
Home » Uncategorized (Page 154)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
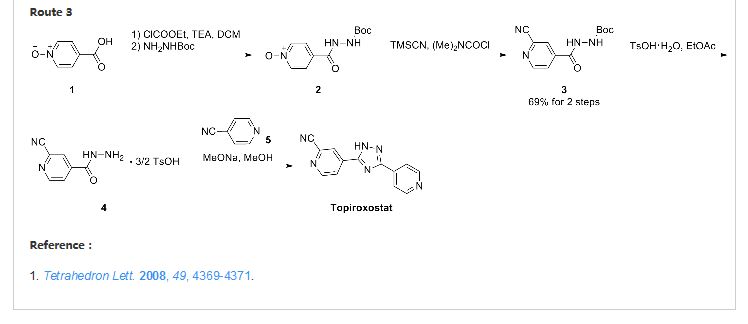
Topiroxostat 托匹司他 for gout and hyperuricemia


Topiroxostat
托匹司他
FUJI YAKUHIN ……..INNOVATOR
Approved in japan PMDA JUNE 28 2013
Xanthine oxidase inhibitor
FOR GOUT AND HYPERURICEMIA
Launched – 2013, Fuji YakuhinSanwa, Topiloric Uriadec
IUPAC Name: 4-(5-pyridin-4-yl-1H-1,2,4-triazol-3-yl)pyridine-2-carbonitrile
CAS Registry Number: 577778-58-6
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1)
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
3-(3-cyano-4-pyridyl)-5-(4-pyridyl)-1,2,4-triazole
Synonyms: 4-(5-PYRIDIN-4-YL-1H-1,2,4-TRIAZOL-3-YL)PYRIDINE-2-CARBONITRILE,
AC1NRB9T, Topiroxostat (JAN/INN), DB01685, D09786, FYX-051
SK-0910
4-[5-PYRIDIN-4-YL-1H-[1,2,4]TRIAZOL-3-YL]-PYRIDINE-2-CARBONITRILE,
C13H8N6 MF,248.2482 MW
 TOPIROXOSTAT
TOPIROXOSTAT
托匹司他
A xanthine oxidase inhibitor used to treat gout and hyperuricemia.

PATENT EXP 3/12/22, US /EU/CN

FYX-051, TOPIROXOSTAT is a xanthine oxidase inhibitor. This agent was approved in Japan by Fuji Yakuhin and Sanwa for the treatment of gout and hyperuricemia in 2013 and launched at the same year. In 2009, the compound was licensed to Sanwa by Fuji Yakuhin in Japan for the codevelopment and commercialization of gout.
The number of patients with hyperuricemia in Japan is reported to be 1.25 million and the number suffering from asymptomatic hyperuricemia is estimated to reach several millions. Hyperuricemia is becoming a popular disease.
Presently, hyperuricemia and gout due to hyperuricemia are treated by improving the living environment and administering various drug therapies for each period when an attack of gout is predicted to occur (presymptomatic period), when an attack of gout occurs, or when an attack of gout subsides. That is, preventive therapy is conducted in the presymptomatic period by administering colchicines as well as controlling the daily living environment. When an attack occurs, drug therapy using non-steroidal or steroidal anti-inflammatory agents is mainly conducted. After the attack subsides, patients are given guidance to improve their lifestyle. When improvement is judged insufficient, an assessment is made as to whether hyperuricemia is caused by reduced excretion of uric acid or by increased production of uric acid followed by treatment with drugs, which exhibit a uricosuric effect, such as probenecid and benzbromarone, those which inhibit resorption of uric acid, such as sulfinpyrazone, those which improve acidurea conditions, such as citrates, and xanthine oxidase inhibitors which inhibit production of uric acid, such as allopurinol. Colchicine is said to be able to prevent about 90% of attacks through inhibiting chemotaxis and phagocytosis of leukocytes, such as neutrophils, if administration thereof has been completed within a few hours before the attack. Since colchicine has various adverse effects, however, the use thereof is limited to the minimum and it is therefore difficult to timely administer it.
Accordingly, drug therapies are mainly adopted, but only allopurinol is available for the treatment of a disease caused by increased production of uric acid. However, a metabolite of allopurinol, oxypurinol, tends to accumulate and may cause calculi formation. Furthermore, this drug has been reported to induce adverse events such as rash, a decreased renal function and hepatitis, and it is not easy to administer.
Examples of compounds having xanthine oxidase inhibiting activity that can be used for treating gout caused by increased production of uric acid and that are effective for hyperuricemia and gout due to hyperuricemia have been described in J. Medicinal Chemistry, 1975, Vol. 18, No. 9, pp. 895–900, Japanese Patent Publication No. 49-46622 and Japanese Patent Publication No. 50-24315, which disclose some 1,3,5-substituted or 3,5-substituted 1,2,4-triazole compounds.
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1) has a xanthine oxidase inhibitory activity and serum uric acid level known as the agent that reduces (Patent Document 1).

The method for producing the compound (1), for example, 2 by Reissert Henze reaction isonicotinic acid methyl N-oxide – is a cyano isonicotinate, and the hydrazide which is then, 4 – this condensed cyanopyridine After obtaining a hydrazide of isonicotinic acid N-oxide (Patent Document 1, Example 12) and method, a cyano group after introduction, 4 by Reissert Henze reaction – method of condensing a cyano pyridine is known (Patent Document 1, Example 39).Further, 4 – as a starting material cyano-N-oxide, a triazole ring after construction (Patent Document 3), Reissert Henze unprotected or (Patent Document 2) to protect the ring condensed with isonicotinic acid hydrazide method of obtaining the compound (1) by introducing a cyano group by the reaction have also been reported.
The crystalline polymorph, yet the same molecule with the same chemical composition, the molecular arrangement in the crystal are different, and are different crystalline states. The pharmaceutical compounds having crystal polymorphism such the differences in physicochemical properties, affect pharmacological activity, solubility, bioavailability, stability and the like are known.Therefore, when the crystal polymorphism is present in a pharmaceutically useful compound, producing compounds of the crystalline form highly useful from polymorphs thereof is desirable.
WO 2003/064410 discloses WO 2005/009991 discloses Japanese Patent Publication No. 2005-41802
However, 4 of the above Patent Document – no description about the presence of crystalline polymorph on carbonitrile – pyridine-2-[yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol] It has not been, to these manufacturing methods, it is disclosed a method for the purpose of improving the chemical purity and yield, there is no description of the crystallographic plane.
Method of producing topiroxostat, useful for preventing or treating gout; and its intermediates. Picks up from WO2012060308, claiming the use of this topiroxostat for treating renal dysfunction. Along with the concurrently published WO2014017515, claiming crystalline Forms I and II of this compound, which, Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017516
Crystalline Forms I and II of topiroxostat, useful for preventing or treating gout. Along with the concurrently published WO2014017516, claiming a method of producing this compound. Picks up from WO2012060308, claiming a method of treating renal dysfunction using topiroxostat, which Fuji Yakuhin, in collaboration with Sanwa Kagaku, has developed and launched for the treatment of gout and hyperuricemia.WO-2014017515
novel 1,2,4-triazole compounds having an optionally substituted 2-cyanopyridin-4-yl group at 3-position and an optionally substituted aromatic group at 5-position inhibit a xanthine oxidase and are useful for treatment of gout and hyperuricemia, and have previously filed a patent application (Patent Document 1). The compounds can be prepared according to a method shown by the following reaction scheme:
-
 wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
wherein TMS represents trimethylsilyl group and Ar represents an aromatic groupAlthough this method can achieve the object in a small-scale production, there were such problems that the process for production of a substituted or unsubstituted 2-cyanoisonicotinic acid hydrazide is complicated, and a reaction solvent must be selected in compliance with the physical property of the product compound in each step, and isolation of a product is required in each step. Furthermore, the overall yield is not sufficiently high, and therefore there is a problem in the production on an industrial scale.
Patent Document 1: JP-A-2002-017825 -
-
A compound represented by formula (1) which is a starting material may be prepared by a method described in, for example, JP-A-47-7120, JP-A-61-152661A, JP-A-62-149673, JP-A-2002-528447, or European Patent Application No. 559363 specification. However, it is preferable to prepare compound (1) according to the following reaction scheme:
-

