
Home » Uncategorized (Page 152)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

FDA Breakthrough Therapy Designation: Fourth Drug Receives FDA Approval
.
The FDA approves on February 12, the fourth drug to have the coveted Breakthrough Therapy Designation (BTD). The approval is for orphan drug Imbruvica (Ibrutinib) as a single agent for the treatment of patients with Chronic Lymphocytic Leukemia (CLL) who have received at least one prior therapy. Imbruvica, a once-daily, oral kinase inhibitor, is developed and commercialized by Pharmacyclics and Janssen Biotech. Imbruvica is the :
• 1st FDA BTD drug to receive approval in 2014
• 1st FDA BTD drug to receive approval for a 2nd indication – Mantle Cell Lymphoma (MCL) on 11.13.13
• 2nd FDA BTD drug to receive approval for the same indication, Chronic Lymphocytic Leukemia (CLL) – Genentech’s Gazyva (Obinutuzumab) receives approval for CLL on 11.01.13
• 3rd FDA BTD drug to receive approval for an oncology indication
• 4th FDA BTD drug to receive approval.
The media, investors, patients, regulatory agencies, and the pharmaceutical industry…
View original post 194 more words
The secret of fertile sperm

Progesterone and other fatty signaling molecules are critical for sperm fertility. Credit: C. Cain
To better understand the causes of male infertility, a team of Bay Area researchers is exploring the factors, both physiological and biochemical, that differentiate fertile sperm from infertile sperm. At the 58th Annual Biophysical Society Meeting, which takes place Feb. 15-19, 2014, in San Francisco, Calif., the team will present its work to identify and characterize proteins known as ion channels, which are crucial for sperm fertility and expressed within a sperm cell’s plasma membrane.
“Any knowledge gained in this area may help create much-needed diagnostic testing and treatments for male infertility, which is in essence an idiopathic disease, because at this time 80 percent of male infertility cases can’t be diagnosed or treated,” said Melissa Miller, a postdoctoral fellow who will present the team’s findings at the meeting. Miller works in the labs of both…
View original post 328 more words
Researchers discover how cancer ‘invisibility cloak’ works

Researchers at National Jewish Health have discovered how a lipid secreted by cancer tumors prevents the immune system from mounting an immune response against it. When lysophosphatidic acid (LPA) binds to killer T cells, it acts almost like an “invisibility cloak,” preventing T cells from recognizing and attacking nascent tumors.
“In recent years, several therapeutic medicines have been developed that spur a person’s own immune system to fight cancer,” said Raul Torres, PhD, professor of immunology at National Jewish Health, and senior author on the paper, published in the October issue of Cancer Immunology Research. “Our findings suggest new targets and strategies for enlisting the immune system’s help in fighting cancer.”
Scientists believe the human immune system recognizes and destroys many cancerous cells before they develop into dangerous tumors. However, tumors also employ strategies to evade detection by the immune system.
Scientists have known that LPA is secreted…
View original post 164 more words
IkB kinase inhibitors , SANOFI, for osteoarthritis

K salt monohydrate, N-[[2-[2-(methylamino)-4-pyrimidinyl]-1H-indol-5-yl]carbonyl]-3-(phenyl-2-pyridinylamino)- L-Alanine,
2-{[2-(2-methylamino-pyrimidin-4-yl)-lH-indole-5- carbonyl]-amino}-3-(phenylpyridin-2-yl-amino)-propionic acid, as the monopotassium monohydrate salt., 899418-66-7 , C28 H25 N7 O3 . H2 O . K
IC 50= 0.4 nm
K SALT
L-Alanine, N-[[2-[2-(methylamino)-4-pyrimidinyl]-1H-indol-5-yl]carbonyl]-3-(phenyl-2-pyridinylamino)-, monopotassium salt , 899418-65-6, C28 H25 N7 O3 . K
Free acid
- C28 H25 N7 O3
- N-[[2-[2-(methylamino)-4-pyrimidinyl]-1H-indol-5-yl]carbonyl]-3-(phenyl-2-pyridinylamino)- L-Alanine,
- 869796-50-9
As an inhibitor of IKB kinase, the compound of the invention, functions via the selective inhibition of IKK, particularly an IKK-2 inhibitor; as well as exhibiting localized activity, as opposed to a systemic activity. Such an inhibitor is particularly useful for treating a patient suffering from or subject to IKK- 2 mediated pathological diseases or conditions, e.g., asthma, rhinitis, chronic obstructive pulmonary disorder (COPD), or COPD exacerbations, that could be ameliorated by the targeted administering of the inhibitor.
Sanofi.. INNOVATOR
SANOFI LISTS http://clinicaltrials.gov/show/NCT01463488 SAR113945 AS IkB kinase inhibitors IN PHASE II…. BUT I AM NOT SURE OF THIS….Protein Kinases as Small Molecule Inhibitor Targets … – ResearchGa click here to see see table 7 (cont)……2227
EMAIL ME amcrasto@gmail.com
WO 2005113554
………………….
Synthesis
EXAMPLES
Example 1, Step 1
Synthesis of 2-{[2-(2-Methylamino-pyrimidin-4-yl)-lH-indole-5-carbonyl]amino}-3-(phenyl-pyridin-
2-yl-amino)-propionic acid

6.04 mmol of the 2-{[2-(2-methylamino-pyrimidin-4-yl)-lH-indole-5-carbonyl]-amino}-3-(phenyl- ρyridin-2-yl-amino)-propionic acid, methyl ester prepared essentially as described in patent application WO2005/113544, is dissolved in 70 mL of ethanol. 24.2 mL of 0.5 N aqueous ΝaOΗ is added and the mixture is stirred at room temperature for 2 h. After the reaction is complete, the pH is adjusted to ~5 using 1 N HCl. Water is added slowly and the resulting precipitate is filtered off and washed with water. After drying under reduced pressure of about 1 mbar at 400C, 2.49 g of 2-{[2-(2-methylamino- pyrimidin-4-yl)-lH-indole-5-carbonyl]-arnino}-3-(phenyl-pyridin-2-yl-amino)-propionic acid is isolated. Empirical formula C28H25N7O3; M. W. = 507.56; MS (M+H) 508.3. 1H NMR (DMSO-^6) 2.95 (s, 3 H), 4.32-4.50 (m, 2 H), 4.65-4.72 (m, 1 H), 6.29-6.36 (d? 1 H), 6.70- 6.79 (m, 1 H), 6.90-7.10 (sb, 1 H), 7.13-7.19 (m, 1 H), 7.22-7.38 (m, 4 H), 7.40-7.48 (m, 3 H), 7.50-7.55 (m, 1 H), 7.57-7.60 (m, 1 H), 7.96 (bs, 1 H), 8.34-8.40(m, 2 H), 8.80-8.90 (d, 1 H), 11.80 (s, 1 H) 12.8 (bs, IH). Chiral HPLC shows 94% ee.
Example 1, Step 2
Enantiomeric Purification of 2-{[2-(2-Methylaminopyrimidin-4-yl)-lH-indole-5-carbonyl]amino}-3-
(phenylpyridin-2-yl-amino)-propionic acid

2- { [2-(2-methylaminopyrimidin-4-yl)- lH-indole-5-carbonyl]amino} -3-(phenylpyridin-2-yl-amino)- propionic acid, prepared essentially according to Example 1, Step 1 above, is heated under reflux for 15 minutes. The insoluble racemic compound is removed by hot filtration. The TΗF of the resulting filtrate is removed by distillation and the residue is precipitated by the addition of isopropanol. After drying under reduced pressure of about 1 mbar at 400C, the desired 2-{[2-(2-methylaminopyrimidin-4- yl)-lH-indole-5-carbonyl]amino}-3-(phenylpyridin-2-yl-amino)-propionic acid is isolated with an ee = 98.5%.
Example 1, Step 3
Synthesis of 2-{[2-(2-Methylamino-pyrimidin-4-yl)-lH-indole-5-carbonyl]-amino}-3-(phenyl-pjτidin- 2-yl-amino)-propionic acid monopotassium monohydrate salt

To a slurry of 2-{[2-(2-methylaminopyrimidin-4-yl)-lH-indole-5-carbonyl]amino}-3-(phenylpyridin- 2-yl-amino)-propionic acid (50.8 mmol from Example 1, Step 2 above) in H2O and EtOH is added 1.02 M KOH (2.00 equiv) with vigorous swirling. The mixture is heated to 670C with swirling on a steam bath to dissolve the starting material, while braking up any remaining clumps. After several minutes the clear orange solution is filtered and the flask containing the filtrate is wrapped in aluminum foil and allowed to cool slowly to room temperature in the hot water remaining in the steam bath. After 17 hours, the mixture is cooled in an ice-bath and the salt is collected by filtration and washed 4 times with ice-cold H2O. The last two washes have a pH of 8. The salt is dried in a vacuum oven at 45 0C with an N2 bleed to yield the desired compound as fine needles:1H NMR (DMSO-«k) 2.95 (s,3 H)5 3.95-4.05 (m, 1 H), 4.35-4.40 (m, IH), 4.55-4.62 (m, 1 H), 6.35-6.39 (d, 1 H), 6.58-6.60 (m, IH), 6.90-7.10 (sb, 1 H), 7.13-7.19 (m, 1 H), 7.22-7.38 (m, 6 H), 7.40-7.48 (m, 3 H), 7.57-7.60 (m,l H), 7.70 (s, 1 H), 8.10-8.15(d, 1 H), 8.30 (bs, 1 H), 11.80 (s, 1 H); LC-MS m/z 509 (M+ + 2), 508 (M+ H- I), 275, 254 (100). Anal. Calcd for C28H24KN7O3-H2O (563.66): C, 59.67; H, 4.65; N, 17.39; K. 6.94; H2O (Karl Fischer), 3.20. Found: C, 59.59; H, 4.66; N, 17.39; K5 6.44; H2O (Karl Fischer), 3.16. Chiral HPLC showed 99.5% S-enantiomer.
Example 2 Synthesis of 2-{[2-(2-Methylammo-pyrimidin-4-yl)-lH-indole-5-carbonyl]-amino}-3-(phenyl-pyridin-
2-yl-amino)-propionic acid monopotassium monohydrate salt

As an alternative procedure for preparing the compound of formula Ha3 (3.8 mmol) of methyl ester 1 is dissolved in ethanol and water and 2 N aqueous KOH is added and the mixture is stirred at room temperature for 4 h. The product starts to crystallize and the mixture is diluted with additional water. The resulting crystalline precipitate is filtered off and washed with water. After drying under reduced pressure of about 1 mbar at 400C, the monopotassium monohydrate salt π is isolated. Empirical formula C28H24KN7O3-H2O M.W. = 563.65; MS (free acid, M+H) 508.3. 1H ΝMR (DMSO-J6) 2.95 (s, 3 H), 3.95-4.05 (m, 1 H), 4.35-4.40 (m, IH), 4.55-4.62 (m, 1 H), 6.35-6.39 (d, 1 H), 6.58-6.60 (m, 1 H), 6.90-7.10 (sb, 1 H), 7.13-7.19 (m, 1 H), 7.22-7.38 (m, 6 H), 7.40-7.48 (m, 3 H), 7.57-7.60 (m, 1 H), 7.70 (s, 1 H), 8.10-8.15(d, 1 H), 8.30 (bs, 1 H), 11.80 (s, 1 H). Water (Karl-Fischer): 3.2% (Monohydrate). XRPD (2 theta): 5.28, 6.45, 7.97, 9.46, 10.18, 10.93, 13.23, 13.66, 14.94, 15.94, 16.71, 18.15, 19.49, 20.38, 21.04, 21.42, 23.76, 24.38, 25.36, 25.71, 26.19, 27.13, 27.67, 28.13, 28.61, 29.12, 29.75, 30.95, 31.37, 32.94. ee: 99.8% (Chiralpak AD-H, 250 x 4.6mm, Heptane : EtOH : MeOH 5 : 1 : 1, RT).
It is known that indole derivatives are used as units for the synthesis of active pharmaceutical ingredients. For example, 2-(2-aminopyrimidin-4-yl)-1H-indole-5-carboxylic acids or their salts are important units for the preparation of IkB kinase inhibitors (see WO 01/30774 A1):

