
Home » Uncategorized (Page 4)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lecufexor
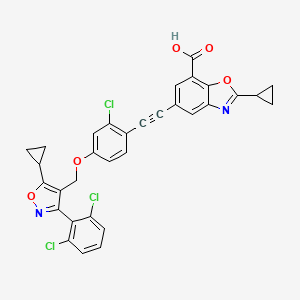
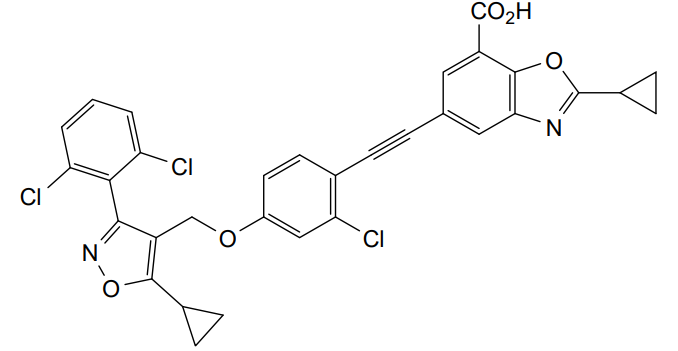
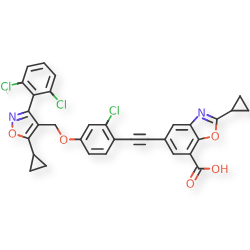
Lecufexor
CAS 2247972-61-6
MF C32H21Cl3N2O5 MW619.88
7-Benzoxazolecarboxylic acid, 5-[2-[2-chloro-4-[[5-cyclopropyl-3-(2,6-dichlorophenyl)-4-isoxazolyl]methoxy]phenyl]ethynyl]-2-cyclopropyl-
5-[2-[2-chloro-4-[[5-cyclopropyl-3-(2,6-dichlorophenyl)-1,2-oxazol-4-yl]methoxy]phenyl]ethynyl]-2-cyclopropyl-1,3-benzoxazole-7-carboxylic acid

farnesoid X receptor (FXR) agonist, K3K2F4N8BY, ID 119031166, ID 166
Lecufexor is a farnesoid X receptor (FXR) agonist.
Lecufexor (also known as ID119031166 or ID166) is a potent, selective, and non-steroidal farnesoid X receptor (FXR) agonist. It is primarily studied for its potential in treating liver diseases, particularly Non-Alcoholic Steatohepatitis (NASH) and liver fibrosis.
Key Characteristics and Research
- Mechanism of Action: It acts as an agonist for the farnesoid X receptor, which is a key regulator of bile acid homeostasis, lipid metabolism, and glucose metabolism.
- Therapeutic Potential: Research indicates it can improve NASH and liver fibrosis by modulating the gut-liver axis.
- Safety Profile: Unlike some other FXR agonists, Lecufexor is designed to be intestine-preferential and does not show activity against potential itch receptors (like MRGPRX4), which may help avoid common side effects like pruritus (itching).
Farnesoid X receptor(FXR, NR1H4)is a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors. FXR is highly expressed in the liver, intestine, kidney, adrenal glands, white adipose tissue and in induced during adipocyte differentiation in vitro . (Cariu B. et al., J. Biol. Chem., 2006, 16, 11039-11049)
Not only FXR regulates various physiological processed such as modulates regulrated of bile acid(BA) regulation, lipids/glucose metabolism, inflammation/fibrosis, but recently it has also been linked to the pathology of FXR receptors
This nuclear receptor is the intracellular bile acid ¡“sensor” and its major physiological role is to protect liver cells from the deleterious effect of bile acids(BA) overload. Intestine is the tissue expressing the first FXR target gene identified. Indeed IBAB-P is expressed in enterocytes and binds bile acids, thus limiting the free concentration of BA intracellularly and consequently their toxicity.(Makishima M, et al., Science, 1999, 284(5418), 1362-1365). FXR is highly expressed in the liver and regulates key genes involved in BA synthesis, metabolism and transport including CYP7A1, UGT2B4, BSEP, MDR3, MRP2, ASBT, NTCP, OST α and OST β in humans. One effect of FXR activation is down regulation of CYP7A1 and thus bile acid synthesis; this is accomplished through induction of SHP(Small Heterodimer Partner) which then represses CYP7A1 transcription(Claude T, et al., Arterioscler. Thromb. Vasc. Biol., 2005, 25, 2020-2031). Altered expression or malfunction of these genes has been described in patients with cholestatic liver disease. FXR agonist 6-ethyl-chenodeoxycholic acid(6EtCDCA)was found to fully reverse the impairment of bile flow and to protect the hepatocytes against liver cell injury caused by the cytotoxic lithocholic acid.(Pelliciari R, et al., J. Med. Chem., 2002, 45(17), 3569-3572).
SYN
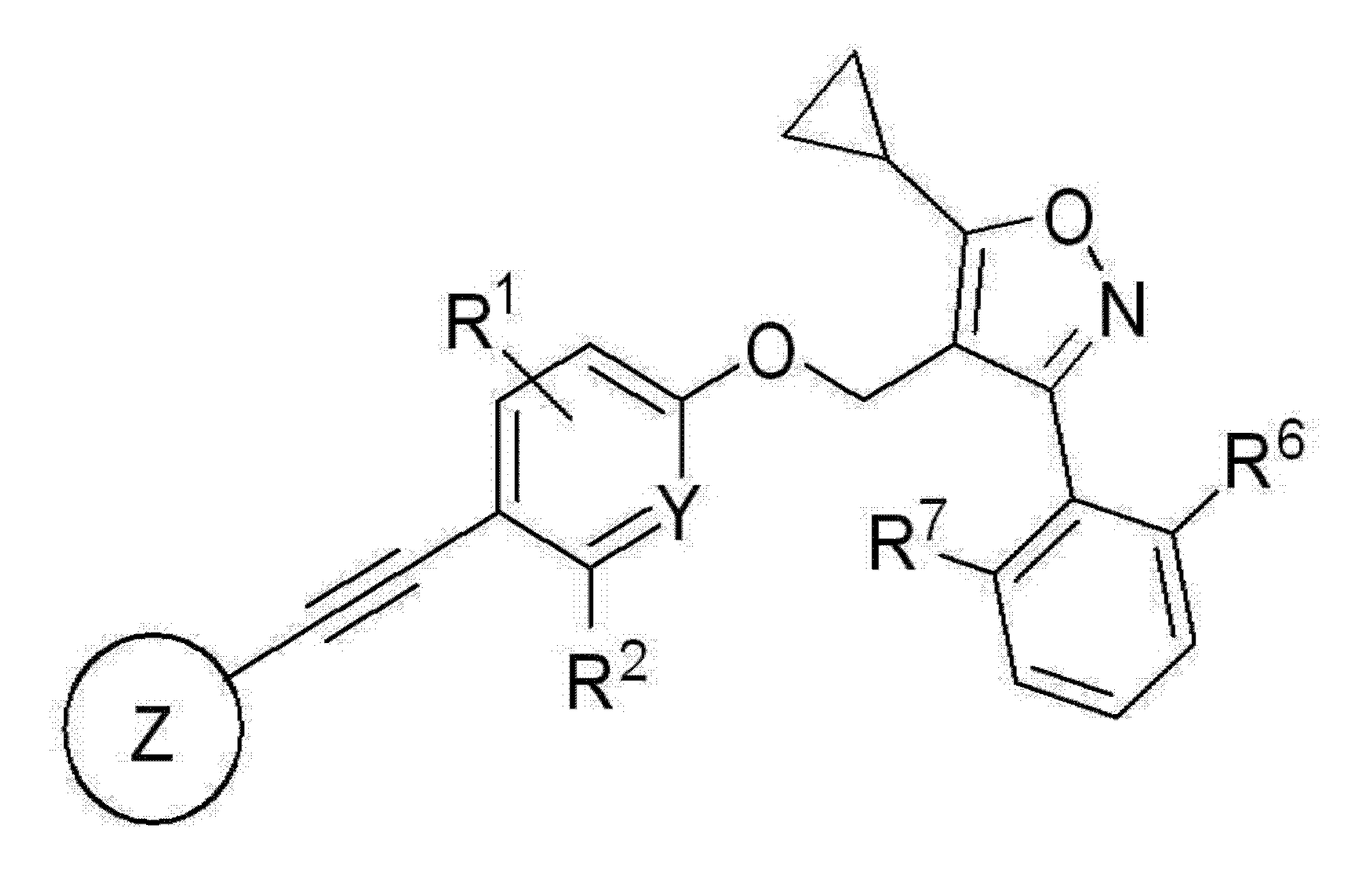
PAT
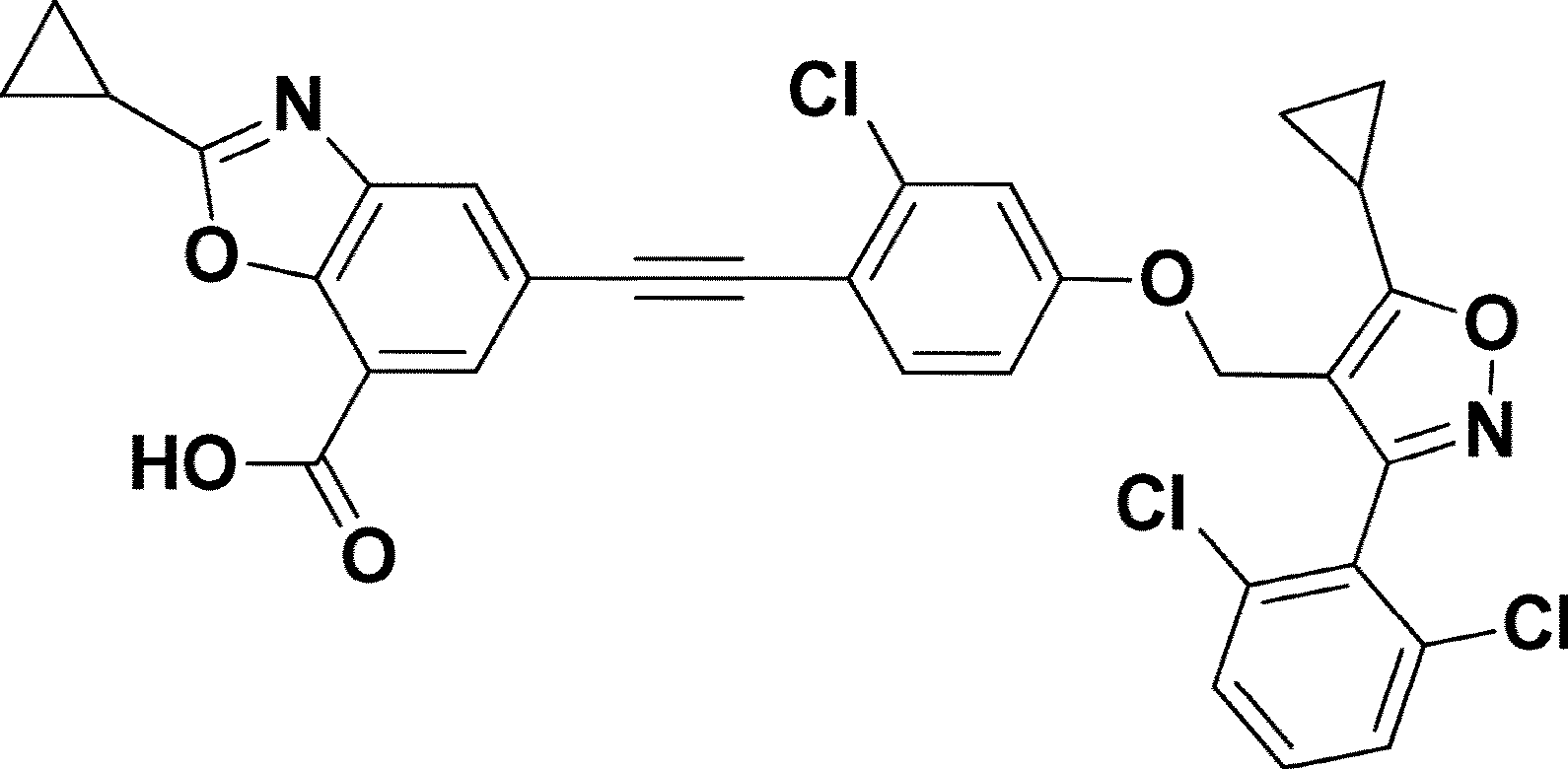
Manufacturing Example 1. Preparation of 5-((2-chloro-4-((5-cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)phenyl)ethynyl)-2-cyclopropylbenzo[d]oxazole-7-carboxylic acid (compound of chemical formula 1)[288] [289]
Step 1: Preparation of methyl 5-((2-chloro-4-((5-cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)phenyl)ethynyl)-2-cyclopropylbenzo[d]oxazole-7-carboxylate[290] [291]4-((3-Chloro-4-ethynylphenoxy)methyl)-5-cyclopropyl-3-(2,6-dichlorophenyl)isoxazole (182 mg, 0.43 mmol), methyl 5-bromo-2-cyclopropylbenzo[d]oxazole-7-carboxylate (117 mg, 0.40 mmol), bis(triphenylphosphine)palladium(II) dichloride (PdCl
2 (PPh
3 )
2 , 14 mg, 0.02 mmol), copper(I) iodide (3.8 mg, 0.02 mmol), and triethylamine (67 μl, 0.48 mmol) were added, and the mixture was stirred at 80°C for 4 hours. The reaction mixture was diluted with ethyl acetate and washed with distilled water. Dried over magnesium sulfate, filtered, concentrated, and purified by silica gel chromatography to obtain the intermediate compound methyl 5-((2-chloro-4-((5-cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)phenyl)ethynyl)-2-cyclopropylbenzo[d]oxazole-7-carboxylate (121 mg, 48%). [292]
1 H-NMR (CDCl
3 , 400MHz): 8.06(d, 1H), 7.90(d, 1H), 7.43-7.40(m, 3H), 7.36-7.31(m, 1H), 6.88(d, 1H), 6.69(dd,1H), 4.82(s, 2H), 4.00(s, 3H), 2.32-2.24(m, 1H), 2.20-2.12(m, 1H), 1.38-1.33(m, 2H), 1.32-1.27(m, 2H), 1.26-1.23(m, 2H), 1.19-1.15(m, 2H). [293] [294]
Step 2: Preparation of 5-((2-chloro-4-((5-cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl)methoxy)phenyl)ethynyl)-2-cyclopropylbenzo[d]oxazole-7-carboxylic acid[295] [296]The intermediate compound (120 mg, 0.19 mmol) prepared in the above step 1 and lithium hydroxide (79.4 mg, 1.9 mmol) were combined in the same manner as in step 6 of Example 1 to obtain the target compound (102 mg, 87.4%). [297]
1 H-NMR (DMSO, 400MHz): 13.6(br s, 1H), 7.99(d, 1H), 7.97(d, 1H), 7.87-7.62(m, 2H), 7.57-7.55(m, 2H), 7.09(d, 1H), 6.83(dd, 1H), 4.98(s, 2H), 2.39-2.30(m, 1H), 1.26-1.12(m, 8H).
