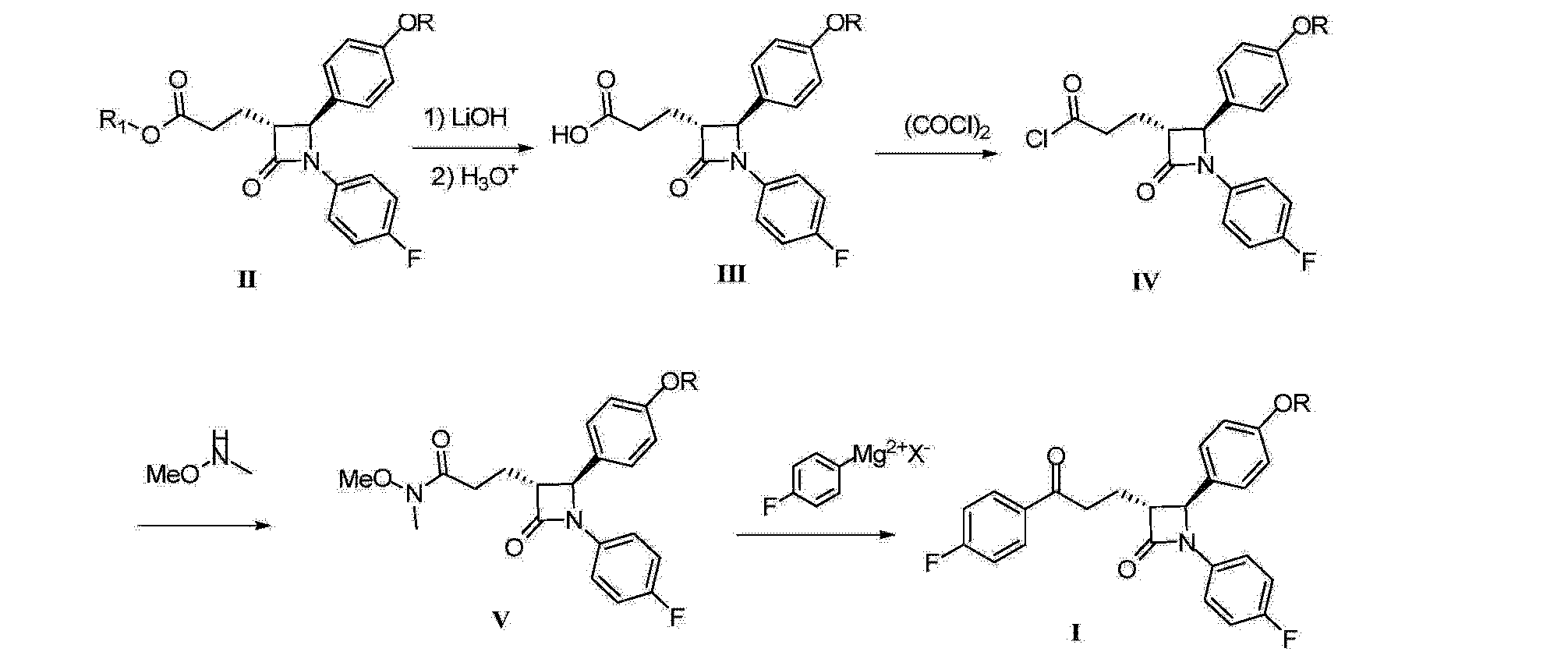
………………….


……………
http://pubs.rsc.org/en/content/articlelanding/2014/ra/c3ra43861a#!divAbstract
The synthesis of four-membered azacycles is of importance because of the chemical and biological relevance of these compounds. Recent progress in copper-catalyzed reactions has been applicable to a variety of research fields, such as heterocyclic synthesis. The aim of the current review is to summarize the synthesis of strained four-membered ring taking advantage of copper catalyzed and mediated processes.


……………
http://www.google.com.tr/patents/WO2006137080A1?cl=en

Taken toluene (250 ml) into cleaned R.B.Flask under nitrogen atmosphere and cooled to 0-5°C. Borane DMS complex and (R)-tetrahydro-l-phenyl-3,3-diphenyl-l H,3H-pyrrol (l,2-c)(l,3,2) oxaza borolidine (R-phenyl CBS) is charged into the reaction mass at 0°C. 25 gm of Keto compound of formula-X is dissolved in toluene(50 ml) and added to the reaction mass at 0-5°C. Maintained the reaction mass for 3 hrs and quenched with methanol and followed by 1 N hydrochloric acid solution. Organic layer separated and washed with 5% hydrogen peroxide solution and 5% sodium sulfate solution and followed by with 10% sodium chloride solution. Distilled the solvent completely under reduced pressure at below 75°C. Product is isolated in diisopropyl ether and dried the product at 60-70°C for 6 hrs. (Yield: 15 gm). Example-2: Preparation of compound of hydroxy compound of formula-XL
Taken toluene (250 nil) into cleaned R.B.Flask under nitrogen atmosphere and cooled to 0-5°C. DIP Chloride (Mole ratio 1:1.5) into the reaction mass at O0C. 25 gm of keto compound of formula-X is dissolved in toluene(50 ml) and added to the reaction mass at 0-50C. Maintained the reaction mass for 3 hrs and quenched with ammonia solution. Organic layer separated and washed with 10% sodium chloride solution. Distilled the solvent completely under reduced pressure at below 750C. Residue is taken for next stage directly without any purification.
Step-h: Preparation of compound of formula-I (Ezetimibe).
Taken compound of formula-XII (10 gm) and isopropanol (100 ml) into a hydrogenation flask, added 5 % Pd/C ( 4gm) at 25°C and maintained at 45-500C for 3 hrs under hydrogen pressure, filtered through hyflow and washed the Pd/C with isopropanol(20 ml). Distilled the solvent completely under vacuum at below 7O0C, product is recrystallised in dichloromethane (Yield: 6 gm).
Purification of Ezetimibe (formula-1).
Ezetimibe (10 gm) is dissolved in 30 ml of methanol and filtered through hyflow and saturated with DM.Water(30 ml) and stirred for 1 hr at 20-250C. Product filtered and dried for 6-8 hrs at 80-850C (Yield:9 gm):
……………….
http://www.google.com.tr/patents/WO2005049592A1?cl=en
scheme A.

The compound of the formula 2

is an useful intermediate for the preparation of ezetimibe. The intermediates represented by the formula 2 can be prepared economically in good yields as represented by the scheme B.

