
Home » Uncategorized (Page 151)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tivorbex (indomethacin); Iroko Pharmaceuticals; For the treatment of acute pain,

Tivorbex (indomethacin); Iroko Pharmaceuticals; For the treatment of acute pain, Approved February of 2014
2-{1-[(4-chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid
cas 53-86-1
PHILADELPHIA—Iroko Pharmaceuticals, LLC, a global specialty pharmaceutical company dedicated to advancing the science of analgesia, today announced that the U.S. Food and Drug Administration (FDA) has approved TIVORBEX™ (indomethacin) capsules, a nonsteroidal anti-inflammatory drug (NSAID), at 20 mg and 40 mg doses for the treatment of mild to moderate acute pain in adults1.
“TIVORBEX is the second NSAID to be approved from Iroko’s lower dose NSAID pipeline that uses proprietary SoluMatrix Fine Particle Technology™.”
TIVORBEX was approved at dosage strengths that are 20 percent lower than the 25 mg and 50 mg indomethacin products currently on the market2. FDA approval of TIVORBEX was supported by data from two Phase 3 multi-center, placebo-controlled trials that demonstrated significant improvement in pain relief in patients with post-surgical acute pain receiving TIVORBEX compared with patients receiving placebo3.
“The FDA approval of TIVORBEX is another significant milestone for Iroko as it validates our strategic approach towards developing a suite of NSAID products that offer pain management at lower doses,” said John Vavricka, President and CEO of Iroko Pharmaceuticals. “TIVORBEX is the second NSAID to be approved from Iroko’s lower dose NSAID pipeline that uses proprietary SoluMatrix Fine Particle Technology™.” read at
Indometacin (INN) or indomethacin (USAN and former BAN) is a non-steroidal anti-inflammatory drug (NSAID) commonly used as a prescriptionmedication to reduce fever, pain, stiffness, and swelling. It works by inhibiting the production of prostaglandins, molecules known to cause these symptoms. It is marketed under more than seventy different trade names.[1]

Indomethacin was discovered in 1963[8] and it was first approved for use in the U.S. by the Food and Drug Administration in 1965. Its mechanism of action, along with several other NSAIDs that inhibit COX, was described in 1971.[9]


References
- Trade names are listed on DrugBank.ca entry DB00328
- Sanders, Lisa (6 January 2012). “Think Like a Doctor: Ice Pick Pain Solved!”. The New York Times.
- Garza, I & Schwedt, TJ. “Hemicrania continua.” UpToDate.http://www.uptodate.com/contents/hemicrania-continua. Accessed 8/27/13.
- Smyth JM, Collier PS, Darwish M et al. (September 2004). “Intravenous indometacin in preterm infants with symptomatic patent ductus arteriosus. A population pharmacokinetic study”. Br J Clin Pharmacol 58 (3): 249–58. doi:10.1111/j.1365-2125.2004.02139.x.PMC 1884560. PMID 15327584.
- “INDOMETHACIN”. Hazardous Substances Data Bank (HSDB). National Library of Medicine’s TOXNET. Retrieved April 4, 2013.
- Giles W, Bisits A (October 2007). “Preterm labour. The present and future of tocolysis”. Best Pract Res Clin Obstet Gynaecol 21 (5): 857–68. doi:10.1016/j.bpobgyn.2007.03.011.PMID 17459777.
- Akbarpour F, Afrasiabi A, Vaziri N (1985). “Severe hyperkalemia caused by indomethacin and potassium supplementation”. South Med J 78 (6): 756–7. doi:10.1097/00007611-198506000-00039. PMID 4002013.
- Hart F, Boardman P (October 1963). “Indomethacin: A New Non-steroid Anti-inflammatory Agent”. Br Med J 2 (5363): 965–70. doi:10.1136/bmj.2.5363.965. PMC 1873102.PMID 14056924.
- Ferreira S, Moncada S, Vane J (Jun 23, 1971). “Indomethacin and aspirin abolish prostaglandin release from the spleen”. Nat New Biol 231 (25): 237–9.doi:10.1038/231237a0. PMID 5284362.
- Effects of Perinatal Indomethacin Treatment on Preterm Infants, academic dissertation (PDF)
- Indomethacin, from MedicineNet
- Indomethacin, from Drugs.com
- Indocin: Description, chemistry, ingredients, from RxList.com
- Indomethacin bound to proteins in the PDB
IBRUTINIB 依鲁替尼 A Btk protein inhibitor.

IBRUTINIB 依鲁替尼
A Btk protein inhibitor.
1-[(3R)-3-[4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]piperidin-1-yl]prop-2-en-1-one
1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one
| CAS number | 936563-96-1 |
|---|
- CRA-032765
- Ibrutinib
- Imbruvica
- Pc-32765
- PCI 32765
- PCI32765
- UNII-1X70OSD4VX
Company: Pharmacyclics
Approval Status: Approved February 2014US FDA:link
Treatment Area: chronic lymphocytic leukemia
Bruton’s tyrosine kinase (Btk) inhibitor
U.S. Patent No: 7,514,444 , 7,718,662
patent validity: December 2026
An orally bioavailable small-molecule inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Ibrutinib binds to and inhibits BTK activity, preventing B-cell activation and B-cell-mediated signaling and inhibiting the growth of malignant B cells that overexpress BTK. BTK, a member of the src-related BTK/Tec family of cytoplasmic tyrosine kinases, is required for B cell receptor (BCR) signaling, plays a key role in B-cell maturation, and is overexpressed in a number of B-cell malignancies.
Imbruvica (ibrutinib) is an orally available, selective inhibitor of Bruton’s tyrosine kinase (Btk), a gene that is disrupted in the human disease X-linked agammaglobulenemia (XLA). BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways.
Imbruvica is specifically approved for chronic lymphocytic leukemia in patients who have received at least one prior therapy.
Imbruvica (Ibrutinib, previously known as PCI-32765) was approved as a “breakthrough therapy” on November 13, 2013 by the US Food and Drug Administration (FDA) for the treatment of mantle cell lymphoma (MCL), a rare and deadly form of blood cancer.
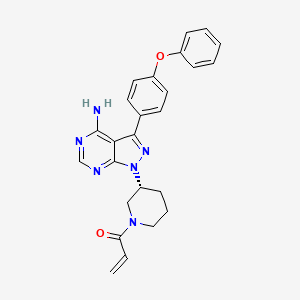
Ibrutinib, a first in class oral Bruton’s tyrosine kinase (Btk) inhibitor, was launched in the U.S. for the treatment of patients with mantle cell lymphoma in 2013, and for the treatment of chronic lymphocytic leukemia in 2014. In the E.U., the product candidate is awaiting registration for both indications. Additional phase III clinical trials are ongoing for the treatment of these indications in combination with bendamustine and rituximab and for the treatment of relapsed or refractory marginal zone lymphoma (MZL). Janssen and Pharmacyclics are conducting phase II clinical trials for the treatment of refractory follicular lymphoma. Early clinical development is also under way at Pharmacyclics for the treatment of recurrent B-cell lymphoma, relapsed/refractory MCL, and relapsed or relapsed and refractory multiple myeloma. The company filed an IND seeking approval to commence clinical evaluation of ibrutinib for the treatment of autoimmune disease. Preclinical studies had been under way for rheumatoid arthritis; however, no recent development has been reported. Ibrutinib is also active against Lyn and LCK tyrosine kinases.
In 2011, a codevelopment agreement was signed between the National Cancer Institute (NCI) and Pharmacyclics for the treatment of hematologic/blood cancer. Also in 2011, a worldwide codevelopment and comarketing agreement was signed by Janssen and Pharmacyclics for the treatment of cancer. In 2012, orphan drug designation was assigned in the U.S. and the E.U. for the treatment of CLL. This designation was also assigned by the FDA in 2012 for the treatment of mantle cell lymphoma. In 2013, several orphan drug designations were assigned in the U.S.; for the treatment of small lymphocytic lymphoma, for the treatment of Waldenstrom’s macroglobulinemia and for the treatment of diffuse large B-cell lymphoma. For this indication, orphan drug designation was assigned also in the E.U. the same year. In 2012, fast track designation was assigned by the FDA for the treatment of CLL. In 2013, breakthrough therapy designations were assigned to the compound in the U.S.: for the treatment (as monotherapy) of patients with chronic lymphocytic leukemia or small lymphocytic lymphoma, for the treatment of relapsed or refractory mantle cell lymphoma who have received prior therapy and for the treatment of Waldenstrom’s macroglobulinemia.
Imbruvica is supplied as a capsule for oral administration. The recommended dose is 420 mg taken orally once daily (three 140 mg capsules once daily). Capsules should be taken orally with a glass of water. Do not open, break, or chew the capsules.
The FDA approval of Imbruvica for chronic lymphocytic leukemia was based on an open-label, multi-center trial of 48 previously treated patients. Imbruvica was administered orally at 420 mg once daily until disease progression or unacceptable toxicity. The overall response rate (ORR) and duration of response (DOR) were assessed using a modified version of the International Workshop on CLL Criteria by an Independent Review Committee. The ORR was 58.3%, all partial responses. None of the patients achieved a complete response. The DOR ranged from 5.6 to 24.2+ months. The median DOR was not reached.
Imbruvica (ibrutinib) is an orally available, selective inhibitor of Bruton’s tyrosine kinase (Btk). Ibrutinib forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. BTK’s crole in signaling through the B-cell surface receptors results in activation of pathways necessary for B-cell trafficking, chemotaxis, and adhesion.
Ibrutinib (USAN,[1] also known as PCI-32765 and marketed in the U.S. under the name Imbruvica) is an anticancer drug targeting B-cell malignancies. It was approved by the US FDA in November 2013 for the treatment of mantle cell lymphoma[2] and in February 2014 for the treatment ofchronic lymphocytic leukemia.[3] It is an orally-administered, selective and covalent inhibitor of the enzyme Bruton’s tyrosine kinase (BTK).[4][5][6]Ibrutinib is currently under development by Pharmacyclics, Inc and Johnson & Johnson‘s Janssen Pharmaceutical division for additional B-cell malignancies including diffuse large B-cell lymphoma and multiple myeloma.[7][8][9]
Mechanism
In preclinical studies on chronic lymphocytic leukemia (CLL) cells, ibrutinib has been reported to promote apoptosis, inhibit proliferation, and also prevent CLL cells from responding to survival stimuli provided by the microenvironment.[12] In this study, treatment of activated CLL cells with ibrutinib resulted in inhibition of Btk tyrosine phosphorylation and also effectively abrogated downstream survival pathways activated by this kinase including ERK1/2, PI3K, and NF-κB. Additionally, ibrutinib inhibited proliferation of CLL cells in vitro, effectively blocking survival signals provided externally to CLL cells from the microenvironment including soluble factors (CD40L, BAFF, IL-6, IL-4, and TNF-α), fibronectin engagement and stromal cell contact.
In early clinical studies, the activity of ibrutinib has been described to include a rapid reduction in lymphadenopathy accompanied by a transient lymphocytosis, suggesting that the drug might have direct effects on cell homing or migration to factors in tissue microenvironments.[13]
Ibrutinib has been reported to reduce CLL cell chemotaxis towards the chemokines CXCL12 and CXCL13, and inhibit cellular adhesion following stimulation at the B cell receptor.[14][15] Together, these data are consistent with a mechanistic model whereby ibrutinib blocks BCR signaling, which drives cells into apoptosis and/or disrupts cell migration and adherence to protective tumour microenvironments.
History
Ibrutinib was first designed and synthesized at Celera Genomics which reported in 2007 a structure-based approach for creating a series of small molecules that inactivate BTK through covalent binding to cysteine-481 near the ATP binding domain of BTK.[4] These small molecules irreversibly inhibited BTK by using a Michael acceptor for binding to the target cysteine. In April 2006, Pharmacyclics acquired Celera’s small molecule BTK inhibitor discovery program, which included a compound, PCI-32765 that was subsequently chosen for further preclinical development based on the discovery of anti-lymphoma properties in vivo.[16] Since 2006, Pharmacyclics’ scientists have advanced the molecule into clinical trials and identified specific clinical indications for the drug. It also has potential effects against autoimmune arthritis.[17] It was approved by the US FDA on November 13, 2013 for the treatment of mantle cell lymphoma.[2] On Feb. 12, 2014, the U.S. Food and Drug Administration expanded the approved use of the drug ibrutinib to chronic lymphocytic leukemia (CLL). [18]
Ibrutinib is an inhibitor of Bruton’s tyrosine kinase (BTK). It is a white to off-white solid with the empirical formula C25H24N6O2 and a molecular weight 440.50. Ibrutinib is freely soluble in dimethyl sulfoxide, soluble in methanol and practically insoluble in water.
The chemical name for ibrutinib is 1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)-1Hpyrazolo[ 3,4-d]pyrimidin-1-yl]-1-piperidinyl]-2-propen-1-one and has the following structure:
 |
IMBRUVICA (ibrutinib) capsules for oral administration are supplied as white opaque capsules that contain 140 mg ibrutinib as the active ingredient. Each capsule also contains the following inactive ingredients: croscarmellose sodium, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate. The capsule shell contains gelatin, titanium dioxide and black ink. Each white opaque capsule is marked with “ibr 140 mg” in black ink.
PCI-32765 (ibrutinib) is disclose d in U.S. Patent No. 7,514,444, issued on April 7, 2009, and has the following structur

