
Home » Uncategorized (Page 14)
Category Archives: Uncategorized
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Brezivaptan
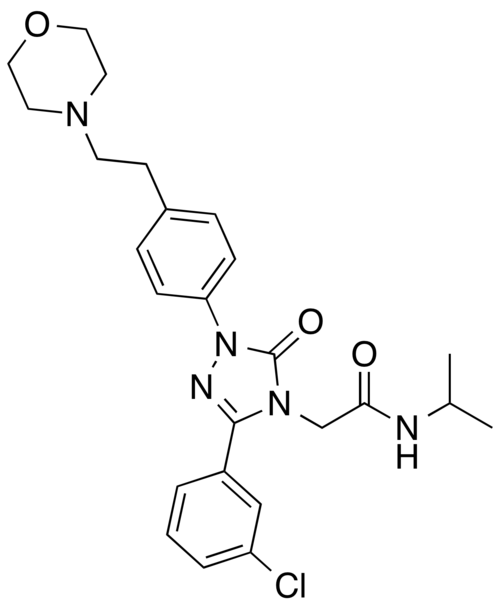
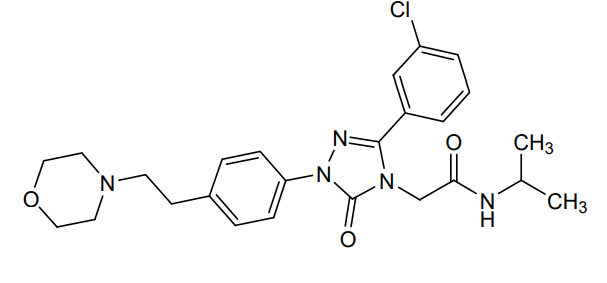
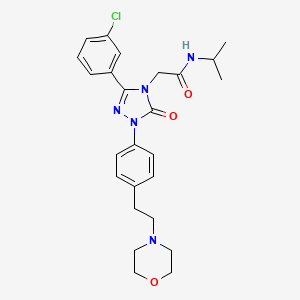
Brezivaptan
CAS 1370444-22-6
ANC-501, THY-1773, TS-121, 575OB1CKN0
MF C25H30ClN5O3 MW 484.0 g/mol
2-[3-(3-chlorophenyl)-1-{4-[2-(morpholin-4-yl)ethyl]phenyl}-5-oxo-1,5-dihydro-4H-1,2,4-triazol-4-yl]-N-(propan-2-yl)acetamide
2-[3-(3-chlorophenyl)-1-[4-(2-morpholin-4-ylethyl)phenyl]-5-oxo-1,2,4-triazol-4-yl]-N-propan-2-ylacetamide
vasopressin receptor antagonist
- ANC-501 in the Treatment of Adults With Major Depressive DisorderCTID: NCT05439603Phase: Phase 2Status: CompletedDate: 2024-12-31
- A Study to Evaluate the Safety and Efficacy of TS-121 as an Adjunctive Treatment for Major Depressive DisorderCTID: NCT03093025Phase: Phase 2Status: TerminatedDate: 2020-07-14
- Exploratory Study Using Positron Emission Tomography With TS-121 and [11C]TASP0410699 in Healthy Adult Male SubjectsCTID: NCT02448212Phase: Phase 1Status: CompletedDate: 2017-02-14
Brezivaptan[1] (developmental code names ANC-501, THY-1773, TS-121) is an orally active, selective vasopressin V1B receptor antagonist which is under development by Taisho Pharmaceutical for the adjunctive treatment of major depressive disorder.[2][3][4] As of November 2022, it is in phase II clinical trials for this indication.[2][3][5]
ANC-501 is under investigation in clinical trial NCT05439603 (ANC-501 in the Treatment of Adults With Major Depressive Disorder).
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US90328697&_cid=P11-MFYT6K-98384-1
Synthesis of Example Aa-1
2-[3-(3-Chlorophenyl)-1-{4-[2-(morpholin-4-yl)ethyl]phenyl}-5-oxo-1,5-dihydro-4H-1,2,4-triazol-4-yl]-N-(propan-2-yl)acetamide
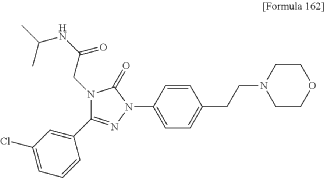
| A mixture of the compound (100 mg) prepared in Reference Example P-I1, morpholine (0.03 mL), N,N-diisopropylethylamine (0.35 mL), and MeCN (3.00 mL) was stirred at an outside temperature of 80° C. overnight. After cooling, the solvent was distilled off under reduced pressure. The residue was purified by column chromatography (SNAP Cartridge HP-Sil: 10 g, mobile phase: CHCl 3/MeOH=98/2 to 85/15 (v/v); and SNAP Cartridge KP-NH: 28 g, mobile phase: n-hexane/CHCl 3=80/20 to 0/100 (v/v)) and preparative thin-layer chromatography (PTLC) (1.0 mm silica gel 60F 254 plate, mobile phase: EtOAc/MeOH=95/5 (v/v)). The resulting crude product was washed with a solvent mixture of EtOAc and n-hexane (EtOAc/n-hexane=1/4 (v/v)) with stirring to yield the title compound (70 mg, colorless solid). |
PAT
- 1,2,4-triazolone derivativePublication Number: NZ-608729-APriority Date: 2010-10-01
- 1, 2, 4-triazolone derivativePublication Number: US-2013197217-A1Priority Date: 2010-10-01
- 1, 2, 4-triazolone derivative and use thereof as an antagonist on the arginine-vasopressin 1B receptorPublication Number: US-9193695-B2Priority Date: 2010-10-01Grant Date: 2015-11-24
- 1,2,4-triazolone derivative, substance and pharmaceutical compositionPublication Number: BR-112013007389-B1Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: EP-2623499-A1Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: EP-2623499-B1Priority Date: 2010-10-01Grant Date: 2015-04-22
- DERIVAT 1,2,4-TRIAZOLONAPublication Number: HR-P20150462-T1Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: HU-E025729-T2Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: IL-225091-APriority Date: 2010-10-01
- Methods of treating depression with 1,2,4-triazolone derivativesPublication Number: WO-2023235785-A1Priority Date: 2022-06-01
- 1,2,4-triazolone derivativePublication Number: AU-2011308403-A1Priority Date: 2010-10-01
- 1,2,4-triazolone derivativePublication Number: AU-2011308403-B2Priority Date: 2010-10-01Grant Date: 2014-08-21
- 1,2,4-Triazolone DerivativesPublication Number: CN-103119028-APriority Date: 2010-10-01
- 1,2,4-Triazolone DerivativesPublication Number: CN-103119028-BPriority Date: 2010-10-01Grant Date: 2016-05-25



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- PubChem. “Brezivaptan”. pubchem.ncbi.nlm.nih.gov. Retrieved 2024-08-15.
- “TS 121 -“. AdisInsight. Springer Nature Switzerland AG.
- “New Drug Pipeline – Taisho Pharmaceutical Holdings”.
- Kamiya M, Sabia HD, Marella J, Fava M, Nemeroff CB, Umeuchi H, Iijima M, Chaki S, Nishino I (September 2020). “Efficacy and safety of TS-121, a novel vasopressin V1B receptor antagonist, as adjunctive treatment for patients with major depressive disorder: A randomized, double-blind, placebo-controlled study”. Journal of Psychiatric Research. 128: 43–51. doi:10.1016/j.jpsychires.2020.05.017. PMID 32521250. S2CID 219587135.
- Inatani S, Mizuno-Yasuhira A, Kamiya M, Nishino I, Sabia HD, Endo H (May 2021). “Prediction of a clinically effective dose of THY1773, a novel V1B receptor antagonist, based on preclinical data”. Biopharmaceutics & Drug Disposition. 42 (5): 204–217. doi:10.1002/bdd.2273. PMC 8252455. PMID 33734452.
External links
- Clinical trial number NCT03093025 for “A Study to Evaluate the Safety and Efficacy of TS-121 as an Adjunctive Treatment for Major Depressive Disorder” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Other names | TS-121; TS121; TS-1211; TS1211; THY1773; THY-1773; ANC-501; ANC501 |
| Routes of administration | By mouth |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1370444-22-6 |
| PubChem CID | 56952080 |
| DrugBank | DB18907 |
| ChemSpider | 129325033 |
| UNII | 575OB1CKN0 |
| ChEMBL | ChEMBL5314910 |
| Chemical and physical data | |
| Formula | C25H30ClN5O3 |
| Molar mass | 484.00 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
////////////Brezivaptan, ANC-501, THY-1773, TS-121, ANC 501, THY 1773, TS 121, 575OB1CKN0
Ateganosine
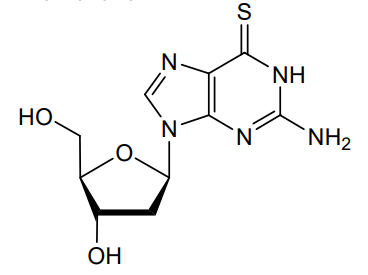
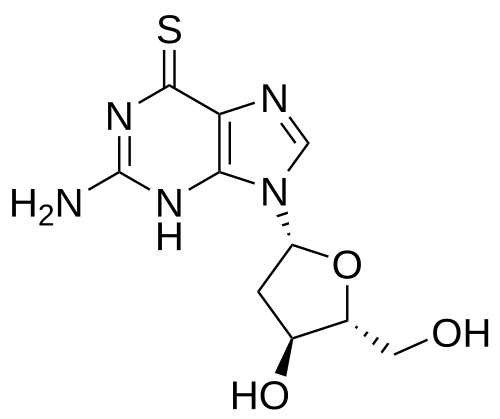
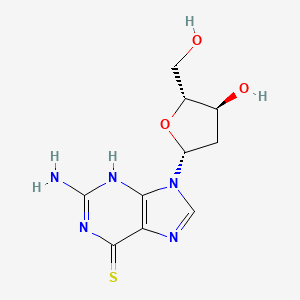
Ateganosine
CAS 789-61-7
MF C10H13N5O3S MW 283.31 g/mol
2′-deoxy-6-thioguanosine
nucleoside analogue, antineoplastic
- 6-THIO-2′-DEOXYGUANOSINE
- 2′-Deoxythioguanosine
- TGdR
- Thioguanine deoxyriboside
- KR0RFB46DF
- NSC-71261
Ateganosine is a telomerase inhibitor[1] and apoptosis inducer currently under investigation for the treatment of various cancers, including non-small cell lung cancer (NSCLC).[2]
Beta-Thioguanine Deoxyriboside is a thiopurine nucleoside derivative with antineoplastic activity. After conversion to the triphosphate, beta-thioguanine deoxyriboside is incorporated into DNA, resulting in inhibition of DNA replication. This agent is cytotoxic against leukemia cell lines and has demonstrated some activity against leukemia cells in vivo. Beta-thioguanine deoxyriboside demonstrates antineoplastic activity against 6-thioguanine-resistant tumor cells. (NCI04)
- THIO Sequenced With Cemiplimab in Advanced NSCLCCTID: NCT05208944Phase: Phase 2Status: RecruitingDate: 2025-05-31
- A Phase III Study With THIO + Cemiplimab vs Chemotherapy as 3rd Line Treatment in Advanced/Metastatic NSCLCCTID: NCT06908304Phase: Phase 3Status: Not yet recruitingDate: 2025-04-08
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Eglenen-Polat B, Kowash RR, Huang HC, Siteni S, Zhu M, Chen K, et al. (January 2024). “A telomere-targeting drug depletes cancer initiating cells and promotes anti-tumor immunity in small cell lung cancer”. Nature Communications. 15 (1) 672. Bibcode:2024NatCo..15..672E. doi:10.1038/s41467-024-44861-8. PMC 10803750. PMID 38253555.
- “Ateganosine”. PatSnap.
| Clinical data | |
|---|---|
| Other names | 2′-Deoxythioguanosine |
| Identifiers | |
| IUPAC name | |
| CAS Number | 789-61-7 |
| PubChem CID | 3000603 |
| DrugBank | DB18117 |
| ChemSpider | 2272164 |
| UNII | KR0RFB46DF |
| KEGG | D13071 |
| ChEMBL | ChEMBL3250476 |
| CompTox Dashboard (EPA) | DTXSID4021345 |
| Chemical and physical data | |
| Formula | C10H13N5O3S |
| Molar mass | 283.31 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
////////Ateganosine, nucleoside analogue, antineoplastic, 6-THIO-2′-DEOXYGUANOSINE, 2′-Deoxythioguanosine, TGdR, Thioguanine deoxyriboside, KR0RFB46DF, fast track designation, NSC-71261, NSC 71261
Bimokalner
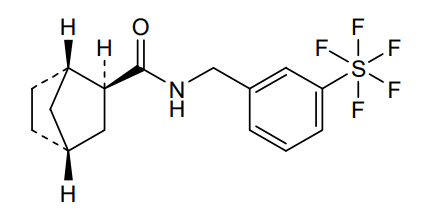
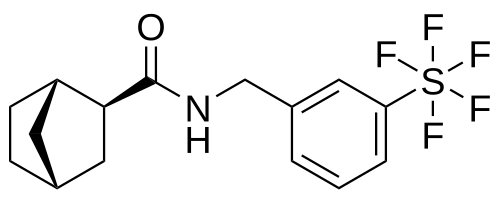
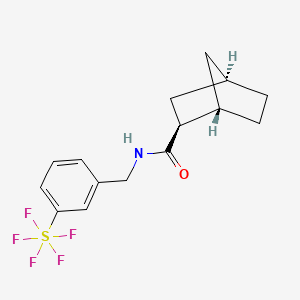
Bimokalner
CAS 2243284-19-5
MF C15H18F5NOS MW 355.4 g/mol
- KEY5KKX6QY
- orb2663976
- (1S,2S,4R)-N-[[3-(pentafluoro-λ6-sulfanyl)phenyl]methyl]bicyclo[2.2.1]heptane-2-carboxamide
(1S,2S,4R)-N-{[3-(pentafluoro-λ6sulfanyl)phenyl]methyl} bicyclo[2.2.1]heptane-2-carboxamide
voltage-gated potassium channel (Kv7.4) agonist
Bimokalner is an investigational new drug under evaluation for preventing and treating hearing loss caused by cisplatin treatment. It is a voltage-gated potassium channel agonist targeting Kv7.4 and is being developed by Acousia Therapeutics GmbH.[1][2]
PAT
Compounds useful as potassium channel openers, Publication Number: US-11884642-B2, Priority Date: 2017-02-28, Grant Date: 2024-01-30
- Novel Compounds Useful As Potassium Channel OpenersPublication Number: KR-20210134826-APriority Date: 2017-02-28
- Novel compounds useful as potassium channel openersPublication Number: KR-102382795-B1Priority Date: 2017-02-28Grant Date: 2022-04-05
- Novel Compounds Useful As Potassium Channel OpenersPublication Number: KR-102443685-B1Priority Date: 2017-02-28Grant Date: 2022-09-15
- Compounds useful as potassium channel openersPublication Number: CN-114105942-BPriority Date: 2017-02-28Grant Date: 2024-07-12
- Novel compounds useful as potassium channel openers.Publication Number: JP-7474289-B2Priority Date: 2017-02-28Grant Date: 2024-04-24
- Compounds useful as potassium channel openersPublication Number: US-11034665-B2Priority Date: 2017-02-28Grant Date: 2021-06-15
- Novel compounds useful as potassium channel openersPublication Number: US-2021261518-A1Priority Date: 2017-02-28
- Novel compounds useful as potassium channel openersPublication Number: AU-2018227005-B2Priority Date: 2017-02-28Grant Date: 2021-11-11
- Compounds useful as potassium channel openersPublication Number: CN-110312710-BPriority Date: 2017-02-28Grant Date: 2022-02-15
- New compounds useful as potassium channel openersPublication Number: CN-114105942-APriority Date: 2017-02-28
- Pentacyclothienyl and indanyl urea derivatives as potassium channel openersPublication Number: EP-3567034-A1Priority Date: 2017-02-28
- New Compounds Useful as Potassium Channel OpenersPublication Number: KR-20190105058-APriority Date: 2017-02-28
- Novel compounds useful as potassium channel openersPublication Number: US-2020157072-A1Priority Date: 2017-02-28
- Novel compounds useful as potassium channel openersPublication Number: WO-2018158256-A2Priority Date: 2017-02-28
- Pentacyclothienyl and indanyl urea derivatives as potassium channel openersPublication Number: EP-3567034-B1Priority Date: 2017-02-28Grant Date: 2020-10-28
PAT
(1R,2R,4S)-rel-N-(3-(pentafluorosulfanyl)benzyl)bicyclo[2.2.1]heptane-2-carboxamide
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Bimokalner”. PatSnap.
- Tavanai E, Rahimi V, Khalili ME, Falahzadeh S, Motasaddi Zarandy M, Mohammadkhani G (2024). “Age-related hearing loss: An updated and comprehensive review of the interventions”. Iranian Journal of Basic Medical Sciences. 27 (3): 256–269. doi:10.22038/IJBMS.2023.72863.15849. PMC 10849199. PMID 38333758.
| Clinical data | |
|---|---|
| Other names | ACOU085 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2243284-19-5 |
| PubChem CID | 135309173 |
| UNII | KEY5KKX6QY |
| Chemical and physical data | |
| Formula | C15H18F5NOS |
| Molar mass | 355.37 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
///////////Bimokalner, Acousia Therapeutics, KEY5KKX6QY, orb 2663976
Abarelix
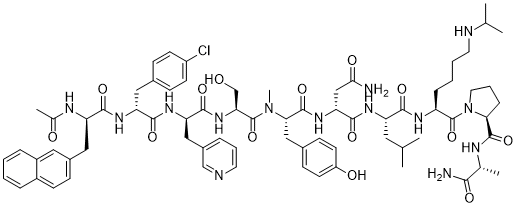
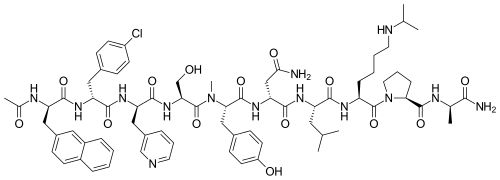
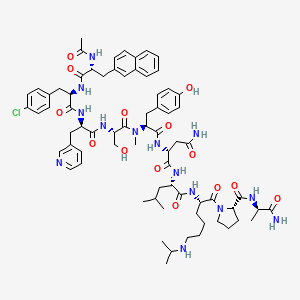
Abarelix
CAS 183552-38-7
785804-17-3 (acetate) 183552-38-7 (free base)
PPI149, PPI-149, PPI 149, R3827, R-3827, R 3827, Abarelix, Abarelix acetate, Plenaxis,
W486SJ5824
Chemical Formula: C72H95ClN14O14
Exact Mass: 1414.6841
Molecular Weight: 1416.06
Ac-D-Nal-[D-(pCl)Phe]-D-Pal-Ser-[Nalpha-Me-Tyr]-D-Asn-Leu-ILys-Pro-DAla-NH2

(2R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-acetamido-3-naphthalen-2-ylpropanoyl]amino]-3-(4-chlorophenyl)propanoyl]amino]-3-pyridin-3-ylpropanoyl]amino]-3-hydroxypropanoyl]-methylamino]-3-(4-hydroxyphenyl)propanoyl]amino]-N-[(2S)-1-[[(2S)-1-[(2S)-2-[[(2R)-1-amino-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxo-6-(propan-2-ylamino)hexan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]butanediamide
(2R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2R)-2-[[(2R)-2-acetamido-3-naphthalen-2-ylpropanoyl]amino]-3-(4-chlorophenyl)propanoyl]amino]-3-pyridin-3-ylpropanoyl]amino]-3-hydroxypropanoyl]-methylamino]-3-(4-hydroxyphenyl)propanoyl]amino]-N-[(2S)-1-[[(2S)-1-[(2S)-2-[[(2R)-1-amino-1-oxopropan-2-yl]carbamoyl]pyrrolidin-1-yl]-1-oxo-6-(propan-2-ylamino)hexan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]butanediamide
Abarelix is a synthetic decapeptide and antagonist of naturally occurring gonadotropin-releasing hormone (GnRH). Abarelix directly and competitively binds to and blocks the gonadotropin releasing hormone receptor in the anterior pituitary gland, thereby inhibiting the secretion and release of luteinizing hormone (LH) and follicle stimulating hormone (FSH). In males, the inhibition of LH secretion prevents the release of testosterone. As a result, this may relieve symptoms associated with prostate hypertrophy or prostate cancer, since testosterone is required to sustain prostate growth.