wherein X is O or S; Y is O, S or N(lower alkyl); and R is alkyl, unsubstituted or substituted phenyl, unsubstituted or substituted naphthyl or lower alkoxy carbonyl, wherein substituents on phenyl and naphthyl are selected from the group consisting of lower alkyl and phenyl. The starting compounds of formula 3 are known or can be obtained from known methods. The reduction may be carried out in a neutral organic solvent or a combination of the neutral organic solvents. Neutral organic solvent means the solvent that is unreactive in the reduction reaction. The preferable organic solvents are chloroalkanes such as methylene dichloride, chloroform, carbon tetrachloride and ethylene dichloride; carbocyclic aromatics such as toluene and benzene; ethers such as methyl tert-butyl ether, diethylether and isopropyl ether; heterocyclic compound such as tetrahydrofuran; dimethylformamide; dimethylsulfoxide; alkanes such as pentane and hexane; and acetonitrile. More preferable solvents are toluene, diethyl ether, isopropyl ether, hexane, methylene dichloride and ethylene dichloride. The preferable reaction temperature is below the boiling temperature of the solvent used, more preferably between about -40°C and the boiling temperature of the solvent, still more preferably between about -20°C and 40°C and most preferably between about -10°C and 10°C. Quantity of (-)-DIP chloride used is preferably at least about 0.3 mole, more preferably about 0.5 to 10 mole, most preferably about 0.8 to 5 mole per mole of the keto compound of formula 3. Yield of the hydroxy compound of formula 2 is usually above 80%, typically between 90 % to 100%. The compounds of formula 2 wherein X is O; Y is O; and R is alkyl, unsubstituted or substituted phenyl are the preferred. Preferable conditions for obtaining a hydroxy compound of formula 2 from the corresponding keto compound of formula is that the keto compound of the formula 3 is mixed with a neutral solvent, reduced with (-)-DIP chloride at a temperature between -40°C and the boiling temperature of the solvent, more preferably between about -20°C and 40°C and most preferably between about -10°C and 10°C. The reaction mass may be subjected to usual work up. The reaction mass may be used directly in the next step to produce finally ezetimibe, or the hydroxy compound may be isolated and used in the next step. The invention will now be further described by the following examples, which are illustrative rather than limiting. Example 1 3-[5-(4-fluorophenyl)-1 ,5-dioxopentyl]-4-phenyl-2-oxazolidinone (100 gm) is dissolved in toluene (750 ml), the mixture of (-)-β- chlorodiisopinocampheylborane ((-)-DIP chloride) in heptane (545 ml, 1.5M) and toluene (750 ml) is added at 0°C to 5°C for 1 hour. The reaction mixture is stirred for 15 hours at 25°C to 30°C and 340 ml of 10% sodium chloride is then added at the same temperature. The layers are separated and the organic layer is washed with 5% sodium bicarbonate (300 ml), 1 N sulfuric acid (300 ml), and 10% sodium chloride (300 ml). Then the organic layer is dried on sodium sulfate to give 3-[(5S)-5-(4-fluorophenyl)-5-hydroxy-1-oxopentyl]-4-phenyI-2- oxazolidinone in 96% yield. Example 2 The organic layer of 3-[(5S)-5-(4-fluorophenyl)-5-hydroxy-1-oxopentyl]-4- phenyl-2-oxazolidinone from example 1 is mixed with 4-fluoro-N-(4- hydroxyphenyl)methylene-benzenamine (121 gm) and cooled to -10°C. Then diisopropylethylamine (260 ml) is added to the reaction mixture for 45 minutes at -10°C to -15°C, trimethylsilylchloride (135 ml) is added and stirred for 1 hour at -20°C to -25°C. The reaction mixture is cooled to -30°C, TiCI4 (35 ml) is slowly added to the reaction mixture at -30°C to -35°C and stirred for 3 hours at the same temperature. 5% Aq. tartaric acid solution (1700 ml) is added to the reaction mixture at 0°C, stirred for 1 hour and allowed the temperature to rise to 25°C. Then 20% Aq. NaHSO3 (350 ml) solution and stirred for 2 hours at 25°C to 30°C. The organic layer is separated and washed with 1000 ml water, concentrated to 250 ml volume and added 100 ml bistrimethylsilylacetamide. Then the reaction mixture is heated to reflux for 30 minutes. The organic layer is concentrated to remove methylene dichloride, crystallized from the mixture of ethyl acetate (250 ml) and n-heptane (250 ml), and filtered and dried to give 135 gm of compound 4 (prot = trimethylsilyl).
……………


| WO2005066120A2 * | 23 Ara 2004 | 21 Tem 2005 | Prosenjit Bose | Process for asymmetric synthesis of hydroxy-alkyl substituted azetidinone derivatives |
| WO2006137080A1 * | 17 Şub 2006 | 28 Ara 2006 | Kumar Muppa Kishore | Improved process for the preparation of ezetimibe |
| WO2008151324A1 * | 9 Haz 2008 | 11 Ara 2008 | Teva Pharma | Reduction processes for the preparation of ezetimibe |
| US7470678 | 1 Tem 2003 | 30 Ara 2008 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| US7842684 | 25 Nis 2007 | 30 Kas 2010 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7863265 | 19 Haz 2006 | 4 Oca 2011 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7871998 | 21 Ara 2004 | 18 Oca 2011 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7893048 | 21 Haz 2006 | 22 Şub 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502 | 21 Haz 2006 | 15 Mar 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US8013150 | 17 Şub 2006 | 6 Eyl 2011 | Msn Laboratories Ltd. | Process for the preparation of ezetimibe |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4044147 * | May 26, 1976 | Aug 23, 1977 | Pfizer Inc. | N-(acyl)-p-amino-N’-(monosubstituted)-benzamide anti-ulcer agents |
| US5248611 * | Jan 3, 1992 | Sep 28, 1993 | Scripps Clinic And Research Foundation | Stereoisomer separation method using antibody combing site-containing molecules |
| US5739321 * | May 31, 1996 | Apr 14, 1998 | Schering Corporation | 3-hydroxy γ-lactone based enantionselective synthesis of azetidinones |
| US5846991 * | Dec 29, 1997 | Dec 8, 1998 | Nissan Chemical Industries, Ltd. | Pyrazole derivatives |
| US5856473 * | Oct 31, 1996 | Jan 5, 1999 | Schering Corporation | Process for preparing 1-(4-fluorophenyl)-3(R)-(3(S)-hydroxy-3-( phenyl or 4-fluorophenyl!)-propyl)-4(S)-(4-hydroxyphenyl)-2-azetidinone |
| US5859035 * | Mar 27, 1997 | Jan 12, 1999 | Merck & Co., Inc. | Arylheteroaryl inhibitors of farnesyl-protein transferase |
| US5886171 * | May 28, 1997 | Mar 23, 1999 | Schering Corporation | 3-hydroxy gamma-lactone based enantioselective synthesis of azetidinones |
| US6194599 * | Apr 8, 1997 | Feb 27, 2001 | Catalytica, Inc. | Process for preparing biaryl compounds |
| US6207822 * | Dec 5, 1999 | Mar 27, 2001 | Schering Corporation | Process for the synthesis of azetidinones |
| US20030013699 * | May 22, 2002 | Jan 16, 2003 | Davis Harry R. | Methods for treating alzheimer’s disease and/or regulating levels of amyloid beta peptides in a subject |
| US20050124808 * | Sep 23, 2004 | Jun 9, 2005 | Dsm I.P. Assets B.V. | Process for preparing unsymmetrical biaryls and alkylated aromatic compounds from arylnitriles |
| US20060247273 * | Sep 24, 2004 | Nov 2, 2006 | Takayuki Kawaguchi | Carbamoyl-type benzofuran derivatives |
| US20090048441 * | Feb 17, 2006 | Feb 19, 2009 | Manne Satyanarayana Reddy | Process for the Preparation of Ezetimibe |
| USRE37721 * | Jun 15, 2000 | May 28, 2002 | Schering Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7470678 | Jul 1, 2003 | Dec 30, 2008 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| US7842684 | Apr 25, 2007 | Nov 30, 2010 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7863265 | Jun 19, 2006 | Jan 4, 2011 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7871998 | Dec 21, 2004 | Jan 18, 2011 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7893048 | Jun 21, 2006 | Feb 22, 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502 | Jun 21, 2006 | Mar 15, 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US8013150 * | Feb 17, 2006 | Sep 6, 2011 | Msn Laboratories Ltd. | Process for the preparation of ezetimibe |
| US8383810 | Dec 12, 2011 | Feb 26, 2013 | Merck Sharp & Dohme Corp. | Process for the synthesis of azetidinones |
| US20050239766 * | Jul 1, 2003 | Oct 27, 2005 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| US20060160785 * | Dec 5, 2005 | Jul 20, 2006 | Judith Aronhime | Ezetimibe polymorphs |
| US20060234996 * | Apr 13, 2006 | Oct 19, 2006 | Itai Adin | Novel crystalline form of ezetimibe and processes for the preparation thereof |
| US20110130378 * | May 26, 2009 | Jun 2, 2011 | Lek Pharmaceuticals D.D. | Ezetimibe process and composition |
| US20110183956 * | Jul 29, 2009 | Jul 28, 2011 | Janez Mravljak | Process for the synthesis of ezetimibe and intermediates useful therefor |
| CN102219803A * | Apr 20, 2011 | Oct 19, 2011 | 浙江大学 | Preparation method of ezetimibe intermediate |
| EP2128133A1 | May 26, 2008 | Dec 2, 2009 | Lek Pharmaceuticals D.D. | Ezetimibe process and composition |
| WO2008096372A2 * | Feb 6, 2008 | Aug 14, 2008 | Pranav Gupta | Process for preparing highly pure ezetimibe using novel intermediates |
| WO2009150038A1 | May 26, 2009 | Dec 17, 2009 | Lek Pharmaceuticals D.D. | Process for the preparation of ezetimibe and composition containing it |
| WO2009157019A2 * | Jun 23, 2009 | Dec 30, 2009 | Ind-Swift Laboratories Limited | Process for preparing ezetimibe using novel allyl intermediates |
| CN1131416A * | Sep 14, 1994 | Sep 18, 1996 | 先灵公司 | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| CN1330721A * | Oct 1, 1999 | Jan 9, 2002 | 先灵公司 | Resolution of trans-2-(alkoxycarbonylethyl)-laitams useful in synthesis of 1-(4-fluorophenyl)-3(R)-[3(s)-hydroxy-3-(flurophenyl)-propyl)]-4(s)-(4-hydroxyphenyl)-2-azetidinone |
| CN1931838A * | 20 Oct 2006 | 21 Mar 2007 | 屠勇军 | Azacyclo butanone derivative and its synthesis process |
| WO2007108007A1 * | 12 Sep 2006 | 27 Sep 2007 | Prabhavalkar Tirtha Suresh | A process for the preparation of ezetimibe via a novel intermediate |
| WO2008096372A2 * | 6 Feb 2008 | 14 Aug 2008 | Pranav Gupta | Process for preparing highly pure ezetimibe using novel intermediates |
| WO2009157019A2 * | 23 Jun 2009 | 30 Dec 2009 | Ind-Swift Laboratories Limited | Process for preparing ezetimibe using novel allyl intermediates |
| WO2008032338A2 * | 10 Eyl 2007 | 20 Mar 2008 | Reddy Manne Satyanarayana | Improved process for the preparation of ezetimibe and its intermediates |
| WO2008089984A2 * | 24 Oca 2008 | 31 Tem 2008 | Krka | Process for the preparation of ezetimibe and derivatives thereof |
| WO2008096372A2 * | 6 Şub 2008 | 14 Ağu 2008 | Pranav Gupta | Process for preparing highly pure ezetimibe using novel intermediates |
| WO2008106900A1 * | 3 Mar 2008 | 12 Eyl 2008 | Zentiva As | Method of manufacturing (3r, 4s) -1- (4-fluorophenyl) -3- [ (3s) -3- (4 -fluorophenyl) -3-hydroxypropyl) ] -4- (4-hyd roxyphenyl) -2-azetidinone |
| WO2008151324A1 * | 9 Haz 2008 | 11 Ara 2008 | Teva Pharma | Reduction processes for the preparation of ezetimibe |
| WO2009067960A2 * | 5 Kas 2008 | 4 Haz 2009 | Zentiva As | A method of manufacturing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone and its intermediates |
| WO2011158052A1 | 17 Haz 2011 | 22 Ara 2011 | Nanoform Cardiovascular Therapeutics Ltd. | Nanostructured ezetimibe compositions, process for the preparation thereof and pharmaceutical compositions containing them |
| WO2012155932A1 | 17 May 2011 | 22 Kas 2012 | Pharmathen S.A. | Improved process for the preparation of ezetimibe |
| CN102531986A * | 23 Şub 2012 | 4 Tem 2012 | 苏州朗科生物技术有限公司 | Preparation method for ezetimibe |
| CN103044305A * | 24 Oca 2013 | 17 Nis 2013 | 上海现代制药股份有限公司 | Preparation method of ezetimibe intermediate |
| EP1922304A2 † | 8 Eyl 2006 | 21 May 2008 | Teva Pharmaceutical Industries Ltd | Processes for the preparation of (3r,4s)-4-((4-benzyloxy)phenyl)-1-(4-fluorophenyl)-3-((s)-3-(4-fluorophenyl)-3-hydroxypropyl)-2-azetidinone, an intermediate for the synthesis of ezetimibe |
| EP1953140A1 * | 24 Oca 2007 | 6 Ağu 2008 | Krka | Process for the preparation of ezetimibe and derivatives thereof |
| US7842684 | 25 Nis 2007 | 30 Kas 2010 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7863265 | 19 Haz 2006 | 4 Oca 2011 | Astrazeneca Ab | 2-azetidinone derivatives and their use as cholesterol absorption inhibitors for the treatment of hyperlipidaemia |
| US7871998 | 21 Ara 2004 | 18 Oca 2011 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7893048 | 21 Haz 2006 | 22 Şub 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US7906502 | 21 Haz 2006 | 15 Mar 2011 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| US8383810 | 12 Ara 2011 | 26 Şub 2013 | Merck Sharp & Dohme Corp. | Process for the synthesis of azetidinones |
| WO2005049592A1 * | 24 Kas 2003 | 2 Haz 2005 | Hetero Drugs Ltd | A novel process for ezetimibe intermediate |
| US5856473 * | 31 Eki 1996 | 5 Oca 1999 | Schering Corporation | Process for preparing 1-(4-fluorophenyl)-3(R)-(3(S)-hydroxy-3-( phenyl or 4-fluorophenyl!)-propyl)-4(S)-(4-hydroxyphenyl)-2-azetidinone |
////
Ciraparantag, Aripazine
Ciraparantag
PER977, Aripazine
CAS Number:1438492-26-2
Chemical Name:N1,N1-[piperazine-1,4-diylbis(propane-1,3-diyl)]bis-L-argininamide
(2S,2’S)-N,N’-(Piperazine-1,4-diyldipropane-3,1-diyl)bis(2-amino-5-carbamimidamidopentanamide)
2S,2’S)-N,N’-(piperazine-1,4-diylbis(propane-3,1-diyl))bis(2-amino-5-guanidinopentanamide)
C22H48N12O2
Mw: 512.40232
Mechanism of Action: an intravenously administered anticoagulant Reversal Agent
Blood coagulation factor modulators; Factor Xa inhibitors
Indication: Anticoagulant Reversal
Development Stage: Phase II
Developer:Perosphere, Inc..Perosphere Inc.
Highest Development Phases
- Phase IIHaemorrhage
Most Recent Events
- 02 Apr 2015Ciraparantag receives Fast Track designation for Haemorrhage [IV] (In volunteers) in USA
- 05 Nov 2014Efficacy and adverse events data from a phase I/II trial in Haemorrhage released by Perosphere
- 06 Oct 2014Aripazine is available for licensing as of 06 Oct 2014. http://www.perosphere.com/
Aripazine(PER977, ciraparantag)