2-(2-Aminopyrimidin-4-yl)-1H-indole-5-carboxylic acids can be prepared by classical Fischer indole synthesis starting from the corresponding 4-acetylpyrimidines (III) and 4-hydrazinobenzoic acid (II) (see scheme 1):

One disadvantage here is the severe reaction conditions which are required for a full conversion. Secondly, the products of this reaction are obtained in a mixture with the corresponding oligomers, which leads to a poor isolability, especially with regard to the filtration times. Moreover, these oligomers, owing to the low solubility of 2-(2-aminopyrimidin-4-yl)-1H-indole-5-carboxylic acids in organic solvents, can only be removed with difficulty and are entrained as an impurity in the further reactions, in some cases up to the active ingredient.
Here are two ways to make a kinase inhibitor intermediates.
http://www.google.com/patents/US8232395
J. Graeser and co-inventors describe indole derivatives such as 4 and 12 as intermediates for preparingIκB kinase inhibitors. Although indoles can be prepared by the classical Fisher synthesis, the inventors state that this method is not satisfactory when it is used for making the desired compounds. Severe reaction conditions are needed, and oligomeric compounds are formed that are difficult to remove.
The inventors describe two routes for preparing the desired compounds. The first route (Figure 1, top) begins with the reaction of indoleboronic acid 1 and chloropyrimidine 2in the presence of (Ph3P)4Pd to form 3, which is isolated in 93% yield and 96% purity. Compound 3 is converted to amine derivative 4 by treating it with MeNH2. The product was isolated in quantitative yield and with 97.6% purity. If desired, the ester group in 4can be hydrolyzed with NaOH to produce sodium salt 5.

Indoleboronic acid 1 is obtained by treating tert-butoxycarbonyl (Boc)–protected indole6 with B(O-i-Pr)3 in the presence of LiN-i-Pr2 (Figure 1, bottom) The reaction initially forms Boc-protected compound 7. After acid hydrolysis, 1 is isolated in 61% yield with 92.7% purity.
The inventors mention the advantage of using unprotected indole 1 in the reaction with2 rather than the N-protected compound. Their explanation is that although some 6 is formed by the loss of the boronate group from 1 during the coupling reaction with 2, 6does not subsequently react with 2. Hence the yield of 3 in the coupling step is not reduced.
The second route to the desired compound is quite different from the first. Figure 2 outlines the process for preparing 12, the methyl ester analogue of 4. This route starts with the preparation of silylated acetylene compound 8, isolated in 90% yield with 99% purity after what is described as an aqueous workup. In the next step, the silyl group is removed, and primary alkyne 9 is isolated in quantitative yield. Alkyne 9 is treated with chloropyrimidine 10 in the presence of CuI and a palladium catalyst in DMF to give 11, which is isolated after aqueous workup in 85% yield and 99.7% purity. The cyclization of 11 to form 12 is carried out with a strong base such as KO-t-Bu. The product is isolated after an aqueous workup in 58% yield and 92.3% purity.
Although the inventors do not provide details for preparing 10, they state that it can be synthesized by the route shown at the bottom of Figure 2. The reaction produces isomers 10 and 13, which can be separated by chromatographic methods or steam distillation.
The inventors describe an alternative route to 4 in which 1 reacts with 10 in place of 2. They point out that 1 reacts with a mixture of 10 and 13 to give 4. Although it may be expected that 13 would react to give an isomer of 4, they claim that this reaction does not take place. No examples of the reaction of 1 and 10 with or without 13 are given Also, the inventors mention “aqueous workup” several times but do not explain what this means.
These processes provide alternative routes to a drug intermediate that overcome product isolation problems. (Sanofi [Paris]. US Patent 8,232,395, July 31, 2012;

EXAMPLE 1 Synthesis of ethyl 2-(2-chloropyrimidin-4-yl)-1H-indole-5-carboxylate

28 g (114 mmol) of 2-borono-5-ethoxycarbonylindole, 12 g (113 mmol) of sodium carbonate and 17.2 g of 2,4-(113 mmol) dichloropyrimidine were initially charged in 412 ml of ethanol. The clear solution was freed of oxygen by vigorous stirring and passing argon through (20 minutes). At RT, 2.67 g of tetrakis(triphenylphosphine)palladium(0) were added. The mixture was heated to from 65° C. to 70° C. for 2 hours (h). Subsequently, 112 ml of water and 112 ml of 30% hydrochloric acid were added and the mixture was cooled to 0° C. After filtration and drying under reduced pressure, 37.3 g (93% of theory) of ethyl 2-(2-chloropyrimidin-4-yl)-1H-indole-5-carboxylate were obtained (HPLC >96%).
The purity was determined by high-pressure liquid chromatography (HPLC):
| Column: | Waters Symetry Shield RP8 3.9 * 150 | ||||
| Temperature: | 40° C. | ||||
| Flow rate: | 1 ml/min | Injection volume: | 10 μl | ||
| Pressure: | 90 bar | UV: | 254 nm | ||
| Eluent: | A: Water/trifluoroacetic acid (0.05%) | ||||
| B: Acetonitrile/trifluoroacetic acid (0.05%) | |||||
| Time (min) | 0 | 15 | 20 | 25 | 30 |
| A (%) | 80 | 25 | 25 | 80 | 80 |
| B (%) | 20 | 75 | 75 | 20 | 20 |
| Retention time of | 12.6 min | ||||
| title compound: | |||||
EXAMPLE 2 Synthesis of ethyl 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylate

30 g (95.4 mmol) of ethyl 2-(2-chloropyrimidin-4-yl)-1H-indole-5-carboxylate were initially charged and suspended in 150 ml of ethanol. 53.9 g of methylamine solution in ethanol (8 M) were added to this suspension which was heated to from 75° C. to 80° C. in an autoclave for 4 h. After concentration and washing with ethanol, 29.7 g of ethyl 2-(2-methylamino-pyrimidin-4-yl)-1H-indole-5-carboxylate were obtained (97.6 HPLC area %). LCMS: [M+H]⊕ 297.12
HPLC method as in example 1; retention time of title compound: 5.8 min
EXAMPLE 3 Synthesis of 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylic acid sodium salt

25 g of ethyl 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylate were admixed with 200 ml of ethanol and 24.5 g of 33% sodium hydroxide solution, and heated to from 65° C. to 70° C. for 4 h. After cooling, the mixture was filtered with suction and the precipitate was washed with 15 ml of ethanol/water (9:1). 24.5 g (87.6% of theory) of 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylic acid sodium salt were obtained (98.1 HPLC area %). LCMS: [M+H]⊕ 269.10
HPLC method as in example 1; retention time of title compound: 3.3 min
EXAMPLE 4 Synthesis of methyl 4-amino-3-trimethylsilylethynylbenzoate

5.83 g (20 mmol) of methyl 4-aminobenzoate, 20.2 g (198 mmol) of triethylamine and 80 ml of toluene were initially charged. The clear solution was freed of oxygen by vigorous stirring and passing argon through (20 minutes). At an internal temperature of 20° C., 3.2 g (33 mmol) of trimethylsilylacetylene, 76 mg of copper(I) iodide and 52 mg of triphenylphosphine were added. After aqueous workup, 5.45 g of 4-amino-3-trimethylsilylethynylbenzoate were obtained (HPLC: >99 area %). HPLC method as in example 1.
EXAMPLE 5 Synthesis of methyl 4-amino-3-ethynylbenzoate

1.9 g (7.7 mmol) of methyl 4-amino-3-trimethylsilylethynylbenzoate were initially charged in 20 ml of tetrahydrofuran (THF). At from 5° C. to 8° C., 8.45 ml (8.5 mmol) of tetrabutylammonium fluoride solution (1 M in THF) were added dropwise within 5 minutes. After 25 min at 2° C., 438 ml of acetic acid were added. After addition of water and extraction with dichloromethane, and after removal of the solvent, 1.35 g of methyl 4-amino-3-ethynylbenzoate were obtained. HPLC method as in example 1.
EXAMPLE 6 Synthesis of methyl 4-amino-3-(1-methylaminopyrimidin-4-yl)-ethynylbenzoate

3.0 g (17 mmol) of methyl 4-amino-3-ethynylbenzoate and 2.6 g (19 mmol) of 4-chloro-2-methylaminopyrimidine were initially charged in 20 ml of dimethylformamide (DMF) and 8.7 g (85 mmol) of triethylamine, and degassed with argon while stirring for 5 min. Subsequently, 65 mg of copper(I) iodide and 20 mg of tetrakis(triphenylamine)palladium(0) were added and the mixture was heated to 71° C. for 3 h. After aqueous workup, 4.1 g of methyl 4-amino-3-(1-methylaminopyrimidin-4-yl)ethynylbenzoate were obtained. (HPLC: 99.7 area %) HPLC method as in example 1.
EXAMPLE 7 Synthesis of methyl 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylate by cyclizing methyl 4-amino-3-(1-methylaminopyrimidin-4-yl)ethynylbenzoate

73 mg (0.7 mmol) of potassium tert-butoxide were dissolved in 1 ml of NMP and admixed with a solution of 140 mg (0.5 mmol) of methyl 4-amino-3-(1-methylaminopyrimidin-4-yl)ethynylbenzoate in 1 ml of NMP. Subsequently, stirring was continued at RT for 24 h. Aqueous workup afforded 115 mg of methyl 2-(2-methylaminopyrimidin-4-yl)-1H-indole-5-carboxylate (HPLC: 92.3 area %).
EXAMPLE 8 Synthesis of 2-borono-5-ethoxycarbonylindole

150 g (519 mmol) of N-Boc-5-ethoxycarbonylindole and 192 ml (833 mmol) of triisopropyl borate in 350 ml of toluene were admixed at from 5° C. to 10° C. with 350 ml of a 1.8 molar solution of LDA in THF. The mixture was stirred for a further 5 min and the reaction mixture was added to a solution of 278 g of 30% hydrochloric acid and 940 ml of water. Subsequently, the mixture was stirred at from 5° C. to 10° C. for 30 min. Thereafter, the mixture was filtered and the filtercake was suspended in 530 ml of ethanol. This suspension was added at 40° C. to a solution of 500 ml of 30% hydrochloric acid and 224 ml of ethanol. Subsequently, the mixture was stirred at from 40° C. to 45° C. for 2.5 h and admixed at 30° C. with 380 ml of water. The mixture was then cooled to from 10° C. to 15° C., stirred at this temperature for 30 min and filtered. Drying under reduced pressure afforded 79.5 g (61% of theory) of 2-borono-5-ethoxycarbonylindole (HPLC: 92.7 area %).
…………………………………………….
C) Synthesis of the heterocyclic base