Abarelix, sold under the brand name Plenaxis, is an injectable gonadotropin-releasing hormone antagonist (GnRH antagonist) which is marketed in Germany and the Netherlands. It is primarily used in oncology to reduce the amount of testosterone made in patients with advanced symptomatic prostate cancer for which no other treatment options are available.[2][3]
It was originally marketed by Praecis Pharmaceuticals as Plenaxis,[2] and is now marketed by Speciality European Pharma in Germany[4] after receiving a marketing authorization in 2005. The drug was introduced in the United States in 2003, but was discontinued in this country in May 2005 due to poor sales and a higher-than-expected incidence of severe allergic reactions.[5] It remains marketed in Germany and the Netherlands however.[6]
Pat
https://patents.google.com/patent/CN107778354B/en
Example 1: synthesis of peptide resin 1
Dissolving 0.15mol of Fmoc-D-Ala and 0.15mol of HOBt by using a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Taking 0.05mol of MOBHA resin (the substitution value is about 0.6mmol/g), swelling with DMF for 25 minutes, washing and filtering, adding the activated solution, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3 times, wherein the washing time is 3min each time, obtaining Fmoc-D-Ala-MOBHA resin, namely the peptide resin 1, removing Fmoc protection with 20% PIP/DMF solution for 25 minutes before carrying out the next coupling reaction, washing and filtering to obtain the D-Ala-MOBHA resin.
Example 2: synthesis of peptide resin 1
Dissolving 0.15mol of Boc-D-Ala and 0.15mol of HOBt with a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Taking 0.05mol of MOBHA resin (the substitution value is about 0.6mmol/g), swelling with DMF for 25 minutes, washing and filtering, adding an activated Fmoc-D-Ala solution, stirring at room temperature for 3 hours, pumping out the reaction solution, washing 3 times with DMF, washing 3 times with DCM, wherein each washing time is 3min, obtaining Boc-D-Ala-MOBHA resin, namely peptide resin 1, deprotecting with 30% TFA/DCM solution for 30 minutes, neutralizing with DIEA/DCM solution, washing and filtering with DMF and DCM, and obtaining D-Ala-MOBHA resin.
Example 3: synthesis of Abarelix peptide resin
Dissolving 0.15mol of Fmoc-Pro and 0.15mol of HOBt in a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Adding the activated Fmoc-Pro solution into the peptide resin 1 obtained in example 1, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3 minutes each time, removing Fmoc protection with 20% PIP/DMF solution for 25 minutes, washing and filtering to obtain Pro-D-Ala-MOBHA resin.
Boc-Lys (iPr, Z), Fmoc-Leu, Fmoc-D-Asn (Trt), Fmoc-N-Me-Tyr (tBu), Fmoc-Ser (tBu), Fmoc-D-Pal, Fmoc-D-Cpa and Ac-D-Nal are sequentially added in the same method, and the Abarelix peptide resin, Ac-D-Nal-D-Cpa-D-Pal-Ser (tBu) -N-Me-Tyr (tBu) -D-Asn (Trt) -Leu-Lys (iPr, Z) -Pro-D-Ala-MOBHA resin are obtained by washing and filtering.
Example 4: synthesis of Abarelix peptide resin
Dissolving 0.15mol of Boc-Pro and 0.15mol of HOBt by using a proper amount of DMF; and adding 0.15mol DIC slowly into the protected amino acid DMF solution under stirring, and reacting for 30 minutes under stirring at room temperature to obtain an activated protected amino acid solution for later use.
Adding the activated Boc-Pro solution into the peptide resin 1 obtained in example 1, stirring at room temperature for reaction for 3 hours, pumping out the reaction solution, washing with DMF for 3 times, washing with DCM for 3min each time, deprotecting with 30% TFA/DCM solution for 30 minutes, neutralizing with DIEA/DCM solution, washing with DMF and DCM, and filtering to obtain Pro-D-Ala-MBHA resin.
Boc-Lys (iPr, Z), Fmoc-Leu, Fmoc-D-Asn (Trt), Fmoc-N-Me-Tyr (tBu), Fmoc-Ser (tBu), Fmoc-D-Pal, Fmoc-D-Cpa and Ac-D-Nal are sequentially added in the same method, and the Abarelix peptide resin, Ac-D-Nal-D-Cpa-D-Pal-Ser (tBu) -N-Me-Tyr (tBu) -D-Asn (Trt) -Leu-Lys (iPr, Z) -Pro-D-Ala-MOBHA resin are obtained by washing and filtering.
Example 5: preparation of crude Abarelix
Taking the abarelix peptide resin prepared in the example 3, adding 8% HBr/TFA solution (acidolysis solution 10mL/g abarelix resin), stirring and reacting for 6 hours, filtering and collecting filtrate, washing the resin with a small amount of TFA for 3 times, combining the filtrates, concentrating under reduced pressure, adding anhydrous ether for precipitation, washing the precipitate with anhydrous ether for 3 times, and draining to obtain white-like powder, namely a crude product of abarelix, wherein the purity of the crude product is 79.3%.
Example 6: preparation of crude Abarelix
Taking the abarelix peptide resin prepared in the example 4, adding 8% HBr/TFA solution (acidolysis solution 10mL/g abarelix resin), stirring and reacting for 6 hours, filtering and collecting filtrate, washing the resin with a small amount of TFA for 3 times, combining the filtrates, concentrating under reduced pressure, adding anhydrous ether for precipitation, washing the precipitate with anhydrous ether for 3 times, and draining to obtain white-like powder, namely a crude product of abarelix, wherein the purity of the crude product is 77.4%.
Example 7: purification and trans-salt conversion of crude Abarelix
Taking the crude Abarelix product obtained in the example 5, dissolving the Abarelix product in 20 percent acetic acid solution, filtering the solution by using a 0.45 mu m microporous membrane, and purifying for later use;
purifying by high performance liquid chromatography, wherein a chromatographic filler is 10 mu m reverse phase C18, a mobile phase system is 0.1% TFA/water solution-0.1% TFA/acetonitrile solution, a chromatographic column with the flow rate of 77mm x 250mm is 90mL/min, eluting by a gradient system, circularly sampling and purifying, sampling a crude product solution in the chromatographic column, starting the mobile phase for elution, collecting a main peak, and evaporating acetonitrile to obtain an abarelix purified intermediate concentrated solution;
taking the Abarelix purified intermediate concentrated solution, and filtering with a 0.45-micrometer filter membrane for later use;
performing salt exchange by high performance liquid chromatography, wherein the mobile phase system is 1% acetic acid/water solution-acetonitrile, the purification is performed by reversed phase C18 with chromatographic packing of 10 μm, the flow rate of a chromatographic column of 77mm × 250mm is 90mL/min, gradient elution and circular sample loading method are adopted, the sample is loaded in the chromatographic column, the mobile phase elution is started, the chromatogram is collected, the change of the absorbance is observed, the main peak of salt exchange is collected and the purity is detected by analyzing the liquid phase, the main peak solutions of salt exchange are combined, the concentration is performed under reduced pressure to obtain the aqueous solution of abarelix acetic acid, and freeze drying is performed to obtain 39.4g abarelix pure product
The total yield was 55.6%, molecular weight: 1417.2, purity: 99.6%, maximum single impurity of 0.13%, no toxic hydantoin degradation products were detected.
Example 8: purification and trans-salt conversion of crude Abarelix
Taking the crude Abarelix product obtained in the example 6, dissolving the Abarelix product by using a purification mobile phase A, and filtering the solution by using a 0.45 mu m microporous filter membrane to purify the Abarelix product for later use;
purifying by high performance liquid chromatography, wherein a chromatographic filler is 10 mu m reverse phase C18, a mobile phase system is 0.1% TFA/water solution-0.1% TFA/acetonitrile solution, a chromatographic column with the flow rate of 77mm x 250mm is 90mL/min, eluting by a gradient system, circularly sampling and purifying, sampling a crude product solution in the chromatographic column, starting the mobile phase for elution, collecting a main peak, and evaporating acetonitrile to obtain an abarelix purified intermediate concentrated solution;
taking the Abarelix purified intermediate concentrated solution, and filtering with a 0.45-micrometer filter membrane for later use;
performing salt exchange by adopting a high performance liquid chromatography, wherein a mobile phase system is 1% acetic acid/water solution-acetonitrile, a chromatographic filler for purification is reversed phase C18 with the diameter of 10 mu m, the flow rate of a chromatographic column with the diameter of 77mm × 250mm is 90mL/min, a gradient elution method and a circular sample loading method are adopted, loading the chromatographic column, starting the mobile phase elution, collecting a spectrum, observing the change of the absorbance, collecting a main salt exchange peak, detecting the purity by using an analysis liquid phase, combining main salt exchange peak solutions, concentrating under reduced pressure to obtain an abarelix acetic acid water solution, and performing freeze drying to obtain 41.7g of an abarelix pure product.
The total yield is 58.9%, molecular weight: 1417.0, purity: 99.5%, maximum single impurity 0.09%, no toxic hydantoin degradation products were detected.
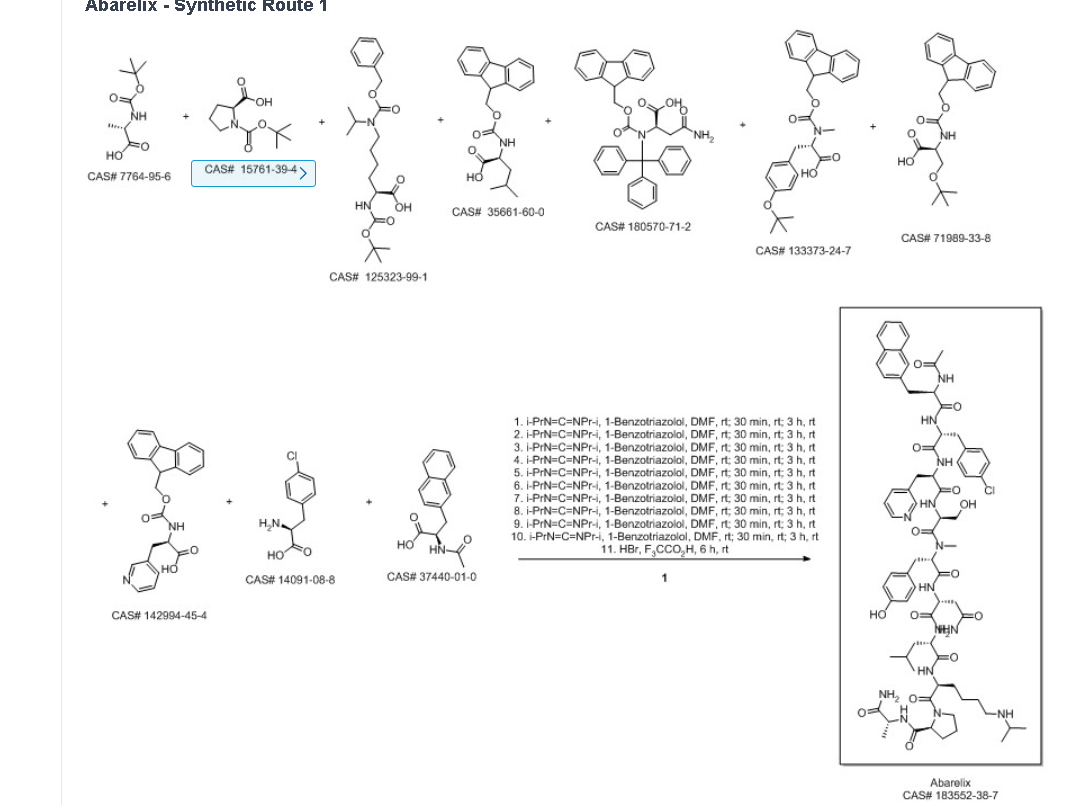
SYN
Ma, Zhonggang; Guo, Dewen; Zeng, Dezhi; Wen, Yongjun. Method for synthesizing abarelix. Assignee Chengdu Shengnuo Biopharm Co., Ltd.. 2018.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
1: Tombal B. New treatment paradigm for prostate cancer: abarelix initiation therapy for immediate testosterone suppression followed by a luteinizing hormone-releasing hormone agonist. BJU Int. 2012 Mar;109(6):E16; author reply E16-7. doi: 10.1111/j.1464-410X.2012.10983.x. PubMed PMID: 22360806.
2: Garnick MB, Mottet N. New treatment paradigm for prostate cancer: abarelix initiation therapy for immediate testosterone suppression followed by a luteinizing hormone-releasing hormone agonist. BJU Int. 2012 Aug;110(4):499-504. doi: 10.1111/j.1464-410X.2011.10708.x. Epub 2011 Nov 16. PubMed PMID: 22093775.
3: Koechling W, Hjortkjaer R, Tankó LB. Degarelix, a novel GnRH antagonist, causes minimal histamine release compared with cetrorelix, abarelix and ganirelix in an ex vivo model of human skin samples. Br J Clin Pharmacol. 2010 Oct;70(4):580-7. doi: 10.1111/j.1365-2125.2010.03730.x. PubMed PMID: 20840449; PubMed Central PMCID: PMC2950992.
4: Retraction statement: Reconstitution of Plenaxis® (Abarelix) 100 mg for injection is more effective with a vortex-like mixer than when performed manually. J Pharm Pract. 2010 Feb;23(1):78. doi: 10.1177/0897190009360369. PubMed PMID: 21507797.
5: Kirby RS, Fitzpatrick JM, Clarke N. Abarelix and other gonadotrophin-releasing hormone antagonists in prostate cancer. BJU Int. 2009 Dec;104(11):1580-4. doi: 10.1111/j.1464-410X.2009.08924.x. Review. PubMed PMID: 20053189.
6: Debruyne F, Bhat G, Garnick MB. Abarelix for injectable suspension: first-in-class gonadotropin-releasing hormone antagonist for prostate cancer. Future Oncol. 2006 Dec;2(6):677-96. Review. PubMed PMID: 17155895.
7: Beer TM, Ryan C, Bhat G, Garnick M; Abarelix Study Group. Dose-escalated abarelix in androgen-independent prostate cancer: a phase I study. Anticancer Drugs. 2006 Oct;17(9):1075-9. PubMed PMID: 17001181.
8: Hogle WP. Abarelix (plenaxis). Clin J Oncol Nurs. 2004 Dec;8(6):663-5. PubMed PMID: 15637961.
9: Mongiat-Artus P, Teillac P. Abarelix: the first gonadotrophin-releasing hormone antagonist for the treatment of prostate cancer. Expert Opin Pharmacother. 2004 Oct;5(10):2171-9. Review. PubMed PMID: 15461552.
10: Wong SL, Lau DT, Baughman SA, Fotheringham N, Menchaca D, Garnick MB. Pharmacokinetics and pharmacodynamics of a novel depot formulation of abarelix, a gonadotropin-releasing hormone (GnRH) antagonist, in healthy men ages 50 to 75. J Clin Pharmacol. 2004 May;44(5):495-502. PubMed PMID: 15102870.
References
- “Abarelix”. PubChem. 2017-07-29.
- “Abarelix”. Drugs.com. Archived from the original on 2018-02-10. Retrieved 2018-01-23.
- Boccon-Gibod L, van der Meulen E, Persson BE (June 2011). “An update on the use of gonadotropin-releasing hormone antagonists in prostate cancer”. Therapeutic Advances in Urology. 3 (3): 127–40. doi:10.1177/1756287211414457. PMC 3159401. PMID 21904569.
- Pharmazeutische Zeitung online: Abarelix (in German)
- Minev B (13 January 2011). Cancer Management in Man: Chemotherapy, Biological Therapy, Hyperthermia and Supporting Measures. Springer Science & Business Media. pp. 182–. ISBN 978-90-481-9704-0.
- “Abarelix”. Drugs.com. Archived from the original on 2019-08-29. Retrieved 2018-08-27.
| Clinical data | |
|---|---|
| Trade names | Plenaxis |
| AHFS/Drugs.com | Monograph |
| Routes of administration | Intramuscular injection |
| Drug class | GnRH analogue; GnRH antagonist; Antigonadotropin |
| ATC code | L02BX01 (WHO) |
| Pharmacokinetic data | |
| Protein binding | 96–99% |
| Identifiers | |
| IUPAC name | |
| CAS Number | 183552-38-7 |
| PubChem CID | 16131215 |
| IUPHAR/BPS | 1188 |
| DrugBank | DB00106 |
| ChemSpider | 10482301 |
| UNII | W486SJ5824 |
| KEGG | D02738 |
| ChEBI | CHEBI:337298 |
| ChEMBL | ChEMBL1252 |
| CompTox Dashboard (EPA) | DTXSID20171443 |
| Chemical and physical data | |
| Formula | C72H95ClN14O14 |
| Molar mass | 1416.09 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
//////Abarelix, PPI149, PPI-149, PPI 149, R3827, R-3827, R 3827, Abarelix, Abarelix acetate, Plenaxis,
W486SJ5824
O=C(N[C@@H](CC(C)C)C(N[C@@H](CCCCNC(C)C)C(N1[C@H](C(N[C@H](C)C(N)=O)=O)CCC1)=O)=O)[C@H](NC([C@@H](N(C([C@@H](NC([C@H](NC([C@H](NC([C@H](NC(C)=O)CC2=CC=C3C=CC=CC3=C2)=O)CC4=CC=C(Cl)C=C4)=O)CC5=CC=CN=C5)=O)CO)=O)C)CC6=CC=C(O)C=C6)=O)CC(N)=O
Tagtociclib
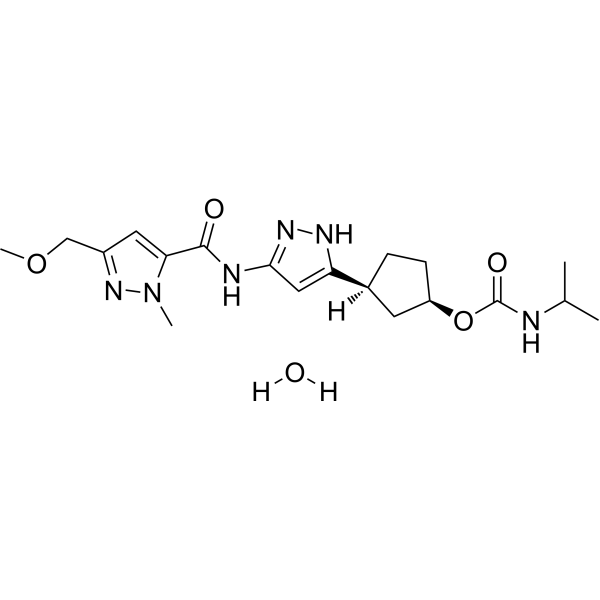
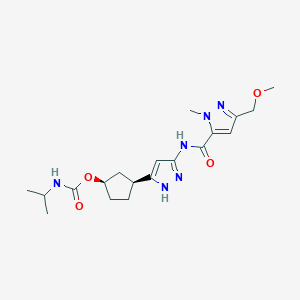
Tagtociclib (PF-07104091), 2460249-19-6, MW 404.5, C19H28N6O4
CAS 2733575-91-0 HYDRATE
| Molecular Weight HYDRATE | 422.48 |
|---|---|
| Formula | C19H30N6O5 |
[(1R,3S)-3-[3-[[5-(methoxymethyl)-2-methylpyrazole-3-carbonyl]amino]-1H-pyrazol-5-yl]cyclopentyl] N-propan-2-ylcarbamate
- (1R,3S)-3-[5-[[[3-(Methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl]amino]-1H-pyrazol-3-yl]cyclopentyl N-(1-methylethyl)carbamate
- (1R,3S)-3-{5-[3-(methoxymethyl)-1-methyl-1H-pyrazole-5carboxamido]-1H-pyrazol-3-yl}cyclopentyl (propan-2yl)carbamate
- (1R,3S)-3-(3-(3-(Methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamido)-1H-pyrazol-5-yl)cyclopentylisopropylcarbamate
- Carbamic acid, N-(1-methylethyl)-, (1R,3S)-3-[5-[[[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl]amino]-1H-pyrazol-3-yl]cyclopentyl ester
PF-07104091 hydrate is a potent and selective CDK2/cyclin E1 and GSK3β inhibitor, with Kis of 1.16 and 537.81 nM, respectively. PF-07104091 hydrate has anti-tumor activity for cyclin E1-amplified cancers. (patent WO2020157652A2).
- OriginatorPfizer
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase 2 inhibitors
Phase IIBreast cancer; Solid tumours
Phase I/IINon-small cell lung cancer; Ovarian cancer; Small cell lung cancer
13 Sep 2024Efficacy, adverse events, pkarmacokinetics and pharmacodynamics data from a phase I/II trial in Solid tumours presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
13 Sep 2024Pharmacodynamics data from a preclinical trial in Breast cancer presented at the 49th European Society for Medical Oncology Congress (ESMO-2024)
05 Apr 2024Pharmacodynamics data form preclinical trial in Breast cancer and Ovarian cancer presented at the 115th Annual Meeting of the American Association for Cancer Research (AACR-2024)
Tegtociclib is an orally bioavailable inhibitor of cyclin-dependent kinase 2 (CDK2), with potential antineoplastic activity. Upon administration, tegtociclib selectively targets, binds to and inhibits the activity of CDK2. This may lead to cell cycle arrest, the induction of apoptosis, and the inhibition of tumor cell proliferation. CDKs are serine/threonine kinases that are important regulators of cell cycle progression and cellular proliferation and are frequently overexpressed in tumor cells. CDK2/cyclin E complex plays an important role in retinoblastoma (Rb) protein phosphorylation and the G1-S phase cell cycle transition. CDK2/cyclin A complex plays an important role in DNA synthesis in S phase and the activation of CDK1/cyclin B for the G2-M phase cell cycle transition.