Ciraparantag, also known as PER977, is a A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. PER977 is water-soluble, cationic molecule that is designed to bind specifically to unfractionated heparin and low-molecular-weight heparin through noncovalent hydrogen bonding and charge–charge interactions.
PER-977 is an intravenous heparin neutralizer in phase II clinical trials at Perosphere to reverse edoxaban’s induced anticoagulation.
In April 2015, fast track designation was assigned in the U.S. as an investigational anticoagulant reversal agent.
WO 2013082210
http://www.google.com/patents/WO2013082210A1?cl=en
In one scheme, the compound of Formula V (DAP)

is synthesized by reacting excess equivalents (e.g., at least about two equivalents) of compound 1

with one equivalent of compound 2

in the presence of a peptide coupling reagent, to obtain a compound 3

wherein PI is a protecting group and P2 is a protecting group or is a hydrogen.
the coupling involved reacting compound 1, wherein PI was Boc and P2 was a hydrogen (depicted as Boc-Arg-OH HCl below), with compound 2 as depicted below:

The resultant crude product was more than 95% pure by thin layer
chromatography (TLC).
Subsequently, the deprotection step was carried out as depicted below:

The deprotected product was purified by preparative HPLC using 1% acetic acid buffer. Product purity of >98% was observed. Residual TFA was removed by low quantity of DOWEX resin. The molecular weight of DAP (the compound of Formula V) is 512.4, and the compound synthesized according to the above scheme exhibited the following primary peak by mass spectroscopy: [M+H]+=513.4.
|
References |
1: Dzik WH. Reversal of oral factor Xa inhibitors by prothrombin complex concentrates: a re-appraisal. J Thromb Haemost. 2015 Jun;13 Suppl 1:S187-94. doi: 10.1111/jth.12949. PubMed PMID: 26149022.
2: Crowther M, Crowther MA. Antidotes for Novel Oral Anticoagulants: Current Status and Future Potential. Arterioscler Thromb Vasc Biol. 2015 Aug;35(8):1736-45. doi: 10.1161/ATVBAHA.114.303402. Epub 2015 Jun 18. PubMed PMID: 26088576.
3: Sullivan DW Jr, Gad SC, Laulicht B, Bakhru S, Steiner S. Nonclinical Safety Assessment of PER977: A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. Int J Toxicol. 2015 Jun 15. pii: 1091581815590667. [Epub ahead of print] PubMed PMID: 26079256.
4: Mo Y, Yam FK. Recent advances in the development of specific antidotes for target-specific oral anticoagulants. Pharmacotherapy. 2015 Feb;35(2):198-207. doi: 10.1002/phar.1532. Epub 2015 Feb 3. PubMed PMID: 25644580.
5: Yates SW. Interrupting anticoagulation in patients with nonvalvular atrial fibrillation. P T. 2014 Dec;39(12):858-80. PubMed PMID: 25516695; PubMed Central PMCID: PMC4264672.
6: Vanden Daelen S, Peetermans M, Vanassche T, Verhamme P, Vandermeulen E. Monitoring and reversal strategies for new oral anticoagulants. Expert Rev Cardiovasc Ther. 2015 Jan;13(1):95-103. doi: 10.1586/14779072.2015.987126. Epub 2014 Nov 28. PubMed PMID: 25431993.
7: Costin J, Ansell J, Laulicht B, Bakhru S, Steiner S. Reversal agents in development for the new oral anticoagulants. Postgrad Med. 2014 Nov;126(7):19-24. doi: 10.3810/pgm.2014.11.2829. Review. PubMed PMID: 25387210.
8: Ansell JE, Bakhru SH, Laulicht BE, Steiner SS, Grosso M, Brown K, Dishy V, Noveck RJ, Costin JC. Use of PER977 to reverse the anticoagulant effect of edoxaban. N Engl J Med. 2014 Nov 27;371(22):2141-2. doi: 10.1056/NEJMc1411800. Epub 2014 Nov 5. PubMed PMID: 25371966.
9: Hankey GJ. Intracranial hemorrhage and novel anticoagulants for atrial fibrillation: what have we learned? Curr Cardiol Rep. 2014 May;16(5):480. doi: 10.1007/s11886-014-0480-9. Review. PubMed PMID: 24643903.
///////
BEXAGLIFLOZIN


Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
D-Glucitol, 1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-, (1S)-
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
(1S)-1,5-anhydro-1-C-[4-chloro-3-({4-[2- (cyclopropyloxy)ethoxy]phenyl}methyl)phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018
Mechanism of Action:SGLT2 inhibitor, Sodium-glucose transporter 2 inhibitors
Indication:Type 2 diabetes
FDA APPROVED
| 1/20/2023 |
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise
Drug Trials Snapshot
Phase II
Developer:Theracos, Inc.
| Conditions | Phases | Recruitment | Interventions | Sponsor/Collaborators |
|---|---|---|---|---|
| Diabetes Mellitus Type 2 | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442 | Theracos |
| Diabetes Mellitus | Phase 2 | Completed | Drug: EGT0001442|Drug: Placebo | Theracos |
| Type 2 Diabetes Mellitus | Phase 3 | Not yet recruiting | Drug: Bexagliflozin|Drug: Placebo | Theracos |
| Diabetes Mellitus, Type 2 | Phase 2|Phase 3 | Recruiting | Drug: Bexagliflozin tablets | Theracos |

 DIPROLINE COMPLEX
DIPROLINE COMPLEX
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)
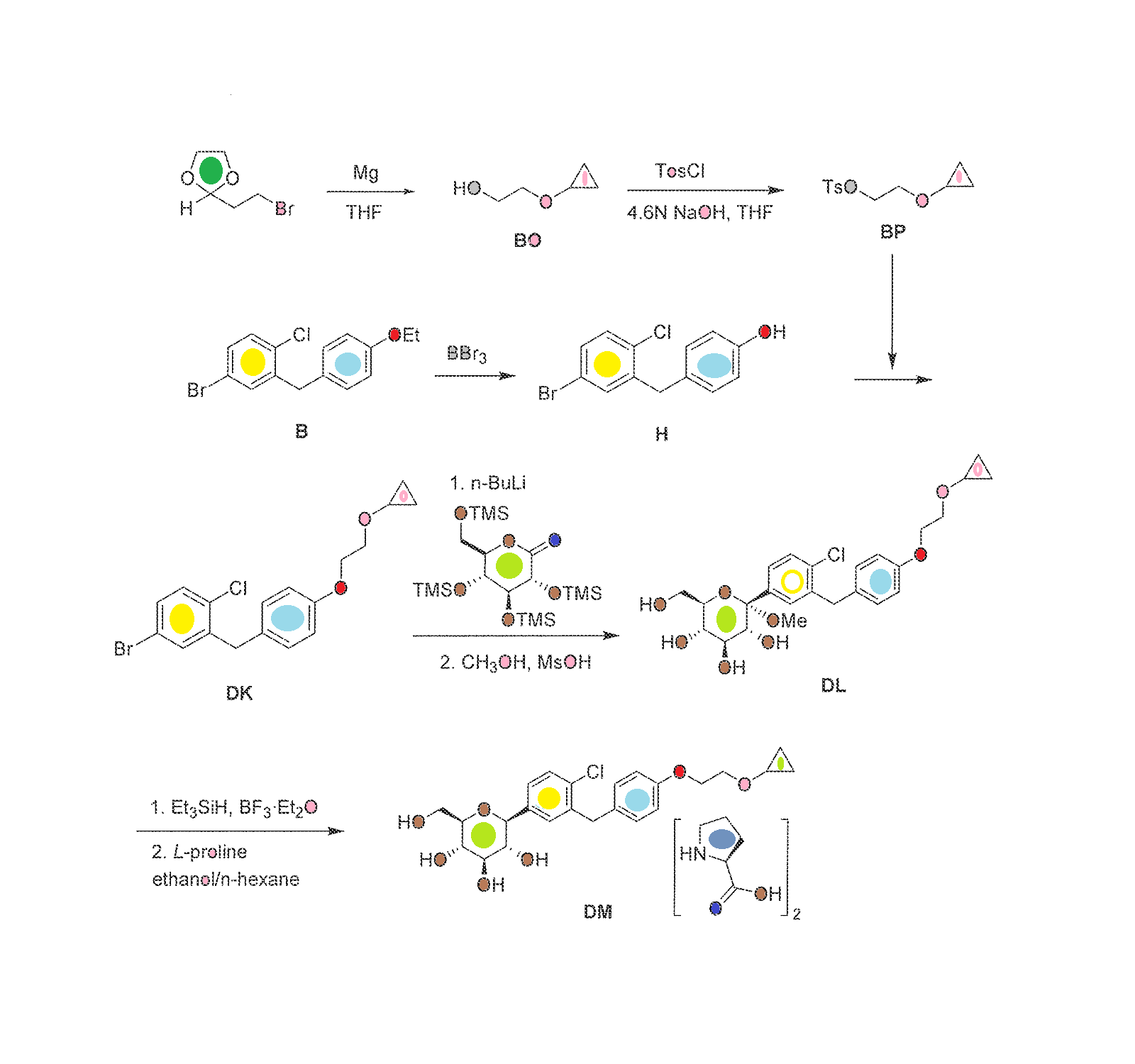

Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).

The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).

PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en


Example 19
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)

To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.

Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)

To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)

A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)

To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)

To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694

Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.

(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.

Preparation of (2-chloro-5-iodophenyl)methan-1-ol

A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol

A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.

Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.

(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex


Example 1A
Preparation of 2-cyclopropoxyethanol (1)

To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)

To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)

To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)

A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)

To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)

To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.

To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7

[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8

A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * | May 15, 2008 | Nov 27, 2008 | Squibb Bristol Myers Co | Crystal structures of sglt2 inhibitors and processes for their preparation | |||||||||||||||
| WO2009026537A1 * | Aug 22, 2008 | Feb 26, 2009 | Theracos Inc | Benzylbenzene derivatives and methods of use | |||||||||||||||
| CN1407990A * | Oct 2, 2000 | Apr 2, 2003 | 布里斯托尔-迈尔斯斯奎布公司 | C-aryl glucoside sgltz inhibitors
|
| WO2010022313A2 * | Aug 21, 2009 | Feb 25, 2010 | Theracos, Inc. | Processes for the preparation of sglt2 inhibitors |
////////BEXAGLIFLOZIN, APPROVALS 2023, FDA 2023
c1cc(ccc1Cc2cc(ccc2Cl)[C@H]3[C@@H]([C@H]([C@@H]([C@H](O3)CO)O)O)O)OCCOC4CC4
SYN
https://doi.org/10.1021/acs.jmedchem.4c02079J.Med.Chem.2025,68,2147−2182
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof
type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting
SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe
reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates,
whichmaybeacommonchallengetootherSGLT2inhibitors.31
Anotherpatent disclosedbyPiramal Enterprises suggesteda
similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34
Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2
whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith
activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal
functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane,
whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was
reported in 92% yield. Details on stereoselectivity of this
approachwerenotdisclosed.
Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol
3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether
3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.