C.1) indole base synthesis. Of 2 – (2-methylamino-pyrimidin-4-yl) -1 H-indole-5-carboxylic acid (20) C.1.1) 1-Dimethylamino-4 ,4-dimethoxy-pent. -1-en-3-one (18)
100 g (0.76 mol) of 3,3-dimethoxy-2-butanone (16) of (17) (0.76 mol) at 120 ° C with stirring 90.2 g of 48 N, N-dimethylformamide dimethyl acetal h. The methanol formed during the reaction was continuously removed from the reaction solution by distillation. On cooling, the solution became a spontaneous crystallization, which was brought by adding a little heptane to completion. This gave 128.24 g of crude 18 (90% yield), which was reacted without further purification. Molecular formula C 9 Hι 7 N0 3, MW = 187.24, MS (M + H) 188.2 i H NMR (DMSO-de) 1.22 (s, 3H), 2.80 (s, 3H), 3.10 (s, 9H), 5.39. (d, J = 15 Hz, 1 H), 7:59 (d, J = 15 Hz, 1 H). . . . . . . .
C.1.2). [4 – (1,1-Dimethoxy-ethyl)-pyrimidin-2-yl]-methyl-amine (19)
1:22 g (53 mmol) of sodium were dissolved in 100 ml absolute ethanol. This was
Stirring 5.8 g (53 mmol) Methylguanidinhydrochlorid and 10 g (53 mmol) of 1-dimethylamino-4,4-dimethoxy-penM-en-3-one (18) and heated to boiling for 4 h. To stop the reaction, the ethanol was evaporated. The product 19 thus obtained was used without further purification for the subsequent reaction. Yield 11.5 g (58 mmol, quantitative) Molecular Formula C9H15N3O2, MW = 197.24, MS (M + H) 198.2 1 H NMR (DMSO-de) 1.45 (s, 3H), 2.78 (s, 3H), 3.10 (s,. 6H), 6.75 (d, J = .3 Hz, 1 H), 7.0 – 7.1 (s (b), 1 H), 8.30 (d, J = 3 Hz, 1 H).
C.1.3) 2 -. (2-methylamino-pyrimidin-4-yl) -1 H-indole-5-carboxylic acid (20) Into 150 ml of 50% sulfuric acid at room temperature 5 g (25 mmol) [4 – ( 1, 1 – dimethoxy-ethyl)-pyrimidin-2-yl]-methyl-amine (19) and, 3.85 g of 4-hydrazinobenzoic acid with stirring and heated 4 h at 130 ° C. The methanol formed during the reaction was continuously removed from the reaction solution by distillation. After cooling to 10 ° C the reaction mixture was poured into 200 mL of ice and adjusted to a pH of about 5.5 with concentrated sodium hydroxide solution. The precipitate formed from sodium sulfate, and the product mixture was filtered and the filter residue was extracted several times with methanol. The combined methanol extracts were concentrated and the product 20 by flash chromatography (DCM / methanol 9:1). Yield: 0.76 g (11%) Molecular formula Oι Hι3 N 4 4 0 2, MW = 268.28, MS (M + H) 269.1.
1 H NMR (DMSO-de) 2.95 (s, 3H), 6.90 – 7.10 (s (b), 1 H), 7.18 (d, J = 3 Hz, 1H), 7.4 (s, 1 H), 7:58 (d, J = 4.5 Hz, 1H), 7.80 (d, J = 4.5 Hz, 1H), 8.30 (s, 1H), 7.80 (d, J = 4.5 Hz, 1H), 8:38 (d, J = 3 Hz, 1H), 11.85 (s, 1H), 12:40 – 12.60 (s (b), 1 H).
| US7285560 | Aug 18, 2003 | Oct 23, 2007 | Sanofi-Aventis Deutschland Gmbh | Indole derivatives or benzimidazole derivatives for modulating IκB kinase |
| US7342029 | Jul 22, 2005 | Mar 11, 2008 | Sanofi-Aventis Deutschland Gmbh | Substituted indoles |
| US7462638 | Aug 18, 2003 | Dec 9, 2008 | Sanofi-Aventis Deutschland Gmbh | Use of IκB-kinase inhibitors in pain therapy |
| US20030119820 | Oct 4, 2002 | Jun 26, 2003 | Aventis Pharma Deutschland Gmbh | Substituted indoles |
| US20040116494 | Aug 18, 2003 | Jun 17, 2004 | Aventis Pharma Deutschland Gmbh | Use of IkappaB-kinase inhibitors in pain therapy |
| US20040209868 | May 11, 2004 | Oct 21, 2004 | Aventis Pharma Deutschland Gmbh | Substituted indoles |
| US20070244139 | Jun 6, 2007 | Oct 18, 2007 | Sanofi-Aventis Deutschland Gmbh | Indole Derivatives or Benzimidazole Derivatives for Modulating IkB Kinase |
| US20090069358 | Nov 6, 2008 | Mar 12, 2009 | Sanofi-Aventis Deutschland Gmbh | Use of IKappaB-Kinase Inhibitors in Pain Therapy |
| JP2003519101A | Title not available | |||
| WO1998040380A1 | Feb 27, 1998 | Sep 17, 1998 | Alessio Roberto D | Indolyl-pyrrolydenemethylpyrrole derivatives and process for their preparation |
| WO2001030774A1 | Oct 17, 2000 | May 3, 2001 | Aventis Pharma Gmbh | Substituted indoles for modulating nfkb activity |
| WO2003066629A2 | Feb 6, 2003 | Aug 14, 2003 | Michael J Arnost | Heteroaryl compounds useful as inhibitors of gsk-3 |
| WO2004022057A1 | Aug 5, 2003 | Mar 18, 2004 | Aventis Pharma Gmbh | USE OF IκB KINASE INHIBITORS FOR THE TREATMENT OF PAIN |
| WO2004022553A1 | Aug 5, 2003 | Mar 18, 2004 | Aventis Pharma Gmbh | INDOLE OR BENZIMIDAZOLE DERIVATIVES FOR MODULATING IκB KINASE |
| WO2004089913A1 | Apr 8, 2004 | Oct 21, 2004 | Novartis Ag | Aminopyrimidine derivatives and their medical use |
| WO2005040133A1 | Oct 11, 2004 | May 6, 2005 | Michael Clare | Pyrimidine compounds for the treatment of inflammation |
| WO2004022553A1 * | Aug 5, 2003 | Mar 18, 2004 | Aventis Pharma Gmbh | INDOLE OR BENZIMIDAZOLE DERIVATIVES FOR MODULATING IκB KINASE |
FDA Approves Vimizim to Treat Mucopolysaccharidosis Type IVA
STRUCTURAL FORMULA
Monomer
APQPPNILLL LMDDMGWGDL GVYGEPSRET PNLDRMAAEG LLFPNFYSAN 50
PLCSPSRAAL LTGRLPIRNG FYTTNAHARN AYTPQEIVGG IPDSEQLLPE 100
LLKKAGYVSK IVGKWHLGHR PQFHPLKHGF DEWFGSPNCH FGPYDNKARP 150
NIPVYRDWEM VGRYYEEFPI NLKTGEANLT QIYLQEALDF IKRQARHHPF 200
FLYWAVDATH APVYASKPFL GTSQRGRYGD AVREIDDSIG KILELLQDLH 250
VADNTFVFFT SDNGAALISA PEQGGSNGPF LCGKQTTFEG GMREPALAWW 300
PGHVTAGQVS HQLGSIMDLF TTSLALAGLT PPSDRAIDGL NLLPTLLQGR 350
LMDRPIFYYR GDTLMAATLG QHKAHFWTWT NSWENFRQGI DFCPGQNVSG 400
VTTHNLEDHT KLPLIFHLGR DPGERFPLSF ASAEYQEALS RITSVVQQHQ 450
EALVPAQPQL NVCNWAVMNW APPGCEKLGK CLTPPESIPK KCLWSH 496
Disulfide bridges
139-139′ 282-393 282′-393′ 463-492 463′-492′ 475-481 475′-481′
Modified residues
C
53 , 53′
3-oxoAla
O
CO2H
H NH2
Glycosylation sites (N)
Asn-178 Asn-178′ Asn-397 Asn-397′
Vimizim (elosufase alfa)
Elosulfase alfa nonproprietary drug name GET STRUCTURE
MOLECULAR FORMULA C5020H7588N1364O1418S34
MOLECULAR WEIGHT 110.8 kDa (peptide)
SPONSOR BioMarin Pharmaceutical Inc.
CODE DESIGNATION BMN 110, rhGALNS
CAS REGISTRY NUMBER 9025-60-9
THERAPEUTIC CLAIM Treatment of Morquio Syndrome
CHEMICAL NAMES
1. Sulfatase, chondroitin
2. Human N-acetylgalactosamine-6-sulfatase (chondroitinsulfatase, galactose-6-sulfate
sulfatase, EC=3.1.6.4) dimer (139-139′)-disulfide glycosylated (produced by CHO cells)
Company: BioMarin Pharmaceutical Inc.
Date of FDA Approval: February 14, 2014
Treatment for: Mucopolysaccharidosis Type IVA
- BMN 110
- Chondroitin 6-sulfatase
- Chondroitin sulfatase
- Chondroitin sulfate sulfatase
- Chondroitinase
- Chondrosulfatase
- E.C. 3.1.6.4
- Elosulfase alfa
- rhGALNS
- UNII-ODJ69JZG85
- Vimizim
CLINICAL….http://clinicaltrials.gov/search/intervention=Elosulfase%20alfa%20OR%20bmn%20110
Vimizim (elosufase alfa) is an enzyme replacement therapy for patients with Mucopolysaccharidosis Type IVA (Morquio A syndrome).
- FDA Advisory Committee Recommends Approval for BioMarin’s Vimizim for the Treatment of Patients With Morquio A Syndrome – November 20, 2013
Feb 16, 2014 Approval FDA Approves Vimizim to Treat Mucopolysaccharidosis Type IVA

The U.S. Food and Drug Administration today approved Vimizim (elosulfase alfa), the first FDA-approved treatment for Mucopolysaccharidosis Type IVA (Morquio A syndrome). Morquio A syndrome is a rare, autosomal recessive lysosomal storage disease caused by a deficiency in N-acetylgalactosamine-6-sulfate sulfatase (GALNS). Vimizim is intended to replace the missing GALNS enzyme involved in an important metabolic pathway. Absence of this enzyme leads to problems with bone development, growth and mobility. There are approximately 800 patients with Morquio A syndrome in the United States.
Vimizim was granted priority review. An FDA priority review provides for an expedited review of drugs for serious diseases or conditions that may offer major advances in treatment. Vimizim is also the first drug to receive the Rare Pediatric Disease Priority Review Voucher – a provision that aims to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases.
“This approval and rare pediatric disease priority review voucher underscores the agency’s commitment to making treatments available to patients with rare diseases,” said Andrew E. Mulberg, M.D., deputy director, Division of Gastroenterology and Inborn Errors Products in the FDA’s Center for Drug Evaluation and Research (CDER). “Prior to today’s approval, patients with this rare disease have had no approved drug treatment options.”
The safety and effectiveness of Vimizim were established in a clinical trial involving 176 participants with Morquio A syndrome, ranging in age from 5 to 57 years. Participants treated with Vimizim showed greater improvement in a 6-minute walk test than participants treated with placebo. On average, patients treated with Vimizim in the trial walked 22.5 meters farther in 6 minutes compared to the patients who received placebo.
The most common side effects in patients treated with Vimizim during clinical trials included fever, vomiting, headache, nausea, abdominal pain, chills and fatigue. The safety and effectiveness of Vimizim have not been established in pediatric patients less than 5 years of age. Vimizim is being approved with a boxed warning to include the risk of anaphylaxis. During clinical trials, life-threatening anaphylactic reactions occurred in some patients during Vimizim infusions.
Vimizim is marketed by Novato, Calif.-based BioMarin Pharmaceutical Inc.
Elosulfase alfa (GALNS), a proposed treatment for Morqio A syndrome. Morquio A syndrome is an inherited, autosomal recessive disease caused by a deficiency of a particular lysosomal enzyme, N- acetylgalactosamine- 6 sulfatase. BioMarin’s experimental drug for Morquio A syndrome is an enzyme replacement of elosulfase alfa (called BMN 110), which is designed to clear keratan sulfate from the lysosome. BMN 110 is being studied to determine if it is safe, if it will slow the progression of the disease and if it will improve some of the symptoms.
BioMarin started BMN 110 clinical studies in humans in 2009 to evaluate safety and efficacy. In a phase III Multicenter, Multinational, Extension Studythe Long-Term Efficacy and Safety of BMN 110 in Patients With Mucopolysaccharidosis IVA (Morquio A Syndrome) MOR-005 was evaluated. Participants will receive 2 mg/kg weekly or every other weekly dosing of study drug via infusion until the MOR- 004 study is unblinded and the optimal dose is selected. All subjects will then be treated with the optimal dose for up to approximately 5 years or until the drug is approved.
Grape Seeds Fight Bowel Cancer
University of Adelaide research has shown for the first time that grape seed can aid the effectiveness of chemotherapy in killing colon cancer cells as well as reducing the chemotherapy’s side effects.
Published in the prestigious journal PLOS ONE, the researchers say that combining grape seed extracts with chemotherapy has potential as a new approach for bowel cancer treatment – to both reduce intestinal damage commonly caused by cancer chemotherapy and to enhance its effect.
Lead author Dr Amy Cheah says there is a growing body of evidence about the antioxidant health benefits of grape seed tannins or polyphenols as anti-inflammatory agents and, more recently, for their anti-cancer properties.
“This is the first study showing that grape seed can enhance the potency of one of the major chemotherapy drugs in its action against colon cancer cells,” says Dr Cheah, researcher in the School of Agriculture, Food and Wine.
“Our research…
View original post 308 more words
Tadalafil (cialis)