SCHEME
COUPLER
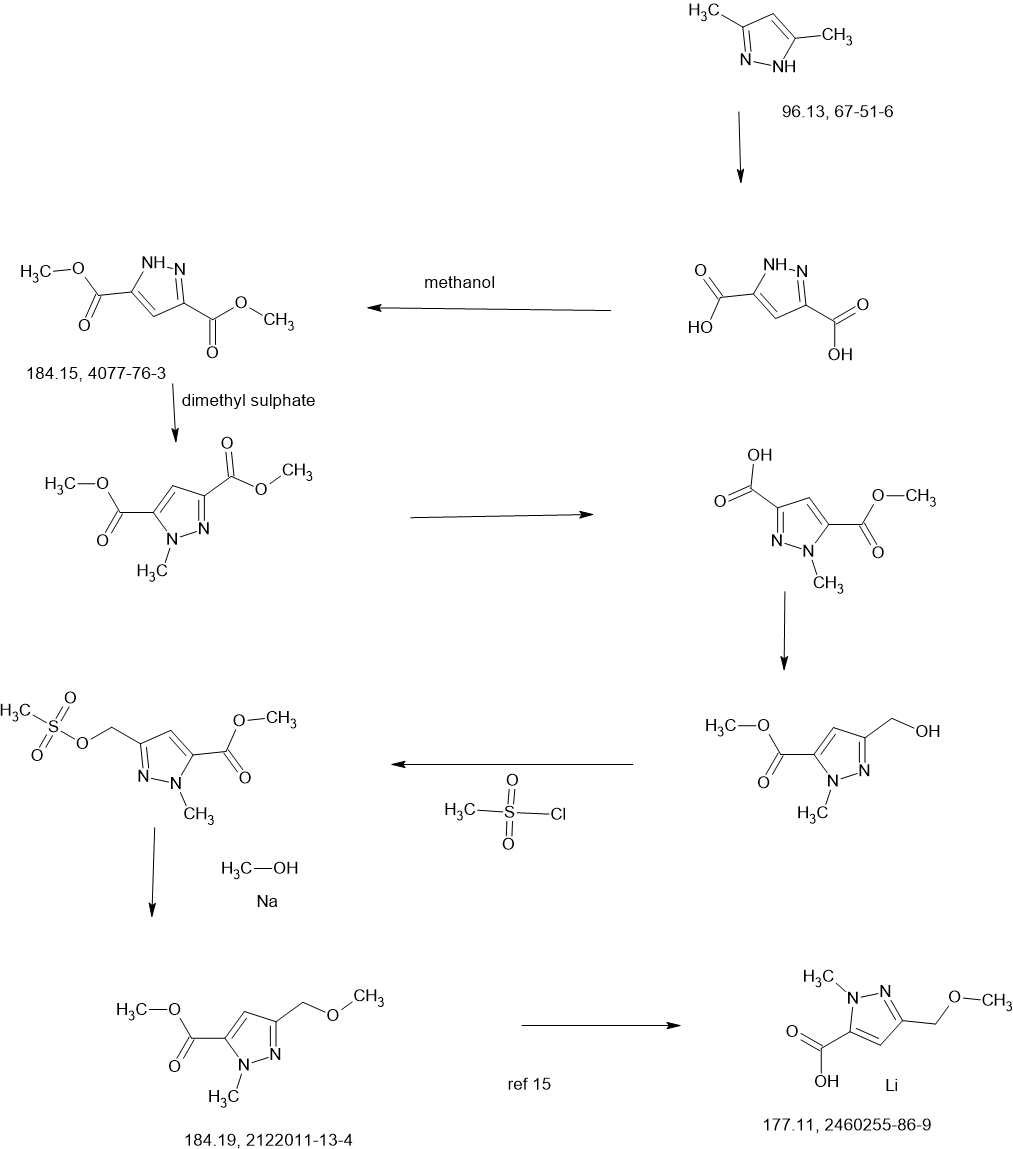
MAIN
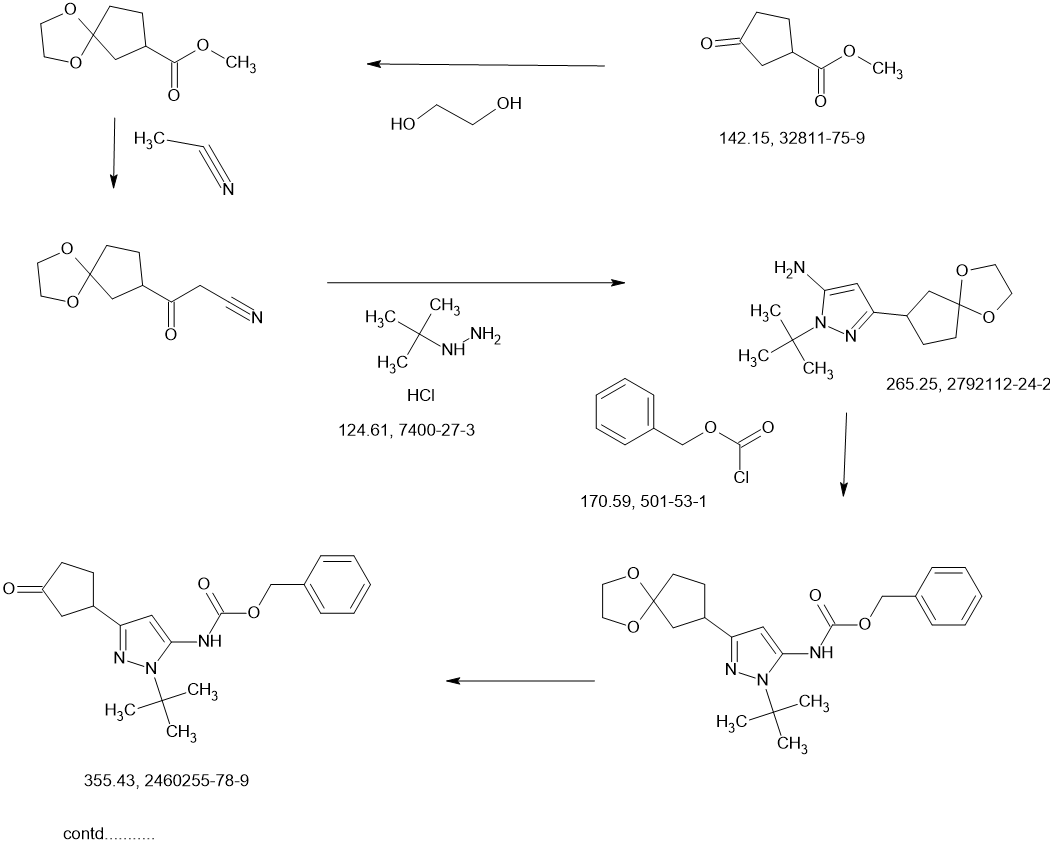
CONTD………….
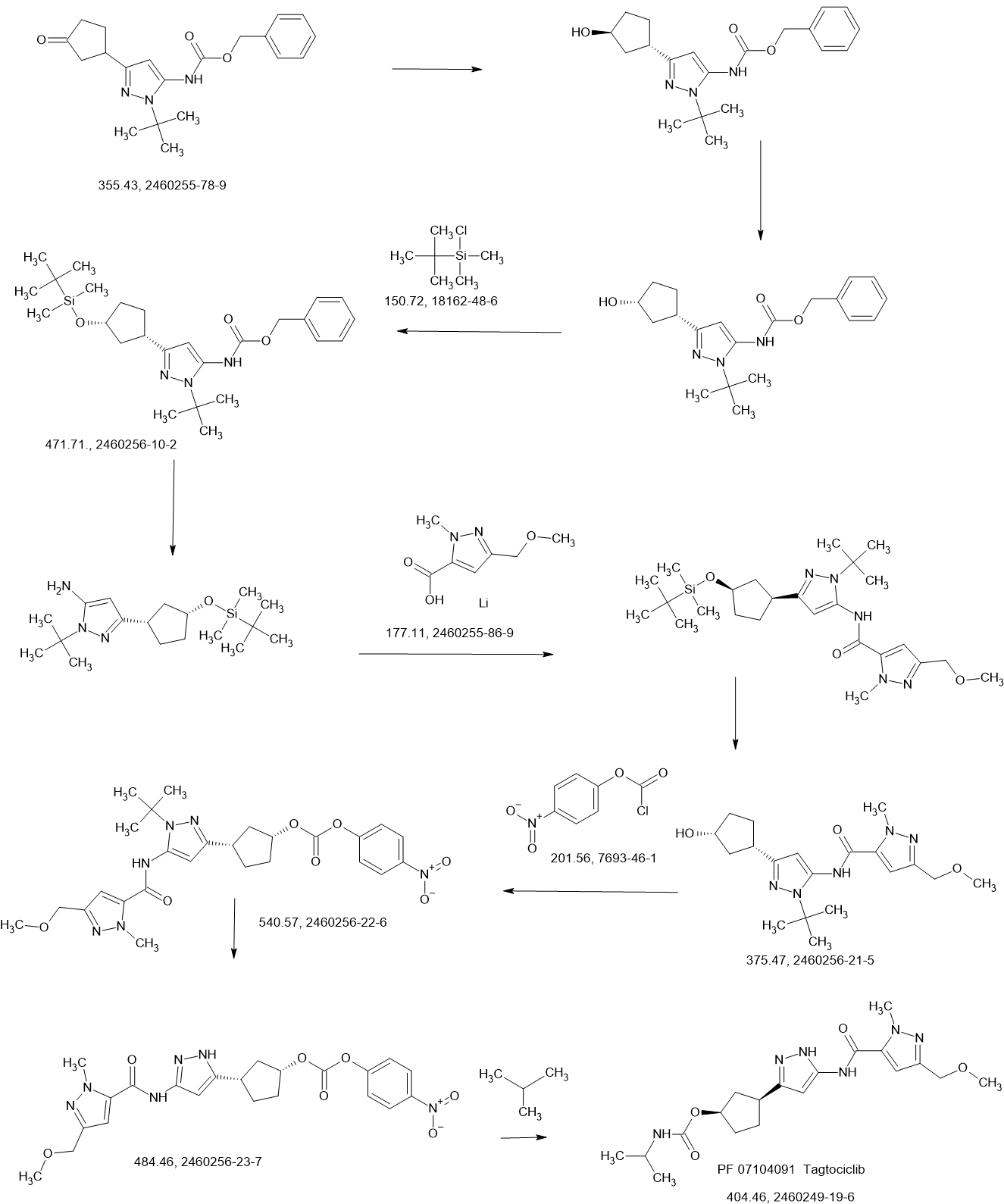
PATENTS
WO2022018596 78%
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022018596&_cid=P22-MDFCVG-44044-1
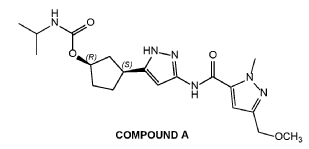
COMPOUND A was prepared as described in Example 13 of U.S. Patent No.
11,014,911.
Preparation of Intermediate 1: benzyl {1-tert-butyl-3-[(1S,3R)-3-hvdroxycvclopentyl]1H-pyrazol-5-yl)carbamate; and Intermediate 2: benzyl {1-tert-butyl-3-[(1R,3S)-3-hydroxycvclopentyl1-1H-pyrazol-5-yl)carbamate.
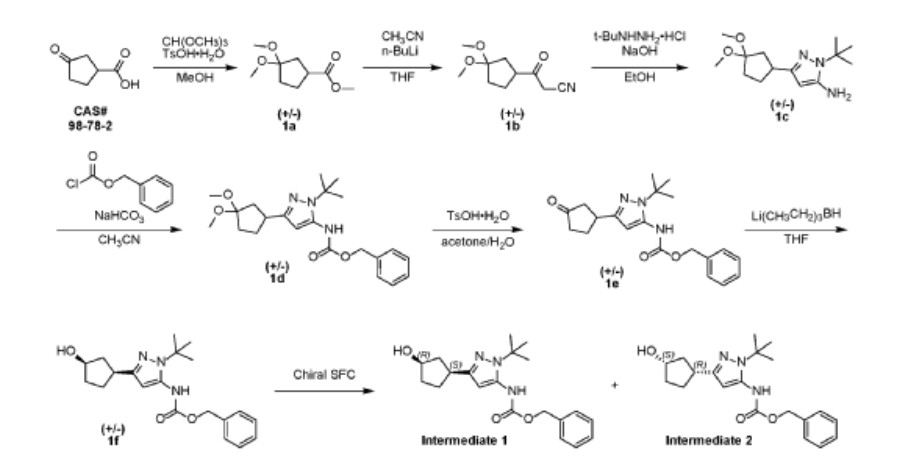
Two parallel reactions, each containing a solution of (±)-3- oxocyclopentanecarboxylic acid (CAS#98-78-2, 900 g, 7.02 mol) in methanol (5 L) at 13 °C were each treated with trimethyl orthoformate (4.47 kg, 42.15 mol, 4.62 L) and 4- toluenesulfonic acid monohydrate (26.72 g, 140.5 mmol). The mixtures were stirred at 13 °C for 25 hours. Each batch was quenched separately with sat. aq NaHCO3 (1 L), then the two batches were combined and concentrated under vacuum to remove most of the methanol. The residue was diluted with ethyl acetate (4 L), and the layers separated. The aqueous layer was further extracted with ethyl acetate (2 x 1 L). The combined organic layers were washed with sat. aq NaCI (3 x 1 L), dried over magnesium sulfate, filtered, and concentrated under vacuum to give (±)-methyl 3,3- dimethoxycyclopentanecarboxylate (1a, 2.5 kg, 13.28 mol, 94%) as a light yellow oil. 1H NMR (400MHz, CHLOROFORM -d) δ = 3.67 (s, 3H), 3.20 (s, 3H), 3.19 (s, 3H), 2.94- 2.82 (m, 1 H), 2.16-2.00 (m, 2H), 1.99-1.76 (m, 4H).
A solution of n-butyllithium (3.44 L of a 2.5 M solution in hexanes, 8.6 mol) was added to a reactor containing THF (3 L) at -65 °C. Anhydrous acetonitrile (453 mL, 353 g, 8.61 mol) was added dropwise, slowly enough to maintain the internal temperature below -55 °C. The mixture was stirred for an additional 1 hour at -65 °C. A solution of (±)-methyl 3,3-dimethoxycyclopentanecarboxylate (1a, 810 g, 4.30 mol) in THF (1 L) was then added dropwise, slowly enough to maintain the internal temperature below -50 °C. After stirring for an additional hour at -65 °C, the reaction was quenched with water (4 L), neutralized with aq HCI (1 M) to pH 7, and extracted with ethyl acetate (3 x 3L). The combined organic layers were washed with sat. aq NaCI (2 x 3L), dried over magnesium sulfate, filtered, and concentrated under vacuum to give crude (±)-3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile (1b, 722 g, 3.66 mol, 85%) as a red oil, which was used without further purification.
Solid sodium hydroxide (131.4 g, 3.29 mol total) was added in portions to a suspension of tert-butylhydrazine hydrochloride (409.4 g, 3.29 mol) in ethanol (3 L) at 16-25 °C. Stirring was continued at 25 °C for 1 hour. A solution of crude (±)-3-(3,3-dimethoxycyclopentyl)-3-oxopropanenitrile (1b, 540 g, 2.74 mol) in ethanol was added at 25 °C, then the mixture was heated to 75 °C internal and stirred for 30 hours. The reaction was filtered, and the filtrate concentrated under vacuum to give crude product as a red oil. This product was combined with crude from three more identically-prepared batches (each starting with 540 g 1b; 2.16 kg, 10.96 mol total for the 4 batches), and purified by silica gel chromatography (eluting with 0-35% ethyl acetate in petroleum ether), affording (±)-1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-amine (1c, 1.60 kg, 5.98 mol, 54% yield) as a red oil. 1H NMR (CHLOROFORM -d) δ = 5.41 (s, 1 H), 3.50 (br. s., 2H), 3.22 (s, 3H), 3.20 (s, 3H), 3.13 (tt, J=7.9, 9.6 Hz, 1H), 2.25 (dd, J=8.0, 13.3 Hz, 1H), 2.09-2.00 (m, 1H), 1.99-1.91 (m, 1H), 1.83 (dd, J=10.8, 12.8 Hz, 2H), 1.78-1.68 (m, 1H), 1.60 (s, 9H).
Benzyl chloroformate (563.6 mL, 676.3 g, 3.96 mol) was added to a chilled (0-5 °C) solution of (±)-1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-amine (1c, 530 g, 1.98 mol) in acetonitrile (3.5 L). The mixture was stirred at 23 °C for 2 hours, and then solid sodium hydrogen carbonate (532.9 g, 6.34 mol) was added in portions. Stirring was continued at 23 °C for 26 hours. The resulting suspension was filtered and the filtrate concentrated under vacuum to give crude (±)-benzyl [1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 d, 980 g, 1.98 mol max) as a red oil, which was used in the next step without further purification.
A solution of the crude (±)-benzyl [1-tert-butyl-3-(3,3-dimethoxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 d, 980 g, 1.98 mol max) in acetone (2 L) and water (2 L) at 18 °C was treated with 4-toluenesulfonic acid monohydrate (48.75 g, 256.3 mmol). The mixture was heated to 60 °C internal for 20 hours. After concentration under vacuum to remove most of the acetone, the aqueous residue was extracted with dichloromethane (3 x 3 L). The combined organic extracts were dried over sodium sulfate, filtered, and concentrated under vacuum to a crude red oil. This crude product was combined with crude from two other identically-prepared batches (each derived from 1.98 mol 1c, 5.94 mol total for the 3 batches), and purified by silica gel chromatography (eluting with 0- 50% ethyl acetate in petroleum ether) to give (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 1.6 kg) as a yellow solid. This solid was stirred in 10:1 petroleum ether/ethyl acetate (1.5 L) at 20 °C for 18 hours. The resulting suspension was filtered, the filter cake washed with petroleum ether ( 2 x 500 mL), and the solids dried under vacuum to give (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 1.4 kg, 3.9 mol, 66% combined for the three batches). 1H NMR (DMSO–d6) δ = 9.12 (br. s., 1H), 7.56-7.13 (m, 5H), 6.03 (s, 1 H), 5.12 (s, 2H), 3.41-3.27 (m, 1H), 2.48-2.39 (m, 1H), 2.34-2.10 (m, 4H), 1.98-1.81 (m, 1 H), 1.48 (s, 9H).
A solution of (±)-benzyl [1-tert-butyl-3-(3-oxocyclopentyl)-1H-pyrazol-5-yl]carbamate (1 e, 320 g, 0.900 mol) in THF (1.5 L) was degassed under vacuum and purged with dry nitrogen (3 cycles), then cooled to -65 °C internal. A solution of lithium triethylborohydride (1.0 M in THF, 1.80 L, 1.80 mol) was added dropwise at a rate which maintained the internal temperature below -55 °C, then stirring was continued at -65 °C for 1.5 hours. The reaction mixture was quenched with sat. aq NaHCO3 (1.5 L) at -40 to -30 °C. Hydrogen peroxide (30% aqueous, 700 g) was added to the mixture dropwise, while the internal temperature was maintained at -10 to 0 °C. The mixture was stirred at 10 °C for 1 hour, then extracted with ethyl acetate (3 x 2 L). The combined organic layers were washed with sat. aq Na2SO3 (2 x 1 L) and sat. aq NaCI (2 x 1 L). The organics were dried over magnesium sulfate, filtered, and concentrated under vacuum to a crude yellow oil. The crude product from this batch was combined with crude from three other, identically-prepared batches (each starting from 0.900 mol 1 e, for a total of 3.60 mol) for purification. Before chromatography, the combined mixture showed ~3.3:1 cis/trans ratio by NMR. The combined crude product was purified twice by silica gel chromatography, eluting with 0-50% ethyl acetate in dichloromethane), affording (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 960 g) as a light yellow solid, which was further purified by trituration, as described below.
A previous batch of 1f had been obtained from smaller-scale reactions, starting from a total of 120 g 1e (0.34 mol). The columned product from this batch was combined with the columned product from the batch above (which had been derived from 3.60 mol 1 e, for a total of 3.94 mol 1e used for all the combined batches), suspended in 10:1 dichloromethane/methanol (1.5 L), and stirred at 20 °C for 16 hours. The suspension was filtered, and the filter cake washed with petroleum ether (2 x 500 mL). The solids were dried under vacuum to give clean (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 840 g, 2.35 mol, 60% total yield for all the combined batches) as a white solid. 1H NMR (400MHz, DMSO-d6) δ = 9.07 (br. s., 1 H), 7.45-7.27 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.5 Hz, 1 H), 4.21-4.07 (m, 1 H), 2.88 (quin, J=8.6 Hz, 1 H), 2.24-2.13 (m, 1 H), 1.92-1.78 (m, 1 H), 1.78-1.62 (m, 2H), 1.61-1.53 (m, 1 H), 1.47 (s, 9H), 1.52-1.43 (m, 1 H). MS: 358 [M+H]+.