PAT
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: US-2024262799-A1Priority Date: 2017-04-12
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: US-10988449-B2Priority Date: 2017-04-12Grant Date: 2021-04-27
- Isoxazole derivatives as nuclear receptor agonists and use thereofPublication Number: RU-2741306-C1Priority Date: 2017-04-12Grant Date: 2021-01-25
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: US-2021188790-A1Priority Date: 2017-04-12
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: CN-110678450-BPriority Date: 2017-04-12Grant Date: 2023-06-20
- An isoxazole derivatives as nuclear receptor agonists and used thereofPublication Number: AU-2018252880-A1Priority Date: 2017-04-12
- ISOXAZOLE DERIVATIVES AS NUCLEAR RECEPTOR AGONISTS AND THEIR USESPublication Number: HR-P20220026-T1Priority Date: 2017-04-12
- Isoxazole derivatives that are nuclear receptor efficacy agents and their usesPublication Number: JP-6886074-B2Priority Date: 2017-04-12Grant Date: 2021-06-16
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: EP-3612520-B1Priority Date: 2017-04-12Grant Date: 2021-11-10
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: US-11912674-B2Priority Date: 2017-04-12Grant Date: 2024-02-27
- Isoxazole derivative which is an effector of nuclear receptor and its usePublication Number: JP-2020516697-APriority Date: 2017-04-12
- An isoxazole derivatives as nuclear receptor agonists and used thereofPublication Number: WO-2018190643-A1Priority Date: 2017-04-12
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: ES-2904294-T3Priority Date: 2017-04-12Grant Date: 2022-04-04
- An isoxazole derivatives as nuclear receptor agonists and used thereofPublication Number: EP-3612520-A1Priority Date: 2017-04-12
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: CN-110678450-APriority Date: 2017-04-12
- Method for preparing isoxazole derivative, and novel intermediate thereforPublication Number: WO-2023090859-A1Priority Date: 2021-11-17
- Preparation method of isoxazole derivatives and intermediates thereofPublication Number: KR-20230072437-APriority Date: 2021-11-17
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: US-2020115349-A1Priority Date: 2017-04-12
- Isoxazole derivatives as nuclear receptor agonists and uses thereofPublication Number: CA-3059869-A1Priority Date: 2017-04-12
- pharmaceutical compound and compositionPublication Number: BR-112019021320-A2Priority Date: 2017-04-12
- Novel use of isoxazole derivates and salts thereofPublication Number: TW-202408498-APriority Date: 2022-06-30
- Novel use of isoxazole derivative or salt thereofPublication Number: WO-2024005587-A1Priority Date: 2022-06-30
- Novel crystalline form of isoxazole derivative or salt thereofPublication Number: WO-2024005586-A1Priority Date: 2022-06-30
- Preparation method of isoxazole derivatives and intermediates thereofPublication Number: KR-102740618-B1Priority Date: 2021-11-17Grant Date: 2024-12-10
- Method for preparing isoxazole derivative, and intermediate thereofPublication Number: WO-2023090858-A1Priority Date: 2021-11-17



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
///////lecufexor, farnesoid X receptor (FXR) agonist, K3K2F4N8BY, ID 119031166, ID 166
Laselipag
Laselipag
CAS 475085-57-5
MF C25H29N3O3 MW 419.52
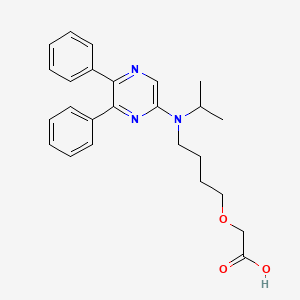
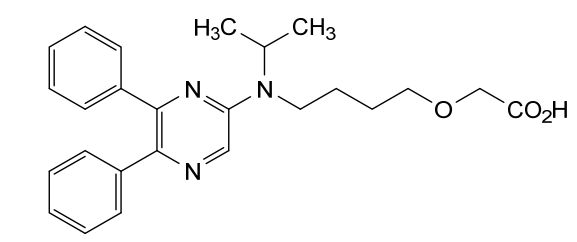
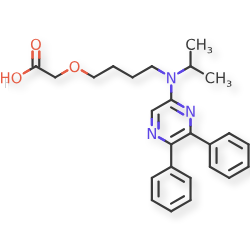
2-[4-[(5,6-diphenylpyrazin-2-yl)-propan-2-ylamino]butoxy]acetic acid
{4-[(5,6-diphenylpyrazin-2-yl)(propan-2-yl)amino]butoxy}acetic acid
prostanoid receptor agonist, ACT-333679, ACT 333679, ACT333679, E9PC7N0DID, MRE 269
ACT-333679 is a member of the class of pyrazines that is {4-[(propan-2-yl)(pyrazin-2-yl)amino]butoxy}acetic acid carrying two additional phenyl substituents at positions 5 and 6 on the pyrazine ring. The active metabolite of selexipag, an orphan drug used for the treatment of pulmonary arterial hypertension. It has a role as an orphan drug, a platelet aggregation inhibitor, a prostacyclin receptor agonist, a vasodilator agent and a drug metabolite. It is an aromatic amine, an ether, a member of pyrazines, a sulfonamide, a tertiary amino compound and a monocarboxylic acid.