(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447−
453.
(29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium
glucose co-transporter-2 inhibitors for the treatment of diabetes
mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79.
(30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive
review of small-molecule drugs for the treatment of type 2 diabetes
mellitus: Synthetic approaches and clinical applications. Eur. J. Med.
Chem. 2024, 267, No. 116185.
(31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F.
Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A.
Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev.
2018, 22, 467−488.
(32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.;
Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′
position as potent and selective renal sodium-dependent glucose co
transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes.
Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470.
(33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the
preparation of benzyl-benzene C-glycosides via coupling reaction as
potential SGLT2 inhibitors. US 20130267694, 2013.
(34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal,
D. Aprocess for the preparation of SGLT2 inhibitor and intermediates
thereof. WO 2018207113, 2018.
(35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu,
B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside
derivatives as antidiabetic agents. WO 2009026537, 2009.
.
European Journal of Medicinal Chemistry
Volume 265, 5 February 2024, 116124
https://doi.org/10.1016/j.ejmech.2024.116124
Bexagliflozin (Brenzavvy)
On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin
can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which
lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108].
The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring.
BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol.
Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453.
[105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng,
B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and
prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293.
[106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M.
P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin.
Pharmacother. 22 (2021) 2095–2103.
[107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am.
Soc. Nephrol. 18 (2023) 279–289.
[108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr.
Drug Saf. 13 (2018) 84–91.
[109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd,
J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives
as Antidiabetic Agents, 2009. WO2009026537A1.
.



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Merestinib

1206799-15-6 (Merestinib)
Chemical Formula: C30H22F2N6O3
Exact Mass: 552.17215
N-(3-fluoro-4-((1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yl)oxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide
- OriginatorEli Lilly…..Eli Lilly And Company
- ClassAmides; Antineoplastics; Dihydropyridines; Pyrazoles; Small molecules
- Mechanism of ActionMKNK1 protein inhibitors; MKNK2 protein inhibitors; Proto oncogene protein c met inhibitors; ROS1-protein-inhibitors
- 29 Jun 2015Immunocore in collaboration with Eli Lilly plans a phase Ib/II trial for Uveal Melanoma (Metastatic disease, Combination therapy)
- 18 Jun 2015Eli Lilly completes a phase I bioavailability trial in healthy volunteers in USA (NCT02370485)
- 01 Feb 2015Eli Lilly initiates enrolment in a phase I bioavailability trial in healthy volunteers in USA (NCT02370485)
| Company | Eli Lilly and Co. |
| Description | C-Met inhibitor |
| Molecular Target | c-Met receptor tyrosine kinase (c-MET) (MET) (HGFR) (c-Met proto-oncogene) |
| Mechanism of Action | c-Met receptor tyrosine kinase inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat advanced cancer |
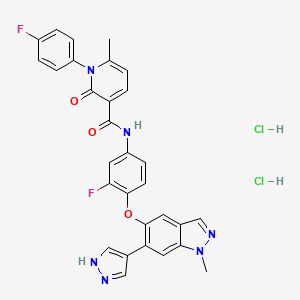 LY2801653 dihydrochloride; UNII-33F79TLF60; LY 2801653 dihydrochloride; LY-2801653 dihydrochloride; 1206801-37-7; 33F79TLF60
LY2801653 dihydrochloride; UNII-33F79TLF60; LY 2801653 dihydrochloride; LY-2801653 dihydrochloride; 1206801-37-7; 33F79TLF60LY2801653 was identified and developed as a novel, potent, and orally active small molecule inhibitor of human c-Met. It demonstrated dose dependent inhibition of c-Met phosphorylation in xenograft tumors with a long lasting PD effect. LY2801653 displayed potent anti-tumor efficacy in a number of non small cell lung, renal, pancreatic, and breast tumor models. Examination of c-Met expression in these tumors by immunohistochemistry (IHC) revealed a good correlation between response and c-Met expression in the tumor tissue. LY2801653 treatment led to increase in functional vessel areas, and decrease in tumor hypoxia. Enhanced anti-tumor efficacy was achieved when Erlotinib was combined with LY2801653. . (source: http://cancerres.aacrjournals.org/cgi/content/meeting_abstract/70/8_MeetingAbstracts/3611).
Patent
MAIN
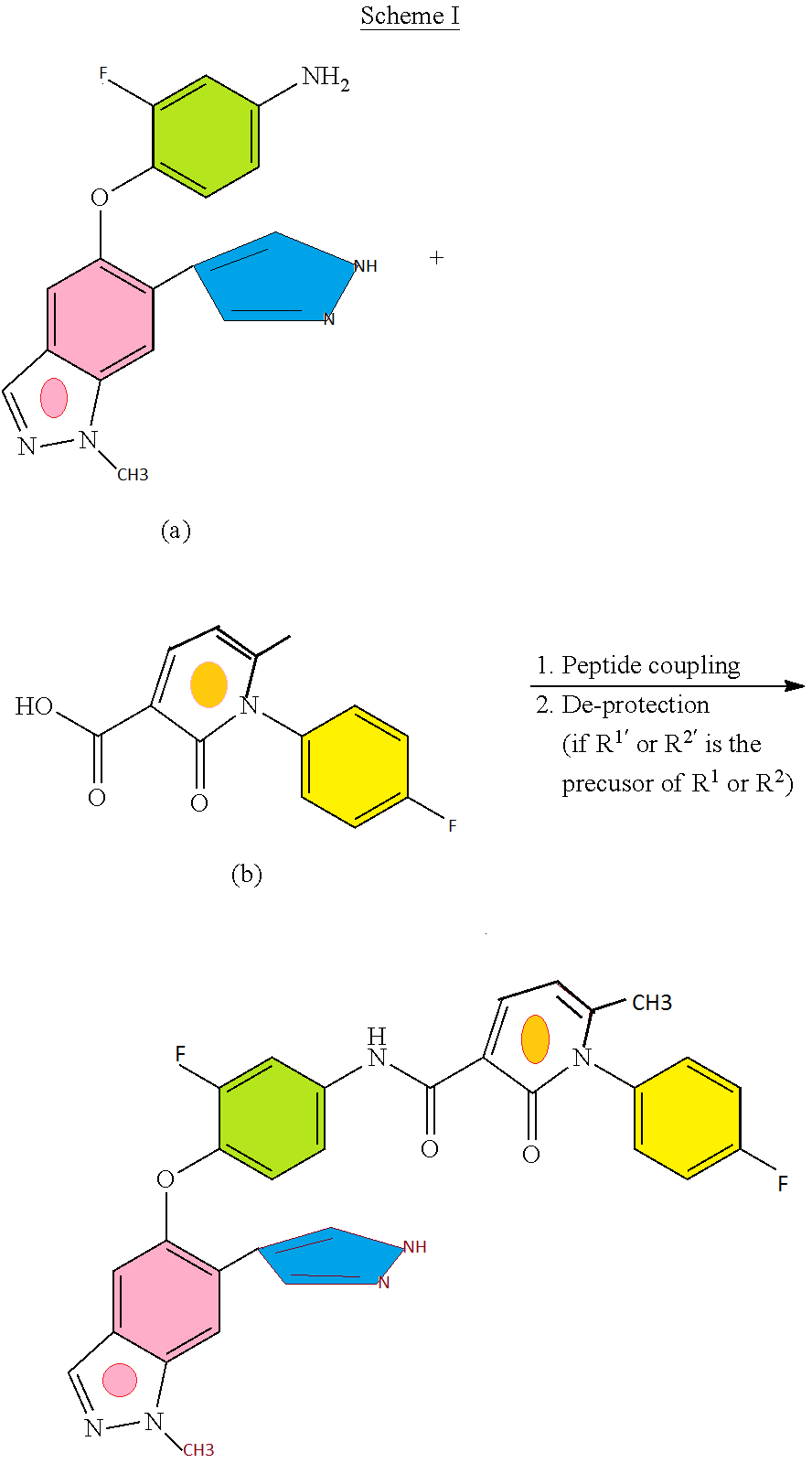
INTERMEDIATES
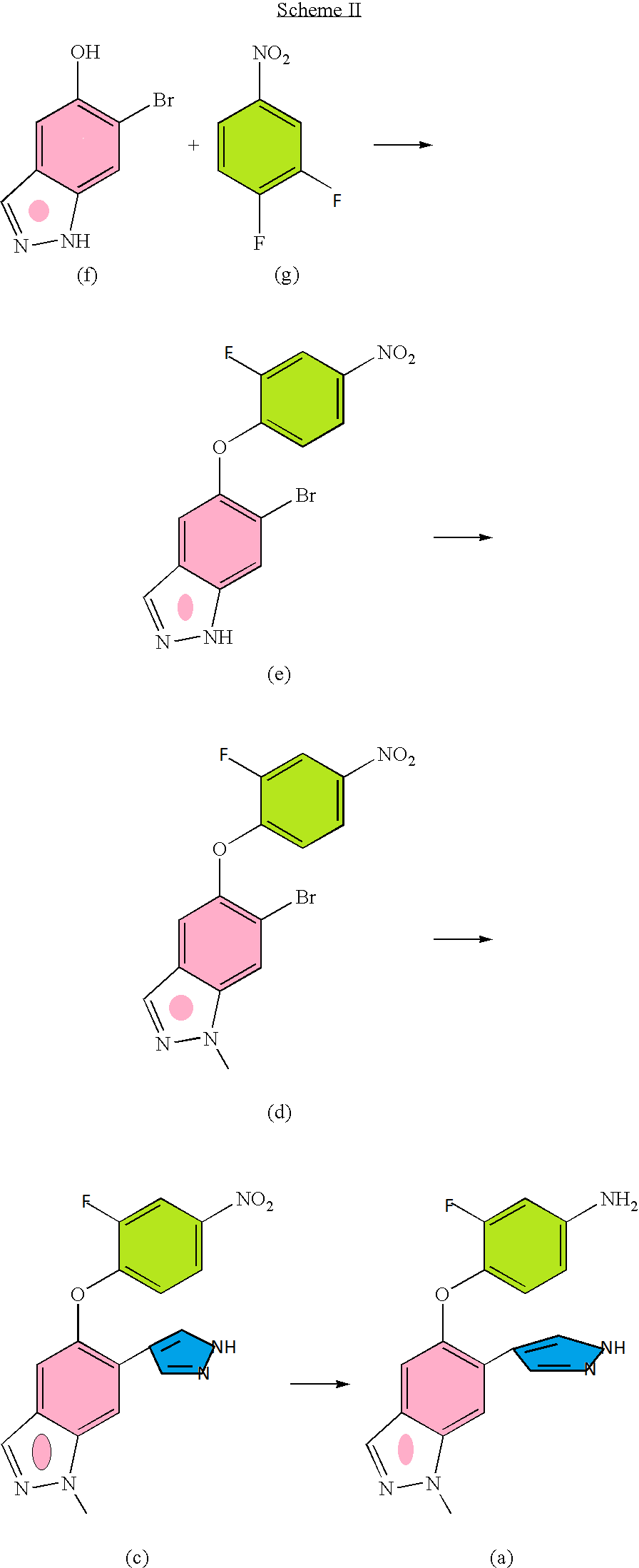
https://www.google.com/patents/US8030302
Example 1 N-(3-Fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide
To a 100 mL round bottom flask is added tert-butyl 4-(5-(4-amino-2-fluorophenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (1.43 g, 3.38 mmol), 1-(4-flurorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxylic acid (1.25 g, 5.07 mmol), EDCI (1.48 g, 7.6 mmol) and HOBt (776 mg, 5.07 mmol) followed by DMF (15 mL, 193.99 mmol) and then DIPEA (1.47 mL, 8.44 mmol). The mixture is allowed to stir at RT overnight. The reaction mixture is diluted into EtOAc (300 mL) and washed with saturated aqueous sodium chloride (5×100 mL). The combined aqueous solution is extracted with EtOAc (1×100 mL) and then the combined organic solutions are dried over N2SO4, filtered, and concentrated to dryness. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in a DCM solution (B), gradient from 100% (A) to 80% (A):20% (B) over 70 min to give tert-butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate as a gold solid (2.20 g, 87% yield). MS (m/z): 653. (M+H), 675 (M+Na).
Example 2 N-(3-Fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide

A 12 L round bottom flask is equipped with overhead agitation, a thermocouple, and a N2 purge. tert-Butyl 4-(5-(4-amino-2-fluorophenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (404 g, 954.08 mmol) is dissolved in DMF (2 L) and charged to the flask. DMF (1 L) is used to rinse the flask. 1-(4-Fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxylic acid (259.46 g, 1.05 mol) and EDCI (228.63 g, 1.19 mol) are added and it is rinse in with DMF (500 mL). Then HOBt (189.94 g, 1.24 mol) is added and it is again rinsed in with DMF (500 mL). Finally, DIPEA is slowly added (184.97 g, 1.43 mol). The dark solution is then stirred at RT over the weekend. To a 20 L bottom outlet flask is added DI water (3 L) and DCM (5 L). The reaction mixture is poured in and it is rinsed in with DCM (1 L). The organic layer is separated, washed with DI water (3×3 L), dried over Na2SO4, filtered, rinsed solids with DCM and concentrated the filtrate. EtOAc (2 L) is added to the residue and the solution is stirred for 1 hour. The product crystallizes out. The mixture is concentrated. Another portion of EtOAc (2 L) is added and concentrated to remove all of the DCM. EtOAc (650 mL) and MTBE (3 L) are added to the residue and the solution is stirred in an ice bath for 1 hour. The tan slurry is filtered using a polypropylene pad. The cake is rinsed with MTBE (2×500 mL). The light tan solid is dried overnight in the vacuum oven at 40° C. to give the crude product (553 g). The crude product is purified by silica gel column chromatography eluting with (50% EtOAc (50%):35% DCM (35%): n-heptane (15%)) to give the pure desired product tert-butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (424 g, 68%). MS (m/z): 651.0 (M−H).
tert-Butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (423.9 g, 649.50 mmol) is dissolved in DCM (4.24 L). HCl in MeOH (5.74 N, 799.99 mL, 4.59 mol) is added and the solution is heated at 30° C. for 1 hour. Then the reaction mixture is heated to 45° C. and DCM (1.5 L) is added. After two hours, the solution is heated to 50° C. and DCM (2 L) is added. After 3 hours, DCM (2 L) is added followed by HCl in MeOH (4.5 N, 721.67 mL, 3.25 mol). After another 45 min, DCM (1 L), HCl in MeOH (4.5 N, 288.67 mL, 1.30 mol), and MeOH (1.5 L) are added. The reaction solution is then heated to 60° C. After 4 hours, MeOH (2 L) is added and 10 min later DCM (1 L) is added followed by HCl in MeOH (4.5 N, 200 mL). After 5 hours, the reaction is complete. The reaction mixture is concentrated to about ⅓ volume. MeOH (2 L) is added and the solution is concentrated to a thick slurry. Again, MeOH (2 L) is added and the mixture is concentrated to a thick slurry. The slurry is cooled to about 10-15° C. and then filtered. The solids are washed with MeOH. The solids are placed in a 55° C. vacuum oven for 2 days to give the desired product N-(3-fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide hydrochloride (377 g, 92.8%). MS (m/z): 551.0 (M−H).
To a 22 L round bottom flask equipped with mechanical stirring under nitrogen is added N-(3-fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide hydrochloride (367 g, 0.62 mol) followed by DCM (7.34 L) and water (7.34 L). Na2CO3 (181.6 g, 1.71 mol) is added and the mixture is stirred at RT for 30 min. The pH is checked and found to be about 9.4. The mixture is filtered over polypropylene. The solids are collected and placed into a 5 L round bottom flask. A 20% water/MeOH solution (2.6 L) is added and the slurry is stirred for 30 min. The slurry is filtered and the solids are washed with 20% water/MeOH (600 mL). The solids are placed in a vacuum oven at 35° C. overnight. The first weighing indicates 394 g (theoretical yield 324.8 g, about 121% mass recovery). TGA (Thermogravimetric analysis)/DSC (differential scanning calorimetry) shows about 17 wt % free water and 10-11 wt% volatile loss at the melt. The solids are dried at 55° C. in a vacuum oven with a N2 sweep for 3.5 hours (354.7 g, about 109% mass recovery, NMR shows about 9.3 wt % DCM). No free water is present according to TGA/DSC. The material is sent for milling.
The jet mill (Aljet™ 0101) in a glove bag is assembled inside a walk in hood and hooked up to N2 to a 100 lb header. The inlet pusher nozzle is adjusted for maximum draw and max nitrogen flow is introduced into the mill. Pressure readings are noted as 90 psi on pusher nozzle and 85 psi on both grind nozzles. The starting material (353.4 g) is slowly fed to the mill inlet, stopping to empty the receiver sock as needed. The total milling time is 22 min and 25 second. The calculated feed rate is 15.8 g/min (353.4 grams divided by 22.42 min). The milled material (335.7 g, 95%) is obtained with 17.7 g loss. Particle size analysis result of the milled material is d90 of 4.6 microns.
TGA/DSC indicates about 11.4 wt % volatiles at the melt and NMR (DMSO) shows about 9.3 wt % DCM. 1H NMR (DMSO) δ 12.94 (br s, 1 H), 11.88 (s, 1H), 8.44 (d,J=7.47 Hz, 1 H), 8.12 (br s, 1 H), 8.00 (br s, 1 H), 7.96 (s, 1 H), 7.94 (d,J=2.2 Hz, 1 H), 7.91 (d,J=2.6 Hz, 1 H), 7.87 (s, 1H), 7.47-7.37 (m, 5 H), 6.82 (t,J=9.26 Hz, 8.82 Hz, 1 H), 6.65 (d,J=7.49 Hz, 1 H), 4.04 (s, 3 H), 2.03 (s, 3 H). LC/MS: (M+H) 553.1.
Anhydrous Crystal Form Preparation
To 10 mL of EtOH is added 120 mg of the above compound into a 20 mL vial. The sample is heated to 70° C. with stirring. Initially the solids start to dissolve and then a suspension forms followed by a white precipitate. The sample is cooled to RT while being stirred. A small sample of the slurry is taken by pipette and allowed to air dry. This material is highly crystalline and proves to be an ethanol solvate by TGA. To the remaining suspension, 10 mL of heptane is added and then heated to boiling. The measured temperature is monitored at 70.8° C. until the volume has been reduced to 10 mL. When the temperature starts to rise, the heat is removed and the slurry stirred at RT overnight. The solid is isolated by vacuum filtration and dried in a vacuum oven at 45° C. for 3 hours, resulting in 77% recovery. The crystalline form shows a weight loss of 0.17% from 25-238° C. by TGA. The form’s onset of melting is 247.8° C.
PAPER
N-(3-Fluoro-4-((1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yl)oxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide (1, LY2801653)