-


SYNTHESIS



PATENT
- Example 2
-
To the toluene solution obtained in Example 1 (2) was added 2-propanol (700 mL), and the mixture was stirred. To the resulting solution was added p-toluenesulfonic acid monohydrate (151.16 g) and the resulting mixture was stirred for 8 hours at an internal temperature of 80°C. The mixture was brought to room temperature, and the precipitated crystals were taken out and washed with 2-propanol (210 mL×2). The white crystals were dried under reduced pressure at 60°C for 15 hours to give 106.0 g of the captioned compound as white crystals. Subsequently, 90.0 g of the crystals was suspended in a mixture of 2-butanol (49 mL) and water (491 mL) and heated to an internal temperature of 80°C for 1 hour. The internal temperature was brought to room temperature, and the crystals were filtered and washed with a mixture of 2-butanol and water (1:10) (270 mL×3). The resulting crystals were dried under reduced pressure at 60°C for 15 hours to give 75.7 g of the captioned compound in a high purity.
-
1H―NMR(DMSO-d6)δppm:2.29(s,3H), 7.11 (m,2H), 7.48 (dd, 2H, J=6.48, 1.62Hz) , 8.32-8.35(m, 3H) , 8.57(dd, 1H, J=1.62, 0.81Hz) , 8.94-8.98(m, 3H)
- Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonate
Example 3
Preparation of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
-
To the white crystals (50.5g) obtained in Example 2 was added 2-propanol (937.5 mL) and water (312.5 mL), and the resulting mixture was heated and dissolved at an internal temperature of 80°C. Immediately thereafter, the solution was filtered and the filtrate was cooled to an internal temperature of 20°C. To the resulting suspension was added dropwise 0.52 mol/l of an aqueous sodium hydrogen carbonate solution (250 mL), and the mixture was stirred at room temperature for 2 hours. Then the crystals were filtered and washed with water (150 mL×3) and 2-butanol (150 mL×2). The crystals were dried under reduced pressure at 80°C for 15 hours to give 29.4 g of the captioned compound as pale yellow crystals.
-
1H―NMR(DMSO-d6)δppm:8.02(dd, 2H, J=4.59, 1.62Hz),8.32(dd, 1H, J=5.13, 1.62Hz), 8.55(dd, 1H, J=1.62, 1.08Hz), 8.80(dd, 2H, J=4.59, 1.62Hz), 8.93 (dd, 1H, J=5.13, 1.08Hz)
SYNTHESIS
Example 12
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of methyl isonicotinate N-oxide
13.9 g of isonicotinic acid N-oxide was added to 209 ml of methylene chloride, 29.7 g of 1-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline was further added thereto, and the mixture was stirred under argon atmosphere at room temperature for one hour. 32.1 g of methanol was added to this mixture, which was stirred at room temperature for 17 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (3:1) was used as an eluent to yield 11.1 g of a white powder.
1H-NMR (CDCl3) δppm: 3.95 (3H, s), 7.88 (2H, d, J=7.25 Hz), 8.22 (2H, J=7.25 Hz)
2) Production of Methyl 2-cyanoisonicotinate
11.1 g of the crystal obtained in 1) was dissolved in 170 ml of acetonitrile, 14.6 g of triethylamine and 21.5 g of trimethylsilylnitrile were added thereto, and the mixture was refluxed under argon atmosphere for 16 hours. After the solvent was evaporated under reduced pressure, the residue was subjected to silica gel column chromatography. Chloroform-acetone (95:5) was used as an eluent to yield 8.44 g of a pale yellow powder.
1H-NMR (CDCl3) δppm: 4.01 (3H, s), 8.08 (1H, d, J=5.45 Hz), 8.24 (1H, s), 8.90 (1H, d, J=5.45 Hz)
3) Production of 2-cyanoisonicotinic acid hydrazide
8.44 g of the crystal obtained in 2) was added to 85 ml of methanol, 1.84 g of hydrazine was further added thereto, and the mixture was stirred under argon temperature for 2 hours. After the solvent was evaporated under reduced pressure, chloroform was added to the residue, which was stirred at room temperature for one hour. The precipitated crystal was filtered, washed with chloroform and dried with a vacuum pump to yield 4.15 g of a pale yellow powder.
1H-NMR (DMSO-d6) δppm: 4.72 (2H, s), 8.05 (1H, d, J=5.12 Hz), 8.31 (1H, s),8.90 (1H, d, J=5.12 Hz), 10.23 (1H, s)
4) Production of the Object Compound
2.67 g of 4-cyanopyridine was dissolved in 40 ml of methanol, 0.83 g of sodium methoxide was added thereto, and the mixture was stirred at room temperature for one hour. Then 4.15 g of the crystal obtained in 3) was added and the mixture was refluxed for 37 hours. After the reaction completed, the precipitated solid was filtered, washed with methanol and dried with a vacuum pump to yield 3.66 g of the object compound as a yellow powder.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54, 1.57 Hz), 8.31 (1H, dd, J=5.11, 1.65 Hz), 8.53 (1H, dd, J=1.65, 0.50 Hz), 8.80 (2H, dd, J=4.54, 1.57 Hz), 8.93 (1H, dd, J=5.11, 0.50 Hz)
Example 39
5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
1) Production of isonicotinic acid (N-2-tert-butoxycarbonyl)hydrazide-1-oxide
585 ml of methylene chloride was added to 39.0 g of isonicotinic acid N-oxide, and after 34.0 g of triethylamine was further added thereto, the mixture was cooled under argon atmosphere to −15° C. 33.5 g of ethyl chlorocarbonate in 117 ml of methylene chloride was added dropwise to this mixture, which was stirred at a temperature from −5 to −10° C. for one hour. Then 44.4 g of tert-butyl ester of carbamic acid in 117 ml of methylene chloride was added dropwise to this mixture and it was allowed to slowly rise to room temperature while it was stirred. The precipitated solid was filtered after 15 hours, washed with methylene chloride, and dried with a vacuum pump to yield 49.7 g of white crystal.
1H-NMR (DMSO-d6) δppm: 1.42 (9H, s), 7.82 (2H, d, J=7.09 Hz), 8.33 (2H, d, J=7.09 Hz), 9.02 (1H, s), 10.44 (1H, s)
Production of 2-cyanoisonicotinic acid hydrazine 1½ P-Toluenesulfonic acid salt
228 ml of dioxane was added to 30.4 g of the crystal obtained in 1), and after 13.1 g of trimethylsilyl cyanide and 38.8 g of N,N-dimethylcarbamoyl chloride were further added thereto, the mixture was stirred under argon atmosphere at 60° C. for 5 hours. After the solvent was evaporated under reduced pressure, the residue was dissolved in ethyl acetate and subsequently washed with 1.5 M sodium carbonate aqueous solution and a saturated saline solution and dried over magnesium sulfate. After the magnesium sulfate was filtered off, the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue, 68.5 g of p-toluenesulfonic acid monohydrate was added thereto, and the mixture was stirred at room temperature for 22 hours. The precipitated crystal was filtered, washed with ethyl acetate, and dried with a vacuum pump to yield 40.3 g of white crystal 2).
1H-NMR (DMSO-d6) δppm: 2.28 (4.5H, s), 7.12 (3H, dd, J=7.92 & 0.66 Hz), 7.48 (3H, dd, J=7.92 & 0.66 Hz), 8.10 (1H, dd, J=5.11 & 1.81 Hz), 8.39 (1H, dd, J=1.81 & 0.33 Hz), 8.99 (1H, dd, J=5.11 & 0.33 Hz)
3) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
9.98 g of 4-cyanopyridine was dissolved in 250 ml of methanol, and after 7.77 g of sodium methoxide was added thereto, the mixture was stirred at room temperature for one hour. Then 40.3 g of the crystal obtained in 2) was added and the mixture was refluxed for 24 hours. After the reaction completed, the precipitated crystal was filtered, washed with methanol, and dried with a vacuum pump to yield 16.3 g of yellow crystal.
1H-NMR (DMSO-d6) δppm: 8.01 (2H, dd, J=4.54 & 1.57 Hz), 8.31 (1H, dd, J=5.11 & 1.65 Hz), 8.53 (1H, dd, J=1.65 & 0.50 Hz), 8.80 (2H, dd, J=4.54 & 1.57 Hz), 8.93 (1H, dd, J=5.11 & 0.50 Hz)
4) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole
45 ml of ethanol and 15 ml of 1-methyl-2-pyrrolidone were added to 3.0 g of the crystal obtained in 3), and the mixture was heated and stirred at 80° C. for 19 hours. The crystal was filtered, subsequently washed with a mixture of ethanol and 1-methyl-2-pyrrolidone (3:1) and ethanol, and dried with a vacuum pump to yield 2.71 g of yellow crystal.
5) Production of 5-(2-cyano-4-pyridyl)-3-(4-pyridyl)-1,2,4-triazole p-toluenesulfonic acid salt
5 ml of ethanol and 30 ml of water were added to 2.48 g of the crystal obtained in 4), and after 3.8 g of p-toluenesulfonic acid monohydrate was further added thereto, the mixture was stirred at room temperature for 5 hours. The precipitated crystal was filtered, subsequently washed with a mixture of ethanol and water (1:6), water and then ethanol, and dried with a vacuum pump to yield 3.5 g of white crystal.
1H-NMR (DMSO-d6) δppm: 2.28 (3H, s), 7.12 (2H, dd, J=7.75 & 0.50 Hz), 7.48 (2H, dd, J=7.75 & 0.50 Hz), 8.33 (1H, dd, J=5.12 & 1.65 Hz), 8.45 (2H, d, J=6.11 Hz), 8.57 (1H, dd, J=1.65 & 0.66 Hz), 8.96˜9.02 (3H, m)
6) Production of the object compound
17 ml of ethanol and 17 ml of water were added to 3.36 g of the crystal obtained in 5), and the mixture was stirred at room temperature for 30 minutes. A solution of sodium carbonate (0.74 g of sodium carbonate in 17 ml of water) was further added, and the mixture was stirred at room temperature for 2 hours. The precipitated crystal was filtered, subsequently washed with water and ethanol, and dried with a vacuum pump to yield 1.89 g of the object compound as a pale yellow crystal.
TOPIROXOSTAT
SYNTHESIS

(First step)
The first step, 4 – is a step of obtaining a compound (3) is reacted in the presence of an alkali metal alkoxide, cyano-N-oxide and (2), and isonicotinic acid hydrazide.
4 used in this reaction – isonicotinic acid hydrazide and (2) a cyano-N-oxide is a known compound both, I can be prepared by known means.
The alkali metal alkoxide is used, 6 alkoxide alkali metal C 1-C are preferred, sodium methylate, sodium ethylate and the like can be given as specific examples. The reaction is preferably carried out in a solvent, as the solvent, alcohol solvents such as methanol, ethanol and the like are preferable.
The reaction is preferably first in a solvent, is treated with an alkali metal alkoxide compound (2) and then to react the isonicotinic acid hydrazide. First, heated to reflux under cooling, at 80 ℃ from 15 ℃ preferably, 30 minutes and 12 hours in general, the reaction temperature in the reaction with an alkali metal alkoxide (2) with the compound is reacted 1-4 hours, preferably about. Under the temperature conditions, using an excess amount or one equivalent of 30 minutes to 12 hours usually, reaction with isonicotinic acid hydrazide Subsequent to reaction for 1 to 5 hours, preferably.
Example 1:
Synthesis 4 oxide (3) – – – (4 – pyridin-carbonyl) -4 – N “pyridine hydrazide imide -1 was suspended in 40mL of methanol cyanopyridine-N-oxide and (2) 5.00g, sodium was added to methylate 22.4mg, and the mixture was stirred for 2 hours under 40 ℃ nitrogen atmosphere. was cooled to room temperature. reaction solution was stirred for 4 hours at 40 ℃ was added isonicotinic acid hydrazide 5.71g at the same temperature, precipitated The filtrated crystals were, washed with methanol 15mL, and dried 15 hours at 80 ℃, N “- to give (3) 9.60g oxide – (4 – pyridin) -4 – pyridine-hydrazide imide -1.
1 H-NMR (DMSO-d 6) δ (ppm): 6.98 (br, 2H), 7.81 (d, 2H, J = 5.77Hz), 7.85 (d, 2H, J = 7 .09 Hz), 8.29 (d, 2H, J = 7.09Hz), 8.73 (d, 2H, J = 5.77Hz), 10.37 (br, 1H)
MS m / z: 256 [M-H] –
(Second step)
The second step is a step of obtaining compound (4) by cyanation agent cyano compound (3).
As the cyanation agent used, trialkyl cyanide alkali metal cyanide, sodium cyanide, potassium cyanide and the like, zinc cyanide, trimethylsilyl cyanide and the like.
The cyanation reaction is preferably, for example, be carried out (Heterocycles, Vol.22, No.5, 1994) by Reissert Henze reaction. This reaction, for example, to give compound (4) by an organic solvent in the compound (3), and after activation with carbamoyl halide, and reacting the cyano agent. The alkylcarbamoyl halide used in the carbamoylation is a first step in Reissert Henze reaction, 6 alkylcarbamoyl halide di C 1-C dimethylcarbamoyl chloride, and di-propyl carbamoyl chloride can be used, preferably, dimethylcarbamoyl is chloride. The solvent used in this reaction, N, N-dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, tetrahydrofuran and acetonitrile can be used, however, N, N-dimethylformamide is preferred. Further, 15 ~ 60 ℃, more preferably 30 ~ 50 ℃ reaction temperature. The reaction time is preferably 1 to 24 hours, more preferably 1 to 3 hours. As the cyanation agent used in the cyanation reaction followed, cyano agents above can be used, sodium cyanide, potassium cyanide, zinc cyanide, and trimethylsilyl cyanide, and more preferably, it is sodium cyanide . -20 ~ 60 ℃ is preferred, more preferably -10 ~ 40 ℃, reaction temperature is 1-4 hours.
Is a novel compound (4) The compound obtained in this second step, it is useful as an intermediate for the production of compound (1). If through Compound (4) can be synthesized in good yield and easily without the need for purification in the second step is also possible, and can be produced (1) Compound industrially efficiently compound (4).
Synthetic N “hydrazide (4) – (4 – pyridine carbonyl) -4 – pyridine carboxylic acid N’-(carboxylic imidoyloxy – 2 – – cyano-4)
Example 2
4 pyridine hydrazide imide -1 – oxide ( was suspended in N, N-dimethylformamide 48mL and 3) 10.0g, under nitrogen atmosphere, followed by stirring for 1 hour was added dimethylcarbamoyl chloride 9.20g at 40 ℃. was added sodium cyanide 2.48g at the same temperature, After cooling to 5 ℃ below. reaction mixture was stirred for 1 hour, the crystals were collected by filtration. precipitate was successively added dropwise a 5% aqueous sodium bicarbonate solution 100mL, and 100mL water, and washed with water 100mL, at 80 ℃ for 15 h and dried under reduced pressure to give 4 – hydrazide (4) 9.28g of pyridine-carboxylic acid N’-(carboxylic imide yl – 2 – cyano-4).
1 H-NMR (DMSO-d 6) δ (ppm): 7.15 (br, 2H), 7.82 (d, 2H, J = 5.61Hz), 8.14 (d, 1H, J = 5 .11 Hz), 8.37 (s, 1H), 8.75 (d, 2H, J = 5.61Hz), 8.86 (d, 1H, J = 5.11Hz), 10.47 (br, 1H )
MS m / z: 265 [M-H] –