SYN
SYN
PAT
- Heterocyclic compound derivatives and medicinesPublication Number: CA-2445344-CPriority Date: 2001-04-26Grant Date: 2011-07-26
- Heterocyclic compound derivatives and medicinesPublication Number: US-2004102436-A1Priority Date: 2001-04-26
- DERIVATIVES OF HETEROCYCLIC COMPOUNDS AND MEDICINAL PRODUCTSPublication Number: DE-60217674-T2Priority Date: 2001-04-26Grant Date: 2007-10-11
- Heterocyclic compound derivatives and medicinesPublication Number: EP-1400518-B1Priority Date: 2001-04-26Grant Date: 2007-01-17
- Heterocyclic compound derivatives and medicinesPublication Number: US-7205302-B2Priority Date: 2001-04-26Grant Date: 2007-04-17
- Heterocyclic compound derivatives and medicinesPublication Number: CA-2445344-A1Priority Date: 2001-04-26
- Heterocyclic derivatives and pharmaceuticalsPublication Number: JP-4479152-B2Priority Date: 2001-04-26Grant Date: 2010-06-09
- DERIVATIVES OF HETEROCYCLIC COMPOUNDS AND MEDICINESPublication Number: RU-2003134190-APriority Date: 2001-04-26
- Heterocyclic derivatives and drugsPublication Number: JP-WO2002088084-A1Priority Date: 2001-04-26
- DERIVATIVES OF HETEROCYCLIC COMPOUNDS AND MEDICINAL PRODUCTS CONTAINING THEMPublication Number: PT-1400518-EPriority Date: 2001-04-26
- Heterocyclic compound derivatives and medicinesPublication Number: EP-1400518-A1Priority Date: 2001-04-26
- DERIVATIVES OF HETEROCICLIC COMPOUNDS AND MEDICINES.Publication Number: ES-2276931-T3Priority Date: 2001-04-26Grant Date: 2007-07-01
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////laselipag, prostanoid receptor agonist, ACT-333679, ACT 333679, ACT333679, E9PC7N0DID, MRE 269
Laroprovstat
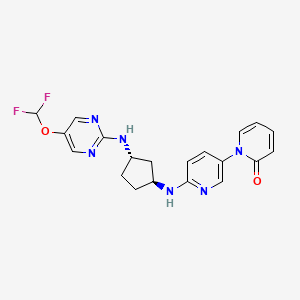

Laroprovstat
CAS 2455427-91-3
MF C20H20F2N6O2 MW414.4 g/mol
1-[6-[[(1S,3S)-3-[[5-(difluoromethoxy)pyrimidin-2-yl]amino]cyclopentyl]amino]-3-pyridinyl]pyridin-2-one
- 1-[6-[[(1S,3S)-3-[[5-(difluoromethoxy)pyrimidin-2-yl]amino]cyclopentyl]amino]pyridin-3-yl]pyridin-2-one
- 6′-(((1S,3S)-3-((5-(Difluoromethoxy)pyrimidin-2-yl)amino)cyclopentyl)amino)-2H-[1,3′-bipyridin]-2-one
- 6′-{[(1S,3S)-3-{[5-(difluoromethoxy)pyrimidin-2-yl]amino}cyclopentyl]amino}-2H-[1,3′-bipyridin]-2-one
6′-{[(1S,3S)-3-{[5-(difluoromethoxy)pyrimidin-2-yl]amino}cyclopentyl]amino}-2H-[1,3′-bipyridin]-2-one
proprotein convertase subtilisin/kexin type 9 (PCSK9), inhibitor, antihyperlipidaemic, 9EEL3YMY29, PCSK9-IN-12, AZD 0780, AZD-0780
- OriginatorDogma Therapeutics
- DeveloperAstraZeneca; Parexel International; Quotient Sciences
- ClassAmines; Antihyperlipidaemics; Cardiovascular therapies; Cyclopentanes; Ethers; Fluorinated hydrocarbons; Hepatoprotectants; Ketones; Pyridines; Pyrimidines; Small molecules; Vascular disorder therapies
- Mechanism of ActionPCSK9 protein inhibitors
- Phase IIIAtherosclerosis; Cardiovascular disorders; Dyslipidaemias; Hyperlipoproteinaemia type II
- Phase ILiver disorders
- 30 Dec 2025AstraZeneca completes a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT07216131),
- 10 Nov 2025AstraZeneca initiates enrolment in a phase I pharmacokinetics trial (In volunteers) in USA (PO)(NCT07216131),
- 29 Oct 2025AstraZeneca completes a phase I trial in Dyslipidaemias (Combination therapy) in USA, United Kingdom (PO) (NCT06742853)
SYN

PAT
PAT
- PCSK9 Inhibitors and Methods of Use ThereofPublication Number: US-2020231584-A1Priority Date: 2019-01-18
- Pcsk9 inhibitors and methods of use thereofPublication Number: US-2022220122-A1Priority Date: 2019-01-18
- Pcsk9 inhibitors and methods of use thereofPublication Number: WO-2024013209-A1Priority Date: 2022-07-13
- PCSK9 Inhibitors and Methods of Use ThereofPublication Number: US-2020291041-A1Priority Date: 2019-01-18
- Pcsk9 inhibitors and methods of use thereofPublication Number: WO-2020150473-A2Priority Date: 2019-01-18
- Pcsk9 inhibitors and methods of use thereofPublication Number: WO-2020150474-A1Priority Date: 2019-01-18
- PCSK9 inhibitors and methods of use thereofPublication Number: US-11248001-B2Priority Date: 2019-01-18Grant Date: 2022-02-15
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////laroprovstat, antihyperlipidaemic, 9EEL3YMY29, PCSK9-IN-12, AZD 0780, AZD-0780
Istisociclib
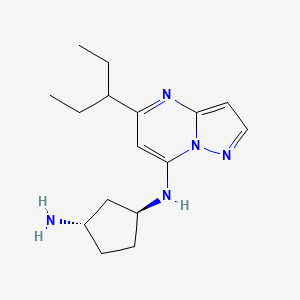
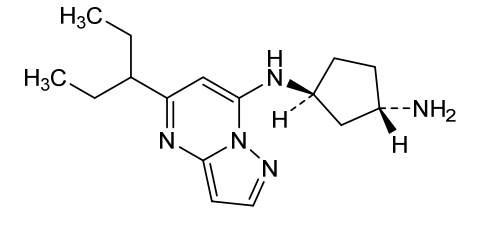
Istisociclib
KB 130742
CAS 2416873-83-9
MF C16H25N5, 287.40 g/mol
trans-(1S,3S)-3-N-(5-pentan-3-ylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine
- (1S,3S)-N1-[5-(1-Ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-1,3-cyclopentanediamine
- 1,3-Cyclopentanediamine, N1-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-, (1S,3S)-
(1S,3S)-N1-[5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine
cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Istisociclib is a small molecule drug. The usage of the INN stem ‘-ciclib’ in the name indicates that Istisociclib is a cyclin dependant kinase inhibitor. Istisociclib is under investigation in clinical trial NCT04718675 (A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)). Istisociclib has a monoisotopic molecular weight of 287.21 Da.
Istisociclib is an orally bioavailable, selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon oral administration, istisociclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins and oncogenic transcription factors including MYC and androgen receptor (AR). This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII), and is an important cofactor for various oncogenic transcription factors. It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
ISTISOCICLIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)
CTID: NCT04718675
Phase: Phase 1/Phase 2
Status: Terminated
Date: 2025-02-17
REF
- Discovery of KB-0742, a Potent, Selective, Orally Bioavailable Small Molecule Inhibitor of CDK9 for MYC-Dependent CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-11-15PMCID: PMC10726352PMID: 37967851DOI: 10.1021/acs.jmedchem.3c01233
- CDK9 inhibitors in cancer researchPublication Name: RSC Medicinal ChemistryPublication Date: 2022-06-22PMCID: PMC9215160PMID: 35814933DOI: 10.1039/d2md00040g
- From Structure Modification to Drug Launch: A Systematic Review of the Ongoing Development of Cyclin-Dependent Kinase Inhibitors for Multiple Cancer TherapyPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-04-29PMID: 35485642DOI: 10.1021/acs.jmedchem.1c02064
- Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery EffortsPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-02PMID: 35235745DOI: 10.1021/acs.jmedchem.1c02190
PAT
(lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine is a pharmaceutically active compound that has been studied for various uses, such as for the treatment of cancer. As used herein, the term “Compound A” is used to refer to both the free base and salt forms of (lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine. The free base of Compound A has the CAS number of 2416873-83-9 and structure of formula (I):
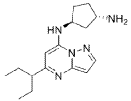
SYN
Example 35: (1S,3S)-N3-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine (35)
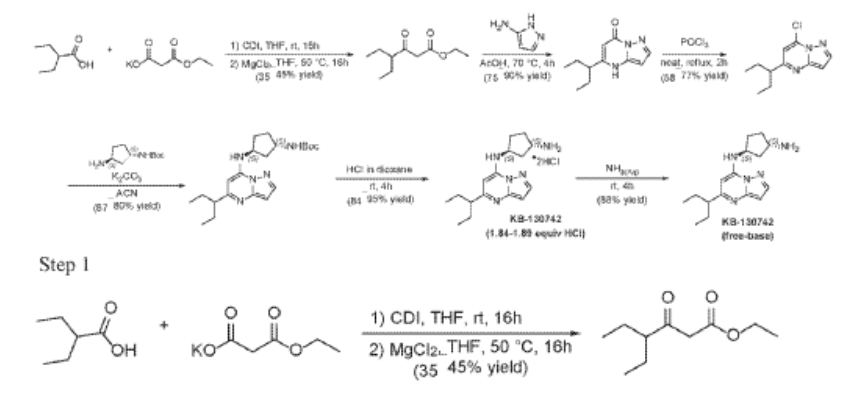
2-Ethylbutanoic acid (7.5 g, 64.57 mmol) was dissolved in THF (150 mL) and cooled to 0 °C. Within 20 min CDI (16.23 g, 100.08 mmol) was added portion-wise. The reaction warmed to room temp (rt) and the mixture was stirred at rt overnight (Solution A). In another flask MgCl2 (6.14 g, 64.57 mmol) and potassium 3-ethoxy-3-oxo-propanoate (17 g, 100.1 mmol) were mixed with THF (150 mL) and stirred under argon overnight at 50 °C. The resultant white suspension was cooled to rt and solution A was added dropwise over 10 min and the reaction mixture (RM) was stirred for 16h at room temperature. After several minutes a sticky, amorphous solid appeared whereupon after several hours the reaction mixture became homogenous in appearance. The RM was concentrated to about a third, taken up in half sat. potassium bisulphate solution and extracted twice with ethyl acetate. The organic layers were subsequently washed with a sat. sodium bicarbonate solution, combined, dried over anhydrous sodium sulfate, filtered and evaporated. Purification by column chromatography gave ethyl 4-ethyl-3-oxo-hexanoate (4.3 g, 23.087 mmol, 35.8% yield) as a transparent liquid. The RM was monitored by TLC (10% EA in Hex, Product Rf=0.6, SM Rf=0.1).
Step 2
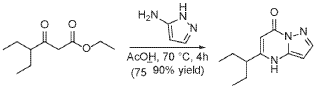
To a suspension of ethyl 4-ethyl-3-oxo-hexanoate (4.4 g, 23.62 mmol) in acetic acid (11 mL) at 70 °C was added 1H-pyrazol-5-amine (4.71 g, 56.7 mmol) in two portions (the second portion was added after 2 hours of stirring the first portion) over a 4 hour period. Upon consumption of SM as indicated by TLC, the reaction was cooled to rt and the solvent was evaporated in a rotary evaporator. The residue was treated with ethyl acetate and filtered to give 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 17.7 mmol, 74.9% yield) as an off-white solid. The reaction mixture was monitored by TLC (5% MeOH in DCM, Product Rf=0.3, SM Rf=0.8).
Step 3

A stirred solution of 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 18.03 mmol) in POCl3 (33.7 mL, 360.52 mmol) was heated to reflux for 4 hours. The reaction mixture was cooled to room temperature, excess reagent was evaporated in a rotary evaporator, and the residue was treated with ice-water. The chlorinated product was extracted from aqueous mixture by DCM. The organic layer was separated, dried over anhydrous Na2SO4, filtered and purified by column chromatography to give 7- chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (3.1 g, 13.9 mmol, 76.9% yield) as a light yellow liquid. The reaction mixture was monitored by TLC (20% EA in Hex, Product Rf=0.6, SM Rf=0.1).
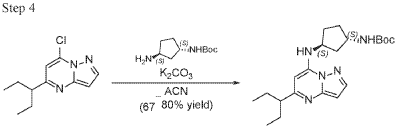
To a stirred solution 7-chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (2.3 g, 10.28 mmol), tert-Butyl ((1S,3S)-3-aminocyclopentyl)carbamate (2.27 g, 11.31 mmol) and K2CO3 (4.26 g, 30.84 mmol) in MeCN (20 mL) were heated to reflux for 16 hours. The reaction mixture was filtered, concentrated under reduced pressure and purified by column chromatography, eluent 30% EA in hexane to give tert-butyl N-[(1S,3S)-3-[[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (4.5 g, 11.6 mmol, 112.8% yield) as an off-white solid. The reaction mixture was monitored by TLC (40% EA in Hex, Product Rf=0.5, SM Rf=0.7).
Step 5
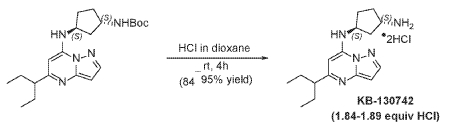
To tert-butyl N-[(1 S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (1.0 g, 2.58 mmol) in l,4-Dioxane (0.2 mL), 4 M HC1 in Dioxane (3.22 mL, 12.9 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give [(lS,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium dichloride (0.9 g, 2.5 mmol, 96.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC ( 100% EA, Product Rf=0.l, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 15.00 (s, 1H), 9.93-9.86 (m, 1H), 8.51 (s, 3H), 8.30 (s, 1H), 6.84 (s, 1H), 6.58 (s, 1H), 4.95 (q, J = 7.8 Hz, 1H), 3.77- 3.66 (m, 1H), 2.84-2.71 (m, 1H), 2.29-2.05 (m, 4H), 1.94-1.63 (m, 6H), 0.81 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.0 [M+H+])
Step 6
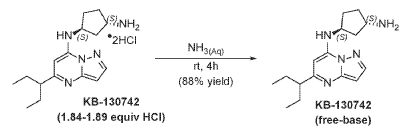
To [(1S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[I,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium-di chloride (0.2 g, 0.5600 mmol) in aq. NH3 (4.0 mL, 0.56 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give (lS,3S)-N3-[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]cyclopentane-l,3-diamine (140 mg, 0.49 mmol, 87.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC (100% EA, Product Rf=0.1, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 7.95 (d, J = 2.2 Hz, 1H), 6.86 (s, 1H), 6.29 (d, J = 2.2 Hz, 1H), 5.95 (s, 1H), 4.31-4.19 (m, 1H), 3.57-3.44 (m, 1H), 2.52-2.44 (m, 1H), 2.36-2.22 (m, 1H),
2.09–1.79 (m, 3H), 1.80–1.59 (m, 5H), 1.58–1.24 (m, 3H), 0.83 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.5 [M+H+]).