1H NMRof Merestinib
PAPER
 1 MERESTENIB
1 MERESTENIBAn NH4Cl-catalyzed ethoxy ethyl deprotection was developed for the synthesis of merestinib, a MET inhibitor. Alternative reactor technologies using temperatures above the solvent boiling point are combined with this mild catalyst to promote the deprotection reaction. The reaction is optimized for flow and has been used to synthesize over 100 kg of the target compound. The generality of the reaction conditions is also demonstrated with other compounds and protecting groups.
1: 1H NMR (500 MHz, DMSO-d6): δ = 13.00 (s, 1 H), 11.93 (s, 1 H), 8.45 (d, J = 7.5 Hz, 1 H), 8.17 (s, 1 H), 8.05 (s, 1 H), 8.01 (s, 1 H), 7.97 (d, J = 2.2 Hz, 1 H), 7.91 (d, J = 2.6 Hz, 1 H), 7.50–7.46 (m, 2 H), 7.43–7.40 (m, 2 H), 7.27 (s, 1 H), 7.26–7.25 (m, 1 H), 6.86 (t, J = 9.0 Hz, 1 H), 6.67–6.65 (m, 1 H), 4.08 (s, 3 H), 2.03 (s, 3 H) ppm; 13C NMR (125 MHz, DMSO-d6): δ = 163.0, 161.5, 161.0, 153.2, 152.2, 147.5, 144.0, 140.1, 138.0, 137.3, 134.5, 134.1, 132.0, 130.1, 127.4, 124.2, 121.8, 119.8, 117.0, 116.8, 116.6, 116.1, 108.9, 108.6, 108.2, 107.8, 35.5, 21.7 ppm; HR-MS [ESI]: Calcd for C30H23F2N6O3+ [M + H+]: 553.1794, found 553.1793.

……….
WO 2010011538
http://www.google.co.in/patents/WO2010011538A1?cl=en
Example 1 N-(3 -Fluoro-4-( 1 -methyl-6-( lH-pyrazol-4-yl)- lH-indazol-5 -yloxy)phenyl)- 1 -(4- fluorophenyl)-6-methyl-2-oxo- 1 ,2-dihydropyridine-3 -carboxamide