The enantiomers of (±)-trans-benzyl [1-tert-butyl-3-(3-hydroxycyclopentyl)-1H-pyrazol-5-yl]carbamate (1 f, 700 g, 1.96 mol) were separated by chiral SFC.
The product from the first-eluting enantiomer peak (310 g solid) was suspended in methanol/petroleum ether (1 :10, 1 L) and stirred at 25 °C for 1 hour. The suspension was filtered, the filter pad washed with petroleum ether (2 x 500 mL), and the solids dried under vacuum to give benzyl {1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}carbamate (Intermediate 1 , 255 g, 713 mmol, 36%, >99% ee) as a white solid. 1H NMR (400MHz, DMSO -d6) δ = 9.08 (br. s., 1 H), 7.58-7.20 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.4 Hz, 1 H), 4.19-4.09 (m, 1 H), 2.88 (quin, J=8.6 Hz, 1 H), 2.24-2.13 (m, 1 H), 1.91-1.79 (m, 1 H), 1.79-1.61 (m, 2H), 1.61-1.53 (m, 1 H), 1.47 (s, 9H), 1.52-1.44 (m, 1 H). MS: 358 [M+H]+. Optical rotation [α]D +3.76 (c 1.0, MeOH). Chiral purity: >99% ee, retention time 3.371 min. Chiral SFC analysis was performed on a ChiralPak AD-3 150 x 4.6 mm ID, 3 pm column heated to 40 °C, eluted with a mobile phase of CO2 and a gradient of 0-40% methanol+0.05%DEA over 5.5 min, then held at 40% for 3 min; flowing at 2.5 mL/min.
The product from the second-eluting enantiomer peak (300 g solid) was suspended in methanol/petroleum ether (1 :10, 1 L) and stirred at 25 °C for 1 hour. The suspension was filtered, the filter pad washed with petroleum ether (2 x 500 mL), and the solids dried under vacuum to give benzyl {1-tert-butyl-3-[(1R,3S)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}carbamate (Intermediate 2, 255 g, 713 mmol, 36%, 94% ee) as a white solid. 1H NMR (400MHz, DMSO-d6) δ = 9.08 (br. s., 1 H), 7.55-7.19 (m, 5H), 5.92 (s, 1 H), 5.11 (s, 2H), 4.57 (d, J=4.4 Hz, 1 H), 4.23-4.07 (m, 1 H), 2.88 (quin, J=8.7 Hz, 1 H), 2.23-2.14 (m, 1 H), 1.90-1.79 (m, 1 H), 1.77-1.61 (m, 2H), 1.61-1.53 (m, 1 H), 1 .47 (s, 9H), 1.52-1 .44 (m, 1 H). MS: 358 [M+H]+. Optical rotation [α]D -2.43 (c 1 .0, MeOH). Chiral purity: 94% ee, retention time 3.608 min. Chiral SFC analysis was performed on a ChiralPak AD-3 150 x 4.6 mm ID, 3 pm column heated to 40 °C, eluted with a mobile phase of CO2 and a gradient of 0-40% methanol+0.05%DEA over 5.5 min, then held at 40% for 3 min; flowing at 2.5 mL/min.
A sample of the second-eluting enantiomer from a previous batch with [α]D -3.1 (c 1.1, MeOH) and 96% ee was crystalized from dichloroethane/pentane. A crystal structure was obtained by small-molecule X-ray crystallography, which showed (1R,3S) geometry. The absolute stereochemistry of Intermediate 2 was thus assigned (1R,3S) based on its comparable optical rotation and order of elution in the analytical method. Intermediate 1, the enantiomer of Intermediate 2, was thus assigned (1S,3R) stereochemistry.
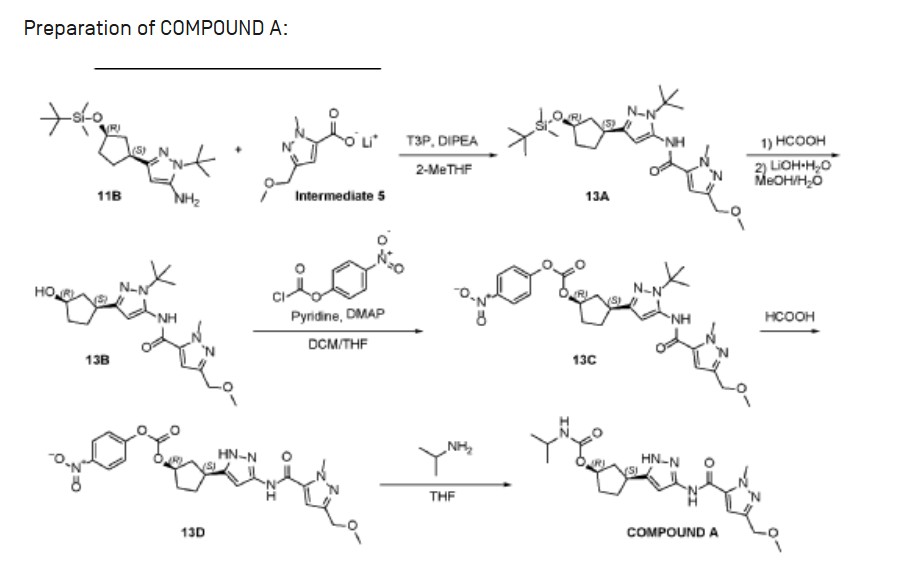
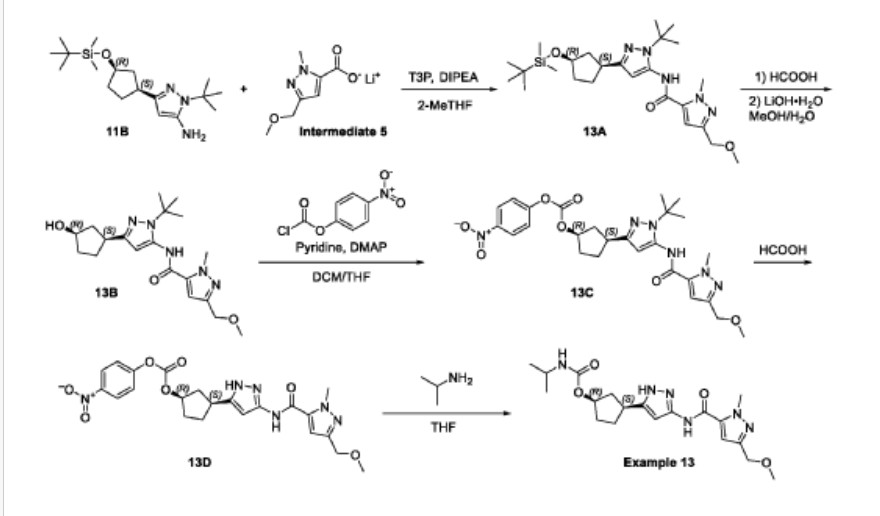
Propylphosphonic anhydride (T3P®, 50 wt% solution in EtOAc, 50.3 g, 79.1 mmol) was added to a room temperature (26 °C) solution of 1-tert-butyl-3-[(1S,3R)-3-{[tert-butyl(dimethyl)silyl]oxy}cyclopentyl]-1H-pyrazol-5-amine (11 B, 8.90g, 26.4 mmol), lithium 3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxylate (Intermediate 5, 5.83 g,
34.3 mmol), and diisopropylethyl amine (10.2 g, 79.1 mmol) in 2-methyltetrahydrofuran (100.0 mL). The resulting mixture was stirred at this temperature for 18 hours. After concentrating the mixture to dryness, the residue was dissolved in dichloromethane (150 mL), and the solution washed sequentially with water (2 x 30 mL), sat. aq NaHCO3 (2 x 30 mL) and sat. aq NaCI (30 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated to give crude N-{1-tert-butyl-3-[(1S,3R)-3-{[tert- butyl(dimethyl)silyl]oxy}cyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H- pyrazole-5-carboxamide (13A, 12.9 g, 100%) as an oil. MS: 490 [M+H]+.
The crude N-{1-tert-butyl-3-[(1S,3R)-3-{[tert-butyl(dimethyl)silyl]oxy}cyclopentyl]- 1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13A, 12.9 g,
26.3 mmol) was dissolved in formic acid (80 mL) and stirred at room temperature (27 °C) for 30 minutes. The mixture was concentrated to dryness, and the residue
dissolved in methanol (80 mL). A solution of lithium hydroxide monohydrate (3.43 g, 81.8 mmol) in water (15 mL) was added, and the mixture stirred at room temperature (27 °C) for 1 hour. The mixture was concentrated to dryness, and the residue was purified by silica gel chromatography (eluting with 0-80% ethyl acetate in petroleum ether) to give N-{1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13B, 8.0 g, 78%) as a yellow gum. MS: 376 [M+H]+.
A solution of N-{1-tert-butyl-3-[(1S,3R)-3-hydroxycyclopentyl]-1H-pyrazol-5-yl}-3-(methoxymethyl)-1-methyl-1H-pyrazole-5-carboxamide (13B, 8.0 g, 21 mmol) in dichloromethane (80 mL) and THF (80 mL) was treated with DMAP (260 mg, 2.13 mmol), pyridine (5.06 g, 63.9 mmol), and 4-nitrophenyl chloroformate (8.59 g, 42.6 mmol). The resulting yellow suspension was stirred at room temperature for 18 hours. The reaction mixture was concentrated to dryness and purified by silica gel chromatography (eluting with 0-45% ethyl acetate in petroleum ether) to give (1R,3S)-3-[1-tert-butyl-5-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-3-yl]cyclopentyl 4-nitrophenyl carbonate (13C, 10.6 g, 92%) as a light brown gum. MS: 541 [M+H]+.
A solution of (1R,3S)-3-[1-tert-butyl-5-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-3-yl]cyclopentyl 4-nitrophenyl carbonate (13C, 10.6 g, 19.6 mmol) in formic acid (80 mL) was stirred at 70 °C for 18 hours. The solution was concentrated to dryness. The residue was dissolved in dichloromethane (150 mL) and the solution neutralized with sat. aq NaHCO3. The organic layer was washed with water (30 mL) and sat. aq NaCI (30 mL), dried over sodium carbonate, filtered, and concentrated to give crude (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl 4-nitrophenyl carbonate (13D, 8.5 g, 90%, 86% pure by LCMS) as a light yellow glass. MS: 485 [M+H]+.
A room temperature (27 °C) solution of crude (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl 4-nitrophenyl carbonate (13D, 1.7 g, 3.5 mmol) and 2-propylamine (1.04 g, 17.5 mmol) in THF (30 mL) was stirred for 6 hours. The solution was concentrated to dryness, and the residue was combined with the residue from a second batch which had been derived from 1.7 g, 3.5 mmol 13D (total 6.27 mmol 13D consumed for the combined two batches) to give 3.2 g crude product. This product was purified by preparative HPLC on a Phenomenex Gemini C18 250*50mm*10 pm column, eluting with 15-45% water (0.05% ammonium
hydroxide v/v) in acetonitrile. After lyophilization, (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl propan-2 -ylcarbamate (COMPOUND A, 2.06 g, 78%) was obtained as a white crystalline solid monohydrate. MS: 405 [M+H]+. 1H NMR (400MHz, DMSO-d6) d = 12.23 (br s, 1H), 10.73 (br s, 1H), 7.11 (s, 1H), 6.96 (br d, J=7.0 Hz, 1H), 6.41 (br s, 1H), 5.00 (br s, 1H), 4.33 (s, 2H), 4.04 (s, 3H), 3.57 (qd, J=6.6, 13.4 Hz, 1H), 3.26 (s, 3H), 3.17-2.96 (m, 1H), 2.48-2.39 (m, 1H), 2.03 (br d, J=6.8 Hz, 1H), 1.95-1.83 (m, 1H), 1.73 (br d, J=8.5 Hz, 2H), 1.61 (br s, 1 H), 1.03 (br d, J=6.3 Hz, 6H). Optical rotation [α]D +4.8 (c 1.0, MeOH). Chiral purity: >99% ee by chiral analytical SFC. Anal. Calcd for C19H28N6O4-H2O: C, 54.02; H, 7.16; N, 19.89. Found: C, 53.94; H, 7.22; N, 19.81.
PATENT
WO2020157652 EX 13
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020157652&_cid=P22-MDFD2U-50269-1
Example 13: (1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}-amino)-1H-pyrazol-5-yl]cyclopentyl propan-2-ylcarbamate
(1R,3S)-3-[3-({[3-(methoxymethyl)-1-methyl-1H-pyrazol-5-yl]carbonyl}amino)-1H-pyrazol-5-yl]cyclopentyl propan-2-ylcarbamate (Example 13, 2.06 g, 78%) was obtained as a white crystalline solid found to be a monohydrate (Form 1) based on elemental analysis. MS: 405 [M+H]+.1H NMR (400MHz, DMSO-d6) d = 12.23 (br s, 1H), 10.73 (br s, 1H), 7.11 (s, 1H), 6.96 (br d, J=7.0 Hz, 1H), 6.41 (br s, 1H), 5.00 (br s, 1H), 4.33 (s, 2H), 4.04 (s, 3H), 3.57 (qd, J=6.6, 13.4 Hz, 1H), 3.26 (s, 3H), 3.17-2.96 (m, 1H), 2.48-2.39 (m, 1H), 2.03 (br d, J=6.8 Hz, 1H), 1.95-1.83 (m, 1H), 1.73 (br d, J=8.5 Hz, 2H), 1.61 (br s, 1H), 1.03 (br d, J=6.3 Hz, 6H). Optical rotation [a]D +4.8 (c 1.0, MeOH). Chiral purity: >99% ee by chiral analytical SFC. Anal. Calcd for C19H28N6O4-H2O: C, 54.02; H, 7.16; N, 19.89. Found: C, 53.94; H, 7.22; N, 19.81.
The white crystalline solid from above (500 mg) was recrystallized from 9: 1 H2O/CH3CN (2 mL) by heating until dissolved and then allowing the resulting solution to stand at room temperature for 18 h. During the 18 h time period, larger crystals of monohydrate (Form 1) formed. Single crystal X-ray diffraction of a selected crystal from this material provided the structure in FIG.1.
PATENTS
WO2022018667
WO2022174031
WO2022137106
[1]. Douglas Carl BEHENNA, et al. Cdk2 inhibitors. WO2020157652A2.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
///////////Tagtociclib, PF-07104091, 2460249-19-6, Tegtociclib, XBD0JF5EHJ, PF 07104091
SPIROBUDIFEN
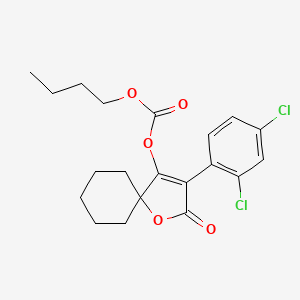
SPIROBUDIFEN
cas 1305319-70-3
413.3 g/mol, C20H22Cl2O5
Butyl 3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl carbonate Butyl 3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl carbonate
- Butyl (3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro(4.5)dec-3-en-4-yl) carbonate
- butyl [3-(2,4-dichlorophenyl)-2-oxo-1-oxaspiro[4.5]dec-3-en-4-yl] carbonate
Spirobudifen is an oxaspiro compound that is 1-oxaspiro[4.5]dec-3-en-2-one substituted by 2,4-dichlorophenyl and (butoxycarbonyl)oxy groups at positions 3 and 4, respectively. It is an acaricide from Zhejiang Udragon Bioscience. It is a dichlorobenzene, an oxaspiro compound, an organochlorine acaricide and a carbonate ester.
SCHEME
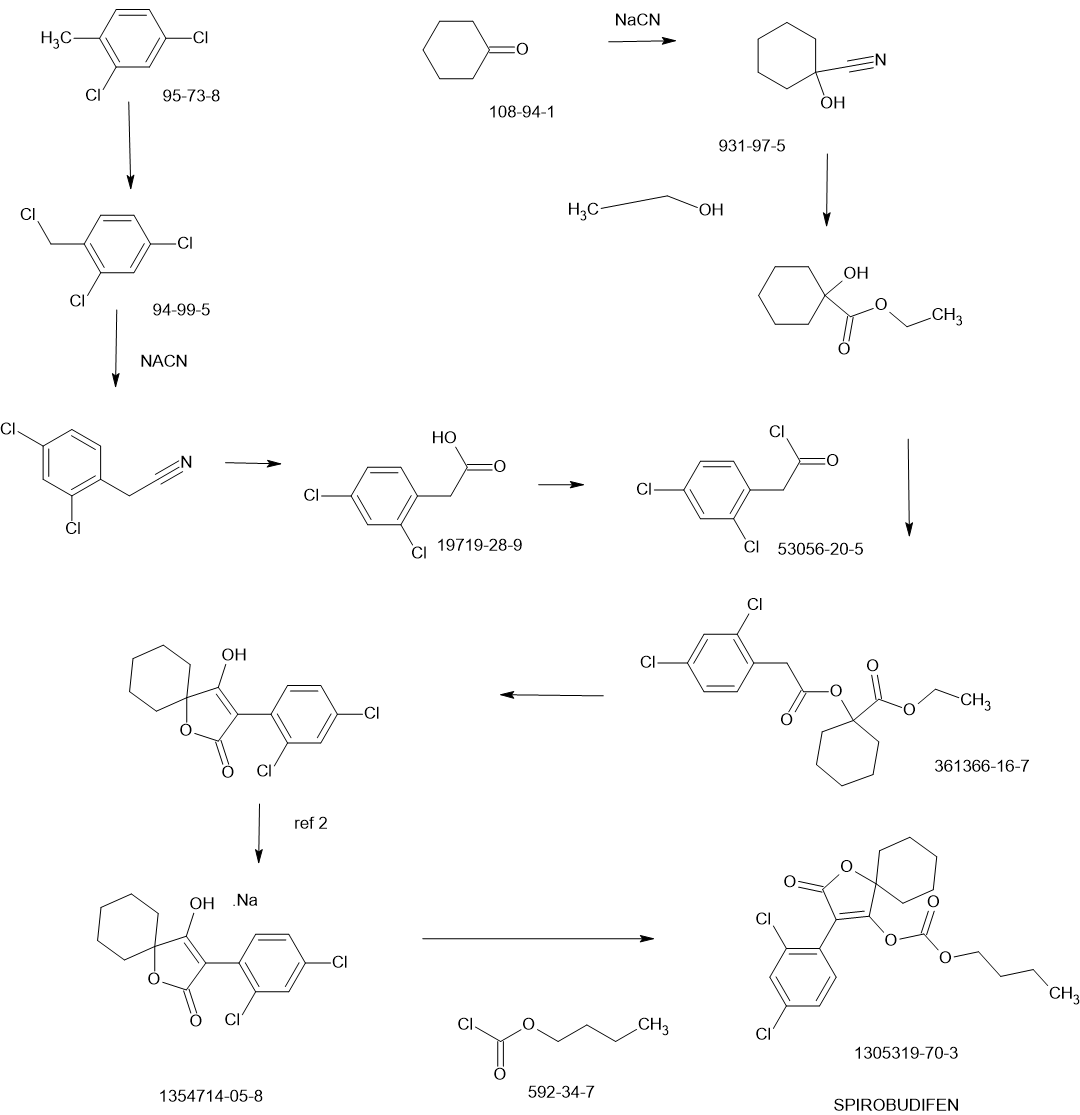
PATENTS
CN112745286
CN102060818
Xiandai Nongyao (2012), 11(1), 15-21
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////SPIROBUDIFEN, 1305319-70-3
SOVESUDIL
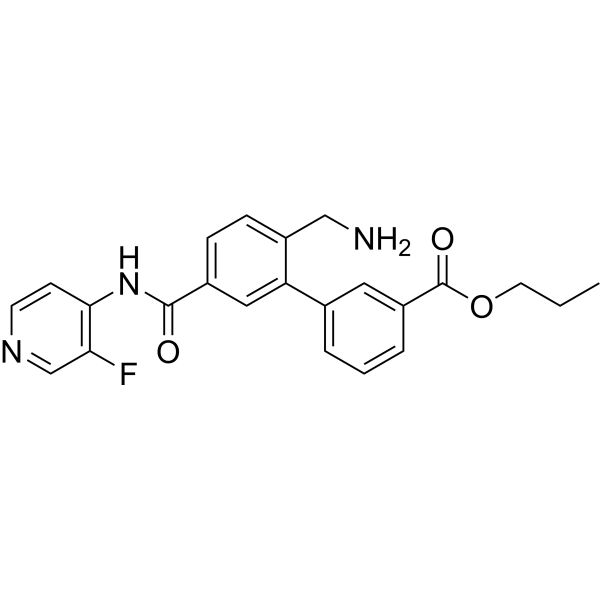
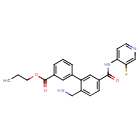
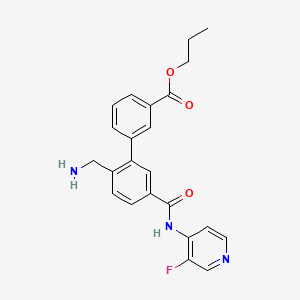
SOVESUDIL
PHP-201; AMA 0076, C23O3R93BM
CAS 1333400-14-8
| Molecular Weight | 407.44 |
|---|---|
| Formula | C23H22FN3O3 |
Sovesudil (PHP-201) is a potent, ATP-competitive, locally acting Rho kinase (ROCK) inhibitor with IC50s of 3.7 and 2.3 nM for ROCK-I and ROCK-II, respectively. Sovesudil lowers intraocular pressure (IOP) without inducing hyperemia.