PAT
- Compounds, compositions and methods for modulating CDK9 activityPublication Number: CN-112996790-BPriority Date: 2018-10-30Grant Date: 2023-11-03
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2024132506-A1Priority Date: 2018-10-30
- Compounds, Compositions, and Methods for Modulating CDK9 ActivityPublication Number: US-2020131189-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2022002305-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: EP-3873911-A1Priority Date: 2018-10-30
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11845754-B2Priority Date: 2018-10-30Grant Date: 2023-12-19
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11155560-B2Priority Date: 2018-10-30Grant Date: 2021-10-26
- Compounds and methods for modulating cdk9 activityPublication Number: EP-4240422-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: WO-2022098843-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: US-2025188084-A1Priority Date: 2020-11-05
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: WO-2021216828-A1Priority Date: 2020-04-24
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: US-2023158159-A1Priority Date: 2020-04-24
- Polymorphic and salt forms of (ls,3s)-n-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: EP-4436569-A1Priority Date: 2021-11-24
- Polymorphs and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-A]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: KR-20240110634-APriority Date: 2021-11-24
- Compositions and methods for enhanced protein productionPublication Number: EP-4412621-A2Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: US-2024252688-A1Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: WO-2023056202-A2Priority Date: 2021-09-22
- A cdk9 inhibitor for use in the treatment of cancer in a subject having an asxl1 mutationPublication Number: WO-2025217597-A1Priority Date: 2024-04-12
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3- yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: WO-2023096922-A8Priority Date: 2021-11-24
- Polymorphic and salt forms of (ls,3s)-n1-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: WO-2023096922-A1Priority Date: 2021-11-24
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl) cyclopentane-1,3-diaminePublication Number: US-2025059193-A1Priority Date: 2021-11-24
- Polymorphic forms and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: CN-118678952-APriority Date: 2021-11-24
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////istisociclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Isomiosamine
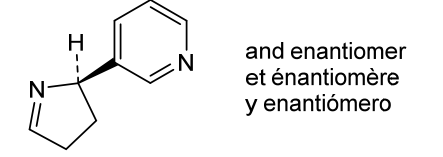
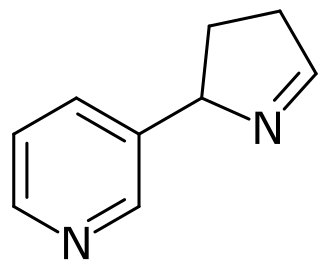
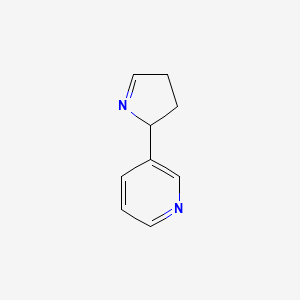
Isomiosamine
CAS 53844-46-5
MF C9H10N2 MW
146.19 g/mol
3-(3,4-dihydro-2H-pyrrol-2-yl)pyridine
rac-(3R)-3-(3,4-dihydro-2H-pyrrol-2-yl)pyridine
tumor necrosis factor alpha (TNFα) inhibitor, MyMD-1, MYMD-1, Isomyosamine, 3A50Y1J4LP, MyMD Pharmaceuticals
synthetic derivative of tobacco alkaloids
Isomyosamine, also known as MyMD-1 or MYMD-1, is a synthetic derivative of tobacco plant alkaloids being developed as a metabolic- and immunomodulator by MyMD Pharmaceuticals. To date, isomyosamine has been shown to suppress the production of IFN-γ, IL-2, IL-10, and TNF-α, and decrease the severity of experimental thyroiditis in a murine model.[1] Trials in humans are being planned, and some are underway, examining the potential benefits of isomyosamine in autoimmune diseases such as rheumatoid arthritis, and in sarcopenia and frailty.[2]
MyMD Pharmaceuticals claim that MYMD-1 is not immunosuppressive, and thus should not be associated with the dangerous side effects such as infections that are seen in currently used TNF-α inhibitors such as adalimumab.[3] While it is true that there currently is no evidence of immunosuppression in isomyosamine recipients, this has not yet been tested in large clinical trials
Safety and Efficacy of Isomyosamine in Reducing Inflammation and Treating Muscle Loss in Older Adults After Hip or Thigh Bone Fractures
CTID: NCT06942182
Phase: Phase 2
Status: Not yet recruiting
Date: 2025-04-24
SYN
Isomyosmine
[08] Isomyosmine (3-(3,4-dihydro-2H-pyrrol-2-yl)-pyridine) is a nicotine related alkaloid present in solanecea plants containing nicotine.
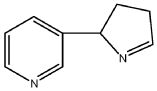
PAT
| Unless otherwise clear from context, all percentages referred to herein are expressed as percent by weight based on the total weight of the composition. Percentages expressed herein as “w/v” refer to mass, in grams, of the component per 100 ml of solvent. For example, a 1% (w/v) composition of isomyosmine contains lg (1000 mg) of isomyosmine per 100 ml of solvent, which is equivalent to 10 mg/ml. |
| Isomyosmine (3-(3,4-dihydro-2H-pyrrol-2-yl)-pyridine) is a nicotine related alkaloid present in solanecea plants containing nicotine. |
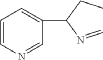
PAT
- Method of Treating Substance AddictionsPublication Number: US-2017333415-A1Priority Date: 2015-02-19
- Method of treating addictions to opioidsPublication Number: US-10471052-B2Priority Date: 2015-02-19Grant Date: 2019-11-12
- Method of treating cocaine addictionPublication Number: US-11331310-B2Priority Date: 2015-02-19Grant Date: 2022-05-17
- Method of treating disorders associated with chronic inflammationPublication Number: US-2021106578-A1Priority Date: 2015-03-31
- Method of Treating Substance AddictionsPublication Number: US-2020215045-A1Priority Date: 2015-02-19
- Compositions for e-cigarettesPublication Number: WO-2016133890-A1Priority Date: 2015-02-19
- Method of treating substance addictionsPublication Number: US-9884055-B2Priority Date: 2015-02-19Grant Date: 2018-02-06
- Method of Treating Substance AddictionsPublication Number: US-2018140590-A1Priority Date: 2015-02-19
- Methods of treating sarcopeniaPublication Number: US-11219620-B2Priority Date: 2015-03-31Grant Date: 2022-01-11
- Methods of Treating Apoptosis and Altering Programmed Cell DeathPublication Number: US-2018021321-A1Priority Date: 2015-03-31
- Methods of reversing normal aging process and extending lifespanPublication Number: US-11179382-B2Priority Date: 2015-03-31Grant Date: 2021-11-23
- Method of treating viral infectionsPublication Number: US-10786493-B2Priority Date: 2015-03-31Grant Date: 2020-09-29
- Methods of treating cancer, autoimmune disorders, and other conditions associated with chronic inflammationPublication Number: CN-107666907-BPriority Date: 2015-03-31Grant Date: 2022-05-13
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Scientific studies
Preclinical studies
One preliminary murine study comparing isomyosamine to rapamycin, the best-characterised drug slowing the progression of aging, reported an increase in lifespan in the isomyosamine cohort, indicating anti-aging activity. Isomyosamine’s anti-proliferative effects were similar to those of rapamycin.[4]
Clinical trials
A phase I randomised double-blind placebo-controlled trial on healthy volunteers examining the safety and pharmacokinetic properties of different amounts of isomyosamine found no serious adverse events, but 3 cases of mild dysgeusia in the highest-dose (600 mg) cohort. A preliminary decrease in TNF-α levels was reported in the lowest-dose (150 mg) cohort, but not in the placebo cohort.[5]
| Identifiers | |
|---|---|
| CAS Number | 53844-46-5 |
| 3D model (JSmol) | Interactive image |
| ChemSpider | 9461533 |
| PubChem CID | 11286546 |
| UNII | 3A50Y1J4LP |
| CompTox Dashboard (EPA) | DTXSID80461155 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C9H10N2 |
| Molar mass | 146.193 g·mol−1 |
| Related compounds | |
| Related compounds | Myosmine Nicotine |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).Infobox references | |
References
- Di Dalmazi, Giulia; Chalan, Paulina; Caturegli, Patrizio (2019-03-01). “MYMD-1, a Novel Immunometabolic Regulator, Ameliorates Autoimmune Thyroiditis via Suppression of Th1 Responses and TNF-α Release”. The Journal of Immunology. 202 (5): 1350–1362. doi:10.4049/jimmunol.1801238. ISSN 0022-1767. PMID 30674573. S2CID 59226562.
- “MyMD Pharmaceuticals® Provides Dosing Update on Phase 2 Multi-Center Clinical Trial of MYMD-1® as a Therapy for Delaying Aging and Extending Healthy Lifespan”. MyMD. Retrieved 2023-08-13.
- “MYMD-1®”. MyMD. Retrieved 2023-08-13.
- Sabini, Elena; O’Mahony, Alison; Caturegli, Patrizio (2023-02-24). Anderson, Rozalyn M (ed.). “MyMD-1 Improves Health Span and Prolongs Life Span in Old Mice: A Noninferiority Study to Rapamycin”. The Journals of Gerontology: Series A. 78 (2): 227–235. doi:10.1093/gerona/glac142. ISSN 1079-5006. PMID 35914953.
- Brager, Jenna; Chapman, Chris; Dunn, Leonard; Kaplin, Adam (2022-11-11). “A Double-blind, Placebo-controlled, Randomized, Single Ascending, and Multiple Dose Phase 1 Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of Oral Dose Isomyosamine Capsules in Healthy Adult Subjects”. Drug Research. 73 (2): 95–104. doi:10.1055/a-1962-6834. ISSN 2194-9379. PMC 9902179. PMID 36368677.
/////////////isomiosamine, tumor necrosis factor alpha (TNFα) inhibitor, MyMD-1, MYMD-1, Isomyosamine, 3A50Y1J4LP, MyMD Pharmaceuticals, ANAX, ADVECT, BLUE JET
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com/
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
ADVERTISEMENT
BLUE JET HEALTHCARE LTD, https://bluejethealthcare.com
Looking for a Reliable SNAC Manufacturer? Let’s Talk.
At Blue Jet Healthcare Ltd, we specialize in the scalable, high-purity production of SNAC—a critical excipient powering the next generation of oral peptide therapeutics.
With increasing demand for SNAC across global pharma pipelines, choosing the right manufacturing partner is essential. Quality, timelines, and consistency matter.

Phone No. +91 (22) 22075307 / +91 (22) 22071691
Business Development/ Contract Manufacturing: marketing1@bluejethealthcare.com, madhu.gautam71@gmail.com
Ismidenon
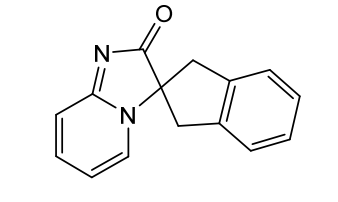
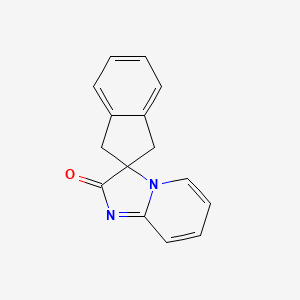
Ismidenon
CAS 887603-94-3
MF C15H12N2O MW236.27 g/mol
spiro(imidazo-(1,2-a)pyridine-3,2-indan)-2(3H)-one
1′,3′-dihydro-2H-spiro[imidazo[1,2-a]pyridine-3,2′-inden]-2-one
voltage-dependent T-type calcium channel modulator, ZSET 1446, ST 101, 7TTT61784C
ST-101 is a small peptide antagonist of C/EBPβ and T-type calcium channel activator. ST101 has been used in trials studying the treatment of Essential Tremor and Alzheimer’s Disease.