Ibrutinib is an orally available drug that targets Bruton’s tyrosine kinase (BTK).
Ibrutinib is an irreversible small molecule BTK inhibitor that is in Ph Ib/II of clinical trials in a variety of B-cell malignancies including chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL) and multiple myeloma (cancer of plasma cells, a type of white blood cell present in bone marrow). At present ibrutinib is administered orally in clinical trials, via the gastrointestinal tract, at high clinical doses (420 mg/day or 840 mg/day) to patients with CLL and SLL to obtain the desired thereapeutic effect. The need for such high doses of ibrutinib may be due to low bioavailability (the oral bioavailability of ibrutinib is reported to be 22.8% in rats) and may be responsible for the adverse side effects associated with the use of ibrutinib such as nausea or emesis, dizziness and diarrhea. Moreover, low bioavailability results in more variable absorption and potential variability of the desired therapeutic response.
As stated above, at present ibrutinib is administered orally, via the gastrointestinal tract, at high clinical doses (420 mg/day or 840 mg/day) to patients to obtain the desired clinical benefit. It is presently disclosed that when ibrutinib is administered intraduodenally versus via the gastrointestinal tract in rats, the oral bioavailability of ibrutinib unexpectedly increased from 21 % to 100% as determined by AUC.
This unexpected increase in oral bioavailability of ibrutinib can translate into a number of desirable practical benefits. The increase in oral bioavailability should enable administration of ibrutinib at a significantly lower therapeutically effective dose than is currently being used. The lower variability associated with this greater bioavailability should lead to a more reliable therapeutic response as well as more predictable drug absorption.
And avoidance of exposure of Ibtrutinib to the stomach and/or use of lower therapeutically effective dose of ibrutinib can reduce or altogether eliminate potential adverse side effects of this drug such as diahrrea, nausea or emesis, and dizziness. U.S. Patent No. 7,514,444, mentioned above, discloses administration of 0.02-5000 mg/kg andl-1500 mg of ibrutinib/per day and in clinical trials 420 or 840 mg/day of ibrutinib is being administered to the patients with CLL and SLL.
There is no reasonable expectation in the art that ibrutinib can be adminstered orally at lower efficacious doses to the patients with CLL and SLL, particularly as evidenced by the 420 or 840 mg/day of ibrutinib being administered in clinical trials to those patients. Moreover, other than for active agents that are unstable in the stomach or at acidic pH delivery of any active agent with low bioavailability further along in the gastrointestinal tract reduces the path length for drug absorption and would be expected to reduce bioavailability. Therefore, it was unexpected to achieve delivery of ibruntinib directly to the small intestine with greater bioavailability.
PC1-32765 (Ibrutinib), chemical name: 1_ [(3R) _3-[4_-3 – (4 – phenoxy-phenyl)-1H-pyrazolo [3,4-d] pyrimidine – 1 – yl] – 1-piperidinyl]-2 – propen-1 – one, and its structural formula is as follows:

PC1-32765 is an oral medication that inhibits B cell as the main receptor tyrosine kinase signaling and promote cell death process, preventing cell migration and adhesion in malignant B cells.
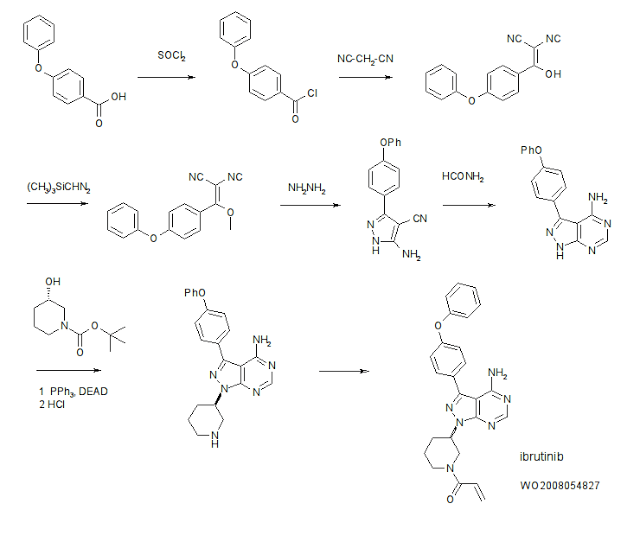
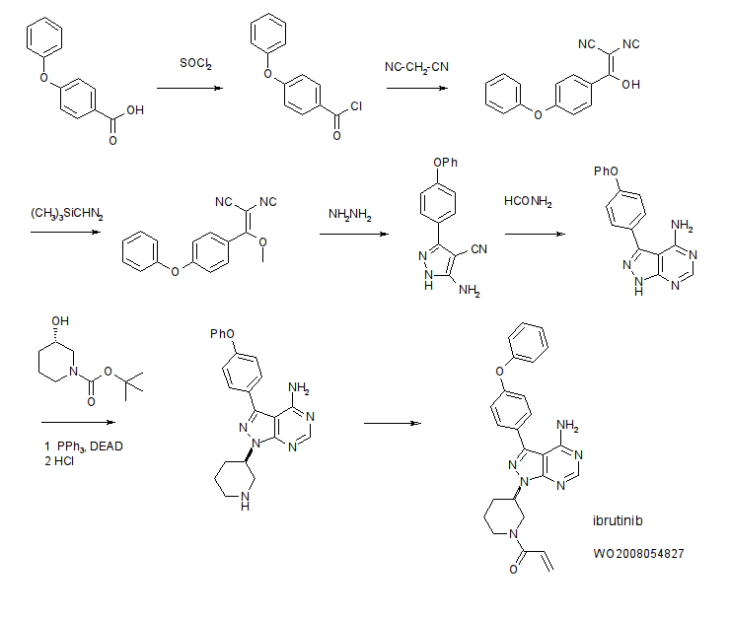
US20080108636 basic patent has been disclosed a synthetic route:
This synthetic route with 4 – phenoxy-benzoic acid as raw material, after eight-step reaction the final product, the following reaction steps:

The above method has the following disadvantages:
1, eight single-step reaction, long route, the economy is bad; i1, to use synthetic intermediates 4:00 trimethylsilyl diazomethane (TMSCHN2), this material easy to blow up, the risk coefficient is large, so large-scale production greatly reduces the possibility;
ii1, synthetic intermediates 7:00, set out to use polymer-supported triphenylphosphine, non-industrial raw materials used, the price is expensive, the cost of smell;
iv, the final step of acylation, the selectivity is poor, a large amount of negative product, purification is difficult, amplification reaction is difficult.
In summary, the route material is not common, expensive step, high costs, the reaction dangerous side reactions, purification difficult, limiting the possibility of industrial production of the route.
………………………
Polymorphs
EXAMPLES
[00438] The following ingredients, formulations, processes and procedures for practicing the methods disclosed herein correspond to that described above. Example 1; Preparation of Crystalline Forms of l-((R)-3-(4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-dlpyrimidin-l-yl)piperidin-l-yl)prop-2-en-l-one (Compound 1)
Form A – Route 1:
[00439] Amorphous Compound 1 (ca. 15 mg) was measured into a vial. Ten volumes (150 μΐ) of solvent [methyl tert-butyl ether (MTBE), diisopropyl ether (DIPE), ethyl acetate, isopropyl acetate, isopropyl alcohol, methyl isobutyl ketone (MIBK), methyl ethyl ketone (MEK), acetone, methanol, nitromethane, 10% aqueous acetone, or 10% aqueous isopropyl alcohol] were added to the vial. The vial was sealed and placed in a shaker at 50 °C for one hour. If a slurry was obtained, an additional thirty volumes (total of 600 μΐ) of solvent was added, then the slurry was returned to 50 °C for another hour. If the sample remained as a slurry at this point, no further solvent was added. The solution/slurry was stirred at 50 °C for one hour, then cooled to 0 °C at 0.1 °C/min, then held at 0 °C overnight. If a slurry was obtained, the solids were filtered under vacuum to provide Compound 1 , Form A; the solution was returned to ambient temperature for slow evaporation through a pin-hole to furnish Compound 1, Form A.
“Compound 1” or “l-((R)-3-(4-amino-3-(4-phenoxyphenyl)-lH-pyrazolo[3,4- d]pyrimidin- 1 -yl)piperidin- 1 -yl)prop-2-en- 1 -one” or “1 – {(3i?)-3-[4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-JJpyrimidin-l-yl]piperidin-l-yl}prop-2-en-l-one” or “2-Propen- 1 -one, 1- [(3R)-3-[4-amino-3-(4-phenoxyphenyl)- lH-pyrazolo[3,4-<f]pyrimidin- 1 -yl] – 1 -piperidinyl-” or ibrutinib or any other suitable name refers to the compound with the following structure:

………….
Synthesis
Synthesis of Compound 3—Btk Activity Probe
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride was removed by distillation, the residual oil was dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride was dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) was added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture was stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue was taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gave a semisolid residue which was treated with a portion of ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gave 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which was sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2).
Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl (Intermediate 4); a) triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) TFA/CH2Cl2.

To a solution of 1-boc-3-(S)-hydroxypiperidine (3.98 g, 19.8 mmol) and triphenylphosphine (5.19 g, 19.8 mmol) in THF (150 ml) was added DIAD (3.9 ml, 19.8 mmol). The yellow solution was stirred 1 minute then Intermediate 2 (4.0 g, 13.2 mmol) was added and the reaction was heated with a heat gun (3-5 minutes) until the solid had dissolved. After stirring for 1 hour at room temperature, the solvent was removed and the resulting brown oil was subjected to flash chromatography (30% then 50% THF/hexanes) to provide 4.45 g (69%) of Intermediate 3 (trace of triphenylphosphine oxide is present) as a light brown foam.
To a solution of Intermediate 3 (4.4 g, 9.0 mmol) in CH2Cl2 (20 ml) was added TFA (2.8 ml, 36.2 mmol). After stirring 2 hrs at room temperature, the solvent was removed and the residue was partitioned between ethyl acetate (250 ml) and dilute aq. K2CO3. The organic layer was dried (MgSO4), filtered and concentrated to 70 ml. The resulting solution was stirred and 4.0M HCl in dioxane (4 ml) was added to provide a thick light orange precipitate. The precipitate was collected by filtration and washed with ethyl acetate (50 ml). The material was then partitioned between ethyl acetate (300 ml) and dilute aq. K2CO3. The organic layer was dried (MgSO4), filtered and concentrated to provide 2.78 g (80%) of Intermediate 4 as a light yellow foam.
……………………
SYNTHESIS
Compounds described herein may be prepared using the synthetic methods described herein as a single isomer or a mixture of isomers.
A non-limiting example of a synthetic approach towards the preparation of compounds of any of Formula (A), (B), (C) or (D) is shown in Scheme I.