Tadalafil
GF-196960, IC-351, Cialis
6R–trans)-6-(1,3-benzodioxol-5-yl)- 2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino [1′, 2′:1,6] pyrido[3,4-b]indole-1,4-dione
Pyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione,6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-, (6R-trans)-; (6R,12aR)-6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-ethylpyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione; GF 196960; Adcirca;
171596-29-5 casno
Molecular Weight:
389.40
Molecular Formula:C22H19N3O4
GlaxoSmithKline (Originator), Lilly Icos (Marketer), Lilly (Licensee), Lilly Icos (Licensee)
Launched-2003
Tadalafil is currently marketed as Cialis. Cialis was developed by Eli Lilly as a treatment for impotence. In this capacity, it is reported that tadalafil functions by inhibiting the formation of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). The inhibition of PDE5 presumably lessens impotence by increasing the amount ot c(iMP, resulting in smooth muscle relaxation and increased blood flow.

Tadalafil is a PDE5 inhibitor marketed in pill form for treating erectile dysfunction (ED) under the name Cialis, and under the name Adcirca for the treatment of pulmonary arterial hypertension. In October 2011 the U.S. Food and Drug Administration (FDA) approved Cialis for treating the signs and symptoms of benign prostatic hyperplasia (BPH) as well as a combination of BPH and erectile dysfunction (ED) when the conditions coincide. It initially was developed by the biotechnology company ICOS, and then again developed and marketed world-wide by Lilly ICOS, LLC, the joint venture of ICOS Corporation and Eli Lilly and Company. Cialis tablets, in 2.5 mg, 5 mg, 10 mg, and 20 mg doses, are yellow, film-coated, and almond-shaped. The approved dose for pulmonary arterial hypertension is 40 mg (two 20-mg tablets) once daily.
Tadalafil can be prepared via a series of intermediates. One synthesis scheme is illustrated in Scheme 1: Scheme 1
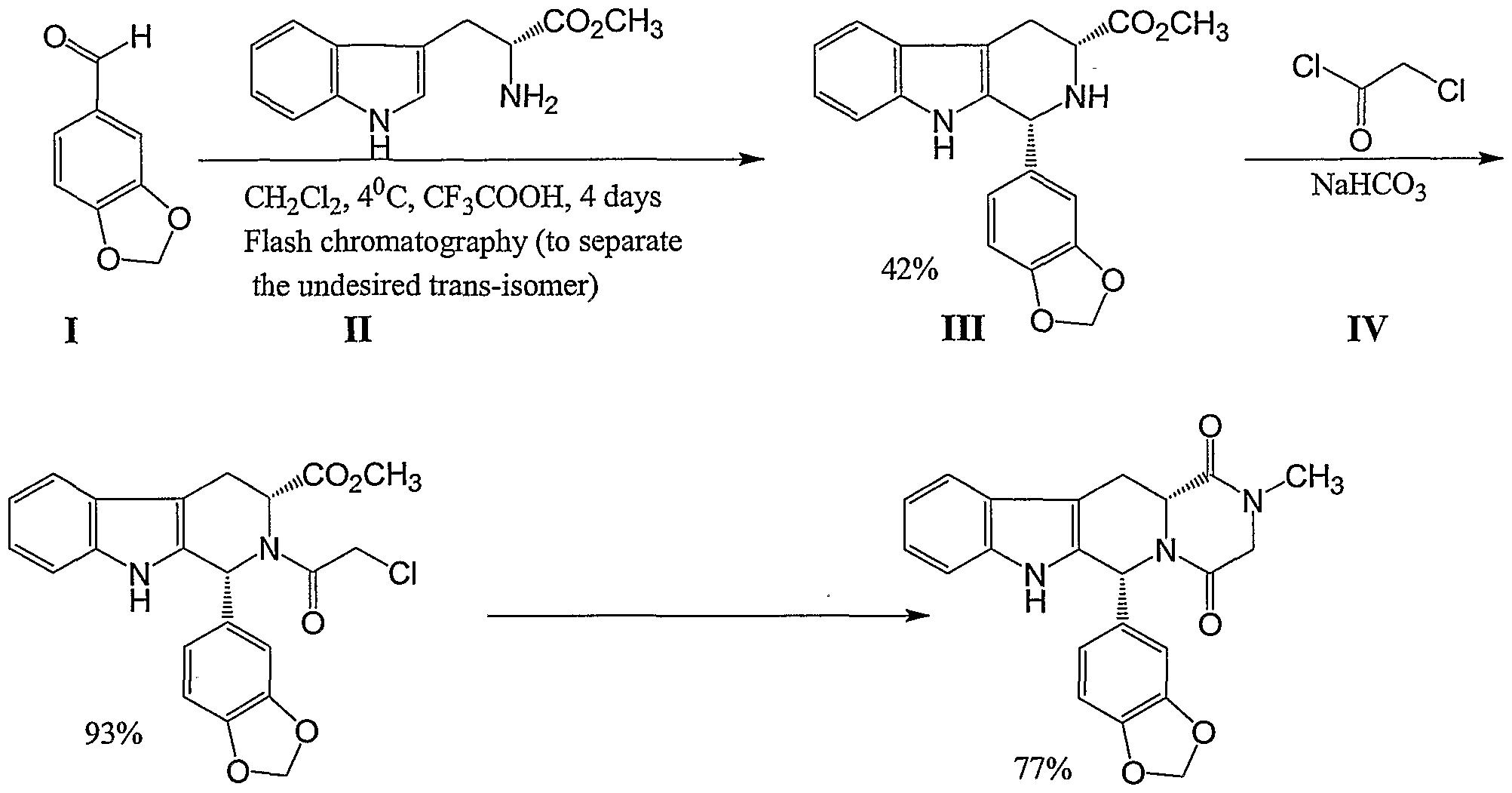
U.S. Patent No. 5,859,006 describes the synthesis of the tadalafil intermediate (Compound III) from D-tryptophan methyl ester (Compound II) and piperonal (Compound I) using trifluoroacetic acid and dichloromethane, a halogenated solvent. Compound III is then reacted with chloroacetyl chloride (Compound IV) and chloroform, providing another intermediate of tadalafil (Compound V). WO 04/011463 describes a process of preparing tadalafil intermediates from D-tryptophan methyl ester HCl salt and piperonal by refluxing the reagents in isopropyl alcohol; the obtained intermediate is reacted with chloroacetyl chloride and THF, resulting in another intermediate of tadalafil.
Tadalafil is also manufactured and sold under the name of Tadacip by the Indian pharmaceutical company Cipla in doses of 10 mg and 20 mg.
On November 21, 2003 the FDA approved tadalafil (as Cialis) for sale in the United States as the third ED prescription drug pill (after sildenafil citrate(Viagra) and vardenafil (Levitra)). Like sildenafil and vardenafil, tadalafil is recommended as an ‘as needed’ medication. Cialis is the only one of the three that is also offered as a once-daily medication.
Moreover, tadalafil was approved in May 2009 in the United States for the treatment of pulmonary arterial hypertension and is under regulatory review in other regions for this condition. In late November 2008, Eli Lilly sold the exclusive rights to commercialize tadalafil for pulmonary arterial hypertension in the United States to United Therapeutics for an upfront payment of $150 million.

The FDA’s approval of Viagra (Sildenafil) on March 27, 1998 was a ground-breaking commercial event for the treatment of ED, with sales exceedingUS$1 billion. Subsequently, the FDA approved Levitra (vardenafil) on August 19, 2003, and Cialis (tadalafil) on November 21, 2003.
Cialis was discovered by Glaxo Wellcome (now GlaxoSmithKline) under a partnership between Glaxo and ICOS to develop new drugs that began in August 1991. [1][2] In 1993, the Bothell, Washington biotechnology company ICOS Corporation began studying compound IC351, a phosphodiesterase type 5 (PDE5) enzyme inhibitor. In 1994, Pfizer scientists discovered that sildenafil, which also inhibits the PDE5 enzyme, caused penile erection in men participating in a clinical study of a heart medicine. Although ICOS scientists were not testing compound IC351 for treating ED, they recognized its potential usefulness for treating that disorder. Soon, in 1994, ICOS received a patent for compound IC351 (structurally unlike sildenafil and vardenafil), and Phase 1 clinical trials began in 1995. In 1997, the Phase 2 clinical studies were initiated for men experiencing ED, then progressed to the Phase 3 trials that supported the drug’s FDA approval. Although Glaxo had an agreement with ICOS to share profits 50/50 for drugs resulting from the partnership, Glaxo let the agreement lapse in 1996 as the drugs developed were not in the company’s core markets.[3]

In 1998, ICOS Corporation and Eli Lilly and Company formed the Lilly ICOS, LLC, joint venture company to further develop and commercialize tadalafil as a treatment for ED. Two years later, Lilly ICOS, LLC, filed a new drug application with the FDA for compound IC351 (under the tadalafil generic name, and the Cialis brand name). In May 2002, Lilly ICOS reported to the American Urological Association that clinical trial testing demonstrated that tadalafil was effective for up to 36 hours, and one year later, the FDA approved tadalafil. One advantage Cialis has over Viagra and Levitra is its 17.5-hour half-life (thus Cialis is advertised to work for up to 36 hours, after which time there remains approximately 25 percent of the absorbed dose in the body) when compared to the four-hour half–life of sildenafil (Viagra).
In 2007, Eli Lilly and Company bought the ICOS Corporation for $2.3 billion. As a result, Eli Lilly owned Cialis and then closed the ICOS operations, ending the joint venture and firing most of ICOS’s approximately 500 employees, except for 127 employees of the ICOS biologics facility, which subsequently was bought by CMC Biopharmaceuticals A/S (CMC).