(Third step)
The third step is a step of obtaining a compound (1) by the presence of an acid catalyst, the cyclization reaction of the compound (4).
As the acid, organic phosphoric acid, p-toluenesulfonic acid, such as hydrochloric acid, inorganic acids can be used, inorganic acids, phosphoric acid is particularly preferable. As the reaction solvent, water, 2 – butanol, 2 – mixed solvent of alcohol and water or alcohol, propanol, ethanol and the like can be used, but water and 2 – I was mixed 5:1 to 10:1 butanol solvent. The reaction temperature and time, 60 ~ 100 ℃, preferably 2 to 12 hours at 70 ~ 90 ℃, I want to 8-10 hours, preferably.
Intermediates and compounds of the present invention the method (1) can be isolated and purified from the washed reaction mixture, recrystallization, by means of various conventional chromatography.
Example 3:
4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile 4 Synthesis of (1) – pyridine-carboxylic acid N’- (2 – cyano-4 – carboxylic imide yl) water 82mL, 2 hydrazide (4) 9.25g – butanol was added 8.2mL, phosphate 4.00g, was stirred for 8 h at 80 ℃. After cooling to room temperature, the reaction mixture was precipitated crystals were collected by filtration, water: 2 – were washed with a mixed solution of 92.5mL butanol = 10:1. The 13 h and dried under reduced pressure at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl) – 1 H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile (1 I got a) 7.89g.
Topiroxostat
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
MS m / z: 247 [M-H] –
PATENT
Synthetic water-carbonitrile p-toluenesulfonate – pyridine Example 1: 4 – [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazol]: 2 – butanol = was added monohydrate 6.62g p-toluenesulfonic acid in a mixed solution of 55mL of 10:1, 4 at 80 ℃ – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl] pyridine-2 – – triazol-3 was added carbonitrile 7.85g, and the mixture was stirred at the same temperature for 1 hour. After cooling to room temperature, the reaction mixture, and the precipitated crystals were collected by filtration, and water: 2 – were washed with a mixed solution of 40mL of butanol = 10:1. The dried under reduced pressure for 10 hours at 80 ℃ crystals obtained 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2 – carbonitrile p-toluene I got a sulfonate 12.6g.
1 H-NMR (DMSO-d 6) δ (ppm): 2.29 (s, 3H), 7.11 (m, 2H), 7.48 (dd, 2H, J = 6.48,1.62 Hz ) ,8.32-8 .35 (m, 3H), 8.57 (dd, 1H, J = 1.62,0.81 Hz) ,8.94-8 .98 (m, 3H)
– [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazole and potassium carbonate 8.22g, 4 in a mixed solution of 80mL of ethanol = 9:1: preparation water of crystal form I: Example 2 I was dissolved carbonitrile p-toluenesulfonate 10.0g – -3 – yl] pyridine-2. After stirring for 5 hours plus 15mL 6M hydrochloric acid at 20 ℃, was the precipitated crystals were collected by filtration, and washed with water 100mL. The 23 h and dried under reduced pressure at 80 ℃, 4 – to obtain carbonitrile 5.78g – pyridin-2 [yl 5 – (pyridin-4 – yl)-1H-1, 2,4 – – -3 triazole. Having a DSC as shown in FIG 4 and the powder X-ray diffraction pattern shown in FIG 1, the resulting crystals were type-I crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
N, N carbonitrile 40.0g – preparation of 4 Form II – [5 – (pyridin-4 – yl)-1H-1, 2,4 – yl – triazol-3]-2: Example 3 – dimethylformamide was added 300mL, and stirred for 25 min at 150 ℃. After cooling to room temperature the solution, and the precipitated crystals were collected by filtration, and washed twice with water 200mL, 4 and dried under reduced pressure overnight at 80 ℃ the crystal – [5 – (pyridin-4 – yl)-1H-1 , 2,4 – I got carbonitrile 30.4g – yl] pyridine-2 – triazole-3. Having a DSC as shown in FIG 5 and powder X-ray diffraction pattern shown in FIG 2, the resulting crystals were type II crystals.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
The 25 ℃, about 2g carbonitrile, – preparation of the hydrate 4 – [5 – (pyridin-4 – yl)-1H-1, 2,4 – triazol-3 – yl] pyridine-2: Example 4 I was stored for 14 days under conditions of relative humidity 97%. Having a DSC as shown in FIG 7 and the powder X-ray diffraction pattern shown in FIG 3, the obtained crystal was a hydrate.
1 H-NMR (DMSO-d 6) δ (ppm): 8.02 (dd, 2H, J = 4.59,1.62 Hz), 8.32 (dd, 1H, J = 5.13,1. 62Hz), 8.55 (dd, 1H, J = 1.62,1.08 Hz), 8.80 (dd, 2H, J = 4.59,1.62 Hz), 8.93 (dd, 1H, 5 .13,1.08 Hz)
Melting point: 327 ℃
Test Example: solubility test Type I crystal by crystal form, II-type crystal, and water solubility of the hydrate was calculated by absorbance measurement method, a saturated solution concentration of each sample. I Figure 8 shows the results.Whereas the 6.2μg/mL water solubility of crystalline Form I, II type crystal 4.2μg/mL, hydrate was 1.9μg/mL.
From Figure 8, the water solubility of Form II and Form I crystals is good, water-soluble type I crystal is particularly good.
NMR
BMCL Volume 19, Issue 21, 1 November 2009, Pages 6225–6229
http://www.sciencedirect.com/science/article/pii/S0960894X09012372?np=y
view compd 39 and ignore rest
 TOPIROXOSTAT, FYX O51
TOPIROXOSTAT, FYX O51
view compd 39 and ignore rest
| 1 | * | Baldwin, J.J., J. Med. Chem.; 1975; 18(9); 895-900, especially p. 898, lines 3-5. |
| 2 | * | Geldard, J.F. et al., J. Org. Chem.; 1965; 30(1); 318-319, especially p. 319, starting line 33. |
| 3 | * | Lever, A.B.P., Inorg. Chem; 1990; 29; 1271-1285, especially p. 1275, line 18 and 19. |
Nucleosides, Nucleotides and Nucleic Acids, 2008 , vol. 27, 6-7 pg. 888 – 893
Inoue, Tsutomu; Sato, Takahiro; Ashizawa, Naoki; Iwanaga, Takashi; Matsumoto, Koji; Nagata, Osamu; Nakamura, Hiroshi
Bioorganic and Medicinal Chemistry Letters, 2009 , vol. 19, 21 pg. 6225 – 6229
WO 2012060308
WO 2007148835
WO 2005009991
| WO2003064410A1 * | Dec 3, 2002 | Aug 7, 2003 | Naoki Ashizawa | Novel 1,2,4-triazole compound |
| US3882134 * | May 21, 1973 | May 6, 1975 | Merck & Co Inc | 1-Substituted-3,5-dipyridyl-1,2,4-triazoles |
| US3947577 * | Jan 8, 1975 | Mar 30, 1976 | Merck & Co., Inc. | Anti-hyperuricemia composition |
| US3984558 * | Nov 29, 1974 | Oct 5, 1976 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds used as bronchodilators |
| US4011218 * | Dec 3, 1974 | Mar 8, 1977 | Merck & Co., Inc. | 1,2,4-triazoles |
| US4104393 * | Sep 2, 1977 | Aug 1, 1978 | Merck & Co., Inc. | 1,3,5-Trisubstituted-1,2,4-triazole compounds |
| US5571897 * | Dec 5, 1991 | Nov 5, 1996 | Wallac Oy | Luminescent lanthanide chelates |
| Publication Number | Publication Date | IPCR Assignee/Applicant | Structure hits | Tools | |
|---|---|---|---|---|---|
|
1.
US-9199970-B2 |
2015-12-01 |
|
|||
|
2.
US-20150322006-A1 |
2015-11-12 |
|
|||
|
3.
US-20150309021-A1 |
2015-10-29 |
|
|||
|
4.
US-20150291543-A1 |
2015-10-15 |
|
|||
|
5.
EP-2927219-A1 |
2015-10-07 |
EN
|
|
||
|
6.
US-20150274680-A1 |
2015-10-01 |
|
|||
|
7.
EP-2913053-A1 |
2015-09-02 |
EN
|
|
||
|
8.
EP-2511844-B1 |
2015-08-12 |
EN
|
|
||
|
9.
EP-2712861-B1 |
2015-07-29 |
EN
|
|
||
|
10.
US-20150203490-A1 |
2015-07-23 |
|
|||
|
11.
US-20150191463-A1 |
2015-07-09 |
|
|||
|
12.
US-20150166510-A1 |
2015-06-18 |
|
|||
|
13.
EP-2878594-A1 |
2015-06-03 |
EN
|
|
||
|
14.
EP-2878598-A1 |
2015-06-03 |
E
|
|
||
|
15.
EP-2878595-A1 |
2015-06-03 |
EN
|
|
||
|
16.
US-20150126558-A1 |
2015-05-07 |
|
|||
|
17.
US-8987473-B2 |
2015-03-24 |
|
|||
|
18.
EP-2842948-A1 |
2015-03-04 |
EN
|
|
||
|
19.
EP-2776028-A1 |
2014-09-17 |
EN
|
|
||
|
20.
US-20140256748-A1 |
2014-09-11 |
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-[5-(4-Pyridinyl)-1H-1,2,4-triazol-3-yl]-2-pyridinecarbonitrile
|
|
| Clinical data | |
| Trade names | Topiloric, Uriadec |
| Legal status |
|
| Identifiers | |
| CAS Number | 577778-58-6 |
| ATC code | None |
| PubChem | CID: 5288320 |
| ChemSpider | 4450517 |
| Chemical data | |
| Formula | C13H8N6 |
| Molecular mass | 248.24 g/mol |
/////////////
C1=CN=CC=C1C2=NC(=NN2)C3=CC(=NC=C3)C#N
Pamicogrel KB 3022 for Coagulation Disorders Therapy


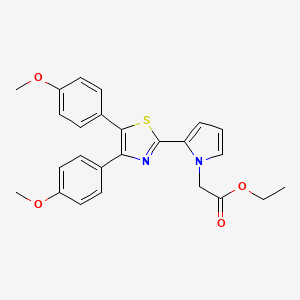 PAMICOGREL
PAMICOGRELPamicogrel (CAS NO.: 101001-34-7), with its systematic name of 1H-Pyrrole-1-acetic acid, 2-(4,5-bis(4-methoxyphenyl)-2-thiazolyl)-, ethyl ester, could be produced through many synthetic methods.
Following is one of the synthesis routes:
alpha-Bromo-4,4-dimethoxidesoxybenzoin (I) is cyclized with pyrrole-2-carbothioamide (II) in hot acetonitrile to produce 4,5-bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole (III), which is then condensed with ethyl bromoacetate (IV) in the prsence of NaOH and tetrabutylammonium bromide in refluxing dichloromethane – water.

- Reaction Scheme-I:
-


wherein R is as defined above, and Y is a halogen such as chlorine, bromine or iodine, or p-toluenesulfonyloxy group.
-
The process of the above reaction scheme-I can be carried out by reacting a compound (II) and an equimolar or excess amount of a compound (III) in the presence of a base or a phase transfer catalyst. In case of using a base such as metallic potassium, metallic sodium, potassium tert-butoxide etc.; the reaction is carried out in a solvent of tetrahydrofuran or dimethoxyethane at a temperature of from room temperature to a boiling point of the solvent for 1 to 24 hours. In case of using a phase transfer catalyst such as a quaternary ammonium salt (e.g. tetra-n-butylammonium bromide, methyltrioctylammonium chloride, etc.), the reaction is carried out in two phases of benzene or dichloromethane and 50 % aqueous sodium hydroxide or 60 % aqeuous potassium hydroxide at a temperature of from 0°C to a boiling point of the solvent for one minute to 24 hours.


- Reaction Scheme-II:
-

wherein X is a halogen such as bromine or chlorine.
-
The above process can be carried out by reacting a compound (IV) and an equimolar amount of a compound (V) in a solvent such as acetonitrile, dimethylformamide (DMF), dimethyl sulfoxide (DMSO), or an alcohol (e.g. ethanol) at a temperature of from 50°C to a boling point of the solvent for 10 minutes to 4 hours.

- Reaction Scheme-III:
-




- Reaction Scheme-IV:
-

wherein R1 is as defined above.
-
The process can be carried out by converting a compound (VII) into an oxime (VIII) by a conventional oxime forming reaction, heating the oxime (VIII) in acetic anhydride to obtain a compound (IX), and treating the compound (IX) with hydrogen sulfide, that is, by blowing hydrogen sulfide gas into a reaction system containing the compound (IX) in a solvent such as DMF, DMSO or pyridine in the presence of 0.5 to 5 equimolar amount of a base such as a tertiary amine (e.g. triethylamine) at a temperature of from 0° to 40°C for 3 to 24 hours

- Reaction Scheme-V:
-


- Reference Example 6
- 4,5-Bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole [compound of the formula (II)]:
-
Pyrrole-2-carbothioamide (cf. J. Org. Chem., 38, 667, 1973) (1.51 g, 12 mmole) and α-bromo-4,4′-dimethoxy- deoxybenzoin (cf. Aust. J. Chem., 8, 385. 1955) (4.02 g, 12 mmole) are dissolved in acetonitrile (120 ml). The mixture is stirred at 60°C for 50 minutes. After the reaction, the reaction mixture is distilled under reduced pressure to remove the solvent. To the resulting residue are added chloroform and aqueous solution of sodium carbonate, and the mixture is shaken. The chloroform layer is taken, and the aqueous layer is further extracted with chloroform. The chloroform layers are combined, dried over anhydrous magnesium sulfate, and distilled under reduced pressure to remove the solvent. The residue is recrystallized from ligroin to give 4,5-bis(4-methoxyphenyl)-2-(pyrrol-2-yl)-thiazole (3.74 g, yield: 86 %).
-
M.p. 131.5 – 134.0°C
-
NMR (CDCl3, δ ppm): 3.7 (6H) , 6.1 (1H, dd) , 6.5-6.9 (6H), 7.1-7.5 (4H), 9.4-9.8 (lH).
-
Elementary analysis for C21H18N2O2S:


- Example 14
-
Ethyl 2-[4,5-bis(4-methoxyphenyl)thiazol-2-yl]-pyrrole-1-acetate (compound of the formula (I) wherein R1 = -CH2-COOC2H5):
- 4,5-Bis(4-methoxyphenyl)-2-(pyrrol-2-yl)thiazole obtained in the same manner as described in Reference Example 6 (3.62 g, 10 mmole), ethyl bromoacetate (1.67 g, 10 mmole), and tetra-n-butylammonium bromide (0.32 g, 1 mmole) are refluxed with vigorous stirring in two phases of dichloromethane (40 ml) and 50 % aqueous sodium hydroxide (40 ml) at room temperature for 2 minutes. To the mixture are added water and dichloromethane under ice-cooling, and the mixture is shaken. The dichloromethane layer is taken, and the aqueous layer is further extracted with dichloromethane. The dichloromethane layers are combined, dried over anhydrous magnesium sulfate, and distilled under reduced pressure to remove the solvent. The residue is recrystallized from ligroin to give ethyl 2-[4,5-bis(4-methoxyphenyl)thiazol-2-yl]pyrrole-1-acetate (3.64 g, yield: 81 %).
-
M.p. 132.5 – 135.5°C
-
NMR (CDCl3, δ ppm): 1.2 (3H, t), 3.8 (6H), 4.15 (2H, q), 5.25 (2H, s), 6.25 (1H, dd), 6.7-6.95 (6H), 7.2-7.55 (4H).
-
Elementary analysis for C25H24N2O4S:


Determination of the antiplatelet agent. KB-3022, and its metabolite by high-performance liquid chromatography.Nakada Y, Ikuta Y, Kawashima T, Awata N.Chem Pharm Bull (Tokyo). 1990 Apr;38(4):1093-5.
 pamicogrel
pamicogrel| EP0037274A1 * | 30 Mar 1981 | 7 Oct 1981 | Eli Lilly And Company | Substituted triaryl thiazole compounds |
| EP0077024A2 * | 7 Oct 1982 | 20 Apr 1983 | Schering Aktiengesellschaft | Imidazole derivatives, process for their preparation and pharmaceutical products containing them |
| US4168315 * | 28 Sep 1977 | 18 Sep 1979 | The Upjohn Company | Dianisyl thiazole compound, compositions and method of antithrombotic treatment |
TAKEDA PHARMACEUTICALS 武田薬品工業株式会社 ON THE RISE
Tadataka Yamada, M.D., Chief Medical & Scientific Officer of Takeda

TAKEDA US CHICAGO OFFICE
TAKEDA PIPELINE SEE LINKS BELOW
1 https://www.takeda.com/investor-information/annual/files/ar2013_10_en.pdf
2. http://www.takeda.com/research/files/pipeline_20131031_en.pdf
3 http://www.takeda.com/research/pipeline/
- 2012 Download Entire File
 PDF 0.4MB 34P
PDF 0.4MB 34P
Takeda’s top executives had frequently pointed to TAK-875 as one of their best shots at coming up with an important new approach to treating diabetes. The drug is designed to spur insulin secretion in the pancreas and Takeda had confidently projected an approval in Japan in 2015 with a follow-up approval in the big U.S. market a year or two later.
The termination of the high-profile program caused some anxiety among investors. Takeda’s shares plunged 8% on the loss as analysts wondered how the pharma company could counter the loss of Actos, a $3.7 billion drug that accounted for about a quarter of its revenue in 2011.
Takeda won an approval on a trio of DPP-4 diabetes drugs–Nesina (alogliptin) and two combos with alogliptin, dubbed Oseni and Kazano–at the beginning of the year. But Takeda suffered some big delays in gaining acceptance, a common fate in this field, where regulators are particularly cautious about new drugs. And Merck had already solidified its lead in the DPP-4 market with Januvia whileOnglyza trailed closely behind it. Takeda had hoped that a combination of TAK-875 and Januvia could help regain some lost market territory–but that dream has clearly vanished as well.
| January 27, 2014 | |
| January 22, 2014 | |
| January 17, 2014 | |
| January 14, 2014 | |
| January 10, 2014 |
2013
| December 27, 2013 | ||
| December 25, 2013 | ||
| December 25, 2013 | ||
| December 24, 2013 | ||
| December 20, 2013 | ||
| December 20, 2013 | ||
| December 19, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 10, 2013 | ||
| December 9, 2013 | ||
| December 5, 2013 | ||
| December 4, 2013 | ||
| November 30, 2013 | ||
| November 21, 2013 | ||
| November 19, 2013 | ||
| November 14, 2013 | ||
| November 12, 2013 | ||
| November 12, 2013 | ||
| October 21, 2013 | ||
| October 7, 2013 | ||
| October 2, 2013 | ||
| October 1, 2013 |
| September 26, 2013 | ||
| September 26, 2013 | ||
| September 24, 2013 | ||
| September 20, 2013 | ||
| September 20, 2013 | ||
| September 13, 2013 | ||
| September 13, 2013 | ||
| September 5, 2013 | ||
| September 2, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 27, 2013 | ||
| August 22, 2013 | ||
| August 13, 2013 | ||
| August 1, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 31, 2013 | ||
| July 30, 2013 | ||
| July 29, 2013 | ||
| July 26, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 19, 2013 | ||
| July 18, 2013 | ||
| July 10, 2013 | ||
| July 1, 2013 |
CLIPPED
Takeda isn’t quite in the top 10 among global drugmakers, but the company boasts the 7th-largest pipeline in the industry, according to its presentation at the conference. Yamada noted that 31% of the pipeline assets are in late-stage trials. Millennium is leading development of three late-stage contenders, TAK-700 for prostate cancer, MLN9708 for multiple myeloma and MLN0002 for ulcerative colitis andCrohn’s disease.
In an effort to revive its diabetes franchise, Takeda is in the final stage of development for a first-of-a-kind GPR40 agonist called TAK-875, designed to provide glucose-dependent insulin secretion.
With a rich late-stage pipeline at Takeda, Yamada wants the company to focus on growing its ranks of earlier-stage drug candidates. To do this the company has landed a variety of deals, including the purchase of Intellikine for $310 million to acquire anti-cancer drugs and more recently the acquisition of Envoy Therapeutics last year for $140 million.
Takeda has formed a New Frontier Science group to scout out the hottest research in academia and elsewhere and form collaborations with scientists behind those innovations. At the J.P. Morgan conference, Yamada said, he was attending many meetings with members of the biotech community.

Takeda Pharmaceutical Company Limited (武田薬品工業株式会社 Takeda Yakuhin Kōgyō Kabushiki-gaisha?) is the largest pharmaceutical company in Japan and Asia and a top 15 pharmaceutical company. The company has over 30,000 employees worldwide and achieved $16.2 billion USD in revenue during the 2012 fiscal year.[1] The company is focused on metabolic disorders, gastroenterology, neurology, inflammation, as well asoncology through its independent subsidiary, Millennium: The Takeda Oncology Company.[2] Its headquarters is located in Chuo-ku, Osaka, and it has an office in Nihonbashi, Chuo, Tokyo.[3][4] In January 2012, Fortune Magazine ranked the Takeda Oncology Company as one the 100 best companies to work for in the United States.
Takeda Pharmaceuticals was founded on June 12, 1781 and was incorporated on January 29, 1925.
Takeda’s Japanese logo
In 1977, Takeda first entered the U.S. pharmaceutical market by developing a joint venture with Abbott Laboratories called TAP Pharmaceuticals.[5]Through TAP Pharmaceuticals, Takeda and Abbott launched the blockbusters Lupron (leuprolide) in 1985 and Prevacid (lansoprazole) in 1995.
One of the firm’s mainstay drugs is Actos, a compound in the thiazolidinedione class of drugs used in the treatment of type 2 diabetes. Launched in 1999, Actos has become the best-selling diabetes drug in the world with $4 billion USD in sales during the 2008 fiscal year.[6]
In February 2005, Takeda announced its acquisition of San Diego, California-based Syrrx, a company specializing in high-throughput X-ray crystallography, for $270 million.[7]
In February 2008, Takeda acquired the Japanese operations of Amgen and rights to a dozen of the California biotechnology company’s pipeline candidates for the Japanese market.[8]
In March 2008, Takeda and Abbott Laboratories announced plans to conclude their 30-year old joint venture, TAP Pharmaceuticals, that had over $3 billion in sales in its final year. The split resulted in Abbott acquiring U.S. rights to Lupron and the drug’s support staff. On the other hand, Takeda received rights to Prevacid and TAP’s pipeline candidates. The move also increased Takeda’s headcount by 3,000 employees.[9]
In April 2008, Takeda announced that it was acquiring Millennium Pharmaceuticals of Cambridge, Massachusetts, a company specializing in cancerdrug research, for $8.8 billion. The acquisition brought in Velcade, a drug indicated for hematological malignancies, as well as a portfolio of pipeline candidates in the oncology, inflammation, and cardiovascular therapeutic areas. Millennium now operates as an independent subsidiary, serving as the global center of excellence in oncology under its new name: “Millennium: The Takeda Oncology Company.” [10]
In May 2008, the company licensed non-exclusively the RNAi technology platform developed by Alnylam Pharmaceuticals, creating a potentially long-term partnership between the companies.[11]
On May 19, 2011, Takeda Pharmaceutical and Nycomed announced that Takeda will acquire Nycomed for € 9.6 billion. The acquisition was completed by September 30, 2011.[12]
On April 11, 2012, Takeda Pharmaceutical and URL Pharma announced that Takeda will acquire URL Pharma for $800 million. The acquisition is expected to be completed within 60 days.
On 25 May 2012, Takeda announced the purchase of Brazilian pharmaceutical company Multilab by R$ 540 million.[13]

Takeda Midosuji Building, headquarters of Takeda Pharmaceutical Company, inChuo-ku, Osaka, Japan
Takeda operates two primary bases in Japan in Osaka and Tokyo. Its United States subsidiary is based in Deerfield, Illinois, and all Global Operations outside of Japan and U.S. are based in Opfikon (Zurich), Switzerland. The company maintains research & development sites in Osaka and Tsukuba, Japan; San Diego andSan Francisco, United States; Cambridge, United Kingdom; and Singapore.[14]
The company has manufacturing facilities in Japan, China, Indonesia, Italy, and Ireland.[15] Following the Nycomed acquisition, the Takeda manufacturing sites have been extended with facilities in Argentina,Austria,Belgium,Brazil,Denmark, Estonia,Germany,Mexico,Norway and Poland. Takeda has overseas marketing presences in the U.S., UK, France, Italy, Germany, Austria, Switzerland, Spain, China, Taiwan, Philippines, Thailand, Indonesia, and Singapore. It has recently[when?] announced its first foray into Canada, Portugal, Spain, Mexico, and Ireland.[15]