SCHEME
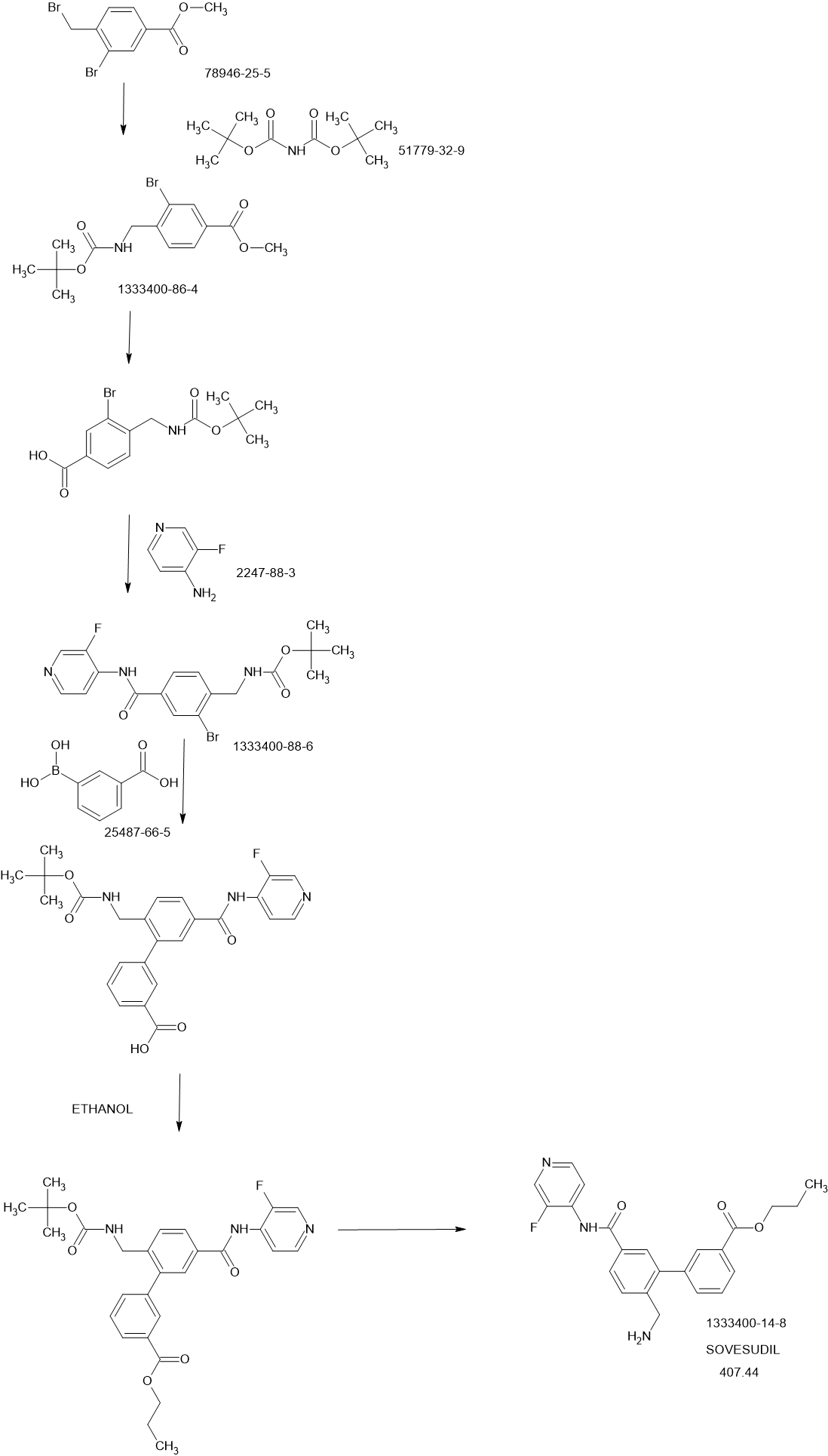
PATENTS
Bioorganic & Medicinal Chemistry Letters (2013), 23(23), 6442-6446
https://www.sciencedirect.com/science/article/abs/pii/S0960894X13011141
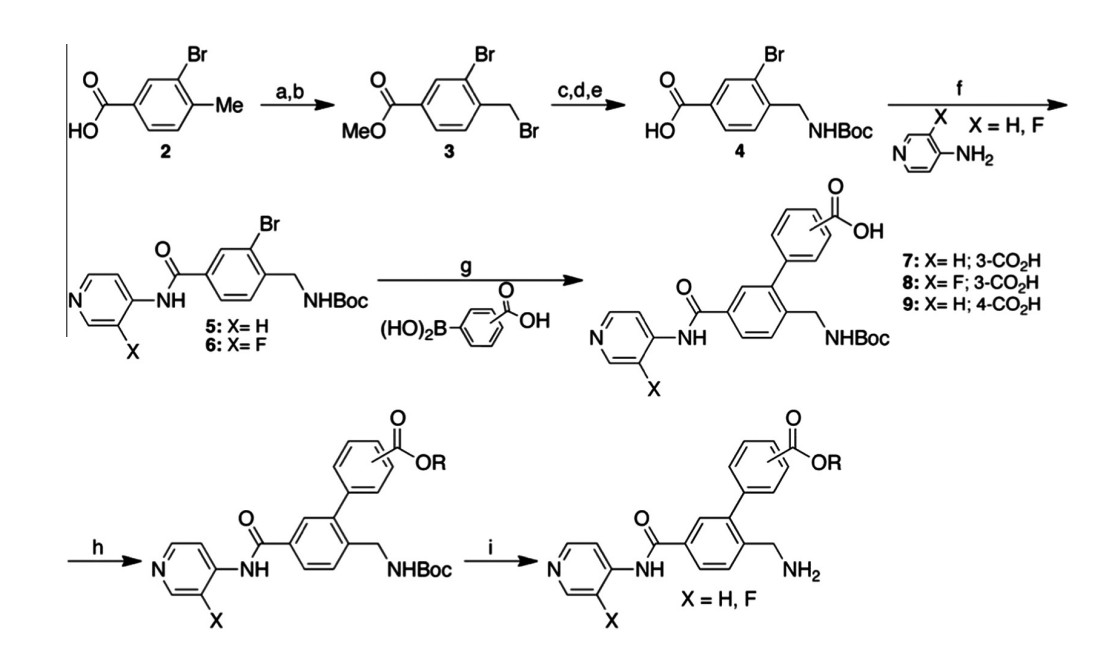
Figure 2. Synthetic scheme for synthesis of compounds 10–35. (a) H2SO4, MeOH, 60 C, 16 h; (b) NBS, AIBN, CCl4, reflux, 16 h; (c) Boc2NH, t-BuOK, DMF, rt, 16 h; (d) DCM/TFA
(50:1), 0 C ? rt, 4 h; (e) NaOH, MeOH, 50 C, 2 h; (f) HATU, DMAP, NEt3, DMA, 30 C, 16 h; (g) Pd(dppf)Cl2, Na2CO3, H2O, DMF, 100 C; 16 h; (h) ROH, TBTU, HOBT, DIEA, DMF,
rt, 16 h or ROH, DCC, DMAP, DCM, rt, 16 h or Me2C@C(Cl)NMe2, THF or DCM, rt, followed by ROH, 16 h; (i) DCM/TFA (7:1), 30 C, 16 h or HCl(g) in DCM, 30 C, 16 h.
PATENT
WO2011107608
- [1]. Van de Velde S, et al. AMA0076, a novel, locally acting Rho kinase inhibitor, potently lowers intraocular pressure in New Zealand white rabbits with minimal hyperemia. Invest Ophthalmol Vis Sci. 2014;55(2):1006-1016. Published 2014 Feb 18. [Content Brief][2]. Ha A, et al. Sovesudil (locally acting rho kinase inhibitor) for the treatment of normal-tension glaucoma: the randomized phase II study [published online ahead of print, 2021 Jul 28]. Acta Ophthalmol. 2021;10.1111/aos.14949. [Content Brief]
////////SOVESUDIL, 1333400-14-8, PHP 201, AMA 0076, C23O3R93BM
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Risevistinel
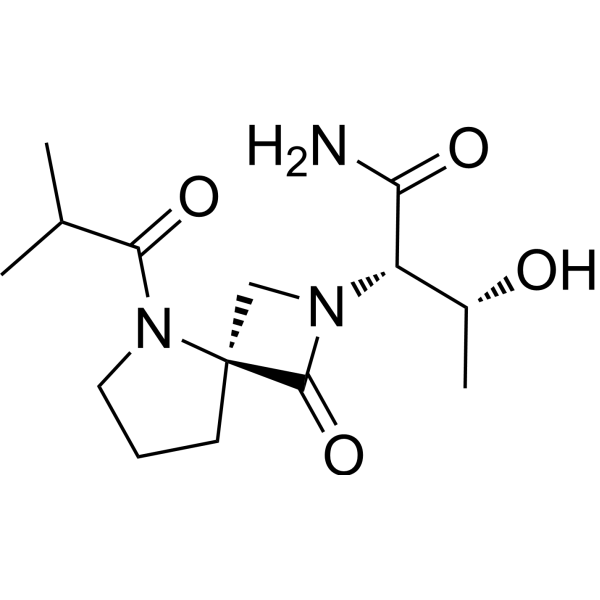
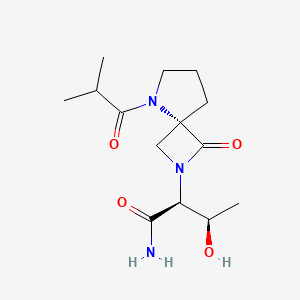
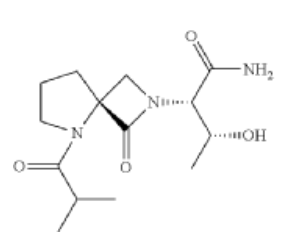
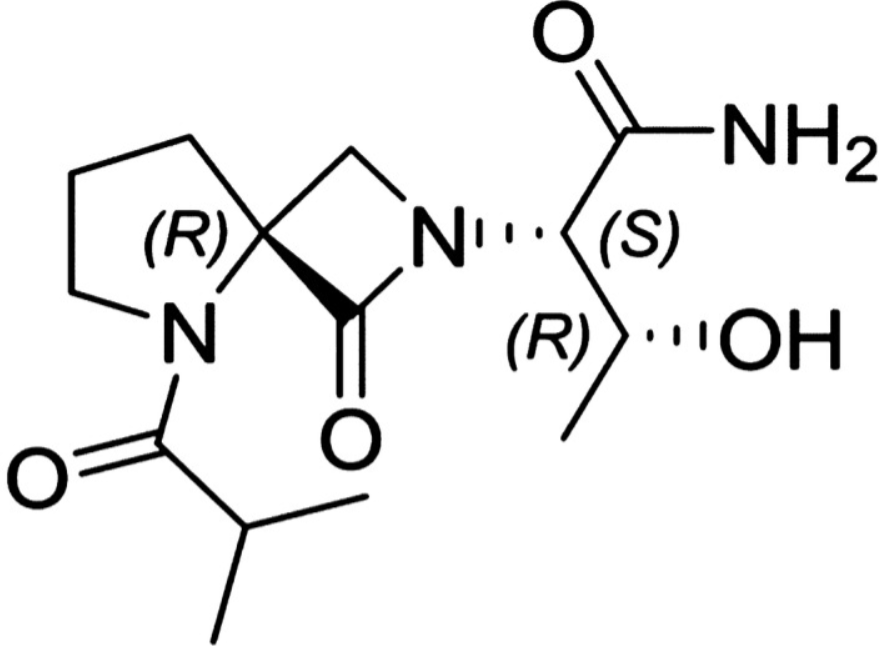
Risevistinel
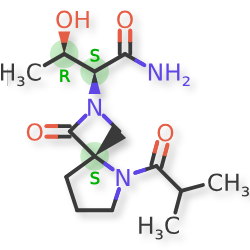
NYX-783 CAS 2591344-26-0, UNII-52TU5MZG22
NYX 2925, 2012536-16-0, X062KF5ZV3
C14H23N3O4, 297.35
UNII-52TU5MZG22,
X062KF5ZV3
(αS,4R)-α-[(1R)-1-Hydroxyethyl]-5-(2-methyl-1-oxopropyl)-1-oxo-2,5-diazaspiro[3.4]octane-2-acetamide
2,5-Diazaspiro[3.4]octane-2-acetamide, α-[(1R)-1-hydroxyethyl]-5-(2-methyl-1-oxopropyl)-1-oxo-, (αS,4S)-
(2S,3R)-3-hydroxy-2-[(4S)-5-(2-methylpropanoyl)-3-oxo-2,5-diazaspiro[3.4]octan-2-yl]butanamide
- NYX-783, NYX 2925
- CS-0113907
- HY-135741
- NYX-2925
Risevistinel (NYX-783) is a positive allosteric modulator of N-methyl-D-aspartate (NMDA) receptor. Nevadistinel can be used to inhibit cognitive impairment associated with neurodegenerative diseases, such as mild cognitive impairment, mild Alzheimer’s disease, Parkinson’s disease, Lewy body disease.
NYX-2925 is an N-methyl-D-aspartate receptor (NMDAR) modulator, and at low concentrations of endogenous agonist (glycine or D-serine) and in the presence of glutamate, NYX-2925 partially activates the NMDAR. NYX-2925 appears to act at a binding site that is distinct from NMDAR agonists or antagonists studied to date, such as D-cycloserine, ketamine, MK-801, or kynurenic acid. The mode of action of NYX-2925 seems to be distinct from that of all existing and emerging drugs that are indicated for the treatment of neuropathic pain. While current medications target individual elements of pain signal transmission or modulation, NYX-2925 can modulate multiple synaptic relays within pain circuits.
NYX-2925 is under investigation in clinical trial NCT04146896 (Efficacy and Safety of NYX-2925 in Subjects With Neuropathic Pain Associated With Diabetic Peripheral Neuropathy (DPN)).
SCHEME
COUPLER
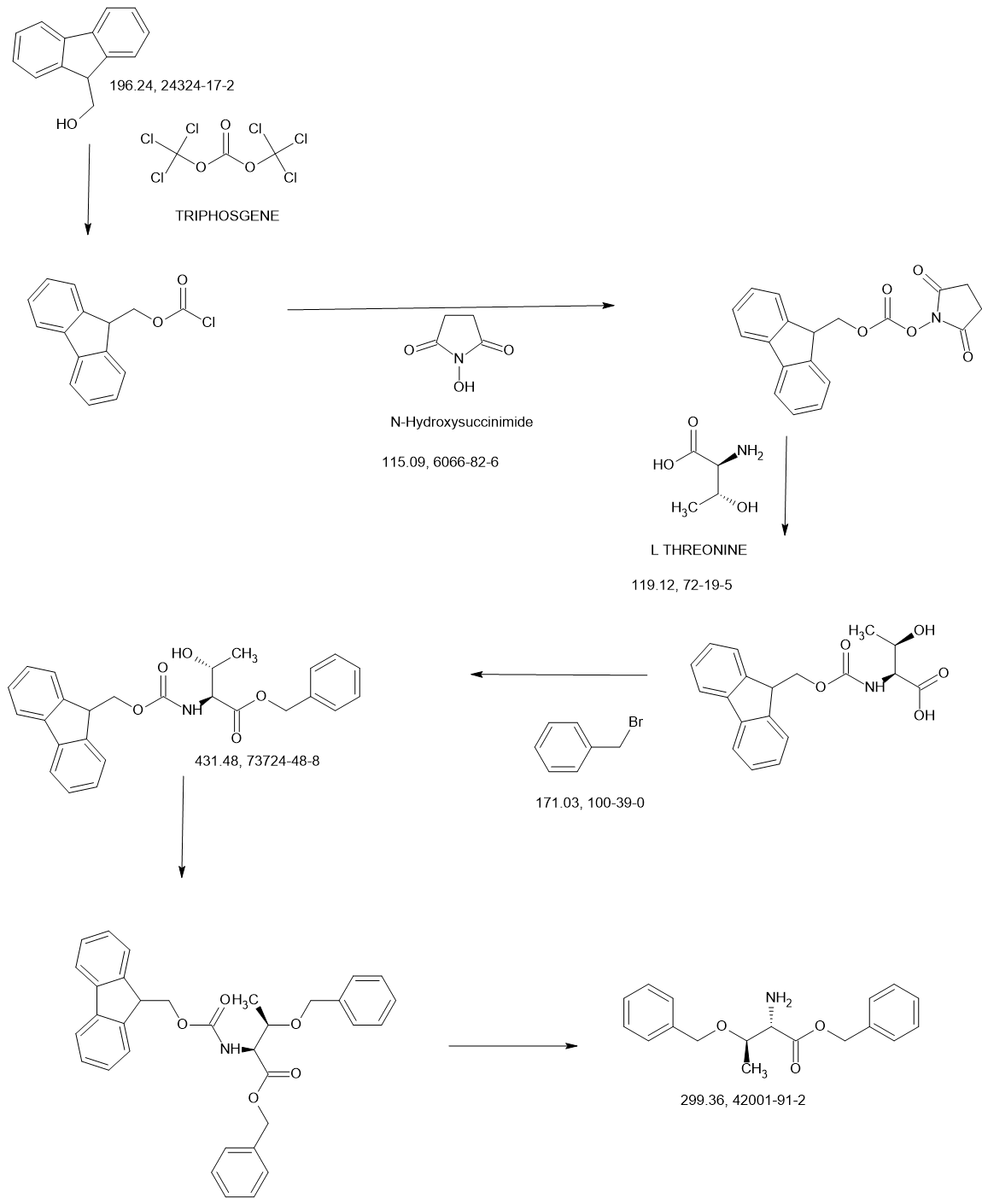
MAIN
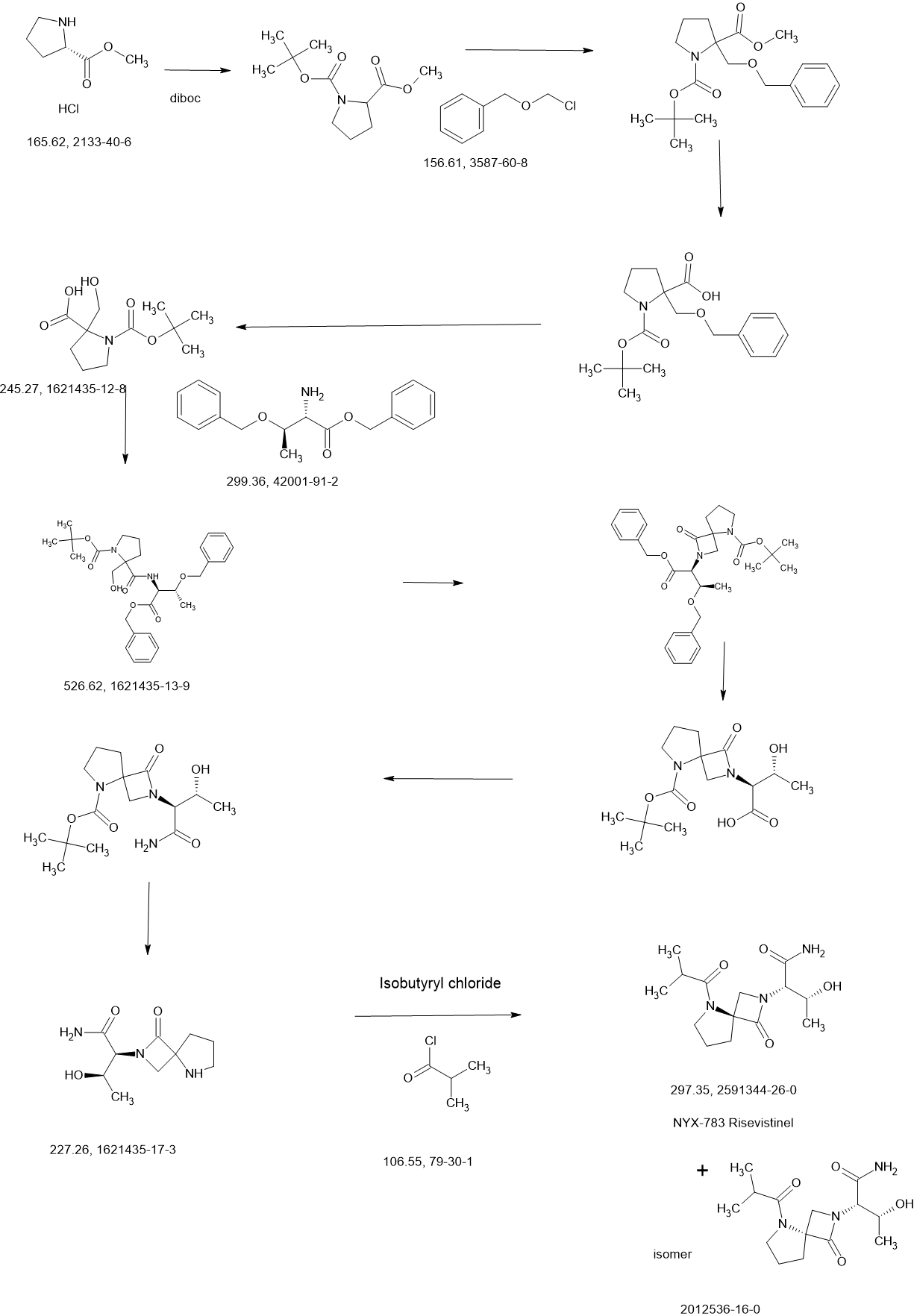
REF
US20210139489 Aptinyx Inc.