Halogenation of commercially available 1H-pyrazolo[3,4-d]pyrimidin-4-amine provides an entry into the synthesis of compounds of Formula (A), (B), (C) and/or (D). In one embodiment, 1H-pyrazolo[3,4-d]pyrimidin-4-amine is treated with N-iodosuccinamide to give 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine. Metal catalyzed cross coupling reactions are then carried out on 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine. In one embodiment, palladium mediated cross-coupling of a suitably substituted phenyl boronic acid under basic conditions constructs intermediate 2. Intermediate 2 is coupled with N-Boc-3-hydroxypiperidine (as non-limiting example) via Mitsunobu reaction to give the Boc (tert-butyloxycarbonyl) protected intermediate 3. After deprotection with acid, coupling with, but not limited to, an acid chloride, such as, but not limited to, acryloyl chloride, completes the synthesis to give compound 4.
Example 1 Synthesis of Compounds Preparation of 4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2)
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) is added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture is stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue is taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gives a semisolid residue which is treated with a little ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gives 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which is sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine.
Example 1a Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4)

-
- Synthesis of compound 4; a) polymer-bound triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) HCl/dioxane; then acryloyl chloride, triethylamine (TEA).
Compounds described herein were synthesized by following the steps outlined in Scheme 1. A detailed illustrative example of the reaction conditions shown in Scheme 1 is described for the synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4).
101 mg of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine and 330 mg of polymer-bound triphenylphosphine(TPP) (polymerlab) were mixed together with 5 mL of tetrahydrofuran (THF). tert-Butyl 3-hydroxypiperidine-1-carboxylate (200 mg; 2.0 equivalents) was added to the mixture followed by the addition of diisopropyl diazodicarboxylate (0.099 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered to remove the resins and the reaction mixture was concentrated and purified by flash chromatography (pentane/ethyl acetate=1/1) to give intermediate 3 (55 mg).
Intermediate 3 (48.3 mg) was treated with 1 mL of 4N HCl in dioxane for 1 hour and then concentrated to dryness. The residue was dissolved in dichloromethane and triethylamine (0.042 mL) was added followed by acryl chloride (0.010 mL). The reaction was stopped after 2 hours. The reaction mixture washed with 5% by weight aqueous citric acid and then with brine. The organic layer was dried with MgSO4, and concentrated. Flash chromatography (with CH2Cl2/MeOH=25/1) gave 22 mg of compound 4 as a white solid. MS (M+1): 441.2; 1H-NMR (400 MHz): 8.26, s, 1H, 7.65, m, 2H, 7.42, m, 2H, 7.1-7.2, m, 5H, 6.7-6.9, m, 1H, 6.1, m, 1H, 5.5-5.7, m, 1H, 4.7, m, 1H, 4.54, m, 0.5H, 4.2, m, 1H, 4.1, m, 0.5H, 3.7, m, 0.5H, 3.2, 1,1H, 3.0, m, 0.5H, 2.3, m, 1H, 2.1, m, 1H, 1.9, m, 1H, 1.6, m, 1H.
Example 1b Synthesis of 1-((R)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 13)

The synthesis of compound 13 was accomplished using a procedure analogous to that described in Example 1a. EM (calc.): 440.2; MS (ESI) m/e (M+1H)+: 441.1, (M−1H)−: 439.2.
Example 1c Synthesis of 1-((S)-3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 14)

The synthesis of compound 14 was accomplished using a procedure analogous to that described for Example 1a. EM (calc.): 440.2; MS (ESI) m/e (M+1H)+: 441.5, (M−1H)−: 439.2.
……………….
Synthesis of Compounds Example 1 Preparation of 4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (2a)
4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine (Intermediate 2) is prepared as disclosed in International Patent Publication No. WO 01/019829. Briefly, 4-phenoxybenzoic acid (48 g) is added to thionyl chloride (100 mL) and heated under gentle reflux for 1 hour. Thionyl chloride is removed by distillation, the residual oil dissolved in toluene and volatile material removed at 80° C./20 mbar. The resulting acid chloride is dissolved in toluene (200 mL) and tetrahydrofuran (35 mL). Malononitrile (14.8 g) is added and the solution and stirred at −10° C. while adding diisopropylethylethylamine (57.9 g) in toluene (150 mL), while maintaining the temperature below 0° C. After 1 hour at 0° C., the mixture is stirred at 20° C. overnight. Amine hydrochloride is removed by filtration and the filtrate evaporated in vacuo. The residue is taken up in ethyl acetate and washed with 1.25 M sulphuric acid, then with brine and dried over sodium sulfate. Evaporation of the solvents gives a semisolid residue which is treated with a little ethyl acetate to give 4.1 g of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a white solid (m.p. 160-162° C.). The filtrate on evaporation gives 56.58 (96%) of 1,1-dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene as a grey-brown solid, which is sufficiently pure for further use.
1,1-Dicyano-2-hydroxy-2-(4-phenoxyphenyl)ethene (56.5 g) in acetonitrile (780 mL) and methanol (85 mL) is stirred under nitrogen at 0° C. while adding diisopropylethylamine (52.5 mL) followed by 2M trimethylsilyldiazomethane (150 mL) in THF. The reaction is stirred for 2 days at 20° C., and then 2 g of silica is added (for chromatography). The brown-red solution is evaporated in vacuo, the residue dissolved in ethyl acetate and washed well with water then brine, dried and evaporated. The residue is extracted with diethyl ether (3×250 mL), decanting from insoluble oil. Evaporation of the ether extracts gives 22.5 g of 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene as a pale orange solid. The insoluble oil is purified by flash chromatography to give 15.0 g of a red-orange oil.
1,1-Dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene (22.5 g) and 1,1-dicyano-2-methoxy-2-(4-phenoxyphenyl)ethene oil (15 g) are treated with a solution of hydrazine hydrate (18 mL) in ethanol (25 mL) and heated on the steambath for 1 hour. Ethanol (15 mL) is added followed by water (10 mL). The precipitated solid is collected and washed with ethanol:water (4:1) and then dried in air to give 3-amino-4-cyano-5-(4-phenoxyphenyl)pyrazole as a pale orange solid.
3-Amino-4-cyano-5-(4-phenoxyphenyl)pyrazole (29.5 g) is suspended in formamide (300 mL) and heated under nitrogen at 180° C. for 4 hours. The reaction mixture is cooled to 30° C. and water (300 mL) is added. The solid is collected, washed well with water, then with methanol and dried in air to give of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine.
Example 1a Synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (4)

Synthesis of compound 4; a) polymer-bound triphenylphosphine (TPP), diisopropyl diazodicarboxylate (DIAD), tetrahydrofuran (THF); b) HCl/dioxane; then acryloyl chloride, triethylamine (TEA)
Compounds described herein were synthesized by following the steps outlined in Scheme III. A detailed illustrative example of the reaction conditions shown in Scheme II is described for the synthesis of 1-(3-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (Compound 4).
101 mg of 4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidine and 330 mg of polymer-bound triphenylphosphine (TPP) (polymerlab) were mixed together with 5 mL of tetrahydrofuran (THF). tert-Butyl 3-hydroxypiperidine-1-carboxylate (200 mg; 2.0 equivalents) was added to the mixture followed by the addition of diisopropyl diazodicarboxylate (0.099 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered to remove the resins and the reaction mixture was concentrated and purified by flash chromatography (pentane/ethyl acetate=1/1) to give intermediate 3a (55 mg).
Intermediate 3a (48.3 mg) was treated with 1 mL of 4N HCl in dioxane for 1 hour and then concentrated to dryness. The residue was dissolved in dichloromethane and triethylamine (0.042 mL) was added followed by acryl chloride (0.010 mL). The reaction was stopped after 2 hours. The reaction mixture was washed with 5% by weight aqueous citric acid and then with brine. The organic layer was dried with MgSO4, and concentrated. Flash chromatography (with CH2Cl2/MeOH=25/1) gave 22 mg of compound 4 as a white solid. MS (M+1): 441.2; 1H-NMR (400 MHz): 8.26, s, 1H, 7.65, m, 2H, 7.42, m, 2H, 7.1-7.2, m, 5H, 6.7-6.9, m, 1H, 6.1, m, 1H, 5.5-5.7, m, 1H, 4.7, m, 1H, 4.54, m, 0.5H, 4.2, m, 1H, 4.1, m, 0.5H, 3.7, m, 0.5H, 3.2, m, 1H, 3.0, m, 0.5H, 2.3, m, 1H, 2.1, m, 1H, 1.9, m, 1H, 1.6, m, 1H.
…………………….
SYNTHESIS
To solve the above problems, the present invention adopts a technical solution is: to provide a tyrosine kinase inhibitor PC1-32765 synthesis method, the reaction steps are as follows:

The beneficial effect of the present invention: The invention relates to a tyrosine kinase inhibitor synthesis of PC1-32765, as the B cell to inhibit the tyrosine kinase receptor signaling key, not only can inhibit the formation of blood cells and less side effects and mild reaction conditions, simple operation, easy purification, low cost, environmentally friendly, suitable for large-scale production.
A tyrosine kinase inhibitor PC1-32765 synthesis method comprising the steps of:
1, the compound 10 and the coupling reaction of compound 15 to give compound 6;
2, the compound 6 obtained by reacting compound 16 with compound 11 in the process, we have chosen a more perfect catalyst;
3, compound 11 to give compound 12 by protecting;
4, selective deprotection of Compound 12 Compound 13; 5, Compound 13 for Compound 17 only attack only remaining position to obtain a very pure compound 14;
6, take off the protecting group to obtain PC1-32765