AT INDONESIA
Products
Some of the key products that Takeda produces on behalf of partners include:[16]
- Actos (pioglitazone) – Type 2 Diabetes
- Amitiza (lubiprostone) – Chronic idiopathic constipation
- Basen (voglibose) – Type 2 Diabetes
- Benet (risedronic acid) – Osteoporosis (Japan)
- Blopress (candesartan) – Hypertension
- Enbrel (etanercept) – Inflammatory diseases (Japan)
- Dexilant (dexlansoprazole) – Gastroesophageal reflux disease – name changed to Dexilant in U.S.
- Lupron/Leuplin (leuprorelin) – GnRH agonist for prostate cancer and endometriosis
- Prevacid/Takepron (lansoprazole) – Gastroesophageal reflux disease
- Rozerem (ramelteon) – Insomnia
- Uloric (febuxostat) – Gout
- Velcade (bortezomib) – Multiple myeloma and mantle cell lymphoma (Millennium Pharmaceuticals)
![]() AT UK
AT UK
References
- “Financial Results for Fiscal 2012” (PDF). Takeda Pharmaceutical Company Limited. May 9, 2013. Retrieved June 13, 2013.
- “Takeda Initiates Cardiovascular Outcomes Trial for Alogliptin, An Investigational Treatment for Type 2 Diabetes”. Newsblaze.com. 2009-08-28. Retrieved 2010-09-18.
- “FAQ.” Takeda Pharmaceutical Company. Retrieved on February 2, 2011. “Q : Where is Takeda located? A : The Head Office is located in Osaka, Japan, and the Tokyo Head Office is located in Tokyo, Japan.”
- “Overview.” Takeda Pharmaceutical Company. Retrieved on February 2, 2011. “Headquarters Head Office 1-1, Doshomachi 4-chome, Chuo-ku, Osaka 540-8645” and “Tokyo Head Office 12-10, Nihonbashi 2-chome, Chuo-ku, Tokyo 103-8668”
- “TAP Pharmaceutical Products, Inc.: Private Company Information – BusinessWeek”. Investing.businessweek.com. 2008-04-30. Retrieved 2010-09-18.
- Decker, Susan (2009-07-06). “Takeda Sues Torrent to Stop Generic Copy of Actos Diabetes Pill”. Bloomberg. Retrieved 2010-09-18.
- Somers, Terri (2005-02-08). “Japanese drug giant taking over Syrrx here | The San Diego Union-Tribune”. Signonsandiego.com. Retrieved 2010-09-18.
- “Takeda, Amgen in exclusive tie-up for Japanese market”. MarketWatch. 2008-02-04. Retrieved 2010-09-18.
- Marrazzo, Amanda (2008-05-15). “Featured Articles From The Chicago Tribune”. Archives.chicagotribune.com. Retrieved 2010-09-18.
- “MILLENNIUM: The Takeda Oncology Company | About Millennium | Our History”. Mlnm.com. Retrieved 2010-09-18.
- staff (2008-06-15). “Takeda Signs On as Alnylam’s Asian Partner for $150M Upfront”. Genetic Engineering & Biotechnology News (print) (Mary Ann Liebert, Inc.). p. 14.
- http://www.takeda.com/press/article_43116.html
- Hirschler, Ben (May 25, 2012). “Farmacêutica Takeda comprará Multilab por até R$ 540 mi”. Grupo Abril (in portuguese). Exame. Retrieved January 27, 2013.
- “Locations | Worldwide | Takeda Pharmaceutical Company Limited”. Takedaism.com. Retrieved 2010-09-18.
- “By Business | Worldwide | Takeda Pharmaceutical Company Limited”. Takedaism.com. Retrieved 2010-09-18.
- “Annual Reports | Investor Information | Takeda Pharmaceutical Company Limited”. Takeda.com. Retrieved 2010-09-18.
 |
|
| Native name | 武田薬品工業株式会社 |
|---|---|
| Type | Public KK |
| Traded as | |
| Industry | Pharmaceuticals |
| Founded | Doshomachi, Osaka, Japan (June 12, 1781) |
| Headquarters | 1-1, Doshomachi Yonchome,Chuo-ku, Osaka, Japan |
| Key people | Yasuchika Hasegawa (President & CEO) |
| Revenue | |
| Operating income | |
| Net income | |
| Total assets | |
| Total equity | |
| Employees | 30,481 (2012) |
| Website | takeda.com (Global website) |
References:
|
|

CMC CENTRE
The Chemistry, Manufacturing and Controls (CMC) Center is a global organization responsible for overall R&D activities ranging from chemical information on development candidates to the processes leading to “manufacturing” of pharmaceutical products.
The main sites are located in Osaka and consist of the following laboratories: the Chemical Development Laboratories in charge of R&D for developing the manufacturing methods of active pharmaceutical ingredients and the manufacturing of drug substances for clinical samples; the Pharmaceutical Technology R&D Laboratories in charge of R&D for dosage forms, manufacturing and packaging, as well as manufacturing of clinical samples; and the Analytical Development Laboratories in charge of R&D for the development of analytical methods and stability studies of clinical samples. In addition, Hikari Bio-Manufacturing Technology Laboratories is located in Hikari (Yamaguchi) and this is where antibody drug substances are manufactured.
As for overseas sites, the Cambridge Biologics CMC Group (Massachusetts) and the Chicago Pharmaceutical Science Group (Illinois) are located in the USA, while the CMC Center Europe is mainly located in Roskilde, Denmark. All research and development activities at Takeda are promoted with the cooperation of these sites.
List of Publications of Takeda Research Laboratories
Trelagliptin succinate (SYR-472) for the treatment of type 2 diabetes.

Trelagliptin succinate (SYR-472)
2-[[6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2, 4-dioxopyrimidin-1-yl]methyl]-4-fluorobenzonitrile; butanedioic acid
2-[6-[3(R)-Aminopiperidin-1-yl]-3-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-1-ylmethyl]-4-fluorobenzonitrile
2- [ [6- [ (3R) -3-amino-l-piperidinyl] -3, 4-dihydro-3- methyl-2, 4-dioxo-l (2H) -pyrimidinyl]methyl] -4-fluorobenzonitrile
succinic acid salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Sponsor/Developer: Takeda Pharmaceuticals and Furiex Pharmaceuticals
Mechanism of action: DPP-4 inhibitor
865759-25-7 cas FREE BASE
1029877-94-8 succinate
- SYR 111472 succinate
- SYR 472
- Syr-472
- Syr111472 succinate
- Trelagliptin succinate
- UNII-4118932Z90
- clinical trials….http://clinicaltrials.gov/search/intervention=SYR+472
Trelagliptin-succinate M. Wt: 475.47
Trelagliptin-succinate Formula: C22H26FN5O6
SYR-472 is an oral dipeptidyl peptidase IV inhibitor originated by Takeda. It is in phase III clinical trials for the treatment of type 2 diabetes.
- Diabetes affects 25.8 million people of all ages, or roughly 8.3 percent of the U.S. population.
- The World Health Organization predicts that there will be 366 million people worldwide affected by diabetes by the year 2030.
- The advent of trelagliptin succinate, a unique once weekly medication for patients with type 2 Diabetes is now the focus of clinical trials and exciting research and development.
- Phase III clinical trials of trelagliptin succinate commenced in September 2011, and are estimated to be complete by the second half of 2013.

Indication (Phase): Japan—Once-weekly oral treatment for type 2 diabetes (Phase III; study expected to be completed in second half of 2013)

trelagliptin succinate
Compound I, A, TRELAGLIPTIN which has the formula:

is a DPP-IV inhibitor that is described in U.S. patent application Ser. No. 11/080,992 filed Mar. 15, 2005 (see Compound 34). Its dosing, administration and biological activities are described in U.S. patent application Ser. No. 11/531,671 filed Sep. 13, 2006. U.S. patent application Ser. No. 11/080,992 and Ser. No. 11/531,671 are incorporated herein by reference in their entirety.
Dipeptidyl peptidase IV (IUBMB Enzyme Nomenclature EC.3.4.14.5) (referred herein as “DPP-IV”) is a type II membrane protein and a non-classical serine aminodipeptidase that removes Xaa-Pro dipeptides from the amino terminus (N-terminus) of polypeptides and proteins. DPP-IV is constitutively expressed on epithelial and endothelial cells of a variety of different tissues (e.g., intestine, liver, lung, kidney and placenta), and is also found in body fluids. DPP-IV is also expressed on circulating T-lymphocytes and has been shown to be synonymous with the cell-surface antigen, CD-26. DPP-IV has been implicated in a number of human disease states, including, but are not limit to, diabetes, particularly type II diabetes mellitus, diabetic dislipidemia, conditions of impaired glucose tolerance (IGT), conditions of impaired fasting plasma glucose (IFG), metabolic acidosis, ketosis, appetite regulation and obesity; autoimmune diseases such as inflammatory bowel disease, multiple sclerosis and rheumatoid arthritis; AIDS; and cancers.
DPP-IV inhibitors are believed to be useful agents for the prevention, delay of progression, and/or treatment of conditions mediated by DPP-IV.
Compound (A) or a salt thereof has been reported as an inhibitor of dipeptidyl peptidase (DPP-IV) , which is an enzyme that decomposes glucagon-like peptide-1 (GLP-1) , a hormone increasing insulin secretion (patent document 1) .
In addition, a method including administering 1 – 250 mg of compound (A) or a salt thereof to a patient once per week (patent documents 2, 3), crystal polymorphs of compound (A) (patent documents 4, 5) , and a preparation of compound (A)
(patent documents 6, 7) have also been reported. Compound (A) and a salt thereof are recommended for oral administration in view of the easiness of self-administration, and a tablet, particularly a tablet in the dosage form for administration once per week, is desired. [0006]
The dosage form of once per week is expected to improve drug compliance of patients, whereas it requires supply of compound (A) or a salt thereof to patients in a high dose as compared to, for example, the dosage form of once per day. Since a solid preparation containing compound (A) or a salt thereof in a high dose increases its size, it may conversely degrade the drug compliance for patients, particularly infants and elderly patients having difficulty in swallowing
……………………..
SYNTHESIS
Compound 34 IS TRELAGLIPTIN

4-Fluoro-2-methylbenzonitrile (31).
A mixture of 2-bromo-5-fluorotoluene (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was refluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 31 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
2-Bromomethyl-4-fluorobenzonitrile (32).
A mixture of 4-fluoro-2-methylbenzonitrile (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
Alternatively, 32 was made as follows.
4-Fluoro-2-methylbenzonitrile (1 kg) in DCE (2 L) was treated with AIBN (122 g) and heated to 75° C. A suspension of DBH (353 g) in DCE (500 mL) was added at 75° C. portionwise over 20 minutes. This operation was repeated 5 more times over 2.5 hours. The mixture was then stirred for one additional hour and optionally monitored for completion by, for example, measuring the amount of residual benzonitrile using HPLC. Additional AIBN (e.g., 12.5 g) was optionally added to move the reaction toward completion. Heating was stopped and the mixture was allowed to cool overnight. N,N-diisopropylethylamine (1.3 L) was added (at <10° C. over 1.5 hours) and then diethyl phosphite (1.9 L) was added (at <20° C. over 30 min). The mixture was then stirred for 30 minutes or until completion. The mixture was then washed with 1% sodium metabisulfite solution (5 L) and purified with water (5 L). The organic phase was concentrated under vacuum to afford 32 as a dark brown oil (3328 g), which was used without further purification (purity was 97% (AUC)).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (33).
A mixture of crude 3-methyl-6-chlorouracil (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) was stirred at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
Alternatively, 33 was made as follows.
To a solution of 6-chloro-3-methyluracil (750 g) and N,N-diisopropylethylamine (998 mL) in NMP (3 L) was added (at <30° C. over 25 min) a solution of 32 (2963 g crude material containing 1300 g of 32 in 3 L of toluene). The mixture was then heated at 60° C. for 2 hours or until completion (as determined, for example, by HPLC). Heating was then stopped and the mixture was allowed to cool overnight. Purified water (3.8 L) was added, and the resultant slurry was stirred at ambient temperature for 1 hour and at <5° C. for one hour. The mixture was then filtered under vacuum and the wet cake was washed with IPA (2×2.25 L). The material was then dried in a vacuum oven at 40±5° C. for 16 or more hours to afford 33 as a tan solid (>85% yield; purity was >99% (AUC)).
TFAsalt OF TRELAGLIPTIN
2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (34).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
FREE BASE NOF TRELAGLIPTIN
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 60° C. until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 60° C. and the mixture was allowed to cool to <25° C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2×3.6 L). The filtrate was concentrated at 45° C. under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34.
HCL salt OF TRELAGLIPTIN
The HCl salt of 34 was prepared from the TFA salt as follows. The TFA salt (34) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0° C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Alternatively, the HCl salt was prepared from the free base as follows. To a solution of free base in CH2Cl2 (12 L) was added (at <35° C. over 18 minutes) 2 M hydrochloric acid (3.1 L). The slurry was stirred for 1 hour and then filtered. The wet cake was washed with CH2Cl2 (3.6 L) and then THF (4.8 L). The wet cake was then slurried in THF (4.8 L) for one hour and then filtered. The filter cake was again washed with THF (4.8 L). The material was then dried in a vacuum oven at 50° C. (with a nitrogen bleed) until a constant weight (e.g., >26 hours) to afford 34 as the HCl salt as a white solid (1423 g, >85% yield).
Succinate salt OF TRELAGLIPTIN