https://patentscope.wipo.int/search/en/detail.jsf?docId=US323750708&_cid=P11-MD3X0E-31059-1
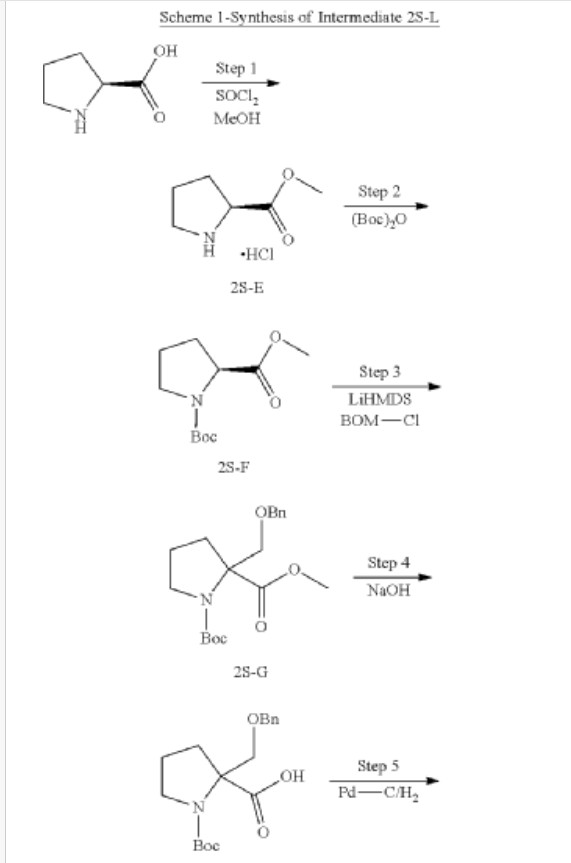
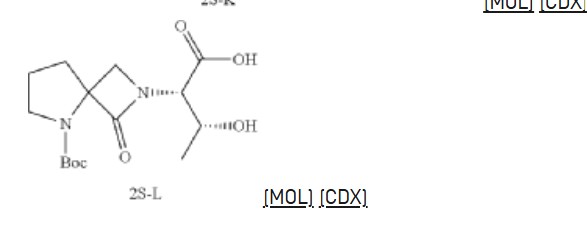
Synthesis of methyl pyrrolidine-2-carboxylate (2S-E)
| 1H-NMR: (500 MHz, DMSO-d 6): δ 4.15-4.13 (m, 1H), 3.65 (s, 3H), 3.35-3.30 (m, 2H), 2.23-2.15 (m, 1H), 1.86-1.78 (m, 3H), 1.41 (s, 9H); |
Synthesis of 1-tert-butyl 2-methyl pyrrolidine-1,2-dicarboxylate (2S-F)
| 1H-NMR: (400 MHz, DMSO-d 6): δ 4.15-4.13 (m, 1H), 3.65 (s, 3H), 3.35-3.30 (m, 2H), 2.23-2.15 (m, 1H), 1.86-1.78 (m, 3H), 1.41 (s, 9H); |
Synthesis of 1-tert-butyl 2-methyl 2-((benzyloxy) methyl) pyrrolidine-1,2-dicarboxylate (2S-G)
| 1H-NMR: (500 MHz, DMSO-d 6): δ 7.36-7.22 (m, 5H), 4.59-4.48 (m, 2H), 4.02-3.88 (m, 1H), 3.63 (s, 3H), 3.49-3.35 (m, 2H), 3.34-3.30 (m, 1H), 2.31-2.23 (m, 1H), 2.04-1.89 (m, 2H), 1.82-1.78 (m, 1H); |
Synthesis of 2-((benzyloxy) methyl)-1-(tert-butoxycarbonyl) pyrrolidine-2-carboxylic acid (2S-H)
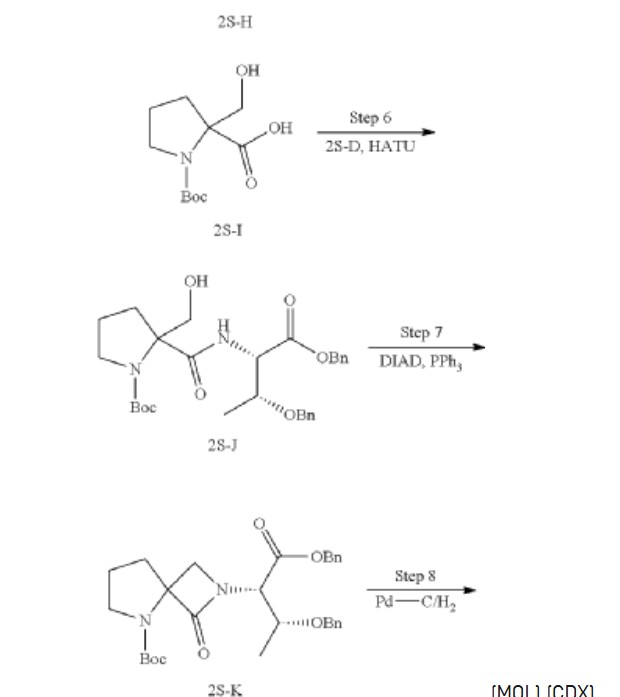
Synthesis of 1-(tert-butoxycarbonyl)-2-(hydroxymethyl) pyrrolidine-2-carboxylic acid (2S-I)
| 1H-NMR: (400 MHz, DMSO-d 6): δ 4.66 (br s, 1H), 3.96-3.83 (m, 1H), 3.63-3.59 (m, 1H), 3.49-3.41 (m, 1H), 3.34-3.25 (m, 1H), 2.30-2.17 (m, 1H), 1.95-1.72 (m, 3H), 1.38 (s, 9H). |
Synthesis of tert-butyl 2-(((2S,3R)-1,3-bis (benzyloxy)-1-oxobutan-2-yl) carbamoyl)-2-(hydroxymethyl) pyrrolidine-1-carboxylate (2S-J)
Synthesis of tert-butyl 2-((2S,3R)-1,3-bis (benzyloxy)-1-oxobutan-2-yl)-1-oxo-2,5-diazaspiro [3.4] octane-5-carboxylate (2S-K)
Synthesis of (2S,3R)-2-(5-(tert-butoxycarbonyl)-1-oxo-2,5-diazaspiro [3.4] octan-2-yl)-3-hydroxybutanoic acid (2S-L)
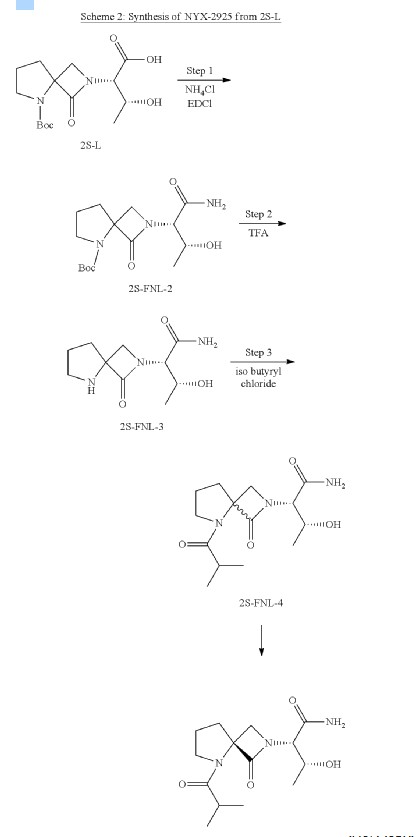
Synthesis of tert-butyl 2-((2S,3R)-1-amino-3-hydroxy-1-oxobutan-2-yl)-1-oxo-2,5-diazaspiro [3.4] octane-5-carboxylate (2S-FNL-2)
Synthesis of (2S,3R)-3-hydroxy-2-(1-oxo-2,5-diazaspiro [3.4] octan-2-yl) butanamide (2S-FNL-3)
| 1H-NMR: (400 MHz, D 2O): δ 4.33-4.29 (m, 2H), 4.09 (d, 1H), 3.95 (d, 1H), 3.57-3.48 (m, 2H), 2.51-2.46 (m, 2H), 2.25-2.19 (m, 2H), 1.31 (d, 3H); |
Synthesis of (2S,3R)-3-hydroxy-2-(5-isobutyryl-1-oxo-2,5-diazaspiro [3.4] octan-2-yl) butanamide (NYX-2925)
PATENT
WO2022086858
WO2021021996
//////Risevistinel, NYX 783, UNII-52TU5MZG22, Aptinyx Inc, NYX 2925, CS 0113907, HY 135741, NYX-2925
Rezatapopt
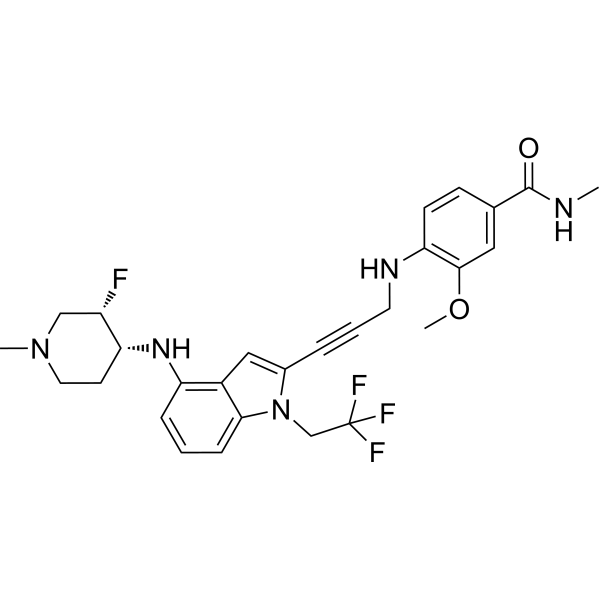
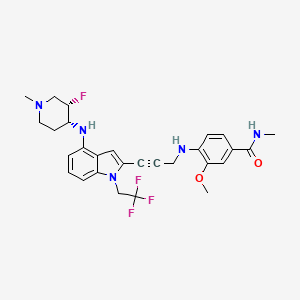
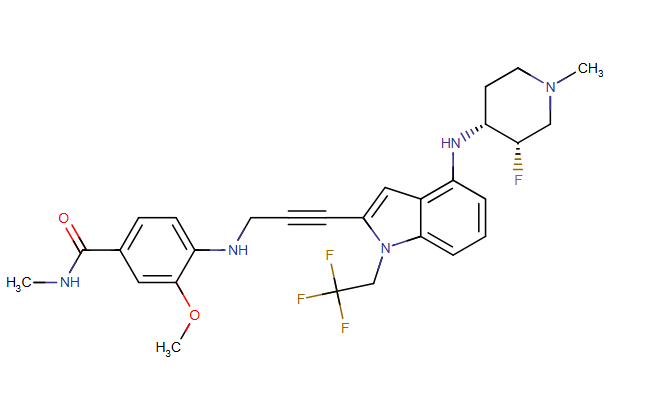
Rezatapopt, PC 14586
CAS 2636846-41-6
| Molecular Weight | 545.57 |
|---|---|
| Synonyms | PC14586 |
| Formula | C28H31F4N5O2 |
| CAS No. | 2636846-41-6 |
4-[3-[4-[[(3S,4R)-3-fluoro-1-methylpiperidin-4-yl]amino]-1-(2,2,2-trifluoroethyl)indol-2-yl]prop-2-ynylamino]-3-methoxy-N-methylbenzamide
- 4-[3-[4-[[(3S,4R)-3-fluoro-1-methylpiperidin-4-yl]amino]-1-(2,2,2-trifluoroethyl)indol-2-yl]prop-2-ynylamino]-3-methoxy-N-methylbenzamide
- Benzamide, 4-[[3-[4-[[(3S,4R)-3-fluoro-1-methyl-4-piperidinyl]amino]-1-(2,2,2-trifluoroethyl)-1H-indol-2-yl]-2-propyn-1-yl]amino]-3-methoxy-N-methyl-
Rezatapopt (PC14586) is an orally active antineoplastic agent. Rezatapopt binds to a pocket created by the TP53 Y220C mutation. Rezatapopt restores p53 tumor suppressor functions by stabilization of the p53 protein structure. Rezatapopt demonstrates tumor inhibition and regression in mouse models with established human tumor xenografts harboring the TP53 Y220C mutation.
SCHEME
COUPLER
COUPLER
MAIN
REF
PAPER
https://pubs.acs.org/doi/10.1021/acsmedchemlett.4c00379
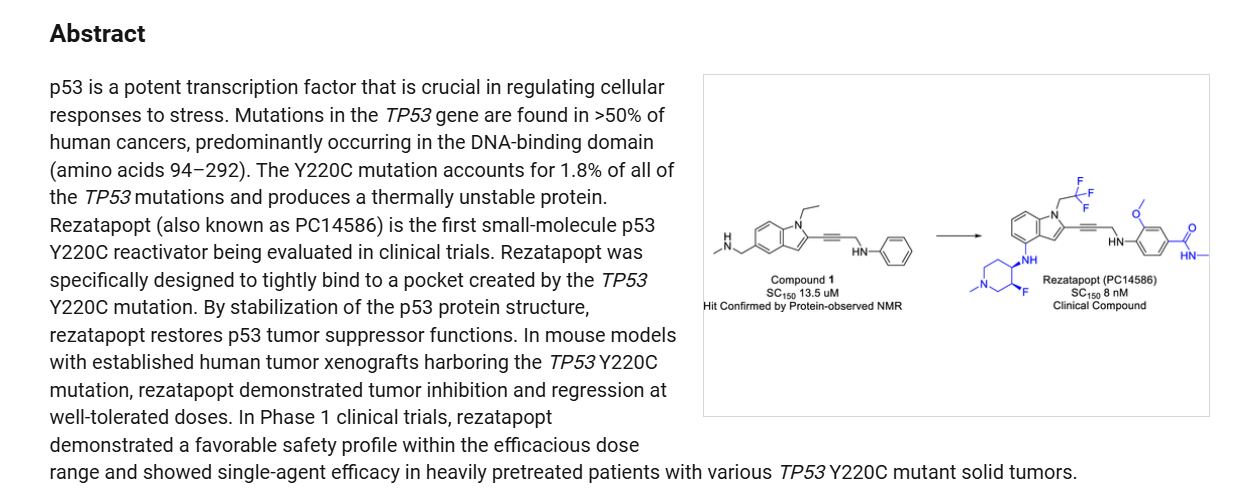
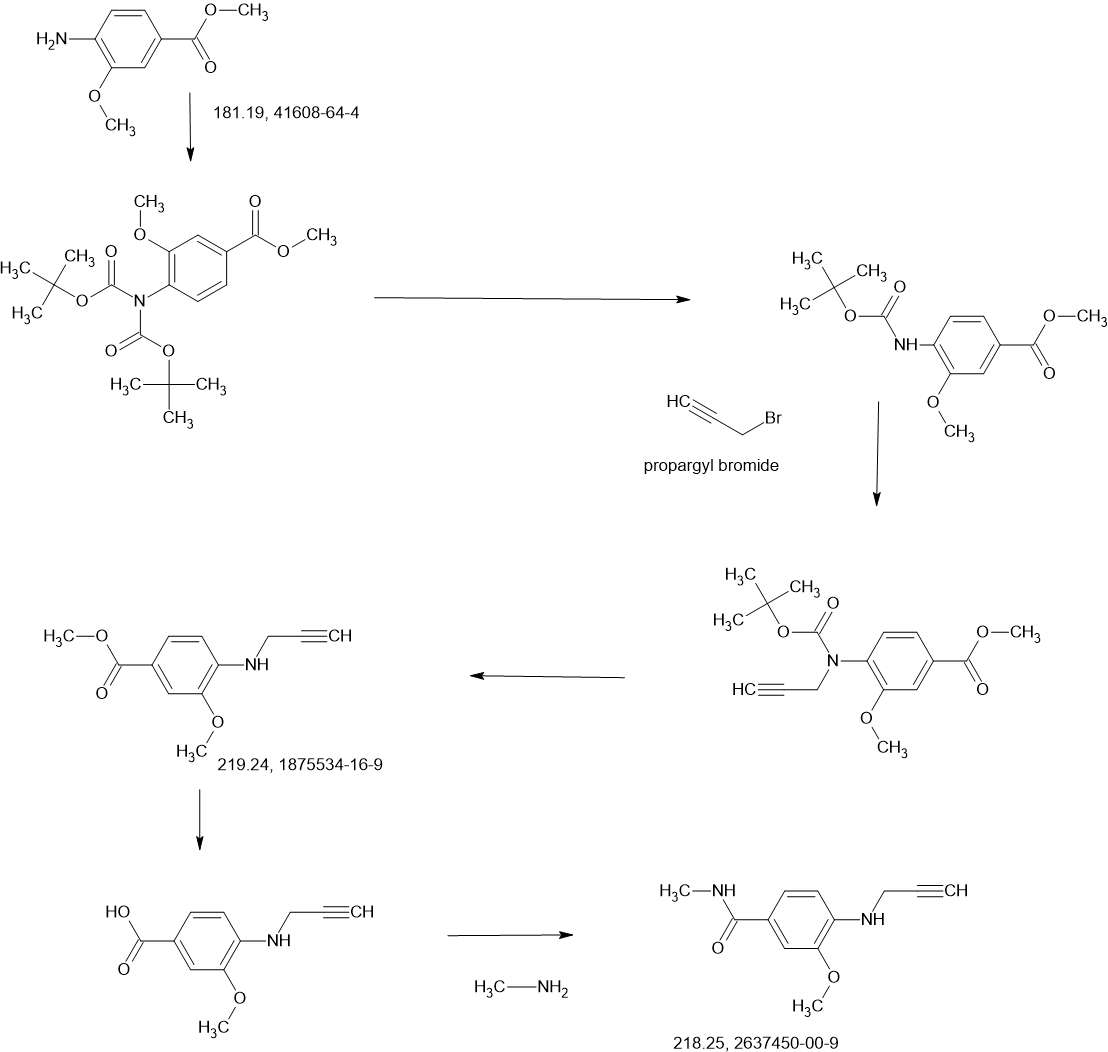
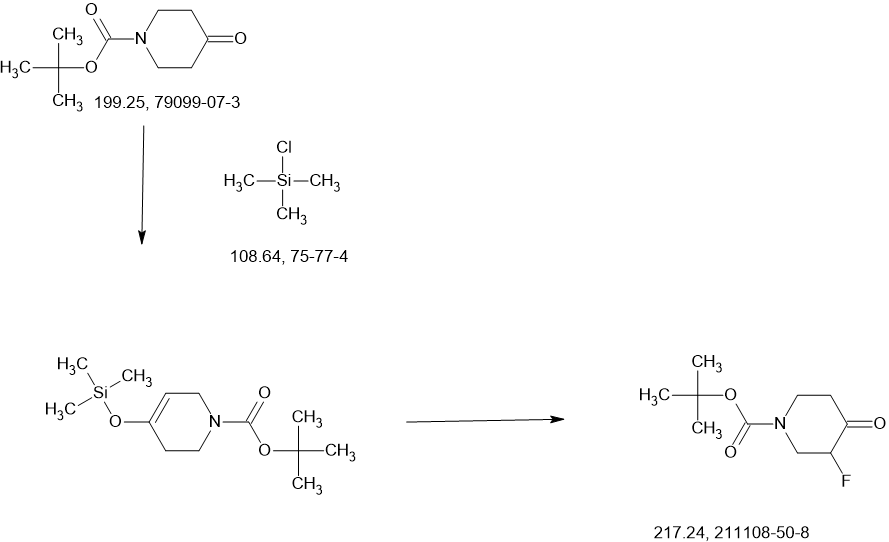
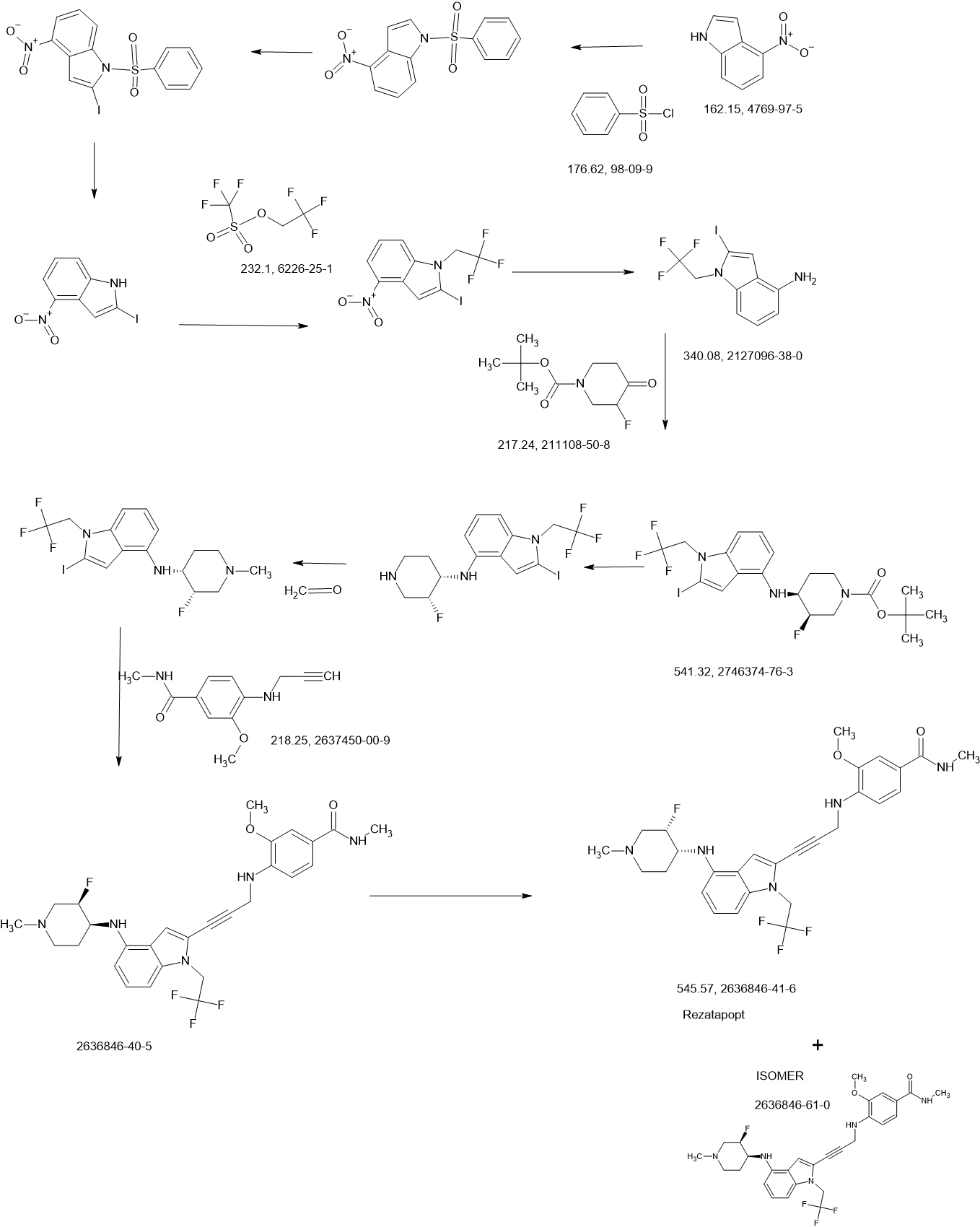
2-Iodo-1-(2,2,2-trifluoroethyl)-1H-indol-4-amine 15 was prepared from 4-nitroindole as described in
WO2017143291. 1
H NMR (400 MHz, dimethylsulfoxide [DMSO]-d6) δ ppm 9.19–10.88 (m, 2 H), 7.63
(d, J=8.34 Hz, 1 H), 7.16–7.25 (m, 1 H), 7.04–7.14 (m, 2 H), 5.14–5.33 (m, 2 H). LCMS (ES+
, m/z):
340.9 [(M+H)+
].