Wherein the compound can 10,15,16,17 agent or industrial grade reagent compound or the use of methods and techniques related to synthesis.
Example 1 Preparation of Compound 6
Under nitrogen and the 0.1moL 1.5 equivalents of compound 10 Compound 15 and 800mL of dioxane was added to 2L reaction flask, and then 1.5 equivalents of sodium acetate was added and the catalyst PdC12 (PPh3) 2 0.2 equivalents, 50_60 ° C for 5 hours , filtered hot and the filter residue was washed three times with ethanol, the combined filtrate was concentrated to give a solid, rinsed with ethanol to give the pure product 16.2 g, yield 60%
Example 2 Preparation of Compound 6
Under nitrogen and the 0.1moL 1.5 equivalents of compound 10 Compound 15 and 2L 800mL DMF was added to the reaction flask, and then 1.5 equivalents of sodium acetate was added and the catalyst PdCl2 (PhCN) 2 0.2 equivalents, 50_60 ° C for 5 hours, hot filtered, the filter residue was washed three times with ethanol, the combined filtrate was concentrated to give a solid, which was rinsed with ethanol to give pure product 21.5 g, yield 71%.
Example 3 Preparation of Compound 11
The compound 0.1moL 1.2 equivalent of compound 6 and 16, and 2L IOOOmL THF was added to the reaction flask, 1.5 equivalents of cesium carbonate was added, refluxed for 24 hours, after the reaction, most of the solvent was concentrated and the remaining water was poured into a large, precipitated solid was filtered, washed with water to afford compound 36.9 g compound 11, yield 76%, used without further purification.
Example 4 Preparation of Compound 12
The compound will be to 0.1moL 11 and 1.2 equivalent of compound IOOOmL THF trifluoroacetyl chloride and the reaction was added to 2L flask, then triethylamine was added 2.5 ,30-40 0C for 24 hours, after the reaction, the solvent was concentrated, diluted with water, extracted with ethyl acetate, washed with water, saturated sodium chloride each time, and concentrated to obtain the product 50.1 g of ethyl acrylate, 86% yield, used directly in the next reaction.
Example 5 Preparation of Compound 13
The compound 0.1moL 12 and 500mL of methanol and 50mL 6N hydrochloric acid was added to IL reaction flask, stirred at room temperature for 3 hours to complete the reaction quickly, and a solid precipitates, filtered and the solid was washed several times with ethyl acetate, obtain 38.5 g of pure compound 13 in 80% yield.
Example 6 Preparation of Compound 14 ‘
The 0.1moL compound 13 and 1.2 equivalents of acrylic acid chloride was added to 2L of methylene chloride IL reaction flask ,20-40 ° C was added dropwise 1.2 equivalents of triethylamine was added dropwise, at room temperature for 3 hours after the reaction with two chloride extraction and concentrated to give the product 47.7 g, yield 89%. Without further purification.
Example 7 PC1-32765 Preparation
Compound 14 with the 0.1moL 160mL 800mL of methanol and a saturated solution of sodium carbonate small, 50_60 ° C for 5 hours,
After completion of the reaction was diluted with water, concentrated and then extracted with methylene chloride, concentrated to obtain crude product was recrystallized from toluene to give the final product 28.6 g, yield 65%. HPLC purity 98.6%, ee%> 98%.
The present invention relates to a tyrosine kinase inhibitor of the synthesis of PC1-32765, as the B cell to inhibit the tyrosine kinase receptor signaling key, not only can inhibit the formation of blood cells and less side effects, and the reaction conditions gentle, simple operation, easy purification, low cost, environmentally friendly, suitable for large-scale production.
…
Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase
ChemMedChem
Volume 2, Issue 1, pages 58–61, January 15, 2007
http://onlinelibrary.wiley.com/doi/10.1002/cmdc.200600221/full
http://www.wiley-vch.de/contents/jc_2452/2007/z600221_s.pdf
SYN OF COMPD 4
To 101 mg of a known intermediate 2 [WO 2001019829] and 330 mg polymer-bound Triphenylphosphine (polymerlab) in 5 ml THF, 200 mg (2.0 eq.) of 3-OH N-Boc piperidine was added followed by 0.099 ml diisopropyl diazodicarboxylate. The reaction mixture stirred at room temperature overnight. After filtered off resins, the reaction mixture was concentrated and purified with flash chromatography (pentane/ethyl acetate = 1/1) to give 55 mg of intermediate 3. This compound (48.3 mg) was treated with 1 ml of 4N HCl in dioxane for 1 hour and concentrated to dryness, which was dissolved in dichloromethane and 0.042 ml of triethylamine, followed by 0.010 ml of acryl chloride. The reaction was stopped after 2 hours. The reaction mixture was washed with 5wt% citric acid (aq.) and brine, dried with MgSO4, and concentrated. Flash chromatography with (CH2Cl2/MeOH = 25/1) gave 22 mg of compound 4 as white solids. MS (M+1): 441.2; 1H-NMR (400MHz): 8.26, s, 1H; 7.65, m, 2H; 7.42, m, 2H; 7.1-7.2, m, 5H; 6.7-6.9, m, 1H; 6.1, m, 1H; 5.5-5.7, m, 1H; 4.7, m, 1H; 4.54, m, 0.5H; 4.2, m, 1H; 4.1, m, 0.5H; 3.7, m, 0.5H; 3.2, m, 1H; 3.0, m, 0.5H; 2.3, m, 1H; 2.1, m, 1H; 1.9, m, 1H; 1.6, m, 1H
……………..
References
- Statement on a Nonproprietary Name Adopted by the USAN Council
- FDA Press Release
- Azvolinsky, PhD, Anna. “FDA Approves Ibrutinib for Chronic Lymphocytic Leukemia”. Cancer Network. Retrieved 14 February 2014.
- Pan, Z; Scheerens, H; Li, SJ; Schultz, BE; Sprengeler, PA; Burrill, LC; Mendonca, RV; Sweeney, MD; Scott, KC; Grothaus, Paul G.; Jeffery, Douglas A.; Spoerke, Jill M.; Honigberg, Lee A.; Young, Peter R.; Dalrymple, Stacie A.; Palmer, James T. (2007). “Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase”. ChemMedChem 2 (1): 58–61.doi:10.1002/cmdc.200600221. PMID 17154430.
- Celera Genomics Announces Sale of Therapeutic Programs to Pharmacyclics
- United States patent 7514444
- Janssen Biotech, Inc. Announces Collaborative Development and Worldwide License Agreement for Investigational Anti-Cancer Drug, PCI-32765
- Clinical trials involve PCI-32765
- Clinical trials involve ibrutinib
- “Imbruvica (Ibrutinib)”. Medscape Reference. WebMD. Retrieved 13 January 2014.
- “IMBRUVICA (ibrutinib) capsule [Pharmacyclics, Inc]”. DailyMed. Pharmacyclics, Inc. November 2013. Retrieved 13 January 2013.
- Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, Flynn J, Jones J, Blum KA, Buggy J.J., Hamdy A, Johnson AJ, Byrd JC, SE (2011). “Bruton’s tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765”. Blood 117 (23): 6287–6296. doi:10.1182/blood-2011-01-328484. PMC 3122947. PMID 21422473.
- The Bruton’s tyrosine kinase (BTK) inhibitor PCI-32765 (P) in treatment-naive (TN) chronic lymphocytic leukemia (CLL) patients (pts): Interim results of a phase Ib/II study” J Clin Oncol 30, 2012 (suppl; abstr 6507)
- Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, Keating MJ, O’Brien S, Chiorazzi N, Burger JA (2012). The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo 119. Blood. pp. 1182–1189.
- de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, Pals ST, Spaargaren M (2012). “The clinically active BTK inhibitor PCI-32765 targets B-cell receptor (BCR)- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia”. Blood 2012: 2590–2594.
- Honigberg, LA; Smith, AM; Sirisawad, M; Verner, E; Loury, D; Chang, B; Li, S; Pan, Z; Thamm, DH; Miller, RA; Buggy, JJ (2010). “The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy”.Proceedings of the National Academy of Sciences of the United States of America 107 (29): 13075–80. doi:10.1073/pnas.1004594107. PMC 2919935. PMID 20615965.
- Chang, BY; Huang, MM; Francesco, M; Chen, J; Sokolove, J; Magadala, P; Robinson, WH; Buggy, JJ (2011). “The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells”. Arthritis Research & Therapy 13 (4): R115.doi:10.1186/ar3400. PMC 3239353. PMID 21752263.
- http://cancer.osu.edu/mediaroom/releases/Pages/Ohio-State-Cancer-Research-Played-a-Significant-Role-in-FDA-Approval-of-Important-New-CLL-Drug.aspx#sthash.3o9uyt78.dpuf
- BTK inhibitor PCI-32765, National Cancer Institute Drug Dictionary
- Official Imbruvica patient website
MORE
1) E · Werner, L · Honeywell Berg, Z · Pan; Bruton tyrosine kinase inhibitor; drugs circulating Company; filing date: 2006.12.28, open) No. (Notice: CN101610676A CN101610676B, CN101805341A, CN101805341B, CN102746305A, CN102887900A
2) Pan, Z ; Scheerens, H; Li, SJ; Schultz, BE; Sprengeler, PA; Burrill, LC; Mendonca, RV; Sweeney, MD; Scott, KC; Grothaus, Paul G.; Jeffery, Douglas A.; Spoerke, Jill M. ..; Honigberg, Lee A.; Young, Peter R.; Dalrymple, Stacie A.; Palmer, James T. (2007) “Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase” ChemMedChem 2 (1): 58-61 ( article link )
3) Celera Genomics Announces Sale of Therapeutic Programs to Pharmacyclics , April 10, 2006
4) Honigberg; Lee, Verner; Erik, Pan; Zhengying; Inhibitors of Bruton’s tyrosine kinase, U.S. Patent 7,514,444 ; US 20080108636 A1; CA2663116A1, CN101610676B, CN101805341A, CN101805341B, CN102746305A, CN102887900A, EP2081435A2, EP2081435A4, EP2201840A1, EP2201840B1, EP2443929A1, EP2526771A1, EP2526933A2, EP2526933A3, EP2526934A2, EP2526934A3, EP2529621A1, EP2529622A1, EP2530083A1, EP2532234A1, EP2532235A1, US7514444, US7732454, US7825118, US7960396, US8008309, US8088781, US8158786, US8232280, US8236812, US8399470, US8476284, US8497277, US8501751, US8552010, US20080076921, US20080108636, US20080139582, US20090181987, US20100004270, US20100022561, US20100041677, US20100324050, US20100331350, US20110008257, US20110039868, US20110184001, US20110257203, US20110281322, US20120088912, US20120095026, US20120108612, US20120115889, US20120122894, US20120129821, US20120129873, US20120135944, US20120214826, US20120252821, US20120252822, US20120277254, US20120283276, US20120283277, US20130005745, WO2008039218A2, WO2008039218A3
5) Buggy, Joseph J. Chang, Betty Y.; Methods and Compositions . for inhibition of Bone resorption, PCT Int Appl, WO2013003629, 03 Jan 2013.
6) Wei Chen, David J. Loury, Tarak D. Mody; Preparation of pyrazolo-pyrimidine Compounds as Inhibitors of Bruton’s tyrosine kinase; U.S. Patent Number 7,718,662 , 18 May 2010; Also published as CA2776543A1, CN102656173A, EP2393816A2, EP2393816A4, EP2650294A1, US7741330, US20110086866, WO2011046964A2, WO2011046964A3
7) Wei Chen, David J. Loury, Tarak D. Mody; Inhibitors of Bruton’s tyrosine kinase; WO2013010136 A3
8) John C. Byrd, et al;. Targeting BTK with Relapsed Ibrutinib in Chronic Lymphocytic Leukemia; N Engl J Med 2013; 369:32-42
9) Michael L. Wang, MD, et al, Targeting with BTK. Ibrutinib in Refractory or Relapsed Mantle-Cell Lymphoma; N Engl J Med 2013; 369:507-516
|
8-8-2012
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
7-32-2012
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
4-18-2012
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
1-4-2012
|
INHIBITORS OF BRUTONS TYROSINE KINASE
|
|
|
11-18-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
9-16-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE FOR THE TREATMENT OF SOLID TUMORS
|
|
|
8-31-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
6-15-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
4-15-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
2-18-2011
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
12-24-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
11-3-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
10-8-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
6-9-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
2-19-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
1-29-2010
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
4-8-2009
|
INHIBITORS OF BRUTON’S TYROSINE KINASE
|
|
|
9-5-2008
|
BRUTON’S TYROSINE KINASE ACTIVITY PROBE AND METHOD OF USING
|
| US7718662 | 16 Oct 2009 | 18 May 2010 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of bruton’s tyrosine kinase |
| US7741330 | 16 Oct 2009 | 22 Jun 2010 | Pharmacyclics, Inc. | Pyrazolo-pyrimidine inhibitors of Bruton’s tyrosine kinase |
| US7982036 | 17 Oct 2008 | 19 Jul 2011 | Avila Therapeutics, Inc. | 4,6-disubstitued pyrimidines useful as kinase inhibitors |
| US7989465 | 20 Apr 2009 | 2 Aug 2011 | Avila Therapeutics, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| US8008309 * | 7 Jul 2009 | 30 Aug 2011 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8088781 | 20 Jan 2009 | 3 Jan 2012 | Pharmacyclics, Inc. | Inhibitors of brutons tyrosine kinase |
| US8158786 | 15 Jun 2011 | 17 Apr 2012 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8232280 * | 21 Jan 2011 | 31 Jul 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’S tyrosine kinase |
| US8236812 | 21 Sep 2010 | 7 Aug 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8329901 | 1 Feb 2011 | 11 Dec 2012 | Celgene Avilomics Research, Inc. | 4,6-disubstitued pyrimidines useful as kinase inhibitors |
| US8338439 | 29 Dec 2009 | 25 Dec 2012 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8377946 | 21 Jun 2012 | 19 Feb 2013 | Pharmacyclics, Inc. | Pyrazolo[3,4-d]pyrimidine and pyrrolo[2,3-d]pyrimidine compounds as kinase inhibitors |
| US8399470 | 30 Jan 2012 | 19 Mar 2013 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8445498 | 1 Feb 2011 | 21 May 2013 | Celgene Avilomics Research, Inc. | 4,6-disubstituted pyrimidines useful as kinase inhibitors |
| US8450335 | 26 Jun 2009 | 28 May 2013 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8476284 | 16 Dec 2011 | 2 Jul 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8481733 * | 19 May 2009 | 9 Jul 2013 | OSI Pharmaceuticals, LLC | Substituted imidazopyr- and imidazotri-azines |
| US8497277 | 6 Dec 2011 | 30 Jul 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8501724 | 30 Apr 2012 | 6 Aug 2013 | Pharmacyclics, Inc. | Purinone compounds as kinase inhibitors |
| US8501751 | 7 Jul 2009 | 6 Aug 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’s tyrosine kinase |
| US8552010 | 18 Jun 2012 | 8 Oct 2013 | Pharmacyclics, Inc. | Inhibitors of Bruton’S tyrosine kinase |
| US8563563 | 30 Jan 2012 | 22 Oct 2013 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US8563568 | 8 Aug 2011 | 22 Oct 2013 | Celgene Avilomics Research, Inc. | Besylate salt of a BTK inhibitor |
| US8609679 | 7 Nov 2012 | 17 Dec 2013 | Celgene Avilomics Research, Inc. | 2,4-diaminopyrimidines useful as kinase inhibitors |
| US20090286768 * | 19 May 2009 | 19 Nov 2009 | Osi Pharmaceuticals, Inc. | Substituted imidazopyr- and imidazotri-azines |
| US20110184001 * | 21 Jan 2011 | 28 Jul 2011 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120088912 * | 29 Sep 2011 | 12 Apr 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120252822 * | 23 May 2012 | 4 Oct 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| US20120277254 * | 5 Jul 2012 | 1 Nov 2012 | Pharmacyclics, Inc. | Inhibitors of bruton’s tyrosine kinase |
| WO2010123870A1 * | 20 Apr 2010 | 28 Oct 2010 | Avila Therapeutics, Inc. | Heteroaryl compounds and uses thereof |
| WO2011031979A1 * | 10 Sep 2010 | 17 Mar 2011 | Cylene Pharmaceuticals Inc. | Pharmaceutically useful heterocycle-substituted lactams |
| WO2013155347A1 | 11 Apr 2013 | 17 Oct 2013 | Izumi Raquel | Bruton’s tyrosine kinase inhibitors for hematopoietic mobilization |
| WO2014004707A1 | 26 Jun 2013 | 3 Jan 2014 | Principia Biopharma Inc. | Formulations comprising ibrutinib |
FDA Approves Monovisc Injection for Knee Pain

Sodium Hyaluronate
9067-32-7 (sodium salt)
| MF: | C14H22NNaO11 |
| MW: | 403.31 |

26 feb 2014
http://www.dddmag.com/news/2014/02/fda-approves-monovisc-injection-knee-pain