Tadalafil Molecule
Persons surnamed “Cialis” objected to Eli Lilly and Company’s so naming the drug, but the company has maintained that the drug’s trade name is unrelated to the surname.[4]
On October 6, 2011, the U.S. FDA approved tadalafil [5] to treat the signs and symptoms of benign prostatic hyperplasia (BPH). BPH is a condition in males in which the prostate gland becomes enlarged, obstructing the free flow of urine. Symptoms may include sudden urges to urinate (urgency), difficulty in starting urination (hesitancy), a weak urine stream, and more frequent urination- especially at night. The FDA has also approved tadalafil for treatment of both BPH and erectile dysfunction (ED) where the two conditions co-exist.
Although available since 2003 in 5, 10, 20 mg dosage, in late 2008/early 2009, the U.S. FDA approved the commercial sale of Cialis in 2.5 mg dosage as a one-a-day treatment for ED. The 2.5 mg dose avoids earlier dispensing restrictions on higher dosages. The price of the 5 mg and 2.5 mg are often similar, so some people score and split the pill.[6] The manufacturer does not recommend splitting.
Moreover, tadalafil (Adcirca) 40 mg was approved in 2009 in the United States and Europe (and 2010 in Canada and Japan) as a once-daily therapy to improve exercise ability in patients withpulmonary arterial hypertension. In patients with pulmonary arterial hypertension, the pulmonary vascular lumen is decreased as a result of vasoconstriction and vascular remodeling, resulting in increased pulmonary artery pressure and pulmonary vascular resistance. Tadalafil is believed to increase pulmonary artery vasodilation, and inhibit vascular remodeling, thus lowering pulmonary arterial pressure and pulmonary vascular resistance. Right heart failure is the principal consequence of pulmonary arterial hypertension.
On October 6, 2011, the U.S. FDA approved tadalafil [6] to treat the signs and symptoms of benign prostatic hyperplasia (BPH). BPH is a condition in males in which the prostate gland becomes enlarged, obstructing the free flow of urine. Symptoms may include sudden urges to urinate (urgency), difficulty in starting urination (hesitancy), a weak urine stream, and more frequent urination- especially at night. The FDA has also approved tadalafil for treatment of both BPH and erectile dysfunction (ED) where the two conditions co-exist.
Tadalafil has been used in approximately 15,000 men participating in clinical trials, and over eight million men worldwide (primarily in the post-approval/post-marketing setting). The most commonside effects when using tadalafil are headache, stomach discomfort or pain, indigestion, burping, acid reflux. back pain, muscle aches, flushing, and stuffy or runny nose. These side effects reflect the ability of PDE5 inhibition to cause vasodilation (cause blood vessels to widen), and usually go away after a few hours. Back pain and muscle aches can occur 12 to 24 hours after taking the drug, and the symptom usually disappears after 48 hours.
In May 2005, the U.S. Food and Drug Administration found that tadalafil (along with other PDE5 inhibitors) was associated with vision impairment related to NAION (nonarteritic anterior ischemic optic neuropathy) in certain patients taking these drugs in the post-marketing (outside of clinical trials) setting. Most, but not all, of these patients had underlying anatomic or vascular risk factors for development of NAION unrelated to PDE5 use, including: low cup to disc ratio (“crowded disc”), age over 50, diabetes, hypertension, coronary artery disease, hyperlipidemia and smoking. Given the small number of NAION events with PDE5 use (fewer than one in one million), the large number of users of PDE5 inhibitors (millions) and the fact that this event occurs in a similar population to those who do not take these medicines, the FDA concluded that they were not able to draw a cause and effect relationship, given these patients underlying vascular risk factors or anatomical defects. However, the label of all three PDE5 inhibitors was changed to alert clinicians to a possible association.
In October 2007, the FDA announced that the labeling for all PDE5 inhibitors, including tadalafil, requires a more prominent warning of the potential risk of sudden hearing loss as the result of postmarketing reports of deafness associated with use of PDE5 inhibitors.[7]
Selectivity compared with other PDE5 inhibitors
Tadalafil, sildenafil, and vardenafil all act by inhibiting the PDE5 enzyme. These drugs also inhibit other PDE enzymes. Sildenafil and vardenafil inhibit PDE6, an enzyme found in the eye, more than tadalafil.[9] Some sildenafil users see a bluish tinge and have a heightened sensitivity to light because of PDE6 inhibition.[3] Sildenafil and vardenafil also inhibit PDE1 more than tadalafil.[9]PDE1 is found in the brain, heart, and vascular smooth muscle.[9] It is thought that the inhibition of PDE1 by sildenafil and vardenafil leads to vasodilation, flushing, and tachycardia.[9] Tadalafil inhibits PDE11 more than sildenafil or vardenafil.[9] PDE11 is expressed in skeletal muscle, the prostate, the liver, the kidney, the pituitary gland, and the testes.[9] The effects on the body of inhibiting PDE11 are not known.[9]

20 mg Cialis tablet
In the United States, the FDA relaxed rules on prescription drug marketing in 1997, allowing advertisements targeted directly to consumers.[10] Lilly-ICOS hired the Grey Worldwide Agency in New York, part of the Grey Global Group, to run the Cialis advertising campaign.[11] Marketers for Cialis has taken advantage of its greater duration compared to its competitors in advertisements for the drug; Stuart Elliot of The New York Times opined: “The continuous presence of women in Cialis ads is a subtle signal that the drug makes it easier for them to set the pace with their men, in contrast to the primarily male-driven imagery for Levitra and Viagra.”[11] Iconic themes in Cialis ads include couples in bathtubs and the slogan “When the moment is right, will you be ready?”[11] Cialis ads were unique among the ED drugs in mentioning specifics of the drug.[12] As a result, Cialis ads were also the first to describe the side effects in an advertisement, as the FDA requires advertisements with specifics to mention side effects. One of the first Cialis ads aired at the 2004 Super Bowl.[12] Just weeks before the Super Bowl, the FDA required more possible side effects to be listed in the advertisement, including priapism.[12] Although many parents objected to the Cialis ad being aired during the Super Bowl, Janet Jackson‘s halftime “wardrobe malfunction” overshadowed Cialis.[12] In January 2006, the Cialis ads were tweaked, adding a doctor on screen to describe side effects and only running ads where more than 90 percent of the audience are adults, effectively ending Super Bowl ads.[10] In 2004, Lilly-ICOS, Pfizer, and GlaxoSmithKline spent a combined $373.1 million to advertise Cialis, Viagra, and Levitra respectively.[12] Cialis has sponsored many golf events, including the America’s Cup and the PGA Tour, once being title sponsor of the PGA Tour Western Open tournament.[13]
CIALIS (tadalafil) is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). Tadalafil has the empirical formula C22H19N3O4 representing a molecular weight of 389.41. The structural formula is:
 |
The chemical designation is pyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione, 6-(1,3-benzodioxol-5-yl)2,3,6,7,12,12a-hexahydro-2-methyl-, (6R,12aR)-. It is a crystalline solid that is practically insoluble in water and very slightly soluble in ethanol.
CIALIS is available as almond-shaped tablets for oral administration. Each tablet contains 2.5, 5, 10, or 20 mg of tadalafil and the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, hypromellose, iron oxide, lactose monohydrate, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, talc, titanium dioxide, and triacetin.
Tadalafil, (6R-trans)-6-(l,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2- methyl-pyrazino[r,2′:l,6]pyrido[3,4-b]indole-l,4-dione, with the structural formula shown below, is a white crystalline powder. (CAS# 171596-29-5). Tadalafil is a potent and selective inhibitor of the cyclic guanosine monophosphate (cGMP) – specific phosphodiesterase enzyme, PDE5. The inhibition of PDE5 increases the amount of cGMP, resulting in smooth muscle relaxation and increased blood flow. Tadalafil is therefore currently used in the treatment of male erectile dysfunction, and is commercially available as CIALIS ®.

Tadalafil U.S. Patent No. 5,859,006 describes the synthesis of tadalafil via the cyclization of TDCL (i.e., cis-methyl l,2,3,4-tetrahydro-2-chloroacetyl-l-(3,4- methylenedioxyphenyl)-9H-pyrido[3,4-b]mdole-3-carboxylate) using methylamine by purification by flash chromatography, followed by subsequent crystallization from methanol. Crude tadalafil typically requires additional purification steps, such as multiple extractions, crystallization, and/or flash chromatography, to remove the impurities present in the compound after synthesis is complete. Such purification processes increase the cost of producing tadalafil. Also, when repeating the US ‘006 process, about 250 volumes of methanol were necessary for the crystallization step
Tadalafil can be prepared via a series of intermediates. One synthesis for preparing tadalafil is illustrated below in Scheme I:
SCHEME I

U.S. Patent No. 5,859,006 discloses the synthesis of a tadalafil intermediate
(Compound III) from D-tryptophan methyl ester (Compound II) and piperonal (Compound
I) using trifluoroacetic acid and dichloromethane, a halogenated solvent. Compound III is then reacted with chloroacetyl chloride (Compound IV) and chloroform to provide another intermediate of tadalafil (Compound V).
WO 2004/011463 discloses a process of preparing tadalafil intermediates from D-tryptophan methyl ester HCl salt and piperonal by refluxing the reagents in isopropyl alcohol, reacting the intermediate thus obtained with chloroacetyl chloride and tetrahydrofuran (THF) to provide another intermediate of tadalafil.
WO 2006/110893 discloses a process for the preparation of methyl ester intermediate (Compound III), and tadalafil using the methyl ester intermediate (CompoundII).
U.S. Patent Application Publication No. 2006/0258865 Al discloses a synthesis of the tadalafil intermediate (Compound III) from D-tryptophan methyl ester
(Compound II) and piperonal (Compound I) using a dehydrating agent selected from Na2SO4, K2SO4, MgSO4, CaSO4, CaCl2, molecular sieve or mixtures thereof and a high boiling solvent such as N,N-Dimethyl acetamide. Compound III is then reacted with chloroacetyl chloride (Compound IV) in the presence of a base such as NaHCO3 and an organic solvent such as dichloromethane, providing another intermediate of tadalafil (Compound V), which is further reacted with aqueous methyl amine solution to provide tadalafil.
………………………………………….
Scheme II and III.



……………………………………………………………………………………………………a compound of .Formula I
SCHEME III

SCHEME IV

EXAMPLE l
The reaction scheme of this example is generally shown below in SchemeIV.
SCHEME IV
Compound – 1 Compound – II
Into a clean dry glass flask charged with ethanol (250 ml) under a nitrogen atmosphere was added Compound 1 (25 g) under stirring. The reaction mass was cooled to 0 to 50C and monomethylamine gas was purged into the reaction mixture for about 2 hours while maintaining the temperature between 0 to 50C. The temperature was raised to 75 to 😯0C and the reaction mixture was stirred under reflux for 2 hours. The reaction mixture was then cooled to 0 to 5°C and monomethylamine gas was again purged into the reaction mixture at 0 to 5°C. The temperature was again raised to 75 to 800C and stirred for about 1 hour. The reaction mixture was concentrated under vacuum to about 1/3 its original volume, cooled to 5 to 1O0C and stirred for 1 hour at this temperature. The solids were filtered and washed with chilled ethanol (50 ml). The wet solids were dried under vacuum for 6 hours.
Yield: 25g; Mp: 202-206.70C
Specific rotation (25°C) :+44.0 ( C=l% in DMSO)
13C NMR, DMSO-D6 : 25.78, 25.92, 57.89, 57.98, 101.17, 108.09, 108.32,
109.08, 111.48, 117.82, 118.62, 122.23, 122.97, 126.97, 135.97, 136.22, 136.55, 146.99,
147.48, 173.13
1H NMR, DMSO-D6, 300 MHz, Delta values: 2.6(m,lH), 2.7(m,3H),
2.8(d,lH), 3.0(d,lH), 3.6(bs,lH), 5.1(m,lH), 6.0(s,3H), 6.9-7.1(m, 5H), 7.2(d,lH),
7.4(d,lH), 7.8(bs, IH), 10.3(s, IH)
EXAMPLE 2
The reaction scheme of this example is generally shown below in SchemeV.
SCHEME V