SnCl2.2H2O (398.11 mg, 1.76 mmol, 0.20 eq.) was added to a solution of 2-iodo-1-(2,2,2-trifluoroethyl)-
1H-indol-4-amine 15 (3.00 g, 8.82 mmol, 1.00 eq.) and 1-methylpiperidin-4-one (1.20 g, 10.61 mmol,
1.20 eq.) in MeOH (10.00 mL). The mixture was stirred at 25 °C for 3 hours (h), and then NaBH3CN
(2.77 g, 44.1 mmol, 5.00 eq.) was added, stirring at 25 °C for 69 h. Thin-layer chromatography (TLC)
indicated that the starting material was consumed, and the reaction mixture was filtered. The filtrate was
poured into H2O (200 mL) and extracted with ethyl acetate ([EtOAc] 200 mL2). The combined organic layers were washed with H2O (200 mL), dried over Na2SO4, and concentrated under reduced pressure to give a residue. The crude material was purified by flash column chromatography (Silica gel, petroleum ether (PE) : EtOAc = 0:1) and then by preparative high performance chromatography ([prep-HPLC] column: Phenomenex Luna C18 10040mm5 um; mobile phase: [H2O (0.2% Formic acid-acetonitrile [ACN])]; gradient: 10%–50% acetonitrile over 8.0 minutes) to yield 2-iodo-N-(1-methylpiperidin-4-yl)-1- (2,2,2-trifluoroethyl)-1H-indol-4-amine 16 (2.50 g, 5.72 mmol, 64.93% yield) as a light-brown solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 8.24 (br s, 1 H, formic acid salt), 7.17 (s, 1 H), 6.85–6.95 (m, 1 H), 6.78 (br d, J = 7.99 Hz, 1 H), 6.16 (br d, J = 7.63 Hz, 1 H), 5.44 (br d, J = 2.62 Hz, 1 H), 4.99 (q, J = 8.54 Hz, 2 H), 3.33 (br s, 1 H), 2.85 (br d, J = 9.66 Hz, 2 H), 2.25 (br s, 3 H), 2.07–2.18 (m, 2 H), 1.94 (br d, J = 12.04 Hz, 2 H), 1.46–1.58 (m, 2 H). LCMS (ES+, m/z): 438.0 [(M+H)+]. Boc2O (26.03 g, 119.26 mmol, 6.00 eq.) was added to a solution of 2-methoxy-4-(methylsulfonyl)aniline 17 (4.00 g, 19.88 mmol, 1.00 eq.) in dioxane (40.00 mL) at 25 o C (room temperature). The reaction mixture was stirred at 110 °C for 16 h. TLC and LCMS indicated that the reaction was completed, and it was concentrated in vacuo. The residue was purified by column chromatography (SiO2, PE/EtOAc = 10/1 to 1:1) to yield tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)carbamate (6.00 g, 19.92 mmol, 72.6% purity, 72% yield) as a yellow gum. 1 H NMR (400 MHz, DMSO-d6) δ ppm 8.33 (s, 1 H), 8.03 (d, J = 8.38 Hz, 1 H), 7.47 (dd, J = 8.38, 2.00 Hz, 1 H), 7.44 (d, J = 2.00 Hz, 1 H), 3.91 (s, 3 H), 3.18 (s, 3 H), 1.47 (s, 9 H). LCMS (ES+, m/z): 324.1 [(M+Na)+ ]. NaH (867.27 mg, 60% purity, 21.69 mmol, 3.00 eq.) was added in portions at 0 °C to a mixture of tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)carbamate (3.00 g, 7.23 mmol, 1.00 eq.) in dimethylformamide ([DMF] 30.00 mL) and stirred at 0 °C for 0.5 h. 3-Bromoprop-1-yne (3.23 g, 21.69 mmol, 3.00 eq.) was added to the reaction mixture, stirring at 0 °C for 2.5 h. TLC (Plate 1: PE : EtOAc = 1:1) and LCMS indicated that the starting material was consumed, and the product was detected. The reaction mixture was poured into a saturated solution of NH4Cl (200 mL) at 0 o C and was extracted with EtOAc (200 mL3). The combined organic phase was dried over Na2SO4, filtered, and concentrated
in vacuo. The residue was purified by column chromatography (SiO2, PE : EtOAc = 5:1 to 1:2) to give
tert-butyl (2-methoxy-4-(methylsulfonyl)phenyl)(prop-2-yn-1-yl)carbamate (3.00 g, 8.85 mmol,
74% purity, 90% yield) as a light-yellow gum.
1
H NMR (400 MHz, DMSO-d6) δ ppm 7.53–7.56 (m, 1 H), 7.46–7.53 (m, 2 H), 4.10–4.51 (m, 2 H), 3.90
(s, 3 H), 3.27 (s, 3 H), 3.17 (t, J = 2.32 Hz, 1 H), 1.27–1.39 (m, 9 H). LCMS (ES+
, m/z): 283.9 [(M+H-tBu)+].
A solution of 4M HCl/EtOAc (20.00 mL) was added to the solution of tert-butyl (2-methoxy-4-
(methylsulfonyl)phenyl)(prop-2-yn-1-yl)carbamate (3.00 g, 6.54 mmol, 1.00 eq.) in EtOAc (1.00 mL).
The reaction mixture was stirred at 25 °C for 2 h. TLC indicated that the starting material was consumed
completely. The reaction mixture was concentrated in vacuo to yield 2-methoxy-4-(methylsulfonyl)-N-
(prop-2-yn-1-yl)aniline 18 (1.80 g, 7.53 mmol, 85.3% yield, HCl salt) as a yellow solid.
1
H NMR (400 MHz, DMSO-d6) δ ppm 7.38 (dd, J = 8.40, 1.60 Hz, 1 H), 7.22 (d, J = 1.60 Hz, 1 H), 6.75
(d, J = 8.80 Hz, 1 H), 3.99 (d, J = 2.4 Hz, 2 H), 3.87 (s, 3 H) 3.10 (s, 3 H), 3.08 (t, J = 2.31 Hz, 1 H).
LCMS (ES+
, m/z): 240.1 [(M+H)+
].
i-Pr2NH (2.08 g, 20.58 mmol, 2.91 mL, 10 eq.), CuI (392.02 mg, 2.06 mmol, 1 eq), 2-iodo-N-(1-
methylpiperidin-4-yl)-1-(2,2,2-trifluoroethyl)-1H-indol-4-amine 16 (0.9 g, 2.06 mmol, 1 eq.) and
Pd(PPh3)4 (475.71 mg, 411.67 μmol, 0.2 eq.) was added to a solution of 2-methoxy-4-(methylsulfonyl)-N-
(prop-2-yn-1-yl)aniline 18 (622.16 mg, 2.47 mmol, 1.2 eq.) in DMSO (10 mL) at 45 °C under N2. The
reaction mixture was stirred at 45 °C for 1 h. TLC (DCM/MeOH=10:1, Rf = 0.3) indicated that the
starting material was consumed completely. It was poured into ethylenediaminetetraacetic acid ([EDTA]
20 mL) and stirred for 1 h, then extracted with EtOAc (40 mL3). The combined organic phase was washed with brine (40 mL), dried with anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography (SiO2, PE : EtOAc = 1:1 to dichloromethane (DCM) / MeOH = 10:1, Rf = 0.3), then by prep-HPLC (column: Phenomenex Luna(2) C18 25050 10u; mobile
phase: [water (0.1% trifluoroacetic acid)-ACN]; B%: 30%–50%, 20 min) to yield compound 13 (0.6 g,
1.09 mmol, 53.08% yield, 99.9% purity) as a light-yellow solid.
1 H NMR (400 MHz, DMSO-d6) δ ppm 1.41–1.54 (m, 2 H), 1.91 (br d, J = 11.00 Hz, 2 H), 1.95–2.08 (m,
2 H) 2.17 (s, 3 H), 2.68–2.80 (m, 2 H), 3.10 (s, 3 H), 3.20–3.29 (m, 1 H), 3.89 (s, 3 H), 4.36 (d,
J = 6.24 Hz, 2 H), 4.92 (q, J = 9.09 Hz, 2 H), 5.49 (d, J = 7.95 Hz, 1 H), 6.15 (d, J = 7.83 Hz, 1 H),
6.50 (t, J = 6.24 Hz, 1 H), 6.68 (d, J = 8.19 Hz, 1 H), 6.89 (d, J = 8.44 Hz, 1 H), 6.99 (t, J = 8.01 Hz,
1 H), 7.09 (s, 1 H), 7.25 (d, J = 1.83 Hz, 1 H), 7.39 (dd, J = 8.31, 1.83 Hz, 1 H). LCMS (ES+, m/z):
549.3 [(M+H)+
]
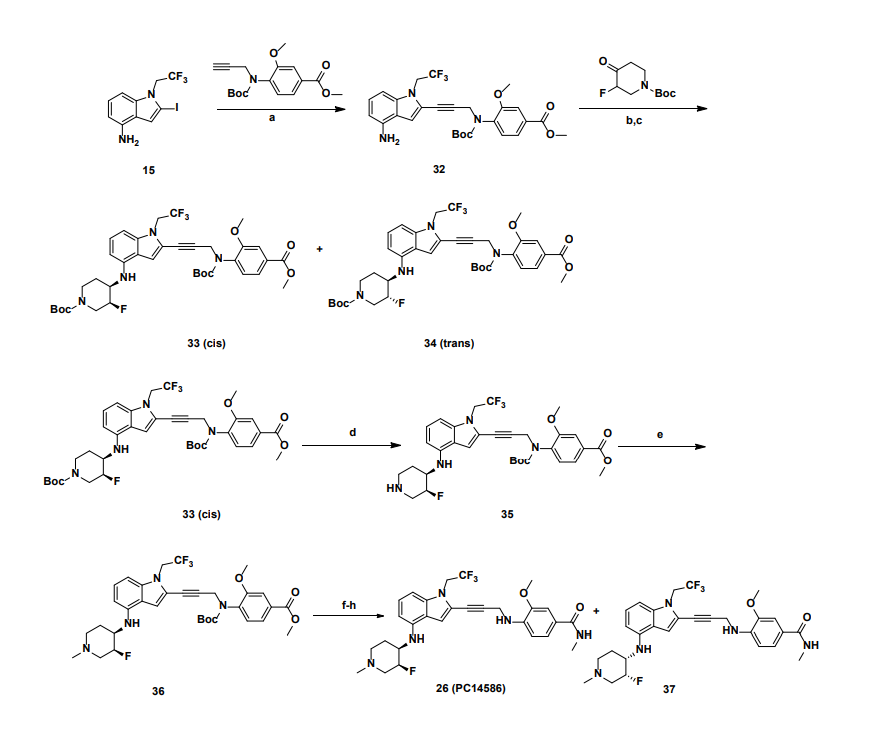
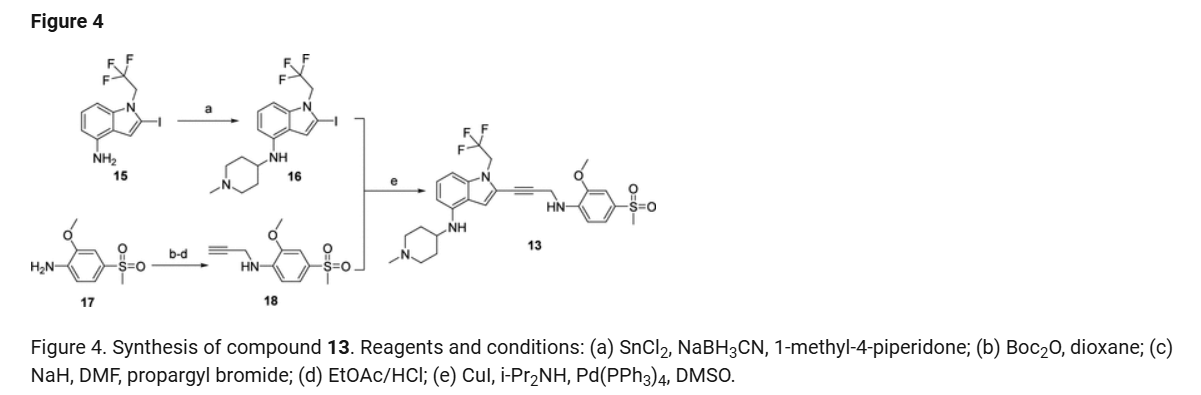
a
Reagents and conditions: (a) Pd(PPh3)4, CuI, diisopropylamine, DMSO, 20 °C, 1 h; (b) TMSCl, DMF, 0 °C, 0.5 h;
(c) BH3.THF, 0 °C, 0.5 h; (d) EtOAc/HCl, 20 °C, 1 h; (e) 10 eq. (CH2O)n, NaBH3CN, MeOH, 20 °C, 16 h; f)
LiOH.H2O, MeOH, 40 °C, 12 h; g) MeNH3Cl, HOBT, EDCI, TEA, DCM, RT, 16 h; h) Chiral SFC separation
PATENTS
WO2023016434 36%
WO2021061643
US20230024905
WO2023016434 Jacobio Pharmaceuticals Co., Ltd.
WO2023225477 PMV Pharmaceuticals, Inc.
US20230024905 PMV Pharmaceuticals, Inc.
WO2021061643 PMV Pharmaceuticals, Inc.
WO2021262483, PMV Pharmaceuticals, Inc.
WO2023196993 PMV Pharmaceuticals, Inc.
WO2021262484 WO2021262541
- [1]. Li Sujing, et al. Heteroarylalkyne compounds for targeting mutant of p53 and their preparation. World Intellectual Property Organization, WO2023016434 A1. 2023-02-16.[2]. Vu BT, et al. Discovery of Rezatapopt (PC14586), a First-in-Class, Small-Molecule Reactivator of p53 Y220C Mutant in Development. ACS Med Chem Lett. 2024 Nov 4;16(1):34-39. [Content Brief][3]. Spiegelberg D, et al. Targeting mutant p53: Evaluation of novel anti-p53R175H monoclonal antibodies as diagnostic tools. Sci Rep. 2025 Jan 6;15(1):1000. [Content Brief][4]. Schram A M, et al. 691TiP PYNNACLE phase II trial of rezatapopt (PC14586) in solid tumors with a TP53 Y220C mutation[J]. Annals of oncology, 2024, 35: S535-S536.