Sodium hyaluronate is the sodium salt of hyaluronic acid, a glycosaminoglycan found in various connective, epithelial, and neural tissues. Sodium hyaluronate, a long-chain polymer containing repeating disaccharide units of Na-glucuronate-N-acetylglucosamine, occurs naturally on the corneal endothelium, bound to specific receptors for which it has a high affinity. The polyanionic form, commonly referred to as hyaluronan, is a visco-elasticpolymer normally found in the aqueous and vitreous humour. As a pharmaceutical, the uses of sodium hyaluronate include:

sodium hyaluronate
Sodium hyaluronate for intra-articular injection (brand names: Euflexxa, Hyalgan, Supartz, Gel-One) is used to treat knee pain in patients withosteoarthritis who have not received relief from other treatments. It is very similar to the lubricating fluid that occurs naturally in the articular capsule of the knee joint. Once injected into the joint capsule, it acts as both a shock absorber and a lubricant for the joint.[1]

Sodium hyaluronate for intraocular viscoelastic injection (brand names: Healon, Provisc, Viscoat) is used as a surgical aid in variety of surgical procedures performed on the eyeball including cataract extraction (intra- and extracapsular), intraocular lens implantation, corneal transplant,glaucoma filtration, and retina attachment surgery. In surgical procedures in the anterior segment of eyeball, instillation of sodium hyaluronate serves to maintain a deep anterior chamber during surgery, allowing for efficient manipulation with less trauma to the corneal endothelium and other surrounding tissues. Its viscoelasticity also helps to push back the vitreous face and prevent formation of a postoperative flat chamber. In posterior segment surgery, sodium hyaluronate serves as a surgical aid to gently separate, maneuver, and hold tissues. It creates a clear field of vision, facilitating intra-operative and post-operative inspection of the retina and photocoagulation.[2]
Sodium hyaluronate is used as a viscosupplement, administered through a series of injections into the knee, increasing the viscosity of the synovial fluid, which helps lubricate, cushion and reduce pain in the joint.[3] It is generally used as a last resort before surgery[4] and provides symptomatic relief, by recovering the viscoelasticity of the articular fluid, and by stimulating new production from synovial fluid.[5] Use of sodium hyaluronate may reduce the need for joint replacement.[6] Injections appear to increase in effectiveness over the course of four weeks, reaching a peak at eight weeks and retaining some effectiveness at six months, with greater benefit for osteoarthritis than oral analgesics.[7] It may also be effective when used with other joints.[8]
Sodium hyaluronate may also be used in plastic surgery to reduce wrinkles on the face or as a filler in other parts of the body.[9] It may be used in ophthalmology to assist in the extraction ofcataracts, the implantation of intraocular lenses, corneal transplants, glaucoma filtration, retinal attachment and in the treatment of dry eyes.[10]
Sodium hyaluronate is also used to coat the bladder lining in treating interstitial cystitis.
 hyaluronan
hyaluronan
cas 9004-61-9
Sodium hyaluronate functions as a tissue lubricant and is thought to play an important role in modulating the interactions between adjacent tissues. Sodium hyaluronate is a polysaccharide which is distributed widely in the extracellular matrix of connective tissue in man. It forms a viscoelastic solution in water which makes it suitable for aqueous and vitreous humor in ophthalmic surgery. Mechanical protection for tissues (iris, retina) and cell layers (corneal, endothelium, and epithelium) are provided by the high viscosity of the solution. Elasticity of the solution assists in absorbing mechanical stress and providing a protective buffer for tissues. This viscoelasticity enables maintenance of a deep chamber during surgical manipulation since the solution does not flow out of the open anterior chamber. In facilitating wound healing, it is thought that it acts as a protective transport vehicle, taking peptide growth factors and other structural proteins to a site of action. It is then enzymatically degraded and active proteins are released to promote tissue repair.[11] Sodium hyaluronate is being used intra-articularly to treat osteoarthritis.
Sodium hyaluronate is an ophthalmic agent with viscoelastic properties that is used in joints to supplement synovial fluid.
Sodium hyaluronate is absorbed and diffuses slowly out of the injection site. It is eliminated via the canal of Schlemm.
Sodium hyaluronate hyaluronan started to be in use to treat osteoarthritis of the knee in year 1986 with the product Hyalart/Hyalgan by Fidia of Italy, in intra-articular injections.
Sodium Hyaluronate
Brand names of Sodium hyaluronate in Market include (alphabetically):
- AMO Vitrax (ocular)
- AMVISIC Plus (ocular)
- CYSTISTAR, Healon (ocular)
- EYEFILL (ocular)
- HYLO-COMOD (Eye Drop)
- OLIXIA Pure (Eye Drop)
- EUFLEXXA, Bio Technology General (Israel)-Meditrina SA (Rx articular), Molecular weight: 2,400,000-3,600,000 Daltons
- GONILERT/Verisfield (UK) (Rx/articular). Molecular weight:1,800,000-2,000,000 Daltons
- HYALGAN/HYALART– Fidia (Italy)(Medical Device/Rx articular)
- MONOVISC– Anika (USA)(MedicalDevice/articular)
- OSTENIL– TRB Chemedica (Switzerland)(articular injection) [1]
- RECOSYN– Merckle Recordati (Germany) Recosyn info leaflet
- SYNOCROM– Croma Pharma (Austria) (articular injection) . Molecular weight:1,600,000 Daltons
- VISCURE– Cube (UK)(Rx/articular), Molecular weight:1,800,000-2,000,000 Daltons
- VISMED– TRB Chemedica (Switzerland)(eye drop)[2]
- YARDEL– Libytec (Impfstoffwerk Dessau-Tornau/Germany,(Rx/articular), Molecular weight:1,800,000-2,000,000 Daltons
Hyaluronic acid (HA) is a glycosaminoglycan which is present in the hyaline cartilage, synovial joint fluid and skin tissues. More particularly, HA is a linear glycosaminoglycan formed by a mixture of chains of different length constituted by the repetition of a regular disaccharide formed by a glucuronic acid unit and a N- acetyl-glucosamine unit linked beta 1-4. Disaccharides are linked beta 1-3 with an average molecular weight up to 6 Md (6×106 Da). Therefore, each chain in said mixture of chains shows the same repetitive sequence of formula (A)

the corresponding cation generally being hydrogen (hyaluronic acid) or sodium (sodium hyaluronate).
In the tissues, the function of hyaluronic acid is mainly to maintain the structural density allowing in the same time the biochemical actions of the natural products in the specific body districts. In fluids like synovia the action of HA is to keep the right viscosity by a lubricant action. To exert these actions, HA needs to be fully biocompatible including a right metabolic balance. Natural HA is continuously degraded and synthesized by the body enzymes. This homeostasis is deviated when pathological situations occur, therefore increases in the HA catabolism can results in wide range of effects from a severe pathology to simple tissue modifications. The application of HA, as sodium hyaluronate, as filler in cosmetic or in viscoelastic replacement in synovitis, requires that the employed HA polymer has enhanced viscoelastic properties. This rheology has to be balanced with an efficient capability to make the production of the injectable product.
The industrial hyaluronic acid is obtained by extraction from animal tissues or by microorganism fermentation and is commonly available as sodium hyaluronate. Concerning molecular weight, it is generally recognized that low molecular weight HA is a mixture of chains having a mean molecular weight below 250 Kd (2.5×105Da). HA is used, generally as sodium hyaluronate, in many applications in cosmetics, ophthalmology, rheumatology and tissues engineering. In particular HA with a mean molecular weight above 1 Md is used as viscosupplement in joint arthrosis or in wrinkle management. The high molecular weight is required to supplement the synovial fluid or to fill skin connective dead spaces thanks to the viscosity of the resulting solution.
Many medicaments based on the above technology are currently available on the market. They have a high biocompatibility but they are subjected to a rather rapid degradation by the body enzymes, in particular by hyaluronidase, with the consequence of a short half-life.
 sodium hyaluronate
sodium hyaluronate
References
- “Hyaluronate sodium: Indications, Side Effects, Warnings” (Web). Drugs.com. Drugs.com. 5 February 2014. Retrieved 25 February 2014.
- “Healon (Sodium Hyaluronate)” [package insert]. (2002). Kalamazo, Michigan: Pharmacia Corporation. (Web). RxList. (Updated 8 December 2004). RxList, Inc. Retrieved 25 February 2014.
- Puhl, W.; Scharf, P. (1997). “Intra-articular hyaluronan treatment for osteoarthritis”. Annals of the rheumatic diseases 56 (7): 441. doi:10.1136/ard.56.7.441. PMC 1752402.PMID 9486013. edit
- Karlsson, J.; Sjögren, L. S.; Lohmander, L. S. (2002). “Comparison of two hyaluronan drugs and placebo in patients with knee osteoarthritis. A controlled, randomized, double-blind, parallel-design multicentre study”. Rheumatology (Oxford, England) 41 (11): 1240–1248.PMID 12421996. edit
- Jubb, R. W.; Piva, S.; Beinat, L.; Dacre, J.; Gishen, P. (2003). “A one-year, randomised, placebo (saline) controlled clinical trial of 500-730 kDa sodium hyaluronate (Hyalgan) on the radiological change in osteoarthritis of the knee”. International journal of clinical practice 57 (6): 467–474. PMID 12918884. edit
- Kotz, R.; Kolarz, G. (1999). “Intra-articular hyaluronic acid: Duration of effect and results of repeated treatment cycles”. American journal of orthopedics (Belle Mead, N.J.) 28 (11 Suppl): 5–7. PMID 10587245. edit
- Bannuru, R. R.; Natov, N. S.; Dasi, U. R.; Schmid, C. H.; McAlindon, T. E. (2011). “Therapeutic trajectory following intra-articular hyaluronic acid injection in knee osteoarthritis – meta-analysis”. Osteoarthritis and Cartilage 19 (6): 611–619. doi:10.1016/j.joca.2010.09.014.PMID 21443958. edit
- Salk, R. S.; Chang, T. J.; d’Costa, W. F.; Soomekh, D. J.; Grogan, K. A. (2006). “Sodium Hyaluronate in the Treatment of Osteoarthritis of the Ankle: A Controlled, Randomized, Double-Blind Pilot Study”. The Journal of Bone and Joint Surgery 88 (2): 295–302.doi:10.2106/JBJS.E.00193. PMID 16452740. edit
- Beasley, K.; Weiss, M.; Weiss, R. (2009). “Hyaluronic Acid Fillers: A Comprehensive Review”.Facial Plastic Surgery 25 (2): 086–094. doi:10.1055/s-0029-1220647. PMID 19415575. edit
- Shimmura, S.; Ono, M.; Shinozaki, K.; Toda, I.; Takamura, E.; Mashima, Y.; Tsubota, K. (1995).“Sodium hyaluronate eyedrops in the treatment of dry eyes”. The British journal of ophthalmology 79 (11): 1007–1011. PMC 505317. PMID 8534643. edit
- Boucher, W. S.; Letourneau, R.; Huang, M.; Kempuraj, D.; Green, M.; Sant, G. R.; Theoharides, T. C. (2002). “Intravesical sodium hyaluronate inhibits the rat urinary mast cell mediator increase triggered by acute immobilization stress”. The Journal of Urology 167 (1): 380–384.doi:10.1016/S0022-5347(05)65472-9. PMID 11743360. edit
Scientists discover new drug targets for aggressive breast cancer

The image shows the aggressive growth of TNBC cells. Credit: Genome Institute of Singapore, A*STAR
Scientists at A*STAR’s Genome Institute of Singapore (GIS) led in a study that has identified genes that are potential targets for therapeutic drugs against aggressive breast cancer. These findings were reported in the July 2013 issue of PNAS.
Out of the 1.5 million women diagnosed with breast cancer in the world annually, nearly one in seven of these is classified as triple negative. Patients with triple-negative breast cancer (TNBC) have tumours that are missing three important proteins that are found in other types of breast cancer. The absence of these three proteins make TNBC patients succumb to a higher rate of relapse following treatment and have lower overall survival rates. There is currently no effective therapy for TNBC.
Using integrated genomic approaches, GIS scientists led by Dr. Qiang Yu, in collaboration with local…
View original post 374 more words
What Does 100% of Your Daily Value of Cholesterol Look Like?
Healthline just published an interesting infograph that gives a visualization of what your daily value of cholesterol looks like. In the graphic, you can see what 300 mg of cholesterol looks like for 20 high cholesterol foods: http://www.healthline.com/health/high-cholesterol/daily-value
This is a very informative resource as it helps us visualize what their cholesterol intake look like
What Does 100% of Your Daily Value of Cholesterol Look Like?
It’s no secret that eating fatty foods raises your bad cholesterol level, also known as LDL. An elevated LDL clogs up your arteries and makes it difficult for your heart to do its job. Potentially, it could lead to heart disease.
The USDA recommends consuming no more than 300 mg of cholesterol a day. While a deep-fried Twinkie at the county fair is an obvious no-no, other high cholesterol culprits may be sneaking into your diet. Check out what that number looks like in terms of everyday food items.
Warning: you may need to revise your grocery list—and your eating habits!