The succinate salt of 34 was prepared from the HCl salt as follows. To a mixture of the HCl salt of 34 (1414 g), CH2Cl2 (7 L) and purified water (14 L) was added 50% NaOH solution (212 mL) until the pH of the mixture was >12. The biphasic mixture was stirred for 30 min and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (5.7 L) and the combined organic layers were washed with purified water (6 L). The organic layer was then passed through an in-line filter and concentrated under vacuum at 30° C. over three hours to afford the free base as an off-white solid. The free base was slurried in prefiltered THF (15 L) and prefiltered IPA (5.5 L). The mixture was then heated at 60° C. until complete dissolution of the free base was observed. A prefiltered solution of succinic acid (446 g) in THF (7 L) was added (over 23 min) while maintaining the mixture temperature at >57° C. After stirring at 60° C. for 15 min, the heat was turned off, the material was allowed to cool, and the slurry was stirred for 12 hours at 25±5° C. The material was filtered under vacuum and the wet cake was washed with prefiltered IPA (2×4.2 L). The material was then dried in a vacuum oven at 70±5° C. (with a nitrogen bleed) for >80 hours to afford the succinate salt of 34 as a white solid (1546 g, >90% yield).
The product was also converted to a variety of corresponding acid addition salts. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of 34 were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(−)-Mandelic and benzenesulfonic. The succinate was found to be crystalline as determined by x-ray powder diffraction analysis.
Methanesulfonate salt
In addition, the methanesulfonate salt was prepared as follows. A 10.5 g aliquot of the benzonitrile product was mixed with 400 mL of isopropylacetate. The slurry was heated to 75° C. and filtered through #3 Whatman filter paper. The solution was heated back to 75° C. and a 1M solution of methanesulfonic acid (30.84 mL) was added slowly over 10 minutes while stirring. The suspension was cooled to room temperature at a rate of about 20° C./hr. After 1 hr at room temperature, the solid was filtered and dried in an oven overnight to obtain the methanesulfonate salt.
…………………………
FORMULATION
COMPD A IS TRELAGLIPTIN
Examples (Comparative Example IA)
Succinate of compound (A) (26.6 mg) was weighed in a glass bottle and used as Comparative Example IA. (Comparative Example 2A)
The succinate of compound (A) and microcrystalline cellulose were uniformly mixed in a mortar at a ratio of 1:10, and the mixture (226.6 mg) was weighed in a glass bottle and used as Comparative Example 2A. (Comparative Example 3A)
The succinate of compound (A) and corn starch were uniformly mixed in a mortar at a ratio of 1:5, and the mixture (126.6 mg) was weighed in a glass bottle and used as Comparative Example 3A. (Example IA) Succinate of compound (A) , mannitol and corn starch according to the formulation of Table IA were uniformly mixed in a fluid bed granulator (LAB-I, POWREX CORPORATION) , and the mixture was granulated by spraying an aqueous solution of dissolved hypromellose 2910, and dried therein. The obtained granules were passed through a sieve -(16M) to give milled granules. To the milled granules were added croscarmellose sodium, microcrystalline cellulose and magnesium stearate, and they were mixed in a bag to give granules for tableting. The granules were punched by a rotary tableting machine (Correct 19K, Kikusui Seisakusho, Ltd.) with a 6.5 mmφ punch to give a plain tablet weighting 121 mg. On the other hand, titanium oxide, yellow ferric oxide and talc were dispersed in a hypromellose 2910 aqueous solution to prepare a film coating liquid. The aforementioned coating liquid was sprayed onto the above-mentioned plain tablet in a film coating machine (Hicoater HCP-75, Freund Corporation), to give 2500 film- coated tablets containing 3.125 mg of compound (A) (free form) per tablet. Table IA

………………………..
POLYMORPHS AND SYNTHESIS
FORM A
Form A may be prepared by crystallization from the various solvents and under the various crystallization conditions used during the polymorph screen (e.g., fast and slow evaporation, cooling of saturated solutions, slurries, and solvent/antisolvent additions). Tables B and C of Example 3 summarize the procedures by which Form A was prepared. For example, Form A was obtained by room temperature slurry of an excess amount of Compound I in acetone, acetonitrile, dichloromethane, 1,4-dioxane, diethyl ether, hexane, methanol, isopropanol, water, ethylacetate, tetrahydrofuran, toluene, or other like solvents on a rotating wheel for approximately 5 or 7 days. The solids were collected by vacuum filtration, and air dried in the hood. Also, Form A was precipitated from a methanol solution of Compound I by slow evaporation (SE).
[0091] Form A was characterized by XRPD, TGA, hot stage microscopy, IR, Raman spectroscopy, solution 1H-NMR, and solid state 13C-NMR.
[0092] Figure 1 shows a characteristic XRPD spectrum (CuKa, λ=1.5418A) of Form A. The XRPD pattern confirmed that Form A was crystalline. Major X-Ray diffraction lines expressed in °2Θ and their relative intensities are summarized in Table 1.
Table 1. Characteristic XRPD Peaks (CuKa) of Form A


Characterization Data of Form A of Compound I

8. Amorphous Form
[0137] The Amorphous Form of Compound I was prepared by lyophilization of an aqueous solution of Compound I (Example 10). The residue material was characterized by XRPD and the resulting XRPD spectrum displayed in Figure 26. The XRPD spectrum shows a broad halo with no specific peaks present, which confirms that the material is amorphous. The material was further characterized by TGA, DSC, hot stage microscopy, and moisture sorption analysis.
Table A. Approximate Solubilities of Compound I



Crystallization Experiments of Compound I from Solvents




a) FE = fast evaporation; SE = slow evaporation; RT = room temperature; SC = slow cool;CC = crash cool, MB = moisture sorption/desorption analysis b) qty = quantity; PO = preferred orientation
…………………………
SYNTHESIS
EXAMPLES
1. Preparation of 2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro- 2H-pyrimidin-l-ylmethyl]-4-fluoro-benzonitrile and pharmaceutically acceptable salts


4-Fluoro-2-methylbenzonitrile (3)
[0166] A mixture of 2-bromo-5fluorotoluene ( 2) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) was re fluxed for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product 3 (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, IH), 6.93-7.06 (m, 2H), 2.55 (s, 3H). 2-Bromomethyl-4-fluorobenzonitrile (4)
[0167] A mixture of 4-fluoro-2-methylbenzonitrile (3) (2 g, 14.8 mmol), NBS (2.64 g, 15 mmol) and AIBN (100 mg) in CCl4 was refluxed under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give crude product as an oil, which was used in the next step without further purification.1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J= 5.2, 8.4 Hz, IH), 7.28 (dd, J= 2.4, 8.8 Hz, IH), 7.12 (m, IH), 4.6 (s, 2H).
2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4-fluoro- benzonitrile (6)
[0168] A mixture of crude 3-methyl-6-chlorouracil (5) (0.6 g, 3.8 mmol), 2- Bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO
(10 mL) was stirred at 60 C for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, 1=12, 8.4Hz, IH), 7.26 (d, J- 4.0Hz, IH), 7.11-7.17 (m, IH), 6.94 (dd, J=2.0, 9.0 Hz, IH), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for Ci3H9ClFN3O2, 293.68; found 293.68.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, TFA salt (1) (TFA salt of Compound I)

[0169] 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l-ylmethyl)-4- fluoro-benzonitrile (5) (300 mg, 1.0 mmol), (i?)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) were stirred in a sealed tube in EtOH (3 mL) at 100 0C for 2 hrs. The final compound was obtained as a TFA salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.16-7.27 (m, 2H), 5.46 (s, IH), 5.17-5.34 (ABq, 2H, J = 35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, IH), 2.67-2.92 (m, 2H), 2.07-2.17 (m, IH), 1.82-1.92 (m, IH), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
2-[6-(3-Amino-piperidin-l-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-l- ylmethyl]-4-fluoro-benzonitrile, HCl salt

[0170] The TFA salt of Compound I was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The residue was dissolved in acetonitrile and HCl in dioxane (1.5 eq.) was added at 0 C. The HCl salt was obtained after removing the solvent. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, IH), 7.12-7.26 (m, 2H), 5.47 (s, IH), 5.21-5.32 (ABq, 2H, J = 32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, IH), 2.69-2.93 (m, 2H), 2.07-2.17 (m, IH), 1.83-1.93 (m, IH), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for Ci8H20FN5O2, 357.38; found, 357.38.
General procedure for the preparation of salts of Compound I.
[0171] The benzonitrile product may be isolated as the free base if desired, but preferably, the product may be further converted to a corresponding acid addition salt. Specifically, the benzonitrile product (approximately 10 mg) in a solution of MeOH (1 mL) was treated with various acids (1.05 equivalents). The solutions were allowed to stand for three days open to the air. If a precipitate formed, the mixture was filtered and the salt dried. If no solid formed, the mixture was concentrated in vacuo and the residue isolated. In this way, salts of Compound I were prepared from the following acids: benzoic, p-toluenesulfonic, succinic, R-(-)-Mandelic and benzenesulfonic. [0172] The isolation and/or purification steps of the intermediate compounds in the above described process may optionally be avoided if the intermediates from the reaction mixture are obtained as relatively pure compounds and the by-products or impurities of the reaction mixture do not interfere with the subsequent reaction steps. Where feasible, one or more isolation steps may be eliminated to provide shorter processing times, and the elimination of further processing may also afford higher overall reaction yields.
…………………..
TABLET
2. Exemplary formulations comprising succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile
Provided are examples of tablet formulations that may be used to administer succinate salt of 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile (Succinate salt of Compound I) according to the present invention. It is noted that the formulations provided herein may be varied as is known in the art.
The exemplary tablet formulations are as follows:
| 12.5 mg of Compound I (weight of free base form) per tablet | ||||
| Core Tablet Formulation | ||||
| (1) | 2-[6-(3-Amino-piperidin-1-yl)-3-methyl-2,4- | 17.0 | mg | |
| dioxo-3,4-dihydro-2H-pyrimidin-1- | ||||
| ylmethyl]-4-fluoro-benzonitrile (succinate salt) | ||||
| (2) | Lactose Monohydrate, NF, Ph, Eur | 224.6 | mg | |
| (FOREMOST 316 FAST FLO) | ||||
| (3) | Microcrystalline Cellulose, NF, Ph, Eur | 120.1 | mg | |
| (AVICEL PH 102) | ||||
| (4) | Croscarmellose Sodium, NF, Ph, Eur | 32.0 | mg | |
| (AC-DO-SOL) | ||||
| (5) | Colloidal Silicon Dioxide, NF, Ph, Eur | 3.2 | mg | |
| (CAB-O-SIL M-5P) | ||||
| (6) | Magnesium Stearate, NF, Ph, Eur | 3.2 | mg | |
| (MALLINCKRODT, Non-bovine Hyqual) | ||||
| TOTAL | 400.0 | mg | ||
| (per tablet) | ||||
…………..
US20080227798 AND US20120197018
POLYMORPHS AND SYNTHESIS
EXAMPLES Example 1 Preparation of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl]-4-fluoro-benzonitrile succinate (Compound I)

Compound I may be prepared by the follow synthetic route (Scheme 1)

A. Preparation of 4-fluoro-2-methylbenzonitrile (Compound B)

Compound B was prepared by refluxing a mixture of 2-bromo-5-fluoro-toluene (Compound A) (3.5 g, 18.5 mmol) and CuCN (2 g, 22 mmol) in DMF (100 mL) for 24 hours. The reaction was diluted with water and extracted with hexane. The organics were dried over MgSO4 and the solvent removed to give product B (yield 60%). 1H-NMR (400 MHz, CDCl3): δ 7.60 (dd, J=5.6, 8.8 Hz, 1H), 6.93-7.06 (m, 2H), 2.55 (s, 3H).
B. Preparation of 2-bromomethyl-4-fluorobenzonitrile (Compound C)

Compound C was prepared by refluxing a mixture of 4-fluoro-2-methylbenzonitrile (Compound B) (2 g, 14.8 mmol), N-bromosuccinimide (NBS) (2.64 g, 15 mmol) and azo-bis-isobutyronitrile (AIBN) (100 mg) in CCl4 under nitrogen for 2 hours. The reaction was cooled to room temperature. The solid was removed by filtration. The organic solution was concentrated to give the crude product the form of an oil, which was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ 7.68 (dd, J=5.2, 8.4 Hz, 1H), 7.28 (dd, J=2.4, 8.8 Hz, 1H), 7.12 (m, 1H), 4.6 (s, 2H).
C. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound D)

Compound E was prepared by stirring a mixture of crude 3-methyl-6-chlorouracil D (0.6 g, 3.8 mmol), 2-bromomethyl-4-fluorobenzonitrile (0.86 g, 4 mmol) and K2CO3 (0.5 g, 4 mmol) in DMSO (10 mL) at 60° C. for 2 hours. The reaction was diluted with water and extracted with EtOAc. The organics were dried over MgSO4 and the solvent removed. The residue was purified by column chromatography. 0.66 g of the product was obtained (yield: 60%). 1H-NMR (400 MHz, CDCl3): δ 7.73 (dd, J=7.2, 8.4 Hz, 1H), 7.26 (d, J=4.0 Hz, 1H), 7.11-7.17 (m, 1H), 6.94 (dd, J=2.0, 9.0 Hz, 1H), 6.034 (s, 2H), 3.39 (s, 3H). MS (ES) [m+H] calc’d for C13H9ClFN3O2, 293.68; found 293.68.
D. Preparation of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound F)

Compound F was prepared by mixing and stirring 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile (Compound E) (300 mg, 1.0 mmol), (R)-3-amino-piperidine dihydrochloride (266 mg, 1.5 mmol) and sodium bicarbonate (500 mg, 5.4 mmol) in a sealed tube in EtOH (3 mL) at 100° C. for 2 hrs. The final compound was obtained as trifluoroacetate (TFA) salt after HPLC purification. 1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.16-7.27 (m, 2H), 5.46 (s, 1H), 5.17-5.34 (ABq, 2H, J=35.2, 15.6 Hz), 3.33-3.47 (m, 2H), 3.22 (s, 3H), 2.98-3.08 (m, 1H), 2.67-2.92 (m, 2H), 2.07-2.17 (m, 1H), 1.82-1.92 (m, 1H), 1.51-1.79 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
E. Preparation of Compound I: the succinic acid salt of 2-(6-Chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluoro-benzonitrile