Formula III Formula II
Into a clean dry flask charged with dichloromethane (200 ml) was added
Compound II (25 g) obtained in Example 1 under stirring at 25 to 300C. Next, triethylamine (16.11 g) was added to the reaction mixture and stirred for 30 minutes at 20 to 300C. The reaction mixture was cooled to 0 to 5°C and a solution of chloroacetyl chloride (12.93 g) in chloroform (50 ml) was added to the reaction mixture while maintaining temperature between -5 to 50C. The reaction mixture was stirred at -5 to 5°C for about 2 hours. Saturated aqueous sodium bicarbonate solution (50 ml) was added to the reaction mass slowly and the temperature of the reaction mixture was raised to 25 to 300C. The lower organic layer was separated and washed twice with water (75 ml). The chloroform extract was dried over anhydrous sodium sulfate. The organic layer was concentrated under vacuum until a thick yellow slurry was obtained. The slurry was cooled to 0 to5°C. The solids obtained were filtered and washed with 50 ml chilled chloroform. The wet product was dried at 750C under vacuum for 6 hours.
Yield: 22.5 g; HPLC Purity: 97%; Mp: 180-1820C
Specific rotation(25°C): -154.3(C=1% in DMSO)
13C. NMR(DMSO-Do, 300 MHZ)= 21.11, 25.88, 44.207, 51.60, 53.95,
101.16,107.66 109.56, 111.38, 118.36, 118.75, 121.58,122.74, 126.30, 130.31, 134.13,
136.57, 146.66, 147.03,167.43, 168.45
1H. NMR (CDC13, 300 MHZ):2.4(bs,3H), 3.1(m,lH), 3.8(m,lH),
4.3(bs,2H), 4.9(m,lH), 5.4(m,lH), 5.9(s,2H), 6.6-6.8(m,3H), 6.9(bs,lH), 7.1-7.3(m,3H),
7.6(d, IH), 7.7(bs,lH)
1H. NMR (DMSO-D6, 300 MHZ): 2.0 (bs,3H), 2.9(m,lH), 3.4(m,lH),
4.5(m,lH), 4.8(m,lH), 4.9(m,lH), 6.0(m,2H), 6.4-6.8(m,4H), 6.9-7.2(m,2H), 7.3(d, IH),
7.4(bs,lH), 7.5(d,lH), 10.8(s,lH)
EXAMPLE 3
The reaction scheme of this example is generally shown below in SchemeVI.
SCHEME VI

Formula II Formula I
Into a clean dry round bottom (RB) flask was charged tetrahydrofuran
(THF) (175 ml) under a nitrogen blanket and then cooled to -35 to -400C. Next 92 ml n- butyllithium (1.6 m solution in hexane) was added while maintaining the temperature between -35 to -400C. After the addition was complete, the reaction mixture was stirred at -35 to -400C for 15 minutes. A solution of compound of formula II (22.5 g) obtained in Example 2 in THF (75 ml) was prepared and slowly added to the reaction mixture while maintaining the temperature between -35 to -400C. After the addition was complete, the reaction mixture was stirred at -35 to -400C for 2.5 hours. Saturated aqueous ammonium chloride solution (25 ml) and 50 ml ethyl acetate was added to the reaction mixture at -35 to -400C. The temperature was raised to 25 to 300C and the two layers formed were separated. The upper organic layer was collected. The lower aqueous layer was thrice extracted with ethyl acetate (25 ml). The organic layers were combined together and washed with water (50 ml). The organic extract was dried over anhydrous sodium sulfate and concentrated under vacuum to obtain crude tadalafil as a dark brown solid. [0058] Yield: 22 g; HPLC Purity: 50%.
EXAMPLE 4
Purification of crude tadalafil
The crude tadalafil (22 g) obtained in Example 3 was suspended in 110 ml methanol and stirred for 1 hour at 25 to 300C. The solids obtained were filtered and washed with 25 ml chilled methanol. The wet product was dried at 600C under vacuum for 6 hours. This was further purified by using isopropyl alcohol. Yield: 9 g; HPLC Purity: >99.5%.
EXAMPLE 5
The reaction scheme of this example is generally shown below in SchemeVII.
Scheme VII

Formula VI where R = -OCH3 Formula VIA [0062] Into a clean dry RB flask charged with methanol (1900 ml) was added D- tryptophan methyl ester (190 g) under stirring at 25 to 300C. The reaction mixture was cooled to 0 to 50C. Monomethylamine gas was purged into the reaction mixture at 0 to 5°C for about 5-7 hours under stirring. The temperature of the reaction mixture was slowly raised to about 25 to 3O0C and stirred at this temperature for 5-7 hours. The reaction mixture was concentrated under vacuum to distill out the solvent. Diisopropyl ether (950 ml) was added and cooled to 25 to 3O0C under stirring for 1-2 hrs. The solids obtained were filtered, washed with Diisopropyl ether and dried under vacuum. [0063] MP: 122.4-1240C; Yield: 150 g (78.9 % w/w).
Specific rotation(25°C): +12.5 (C=I % in DMSO)
13 C NMR (300 MHZ,DMSO-D6): 25.71, 31.40, 55.67, 110.93, 111.55,
118.42, 118.73, 121.09, 123.95, 127.66,136.44, 175.39.
1H NMR (300 MHZ,DMSO-D6): 1.6(bs,2H), 2.5(m,3H), 2.8(m,lH),
3.1(m,lH), 3.4(m, IH), 6.9-7.2(m,3H), 7.3(d,lH), 7.5(d,lH), 7.8(bs,lH), 10.8(bs,lH)
EXAMPLE 6
The reaction scheme of this example is generally shown below in Scheme VIII.
SCHEME VIII

Formula VIA Formula VII

Into a clean, dry flask charged with methylene dichloride (MDC) (1000 ml) was added D-tryptophan methyl amide, the compound of Formula VIA (50 g), and piperonal, the compound of Formula VII (31.09 g), under stirring at 25 to 300C. The reaction mixture was cooled to 0 to 5°Cunder nitrogen atmosphere. Trifluoroacetic acid (85.3 g) was dissolved in MDC (250 ml) and the solution was slowly added to the reaction mixture at 0 to 5°C. The temperature of the reaction mixture was raised to 20 to 300C and stirred at this temperature for 14-16 hours. The reaction was monitored by TLC, workup was done as follows, the pH of the reaction mixture was adjusted to 8-9 using sodium carbonate solution under stirring, the two layers were settled, separated and the lower MDC layer was washed with water. The MDC layer was then dried over anhydrous sodium sulfate. The reaction mass was concentrated under vacuum at 40 to 5O0C to remove the solvent. The compound was precipitated using ethyl acetate, the solids were filtered, washed with ethyl acetate and dried.
Yield: 52.5 g; Yield: 105% w/w, HPLC Purity: 71% cis and 27% trans isomer (HPLC).
EXAMPLE 7
The reaction scheme of this example is generally shown below in Scheme IX.
SCHEME IX
1]CICOCH2C1 2]crystn


Formula H
Into a clean dry flask charge with dichloromethane (400 ml) under a nitrogen atmosphere was added the compound of Formula III obtained in Example 6 and triethylamine (28.96 g) under stirring at 20 to 3O0C. The reaction mixture was then cooled to 0 to 50C. A mixture of chloroacetyl chloride (25.85 g) in dichloromethane (100 ml) was prepared and slowly added to the reaction mixture while maintaining the temperature between -5 to 50C in 1-2 hrs. The reaction mixture was stirred at 0 to 50C for 30 min and then saturated sodium bicarbonate solution (100 ml) was added at 5 to 100C under stirring. The temperature of the reaction mixture was raised to 25 to 300C and stirred at this temperature for 15 minutes. The layers were then separated. The lower MDC layer was collected, washed twice with 100 ml water and dried over anhydrous sodium sulfate. The
MDC layer was concentrated to distill out MDC until a stirrable mass was left behind. The mass was cooled to 25-3O0C and filtered, washed, to yield off-white to light yellow colored solids. The resulted product was the cis isomer, the trans isomer left behind in the mother liquor.
Yield = 25.5 g (50%w/w); HPLC Purity: > 97%.
The physical and spectral data was similar to that obtained in Example 2.
EXAMPLE 8
The reaction scheme of this example is generally shown below in SchemeX.
SCHEME X

Formula II
Into a clean dry round bottom (RB) flask was charged THF (1625 ml) under a nitrogen blanket and then cooled to -35 to -400C. Next, 505 ml n-butyllithium (1.6 m solution in hexane) was added while maintaining the temperature between -35 to -4O0C. After the addition was complete, the reaction mixture was stirred at -35 to -4O0C for 15 minutes. 72 ml diisopropyl amine was then added at -35 to -400C and then stirred at 0-50C for 1 hr. A solution of Compound of formula II (125 g) obtained in Example 7 in THF (625 ml) was prepared and slowly added to the reaction mixture while maintaining the temperature between -40 to -5O0C. After the addition was complete, the reaction mixture was stirred at -35 to -400C for 2-6 hours. Saturated aqueous ammonium chloride solution (250 ml) and ethyl acetate (125 ml) was added to the reaction mixture at -35 to -400C. The temperature was raised to 25 to 300C and the two layers formed were separated. The upper organic layer was collected. The lower aqueous layer was extracted with ethyl acetate (65 ml). The organic layers were combined together and distilled. Isopropyl alcohol (1250 ml) was added and the distillation was continued. A mixture of methanol (250 ml) and isopropanol (375 ml) were added and crude tadalafϊl was obtained upon cooling. The crude product was filtered, washed with water and dried. [0076] Yield: 60 g; (48% w/w); HPLC Purity: >99%.
EXAMPLE 9
Purification of crude Tadalafil
The crude tadalafil obtained in Example 8 was suspended in methanol (600 ml) and stirred for 1 hour at reflux. The mixture was cooled and the solids obtained were filtered and washed with chilled methanol (60 ml). The wet product was dried at under vacuum.
Yield: 56 g; HPLC Purity: 99.8%.
……………………………………………………………………………………
Beilstein J. Org. Chem. 2011, 7, 442–495.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-7-57#S28
A different approach was used in the synthesis of the phosphodiesterase inhibitor tadalafil (132, Cialis) starting from commercially available (D)-tryptophan methyl ester to form the indolopiperidine motif 135 via a Pictet–Spengler reaction followed by a double condensation to install the additional diketopiperazine ring (Scheme 28) [38,39].
![[1860-5397-7-57-i28]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i28.png?scale=2.0&max-width=1024&background=FFFFFF)
To achieve the high levels of cis selectivity required from the Pictet–Spengler reaction, an extensive investigation of solvents and the influence of additives was undertaken [40]. It was identified that the use of a specific 23 mol % of benzoic acid significantly increased the cis/trans ratio from a base level of 55:45 to 92:8 (16 h reaction time at ambient temperature) in an overall yield of 86%. It was also determined that more polar solvents such as acetonitrile and nitromethane preferentially solvated the trans product and thereby allowed the isolation of the ciscompound by precipitation. It was also shown that by heating the reaction mixture under reflux the product distribution could be driven to the thermodynamically more favoured cis isomer having both the ester and the piperonyl moiety in equatorial positions. Hence, after heating under reflux for 8 h the cis/trans ratio was found to be 99:1 and the product could be isolated in an overall yield of 91%. This work represents an impressive example of a well considered and executed process optimisation study.
………………………………
The process disclosed in the patent US 5 859 006 (Scheme 1) involves condensation of D-tryptophan methyl ester with a piperonal derivative to yield a compound of formula (II). After conversion into a thioamide derivative of formula (III), cyclization occurs in presence of both an alkylating and reducing agents to provide a tetrahydro-β-carboline derivative of formula (IV), which on treatment with chloroacetyl chloride and methyl amine, gives Tadalafil. The compound of formula (IV) can also be obtained in one step, after separation of the other diastereoisomer, by a Pictet Spengler reaction between D-tryptophan methyl ester and piperonal in presence of an acid, such as trifluoroacetic acid.
-

-
The patent application WO2007/10038 discloses the reaction of D-tryptophan with piperonal to provide a tetrahydro-β-carboline acid that was cyclised to Tadalafil in presence of a sarcosine derivative.
The patent application WO2007/1107 discloses the reaction of D-tryptophan methyl amide with piperonal, to provide an intermediate that after reaction with chloroacetyl chloride cyclises to Tadalafil in presence of butyllithium.
Thus, the active substance prepared by the processes known up till now can only be obtained in a satisfactory quality after running through a large number of process steps. Moreover a toxic alkylating agent, such as methylamine, is often used.