Ismidenon is an orally bioavailable azaindolizinone derivative and acetylcholine releasing agent, with potential neurocognitive-enhancing activity. Upon oral administration, ismidenon increases acetylcholine release in the central nervous system (CNS), thereby enhancing cholinergic neurotransmission. Ismidenon also decreases amyloid beta peptide production. This may improve neurocognitive function in Alzheimer’s disease.
- Evaluating the Pharmacokinetic Characteristics of AD-101 in Healthy VolunteersCTID: NCT03764462Phase: Phase 1Status: CompletedDate: 2019-08-30
- A Pilot Efficacy and Safety Study of ST101 in Essential TremorCTID: NCT01332695Phase: Phase 2Status: CompletedDate: 2012-01-06
- A Phase 1-2 Study of ST101 in Patients With Advanced Solid TumorsCTID: NCT04478279Phase: Phase 1/Phase 2Status: Active, not recruitingDate: 2025-05-11
- Preliminary Efficacy and Safety Study of ST101 in Alzheimer’s DiseaseCTID: NCT00842673Phase: Phase 2Status: CompletedDate: 2012-06-07
- Preliminary Efficacy and Safety Study of ST101 Plus Aricept in Alzheimer’s DiseaseCTID: NCT00842816Phase: Phase 2Status: CompletedDate: 2012-06-07
SYN
PAT
[ 0149 ]
spiro[imidazo[l,2-a]pyridin-2(3H)-one-3,2′-indan] (Compound 24)
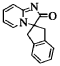
[ 0150 ]
Melting Point: 206 0C (decomposition);
NMR (CDCh) δ: 3.16 (2H, d, J=16Hz), 3.89 (2H, d, J=16Hz), 6.49 (IH, t, J=7Hz), 7.1-7.2
(2H, m), 7.2-7.3 (4H, m), 7.61 (IH, t, J=7Hz);
MS m/z: 236 (M+).
PAT
- Heterocyclic compounds and cerebral function improvers containing the same as the active ingredientPublication Number: US-7767824-B2Priority Date: 2001-01-30Grant Date: 2010-08-03
- Azaindolizinone derivatives and cognitive enhancers comprising the same as effective componentsPublication Number: EP-1219621-B1Priority Date: 1999-07-30Grant Date: 2003-10-22
- Azaindolizinone derivatives and cerebral function improvers containing the same as the active ingredientPublication Number: US-6635652-B1Priority Date: 1999-07-30Grant Date: 2003-10-21
- Azaindolizinone derivatives and cerebral function improvers containing the same as the active ingredientPublication Number: EP-1219621-A1Priority Date: 1999-07-30
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////ismidenon, voltage-dependent T-type calcium channel modulator, ZSET 1446, ST 101, 7TTT61784C
Copper histidinate
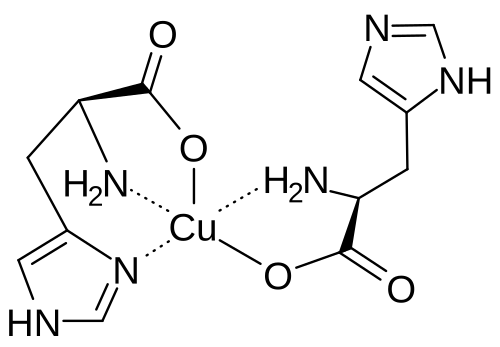
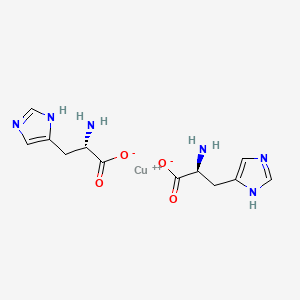
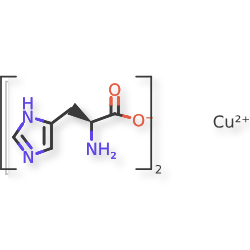
Copper histidinate
CAS 12561-67-0 AND 13870-80-9
MF C12H16CuN6O4
FDA 2026, JAN/12/26, Zycubo, To treat Menkes disease, APPROVALS 2026, 9078K3MO9U, MN 88, CUTX 101
copper bis((2S)-2-amino-3-(1H-imidazol-5-yl)propanoate)
Copper histidinate, sold under the brand name Zycubo, is a medication used for the treatment of Menkes disease.[1] Copper histidinate is a copper replacement therapy given by subcutaneous injection.[1][2]
The most common side effects include infections, respiratory problems, seizures, vomiting, fever, anemia and injection site reactions.[2]
Copper histidinate was approved for medical use in the United States in January 2026.[2]
Medical uses
Copper histidinate is indicated for the treatment of Menkes disease in children.[1]
Menkes disease is a neurodegenerative disorder caused by a genetic defect that impairs a child’s ability to absorb copper.[2] The disease is characterized by seizures, failure to gain weight and grow, developmental delays, and intellectual disability.[2] It leads to abnormalities of the vascular system, bladder, bowel, bones, muscles, and nervous system.[2]
SYN
A275388 — Flores-Pulido AA, Jimenez-Perez VM, Garcia-Chong NR: Sintesis y uso de histidinato de cobre en ninos con enfermedad de Menkes en Mexico. Gac Med Mex. 2019;155(2):191-195. doi: 10.24875/GMM.18004310. [PubMed:31056589]
PAT
PAT
Publication Number: US-5576326-A
Priority Date: 1989-12-20
Grant Date: 1996-11-19
- Wilson disease genePublication Number: CA-2108927-CPriority Date: 1993-09-21Grant Date: 2008-09-02
- Wilson disease genePublication Number: CA-2106602-A1Priority Date: 1993-09-21
- Wilson disease genePublication Number: WO-9508641-A1Priority Date: 1993-09-21
- Process to obtain new mixed copper aminoacidate complexes from phenanthrolines and their alkyl derivatives to be used as anticancerigenic agentsPublication Number: EP-0434445-A2Priority Date: 1989-12-20
- Method for producing a novel mixed copper amino acid complex used in anticancer drugsPublication Number: JP-H04316581-APriority Date: 1989-12-20METHOD FOR DETECTION OF BIOLOGICAL AGENTSPublication Number: EP-1097242-A4Priority Date: 1999-05-05
- Method for detecting biological agentsPublication Number: US-2004023272-A1Priority Date: 1999-05-05
- Method for detecting biological agentsPublication Number: WO-0066790-A1Priority Date: 1999-05-05
- Method for detecting biological agentsPublication Number: EP-1097242-A1Priority Date: 1999-05-05
- Process for producing nitrosonium ionsPublication Number: AU-773835-B2Priority Date: 1999-02-24Grant Date: 2004-06-10
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Side effects
The most common side effects include infections, respiratory problems, seizures, vomiting, fever, anemia and injection site reactions.[2]
Society and culture
Legal status
Copper histidinate was approved for medical use in the United States in January 2026.[2] The US Food and Drug Administration (FDA) granted the application for copper histidinate priority review, fast track, breakthrough therapy, and orphan drug designations.[2] The FDA granted approval of Zycubo to Sentynl Therapeutics.[2]
Names
Copper histidinate is the international nonproprietary name[3] and the United States Adopted Name.[4]
Copper histidinate is sold under the brand name Zycubo.[5]
References
- Sentynl Therapeutics (12 January 2026). “Zycubo (copper histidinate) for injection, for subcutaneous use” (PDF). Retrieved 15 January 2026.
- “FDA Approves First Treatment for Children With Menkes Disease”. U.S. Food and Drug Administration (FDA) (Press release). 12 January 2026. Retrieved 15 January 2026.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - World Health Organization (2025). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 94”. WHO Drug Information. 39 (3). hdl:10665/383022.
- “Copper histidinate”. American Medical Association. Retrieved 15 January 2026.
- “Sentynl Therapeutics Inc. Announces FDA Approval of Zycubo (copper histidinate)”. Sentynl Therapeutics. 13 January 2026. Retrieved 15 January 2026 – via PR Newswire.
Further reading
- Scanga R, Scalise M, Marino N, Parisi F, Barca D, Galluccio M, et al. (October 2023). “LAT1 (SLC7A5) catalyzes copper(histidinate) transport switching from antiport to uniport mechanism”. iScience. 26 (10) 107738. Bibcode:2023iSci…26j7738S. doi:10.1016/j.isci.2023.107738. PMC 10492218. PMID 37692288.
External links
- Clinical trial number NCT00001262 for “Copper Histidine Therapy for Menkes Diseases” at ClinicalTrials.gov
- Clinical trial number NCT00811785 for “Molecular Bases of Response to Copper Treatment in Menkes Disease, Related Phenotypes, and Unexplained Copper Deficiency” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zycubo |
| Other names | Copper(II) bis(histidinate) |
| AHFS/Drugs.com | zycubo |
| License data | US DailyMed: Copper histidinate |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 13870-80-9 |
| PubChem CID | 151722 |
| DrugBank | DB32041 |
| ChemSpider | 133722 |
| UNII | 9078K3MO9U |
| KEGG | D13117 |
| CompTox Dashboard (EPA) | DTXSID30154803 |
| Chemical and physical data | |
| Formula | C12H16CuN6O4 |
| Molar mass | 371.844 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Copper histidinate, FDA 2026, JAN/12/26, Zycubo, To treat Menkes disease, APPROVALS 2026,
9078K3MO9U, 9078K3MO9U, MN 88, CUTX 101
Idrebormilast
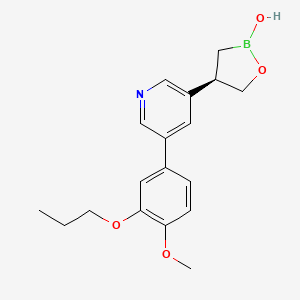
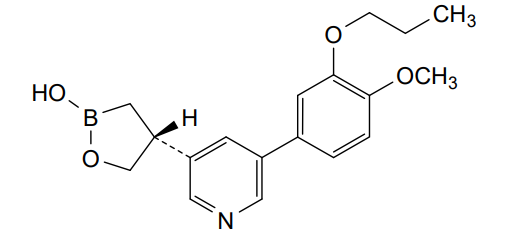
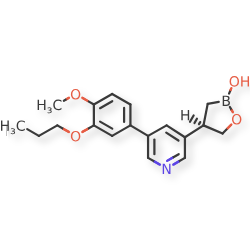
Idrebormilast
CAS 2415085-44-6
MF C18H22BNO4, MW 327.18
Pyridine, 3-[(4R)-2-hydroxy-1,2-oxaborolan-4-yl]-5-(4-methoxy-3-propoxyphenyl)-
(4R)-4-[5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl]-1,2-oxaborolan-2-ol
phosphodiesterase 4 (PDE4) inhibitor, non-steroidal anti-inflammatory, M6ZU548FWD, PF07038124, PF 07038124
PF-07038124 is under investigation in clinical trial NCT05298033 (Study of Efficacy, Safety and Tolerability of Crisaborole and PF-07038124 With and Without NBUVB in Vitiligo).
IDREBORMILAST is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 2 investigational indications.