The TFA salt prepared in the above step (Example 1, Step D) was suspended in DCM, and then washed with saturated Na2CO3. The organic layer was dried and removed in vacuo. The benzonitrile product (approximately 10 mg) was dissolved in MeOH (1 mL) and to which succinic acid in THF (1.05 equivalents) was added. The solutions were allowed to stand for three days open to the air. If a precipitate formed, the solid was collected by filtration. If no solid formed, the mixture was concentrated in vacuo, and the succinate salt was obtained after removing the solvent.
SUCCINATE SALT OF TRELAGLIPTIN
1H-NMR (400 MHz, CD3OD): δ. 7.77-7.84 (m, 1H), 7.12-7.26 (m, 2H), 5.47 (s, 1H), 5.21-5.32 (ABq, 2H, J=32.0, 16.0 Hz), 3.35-3.5 (m, 2H), 3.22 (s, 3H), 3.01-3.1 (m, 1H), 2.69-2.93 (m, 2H), 2.07-2.17 (m, 1H), 1.83-1.93 (m, 1H), 1.55-1.80 (m, 2H). MS (ES) [m+H] calc’d for C18H20FN5O2, 357.38; found, 357.38.
Compound I such prepared was found to be crystalline as determined by x-ray powder diffraction analysis (FIG. 1). The crystal material was designated Form A.
……………
patents
1. US 2013172377
2. WO 2011013639
3. WO 2009099172
4.WO 2009099171
5. WO 2008114807
6.WO 2008114800
7. WO 2008033851
8. WO 2007074884
9WO 2007035629
patent document 1: US2005/0261271
patent document 2: US2007/0060530
patent document 3: US2008/0287476
patent document 4: US2008/0227798
patent document 5: US2008/0280931
patent document 6: WO2008/114800
patent document 7: WO2011/013639
| US7906523 * | Oct 30, 2007 | Mar 15, 2011 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605 * | Nov 29, 2007 | Dec 27, 2011 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8188275 * | Oct 30, 2007 | May 29, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8222411 * | Sep 15, 2006 | Jul 17, 2012 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US20090275750 * | Sep 15, 2006 | Nov 5, 2009 | Jun Feng | Dipeptidyl peptidase inhibitors |
| WO2013183784A1 | Jun 4, 2013 | Dec 12, 2013 | Takeda Pharmaceutical Company Limited | Solid preparation |
| US20080227798 * | Nov 29, 2007 | Sep 18, 2008 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US20120197018 * | Feb 15, 2012 | Aug 2, 2012 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2h-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| WO2007033265A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033266A2 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetis |
| WO2007033350A1 * | Sep 13, 2006 | Mar 22, 2007 | Takeda Pharmaceutical | Dipeptidyl peptidase inhibitors for treating diabetes |
| EP1586571A1 * | Dec 21, 2004 | Oct 19, 2005 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
13 NMR TRELAGLIPTIN SUCCINATE

1H NMR TRELAGLIPTIN SUCCINATE

VARDENAFIL

Mirodenafil 米罗那非 标准品 ………..An erectogenic agent.

862189-95-5 (free base)
SK Chemicals (Originator)
Treatment of Erectile Dysfunction , hypertention
Mirodenafil belongs to a class of drugs called PDE5 inhibitors, which many other erectile dysfunction drugs such as sildenafil, tadalafil, andvardenafil also belong to. It was developed by SK Chemicals Life Science and is marketed under the trade name of Mvix tab which comes in different doses (50 mg, 100 mg).
Mirodenafil is also available under the name of Mvix S ODF 50 mg as an orally dissolving film (ODF) which dissolves on the tongue without water. It is the first licensed medicine for the treatment of erectile dysfunction as a dosage form of film.
Mirodenafil is a newly developed oral phosphodiesterase type 5 inhibitor, currently under investigation as a treatment for erectile dysfunction (ED).
MIRODENAFIL米罗那非 标准品
Mirodenafil hydrochloride is a high selective PDE5 inhibitor commercialized by SK Chemicals which had been in early clinical development for the treatment of erectile dysfunction (ED). Early clinical studies had also been ongoing for the treatment of hypertension in patients taking amlodipine; however, no recent development has been reported for this research. The development of compound started in 1998 jointly by SK Chemicals and a bio-venture In2Gen.
Several clinical trials were conducted,[1][2][3] but mirodenafil has not been approved for use in the United States by the U.S. Food and Drug Administration.

Mirodenafil dihydrochloride
|
||||||||||||
Korean Patent No. 358083 discloses pyrrolopyrimidinone derivatives having good inhibition activity against PDE-5, a method of its preparation thereof, an intermediate compound used to prepare the same and their use for prevention and treatment of erectile dysfunction, pulmonary arterial hypertension, chronic obstructive pulmonary disease, benign prostatic hypertrophy and lower urinary tract diseases.
Of the pyrrolopyrimidinone derivatives disclosed in Korean Patent No. 358083, 5-ethyl-2-{5-[4- (2-hydroxyethyl)piperazin-1-ylsulfonyl]-2-n-propoxyphenyl}-7-n-propyl-l-3,5-dihydro-4 H-pyrrolo[3,2-d]pyrimidin-4-one (hereinafter, “SK-3530”) represented by the following formula (1 ) is an excellent selective inhibitor PDE-5 over other PDEs and is under clinical trial for the treatment of erectile dysfunction after passing through the preclinical stage.

The dihydrochloride salt (2HCI) of SK-3530 has been under investigation through the preclinical and clinical stages.
The SK-3530 dihydrochloride salt has good solubility and can be easily stabilized for pharmaceutical preparation. But, it has the following drawbacks.
First, because the SK-3530 dihydrochloride salt is hygroscopic, it easily absorbs moisture from the atmosphere and becomes discolored when the moisture content is high. And, due to the hygroscopic property, an anhydrous solvent condition and a dry air condition have to be provided to obtain a stable product. Second, the SK-3530 dihydrochloride salt should be kept at a temperature lower than room temperature because it does not show enough stability at room temperature. In particular, the SK-3530 dihydrochloride salt is labile to heat or light, and thus any prolonged exposure to heat or light results in various impurities.
Third, the SK-3530 dihydrochloride salt could corrode the punch during tablet ting due to its somewhat corrosive properties. This is because the SK-3530 dihydrochloride salt is a simple amorphous salt rather than being a stable crystalline acid addition salt or hydrate form. Thus, one of the two hydrochloric acid groups with a relatively weak ionic bond character may leave the molecule under severe conditions. As aforementioned, the SK-3530 dihydrochloride salt may be endowed with a sufficient stability for pharmaceutical preparation. But, some additional techniques and costs are needed due to the deficiency in intrinsic physicochemical property and stability of the compound.


…………………………

The invention relates to a series of pyrrolopyrimidinone derivatives of the formula (1):

R1 ETHYL
R2=H
R3= PROPYL
R4 = PROPYL
R5=R5=SO2NR6R7, NR6R7 is 4-(3-hydroxypropyl)piperazinyl) IS MIRODENAFIL
ANALOGOUS METHOD
BELOW IS CUT PASTE OF R1 METHYL ANALOGUE ……………..R1 =METHYL AND NOT ETHYL ….CAUTION
Example 39 Preparation of
5-(5-(4-(2-hydroxyethyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one hydrochloride (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(2-hydroxyethyl)piperazinyl)
The titled compound was prepared as described in Example 2 by using 5-(5-(4-(2-hydroxyethyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one in place of 5-(2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one.
yield: 99%
mp 66.5° C. dec;
IR (neat) 3332 (NH and OH), 1676 (C═O), 1166 (SO2) cm−1;
1H NMR (DMSO-d6) δ 0.92 (t, J=7.2 Hz, 3H, CH2CH2CH3), 0.96 (t, J=7.2 Hz, 3H, OCH2CH2CH3), 1.56-1.80 (m, 4H, 2 CH2CH2CH3), 2.59 (t, J=7.5 Hz, 2H, CH2CH2CH3), 2.91 (br t, J=11.7 Hz, 2H, 2 SO2NCHax), 3.12-3.27 (m, 4H, NCH2CH2 and 2 SO2NCHeq), 3.58 (br d, J=11.7 Hz, 2H, 2 +HNCHax), 3.68-3.85 (m, 4H, CH2CH2OH and 2 +HNCHeq), 4.00 (s, 3H, NCH3), 4.15 (t, J=6.3 Hz, 2H, OCH2CH2CH3), 4.66 (br s, 1H, OH), 7.28 (s, 1H, H-2), 7.44 (d, J=9.0 Hz, 1H, H-3′), 7.89 (dd, J=9.0 Hz, 2.4 Hz, 1H, H-4′), 8.01 (d, J=2.4 Hz, 1H, H-6′), 10.85 (br s, 1H, NH+), 12.01 (br s, 1H, NH).
Example 42 Preparation of
5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(3-hydroxypropyl)piperazinyl)
The titled compound was prepared as described in Example 1 by using 5-(5-chlorosulfonyl-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one and 1-(3-hydroxypropyl)piperazine in place of 5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one and 1-methylpiperazine.
yield: 94%
mp 162.5° C. dec (EtOAc/hexanes);
IR (neat) 3484, 3302 (NH and OH), 1669 (C═O), 1170 (SO2) cm−1;
1H NMR (CDCl3/TMS) δ 1.00 (t, J=7.5 Hz, 3H, CH2CH2CH3), 1.20 (t, J=7.5 Hz, 3H, OCH2CH2CH3), 1.64-1.80 (m, 4H, CH2CH2CH2OH and CH2CH2CH3), 1.99-2.11 (m, 2H, OCH2CH2CH3), 2.58-2.64 (m, 6H, NCH2CH2 and 2 NCH2), 2.71 (t, J=7.5 Hz, 2H, CH2CH2CH3), 3.08 (br s, 4H, 2 SO2NCH2), 3.71 (t, J=5.4 Hz, 2H, CH2CH2OH), 4.08 (s, 3H, NCH3), 4.26 (t, J=6.3 Hz, 2H, OCH2CH2CH3), 4.28 (br s, 1H, OH), 6.88 (s, 1H, H-2), 7.14 (d, J=8.7 Hz, 1H, H-3′), 7.77 (dd, J=8.7 Hz, 2.7 Hz, 1H, H-4′), 8.87 (d, J=2.7 Hz, 1H, H-6′), 10.69 (br s, 1H, NH); MS (FAB) m/z 532 (MH+).
Example 43 Preparation of
5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one hydrochloride (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(3-hydroxypropyl)piperazinyl)
The titled compound was prepared as described in Example 2 by using 5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one in place of 5-(2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one.
yield: 99%
mp 62.5° C. dec;
IR (neat) 3347, 3321 (NH and OH), 1689 (C═O), 1168 (SO2) cm−1;
1H NMR (DMSO-d6) δ 0.93 (t, J=7.5 Hz, 3H, CH2CH2CH3), 0.96 (t, J=7.5 Hz, 3H, OCH2CH2CH3), 1.57-1.87 (m, 6H, CH2CH2CH2OH and 2 CH2CH2CH3), 2.59 (t, J=7.5 Hz, 2H, CH2CH2CH3), 2.89 (br t, J=11.7 Hz, 2H, 2 SO2NCHax), 3.01-3.19 (m, 4H, NCH2CH2 and 2 SO2NCHeq), 3.44 (t, J=6.0 Hz, 2H, CH2CH2OH), 3.52 (br d, J=11.7 Hz, 2H, 2 +HNCHax), 3.79 (br d, J=11.7 Hz, 2H, 2 +HNCHeq), 4.00 (s, 3H, NCH3), 4.15 (t, J=6.6 Hz, 2H, OCH2CH2CH3), 4.71 (br s, 1H, OH), 7.29 (s, 1H, H-2), 7.44 (d, J=8.7 Hz, 1H, H-3′), 7.89 (dd, J=8.7 Hz, 2.4 Hz, 1H, H-4′), 8.02 (d, J=2.4 Hz, 1H, H-6′), 11.13 (br s, 1H, NH+), 12.05 (br s, 1H, NH).
……………………………
Synthesis from patent and some construction by me
you can synthesize as follows, A CHEMIST CAN PICK THIS UP, this is not available clearly anywhere
Chlorosulfonation of the methyl salicylate with ClSO3H in SOCl2 affords the Methyl 3-Chlorosulfonyl-6-hydroxybenzoate described below
THESE INTERMEDIATES FROM PATENT MAY HELP YOU
 methyl salicylate
methyl salicylate
 X=CL, R8=ME
X=CL, R8=ME
- Methyl 3-Chlorosulfonyl-6-hydroxybenzoate
Example 1 EP1362858A1
-
To a cooled solution of SOCl2 (156 g, 1. 31 mol) and ClSO3H (460 g, 3.94 mol) at 0°C was added slowly methyl salicylate (200 g, 1.31 mol) for 30 minutes, and the mixture was stirred at room temperature for 20 hours. The reaction mixture was poured slowly into the ice (2 kg) and H2O (3 L) mixture, and the resulting white precipitates were collected by filtration. The filtered solid was washed with H2O (3 L), air-dried for 2 days and then dried under vacuum at 40°C for 2 days to afford the titled product (232 g, 93%) as a white solid.
mp 76.5-77.5 °C (toluene/hexanes);
IR (neat) 1699 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 3. 90 (s, 3 H, OCH3), 6. 93 (d, J= 8. 7 Hz, 1 H, H-3), 7. 70 (dd, J= 8. 7 Hz, 2. 4 Hz, 1 H, H-4), 8. 03 (d, J= 2. 4 Hz, 1 H, H-6).
- Methyl 3-Chlorosulfonyl-6-hydroxybenzoate
Example 2 EP1362858A1
-
 1-(2-hydroxyethyl)piperazine
1-(2-hydroxyethyl)piperazine  R8=ME, W=N, n=2
R8=ME, W=N, n=2-
- Methyl 2-Hydroxy-5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]benzoate
-
To a mixture of 1-(2-hydroxyethyl)piperazine (27 mg, 0. 21 mmol) and K2CO3 (33 mg, 0. 24 mmol) in DMF (5 mL) was added methyl 3-chlorosulfonyl-6-hydroxybenzoate (50 mg, 0. 20 mmol), and the mixture was stirred at room temperature for 1 hour. The reaction mixture was washed with H2O (10 mL), and the aqueous layer was further extracted with 5% MeOH in CH2Cl2 (20 mL). The combined organic layer was dried (MgSO4), filtered, and the filtrate was evaporated to dryness under reduced pressure. The crude residue was purified by MPLC on silica gel (5% MeOH in CH2Cl2) to afford the titled compound (59 mg, 86%) as white solid.
mp 152 °C (dec) (CH2Cl2/ether);
IR (neat) 1685 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 2. 30 (br s, 1 H, CH2OH), 2. 63 (t, J = 5. 4 Hz, 2 H, NCH 2CH2O), 2. 70 (m, 4 H, 2 NCH2), 3. 12 (m, 4 H, 2 SO2NCH2), 3. 64 (t, J= 5. 4 Hz, 2 H, NCH2CH 2O), 4. 01 (s, 3 H, OCH3), 7. 12 (d, J= 8. 7 Hz, 1 H, H-3), 7. 81 (dd, J= 8. 7 Hz, 2. 4 Hz, 1 H, H-4), 8. 26 (d, J = 2. 4 Hz, 1 H, H-6), 11. 26 (br s, 1 H, OH);
MS (FAB) m/z 345 (MH+).
- Methyl 2-Hydroxy-5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]benzoate
Example 3 EP1362858A1
Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate
-
To a mixture of methyl 2-hydroxy-5-(4-(2-hydroxyethyl)piperazin-1-ylsulfonyl)benzoate (800 mg, 2. 32 mmol) and K2CO3 (482 mg, 3. 49 mmol) in DMF (5 mL) was added 1-bromopropane (253 µL, 2.79 mmol), and the mixture was stirred at 60°C overnight. The reaction mixture was evaporated to dryness under reduced pressure, washed with H2O (10 mL), and the aqueous layer was further extracted with CH2Cl2 (50 mL x 2). The combined organic layer was dried (MgSO4), filtered, and the filtrate was evaporated to dryness under reduced pressure. The crude residue was purified by MPLC on silica gel (3% MeOH in CHCl3) to afford the titled compound (309 mg, 80%) as a white solid.
mp 88-89 °C (EtOAc/hexanes);
IR (neat) 3242 (OH), 1741 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 1. 09 (t, J = 7. 5 Hz, 3 H, OCH2CH2CH 3), 1. 84-1. 95 (m, 2 H, OCH2CH 2CH3), 2. 23 (br s, 1 H, CH2OH), 2. 54 (t, J= 5. 4 Hz, 2 H, NCH 2CH2O), 2. 60 (m, 4 H, 2 NCH2), 3. 04 (m, 4 H, 2 SO2NCH2), 3. 58 (t,J = 5. 4 Hz, 2 H, NCH2CH 2O), 3. 91 (s, 3 H, OCH3), 4. 08 (t, J= 6. 6 Hz, 2 H, OCH 2CH2CH3), 7. 07 (d, J = 9. 0 Hz, 1 H, H-3), 7. 82 (dd, J = 9. 0 Hz, 2. 4 Hz, 1 H, H-4), 8. 15 (d, J = 2. 4 Hz, 1 H, H-6);
MS (FAB) m/z 387 (MH+).
- FURTHER INFO OTHER THAN ABOVE PATENT
- HYDROLYSE Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate TO -COOLi SALT using LiOH
- CONDENSE WITH 3-amino-1-ethyl-4-propyl-1H-pyrrole-2-carboxamide USING HOBt AND DMAP/ PYRIDINE