//////////Rezatapopt, PC 14586, 5W59S33KC9
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Resigratinib
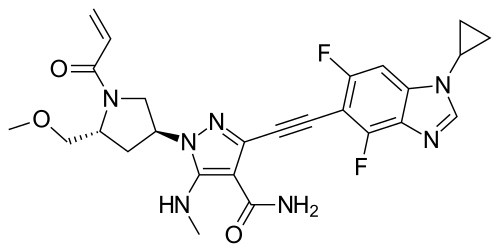
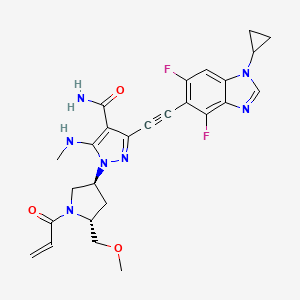
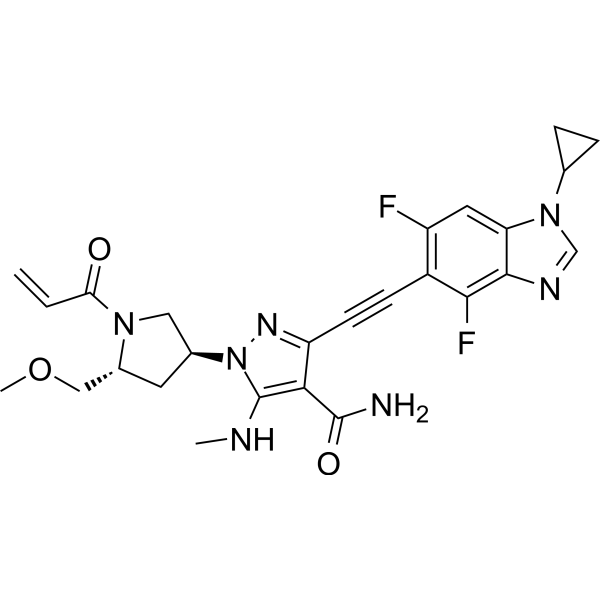
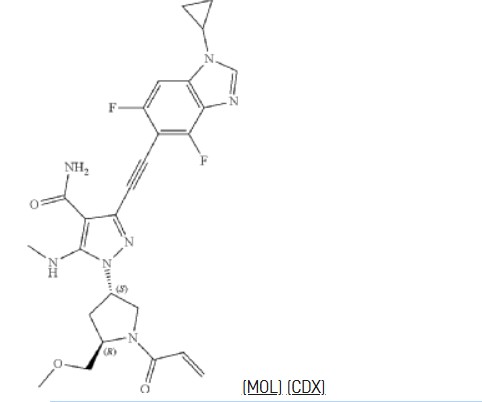
Resigratinib, KIN 3248
CAS 2750709-91-0
C26H27F2N7O3
523.5 g/mol
3-[2-(1-cyclopropyl-4,6-difluorobenzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-prop-2-enoylpyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
- 3-[2-(1-Cyclopropyl-4,6-difluoro-1H-benzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(1-oxo-2-propen-1-yl)-3-pyrrolidinyl]-5-(methylamino)-1H-pyrazole-4-carboxamide
- 3-[2-(1-cyclopropyl-4,6-difluorobenzimidazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-prop-2-enoylpyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
Resigratinib (KIN-3248) is an experimental anticancer medication which acts as a fibroblast growth factor receptor inhibitor (FGFRi) and is in early stage human clinical trials.[1][2][3]
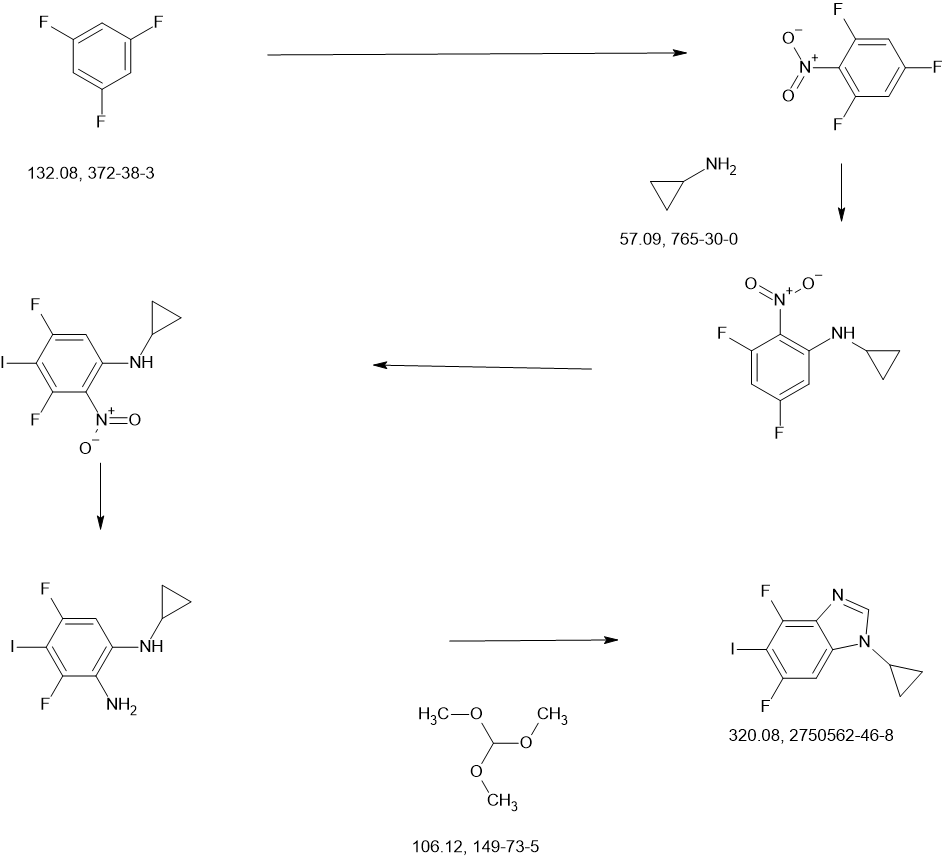
KIN-3248 is a small molecule that targets and inhibits oncogenic fibroblast growth factor receptors (FGFRs). It was designed to mainly target FGFR2 and FGFR3 alterations, which act as oncogenic drivers in 10-20% of cholangiocarcinoma and 20-35% of urothelial cancers, respectively. While effective, disease progression may occur 6 to 8 months after treatment with currently approved FGFR inhibitors is started, and this effect is usually associated with on-target resistance mutations in the kinase domain of FGFR. Therefore, the broad inhibition of FGFR isoforms may be effective against different types of tumors. The safety, tolerability, pharmacokinetics, and preliminary efficacy of KIN-3248 are currently being evaluated in adults with advanced tumors harboring FGFR2 and/or FGFR3 gene alterations. In February 2023, Kinnate Biopharma received Fast Track designation from the FDA for KIN-3248 to treat unresectable, locally advanced or metastatic cholangiocarcinoma (CCA).
SCHEME
COUPLER
COUPLER
MAIN
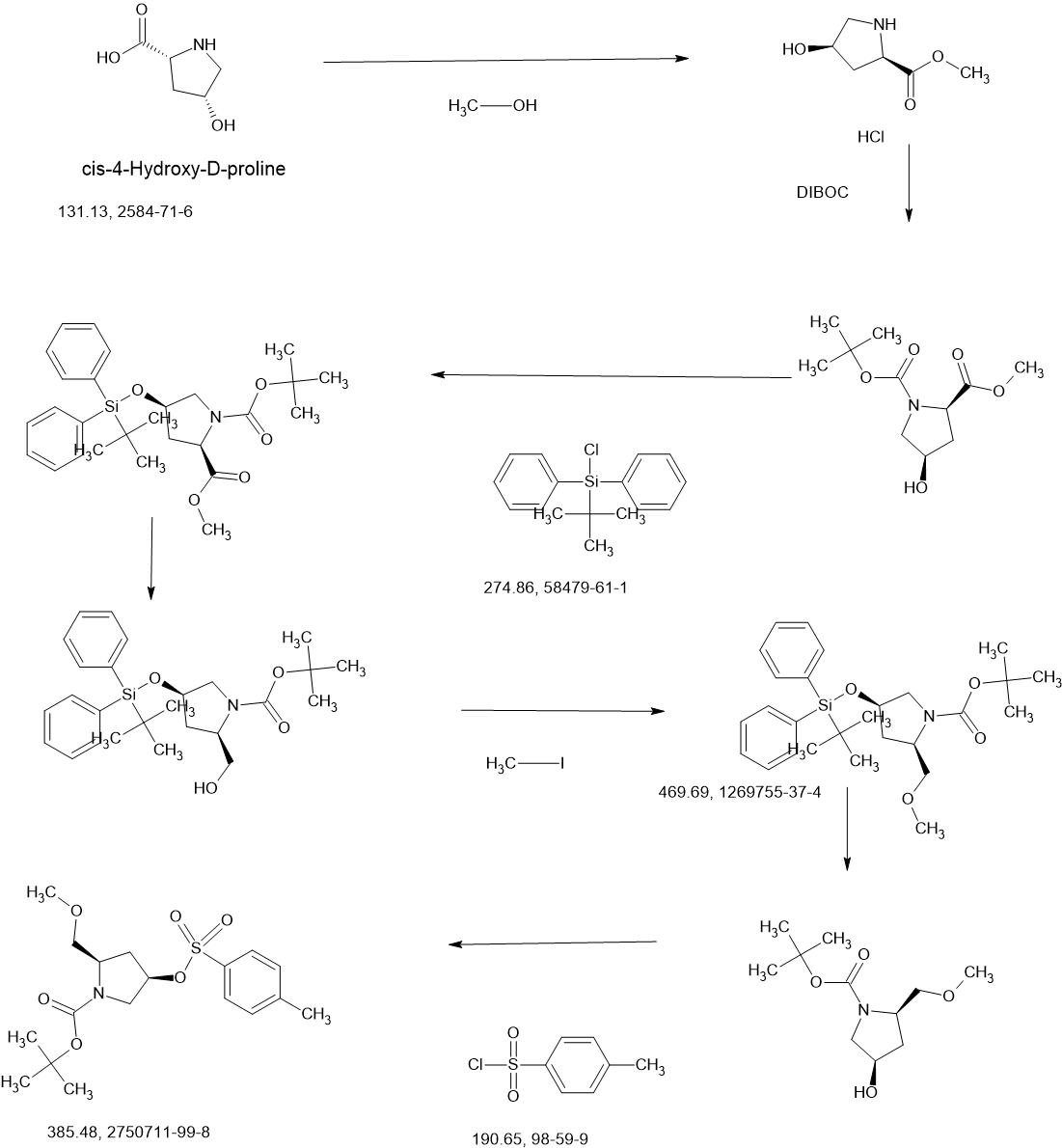
CONTINUED………….
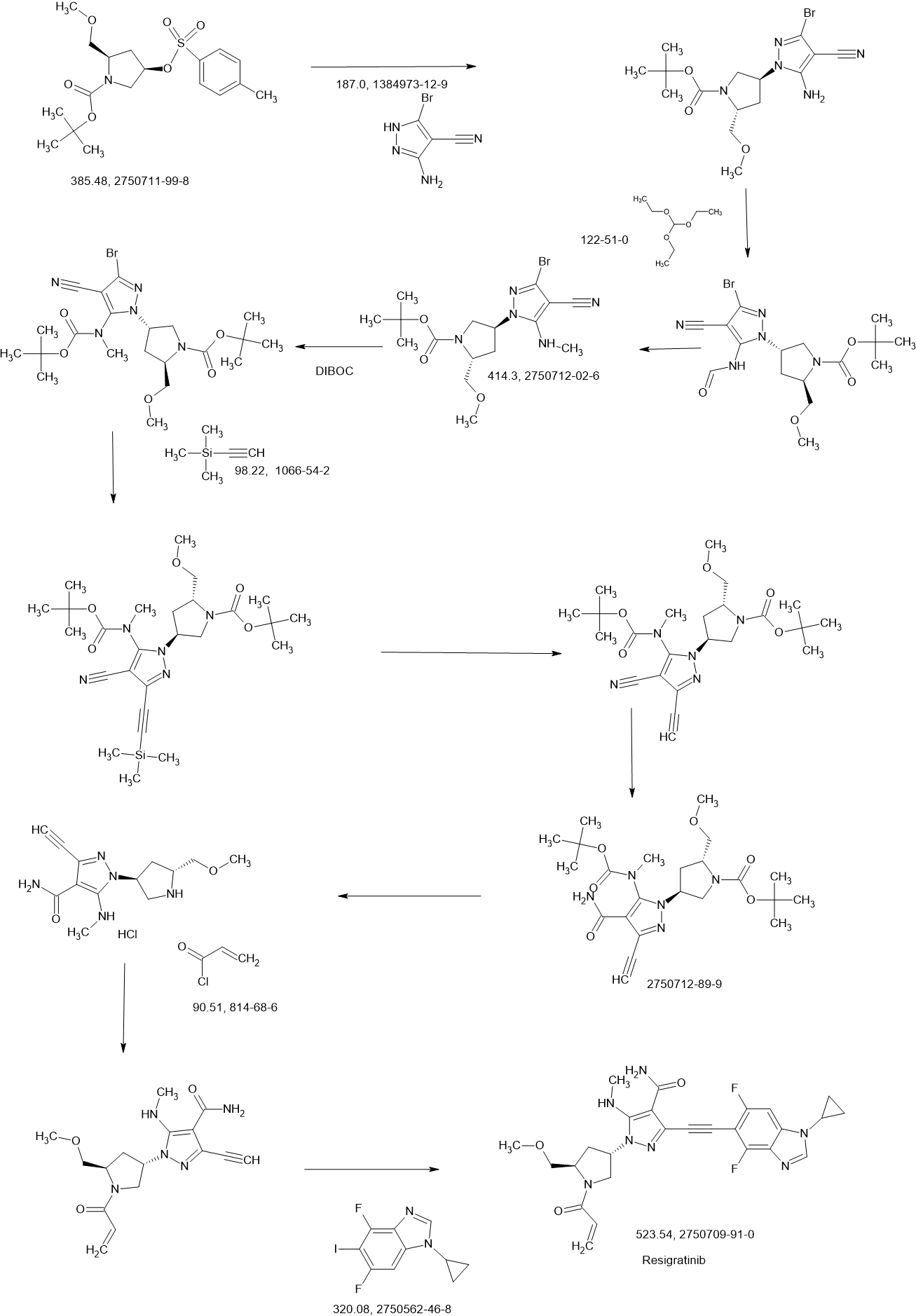
REF
https://patents.google.com/patent/US11345681B1/en
Example 78
3-[2-(1-Cyclopropyl-4,6-difluoro-1,3-benzodiazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide
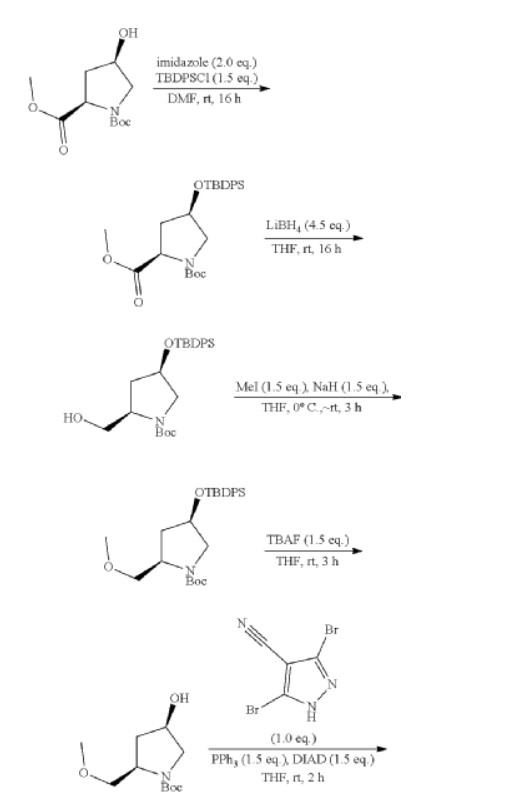


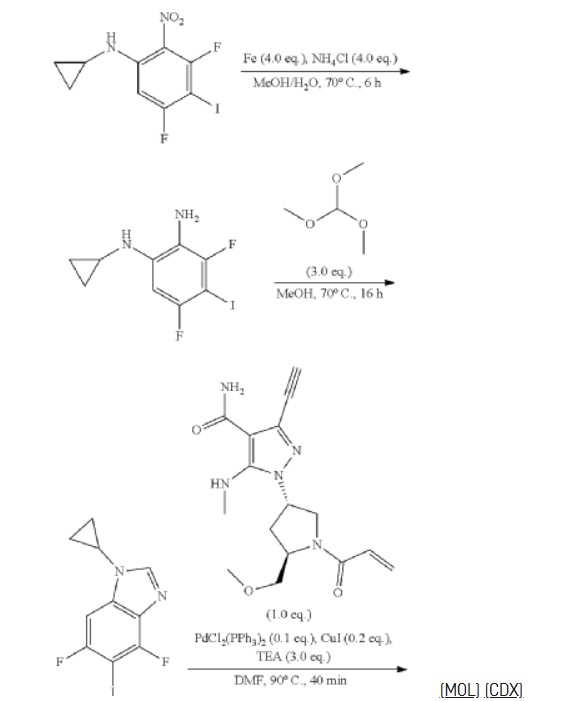
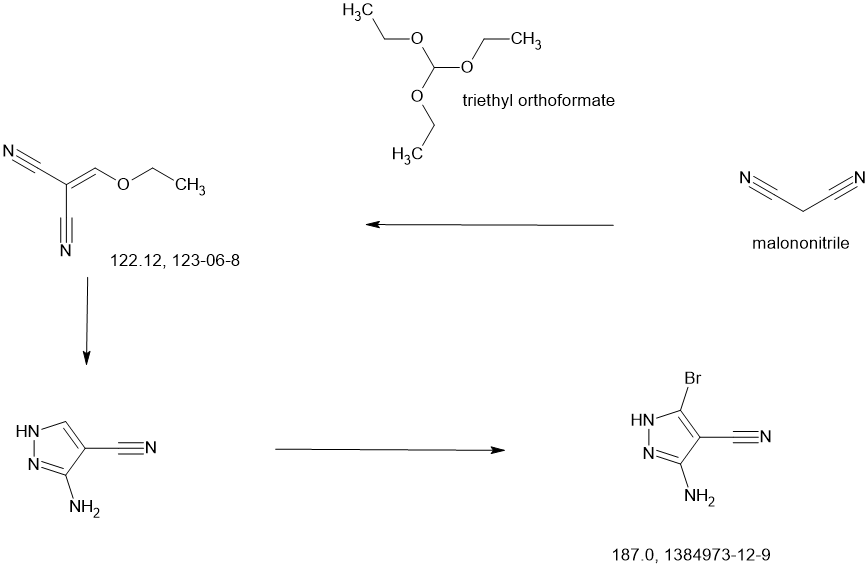
| Step 1: 1-(Tert-butyl) 2-methyl (2R,4R)-4-((tert-butyldiphenylsilyl)oxy)pyrrolidine-1,2-dicarboxylate |
| Step 2: Tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(hydroxymethyl)pyrrolidine-1-carboxylate |
| Step 3: Tert-butyl (2R,4R)-4-[(tert-butyldiphenylsilyl)oxy]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 5: Tert-butyl (2R)-4-(3,5-dibromo-4-cyanopyrazol-1-yl)-2-methoxymethyl)pyrrolidine-1-carboxylate |
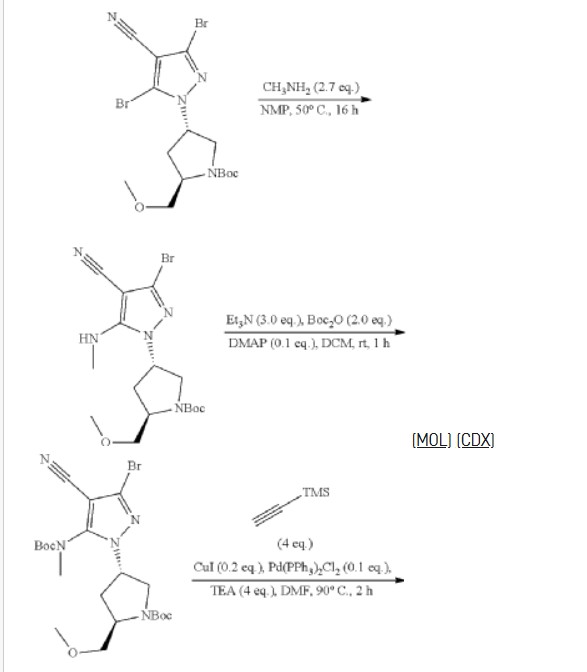
| Step 6: Tert-butyl (2S,4R)-4-[3-bromo-4-cyano-5-(methylamino)pyrazol-1-yl]-2-(methoxymethyl)pyrrollidine-1-carboxylate |
| Step 7: (2R,4S)-4-[3-bromo-5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyanopyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 8: Tert-butyl (2R,4S)-4-(5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-[2-(trimethylsilyl)ethynyl]pyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 9: Tert-butyl (2R,4S)-4-(5-[(tert-butoxycarbonyl)(methyl)amino]-4-cyano-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 10: Tert-butyl (2R,4S)-4-[5-[(tert-butoxycarbonyl)(methyl)amino]-4-carbamoyl-3-ethynylpyrazol-1-yl]-2-(methoxymethyl)pyrrolidine-1-carboxylate |
| Step 11: 3-Ethynyl-1-[(3S,5R)-5-(methoxymethyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide dihydrochloride |
| Step 12: 3-Ethynyl-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide |
| Step 17: 3-[2-(1-Cyclopropyl-4,6-difluoro-1,3-benzodiazol-5-yl)ethynyl]-1-[(3S,5R)-5-(methoxymethyl)-1-(prop-2-enoyl)pyrrolidin-3-yl]-5-(methylamino)pyrazole-4-carboxamide |
PATENT
WO2021247969 Kinnate Biopharma Inc EG78
WO2023107980 solid state forms, Kinnate Biopharma Inc
WO2023107979 FGFR kinase inhibitor, Kinnate Biopharma Inc
References
- Franovic A, Mohan A, Uryu S, Wu Q, Jiang P, Miller N, et al. (February 2022). “Activity of KIN-3248, a next-generation pan-FGFR inhibitor, against acquired FGFR-gatekeeper and molecular-brake drug resistance mutations”. Journal of Clinical Oncology. 40 (4_suppl): 461. doi:10.1200/JCO.2022.40.4_suppl.461.
- Harding JJ, Perez CA, Kato S, Sharma M, Garmezy B, Quah CS, et al. (February 2023). “First in human (FIH) phase 1/1b study evaluating KIN-3248, a next-generation, irreversible pan-FGFR inhibitor (FGFRi), in patients (pts) with advanced cholangiocarcinoma (CCA) and other solid tumors harboring FGFR2 and/or FGFR3 gene alterations”. Journal of Clinical Oncology. 41 (4_suppl): TPS637-TPS637. doi:10.1200/JCO.2023.41.4_suppl.TPS637. S2CID 256257314.
- Wang Z, Anderson KS (2022). “Therapeutic Targeting of FGFR Signaling in Head and Neck Cancer”. Cancer Journal (Sudbury, Mass.). 28 (5): 354–362. doi:10.1097/PPO.0000000000000615. PMC 9523489. PMID 36165723.
| Identifiers | |
|---|---|
| CAS Number | 2750709-91-0 |
| PubChem CID | 162381323 |
| UNII | W728TB393W |
| Chemical and physical data | |
| Formula | C26H27F2N7O3 |
| Molar mass | 523.545 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
{kind=link}
- [1]. Tyhonas JS, et al. Discovery of KIN-3248, An Irreversible, Next Generation FGFR Inhibitor for the Treatment of Advanced Tumors Harboring FGFR2 and/or FGFR3 Gene Alterations. J Med Chem. 2024 Feb 8;67(3):1734-1746. [Content Brief][2]. Balasooriya ER, et al. The Irreversible FGFR Inhibitor KIN-3248 Overcomes FGFR2 Kinase Domain Mutations. Clin Cancer Res. 2024 May 15;30(10):2181-2192. [Content Brief]
/////////Resigratinib, Pan-FGFR Inhibitor KIN-3248, KIN 3248, Pan-fibroblast Growth Factor Receptor Inhibitor KIN-3248, W728TB393W
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com