- Study of Efficacy, Safety and Tolerability of Crisaborole and PF-07038124 With and Without NBUVB in VitiligoCTID: NCT05298033Phase: Phase 2Status: CompletedDate: 2024-06-12
- PDE4 Inhibition in Seborrheic Dermatitis and Papulopustular RosaceaCTID: NCT06013371Phase: Phase 2Status: TerminatedDate: 2025-04-24
- A Study To Determine The Safety, Tolerability, Skin Irritation Potential, And PK Following Topical Application Of PF-07038124 In Healthy ParticipantsCTID: NCT04135560Phase: Phase 1Status: CompletedDate: 2020-05-14
- Study to Evaluate the Safety, Local and Systemic Tolerability, and Pharmacokinetics of Multiple-Dose Topical Administration of PF-07038124 in Japanese Healthy ParticipantsCTID: NCT04863417Phase: Phase 1Status: CompletedDate: 2024-01-25
- Study To Assess Efficacy, Safety, Tolerability And Pharmacokinetics Of PF-07038124 Ointment In Participants With Atopic Dermatitis Or Plaque PsoriasisCTID: NCT04664153Phase: Phase 2Status: CompletedDate: 2022-08-26
SYN
PAT
Example 4: (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol
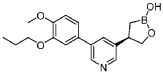
Method A:
To a mixture of (R)-(3-((tert-butyldimethylsilyl)oxy)-2-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)propyl)boronic acid (Preparation 6, 55 g, 120 mmol) in IPA (247 mL) was added 5 M hydrogen chloride in IPA (37 mL, 185 mmol) at about 20 °C. The mixture was stirred for about 3 h and concentrated. The residue was diluted with EtOAc (500 mL) and 1 N
HCI (500 mL) was added. The layers were separated and the EtOAc layer was extracted with 0.5 N HCI (2 x 200 mL). The aqueous extracts were combined with the separated acidic aqueous layer and washed with EtOAc (3 x 250 mL). The combined acidic aqueous layers were treated with K3PO4 to pH 5-6. The mixture was extracted with EtOAc (1 x 500 mL, 2 x 200 mL). The combined EtOAc extracts were washed with brine, dried over Na2S04, filtered and concentrated to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol (34.5 g, 88%). This was further purified by preparative SFC (Prep SFC Method C) to afford 29 g as a crude product. The crude product was dissolved in methanol (250 mL) and water (50 mL) and stirred at 20 °C for about 30 min before concentrating. The concentrated solution was partitioned between brine and EtOAc. The aqueous layer was separated and extracted with EtOAc. The EtOAc extracts were combined with the separate EtOAc layer and were washed with brine, dried over Na2S04 and concentrated. The residue was dissolved in degassed EtOAc (200 mL) and degassed heptane (100 mL) was added slowly. Heptane was added until a precipitate was observed and and the resulting mixture was stirred overnight under N2. The solid was filtered to afford 8.08 g of product. The filtrate was concentrated and the residue dissolved in EtOAc (50 mL). Heptane (25 mL) was slowly added and the mixture stirred overnight open to air. The solid was filtered to afford a second batch (6.16 g). This was repeated a second time to afford 3.0 g. The filtrate was stirred overnight to afford additional batches (2.09 g and 3.1 g) respectively. The solid batches were combined to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol (22.3 g, 57%) as a crystalline solid. Ή NMR (DMSO-cfe, 400MHz): d 8.70 (d, J = 2.3 Hz, 1 H), 8.68 (s, 1 H), 8.42 (d, J = 2.0 Hz, 1 H), 7.93 (s, 1 H), 7.27 (d, J = 2.0 Hz, 1 H), 7.23-7.26 (m, 1 H), 7.06 (d, J = 8.2 Hz, 1 H), 4.28 (t, J = 8.2 Hz, 1 H), 4.03 (t, J = 6.4 Hz, 2H), 3.86 (t, J = 9.0 Hz, 1 H), 3.81 (s, 3H), 3.46-3.54 (m, 1 H), 1 .71 -1 .80 (m, 2H), 1 .28-1 .35 (m, 1 H), 1 .15 (dd, J = 10.5, 16.4 Hz, 1 H), 1 .00 (t, J = 7.4 Hz, 3H). LCMS m/z = 328 [MH]+; RT [Analytical SFC Method B] = 7.30 min. [a]20D -23.7 (c = 0.9, EtOH).
Elemental analysis calculated (%) for Ci8H22BN04: C 66.08, H 6.78, N 4.28. Found: C 65.86, H 6.59, N 4.18.
Method B:
Step 1 : To THF (18.0 mL) was added 3-(3-((tert-butyldimethylsilyl)oxy)prop-1 -en-2-yl)-5-(4-methoxy-3-propoxyphenyl)pyridine (Preparation 50, 3.0 g, 7.25 mmol), [lr(COD)CI]2 (CAS 121 12-67-3, 36.9 mg, 0.054 mmol) and (S)[(Sp)-2-(diphenylphosphino)ferrocenyl]-4-isopropyloxazoline (CAS 163169-29-7, 52.4 mg, 0.109 mmol). Additional THF (6.0 mL) was added to the mixture which was warmed to about 50 °C for about 5 min. Catecholborane (10.9 mL, 1 0M in THF) was added to the mixture and stirred at about 50 °C for about 1 h. The mixture was cooled to about 20 °C and treated with HCI (12.2 M, 1 .51 mL) over 1 min. The mixture was held at about 20 °C for about 1 h, afterwhich a precipitate had formed. The mixture was cooled to about 10 °C and filtered. The filtered solid was washed with THF (6.0 mL) and
dried overnight at 35°C under vacuum to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridine-3-yl)-1 ,2-oxaborolan-2-ol hydrochloride monohydrate (3.98 g, 91 %) as a crystalline solid. 1H NMR (CD3OD, 400MHz): d 8.98 (d, J = 1 .5 Hz, 1 H), 8.75 (s, 1 H), 8.67 (d, J = 1 .3 Hz, 1 H), 7.37-7.43 (m, 2H), 7.15 (d, J = 8.3 Hz, 1 H), 4.09 (t, J = 6.5 Hz, 2H), 3.89-3.92 (m, 1 H), 3.86-3.95 (m, 5H), 3.46 (br s, 1 H), 1 .85 (m, 2H), 1 .31 -1 .42 (m, 2H), 1 .08 (t, J = 7.4 Hz, 3H). LCMS m/z = 328 [MH]+.
Step 2: To a solution of (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridine-3-yl)-1 ,2-oxaborolan-2-ol hydrochloride monohydrate (2.0 g, 5.24 mmol) in water (60 ml_) was added EtOAc (20 ml_). To the stirred mixture was added NaOH (1 N) dropwise to adjust the pH of the aqeous layer to 7-8. The mixture was stirred at about 20 °C for about 5 min. The layers were separated and the aqueous layer was extracted with EtOAc (2 x 10 ml_). The combined EtOAc extracts were concentrated. The residue was dissolved in THF/MTBE (1 :3, 22 ml_) and stirred at about 20 °C overnight. The precipitate was filtered and dried under vacuum to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridine-3-yl)-1 ,2-oxaborolan-2-ol (1 .17 g, 68%) as a crystalline solid. Ή NMR (DMSO-cfe, 400MHz): d 8.70 (d, J = 2.3 Hz, 1 H), 8.68 (s, 1 H), 8.42 (d, J = 2.0 Hz, 1 H), 7.93 (s, 1 H), 7.27 (d, J = 2.0 Hz, 1 H), 7.23-7.26 (m, 1 H), 7.06 (d, J = 8.2 Hz, 1 H), 4.28 (t, J = 8.2 Hz, 1 H), 4.03 (t, J = 6.4 Hz, 2H), 3.86 (t, J = 9.0 Hz, 1 H), 3.81 (s, 3H), 3.46-3.54 (m, 1 H), 1 .71 -1 .80 (m, 2H), 1 .28-1 .35 (m, 1 H), 1 .15 (dd, J = 10.5, 16.4 Hz, 1 H), 1 .00 (t, J = 7.4 Hz, 3H). LCMS m/z = 328 [MH]+.
Method C:
To a solution of (R)-(3-((tert-butyldimethylsilyl)oxy)-2-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)propyl)boronic acid (Preparation 6, 29.0 g, 63.1 mmol) in THF (66 mL) was added aqueous HCI (84.2 mL, 252 mmol, 3.0 M) and stirred at 20 °C for about 1 .5 h. The mixture was concentrated. The mixture was diluted with 1 M HCI and extracted with EtOAc (3 x 100 mL). The combined EtOAc extracts were washed with 1 M HCI (3 x 50 mL). The combined aqueous extracts were neutralized with K3PO4 to pH 7-8 and extracted with EtOAc (3 x 100 mL). The combined EtOAc extracts were dried over Na2S04, filtered and concentrated to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol (19.0 g, 92%).
This was further purified by preparative SFC (Prep SFC Method C) to afford 18 g of the crude product. The crude product was dissolved in MeOH (100 mL) and water (50 mL). The mixture was partitioned between brine and EtOAc. The layers were separated and the aqueous layer was extracted with EtOAc. The combined EtOAc extracts were washed with brine, dried over Na2S04 and concentrated to afford 15 g of product. The residue was dissolved in EtOAc (60 mL) and heptane (30 mL) was slowly added over about 3 h. The mixture was stirred at about 20 °C overnight. The precipitate was filtered and dried to afford (8.08 g). This process was repeated 2 more times to afford additional batches (2.01 g and 1 .03 g), respectively. The three batches were combined in heptane (100 mL), chilled to about -78 °C for about 10 min, filtered
and dried to afford (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1 ,2-oxaborolan-2-ol (10.4 g, 51 %) as a crystalline solid. Ή NMR (DMSO -d6, 400MHz): d 8.70 (d, J = 2.3 Hz, 1 H), 8.68 (s,
1 H), 8.42 (d, J = 2.0 Hz, 1 H), 7.93 (s, 1 H), 7.27 (d, J = 2.0 Hz, 1 H), 7.23-7.26 (m, 1 H), 7.06 (d, J = 8.2 Hz, 1 H), 4.28 (t, J = 8.2 Hz, 1 H), 4.03 (t, J = 6.4 Hz, 2H), 3.86 (t, J = 9.0 Hz, 1 H), 3.81 (s, 3H), 3.46-3.54 (m, 1 H), 1 .71 -1 .80 (m, 2H), 1 .28-1 .35 (m, 1 H), 1 .15 (dd, J = 10.5, 16.4 Hz, 1 H),
1 .00 (t, J = 7.4 Hz, 3H). LCMS m/z = 328 [MH]+.