Fried Chicken:
4 pieces=300mg cholesterol

Croissants:
6 2/3 rolls=300mg cholesterol

Cheddar Cheese:
12 3/4 slices=300mg cholesterol

Prosciutto:
28 slices=300mg cholesterol

Corned Beef:
14 thin slices=300mg cholesterol

Butter:
1 1/5 sticks=300mg cholesterol
read at
http://www.healthline.com/health/high-cholesterol/daily-value
FDA Approves BMS Drug for Rare Fat Disorder
1. Leptin (human), N-methionyl-
2. N-methionylleptin (human)
STRUCTURAL FORMULA
MVPIQKVQDD TKTLIKTIVT RINDISHTQS VSSKQKVTGL DFIPGLHPIL 50
TLSKMDQTLA VYQQILTSMP SRNVIQISND LENLRDLLHV LAFSKSCHLP 100
WASGLETLDS LGGVLEASGY STEVVALSRL QGSLQDMLWQ LDLSPGC 147
Disulfide bridge location
97-147
http://www.ama-assn.org/resources/doc/usan/metreleptin.pdf
MOLECULAR FORMULA C714H1167N191O221S6
MOLECULAR WEIGHT 16.16 kDa
MANUFACTURER Amylin Pharmaceuticals, Inc.
CODE DESIGNATION r-metHuLeptin
An analog of human leptin, metreleptin, has been approved in Japan and is currently under review by the FDA in the US for the treatment of diabetes and/or hypertriglyceridemia, in patients with rare forms of lipodystrophy, syndromes characterized by abnormalities in adipose tissue distribution, and severe metabolic abnormalities. Bristol-Myers Squibb has submitted a New Drug Approval (NDA) for metreleptin to the US Food and Drug Administration (FDA) Office of Orphan Products Development. In a three-year study of metreleptin in patients with lipodystrophy organized by the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health, metreleptin treatment was associated with a significant decrease in blood glucose (A1c decreased from 9.4% at baseline to 7.0% at study end) and triglyceride concentration (from 500 mg/dl at baseline to 200 mg/dl at study end). The Juvenile Diabetes Research Foundation has also partnered with Amylin Pharmaceuticals and researchers at the University of Texas Southwestern Medical Center to study whether metreleptin can be used to improve the treatment of type 1 diabetes.
N-Methionylleptin (human)
Recombinant human OB protein, purified to homogenicity as a 16-kDa monomer
Treatment of obesity and related disorders (metabolic homeostasis regulator)
- Brand name: Myalept
- Generic name: metreleptin
- Company: Amylin Pharmaceuticals, Inc.
- Treatment for: Lipodystrophy
| Feb 25, 2014 | FDA Approves Myalept to Treat Generalized Lipodystrophy |
| Dec 12, 2013 | FDA Advisory Committee Votes on Investigational Medicine Metreleptin |
| Apr 3, 2012 | Amylin Completes Biologics License Application for Metreleptin to Treat Diabetes and/or Hypertriglyceridemia in Patients With Rare Forms of Lipodystrophy |
| Dec 20, 2010 | Amylin Submits Clinical and Nonclinical Sections of Rolling Biologics License Application for Metreleptin to Treat Rare Forms of Lipodystrophy |
| Leptin | |
|---|---|

Structure of the obese protein leptin-E100.
|
“Myalept is the first approved therapy indicated for treating the complications associated with congenital or acquired generalized lipodystrophy and provides a needed treatment option for patients with this orphan disease,” said Mary Parks, M.D., deputy director of the Office of Drug Evaluation II in the FDA’s Center for Drug Evaluation and Research.
The safety and effectiveness of Myalept, an analog of leptin made through recombinant DNA technology, were evaluated in an open-label, single-arm study that included 48 patients with congenital or acquired generalized lipodystrophy who also had diabetes mellitus, hypertriglyceridemia, and/or elevated levels of fasting insulin. The trial showed reductions in HbA1c (a measure of blood sugar control), fasting glucose, and triglycerides.
Anti-drug antibodies with neutralizing activity to leptin and/or Myalept may develop, which could result in severe infections or loss of treatment effectiveness. T-cell lymphoma has been reported in patients with acquired generalized lipodystrophy, both treated and not treated with Myalept, so healthcare professionals should carefully consider the benefits and risks of treatment with Myalept in patients with significant hematologic abnormalities and/or acquired generalized lipodystrophy. Myalept is contraindicated in patients with general obesity. Myalept is not approved for use in patients with HIV-related lipodystrophy or in patients with metabolic disease, including diabetes mellitus and hypertriglyceridemia, without concurrent evidence of generalized lipodystrophy.
Because of the risks associated with the development of neutralizing antibodies and lymphoma, Myalept is available only through the Myalept Risk Evaluation and Mitigation Strategy (REMS) Program. Under this REMS program, prescribers must be certified with the program by enrolling in and completing training. Pharmacies must be certified with the program and only dispense Myalept after receipt of the Myalept REMS Prescription Authorization Form for each new prescription.
Myalept is also approved with a Medication Guide and instructions for use that provides patients with important information about the medication. The guide will be distributed each time a patient fills a prescription.
The FDA is requiring seven studies (post-marketing requirements) for Myalept, including a long-term prospective observational study (product exposure registry) of patients treated with Myalept, a study to assess for the immunogenicity (antibody formation) of Myalept, and an assessment and analysis of spontaneous reports of potential serious risks related to the use of Myalept. Eight additional studies are being requested as post-marketing commitments.
In clinical trials, the most common side effects observed in patients treated with Myalept were low blood sugar (hypoglycemia), headache, decreased weight, and abdominal pain.
Myalept is marketed by San Diego-based Amylin Pharmaceuticals, L.L.C.
For more information:
Metreleptin is an analogue of the human hormone leptin being developed by Amylin Pharmaceuticals (a subsidiary of Bristol-Myers Squibb) for the subcutaneous treatment of metabolic disorders including lipodystrophy. The compound is expected to improve insulin sensitivity, hypertriglyceridaemia and hyperglycaemia in patients with lipodystrophy who are unresponsive to conventional treatment.
Metreleptin has been approved in Japan as a leptin therapy for the treatment of lipodystrophy. Amylin has also completed a submission for regulatory approval to the US FDA for metreleptin in the treatment of diabetes mellitus and/or hypertriglyceridaemia in patients with rare forms of lipodystrophy.
Clinical development of the drug is also underway in the USA for the treatment of type 1 diabetes. Amgen was previously assessing the use of metreleptin as a treatment for amenorrhoea; however, it appears that development in this indication has been discontinued. This article summarizes the milestones in the development of metreleptin leading to this first approval for lipodystrophy.
Metreleptin is a leptin replacement therapy first launched in Japan in 2013 for the treatment of congenital lipodystrophy. Amylin filed for approval in the U.S. in 2010 for the treatment of diabetes and/or hypertriglyceridemia in patients with rare forms of lipodystrophy. In 2013, the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) recommended the approval for the treatment of pediatric and adult patients with generalized lipodystrophy , but not for partial lipodystrophy.
Phase II clinical studies are also under way at Beth Israel Deaconess Medical Center for the treatment of lipodystrophy syndrome associated with AIDS. The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) is conducting phase II clinical trials for the treatment of nonalcoholic steatohepatitis. Phase II are ongoing at the National Institute for Diabetes and Digestive and Kidney Diseases for the treatment of non-alcoholic fatty liver disease (NAFLD) associated with lipodystropy. Early clinical studies had also been ongoing for the treatment of leptin deficiencies.
The University Texas Southwestern Medical Center at Dallas is evaluating metreleptin for the treatment of type 1 diabetes. Beth Israel Deaconess Medical Center is conducting phase II clinical trials for the treatment of amenorrhea. Amgen had been conducting clinical trials for this indication and for the treatment of type 1 diabetes and depression; however no recent development has been reported for this research.
In 2011, Amylin and Takeda put on hold their clinical trials with metreleptin in combination with pramlintide for the treatment of obesity in order to investigate an antibody-related laboratory finding. Amylin is currently evaluating the compound as monotherapy for the treatment of obesity. The companies had been conducting phase II clinical trials of metreleptin not in combination with pramlintide for the treatment of obesity; however, no recent development has been reported for this research.
Originally developed at the Rockefeller University, an exclusive license to metreleptin was granted to Amgen in 1995. In 2009, the drug candidate was licensed to Takeda by Amylin worldwide for the treatment of obesity. In 2010, orphan drug designation was assigned in the U.S. for the treatment of metabolic disorders secondary to lipodystrophy and for the treatment of leptin deficiency secondary to generalized lipodystrophy and partial familial lipodystrophy.
In 2012, orphan drug designation was assigned in Japan for the treatment of diabetes or hyperlipidemia due to lipoatrophy. In 2012, orphan drug designation was assigned in the E.U. for the treatment of Barraquer-Simons syndrome, Berardinelli-Seip syndrome, familial partial lipodystrophy and Lawrence syndrome. In 2014, AstraZeneca acquired the global rigths for development, manufacture and commercialization of the product.
……………
Other exemplary leptins for use in the methods and compositions described herein include, but are not limited to, the amino acid sequence for mature, recombinant methionyl human leptin (herein called rmetHu-Leptin 1-146 or Metreleptin) having the amino acid sequence:
MVPIQKVQDDTKTLIKTΓVTRINDISHTQSVSSKQKVTGLDFIPGLHPILTLSKMDQTLA VYQQILTSMPSRNVIQISNDLENLRDLLHVLAFSKSCHLPWASGLETLDSLGGVLEASG YSTEWALSRLQGSLQDMLWQLDLSPGC (SEQ ID NO:274).
Vertex Pharmaceuticals: Another Step Forward For Kalydeco
.
On February 21st, Vertex Pharmaceuticals announces that the FDA approves a supplemental New Drug Application (sNDA) for orphan drug Kalydeco (Ivacaftor) for people with Cystic Fibrosis (CF), ages 6 and older, who have one of the 8 additional mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene :
• G178R
• S549N
• S549R
• G551S
• G1244E
• S1251N
• S1255P
• G1349D.
Kalydeco receives approval from the FDA in January 2012 for CF patients, ages 6 and older who have at least one copy of the G551D mutation. Thus, Kalydeco is currently approved for 9 mutations. The new approval affects approximately 150 in the United States.
The sNDA approval is based on previously announced data from a Phase III, 2-part, randomized, double-blind, placebo-controlled, cross-over study of 39 CF patients who have one of the above listed 8 mutations + the G970R mutation. Based on this…
View original post 262 more words
Immune cells regulate blood stem cells

Blood stem cell cultures: Blood stem cells from colonies (cell clusters) in vitro consisting of different blood cells. Nine blood stem cell colonies are illustrated in the image, which have developed into differentiated cell types, particularly into white blood cells (leukocytes).Credit: Department of Clinical Research of the University of Bern, Tumor-Immunology research group
Researchers in Bern, Switzerland have discovered that, during a viral infection, immune cells control the blood stem cells in the bone marrow and therefore also the body’s own defences. The findings could allow for new forms of therapy, such as for bone marrow diseases like leukaemia.
During a viral infection, the body needs various defence mechanisms – amongst other things, a large number of white blood cells (leukocytes) must be produced in the bone marrow within a short period of time. In the bone marrow, stem cells are responsible for this task: the blood stem cells. In…
View original post 390 more words
KW-4490 A PDE4 inhibitor from Kyowa Hakko Kirin
 KW 4490
KW 4490- cis-4-Cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxin-5-yl)cyclohexanecarboxylic Acid
- Cyclohexanecarboxylic acid, 4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxin-5-yl)-, cis–
- cis-4-Cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxin-5-yl)cyclohexane-1-carboxylic acid;
- cis-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanecarboxylic acid
KF 66490; KW 4490;
MF C17 H19 N O5
A phosphodiesterase type 4 inhibitor, commonly referred to as a PDE4 inhibitor, is a drug used to block the degradative action ofphosphodiesterase 4 (PDE4) on cyclic adenosine monophosphate (cAMP). It is a member of the larger family of PDE inhibitors. The PDE4 family of enzymes are the most prevalent PDE in immune cells. They are predominantly responsible for hydrolyzing cAMP within both immune cells and cells in the central nervous system
PDE4 hydrolyzes cyclic adenosine monophosphate (cAMP) to inactive adenosine monophosphate (AMP). Inhibition of PDE4 blocks hydrolysis of cAMP, thereby increasing levels of cAMP within cells.