Example 1
-
(1R,3R)-methyl-1,2,3,4-tetrahydro-2-(2-(benzyl(methyl)amino)acetyl)-1-(3,4-methylenedioxyphenyl)-9H-pyrido[3,4-b]indole-3-carboxylate (VII)

-
A 50 mL three-necked flask fitted with thermometer and reflux condenser was charged with (1R,3R)-methyl 1,2,3,4-tetrahydro-2-chloroacetyl-1-(3,4-methylenedioxyphenyl)-9H-pyrido [3,4-b] indole-3-carboxylate (VI) (1.39 g, 3.26 mmol), DMA (5.33 mL), K2CO3 (0.5 g, 3.6 mmol) and N-benzylmethylamine (0.41 mL, 3.26 mmol). The resultant solution was stirred at room temperature. After 2 hours, the mixture was poured in brine (20 mL) and extracted with isopropyl acetate. The combined organic phases were washed with brine (3 x 5 mL), dried over sodium sulfate and concentrated to a residue under reduced pressure, affording 1.5 g of the desired product (VII), as a white solid. Yield: 70%.
1H NMR (d6-DMSO 300 MHz, 298K) 2.24 (s, 3H), 2.94-3.00 (m, 5H), 3.44-3.68 (m, 3H), 5.56 (bd, J = 6.4, 1H), 5.95 (s, 1H), 5.96 (s, 1H), 6.55 (bd, J = 7.4, 1H), 6.75 (bs, 1H), 6.77 (d, J = 8.0, 1H), 6.84 (bs, 1H), 7.05 (td, J = 7.4, 0.9, 1H), 7.12 (td, J = 7.5, 1.2, 1H), 7.17-7.32 (m, 6H), 7.56 (d, J = 7.7, 1H), 10.76 (bs, 1H)
13C NMR (d6-DMSO 75.4 MHz, 298K) 21.9, 42.5, 51.3, 51.9, 52.4, 61.0, 61.7, 101.5, 107.0, 108.0, 109.8, 111.8, 118.5, 119.2, 122.0, 123.0, 126.7, 127.7, 128.7, 129.6, 131.1, 134.7, 137.1, 138.6, 147.1, 147.5, 170.6, 171.5

Example 2
-
(6R-trans)-6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino [1′,2′:1,6] pyrido [3,4-b] indole-1,4-dione (Tadalafil) (I)

Under H2 atmosphere (3 atm) and magnetic stirring, Raney® Ni (2800 slurry in water, 0.0276 g, 0.47 mmol), previously washed with methanol (3 times), was added to a solution of (1R,3R)-methyl-1,2,3,4-tetrahydro-2-(2-(benzyl(methyl)amino)acetyl)-1-(3,4-methylenedioxyphenyl)-9H-pyrido[3,4-b]indole-3-carboxylate (VII) (3.00 g, 4.70 mmol) in DMA (21.3 mL). The mixture was heated at 90°C for 17 hours and then cooled to room temperature. The suspension was filtered over a pad of Celite® and the resulting solutionand the resulting solution was concentrated until 6 mL. Methanol (12 mL) was added and the solid which was so obtained was filtered over Buchner, washed with methanol (4 mL) and oven-dried under reduced pressure for 2 hours, affording 1.3 g of the title compound, as a white solid. Yield: 70%
1H NMR (d6-DMSO 300 MHz, 298K): 2.91-3.00 (m, 4H), 3.32 (s, 1H), 3.47-3.54 (dd, J = 4.6, 11.3, 1H), 3.93 (d, J = 17.1, 1H), 4.17 (d, J = 17.1, 1H), 4.35-4.40 (dd, J = 4.27, 11.6, 1H), 5.91 (s, 2H), 6.11 (s, 1H), 6.76 (s, 2H), 6.85 (s, 1H), 6.98-7.06 (m, 2H), 7.28 (d, J = 7.9, 1H), 7.52 (d, J = 7.3, 1H), 11.0 (s, 1H)
13C NMR (d6-DMSO 75.4 MHz, 298K) 23.8, 33.4, 52.0, 55.9, 56.1, 101.5, 105.3, 107.6, 108.6, 111.9, 118.7, 119.5, 119.9, 121.8, 126.4, 134.5, 136.8, 137.6, 146.7, 147.6, 167.1, 167.5………………………………………………………….
Synthesis pathway
Synthesis a) 

Trade Names
Country Trade name Manufacturer Germany Cialis Lilly France Cialis – “- United Kingdom – “- – “- Italy – “- Eli Lilly USA – “- Lilly ICOS Ukraine Cialis Lilly del Caribe, Inc.., Puerto Rico (USA) Lilly SA (Packing), Spain Tadalafil Aurohem Laboratories Pvt. Ltd.., India Formulations
-
Tablets 10 mg, 20 mg
Links
-
EP 740 668 (Lab. Glaxo SA, Fr .; GB -prior. 21.01.1994).
-
US 6,140,329 (Lab. Glaxo SA, Fr .; GB -prior. 14/07/1995).
-
US 6,143,746 (Icos Corp .; 07/11/2001; GB -prior. 01.21.1994).
-
US 6,821,975 (Lilly ICOS; 23.11.2004; appl. 19.7.2002; USA-prior. 3.8.1999).
-
US 6,943,166 (Lilly ICOS; 13.9.2005; appl. 19.10.2001; USA-prior. 30.4.1999).
-
US 7,182,958 (Lilly ICOS; 27.2.2007; appl. 26.4.2000; USA-prior. 3.8.1999).
-



References
- Daugan, A; Grondin P, Ruault C, Le Monnier de Gouville AC, Coste H, Kirilovsky J, Hyafil F, Labaudinière R (October 9, 2003). “The discovery of tadalafil: a novel and highly selective PDE5 inhibitor. 1: 5,6,11,11a-tetrahydro-1H-imidazo[1′,5′:1,6]pyrido[3,4-b]indole-1,3(2H)-dione analogues”. Journal of Medicinal Chemistry 46 (21): 4525–32. doi:10.1021/jm030056e . PMID 14521414.
- Richards, Rhonda (September 17, 1991). “ICOS At A Crest On Roller Coaster”. USA Today. p. 3B.
- Ervin, Keith (June 21, 1998). “Deep Pockets + Intense Research + Total Control = The Formula — Bothell Biotech Icos Keeps The Pipeline Full Of Promise”. The Seattle Times. p. F1. Retrieved January 10, 2009.
- Revill, Jo (February 2, 2003). “Drugs giant says its new pill will pack more punch than rival Viagra”. The Observer. Retrieved 2007-04-06.
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm274642.htm
- https://www.consumerreports.org/health/resources/pdf/best-buy-drugs/money-saving-guides/english/PillSplitting-FINAL.pdf
- “FDA Announces Revisions to Labels for Cialis, Levitra and Viagra”. Food and Drug Administration. 2007-10-18. Retrieved 2009-09-28.
- “Cialis: Warnings, Precautions, Pregnancy, Nursing, Abuse”. RxList. 2007. Retrieved 2007-04-06.
- Bischoff, E (June 2004). “Potency, selectivity, and consequences of nonselectivity of PDE inhibition”. International Journal of Impotence Research 16: S11–4.doi:10.1038/sj.ijir.3901208 . PMID 15224129. Retrieved January 19, 2009.
- Elliott, Stuart (January 10, 2006). “For Impotence Drugs, Less Wink-Wink”. The New York Times. p. C2. Retrieved January 15, 2009.
- Elliott, Stuart (April 25, 2004). “Viagra and the Battle of the Awkward Ads”. The New York Times. p. 1. Retrieved January 15, 2009.
- McCarthy, Shawn (March 5, 2005). “First they tried to play it safe; Ads for erectile dysfunction drug Cialis bared all – including a scary potential side effect. It was risky but it has paid off”. The Globe and Mail. p. B4.
- Loyd, Linda (July 6, 2003). “Two Pills Look to Topple Viagra’s Reign in Market; Levitra Expects Approval Next Month, Cialis Later This Year”. The Philadelphia Inquirer. p. E01.
- 38 is 1 below
- 39 is 2 below
- 40 is 3 below
-
- daugan, A. C.-M. Tetracyclic Derivatives; Process of Preparation and Use. U.S. Patent 5,859,006, Jan 12, 1999.
- Daugan, A. C.-M. Tetracyclic Derivatives, Process of Preparation and Use. U.S. Patent 6,025,494, Feb 15, 2000.
- Shi, X.-X.; Liu, S.-L.; Xu, W.; Xu, Y.-L. Tetrahedron: Asymmetry 2008, 19, 435–442.doi:10.1016/j.tetasy.2007.12.017
DAUGAN A ET AL: “THE DISCOVERY OF TADALAFIL: A NOVEL AND HIGHLY SELECTIVE PDE5 INHIBITOR. 2: 2,3,6,7,12,12A-HEXAHYDROPYRAZINO[1′,2′:1,6 ÜPYRIDO[3,4-B ÜINDOLE-1,4-DIONE” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 46, no. 21, 2003, pages 4533-4542, XP008052656 ISSN: 0022-2623
- Tadalafil bound to proteins in the PDB
- National Institutes of Health – Medlineplus
- Material Safety Data Sheet PDF file
- Official Cialis (Tadalafil) Website
- U.S. National Library of Medicine: Drug Information Portal – Tadalafil
| WO2009004557A2 * | Jun 28, 2008 | Jan 8, 2009 | Ranbaxy Lab Ltd | A process for the preparation of intermediates of tetracyclic compounds |
| WO2009148341A1 | Jun 3, 2009 | Dec 10, 2009 | Zaklady Farmaceutyczne Polpharma Sa | Process for preparation of tadalafil |
| WO2012107549A1 | Feb 10, 2012 | Aug 16, 2012 | Interquim, S.A. | PROCESS FOR OBTAINING COMPOUNDS DERIVED FROM TETRAHYDRO-ß-CARBOLINE |
| EP2107059A1 | Mar 31, 2008 | Oct 7, 2009 | LEK Pharmaceuticals D.D. | Conversion of tryptophan into ß-carboline derivatives |
| US8445698 | Jun 28, 2008 | May 21, 2013 | Ranbaxy Laboratories Limited | Process for the preparation of an intermediate of tadalafil |
Flow chemistry approaches directed at improving chemical synthesis

The true potential of flow chemistry as an enabling technology can really only be fully appreciated when seen in the context of a target driven multi-step synthesis, aimed at the delivery of advanced chemical structures such as active pharmaceutical ingredients (APIs) .
As most pharmaceutical syntheses typically require between 8 and 10 chemical transformations (this is often somewhat reduced to 5/6 steps when analogue/library syntheses are being conducted), excluding protecting group manipulations, to realize the target molecule, this is a good foundation from which to explore the advantages of flow chemistry. We have generated a flow protocol for the synthesis of imatinib, the API of the Novartis block buster anticancer therapeutic Gleevec (imatinib mesylate), including a series of analogues (Scheme 11)
Furthermore, we aimed to create a route which would allow each of the three main fragments to be exchanged to address maximum variation in subsequent analogue synthesis. This requires additional planning to build flexibility into the sequence where this desired diversity can be easily introduced. Again, prior consideration of the generated intermediates, and any potential by-products that may arise, is critical and should be addressed prior to embarking on the synthesis.
Consequently, the extensive profiling of the reaction in terms of its purity profile is more closely analogous to process chemistry than traditional Medicinal Chemistry, even at the development stage. So, although more time consuming in the planning stage, having a greater understanding of the chemistry, does then enable a smoother up scaling and more rapid optimization of the route.