PAT
- Substituted 1,2-oxaborolan-2-ols as PDE4 inhibitorsPublication Number: US-11559538-B2Priority Date: 2018-10-05Grant Date: 2023-01-24
- Boron-containing PDE4 inhibitorsPublication Number: CN-113166177-BPriority Date: 2018-10-05Grant Date: 2024-09-03
- Boron Containing PDE4 InhibitorsPublication Number: US-2021069219-A1Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: CA-3114702-CPriority Date: 2018-10-05Grant Date: 2023-08-08
- PDE4 INHIBITORS CONTAINING BORONPublication Number: BR-112021005870-B1Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: WO-2020070651-A1Priority Date: 2018-10-05
- Boron-containing PDE4 inhibitorsPublication Number: CN-113166177-APriority Date: 2018-10-05
- Boron Containing PDE4 InhibitorsPublication Number: US-2023148402-A1Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: EP-3861001-A1Priority Date: 2018-10-05
- Boron-containing PDE4 inhibitorsPublication Number: ES-2974208-T3Priority Date: 2018-10-05Grant Date: 2024-06-26
- PDE4 inhibitor (R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: US-10946031-B2Priority Date: 2018-10-05Grant Date: 2021-03-16
- Boron-containing PDE4 inhibitorsPublication Number: KR-102576125-B1Priority Date: 2018-10-05Grant Date: 2023-09-08
- Boron-containing PDE4 inhibitorsPublication Number: KR-20210068532-APriority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: EP-3861001-B1Priority Date: 2018-10-05Grant Date: 2023-12-13
- Boron Containing PDE4 InhibitorsPublication Number: US-2020108083-A1Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: CA-3114702-A1Priority Date: 2018-10-05
- boron containing pde4 inhibitorsPublication Number: BR-112021005870-A2Priority Date: 2018-10-05
- Boron containing pde4 inhibitorsPublication Number: TW-202027755-APriority Date: 2018-10-05
- Stable topical formulations of 1(r)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: CA-3191886-A1Priority Date: 2020-08-20
- STABLE TOPICAL FORMULATIONS OF 1(R)-4-(5-(4-METHOXY-3-PROPOXYPHEN IL)PYRIDIN-3-IL)-1,2-OXABOROLAN-2-OL.Publication Number: MX-2023002086-APriority Date: 2020-08-20
- Stable topical formulations of 1(r)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: WO-2022038485-A1Priority Date: 2020-08-20
- 1(R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-ol is a stable partial preparationPublication Number: CN-115916260-APriority Date: 2020-08-20
- Stable Topical Formulations of 1(R)-4-(5-(4-Methoxy-3-Propoxphenyl)Pyridin-3-YL)-1,2-OX-Aborolan-2-OLPublication Number: US-2023310472-A1Priority Date: 2020-08-20
- Combination therapyPublication Number: WO-2024231312-A2Priority Date: 2023-05-05
- Borate derivative and uses thereofPublication Number: EP-4269418-A1Priority Date: 2020-12-25
- Stable topical formulations of 1(r)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: EP-4199902-A1Priority Date: 2020-08-20
- Stable topical formulation of 1(R)-4-(5-(4-methoxy-3-propoxyphenyl)pyridin-3-yl)-1,2-oxaborolan-2-olPublication Number: JP-2023538362-APriority Date: 2020-08-20
- STABLE TOPICAL FORMULATIONS OF 1(R)-4-(5-(4-METHOXY-3-PROPOXYPHENYL)PYRIDIN-3-IL)-1,2-OXABOROLAN-2-OLPublication Number: BR-112023002533-A2Priority Date: 2020-08-20
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////////idrebormilast, phosphodiesterase 4 (PDE4) inhibitor, non-steroidal anti-inflammatory, M6ZU548FWD, PF07038124, PF 07038124
Gridegalutamide
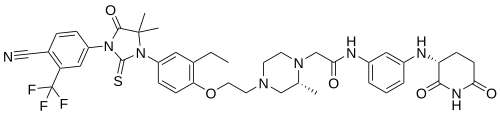
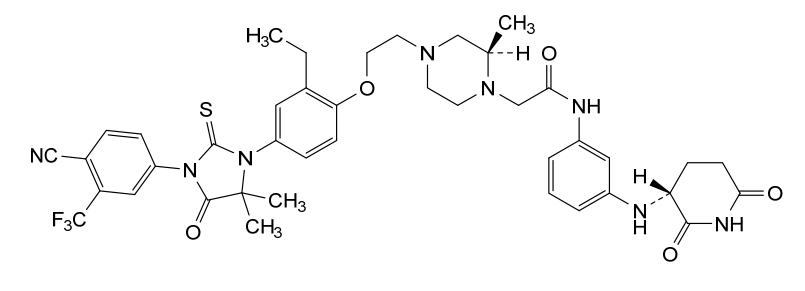
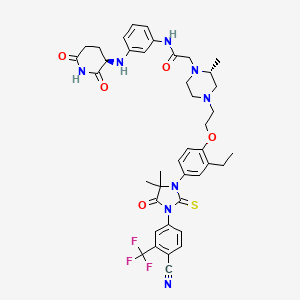
Gridegalutamide
CAS 2446929-86-6
MF C41H45F3N8O5S MW818.9 g/mol
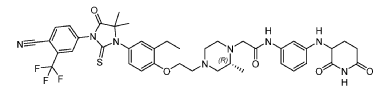
2-[(2R)-4-[2-[4-[3-[4-cyano-3-(trifluoromethyl)phenyl]-5,5-dimethyl-4-oxo-2-sulfanylideneimidazolidin-1-yl]-2-ethylphenoxy]ethyl]-2-methylpiperazin-1-yl]-N-[3-[[(3R)-2,6-dioxopiperidin-3-yl]amino]phenyl]acetamide

antiandrogen, antineoplastic, BMS 986365, CC 94676, BMS-986365, CC-94676, CEL 010355,
VA228VR2DI,
Gridegalutamide is an investigational oral androgen receptor (AR) degrader being developed for the treatment of metastatic castration-resistant prostate cancer (mCRPC). It belongs to a class of drugs called proteolysis targeting chimeras (PROTACs), which are designed to selectively degrade specific proteins by hijacking the ubiquitin-proteasome system.[1][2] CC-94676 employs a unique dual mechanism of action, combining AR degradation with AR antagonism, potentially offering advantages over traditional AR inhibitors in overcoming resistance mechanisms.[3] Initially developed by Celgene and now under Bristol Myers Squibb, CC-94676 has demonstrated AR protein degradation and suppression of tumor growth in CRPC mouse models.[2] As of 2024, CC-94676 is being evaluated in phase I clinical trials for patients with mCRPC who have progressed on androgen deprivation therapy and at least one prior secondary hormonal therapy.[1][2]
Gridegalutamide is a small molecule drug. The usage of the INN stem ‘-lutamide’ in the name indicates that Gridegalutamide is a non-steroid antiandrogen. Gridegalutamide is under investigation in clinical trial NCT04428788 (Study to Evaluate the Safety and Tolerability of CC-94676 in Participants With Metastatic Castration-Resistant Prostate Cancer). Gridegalutamide has a monoisotopic molecular weight of 818.32 Da.
GRIDEGALUTAMIDE is a small molecule drug with a maximum clinical trial phase of II (across all indications) and has 3 investigational indications.
Gridegalutamide is an orally bioavailable androgen receptor (AR) degrader, with potential antineoplastic activity. Upon administration, gridegalutamide causes degradation of AR, prevents AR-mediated signaling and inhibits the proliferation of AR-overexpressing tumor cells. AR plays a key role in tumor cell proliferation in castration-resistant prostate cancer (CRPC).
- A Study to Evaluate the Drug Levels, Metabolism and Excretion, and Absolute Bioavailability of BMS-986365 in Healthy Male ParticipantsCTID: NCT06433505Phase: Phase 1Status: CompletedDate: 2025-03-26
- Study to Evaluate the Safety and Tolerability of CC-94676 in Participants With Metastatic Castration-Resistant Prostate CancerCTID: NCT04428788Phase: Phase 1Status: CompletedDate: 2025-12-22
SYN
DRUGHUNTER
https://drughunter.com/molecule/gridegalutamide-bms-986365-cc-94676
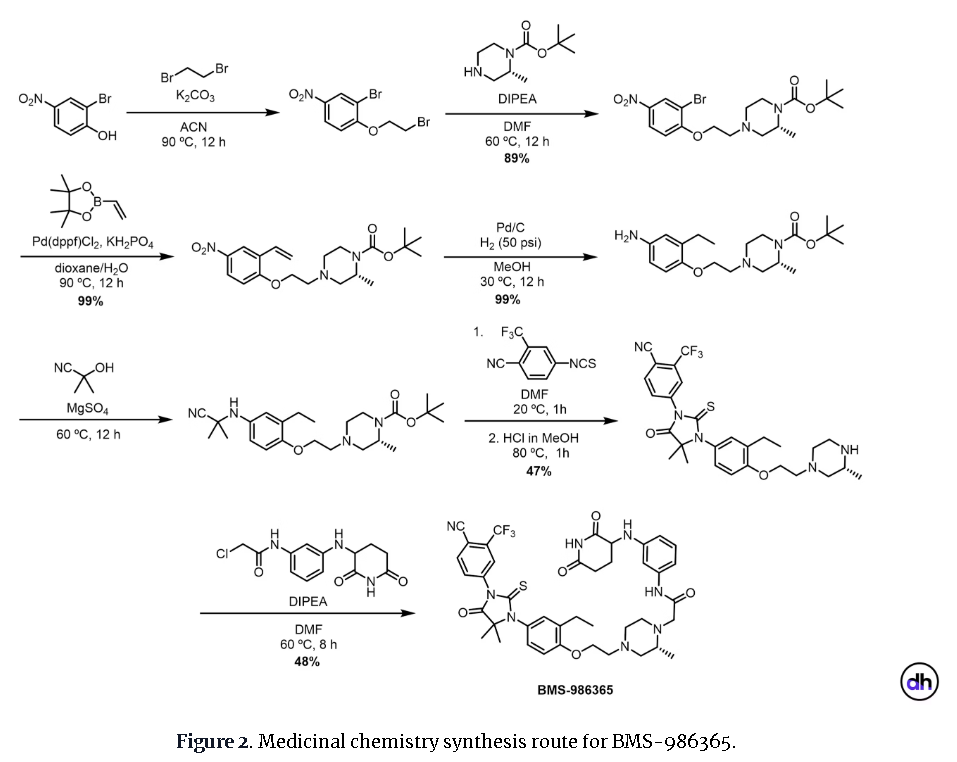
PAT
Example 17: 2-((R)-4-(2-(4-(3-(4-Cyano-3-(trifluoromethyl)phenyl)-5,5-dimethyl-4-oxo-2- thioxoimidazolidin-1-yl)-2-ethylphenoxy)ethyl)-2-methylpiperazin-1-yl)-N-(3-((2,6- dioxopiperidin-3-yl)amino)phenyl)acetamide hydrochloride



PAT
- Combination therapy with substituted 3- ((3-aminophenyl) amino) piperidine-2, 6-dione compoundsPublication Number: CN-120152718-APriority Date: 2022-11-09
- Combination therapy with substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compoundsPublication Number: WO-2024102706-A1Priority Date: 2022-11-09
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-2020199073-A1Priority Date: 2018-12-19
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-11149007-B2Priority Date: 2018-12-19Grant Date: 2021-10-19
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-11873283-B2Priority Date: 2018-12-19Grant Date: 2024-01-16
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-2024368083-A1Priority Date: 2018-12-19
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-12404241-B2Priority Date: 2018-12-19Grant Date: 2025-09-02
- Substituted 3-((3-aminophenyl)amino)piperidine-2,6-dione compounds, compositions thereof, and methods of treatment therewithPublication Number: US-2023002321-A1Priority Date: 2018-12-19
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com

ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
References
- Salama AK, Trkulja MV, Casanova E, Uras IZ (December 2022). “Targeted Protein Degradation: Clinical Advances in the Field of Oncology”. International Journal of Molecular Sciences. 23 (23) 15440. doi:10.3390/ijms232315440. PMC 9741350. PMID 36499765.
- Xie H, Liu J, Alem Glison DM, Fleming JB (2021). “The clinical advances of proteolysis targeting chimeras in oncology”. Exploration of Targeted Anti-Tumor Therapy. 2 (6): 511–521. doi:10.37349/etat.2021.00061. PMC 9400722. PMID 36046114.