Practical synthesis of the PDE4 inhibitor, KW-4490
ORGN 699 |
|
Arata Yanagisawa, arata.yanagisawa@kyowa.co.jp1, Koichiro Nishimura2, Tetsuya Nezu2, Kyoji Ando2, Ayako Maki2, Eiichiro Imai2, and Shin-ichiro Mohri2. (1) Pharmaceutical Research Center, Medicinal Chemistry Research Laboratories, Kyowa Hakko Kogyo Co., Ltd, 1188 Shimotogari, Nagaizumi-cho, Sunto-gun, Shizuoka, Japan, (2) Pharmaceutical Research Center, Sakai Research Laboratories, Kyowa Hakko Kogyo Co., Ltd, 1-1-53 Takasu-cyo, Sakai-ku, Sakai, Osaka, Japan
|
A practical and scaleable synthesis of the PDE4 inhibitor, KW-4490 (1), was developed for the multi-kilogram preparation. This improved synthesis features construction of the 1-arylcyclohexene by Diels-Alder reaction, followed by a newly established acid-mediated hydrocyanation. The synthesis was achieved in 7 steps in 38% overall yield. Efforts toward increasing the regioselectivity in the Diels-Alder reaction, optimization of crystallization-induced dynamic resolution of the hydrocyanation product, and investigation of other synthetic routes will be presented. |
|
New Reactions and Methodology, Metal-Mediated Reactions, Physical Organic Chemistry, Molecular Recognition and Self-Assembly
7:00 PM-9:00 PM, Wednesday, August 20, 2008 Pennsylvania Convention Center — Blrm A/B, PosterDivision of Organic ChemistryThe 236th ACS National Meeting, Philadelphia, PA, August 17-21, 2008 |
A team at Kyowa Hakko Kirin in Japan has used a crystallisation-induced dynamic resolution in the synthesis of KW-4490, a PDE-4 inhibitor being developed for asthma and chronic obstructive pulmonary disease.6 Towards the end of the synthesis, they were faced with a mixture of cis and trans diastereomers of an intermediate derived from a hydrocyanation reaction, which was about 62:38 cis:trans; altering the conditions of the reaction did not give a selective process. The desired isomer was the cis, so they wanted to convert the unwanted trans isomer to cis to improve the yield (Scheme 2).
They first tried using a base-induced isomerisation using a base such as potassium t-butoxide, but although this worked to a degree the best ratio of products obtained was 75:25. The same result was obtained when they tested the system on both pure cis and trans isomers, indicating that this ratio represented the thermodynamic equilibrium. However, they realised that the cis isomer was less soluble in ethanol, so they thought the answer might lie in crystallisation-induced dynamic resolution.
They therefore suspended a crude mixture of the two isomers in ethanol and added a catalytic amount of potassium t-butoxide to effect the isomerisation. It was stirred and warmed, and hexane added portion-wise to crash the cis isomer out of solution. The group managed to increase the ratio of isomers to 99:1 by continuous isomerisation, with a 90% isolated yield.
Scheme 2: Kyowa Hakko Kirin found a way to improve the yield of the cis isomerA Practical Synthesis of the PDE4 Inhibitor, KW-4490
http://pubs.acs.org/doi/abs/10.1021/op1001287?prevSearch=KW%2B4490&searchHistoryKey=



Compound (XIII) is disclosed in WO00/14085 as being useful as a PDE-IV inhibitor. A method for the preparation of a typical compound among compounds (XIII) disclosed in WO00/14085 is as follows:


However, this method is not practically satisfactory as a industrially applicable preparation method, because of (1) requiring multiple steps, (2) low overall yield, (3) requiring purification by silica-gel column chromatography, and the like.
REFERENCE EXAMPLE 1
Synthesis of cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid
(1) Synthesis of cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid ethyl ester
Under a nitrogen atmosphere, trifluoromethanesulfonic acid (2.25 g) and trimethylsilylcyanide (1.57 mL) were dissolved in benzotrifluoride (10 mL), and a solution of 4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)-3-cyclohexenecarboxylic acid ethyl ester (0.79 g) prepared according to the method described in EXAMPLE 1 in benzotrifluoride (10 mL) was added dropwise at −25° C. After being stirred for for one hour at −20° C., an aqueous saturated sodium hydrogen carbonate was added and the mixture was extracted with ethyl acetate. The organic layer was washed with brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was crystallized from ethanol (1 mL) to give a solid substance (0.64 g). The solid substance (0.030 g) was crystallized from a mixed solvent of diisopropyl ether and ethyl acetate (0.36 mL, diisopropyl ether/ethyl acetate=4/1) to give cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid ethyl ester (0.019 g, 47.3%) as a solid.
Melting point 131° C.
1H-NMR (CDCl3, δ ppm) 6.84 (d, J=8.9 Hz, 1H), 6.49 (d, J=8.9 Hz, 1H), 4.39−4.33 (m, 4H), 4.17 (q, J=7.1 Hz, 2H), 3.88 (s, 3H), 2.44 (brd, J=12.6 Hz, 2H), 2.32 (tt, J=11.8, 3.8 Hz, 1H), 2.18−1.95 (m, 4H), 1.86 (dt, J=3.6, 12.6 Hz, 2H), 1.28 (t, J=7.1 Hz, 3H).
[0184] IR (KBr, cm−1) 2953, 2228, 1722, 1607, 1504, 1460, 1381, 1325, 1281, 1117, 1043, 953, 787.
MS (m/z) 346(M+H)+.
(2) Synthesis of cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid
To a suspension of cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid ethyl ester (397 g) prepared according to the method described (1) of REFERENCE EXAMPLE 1 in ethanol (1.99 L) was added a 6 ml/L aqueous potassium hydroxide (377 mL), and the mixture was stirred for 4 hours at room temperature. After water (2.03 L) was added to the reaction mixture, a 6 mol/L aqueous hydrochloric acid (576 mL) was added to crystallize and to give cis-4-cyano-4-(2,3-dihydro-8-methoxy-1,4-benzodioxine-5-yl)cyclohexanecarboxylic acid (366 g, 98.1%) as a solid.
Melting point 245° C.
1H-NMR (DMSO-d6, δ ppm) 12.26 (brs, 1H), 6.79 (d, J=8.9 Hz, 1H), 6.59 (d, J=8.9 Hz, 1H), 4.27 (dd, J=11.9, 5.0 Hz, 4H), 3.75 (s, 3H), 2.34−2.26 (m, 3H), 2.05−2.00 (m, 2H), 1.86−1.63 (m, 4H).
IR (KBr, cm−1) 3287, 2932,1728, 1609, 1508, 1454, 1285, 1119, 953, 802, 764.
MS (m/z) 318(M+H)+.