read all this at
http://www.degruyter.com/view/j/gps.2013.2.issue-3/gps-2013-0029/gps-2013-0029.xml
Flow chemistry approaches directed at improving chemical synthesis
1Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
Corresponding author: Ian R. Baxendale, Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
Citation Information: Green Processing and Synthesis. Volume 2, Issue 3, Pages 211–230, ISSN (Online) 2191-9550, ISSN (Print) 2191-9542, DOI: 10.1515/gps-2013-0029, May 2013

Fiduxosin ….An α1-Adrenoceptor antagonist
Fiduxosin hydrochloride, 208992-74-9, NCGC00162178-02, AC1L58WW,
208993-54-8 (free base)
 Fiduxosin
Fiduxosin-

-

-

-

-

-

-

-

-
Example 108
- 3-[4-((3aR,9bR)-cis -9-Methoxy-1,2,3,3a,4,9b-hexahydro-[1]-benzopyrano[3,4-c]pyrrol-2-yl)butyl]-8-(4-hydroxyphenyl)-pyrazino[2′,3′:4,5]thieno[3,2-d]pyrimidine-2,4(1H,3H)-dione
-
The product of Example 16 (0.07 g,0.105 mmol) and 4-(methoxymethyloxy) phenyl boronic acid (0.02 g, 0.11 mmol) prepared by the procedure described in Tetr.Lett., 31, 27, (1990) were treated as described in Example 106 to yield 0.029g(45%) of MOM-protected product. To the solution of this product (0.11g, 0.17 mmol) in CH3OH/THF was added 2N HCl (0.2ml) and the reaction mixture was refluxed for 1 hour. The reaction was evaporated and partitioned in NaHCO3 sol. and CH2Cl2/CH3OH to yield 0.005 g (51%) of the title compound.
-
1H NMR (500 MHz, CDCl3) d 1.81 (m, 2H), 1.98 (m, 2H), 2.25 (m, 1H), 2.65 (m, 1H), 2.88 (m, 1H), 3.08 (m, 2H), 3.22(m, 2H), 3.65 (m, 1H), 3.73 (m, 1H), 3.82 (s, 3H), 3.9 (m, 1H), 4.25 (m, 1H), 4.42 (m, 1H), 6.52 (m, 2H), 7.38 (m, 2H),7.49(m, 1H), 7.9 (t, 1H), 8.09 (d, 1H),9.12 (s, 1H);
-
MS(ESI)m/e 572 (M+H)+.
-








3-[4-((3aR,9bR)- cis -9-Methoxy-1,2,3,3a,4,9b-hexahydro-[1]-benzopyrano[3,4-c]pyrrol-2-yl)butyl]-8-chloro-pyrazino[2′,3′:4,5]thieno[3,2-d]pyrimidine-2,4(1H,3H)-dione hydrochloride
-
The product from Example 10 C (0.27 g, 1.0 mmol) and the product from Example 1E (0.20 g, 0.73 mmol) were treated as described in Example 1F to yield 0.29 g (77%) of the title compound: m.p. 220-222°;
-
1H NMR (300 MHz, CDCl3(free base)) δ 8.68 (s, 1H), 7.0 (t, 1H), 6.48 (d, 1H), 6.45 (d, 1H), 4.28 (m, 1H), 4.12 (m, 3H), 4.0 (m, 2H), 3.75 (s, 3H), 3.6 (m, 1H), 3.08 (m, 3H), 2.9 (m, 2H), 1.75 (m, 4H); MS (DCI/NH3) m/e 514(M+H)+;
-
Analysis calc’d for C24H24ClN5O4S·HCl·0.75H2O: C, 51.11; H, 4.74; N, 12.42; found: C, 51.09; H, 4.75; N, 12.43.

Fiduxosin (ABT-980), α1a-adrenoreceptor antagonist, a development compound at Abbot for the treatment of benign prostate hyperplasia, is disclosed in Organic Process Research & Development 2004, 8, 897-902 and references cited therein.
The synthetic route for preparation of Fiduxosin is as follows:


Fiduxosin (1) has been under development at Abbott Laboratories for the treatment of benign prostatic hyperplasia. A convergent strategy required methodologies for preparation of an enantiomerically pure 3,4-cis-disubstituted pyrrolidine and a 2,3,5-trisubstituted thienopyrazine in a regiospecific manner.
A [3+2] cycloaddition of an enantiopure azomethine ylide followed by a diastereoselective crystallization was employed to prepare the benzopyranopyrrolidine in high diastereomeric and enantiomeric purity. Conditions for reduction of an O-aryl lactone susceptible to epimerization were developed, and cyclization of the alcohol/phenol to the ether was accomplished in high yield.
The thienopyrazine was prepared by condensation of methyl thioglycolate and a regiospecifically prepared 2-bromo-3-cyano-5-phenylpyrazine. Conditions for effective halogen substitutive deamination to prepare regiospecific trisubstituted pyrazines will be described.
The mixture of 5 – and 6-phenyl regioisomers of 2-hydroxy-3-carboxamidopyrazine (IX) and (X), prepared by a known method, was treated with POCl3 and Et3N to produce the corresponding chloro nitriles (XI) and (XII ). Condensation of this mixture with methyl thioglycolate in the presence of NaOMe, followed by chromatographic separation of isomers furnished the desired thienopyrazine intermediate (XIII).
http://pubs.acs.org/doi/suppl/10.1021%2Fop049889k
…………………………………………………..
 Fiduxosin
Fiduxosin
……………………………………………………….
SYNTHESIS

Cycloaddition of the azomethine ylide resulting from N-trimethylsilylmethyl-N-methoxymethyl-(R)-alpha-methylbenzylamine (II) to 5-methoxycoumarin (I) produced the chiral cis-benzopyranopyrrole system (III). Lactone reduction by means of LiAlH4 or LiBH4 afforded diol (IV). After conversion of the primary alcohol of (IV) to either the corresponding chloride or the mesylate, cyclization in the presence of potassium tert-butoxide generated the tricyclic compound (V).
The alpha-methylbenzyl group of ( V) was removed by catalytic hydrogenation to give amine (VI), which was alkylated with 4-bromobutyronitrile yielding (VII). Reduction of the cyano group of (VII) using LiAlH4 in the presence AlCl3 or by catalytic hydrogenation in the presence of Raney -Ni produced the primary amine (VIII).
…………………………………………………

The mixture of 5 – and 6-phenyl regioisomers of 2-hydroxy-3-carboxamidopyrazine (IX) and (X), prepared by a known method, was treated with POCl3 and Et3N to produce the corresponding chloro nitriles (XI) and (XII ). Condensation of this mixture with methyl thioglycolate in the presence of NaOMe, followed by chromatographic separation of isomers furnished the desired thienopyrazine intermediate (XIII).
………………………………………………………….

In a regioselective synthetic method, phenyl glyoxime (XIV) was condensed with aminomalononitrile to produce the pyrazine N-oxide (XV). Reduction of the N-oxide of (XV) with triethyl phosphite yielded (XVI). Diazotization of the amino group of (XVI), followed by diazo displacement with CuBr2, furnished bromo pyrazine (XVII). This was then cyclized with methyl thioglycolate as above to yield the desired thienopyrazine intermediate (XIII).
………………………………………………….

In an alternative synthesis, phenylacetaldehyde (XVIII) was condensed with pyrrolidine (XIX) to give enamine (XX). Nitrosation of malononitrile (XXI), followed by treatment with tosyl chloride, produced the O-tosyl oxime (XXII). This was condensed with enamine (XX), and to the intermediate adduct (XXIII) was added thiophenol producing the phenylthiopyrazine (XXIV). Subsequent oxidation of the sulfide group of (XXIV) to sulfone (XXV), followed by condensation with methyl thioglycolate, gave the desired thienopyrazine (XIII).
……………………………………………………………..

The amino ester intermediate (XIII) was treated with phosgene and Et3N, and to the resulting isocyanate (XXVI) was added the primary amine (VIII), producing urea (XXVII). Then, cyclization of (XXVII) in refluxing toluene generated the desired compound,
fiduxosin
|
2-1-2002
|
Effect of fiduxosin, an antagonist selective for alpha(1A)- and alpha(1D)-adrenoceptors, on intraurethral and arterial pressure responses in conscious dogs.
|
The Journal of pharmacology and experimental therapeutics
|
|
|
2-1-2002
|
Modeling of relationships between pharmacokinetics and blockade of agonist-induced elevation of intraurethral pressure and mean arterial pressure in conscious dogs treated with alpha(1)-adrenoceptor antagonists.
|
The Journal of pharmacology and experimental therapeutics
|
|
|
1-1-2002
|
Effect of food on the pharmacokinetics of fiduxosin in healthy male subjects.
|
European journal of drug metabolism and pharmacokinetics
|
|
9-1-2012
|
Identification and analysis of hepatitis C virus NS3 helicase inhibitors using nucleic acid binding assays.
|
Nucleic acids research
|
|
|
3-1-2012
|
Small molecule screening identifies targetable zebrafish pigmentation pathways.
|
Pigment cell & melanoma research
|
|
|
7-1-2010
|
A small molecule inverse agonist for the human thyroid-stimulating hormone receptor.
|
Endocrinology
|
|
|
11-1-2009
|
A new homogeneous high-throughput screening assay for profiling compound activity on the human ether-a-go-go-related gene channel.
|
Analytical biochemistry
|
|
|
10-1-2009
|
Genetic mapping of targets mediating differential chemical phenotypes in Plasmodium falciparum.
|
Nature chemical biology
|
|
|
5-1-2007
|
Chemical genetics reveals a complex functional ground state of neural stem cells.
|
Nature chemical biology
|
|
|
5-1-2006
|
Microsphere-based protease assays and screening application for lethal factor and factor Xa.
|
Cytometry. Part A : the journal of the International Society for Analytical Cytology
|
|
|
5-1-2002
|
Single- and multiple-dose pharmacokinetics of fiduxosin under nonfasting conditions in healthy male subjects.
|
Journal of clinical pharmacology
|
|
|
5-1-2002
|
Multiple dose pharmacokinetics of fiduxosin under fasting conditions in healthy elderly male subjects.
|
The Journal of pharmacy and pharmacology
|
|
|
2-1-2002
|
Preclinical pharmacology of fiduxosin, a novel alpha(1)-adrenoceptor antagonist with uroselective properties.
|
The Journal of pharmacology and experimental therapeutics
|
Scientists develop potential new drug treatment to tackle viruses

An international team of scientists have successfully developed a novel compound which early signs suggest might prevent a range of viruses from infecting humans. Researchers from Oxford, Beijing, Leeds and Innsbruck collaborated on the new inhibitor. It targets a group of viruses responsible for hand, foot and mouth disease, especially the EV71 virus. This viral group causes numerous epidemics in children, mainly in Asia, with roughly 10 million cases reported every year in China alone. Symptoms are usually mild but in some cases the disease can prove fatal – the Chinese government reported over 900 deaths in 2010. The disease is currently untreatable and is a major global threat to public health.
This discovery, published in Nature Structural and Molecular Biology, may also have important implications for combating other diseases. Hand, foot and mouth disease is caused by several closely related viruses, and the new compound is effective…
View original post 510 more words