- Rathkopf DE, Patel MR, Choudhury AD, Rasco D, Lakhani N, Hawley JE, et al. (September 2024). “Safety and clinical activity of BMS-986365 (CC-94676), a dual androgen receptor ligand-directed degrader and antagonist, in heavily pretreated patients with metastatic castration-resistant prostate cancer”. Annals of Oncology. 36 (1): 76–88. doi:10.1016/j.annonc.2024.09.005. PMC 12094577. PMID 39293515.
| Clinical data | |
|---|---|
| Other names | BMS-986365; CC-94676 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2446929-86-6 |
| PubChem CID | 153513643 |
| ChemSpider | 133326102 |
| UNII | VA228VR2DI |
| KEGG | D12866 |
| ChEMBL | ChEMBL6068413 |
| Chemical and physical data | |
| Formula | C41H45F3N8O5S |
| Molar mass | 818.92 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
//////////gridegalutamide, ANAX, ADVECT, antiandrogen, antineoplastic, BMS 986365, CC 94676, BMS-986365, CC-94676, CEL 010355, VA228VR2DI,
Frespaciguat



Frespaciguat
CAS 2101645-33-2
MF C27H22ClF5N6O3 MW 608.9 g/mol
3-[4-[(5S)-4-amino-2-[6-chloro-1-(3,3,4,4,4-pentafluorobutyl)indazol-3-yl]-5-methyl-6-oxo-7H-pyrrolo[2,3-d]pyrimidin-5-yl]phenyl]propanoic acid
3-{4-[(5S)-4-amino-2-[6-chloro-1-(3,3,4,4,4-pentafluorobutyl)-1H-indazol-3-yl]-5-methyl-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidin-5-yl]phenyl}propanoic acid
guanylate cyclase activator, MK5475, MK 5475, sGC activator 1, 6DXN080KGB
Frespaciguat (development code MK-5475) is an experimental inhaled soluble guanylate cyclase stimulator developed by Merck for pulmonary arterial hypertension.[1][2][3][4]
Frespaciguat is a small molecule drug. The usage of the INN stem ‘-ciguat’ in the name indicates that Frespaciguat is a guanylate cyclase activator and stimulator. Frespaciguat is under investigation in clinical trial NCT05612035 (Frespaciguat (MK-5475) INSIGNIA-PH-COPD: A Study of the Efficacy and Safety of Frespaciguat (an Inhaled sGC Stimulator) in Adults With PH-COPD). Frespaciguat has a monoisotopic molecular weight of 608.14 Da.
- Frespaciguat (MK-5475) in Participants With Pulmonary Hypertension Associated With Chronic Obstructive Pulmonary Disease (PH-COPD) (MK-5475-006)CTID: NCT04370873Phase: Phase 1Status: CompletedDate: 2025-05-28
- A Study of the Efficacy and Safety of Frespaciguat (MK-5475) in Participants With Pulmonary Arterial Hypertension (INSIGNIA-PAH: Phase 2/3 Study of an Inhaled sGC Stimulator in PAH) (MK-5475-007)CTID: NCT04732221Phase: Phase 2/Phase 3Status: CompletedDate: 2025-05-25
- Frespaciguat (MK-5475) in Participants With Hypoxemia Due to COVID-19 Pneumonia (MK-5475-009)CTID: NCT04425733Phase: Phase 1Status: WithdrawnDate: 2025-05-15
- Frespaciguat (MK-5475) INSIGNIA-PH-COPD: A Study of the Efficacy and Safety of Frespaciguat (an Inhaled sGC Stimulator) in Adults With PH-COPDCTID: NCT05612035Phase: Phase 2Status: Active, not recruitingDate: 2025-10-07
- A Study of Single Doses of Frespaciguat (MK-5475) on Pulmonary Vascular Resistance (MK-5475-002)CTID: NCT03744637Phase: Phase 1Status: CompletedDate: 2025-06-04
SYN
US10030027,
https://patentscope.wipo.int/search/en/detail.jsf?docId=US199416000&_cid=P12-MKUJSJ-89968-1
Example 10B
(S)-3-(4-{4-Amino-2-[6-chloro-1-(3,3,4,4,4-pentafluorobutyl)-1H-indazol-3-yl]-5-methyl-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidin-5-yl}phenyl)propanoic acid

Step A—(S)-Methyl 3-(4-{4-Amino-2-[6-chloro-1-(3,3,4,4,4-pentafluorobutyl)-1H-indazol-3-yl]-5-methyl-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidin-5-yl}phenyl)propanoate
Step B—(S)-3-(4-{4-Amino-2-[6-chloro-1-(3,3,4,4,4-pentafluorobutyl)-1H-indazol-3-yl]-5-methyl-6-oxo-6,7-dihydro-5H-pyrrolo[2,3-d]pyrimidin-5-yl}phenyl)propanoic acid
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2025006295&_cid=P12-MKUJNS-84717-1
EXAMPLE 2

[0038] To an autoclave was charged anisole (12.78 L), indazole ester (IV) (2.13 kg, 5.64 mol), and hexamethyldisilazane (4.55 kg, 28.2 mol). The mixture was cooled to 0-5 oC and the vessel was placed under slight positive pressure with no N2 sweeping. A solution of H2O (203.21 g, 11.27 mol) in sulfolane (6.39 L) was added while keeping at < 10 oC in order to minimize NH3 gas escaping. The resulting mixture was cooled to -10 oC, then TfOH (1.692 kg, 11.27 mol) was slowly added at < 22 oC. The vessel was sealed and the mixture was heated at 120-130 °C for 24 h. The upper vessel was kept warm so that solid ammonium triflate did not deposit there.
[0039] After cooling the mixture to rt, the batch (biphasic) was further cooled to 0-10 oC.2.4 equiv 1N KOH (13.53 L, 14.207 kg, 13.53 mol) was slowly added at < 25 oC. After agitating for 30 min, and letting settle at rt, the bottom aqueous layer was removed (pH~14). The organic layer was washed with 18% brine (10.5 L). The aqueous layer was removed (pH ~12). To the organic phase was added ¼ (101 mL, 149 g) of 1.1 equiv methanesulfonic acid (402 mL, 595 g, 6.20 mol), then seeded with 0.2 wt% amidine MSA type A (8.5 g). The rest of MSA (447 g, 302 mL) was then slowly added over 1 h. During MSA addition, the temperature was controlled at < 25 oC. The resulting slurry, after aging at 22 oC for 15 h, was filtered, then displacement washed with 2 x 3 vol 2-MeTHF (2 x 6.4 L), and vacuum dried under N2 at < 30 °C for 24 h. The product (III) was obtained (4.62 kg, 10.58 mol, 93 % yield) as an off-white to light beige solid.
PAT
- 4-amino-2-(1h-pyrazolo[3,4-b]pyridin-3-yl)-6-oxo-6,7-dihydro-5h-pyrrolo[2,3-d]pyrimidine derivatives and the respective (1h-indazol-3-yl) derivatives as cgmp modulators for treating cardiovascular diseasesPublication Number: EP-3394067-B1Priority Date: 2015-12-22Grant Date: 2020-04-01
- Soluble guanylate cyclase stimulatorPublication Number: TW-201734017-APriority Date: 2015-12-22
- 4-Amino-2- (1h-pyrazolo [3,4-b] pyridin-3-yl) -6-oxo-6,7-dihydro-5h-pyrrolo [2,3-d] pyrimidine derivatives and respectively (1h- indazol-3-yl) are derived as cgmp activators for the treatment of cardiovascular diseasePublication Number: IL-260020-APriority Date: 2015-12-22
- Soluble guanylate cyclase stimulatorsPublication Number: TW-I724079-BPriority Date: 2015-12-22Grant Date: 2021-04-11
- cGMP modulators for the treatment of cardiovascular diseasePublication Number: CN-108738320-BPriority Date: 2015-12-22Grant Date: 2021-11-19
- 4-Amino-2-(1H-pyrazolo[3,4-b]pyridin-3-yl)-6-oxo-6,7-dihydro- 5H-pyrrolo[2,3-d]pyrimidine derivatives and their respective (1H-indazol-3-yl) derivativesPublication Number: CN-108738320-APriority Date: 2015-12-22
- 4-amino-2-(1H-pyrazolo[3,4-B]pyridin-3-yl)-6-oxo-6,7-dihydro-5H-pyrrolo[ as a cGMP modulator for the treatment of cardiovascular disease 2,3-D]pyridine derivatives and respective (1H-indazol-3-yl) derivativesPublication Number: KR-102191312-B1Priority Date: 2015-12-22Grant Date: 2020-12-15
- Soluble guanylate cyclase stimulatorsPublication Number: US-10030027-B2Priority Date: 2015-12-22Grant Date: 2018-07-24
- 4-Amino-2- (1H-pyrazolo [3,4-b] pyridin-3-yl) -6-oxo-6,7-dihydro-5H-pyrrolo [2,3-d] pyrimidine derivatives and cardiovascular disease (1H-indazol-3-yl) derivatives as cGMP modulators for treatingPublication Number: JP-2018538333-APriority Date: 2015-12-22
- Soluble guanylate cyclase stimulatorsPublication Number: US-2018305366-A1Priority Date: 2015-12-22
- Processes for preparing crystalline soluble guanylate cyclase stimulatorsPublication Number: WO-2025006294-A2Priority Date: 2023-06-26
- Processes for preparing soluble guanylate cyclase stimulatorsPublication Number: WO-2025006295-A1Priority Date: 2023-06-26
- 4-Amino-2- (1H-pyrazolo [3,4-b] pyridin-3-yl) -6-oxo-6,7-dihydro-5H-pyrrolo [2,3-d] pyrimidine derivatives and cardiovascular disease (1H-indazol-3-yl) derivatives as cGMP modulators for treatingPublication Number: JP-6454448-B2Priority Date: 2015-12-22Grant Date: 2019-01-16
- Soluble guanylate cyclase stimulatorsPublication Number: US-10428076-B2Priority Date: 2015-12-22Grant Date: 2019-10-01
- Soluble guanylate cyclase stimulatorsPublication Number: US-2017174693-A1Priority Date: 2015-12-22
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
ADVERTISEMENT
ANAX LABORATORIES, WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
ADVERTISEMENT
Advect Process Systems Ltd. https://advectprocess.com
ADVECT PROCESS SYSTEMS CANADA LTD
51 Beechwood Rd., Cambridge, ON Canada N1S 3S1, Call Now +1 306 850 6737, Mail Now, ask@advectprocess.com
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2101645-33-2 |
| PubChem CID | 129242560 |
| ChemSpider | 129394387 |
| UNII | 6DXN080KGB |
| ChEMBL | ChEMBL5944803 |
| Chemical and physical data | |
| Formula | C27H22ClF5N6O3 |
| Molar mass | 608.95 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Bajwa, Ednan K.; Cislak, Dawn; Palcza, John; Feng, Hwa-ping; Messina, Eric J.; Reynders, Tom; Denef, Jean-François; Corcea, Vasile; Lai, Eseng; Stoch, S. Aubrey (January 2023). “Effects of an inhaled soluble guanylate cyclase (sGC) stimulator MK-5475 in pulmonary arterial hypertension (PAH)”. Respiratory Medicine. 206 107065. doi:10.1016/j.rmed.2022.107065. PMID 36521262.
- Patel, Mahesh J.; Bajwa, Ednan K.; Cislak, Dawn; Palcza, John; Reynders, Tom; Barthson, Jenny; Lai, Eseng; Stoch, S. Aubrey (9 September 2023). “A randomized study to evaluate the effects of single-dose MK-5475 co-administered with sildenafil on systemic hemodynamics”. European Respiratory Journal PA1208. doi:10.1183/13993003.congress-2023.PA1208.
- El-Kersh, Karim; Jalil, Bilal A. (July 2023). “Pulmonary hypertension inhaled therapies: An updated review”. The American Journal of the Medical Sciences. 366 (1): 3–15. doi:10.1016/j.amjms.2023.03.002. PMID 36921672.
- Tawa, Masashi; Okamura, Tomio (August 2022). “Factors influencing the soluble guanylate cyclase heme redox state in blood vessels”. Vascular Pharmacology. 145 107023. doi:10.1016/j.vph.2022.107023. PMID 35718342.
/////////frespaciguat, ANAX, ADVECT, guanylate cyclase activator, MK5475, MK 5475, sGC activator 1, 6DXN080KGB

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}