9……….Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate R8= ME, R4=PROPYL, W=N, n=2
10……….3-amino-1-ethyl-4-propyl-1H-pyrrole-2-carboxamide R1=ETHYL, R2=H, R3=PROPYL, IN ABOVE
YOU WILL GET A COMPD

R1 ETHYL
R2=H
R3= PROPYL
R4 = PROPYL
W=N
n=2
IS MIRODENAFIL precursor ie n-1 compund
- CYCLIZE THIS WITH BuOK/tBuOH AND USE ACID TO GET FINAL PRODUCT MIRODENAFIL
- A cyclization reaction is generally carried out by heating at an elevated temperature, for example 50-150° C., in the presence of an acid or a base in a suitable solvent such as an aqueous C1-C4 alkanol, water, a halogenated hydrocarbon, or acetonitrile. Thus, for example, the cyclization may be affected by treatment of a compound with an inorganic or organic base such as sodium hydroxide, potassium carbonate or potassium tert-butoxide, in an alcoholic aqueous medium, preferably potassium tert-butoxide in tert-butanol at 60° C. to reflux temperature.
SYNTHESIS OF 1-(2-hydroxyethyl)piperazine needed for MIRODENAFIL SYNTHESIS
Compounds of the formula (29) can be prepared from the compounds of the formula (30):

wherein X and P are as previously defined.
note X=N ATOM, n = 2
…………………………….
 MIRODENAFIL
MIRODENAFIL
Two methods were published for the determination of mirodenafil in biological fluids. Choi et al. (2009) describe an isocratic reversed-phase liquid chromatographic method for simultaneous analysis of mirodenafil and its two main metabolites, SK3541 and SK3544, in rat plasma, urine, and tissue homogenates. The authors used a simple deproteinization procedure for sample preparation, and the compounds were separated on a C18 column (250 mm x 4.6 mm, i.d.; 5 µm particle size; Shiseido, Tokyo, Japan). The mobile phase was constituted with 0.02 M ammonium acetate buffer (pH 6):acetonitrile (52:48, v/v) at a flow rate of 1.4 mL/min. UV detection was at 254 nm.
Lee et al. (2009) developed a study with the proposed method to determine sildenafil and mirodenafil in the plasma and corpus cavernosum tissue of rats using LC–MS/MS. A CapcellPak phenyl column (2.1mm x 150 mm, 5µm) maintained constant at 40 ºC was used for the separation. The mobile phase consisted of 90% acetonitrile in 5 mM ammonium formate (pH 6.0). A gradient program was used for the LC separation with a flow rate of 0.2 mL/min.
References
- Paick JS, Ahn TY, Choi HK, Chung WS, Kim JJ, Kim SC, Kim SW, Lee SW, Min KS, Moon KH, Park JK, Park K, Park NC, Suh JK, Yang DY, Jung HG (November 2008). “Efficacy and safety of mirodenafil, a new oral phosphodiesterase type 5 inhibitor, for treatment of erectile dysfunction”. The Journal of Sexual Medicine 5 (11): 2672–80. doi:10.1111/j.1743-6109.2008.00945.x. PMID 18638004.
- Kim BH, Yi S, Kim J, Lim KS, Kim KP, Lee B, Shin SG, Jang IJ, Yu KS (June 2009). “Influence of alcohol on the hemodynamic effects and pharmacokinetic properties of mirodenafil: a single-dose, randomized-sequence, open-label, crossover study in healthy male volunteers in Korea”.Clinical Therapeutics 31 (6): 1234–43. doi:10.1016/j.clinthera.2009.06.008. PMID 19695390.
- Shin KH, Kim BH, Kim TE, Kim JW, Yi S, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS (December 2009). “The effects of ketoconazole and rifampicin on the pharmacokinetics of mirodenafil in healthy Korean male volunteers: an open-label, one-sequence, three-period, three-treatment crossover study”.Clinical Therapeutics 31 (12): 3009–20. doi:10.1016/j.clinthera.2009.12.012. PMID 20110038.
- Synthesis of 5-ethyl-2-[5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]-2-n-propoxyphenyl]-7-n-propyl-3,5-dihydro-4H-pyrrolo[3,2-d]-[2-14C]pyrimidin-4-one·2 HCl (14C-SK3530·2 HCl)J Label Compd Radiopharm 2006, 49(13): 1141
- More information about mirodenafil can be found at Paick J S et al., (2008) The Journal of Sexual Medicine, 5 (11): 2672-80.
- PDE-5 inhibitor that came into the market recently (Choi et al., 2009; Lee et al., 2009).not currently approved for use in the United States but clinical trials are being conducted.
- Crystal forms of SK-3530.
Song HO, Sohn YT.Arch Pharm Res. 2010 Dec;33(12):2033-6. doi: 10.1007/s12272-010-1220-3. Epub 2010 Dec 30. - Looking to the future for erectile dysfunction therapies.Hatzimouratidis K, Hatzichristou DG.Drugs. 2008;68(2):231-50. Review.
-
- Paick JS, Ahn TY, Choi HK, Chung WS, Kim JJ, Kim SC, Kim SW, Lee SW, Min KS, Moon KH, Park JK, Park K, Park NC, Suh JK, Yang DY, Jung HG (November 2008). “Efficacy and safety of mirodenafil, a new oral phosphodiesterase type 5 inhibitor, for treatment of erectile dysfunction”. The Journal of Sexual Medicine 5 (11): 2672–80. doi:10.1111/j.1743-6109.2008.00945.x. PMID 18638004.
- Kim BH, Yi S, Kim J, Lim KS, Kim KP, Lee B, Shin SG, Jang IJ, Yu KS (June 2009). “Influence of alcohol on the hemodynamic effects and pharmacokinetic properties of mirodenafil: a single-dose, randomized-sequence, open-label, crossover study in healthy male volunteers in Korea”. Clinical Therapeutics 31 (6): 1234–43. doi:10.1016/j.clinthera.2009.06.008. PMID 19695390.
- Shin KH, Kim BH, Kim TE, Kim JW, Yi S, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS (December 2009). “The effects of ketoconazole and rifampicin on the pharmacokinetics of mirodenafil in healthy Korean male volunteers: an open-label, one-sequence, three-period, three-treatment crossover study”. Clinical Therapeutics 31 (12): 3009–20. doi:10.1016/j.clinthera.2009.12.012. PMID 20110038.
- Matheny, C., et al., Drug Metab. Dispos., 32, 1008 (2004)
Gupta, M., et al., J. Clin. Pharmacol., 45, 987 (2005)
Ek, M., et al., Biochem. Pharmacol., 74, 496 (2007)
Lee, H., et al., Xenobiotica, 38, 21 (2008)

| WO2006018088A1 * | Jul 15, 2005 | Feb 23, 2006 | Switch Biotech Ag | Use of a pde 5 inhibitor for treating and preventing hypopigmentary disorders |
| KR20010083637A * | Title not available |
| US6962911 * | Feb 15, 2001 | Nov 8, 2005 | Sk Chemicals Co., Ltd. | Pyrrolopyrimidinone derivatives, process of preparation and use |
| US20100069632 * | Jul 3, 2007 | Mar 18, 2010 | Sk Chemicals Co., Ltd | Salts of pyrrolopyrimidinone derivatives and process for preparing the same |
| EP2038282A1 * | Jul 3, 2007 | Mar 25, 2009 | SK Chemicals, Co., Ltd. | Salts of pyrrolopyrimidinone derivatives and process for preparing the same |