To a 100 mL round bottom flask is added tert-butyl 4-(5-(4-amino-2- fluorophenoxy)-l -methyl- IH- indazol-6-y I)- lH-pyrazole-1-carboxylate (1.43 g, 3.38 mmol), l-(4-fluorophenyl)-6-methyl-2-oxo-l,2-dihydropyridine-3-carboxylic acid (1.25 g, 5.07 mmol), EDCI (1.48 g , 7.6 mmol) and ΗOBt (776 mg, 5.07 mmol) followed by DMF (15 mL, 193.99 mmol) and then DIPEA (1.47 mL, 8.44 mmol). The mixture is allowed to stir at RT overnight. The reaction mixture is diluted into EtOAc (300 mL) and washed with saturated aqueous sodium chloride (5 x 100 mL). The combined aqueous solution is extracted with EtOAc (1 x 100 mL) and then the combined organic solutions are dried over Na2SO4, filtered, and concentrated to dryness. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in a DCM solution (B), gradient from 100% (A) to 80% (A):20% (B) over 70 min to give tert-butyl 4-(5-(2- fluoro-4-(l-(4-fluoroplienyl)-6-metliy 1-2-oxo- 1,2-dihy dropyridine-3- carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 -carboxylate as a gold solid (2.20 g, 87% yield). MS (m/z): 653. (M+Η), 675 (M+Na).
To a round bottom flask is added tert-butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)- 6-methyl-2-oxo- 1 ,2-dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6- yl)-lΗ-pyrazole-l -carboxylate (1.92 g, 2.94 mmol) and DCM (50 mL) followed by triethylsilane (1.88 mL, 11.77 mmol) and TFA (17.8 mL, 235.35 mmol). The reaction mixture is allowed to stir at RT for 1.5 hours. The solvent is removed and diluted into DCM (150 mL) and washed with saturated aqueous NaHCθ3 solution (2 x 100 mL). The organic solution is dried with Na2SO4, and concentrated under reduced pressure to give a solid material. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in DCM solution (B), gradient from 100% (A) to 75%(A):25%(B) over 70 min, held at this 75:25 ratio for 15 min to give the title compound as an off-white solid. The solid is dissolved in hot EtOH (50 mL) followed by a portion-wise addition of distilled water (250 mL) causing a white solid to precipitate. The solid is filtered over a Buchner funnel and washed with distilled water (3 x 15 mL), air dried, and vacuum dried at 60 0C for 15 hours to give the title compound as an off-white solid (1.27 g, 78% yield). MS (m/z): 552.8 (M+H).
Example 2
N-(3-Fluoro-4-(l-methyl-6-(lH-pyrazol-4-yl)-lH-indazol-5-yloxy)phenyl)-l-(4- fluorophenyl)-6-methyl-2-oxo-l,2-dihydropyridine-3-carboxamide
A 12 L round bottom flask is equipped with overhead agitation, a thermocouple, and a N2 purge. tert-Butyl 4-(5-(4-amino-2-fluorophenoxy)-l -methyl- lH-indazol-6-yl)- lH-pyrazole-1-carboxylate (404 g, 954.08 mmol) is dissolved in DMF (2 L) and charged to the flask. DMF (1 L) is used to rinse the flask. l-(4-Fluorophenyl)-6-methyl-2-oxo- l,2-dihydropyridine-3-carboxylic acid (259.46 g ,1.05 mol) and EDCI (228.63 g , 1.19 mol) are added and it is rinse in with DMF (500 mL). Then ΗOBt (189.94 g, 1.24 mol) is added and it is again rinsed in with DMF (500 mL). Finally, DIPEA is slowly added (184.97 g, 1.43 mol). The dark solution is then stirred at RT over the weekend. To a 20 L bottom outlet flask is added DI water (3 L) and DCM (5 L). The reaction mixture is poured in and it is rinsed in with DCM (1 L). The organic layer is separated, washed with DI water (3 X 3 L), dried over Na2SO4, filtered, rinsed solids with DCM and concentrated the filtrate. EtOAc (2 L) is added to the residue and the solution is stirred for 1 hour. The product crystallizes out. The mixture is concentrated. Another portion of EtOAc (2 L) is added and concentrated to remove all of the DCM. EtOAc (650 mL) and MTBE (3 L) are added to the residue and the solution is stirred in an ice bath for 1 hour. The tan slurry is filtered using a polypropylene pad. The cake is rinsed with MTBE (2 x 500 mL). The light tan solid is dried overnight in the vacuum oven at 40 0C to give the crude product (553 g). The crude product is purified by silica gel column chromatography eluting with (50% EtOAc (50%):35% DCM (35%): n-heptane (15%)) to give the pure desired product tert-butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)-6-methyl-2-oxo-l,2- dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 – carboxylate (424 g, 68%). MS (m/z): 651.0 (M-H). tert-Butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)-6-methyl-2-oxo-l,2- dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 – carboxylate (423.9 g, 649.50 mmol) is dissolved in DCM (4.24 L). HCl in MeOH (5.74 N, 799.99 mL, 4.59 mol) is added and the solution is heated at 30 0C for 1 hour. Then the reaction mixture is heated to 45 0C and DCM (1.5 L) is added. After two hours, the solution is heated to 50 0C and DCM (2 L) is added. After 3 hours, DCM (2 L) is added followed by HCl in MeOH (4.5 N, 721.67 mL, 3.25 mol). After another 45 min, DCM (1 L), HCl in MeOH (4.5 N, 288.67 mL, 1.30 mol), and MeOH (1.5 L) are added. The reaction solution is then heated to 60 0C. After 4 hours, MeOH (2 L) is added and 10 min later DCM (1 L) is added followed by HCl in MeOH (4.5 N, 200 mL). After 5 hours, the reaction is complete. The reaction mixture is concentrated to about 1/3 volume. MeOH (2 L) is added and the solution is concentrated to a thick slurry. Again, MeOH (2 L) is added and the mixture is concentrated to a thick slurry. The slurry is cooled to about 10- 15 0C and then filtered. The solids are washed with MeOH. The solids are placed in a 55 0C vacuum oven for 2 days to give the desired product N-(3-fluoro-4-(l-methyl-6-(lH- pyrazol-4-yl)- lH-indazol-5-yloxy)phenyl)- 1 -(4-fluorophenyl)-6-methyl-2-oxo- 1,2- dihydropyridine-3-carboxamide hydrochloride (377 g, 92.8%). MS (m/z): 551.0 (M-H).
To a 22 L round bottom flask equipped with mechanical stirring under nitrogen is added N-(3-fluoro-4-(l-methyl-6-(lH-pyrazol-4-yl)-lH-indazol-5-yloxy)phenyl)-l-(4- fluorophenyl)-6-methyl-2-oxo-l,2-dihydropyridine-3-carboxamide hydrochloride (367 g, 0.62 mol) followed by DCM (7.34 L) and water (7.34 L). Na2CO3 (181.6 g, 1.71 mol) is added and the mixture is stirred at RT for 30 min. The pΗ is checked and found to be about 9.4. The mixture is filtered over polypropylene. The solids are collected and placed into a 5 L round bottom flask. A 20% water/MeOΗ solution (2.6 L) is added and the slurry is stirred for 30 min. The slurry is filtered and the solids are washed with 20% water/MeOΗ (600 mL). The solids are placed in a vacuum oven at 35 0C overnight. The first weighing indicates 394 g (theoretical yield 324.8 g, about 121% mass recovery).
TGA (Thermogravimetric analysis)/DSC (differential scanning calorimetry) shows about 17 wt % free water and 10-11 wt% volatile loss at the melt. The solids are dried at 55 0C in a vacuum oven with a N2 sweep for 3.5 hours (354.7 g, about 109% mass recovery, NMR shows about 9.3 wt % DCM). No free water is present according to TGA/DSC. The material is sent for milling. The jet mill (AIj et™ 0101) in a glove bag is assembled inside a walk in hood and hooked up to N2 to a 100 Ib header. The inlet pusher nozzle is adjusted for maximum draw and max nitrogen flow is introduced into the mill. Pressure readings are noted as 90 psi on pusher nozzle and 85 psi on both grind nozzles. The starting material (353.4 g) is slowly fed to the mill inlet, stopping to empty the receiver sock as needed. The total milling time is 22 min and 25 second. The calculated feed rate is 15.8 g/min (353.4 grams divided by 22.42 min). The milled material (335.7 g, 95%) is obtained with 17.7 g loss. Particle size analysis result of the milled material is d90 of 4.6 microns.
TGA/DSC indicates about 11.4 wt % volatiles at the melt and NMR (DMSO) shows about 9.3 wt % DCM. 1H NMR (DMSO) δ 12.94 (br s, 1 H), 11.88 (s, IH), 8.44 (d, J= 7.47 Hz, 1 H), 8.12 (br s, 1 H), 8.00 (br s, 1 H), 7.96 (s, 1 H), 7.94 (d, J= 2.2 Hz, 1 H), 7.91 (d, J= 2.6 Hz, 1 H), 7.87 (s, IH), 7.47-7.37 (m, 5 H), 6.82 (t, J= 9.26 Hz, 8.82 Hz, 1 H), 6.65 (d, J= 7.49 Hz, 1 H), 4.04 (s, 3 H), 2.03 (s, 3 H). LC/MS: (M + H) 553.1.
Anhydrous Crystal Form Preparation To 10 mL of EtOH is added 120 mg of the above compound into a 20 mL vial.
The sample is heated to 70 0C with stirring. Initially the solids start to dissolve and then a suspension forms followed by a white precipitate. The sample is cooled to RT while being stirred. A small sample of the slurry is taken by pipette and allowed to air dry. This material is highly crystalline and proves to be an ethanol solvate by TGA. To the remaining suspension, 10 mL of heptane is added and then heated to boiling. The measured temperature is monitored at 70.8 0C until the volume has been reduced to 10 mL. When the temperature starts to rise, the heat is removed and the slurry stirred at RT overnight. The solid is isolated by vacuum filtration and dried in a vacuum oven at 45 0C for 3 hours, resulting in 77% recovery. The crystalline form shows a weight loss of 0.17% from 25-238 0C by TGA. The form’s onset of melting is 247.80C.
|
References |
1: Yan SB, Peek VL, Ajamie R, Buchanan SG, Graff JR, Heidler SA, Hui YH, Huss KL, Konicek BW, Manro JR, Shih C, Stewart JA, Stewart TR, Stout SL, Uhlik MT, Um SL, Wang Y, Wu W, Yan L, Yang WJ, Zhong B, Walgren RA. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs. 2012 Dec 29. [Epub ahead of print] PubMed PMID: 23275061.
-
Liu, X.; Newton, R. C.; Scherle, P. A. Expert Opin. Invest. Drugs 2011, 20, 1225,DOI: 10.1517/13543784.2011.600687
-
Yan, S. B.; Peek, V. L.; Ajamie, R.; Buchanan, S. G.; Graff, J. R.; Heidler, S. A.; Hui, Y.;Huss, K. L.; Konicek, B. W.; Manro, J. R.; Shih, C.; Stewart, J. A.; Stewart, T. R.; Stout, S. L.; Uhlik, M. T.; Um, S. L.; Wang, Y.; Wu, W.; Yan, L.; Yang, W. J.; Zhong, B.; Walgren, R. A. Invest. New Drugs 2013, 31, 833, DOI: 10.1007/s10637-012-9912-9
-
Kallman, N. J.; Liu, C.; Yates, M. H.; Linder, R. J.; Ruble, J. C.; Kogut, E. F.; Patterson, L. E.; Laird, D. L. T.; Hansen, M. M. Org. Process Res. Dev. 2014, 18, 501,DOI: 10.1021/op400317zKallman, N.J.; Yates, M.H.; Linder, R.J.; Hansen, M.M.
Route design and development of c-Met inhibitor LY2801653
244th Am Chem Soc (ACS) Natl Meet (August 19-23, Philadelphia) 2012, Abst ORGN 212
////////
UPDATE…..

A new synthesis of a key indazole-containing building block for the MET kinase inhibitor merestinib was designed and demonstrated. Crucial to the successful construction of the challenging indazole is an SNAr reaction, which forges the heterocyclic ring. Continuous processing was applied to two of the five steps: nitration of a benzaldehyde and high-temperature hydrolysis of an aniline to phenol. Compared to a highly developed historical route, the new route shows clear benefits in terms of product quality and potentially manufacturability and robustness.
An Alternative Indazole Synthesis for Merestinib
 , Kevin P. Cole† , Jared W. Fennell†, Todd D. Maloney†, David Mitchell†§ , Ramesh Subbiah‡, and Balakumar Ramadas‡
, Kevin P. Cole† , Jared W. Fennell†, Todd D. Maloney†, David Mitchell†§ , Ramesh Subbiah‡, and Balakumar Ramadas‡6-Bromo-5-(2-fluoro-4-nitrophenoxy)-1-methyl-1H-indazole (2)