-
To a solution of 12 g (62 mmol) of 8-methoxy-1,4-benzodioxane-5-carbaldehyde in 140 ml of acetonitrile was added 12 g (110 mmol) of lithium bromide, and then 12 ml (95 mmol) of trimethylsilyl chloride was dropwise added thereto. After 15 minutes, the mixture was ice-cooled, and 19 ml (110 mmol) of 1,1,3,3-tetramethyldisiloxane was dropwise added thereto, followed by stirring at room temperature for 2 hours. The mixture was diluted with methylene chloride, and then was filtered through Celite. The solvent was evaporated in vacuo from the filtrate to give a pale yellow oily substance. To a solution of the obtained crude 5-bromomethyl-8-methoxy-1,4-benzodioxane in 180 ml of DMF was added 9.2 g (190 mmol) of sodium cyanide, followed by stirring at room temperature for 60 hours. To the mixture was added water under ice-cooling, and a solid separated out therefrom was collected by filtration to give 6.8 g (53%) of Compound 1a as an ash-colored solid.Melting Point: 121 – 125 °C
1H-NMR (CDCl3, δ, ppm) 3.60 (s, 2H), 3.88 (s, 3H), 4.33 (s, 4H), 6.50 (d, J = 8 Hz, 1H), 6.86 (d, J = 8 Hz, 1H).
MASS (m/z) 205 (M+).
- Example 1.
- 4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl) cyclohexanone (Compound 1)(Step A)
- Synthesis of 2-(8-methoxy-1,4-benzodioxan-5-yl)acetonitrile (Compound 1a)
-
To a solution of 6.2 g (30 mmol) of Compound 1a obtained in Step A in 94 ml of acetonitrile were added 1.4 ml (3.0 mmol) of a 40% methanolic solution of Triton B and 27 ml (300 mmol) of methyl acrylate, followed by heating under reflux for 5 hours. The mixture was allowed to stand for cooling, and then poured into water, followed by extraction with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate, and the solvent was evaporated in vacuo. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 2/1) to give 6.4 g (56%) of Compound 1b as a pale yellow oily substance.
1H-NMR (CDCl3, δ, ppm) 2.05-2.37 (m, 4H), 2.39-2.59 (m, 2H), 2.62-2.82 (m, 2H), 3.60 (s, 6H), 3.87 (s, 3H), 4.20-4.40 (m, 4H), 6.48 (d, J = 9 Hz, 1H), 7.01 (d, J = 9 Hz, 1H).
MASS (m/z) 377 (M+).
- (Step B) Synthesis of dimethyl 4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)pimelate (Compound 1b)
-
To a solution of 6.4 g (17 mmol) of Compound 1b obtained in Step B in 96 ml of 1,2-dimethoxyethane was added 2.0 g (50 mmol) of 60% sodium hydride. After heating under reflux for 3 hours, the mixture was allowed to stand for cooling, poured into ice water, acidified with a 6 mol/liter aqueous hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 2/1) to give 5.0 g (86%) of Compound 1c as a white solid.
Melting Point: 129 – 132 °C
1H-NMR (CDCl3, δ, ppm) 2.21-2.50 (m, 3H), 2.61-2.89 (m, 2H), 3.11(d, J = 15 Hz, 1H), 3.79 (s, 3H), 3.89 (s, 3H), 4.37 (s, 4H), 6.49 (d, J = 9 Hz, 1H), 6.84 (d, J = 9 Hz, 1H), 12.2 (s, 1H).
MASS (m/z) 345 (M+).
- (Step C) Synthesis of 4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)-2-methoxycarbonylcyclohexanone (Compound 1c)
-
A mixture of 5.0 g (15 mmol) of Compound 1c obtained in Step C, 50 ml of DMSO, 5 ml of water, and 5.0 g of sodium chloride was stirred at 150°C for 5 hours. The mixture was allowed to stand for cooling, and water was added thereto, followed by extraction with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate, and the solvent was evaporated in vacuo. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 3/1) to give 3.6 g (86%) of Compound 1 as a white solid.
Melting Point: 157 – 161 °C
1H-NMR (CDCl3, δ, ppm) 2.21-2.41 (m, 2H), 2.45-2.72 (m, 4H), 2.81-3.00 (m, 2H), 3.89 (s, 3H), 4.37 (s, 4H), 6.51 (d, J = 9 Hz, 1H), 6.88 (d, J = 9 Hz, 1H).
MASS (m/z) 287 (M+).
- (Step D) Synthesis of Compound 1
-
In 65 ml of THF was dissolved 10 g (41 mmol) of 5-bromo-8-methoxy-1,4-benzodioxane, and 28 ml (45 mmol) of a 1.59 mol/liter solution of n-butyl lithium in hexane was dropwise added thereto at -78°C. After 15 minutes, a solution of 9.6 g (61 mmol) of 1,4-cyclohexadione monoethyleneketal in 50 ml of THF was dropwise added thereto. The mixture was stirred for 1 hour, followed by stirring at room temperature for 20 minutes. Water was added thereto, the mixture was extracted with ethyl acetate, and the extract was washed with brine and dried over sodium sulfate. The solvent was evaporated therefrom, and the residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 1/1) to give 9.0 g (68%) of Compound 2a as a white solid.
Melting Point: 94 – 96 °C
1H-NMR (CDCl3, δ, ppm) 1.58-1.72 (m, 2H), 1.88-2.28 (m, 6H), 3.57 (s, 1H), 3.86 (s, 3H), 3.90-4.07 (m, 4H), 4.35 (s, 4H), 6.46 (d, J = 9 Hz, 1H), 6.82 (d, J = 9 Hz, 1H).
MASS (m/z) 322 (M+). -
- (Step B) Synthesis of Compound 2
-
In 4.9 ml of methylene chloride was dissolved 0.49 g (1.5 mmol) of Compound 2a obtained in Step A, 0.26 ml (1.9 mmol) of trimethylsilyl cyanide was added thereto at -78°C, then 0.20 ml (1.6 mmol) of a boron trifluoride-ethyl ether complex was dropwise added thereto, and the mixture was stirred for 10 minutes, followed by stirring at room temperature for 10 minutes. A saturated aqueous solution of sodium bicarbonate was added thereto and the mixture was extracted with ethyl acetate. The extract was washed with brine and dried over sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 2/1) to give 0.30 g (61%) of Compound 2 as a colorless oily substance.
1H-NMR (CDCl3, δ, ppm) 1.79-1.95 (m, 2H), 2.06-2.20 (m, 4H), 2.30-2.46 (m, 2H), 3.87 (s, 3H), 3.90-4.07 (m, 4H), 4.36 (s, 4H), 6.48 (d, J = 9 Hz, 1H), 6.82 (d, J = 9 Hz, 1H).
MASS (m/z) 331 (M+).
-
- Example 2. 4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanone ethyleneketal (Compound 2)
- (Step A)Synthesis of 4-hydroxy-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanone ethyleneketal (Compound 2a)
-
In 2.9 ml of acetone was dissolved 0.29 g (0.87 mmol) of Compound 2 obtained in Example 2, 1.2 ml (7.2 mmol) of a 6 mol/liter aqueous hydrochloric acid was added thereto, and the mixture was heated under reflux for 3 hours. The mixture was allowed to stand for cooling and poured into a saturated aqueous solution of sodium bicarbonate, the mixture was extracted with ethyl acetate, and the extract was washed with brine. The mixture was dried over sodium sulfate, and the solvent was evaporated to give 0.23 g (92%) of Compound 1 as a white solid.
- Example 3. Compound 1
- Example 4. Methyl
cis-4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanecarboxylate (Compound 3) and methyltrans-4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanecarboxylate (Compound 4)(Step A) Synthesis of 2-[4-cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexylidene]-1,3-dithiane (Compound 3a)
-
To a solution of 5.0 ml (26 mmol) of 2-trimethylsilyl-1,3-dithiane in 50 ml of THF was added dropwise 17 ml (26 mmol) of a 1.54 mol/liter solution of n-butyl lithium in hexane under ice-cooling. After 10 minutes, the mixture was cooled to -78°C, and a solution of 3.6 g (13 mmol) of Compound 1 obtained in Example 1 in 40 ml of THF was dropwise added thereto. After 10 minutes, to the mixture was added brine, followed by addition of water at room temperature. The mixture was extracted with ethyl acetate, the extract was dried over sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 4/1) to give 3.9 g (79%) of Compound 3a as a white solid.
Melting Point: 164 – 166 °C
1H-NMR (CDCl3, δ, ppm) 1.70-1.92 (m, 2H), 2.05-2.24 (m, 2H), 2.28-2.53 (m, 4H), 2.89 (t, J = 6 Hz, 4H), 3.18-3.38 (m, 2H), 3.87 (s, 3H), 4.36 (s, 4H), 6.47 (d, J = 9 Hz, 1H), 6.79 (d, J = 9 Hz, 1H).
MASS (m/z) 389 (M+).
-
In 120 ml of methanol was suspended 3.9 g (10 mmol) of Compound 3a obtained in Step A, 1.7 ml (20 mmol) of 70% perchloric acid, and 4.3 g (16 mmol) of mercury chloride (HgCl2) were added thereto, and the mixture was stirred for 4 hours. The mixture was diluted with methylene chloride and was filtered through Celite, the filtrate was poured into a saturated aqueous solution of sodium bicarbonate, and the mixture was extracted with methylene chloride. The organic layer was washed with brine and dried over sodium sulfate, and the solvent was evaporated. The residue was purified by silica gel column chromatography (eluted with hexane/ethyl acetate = 1/1) to give the crude Compound 3 as a white solid and also to give 0.18 g (5.5%) of Compound 4 as a colorless transparent oily substance. Compound 3 was further recrystallized from ethyl acetate to give 0.57 g (17%) of white crystals.
Compound 3
Melting Point: 123 – 124 °C
1H-NMR (CDCl3, δ, ppm) 1.75-2.22 (m, 6H), 2.27-2.51 (m, 3H), 3.71 (s, 3H), 3.88 (s, 3H), 4.36 (s, 4H), 6.48 (d, J = 9 Hz, 1H), 6.84 (d, J = 9 Hz, 1H).
MASS (m/z) 331 (M+).
Compound 4
1H-NMR (CDCl3, δ, ppm) 1.92-2.38 (m, 8H), 2.70-2.88 (m, 1H), 3.69 (s, 3H), 3.87 (s, 3H), 4.36 (s, 4H), 6.48 (d, J = 9 Hz, 1H), 6.81 (d, J = 9 Hz, 1H).
MASS (m/z) 331 (M+).
- (Step B) Synthesis of Compound 3 and Compound 4
Example 5.
cis-4-Cyano-4-(8-methoxy-1,4-benzodioxan-5-yl)cyclohexanecarboxylic acid (Compound 5)
-
To a mixture of 0.55 g (1.7 mmol) of Compound 3 obtained in Example 4 and 3.3 ml of methanol was added 3.3 ml of THF to dissolve them. To the mixture was dropwise added 2.6 ml of a 1.3 mol/liter aqueous solution of potassium hydroxide, followed by stirring at room temperature for 1 hour. The mixture was poured into water, ethyl acetate was added thereto, and an aqueous layer was extracted. The aqueous layer was acidified with a 1 mol/liter aqueous hydrochloric acid, and the precipitated solid was collected by filtration and re-slurried with ethanol to give 0.45 g (86%) of Compound 5 as a white solid.
Melting Point: 228 – 230 °C
1H-NMR (DMSO-d6 , δ, ppm) 1.59-1.90 (m, 4H), 1.94-2.10 (m, 2H), 2.20-2.45 (m, 3H), 3.75 (s, 3H), 4.27 (dd, J = 5, 12 Hz, 4H), 6.60 (d, J = 9 Hz, 1H), 6.79 (d, J = 9 Hz, 1H), 12.2 (br s, 1H).
MASS (m/z) 317 (M+).Elemental analysis: C17H19NO5 Found (%) C 64.09, H : 6.01, N : 4.51 Calcd. (%) C 64.34, H : 6.03, N : 4.41
-
Teixeira, M. M.; Gristwood, R. W.; Cooper, N.; Hellewell, P. G. Trends Pharmacol. Sci.1997, 18, 164– 170, [PubMed],
-
Dyke, H. J.; Montana, J. G. Exp. Opin. Investig. Drugs 2002, 11, 1– 13Lipworth, B. J. Lancet 2005, 365 ( 9454) 167– 175Kroegel, C.; Foerster, M. Exp. Opin. Investig. Drugs 2007, 16, 109– 124
-
Yanagawa, K. The 26th Medicinal Chemistry Symposium, 2007, 2P-29.Ohshima, E.;Yanagawa, K.; Manabe, H.; Miki, I.; Masuda, Y. PCT Int. Appl. WO0164666 A1, 2001.
-
Christensen, S. B.; Guider, A.; Forster, C. J.; Gleason, J. G.; Bender, P. E.; Karpinski, J. M.; DeWolf, W. E., Jr.; Barnette, M. S.; Underwood, D. C.; Griswold, D. E.; Cieslinski, L. B.; Burman, M.; Bochnowicz, S.; Osborn, R. R.; Manning, C. D.; Grous, M.; Hillegas, L. M.; O’Leary-Bartus, J.; Ryan, M. D.; Eggleston, D. S.; Haltiwanger, R. C.; Torphy, T. J. J. Med. Chem. 1998, 41, 821– 835
-
Caron, S.; Vazquez, E.; Wojcik, J. M. J. Am. Chem. Soc. 2000, 122, 712– 713Culkin, D. A.; Hartwig, J. F. Acc. Chem. Res. 2003, 36, 234– 245You, J.; Verkade, J. G. Angew. Chem., Int. Ed. 2003, 42, 5051–5053
-
Muratake, H.; Natsume, M. Tetrahedron 1990, 46, 6331– 6342Caron, S.; Vazquez, E. Org. Process Res. Dev. 2001, 5, 587– 592
-
North, M. In Comprehensive Organic Functional Group Transformations; Katritzky, A. R.;Meth-Cohn, O.; Rees, C. W., Eds.; Pergamon: Oxford, 1995; Vol. 3, p 614.
-
Lemaire, M.; Doussot, J.; Guy, A. Chem. Lett. 1988, 1581– 1584Guy, A.; Doussot, J.; Guette, J.-P.; Garreau, R.; Lemaire, M. Synlett 1992, 821– 822Kurti, L.; Czako, B.; Corey, E. J. Org. Lett. 2008, 10, 5247– 5250
-
Yanagisawa, A.; Nezu, T.; Mohri, S. Org. Lett. 2009, 11, 5286– 5289
-
Brown, H. C.; Cole, T. E. Organometallics 1983, 2, 1316– 1319
-
Kuivila, H. G.; Nahabedian, K. V. J. Am. Chem. Soc. 1961, 83, 2159– 2163Kuivila, H. G.; Nahabedian, K. V. J. Am. Chem. Soc. 1961, 83, 2164– 2166Nahabedian, K. V.; Kuivila, H. G. J. Am. Chem. Soc. 1961, 83, 2167–2174
-
Stang, P. J.; Fisk, T. E. Synthesis 1979, 438– 440Stang, P. J.; Treptow,W. Synthesis 1980, 283– 284Saulnier, M. G.; Kadow, J. F.; Tun, M. M.; Langley, D. R.; Vyas, D. M. J. Am. Chem. Soc. 1989, 111, 8320– 8321
-
Kato, S.; Chujo, I.; Suzuki, K. PCT Int. Appl. WO04000795 A1, 2004.
-
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457– 2483O’Keefe,D. F.; Dannock, M. C.; Marcuccio, S. M. Tetrahedron Lett. 1992, 33, 6679– 6680Segelstein, B. E.; Bulter, T. W.; Chenard, B. L. J. Org. Chem. 1995, 60, 12– 13Kong, K.-C.; Cheng, C.-H. J. Am. Chem. Soc. 1991, 113, 6313– 6315
-
Brands, K. M. J.; Davies, A. J. Chem. Rev. 2006, 106, 2711– 2733
-
Baker, W.; Jukes, E. H. T.; Subrahmanyam, C. A. J. Chem. Soc. 1934, 1681–1684Dallacker, F.; Van Wersh, J. Chem. Ber. 1972, 105, 3301–3305
- Yanagisawa, Arata; Organic Process Research & Development 2010, 14(5), P1182-1187
- Yanagisawa, Arata; Organic Letters 2009, 11(22), P5286-5289
- US 20010056117
- WO 2002059105
- WO 2000014085
| WO1998022455A1 * | Nov 19, 1997 | May 28, 1998 | Michio Ichimura | Oxygenic heterocyclic compounds |
| JP10147585A * | Title not available |
| WO2001064666A1 * | Mar 2, 2001 | Sep 7, 2001 | Kyowa Hakko Kogyo Kk | Oxygen-containing heterocyclic compounds |
| WO2002059105A1 * | Jan 25, 2002 | Aug 1, 2002 | Kyowa Hakko Kogyo Kk | Styrene derivatives and process for production thereof |
| WO2006041120A1 * | Oct 13, 2005 | Apr 20, 2006 | Daisuke Harada | Pharmaceutical composition |
| WO2006041121A1 * | Oct 13, 2005 | Apr 20, 2006 | Daisuke Harada | Remedies/preventives for chronic skin disease |
| WO2006123726A1 * | May 18, 2006 | Nov 23, 2006 | Daisuke Harada | Pharmaceutical composition |
| WO2011134468A1 | Apr 28, 2011 | Nov 3, 2011 | Leo Pharma A/S | Biaryl phosphodiesterase inhibitors |
| EP1362853A1 * | Jan 25, 2002 | Nov 19, 2003 | Kyowa Hakko Kogyo Co., Ltd | Styrene derivatives and process for production thereof |
Cinnamon cuts blood glucose levels in diabetes patients
Consumption of cinnamon is associated with favorable reductions in plasma glucose and lipid levels, according to research published in the September/October issue of the Annals of Family Medicine.
Robert W. Allen, Pharm.D., of the Western University of Health Sciences in Pomona, Calif., and colleagues used data from 10 randomized, controlled trials involving 543 patients with type 2 diabetes to conduct an update of a previous systematic review and meta-analysis examining the effect of cinnamon consumption on glucose and lipid levels.
The researchers found that cinnamon, in daily doses of 120 mg/d to 6 g/d for four to 18 weeks, was associated with a significant reduction in levels of fasting plasma glucose (?24.59 mg/dL), but no significant effect on glycosylated hemoglobin. Cinnamon intake also was linked to significant changes in lipid levels, including decreases in levels of total cholesterol (?15.60 mg/dL), low-density lipoprotein cholesterol (LDL-C) (?9.42 mg/dL), and triglycerides…
View original post 89 more words
